
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and chemical recycling of biobased poly(acetal-ester)s with a non-cyclic acetal unit†
Niklas
Warlin
 ab,
Sathiyaraj
Subramaniyan
a,
Maria Nelly
Garcia Gonzalez
c,
Rafael N. L.
de Menezes
a,
Smita V.
Mankar
ad,
Nitin G.
Valsange
a,
Nicola
Rehnberg
ae,
Patric
Jannasch
*a and
Baozhong
Zhang
*a
ab,
Sathiyaraj
Subramaniyan
a,
Maria Nelly
Garcia Gonzalez
c,
Rafael N. L.
de Menezes
a,
Smita V.
Mankar
ad,
Nitin G.
Valsange
a,
Nicola
Rehnberg
ae,
Patric
Jannasch
*a and
Baozhong
Zhang
*a
aCentre for Analysis and Synthesis, Department of Chemistry, Lund University, P.O. Box 124, SE-22100 Lund, Sweden. E-mail: baozhong.zhang@chem.lu.se, patric.jannasch@chem.lu.se
bDepartment of Chemistry, Stanford University, Stanford, California 94305-5080, USA
cEnvironmental and Energy Systems Studies, Department of Technology and Society, Lund University, PO Box 118, SE-221 00 Lund, Sweden
dDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA
eBona Sweden AB, R&D, Box 210 74, SE-200 21 Malmö, Sweden
First published on 4th February 2025
Abstract
There is a high demand to design and develop recyclable biobased polymers using eco-friendly synthetic approaches. In this work, we present the facile synthesis of two dicarboxylate monomers with a central non-cyclic acetal unit (dimethyl 4,4′-(methylenebis(oxy))dibenzoate and dimethyl 4,4′-(methylenebis(oxy))bis(3-methoxybenzoate)), using potentially bio-sourced methyl paraben and methyl vanillate. The synthetic parameters were evaluated by a cradle to gate life cycle greenhouse gas emissions aiming to understand the environmental impacts. The two monomers were subjected to melt polycondensation with three linear aliphatic diols (ethylene glycol, 1,4-butanediol, and 1,6-hexanediol) with varied lengths and flexibility to yield six polyesters with molecular weights (Mn) ranging from 10–19 kDa, tunable glass transition (Tg ∼ 41–83 °C), and relatively high thermal stability. Most of the obtained biobased polyesters were amorphous, except for the polymer derived from butanediol and methyl paraben, which showed slow crystallization according to differential scanning calorimetry results. We discovered that the non-cyclic acetal bonds in the poly(acetal-ester)s could be selectively hydrolyzed under mild temperature (70 °C) by varying the acid concentration, offering a potential strategy for chemical recycling.
Green foundation1. Biobased polyesters containing non-cyclic acetal units were designed and synthesized, which showed decent thermal stability and tunable glass transition temperatures, and they could be conveniently depolymerized under mild acidic conditions to yield monomers that are suitable for repolymerization.2. The molecular design towards biobased polyesters with chemical recyclability based on non-cyclic acetal structures was demonstrated. The environmental impacts of the synthetic factors were preliminarily understood by LCA. 3. In the future, various possibilities of repolymerization of the monomers obtained from acidic depolymerization of the new polyesters with non-cyclic acetals could be investigated in detail, which is expected to provide insights into closed-loop recycling or upcycling. |
Introduction
Conventional plastics have received growing environmental concerns, such as plastics littering, greenhouse gas emissions, and global warming.1 The transition toward sustainable biobased plastics has been considered a solution to some of these challenges.2–5 However, biobased polymers have been frequently criticized for imposing challenges in the current recycling process.6,7 Today, most polymers are recycled by thermomechanical processes (i.e., mechanical recycling), which requires perfect sorting and separation of different types of polymers.8,9 Any new polymer (even biobased ones) may further complicate the sorting of mixed plastic waste and thus hinder mechanical recycling.10 Therefore, alternative recycling methods based on chemical or biological processes have been under intensive development,11–18 aiming to enhance the recyclability of new biobased polymers. In this context, biobased polymers with acetal units (namely polyacetals) have received growing attention because acetal bonds can be conveniently cleaved under acidic conditions, which may facilitate chemical recycling.19–24The outcome of acidic hydrolysis of polyacetals can be different if their backbones contain other acid labile functional groups (e.g., esters, amides, urethanes, etc.). For instance, our group has recently synthesized lignin-based poly(acetal-ester)s that can be selectively depolymerized by acetal hydrolysis, while leaving the ester bonds completely intact.19 This enabled energy-efficient short-loop chemical recycling via the formation and repolymerization of telechelic polymers. Interestingly, the ester bonds in a different xylose-based poly(acetal-ester) could be selectively hydrolyzed under acidic conditions, leaving the cyclic acetal bonds intact.25 Other cyclic acetal units have also been used for the design and synthesis of biobased chemically-recyclable polyacetals, which yielded different building blocks upon acidic hydrolysis.15,19,26 In addition, noncyclic acetals have been investigated in the development of drug delivery vehicles27–30 and crosslinked thermoset resins31–33 which are generally easier to hydrolyze compared to cyclic acetals.23,34 Surprisingly, noncyclic acetals have received little attention in the development of chemical recyclable thermoplastic polymers, and the structure–property relationship concerning their acidic hydrolysis remains largely unknown.
Another frequently encountered criticism of bioplastics is whether they are truly eco-friendly simply because they are made from renewable carbon.35 Although the use of renewable biomass sources can be generally perceived as sustainable, the production of raw materials and polymers often involves many different parameters such high temperature, and the use of solvents and catalysts. The full environmental impacts of bioplastic synthesis can thus be difficult to predict. As such, preliminary life cycle assessments (LCA) have recently started gaining popularity in the development of new bioplastics. For instance, the design and synthesis of plant oil-based acrylic monomers were optimized to minimize their environmental impacts according to LCA.36 In another approach, a prospective LCA was used to assess the chemical and biocatalytic synthesis of a lactone-based monomer.37 In our own efforts to develop new sugar- or lignin-based polymers, a preliminary LCA was used to confirm the low greenhouse gas (GHG) emissions of diol – and dicarboxylate monomers containing spirocyclic acetal structures.15,22,38,39 However, the use of LCA in the synthesis of new bioplastics is still rare, presumably due to the lack of LCA data for most new molecules.
Herein, we report the molecular design and synthesis of two new dicarboxylate monomers with a non-cyclic formaldehyde-based acetal unit using the potentially bio-based monomeric building blocks methyl vanillate and methyl paraben (Scheme 1).40,41 Various synthetic parameters were optimized according to their preliminary GHG emissions. The monomers were reacted with linear aliphatic diols to produce two series of poly(acetal-ester)s, of which the molecular, thermal characteristics and chemical recyclability under acidic conditions were preliminarily investigated. The impact of methoxy groups on the acidic stability of the obtained polymers was also investigated.
 | ||
| Scheme 1 Synthesis of dicarboxylate monomers 1 and 2 and their polymerization with three aliphatic diols 3a–c to prepare polyesters P1a–c and P2a–c (a–c in the polymer names indicate the use of aliphatic diols 3a–c for polymerizations, respectively). | ||
Experimental
Materials
Potassium carbonate (K2CO3, 99%), dibutyltinoxide (DBTO, 98%), ethylene glycol (99%), mesitylene (98%), CDCl3 (99.8% atom D, 1% TMS), 1,6-hexanediol (97%), dimethylsulfoxide (DMSO, 99.9%), DMSO-d6 (99.9% atom D), KI (99%), methyl vanillate (99%), methyl paraben (99%), and dibromomethane (99%) were purchased from Sigma Aldrich. Acetone (99.8%) was purchased from Acros. NaHCO3 (99.7%) and 2-propanol (99.8%) were purchased from Merck. 1,4-Butanediol (99%) was purchased from Riedel de haen. Xylenes was purchased from Scharlau. NaOH and chloroform (99.8%) were purchased from Fischer. Methanol, dichloromethane (DCM), tert-butanol (t-BuOH), and acetonitrile (ACN) were purchased from VWR chemicals. All solvents were of analytical grade or higher, and all reagents and chemicals were used without further purification.Analytical methods
1H and 13C NMR measurements were performed on a Bruker DR X400 spectrometer at 400.13 MHz and 100.61 MHz, respectively. Chemical shifts were reported as δ values (ppm). HRMS (high-resolution mass spectrometry) was performed on a Micromass QTOF mass spectrometer (ESI). Size exclusion chromatography (SEC) measurements for the polyesters were carried out using Malvern Viscotek TDAmax instrument with a 2 × PL-Gel Mix-B LS column set (2 × 30 cm) equipped with OmniSEC triple detectors (refractive index, viscosity and light scattering) and chloroform as eluent at 35 °C at a flow rate of 1 mL min−1. Calibration was performed with a narrow polystyrene standard Mp = 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, Đ = 1.03 (Polymer Laboratories Ltd, Agilent Technologies and Water Associates). Fourier transform infrared (FTIR) spectra were measured with an attenuated total reflection (ATR) setup using a Bruker Alpha II FT-IR spectrometer. TGA (thermogravimetric analysis) measurements were carried out on a TA instrument mode TGA Q500. The measurement was performed by heating from 50–600 °C with a heating rate of 10 °C min−1. DSC (differential scanning calorimetry) measurements were performed on a DSC Q2000 analyzer from TA instruments. Data was recorded from −50 to 200 °C, and Tg was determined from the second heating cycle. Annealing was performed at 10 °C below the Tm observed in the first heating cycle. The synthesized polyesters were made into films of dimensions (17.5 × 5.5 × 1 mm) for dynamic mechanical analysis (DMA). DMA measurements were performed in a stretching mode using TA instruments Q800 analyzer. The measured samples were heated from 20 °C to 100 °C at a heating rate of 3 °C min−1 and frequency of 1 Hz with strain of 0.1%.
000 Da, Đ = 1.03 (Polymer Laboratories Ltd, Agilent Technologies and Water Associates). Fourier transform infrared (FTIR) spectra were measured with an attenuated total reflection (ATR) setup using a Bruker Alpha II FT-IR spectrometer. TGA (thermogravimetric analysis) measurements were carried out on a TA instrument mode TGA Q500. The measurement was performed by heating from 50–600 °C with a heating rate of 10 °C min−1. DSC (differential scanning calorimetry) measurements were performed on a DSC Q2000 analyzer from TA instruments. Data was recorded from −50 to 200 °C, and Tg was determined from the second heating cycle. Annealing was performed at 10 °C below the Tm observed in the first heating cycle. The synthesized polyesters were made into films of dimensions (17.5 × 5.5 × 1 mm) for dynamic mechanical analysis (DMA). DMA measurements were performed in a stretching mode using TA instruments Q800 analyzer. The measured samples were heated from 20 °C to 100 °C at a heating rate of 3 °C min−1 and frequency of 1 Hz with strain of 0.1%.
Assessing global GHG emissions
The comparative analysis between Monomers 1 and 2 was performed following LCA methodology, which is standardized in the ISO 14040-14044 series by the International Organization of Standardization. The lab data was incorporated into the SimaPro LCA software, and the Ecoinvent database version 3.8. (allocation at the point of substitution (APOS) system model) was used as a background source. The foreground data was then evaluated. The functional unit was “kg of monomer”. The system boundary was set up following a cradle-to-factory gate approach. At this early stage, the study was based on the IPCC 2021 method, which contains the Global Warming Potential (GWP100) climate change factors of IPCC with a timeframe of 100 years. The most important precursor emissions originating from this impact category are carbon dioxide (CO2), nitrous oxide (N2O), and methane (CH4). The characterization factors needed to quantify how much impact the new monomers have in GHG emissions are taken from the IPCC 2021 method.We assumed that the energy required to produce 1 kg of monomer was equal to the energy required to produce 1 kg of polyvinylchloride by emulsion polymerization at 70–90 °C. The energy required for heating the reaction was calculated according to eqn (1), ESI.† The energy costs and reagents were taken from the Ecoinvent database, version 3.8. The energy cost to produce the solvent was excluded. LCA data for dibromomethane was unavailable, so its analogous molecule dichloromethane was used for calculations. The LCA data on 4-hydroxybenzoic acid (4-HBA) based on cane sugar was taken from Krömer et al. (6.57 kg CO2-eq per kg 4-HBA).42 In this study, results are given in a range of 5.96–12.25 kg CO2-eq per kg 4-HBA. These values depended on recycling of the biomass, the type of feedstock etc. Our choice was based on the scaled up base case for non-recycled biomass, and the intermediate value for the available feedstocks was taken as an average.42 Transportation of the feedstock and solvents from one factory to the other for the monomer production was excluded.
The LCA data on lignin-derived vanillin was taken from Borregaard (0.7 kg CO2-eq per kg DM).43 To this value was added the estimated GHG emissions corresponding to the process of oxidizing vanillin to vanillic acid. Since LCA data for the exact oxidation of vanillin to vanillic acid is unavailable in the Sima Pro software, the GHG emissions for the oxidation of a similar molecule, toluene, into benzoic acid was used as an approximation (0.41 kg CO2-eq per kg molecule). The total GHG emissions to produce vanillic acid were therefore estimated as 1.11 kg CO2-eq per kg vanillic acid.
Monomer synthesis
To a 250 mL round bottom flask equipped with magnetic stirrer, dibromomethane (5.00 g, 0.0285 mol), methyl paraben (8.75 g, 0.0575 mol), potassium carbonate (8.74 g, 0.0632 mol) and potassium iodide (0.477 g, 0.0028 mol) were added. Afterwards, acetonitrile (100 mL) was added until the reagents were completely dissolved. The reaction was heated with reflux for 24 h, and then poured into cold water (1000 mL) to form a white precipitate. The precipitate was collected by vacuum filtration, washed with cold water (200 mL), and dried at 50 °C in vacuum oven to give monomer 1 as a white powder (8.0 g, 88%). 1H NMR (400.13 MHz, CDCl3, δ, ppm): 8.00 (d, 4H, J = 8.93 Hz, Ar–H), 7.12 (d, 4H, J = 8.89 Hz, Ar–H), 5.81 (s, 2H, acetal CH2), 3.88 (s, 6H, –COOCH3) 13C NMR (100.61 MHz, CDCl3, δ, ppm): 166.56, 160.26, 131.64, 124.51, 115.75, 90.03, 51.99. HR-MS (ESI+, m/z): exact calculated for [C17H16O6 + Na]+: 339.0844 found 339.0845.To a 250 mL round bottom flask equipped with a magnetic stirrer, dibromomethane (5.00 g, 0.0285 mol), methyl vanillate (10.5 g, 0.0575 mol), potassium carbonate (8.74 g, 0.0632 mol) and potassium iodide (0.477 g, 2.80 mmol) were added. Afterwards, acetonitrile (100 mL) was added until the reagents were completely dissolved. The reaction was heated with reflux for 24 h, and then poured into cold water (1000 mL) to form a white precipitate. The precipitate was collected by vacuum filtration, washed with cold water (200 mL), and dried at 50 °C in vacuum oven to give monomer 2 as a white powder (9.8 g, 91%). 1H NMR (400.13 MHz, CDCl3, δ, ppm): 7.63 (dd, 2H, J = 8.45 Hz, J = 1.98 Hz, Ar–H), 7.57 (d, 2H, J = 1.93 Hz, Ar–H), 7.28 (d, 2H, J = 8.49, Ar–H), 5.87 (s, 2H, acetal CH2), 3.889 (s, 6H, –OCH3), 3.886 (s, 6H, –COOCH3). 13C NMR (400.13 MHz, CDCl3, δ, ppm): 166.65, 149.78, 149.57, 125.19, 123.26, 116.29, 112.93, 92.13, 56.06, 52.13. HR-MS (ESI+, m/z): exact calculated for [C19H20O8 + Na]+: 399.1052 found 399.1056.
Optimization of monomer synthesis
The synthetic procedure of reaction F was described as a typical example:To a 25 mL round bottom flask equipped with magnetic stirrer, dibromomethane (171 mg, 0.980 mmol), methyl paraben (302 mg, 1.98 mmol), KI catalyst (34 mg, 0.2 mmol), and NaOH (88 mg, 2.2 mmol) were added. Afterwards, DMSO (5 mL) was added. The reaction was stirred at 56 °C for 24 hours after which a droplet was taken out, dried for 3 minutes under vacuum, and then analyzed by 1H-NMR spectroscopy.
Polymer synthesis
The synthetic procedure of P1b was described as a typical example:To a 25 mL round bottom flask equipped with mechanical stirrer, 1,4-butanediol (0.860 g, 9.54 mmol), DBTO (20 mg, 0.080 mmol), monomer 1 (2.00 g, 6.32 mmol) and 2 mL of xylenes were added. The reaction mixture was heated to 200 °C and stirred under nitrogen for 4 h. Afterwards, the temperature was increased to 220 °C with mesitylene (10 mL) added. A faster nitrogen flow was applied for 12 h to facilitate removal of the diol. Afterwards, the polymer was dissolved in chloroform and precipitated into methanol to give the polymer (P1b) as a white solid (1.90 g, 88%). 1H NMR (400.13 MHz, CDCl3, δ, ppm): 8.00 (d, 4 H, J = 8.92 Hz, Ar–H), 7.11 (d, 4 H, J = 8.91 Hz, Ar–H), 5.80 (s, 2 H, acetal CH2), 4.35 (s, 4 H, O–CH2–), 1.91 (s, 4 H, –CH2–). 13C NMR (100.61 MHz, CDCl3, δ, ppm): 166.02, 160.26, 131.63, 124.61, 115.77, 90.02, 64.34, 25.58.
Chemical recycling
Acid hydrolysis of P1a was described as a typical example:P1a (158 mg) powder was added to a 25 mL round bottom flask equipped with a magnetic stirrer. HCl (12 M, 15 mL) was added, and the reaction was stirred at 70 °C for 24 h. Afterwards, the solution was diluted with water (30 mL), and K2CO3 (9.16 g) was added until pH ≈ 1. Afterward, the slurry was filtered to give a solid as the main crude product (107 mg), which was the hydrolyzed bis-phenol structure. The aqueous phase was extracted with DCM (3 × 50 mL) and the combined organic phase was concentrated in vacuo to give another crude solid (3.5 mg), which was a mixture of small molecules formed by hydrolysis of the ester bonds in the polymer.
Ester hydrolysis
A typical example is described below.Methyl vanillate (180 mg) was added to a 40 mL vial equipped with a magnetic stirrer. HCl (12 M, 5 mL) was added to the vial and the reaction was stirred at 70 °C for 1 h. Afterwards, the solution was diluted with water (10 mL), and K2CO3 (3.0 g) was added until pH ≈ 1. After this, the slurry was filtered to give a solid as the first crude product consisting primarily of methyl vanillate and vanillic acid (35.7 mg). The aqueous phase was extracted with DCM (3 × 15 mL) and the combined organic phase was concentrated in vacuo to give another crude solid consisting of methyl vanillate and vanillic acid (89.8 mg). For detailed calculations of the methyl vanillate content please see the ESI.†
Results and discussion
Synthesis and environmental analysis of monomers
The dicarboxylate ester Monomers 1 and 2 were synthesized by a straightforward SN2 reaction of methyl vanillate or methyl paraben using dibromomethane according to a modified procedure from a patent (Scheme 1).44 A mild base (K2CO3) was used to enhance the nucleophilicity of the phenol groups, and potassium iodide (KI) was used as catalyst. The reaction was completed in 24 h, which afforded Monomer 1 and 2 in good yields (88–91%) after a straightforward purification by precipitation in water. The monomers were unambiguously characterized by NMR, as shown in Fig. S1–8.†The key reaction parameters influencing GHG emissions and conversion were evaluated for the synthesis of Monomer 1 from an environmental impact perspective. The Global Warming Potential (GWP), an index measuring the infrared thermal radiation absorption of a greenhouse gas over a given time frame, was calculated for various conditions. These included different solvents (ACN, acetone, DMSO, and t-BuOH), bases (NaOH and K2CO3), and reaction temperatures (20 °C to 82 °C), as detailed in Table S2 (ESI†). In general, the lowest GWP values coincided with the highest conversions, as this resulted in minimal waste of the reagents. Specifically, the optimal conditions were achieved using DMSO as the solvent, NaOH as the base, and a reaction temperature of 56 °C–82 °C. Similar GWP and yields were observed when the reaction was performed in ACN at 82 °C with NaOH as the base. Detailed results for all tested conditions and the individual impacts of each parameter on the GWP are provided in the ESI.†
Polyester synthesis and characterization
The two monomers (1,2) were polymerized by melt polycondensation with three linear aliphatic diols 3a–c with varied lengths, yielding two series of polyesters (P1a–c and P2a–c, Scheme 1) with moderately high molecular weights (10.2–18.8 kDa) according to SEC results (Table 1 and Fig. S9–14†). Excess diol (1.5 eq.) was used to compensate for losses due to evaporation during the polycondensations. A small volume (2 mL) of xylene was added to reduce the viscosity and thus facilitate the polycondensations.45–47 The reaction was initially performed at 200 °C under nitrogen to form oligomers by transesterification. After 4 h, the temperature was raised to 220 °C with a faster nitrogen flow to facilitate removal of the condensed diol and shift the reaction equilibrium towards higher molecular weight polymers. The reaction was completed after an additional 12 h, and the resulting crude was dissolved in chloroform and precipitated in methanol to yield the polyesters as white powders. The chemical structures of the polyesters were characterized using FTIR and 1H NMR spectroscopy (Fig. S15–32, ESI†).| Structure | Sample | M n (kDa) | M w (kDa) | Ð | T g (°C) | T 5 (°C) | T d (°C) | CY (%) |
|---|---|---|---|---|---|---|---|---|
| a M n and Mw was measured by SEC in chloroform. Tg was measured from the second heating curve in DSC. T5, Td and CY (char yield at 600 °C) were measured by TGA. | ||||||||

|
P1a | 14.7 | 24.1 | 1.64 | 79 | 359 | 385 | 23 |

|
P1b | 13.0 | 21.4 | 1.65 | 53 | 353 | 377 | 23 |
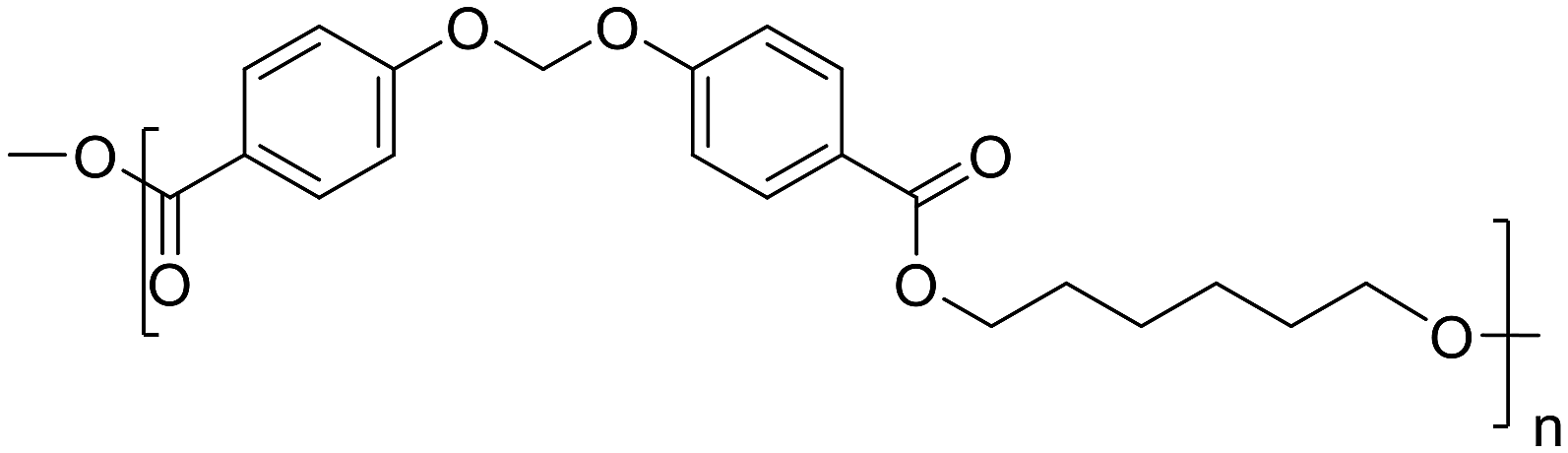
|
P1c | 18.8 | 28.1 | 1.50 | 41 | 348 | 384 | 22 |

|
Monomer 1 | — | — | — | — | 200 | 258 | 2 |

|
P2a | 10.2 | 14.8 | 1.41 | 83 | 352 | 378 | 28 |
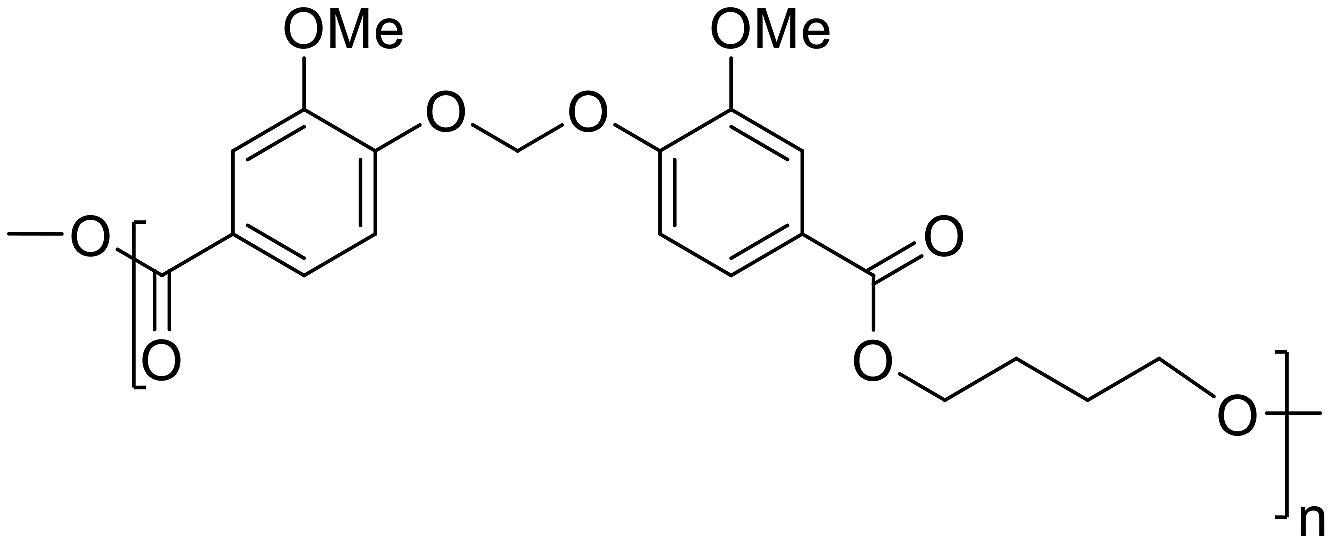
|
P2b | 16.0 | 23.8 | 1.50 | 65 | 334 | 356 | 31 |

|
P2c | 16.0 | 24.1 | 1.58 | 52 | 332 | 363 | 15 |

|
Monomer 2 | — | — | — | — | 232 | 278 | 1 |
Thermal properties
The thermal stability of the polyesters was investigated by TGA (Fig. 1). All the polyesters exhibited an initial thermal decomposition temperature (T5) above 330 °C, which is comparable to many other bio-based polyesters.48,49 A single decomposition step was observed for all polymers (Fig. 1B), which could be due to the degradation of the ester bond by the typical beta hydrogen transfer mechanism.50,51 | ||
| Fig. 1 Thermograms (A) and 1st derivative curves (B) of the monomers and polymers. | ||
Polyesters P1a–c and P2a–c were further characterized by DSC measurements (Fig. 2). As shown in the second heating curves (Fig. 2A), most polyesters were fully amorphous without any melting endotherm. The only exception was P1b, which showed a small melting endotherm at ∼150 °C. However, no corresponding crystallization exotherm was observed in its 1st cooling curve (Fig. 2B), which indicated a slow crystallization process. The Tg values of the polyesters ranged from 41 to 83 °C (Fig. 2A and Table 1), and decreased with an increased length of the diol, which was consistent with the increased backbone flexibility.52,53 This Tg is within the range for many commercial polyesters used in packaging, such as PET, PLA, and PETG, and slightly higher than other biobased polyesters with spirocyclic acetal units reported in the literature.54–57 Furthermore, the Tg values for P2a–c were higher compared to that of the corresponding member in the P1a–c series (by 4–12 °C). This indicated that the additional methoxy groups in P2a–c increased the bulkiness and reduced the segmental mobility of the backbones, which was consistent with other reported polymers with methoxy side groups.58,59 The glass transition of all these polymers was also observed in the 1st cooling curves (Fig. 2B).
 | ||
| Fig. 2 DSC (A) 2nd heating and (B) 1st cooling cycle of P1a–c and P2a–c. | ||
Because P1b showed slow crystallization in the DSC measurements, all the polymers were thermally annealed (during the second cooling cycle) at a temperature that was 20 °C above their respective Tg for 3 h. Afterward, the samples were cooled to −50 °C to start the third heating cycle (Fig. 3). Consequently, the melting endotherm of P1b was notably larger during the third heating cycle, indicating a significant increase in crystallinity due to the thermal annealing process. However, all the other polymers remained completely amorphous after annealing (Fig. 3). The reason for the unique crystallization behavior of P1b remains to be explored, but it may be related to the absence of a methoxy group in P1b (compared to P2a–c) which increases the regularity and packing of the chains. In addition, it also has relatively low molecular weight among the series (compared to P1a and P1c), which can increase the crystallization rate.60,61
 | ||
| Fig. 3 DSC third heating cycle of polyesters after thermal annealing for 3 h during the second cooling cycle (curves not shown). | ||
Thermomechanical properties
The thermomechanical properties of the polyesters were analyzed by DMA (Fig. 4). The samples were subjected to an oscillatory deformation while heated from room temperature to 100 °C with a rate of 3 °C min−1. As a result, the storage moduli (E′) of all the samples exhibited a characteristic 2 orders of magnitude reduction, followed by a rubber plateau (Fig. 4A). The E′ was in the range of 1–3 GPa at the glassy plateau (at 30 °C), which was comparable to that of commercial polyesters like PET and PLA.48,62–64 The E′ curves for P2b and P2c showed a slight increasing trend after the glass transition, which might be due to cold crystallization.65–67 The Tg values (taken from E′′ data as the temperature at the peak value, Fig. 4B and Table 2) also showed a decreasing trend with increased length of the alkylene units, which is consistent with the DSC results (Table 1). The slightly different Tg values according to DMA and DSC results were due to the differences in the mechanical and thermal response in connection with the glass transition, which is a commonly observed effect.68 | ||
| Fig. 4 DMA temperature sweep showing the (A) storage moduli, E′, and (B) loss moluli, E′′ of the obtained polyesters. | ||
| Polymer |
T
ga (°C) |
T
gb (°C) |
E′c (MPa) |
|---|---|---|---|
| a T g was measured as the peak temperatures in the E′′ curves. b T g was measured by DSC. c E′ was measured as the storage modulus at the glassy plateau at 30 °C for the polyesters. | |||
| P1a | 69 | 79 | 1720 |
| P1b | 49 | 53 | 1340 |
| P1c | 38 | 41 | 1070 |
| P2a | 78 | 83 | 1100 |
| P2b | 60 | 65 | 3080 |
| P2c | 46 | 52 | 1930 |
Acidic hydrolysis
Two polyesters from each series (P1a and P2a) were selected for a preliminary investigation of their chemical recyclability under acidic conditions. The polymer powders were suspended in concentrated HCl (12 M) and stirred at 70 °C for 24 h while being constantly monitored by visual inspection (Fig. 5). For both samples, the initial polymer powders gradually turned into finer particles, which formed a heterogeneous slurry after 24 h (Fig. S33, ESI†). Interestingly, the P1a slurry remained white throughout the hydrolysis experiment, while the P2a slurry turned brown after 8 h, and then black after 24 h. The coloration during the hydrolysis of P2a was consistent with side reactions commonly observed when working with lignin and the formation of humic substances.69–72 | ||
| Fig. 5 Acidic hydrolysis of P1a and P2a in HCl (12.0 and 10.5 M, respectively), yielding a solid residue and a liquid filtrate phase after filtration. | ||
After the hydrolysis, the slurry was diluted with water, and the pH was adjusted to ∼1 by the addition of K2CO3 powder. The resulting slurry was then filtered, and the aqueous phase was extracted with DCM. The filtered crude solid and the concentrated organic phase were both analyzed by 1H NMR spectroscopy (Fig. S34 and 35†). In the case of P1a, the solid was identified as a bisphenol BP1 (Fig. 5), with 71% yield and relatively high purity according to the 1H NMR spectrum (Fig. S34B, ESI†). This recovered BP1 structure has been previously investigated in the production of liquid crystalline polyesters,73 and it could act as a potential monomer for other polymers like polycarbonates, polyesters, or polyurethanes. In addition, this bisphenol may also have potential in biomedical applications.74 However, direct repolymerization (i.e. polyacetalization) of the obtained BP1 with dibromomethane was not possible, due to incomplete conversion of the phenol groups (Fig. S36, ESI†). The filtrate (from the organic phase) contained only negligible amounts (∼2%) of dry substances, which were presumably small molecules formed by the hydrolysis of ester bond according to the 1H NMR spectrum (Fig. S34C, ESI†). This indicated that the acidic hydrolysis mainly took place at the acetal bonds while the ester bonds remained largely intact under these conditions.
Acidic hydrolysis of P2a under the same conditions led to significantly different results, despite the seemingly similar structures of the two polymers (only the presence/absence of methoxy groups). After the hydrolysis of P2a, a black insoluble solid was isolated (in 21% yield), of which the structure could not be identified. In the meantime, vanillic acid was recovered (in 31% yield) from the organic phase (Fig. S35, ESI†). This indicated that both the acetal and ester bonds in P2a were hydrolyzed. To explore the possibility to gain better control over the hydrolysis process and achieve selective hydrolysis of the acetal bonds in P2a, different experimental conditions were investigated (Table S3 and Fig. S37–47, ESI†). The best result was observed when 10.5 M of HCl was used at 70 °C. Under this condition, the corresponding bis-phenol (BP2) was recovered in 45% yield as a solid residue. However, we also observed 15% of vanillic acid in the organic phase, which indicated that some ester bonds were still hydrolyzed. The use of even lower concentrations of HCl (3–9 M), or milder organic acids (formic acid and oxalic acid) did not result in any significant hydrolysis, and the polymer remained intact as the solid residue (Fig. S45–47, ESI†).
According to our observations, the hydrolysis of P2a was significantly faster than that of P1a, which indicated the effect of the methoxy groups in the ortho position to the acetal. To gain a further insight, a model hydrolysis of the two monomers (methyl paraben and methyl vanillate) was carried out in 12 M HCl at 70 °C for 4 h, which was basically ester hydrolysis. The production of carboxylic acids (i.e., 4-hydroxylbenzoic acid and vanillic acid, respectively) and the mass of the residual solid weight were monitored (Fig. S48–49, eqn (S2)–(S5), ESI†). As a result, the consumption of methyl vanillate and formation of vanillic acid was both faster than that of methyl paraben and 4-hydroxylbenzoic acid, which indicated that the presence of methoxy groups could enhance the acidic hydrolysis of the ester bonds. Although the exact reason remained to be explored, the enhanced hydrolysis rate by the presence of methoxy group could be related to the electron donating effect of methoxy group.75 In principle, the methoxy group can stabilize the positively charged transition state,34 which will thus facilitate the hydrolysis. However, other factors such as steric hindrance (by the methoxy groups) and the acidity of the leaving group may also play a role. In the literature, decreased thermal stability of molecules and polymers by the presence of methoxy groups have also been reported.76–78
We also noticed that there was no color change during the acidic hydrolysis of both monomers. However, the solution obtained from the hydrolysis of methyl vanillate turned black after the addition of one drop of formaldehyde (Fig. S49, ESI†). This observation suggested that the color change during the hydrolysis of P2a could be attributed to formaldehyde-related side reactions. To gain a further insight, a model reaction was carried out by reacting formaldehyde and vanillic acid (equimolar amounts in 12 M HCl) at 70 °C until it started to turn dark (ca. 2 h). Afterwards, a pale-yellow solid residue and dark-brown concentrated filtrate were obtained using the same separation procedure adopted for the hydrolysis of P2a. 1H-NMR analysis revealed that the solid residue consisted of vanillic acid (Fig. S51, ESI†), whereas the concentrated filtrate presented the expected peaks for vanillic acid, but also a series of peaks at 3.8–3.9, 4.2–5.5, 6.5–8.0 and 9.3–10.4 ppm that could not be elucidated. Some of these peaks (e.g. those observed at 5.2, 7.2–7.4 and 9.4–9.9 ppm) were also present in the spectra of the analogous filtrate obtained from the acidic hydrolysis of P2a in HCl 12 M (Fig. S35 and S51, ESI†), indicating that the black coloration observed during the acidic hydrolysis of P2a could be related to the side reactions involving vanillic acid and formaldehyde (which could form during the hydrolysis of the acetal bonds).
Conclusions
Two series of novel biobased poly(acetal-ester)s with non-cyclic acetal units, tunable Tgs, and reasonably high molecular weights were synthesized, which provided an insight into the fundamental structure–property relationship of this new class of polymers. The key parameters for the monomer synthesis were evaluated according to their GHG emissions, which provided a preliminary environmental insight for the further development of such polymers in the future. Furthermore, the non-cyclic acetals in this class of polymers could be selectively cleaved under acidic hydrolysis conditions, which could facilitate chemical recycling by the molecular design. We also observed that the presence of methoxy groups on the polymer backbone accelerated the hydrolysis, which could enable hydrolysis under milder conditions. However, this also increased the risk of coloration due to side reactions. The nature of the coloration and side reaction was preliminarily investigated by model reactions of the corresponding small molecules. In general, this work provided an effective method to design and fabricate recyclable polyesters by utilizing bio-sourced (e.g. lignin-based) monomeric hydroxybenzoic acids with or without methoxy groups. In the future, synthetic investigations of more structural varieties (e.g. the substitution on the acetal carbon, or other substitutions on the phenolic rings) are expected to provide deeper knowledge about how to utilize non-cyclic acetals in the design of chemically recyclable biobased polymers.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Mistra Foundation (the “STEPS” project, No. 2016/1489), Swedish Research Council for Sustainable Development (Formas, No. 2021-01107), Carl-Trygger Foundation (No. 18:435), the Crafoord Foundation (No. 20160774 and 20180939), and the Royal Physiographic Society in Lund. We thank John P. Jensen from Nordzucker Technology for valuable discussions, Sofia Essén for the mass spectrometry measurements.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- S. A. Park, H. Jeon, H. Kim, S. H. Shin, S. Choy, D. S. Hwang, J. M. Koo, J. Jegal, S. Y. Hwang, J. Park and D. X. Oh, Nat. Commun., 2019, 10, 2601 CrossRef PubMed.
- J. G. Rosenboom, D. K. Hohl, P. Fleckenstein, G. Storti and M. Morbidelli, Nat. Commun., 2018, 9, 2701 CrossRef PubMed.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 1–7 CrossRef PubMed.
- R. Hatti-Kaul, L. J. Nilsson, B. Zhang, N. Rehnberg and S. Lundmark, Trends Biotechnol., 2020, 38, 50–67 CrossRef CAS PubMed.
- J. G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- T. P. Kainulainen, T. I. Hukka, H. D. Özeren, J. A. Sirviö, M. S. Hedenqvist and J. P. Heiskanen, Biomacromolecules, 2020, 21, 743–752 CrossRef CAS PubMed.
- C. Pang, J. Zhang, Q. Zhang, G. Wu, Y. Wang and J. Ma, Polym. Chem., 2015, 6, 797–804 RSC.
- S. Subramaniyan, N. Najjarzadeh, S. R. Vanga, A. Liguori, P. O. Syrén and M. Hakkarainen, ACS Sustainable Chem. Eng., 2023, 11, 3451–3465 CrossRef CAS.
- M. Moroni and A. Mei, Appl. Sci., 2020, 10, 2800–2818 CrossRef CAS.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS PubMed.
- A. R. Rahimi and J. M. Garciá, Nat. Rev. Chem., 2017, 1, 1–11 CrossRef.
- Z. Guo, M. Eriksson, H. de la Motte and E. Adolfsson, J. Cleaner Prod., 2020, 124579 Search PubMed.
- S. Thiyagarajan, E. Maaskant-Reilink, T. A. Ewing, M. K. Julsing and J. Van Haveren, RSC Adv., 2022, 12, 947–970 RSC.
- N. Warlin, E. Nilsson, Z. Guo, S. V. Mankar, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, Polym. Chem., 2021, 12, 4942–4953 RSC.
- N. G. Valsange, R. N. L. de Menezes, N. Warlin, S. V. Mankar, N. Rehnberg, B. Zhang and P. Jannasch, Macromolecules, 2024, 57, 2868–2878 CrossRef CAS.
- R. N. L. de Menezes, O. Gordivska, T. T. Nguyen, N. Warlin, N. Rehnberg and B. Zhang, React. Funct. Polym., 2024, 205, 106062 CrossRef CAS.
- N. Warlin, M. N. Garcia Gonzalez, R. Menezes, A. Karajos, E. Olsson, C. Almqvist, M. Sayed, S. V. Mankar, N. Valsange, O. Y. Abdelaziz, C. P. Hulteberg, F. G. Bäcklund, Z. Guo, N. Rehnberg, S. Lundmark, R. Hatti-Kaul, P. Jannasch and B. Zhang, ChemSusChem, 2024, e202402067 CrossRef PubMed.
- S. V. Mankar, J. Wahlberg, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, ACS Sustainable Chem. Eng., 2023, 11, 5135–5146 CrossRef CAS.
- N. G. Valsange, N. Warlin, S. V. Mankar, N. Rehnberg, B. Zhang and P. Jannasch, Green Chem., 2024, 26, 2858–2873 RSC.
- R. Sedrik, O. Bonjour, S. Laanesoo, I. Liblikas, T. Pehk, P. Jannasch and L. Vares, Biomacromolecules, 2022, 23, 2685–2696 CrossRef CAS PubMed.
- N. G. Valsange, M. N. Garcia Gonzalez, N. Warlin, S. V. Mankar, N. Rehnberg, S. Lundmark, B. Zhang and P. Jannasch, Green Chem., 2021, 23, 5706–5723 RSC.
- A. Hufendiek, S. Lingier and F. E. Du Prez, Polym. Chem., 2019, 10, 9–33 RSC.
- K. Saito, F. Eisenreich and Ž. Tomović, Macromolecules, 2024, 57, 8690–8697 CAS.
- L. P. Manker, G. R. Dick, A. Demongeot, M. A. Hedou, C. Rayroud, T. Rambert, M. J. Jones, I. Sulaeva, M. Vieli, Y. Leterrier, A. Potthast, F. Maréchal, V. Michaud, H.-A. Klok and J. S. Luterbacher, Nat. Chem., 2022, 14, 976–984 CrossRef CAS PubMed.
- A. G. Pemba, M. Rostagno, T. A. Lee and S. A. Miller, Polym. Chem., 2014, 5, 3214–3221 RSC.
- B. Kwon, E. Han, W. Yang, W. Cho, W. Yoo, J. Hwang, B. M. Kwon and D. Lee, ACS Appl. Mater. Interfaces, 2016, 8, 5887–5897 CrossRef CAS PubMed.
- W. Yang, J. Noh, H. Park, S. Gwon, B. Singh, C. Song and D. Lee, Biomaterials, 2018, 154, 48–59 CrossRef CAS PubMed.
- S. Park, B. Kwon, W. Yang, E. Han, W. Yoo and D. Lee, J. Controlled Release, 2014, 196, 19–27 CrossRef CAS PubMed.
- S. Samanta, C. C. De Silva, P. Leophairatana and J. T. Koberstein, J. Mater. Chem. B, 2018, 6, 666–674 RSC.
- T. Hashimoto, H. Meiji, M. Urushisaki, T. Sakaguchi, K. Kawabe, C. Tsuchida and K. Kondo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3674–3681 CrossRef CAS.
- L. Zhao, L. Zhang and Z. Wang, RSC Adv., 2015, 5, 95126–95132 RSC.
- Q. Li, S. Ma, S. Wang, Y. Liu, M. A. Taher, B. Wang, K. Huang, X. Xu, Y. Han and J. Zhu, Macromolecules, 2020, 53, 1474–1485 CrossRef CAS.
- B. Liu and S. Thayumanavan, J. Am. Chem. Soc., 2017, 139, 2306–2317 CrossRef CAS PubMed.
- L. Zimmermann, A. Dombrowski, C. Völker and M. Wagner, Environ. Int., 2020, 145, 106066 CrossRef CAS PubMed.
- Z. Demchuk, N. Wu, G. Pourhashem and A. Voronov, ACS Sustainable Chem. Eng., 2020, 8, 12870–12876 CrossRef CAS.
- M. A. F. Delgove, A. B. Laurent, J. M. Woodley, S. M. A. De Wildeman, K. V. Bernaerts and Y. van der Meer, ChemSusChem, 2019, 12, 1349–1360 CrossRef CAS PubMed.
- N. Warlin, M. N. Garcia Gonzalez, S. Mankar, N. G. Valsange, M. Sayed, S. H. Pyo, N. Rehnberg, S. Lundmark, R. Hatti-Kaul, P. Jannasch and B. Zhang, Green Chem., 2019, 21, 6667–6684 RSC.
- S. V. Mankar, M. N. Garcia Gonzalez, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 19090–19103 CrossRef CAS.
- S. G. Yao, J. K. Mobley, J. Ralph, M. Crocker, S. Parkin, J. P. Selegue and M. S. Meier, ACS Sustainable Chem. Eng., 2018, 6, 5990–5998 CrossRef CAS.
- T. T. Nguyen, E. Olsson, S. V. Mankar, N. Warlin, N. G. Valsange, J. Engqvist, N. Rehnberg, P. Jannasch and B. Zhang, Macromolecules, 2024, 57, 9289–9301 CrossRef CAS.
- J. O. Krömer, R. G. Ferreira, D. Petrides and N. Kohlheb, Front. Bioeng. Biotechnol., 2020, 8, 403 CrossRef PubMed.
- I. S. Modahl and E. Soldal, The 2019 LCA of products from Borregaard, Sarpsborg, 2019 Search PubMed.
- Adesso Advanced Materials Wuxi Co., Ltd, WO2016177305A1, 2016.
- P. Wang, C. R. Arza and B. Zhang, Polym. Chem., 2018, 9, 4706–4710 RSC.
- X. Li, X. Wang, S. Subramaniyan, Y. Liu, J. Rao and B. Zhang, Biomacromolecules, 2022, 23, 150–162 CrossRef CAS PubMed.
- H. Köpnick, M. Schmidt, W. Brügging, J. Rüter and W. Kaminsky, Polyesters, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000 Search PubMed.
- O. Y. Abdelaziz, M. B. Vives, S. V. Mankar, N. Warlin, T. T. Nguyen, B. Zhang, C. P. Hulteberg and A. Khataee, iScience, 2024, 27, 109418 CrossRef CAS PubMed.
- C. R. Arza and B. Zhang, ACS Omega, 2019, 4, 15012–15021 CrossRef CAS PubMed.
- F. Samperi, C. Puglisi, R. Alicata and G. Montaudo, Polym. Degrad. Stab., 2004, 83, 11–17 CrossRef CAS.
- H. Ohtani, T. Kimura and S. Tsuge, Anal. Sci., 1986, 2, 179–182 CrossRef CAS.
- P. Wang, J. A. Linares-Pastén and B. Zhang, Biomacromolecules, 2020, 21, 1078–1090 CrossRef CAS PubMed.
- P. Wang and B. Zhang, RSC Adv., 2021, 11, 16480–16489 RSC.
- N. G. Valsange, N. Warlin, S. V. Mankar, N. Rehnberg, B. Zhang and P. Jannasch, Green Chem., 2024, 26, 2858–2873 RSC.
- N. Warlin, M. N. Garcia Gonzalez, S. Mankar, N. G. Valsange, M. Sayed, S.-H. Pyo, N. Rehnberg, S. Lundmark, R. Hatti-Kaul, P. Jannasch and B. Zhang, Green Chem., 2019, 21, 6667–6684 RSC.
- S. V. Mankar, J. Wahlberg, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, ACS Sustainable Chem. Eng., 2023, 11, 5135–5146 CrossRef CAS.
- Omnexus, Glass Transition Temperature, https://omnexus.specialchem.com/polymer-property/glass-transition-temperature, (accessed 20 January 2025).
- O. Bonjour, H. Nederstedt, M. V. Arcos-Hernandez, S. Laanesoo, L. Vares and P. Jannasch, ACS Sustainable Chem. Eng., 2021, 9, 16874–16880 CrossRef CAS PubMed.
- J. A. Emerson, N. T. Garabedian, D. L. Burris, E. M. Furst and T. H. Epps, ACS Sustainable Chem. Eng., 2018, 6, 6856–6866 CrossRef CAS.
- X. Chen, G. Hou, Y. Chen, K. Yang, Y. Dong and H. Zhou, Polym. Test., 2007, 26, 144–153 CrossRef CAS.
- J. R. Fried, Polymer Science and Technology, Prentice Hall, Westford, 3rd edn, 2014 Search PubMed.
- C. W. Chen, P. H. Liu, F. J. Lin, C. J. Cho, L. Y. Wang, H. I. Mao, Y. C. Chiu, S. H. Chang, S. P. Rwei and C. C. Kuo, J. Polym. Environ., 2020, 28, 2880–2892 CrossRef CAS.
- B. A. Alshammari, M. Hossain, A. M. Alenad, A. G. Alharbi and B. M. Alotaibi, Polymers, 2022, 14, 1–14 Search PubMed.
- X. Wang, S. Peng, H. Chen, X. Yu and X. Zhao, Composites, Part B, 2019, 173, 107028 CrossRef CAS.
- F. Ali, Y. W. Chang, S. C. Kang and J. Y. Yoon, Polym. Bull., 2009, 62, 91–98 CrossRef CAS.
- J. S. Lee, G. H. Hwang, Y. S. Kwon and Y. G. Jeong, Polymer, 2020, 202, 122677 CrossRef CAS.
- M. Cristea, D. Ionita and M. M. Iftime, Materials, 2020, 13, 1–24 CrossRef PubMed.
- K. Whitcomb , Measurement of Glass Transition Temperatures by Dynamic Mechanical Analysis and Rheology, https://www.tainstruments.com/applications-notes/measurement-of-glass-transition-temperatures-by-dynamic-mechanical-analysis-and-rheology/, (accessed 20 January 2025).
- J. P. Martin and K. Haider, Soil Sci. Soc. Am. J., 1976, 40, 377–380 CrossRef CAS.
- S. Tang, F. Li, N. T. Tsona, C. Lu, X. Wang and L. Du, ACS Earth Space Chem., 2020, 4, 862–872 CrossRef CAS.
- Y. Zhang and M. Naebe, ACS Sustainable Chem. Eng., 2021, 9, 1427–1442 CrossRef CAS.
- A. Burges, H. M. Hurst, S. B. Walkden, F. M. Dean and M. Hirst, Nature, 1963, 199, 696–697 CrossRef CAS PubMed.
- J. A. Moli, S. Vasanthi, N. Prakash and D. R. Singh, High Perform. Polym., 2012, 24, 507–520 CrossRef.
- Rutgers, The State University of New Jersey, US10087285B2, 2018.
- I. Meloche and K. J. Jaidler, J. Am. Chem. Soc., 1951, 73, 1712–1714 CrossRef CAS.
- M. Hasegawa, Y. Tsujimura, K. Koseki and T. Miyazaki, Polym. J., 2008, 40, 56–67 CrossRef CAS.
- C. R. Arza, P. Wang, J. Linares-Pastén and B. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2314–2323 CrossRef CAS.
- B. G. Harvey, A. J. Guenthner, W. W. Lai, H. A. Meylemans, M. C. Davis, L. R. Cambrea, J. T. Reams and K. R. Lamison, Macromolecules, 2015, 48, 3173–3179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05778c |
| This journal is © The Royal Society of Chemistry 2025 |