
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient continuous flow oxidation of furfural to maleic anhydride using O2 as a green oxidant†
Jonas
Mortier
 ,
Christian V.
Stevens
and
Thomas S. A.
Heugebaert
*
,
Christian V.
Stevens
and
Thomas S. A.
Heugebaert
*
Department of Green Chemistry and Technology, Faculty of Bioscience Engineering, Ghent University, Coupure Links 653, 9000 Ghent, Belgium. E-mail: Thomas.Heugebaert@UGent.be
First published on 15th April 2025
Abstract
This research investigates different oxidative pathways to obtain maleic anhydride starting from the biorenewable furfural in continuous flow. A single step O21 oxidation towards maleic anhydride was investigated but found to lack selectivity. Nonetheless, the resulting product analysis provided valuable insight into the possible reaction mechanism, uncovering an interesting solvent effect. Building on this, an existing slow reacting two-step pathway was translated towards a continuous one, achieving a fast and highly productive conversion towards maleic anhydride while aligning with green chemistry principles.
Green foundation1. In this work, n-butane and/or benzene were replaced with biorenewable furfural as feedstock for maleic anhydride. Previous such valorization pathways suffer from low productivity and selectivity. We have identified the underlying mechanistic origins of the selectivity issue, which inherently dictates a two-step catalytic approach.2. By leveraging continuous flow chemistry a more efficient and competitive process was developed with high selectivity (98%), and facile scalability. We significantly reduced catalyst loadings (2%) compared to the existing batch process (5 & 10%) and replaced acetonitrile with ethyl acetate in the second reaction step; while also drastically reducing reaction time (30 min vs. 480 min). 3. Future research should prioritize exploring the use of heterogeneous, easily removed and recycled catalysts, such as polymer-immobilized TEMPO (PIPO). However, this topic falls outside the scope of the present study. |
Introduction
The global chemical industry, traditionally reliant on petrochemical processes, faces increasing pressure to transition to more sustainable practices due to environmental concerns and the finite nature of fossil resources. This shift is critical not only for reducing greenhouse gas emissions but also for minimizing the environmental footprint of chemical production.1 A promising approach to achieving this goal is through the principles of green chemistry, which emphasize the design of chemical products and processes that reduce or eliminate the use and generation of hazardous substances. Key principles of green chemistry include the use of renewable feedstocks, energy efficiency, and waste minimization. By adhering to these principles, the industry can move towards a more sustainable, bio-based economy.2Furfural (FUR), a valuable platform chemical derived from lignocellulosic biomass, has emerged as a key renewable feedstock with the potential to replace petrochemical precursors in various industrial processes.3 Lignocellulosic biomass, which includes agricultural residues and non-food crops, offers a renewable and abundant source of carbon. FUR is obtained via the acid-catalyzed dehydration of pentoses present in hemicellulose and serves as an intermediate for the synthesis of numerous chemicals.4 One particularly valuable transformation of FUR is its conversion to maleic anhydride (MA), a versatile chemical used in the production of resins, plastics, and agrochemicals.5,6 Traditionally, MA is produced from benzene or n-butane through an energy-intensive process employing a phosphorus vanadium oxide catalyst at high temperatures (Fig. 1).7–10
 | ||
| Fig. 1 A comparison between the traditional synthesis of maleic anhydride starting from benzene or n-butane and the biorenewable route starting from furfural. | ||
Thus, developing a green and efficient method to convert FUR to MA is highly desirable. In recent years, various one-step oxidations producing MA directly from FUR have been explored. Among these methods, the vanadium-catalyzed aerobic oxidation of furfural has been the most widely studied. Using the same catalysts as the traditional petroleum-based process, medium to high yields are only obtained after exposing gas-phase furfural to temperatures of 300–340 °C.11–14 Besides the low productivity and the risk of overoxidation, catalyst deactivation at these temperatures also poses a major problem. These problems could be minimized by performing the oxidations in solution-phase at lower temperature, but long reaction times, low selectivity and the hydrolysis of MA to maleic acid make this approach non-optimal as well (Table S1, ESI†).15–17
While maleic acid remains valuable-since it can be isomerized into fumaric acid,18,19 a key precursor for bio-based polyesters20,21-maleic anhydride is generally preferred as a building block due to its higher reactivity, particularly in Diels–Alder-type reactions22–24 and photochemical [2 + 2] cycloadditions.25,26 Moreover, maleic anhydride can readily hydrolyze in water under mild conditions,27 whereas reversing this process typically requires high temperatures and additional catalysts and additives,28–30 making it the more reactive and energetically enriched form of maleic acid.
A more convenient method to oxidize furfural has been examined by Zhang et al.31 where a solution of formic acid/H2O2 and furfural is heated at 60 °C to achieve 95% conversion after 4 hours of reaction time. However, due to the choice of solvent, complete hydrolysis occurred resulting in the formation of the ring-opened maleic acid as final product. It should also be noted that in this reaction, oxygen has been replaced by the less desired hydrogen peroxide as oxidant.
To step away from high temperatures and protic solvents that might catalyze the hydrolysis of MA, two direct photocatalytic aerobic oxidations were examined (Table 1). In both methods, the authors reason that the photocatalyst generates multiple reactive oxygen species, which subsequently lead to the oxidation of furfural. Although these reactions can be operated at room temperature, the reactive oxygen species give rise to many byproducts with 5-hydroxy-2(5H)-furanone 3 (HFO) being the most prominent one. The low selectivity and long reaction time currently make this method unsuitable for industrial-scale applications.

Intrigued by these results showing the simultaneous formation of HFO 3 and MA 2, our aim was to determine the cause of this unusual outcome and explore whether continuous flow intensification strategies could enhance the reaction selectivity. Upon reviewing these two previous photocatalytic methods, several key similarities emerge. In both approaches, the final products consist of a mixture of MA 2 and HFO 3, with similar ratios (1.7 vs. 1.3). Moreover, singlet oxygen (O21) was identified as the primary reactive oxygen species driving the reactions. Both studies also suggested that superoxide radicals (O2˙−) play a role, although when superoxide radical quencher benzoquinone (BQ) was introduced, the reaction only experienced a slight decrease in conversion, rather than ceasing entirely. This evidence suggests that singlet oxygen may be the sole active species. However, if singlet oxygen were the only reactive species, previous literature indicates that furfural should fully convert to HFO under similar conditions.27,34–37 It is worth noting that in previously cited literature, the conversion of FUR to HFO was consistently carried out in protic, nucleophilic solvents such as EtOH, MeOH, or H2O. These mildly nucleophilic solvents help resolve the intermediate endoperoxide through nucleophilic attack on the aldehyde moiety, leading to the formation of HFO 3 (Fig. 2). In contrast, when non-nucleophilic solvents like MeCN or DCM are used, the inability to efficiently resolve the endoperoxide intermediate may result in the production of various other products, such as MA 2.
 | ||
| Fig. 2 Mechanistic pathway for the oxidation of furfural 1 towards HFO 3 in solvent ROH (R = H, Me, Et). | ||
Results and discussion
One-step continuous flow O21 oxidation of FUR towards various reaction products
To test the hypothesis that singlet oxygen oxidation of furfural in a non-nucleophilic solvent like acetonitrile leads to a combination of MA and HFO, methylene blue was used as photosensitizer to generate singlet oxygen. To overcome the attenuation effect which limits the efficient penetration of light throughout the reaction mixture, a continuous flow reactor is used. The reaction rate was remarkably high, achieving almost full conversion after only 6.5 minutes. Interestingly, a very similar 2/3 ratio of around 1.3 was achieved, thus confirming their inherent co-production under singlet oxygen mediated oxidation (Table 2, entries 1–3). When initially conducting the reaction however, we observed variability in the 2/3 ratio across duplicate experiments, along with a poor mass balance. We later determined that maleic anhydride 2 had sublimated during the evaporatory work-up process. To prevent this issue, we opted to directly analyze the reaction mixture using 1H-NMR without performing solvent evaporation. This approach revealed several additional signals that had gone undetected in previous literature reports. The volatility and/or fragility of these side products made it challenging to fully characterize all of them. However, cis-β-formylacrylic acid (CBF) 6 was identified as the major side product. trans-β-Formylacrylic acid (TBF) 7 was also formed in smaller quantities along with, interestingly, the 5-(formyloxy)-2(5H)-furanone (FOF) 8 and formic acid. The formation of CBF 6 was also reported in the work of Kailasam et al.,33 and is a known tautomeric product of HFO 3.38 As the MeCN/H2O solvent mixture shifts more towards H2O, it is seen that HFO emerges as the sole product (Table 2, entries 5–9). This emphasizes the critical importance of solvent effects on product formation, as increasing the water content directs the oxidation to proceed via the well-known nucleophilic solvent induced pathway delivering formic acid and HFO (Fig. 2). As more water is added to the reaction medium, the nucleophilic attack pathway will become the more dominant one resulting in more HFO 3 formation. This trend continues until a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O/MeCN ratio is reached, where HFO 3 becomes the sole product (Table 2, entry 9).
:1 H2O/MeCN ratio is reached, where HFO 3 becomes the sole product (Table 2, entry 9).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Pump flow rate (ml min−1) | O2 flow rate (mlN min−1) | Residence timea (min) | Conv. 1b (%) |
Yieldc2/3/6/7/8 (%) |
| a Residence time determined experimentally. b Conversion determined via quantitative HPLC analysis. c Yield determined via quantitative 1H-NMR analysis with mesitylene as internal standard, unless stated otherwise. d Yield determined via quantitative GC-MS analysis (2) and quantitative 1H-NMR analysis (3, 6, 7 and 8). | ||||||
| 1 | MeCN | 0.3 | 10 | 45 | 100 | 29/23/20/4/7d |
| 2 | MeCN | 1.5 | 20 | 6.5 | 98 | 27/21/19/2/5d |
| 3 | MeCN | 3 | 40 | 3.5 | 86 | 25/19/18/2/4d |
| 4 | Dry MeCN | 3 | 40 | 3.5 | 87 | 24/19/19/2/5d |
| 5 | MeCN/H2O (24/1) | 3 | 40 | 3.5 | 96 | 23/44/9/1/3 |
| 6 | MeCN/H2O (14/1) | 3 | 40 | 3.5 | 93 | 17/54/1/1/3 |
| 7 | MeCN/H2O (9/1) | 3 | 40 | 3.5 | 96 | 13/63/0/0/2 |
| 8 | MeCN/H2O (4/1) | 3 | 40 | 3.5 | 95 | 2/89/0/0/0 |
| 9 | MeCN/H2O (1/1) | 3 | 40 | 3.5 | 81 | 0/77/0/0/0 |

In the absence of water though, the mechanism in which HFO and formic acid, but also MA are formed remains unclear. To rule out the possibility that HFO and formic acid formation in MeCN was caused by trace amounts of water present in the solvent, the experiment was repeated using strictly anhydrous MeCN, yielding the same result (Table 2, entry 4). Furthermore, when the reaction was conducted with 4% water intentionally added to the mixture, the HFO yield increased slightly but not dramatically (Table 2, entry 5). This observation confirms that trace amounts of water cannot fully account for the observed amounts of HFO and formic acid formation, and the classical nucleophilic mechanism (Fig. 2) is not the predominant one. A closer evaluation of the newly found side products however reveals that 5-(formyloxy)-2(5H)-furanone 8 could be the result of a Baeyer–Villiger rearrangement, which has been proposed in several H2O2-mediated pathways towards maleic acid.31,39,40 Interestingly, Yin and co-workers17 observed the very closely related compound 5-acetoxyl-2(5H)-furanone when performing a H5PV2Mo10O40 catalyzed oxidation of furfural using acetic acid as solvent.
To evaluate whether 8, or any of the other products might act as intermediates for MA formation, the reaction was carried out in batch and monitored over time. There is no indication that any of the products act as intermediates, as their yields increase throughout the reaction without any subsequent consumption (Fig. S14, ESI†). Similar results were observed in the continuous flow experiments, where nearly full conversion was achieved within 6.5 minutes, and extending the reaction time to 45 minutes did not significantly alter the product ratios (Table 2, entries 1 and 2). Unfortunately, this indicates that, with this approach, the yield of maleic anhydride peaks at around 30%.
Mechanism
A plausible mechanism to explain the formation of all reaction products begins with the generation of endoperoxide intermediate 9via O21 oxidation (Fig. 3). In nucleophilic solvents, this endoperoxide would be resolved by a nucleophilic attack by the solvent resulting in HFO 3 formation (Fig. 2). When the reaction is performed in an anhydrous, non-nucleophilic solvent like acetonitrile, it is proposed that a Baeyer–Villiger type of rearrangement will result in 5-hydroxy intermediate 10. This intermediate can either tautomerize to its more stable keto-form 8via a 1,4-rearrangement, which is detected in the reaction mixture, or alternatively will react with O21 to form a secondary endoperoxide intermediate 11. The subsequent endoperoxide will open, resulting in hydroperoxide intermediate 12, which rearranges into maleic anhydride 2 and performic acid. The performic acid formed in this step will then oxidize another molecule of furfural towards 2-furoic acid 13, releasing formic acid as byproduct, as observed in the 1H-NMR spectrum. The resulting furoic acid will then react with O21, followed by decarboxylation, to produce HFO 3 without requiring a nucleophilic solvent to resolve the endoperoxide 14. HFO can then tautomerize into CBF 6, which, in turn, can undergo cis–trans isomerization to form TBF 7. In this way, for every molecule of maleic anhydride 2 formed, one molecule of performic acid would also be generated. This performic acid oxidizes furfural to produce one equivalent of 2-furoic acid 13, which subsequently yields one equivalent of HFO 3. HFO could then isomerize to form TBF 6 and CBF 7. Based on this reasoning, the expected ratio of 2 to (3 + 6 + 7) in acetonitrile should be 1:1. However, in our experiments, the observed ratio is around 1:1.6 (Table 2, entries 1–4). While the hydrolysis of FOF 8 could explain the excess HFO, this is unlikely due to the very low water content. Instead, the surplus HFO and its isomers is attributed to a minor, as-yet-undiscovered pathway. Numerous studies suggest that 2-furoic acid 13 acts as a direct intermediate in furfural oxidation processes.31,33,40,41 However, we did not detect this product in the 1H-NMR analysis of our reaction mixture. This absence could be explained if the subsequent reaction of 2-furoic acid with O21 to form HFO 3 occurs at a rate significantly faster—potentially an order of magnitude—than the overall reaction rate, making this intermediate undetectable during the reaction follow-up (Fig. S14, ESI†). Supporting this hypothesis, the reaction was conducted using 2-furoic acid 13 as the starting material, an exceptionally high reaction rate was observed, with nearly complete conversion to HFO 3 occurring within just 20 seconds (Table 3). In the reaction mixture of this experiment, no formic acid was detected, consistent with the decarboxylation observed by Wang et al., who reported a 1:1 ratio of HFO to CO2 during a CuOx/Nb2O5-catalyzed furfural oxidation.32 These experiments suggest that a parallel, direct oxidation of furfural to the intermediate 2-furoic acid 13 could account for the observed excess in HFO formation.
 | ||
| Fig. 3 Plausible pathway for the O21 oxidation of furfural towards various reaction products. | ||
|
|
|||
|---|---|---|---|
| Entrya | Residence timeb | Conv. 1c (%) |
Yield 3c (%) |
| a All reactions were performed with a starting concentration of 0.2 M. b Residence time determined experimentally. c Conversion and yield determined via quantitative 1H-NMR analysis with mesitylene as internal standard. | |||
| 1 | 6.5 min | 100 | 100 |
| 2 | 1.5 min | 100 | 100 |
| 3 | 1 min | 100 | 100 |
| 4 | 20 s | 96 | 96 |

In conclusion, a one-step conversion of FUR to MA is subjected to various limitations. In literature it was seen that the need for high reaction temperatures in the gas phase vanadium-catalyzed oxidations gives rise to overoxidation resulting in lower yields, while solution-state reactions with the latter catalyst are plagued by long reaction times and only medium selectivity. The singlet oxygen oxidation of FUR to MA, on the other hand, proceeds at room temperature but exhibits low selectivity due to the inherent co-production of HFO.
Two-step continuous flow oxidation of FUR towards MA
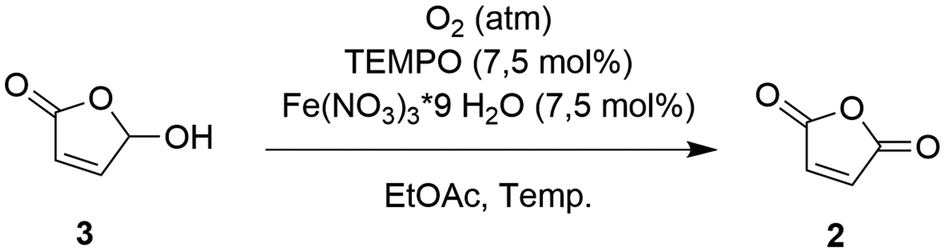
A more recent study by Feringa and co-workers27 has suggested that a two-step pathway – first converting FUR to HFO, followed by the transformation of HFO to MA – may be more convenient. The photocatalytic conversion of FUR to HFO has been demonstrated in various studies and results in quantitative yield with short reaction times.27,36,37,42 The bottleneck, however, is the TEMPO-mediated aerobic oxidation of HFO 3 to MA 2, which takes 8 hours to complete resulting in a low productivity, in spite of the high catalyst loadings (Fig. 4). | ||
| Fig. 4 TEMPO-mediated aerobic oxidation from HFO to MA (Feringa et al., 2022).27 | ||
By transitioning from a batch process to a continuous flow system, the key challenges of extended reaction times and high catalyst loadings can be effectively addressed, creating a truly efficient and productive pathway for converting FUR to MA. We will therefore focus on creating a straightforward, productive, and selective two-step synthesis in continuous flow, utilizing the TEMPO-mediated oxidation to convert the platform molecule FUR to MA previously described by Feringa and co-workers (Fig. 5).27
 | ||
| Fig. 5 This work: a two-step continuous flow process to synthesize MA 2. | ||
Continuous flow O21 of FUR to HFO
The continuous flow oxidation of furfural to HFO 3 by singlet oxygen has been well-documented in literature.36,37 With methylene blue as the photosensitizer in MeOH, a residence time of 20 minutes was sufficient to achieve complete conversion with high selectivity (Table 4). From a sustainability standpoint, it is highly recommended to minimize purification steps between reaction steps while also paying attention to solvent recovery. In this instance, the only byproduct formed is methyl formate, which has a boiling point of 32 °C – significantly different from methanol, making it easier to separate during solvent recovery. This simplifies the purification process, lowering operational complexity and minimizing the environmental impact.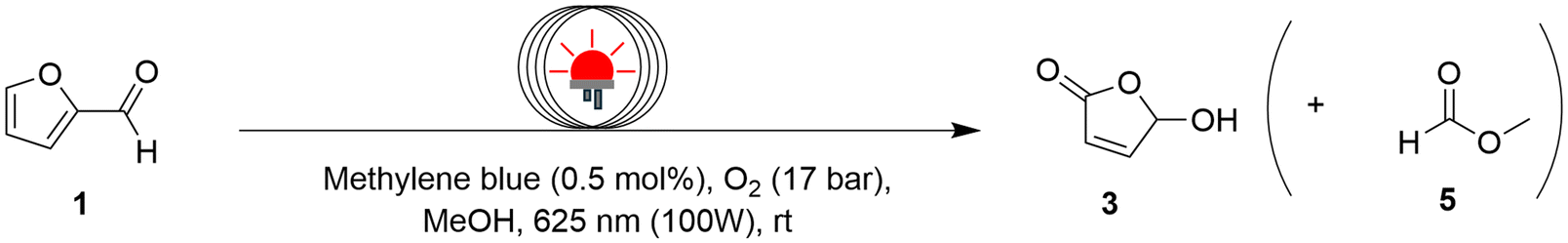
Batch optimization of TEMPO-mediated oxidation of HFO to MA
The final step in the pathway consists of a TEMPO-catalyzed aerobic oxidation from HFO to MA. In 2011, Yu et al.43 described a general method for the oxidation of alcohols to aldehydes/ketones, employing oxygen and catalytic amounts of TEMPO, Fe(NO3)3 and table salt. In 2022, Feringa and co-workers27 displayed how this method could convert HFO to MA quantitatively after 8 hours of stirring (Fig. 4). Before translating this reaction to continuous flow, the first optimization reactions were carried out in batch starting with the reaction conditions that were already established for this method. Although it is preferable to choose a solvent that can be utilized in both reaction steps, adhering to the principle of reaction telescoping,44 it was quickly realized that this would be impossible. A review of the literature reveals that all high-yielding singlet oxygen oxidations of FUR occur in protic solvents like MeOH, EtOH, H2O or a combination of prior solvents.27,34–37 In contrast, for the aerobic oxidation of HFO 3 to MA 2, even trace amounts of MeOH completely inhibited conversion, likely due to this method's tendency to oxidize MeOH itself (Table 5, entries 1 and 2). When performing the reaction in water on the other hand, 1H-NMR analysis revealed that the desired conversion takes place, but maleic anhydride is immediately hydrolyzed to maleic acid (Table 5, entry 3). Furthermore, MeCN as a solvent performed poorly unless NaCl was added (Table 5, entries 4 and 5). EtOAc showed the best performance, and the presence of NaCl had no effect (Table 5, entries 6 and 7). Other non-alcohol solvents like THF, DMSO and cyrene were also tested, but resulted in low yields (Table 5, entries 8–10).|
|
||||
|---|---|---|---|---|
| Entry | Solvent | NaCl (mol%) | Reaction time (h) | Yield 2a (%) |
| a Based on 1H-NMR analysis, no by-products were detected, and the yield of MA was found to be equal to the conversion of furfural when verified using an internal standard. Thus, the yield was estimated using the following formula: yield = MA/(FUR + MA) via1H-NMR analysis. | ||||
| 1 | MeOH | — | 2 | 0 |
| 4,5 | 0 | |||
| 2 | EtOAc/MeOH (99/1) | — | 2 | 4 |
| 4,5 | 8 | |||
| 3 | H2O | — | 2 | 0 |
| 4,5 | 0 | |||
| 4 | MeCN | — | 2 | 14 |
| 4,5 | 23 | |||
| 5 | MeCN | 25 | 2 | 45 |
| 4,5 | 62 | |||
| 6 | EtOAc | — | 2 | 60 |
| 4,5 | 80 | |||
| 7 | EtOAc | 25 | 2 | 63 |
| 4,5 | 78 | |||
| 8 | THF | — | 2 | 10 |
| 4,5 | 21 | |||
| 9 | DMSO | — | 2 | 0 |
| 4,5 | 0 | |||
| 10 | Cyrene | — | 2 | 0 |
| 4,5 | 0 | |||

After identifying the optimal solvent, we focused on determining the ideal catalyst loading, particularly the optimal TEMPO/Fe(NO3)3 ratio. Interestingly, previous research27 indicated that a 1:1 ratio of TEMPO to Fe(NO3)3 was not always optimal, with a 5 mol%/2.5 mol% ratio yielding better results than a 5 mol%/5 mol% ratio in acetonitrile. To address this issue, a full catalyst ratio screening was conducted in EtOAc. Starting with a 5 mol%/5 mol% ratio, we observed that increasing the loading to either 5 mol%/10 mol% or 10 mol%/5 mol% did not lead to a significant increase in yield. Therefore, we concluded that in EtOAc, a catalyst ratio of 1:1 is optimal and a 7.5 mol%/7.5 mol% loading was used in the following experiments (Fig. S15, ESI†).
In the next step of the optimization process, temperature variation was evaluated. It was hypothesized that, if the reaction is O2 limited, lowering the temperature might positively affect the oxidation reaction due to an increased oxygen solubility. However, the reaction was significantly attenuated at 0 °C. In TEMPO-mediated oxidations of primary alcohols to aliphatic aldehydes, careful temperature control is recommended due to the potential risk of overoxidation to carboxylic acids at higher temperatures.45 However, the butanolide system 3 displays little risk of overoxidation in its ring-closed form. As such, we found that increasing the temperature significantly accelerated the reaction rate, resulting in a 90% yield in just 30 minutes with little to no side product formation (Table 6). This significant decrease in reaction time paves the way for implementing a continuous flow process.
|
|
|||
|---|---|---|---|
| Entry | Temperature (°C) | Reaction time (h) | Yield 2a (%) |
| a Based on 1H-NMR analysis, no by-products were detected, and the yield of MA was found to be equal to the conversion of furfural when verified using an internal standard. Thus, the yield was estimated using the following formula: yield = MA/(FUR + MA) via1H-NMR analysis. | |||
| 1 | 0 | 2 | 14 |
| 4,5 | 15 | ||
| 2 | 25 | 2 | 73 |
| 4,5 | 97 | ||
| 3 | 50 | 0,5 | 90 |
| 1 | 100 | ||
Continuous flow optimization of TEMPO-mediated oxidation of HFO to MA
Working with oxygen at higher temperatures presents several challenges. Since oxygen solubility decreases with increasing temperature, maintaining a constant supply of oxygen in a batch setup can be difficult due to the limited headspace and the limited interphase surface area. In contrast, a continuous flow setup offers a more elegant solution. By using a reaction mixture-oxygen Taylor flow, it ensures a consistent supply of oxygen which is readily taken up by the mixture due to the high interphase surface area while also allowing for safe operation at higher pressures and temperatures. In a continuous flow system, temperatures can be substantially increased compared to batch processes because the back pressure exerted on the reaction mixture raises the solvent's boiling point, allowing it to remain in a liquid state even above its atmospheric boiling temperature. It was observed that at 70 °C, a 98% conversion could be achieved in just 15 minutes, while at 50 °C it takes 30 minutes to achieve 98% conversion. Increasing the temperature to 100 °C further accelerated the reaction rate, but this temperature could not be utilized due to reproducibility issues (Table 7, entries 1–3). The final step was to increase the product concentration to enhance productivity. It was observed that higher concentrations led to improved yields within the same reaction time (Table 7, entry 4). At a concentration of 0.2 M, it was observed that the catalyst loading could be lowered to 2 mol% while still maintaining near-quantitative yield after 15 minutes (Table 7, entries 5–7). However, further reducing the catalyst loading resulted in incomplete conversion (Table 7, entry 7).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Conc. (M) | Cat. loading (mol%) | Reaction mixture flow (ml min−1) | Oxygen flow (mlN min−1) | Temp. (°C) | RT (min) | Conversion 3a (yield 2) (%) |
| a Yield and conversion are determined via1H-NMR analysis with mesitylene as internal standard. | |||||||
| 1 | 0.05 | TEMPO (7.5) | 1.4 | 16 | 50 | 5 | 58 (58) |
| Fe(NO3)3 (7.5) | 0.7 | 8 | 10 | 68 (68) | |||
| 0.23 | 2.66 | 30 | 98 (96) | ||||
| 2 | 0.05 | TEMPO (7.5) | 1.4 | 16 | 70 | 5 | 69 (69) |
| Fe(NO3)3 (7.5) | 0.7 | 8 | 10 | 86 (84) | |||
| 0.47 | 5.33 | 15 | 98 (98) | ||||
| 3 | 0.05 | TEMPO (7.5) | 0.7 | 8 | 100 | 10 | 92 (92) |
| Fe(NO3)3 (7.5) | |||||||
| 4 | 0.1 | TEMPO (7.5) | 1.4 | 16 | 70 | 5 | 99 (98) |
| Fe(NO3)3 (7.5) | 0.7 | 8 | 10 | 100 (100) | |||
| 5 | 0.2 | TEMPO (3.75) | 2.8 | 32 | 70 | 2.5 | 72 (72) |
| Fe(NO3)3 (3.75) | 1.4 | 16 | 5 | 98 (97) | |||
| 6 | 0.2 | TEMPO (2) | 1.4 | 16 | 70 | 5 | 57 (57) |
| Fe(NO3)3 (2) | 0.47 | 5.33 | 15 | 97 (97) | |||
| 0.23 | 2.66 | 30 | 100 (99) | ||||
| 7 | 0.2 | TEMPO (1.5) | 0.23 | 2.66 | 70 | 30 | 93 (92) |
| Fe(NO3)3 (1.5) | |||||||

Continuous flow oxidation of FUR to MA without intermediate purification
A final step to minimize waste production is to directly link the two reaction steps without performing intermediate purification. Since the first oxidation step from FUR to HFO does not produce any side products, the only potential impurity in the second oxidation is methylene blue, which should remain inactive if shielded from light. However, it is crucial to completely evaporate MeOH before dissolving the reaction mixture in EtOAc for the second reaction step, as even small amounts of MeOH were found to completely halt the reaction (Table 5, entry 2). It was observed that the reaction with the unpurified intermediate still resulted in a high yield of 98% maleic anhydride (Fig. 6). | ||
| Fig. 6 Continuous flow oxidation of FUR 1 to MA 2 without intermediate purification. aYield is determined via quantitative HPLC analysis after first reaction step. bYield and conversion are determined via1H-NMR analysis with mesitylene as internal standard which was added after the first reaction step. | ||
Conclusions
This study provides valuable insights into the O21 oxidation of furfural, underscoring the critical role of solvent choice in product selectivity and offering a detailed overview of the product range. It concludes that a one-step singlet oxygen oxidation of furfural to maleic anhydride is inefficient due to low selectivity, but in the process, valuable insights in the potential reaction mechanism of O21 furfural oxidation were found. Instead, an existing two-step pathway has been optimized to create a highly selective, productive, and scalable route towards maleic anhydride in a green manner. It should be noted that the solvents used in the pathway, are both classified as ‘recommended’ according to the CHEM21 selection guide of classical- and less classical-solvents.46 In the process, both reaction steps utilize catalysts, with oxygen being the only reagent used in stoichiometric amounts. Unlike previous methods, this approach avoids excessive heating, as the first step is conducted at room temperature and the second step at just 70 °C. This method minimizes waste by eliminating the need for intermediate purification between the reaction steps. A final enhancement achieved by utilizing continuous flow is the improved safety when operating under higher pressures.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Jonas Mortier gratefully acknowledges financial support for this publication by the Ghent University Special Research Fund; grant nr. BOF/STA/202102/003.References
- E. Gawel, N. Pannicke and N. Hagemann, Sustainability, 2019, 11, 3005 Search PubMed.
- T. L. Chen, H. Kim, S. Y. Pan, P. C. Tseng, Y. P. Lin and P. C. Chiang, Sci. Total Environ., 2020, 716, 136998 Search PubMed.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. L. López Granados, Energy Environ. Sci., 2016, 9, 1144–1189 Search PubMed.
- A. S. Mamman, J. M. Lee, Y. C. Kim, I. T. Hwang, N. J. Park, Y. K. Hwang, J. S. Chang and J. S. Hwang, Biofuels, Bioprod. Biorefin., 2008, 2, 438–454 Search PubMed.
- K. Lohbeck, H. Haferkorn, W. Fuhrmann and N. Fedtke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2000 Search PubMed.
- E.-R. Kenawy and M. A. Sakran, J. Appl. Polym. Sci., 2001, 80, 415–421 CrossRef CAS.
- G. Cent, F. Trifirb, J. R. Ebner’ and V. M. Franchetti, Chem. Rev., 1988, 88, 55–80 CrossRef.
- B. K. Hodnett, Catal. Rev., 1985, 27, 373–424 CrossRef.
- N. F. Dummer, J. K. Bartley and G. J. Hutchings, Adv. Catal., 2011, 54, 189–247 CAS.
- N. Ballarini, F. Cavani, C. Cortelli, S. Ligi, F. Pierelli, F. Trifirò, C. Fumagalli, G. Mazzoni and T. Monti, Top. Catal., 2006, 38, 147–156 CrossRef CAS.
- P. Santander, L. Bravo, G. Pecchi and A. Karelovic, Appl. Catal., A, 2020, 595, 117513 CrossRef CAS.
- X. Li, J. Ko and Y. Zhang, ChemSusChem, 2018, 11, 612–618 CrossRef CAS PubMed.
- N. Alonso-Fagúndez, M. Ojeda, R. Mariscal, J. L. G. Fierro and M. López Granados, J. Catal., 2017, 348, 265–275 CrossRef.
- N. Alonso-Fagúndez, M. López Granados, R. Mariscal and M. Ojeda, ChemSusChem, 2012, 5, 1984–1990 CrossRef PubMed.
- X. Li, B. Ho and Y. Zhang, Green Chem., 2016, 18, 2976–2980 RSC.
- G. Lv, C. Chen, B. Lu, J. Li, Y. Yang, C. Chen, T. Deng, Y. Zhu and X. Hou, RSC Adv., 2016, 6, 101277–101282 RSC.
- J. Lan, Z. Chen, J. Lin and G. Yin, Green Chem., 2014, 16, 4351–4358 RSC.
- Md. M. Baag, A. Kar and N. P. Argade, Tetrahedron, 2003, 59, 6489–6492 CrossRef CAS.
- H. Tateno, Y. Miseki, H. Kusama, S.-Y. Chen, T. Mochizuki and K. Sayama, ACS Sustainable Chem. Eng., 2021, 9, 6886–6893 CrossRef CAS.
- M. Hofmann, M. Garrido, M. Machado, J. R. Correia and J. C. Bordado, J. Appl. Polym. Sci., 2022, 139, e53029 CrossRef CAS.
- Y. Yu, H. Xiong, J. Xiao, X. Qian, X. Leng, Z. Wei and Y. Li, ACS Sustainable Chem. Eng., 2019, 7, 6859–6869 CrossRef CAS.
- D. Sun, F. Sato, Y. Yamada and S. Sato, Bull. Chem. Soc. Jpn., 2013, 86, 276–282 CrossRef CAS.
- J. Agarwal, R. Rani and R. Peddinti, Synlett, 2017, 1336–1340 CAS.
- D. A. Kornilov, I. F. Dinikaev and V. D. Kiselev, High Pressure Res., 2019, 39, 640–654 CrossRef CAS.
- J. D. Williams, M. Nakano, R. Gérardy, J. A. Rincón, Ó. de Frutos, C. Mateos, J.-C. M. Monbaliu and C. O. Kappe, Org. Process Res. Dev., 2019, 23, 78–87 CrossRef CAS.
- E. Torres, E. Gorrea, K. K. Burusco, E. Da Silva, P. Nolis, F. Rúa, S. Boussert, I. Díez-Pérez, S. Dannenberg, S. Izquierdo, E. Giralt, C. Jaime, V. Branchadell and R. M. Ortuño, Org. Biomol. Chem., 2010, 8, 564–575 RSC.
- J. G. H. Hermens, A. Jensma and B. L. Feringa, Angew. Chem.,– Int. Ed., 2022, 61, e202112618 CrossRef CAS PubMed.
- C. Robert, F. de Montigny and C. M. Thomas, ACS Catal., 2014, 4, 3586–3589 CrossRef CAS.
- W. Jia, Z. Si, Y. Feng, X. Zhang, X. Zhao, Y. Sun, X. Tang, X. Zeng and L. Lin, ACS Sustainable Chem. Eng., 2020, 8, 7901–7908 CrossRef CAS.
- W. Jia, Y. Sun, M. Zuo, Y. Feng, X. Tang, X. Zeng and L. Lin, ChemSusChem, 2020, 13, 640–646 CrossRef CAS PubMed.
- X. Li, B. Ho, D. S. W. Lim and Y. Zhang, Green Chem., 2017, 19, 914–918 RSC.
- P. Ren, Y. Zhou, K. Su, L. Sun, N. Luo and F. Wang, Chem. – Asian J., 2023, 18, e202300732 CrossRef CAS.
- D. K. Chauhan, V. R. Battula, A. Giri, A. Patra and K. Kailasam, Catal. Sci. Technol., 2022, 12, 144–153 RSC.
- J. de Jong and B. Feringa, Tetrahedron:Asymmetry, 1991, 2, 1247–1262 CrossRef CAS.
- X. Zhang, H. H. Huang and Q. H. Chen, J. Asian Nat. Prod. Res., 2005, 7, 711–721 CrossRef CAS PubMed.
- M. D. Edwards, M. T. Pratley, C. M. Gordon, R. I. Teixeira, H. Ali, I. Mahmood, R. Lester, A. Love, J. G. H. Hermens, T. Freese, B. L. Feringa, M. Poliakoff and M. W. George, Org. Process Res. Dev., 2024, 28, 1917–1928 CrossRef CAS PubMed.
- J. G. H. Hermens, M. L. Lepage, A. Kloekhorst, E. Keller, R. Bloem, M. Meijer and B. L. Feringa, React. Chem. Eng., 2022, 7, 2280–2284 RSC.
- S. Thiyagarajan, D. Franciolus, R. J. M. Bisselink, T. A. Ewing, C. G. Boeriu and J. Van Haveren, ACS Sustainable Chem. Eng., 2020, 8, 10626–10632 CAS.
- A. M. S. Pembere, H. Louis and H. Wu, J. Mol. Model, 2023, 29, 359 CrossRef CAS.
- N. Alonso-Fagúndez, I. Agirrezabal-Telleria, P. L. Arias, J. L. G. Fierro, R. Mariscal and M. López Granados, RSC Adv., 2014, 4, 54960–54972 RSC.
- M. R. Ebrahimian, M. Tavakolian and M. Hosseini-Sarvari, J. Environ. Chem. Eng., 2023, 11, 109347 CrossRef CAS.
- J. G. H. Hermens, T. Freese, G. Alachouzos, M. L. Lepage, K. J. van den Berg, N. Elders and B. L. Feringa, Green Chem., 2022, 24, 9772–9780 RSC.
- S. Ma, J. Liu, S. Li, B. Chen, J. Cheng, J. Kuang, Y. Liu, B. Wan, Y. Wang, J. Ye, Q. Yu, W. Yuan and S. Yu, Adv. Synth. Catal., 2011, 353, 1005–1017 CrossRef CAS.
- D. E. Fitzpatrick and S. V. Ley, Tetrahedron, 2018, 74, 3087–3100 CrossRef CAS.
- B. L. Ryland and S. S. Stahl, Angew. Chem.,– Int. Ed., 2014, 53, 8824–8838 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc06305h |
| This journal is © The Royal Society of Chemistry 2025 |