
Photo-enzyme-coupled catalysis for selective oxidation of 2,5-diformylfuran into 2,5-furandicarboxylic acid†
Chenxi
Zhang
ab,
Hongqing
Zhao
a,
Peng
Zhan
b,
Houchao
Shan
c,
Yanou
Qi
de,
Wenqiang
Ren
f,
Xiangshi
Liu
a,
Peiyong
Qin
 *b,
Di
Cai
*a and
Tianwei
Tan
*a
*b,
Di
Cai
*a and
Tianwei
Tan
*a
aNational Energy R&D Center for Biorefinery, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: twtan@buct.edu.cn; caidibuct@163.com
bCollege of Life Science and Technology, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: qinpeiyong@tsinghua.org.cn
cHebei Key Laboratory of Applied Chemistry, College of Environment and Chemical Engineering, Yanshan University, Qinhuangdao 066004, China
dSchool of International Education, Beijing University of Chemical Technology, Beijing 100029, China
eCollege of Chemistry, Liaoning University, Shenyang 110036, PR China
fResearch Center for Eco-Environmental Sciences, Chinese Academy of Science, Beijing 100085, China
First published on 2nd January 2025
Abstract
The transformation of 2,5-diformylfuran (DFF) into renewable biomass-derived 2,5-furandicarboxylic acid (FDCA) is an attractive reaction. In this study, a novel photo-enzymatic catalysis system was established for selective oxidation of DFF into FDCA under mild conditions and through a one-pot process, in which 2-ethylanthraquinone served as the homogeneous photocatalyst for in situ H2O2 production under visible light irradiation, while H2O2 served as the oxidant for the organic peracid-assisted oxidation of DFF into FDCA by commercial lipase (Lipozyme 435). Results indicated that the constitution of the buffering system evidently influenced both photocatalyst and lipase activities in the coupling system, which further affected the kinetics of the tandem reactions and FDCA yield. Under optimized conditions (2 mg mL−1 of 2-ethylanthraquinone, 3 mg mL−1 of lipase, 7 W and λ > 420 nm of the light source, and tert-butanol/ethanol ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v:v)), 84.14% ± 4.68% of FDCA yield with complete DFF conversion was realized. Mechanism investigation using molecular docking simulation indicated that H2O2 produced from the light reaction was consumed by lipase to generate peracetic acid from ethyl acetate that drove the selective oxidation of aldehyde groups in the DFF molecule. The novel visible-light-driven photo-enzyme coupling system successfully addressed the challenges associated with manually adding, storing, and transporting H2O2 in conventional DFF oxidation, thus showing promise in valorization of a broader range of aldehyde group-containing biomass-derived molecules into renewable chemicals.
:1 (v:v)), 84.14% ± 4.68% of FDCA yield with complete DFF conversion was realized. Mechanism investigation using molecular docking simulation indicated that H2O2 produced from the light reaction was consumed by lipase to generate peracetic acid from ethyl acetate that drove the selective oxidation of aldehyde groups in the DFF molecule. The novel visible-light-driven photo-enzyme coupling system successfully addressed the challenges associated with manually adding, storing, and transporting H2O2 in conventional DFF oxidation, thus showing promise in valorization of a broader range of aldehyde group-containing biomass-derived molecules into renewable chemicals.
Green Foundation1. Our research developed efficient, sustainable catalytic systems for converting biomass compounds. Using an enzyme-coupled system, we selectively oxidized DFF to FDCA without exogenous hydrogen peroxide. This method avoids high-temperature, high-pressure conditions, minimizes byproducts, and contributes to advancing green chemistry.2. Without external hydrogen peroxide, we achieved an 84.14 ± 4.68% yield of FDCA. Molecular docking and kinetic analyses confirmed the catalytic feasibility of converting ethyl acetate to peracetic acid. 3. Adopting an aqueous system: employing a photo-enzyme coupling approach to achieve the conversion of HMF to FDCA in a water-based system. Improving enzyme stability and reusability: enhancing enzyme stability and recyclability through modification or immobilization, thereby reducing enzyme consumption. Leveraging photocatalytic technology: utilizing photocatalysis to activate specific reaction pathways, thus minimizing the use of oxidizing agents. These improvements are expected to lower production costs while significantly reducing the environmental footprint of the process. |
Introduction
With the rapid growth of the global economy, the increased consumption of non-renewable fossil fuels led to global warming and more frequent natural disasters.1,2 To address these issues, in recent decades, the proper substitution of traditional hydrocarbon-based petroleum refining with biomass-based biorefineries has gained increasing interest.3,4 Lignocelluloses, which are mainly composed of cellulose, hemicellulose, and lignin and are the most abundant biomass resources on the earth,5,6 have long been considered the primary raw materials for the production of various renewable biochemical, fuel, and materials.7–9 In this regard, to valorize carbohydrate fractions in lignocellulose after hydrolysis into mono-sugars, there are basically two-parallel transformation strategies.10 In addition to biologically upgrading sugars via enzymatic catalysis or fermentation,11 the exploration of chemical catalytic transformation for chemical production, for example, 5-hydroxymethylfurfural (HMF), the furan derivate derived from cellulose, is in the ascendant.12 In fact, HMF can be treated as the precursor and further valorized into various fine chemicals in downstream processes.13 Among the candidate valorization routes, the selective oxidation of HMF into 2,5-furandicarboxylic acid (FDCA), the basic building block of a bio-composite, has become a hotpot.14As summarized in Scheme 1, there are primarily three routes for FDCA production from HMF. In addition to the extensively investigated thermo-chemical and electro-chemical routes,15,16 several studies have been devoted to constructing biocatalysis processes for FDCA production under relatively mild and ‘green’ conditions.17,18 Biological catalysis involved in FDCA production from HMF is essentially a two-stage process.19 In the first stage, the hydroxy group in the HMF molecule is oxidized under photo-catalysis or enzymatic catalysis,20,21 eventually forming the 2,5-diformylfuran (DFF) intermediate.22 Next, in the second stage, the aldehyde group in DFF is selectively oxidized into carboxyl groups and finally converted to FDCA.23 In contrast to the well-developed DFF production in the first stage, the oxidation of DFF into FDCA in the second stage is much more difficult due to the presence of active aldehyde groups in the DFF molecule. Therefore, in the oxidation process, other competing reactions, such as over-oxidation, may occur and cause the FDCA yield to decrease.
 | ||
| Scheme 1 Various methods for the preparation of FDCA from HMF. | ||
To this end, in recent years, several studies have focused on developing alternative processes for the biological selective oxidation of DFF into FDCA.24 For instance, Marcelo A. do Nascimento et al. suggested to use LacTV for the selective oxidation of DFF.25 In another work, Li et al. demonstrated good performances in selective DFF oxidation by using CAL-B as a bio-catalyst.26 In addition to the above-mentioned enzymes, literature has also indicated that lipase, the versatile enzyme with high stability in organic solvents, could be used as the biocatalyst for FDCA production.27 In a typical FDCA process catalyzed by lipase, the alkyl ester in the buffer is activated by lipase and oxidized and hydrolyzed into peracids by H2O2, the nucleophile. Then, the peracids served as the oxidant, which drove the selective oxidation of the aldehyde group into carboxyl groups in the DFF molecule.28 A prerequisite to realize effective FDCA production by lipase catalysis is the external addition of excessive H2O2. However, H2O2 is a highly oxidative molecule and poses potential safety hazards, particularly at high concentrations, with risks of explosion and corrosion.29,30
To eliminate the risks and challenges associated with H2O2 storage and application, photocatalytic production of H2O2 is considered a promising solution. This approach enables the green and sustainable synthesis of H2O2 by utilizing visible light sources. Moreover, the process is environmentally friendly, does not need additional energy input, and could significantly reduce the safety hazards and complex steps in conventional H2O2 production.31 To date, various homogeneous and heterogeneous photocatalysts have been developed and employed in H2O2 production. Among them, modeling the industrial anthraquinone oxidation, 2-ethylanthraquinone was suggested as an effective homogeneous photocatalyst capable of producing H2O2 efficiently using ethanol as an electron donor.32
Thus, in this study, inspired by the photocatalytic production of H2O2, we propose to develop a novel chemical-enzymatic catalysis system to produce FDCA from DFF, using inexhaustible solar energy as a driving force, overcoming the series of technical barriers and highlighting the superiorities of the biological transformations; for instance, the mild reaction conditions, high selectivity, and the substrate specificity. In addition, the mechanism of the tandem reactions involved in the one-pot process was clarified by characterizing the photocatalytic performances by performing free radical scavenging experiments and adopting molecular docking simulations. The attractive results obtained in this study suggested that the photo-enzymatic catalysis process exhibited a great perspective for selective catalytic oxidation of bio-based platform compounds in a mild, energy-saving, and ‘green’ way.
Experimental
Materials
Lipase was purchased from Novozymes (Lipozyme 435). 2-Ethylanthraquinone, 2,5-diformylfuran, 5-formylfuran-2-carboxylic acid, 2,5-furandicarboxylic acid, CuSO4·5H2O, 2,9-dimethyl-1,10-phenanthroline, silver nitrate, 1,4-benzoquinone (PBQ), tert-butanol, ethyl acetate, dehydrated ethanol, and ethylenediaminetetraacetic acid disodium salt (EDTA-2Na) were purchased from Macklin Biochemical Co. Ltd. H2O2 (30%, v/v) was produced from Beijing Chemical Work.Photocatalytic H2O2 production
The photocatalytic production of H2O2 was conducted in a 20 mL photo-bioreactor (WATTCAS ChemTech Co. Ltd, China) that was equipped with an LED lamp (λ > 420 nm) as a light source (Fig. S1†). Generally, different concentrations of 2-ethyl anthraquinone as a photocatalyst were dissolved in 10 mL of the solvent composed of tert-butanol and ethanol with different ratios. After stabilizing the mixture in the dark for 0.5 h, batch photoreaction was conducted at room temperature (∼20 °C) with aeration (0.8 mL min−1). During the photo-reaction process, H2O2 concentration in the reactant was detected. All experiments were repeated in duplicate, and the average results are listed. Capturing of active species was conducted to investigate the mechanism of photocatalysis. Before the photocatalysis was carried out, 5 mM of the active species scavenger (PBQ for superoxide anion (˙O2−), AgNO3 for electron (e−), or EDTA-2Na for the hole (h+)) was added into 10 mL of pure tert-butanol, together with 2 mg mL−1 of 2-ethylanthraquinone.Selective oxidation of 2,5-diformylfuran by photo-enzymatic catalysis
In a typical process for selective oxidation of DFF by photo-enzymatic catalysis, 2 mg mL−1 of 2-ethylanthraquinone was dissolved in10 mL of tert-butyl alcohol/ethanol mixture, followed by stabilization at 40 °C for 0.5 h in the dark. The aeration rate was 0.8 mL min−1. A condenser was equipped to recycle solvents from the photoreactor. After 12 h of the photoreaction, 10 mL of ethyl acetate, 3 mg mL−1 of lipase, and 1 mM of DFF were further added into the reactant, and the photo-enzyme coupled reaction was conducted at 40 °C. A control group using 50 mM of H2O2 as an oxidant was also used. In this process, 10 mL of tert-butanol/ethanol mixture was mixed with 10 mL of ethyl acetate as the initial solvent, and the dark reaction was carried out at 40 °C after adding 3 mg mL−1 of lipase.Molecular docking simulations
The feasibility of lipase binding with ethyl acetate was investigated through molecular docking simulations. The lipase structure was obtained from the Protein Data Bank (PDB ID: 1IBS), and the ethyl acetate ligand was sourced from PubChem. Auto dock 4.0 was used for semi-flexible docking studies, and PyMOL 2.5.4 was employed to analyze the active sites. For detailed information, please refer to the ESI.†Assay
H2O2 was determined according to the method in the literature.33 In the first step, two solutions including a 10 g L−1 of 2,9-dimethyl-1,10-phenanthroline (DMP) were dissolved in ethanol and 0.01 M of copper sulfate was prepared. Then, the diluted samples were mixed well with the above two solutions under a volume ratio of 1:1:1. The absorbance at 454 nm was measured after standing the mixture for 10 min in the dark. H2O2 concentration was calculated by an external standard method.
The concentrations of DFF, FFCA, and FDCA were determined by HPLC (Hitachi, Ltd, Japan) equipped with a Thermo C18 column (250 × 4.6 mm, 5 μm) and an ultraviolet detector (265 nm). The mobile phase was a methanol/ammonium formate (3/7, v/v) mixture. The flow rate was 0.5 mL min−1, and the column temperature was 40 °C. Quantitative analysis was conducted by the external standard method (the standard curves are shown in Fig. S2–S4†). All the samples were detected in triplicate.
Results and discussion
Photocatalytic production of H2O2
Literature data suggest that the effective photocatalytic production of H2O2 is the prerequisite to realizing high selectivity for DFF oxidation in tandem lipase catalysis.34,35 Hence, at first, the H2O2 production by homogeneous 2-ethylanthraquinone photocatalyst was conducted in pure tert-butanol under visible light irradiation. As shown in Fig. 1a, the H2O2 concentration in the reactant first increased with the increase of 2-ethylanthraquinone dosage in buffer, and the highest H2O2 concentration of 20.7 ± 1.38 mM was obtained in the group with 2 mg mL−1 of 2-ethylanthraquinone. As 2-ethyl anthraquinone is a homogeneous photocatalyst in the photocatalysis process, the block of active sites in the photocatalyst can be excluded. However, once there were excessive photocatalysts involved in the photo-reaction, the H2O2 yield was decreased. This can be explained by the –OH groups in the tert-butanol molecule; it was difficult for the tertiary alcohol in the buffer to release active hydrogen in the photocatalysis process. This limited proton availability subsequently negatively affected the H2O2 production.36 The effect of light intensity in the photocatalytic H2O2 production was also investigated. Fig. 1b illustrates that H2O2 production increased with the increase of light power. Therefore, for a given amount of photocatalyst dissolved in buffer, a higher light intensity provided more energy, thus, producing more photogenerated carriers.37 However, when the power of the light source exceeded 7 W, the increase in H2O2 concentration became insignificant.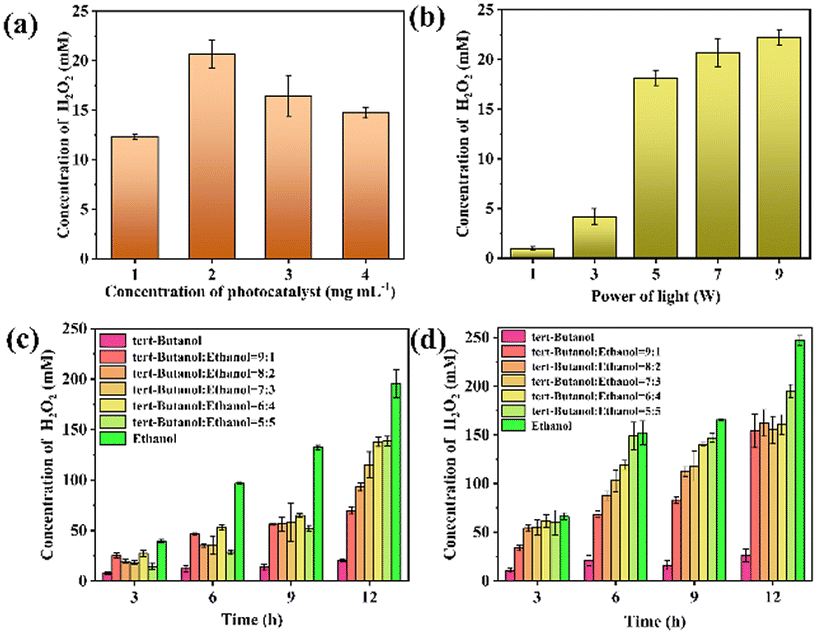 | ||
| Fig. 1 Optimization of photocatalytic H2O2 production using tert-butanol/ethanol mixture as a buffer and 2-ethylanthraquinone as a homogeneous photocatalyst. (a) Photocatalyst concentration (light power was 7 W, and pure tert-butanol was treated as a buffer), and (b) light power (2-ethylanthraquinone dosage was 2 mg mL−1 in pure tert-butanol). Photocatalytic H2O2 production in tert-butanol and ethanol mixture with different volume ratios (2-ethylanthraquinone dosage was 2 mg mL−1, and the light power was 7 W). The reactions (c) without aeration and (d) with aeration. | ||
The above results demonstrated that the H2O2 obtained by the photoreaction in the pure tert-butanol was not as high as the demand for selective catalysis of the aldehyde groups in DFF by lipase. To further improve the photocatalytic production of H2O2, one effective strategy is to use a solution with high hydrogen activity. It has been reported that more H2O2 can be produced by 2-ethylanthraquinone photocatalysis in a buffer consisting of ethanol as an electron donor, because ethanol was easier to oxidize and form H+.32 Whereas, literature also indicated that tert-butanol as a buffer is essential, in order to guarantee the enzyme's stability, reduce side reactions, and improve the selectivity in cascade bio-oxidation.38 Thus, under comprehensive consideration, we propose a compromise solution for the photo-enzymatic catalysis coupling process that could be a tert-butanol/ethanol mixture as the buffer to balance the above two demands.
Therefore, to improve the photocatalytic H2O2 yield, the solvent constitution was further optimized. On this basis, a series of experiments was conducted emphasizing the impact of the tert-butanol/ethanol ratio on H2O2 production. As expected, the addition of ethanol into the pure tert-butanol did promote H2O2 production, and the H2O2 yield was gradually increased with the increase of ethanol ratio in the buffer. A maximum of 195.7 ± 13.8 mM of H2O2 was obtained when using pure ethanol as a buffer (Fig. 1c).
Another effective solution for improved photocatalytic H2O2 production is aeration. Literature indicated that artificially introducing air into the photocatalysis system benefited the H2O2 production, as the oxygen in the air facilitated the oxidation in the photocatalytic reaction.37 The results in Fig. 1d confirmed the above statement. All the tested groups with aeration exhibited higher H2O2 productivity compared to the simple open-air conditions. More specifically, the H2O2 production in 9:1 tert-butanol/ethanol mixture (154.0 ± 17.2 mM) was not much different from the group in 6:4 (160.5 ± 10.0 mM) buffer after 12 h of light irradiation, and both showed much higher production than those obtained in the pure tert-butanol buffer.
Selective oxidation of DFF into FDCA by photo-enzymatic catalysis
The improved photocatalysis of H2O2 production catalyzed by 2-ethylanthraquinone under the optimized conditions was attractive, which promoted further coupling of the photocatalysis with the selective oxidation of DFF into FDCA by lipase. To determine the biocatalysis performances by lipase and guarantee the suitability, the uncoupled enzymatic catalysis reactions in the dark by externally adding H2O2 as an oxidant were performed before carrying out the one-pot photo-enzyme catalysis. The results indicate that in dark reactions, the highest yield of FDCA was achieved when 3 mg mL−1 of lipase was added into a solution containing 10 mL of tert-butanol, 10 mL of ethyl acetate, and 30% H2O2 (10 M) (Fig. S5†).Meanwhile, different from the promotion effect of ethanol in buffer towards the photocatalysis H2O2 production, the presence of excessive ethanol in the reactant always exhibited severe inhibition to the activity of lipase.39 Therefore, there would be a compromise between the satisfied buffering conditions for photocatalysis, which favored a high ethanol proportion, and biocatalysis, which favored a high tert-butyl alcohol proportion. Hence, as aforementioned, the impact of the chemical constitution of the buffer could be a critical issue to be concerned with when coupling photo-catalysis and enzymatic catalysis in a one-pot process.
On this basis, the inhibitory effect of ethanol in buffer on the lipase activity in dark reactions was further investigated. As expected, the FDCA yield from DFF gradually decreased with the increase of ethanol proportion in the buffer. The highest FDCA yield of 90.80% ± 1.21% was obtained when the biocatalysis was carried out in pure tert-butanol, which was 7.1 times higher than that in pure ethanol (Fig. 2). Whereas, the yield of the intermediate product, FFCA, first tended to increase followed by a decrease with the increase of ethanol proportion in the buffer. Among all the candidate buffers, a relatively high FDCA yield (88.83%) can be obtained in 9:1 of tert-butanol/ethanol mixture.
 | ||
| Fig. 2 The impact of buffer constitutions on the selective oxidation of DFF by lipase in the dark reaction. The initial buffer included 30% of external H2O2 (10 M). | ||
After solving the problem of buffer compatibility, we attempted to construct the one-pot photo-enzymatic catalysis system for the selective oxidation of DFF into FDCA, in which 2-ethylanthraquinone served as the photocatalyst to produce H2O2, while lipase served as a biocatalyst for selective oxidation of the aldehyde group in the DFF molecule. Table 1 summarizes the experimental results under various conditions.
| Entry | tert-Butanol/ethanol ratio | Reaction time (h) | Aeration | Catalyst (mg mL−1) | FDCA yield (%) |
|---|---|---|---|---|---|
| Standard reaction conditions: 1 mM DFF as a substrate, 10 mL buffer and 10 mL ethyl acetate, and 40 °C. The aeration rate was 0.8 mL min−1. | |||||
| 1 | Pure tert-butanol | 96 | No | 3 | 35.94 |
| 2 | 9:1 |
96 | No | 3 | 54.42 |
| 3 | 8:2 |
96 | No | 3 | 28.23 |
| 4 | 7:3 |
96 | No | 3 | 22.37 |
| 5 | 6:4 |
96 | No | 3 | 18.10 |
| 6 | 5:5 |
96 | No | 3 | 19.15 |
| 7 | Pure ethanol | 96 | No | 3 | 21.08 |
| 8 | Pure tert-butanol | 96 | Yes | 3 | 53.60 |
| 9 | 9:1 |
96 | Yes | 3 | 65.76 |
| 10 | 8:2 |
96 | Yes | 3 | 50.73 |
| 11 | 7:3 |
96 | Yes | 3 | 25.41 |
| 12 | 6:4 |
96 | Yes | 3 | 31.20 |
| 13 | 5:5 |
96 | Yes | 3 | 32.08 |
| 14 | Pure ethanol | 96 | Yes | 3 | 27.84 |
| 15 | 9:1 |
120 | Yes | 3 | 88.83 |
Specifically, the effect of aeration on FDCA yield in the coupled system was investigated. Fig. 3a shows that FDCA production was significantly benefited by aeration, owing to the promotion of H2O2 production. The highest FDCA yield of 65.8 ± 0.06% was obtained in a 9:1 tert-butanol/ethanol mixture, which is mainly attributed to the relatively high activity of lipase and acceptable H2O2 yield. Besides, although the photocatalytic production of H2O2 in pure tert-butanol was much lower than in the ethanol-containing buffers, a relatively high FDCA yield (53.60 ± 2.12%) was obtained due to the effective preservation of lipase activity in pure tert-butanol. Under these conditions, we hypothesize that the H2O2 productivity by photocatalysis would be the rate-limiting step in the coupled system.
 | ||
| Fig. 3 The performances of a one-pot photo-enzymatic catalysis system for the selective oxidation of DFF into FDCA. (a) The impact of tert-butanol and ethanol ratio in buffer towards the FDCA production under aeration. Samples were taken after 96 h of reactions. The time course of the FDCA production from DFF by photo-enzymatic catalysis in buffer consisted of (b) pure tert-butanol, (c) pure ethanol, and (d) 9:1 of tert-butanol/ethanol. | ||
To further confirm the speculation and better understand the coupling effect of photocatalysis and biocatalysis in one-pot processes, the time course of the FDCA production in photo-enzymatic catalysis systems using pure tert-butanol, pure ethanol, and 9:1 of tert-butanol/ethanol as buffer were compared. In these processes, the yield of FFCA, the intermediate product, was also determined to better understand and compare the kinetics of reactions.
Fig. 3b shows the time course of the coupled system in pure tert-butanol. It illustrated that the FFCA intermediate yield gradually decreased with the increase of the reaction time, demonstrating that lipase catalysis was more effective than the photocatalytic production of H2O2. Indeed, because of the relatively low productivity of H2O2, still ∼13% of FFCA remained in the reactants after 96 h. As expected, in the group with pure ethanol, the lipase activity was largely inhibited though the productivity of H2O2 could be high. Consequently, an extremely low FDCA yield was obtained after 96 h of reaction (Fig. 3c). It is interesting that the FFCA yield gradually increased with the increase of time, indicating that the ethanol buffer was less negatively influenced by the oxidation of DFF (containing two active aldehyde groups) into FFCA (containing one aldehyde group and one carboxyl groups).
However, there was severe inhibition towards the FFCA biotransformation into FDCA, the final product containing two carboxyl groups in a molecule. This phenomenon could be attributed to the more obvious negative impact of ethanol on enzymes or reactive substances, thereby hindering the conversion of FFCA to FDCA. In contrast, in the group that used a tert-butanol/ethanol mixture, the low concentration of ethanol in the buffer realized much more efficient bioconversion of FFCA into FDCA by lipase, while there was an acceptable H2O2 yield. Consequently, the FFCA intermediate can be completely converted into FDCA within 48 h. After 120 h of reaction, the yield of FDCA reached 84.14% ± 4.68%, while the DFF in the initial substrate was completely converted (Fig. 3d).
To investigate whether the buffer has adverse effects on lipase or not, we further recovered the lipase after the reaction through simple filtration and vacuum drying. The elemental analysis results of the recycled lipase were almost unchanged compared to the fresh ones, indicating that the lipase was stable in the coupled reaction processes (the commercial Lipozyme 435 used in this study is an immobilized enzyme) (Table S1†).
Reaction mechanism
The above-mentioned attractive results led us to further explore the synergistic effect of the photocatalysis by 2-ethylanthraquinone and enzymatic catalysis by lipase in the one-pot system. Here, we tried to take a deeper insight into the mechanisms by investigating the uncouple processes. For the photocatalysis process, Fig. 4a shows the visible light absorption capacity of 2-ethylanthraquinone. The absorption edge of the photocatalyst for light lies around 460 nm, and the wider spectral region inferred that the photocatalyst has better absorption and utilization of visible light, i.e., generation of more photogenerated electrons and holes. Plotting (αhv)2versus energy (hv) according to the Kubelka–Munk formula yields a forbidden bandwidth Eg of 2.8 eV. From the M–S diagram (Fig. 4b), it can be further deduced that the conduction band (CB) and valence band (VB) values of 2-ethylanthraquinone were −0.49 V and 2.31 V, respectively (vs. NHE, pH = 7), which satisfied the redox potential of O2/˙O2− (−0.33 V). Meanwhile, the transient photocurrent response in Fig. 4c reflects the charge generation and transfer behavior and demonstrates the reversibility. Therefore, under visible light irradiation, the photocatalyst 2-ethylanthraquinone was excited to produce electrons (e−) and holes (h+). Meanwhile, ˙O2− with oxidizing ability was co-generated. | ||
| Fig. 4 Characterization of the photocatalytic performances by 2-ethylanthraquinone and free radical scavenging experiments. (a) UV-vis DRS, (b) Mott-Schottky (M–S), and (c) transient photocurrent responses. (d) H2O2 production using 2-ethylanthraquinone as a photocatalyst in the presence of reactive species scavengers. | ||
The mechanism of the radical effect on photocatalytic H2O2 production was further investigated by adding scavengers for the reactive species, where pure tert-butanol was used as the buffer and the system without the addition of the scavenger as a blank. As shown in Fig. 4d, the addition of PBQ, a trapping agent for ˙O2−, could reduce the H2O2 yield by 64.3%. Besides, the H2O2 yield also decreased by 27.7% after adding AgNO3 into the buffer, the trapping agent for e−. The above results suggest that ˙O2− and e− played an important role in the photocatalytic generation of H2O2. Moreover, CB and VB values of 2-ethylanthraquinone inferred that the photocatalytic generation of H2O2 can be realized through an indirect pathway of two consecutive steps of single-electron reduction of O2 and a direct pathway of one-step two-electron reduction.40 The addition of an h+ trapping agent (EDTA-2Na) reduced the recombination of electron–hole pairs, improving the electron separation efficiency and facilitating H2O2 generation. However, since h+ is an important driving force for H+ production from oxidized electron donors,41 there was also a certain decrease in H2O2 production after adding a large amount of EDTA-2Na, due to insufficient H+ supplementation. Overall, the importance of the active species involved in photocatalytic H2O2 production ranked as follows ˙O2− > e− > h+.
To further investigate the mechanism of FDCA production in the one-pot process, we employed molecular docking simulations to determine the influence of lipase on the ethyl acetate (as a substrate). The docking results indicate that the binding site of the ethyl acetate molecule was located within the active pocket of lipase (Fig. S6†). This pocket was formed by several hydrophobic residues with substantial side chains: PRO-38, GLY-41, THR-42, THR-43, GLY-44, SER-67, PRO-68, PRO-70, PHE-71, and MET-72. The residues create a cavity that accommodates the binding of ethyl acetate (Fig. 5). In the catalytic state, the distance between the hydrogen atom of PHE-71 in the lipase molecule structure and the two oxygen atoms in the ester bond of the ethyl acetate molecule was 2.1 Å, which was conducive to the formation of intermolecular hydrogen bond and made the C atom in the ester bond more vulnerable to the attack of nucleophiles.42 H2O2 acted as a nucleophile attack to the carbon atom in the ester bond, leading to the conversion of ethyl acetate into peracetic acid, the driving force for the DFF oxidation.
 | ||
| Fig. 5 Molecular docking simulation of lipase with ethyl acetate. | ||
Combining the mechanisms for photocatalysis and biocatalysis, the mechanistic diagram of coupling the photocatalytic production of H2O2via 2-ethylanthraquinone and the selective oxidation of DFF into FDCA via lipase in a one-pot process is presented in Fig. 6. First, 2-ethylanthraquinone produces electron–hole pairs under visible light irradiation. The electrons can reduce the O2 into ˙O2−, while the holes oxidize the electron donors (tert-butanol and ethanol) in the system and produce H+. Then, ˙O2− combines with H+, and continues to be reduced by another electron as well as combining with another H+, forming H2O2 by an indirect pathway. Alternatively, it can be generated by a direct pathway via a one-step two-electron reduction of O2. Subsequently, the H2O2 generated by these two pathways will be utilized by lipase for the biocatalysis conversion of ethyl acetate into peroxyacetic acid, which eventually relies on the strong oxidative properties of peroxyacetic acid to achieve a stepwise oxidation from DFF to FDCA.34
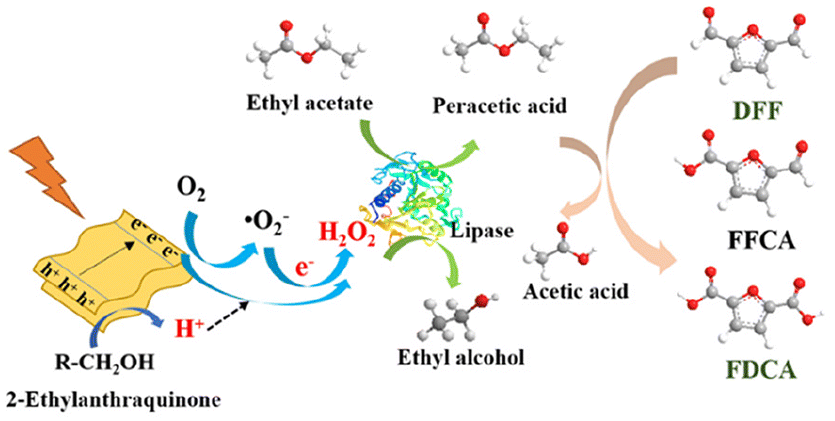 | ||
| Fig. 6 Proposed reaction mechanism of the one-pot photo-enzymatic catalysis for selective oxidation of DFF into FDCA. In the depiction, white represents hydrogen atoms, red represents oxygen atoms, and gray represents carbon atoms. | ||
Therefore, based on a comprehensive analysis of the mechanism of photocatalysis and enzymatic catalysis, the reactions involved in the highly selective oxidation of DFF into FDCA in a one-pot process were clarified. It is not debated that the novel process could successfully avoid the exogenous H2O2 addition. Compared to the traditional methods (as mentioned in Scheme 1), this system was not required to be heated and the addition of any other environmentally unfriendly agents, making it greener and more sustainable.
Conclusions
In this study, for the first time, a one-pot photo-enzymatic catalysis process was constructed for selective DFF oxidation into FDCA, an important building block of bio-composites. In this process, 2-ethyl anthraquinone was employed as a homogeneous photocatalyst, while lipase was used as the biocatalyst. The influence of the buffer constitution and aeration conditions on the substrate H2O2 and FDCA productions were highlighted. Under the optimized conditions, 84.14% ± 4.68% of FDCA yield was obtained after 120 h of the reaction. The mechanisms for the coupled reactions involved in the one-pot process were further clarified by carrying out the free radical scavenging experiments and molecular docking simulations. During the process, H2O2 produced from the light reaction converted ethyl acetate into peracetic acid by lipase, which served as the oxidant and enabled the highly selective oxidation of the aldehyde group in DFF into carboxyl groups in FDCA. This visible-light-driven photo-enzymatic catalysis system expands the reaction pathways of artificial photosynthesis, addresses technical barriers such as the low selectivity in conventional processes, and the difficulties in H2O2 storage and transportation, and holds great promise in extending to valorize a broader range of aldehyde-containing biomass-derived compounds.Author contributions
Chenxi Zhang and Hongqing Zhao completed the main study design and data collection. Chenxi Zhang, Hongqing Zhao and Peng Zhan performed the data analysis and were primarily responsible for writing the paper. All authors actively participated in the discussion, providing valuable feedback and suggestions that enhanced the direction of the research. Cai Di, Qin Peiyong and Tianwei Tan provided the necessary guidance throughout the research process, especially in the experimental design and methods, ensuring the rigor and scientific validity of the research.Data availability
The data that support the findings of this study are available from the corresponding author, [Di Cai], upon reasonable request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is funded by the National Natural Science Foundation of China (22078018; 22478021), and the Beijing Natural Science Foundation (2222016).References
- T. Sharma and A. Kumar, Process Biochem., 2021, 100, 171–177 CrossRef CAS.
- Q. Liu, L. P. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef.
- R. Sindhu, P. Binod and A. Pandey, Bioresour. Technol., 2016, 199, 76–82 CrossRef CAS PubMed.
- T. P. Vispute, H. Y. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS PubMed.
- H. Zu, Y. Wu, Z. Liao, Y. Wang, B. Wang, P. Qin, W. Ren, J. Zhao and D. Cai, Biomass Bioenergy, 2024, 182, 107067 CrossRef CAS.
- Y. Yang, Y. Wang, M. Zhu, J. Zhao, D. Cai and H. Cao, Mater. today Sustain., 2023, 22, 100367 Search PubMed.
- Y. Wang, D. Cai, Y. Jiang, X. Mei, W. Ren, M. Sun, C. Su, H. Cao, C. Zhang and P. Qin, Biotechnol. Biofuels Bioprod., 2024, 17, 62 CrossRef CAS.
- Y. Yang, H. Cao, R. Liu, Y. Wang, M. Zhu, C. Su, X. Lv, J. Zhao, P. Qin and D. Cai, Ind. Crops Prod., 2023, 193, 116213 CrossRef CAS.
- Y. Wu, J. Wen, C. Su, C. Jiang, C. Zhang, Y. Wang, Y. Jiang, W. Ren, P. Qin and D. Cai, Chem. Eng. J., 2023, 452, 139267 CrossRef CAS.
- Y. Wu, C. Su, Z. Liao, G. Zhang, Y. Jiang, Y. Wang, C. Zhang, D. Cai, P. Qin and T. Tan, Biotechnol. Biofuels Bioprod., 2024, 17, 8 CrossRef CAS.
- C. Su, D. Cai, H. Zhang, Y. Wu, Y. Jiang, Y. Liu, C. Zhang, C. Li, P. Qin and T. Tan, Green Carbon, 2024, 2, 81–93 CrossRef.
- J. Zhao, Z. Si, H. Shan, D. Cai, S. Li, G. Li, H. Lin, J. Baeyens, G. Wang, H. Zhao and P. Qin, Ind. Eng. Chem. Res., 2020, 59, 14569–14577 CrossRef CAS.
- J. Zhang, D. Cai, Y. Qin, D. Liu and X. Zhao, Biofuels, Bioprod. Biorefin., 2020, 14, 371–401 CrossRef CAS.
- J. Li, G. Wang, X. Wang, Y. Zhao, Y. Zhao, W. Sui, D. Wang and C. Si, ACS Catal., 2024, 16003–16013 CrossRef CAS.
- J. Xu, Z. Zhu, Z. Yuan, T. Su, Y. Zhao, W. Ren, Z. Zhang and H. Lü, J. Taiwan Inst. Chem. Eng., 2019, 104, 8–15 CrossRef CAS.
- C. V. Nguyen, Y.-T. Liao, T.-C. Kang, J. E. Chen, T. Yoshikawa, Y. Nakasaka, T. Masuda and K. C. W. Wu, Green Chem., 2016, 18, 5957–5961 RSC.
- W. P. Dijkman, D. E. Groothuis and M. W. Fraaije, Angew. Chem., Int. Ed., 2014, 53, 6515–6518 CrossRef CAS.
- Z.-W. Wang, C.-J. Gong and Y.-C. He, Bioresour. Technol., 2020, 303, 122930 CrossRef CAS PubMed.
- L. Hu, A. He, X. Liu, J. Xia, J. Xu, S. Zhou and J. Xu, ACS Sustainable Chem. Eng., 2018, 6, 15915–15935 CrossRef CAS.
- Q. Li, C.-L. Ma and Y.-C. He, Bioresour. Technol., 2023, 378, 128965 CrossRef CAS PubMed.
- D. Yang, N. Zhao, S. Tang, X. Zhu, C. Ma, B. Fan, J. Liang, B. Yu, L. Yang and Y.-C. He, Ind. Crops Prod., 2022, 177, 114434 CrossRef CAS.
- Q. Zhu, Y. Zhuang, H. Zhao, P. Zhan, C. Ren, C. Su, W. Ren, J. Zhang, D. Cai and P. Qin, Chin. J. Chem. Eng., 2023, 54, 180–191 CrossRef CAS.
- X. Zhu, C. Ma, J. Xu, J. Xu and Y.-C. He, Energy Fuels, 2020, 34, 14573–14580 CrossRef CAS.
- M. M. Cajnko, M. Grilc and B. Likozar, Chem. Eng. Sci., 2021, 246, 116982 CrossRef CAS.
- M. A. do Nascimento, B. Haber, M. R. B. P. Gomez, R. A. C. Leão, M. Pietrowski, M. Zieliński, R. O. M. A. de Souza, R. Wojcieszak and I. Itabaiana, Green Chem., 2024, 26, 8211–8219 RSC.
- Y.-Z. Qin, Y.-M. Li, M.-H. Zong, H. Wu and N. Li, Green Chem., 2015, 17, 3718–3722 RSC.
- P. Reis, K. Holmberg, H. Watzke, M. E. Leser and R. Miller, Adv. Colloid Interface Sci., 2009, 147–148, 237–250 CrossRef CAS.
- M. Krystof, M. Pérez-Sánchez and P. Domínguez de María, ChemSusChem, 2013, 6, 826–830 CrossRef CAS.
- M. Dan, R. Zhong, S. Hu, H. Wu, Y. Zhou and Z.-Q. Liu, Chem Catal., 2022, 2, 1919–1960 CrossRef CAS.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS.
- Ajay, Dimple, P. Verma and H. Yamashita, Chem Catal., 2024, 4, 100870 CrossRef CAS.
- G. Q. Xu, Y. Liang and F. Chen, J. Mol. Catal. A: Chem., 2016, 420, 66–72 CrossRef CAS.
- X. K. Zeng, Y. Liu, Y. Kang, Q. Y. Li, Y. Xia, Y. L. Zhu, H. L. Hou, M. H. Uddin, T. R. Gengenbach, D. H. Xia, C. H. Sun, D. T. McCarthy, A. Deletic, J. G. Yu and X. W. Zhang, ACS Catal., 2020, 10, 3697–3706 CrossRef CAS.
- M. Krystof, M. Perez-Sanchez and P. D. de Maria, ChemSusChem, 2013, 6, 826–830 CrossRef CAS PubMed.
- Y. Z. Qin, Y. M. Li, M. H. Zong, H. Wu and N. Li, Green Chem., 2015, 17, 3718–3722 RSC.
- S. Dutta Banik, M. Nordblad, J. M. Woodley and G. H. Peters, ACS Catal., 2016, 6, 6350–6361 CrossRef CAS.
- Y. Shao, J. Hu, T. Yang, X. Yang, J. Qu, Q. Xu and C. M. Li, Carbon, 2022, 190, 337–347 CrossRef CAS.
- M. Rospiccio, P. Casucci, A. Arsiccio, C. Udrescu and R. Pisano, Mol. Pharmaceutics, 2023, 20, 3975–3986 CrossRef CAS.
- F. T. T. Cavalcante, F. S. Neto, I. R. D. Falca, J. E. D. Souza, L. S. de Moura, P. D. Sousa, T. G. Rocha, I. G. de Sousa, P. H. D. Gomes, M. C. M. de Souza and J. C. S. dos Santos, Fuel, 2021, 288, 119577 CrossRef CAS.
- X. S. Zhao, Y. Y. You, S. B. Huang, Y. X. Wu, Y. Y. Ma, G. Zhang and Z. H. Zhang, Appl. Catal., B, 2020, 278, 119251 CrossRef CAS.
- J. Luo, C. Z. Fan, L. Tang, Y. N. Liu, Z. X. Gong, T. S. Wu, X. L. Zhen, C. Y. Feng, H. P. Feng, L. L. Wang, L. Xu and M. Yan, Appl. Catal., B, 2022, 301, 120757 CrossRef CAS.
- S. C. C. van der Lubbe and C. Fonseca Guerra, Chem. – Asian J., 2019, 14, 2760–2769 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05475j |
| This journal is © The Royal Society of Chemistry 2025 |