
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the use of propylene carbonate and dimethyl carbonate as green solvents in organic electrosynthesis†
Adrian
Prudlik
a,
Alexandra
Matei
 b,
Anton
Scherkus
a,
Javier Ivan
Bardagi
cd,
Sebastian B.
Beil
*be and
Robert
Francke
*a
b,
Anton
Scherkus
a,
Javier Ivan
Bardagi
cd,
Sebastian B.
Beil
*be and
Robert
Francke
*a
aLeibniz Institute for Catalysis, Albert-Einstein-Straße 29a, Rostock, Germany. E-mail: robert.francke@catalysis.de
bStratingh Institute for Chemistry, University of Groningen, 9747 AG Groningen, The Netherlands
cInstituto de Investigaciones en Fisicoquímica de Córdoba (INFIQC-CONICET), Córdoba, Argentina
dDepartment of Organic Chemistry, Faculty of Chemical Sciences, National University of Cordoba, University City, X5000HUA, Cordoba, Argentina
eMax Planck Institute for Chemical Energy Conversion, Stiftstraße 34–36, 45470 Mülheim an der Ruhr, Germany. E-mail: sebastian.beil@cec.mpg.de
First published on 20th March 2025
Abstract
Electroorganic syntheses are often carried out in polar aprotic solvents such as DMF, acetonitrile, or dichloromethane, which exhibit excellent electrochemical properties, but are highly problematic in terms of sustainability. The propylene carbonate–dimethyl carbonate (PC–DMC) system is a promising alternative with enhanced environmental, health, and safety parameters, and has already found numerous applications in electrochemical energy storage systems. Herein, we present a systematic study on the PC–DMC system as reaction medium for organic electrosyntheses, spanning from the characterization of electrolyte properties to representative test reactions on a preparative scale. Anodic synthesis of diaryliodonium salts, cathodic reduction of ketones, and TEMPO-mediated alcohol oxidations serve as use cases, showing that yields are comparable to the ones obtained in conventional solvents. An interesting feature is the possibility for tuning the physicochemical properties of the reaction medium by varying the PC–DMC ratio, which was shown to impact the catalytic rate of TEMPO-mediated alcohol oxidations and the yield of diaryl iodonium synthesis.
Green foundation1. Organic electrosynthesis frequently involves harmful and problematic solvents, which does not do justice to the ‘green potential’ of the method. Our work adds a binary solvent system with excellent electrochemical properties and good sustainability ratings to the synthetic electrochemist's solvent portfolio.2. Our approach includes a detailed analysis of physico- and electrochemical electrolyte properties as well as synthetic studies, including the development of strategies for convenient product isolation. Evaluation in three representative electrochemical applications showed similar or improved yields compared to the literature, along with further improvements of reaction control and process mass intensity. 3. Further progress is expected upon studying the PC–DMC system under process-relevant conditions including a life cycle analysis. Our conceptual approach may serve as a blueprint for future studies into sustainable solvents for organic electrochemistry and thereby lead to further innovations. |
1 Introduction
Organic electrochemistry has frequently been referred to as inherently “green”, since it offers the opportunity to address most of the 12 principles of green chemistry,1 such as optimizing atom economy, lowering energy consumption, or developing less hazardous syntheses. By using electric current, dangerous and expensive redox agents can be avoided, thereby improving atom economy, and reducing waste generation as well as energy consumption.2 Through the electrode potential as the continuously variable driving force, reactions can be carried out under mild conditions, leading to reactive intermediates that are not (or hardly) accessible by conventional means.3–6 Although the use of organic electrochemistry can indeed lead to “greener” conversions, it is not possible to generalize this assessment, since the actual sustainability depends very much on how the method is applied.7–9 The abovementioned benefits are offset by inherent features that can have a detrimental effect on sustainability and therefore require particular attention, one being the necessity of using a supporting electrolyte additive, which requires additional separation steps and represents a potential source of waste.7,9–11 A further separation issue is introduced by mediators, which are often necessary to control the selectivity.12,13 A possible solution for reducing waste originating from mediators and supporting electrolytes is simplifying the recycling, which can be achieved by attaching the components to suspended particles14 or soluble polymers,11,15,16 and in the case of mediators by immobilization on the electrode surface.17–19In contrast to the extensive work on sustainable solutions for the supporting electrolyte and mediator issues, there has been little research into alternative and more sustainable solvents for electrosynthesis. Consequently, problematic solvents are used frequently, especially when aprotic reaction media are needed.7 A possible explanation is that solvents must fulfil a broader array of criteria in electrosynthesis, rendering the establishment of new candidates more challenging. Thus, the solvent is not only one of the key parameters influencing all (electro)chemical steps of the desired conversions in the electrolysis cell, but must also provide a high electrochemical stability, the ability to dissolve supporting electrolytes, and sufficient conductivity.
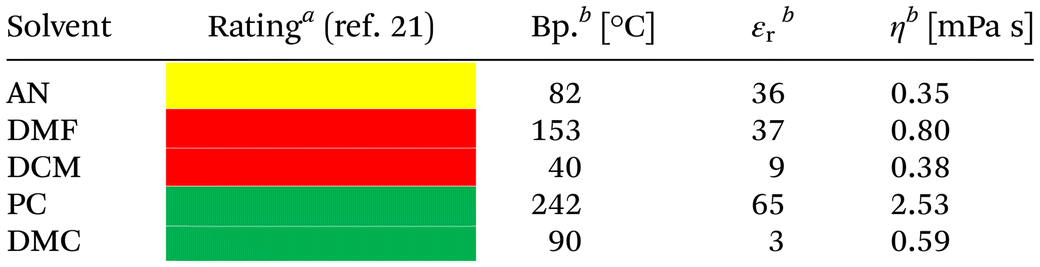
In view of the specific demands of electrochemical reactions, alcohols and occasionally water are the solvents of choice when a protic medium is desired. On the other hand, acetonitrile (AN), dimethyl formamide (DMF), or dichloromethane (DCM) are the go-to aprotic solvents for organic electrochemistry. Particularly these aprotic representatives have drawbacks in terms of sustainability.20–24 This is illustrated, for example, by their comprehensive assessment in GSK's solvent selection guide (see Table 1).21 The color coding corresponds to the traffic light system and is derived by a comprehensive evaluation of waste potential as well as environmental, health, and safety aspects.
Substitutes for the abovementioned solvents should be used to reduce the environmental impact of current processes and the ones under development. In this context, a study has been recently published on the use of Cyrene™,33 a bio-renewable aprotic solvent and potential replacement to DMF,34,35 highlighting the possibility for improved sustainability of electrochemical conversions through alternative solvents. Electrochemical ketone reduction was investigated as a test reaction, with good performance reported in combination with specific salts and co-solvents. However, the work also identified challenges, such as the relatively easy decomposition and high viscosity, in addition to the limited availability of Cyrene™. Not least for these reasons, research into further aprotic solvent alternatives for electrosynthesis appears essential.
A look outside the box of electrosynthesis shows that alkyl carbonates such as propylene carbonate (PC), ethylene carbonate (EC), or dimethyl carbonate (DMC) are frequently used as solvents in electrochemical energy storage media.36–39 They provide high electrochemical stability as well as good solubility and dissociation of supporting electrolytes. For example, binary mixtures of cyclic and linear carbonates are employed in lithium-ion batteries to overcome drawbacks of the individual solvent components such as high viscosity or low polarity, enabling a tuning of the electrolyte properties via the composition of the binary mixture. Furthermore, alkyl carbonate solvents have been frequently used in organic chemistry, including reactions promoted by homogeneous catalysts or enzymes.40–42 Due to the advantageous properties of organic carbonates as sustainable solvents,21,37,43,44 exploring their potential for organic electrosynthesis appears particularly promising. However, the use of alkyl carbonates in electrosynthesis has not yet been systematically investigated. To the best of our knowledge, there are only a few scattered examples in which tests have been carried out as part of solvent screenings. For example, the electrochemical synthesis of α-hydroxy acids from benzaldehyde and CO2 was reported to proceed more efficiently in PC than in AN or DMF.45
Herein, we present a systematic study on the use of carbonates as solvents in organic electrochemistry. For this purpose, we have selected the binary PC–DMC solvent system due to its promising performance in energy storage electrolytes and the possibility for tuning the properties with a low-viscosity (DMC) and a high-polarity (PC) component (compare values for viscosity, η, and dielectric permittivity, εr, in Table 1). A major argument in favor of PC and DMC is the significantly improved sustainability compared to standard solvents (see Table 1). On the one hand, the high boiling point of PC poses a challenge for product separation. On the other hand, high boiling points are usually associated with inferior flammability (higher flash points), which is why PC has received excellent ratings with respect to process safety and environmental impact in GSK's solvent selection guide (for details, see Table S9†). Taking these and other criteria together, PC and DMC receive a favorable sustainability rating, which is why a study on their use in electrosynthesis appears promising.
To evaluate the potential of the PC–DMC solvent system, we have selected representative model reactions, i.e., a mediated process (TEMPO-catalyzed alcohol oxidation), a direct reduction (cathodic conversion of benzophenone), and a direct oxidation (anodic synthesis of diaryliodonium salts).
2. Results and discussion
2.1 Electrochemical properties of PC–DMC electrolyte systems
For applications in electrosynthesis, electrolyte properties such as electrochemical stability and conductivity are of the utmost importance. We therefore initiated our study by investigating the key-properties of the binary solvent system in combination with a supporting electrolyte frequently used in organic electrochemistry, i.e., Bu4NBF4. First, the electrochemical stability window was determined by cyclic voltammetry (CV) for a PC–DMC mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) at 10 mV s−1 and compared to pure PC, DMF, DCM, and AN (Fig. 1, for experimental details see the ESI†). As threshold current density, 0.1 mA cm−2 was defined, and the potentials read out at this position form the outer edges of the bars in the diagram.
:4) at 10 mV s−1 and compared to pure PC, DMF, DCM, and AN (Fig. 1, for experimental details see the ESI†). As threshold current density, 0.1 mA cm−2 was defined, and the potentials read out at this position form the outer edges of the bars in the diagram.
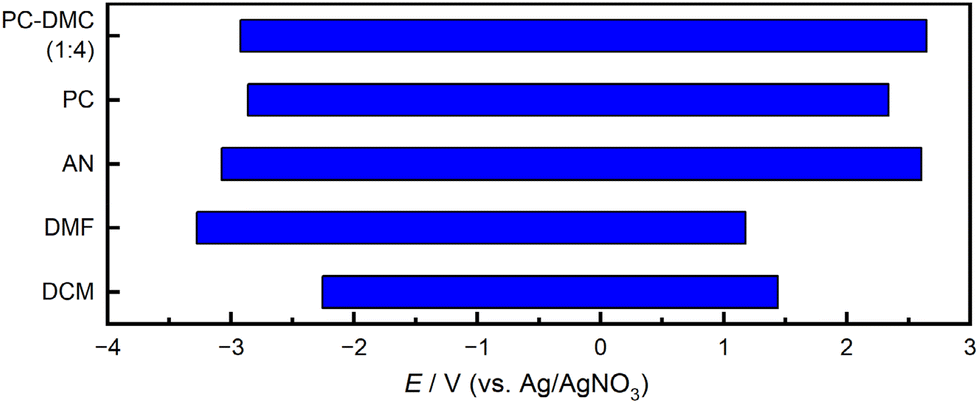 | ||
| Fig. 1 Electrochemical windows of PC and a PC/DMC mixture (1:4, w/w) compared to common solvents used in organic electrosynthesis. Determined using cyclic voltammetry at v = 10 mV s−1 using a glassy carbon working electrode (threshold current density: 0.1 mA cm−2). Supporting electrolyte: 0.1 M Bu4NBF4.‡ | ||
The measurements show for PC–DMC, pure PC, and AN comparable electrochemical windows, reaching from around −3.0 V to approx. 2.5 V vs. Ag/AgNO3. Interestingly, the PC–DMC mixture shows a slightly broader window than pure PC. DMF is most stable towards negative potentials, but much easier to oxidize, while DCM renders the lowest stability toward both anodic oxidation and cathodic reduction. Taken together, PC and DMC provide a window comparable to AN and superior to DMF and DCM, which should enable selective oxidation and reduction of a broad array of organic molecules even at strongly positive or negative potentials.
As another critical parameter influencing not only the ionic conductivity of the electrolyte, but also the diffusion rate of the substrate in the boundary layer between electrode and solution, we examined the dynamic viscosity (η) of PC–DMC mixtures as a function of the mass fraction of PC (Fig. 2a) with and without supporting electrolyte. As shown in Table 1, the viscosity of pure PC is significantly higher than AN, DMF, and DCM, while DMC is comparable to DCM. As expected, the viscosity of PC–DMC increases with increasing PC content, whereby below approx. 40% w/w, the η values are in the same range as DMF and DCM. Not surprisingly, addition of Bu4NBF4 (0.1 M) leads to slightly increasing η values.
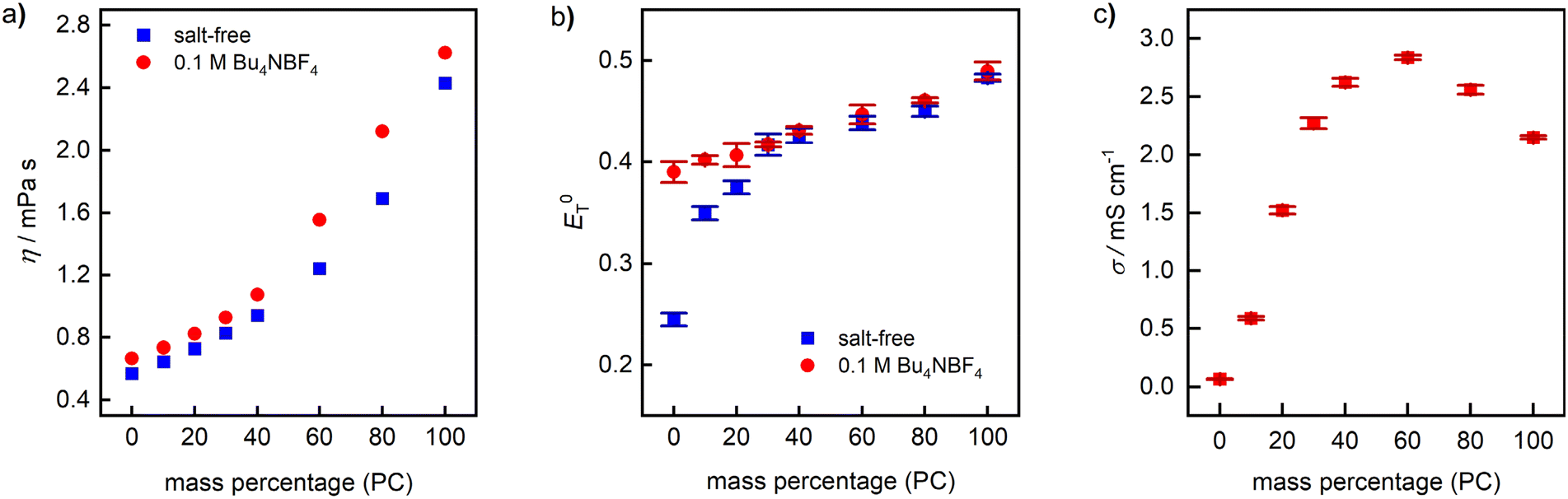 | ||
Fig. 2 Physical properties of PC–DMC-based electrolyte systems at 25 °C. (a) Dynamic viscosity (η). (b) Polarity  determined photometrically using Reichhardt's dye. (c) Ionic conductivity (σ) of a 0.1 M solution of Bu4NBF4. Reference values of 0.1 M solutions of Bu4NBF4 are 10.25 ± 0.02 mS cm−1 in AN, 5.15 ± 0.03 mS cm−1 in DMF, and 1.33 ± 0.03 mS cm−1 in DCM.‡ determined photometrically using Reichhardt's dye. (c) Ionic conductivity (σ) of a 0.1 M solution of Bu4NBF4. Reference values of 0.1 M solutions of Bu4NBF4 are 10.25 ± 0.02 mS cm−1 in AN, 5.15 ± 0.03 mS cm−1 in DMF, and 1.33 ± 0.03 mS cm−1 in DCM.‡ | ||
For the determination of the polarity (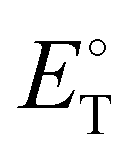 , Fig. 2b) on an empirical scale from 0 to 1, we applied a well-established spectroscopic method using Betaine 30 as solvatochromic dye.46,47 Generally, a high polarity is desirable, as it ensures good solubility and dissociation of supporting electrolytes. Since PC is the more polar solvent, an increase of its content leads to rising
, Fig. 2b) on an empirical scale from 0 to 1, we applied a well-established spectroscopic method using Betaine 30 as solvatochromic dye.46,47 Generally, a high polarity is desirable, as it ensures good solubility and dissociation of supporting electrolytes. Since PC is the more polar solvent, an increase of its content leads to rising  values,48 just as the addition of Bu4NBF4. The
values,48 just as the addition of Bu4NBF4. The  values of salt-free mixtures range from 0.24 to 0.49, which is comparable to DCM (0.31) and AN (0.46).25
values of salt-free mixtures range from 0.24 to 0.49, which is comparable to DCM (0.31) and AN (0.46).25
Finally, the ionic conductivity (σ, Fig. 2c) of the binary mixture was determined in the presence of 0.1 M Bu4NBF4. The ionic conductivity depends on the ion mobility and the degree of salt dissociation, whereby the former is influenced by the viscosity and the latter by the polarity of the medium.49 Consequently, only poor conductivity is observed in pure DMC, whereby σ strongly increases with increasing PC content. With 2.84 mS cm−1, the highest σ value was found at 60% w/w, followed by a decrease that is caused by the high viscosity of PC. Reference values of 0.1 M Bu4NBF4 solutions are 10.25 mS cm−1 in AN, 5.15 mS cm−1 in DMF, and 1.33 mS cm−1 in DCM. For electro-synthetic applications, σ of the PC–DMC system is in an acceptable regime at a PC content of approx. 20% w/w or higher. It should be noted that different supporting electrolytes will render different results and the concentration of the salt can also be varied to achieve better conductivity. Overall, pure DMC and mixtures with a high DMC content (>90%) seem to be unsuitable due to low conductivity, whereas pure PC or PC-rich mixtures provide acceptable values.
In general, the results shown in Fig. 1 and 2 are promising with respect to applications in electrochemical synthesis. The PC–DMC system should be suitable for anodic oxidations and cathodic reductions, both with respect to electrochemical stability and ionic conductivity. An interesting aspect is that the key-features are tunable via the composition of the binary solvent mixture, overcoming the drawbacks of the individual components.
2.2 TEMPO-mediated oxidation of alcohols
With encouraging electrochemical properties at hand, we investigated the behavior in applications using representative model cases starting with a mediated process. As an example for a mediated reaction, we chose the TEMPO-catalyzed alcohol oxidation (TEMPO = 2,2,6,6-tetramethylpiperidinyl-1-oxyl).50 In this reaction, TEMPO is anodically oxidized to the corresponding oxoammonium cation, followed by reaction with an alcohol to the corresponding carbonyl compound and the hydroxylamine. This hydroxylamine is then converted back to TEMPO by reaction with a second oxoammonium cation to close the catalytic cycle.51Prior to the synthetic work, CV studies were carried out with TEMPO in different mixtures of PC and DMC using 0.1 M Bu4NBF4 as a supporting electrolyte (Fig. 3). The latter was selected due to its good solubility at all PC–DMC ratios. It should be noted that although experiments were in principle feasible in pure DMC under non-catalytic conditions applying Ohmic drop compensation, the high electrolyte resistance led to unreliable results and made the detection of catalytic responses impossible. The test range was therefore limited to 20–100% PC w/w.
 | ||
| Fig. 3 (a) Diffusion coefficient D of TEMPO determined by CV of a 2.5 mM solution in varying PC–DMC compositions (top). Exemplary CVs recorded at 100 mV s−1 are shown at the bottom. (b) Maximum current densities (jmax, top) obtained from the catalytic responses in the presence of 4-MBA and 1-NMI under pure kinetic conditions (no substrate consumption) and of TEMPO in the presence of 0.1 M 4-MBA and 0.45 M 1-NMI (exemplary catalytic CVs recorded at 100 mV s−1 are shown at the bottom). (c) Shift of the equilibrium redox potential of TEMPO (E0, top) with varying composition of the PC–DMC (exemplary CVs are shown at the bottom).‡§ | ||
Initially, it was of interest to measure the diffusion coefficient of TEMPO depending on the solvent composition, as this plays an important role in determining the rate of the catalytic reaction.52 The diffusion rate of TEMPO should be governed by the viscosity of the medium. Indeed, as Fig. 3a shows, D decreases with increasing PC content and thereby with increasing viscosity (compare Fig. 2a). A similar behavior was also observed for ferrocene (see Fig. S20–S26†).
After addition of 0.45 M 1-methylimidazole (1-NMI) as a base and 0.1 M 4-methoxybenzylalcohol (4-MBA) as a substrate, the catalytic response of TEMPO was analyzed (Fig. 3b). The catalytic rate decreases with increasing PC content, as reflected by the representative CVs (bottom) and the maximum current densities (jmax, top) obtained under pure kinetic conditions (no substrate consumption, for details see the ESI†).52 Three explanations for this trend appear plausible: First, the decrease of the diffusion rate of TEMPO with increasing PC content affects the rate of the catalytic process, which would align well with the theory of homogeneous electrocatalysis.52 Second, a change of the polarity may have an impact on the activation barrier of the rate-limiting step of the homogeneous process. Third, a changing composition of the binary mixture alters the equilibrium redox potential (E0) of TEMPO and thereby the driving force for alcohol oxidation. The effect of the polarity of liquid media on E0 of redox couples is well known and described elsewhere.53–55 Indeed, a decrease of E0 at higher PC content is observed (Fig. 3c), suggesting that the equilibrium redox potential may also contribute to the observed kinetic trend. A similar shift of E0 can also be observed for the ferrocene/ferrocenium redox couple (see Fig. S20–S26†).
The effect of the composition of the PC–DMC mixture on the catalytic rate shown in Fig. 3b has implications for the conversion of alcohols in preparative-scale electrolysis, although other factors also play a role here and the situation is much more complex than in the CV experiment. The standard method for TEMPO oxidations in aprotic media involves an H-type divided cell and potentiostatic reaction control.50 To investigate the performance of the PC–DMC system, we aimed to develop a straightforward protocol that is easy to adopt by others and as resource-efficient as possible. The reaction was therefore optimized in a commercially available setup (IKA ElectraSyn 2.0) using an undivided cell, graphite electrodes, and NaClO4 as a cheap supporting electrolyte (for details of the optimization, see the ESI†). 4-MBA was chosen as the substrate and 1-NMI as the base. During these studies, it was possible to reduce the salt loading to 40 mM while maintaining sufficient conductivity. The optimized conditions further comprise a PC–DMC ratio of 1:4 w/w, a current density of 6 mA cm−2, and application of three charge equivalents (see Scheme 1).
 | ||
| Scheme 1 Preparative-scale TEMPO-mediated oxidation of different alcohols. Yields determined with 1H NMR spectroscopy using mesitylene as an internal standard. a0.2 M NaClO4. bIsolated yield. | ||
Under optimized conditions, a 74% yield of 2a was obtained. A comparative experiment conducted with a fivefold enhanced salt loading (0.2 M NaClO4) showed that the yield can be slightly improved to 82%, albeit at the expense of resource efficiency. Further experiments carried out in different standard solvents (DMF, AN, DCM) under otherwise identical conditions showed significantly lower yields in 2a (see Scheme S3†).
For a brief exploration of the substrate scope, 4-bromo benzyl alcohol, citronellol, and geraniol were converted to the corresponding aldehydes 2b–d under optimized conditions in PC–DMC (1:4), resulting in 1H NMR yields between 67 and 78%. Noteworthy, 2b could be isolated without significant loss of yield. For this purpose, a convenient work-up procedure was developed that involved partitioning of the reaction mixture followed by extraction (for details, see the ESI†).
2.3 Reduction of benzophenone
As an example for a direct (uncatalyzed) cathodic reduction, the conversion of benzophenone (3) to diphenyl carbinol (4) was selected (Scheme 2). Recently, two electrochemical methods were reported for the same transformation that used DMF and a 1:1 mixture of Cyrene™/ethanol, respectively (entries 1 and 2 in Table 2).33,56 In these cases, 4 was obtained in 79 and 85% yield, respectively, under optimized conditions. The protocols each comprise an undivided cell, galvanostatic conditions, Bu4NBF4 as the supporting electrolyte, and a glassy carbon (GC) cathode. In both examples, 1,4-diazabicyclo[2.2.2]octane (DABCO) was added as a sacrificial agent to be oxidized at the counter electrode.
 | ||
| Scheme 2 Direct reduction of benzophenone (3) to 4 in PC–DMC with possible by-product 5. | ||
| Entry | j [mA cm−2] | Reaction medium | Q [F] |
3a [%] |
4a [%] |
5a [%] |
Ref. |
|---|---|---|---|---|---|---|---|
| Conditions: WE = GC, n(substrate) = 0.5 mmol, c(NBu4BF4) = 0.12 M, n(DABCO) = 1.5 mmol (3 eq.).a Yields determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard.b Exclusion of O2 by purging with Ar.c 5 μL H2O added per mL electrolyte solution.d Potentiostatic electrolysis, E = −2.20 V vs. Ag/AgNO3.e n(substrate) = 1 mmol.f n(substrate) = 5 mmol.g Isolated yield. | |||||||
| 1 | 10 | DMF | 6.2 | — | 79g | — | 56 |
| 2 | 5 | Cyrene™/EtOH (1:1) |
3.5 | — | 85g | — | 33 |
| 3b | 7.5 | PC–DMC (1:4) |
2.2 | 10 | 45 | 32 | This work |
| 4b,c | 7.5 | PC–DMC (1:4) |
2.2 | 2 | 83 | 0 | This work |
| 5b,c | PC–DMC (1:4) |
2.0 | 2 | 95 | 2 | This work | |
| 6b,c,e | PC–DMC (1:4) |
2.0 | 12 | 88 (87)g | 0 | This work | |
| 7b,c,f | PC–DMC (1:4) |
2.0 | 8 | 89 (82)g | 0 | This work | |
Interestingly, reduction was carried out under aerobic conditions in both reports, whereby excess amount of charge was needed. We initiated our study with the same approach, but experienced unsatisfactory selectivity and irreproducible results. A possible explanation is cathodic reduction of O2 at −1.2 V, which proceeds much easier than conversion of 3 (−2.1 V). Since variation of current density and charge equivalents did not improve the results, we decided to exclude O2 by purging the electrolyte with argon prior to electrolysis. As a result, only 2.2 F of charge were needed for nearly full conversion (entry 3). However, under these conditions, monomethylcarbonate 5 was obtained in 32% yield. Such reductive transesterifications are known from conventional carbonyl reductions, for example when using NaBH4 in pure DMC.57 The selectivity was improved in favor of 4 by adding a small amount of water, resulting in a yield of 83% (entry 4). Further improvement of the yield to 95% was achieved by controlled potential electrolysis, whereby only 2.0 F were required per mole starting material (entry 5). To determine the isolated yield, another experiment was carried out with doubled batch size (entry 6), generating 4 in 88% NMR yield with 12% unconverted starting material. After removal of the solvent mixture by vacuum distillation and purification by column chromatography, 4 was obtained in 87% yield. To lower the process mass intensity (PMI),58 the substrate concentration was increased fivefold while maintaining the electrolyte volume and conditions of entry 6. As a result, 4 was isolated in 82% yield (5 mmol scale, entry 7).
The results in Table 2 show that harmful DMF can easily be replaced by PC–DMC while slightly increasing the efficiency and considerably reducing the required charge equivalents. To make further assertions on the sustainability of the reaction, the PMI was calculated and compared to literature examples (Table 3; for details, see the ESI†). The parameter describes the ratio between the mass of all components used (reactants, reagents, catalysts, and solvent) and the mass of the isolated product. Further metrics summarized in Table 3 are PMIsolv (mass of solvent vs. mass of isolated product) and PMIRRC (total mass of reactants, reagents and catalysts vs. mass of product). Compared to cathodic reduction of 3 in DMF,56 the values obtained in PC–DMC are significantly better, which can be ascribed to a higher concentration of 3. In comparison to a reported chemical reduction using NaBH4,59 our PMI is considerably lower, again resulting from a higher concentration of starting material. However, our PMIRRC value is considerably worse, which can be attributed to the use of DABCO as depolarizer and Bu4NBF4 as supporting electrolyte.
Comparing the PMIsolv and PMIRRC values of the two electrochemical examples highlights the importance of substrate concentration and supporting electrolyte loading for optimizing organic electrosyntheses. Processes at the counter electrode, in our case the anodic conversion of depolarizer, must also be included in the overall assessment. The data in Table 3 puts into question the often postulated “inherent greenness” of electrosynthesis, and clearly shows how much sustainability and effectiveness depend on the way the method is used.
2.4 Synthesis of a diaryl iodonium salt
As a test scenario for probing the suitability of the PC–DMC system in a direct anodic process, we selected the anodic synthesis of diaryl iodonium compound 7 (Scheme 3). In general, diaryliodonium salts have gained a growing interest as metal-free, easy-to-handle, and highly selective arylation reagents.60–62 Since many existing conventional procedures for their synthesis are waste-intensive, time-consuming, and involve hazardous reagents, electrochemical approaches have been developed that are based on anodic coupling between aryl iodide and a second arene.63–65 However, these methods involve either the use of strong acids and/or fluorinated solvents. In this context, we have recently developed a low-cost and safe method, in which an acid-free solution of a lithium salt in AN was used as the electrolyte, enabling the introduction of various counter ions to the product by choice of the appropriate Li salt.66 This protocol served as the starting point for our studies on anodic oxidation in the PC–DMC system. We chose 4-bromo-iodobenzene (6) as a test substrate, as it rendered excellent results in AN with an isolated yield of 97%.66 The reactions were carried out in a divided cell using a Pt anode and 1 M LiClO4 as the supporting electrolyte. After optimizing the PC–DMC ratio, current density, and number of charge equivalents, 7 was obtained in 89% isolated yield. The optimized conditions comprise a PC–DMC ratio of 4:1, a current density of 5 mA cm−2, and the application of 4 F per mole precursor 6.
 | ||
| Scheme 3 Electrochemical synthesis of diaryliodonium salt 7 (isolated yield). | ||
Interestingly, it turned out during the optimization that conversion and yield strongly depend on the PC–DMC ratio (see Fig. 4). While conversion of 6 decreases continuously from 92 to 75%, yields varied between 66% and 82%, with a maximum at a PC–DMC ratio of 4:1 w/w. These trends highlight the flexibility and possibilities for tuning the electrolyte properties for synthetic applications that arise when using the PC–DMC system.
 | ||
| Fig. 4 Anodic synthesis of 7 from 6 and benzene: relationship between yield, conversion, and the composition of the PC–DMC system. Except for the current density, the conditions are the same as in Scheme 3 (here: j = 10 mA cm−2). | ||
3 Conclusions
In summary, the present study shows that the PC–DMC system can be a sustainable replacement for problematic aprotic solvents frequently used in electrosynthesis, i.e., DMF, AN, and DCM. Both PC and DMC are highly stable to both anodic oxidation and cathodic reduction, which makes them versatile for a wide range of transformations. Using the examples of cathodic ketone reduction and anodic diaryl iodonium synthesis, it has been demonstrated that conversions requiring very negative or positive potentials can proceed selectively in PC–DMC on a preparative scale. The obtained yields are comparable to those reported in the literature for DMF and AN, respectively.An interesting feature of the binary solvent system is the possibility of modifying the physicochemical properties such as viscosity, polarity, and conductivity by simply changing the PC–DMC ratio. Electroanalytical studies on TEMPO-catalyzed alcohol oxidation showed how this ratio affects the diffusion rate of the catalyst and its redox potential. As a result of these influences, a clear dependence of the current density on the PC–DMC composition was observed, whereby the rate of alcohol oxidation decreases continuously with increasing PC content. Based on these results, a new protocol for TEMPO-catalyzed alcohol oxidation was developed, which stands out due to its simplicity and resource efficiency (i.e., undivided cell, galvanostatic mode, low supporting electrolyte loading), and is applicable to benzylic, allylic, and even aliphatic alcohols.
The remaining challenge is the high boiling point of PC, which may complicate the workup of the reaction mixture. Therefore, three strategies were developed for convenient isolation of the products from PC–DMC electrolyte mixtures without significant losses in yield. The first strategy, applied in the TEMPO-catalyzed alcohol oxidation, is based on partitioning of the product mixture, followed by extraction. The second strategy, applied to the ketone reduction, features vacuum distillation for removing the solvent mixture. The third strategy was developed for our synthesis of the diaryl iodonium salt and includes separation of the product from the PC–DMC electrolyte by adsorption on silica gel. Thus, at least in our three test cases, the high boiling point of PC does not pose a particular problem when separating the product mixture on the laboratory scale. However, it should be noted that the three presented approaches are challenging with respect to upscaling. While the partitioning/extraction approach requires considerable amounts of extractant, distillation in vacuum exhibits an increased energy demand for removal of PC–DMC. The third approach, product adsorption on silica gel, depends on the use of a high quantity of adsorbent. Consequently, developing new separation strategies (and improving existing ones) is an important area for future research, as many green solvents struggle with separation due to higher boiling points.
Moreover, in view of possible developments of new electrochemical processes on an industrial scale, it should be noted that PC receives excellent ratings in terms of process safety and environmental compatibility precisely because of its low volatility (high flash point).21
In summary, our study reveals promising properties and performance of the PC–DMC system in electrosynthesis applications. Consequently, we encourage readers with a focus on organic electrochemistry to include the PC–DMC system in solvent screenings for future reaction developments. Likewise, physical and theoretical chemists are urged to support with a better understanding of solvation properties of the PC–DMC system.
Author contributions
Investigations in the reactions, development of methodology and writing of the original draft were done by A. Prudlik. J. I. Bardagí, A. Matei. A. Scherkus assisted in conducting the experiments and writing. S. Beil and R. Francke supervised the research and reviewed and edited the draft. Conceptualization was carried out by R. Francke.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the German Research Foundation (DFG Heisenberg Program, FR 3848/4-1 and Individual Research Grant, FR 3848/2-2) is gratefully acknowledged. The authors thank Anika Wilhelms (Institute of Chemistry, Rostock University) for conducting the viscosity measurements and Stefanie Perdomo (Stratingh Institute for Chemistry, University of Groningen) for initial testing of alcohol oxidation conditions. JIB acknowledges financial support by the Alexander von Humboldt Foundation. SB and AM are grateful for financial support from the Netherlands Organization for Scientific Research (NWO) through the Advanced Research Center Chemical Building Blocks Consortium (ARC-CBBC, 2021.038.C.RUG.9).References
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 CAS.
- H. J. Schäfer, C. R. Chim., 2011, 14, 745–765 Search PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CAS.
- A. Wiebe, T. Gieshoff, S. Mohle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CAS.
- S. B. Beil, D. Pollok and S. R. Waldvogel, Angew. Chem., Int. Ed., 2021, 60, 14750–14759 CAS.
- R. Francke, Curr. Opin. Electrochem., 2022, 36, 101111 CAS.
- R. Francke, Chimia, 2020, 74, 49 CAS.
- Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CAS.
- T. Broese, A. F. Roesel, A. Prudlik and R. Francke, Org. Lett., 2018, 20, 7483–7487 CAS.
- B. Schille, N. O. Giltzau and R. Francke, Angew. Chem., Int. Ed., 2018, 57, 422–426 CAS.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 CAS.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- T. Sawamura, S. Kuribayashi, S. Inagi and T. Fuchigami, Adv. Synth. Catal., 2010, 352, 2757–2760 CrossRef CAS.
- N. Mohebbati, A. Prudlik, A. Scherkus, A. Gudkova and R. Francke, ChemElectroChem, 2021, 8, 3837–3843 CAS.
- A. Prudlik, N. Mohebbati, L. Hildebrandt, A. Heck, L. Nuhn and R. Francke, Chem. – Eur. J., 2023, 29, e202202730 CAS.
- B. Johnson, R. Francke, R. D. Little and L. A. Berben, Chem. Sci., 2017, 8, 6493–6498 CAS.
- A. Das and S. S. Stahl, Angew. Chem., Int. Ed., 2017, 56, 8892–8897 CrossRef CAS PubMed.
- D. P. Hickey, R. D. Milton, D. Chen, M. S. Sigman and S. D. Minteer, ACS Catal., 2015, 5, 5519–5524 CAS.
- M. C. Leech and K. Lam, Nat. Rev. Chem., 2022, 6, 275–286 Search PubMed.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 CAS.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 Search PubMed.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 Search PubMed.
- A. Wypych and G. Wypych, Databook of Solvents, ChemTec Publishing, 2019 Search PubMed.
- M. N. Roy, L. Sarkar and R. Dewan, Bull. Chem. Soc. Ethiop., 2010, 24, 103–114 CAS.
- S. Schrödle, G. Annat, D. R. MacFarlane, M. Forsyth, R. Buchner and G. Hefter, Chem. Commun., 2006, 1748–1750 Search PubMed.
- A. G. Kontos, M. Fardis, M. I. Prodromidis, T. Stergiopoulos, E. Chatzivasiloglou, G. Papavassiliou and P. Falaras, Phys. Chem. Chem. Phys., 2006, 8, 767–776 CAS.
- C. Mialkowski, A. Chagnes, B. Carré, D. Lemordant and P. Willmann, J. Chem. Thermodyn., 2002, 34, 1847–1856 CAS.
- J. Barthel and M. Kleebauer, J. Solution Chem., 1991, 20, 977–993 CrossRef CAS.
- E. Hawlicka and R. Grabowski, Ber. Bunsenges. Phys. Chem., 1990, 94, 486–489 CrossRef CAS.
- R. Schmid, J. Solution Chem., 1983, 12, 135–152 CrossRef CAS.
- J. M. Ramos-Villasenor, J. Sotelo-Gil, S. E. Rodil and B. A. Frontana-Uribe, Faraday Discuss., 2023, 247, 182–194 RSC.
- K. L. Wilson, J. Murray, C. Jamieson and A. J. B. Watson, Org. Biomol. Chem., 2018, 16, 2851–2854 RSC.
- K. L. Wilson, A. R. Kennedy, J. Murray, B. Greatrex, C. Jamieson and A. J. B. Watson, Beilstein J. Org. Chem., 2016, 12, 2005–2011 CAS.
- G. Moumouzias, G. Ritzoulis, D. Siapkas and D. Terzidis, J. Power Sources, 2003, 122, 57–66 CrossRef CAS.
- S. H. Pyo, J. H. Park, T. S. Chang and R. Hatti-Kaul, Curr. Opin. Green Sustainable Chem., 2017, 5, 61–66 Search PubMed.
- T. R. Jow, K. Xu, O. Borodin and M. Ue, Electrolytes for Lithium and Lithium-Ion Batteries, Springer, 2014 Search PubMed.
- E. Lust, A. Jänes and M. Arulepp, J. Electroanal. Chem., 2004, 562, 33–42 CAS.
- C. C. Truong, D. K. Mishra and V. Mishra, in Green Sustainable Process for Chemical and Environmental Engineering and Science, ed. Inamuddin, R. Boddula, M. I. Ahamed and A. M. Asiri, Elsevier, 2021, pp. 253–275 Search PubMed.
- B. Schaffner, F. Schaffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CAS.
- J. S. Bello Forero, J. A. Hernández Muñoz, J. Jones Jr. and F. M. da Silva, Curr. Org. Synth., 2016, 13, 834–846 CAS.
- M. A. Rasool, P. P. Pescarmona and I. F. J. Vankelecom, ACS Sustainable Chem. Eng., 2019, 7, 13774–13785 CAS.
- H. L. Parker, J. Sherwood, A. J. Hunt and J. H. Clark, ACS Sustainable Chem. Eng., 2014, 2, 1739–1742 CrossRef CAS.
- J. Seidler, A. Roth, L. Vieira and S. R. Waldvogel, ACS Sustainable Chem. Eng., 2023, 11, 390–398 CAS.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CAS.
- V. Saini and R. Kumar, New J. Chem., 2022, 46, 16981–16989 CAS.
- H. Langhals, Angew. Chem., Int. Ed. Engl., 1982, 21, 724–733 CrossRef.
- G. Wittstock, Lehrbuch der Elektrochemie, Wiley-VCH, 2023 Search PubMed.
- M. F. Semmelhack, C. S. Chou and D. A. Cortes, J. Am. Chem. Soc., 1983, 105, 4492–4494 CAS.
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed.
- C. Costentin and J.-M. Savéant, Elements of molecular and biomolecular electrochemistry: An electrochemical approach to electron transfer chemistry, Wiley, 2019 Search PubMed.
- J. Zhang, H. Ying, Y. Zhang, J. Yao and H. Li, J. Phys. Chem. B, 2023, 127, 5899–5904 CrossRef CAS PubMed.
- I. Noviandri, K. N. Brown, D. S. Fleming, P. T. Gulyas, P. A. Lay, A. F. Masters and L. Phillips, J. Phys. Chem. B, 1999, 103, 6713–6722 CrossRef CAS.
- N. G. Tsierkezos and U. Ritter, J. Appl. Electrochem., 2010, 40, 409–417 CrossRef CAS.
- L. Wang, X. Zhang, R. Y. Xia, C. Yang, L. Guo and W. J. Xia, Synlett, 2022, 1302–1308 CrossRef CAS.
- A. Osumah, J. Magolan and K. V. Waynant, Tetrahedron Lett., 2019, 60, 151203 Search PubMed.
- E. R. Monteith, P. Mampuys, L. Summerton, J. H. Clark, B. U. W. Maes and C. R. McElroy, Green Chem., 2022, 22, 123–135 Search PubMed.
- J. Desroches, P. A. Champagne, Y. Benhassine and J.-F. Paquin, Org. Biomol. Chem., 2015, 13, 2243–2246 RSC.
- E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- P. Villo and B. Olofsson, in Patai's Chemistry of Functional Groups - Chemistry of Hypervalent Halogen Compounds, ed. B. Olofsson, I. Marek, Z. Rappoport, Wiley, 2018, pp. 461–522 Search PubMed.
- M. J. Peacock and D. Pletcher, J. Electrochem. Soc., 2001, 148, D37–D42 CrossRef CAS.
- K. Watts, W. Gattrell and T. Wirth, Beilstein J. Org. Chem., 2011, 7, 1108–1114 CrossRef CAS PubMed.
- M. Elsherbini and W. J. Moran, Org. Biomol. Chem., 2021, 19, 4706–4711 RSC.
- A. Scherkus, A. Gudkova, B. Müller, T. Bystron and R. Francke, J. Org. Chem., 2024, 89, 14129–14134 CrossRef CAS PubMed.
- K. Izutsu, Anal. Sci., 2011, 27, 685–694 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc06199c |
| ‡ Shown results in Fig. 1–3 are average values of at least 2 measurements. Since the statistic error in the determination of the potential window (Fig. 1) is <0.025 V in all cases, error bars have been omitted. In the case of viscosity determinations (Fig. 2), the measurements were automatically repeated until the results are within a fixed error limit. |
| § Deviations may be caused by liquid junction potentials (LJPs) occurring at the interface between main cell compartment and reference electrode (PC–DMC|AN). Since for analyte and reference electrode solutions, the same supporting electrolyte has been used in the same concentration, the effect is expected to be minor. Under these circumstances, LJPs between polar aprotic solvents are typically <20 mV (ref. 67). |
| This journal is © The Royal Society of Chemistry 2025 |