
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
KuQuinone-sensitized cobalt oxide nanoparticles for photoelectrocatalytic oxygen evolution with visible light†
Ruggero
Bonetto
 ab,
Nuria
Romero
*c,
Federica
Sabuzi
*d,
Mattia
Forchetta
d,
Mirco
Natali
e,
Raffaella
Signorini
b,
Roger
Bofill
a,
Laia
Francàs
a,
Marcos
Gil-Sepulcre
*af,
Olaf
Rüdiger
f,
Serena
DeBeer
f,
Jordi
García-Antón
a,
Karine
Philippot
c,
Pierluca
Galloni
d,
Andrea
Sartorel
*b and
Xavier
Sala
*a
ab,
Nuria
Romero
*c,
Federica
Sabuzi
*d,
Mattia
Forchetta
d,
Mirco
Natali
e,
Raffaella
Signorini
b,
Roger
Bofill
a,
Laia
Francàs
a,
Marcos
Gil-Sepulcre
*af,
Olaf
Rüdiger
f,
Serena
DeBeer
f,
Jordi
García-Antón
a,
Karine
Philippot
c,
Pierluca
Galloni
d,
Andrea
Sartorel
*b and
Xavier
Sala
*a
aDepartment of Chemistry, Faculty of Sciences, Autonomous University of Barcelona, 08193 Bellaterra, Catalonia, Spain. E-mail: xavier.sala@uab.cat
bDepartment of Chemical Sciences, University of Padova, Via Francesco Marzolo 1, 35131 Padova, Italy. E-mail: andrea.sartorel@unipd.it
cCNRS, LCC (Laboratoire de Chimie de Coordination), UPR8241, University of Toulouse, UPS, INPT, Toulouse cedex 4 F-31077, France. E-mail: nuria.romero@lcc-toulouse.fr
dDepartment of Chemical Science and Technologies, University of Rome Tor Vergata, Via Della Ricerca Scientifica, snc 001133 Rome, Italy. E-mail: federica.sabuzi@uniroma2.it
eDepartment of Chemical, Pharmaceutical and Agricultural Sciences, Via L. Borsari 46, 44121 Ferrara, Italy
fMax Planck Institute Chemical Energy Conversion, Stiftstrasse 34-36, D-45470 Mülheim an der Ruhr, Germany. E-mail: marcos.gil-sepulcre@cec.mpg.de
First published on 18th March 2025
Abstract
Photocatalytic nanomaterials offer promising solutions for conducting chemical transformations under safe, green and sustainable conditions. In particular, the storage of solar energy into chemical bonds is an appealing but challenging goal in the field of artificial photosynthesis. Using water as the source of electrons and protons through the photodriven water oxidation (WO) reaction is at the core of this endeavour. In this work, we disclose photoactive hybrid nanomaterials designed through a dyadic approach. We exploit Co3O4 nanoparticles (NPs) covalently functionalized with a fully organic pentacyclic polyquinoid KuQuinone (KuQ) dye, providing a rare example of a noble metal-free photocatalytic dyadic nanomaterial (hereafter denoted as KuQ3Pn@Co3O4). KuQ3Pn@Co3O4 NPs have been characterized by electron microscopy and optical and core-level spectroscopy studies. When cast onto a SnO2 photoanode, they are active towards WO upon visible light irradiation (400–580 nm) with a faradaic efficiency for O2 evolution of ca. 90%. This work provides a novel contribution to the rational design and mechanistic understanding of hybrid photocatalytic nanomaterials relevant for energy and sustainable synthesis applications.
 Nuria Romero | Nuria Romero has been an Associate Professor at the University of Toulouse and the Laboratory of Coordination Chemistry in France since 2022. After receiving her Ph.D. in organometallic and coordination chemistry, she completed a postdoctoral fellowship at the Institute of Chemical Sciences of Rennes (France), working on photoswitchable polymerization catalysts. She then held a position as a contractual teacher-researcher at the Autonomous University of Barcelona, where she began her research in energy, focusing on electro- and photo-catalytic water splitting. Her current work focuses on developing new materials for artificial photosynthesis catalysts based on metal nanoparticles. |
Green foundation1. Our work advances the field of green chemistry by designing and characterizing a novel hybrid photocatalytic nanomaterial (KuQn@Co3O4) that enables solar-driven water oxidation using sustainable, noble metal-free components. This approach supports the development of artificial photosynthetic systems to store solar energy in chemical bonds, addressing energy sustainability challenges.2. The KuQ3Pn@Co3O4 nanomaterial achieves photoelectrochemical water oxidation under visible light irradiation (400–580 nm) with a faradaic efficiency of ∼90%. This highlights its high efficiency as a green catalytic system for solar energy conversion without reliance on precious metals. 3. These findings demonstrate the potential of our dyadic approach for the catalytic activation of small molecules and open the path to greener organic dyes/Earth-abundant metal hybrids. Future research could enhance the sustainability of this work by incorporating naturally derived organic dyes to minimize environmental impact. |
Introduction
Artificial photosynthesis aims at the efficient storage of solar energy into chemical bonds and is considered a primary goal in the scope of a sustainably powered society. The core target is the utilization of naturally abundant raw materials such as water, carbon dioxide or dinitrogen to produce energy-dense chemical vectors and valuable chemicals.1 These reductive transformations are typically coupled to the oxidation of readily available water to oxygen (WO, vs. the reversible hydrogen electrode, RHE; WO is also reported in the literature as the oxygen evolution reaction, OER), providing the reducing equivalents required to feed fuel synthesis.2,3 A general approach to drive sunlight-induced redox reactions is the integration of a light harvesting unit (LHU) and a water oxidation catalyst (WOC) through chemical interactions.4–9 This dyadic approach allows the attainment of a controllable LHU/WOC ratio and interaction to promote the desired photoredox events: light absorption by the LHU followed by charge separation to funnel oxidizing equivalents to the WOC, enabling the 4e−/4H+ WO reaction.10
vs. the reversible hydrogen electrode, RHE; WO is also reported in the literature as the oxygen evolution reaction, OER), providing the reducing equivalents required to feed fuel synthesis.2,3 A general approach to drive sunlight-induced redox reactions is the integration of a light harvesting unit (LHU) and a water oxidation catalyst (WOC) through chemical interactions.4–9 This dyadic approach allows the attainment of a controllable LHU/WOC ratio and interaction to promote the desired photoredox events: light absorption by the LHU followed by charge separation to funnel oxidizing equivalents to the WOC, enabling the 4e−/4H+ WO reaction.10
Over several decades, the principles of natural photosynthesis have been followed in the design of biomimetic LHU/WOC assemblies, starting with dyads based on covalently bound molecular components.11–16 However, covalently assembled molecular dyads suffer from heavily demanding synthetic routes that may afford only a limited degree of tuning of the properties of the resulting systems, especially of the LHU/WOC ratio, compared to assemblies produced via supramolecular interactions.17 Furthermore, stability issues may impair the outcome of elegant design work required for the covalent bridging of the dye and catalyst fragments. Notable recent examples of LHU/WOC integration deal with the supramolecular electrostatic assembly of stable cationic perylene bisimide chromophores and a ruthenium polyoxometalate WOC (quantasome),18,19 providing more convenient access to photoelectrochemical WO on semiconductor slides. A related strategy was followed in the self-assembly of a LHU/WOC layer, driven by hydrophobic interactions or inclusion complex formation on the surface of nano-SnO2 or TiO2 photoanodes, respectively. The devices featured a RuII tris-bipyridine dye and a Ru bipyridine dicarboxylate WOC modified with aliphatic chains20–22 or β-cyclodextrin.23
The literature addressing the design of photocatalytic assemblies also reports the combination of molecular dyes and metal oxide nanoparticles (NPs) as LHU and WOC units, respectively.24 However somewhat scarcer, such examples are appealing since nanostructured catalysts can be easily prepared and display higher density of active sites, resulting in higher current densities than molecular complexes in operational devices, along with improved stability.25–31 Seminal studies by Mallouk and co-workers took advantage of Ru(bpy)3-type derivatives as LHUs and colloidal hydrated iridium oxide (IrOx) as a WOC. A tailored synthesis method employed RuII polypyridine complexes bearing dicarboxylic acid groups in the periphery of the bipyridine ligands, acting as capping agents in the synthesis of IrOx NPs32 (Scheme 1A). The advantage of chemically linking the LHU and WOC moieties was supported by an enhancement of ca. two orders of magnitude of the electron transfer (ET) rate from IrOx to the photochemically generated oxidized RuIII dye in colloidal suspensions with respect to the “unbound” [Ru(bpy)3]2+ dye.33 Based on these results, heteroleptic RuII polypyridine complexes were devised, bearing both malonate stabilizing groups during the IrOx NP synthesis and a phosphonate anchor for post-synthetic connection to TiO2 films.34–36 Such a strategy allowed the integration of these materials within a dye-sensitized photoelectrochemical cell (DSPEC)37 photoanode (Scheme 1B).38 Later developments exploited IrOx and RuO2 WOCs functionalized on their surface by either free-base or metalated (with either Zn or Pd) porphyrin dyes.39–42 Despite the encouraging results of these preliminary studies, these systems based on organic dyes were not further investigated in photoinduced WO.
 | ||
| Scheme 1 Photo(electro)catalytic dyadic systems for water oxidation (WO) based on transition metal oxide NPs functionalized with Ru-based molecular dyes (LHU) (top, A–D) and the KuQ3P@Co3O4 hybrid nanomaterials presented in this work (bottom). | ||
A key step in the development of photoactive hybrid nanomaterials for WO was published by some of us.43 We reported the synthesis of cobalt spinel-structured NPs sensitized via surface decoration with phosphonated analogues of [Ru(bpy)3]2+ (RuPS@Co3O4) (Scheme 1C). These NPs were studied under visible light irradiation, catalyzing WO through light absorption by the ruthenium complex in the presence of persulfate anions as electron acceptors. The direct electronic interaction between the dye and the catalyst was clearly demonstrated, alongside the superior catalytic performance of the dyadic RuPS@Co3O4 NPs compared to the “unbound” system, i.e., co-suspended RuPS and Co3O4 NPs, with photocatalytic WO being relevant only in the former, dyadic case.
Finally, in 2021, Amiens and co-workers reported an analogous strategy applied to core–shell iron–iron oxide (Fe–FeOx) NPs stabilized by a phosphonate-bound Ru phenanthroline dye44 (Scheme 1D). These composites were employed under photoelectrochemical conditions upon deposition on fluorine-doped tin oxide (FTO). They displayed photocurrent activity associated with O2 evolution, thereby reinforcing the applicability of nanostructured WOC–molecular LHU combinations to both colloidal and DSPEC artificial photosynthetic reaction architectures.
These last two works were the first examples of photocatalytic dyadic materials for WO exploiting a first row-based metal oxide WOC, although still taking advantage of a LHU based on a scarce transition metal like ruthenium. This drawback, together with the need to exploit harness photons beyond 500 nm, prompted us to investigate the use of highly absorbing organic dyes for the design of LHU/WOC composites.45–47
Nanostructured Co3O4 is a promising electrocatalyst, able to oxidize water to O2 with overpotentials as low as 350 mV and operating in a broad pH range, including acidic conditions.48–52 Thus, Co3O4 represents a viable alternative to precious Ir and Ru-based WOCs. Furthermore, the solid mechanistic framework on Co3O4 as the WOC built by means of spectroscopic, spectroelectrochemical and computational investigations provides a foundation for system design and optimization.53–60 Herein, we employed Co3O4 NPs synthesized via the organometallic approach,43,61,62i.e., the controlled decomposition of an organometallic Co compound under H2 pressure in the presence of a stabilizing ligand followed by air exposure. This routine allows control over particle size, shape, and surface composition with high reproducibility, affording well-defined NPs whose catalytic properties can be fine-tuned through post-synthetic modifications. Subsequently, we undertook the synthesis of photocatalytic dye–catalyst hybrid nanomaterials incorporating several chromophore units bonded to individual catalyst NPs, designed with the aim of providing a controllable catalyst/dye ratio and a fast ET pathway between the two components.
We employed organic dyes denoted as KuQuinones (KuQs),63,64 since they are well-established active units in photo(electro)catalytic systems that are suited for performing photoinduced WO.65–70 Indeed, they manage proton-coupled electron-transfer (PCET) events both in their ground and excited states, which are highly valuable in the scope of multi-electron, multi-proton photoinduced reactions for artificial photosynthesis applications.63–70 Incidentally, the structural quinoid motif is privileged within the field of functional mimetics of natural photosynthetic systems, employing quinones as photoinduced ET relays. Furthermore, KuQ dyes can partake in keto–enol tautomerism and enol–enolate equilibria at the level of the pentacyclic core depending on their protonation state, with the enol and enolate forms displaying different light harvesting features, both exhibiting intense absorption in the visible region up to 600 nm71,72 (vide infra). Their singlet excited states (1*KuQ) are highly oxidizing67 (>2.6 V vs. RHE), and they can inject electrons from their singly reduced form (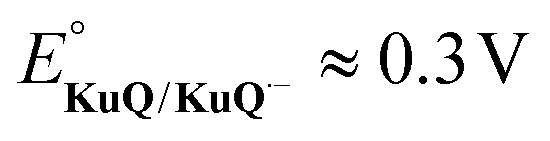 vs. RHE) into the conduction band of mesoporous SnO2 (conduction band edge potential ECB = 0.50 V vs. RHE). Lastly, their synthetic space allows for the introduction of side chains that do not impact the chromophore core, while imparting different acid-base, solubility, and anchoring properties. These features contribute to the appeal of KuQ dyes as cutting-edge molecular LHUs for WO applications. Based on these premises, a KuQ dye with a pendant phosphonate moiety, 1-[3-(dihydroxyphosphonyl)propyl]KuQuinone (KuQ3P),68,73 was used, exploiting their side chains to act as ligands to the surface of the pre-synthesized Co3O4 NPs via a highly affine and hydrolytically stable anchor.
vs. RHE) into the conduction band of mesoporous SnO2 (conduction band edge potential ECB = 0.50 V vs. RHE). Lastly, their synthetic space allows for the introduction of side chains that do not impact the chromophore core, while imparting different acid-base, solubility, and anchoring properties. These features contribute to the appeal of KuQ dyes as cutting-edge molecular LHUs for WO applications. Based on these premises, a KuQ dye with a pendant phosphonate moiety, 1-[3-(dihydroxyphosphonyl)propyl]KuQuinone (KuQ3P),68,73 was used, exploiting their side chains to act as ligands to the surface of the pre-synthesized Co3O4 NPs via a highly affine and hydrolytically stable anchor.
The resulting noble metal-free KuQ3P@Co3O4 hybrid nanomaterials operate in photoelectrochemical WO with visible light (400–580 nm), and the covalent anchoring of the two components is crucial in achieving a high faradaic efficiency for WO. Spectroscopic evidence confirms the photoelectrochemical formation of Co-oxo/hydroxo active sites.
Results and discussion
Synthesis and characterization of KuQ3Pn@Co3O4
The synthesis of spherical cobalt(II,III) oxide NPs was carried out via a two-step method (Scheme 2) following adaptation of a previous report.43,61,62 The first step involved the decomposition of (cyclooctadienyl)(1,5-cyclooctadiene)cobalt(I), [Co(η3-C8H13)(η4-C8H12)] under H2 at 3 bars in 1-heptanol (heptOH), which acted both as the solvent and stabilizing ligand. This yielded spherical Co0 NPs with a diameter of 2.9 ± 0.6 nm, stabilized by heptOH (CoheptOH). Subsequently, air diffusion allowed for a controlled oxidation process of the isolated NPs to generate Co3O4heptOH, with no substantial change in the particle size (3.2 ± 0.6 nm) and shape. High-resolution transmission electron microscopy (HR-TEM) images of CoheptOH and Co3O4heptOH NPs are provided in Fig. S1.† Considering spherical NPs with a ca. 3.0 nm diameter, it was estimated that 39% of total Co3O4 units are surface-exposed (see the ESI, Section 1.4† for details).‡74 | ||
| Scheme 2 Synthesis of KuQ3Pn@Co3O4 dyads via (1) the organometallic approach, (2) controlled air oxidation of CoheptOH NPs and (3) Co3O4heptOH NP sensitization with KuQ3P. | ||
The synthesis protocol for producing KuQ3P-sensitized NPs (Scheme 2) consisted of stirring Co3O4heptOH NPs in the presence of different amounts of KuQ3P (0.1 and 0.2 equivalents of KuQ3P with respect to Co3O4heptOH) for 4 days in a heptOH/MeOH/H2O mixture, followed by washing and drying (see the Methods section for further details). The phosphonate anchoring group was selected based on a previous report by some of us,43 which showed a better stability of hybrids with this group compared to the carboxylate one. The complete discolouration of the reaction mixtures (Fig. S2†) and the elemental composition of the resulting products afforded by inductively coupled plasma optical emission spectroscopy (ICP-OES) and ICP coupled to mass spectrometry (ICP-MS, see the ESI and Tables S1 and S2†) were consistent with the quantitative chemisorption of the dye on the NP surface for both materials. The resulting dye-sensitized NPs are hereafter denoted as KuQ3Pn@Co3O4 (n = 0.1 and 0.2, respectively). Based on the fraction of surface Co3O4 sites, this results in a surface ratio between KuQ3P and Co3O4 units of 0.25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 0.5:1 for KuQ3P0.1@Co3O4 and KuQ3P0.2@Co3O4, respectively (ESI, Section 1.4†).
:1 and 0.5:1 for KuQ3P0.1@Co3O4 and KuQ3P0.2@Co3O4, respectively (ESI, Section 1.4†).
The dyads were then characterized by means of TEM, X-ray absorption spectroscopy (XAS), X-ray photoelectron spectroscopy (XPS), resonance Raman spectroscopy and attenuated internal reflection Fourier transform infrared spectroscopy (ATR-FTIR) (Fig. 1 and Fig. S3–S16†).
 | ||
| Fig. 1 (A) HR-TEM and (B) STEM-HAADF images of KuQ3P0.1@Co3O4 NPs with (C) their corresponding size histogram. (D) Normalized Co K-edge XANES spectra of Co3O4heptOH NPs (black line), KuQ3P0.1@Co3O4 NPs (blue line), CoO (orange line) and Co(OH)2 (dark red line) references. Inset: zoom of the pre-edge region. (E) Fourier transforms of the k3-weighted Co EXAFS spectra for Co3O4heptOH NPs, KuQ3P0.1@Co3O4 NPs, CoO and Co(OH)2, same colour code as in (D). Inset: corresponding k3-weighted Co EXAFS spectra. (F) Resonance Raman spectra of Co3O4heptOH NPs (black line), KuQ3P0.1@Co3O4 NPs (blue line) and KuQ3P dye (magenta line). Spectra were recorded on powder samples. | ||
TEM analysis of the KuQ3Pn@Co3O4 NPs revealed no significant changes in the mean particle size with respect to the initial Co3O4heptOH NPs, and a certain degree of particle aggregation resulting from KuQ3P functionalization (Fig. S3†). HR-TEM images (Fig. 1A and Fig. S4 and S5†) and electron diffraction patterns (Fig. S6†) of KuQ3P0.1@Co3O4 evidenced interplanar distances of 0.249, 0.211, 0.148 and 0.145 nm, with values in agreement with those of the (111), (004), (044) and (044) planes of Co3O4, respectively, despite a certain degree of surface amorphization. Additionally, a punctiform envelope surrounding the NPs was detected by High Angle Annular Dark Field-Scanning Transmission Electron Microscopy (STEM-HAADF) (Fig. 1B and Fig. S3†). EDX (energy-dispersive X-ray spectroscopy, Fig. S4 and S5†) analyses indicated the presence of Co in this envelope, its formation being observed upon treatment of Co3O4heptOH NPs with the solvent mixture used for the dyad preparation (Scheme 2), irrespective of the presence of KuQ3P (see Fig. S7†).
The electronic and structural properties of the KuQ3Pn@Co3O4 dyads were investigated by XAS, a sensitive probe of the oxidation state, geometry and coordination environment of the absorbing metal center. The X-ray absorption near-edge structure (XANES) of the initial Co3O4hepOH NPs aligns with a spinel structure, containing a mixture of CoII (Td sites) and CoIII (Oh sites) in a 1:2 ratio, exhibiting an intense 1s-to-3d pre-edge feature and a shifted edge position with respect to the purely Oh CoII references CoO and Co(OH)2, consistent with the expected CoII/CoIII ratio (Fig. 1D). This was confirmed by the extended X-ray absorption fine structure (EXAFS) region of the spectra, which provided additional information about the coordination environment of the Co absorber. The R-space spectrum shown in Fig. 1E is dominated by contribution from Co–O scattering vectors at 1.92 Å, and two additional intense peaks at 2.86 and 3.36 Å, attributed to remote Co–Co[Oh] and Co–Co[Td] distances, respectively (Fig. S8, S10, and Tables S3, S5†). The bond lengths obtained by EXAFS fitting analysis are in the range of the expected values for Co3O4 X-ray structures and related EXAFS data in the literature.75
Although Co3O4heptOH NPs display a textbook spectrum for a cobalt spinel structure, significant changes in both XANES and EXAFS regions have been observed for their functionalized KuQ3P0.1@Co3O4 (Fig. 1D and S8, 9†) and KuQ3P0.2@Co3O4 (Fig. S8†) counterparts. The edge and white-line features are shifted to lower energies, indicating a slight reduction of the metal center. Additionally, the quadrupole-allowed but dipole-forbidden 1s-to-3d transitions of the pre-edge are characterized by a clear intensity decrease, suggesting the presence of more centrosymmetric Oh cobalt sites.75–77 On the other hand, the XANES spectrum is clearly different from the spectra of CoO or Co(OH)2. The absence of CoO/Co(OH)2 was further reinforced by XPS (Fig. S12†) and resonance Raman spectroscopy measurements (Fig. 1F), vide infra.
The R-space EXAFS spectra showed a clear amorphization of the Co3O4 NPs, as reflected by the loss of intensity in the EXAFS features relative to the pristine Co3O4heptOH NPs (Fig. S11,† right). Further fitting of the KuQ3P0.1@Co3O4 EXAFS spectrum to a simple spinel model showed a decrease of the Co–O and Co–Co distances relative to a spinel structure to values of 1.90 and 2.82 Å, respectively (Fig. S10†). This trend is in good accordance with the HR-TEM images of KuQ3P0.1@Co3O4, which suggest partial surface fragmentation of Co3O4, leading to the formation of new single-atomic Co species ascribed to CoII and CoIII sites in accordance with XAS data. The influence of KuQ3P on the evolution of the initial Co3O4heptOH NPs into a more amorphous material was ruled out, as a similar transformation of both XANES and EXAFS regions was also observed in the absence of KuQ3P (Fig. S11†), in accordance with the HR-TEM data, vide supra (Fig. S7†).
Therefore, we can hypothesize that under the conditions applied for the post-functionalization of their surface, the pristine Co3O4heptOH NPs evolved into a complex mixture of Co3O4 and single/multi-atomic octahedral CoII and CoIII sites, without the direct involvement of KuQ3P in the transformation. Single/multi-atomic octahedral CoII and CoIII species have been reported in the literature, and present short Co–O and Co–Co distances (1.89 and 2.82 Å, respectively) that could explain the decrease of the overall Co–O and Co–Co distances observed in FT-EXAFS.78–82 A pre-edge fitting of the KuQ3P0.1@Co3O4 feature could be performed to roughly estimate the loss of the Co3O4Td sites by combining the spectra of Co3O4 and [CoII(MeOH)6]2+ as a purely octahedrally coordinated CoII model in a 6:4 ratio, suggesting that ca. 40% of the Co3O4 surface atoms have been fragmented forming the single/multi-atomic species (Fig. S9 and S10†).
Resonance Raman spectroscopy was employed to probe the KuQ3P–Co3O4 surface interaction; spectra recorded for Co3O4heptOH, KuQ3P, and KuQ3P0.1@Co3O4 are reported in Fig. 1F (additional spectroscopic data are presented in the ESI, Fig. S13–S15†). The KuQ3P signals collected in the 500–1700 cm−1 region are mostly ascribable to the characteristic vibrational signature of the KuQ chromophore core motions based on comparison of KuQ3P to other dyes in the KuQ category (specifically 1-(3-carboxylpropyl)KuQuinone, KuQ3C, and 1-hexylKuQuinone KuQ-Hex, Fig. S14†). The spectrum of Co3O4heptOH NPs (black line in Fig. 1F) displays the expected bands at 485, 524 and 691 cm−1 (blue line in Fig. 1F), ascribed to the Eg, F2g and A1g transitions in Co3O4.48,60,83 The spectra of KuQ3Pn@Co3O4 (blue line in Fig. 1F for KuQ3P0.1@Co3O4, while the spectrum of KuQ3P0.2@Co3O4 is reported in Fig. S13†) show an overlap of the signatures of the Co3O4heptOH NPs and of the KuQ3P dye (magenta line in Fig. 1F), with two relevant features: (i) in KuQ3Pn@Co3O4, the A1g band peaking at 680 cm−1 is slightly redshifted with respect to Co3O4heptOH, where it is observed at 691 cm−1. Such a redshift may be attributed to crystal disorder induced by the functionalization step83 and (ii) the signals of KuQ3P are retained in KuQ3Pn@Co3O4, except for the band at 934 cm−1; the latter is attributed to a vibration of the phosphonate group,84–87 and thus supports the involvement of phosphonate in the covalent bonding to the NPs. Lastly, the resonance Raman spectrum of free KuQ3P is characterized by the onset of the expected intense emission in the 2000–4000 cm−1 range, whereas the spectra are instead strongly quenched in KuQ3Pn@Co3O4 (Fig. S15†), suggesting that a photoinduced event is occurring in the latter (vide infra).
A comparison of the ATR-FTIR spectra recorded for Co3O4heptOH, KuQ3P and KuQ3P0.1@Co3O4 (as a representative of the hybrid nanomaterials) is reported in the ESI (Fig. S16†). These data correlate well with the information provided by resonance Raman spectroscopy. The vibrations of KuQ3P observed in the “fingerprint” region were consistently identified also in KuQ3P0.1@Co3O4. Furthermore, the motion of the phosphonic acid group at 934 cm−1 for the free KuQ3P is absent in the spectrum of KuQ3P0.1@Co3O4, further supporting the involvement of phosphonate in surface binding to the NPs. ATR-FTIR spectroscopy, not limited by the insurgence of KuQ3P emission, allowed us to characterize the NPs in the region above 2500 cm−1. Co3O4heptOH was found to display characteristic peaks at 2925 and 2855 cm−1, attributable to the C–H stretching of the alkyl chain of heptOH; likewise, the broad feature at 3320 cm−1 is ascribed to the O–H stretching of the heptOH alcohol moiety, consistent with a previous report.43 Such spectroscopic signatures were found significantly abated in KuQ3P0.1@Co3O4, thus supporting the expected, nearly quantitative ligand substitution of heptOH by KuQ3P during Co3O4heptOH post-functionalization, also in coherence with the ICP-OES results.
Once the successful synthesis of the KuQ3Pn@Co3O4 hybrid nanomaterials was confirmed, their photophysical properties were evaluated. In aqueous solution, the KuQ3P dye shows a broad fluorescence emission band from the singlet excited state 1*KuQ3P, centred at 620 nm and characterized by a lifetime of 0.51 ns. The fluorescence was strongly abated in KuQ3P0.2@Co3O4 with respect to that of the KuQ3P dye, suggesting a fast quenching of 1*KuQ3P by the Co3O4 NPs (Fig. S17†).88 Time-resolved luminescence analysis also suggested ultrafast kinetics (Fig. S17†), with a time-constant τ < 0.2 ns, i.e., below the instrumental limit of the time-correlated single photon counting (TCSPC) apparatus, corresponding to a quenching constant lower limit of k > 5 × 109 s−1. Considering the high oxidizing power of the 1*KuQ3P excited state (E1*KuQ3P/KuQ3P˙– = 2.62–2.79 V vs. RHE), estimated from the combination of the redox potential of the ground state and from the E0–0 of 1*KuQ3P (see the ESI and Fig. S18 and S19†), and considering previous investigations on KuQ dyes under homogeneous conditions70 and at the surface of photoelectrodes,67 a plausible hypothesis for the observed emission quenching in KuQ3Pn@Co3O4 is ET from Co3O4 to the photoexcited KuQ3P, leading to oxidized Co3O4, Co3O4(h+), and KuQ3P˙− (reductive quenching of the dye, as depicted in Scheme 3).67 This proposed preferential photoinduced ET pathway is exergonic by ca. 1 eV, as estimated from the difference between the potential of 1*KuQ/KuQ˙− and the potential threshold to access the catalytic WO regime by Co3O4 (ca. 1.75 V vs. RHE, as estimated by cyclic voltammetry, CV, see Fig. S20†). The saturated chain linker of the phosphonate anchoring group is not expected to mediate the photoinduced ET.
 | ||
| Scheme 3 (A) Proposed mechanism for photoinduced WO by KuQ3Pn@Co3O4 in a colloidal suspension, employing Na2S2O8 as an irreversible electron acceptor. The preferred reductive quenching of KuQ dyes is further supported by the negligible direct reactivity of both singlet and triplet (3*KuQ3P) excited states of the dye with the Na2S2O8 electron acceptor (Fig. S21 and S22†), thus excluding the photoinduced oxidation of KuQ3P (oxidative quenching pathway). The KuQ3P oxidative radical decomposition pathway likely involved the methylene site on the side chain, at the α position to the five-membered central ring. (B) Energy levels involved in the initial steps of the WO photocatalytic cycle under our conditions.90Note: potentials are not to scale. | ||
Photochemical water oxidation and evolution of Co3O4 to highly oxidized states
The activity of KuQ3Pn@Co3O4 towards photoinduced WO was assayed under 1 Sun illumination using a colloidal suspension of the materials (0.56 mg mL−1) in an aqueous NaHCO3/Na2SiF6 (pH 5.6) buffer containing 84 mM Na2S2O8 as the sacrificial electron acceptor (see details in the ESI, and Fig. S23–S28†). Oxygen evolution was detected during the first 3 h of irradiation for KuQ3P0.1@Co3O4 (0.85 μmol mgmaterial−1, with turnover numbers TONO2/NP = 443 and TONO2/KuQ = 3.8, and turnover frequency TOFmax = 1.0 molO2 h−1 molCo3O4−1). Based on the our photophysical and electrochemical studies, the proposed photochemical cycle involves reductive quenching of the singlet 1*KuQ3P by the appended Co3O4, followed by reconversion of KuQ3P˙− to KuQ3P upon ET to the persulfate anion (Scheme 3A). The amount of O2 evolved by the KuQ3P0.1@Co3O4 dyad was also greater than that for KuQ3P0.2@Co3O4, suggesting that an excess of KuQ3P bound at the Co3O4 NP surface may be precluding the WO process by blocking the active sites.43 Incidentally, blank experiments conducted with Co3O4heptOH in the absence of KuQ3P evolved a modest but non-negligible amount of O2 (up to 0.2 μmol, compared with 0.5 μmol evolved by KuQ3P0.1@Co3O4), consistent with a low photooxygenic activity of unsensitised Co3O4.51,89XAS investigation allowed the study of the evolution of KuQ3P0.1@Co3O4 along photoirradiation. Interestingly, the XANES data recorded on the photoirradiated material indicated similar features to those for the initial Co3O4heptOH NPs (Fig. 2A). However, a shift of the edge to higher energies was observed, suggestive of a higher oxidation degree of the cobalt centres in comparison with those of Co3O4, together with a partial recovery of the intensity in the pre-edge feature. This transformation was found to occur very rapidly (15 s) after starting the irradiation, indicating that the single-atomic Co species quickly evolved under photooxidative conditions. The resulting XANES spectra after irradiation fall between those of Co3O4 and CoO(OH),91,92 the latter being a widely proposed steady-state intermediate for the WO catalytic cycle (Fig. S32†).93–95
 | ||
| Fig. 2 (A) Normalized Co K-edge XANES spectra of Co3O4heptOH (black line), pristine (blue line) KuQ3P0.1@Co3O4 NPs, and KuQ3P0.1@Co3O4 NPs after 15 seconds (orange line), 1.5 h (green line) and 4.5 h (purple line) of photoirradiation. Conditions: NaHCO3/Na2SiF6 (pH 5.6) electrolyte containing 84 mM Na2S2O8; irradiation was performed using 100 mW cm−2 simulated visible solar light (λ > 400 nm). Inset: zoom of the pre-edge region. (B) Fourier transforms of the k3-weighted Co EXAFS spectra for the same samples. Inset: corresponding k3-weighted Co EXAFS spectra. (C) HR-TEM images of KuQ3P0.1@Co3O4 NPs recovered after 5 h of irradiation in the colloidal suspension (NaHCO3/Na2SiF6, pH 5.6, 0.84 mM Na2S2O8). (D) STEM-HAADF images and (E) size distribution histogram of the KuQ3P0.1@Co3O4 NPs recovered after 5 h of irradiation. | ||
The description of the photoirradiated KuQ3P0.1@Co3O4 NPs is further integrated by the recovery of the intensity in the EXAFS spectra, indicating a higher degree of crystallinity of the sample after irradiation (Fig. 2B). Whereas XANES spectra indicate a partial recovery of the Co3O4 structure, the fittings of the FT-EXAFS spectra revealed that after 15 s of irradiation, the Co–O and Co–Co distances slightly decreased to 1.91 Å and 2.84/3.36 Å, respectively, and further decreased to 1.89 and 2.83/3.34 Å after 4.5 h (Fig. S33, S34 and Tables S5, S6†). These slightly shortened Co–O and Co–Co bond distances with respect to the 1.92 and 2.86/3.36 Å expected for the pristine Co3O4 supports the partial transformation of Co3O4 into octahedrally coordinated CoO(OH) species under photochemical conditions.93
Visual examination of the reaction mixture at the end of KuQ3P0.1@Co3O4 irradiation (after 5 h) showed that the material precipitated during the process to give a black solid. ICP-OES analysis was carried out both on the recovered solid and on the liquid phase. The results indicated a modest leaching of cobalt from Co3O4 to the solution (6.3%), but a high (67.8%) leaching of phosphorus from KuQ3P (Table S4†). Given (i) the colorless nature of the final reaction mixture and (ii) the absence of spectroscopic features of the KuQ chromophore upon dichloromethane extraction (Fig. S28†), simple decoordination or loss of the phosphonate moiety by the deeply colored water-soluble KuQ3P could be excluded.§ The experimental evidence instead suggests the chemical disruption of the KuQ3P chromophore core, which could originate from the aggressive reaction environment. Indeed, SO4˙− radicals generated by S2O82− reductive peroxo cleavage at each photoinduced ET event96 are powerful oxidizing agents (E° ranging from 2.6 to 3.1 V vs. the normal hydrogen electrode, NHE, for SO4˙−/SO42−), which can react with organic compounds and generate carbon-based radicals. Furthermore, SO4˙− can react with water to activate aggressive Fenton chemical processes.96–99¶ These considerations support the attribution of the fast deactivation of the colloidal system in photoinduced WO to the intrinsic persulfate reactivity.
Consistent examination by TEM and STEM-HAADF of the solid recovered after 5 h of irradiation revealed the absence of isolated atoms around the Co3O4 NPs (vide supra) accompanied by a diameter increase from 3.9 ± 0.6 nm to 5.3 ± 0.8 nm (Fig. 2C–E and Fig. S29–S31 in the ESI†). This size increase may arise from an Ostwald ripening process prompted by the photoirradiation conditions that induced progressive disruption of KuQ3P, together with the incorporation of the single-atomic Co species initially observed in the images of KuQ3Pn@Co3O4 hybrid nanomaterials.100
Overall, XAS analysis of the hybrid nanomaterials after photoirradiation in the presence of persulfate supports the achievement of a higher overall oxidation state of the Co3O4-based NPs, coherent with oxidative activation of the Co3O4 WOC along with chemical and structural transformations highlighted by HR-TEM and ICP-OES (i.e., disruption of the KuQ3P chromophore and NP size increase) during the photoinduced process.
KuQ3Pn@Co3O4 on meso-SnO2 for photoelectrochemical water oxidation
Based on the competence of KuQ3Pn@Co3O4 dyads in photoinduced WO, we sought to steer their oxygen evolving reactivity in a DSPEC configuration. Thus, we translated the dyadic systems into a regenerative photoanode by embedding them on a SnO2 semiconductor film, avoiding the need for the S2O82− electron acceptor and the generation of highly oxidizing radicals. Mesoporous tin oxide (m-SnO2) was chosen as the semiconductor due to its low conduction band edge energy, providing more favorable charge injection with respect to other n-type semiconductors.16,101 Electrodes with a 2 μm m-SnO2 film thickness and 10–40 nm pore size, blade-coated onto FTO conductive slides, were employed in the present work, as they were previously optimized in photoelectrochemical applications of KuQ dyes.67,68SnO2 photoelectrodes functionalized with KuQ3Pn@Co3O4, hereafter indicated as SnO2|KuQ3Pn@Co3O4, were obtained by dropcasting a colloidal suspension of the KuQ3Pn@Co3O4 NPs in distilled tetrahydrofuran (THF), followed by air drying. Analysis of the XAS and resonance Raman spectra of SnO2|KuQ3P0.2@Co3O4 showed consistency with those of the as-prepared KuQ3P0.2@Co3O4 sample (Fig. S35–S37†).
In order to allow a comparison among the different materials, we deposited an amount of NPs to attain the same nominal KuQ3P loading of 140 nmoldye cm−2.67 The photoelectrodes were then tested in a three-electrode photoelectrochemical cell configuration (Fig. S38†), integrating a glassy carbon auxiliary electrode and a Ag/AgCl (3 M NaCl) reference electrode (potentials were then converted vs. RHE: E (V vs. RHE) = E (V vs. Ag/AgCl) + 0.197 + 0.0592 × pH).
The photoelectrochemical response was analyzed by comparing linear sweep voltammograms (LSV traces) under dark, intermittent (chopped), and continuous illumination, upon irradiation of the electrodes through their back side with the solar spectrum (AM 1.5G, 100 mW cm−2) cut with a long-pass filter (λ > 400 nm) to avoid direct band gap excitation of SnO2. Fig. 3 displays the photoelectrochemical characterization of SnO2|KuQ3P0.2@Co3O4 as a representative case (additional data are reported in Fig. S39 in the ESI†). Under continuous illumination, an anodic photocurrent response associated with photoelectrochemical WO (vide infra) was observed, rising at ca. 0.54 V vs. RHE, below the thermodynamic value for oxygen evolution,  vs. RHE (underpotential regime; a similar onset potential of 0.55 V vs. RHE was previously observed for KuQ-sensitized SnO2 electrodes embedding a tetraruthenium polyoxometalate catalyst, Ru4POM).67 The photocurrent density then flattens to a plateau value of ca. 10 μA cm−2 above 0.84 V vs. RHE; the photocurrent profile of chopped-light LSVs resembles the one observed under continuous illumination, with cathodic current spikes being observed upon turning the light off up to 0.84 V vs. RHE, indicative of recombination events from the photoinduced charge-separated states.102 At higher potentials, the spikes disappear due to a higher bias-related driving force for photoinjected electrons collection by the FTO contact.67,103
vs. RHE (underpotential regime; a similar onset potential of 0.55 V vs. RHE was previously observed for KuQ-sensitized SnO2 electrodes embedding a tetraruthenium polyoxometalate catalyst, Ru4POM).67 The photocurrent density then flattens to a plateau value of ca. 10 μA cm−2 above 0.84 V vs. RHE; the photocurrent profile of chopped-light LSVs resembles the one observed under continuous illumination, with cathodic current spikes being observed upon turning the light off up to 0.84 V vs. RHE, indicative of recombination events from the photoinduced charge-separated states.102 At higher potentials, the spikes disappear due to a higher bias-related driving force for photoinjected electrons collection by the FTO contact.67,103
 | ||
| Fig. 3 (A) LSVs recorded for SnO2|KuQ3P0.2@Co3O4 under darkness (black, dashed line) and under chopped (red, full line) or continuous (blue, dotted line) illumination, in the NaHCO3/Na2SiF6 (pH 5.8) electrolyte, scan rate 0.020 V s−1. (B) Sustained (1 h) CA curves of SnO2|KuQ3P0.2@Co3O4 recorded at 1.14 V vs. RHE in the NaHCO3/Na2SiF6 (pH 5.8) electrolyte. Inset: 20 s CA curves recorded for the same samples. Multiple CA curves were collected, preceded by 30 s electrode de-polarization at 0.54 V vs. RHE. (C) CA curves recorded on SnO2|KuQ3P0.2@Co3O4 electrodes in a two-plate generator–collector setup to detect evolved O2. The generator was poised at 1.14 V vs. RHE, the collector was poised at −0.36 V vs. RHE; experiments were performed in the NaHCO3/Na2SiF6 (pH 5.8) electrolyte. (D) IPCE photo-action spectrum (red squares) for SnO2|KuQ3P0.2@Co3O4 photoanodes, constructed using monochromatic light, superimposed to the absorption spectra of SnO2|KuQ3C electrodes with the dye in the enolate (orange line) and enol (magenta line) forms. CA curves were recorded at 1.14 V vs. RHE in the NaHCO3/Na2SiF6 (pH 5.8) electrolyte. In (A)–(C), irradiation was performed using 100 mW cm−2 simulated visible solar light, λ > 400 nm. | ||
Chronoamperometry (CA) analyses performed with SnO2|KuQ3P0.2@Co3O4 at 1.14 V vs. RHE under continuous illumination produced an initial photocurrent density of ca. 35 μA cm−2, evolving to a steady-state photocurrent density (JSS) of 5–10 μA cm−2 after a few seconds. The latter was shown to be stable for at least 1 h; after 5.5 h, a 60% photocurrent abatement was observed, mainly due to material detachment from the electrode surface (Table 1 and Fig. 3B and S40†). For the sake of comparison, a JSS value of 20 μA cm−2 was obtained by combining KuQ3C with the Ru4POM catalyst, although the latter value was registered at 2 Sun intensity. Concerning the photocurrent behaviour in the early few seconds of irradiation, superimposable CA curves could be achieved in consecutive experiments either by allowing the photoanode to rest until its original open circuit potential (OCP) value (ca. 0.74 V vs. RHE) or by performing a 60 s CA in the dark at 0.54 V vs. RHE (inset of Fig. 3B). Therefore, the initial photocurrent decay may be ascribed to photoanode polarization upon light irradiation rather than physical or chemical degradation of the device.104
| # | Electrode | KuQ/Co3O4 loading (nmol cm−2) | J ss@1.14 V vs. RHE (μA cm−2) | FEO2 (%) |
|---|---|---|---|---|
| a The value for FEO2 is reported for experiments run with a 150 s illumination phase. b See Fig. S39,† bottom right. The higher value of 95% was observed for longer experiments with SnO2|KuQ3P0.2@Co3O4, Fig. S42.† The lower FEO2 registered at 150 s with respect to the almost quantitative value at longer times may be explained by the initial need for multi-electron WOC activation to reach their active state before evolving oxygen. c See Fig. S45,† right. d See Fig. S49.† e See Fig. S50† for repeated experiments and footnote ¶. | ||||
| 1 | SnO2|KuQ3P0.1@Co3O4 | 140/1400 | 11 | 88a |
| 2 | SnO2|KuQ3P0.2@Co3O4 | 140/700 | 8 | 87–95b |
| 3 | SnO2|KuQ3C0.1@Co3O4 | 140/1400 | 5 | 87c |
| 4 | SnO2|KuQ3C|Co3O4heptOH | 140/1400 | 14 | 45d |
| 5 | SnO2|KuQ3P|Co3O4heptOH | 140/1400 | 8 | 45d,e |
Detection and quantification of the evolved oxygen was performed through a two-plate generator–collector chronoamperometric method19,67,105,106 (Fig. 3C, see details in the ESI and Fig. S41–S43†). When held at 1.14 V vs. RHE, irradiation of the SnO2|KuQ3P0.2@Co3O4 electrodes provided a faradaic efficiency for oxygen evolution (FEO2) of 87% (Table 1) (red line in Fig. 3C, and Fig. S43†). The steady-state photocurrent densities (JSS) in CA and FEO2 were exploited as key performance indicators to compare the reactivity among the different materials (Table 1 and Fig. S42†). This included a hybrid nanomaterial with the KuQ dye bearing a carboxylate linker (KuQ3C0.1@Co3O4, entry 3 in Table 1; characterization studies are presented in the ESI, Fig. S44–S46†) and “unbound” KuQ|Co3O4heptOH systems (entries 4 and 5 in Table 1), prepared by dropcasting a suitable amount of Co3O4heptOH NPs onto pre-sensitized SnO2|KuQ electrodes, using both KuQ3C and KuQ3P (preparation and study of the “unbound” photoanodes are described in the ESI, Section 11 and Fig. S47–S51†).
The data in Table 1 can be discussed according to the following points:
(i) The photocurrent density performance indicator suggests a higher activity for KuQ3P0.1@Co3O4 with respect to KuQ3P0.2@Co3O4 (entries 1 and 2 in Table 1), while both materials are characterized by a high and consistent FEO2 of 88 and 87%, respectively (see Section 12 in the ESI† for additional discussions).
(ii) In the KuQ0.1@Co3O4 hybrid nanomaterials, the phosphonate anchoring group provides better results than the carboxylate one in terms of photocurrent density (entries 1 and 3 in Table 1: 11 vs. 5 μA cm−2 for KuQ3P0.1@Co3O4 and KuQ3C0.1@Co3O4, respectively), which can be ascribed in part to the lower stability of the bond between Co3O4 and the carboxylate pendant group with respect to the phosphonate one.107,108
(iii) A key comparison comes with the “unbound” systems (entries 4 and 5 in Table 1), for which the most striking difference is a drop of the FEO2 value to <50%, indicating a much lower efficiency in translating the photo-accumulated holes to the WO process.|| These results indicate that the direct chemical interaction between the Co3O4 NPs and the dye shell surrounding their surface drives more efficient management of the ET chain in the dyadic materials.109,110** This latter point is particularly relevant in the case of the postulated reductive quenching mechanism (vide supra), where the Co3O4 WOC is oxidized by the excited state of the dye, 1*KuQ3P.111,112
The response of SnO2|KuQ0.2@Co3O4 photoanodes was further analyzed in terms of their photo-action spectrum to unequivocally ascribe the photocurrent to the presence of the KuQ3P chromophore. The photoelectrodes were thus irradiated with monochromatic light (wavelength intervals of 10 nm) while recording CA curves at 1.14 V vs. RHE. The photo-action spectrum of the SnO2|KuQ3P0.2@Co3O4 electrodes was constructed in terms of their incident photon-to-current conversion efficiency (IPCE)113 (see the ESI† for a detailed procedure; experimental setup and raw CA curves are reported in Fig. S53 and S54†). An IPCE value of up to 0.44% (at 510 nm, the absorption maximum of KuQ3P, see Fig. 3D and Fig. S18, S47†) was determined for SnO2|KuQ3P0.2@Co3O4; this value is more than four times higher than the one reported for SnO2|KuQ3C|Ru4POM photoanodes, reaching an IPCE value of up to 0.09% at 490 nm.67 Additionally, the operational chemical state of the KuQ dye in the dyad could be determined from the photo-action spectrum: KuQ dyes can in fact partake in enol–enolate equilibria (pKa ≈ 5),72 with the enol and enolate forms displaying different optical spectra.67,72 Indeed, the IPCE spectrum suggests that the KuQ3P chromophore within the dyad is in its enolate form, supported by a comparison with the absorption spectra of SnO2|KuQ3C electrodes with the dye in the enolate (Fig. 3D, orange line) and enol (magenta line) forms.††
State-of-the-art systems based on organic dyes for photoelectrochemical WO have been recently surveyed (Table S6†).3,4,114 These systems are often characterized by photocurrent densities from tenths up to a few hundreds of μA cm−2, associated with FEO2 in the range of 30–95%, while systems operating at wavelengths above 500 nm are rare. More specifically, in the case of dyadic hybrid nanomaterials, the most pertinent comparisons deal with: (i) the Ru(bpy)3–IrOx hybrids proposed by Mallouk and coworkers33–36,38 (systems A and B in Scheme 1) bound to nanostructured TiO2, displaying photocurrent densities between 15 and 80–100 μA cm−2, FEO2 between 20 and 100% and IPCE up to 0.7% at 464 nm at pH 5.8, and (ii) the Ru(phenanthroline)-Fe/FeOx material44 (system D in Scheme 1) onto flat FTO slides, for which photocurrent densities of 10–30 μA cm−2 were registered in NaOH solutions at pH 13, although no FEO2 or IPCE values were reported. In this broader context, the high FEO2 and notable IPCE beyond 500 nm for the Ru-free SnO2|KuQ0.2@Co3O4 photoanodes are significant results. An improvement in the performance, and in particular of the photocurrent density, should mainly target appropriate design of the KuQ3P@Co3O4 interface with the surface of the SnO2 semiconductor. Efforts in this direction should follow literature indications, such as: (i) a further nanostructuring of the semiconductor115 to increase the hybrid loading, (ii) the use of redox mediators116,117 – also in polymeric forms118 – to favour photoinduced electron injection into the semiconductor (a 50-fold increase of the maximum photocurrent was observed for photosystem II-functionalized photoanodes in the presence of osmium-based polymeric redox mediators), and (iii) the use of an insulating layer to limit back electron transfer.119 The use of surface coatings (i.e., polymeric or hydrogel coatings)120 was reported to be beneficial for the stability of molecularly-designed photoanodes.
The evolution of KuQ3P0.2@Co3O4 during photoelectrochemical WO was investigated by complementary resonance Raman and XAS spectroscopy analyses. The resonance Raman spectrum (Fig. 4A) shows that the KuQ3P bands are reasonably intact after 1 h of photoelectrolysis, consistent with the stable photocurrent and preserved oxygen evolution ability of the regenerative photoelectrodes. Therefore, under these operation conditions, the KuQ dye shows good stability; KuQ dyes were indeed shown to be recyclable up to 20 times in aerobic homogeneous photo-oxidation of thioethers.70
 | ||
| Fig. 4 (A) Resonance Raman spectra of SnO2|KuQ3P0.2@Co3O4 electrodes before (red line) and after (purple line) 1 h of photoelectrolysis at 1.14 V vs. RHE in the NaHCO3/Na2SiF6 (pH 5.8) electrolyte. The full spectral region is displayed. Inset: zoom of the resonance Raman spectra in the 400–750 cm−1 region, displaying changes in signals pertaining to Co3O4 and CoO(OH). (B) Normalized Co K-edge XANES spectra of Co3O4heptOH NPs (black line) and SnO2|KuQ3P0.2@Co3O4, before (red line) and after 15 s (green line) and 30 min (purple line) of photoelectrocatalysis at 1.14 V vs. RHE in the NaHCO3/Na2SiF6 (pH 5.8) electrolyte. Inset: zoom of the edge (top) and pre-edge (bottom) regions. (C) Fourier transforms of the k3-weighted Co EXAFS spectra for the same samples (k-range of 3–11 Å−1). Inset: corresponding k3-weighted Co EXAFS spectra; same color code as in (B). | ||
The major changes in Raman spectra are associated with the A1g band of Co3O4, which undergoes a blue shift from 680 cm−1 in the pristine KuQ3P0.2@Co3O4 material to 688 cm−1 in the sample subjected to 1 h of photoelectrolysis. No further changes ascribable to a disorder increase in the material could be identified, thus supporting the absence of particle disruption as a consequence of photoelectrocatalysis. The observed blue shift of the A1g mode, on the other hand, could be attributed to an increasing component of the A1g mode of CoO(OH) formed during photoelectrolysis. This hypothesis is supported by the presence of the bands at 471 cm−1 and 508 cm−1, attributed to the Co3O4F2g and Eg modes, respectively, that evolve into a weaker signal centered at 485 cm−1. Albeit ill-defined, this novel feature is coherent with the Eg mode of CoO(OH).48 Therefore, resonance Raman surface probing of the dyads after extensive photoelectrochemical oxygen evolution has led to the identification of a modification of the Co-based WOC, evolving from a preeminently Co3O4 structure towards a mixture of Co3O4 and CoO(OH) coexisting on the surface of the NPs, consistent with XAS data analysis (vide infra).
XAS spectra were acquired ex situ on SnO2|KuQ3P0.2@Co3O4 subjected to photoelectrocatalysis for up to 30 min (Fig. S55 and 56†), under the same conditions reported above (Fig. 4B and C). Monitoring of XAS features of SnO2|KuQ3P0.2@Co3O4 photoanodes subjected to increasing photoelectrolysis time revealed significant changes that can be summarized as follows:
(i) In the XANES spectra, the edge is gradually shifted towards higher energy values, consistent with an enrichment of CoIII over CoII sites.
(ii) The intensity of the pre-edge region of the spectra remains unchanged after the catalytic process, indicating that the species maintained a centrosymmetric Oh geometry around the metal center.
(iii) In the FT-EXAFS spectra, a recovery of the Co3O4 structure can be observed, accompanied by a very minor shortening of the Co–Co distances (Fig. S4A and S56;† fitting parameters are collected in Table S7†).
Overall, XAS analysis shows a similar trend in comparison with the colloidal system, indicating a partial evolution of the spinel Co3O4 material into a Co3O4/CoO(OH) species under catalytic conditions, consistent with our Raman data and existing reports.95,121 Additional support for the formation of CoO(OH) as a result of WO is provided by CV experiments (Fig. S57†).122 The role of the CoO(OH) species in WO has been indeed documented in the literature.56,123
Conclusions
This work describes the preparation of the first fully noble metal-free dyadic hybrid nanomaterial constituted by Co3O4 nanoparticles functionalized with a fully organic molecular dye, able to catalyze visible light-induced WO. Our strategy takes advantage of the organometallic synthesis to achieve nanometer-sized, monodisperse Co NPs, which were controllably oxidized and covalently post-functionalized with a KuQ dye via a phosphonate anchoring group. KuQ3P was confirmed to act as a LHU within the coordination sphere of the Co3O4 NPs, driving WOC activation via a mechanism suggestive of directional photoinduced ET. SnO2 photoanodes then provided a suitable platform for the dyads, displaying selective O2 evolution in a visible light-operated DSPEC. Spectroscopic evidence allowed us to assess the stability of both the Co3O4 WOC unit and the KuQ3P LHU, and to gain insight into the WO mechanism.Current studies are being devoted to the implementation of KuQ3Pn@Co3O4 dyads into higher complexity DSPEC devices tackling greater oxygenic photocurrents by means of suitably designed redox mediators or semiconductor–molecule heterojunctions. The approach herein described introduces to a wider scenario, where the principles relevant to artificial photosynthesis, such as photoinduced ET preferential pathways, could be routinely interpreted by modular design strategies applied to functional nanomaterials starting at the molecular level.
Experimental
General methods
For the synthesis of the CoheptOH NPs, operations were conducted using Schlenk-line techniques or in a glovebox, employing Ar as the inert gas. (Cyclooctadienyl)(1,5-cyclooctadiene)cobalt(I), [Co(COD)(COE)], was purchased from Nanomeps. Sodium persulfate, sodium hydroxide, sodium hexafluorosilicate, and sodium hydrogen carbonate were acquired from Merck. 1-Heptanol (heptOH) was dried on 4 Å molecular sieves, filtered using a cannula, and degassed by Ar bubbling before the synthesis. Pentane was dried using an MBraun solvent purifier, passed through molecular sieve columns, and degassed by three freeze–pump–thaw cycles before use. Ultrapure argon was purchased from Abelló Linde. Tetrahydrofuran (THF) was purified by distillation over sodium-benzophenone under an Ar atmosphere, subsequently degassed by three freeze–pump–thaw cycles and stored in an Ar-filled glovebox. 1-Heptanol-stabilized Co3O4 NPs (Co3O4heptOH) and KuQ dyes were prepared following previously reported protocols.43,66–68,70Synthesis of KuQ3Pn@Co3O4 NPs
A colloidal suspension of Co3O4heptOH NPs (10 mg) was prepared in heptOH (0.1 mL) by sonication. Independently, a suspension of 1-[3-(dihydroxyphosphonyl)propyl]KuQuinone (KuQ3P) (0.78 mg or 1.55 mg, corresponding to 0.10 or 0.20 equivalents, respectively) was prepared in methanol (2.4 mL) by sonication. Both suspensions were mixed, and water (0.4 mL) was added. Each mixture was stirred for 4 days in the dark, then centrifuged and washed with methanol and hexane, and dried under vacuum, yielding a brown powder corresponding to KuQ3P0.1@Co3O4 NPs or KuQ3P0.2@Co3O4 NPs, respectively. All materials were characterized by TEM in order to determine the NP mean size. ICP-OES or ICP-MS allowed the determination of the composition. KuQ3P0.1@Co3O4 NPs: 3.9 ± 0.6 nm, 35% Co, 0.52% P. KuQ3P0.2@Co3O4 NPs: 3.0 ± 0.6 nm, 35% Co, 1.02% P.The same protocol was applied using 0.10 equivalents of 1-(3-carboxylpropyl)KuQuinone (KuQ3C) to obtain KuQ3C0.1@Co3O4 NPs. The only difference registered was an orange intermediate suspension, observed after 1 h, which then turned into a brown suspension. KuQ3C0.1@Co3O4 NPs: 3.7 ± 0.6 nm, 35% Co.
Full details of microscopic and spectroscopic characterization of the systems, procedures for the preparation and characterization of the photoanodes, and photocatalytic and photoelectrochemical experiments are presented in the ESI.†
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
Data will be made available upon reasonable request to the corresponding authors. The raw synchrotron data are available at the Edmond repository.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
N. R. and F. S. acknowledge the YERUN-YRMA projects for financial support. X. S. and J. G.-A. thank MINECO/FEDER (PID2019-104171RB-I00) and MICIU (PID2023-146787OB-I00) for financial support. L. F. thanks MINECO/FEDER for RyC (RYC2018-025394-I Fellowship) and PID project (PID2021-128197NA-I00) and the Royal Society of Chemistry for the research fund (R20-8077). The authors acknowledge Vincent Colliere (Université de Toulouse and Centre de MicroCaractérisation Raimond Castaing) for technical support in HR-TEM and STEM-HAADF investigations. CNRS and Université de Toulouse are acknowledged for financial support. Financial support from the Italian MUR (PRIN 2022KPK8WM PROMETEO, to A. S. and P. G.) and from the European Union – Next Generation EU (PRIN2022 PNRR PHOTOCORE, P2022ZSPWF to A. S. and M. N.) is gratefully acknowledged. R. B. acknowledges the Department of Chemical Sciences of the University of Padova for funding. O. R., S. D. and M.G.-S. acknowledge the Max Planck Society for funding. M.G.-S. acknowledges the support from the HORIZON-MSCA-2021-PF project TRUSol No. 101063820. Dr Giulia Alice Volpato and Dr Elena Colusso (University of Padova) are gratefully acknowledged for useful scientific discussion. The authors acknowledge Rafael Moraira and Ramon Violeta (technical service of the Physics Department of the Autonomous University of Barcelona) for support in the Si photodiode maintenance. Prof. Jordi Hernando (Autonomous University of Barcelona) is gratefully acknowledged for providing the equipment used for IPCE determination. XAS experiments were performed at the CLÆSS beamline at the ALBA Synchrotron under proposals No. 2021095409 and 2022086973 with the collaboration of ALBA staff. The authors thank Dr. Giulio Gorni and Dr. Vlad Martin-Diaconescu (CLÆSS staff at ALBA) for their assistance during the experiments. F. S. and P. G. thank Grant MUR Dipartimento di Eccellenza 2023-27 X-CHEM project “eXpanding CHEMistry: implementing excellence in research and teaching”.References
- H. Lu and L. Wang, Appl. Catal., B, 2024, 345, 123707, DOI:10.1016/j.apcatb.2024.123707.
- P. Hunter, EMBO Rep., 2022, 23(11), e56149, DOI:10.15252/embr.202256149.
- T. Gobbato, G. A. Volpato, A. Sartorel and M. Bonchio, Chem. Sci., 2023, 14, 12402–12429, 10.1039/d3sc03780k.
- J. T. Kirner and R. G. Finke, J. Mater. Chem. A, 2017, 5, 19560–19592, 10.1039/c7ta05709a.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875, DOI:10.1021/acs.chemrev.8b00392.
- P. D. Tran, L. H. Wong, J. Barber and J. S. C. Loo, Energy Environ. Sci., 2012, 5, 5902–5918, 10.1039/c2ee02849b.
- F. Li, H. Yang, W. Li and L. Sun, Joule, 2018, 2, 36–60, DOI:10.1016/j.joule.2017.10.012.
- L. Alibabaei, H. Luo, R. L. House, P. G. Hoertz, R. Lopez and T. J. Meyer, J. Mater. Chem. A, 2013, 1(13), 4133–4145, 10.1039/c2ta00935h.
- A. Charisiadis, E. Glmenaki, A. Planchat, S. Margiola, A.-C. Lavergne-Bril, E. Nikoloudakis, V. Nikolaou, G. Charalamidis, A. G. Coutsolelosand and F. Odobel, Dyes Pigm., 2021, 185, 108908, DOI:10.1016/j.dyepig.2020.108908.
- E. Sundin and M. Abrahamsson, Chem. Commun., 2018, 54, 5289–5298, 10.1039/c8cc01071d.
- C. Herrero, B. Lassalle-Kaiser, W. Leibl, A. W. Rutherford and A. Aukauloo, Coord. Chem. Rev., 2008, 252, 456–468, DOI:10.1016/j.ccr.2007.09.002.
- C. W. Cady, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2008, 252, 444–455, DOI:10.1016/j.ccr.2007.06.002.
- A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensjö, S. Styring, V. Sundström and L. Hammarström, Acc. Chem. Res., 2009, 42(12), 1899–1909, DOI:10.1021/ar900127h.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115(23), 13006–13049, DOI:10.1021/acs.chemrev.5b00229.
- D. Wang, R. N. Sampaio, L. Troian-Gautier, S. L. Marquard, B. H. Farnum, B. D. Sherman, M. V. Sheridan, C. J. Dares, G. J. Meyer and T. J. Meyer, J. Am. Chem. Soc., 2019, 141(19), 7926–7933, DOI:10.1021/jacs.9b02548.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519, 10.1039/C3CS60405E.
- T. Keijer, T. Bouwens, J. Hessels and J. N. H. Reek, Chem. Sci., 2021, 12, 50–70, 10.1039/D0SC03715J.
- M. Bonchio, Z. Syrgiannis, M. Burian, N. Marino, E. Pizzolato, K. Dirian, F. Rigodanza, G. A. Volpato, G. La Ganga, N. Demitri, S. Berardi, H. Amenitsch, D. M. Guldi, S. Caramori, C. A. Bignozzi, A. Sartorel and M. Prato, Nat. Chem., 2019, 11(2), 146–153, DOI:10.1038/s41557-018-0172-y.
- T. Gobbato, F. Rigodanza, E. Benazzi, P. Costa, M. Garrido, A. Sartorel, M. Prato and M. Bonchio, J. Am. Chem. Soc., 2022, 144, 14021–14025, DOI:10.1021/jacs.2c05857.
- L. Wang, D. E. Polyansky and J. J. Concepcion, J. Am. Chem. Soc., 2019, 141, 8020–8024, DOI:10.1021/jacs.9b01044.
- D. Wang, L. Wang, M. D. Brady, C. J. Dares, G. J. Meyer, T. J. Meyer and J. J. Concepcion, J. Phys. Chem. C, 2019, 123, 30039–30045, DOI:10.1021/acs.jpcc.9b07125.
- Y. Li, S. Zhan, Z. Deng, M. Chen, Y. Zhao, C. Liu, Z. Zhao, H. Ning, W. Li, F. Li, L. Sunand and F. Li, J. Energy Chem., 2024, 93, 526–537, DOI:10.1016/j.jechem.2024.02.017.
- H. Li, F. Li, Y. Wang, L. Bai, F. Yu and L. Sun, ChemPlusChem, 2016, 81, 1056–1059, DOI:10.1002/cplu.v81.10.
- D. A. Kader and S. J. Mohammed, RSC Adv., 2023, 13, 26484–26508, 10.1039/d3ra05098j.
- T. T. T. Toan, D. M. Nguyen, A. Q. Dao, V. T. Le and Y. Vasseghian, Mol. Catal., 2023, 538, 113001, DOI:10.1016/j.mcat.2023.113001.
- J. Wang, H.-Y. Tan, M.-Y. Qi, J.-Y. Li, Z.-R. Tang, N.-T. Suen, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2023, 52, 5013–5050, 10.1039/D2CS00441K.
- Y.-F. Wang, M.-Y. Qi, M. Conte, Z.-R. Tang and Y.-J. Xu, Angew. Chem., Int. Ed., 2024, 63, e202407791, DOI:10.1002/anie.202407791.
- G. Martí, L. Mallón, N. Romero, L. Francàs, R. Bofill, K. Philippot, J. García-Antón and X. Sala, Adv. Energy Mater., 2023, 13, 202300282, DOI:10.1002/aenm.202300282.
- J. K. Hurst, Science, 2010, 328(5976), 315–316, DOI:10.1126/science.1187721.
- A. Singh and L. Spiccia, Coord. Chem. Rev., 2013, 257(17–18), 2607–2622, DOI:10.1016/j.ccr.2013.02.027.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116(22), 14120–14136, DOI:10.1021/acs.chemrev.6b00398.
- P. G. Hoertz, Y. II. Kim, W. J. Youngblood and T. E. Mallouk, J. Phys. Chem. B, 2007, 111(24), 6845–6856, DOI:10.1021/jp070735r.
- N. D. Morris, M. Suzuki and T. E. Mallouk, J. Phys. Chem. A, 2004, 108(42), 9115–9119, DOI:10.1021/jp0480145.
- J. W. Youngblood, S. H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131(3), 926–927, DOI:10.1021/ja809108y.
- W. J. Youngblood, S. H. Anna Lee, K. Maeda and T. E. Mallouk, Acc. Chem. Res., 2009, 42(12), 1966–1973, DOI:10.1021/ar9002398.
- S. H. A. Lee, Y. Zhao, E. A. Hernandez-Pagan, L. Blasdel, W. J. Youngblood and T. E. Mallouk, Faraday Discuss., 2012, 155, 165–176, 10.1039/c1fd00083g.
- A. B. Muñoz-García, I. Benesperi, G. Boschloo, J. J. Concepcion, J. H. Delcamp, E. A. Gibson, G. J. Meyer, M. Pavone, H. Pettersson, A. Hagfeldt and M. Freitag, Chem. Soc. Rev., 2021, 50, 12450–12550, 10.1039/D0CS01336F.
- P. Xu, N. S. McCool and T. E. Mallouk, Nano Today, 2017, 14, 42–58, DOI:10.1016/j.nantod.2017.04.009.
- U. Reschand and M. A. Fox, J. Phys. Chem., 1991, 95(16), 6316, DOI:10.1021/j100169a045.
- B. D. Sherman, S. Pillai, G. Kodis, J. Bergkamp, T. E. Mallouk, D. Gust, T. A. Moore and A. L. Moore, Can. J. Chem., 2011, 89(2), 152–157, DOI:10.1139/V10-118.
- L. A. Fontana, V. H. Rigolin, C. B. Braga, C. Ornelas and J. D. Megiatto, Jr., Chem. Commun., 2021, 57, 7398–7401, 10.1039/D1CC02931B.
- J. D. Megiatto, Jr. and C. Ornelas, in Photosynthesis. Methods in Molecular Biology, ed. S. Covshoff, Humana Press, New York, NY, USA, 2018, Vol. 1770, Ch. 19 Search PubMed.
- J. De Tovar, N. Romero, S. A. Denisov, R. Bofill, C. Gimbert-Suriñach, D. Ciuculescu-Pradines, S. Drouet, A. Llobet, P. Lecante, V. Colliere, Z. Freixa, N. McClenaghan, C. Amiens, J. García-Antón, K. Philippot and X. Sala, Mater Today Energy, 2018, 9, 506–515, DOI:10.1016/j.mtener.2018.07.008.
- Q. T. Nguyen, E. Rousset, V. T. H. Nguyen, V. Colliere, P. Lecante, W. Klysubun, K. Philippot, J. Esvan, M. Respaud, G. Lemercier, P. D. Tran and C. Amiens, ACS Appl. Mater. Interfaces, 2021, 13(45), 53829–53840, DOI:10.1021/acsami.1c15051.
- C. Decavoli, C. L. Boldrini, N. Manfredi and A. Abbotto, Eur. J. Inorg. Chem., 2020, 11–12, 978–999, DOI:10.1002/ejic.202000026.
- N. Manfredi, C. L. Boldrini and A. Abbotto, ChemElectroChem, 2018, 5(17), 2395–2402, DOI:10.1002/celc.201800592.
- B. Cecconi, N. Manfredi, T. Montini, P. Fornasiero and A. Abbotto, Eur. J. Org. Chem., 2016, 5194–5215, DOI:10.1002/ejoc.201600653.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, J. Phys. Chem. C, 2010, 114(1), 111–119, DOI:10.1021/jp908548f.
- X. Deng and H. Tüysüz, ACS Catal., 2014, 4, 3701–3714, DOI:10.1021/cs500713d.
- N. H. Chou, P. N. Ross, A. T. Bell and T. D. Tilley, ChemSusChem, 2011, 4(11), 1566–1569, DOI:10.1002/cssc.201100075.
- L. Reith, C. A. Triana, F. Pazoki, M. Amiri, M. Nyman and G. R. Patzke, J. Am. Chem. Soc., 2021, 143(37), 15022–15038, DOI:10.1021/jacs.1c03375.
- F. Jiaoand and H. Frei, Energy Environ. Sci., 2010, 3, 1018–1027, 10.1039/c002074e.
- J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt and S. S. Stahl, J. Am. Chem. Soc., 2011, 133(36), 14431–14442, DOI:10.1021/ja205647m.
- M. R. Mohammadi, S. Loos, P. Chernev, C. Pasquini, I. Zaharieva, D. González-Flores, P. Kubella, K. Klingan, R. D. L. Smith and H. Dau, ACS Catal., 2020, 10(14), 7990–7999, DOI:10.1021/acscatal.0c01944.
- T. Wiegmann, I. Pacheco, F. Reikowski, J. Stettner, C. Qiu, M. Bouvier, M. Bertram, F. Faisal, O. Brummel, J. Libuda, J. Drnec, P. Allongue, F. Maroun and O. M. Magnussen, ACS Catal., 2022, 12(6), 3256–3268, DOI:10.1021/acscatal.1c05169.
- M. Zhang, M. De Respinis and H. Frei, Nat. Chem., 2014, 6(4), 362–367, DOI:10.1038/nchem.1874.
- D. K. Bediako, A. M. Ullman and D. G. Nocera, in Top. Curr. Chem, ed. H. Tüysüz and C. Chan, Springer Verlag, 2015, Vol. 371, pp. 173–214. DOI:10.1007/128_2015_649.
- C. N. Brodsky, D. K. Bediako, C. Shi, T. P. Keane, C. Costentin, S. J. L. Billinge and D. G. Nocera, ACS Appl. Energy Mater., 2019, 2(1), 3–12, DOI:10.1021/acsaem.8b00785.
- F. T. Haase, A. Bergmann, T. E. Jones, J. Timoshenko, A. Herzog, H. S. Jeon, C. Rettenmaier and B. R. Cuenya, Nat. Energy, 2022, 7(8), 765–773, DOI:10.1038/s41560-022-01083-w.
- M. Wang, Q. Wa, X. Bai, Z. He, W. S. Samarakoon, Q. Ma, Y. Du, Y. Chen, H. Zhou, Y. Liu, X. Wang and Z. Feng, JACS Au, 2021, 1(12), 2216–2223, DOI:10.1021/jacsau.1c00346.
- C. Amiens, B. Chaudret, D. Ciuculescu-Pradines, V. Collière, K. Fajerwerg, P. Fau, M. Kahn, A. Maisonnat, K. Soulantica and K. Philippot, New J. Chem., 2013, 37(11), 3374–3401, 10.1039/c3nj00650f.
- C. Amiens, D. Ciuculescu-Pradines and K. Philippot, Coord. Chem. Rev., 2016, 308, 409–432, DOI:10.1016/j.ccr.2015.07.013.
- A. Coletti, S. Lentini, V. Conte, B. Floris, O. Bortolini, F. Sforza, F. Grepioni and P. Galloni, J. Org. Chem., 2012, 77, 6873–6879, DOI:10.1021/jo300985x.
- F. Valentini, F. Sabuzi, M. Forchetta, V. Conte and P. Galloni, RSC Adv., 2023, 13, 9065–9077, 10.1039/d3ra00539a.
- F. Sabuzi, V. Armuzza, V. Conte, B. Floris, M. Venanzi, P. Galloni and E. Gatto, J. Mater. Chem. C, 2016, 4(3), 622–629, 10.1039/c5tc03363b.
- M. Bonomo, F. Sabuzi, A. Di Carlo, V. Conte, D. Dini and P. Galloni, New J. Chem., 2017, 41, 2769–2779, 10.1039/c6nj03466g.
- G. A. Volpato, M. Marasi, T. Gobbato, F. Valentini, F. Sabuzi, V. Gagliardi, A. Bonetto, A. Marcomini, S. Berardi, V. Conte, M. Bonchio, S. Caramori, P. Galloni and A. Sartorel, Chem. Commun., 2020, 56, 2248–2251, 10.1039/c9cc09805d.
- G. A. Volpato, E. Colusso, L. Paoloni, M. Forchetta, F. Sgarbossa, V. Cristino, M. Lunardon, S. Berardi, S. Caramori, S. Agnoli, F. Sabuzi, P. Umari, A. Martucci, P. Galloni and A. Sartorel, Photochem. Photobiol. Sci., 2021, 20, 1243–1255, DOI:10.1007/s43630-021-00097-9.
- V. Mazzaracchio, R. Marrone, M. Forchetta, F. Sabuzi, P. Galloni, M. Wang, A. Nazligul, K. L. Choy, F. Arduini and D. Moscone, Electrochim. Acta, 2022, 426, 140766, DOI:10.1016/j.electacta.2022.140766.
- M. Forchetta, F. Sabuzi, L. Stella, V. Conte and P. Galloni, J. Org. Chem., 2022, 87(21), 14016–14025, DOI:10.1021/acs.joc.2c01648.
- F. Valentini, F. Sabuzi, V. Conte, V. N. Nemykin and P. Galloni, J. Org. Chem., 2021, 86, 5680–5689, DOI:10.1021/acs.joc.1c00165.
- F. Sabuzi, S. Lentini, F. Sforza, S. Pezzola, S. Fratelli, O. Bortolini, B. Floris, V. Conte and P. Galloni, J. Org. Chem., 2017, 82, 10129–10138, DOI:10.1021/acs.joc.7b01602.
- M. Forchetta, V. Conte, G. Fiorani, P. Galloni and F. Sabuzi, Organics, 2021, 2, 107–117, DOI:10.3390/org2020010.
- I. Bazzan, A. Volpe, A. Dolbecq, M. Natali, A. Sartorel, P. Mialane and M. Bonchio, Catal. Today, 2017, 290, 39–50, DOI:10.1016/j.cattod.2017.03.027.
- M. Yu, C. Weidenthaler, Y. Wang, E. Budiyanto, E. Onur Sahin, M. Chen, S. DeBeer, O. Rüdiger and H. Tüysüz, Angew. Chem., Int. Ed., 2022, 61(42), e202211543, DOI:10.1002/anie.202211543.
- T. E. Westre, P. Kennepohl, J. G. Dewitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314, DOI:10.1021/ja964352a.
- M. Risch, D. M. Morales, J. Villalobos and D. Antipin, Angew. Chem., Int. Ed., 2022, 61, e202211949, DOI:10.1002/anie.202211949.
- T. Zhou, D. Wang, S. C.-K. Goh, J. Hong, J. Han, J. Mao and R. Xu, Energy Environ. Sci., 2015, 8, 526–534, 10.1039/C4EE03234A.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dincǎ, V. K. Yachandra and D. G. Nocera, J. Am. Chem. Soc., 2010, 132(39), 13692–13701, DOI:10.1021/ja1023767.
- E. M. Sproviero, J. A. Gascón, J. P. McEvoy, G. W. Brudvig and V. S. Batista, J. Am. Chem. Soc., 2008, 130(21), 6728–6730, DOI:10.1021/ja801979n.
- M. Risch, K. Klingan, F. Ringleb, P. Chernev, I. Zaharieva, A. Fischer and H. Dau, ChemSusChem, 2012, 5(3), 542–549, DOI:10.1002/cssc.201100574.
- C. H. M. Van Oversteeg, H. Q. Doan, F. M. F. De Groot and T. Cuk, Chem. Soc. Rev., 2017, 46, 102–125, 10.1039/c6cs00230g.
- R. Zhang, L. Pan, B. Guo, Z. F. Huang, Z. Chen, L. Wang, X. Zhang, Z. Guo, W. Xu, K. P. Loh and J. J. Zou, J. Am. Chem. Soc., 2023, 145(4), 2271–2281, DOI:10.1021/jacs.2c10515.
- N. Piergies and E. Proniewicz, J. Spectrosc., 2014, 2014, 47237, DOI:10.1155/2014/247237.
- C. Schuschke, M. Schwarz, C. Hohner, T. N. Silva and J. Libuda, J. Phys. Chem. C, 2018, 122(28), 16221–16233, DOI:10.1021/acs.jpcc.8b06147.
- C. Schuschke, M. Schwarz, C. Hohner, T. N. Silva, L. Fromm, T. Döpper, A. Görling and J. Libuda, J. Phys. Chem. Lett., 2018, 9(8), 1937–1943, DOI:10.1021/acs.jpclett.8b00668.
- M. Bertram, C. Schuschke, F. Waidhas, M. Schwarz, C. Hohner, M. A. Montero, O. Brummel and J. Libuda, Phys. Chem. Chem. Phys., 2019, 21, 23364–23374, 10.1039/C9CP03779A.
- F. Dumur, A. Guerlin, A. Lehoux, P. R. Selvakannan, F. Miomandre, R. Méallet-Renault, M. Rebarz, M. Sliwa, E. Dumas, L. Le Pleux, Y. Pellegrin, F. Odobel and C. R. Mayer, Appl. Surf. Sci., 2020, 499, 143847, DOI:10.1016/j.apsusc.2019.143847.
- E. A. Sprague-Klein, X. He, M. W. Mara, B. J. Reinhart, S. Lee, L. M. Utschig, K. L. Mulfort, L. X. Chen and D. M. Tiede, ACS Energy Lett., 2022, 7(9), 3129–3138, DOI:10.1021/acsenergylett.2c01560.
- Standard Potentials in Aqueous Solution, ed. A. J. Bard, R. Parsons and J. Jordan, Marcel Dekker Inc., New York, 1985 Search PubMed.
- J. Zhou, Y. Wang, X. Su, S. Gu, R. Liu, Y. Huang, S. Yan, J. Li and S. Zhang, Energy Environ. Sci., 2019, 12, 739–746, 10.1039/c8ee03208d.
- Z. Wang, Y. Tan, X. Duan, Y. Xie, H. Jin, X. Liu, L. Ma, Q. Gu and H. Wei, Chemosphere, 2023, 313, 137346, DOI:10.1016/j.chemosphere.2022.137346.
- H. Y. Wang, S. F. Hung, H. Y. Chen, T. S. Chan, H. M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138(1), 36–39, DOI:10.1021/jacs.5b10525.
- A. Bergmann, E. Martinez-Moreno, D. Teschner, P. Chernev, M. Gliech, J. F. De Araújo, T. Reier, H. Dau and P. Strasser, Nat. Commun., 2015, 6, 8625, DOI:10.1038/ncomms9625.
- M. Favaro, J. Yang, S. Nappini, E. Magnano, F. M. Toma, E. J. Crumlin, J. Yano and I. D. Sharp, J. Am. Chem. Soc., 2017, 139(26), 8960–8970, DOI:10.1021/jacs.7b03211.
- J. Schneider and D. W. Bahnemann, J. Phys. Chem. Lett., 2013, 4, 3479–3483, DOI:10.1021/jz4018199.
- T. N. Myers, Encyclopedia of Polymer Science and Technology, John Wiley & Sons, 2002, Vol. 6, pp. 563–600 Search PubMed.
- T. Nawaz and S. Sengupta, Handbook of Water Purity and Quality, 2021, pp. 293–337. DOI:10.1016/B978-0-12-821057-4.00015-X.
- L. M. Alrehaily, J. M. Joseph and J. C. Wren, Phys. Chem. Chem. Phys., 2015, 17(37), 24138–24150, 10.1039/c5cp02828k.
- D. Hochfilzer, I. Chorkendorff and J. Kibsgaard, ACS Energy Lett., 2023, 8(3), 1607–1612, DOI:10.1021/acsenergylett.3c00021.
- R. Gong, D. Mitoraj, R. Leiter, M. Mundszinger, A. K. Mengele, I. Krivtsov, J. Biskupek, U. Kaiser, R. Beranek and S. Rau, Front. Chem., 2021, 9, 709903, DOI:10.3389/fchem.2021.709903.
- C. F. Jewel, A. Subramanian, C.-Y. Nam and R. G. Finke, Sustainable Energy Fuels, 2021, 5, 5257–5269, 10.1039/d1se00908g.
- E. Nikoloudakis, P. B. Pati, G. Charalambidis, D. S. Budkina, S. Diring, A. Planchat, D. Jacquemin, E. Vauthey, A. G. Coutsolelos and F. Odobel, ACS Catal., 2021, 11(19), 12075–12086, DOI:10.1021/acscatal.1c02609.
- J. R. Swierk, N. S. McCool, T. P. Saunders, G. D. Barber and T. E. Mallouk, J. Am. Chem. Soc., 2014, 136(31), 10974–10982, DOI:10.1021/ja5040705.
- B. D. Sherman, M. V. Sheridan, C. J. Dares and T. J. Meyer, Anal. Chem., 2016, 88, 7076–7082, DOI:10.1021/acs.analchem.6b00738.
- S. Grau, S. Berardi, A. Moya, R. Matheu, V. Cristino, J. J. Vilatela, C. A. Bignozzi, S. Caramori, C. Gimbert-Suriñach and A. Llobet, Sustainable Energy Fuels, 2018, 2(9), 1979–1985, 10.1039/c8se00146d.
- K. L. Materna, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2017, 46, 6099–6110, 10.1039/C7CS00314E.
- L. Fusek, M. Kastenmeier, E. Franz, L. Fromm, A. Gorling, O. Brummel and J. Libuda, Surf. Sci., 2022, 718, 122013, DOI:10.1016/j.susc.2021.122013.
- J. T. Kirner, J. J. Stracke, B. A. Gregg and R. G. Finke, ACS Appl. Mater. Interfaces, 2014, 6, 13367–13377, DOI:10.1021/am405598w.
- J. T. Kirner and R. G. Finke, ACS Appl. Mater. Interfaces, 2017, 9, 27625–27637, DOI:10.1021/am405598w.
- D. Ung and B. M. Cossairt, ACS Appl. Energy Mater., 2019, 2(3), 1642–1645, DOI:10.1021/acsaem.9b00240.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136, DOI:10.1021/acs.chemrev.6b00398.
- Y.-H. Lai, M. Kato, D. Mersch and E. Reisner, Faraday Discuss., 2014, 176, 199–211, 10.1039/c4fd00059e.
- M.-N. Collomb, D. V. Morales, C. N. Astudillo, B. Dautreppe and J. Fortage, Sustainable Energy Fuels, 2020, 4, 31–49, 10.1039/C9SE00597H.
- D. Mersch, C.-Y. Lee, J. Z. Zhang, K. Brinkert, J. C. Fontecilla-Camps, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2015, 137, 8541–8549, DOI:10.1021/jacs.5b03737.
- Y. Zhao, J. R. Swierk, J. D. Megiatto Jr, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612–15616, DOI:10.1073/pnas.1118339109.
- D. Wang, R. N. Sampaio, L. Troian-Gautier, S. L. Marquard, B. H. Farnum, B. D. Sherman, M. V. Sheridan, C. J. Dares, G. J. Meyer and T. J. Meyer, J. Am. Chem. Soc., 2019, 141, 7926–7933, DOI:10.1021/jacs.9b02548.
- K. P. Sokol, D. Mersch, V. Hartmann, J. Z. Zhang, M. M. Nowaczyk, M. Rögner, A. Ruff, W. Schuhmann, N. Plumeré and E. Reisner, Energy Environ. Sci., 2016, 9, 3698–3709, 10.1039/c6ee01363e.
- M. S. Eberhart, D. Wang, R. N. Sampaio, S. L. Marquard, B. Shan, M. K. Brennaman, G. J. Meyer, C. Dares and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 16248–16255, DOI:10.1021/jacs.7b08317.
- D. F. Bruggeman, A. A. H. Laporte, R. J. Detz, S. Mathew and J. N. H. Reek, Angew. Chem., Int. Ed., 2022, 61, e202200175, DOI:10.1002/anie.202200175.
- A. Moysiadou, S. Lee, C.-C. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914, DOI:10.1021/jacs.0c04867.
- K. Fan, D. Zhou, H. Yang, L. Wang, Y. Shan, M. Wan, A. Zheng and L. Sun, ACS Catal., 2025, 15(4), 3256–3266, DOI:10.1021/acscatal.4c07903.
- J. T. Mefford, A. R. Akbashev, M. Kang, C. L. Bentley, W. E. Gent, H. D. Deng, D. H. Alsem, Y.-S. Yu, N. J. Salmon, D. A. Shapiro, P. R. Unwin and W. C. Chueh, Nature, 2021, 593, 67–73, DOI:10.1038/s41586-021-03454-x.
Footnotes |
| † Electronic supplementary information (ESI) available: Complete description of experimental procedures and graphic materials, including photographs of experimental setups and spectral, microscopic and electrochemical data. See DOI: https://doi.org/10.1039/d4gc06606e |
| ‡ Comparable results were obtained by adopting the approach reported in ref. 74, as described in the ESI, Section 1.4.† |
| § Assembly of a dyad equivalent to KuQ3P0.1@Co3O4, could be achieved by co-suspending KuQ3P (0.1 eq.) and Co3O4heptOH in the aqueous buffer used for the photoreactions; see Tables S1, S4 and Fig. S26, S27.† In contrast, a co-suspension of Co3O4heptOH and KuQ-Hex (incapable of binding to cobalt(II,III) oxide due to the lack of a suitable anchoring group) allows for complete recovery of KuQ-Hex upon extraction with dichloromethane, even after photoirradiation in the presence of persulfate (Fig. S28†). |
| ¶ When considering the oxidation of Co3O4 through a photoinduced oxidative cycle, the release of highly oxidizing sulfate radicals can also contribute to the oxidation of CoII sites. |
| || A generally poorly reproducible FEO2 was observed in the case of “unbound” photoanodes, with average values of 29%; furthermore, repeated CA curves recorded for SnO2|KuQ3P|Co3O4heptOH photoelectrodes provided nearly constant FEO2 values, thus not encouraging the hypothesis of in situ assembly of the dyads during photoelectrocatalysis (Fig. S51 and S52†). |
| ** Additionally, the KuQ3P shell on the surface of KuQ3Pn@Co3O4 is expected to provide a platform for better control over proton exchange in photo-driven WO than the hydrophobic heptOH shell in Co3O4heptOH NPs. |
| †† KuQ3C was chosen to compare the optical features of KuQ dyes on SnO2 films, since its enolate and enol forms are both accessible by chemical means once chemisorbed. KuQ3P, on the other hand, directly provides sensitized SnO2 featuring the enol form of the dye, as described in the ESI (Sections 2 and 11).† |
| This journal is © The Royal Society of Chemistry 2025 |