
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimized synthesis of a high oleic sunflower oil derived polyamine and its lignin-based NIPUs†
Francesca C.
Destaso‡
ab,
Celeste
Libretti‡
ac,
Cédric
Le Coz
 b,
Etienne
Grau
b,
Henri
Cramail
b and
Michael A. R.
Meier
*ac
b,
Etienne
Grau
b,
Henri
Cramail
b and
Michael A. R.
Meier
*ac
aInstitute of Biological and Chemical Systems – Functional Molecular Systems (IBCS-FMS), Karlsruhe Institute of Technology (KIT), Kaiserstraße 12, 76131 Karlsruhe, Germany
bUniv. Bordeaux, CNRS, Bordeaux INP, LCPO, 16 avenue Pey-Berland, 33600 Pessac, France
cInstitute of Organic Chemistry (IOC), Materialwissenschaftliches Zentrum für Energiesysteme (MZE), Karlsruhe Institute of Technology (KIT), Kaiserstraße 12, 76131 Karlsruhe, Germany. E-mail: m.a.r.meier@kit.edu; Web: https://www.meier-michael.com
First published on 26th December 2024
Abstract
In the continuing effort towards reducing reliance on fossil resources, the transition to biobased materials is of utmost importance, together with the adoption of more sustainable and safe production processes. This work focuses on the implementation of renewable resources and greener protocols for the synthesis of a bio-based amino crosslinker and its subsequent use in non-isocyanate polyurethane (NIPU) synthesis. NIPUs are a new class of materials, analogous to isocyanate derived polyurethanes (PUs), that avoid hazardous chemicals in their production process, such as phosgene and isocyanates, that raise high concern in the PU manufacturing processes. Vegetable oils and lignin are two abundant renewable feedstocks largely investigated during the last decade. In this paper, we study the synthesis of a high oleic sunflower oil derived polyamine (PA) via thiol–ene photoreaction in batch and in flow. Additionally, cyclic carbonate functionalized lignin (CCLF) was synthesized and reacted with PA to create fully bio-based NIPU networks with different lignin contents. The curing behavior as well as the characterization of the obtained thermosets is described.
Green Foundation Box1. This work focuses on non-isocyanate polyurethane (NIPUs) synthesis from renewable polyamine and cyclic carbonate sources, using lignin, a plant oil and erythritol. Green Chemistry principles form the foundation of our work and are highlighted throughout our work.2. The synthesis pathways for both the polyamine and cyclic carbonate monomers were carefully designed to minimize solvent usage and avoid commonly employed toxic routes. Additionally, the obtained NIPUs are fully renewable and further contribute to the field of renewable-based alternatives to conventional polyurethanes. 3. Future research effort could focus on the investigation of upscaling the production of the polyamine in continuous flow, reducing further the monomer costs and energy consumption, advancing toward the potential industrial application of this monomer for renewable NIPUs and other thermosets, such as expoxies. |
Introduction
With a global market production of 25 million tons,1 polyurethanes (PU) are a class of polymers useful for many applications, such as coatings, adhesives, foams, sealants, elastomers, and more.2,3 Among their relevance in everyday human life, PU production faces significant obstacles, mainly due to the use of isocyanates. Isocyanates are known to be irritant and sensitizing agents.4,5 Even worse, their production relies on the use of phosgene, a highly toxic gas. Both their precursor, phosgene, and isocyanates themselves present several concerns correlated to toxicity6 and carcinogenicity.7 Moreover, the most commonly used diisocyanates, (toluene diisocyanate, TDI, LD50,oral,rat = 5130 mg kg−1 and methylenediphenyl diisocyanate, MDI, LD50,oral,rat = 4130 mg kg−1)8 have been restricted in their use by REACH regulations.9,10 The most updated REACH regulation limits the presence of isocyanates in formulations to a maximum of 0.1% by weight.11 For all these reasons, current research efforts in the area of PUs concern the development of isocyanate-free routes to substitute isocyanate derived PU and generate a new class of materials named non-isocyanate polyurethanes (NIPUs).8,12 Common pathways may involve, for instance, transurethanization reactions or aminolysis of cyclic carbonates, generating, respectively, polyurethanes and polyhydroxyurethanes (PHU). In a world increasingly driven by the need for more sustainable solutions, shifting towards greener protocols that adhere to the principles of Green Chemistry is imperative.13 This involves, for instance, the utilization and valorization of renewable resources to increase the bio-content of materials and reduce waste. Numerous studies concerning the development of bio-based NIPUs using various renewable feedstocks, such as terpenes, biomass or oleochemicals, can be found in the literature.14–20Lignocellulosic biomass contains three main components, cellulose, hemicelluloses and lignins, with percentages varying depending on, for instance, the type of renewable resource used and its growing conditions. These three biopolymers are biodegradable and mainly derived from woody biomass, i.e. they are non-food competitive. Lignin, accounting for up to 40 wt% of the biomass content,21 is the most abundant renewable source of aromatic compounds. However, lignin is usually treated as a low-value by-product of the pulp and paper industry, and is often thermally utilized as bio-fuel.21,22 A recent review by Meier et al.23 summarizes advancements on lignin modifications, quantitatively comparing synthesis protocols in terms of sustainability and toxicity. The utilization of lignin as an attractive macromonomer in NIPU formulations is yet scarcely researched. A study from 2017 reports the use of cyclic carbonate-functionalized lignin as cross-linker in different PHU formulations in the presence of 1,12-diaminododecane and poly(ethylene glycol) bis-cyclic carbonate as chain extender.24 However, the protocol employs an epichlorohydrin (LD50,oral,rat = 175 mg kg−1) route followed by CO2 insertion for the synthesis of the lignin monomer. Additionally, the final materials were too brittle for mechanical characterization. More recently, several works describe the development of more sustainable protocols based on a two-step modification with organic carbonates, the thereof derived PHUs were characterized in detail.25–27
While bio-based cyclic carbonate monomers are well-researched, bio-based polyamines are still required as important building blocks to achieve fully bio-based NIPUs. Such renewable polyamines are far less frequently described.
With a global annual production of 210.3 MMT (million metric tons), vegetable oils are an important class of renewable feedstock.28 Their abundance and structural variety render them suitable sources for the synthesis of many different bio-based monomers and polymers.29 The key characteristic of triglycerides is the presence of one or multiple unsaturations, facilitating easy functionalization or cleavage of the structure.30–32 In the field of NIPUs, different examples of polycyclic carbonate monomers obtained from triglycerides are reported.33,34 Moreover, the presence of double bonds in triglycerides allowed the introduction of primary amine moieties via thiol–ene click chemistry.35 Following this approach, also a variety of (bio)polyols were synthesized employing mercapto alcohols. Furthermore, Meier et al. reported the first procedure to achieve a difunctional amine from limonene via thiol–ene click-reaction with cysteamine hydrochloride as the sulfur-containing molecule, bearing a primary amine moiety. Following this work, the application of this route was extended to vegetable oils, such as grapeseed oil and canola oil, more recently by others.16,36,37 Here, a bio-based polyamine derived from high oleic sunflower oil was synthesized via thiol–ene chemistry. Reaction conditions were thoroughly investigated and optimized, testing two different reaction systems, in batch and continuous flow. Subsequently, the polyamine was utilized in the formulation of fully biobased PHUs, together with cyclic carbonate functionalized lignin and erythritol bis-cyclic carbonate, obtaining flexible materials with tunable lignin content.
Results and discussion
Synthesis of high oleic sunflower oil derived polyamine (PA) in batch
High oleic sunflower oil was chosen to be modified with cysteamine due to its quite homogeneous fatty acid composition to yield a renewable polyamine (PA) (≥85% oleic acid, see Scheme 1).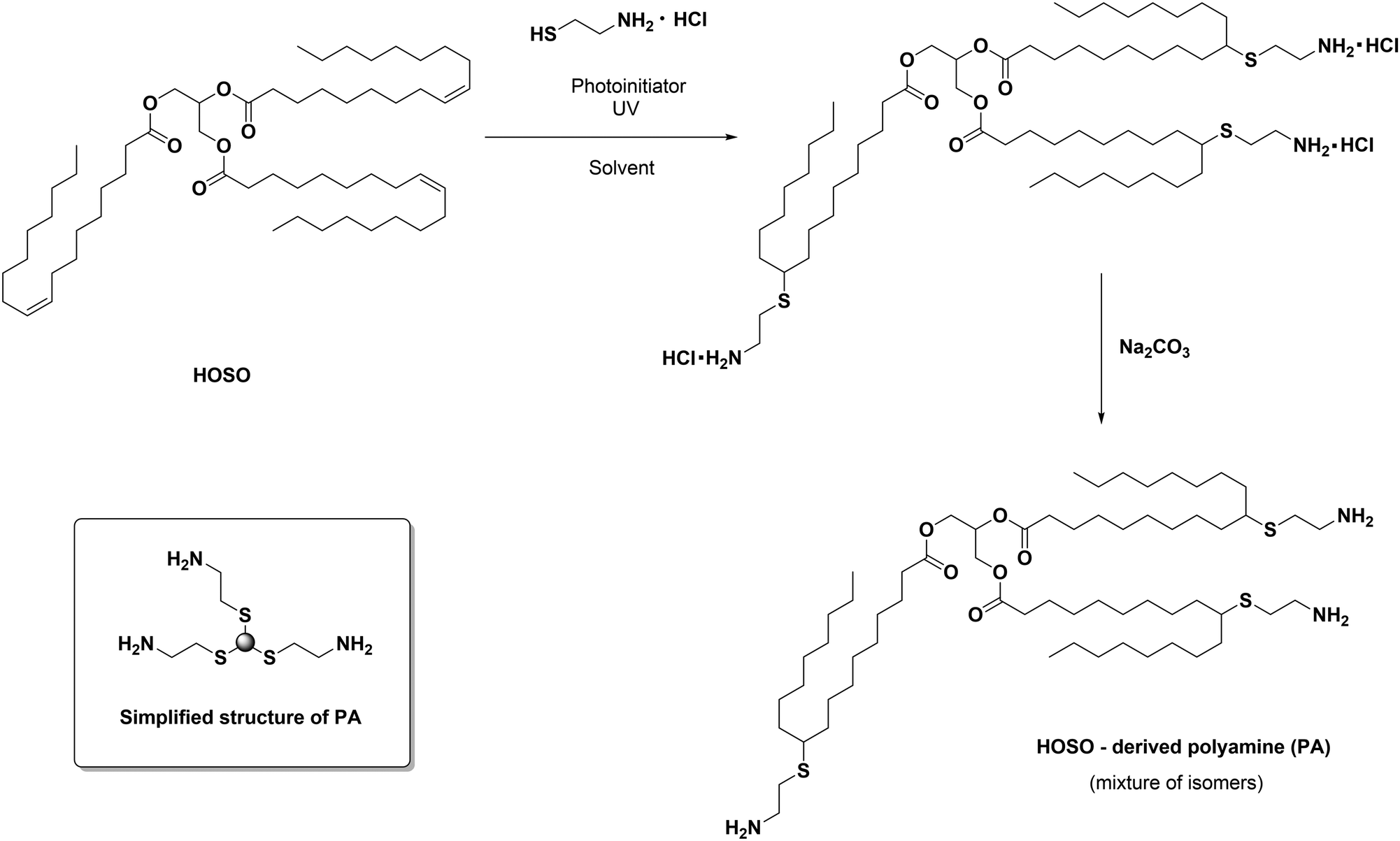 | ||
| Scheme 1 General reaction scheme for the synthesis of PA. On the bottom left (box inset) of the scheme, a simplified structure of PA is provided and will be used throughout the manuscript. | ||
To understand and optimize the reaction, the solvent and photoinitiator concentration, wavelength and light intensity were investigated. First, 2,2-dimethoxy-2-phenyl-acetophenone (DMPA) was chosen as it is a widely used benzoin ether photoinitiator. Because the oil and the cysteamine hydrochloride salt (CAHC) were not soluble in the same medium, a solvent mixture of dichloromethane and ethanol was initially tested, but also in this mixture, due to the excess of the cysteamine salt used, it was never fully solubilized. An initial test was performed irradiating the reaction mixture in DCM/EtOH (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) with a UV lamp (365 nm, 45 W) for 24 h with a HOSO concentration of 0.04 g mL−1 (setup: see ESI, Fig. S1† and Table 1, entry 1), leading to 40% double bond conversion. Next, the equivalents of cysteamine salt and photoinitiator were increased and a longer reaction time was applied, resulting in an increased conversion of 55% after 48 h (Table 1, entry 2).
:3) with a UV lamp (365 nm, 45 W) for 24 h with a HOSO concentration of 0.04 g mL−1 (setup: see ESI, Fig. S1† and Table 1, entry 1), leading to 40% double bond conversion. Next, the equivalents of cysteamine salt and photoinitiator were increased and a longer reaction time was applied, resulting in an increased conversion of 55% after 48 h (Table 1, entry 2).
| Photoinitiator | Solvent | Wavelength (nm) | Concentration (g mL−1) | Conversion % | Time (h) | |
|---|---|---|---|---|---|---|
| Entry 1 | DMPA | DCM:EtOH (7:3) |
365 lamp | 0.04 g mL−1 | 40% | 24 |
| Entry 2 | DMPA | DCM:EtOH (7:3) |
365 lamp | 0.04 g mL−1 | 55% | 48 |
| Entry 3 | DMPA | Dioxane:EtOH (7:3) |
365 LED | 0.04 g mL−1 | 94 | 48 |
| Entry 4 | DMPA | Dioxane:EtOH (7:3) |
405 LED | 0.04 g mL−1 | 100 | 48 |
| Entry 5 | DMPA | Dioxane:EtOH (7:3) |
405 LED | 0.28 | 100 | 7 |
| Entry 6 | TPO-L | Dioxane:EtOH (7:3) |
405 LED | 0.28 | 100 | 7 |
| Entry 7 | DMPA | Isopropanol:EtOH (1:1) |
405 LED | 0.42 | 100 | 7 |
| Entry 8 | TPO-L | Isopropanol:EtOH (1:1) |
405 LED | 0.28 | >99 | 7 |
In order to find a better suitable solvent mixture, we started our investigations by repeating a literature known procedure from Stemmelen et al.36 Thus, the solvent mixture was changed to 1,4-dioxane:ethanol (7:3). Moreover, an irradiation system consisting of a metallic plate with LEDs installed was tested, one plate consisting of 365 nm LEDs (2 W) and one using 405 nm LEDs (2 W) (see ESI, Fig. S2†). The LEDs have the advantage of not producing heat and therefore the distance between light and the sample can be reduced. A considerable advantage of LEDs is their lowered energy consumption. Two otherwise identical reactions were irradiated by the two different wavelengths and run for 48 h (Table 1, entries 4 and 5). In their work, Caillol et al. reached 87% conversion for grapeseed oil. In our case, 94% and 100% conversion of HOSO double bonds were achieved for the different investigated wavelengths after 48 h (Table 1, entry 3) (see ESI†).
The concentration of the reaction mixture is another relevant parameter to tune. A high turbidity of the reaction mixture is disadvantageous for a homogeneous and effective light penetration, which is a crucial factor for photoreactions. Meanwhile, the minimum amount of solvent is desirable from a Green Chemistry perspective and typically leads to higher conversions. Different experiments were carried out increasing the concentration of HOSO. For the highest concentration (0.28 g mL−1), the reaction reached full conversion after 7 hours and 89% conversion after only 1 h (Table 1, entry 5).
Further following the principles of Green Chemistry to avoid unsafe chemicals, dioxane was replaced in the solvent mixture by isopropanol, as reported by Rios et al.16 As in the previous case, the reaction reached completion within 7 h (Table 1, entry 7) using the LED setup. TPO-L (diphenyl(2,4,6-trimethylbenzoyl)-phosphine oxide) was investigated as an alternative photoinitiator. The wide absorption range (absorption maxima: 272 nm, 382 nm) of this initiator makes it suitable to be excited under 405 nm wavelength irradiation.38 Similar results were achieved with this photoinitiator, reaching quantitative conversions within 7 h of reaction time for both solvent systems (Table 1, entry 6 and 8).
To allow for an increased scale of production of PA, upscaling was tested. Indeed, the use of three UV lamps and the solvent mixture iPrOH:EtOH (setup: see ESI, Fig. S3†) led to 95% double bond conversion in 7 h for a 2 g scale reaction, a good compromise to obtain useful quantities of PA.
Furthermore, a series of experiments were performed to investigate the effect of the addition of CAHC over time (see Fig. 1). A stepwise cysteamine salt addition was compared with a single-step addition method, revealing that the stepwise addition led to better performance. Initially, a solvent mixture of EtOH:iPrOH (1:1) was used, with a single-step addition of CAHC under a 365 nm, 45 W irradiation system (setup: see ESI, Fig. S1†), leading to a conversion of 63% after 48 h. Switching the solvent to iPrOH only showed an improvement in the conversion to 84% under the same reaction conditions. Finally, by adding another source of irradiation at the same wavelength of 365 nm (12 W) to the flask and keeping the stepwise addition of CAHC method, a conversion of 97% after 24 h was observed.
 | ||
| Fig. 1 Overview of the different experiments conducted to investigate the influence of CAHC addition on the synthesis of PA. Entry 9: single step addition of CAHC, in EtOH:iPrOH 1:1; entry 10: single step addition of CAHC in iPrOH; entry 11a: stepwise addition of CAHC (half of the calculated total amount at the starting time, then a quarter of the total amount after 1.5 h, the remaining quarter after 4 h) in iPrOH. DMPA was used as photoinitiator for each entry. For all entries the scale of the reaction corresponds to 1 g of oil. Concentration is 0.2 g ml−1 for each entry.16 | ||
Synthesis of high oleic sunflower oil derived polyamine (PA) in flow
As a further promising path to scale up the production of the desired polyamines, the reaction depicted in Scheme 1 was also investigated using a flow reactor equipped with a 365 nm UV lamp (see ESI, Fig. S4†). Due to the long reaction time necessary to achieve complete conversion, the mixture was run in a closed loop system, and aliquots were collected to monitor the process.Isopropanol was selected as a solvent due to its good performance in bulk reactions and different flow rate values were screened. It was noticed, as could be expected in the used closed loop setup, that increasing the flow rate led to improved conversion at the same reaction time. The best outcome was achieved with 9 ml min−1 with a conversion of 99% after 6 h (Table 2). For a better comparison of the different flow rates, conversions after 3 h are summarized in Fig. 2. Thus, at high flow rates, higher conversions can be achieved, at the same increased scale of 2 g. In summary of all reported optimization reactions, PA can now be prepared in larger scales at shorter reaction times and by using less environmentally problematic solvents.
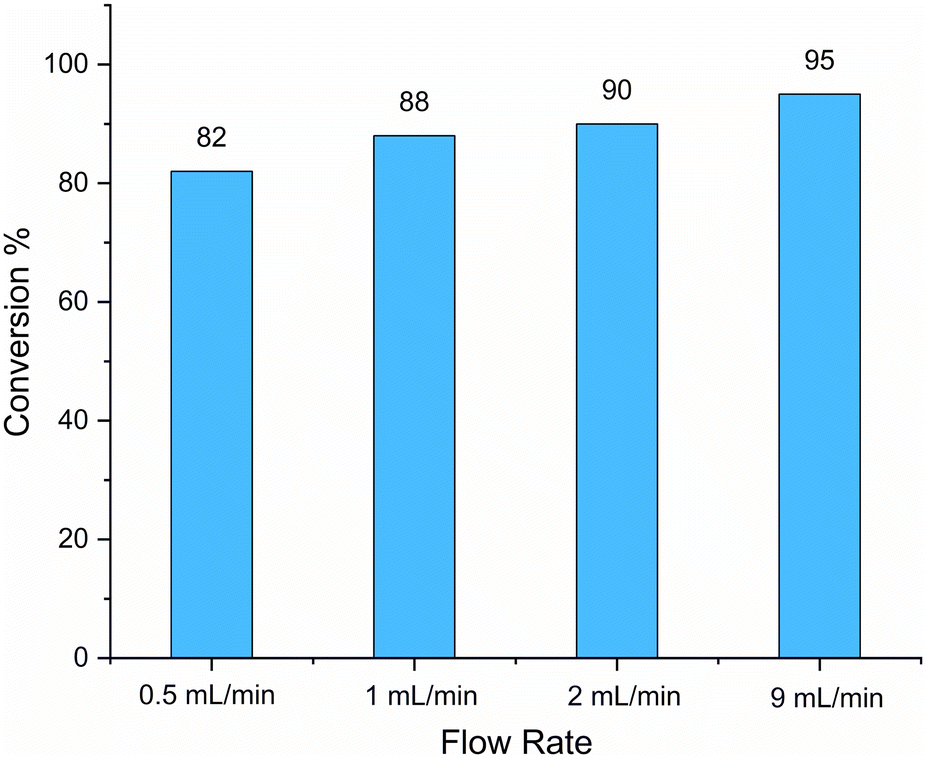 | ||
| Fig. 2 Conversions of HOSO via thiol–ene reaction in flow at different flow rates after 3 h. To evaluate the conversion, a 100 μL sample was taken at different reaction times and 1H-NMR was recorded. Reactions in isopropanol with a flow rate 0.5 mL min−1, 1 mL min−1, 2 ml min−1 and 9 mL min−1 were performed. | ||
Thermosets
As a possible application, PA was employed as a crosslinking agent for the formation of non-isocyanate polyurethane (NIPU) networks together with cyclic carbonate functionalized lignin (CCFL) and erythritol-bis cyclic carbonate (EBC), to afford renewable thermosets as shown in Scheme 2. | ||
| Scheme 2 General reaction scheme for lignin functionalization to achieve CCFL, and subsequent reaction with EBC and crosslinker PA to form NIPU networks. For simplicity, only the main reactions are shown, for further details and discussion see ESI.† | ||
CCFL was synthesized according to the protocol originally proposed by Lehnen et al.,25 with minor modifications, as detailed in the experimental section. Briefly, a two-step procedure was employed. First, glycerol carbonate (GC) was utilized to introduce further 1,2-diol moieties to the structure of lignin. Subsequently, these 1,2-diols underwent ring closure to 5-membered cyclic carbonates by further treatment with dimethyl carbonate (DMC), as shown in Scheme 2. In the first step of the synthesis, several side reactions are possible depending on the type of hydroxyl groups reacting. A more detailed discussion highlighting different reactivities is reported in the ESI,† as well as full characterization of CCFL and its precursors. The first step of the modification is solventless, as glycerol carbonate acts both as reactant and solvent, the second step was further optimized proving that a solventless procedure is also possible, achieving similar results compared to the one using solvent.
Curing behavior of lignin-NIPUs thermosets
Having both monomers in hand, the curing behavior of the lignin-NIPU thermosets (Scheme 2) was followed by IR spectroscopy. Initially, thermosets with only cyclic carbonate functionalized lignin (CCFL, 0.89 mmol g−1 carbonate moieties) and PA were synthesized, with a CC:NH2 ratio of 1:1 and 1:1.5. For both formulations, the samples resulted in a very brittle material after curing, breaking instantly when recording IR spectra (see ESI, Fig. S25†). Therefore, a third bifunctional component was added to the thermoset formulation to achieve more flexible materials, decreasing the crosslinking density inherently brought by the lignin macromolecular structure. Thus, erythritol bis-cyclic carbonate (EBC) was synthesized following a more sustainable procedure developed by Meier et al.39 starting from erythritol, a naturally occurring polyalcohol present in fruit and used as sweetener. Initial experiments were conducted with PA and EBC only. However, without the contribution of lignin, a network was not successfully formed, resulting in materials lacking structural integrity and exhibiting very high viscosity, making them difficult to handle. This outcome reinforces the fact that the optimal formulation for the thermosets relies on the contribution of all three components (CCFL, EBC and PA) to form a PHU network efficiently. Considering the chemical nature of the three components, their combination can overcome the typical brittleness of lignin-containing materials.
Due to the solid nature of lignin and EBC, a minimal amount of DMSO was employed to aid the mixing of the monomers. The viscous-liquid nature of PA also contributed an efficient and homogeneous mixing of all the components. Different thermoset compositions were prepared, an overview is reported in Table 3.
| Entry | cyclic carbonate component (equiv.) | amine component | Catalyst | Lignin contenta (%mol) | Lignin contentb (%wt) | ||
|---|---|---|---|---|---|---|---|
| EBC | CCFL | Total equiv. | PA (equiv.) | TBD (equiv.) | |||
| a Molar content of CCFL (in percent) with respect to the total moles of cyclic carbonate component. b Weight content of lignin (in percent) with respect to the total weight of all components, not considering the weight of the solvent. | |||||||
| 1 | 0.85 | 0.15 | 1 | 1 | 0.1 | 15 | 26 |
| 2 | 0.75 | 0.25 | 1 | 1 | 0.1 | 25 | 38 |
| 3 | 0.50 | 0.50 | 1 | 1 | 0.1 | 50 | 56 |
| 4 | 0.25 | 0.75 | 1 | 1 | 0.1 | 75 | 67 |
Initially, a 38 wt% lignin formulation with EBC, CCFL and PA was prepared. Its curing behavior was followed in time at 150 °C via IR spectroscopy, revealing an optimal curing time of 2.5 days, as after this time the signal ascribed to the cyclic carbonate v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 1792 cm−1 was no longer observed. Next, applying these conditions, four different lignin weight percentages were tested, 26, 38, 56 and 67 wt% (see Table 4). IR spectroscopy of the cured thermosets is shown in Fig. 3. Comparing the IR spectrum of CCFL (Fig. 3, left, top) with the fully-cured thermoset with 38 wt% lignin (Fig. 3, left, bottom), different signals show the successful formation of the PHU thermosetting network. In particular, a strong increase in the stretching vibration signal ascribed to C–H bonds (2760–3000 cm−1) relates to the incorporation of the aliphatic fatty acid chains into the structure of the thermoset. Also, v(O–H) and v(N–H) stretching vibration signals (3100–3600 cm−1) increased as a consequence of the formation of hydroxyl groups due to ring opening of cyclic carbonate and urethane moiety formation, respectively. The disappearance of the cyclic carbonate v(CO) signal (1792 cm−1) confirms the effective curing of the material, while a new signal at 1728 cm−1 is associated with the v(CO) of both ester and urethane moieties overlapping with each other. In Fig. 3, right, an overlay of all IR spectra of the thermosets with different lignin compositions is shown. The absence of the cyclic carbonate v(CO) stretching absorbance reveals an effective reaction in all compositions.
O) at 1792 cm−1 was no longer observed. Next, applying these conditions, four different lignin weight percentages were tested, 26, 38, 56 and 67 wt% (see Table 4). IR spectroscopy of the cured thermosets is shown in Fig. 3. Comparing the IR spectrum of CCFL (Fig. 3, left, top) with the fully-cured thermoset with 38 wt% lignin (Fig. 3, left, bottom), different signals show the successful formation of the PHU thermosetting network. In particular, a strong increase in the stretching vibration signal ascribed to C–H bonds (2760–3000 cm−1) relates to the incorporation of the aliphatic fatty acid chains into the structure of the thermoset. Also, v(O–H) and v(N–H) stretching vibration signals (3100–3600 cm−1) increased as a consequence of the formation of hydroxyl groups due to ring opening of cyclic carbonate and urethane moiety formation, respectively. The disappearance of the cyclic carbonate v(CO) signal (1792 cm−1) confirms the effective curing of the material, while a new signal at 1728 cm−1 is associated with the v(CO) of both ester and urethane moieties overlapping with each other. In Fig. 3, right, an overlay of all IR spectra of the thermosets with different lignin compositions is shown. The absence of the cyclic carbonate v(CO) stretching absorbance reveals an effective reaction in all compositions.
 | ||
| Fig. 3 Left: overlayed IR spectra of CCFL (blue, top) and the thermoset with 38 wt% lignin content (Table 4, entry 2, black, bottom). Right: overlay of IR spectra of CCFL (violet, top) and the thermosets with different lignin compositions (see Table 4). Relevant signal changes are highlighted. | ||
| wt.% lignin |
T
g
(°C) |
T d,5% (°C) | T d,50% (°C) | Residue (%) | WCA (°) | Swelling (%) | Gel content (%) | Aspect |
|---|---|---|---|---|---|---|---|---|
| a T g ranges obtained from the DSC thermograms, precise assignation is not possible. | ||||||||
| 26 | −30 to 60 | 287 | 400 | 21.0 | 81.13 | 96 | 79 | Very flexible |
| 38 | 20–80 | 290 | 404 | 21.3 | 87.99 | 48 | 87 | Flexible |
| 56 | 80–120 | 301 | 421 | 33.9 | 101.02 | 44 | 93 | Fragile |
| 67 | 90–130 | 307 | 422 | 33.3 | 95.66 | 41 | 98 | Fragile |
Gel content and swelling properties
Gel content and swelling behavior of the thermosets were investigated according to the procedure described in the ESI† and are reported in Table 4. Typically, these tests provide valuable insights into the material's crosslinking density: the higher the gel content of the material, the higher the crosslinking degree. The soluble fraction typically consists of low molecular weight impurities, unreacted starting materials, or products of low crosslinking density. Here, the presence of soluble oligomeric structures can be excluded as indicated by GPC analysis (see ESI, Fig. S26†).A clear trend can be observed in both parameters related to lignin content of the PHUs. As the lignin content increases, the swelling percentage decreases, while the gel content exhibits the opposite trend. For the lowest lignin content (26 wt%), the crosslinking density is the lowest among all compositions. This is because EBC, which does not contribute to cross-linking, accounts for the majority of cyclic carbonate moieties. As the lignin percentage increased, the crosslinking density also increased. Consequently, the gel content rises, reaching a maximum of 98% for 67 wt% lignin, and the swelling decreased to 41% for the highest lignin composition. Generally, the results show a satisfactory gel content in all compositions, demonstrating the efficient formation of a network.
Thermal properties of the thermosets
The thermal properties of the thermosets were also investigated. Initially, thermal stability was analyzed by TGA (see Fig. 4, right). All thermosets showed a higher thermal stability compared to the precursor CCFL. | ||
| Fig. 4 Left: DSC thermograms of thermosets with different lignin weight percentages (compare Table 3), right: TGA decomposition curves for the CCFL (black) and the thermosets with different lignin weight percentages. | ||
Moreover, they all showed a single-step decomposition curve, corresponding to the degradation of the thermoset structure. In contrast, CCFL showed a two-step decomposition curve, where the first step could be ascribed to the cyclic carbonate moieties and the second one to the aromatic structure of lignin. Increased lignin content contributed to improve thermal stability (Td,5% = 287 °C, Td,50% = 400 °C for 26 wt% lignin; Td,5% = 307 °C, Td,50% = 422 °C for 67 wt% lignin), because a higher amount of lignin increases the cross-linking density. Furthermore, lignin is generally known to enhance thermal stability.40,41 A trend in the char residue percentage (see Table 4) was observed, where the residue percentage increased with a higher lignin content (from 21.0 to 33.3%), as lignin contributes to a higher char content.42
DSC thermograms of the different thermosets were also recorded to identify the possible presence of a Tg. Fig. 4, left, shows the thermograms in a temperature range −30 to 180 °C. Assignment of a clear Tg value was not possible, as the range was too broad to identify it. This is probably a result of the complex and heterogenous networks formed. An increase in the heating rate from 10 to 30 K min−1 did not improve the quality of the thermograms. Nevertheless, a Tg is certainly present, as evidenced by the material's behavior when heated. Upon heating, in fact, the material's elasticity changed, making the thermosets appear more flexible, changing back to a more brittle state when cooled back to room temperature (RT). The only thermoset that remained very flexible at RT is the one with a 26 wt% lignin composition, which has a Tg range near RT, as indicated by the thermogram.
DMA analysis
Amongst the different thermoset compositions that were not fragile (see Table 4), the one with the highest lignin content (38 wt%) was selected for DMA analysis. A representative curve of the storage modulus E′ and the tan δ curve is shown in Fig. 5 and the results are reported in Table 5. The material presents a storage modulus below Tg ranging from 1970 MPa at - 30 °C to 1220 MPa at 25 °C (average values, see Table 5 and Table S8†), which dropped reaching a minimum around a temperature of 150 °C (E′150 °C = 5.72 ± 2.41 MPa). The glass transition (or α relaxation temperature, Tα) causes a large change in the material's elasticity, resulting in a decrease of the storage modulus and a peak in the tan(δ) curve. Lignin's aromatic backbone structure and crosslinking density are the main influencing factors for this change in the thermosets.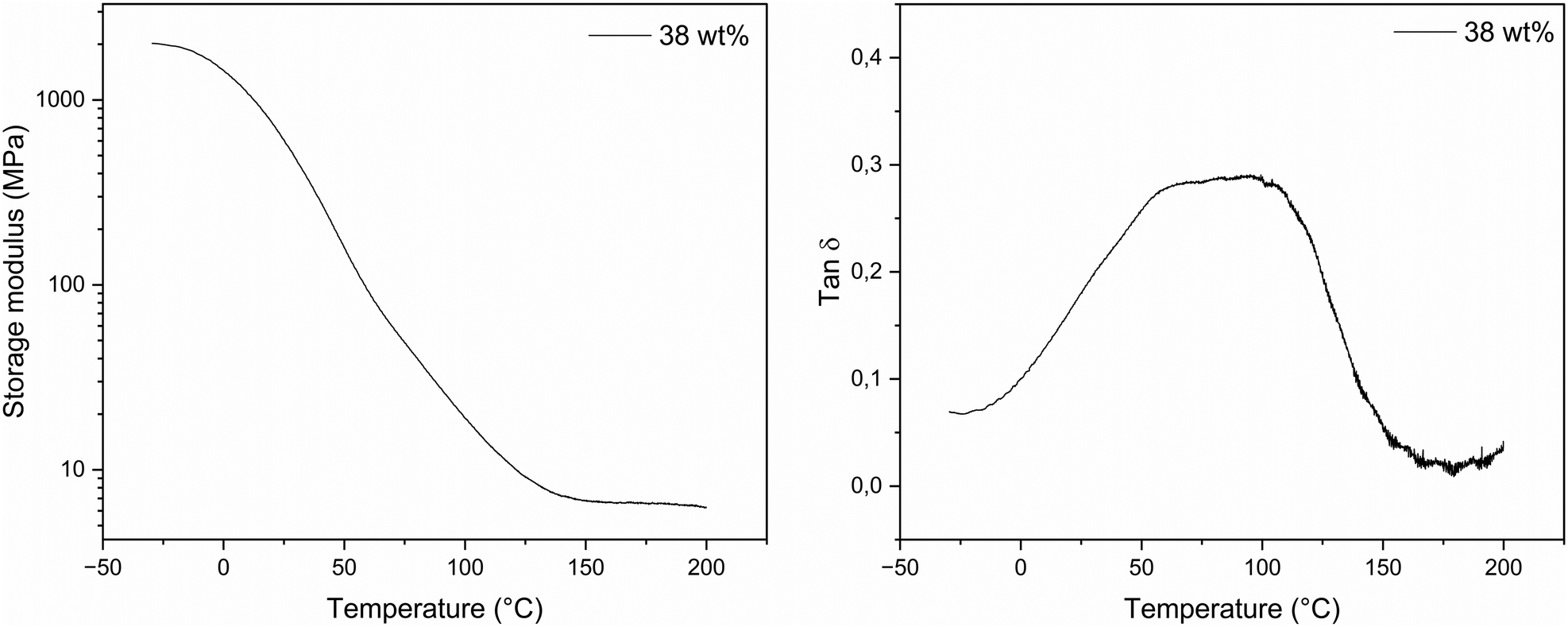 | ||
| Fig. 5 Left: representative curve of the storage modulus (E′) curve against temperature for the 38 wt% lignin content thermoset. Right: representative curve of the tan (δ) curve against temperature for the 38 wt% lignin content thermoset. | ||
| wt% lignin | E′−30 °C (MPa) | E′25 °C (MPa) | E′150 °C (MPa) | T g (°C) Onset E′ | T g (°C) max tan δ |
|---|---|---|---|---|---|
| a Measurements performed in triplicate. | |||||
| 38 | 1970 ± 386 | 1220 ± 223 | 5.72 ± 2.41 | 55.2 ± 2.4 | 97.3 ± 4.1 |
The glass transition can be measured from the onset of the storage modulus or the temperature at the maximum of the tan(δ) peak. The first method usually gives the lowest value of Tg and it is often a good indicator of when the mechanical strength of the material starts to fail. This value may be useful to consider for possible applications. For the composition with 38 wt% lignin content, a maximum Tg of 97.3 °C was reached (peak tan(δ)), average value of three measurements, (see Table 5 and Table S8†), showing a value aligning with literature reported results for lignin-based NIPUs.26
Water contact angle (WCA) measurements
Hydrophobicity and wettability of the thermosets were investigated via static single-drop water contact angle (WCA) measurements. Usually, to be considered hydrophobic, a material should present a WCA higher than 90°. A material is considered superhydrophobic when the WCA is higher than 150°.43 Due to its complex structure, rich in carbon and aromatics, lignin usually presents a more hydrophobic behavior compared to other bioderived polymers, i.e. cellulose or hemicellulose.44 However, hydrophobic/hydrophilic behavior is strongly influenced by the type of functional groups present in the structure. The measurements of the different thermoset compositions reveal a gradual increase in hydrophobicity in concomitance with increasing the lignin content (see Fig. 6), especially when higher than 38 wt%. This trend suggests that the hydrophobic nature of the modified lignin contributes to the final wettability of the material, considering that the molar amount of the PA is the same in every composition. Although the average value of the WCA for the 67 wt% formulation appears to be lower than the value for the 56 wt% formulation, the two resins behave quite similar within error margins. | ||
| Fig. 6 Water contact angle measurements for thermosets with different lignin composition. Measurements were performed 5 times per sample to ensure reproducibility and average values are reported, as well as measurement errors. | ||
To compare with literature data, a study showed that the inherently hydrophilic behavior of linear PHU coatings (due to the pendant hydroxyl groups) can be influenced by varying the chain length of the diamine.45 However, contact angle measurements were still below 90° for all the formulations tested. Detrembleur et al.46 on the other hand investigated the wettability for crosslinked PHUs obtained from cyclocarbonated soybean oil and different diamines revealing a high WCA of 103° for the formulation obtained with m-xylylenediamine (MXDA). This suggests a higher hydrophobicity for aromatic structures, compared to cycloaliphatic (isophorone diamine, 99°) and aliphatic (hexamethylenediamine, 95°) ones.
Conclusions
This work represents the synthesis of a high oleic sunflower oil-based amino crosslinker via thiol–ene click chemistry and its subsequent use in the production of lignin-based non-isocyanate polyurethanes (NIPUs). Although bio-based amines are a crucial component in NIPU formulations, they are rarely documented in the literature. The crosslinker structure was characterized in detail and reaction conditions were investigated both in batch and under continuous flow processes, resulting in a conversion of 99% obtained in 6 hours and in a 2 grams scale, demonstrating the feasibility of scaling up the reaction. Furthermore, the plant oil based amino crosslinker was employed for the synthesis of fully bio-based NIPUs together with cyclic carbonate functionalized lignin and erythritol bis-cyclic carbonate. The lignin content of the resulting thermosets was varied between 26 and 67 wt%, showing how lignin influences the properties of the final materials. IR spectroscopy confirmed the successful and complete curing in all formulations, which was further validated by high gel content values that increased proportionally with lignin content. Formulations up to 38 wt% of lignin were still flexible at room temperature conditions, whereas higher lignin contents resulted in fragile materials with poor mechanical performance. All formulations exhibited thermal stability up to 250 °C, with the 38 wt% lignin formulation showing a Tg of 97.3 °C, as determined by DMA analysis, indicating suitability for applications requiring both high thermal resistance and elevated Tg. The wettability of the materials was also examined, showing an increase in hydrophobicity with higher lignin content. In summary, this work highlights the potential of the bio-based amino crosslinker in the formulation of sustainable NIPUs and underscores the significant role of lignin content in tailoring the mechanical, thermal, and surface properties of the resulting materials.Author contributions
F. C. Destaso: conceptualization, methodology, analyses, investigation, writing. C. Libretti: conceptualization, methodology, analyses, investigation, writing; C. Le Coz: analyses; E. Grau: validation, supervision, writing; M. A. R. Meier: conceptualization, validation, supervision, writing. H. Cramail: validation, supervision, writing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We would like to thank the financial support provided by the NIPU-EJD project; this project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 955700. The authors would like to thank Mathis Mitha and the Wagenknecht group for providing the means and expertise to perform the flow reactor experiments, the working group of Prof. Théato for the TGA and SEC measurements and the Bordeaux University for DMA measurements. Additionally, the authors would like to thank the Fraunhofer Center for Chemical-Biotechnological Processes CBP for kindly donating the lignin used in this work.References
- Statista Research Department . Market volume of polyurethane worldwide from 2015 to 2022, with a forecast for 2023 to 2030. Statista. https://www.statista.com/statistics/720341/global-polyurethane-market-size-forecast (accessed 2024-07-02).
- A. Das and P. Mahanwar, A Brief Discussion on Advances in Polyurethane Applications, Adv. Ind. Eng. Polym. Res., 2020, 3(3), 93–101, DOI:10.1016/j.aiepr.2020.07.002.
- A. Brito, How Polyurethane Can Be Used in Today's Manufacturing Industry, Reinf. Plast., 2020, 64(5), 268–270, DOI:10.1016/j.repl.2020.06.001.
- D. Bello, C. A. Herrick, T. J. Smith, S. R. Woskie, R. P. Streicher, M. R. Cullen, Y. Liu and C. A. Redlich, Skin Exposure to Isocyanates: Reasons for Concern, Environ. Health Perspect., 2007, 115(3), 328–335, DOI:10.1289/ehp.9557.
- M. H. Karol and J. A. Kramarik, Phenyl Isocyanate Is a Potent Chemical Sensitizer, Toxicol. Lett., 1996, 89(2), 139–146, DOI:10.1016/S0378-4274(96)03798-8.
- PHOSGENE. In National Research Council (US) Committee on Toxicology. Emergency and Continuous Exposure Limits for Selected Airborne Contaminants, National Academies Press (US), Washington (DC), 1984, vol. 2 Search PubMed.
- Isocyanates . Occupational Safety and Health Administration (OSHA). https://www.osha.gov/isocyanates (accessed 2024-07-02).
- F. Mundo, S. Caillol, V. Ladmiral and M. A. R. Meier, On Sustainability Aspects of the Synthesis of Five-Membered Cyclic Carbonates, ACS Sustainable Chem. Eng., 2024, 12(17), 6452–6466, DOI:10.1021/acssuschemeng.4c01274.
- D. Rother and U. Schlüter, Occupational Exposure to Diisocyanates in the European Union, Ann. Work Exposures Health, 2021, 65(8), 893–907, DOI:10.1093/annweh/wxab021.
- ANNEX XVII TO REACH – Conditions of restriction – Restrictions on the manufacture, placing on the market and use of certain dangerous substances, mixtures and articles. https://echa.europa.eu/documents/10162/503ac424-3bcb-137b-9247-09e41eb6dd5a (accessed 2024-07-03).
- PDF.Pdf. https://eur-lex.Europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32020R1149 (accessed 2024-07-18).
- A. Gomez-Lopez, F. Elizalde, I. Calvo and H. Sardon, Trends in Non-Isocyanate Polyurethane (NIPU) Development, Chem. Commun., 2021, 57(92), 12254–12265, 10.1039/D1CC05009E.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- F. C. M. Scheelje and M. A. R. Meier, Non-Isocyanate Polyurethanes Synthesized from Terpenes Using Thiourea Organocatalysis and Thiol-Ene-Chemistry, Commun. Chem., 2023, 6(1), 239, DOI:10.1038/s42004-023-01041-x.
- F. C. M. Scheelje, F. C. Destaso, H. Cramail and M. A. R. Meier, Nitrogen–Containing Polymers Derived from Terpenes: Possibilities and Limitations, Macromol. Chem. Phys., 2023, 224(3), 2200403, DOI:10.1002/macp.202200403.
- D. A. Echeverri, H. C. Inciarte, N. Cortés, D. A. Estenoz, M. L. Polo and L. A. Rios, Synthesis of a Renewable Polyamine from Canola Oil by Photocatalyzed Thiol–ene Addition of Cysteamine under Green Conditions, J. Appl. Polym. Sci., 2023, 140(28), e54036, DOI:10.1002/app.54036.
- M. Stemmelen, V. Lapinte, J.-P. Habas and J.-J. Robin, Plant Oil-Based Epoxy Resins from Fatty Diamines and Epoxidized Vegetable Oil, Eur. Polym. J., 2015, 68, 536–545, DOI:10.1016/j.eurpolymj.2015.03.062.
- I. Javni, D. P. Hong and Z. S. Petrović, Soy–based Polyurethanes by Nonisocyanate Route, J. Appl. Polym. Sci., 2008, 108(6), 3867–3875, DOI:10.1002/app.27995.
- M. A. R. Meier, Plant–Oil–Based Polyamides and Polyurethanes: Toward Sustainable Nitrogen–Containing Thermoplastic Materials, Macromol. Rapid Commun., 2019, 40(1), 1800524, DOI:10.1002/marc.201800524.
- N. Mattar, V. Langlois, E. Renard, T. Rademacker, F. Hübner, M. Demleitner, V. Altstädt, H. Ruckdäschel and A. Rios de Anda, Fully Bio-Based Epoxy-Amine Thermosets Reinforced with Recycled Carbon Fibers as a Low Carbon-Footprint Composite Alternative, ACS Appl. Polym. Mater., 2021, 3(1), 426–435, DOI:10.1021/acsapm.0c01187.
- J. Liu, X. Li, M. Li and Y. Zheng, Lignin Biorefinery: Lignin Source, Isolation, Characterization, and Bioconversion, in Advances in Bioenergy, Elsevier, 2022, vol. 7, pp. 211–270. DOI:10.1016/bs.aibe.2022.05.004.
- E. I. Evstigneyev and S. M. Shevchenko, Lignin Valorization and Cleavage of Arylether Bonds in Chemical Processing of Wood: A Mini-Review, Wood Sci. Technol., 2020, 54(4), 787–820, DOI:10.1007/s00226-020-01183-4.
- C. Libretti, L. Santos Correa and M. A. R. Meier, From Waste to Resource: Advancements in Sustainable Lignin Modification, Green Chem., 2024, 26(8), 4358–4386, 10.1039/D4GC00745J.
- A. Salanti, L. Zoia, M. Mauri and M. Orlandi, Utilization of Cyclocarbonated Lignin as a Bio-Based Cross-Linker for the Preparation of Poly(Hydroxy Urethane)s, RSC Adv., 2017, 7(40), 25054–25065, 10.1039/C7RA03416D.
- I. Kühnel, B. Saake and R. Lehnen, A New Environmentally Friendly Approach to Lignin-Based Cyclic Carbonates, Macromol. Chem. Phys., 2018, 219 (7), 1700613, DOI:10.1002/macp.201700613.
- J. Sternberg and S. Pilla, Materials for the Biorefinery: High Bio-Content, Shape Memory Kraft Lignin-Derived Non-Isocyanate Polyurethane Foams Using a Non-Toxic Protocol, Green Chem., 2020, 22(20), 6922–6935, 10.1039/D0GC01659D.
- J. Sternberg and S. Pilla, Chemical Recycling of a Lignin-Based Non-Isocyanate Polyurethane Foam, Nat. Sustain., 2023, 6(3), 316–324, DOI:10.1038/s41893-022-01022-3.
- Global vegetable oil production 2023/24. Statista. https://www.statista.com/statistics/263978/global-vegetable-oil-production-since-2000-2001/(accessed 2024-07-18).
- U. Biermann, U. T. Bornscheuer, I. Feussner, M. A. R. Meier and J. O. Metzger, Fatty Acids and Their Derivatives as Renewable Platform Molecules for the Chemical Industry, Angew. Chem., Int. Ed., 2021, 60(37), 20144–20165, DOI:10.1002/anie.202100778.
- M. Ionescu, D. Radojčić, X. Wan, Z. S. Petrović and T. A. Upshaw, Functionalized Vegetable Oils as Precursors for Polymers by Thiol-Ene Reaction, Eur. Polym. J., 2015, 67, 439–448, DOI:10.1016/j.eurpolymj.2014.12.037.
- D. Myriam, A. Rémi, B. Bernard and C. Sylvain, Synthesis of Bio-Based Building Blocks from Vegetable Oils: A Platform Chemicals Approach, OCL, 2013, 20, 16–22, DOI:10.1051/ocl.2012.0489.
- N. D. Vu, S. Bah, E. Deruer, N. Duguet and M. Lemaire, Robust Organocatalysts for the Cleavage of Vegetable Oil Derivatives to Aldehydes through Retrobenzoin Condensation, Chem. – Eur. J., 2018, 24 (32), 8141–8150, DOI:10.1002/chem.201800091.
- H. Cramail, L. Maisonneuve, E. Grau, C. Alfos and A. Wirotius, Fatty Acid-Based (Bis) 6-Membered Cyclic Carbonates as Efficient Isocyanate Free Poly(Hydroxyurethane) Precursors, Polym. Chem., 2014, 5, 6142–6147, 10.1039/C4PY00922C.
- D. Miloslavskiy, E. Gotlib, O. Figovsky and D. Pashin, Cyclic Carbonates Based on Vegetable Oils, Int. Lett. Chem., Phys. Astron., 2014, 27, 20–29, DOI:10.18052/www.scipress.com/ILCPA.27.20.
- C. E. Hoyle and C. N. Bowman, Thiol-Ene Click Chemistry, Angew. Chem., Int. Ed., 2010, 49 (9), 1540–1573, DOI:10.1002/anie.200903924.
- M. Stemmelen, F. Pessel, V. Lapinte, S. Caillol, J.-P. Habas and J.-J. Robin, A Fully Biobased Epoxy Resin from Vegetable Oils: From the Synthesis of the Precursors by Thiol-Ene Reaction to the Study of the Final Material: A Fully Biobased Epoxy Resin, J. Polym. Sci., Part A: Polym. Chem., 2011, 49(11), 2434–2444, DOI:10.1002/pola.24674.
- M. Firdaus and M. A. R. Meier, Renewable Polyamides and Polyurethanes derived from Limonene, Green Chemistry, 2013, 15(2), 370–380, 10.1039/C2GC36557J.
- B. Rongxia, L. Shiyong, X. Wencai, M. Ruiqiang, H. Di, M. Kelin, H. Jiangwei, X. Yong and L. Caichang, characterization of the uv – visible absorption spectra of commonly used photoinitiators, 2017, pp 18–20. DOI:10.26480/wsmce.01.2017.18.20.
- P.-K. Dannecker and M. A. R. Meier, Facile and Sustainable Synthesis of Erythritol Bis(Carbonate), a Valuable Monomer for Non-Isocyanate Polyurethanes (NIPUs), Sci. Rep., 2019, 9(1), 9858, DOI:10.1038/s41598-019-46314-5.
- N. Zhang, P. Tao, Y. Lu and S. Nie, Effect of Lignin on the Thermal Stability of Cellulose Nanofibrils Produced from Bagasse Pulp, Cellulose, 2019, 26(13–14), 7823–7835, DOI:10.1007/s10570-019-02657-w.
- G. Hasan, D. Musajan, G. Hou, M. He, Y. Li and M. Yimit, Role of Different Lignin Systems in Polymers: Mechanical Properties and Thermal Stability, Pol. J. Chem. Technol., 2020, 22(4), 10–16, DOI:10.2478/pjct-2020-0032.
- Y. Xu, K. Odelius and M. Hakkarainen, One-Pot Synthesis of Lignin Thermosets Exhibiting Widely Tunable Mechanical Properties and Shape Memory Behavior, ACS Sustainable Chem. Eng., 2019, 7(15), 13456–13463, DOI:10.1021/acssuschemeng.9b02921.
- A. Lisý, A. Ház, R. Nadányi, M. Jablonský and I. Šurina, About Hydrophobicity of Lignin: A Review of Selected Chemical Methods for Lignin Valorisation in Biopolymer Production, Energies, 2022, 15(17), 6213, DOI:10.3390/en15176213.
- A. Elniski, P. Dongre and B. M. Bujanovic, Lignin Use in Enhancing the Properties of Willow Pellets, Forests, 2023, 14(10), 2041, DOI:10.3390/f14102041.
- M. Tryznowski, J. Izdebska-Podsiadły and Z. Żołek-Tryznowska, Wettability and Surface Free Energy of NIPU Coatings Based on Bis(2,3-Dihydroxypropyl)Ether Dicarbonate, Prog. Org. Coat., 2017, 109, 55–60, DOI:10.1016/j.porgcoat.2017.04.011.
- S. Panchireddy, B. Grignard, J.-M. Thomassin, C. Jerome and C. Detrembleur, Bio-Based Poly(Hydroxyurethane) Glues for Metal Substrates, Polym. Chem., 2018, 9(19), 2650–2659, 10.1039/C8PY00281A.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05645k |
| ‡ These authors contributed equally to the research work. |
| This journal is © The Royal Society of Chemistry 2025 |