
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amine-functionalized lignin hydrogels for high-performance N-type ionic thermoelectric materials†
Nazish
Jabeen
a,
Clara M.
Gómez
a,
Rafael
Muñoz-Espí
 a,
Andrés
Cantarero
b,
Maurice N.
Collins
c and
Mario
Culebras
*a
a,
Andrés
Cantarero
b,
Maurice N.
Collins
c and
Mario
Culebras
*a
aInstitute of Materials Science (ICMUV), University of Valencia, PO Box 22085, 46071 Valencia, Spain. E-mail: Mario.Culebras@uv.es
bInstitute of Molecular Science (ICMol), Universitat de Valencia, PO Box 22085, 46071 Valencia, Spain
cStokes Laboratories, School of Engineering, Bernal Institute, University of Limerick, Limerick, Ireland
First published on 19th June 2025
Abstract
Over the past decade, the escalating global energy crisis has necessitated innovative solutions for sustainable development. This study explores a novel approach to address this challenge by harnessing abundant and ubiquitous low-grade thermal energy for electricity generation. Here, we report an efficient method utilizing lignin, a byproduct of the paper and pulp industry, to develop high-performance ionic thermoelectric (i-TE) materials. This research demonstrates the transformation of lignin-based hydrogels from p-type to n-type materials through a green synthesis approach involving cross-linking and amine functionalization. The optimized functionalized lignin-based hydrogel when infiltrated with a 1 M KCl electrolyte, exhibits a negative ionic thermopower (−7.48 mV K−1) and high ionic conductivity (39.9 mS cm−1). These properties result in an exceptional power factor (223.52 μW m−1 K−2) and a figure of merit (iZT) of (0.145), surpassing many existing state-of-the-art materials in sustainable n-type ionic thermoelectric performance. Our findings highlight the potential of functionalized hydrogels (n-type material) for low-grade waste energy harvesting. This work advances the performance of n-type i-TE materials and opens avenues for further practical applications in n–p type connected devices. These novel materials offer a promising pathway for self-powered flexible wearable electronics, marking a significant step towards sustainable energy solutions and energy storage devices for efficient utilization of low-grade thermal energy.
Green foundation1. Our work transforms lignin, a waste product from the paper and pulp industry, into high-performance ionic thermoelectric materials using a water-based, organic solvent-free functionalization process. This aligns with green chemistry principles by valorizing waste, eliminating hazardous reagents, and promoting sustainable energy via waste heat recovery.2. We demonstrate the use of lignin as a sustainable feedstock, achieving high ionic conductivity (39.9 mS cm−1) and negative thermopower (−7.48 mV K−1), surpassing most n-type materials. Our solvent-free synthesis and waste utilization significantly reduce environmental impact. 3. Future research could explore broader feedstocks, for example agricultural and forestry waste not only the lignin from the paper and pulp industry. Also the scalability and the further implementation in functional thermoelectric devices that can recover energy and storage it at the same time, will contribute to power our society in a greener way. |
1. Introduction
Low-grade heat (<100 °C) presents a promising and consistent energy source. It is estimated that over 30% of global primary energy consumption is lost as low-grade heat below 100 °C. Thus, capturing this abundant and widespread low-grade heat and converting it into electrical energy is crucial for addressing the global energy crisis.1 Over the past few decades, thermoelectric (TE) technology has been widely recognized for its remarkable ability to convert heat into electricity, making it a promising solution for low-grade waste heat recovery.2,3 Traditional thermoelectric generators (TEGs) primarily utilize a variety of inorganic materials, including semiconductors such as bismuth telluride (Bi2Te3), lead telluride (PbTe), and silicon–germanium (SiGe) alloys.4,5 The thermoelectric conversion in these type of semiconductor alloys is based on the Seebeck effect, which involves the thermodiffusion of electrons or holes. Despite their potential, these devices face significant challenges in harvesting low-grade including high cost of raw materials, toxicity, limited design flexibility, and a low Seebeck coefficient (S = ΔV/ΔT, typically in the range of hundreds of μV K−1).6,7 To facilitate the practical application of these devices and meet the rising demand for efficient low-grade heat capture, ionic thermoelectric (i-TE) materials that utilize the Soret effect have emerged as a promising research domain. These materials are typically based on liquid electrolytes, and solid-state electrolytes exhibiting remarkably high ionic Seebeck coefficient.8 The operating mechanism of i-TE materials involves generating a voltage difference due to the varied mobility of cations and anions within the electrolyte under a temperature gradient.9 The efficiency of these materials is measured by dimensionless ionic figure of merit (iZT), iZT = (S2σi)/κT where ionic Seebeck coefficient (S), ionic conductivity (σi), thermal conductivity (κ), and absolute temperature (T). One key advantage of i-TEs is the possibility to produce substantial voltages from minimal temperature differences, with ionic Seebeck coefficients typically in the range of mV K−1, which are 2–3 orders of magnitude greater than traditional electronic conductors.10,11 However, these i-TE materials face significant hurdles such as electrolyte leakage, limited stretchability, and high resistance. To overcome these limitations, researchers have focused on developing quasi-solid electrolytes by creating gels where the liquid phase is immobilized within the solid networks particularly hydrogels.12,13 Hydrogels, which are three-dimensional networks of cross-linked, hydrophilic polymers, are emerging as promising candidates for i-TE devices. These versatile materials boast tunable mechanical properties, including elasticity and strength, alongside a remarkable capacity for electrolyte absorption and retention.14 This unique characteristic enables hydrogels to function as quasi-solid electrolytes, effectively addressing the leakage issues associated with conventional liquid electrolytes in i-TE devices. Moreover, hydrogels offer the additional advantage of mechanical adaptability, making them a prominent material choice for the design of flexible and robust i-TE devices.15,16On the other side, our growing dependence on non-renewable and synthetic polymers also presents a significant challenge. To address this concern and unlock the full potential of biorefineries, researchers are exploring the use of biopolymers (derived from biomass) to develop sustainable and eco-friendly i-TE materials. This approach leverages the inherent renewability and unique properties of biomass-derived biopolymers, including carbon-based materials, polysaccharides, and lignocellulosic biomass. Carbon-based materials, including graphene, carbon nanotubes, activated carbon, and sustainable biochar (BC), are particularly notable for their excellent biocompatibility and sustainability, making them highly suitable for applications in environmental remediation and i-TE devices.17 Among polysaccharides, chitosan stands out as a prominent biopolymer derived from chitin through deacetylation. Chitin, the second most abundant renewable bio-waste, is naturally present in the exoskeletons of crustaceans, insect cuticles, and fungal cell walls. Chitosan is distinguished by its natural amino groups, which impart valuable properties such as biocompatibility, non-toxicity, biodegradability, and cost-effectiveness.18 Additionally, lignocellulosic biomass, a widely available bioresource, has gained attention as a promising candidate due to its high thermoelectric performance and biodegradability. Lignin is a major component of lignocellulose biomass found alongside cellulose and hemicellulose in wood and fibers and a readily available byproduct of paper pulping and biorefinery industries.19,20 This complex aromatic polymer, primarily composed of phenylpropanoid units, is rich in phenolic and aliphatic hydroxyl groups. These inherent functionalities make lignin highly suitable for chemical modifications and functionalization, offering a compelling alternative to traditional thermoelectric materials.21,22 Lignin exhibits excellent biodegradability, biocompatibility, low toxicity, and environmental friendliness, making it an ideal candidate for hydrogel development. The utilization of lignin in hydrogel synthesis aligns well with a wide range of applications, from sustainable energy generation to biomedicine, where eco-friendly and functional materials are in increasing demand.23–27 A recent review reported the lignin-based hydrogels infiltrated with 6 M KOH electrolyte, which exhibited exceptional thermoelectric properties. These hydrogels demonstrated a high ionic conductivity of 226.5 mS cm−1, a remarkably low thermal conductivity of 0.29 W m−1 K−1, and an impressive ionic Seebeck coefficient of 13 mV K−1, resulting in a superior ionic figure of merit (iZT) of 3.75.28 Notably, in i-TE materials, the thermodiffusion effect can be categorized into p-type and n-type based on the differences in ion migration rates during thermal diffusion. When cations in the electrolyte migrate more quickly and accumulate at the cold end, a p-type thermal potential is observed.10 Recent research reported the p-type polarity in TcB9–2.5% lignin-derived infiltrated hydrogels with positive ionic Seebeck coefficients.29 Conversely, an n-type thermal potential occurs under opposite condition (negative S).30 Despite advancements in p-type i-TE materials, the development of comparable n-type lignin-based materials has lagged, hindering the realization of efficient thermoelectric devices. This scarcity of n-type materials restricts design strategies for TE modules, impeding overall progress in the field. Because functional TE devices require both p-type and n-type electrodes to form a complete circuit and generate electricity effectively. Therefore, prioritizing research efforts towards the development of high-performance n-type i-TE materials is critical to overcome this bottleneck and unlock the full potential of TE technology.31
In recent years, previous studies have reported several examples of n-type i-TE materials, such as alkylmethylimidazolium chloride (Rmim:Cl)/PVA hydrogels using poly(3,3-ethylenedioxythiophene):poly(styrenesulfonyl) (PEDOT:PSS) electrodes, exhibiting a Seebeck coefficient of approximately −1 mV K−1. Other promising candidates include Hmim:Cl/PEDOT:PSS (P.F of 0.189 μW m−1 K−2) and Dmim:Cl/PEDOT:PSS (P.F of 0.194 μW m−1 K−2). However, these materials often face limitations in practical applications due to their low thermoelectric performance.32 Additionally, Xu et al.33 designed a gelatin-based hydrogel, exhibiting a low value of S (−2.89 mV K−1) as well as σi (2.1 mS cm−1). Regarding n-type ionogels, a poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP) based i-TE material was recently demonstrated.34 Although this material yielded stretchable and self-healing ionogels with a negative S of −11.5 mV K−1 at 50% relative humidity, their σi was as low as 3 × 10−2 mS cm−1.35 Additionally, a composite hydrogel based on polyacrylic acid (PAA) and bacterial cellulose (BC), utilizing the K3[Fe(CN)6]/K4[Fe(CN)6] redox couple, with a power factor (PF) of 1.83 μW m−1 K−2.31 However, further exploration is needed to ensure long-term stability of this system. In contrast, several n-type organic TE candidates exhibit degradation in everyday environments while other inorganic and carbon-based n-type materials have inherently high thermal conductivity.36 However, a major obstacle hindering the widespread adoption of these materials lies in their limited stability under ambient conditions, hindering their overall performance.30 A recent study by Liu et al.37 demonstrated a promising n-type TE material based on a tetramethylene sulfone Fe2+/Fe3+ hydrogel. However, its practical application is hampered by the potential health risks associated with the strong toxicity of tetramethylene sulfone. These limitations highlight the critical need for novel n-type i-TE materials with desirable properties like leak-free, non-toxic, and capable of achieving high iZT values.31
This study presents a novel approach to developing sustainable and efficient p-type i-TE materials through the chemical cross-linking of lignin-based hydrogel with PVA. The p-type nature of the cross-linked hydrogel was strategically inverted to n-type through an amination reaction using 2-chloroethylamine hydrochloride as a functionalizing reagent. Polymers with ionic side chains were utilized as a medium for mobile, bulky counter ions. In this medium, a single type of ion (either cation or anion) diffuses selectively under a temperature gradient. These functionalized hydrogels were subsequently infiltrated with a 1 M KCl electrolyte to significantly enhance their performance, leading to a negative Seebeck coefficient (−7.48 mV K−1), superior ionic conductivity (39.9 mS cm−1), and a remarkable power factor of 223.52 μW m−1 K−2. This work demonstrates the transformation of lignin into high-performance i-TE materials through a water-based, solvent-free functionalization process. This approach aligns with green chemistry principles by valorizing waste, eliminating hazardous reagents, and promoting sustainable energy via waste heat recovery. Furthermore, these materials present a compelling avenue for developing energy storage devices due to its low thermal conductivity, a crucial factor in minimizing energy losses. These findings highlight the potential of functionalized hydrogels as promising n-type i-TE materials, offering a sustainable and environmentally friendly alternative for applications in temperature sensing and low-grade thermal energy harvesting. Future research endeavors could further explore the versatility and potential of these ionic conductive hydrogels in various energy-related fields.
2. Experimental
2.1. Materials
Kraft lignin with Mw of 3715 g mol−1 (Đ ≈ 4), was supplied by ENCE Energía y Celulosa SA (Spain). Sodium hydroxide (NaOH) pellets having 98% purity, epichlorohydrin (ECH) with 99% purity, and poly(vinyl alcohol) (PVA) having 99% hydrolyzed purity of Mw = 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–124000 g mol−1, 2-chloroethylamine, hydrochloride with 99% purity were purchased from Sigma-Aldrich (Spain). Potassium chloride (KCl, ≥99%), sodium chloride (NaCl, ≥99%), calcium chloride (CaCl2, granular, ≥93.0%), and iron(III) chloride (FeCl3, ≥99%) were purchased from Sigma-Aldrich. Sulfuric acid (H2SO4, 95–98%), acetic acid glacial (CH3COOH, ≥99.8%), sodium acetate trihydrate (CH3COONa·3H2O, ≥99.5%), sodium dihydrogen phosphate (NaH2PO4, ≥99%), di-sodium hydrogen phosphate dihydrate (Na2HPO4·2H2O, ≥99.5%), were purchased from Scharlau.
000–124000 g mol−1, 2-chloroethylamine, hydrochloride with 99% purity were purchased from Sigma-Aldrich (Spain). Potassium chloride (KCl, ≥99%), sodium chloride (NaCl, ≥99%), calcium chloride (CaCl2, granular, ≥93.0%), and iron(III) chloride (FeCl3, ≥99%) were purchased from Sigma-Aldrich. Sulfuric acid (H2SO4, 95–98%), acetic acid glacial (CH3COOH, ≥99.8%), sodium acetate trihydrate (CH3COONa·3H2O, ≥99.5%), sodium dihydrogen phosphate (NaH2PO4, ≥99%), di-sodium hydrogen phosphate dihydrate (Na2HPO4·2H2O, ≥99.5%), were purchased from Scharlau.
2.2. Synthesis of cross-linked hydrogels (CLHs)
Fig. 1a depicts the schematic representation of the method used to synthesize the cross-linked hydrogel, designated as CLH hydrogel. Firstly, a homogenized PVA solution was prepared by adding 0.8 g of PVA into 5 mL of deionized water under constant stirring at room temperature. The mixture was then vigorously stirred at 90 °C under reflux for 30 min to achieve complete PVA dissolution and cooled down at room temperature. The cooled PVA solution was ultrasonicated for 5 min to eliminate air bubbles. Subsequently, 5 mL of 2.5 M NaOH solution was incorporated into the PVA solution while continuous stirring. This was followed by the gradual addition of 0.9 g of lignin. The entire mixture was magnetically stirred at room temperature for 5 h, resulting in a homogenous lignin/PVA mixture. A specific volume (5%, v/v) of the epichlorohydrin (ECH) cross-linker was introduced into the mixture. Vigorous stirring for 30 min ensured a uniform blend. The well-defined blend was carefully poured into polytetrafluoroethylene molds and left undisturbed at room temperature overnight. This allowed for complete cross-linking, resulting in the formation of cross-linked lignin-based hydrogels. | ||
| Fig. 1 (a) Schematic representation for the synthesis of cross-linked hydrogels (CLHs), (b) molecular interactions of lignin with the cross-linking agent, (c) green synthesis approach for fabricating functionalized hydrogel (FLH). | ||
2.3. Functionalization of lignin-based hydrogel (FLH)
Before functionalization (amination), CLHs were thoroughly rinsed with deionized water to remove any unreacted lignin. The rinsed hydrogels underwent functionalization to introduce amine groups. This involved adding 20 ml of 2.5 M NaOH solution to a 100 ml round-bottom flask containing the hydrogels and an excess of 2-chloroethylamine, hydrochloride (functionalized reagent). The reaction mixture was heated at 80 °C under a reflux condenser with magnetic stirring for 72 h. Following the reaction, the functionalized lignin-based hydrogel (abbreviated as FLH) was thoroughly rinsed multiple times with deionized water to remove any unreacted functionalizing reagents. The resulting hydrogel was then subjected to various characterization techniques for further investigation.2.4. Characterization
Freeze-dried cross-linked and functionalized hydrogels were investigated for functional group analysis using FT-IR spectroscopy. Measurements were taken in transmittance mode on an Agilent Cary 630 FT-IR spectrophotometer (Agilent Technologies) across a wavenumber range of 500 to 4000 cm−1 with 32 scanning times. To gain deeper insight into the chemical structure of the FLH, we utilized X-ray photoelectron spectroscopy (XPS). The XPS analysis was performed using a K-alpha X-ray photoelectron spectrometer (Thermo Scientific) equipped with a monochromatized Al K-α ray (hv = 1486.6 eV) at 4 × 10−9 mbar. Surface morphology study was performed using a scanning electron microscope SCIOS 2 (field emission electron microscope, SCIOS 2 FIB-SEM). Prior to analysis, the samples were sputter-coated with a thin layer of gold to enhance their conductivity for SEM imaging. An accelerating voltage of 20 kV was used during SEM observation. The thermal behavior of the hydrogel samples (5–10 mg) was examined using two complementary techniques: differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). For the DSC analysis (TA Instruments DSA Q20 instrument, USA), the sample was heated from 0 to 220 °C at a rate of 10 °C min−1 under a nitrogen purge gas flow. Similarly, during TGA analysis (TA Instruments TGA 550 instrument, USA), the sample was subjected to a temperature program ranging from room temperature to 800 °C at 10 °C min−1 under a nitrogen flow rate of 10 mL min−1.The swelling behavior of the hydrogel samples was evaluated by immersing them in deionized water (DI), acetate buffer, and phosphate buffer at room temperature. Prior to testing, the hydrogels were dried in a vacuum oven overnight at 60 °C to remove any residual moisture. The dry weight of hydrogel (Wd) was then carefully measured using a weighing scale. The hydrogels were subsequently placed in the water bath, and their weight (Ws) was monitored periodically over 72 h. To ensure accurate weight measurements, any surface water adhering to the hydrogels was gently removed using blotting paper before each weighing. The percentage swelling (%swelling) of the hydrogels was calculated using the following eqn (1):
 | (1) |
2.5. Ionic thermoelectric measurements
Prior to ionic thermoelectric characterization, hydrogels were encapsulated by sandwiching them between two stainless steel plates and sealing the assembly with epoxy to ensure good thermal and electrical contact. The Seebeck coefficient (S) was determined using a Laboratory-designed setup comprising two Peltier cells (heating/cooling), two K-type thermocouples (monitoring the temperature gradient across the sample), and a Keithley 2400 voltmeter (recording the induced voltage difference). The hydrogels were precisely placed within the apparatus, ensuring each end made firm contact with a Peltier cell, a thermocouple, and a voltage probe. The Seebeck coefficient (S) for each hydrogel was calculated using the following eqn (2): | (2) |
 | (3) |
For the thermal conductivity measurements, a foil heat flux sensor (FHF05-50X50-02) monitored the rate of heat flux through the hydrogels by detecting voltage changes and recorded with an Agilent 344420A digital multimeter. While the heat flux sensor measures temperature, a separate setup was used to establish a controlled temperature gradient across the sample. The hot side of the setup was maintained at a constant 308 K (simulating body temperature) using an Omega model CN7500 temperature controller connected to a Peltier TEC1-12706 element and a Pt100 temperature sensor. An additional Peltier cooler (ATS-TEC30-36-017) was used on the cold side to simulate ambient temperature. The heat flux (Q) was calculated using the specific eqn (4) developed by Hukseflux for the particular sensor model:38
 | (4) |
 | (5) |
The efficiency of i-TEs is evaluated using a dimensionless parameter called ionic figure of merit (iZT), as expressed in eqn (6):
 | (6) |
3. Results and discussion
ECH is introduced as a cross-linking agent between PVA and lignin, forming a stable network within the hydrogel producing robust samples facilitating their handling and encapsulation. In the reaction mixture, the addition of NaOH plays a crucial role through the deprotonation of lignin, which increases its reactivity with both PVA and ECH. This enhanced reactivity facilitates the formation of chemical cross-linking. As illustrated in Fig. 1b, the cross-linking reaction involves the creation of ether bonds between hydroxyl groups on PVA or lignin and the epoxy group present in ECH. During the cross-linking reaction, HCl molecules are removed simultaneously from the opposite side of ECH.29 The CLH undergoes further functionalization with amine groups to broaden their range of utilization and tailor lignin for several applications such as wearable electronics, batteries, biosensors, and energy conversion devices. Jiansong Chen et al.39 reported a novel approach for lignin amination. This approach utilizes a green synthesis method composed solely of water–NaOH, eliminating the need for organic solvents. Due to the presence of abundant hydroxyl groups, the CLH undergoes a substitution reaction with 2-CEH for 72 h (Fig. 1c). The amination of CLH with 2-CEH was highly affected by the concentration of 2-CEH and the optimized reaction condition. Building upon these fundamental principles of lignin animation, the functionalization process of lignin-based hydrogels involves heating the reaction mixture at 80 °C under reflux for 72 hours. Reflux maintains a constant temperature and efficient heat distribution, reducing the need for repeated heating and thus optimizing energy consumption. While specific energy usage can vary based on reactor design and scale, similar lignin modification processes have reported energy consumptions ranging from 1.2 to 2.5 kWh per kilogram of product, which is relatively low compared to conventional petrochemical-based polymer syntheses.40 The synthesis process includes rinsing the hydrogels with deionized water to remove any unreacted lignin and functionalizing reagents. Utilizing deionized water is environmentally favorable as it avoids introducing additional contaminants into the waste stream. Moreover, the use of 2-chloroethylamine hydrochloride as a functionalizing reagent is considered sustainable, as it can be handled safely and efficiently in aqueous solutions, minimizing the need for hazardous organic solvents. Regarding waste emissions, the overall process exhibits a low environmental footprint. The E-factor (mass ratio of waste to desired product) for lignin-based hydrogel synthesis has been reported to range between 0.5 and 1.5, signifying minimal waste generation compared to many conventional synthetic polymer systems.40 Aminated lignin-based hydrogels also offer promising reuse possibilities. The functionalized hydrogels can be applied in various domains, notably in water purification, where aminated lignin has enhanced adsorption capabilities for heavy metals. Notably, these hydrogels can maintain over 90% adsorption efficiency after multiple reuse cycles, underlining their recyclability and supporting a circular economy by extending material lifespan and reducing reliance on virgin resources.41
Fig. 2a presents the FTIR spectra of CLH and FLH hydrogel. These spectra offer valuable insights into the chemical structure of the cross-linked network and the presence of functional groups. The broad peak observed around 3550–3200 cm−1 is attributed to the O–H stretching vibrations of bonded hydroxyl groups.42 The spectral peak at 3150–3000 cm−1 is assigned to the –C–H stretching associated with the aromatic structure, while 3000–2840 cm−1 is attributed to the aliphatic –CH2–CH3 stretching modes.29 CLH exhibits characteristic peaks corresponding to the aromatic skeleton vibrations of lignin at 1584 cm−1 and 1455 cm−1, indicative of C–H bending. The band at 834 cm−1 is attributed to the bending vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C. A slight shift in the 1584 cm−1 peak suggests successful cross-linking between lignin and PVA.43 Additionally, a characteristic peak for hydrogels at 1082 cm−1 indicates the presence of alcoholic/phenolic C–O stretching vibrations.29
C. A slight shift in the 1584 cm−1 peak suggests successful cross-linking between lignin and PVA.43 Additionally, a characteristic peak for hydrogels at 1082 cm−1 indicates the presence of alcoholic/phenolic C–O stretching vibrations.29
 | ||
| Fig. 2 (a) FTIR spectra of cross-linked and functionalized hydrogel, (b) zoom plot to show the appearance of amine bend. | ||
In comparison to CLH, the FLH spectrum exhibits a broadening of the peaks between 3400 and 3200 cm−1, attributed to the presence of an amine (–NH2) stretching band at approximately 3300 cm−1. This peak broadening is attributed to the overlap between the amine (–NH2) stretching band and the hydroxyl (–OH) group absorption band (in the wavenumber range of 3200–3500 cm−1).44 Furthermore, the substitution reaction between CEA and the hydroxyl groups of lignin leads to a significant decrease in the intensity of the –OH peak. A significant increase in the intensity of peaks observed around 1031 cm−1, attributed to the formation of C–O–C ether linkages during the reaction.39 The double peaks at 3300–3400 cm−1 indicate the presence of primary amine groups in the aminated lignin molecule. The enhanced band at 1085 cm−1 corresponds to C–N and CO bonds.45 Similarly, the intensity of peaks at 2920 cm−1 increased, suggesting successful C–H grafting by CEH. Furthermore, the presence of an acylamino group, indicative of amine incorporation, was confirmed by the appearance of a new adsorption peak at 1645 cm−1.39,46 These observations confirm the successful amination of FLH, as evidenced by the introduction of amine groups into the modified lignin structure (Fig. 2b).
The chemical composition of the synthesized aminated lignin was analyzed using X-ray photoelectron spectroscopy (XPS). High-resolution XPS spectra were acquired for hydrogel before and after functionalization (amination) to assess the changes in elemental composition.45 This technique provides a detailed elemental fingerprint, identifying the elements present and their chemical states on the hydrogel surface. As both hydrogels are lignin-based, the XPS spectra are dominated by peaks corresponding to carbon (C 1s) and oxygen (O 1s). These peaks offer valuable information regarding the specific carbon bonding environments within the lignin structure, including C–C aromatic bonds, C–O bonds in alcohols or ethers, and potentially some carbonyl groups (CO). Fig. 3a–c revealed strong carbon (C 1s) and oxygen (O 1s) peaks in CLH, along with a minor nitrogen (N 1s) peak likely attributable to inherent lignin impurities (whose content varies with source and purification method). High-resolution C 1s spectra provided further insights. CLH exhibited four deconvoluted peaks at around 288.92, 287.19, 286.11, and 284.77 eV, corresponding to C–O–C, CO, C–O, and C–C bonds in the lignin structure, respectively.39 Amine functionalization resulted in key spectral differences (shown in Fig. 3d). FLH displayed a distinct N 1s peak at 400.61 eV, accompanied by a significant increase in nitrogen content 1.22% (compared to 0.27% for CLH) as illustrated by Fig. 3f. This rise confirms successful attachment of amino groups to the lignin network. Interestingly, the C 1s spectra of the FLH displayed similar peaks at slightly shifted positions (around 288.78 eV, 287.06 eV, 286.08 eV, and 284.8 eV), reflecting the preserved lignin structure (Fig. 3e). However, a crucial difference lies in peak intensity at 286.08 eV.45 This peak corresponds to C–N bonds formed during amination, indicating a significant increase in nitrogen content within the FLH as a result of C–N peak that partially overlaps with the C–O peak.39,47 The distinct N 1s peak observed in the XPS spectra of FLH confirms the successful attachment of amine groups to the lignin-based network (Fig. 3f).48
 | ||
| Fig. 3 XPS scan spectra for (a–c) cross-linked hydrogel, and (d–f) functionalized hydrogel. | ||
SEM demonstrates the morphological differences between the cross-linked and functionalized hydrogels. Fig. 4a presents SEM images of CLH, revealing a network-like structure formed by lignin (a biopolymer) within the hydrogel, characterized by interconnected pores.29 The CLH exhibited a porous network with an estimated porosity of 50–65%, and pore sizes ranging from 20 to 110 μm, with a more balanced size distribution. Functionalization introduces new amine groups onto the lignin network, significantly altering morphology as illustrated in Fig. 4b. SEM images of the FLH revealed a smoother surface compared to the CLH. Additionally, the porosity, or interconnected spaces within the network, appeared different. The quantitative metrics also demonstrate that hydrogel structure became more open, with an estimated increase in porosity to 70–85%. Furthermore, the pore size distribution shifted towards larger dimensions, with many pores in the range of 80–200 μm or greater. The significant increase in pores exceeding 100 μm was observed post-functionalization. The enhanced porosity suggests successful surface modification.49 The highly porous structure of FLH provides organized pathways for ion migration during thermal–electrical measurements. This is attributed to the highly ordered arrangement of polymer molecular chains within the FLH, facilitating ion migration and enhancing its conductivity. It is important to mention that observed morphology in SEM images results from the freeze-drying process required for SEM sample preparation. In their actual application, the hydrogels are used in a hydrated state, where their hydrated structure is more uniform. All thermoelectric testing was conducted on hydrated samples, ensuring that the observed SEM morphology does not impact the functional properties of the hydrogels.
 | ||
| Fig. 4 SEM images of (a) the cross-linked and (b) functionalized hydrogel. | ||
The thermal stability of lignin derivatives is crucial for their application in heat harvesting. To gain a better understanding of how amination affects the thermal stability of lignin derivatives, thermal gravimetric analysis (TGA) was conducted on hydrogels before and after functionalization. As illustrated in Fig. 5a, the decomposition process occurred over a wide temperature range, reflecting the complex nature of lignin derivatives. A minimal weight loss was observed for both the cross-linked and functionalized hydrogels in the temperature range of 50–195 °C, which can be attributed to the evaporation of physically adsorbed moisture.45 Both hydrogels remained relatively stable up to 195 °C, with an overall weight loss of less than 10%. The CLH exhibited significant thermal decomposition within a temperature range of 200 to 460 °C, characterized by the cleavage of C–C bonds between lignin units and the breakdown of aliphatic side chains attached to aromatic rings. This broad range of thermal decomposition is primarily attributed to the presence of PVA polymeric templates, which are susceptible to thermal degradation at elevated temperatures. The major thermal degradation of PVA commences with the elimination of side chains around 342 °C, followed by the degradation of the main chain at approximately 460 °C in the CLH. Additionally, the significant weight loss is associated to the thermal cleavage of oxygen-containing groups and carbon–carbon linkages within lignin at higher temperatures (320–370 °C).50,51 After amination, FLH exhibited a distinct decomposition pattern. Compared to the CLH, the maximum decomposition temperature of the FLH slightly increased, occurring around 500 °C. The final residual mass at 777 °C was higher in the FLH (2.56%) compared to the CLH (0.75%).48,52
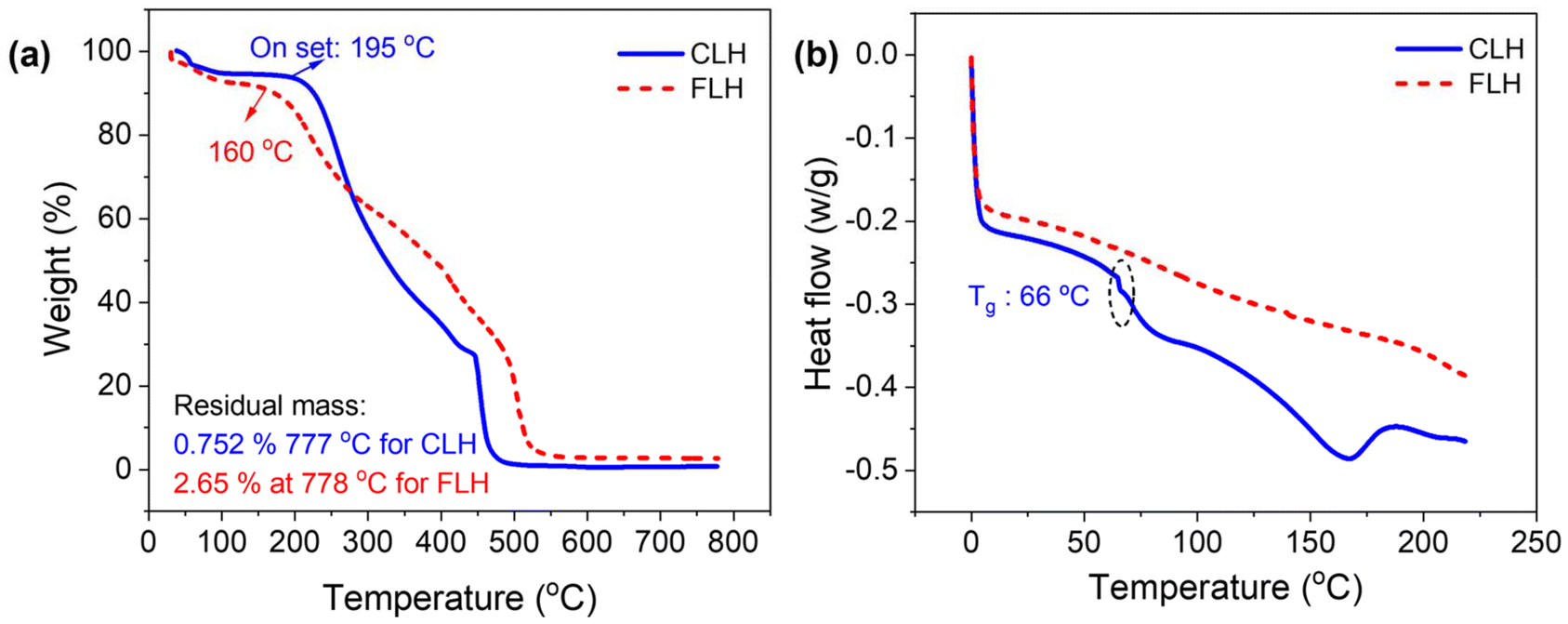 | ||
| Fig. 5 (a) TGA isotherm, and (b) DSC isotherm of both cross-linked and functionalized hydrogel. | ||
The possible thermal transitions of the hydrogels were further investigated using Differential Scanning Calorimetry (DSC). As shown in Fig. 5b, the DSC thermogram revealed a lower glass transition temperature (Tg) for CLH hydrogel at approximately 66 °C compared to pure PVA (75–85 °C).53 Because the presence of lignin disrupts the regular packing of PVA chains and introduces additional free volume due to its long side chains within the hydrogel matrix. Consequently, low Tg caused the vibrant segmental motion which facilitates the ion moieties within the matrix.54 After functionalization, a significant change in phase behavior was observed, attributed to the incorporation of amine groups (–NH2) into the hydrogel matrix, which disrupted the uniformity of the cross-linked network. FLH hydrogel demonstrated additional hydrogen bonding and electrostatic interactions between the functional groups and the polymer chains. Consequently, the thermal transition of FLH appeared less defined compared to CLH, indicating that the presence of amine groups significantly influenced the mobility and thermal stability of the hydrogel network.
The swelling capacity of hydrogel samples was examined at room temperature in deionized water (DI) (1009% for CLH and 213% for FLH), phosphate buffer (550% for CLH), acetate buffer (311% for FLH and 225% for CLH) and the results are plotted in Fig. 6. CLH represent the maximum swelling capacity of 1009% (DI at pH 7.0) attributed to the presence of hydrophilic hydroxyl groups within the lignin structure that readily form hydrogen bonds with water molecules.51 In contrast, the swelling capacity in phosphate buffer at pH 7.6 is lower (550%) due to the presence of ions in the buffer. The phosphate ions in the solution can interact with the hydroxyl groups and other charged sites within the hydrogel through ionic interactions and electrostatic shielding. This reduces the availability of hydrogen bonding between the hydrogel and water molecules, limiting water absorption and decreasing the swelling capacity. Furthermore, at a pH of 7.6, ionization of acidic groups (such as carboxyl or phenolic groups) in the hydrogel structure may be reduced, further influencing the electrostatic interactions and causing the hydrogel to swell less in the buffer compared to distilled water. In acidic media (pH 3.8), the CLH hydrogel exhibits a significantly reduced swelling capacity of 225%. This reduction occurs due to the lack of ionization, which eliminates the repulsive forces between acidic groups. Consequently, the polymer chains pack more closely together, limiting the available space for water molecules and resulting in decreased swelling.55 However, FLH demonstrated a huge reduction in swelling capacity 213% due to the introduction of amine group (–NH2) into hydrogel network as compared to CLH (DI). This reduction is associated with the hydrophobicity of amine groups which reduces water imbibition in the hydrogel network and results in lower swelling capacities.49 The swelling capacity of FLH increased from 213% (in distilled water) to 311% in acetate buffer (pH 3.8), due to the behavior of the introduced amine groups (–NH2), which become ionizable and positively charged under acidic conditions.52 At pH 3.8, which is below the pKa of most amine groups, the amine groups become protonated, forming –NH3+. This protonation introduces positive charges into the hydrogel network, leading to electrostatic repulsion between the positively charged amine groups. This repulsion causes the hydrogel matrix to expand and take up more water, resulting in a higher swelling capacity. The observed swelling capacity of the FLH in acetate buffer (pH 3.8) demonstrates its potential for enhanced electrolyte infiltration, which is crucial for optimizing ionic thermoelectric performance. The increased swelling can create more accessible pathways for ion diffusion and selective ion transport, which are key factors in designing efficient ionic thermoelectric materials.
 | ||
| Fig. 6 Swelling behavior of both cross-linked and functionalized hydrogel in different media. | ||
The ionic thermoelectric (i-TE) properties of hydrogels have been investigated to assess their potential as energy harvesting devices. When subjected to a temperature gradient (ΔT), mobile ions within the hydrogel undergo diffusion in a specific direction due to the Soret effect, resulting in the generation of a thermovoltage (ΔV). This ion migration is characterized by the selective thermo-diffusion of only single-type ions because the other charges remain attached to polymers as ionic side chains and immobilized within the matrix54 as illustrated in Fig. 7a. The efficiency of i-TE materials is primarily evaluated using the ionic Seebeck coefficient (S). The S is crucial in determining the overall performance of the i-TE system, which quantifies the amount of heat converted into electricity expressed as the dimensionless figure of merit (iZT).28 Therefore, similar to traditional thermoelectric generators based on electronic conductors, the ionic Seebeck coefficient derived from the slope of the eqn (7):
 | (7) |
 | ||
| Fig. 7 (a) Schematic representation of selective ion migration under thermodiffusion effect, (b) p-type material, (c) n-type material, (d) ionic Seebeck coefficient (S) of CLH, (e) functionalized hydrogel under different time intervals, (f) ionic Seebeck coefficient of FLH-72 h, (g) ionic conductivity, (h) thermal conductivity and (i) power factor of hydrogel. | ||
The sign of the S is fundamentally determined by charge carrier in the thermoelectric material that exhibits higher thermal mobility.56 Under isothermal conditions, both cations and anions are uniformly distributed throughout the i-TE material, leading to equal potentials at both electrodes (φh = φc). However, when a temperature gradient is applied, the ion with a faster thermal diffusion rate dominates the charge transport. If cations exhibit a higher thermal diffusion rate than anions, they are preferentially driven toward the cold electrode due to the temperature differential. This accumulation of cations at the cold electrode raises its potential relative to the hot electrode (φh < φc), ultimately resulting in a positive S (known as p-type i-TE material) as illustrated in Fig. 7b. Conversely, if anions possess a faster thermal migration rate, they become enriched at the cold electrode, leading to a negative S (n-type material shown in Fig. 7c).8 Thus, the sign of S directly reflects the dominant charge carrier and its thermal mobility within the i-TE material.
The i-TE properties of samples were measured under ambient conditions, and the results are presented below. The voltage verses temperature difference profile of the CLH hydrogel was presented in Fig. 7d and exhibited 1.61 mV K−1 (positive S), indicating that functional groups like carboxylic acids or hydroxyl groups within the hydrogel network, release protons (H+) under the thermal diffusion effect (Fig. S1†). For impedance measurements, specifically Nyquist plots (Fig. S2†), were obtained to evaluate the ionic resistance within the CLH network, and to measure their ionic conductivity (σi).29 A σi value was determined by the charge carrier, density of charge carriers, and average speed of carriers, which are affected by molecular weight.54 In CLH, protons serve as the primary mobile charge carriers, contributing to σi of 6.26 mS cm−1 and a positive S of 1.61 mV K−1, demonstrating the p-type ionic thermoelectric behavior. The power factor of 1.63 μW m−1 K−2 was described the performance of i-TE materials using the measured S and σi. The thermal conductivity (k) of CLH hydrogel was obtained using a hot disk method to evaluate iZT. The lower value of k (0.33 W m−1 K−1) obtained for CLH, attributed to the large free volume in the hydrogel matrix caused by the long ionic side chains.54
The functionalization of the CLH hydrogel involves introducing amine groups (–NH2) onto the hydrogel network by reacting 2-chloroethylamine hydrochloride in the presence of 2.5 M NaOH.39 This process can be explained by a mechanism where the terminating alcoholic or phenolic groups of the cross-linked framework (C–OH) are transformed into anionic alkoxide groups (C–O−) during the functionalization reaction. This transformation facilitates the nucleophilic substitution of chlorine atoms, resulting in covalent attachment of –NH2 group on the hydrogel surface through an oxygen–carbon bond.29,57 Following rinsing, the attached –NH2 group become protonated to form –NH3+ within the hydrogel matrix. These protonated amine group (–NH3+) are immobilized within the polymer network, resulting in the formation of bonded cations and free anions. The electrostatic interaction immobilized the positive charges within the hydrogel network due to protonation of –NH3+, which facilitates the diffusion of anions across the thermal gradient within the hydrogel.58 Under a temperature gradient, mobile anions migrate from the hot side to the cold side of the hydrogel. This migration generates a negative thermoelectric voltage across the hydrogel framework, resulting in a negative S.54 The anion migration induces n-type behavior in the FLH hydrogel, contrasting with p-type behavior driven by cation migration in CLH hydrogel. The reaction was monitored at 24, 48, and 72 h to determine the optimal level of amine group substitution during functionalization (illustrated in Fig. S1†). As shown in Fig. 7e, increasing reaction time allowed for greater amine group incorporation into the hydrogel network as investigated by increasing value of S such as −0.90, −5.70, and −7.87 mV K−1 for 24, 48, and 72 h respectively. The results indicate that a 72 h reaction time is optimal for achieving the highest degree of amine group substitution, leading to the superior negative S for n-type i-TE materials (as presented in Fig. 7f).
FLH hydrogel exhibited σi values of 7.93, 10.57, and 18.58 mS cm−1 after 24, 48, and 72 h of functionalization, respectively. As the reaction time increases, the ionic resistance decreases (σi increases) as a greater number of amine groups (–NH2) are substituted onto the hydrogel network, which provided an organized channel for ion migration transport.58 The higher composition of ionic side chains during functionalization enhances ion migration efficiency within the hydrogel, thereby increasing ionic conductivity by making it more conducive to ion transport54 as shown in Fig. 7g (Fig. S2†). This results in an exceptionally high ionic conductivity of 18.58 mS cm−1, as shown in Fig. 7h. FLH hydrogel also demonstrated thermal conductivity (k) values of 0.52, 0.43, and 0.41 after 24, 48, and 72 h of functionalization reaction, respectively. The results indicate that FLH hydrogel absorbs slightly more amount of water than CLH during rinsing, which marginally affects the high value of k for n-type hydrogel due to high k of water (0.6 W m−1 K−1).54 During functionalization, a greater number of amino groups are substituted within the hydrogel matrix. This progressive substitution leads to the formation of organized channels with free volume, resulting in a decrease k of the hydrogel over time. These parameters resulted in a power factor of 115.10 μW m−1 K−2 as illustrated in Fig. 7i. The enhanced n-type ionic thermoelectric performance is attributed to the increased presence of amine groups, resulting from the extended functionalization process. This higher degree of amine substitution likely contributes to both the elevated σi and the more pronounced negative S observed in FLH hydrogel, demonstrated superior n-type thermoelectric behavior.
Subsequently, FLH-72 h were infiltrated with various electrolytes such as 1 M NaCl, 1 M KCl, and 0.1 M H2SO4 to investigate the most suitable electrolyte based on negative S and σi, as shown in Fig. 8a and b. Upon electrolyte infiltration, the ionic thermophoretic mobility is directly influenced by the microstructure of the hydrogel matrix and its interactions with ions and the polymer network.58 Acidic electrolytes such as H2SO4, are unsuitable due to their acidic property and salting-out effects on the polymer matrix due to the excess of protons. In contrast, alkali metal ions like Na+ and K+ are preferred as neutral electrolytes because they are inexpensive, readily available, and environmentally friendly.59 The S and σi were utilized to evaluate the most suitable electrolyte as illustrated in Fig. 8a. The infiltrated hydrogels (FLH-72 h) containing KCl and NaCl electrolytes displayed negative S of −7.48 mV K−1 and −4.80 mV K−1, respectively. Furthermore, σi of KCl and NaCl electrolytes were measured at 39.9 mS cm−1 and 30.3 mS cm−1, respectively (Fig. S3†). Results shows that KCl-infiltrated hydrogel (1 M KCl/FLH-72 h) exhibited a higher negative S, representing a more pronounced effect on the hydrogel compared to NaCl (Fig. S4†). This enhancement is attributed to the ability of KCl electrolyte to disrupt hydrogen bonds between water molecules and the hydrogel polymer matrix, leading to stronger interactions between K+ ions and PVA macromolecules.60,61 Consequently, K+ ions become immobilized within the polar regions of PVA reducing their mobility and enhancing anion mobility.58,62 In contrast, Na+ cations demonstrate weaker interactions with the polymer matrix, which is already engaged in hydrogen bonding with water molecules. This results in reduced retention of Na+ ions within the hydrogel structure.60 However, dominant role of Cl− ions (anions) in generating negative thermoelectric voltage is attributed to their efficient migration from the hot side to the cold side under a thermal gradient, demonstrating a Soret effect, as illustrated in Fig. 8c. This mechanism contributes to the increased negative S in KCl-infiltrated hydrogels, a crucial factor in developing n-type thermoelectric materials.
 | ||
| Fig. 8 (a and b) Effect of n-type i-TE behavior in FLH-72 h hydrogel infiltrated with different electrolyte (c) schematic representation of Soret effect for selective ion-diffusion within hydrogel network. | ||
Following the optimization of reaction time and electrolyte selection, functionalized hydrogel (FLH-72 h) was further investigated based on electrolyte concentration. FLH-72 h hydrogel was infiltrated with KCl electrolyte solutions of varying molar concentrations (0.5 to 4 M) to evaluate their n-type i-TE properties. Fig. 9a–d illustrates the ionic Seebeck coefficient, ionic conductivity, thermal conductivity, power factor, and figure of merit for the FLH-72 h hydrogels subjected to a thermal gradient. A positive correlation was observed between increasing electrolyte concentration and the magnitude of both σi and negative S for 0.5 and 1 M. The maximum negative thermal voltage of −7.48 mV K−1, was achieved with a 1 M KCl concentration (Fig. S5†). The enhancement in thermal voltage can be attributed to increased ionic selectivity within the hydrogel framework, a consequence of the elevated electrolyte concentration.63 In 1 M KCl infiltrated hydrogel, the K+ ions are immobilized inside the polymer chains through the interaction of the hydroxyl group of the PVA and the carboxyl group of lignin, as a result the chloride ion (Cl−) remains the dominant carrier influencing the negative thermal voltage. When temperature gradient is introduced, Cl− ions migrate from the hot end to the cold end by thermal diffusion effect, establishing a difference in ionic concentration between the two ends of the hydrogel, which ultimately generates a negative S.58 It is important to report that negative thermal voltage exhibited a significant increase with negative S from −4.85 mV K−1 to −7.48 mV K−1 as the KCl electrolyte concentration was raised 0.5 M to 1 M, respectively as illustrated in Fig. 9a. However, at concentrations exceeding 2 M, the negative thermovoltage showed no substantial negative S (as illustrated in Fig S5†). Therefore, it can be explained that at the highest concentration, all the available binding sites on PVA for K+ ions may become saturated, affecting the further association of K+ ions into hydrogel matrix via electrostatic interaction. As a result, the incoming K+ ions participate in ion mobility under the thermal gradient rather than being immobilized inside the polymer chains. However, higher KCl concentrations (2, 3, and 4 M), do not necessarily increase the negative thermopower since potassium ions (K+) become more influential in the hydrogel network towards the i-TE behavior. Under a thermal gradient, free K+ ions tend to migrate towards the negative electrode. While Cl− ions continue to migrate from the hot to the cold region, the increasing influence of K+ ions moving in the opposite direction begins to affect the overall ionic thermopower. This countermovement contributes to a positive S, despite the presence of some negative thermal voltage generating from Cl− migration (as shown in Fig. S5†). Similarly, the ionic conductivity of the FLH-72 h hydrogel infiltrated with KCl electrolyte increases consistently with electrolyte concentration up to 2 M before reaching the saturation point (Fig. S6†). Remarkably, the measured values of σi were 23.7, 39.9, 85.1, 71.6, and 41.3 mS cm−1 for 0.5 M, 1 M, 2 M, 3 M, and 4 M KCl concentrations, respectively as shown in Fig. 9b. Notably, in more concentrated solutions such as 3 and 4 M KCl, the temperature-driven diffusion of ions becomes less efficient due to ion crowding and pairing. This could explain by ionic paring formation and other effects since electrolyte becomes more viscous and selective ion thermodiffusion may be hindered, effectively reducing the number of free ions available for ion conduction, and decreasing σi within the hydrogel framework.34 As a result, the n-type functionalized hydrogel infiltrated with 0.5 and 1 M KCl electrolyte exhibited an exceptionally high-power factor of 55.88 and 223.52 μW m−1 K−2, respectively (illustrated in Fig. 9d) surpassing the performance of most existing state-of-the-art n-type ionic thermoelectric materials.
 | ||
| Fig. 9 Effect of electrolyte concentration in terms of (a) Seebeck coefficient, (b) ionic conductivity, (c) thermal conductivity, and (d) power factor and figure of merit of functionalized hydrogel FLH-72 h. | ||
The thermal conductivity of infiltrated FLH-72 h hydrogel was also investigated as a function of electrolyte concentration, as illustrated in Fig. 9c. The observed thermal conductivity values ranged from 0.25 to 0.43 W m−1 K−1. The FLH-72 h hydrogel infiltrated with 0.5 M KCl electrolyte exhibited the highest thermal conductivity of 0.43 W m−1 K−1, while the lowest value of 0.25 W m−1 K−1 was recorded for the hydrogel containing 4 M electrolyte. The observed thermal conductivity values were comparable to those of the precursor materials, including pure PVA (0.2–0.3 W m−1 K−1)53 and other lignin-based materials (0.3–0.5 W m−1 K−1).29,64 Additionally, the low thermal conductivity of functionalized hydrogel, a key characteristic for enhancing the performance of ionic thermoelectric materials and provide a promising foundation for practical applications. In, addition, the sample FLH-72 h: 1 M KCl was evaluated as ionic thermoelectric capacitor (see Fig. S8†). The results from the Nyquist plot and the cyclic voltammetry confirms capacitive characteristics of the hydrogel.
It is important to mention that these results establish lignin as a pivotal, multifunctional component for advanced ionic thermoelectric devices, moving beyond its conventional application as a renewable filler. Its unique combination of ionizable groups, a rigid aromatic backbone, and chemical functionalization capabilities synergistically addresses critical challenges such as: (1) enabling selective ion transport for significantly enhanced Seebeck coefficients through specific ion interactions within its structure, (2) providing essential mechanical robustness while maintaining a highly porous network that facilitates efficient ion mobility due to its inherent rigidity and tailored processing, and (3) permitting eco-friendly and cost-effective processing through benign methods leveraging lignin's natural properties. These interconnected advantages, validated by our structural and thermoelectric characterization, position lignin-based TE materials as a promising sustainable alternative for advanced thermoelectric application.
As illustrated in Fig. 10a maximum ionic Seebeck coefficient of −7.84 mV K−1 is observed in the amine-lignin/PVA/KCl (FLH-72 h/1 M KCl) hydrogel, which is only below the PEO/LiTFSI/EmimBF4 ionogel among reported n-type i-TE materials.35 Despite their promising thermoelectric properties, these ionogels face limitations. The high crystallinity of PEO and its vulnerability to decomposition under high voltage result in low ionic conductivity, constraining their practical applications.65Fig. 10b presents a comparison of ionic conductivity and ionic Seebeck coefficient for various i-TE materials reported in the literature, based on thermodiffusive effects.8,31–33,35,54 The results demonstrate that amine-lignin/PVA/KCl hydrogel exhibits significantly superior overall thermoelectric performance compared to previously reported i-TE materials. The power factor (PF) of the functionalized hydrogel was determined, as shown in Fig. 9d. The results shows that 1 M KCl infiltrated FLH-72 h hydrogel achieved its highest PF of 223.52 μW m−1 K−2, resulting from its excellent σi of 39.9 mS cm−1 and a S of −7.48 mV K−1. The n-type FLH-72 h/1 M KCl hydrogel exhibit a synergistic combination of impressive power factor 223.52 μW m−1 K−2 and 0.145 ionic figure of merit (iZT) (Fig. 9d). These n-type i-TE materials represent a significant advancement in green chemistry and sustainable materials science. By utilizing water-based solvents for functionalization, we have developed a safer and more environmentally friendly approach compared to traditional organic solvent-based methods.66 This aligns with the principles of green chemistry by reducing the use of hazardous substances and minimizing environmental impact. Furthermore, our innovative preparation of n-type materials from biomass not only contributes to the recycling of environmental waste but also offers a non-toxic alternative to conventional thermoelectric material, many of which rely on scarce or toxic elements (as shown in Table 1). This work exemplifies a holistic green approach, encompassing sustainable sourcing, eco-friendly processing, and the potential for end-of-life recycling. These green chemistry principles are further exemplified by the promising applications of these functionalized/lignin-based n-type ionic thermoelectric materials in various low-grade heat energy harvesting domains. For instance, their integration into solar thermal devices, supercapacitors, textile electronics for body heat recovery, microelectronic processors, and biomedical equipment can contribute to a more sustainable energy landscape by harnessing waste heat and reducing reliance on fossil fuels. This research not only advances the field of ionic thermoelectricity but also demonstrates the feasibility of developing high-performance materials that are in harmony with environmental stewardship principles. By harnessing the inherent properties of lignin, an abundant and renewable resource, we are paving the way for a new generation of sustainable thermoelectric materials and contributing to the advancement of green chemistry.
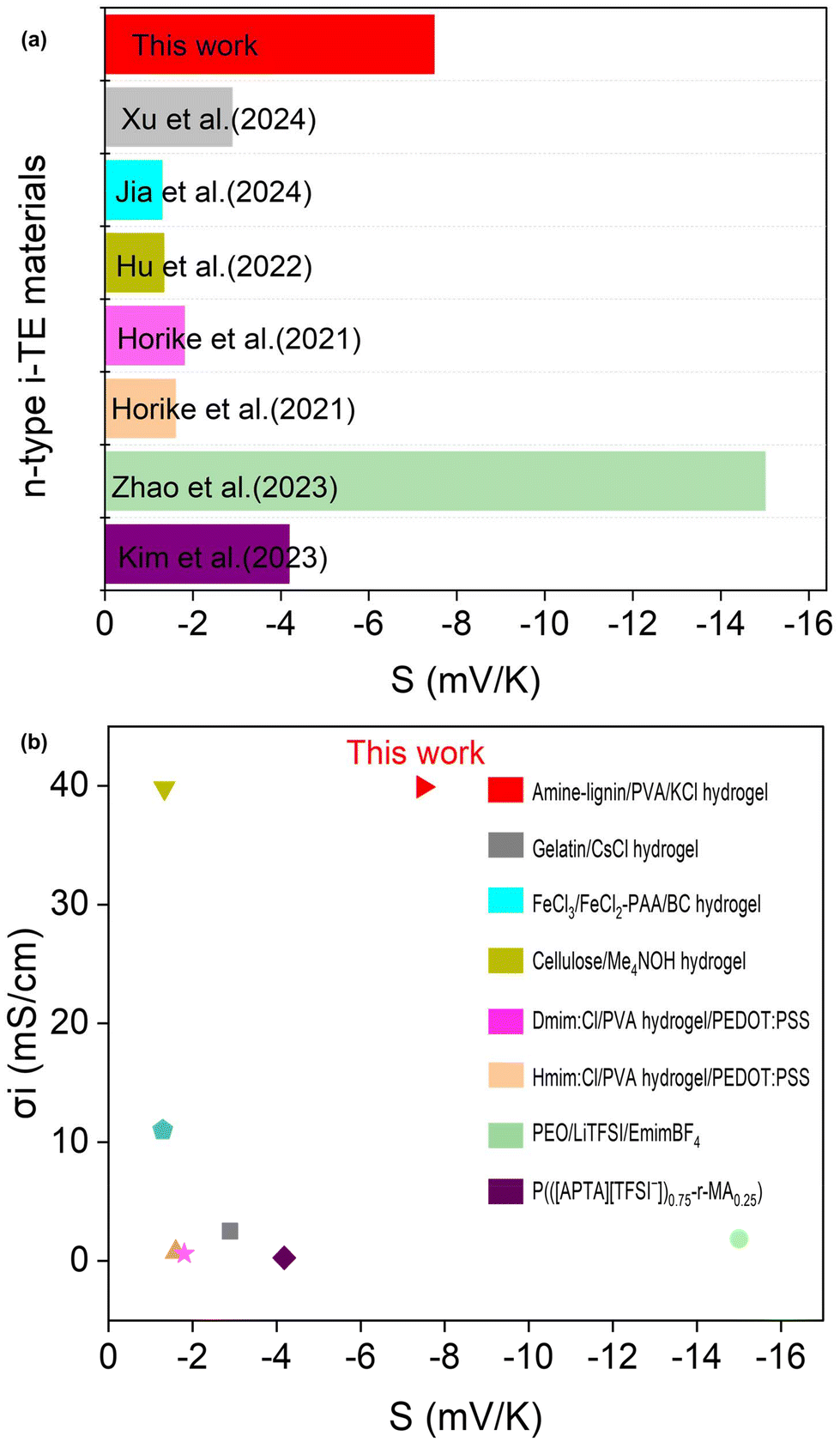 | ||
| Fig. 10 (a) Ionic Seebeck coefficient of different type of n-type i-TE materials, (b) comparison of ionic conductivity and ionic Seebeck coefficient between amine-lignin/PVA/KCl (FLH-72 h/1 M KCl) hydrogel and other reported n-type i-TE materials. | ||
| Material | Strategies used | Differences/limitation | Green chemistry aspects | Ref. |
|---|---|---|---|---|
| Conductive polymer films | PEDOT:PSS films doped with CuCl2, polymer film preparation, n-type material | Sensitive to humidity and temperature variations, moderate chemical stability | Not biodegradable, potential to contribute to microplastic pollution, often requires toxic dopants or solvents for processing | 30 |
| Carbon nanotube/PEDOT hybrids | Hybridizing carbon nanotubes with PEDOT and organic compounds, n-type material | Limited mechanical stability, complex synthesis, performance degradation under prolonged thermal cycling | Carbon nanotube synthesis is energy-intensive, potential for nanomaterial toxicity, non-biodegradable components | 67 |
| Poly(nickel-ethylenetetrathiolate) thin films | Electrochemical synthesis, flexible and high-performance thin films | Sensitive to moisture, potential degradation in harsh environments, use of nickel reduces green impact | Uses nickel, a heavy metal with environmental concerns, synthesis may involve toxic precursors or solvents | 68 |
| PEDOT:PSS ionic hydrogels | Ionic intercalation, anomalous polarity conversion, n-type TE materials | Limited thermal stability, sensitive to environmental changes like humidity | Non-biodegradable synthetic polymer base, potential leaching of ionic components | 32 |
| Layered TiS2 with organic intercalation | Organic intercalation into layered transition metal dichalcogenides, n-type TE materials | TiS2 synthesis involves hazardous chemicals, moderate thermal stability | Resource-intensive and potentially toxic synthesis of TiS2, non-renewable inorganic component | 69 |
| Gel-based thermocells (coordination enhanced) | Coordination-enhanced ion mobility, n-type material | Sensitivity to environmental conditions (e.g., drying or cracking of gels), moderate mechanical robustness | Gel-based materials highlight renewable and biodegradable options | 31 |
| Gelatin-based ionic thermoelectric supercapacitors | Hydrogel synergy with electrodes, n-type material | Gelatin has limited durability, issues with long-term thermal stability in extreme conditions | Use of gelatin aligns with biodegradable and renewable principles | 33 |
| Lignin-derived hydrogels | Sustainable synthesis, crosslinking strategies, efficient for p-type components | Derived from renewable biomass, recyclability of industrial waste, efficient for heat harvesting application | Use of renewable feedstocks, waste valorization, sustainable and biodegradable materials, waste heat recovery | 29 |
| Amine-lignin/PVA hydrogel | Crosslinking synthesis, functionalization strategies, efficient for n-type components | Derived from renewable biomass, recyclability of industrial waste, reliability and scalability of the n–p junctions | Use of renewable feedstocks, waste valorization, sustainable and biodegradable materials, waste heat recovery, eco-friendly material, water-based solvent system, eliminating hazardous reagents | This work |
4. Conclusion
This study investigates the innovative use of lignin-based hydrogels for harnessing low-grade thermal energy, offering a promising solution to address challenges in energy conservation and waste heat recovery. Here, we developed efficient p-type ionic thermoelectric (i-TE) materials through cross-linking and then subsequently converted them to n-type materials through functionalization. It is of great significance to develop high-performance n-type i-TE behavior by introducing amine group through a green synthesis approach. The optimized functionalized lignin-based hydrogel, infiltrated with a 1 M KCl electrolyte exhibited −7.48 mV K−1 negative Seebeck coefficient, enhanced ionic conductivity 39.9 mS cm−1 and lower value of thermal conductivity 0.41 W m−1 K−1. These characteristics resulted in a remarkable power factor of 223.52 μW m−1 K−2 and an ionic figure of merit (iZT) of 0.145, establishing this material as the most efficient sustainable n-type ionic thermoelectric. Our findings underscore the exceptional performance of functionalized lignin-based hydrogels in low-grade waste energy harvesting applications. This approach demonstrates significant potential for developing more efficient and practical thermoelectric devices for energy harvesting and power generation. Future research should focus on optimizing the performance of both n-type and p-type thermoelectric components and enhancing the reliability and scalability of the n–p junctions. In conclusion, this work represents a substantial advancement in sustainable ionic thermoelectric materials and paves the way for further innovations in waste heat recovery technologies.Author contributions
Nazish Jabeen: conceptualization, methodology, writing – original draft, visualization, software, formal analysis, writing – review & editing. Mario Culebras: conceptualization, supervision, investigation, visualization, writing – review & editing, funding acquisition, Rafael Muñoz Espí, Clara M. Gómez, Andrés Cantarero, & Maurice Collins: writing – review & editing.Conflicts of interest
The authors declare that there is no conflict of interest.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This research was supported through the grant PID2021-124845OA-I00 funded by MCIN/AEI/10.13039/501100011033 and by the “European UnionNextGenerationEU/PRTR”. NJ and MC acknowledge the Santiago Grisolia program CIGRIS/2022/132 funded by the Generalitat Valenciana.References
- Z. Wu, B. Wang, J. Li, R. Wu, M. Jin, H. Zhao, S. Chen and H. Wang, Nano Lett., 2022, 22, 8152–8160 CrossRef CAS PubMed.
- X.-L. Shi, J. Zou and Z.-G. Chen, Chem. Rev., 2020, 120, 7399–7515 CrossRef CAS PubMed.
- N. Kim, S. Lienemann, I. Petsagkourakis, D. Alemu Mengistie, S. Kee, T. Ederth, V. Gueskine, P. Leclère, R. Lazzaroni and X. Crispin, Nat. Commun., 2020, 11, 1424 CrossRef CAS PubMed.
- G. Tan, L.-D. Zhao and M. G. Kanatzidis, Chem. Rev., 2016, 116, 12123–12149 CrossRef CAS PubMed.
- J. He and T. M. Tritt, Science, 2017, 357, eaak9997 CrossRef PubMed.
- M. Culebras, G. Ren, S. O'Connell, J. J. Vilatela and M. N. Collins, Adv. Sustainable Syst., 2020, 4, 2000147 CrossRef CAS.
- M. Culebras, Y.-y. Byun, J. Jang, T. K. Lee, D. Choi, A. Serafin, M. N. Collins, J. S. Cho and C. Cho, ACS Appl. Energy Mater., 2024, 7, 2351–2361 CrossRef CAS.
- Y. Hu, M. Chen, C. Qin, J. Zhang and A. Lu, Carbohydr. Polym., 2022, 292, 119650 CrossRef CAS PubMed.
- H. Cheng and J. Ouyang, J. Phys. Chem. Lett., 2022, 13, 10830–10842 CrossRef CAS PubMed.
- Z. Zhou, Y. Wan, J. Zi, G. Ye, T. Jin, X. Geng, W. Zhuang and P. Yang, Mater. Today Sustainability, 2023, 21, 100293 CrossRef.
- D.-H. Kim, Z. A. Akbar, Y. T. Malik, J.-W. Jeon and S.-Y. Jang, Nat. Commun., 2023, 14, 3246 CrossRef CAS PubMed.
- M. Fu, Z. Sun, X. Liu, Z. Huang, G. Luan, Y. Chen, J. Peng and K. Yue, Adv. Funct. Mater., 2023, 33, 2306086 CrossRef CAS.
- H. Cheng, Q. Le, Z. Liu, Q. Qian, Y. Zhao and J. Ouyang, J. Mater. Chem. C, 2022, 10, 433–450 RSC.
- N. Sarkar, G. Sahoo, A. Sahoo and B. R. Jali, Biodegradable Materials and Their Applications, 2022, pp. 29–54 Search PubMed.
- J. Chen, L. Zhang, Y. Tu, Q. Zhang, F. Peng, W. Zeng, M. Zhang and X. Tao, Nano Energy, 2021, 88, 106272 CrossRef CAS.
- Z. Dong, Q. Xiang, Z. Zhang, J. Tang, F. Li, K. Sun and S. Chen, CCS Chem., 2024, 6, 1–37 CrossRef.
- H. Zhou, R. Zhang, C. Yue, X. Wu, Q. Yan, H. Wang, H. Zhang and T. Ma, Chin. J. Catal., 2024, 59, 169–184 CrossRef CAS.
- B. Zheng, L. Chen, L. He, H. Wang, H. Li, H. Zhang and S. Yang, Ind. Crops Prod., 2024, 210, 118058 CrossRef CAS.
- D. R. Lobato-Peralta, E. Duque-Brito, H. I. Villafan-Vidales, A. Longoria, P. Sebastian, A. K. Cuentas-Gallegos, C. A. Arancibia-Bulnes and P. U. Okoye, J. Cleaner Prod., 2021, 293, 126123 CrossRef CAS.
- Y. Feng, J. Yu, C. Shao and R. Sun, Pap. Biomater., 2023, 8, 20 CrossRef CAS.
- S. Shah, Q. Xu, M. Ullah, S. S. Zahoor, G. Morales, J. Sun and D. Zhu, Addit. Manuf., 2023, 74, 103711 CAS.
- M. Norgren and H. Edlund, Curr. Opin. Colloid Interface Sci., 2014, 19, 409–416 CrossRef CAS.
- S. Chandna, S. K. Bhardwaj, S. Paul and J. Bhaumik, Lignin: Biosynthesis and Transformation for Industrial Applications, 2020, pp. 231–252 Search PubMed.
- D. Ciolacu and G. Cazacu, J. Nanosci. Nanotechnol., 2018, 18, 2811–2822 CrossRef CAS PubMed.
- Y. Meng, J. Lu, Y. Cheng, Q. Li and H. Wang, Int. J. Biol. Macromol., 2019, 135, 1006–1019 CrossRef CAS PubMed.
- A. K. Mondal, M. T. Uddin, S. Sujan, Z. Tang, D. Alemu, H. A. Begum, J. Li, F. Huang and Y. Ni, Int. J. Biol. Macromol., 2023, 245, 125580 CrossRef CAS PubMed.
- D. Rico-García, L. Ruiz-Rubio, L. Pérez-Alvarez, S. L. Hernández-Olmos, G. L. Guerrero-Ramírez and J. L. Vilas-Vilela, Polymers, 2020, 12, 81 CrossRef PubMed.
- N. Jabeen, M. Muhammad, N. Menéndez, M. A. Nasiri, C. Gomez, M. Collins, R. Munoz-Espi, A. Cantarero and M. Culebras, Chem. Sci., 2024, 15, 14122–14153 RSC.
- M. Muddasar, N. Menéndez, Á. Quero, M. A. Nasiri, A. Cantarero, J. García-Cañadas, C. M. Gómez, M. N. Collins and M. Culebras, Adv. Compos. Hybrid Mater., 2024, 7, 47 CrossRef CAS.
- B. Kim, J. U. Hwang and E. Kim, Energy Environ. Sci., 2020, 13, 859–867 RSC.
- Y. Jia, S. Zhang, J. Li, Z. Han, D. Zhang, X. Qu, S. Chen and H. Wang, J. Power Sources, 2024, 602, 234400 CrossRef CAS.
- S. Horike, Q. Wei, K. Kirihara, M. Mukaida, Y. Koshiba and K. Ishida, J. Mater. Chem. C, 2021, 9, 15813–15819 RSC.
- Z. Xu, S. Lin, Y. Yin and X. Gu, Chem. Eng. J., 2024, 493, 152734 CrossRef CAS.
- S. Liu, Y. Yang, S. Chen, J. Zheng, D. G. Lee, D. Li, J. Yang and B. Huang, Nano Energy, 2022, 100, 107542 CrossRef CAS.
- W. Zhao, Y. Zheng, M. Jiang, T. Sun, A. Huang, L. Wang, W. Jiang and Q. Zhang, Sci. Adv., 2023, 9, eadk2098 CrossRef CAS PubMed.
- C. J. An, Y. H. Kang, H. Song, Y. Jeong and S. Y. Cho, J. Mater. Chem. A, 2017, 5, 15631–15639 RSC.
- Y. Liu, Q. Zhang, G. O. Odunmbaku, Y. He, Y. Zheng, S. Chen, Y. Zhou, J. Li, M. Li and K. Sun, J. Mater. Chem. A, 2022, 10, 19690–19698 RSC.
- J. F. Serrano-Claumarchirant, M. A. Nasiri, C. Cho, A. Cantarero, M. Culebras and C. M. Gómez, Adv. Mater. Interfaces, 2023, 10, 2202105 CrossRef CAS.
- J. Chen, L. An, J. H. Bae, J. W. Heo, S. Y. Han and Y. S. Kim, Ind. Crops Prod., 2021, 173, 114102 CrossRef CAS.
- C. Libretti, L. S. Correa and M. A. Meier, Green Chem., 2024, 26, 4358–4386 RSC.
- J. Díaz, K. Roa, A. Boulett, L. Azócar and J. Sánchez, Int. J. Biol. Macromol., 2025, 304, 140842 CrossRef PubMed.
- D. L. Pavia, G. M. Lampman, G. S. Kriz and J. R. Vyvyan, Introduction to Spectroscopy, Stamford, Conn: Cengage Learning, 2015, vol. 3. XVII Search PubMed.
- L. Wu, S. Huang, J. Zheng, Z. Qiu, X. Lin and Y. Qin, Int. J. Biol. Macromol., 2019, 140, 538–545 CrossRef CAS PubMed.
- A. Abou-Okeil, A. Amr and F. Abdel-Mohdy, Carbohydr. Polym., 2012, 89, 1–6 CrossRef CAS PubMed.
- H. Pan, G. Sun and T. Zhao, Int. J. Biol. Macromol., 2013, 59, 221–226 CrossRef CAS PubMed.
- Y. H. Yu, L. An, J. H. Bae, J. W. Heo, J. Chen, H. Jeong and Y. S. Kim, Front. Bioeng. Biotechnol., 2021, 9, 682070 CrossRef PubMed.
- H. Maachou, M. J. Genet, D. Aliouche, C. C. Dupont-Gillain and P. G. Rouxhet, Surf. Interface Anal., 2013, 45, 1088–1097 CrossRef CAS.
- Q. Zheng, G. Nong and N. Li, Polymers, 2024, 16, 1237 CrossRef CAS PubMed.
- J. Zhu, Y. Luo, J. Wang, J. Yu, Q. Liu, J. Liu, R. Chen, P. Liu and J. Wang, J. Mol. Liq., 2022, 368, 120744 CrossRef CAS.
- M. Muddasar, M. A. Nasiri, A. Cantarero, C. Gómez, M. Culebras and M. N. Collins, Adv. Funct. Mater., 2024, 34, 2306427 CrossRef CAS.
- A. Zerpa, L. Pakzad and P. Fatehi, ACS Omega, 2018, 3, 8233–8242 CrossRef CAS PubMed.
- J. Chen, L. An, J. W. Heo, J. H. Bae, H. Jeong and Y. S. Kim, J. Wood Chem. Technol., 2022, 42, 114–124 CrossRef CAS.
- E. Corradini, E. A. G. Pineda and A. A. W. Hechenleitner, Polym. Degrad. Stab., 1999, 66, 199–208 CrossRef CAS.
- S. Kim, M. Ham, J. Lee, J. Kim, H. Lee and T. Park, Adv. Funct. Mater., 2023, 33, 2305499 CrossRef CAS.
- M. U. Minhas, M. Ahmad, L. Ali and M. Sohail, Daru, J. Pharm. Sci., 2013, 21, 1–9 Search PubMed.
- C.-G. Han, X. Qian, Q. Li, B. Deng, Y. Zhu, Z. Han, W. Zhang, W. Wang, S.-P. Feng and G. Chen, Science, 2020, 368, 1091–1098 CrossRef CAS PubMed.
- Y. Xiao, J. Jiang and H. Huang, Sci. Rep., 2014, 4, 6305 CrossRef CAS PubMed.
- L. Chen, X. Rong, Z. Liu, Q. Ding, X. Li, Y. Jiang, W. Han and J. Lou, Chem. Eng. J., 2024, 481, 148797 CrossRef CAS.
- H. Li, X. Wang, W. Jiang, H. Fu, X. Liang, K. Zhang, Z. Li, C. Zhao, H. Feng and J. Nie, Adv. Mater. Interfaces, 2018, 5, 1701648 CrossRef.
- S. Patachia, A. J. Valente and C. Baciu, Eur. Polym. J., 2007, 43, 460–467 CrossRef CAS.
- K. Kazimierska-Drobny and M. Kaczmarek, Polimery, 2020, 65, 44–50 CrossRef CAS.
- Y. Zhou, C. Yao, X. Lin, J. Oh, J. Tian, W. Yang, Y. He, Y. Ma, K. Yang and B. Ai, Adv. Funct. Mater., 2023, 33, 2214563 CrossRef CAS.
- T. Li, X. Zhang, S. D. Lacey, R. Mi, X. Zhao, F. Jiang, J. Song, Z. Liu, G. Chen and J. Dai, Nat. Mater., 2019, 18, 608–613 CrossRef CAS PubMed.
- C. Huang, X. Qian and R. Yang, Mater. Sci. Eng., R, 2018, 132, 1–22 CrossRef.
- S. J. Li, G. T. Fei, S. H. Xu, W. C. Chen, H. Y. Xu, X. X. Qu, X. F. Li and H. M. Ouyang, J. Power Sources, 2024, 623, 235376 CrossRef CAS.
- F. Jiang, J. Xiong, W. Zhou, C. Liu, L. Wang, F. Zhao, H. Liu and J. Xu, J. Mater. Chem. A, 2016, 4, 5265–5273 RSC.
- H. Wang, J. H. Hsu, S. I. Yi, S. L. Kim, K. Choi, G. Yang and C. Yu, Adv. Mater., 2015, 27, 6855–6861 CrossRef CAS PubMed.
- Y. Sun, L. Qiu, L. Tang, H. Geng, H. Wang, F. Zhang, D. Huang, W. Xu, P. Yue and Y. s. Guan, Adv. Mater., 2016, 28, 3351–3358 CrossRef CAS PubMed.
- C. Wan, X. Gu, F. Dang, T. Itoh, Y. Wang, H. Sasaki, M. Kondo, K. Koga, K. Yabuki and G. J. Snyder, Nat. Mater., 2015, 14, 622–627 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc06542e |
| This journal is © The Royal Society of Chemistry 2025 |