
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel arylindane diols as sustainable primary antioxidants from lignin†
Tessy
Hendrickx
 a,
Laura
Trullemans
*a,
Alexander J.
Heyer
ab,
Imke
Boonen
c,
Marko
Turkalj
d,
Fatima
Rammal
a,
Yiqi
Su
c,
Besarta
Matranxhi
c,
Durgasruthi
Pully
d,
Bart
Van Meerbeek
d,
Peter
Van Puyvelde
e,
Marc
Elskens
c,
Kirsten L.
Van Landuyt
d and
Bert F.
Sels
*a
a,
Laura
Trullemans
*a,
Alexander J.
Heyer
ab,
Imke
Boonen
c,
Marko
Turkalj
d,
Fatima
Rammal
a,
Yiqi
Su
c,
Besarta
Matranxhi
c,
Durgasruthi
Pully
d,
Bart
Van Meerbeek
d,
Peter
Van Puyvelde
e,
Marc
Elskens
c,
Kirsten L.
Van Landuyt
d and
Bert F.
Sels
*a
aDepartment of Microbial and Molecular Systems (M2S), Centre for Sustainable Catalysis and Engineering (CSCE), KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: laura.trullemans@kuleuven.be; bert.sels@kuleuven.be
bDepartment of Chemistry, Stanford University, Stanford, California 94305, USA
cDepartment of Analytical, Environmental and Geo-Chemistry, Vrije Universiteit Brussel, Ixelles, Belgium
dDepartment of Oral Health Sciences, BIOMAT & UZ Leuven, Dentistry, Leuven, Belgium
eDepartment of Chemical Engineering, Soft Matter, Rheology and Technology (SMaRT), KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
First published on 7th May 2025
Abstract
Synthetic phenolic antioxidant additives are commonly used to prevent oxidative degradation in various materials, but they present significant challenges due to their non-renewable origins and potential health risks. This study explores the synthesis of novel arylindane diols—specifically diisoeugenol (DiE) and diisoallylsyringol (DiAS)—and evaluates their potential as safer and more sustainable alternatives. Using zeolite catalysis, a highly selective pathway for synthesizing DiE through the dimerization of the lignin-derived monomer isoeugenol (IE) was demonstrated. The synthesis of DiAS and diisoallylphenol (DiAP) enabled further exploration of how structural differences, such as o-methoxy groups, affect the physicochemical and toxicological properties of these arylindanes. The antioxidant activity of the compounds was tested using ABTS and DPPH assays, revealing strong radical-scavenging capabilities. Furthermore, oxidation onset temperature (OOT) measurements in polypropylene (PP) formulations containing these antioxidants showed improved thermal stability, matching or surpassing that of commercial antioxidants. Toxicological evaluations, including cytotoxicity tests on human gingival fibroblasts and an estrogenic activity (EA) screening using the CALUX assay, indicated low levels of EA and cytotoxicity. These results highlight the potential of DiE and DiAS as effective, renewable, and safe lignin-derived antioxidants for industrial applications.
Green foundation1. This work advances green chemistry by demonstrating the sustainable synthesis of lignin-derived antioxidants, DiE and DiAS, through a selective, zeolite-catalyzed process. It is designed to minimize waste, utilize renewable feedstocks, and align with the EU's Safe and Sustainable-by-Design (SSbD) framework.2. The catalytic approach used in this work results in a high yield and selectivity for DiE, achieving an impressive atom economy of >85%. It eliminates stoichiometric reagents, and the recyclable catalyst enhances sustainability. Toxicological evaluations of the antioxidants confirm low estrogenic and cytotoxic activity, ensuring safety and avoiding regrettable substitutions. 3. Additional detailed toxicity studies and life-cycle analysis (LCA) are recommended to better understand the safety, environmental impact, and overall sustainability of the process. These future steps would strengthen the alignment with the SSbD principles, promoting its potential as a valuable contribution to the advancement of sustainable chemistry. |
Introduction
The degradation of organic materials, lubricants, and plastics due to oxidation is a persistent challenge. Antioxidants play a crucial role in polymer chemistry by delaying oxidation and protecting materials from environmental factors such as UV light, heat, and atmospheric oxygen.1–3 Free radicals generated by heat, shear, or radiation can lead to polymer degradation, especially in the presence of tertiary C atoms, resulting in chain scission. In Fig. 1, the polymer degradation process is represented,4,5 involving the reaction of hydrocarbon compounds with molecular oxygen, forming oxidation products through autoxidation.6 Free radicals react with oxygen to generate peroxy radicals, which further react with organic material to form hydroperoxides (ROOH). These hydroperoxides trigger both thermal and photo-oxidation, which alters the molecular structure and molar mass of polymers. This degradation results in the loss of mechanical properties, such as impact resistance, flexibility, tensile strength, and elongation, as well as changes to the polymer surface, including reduced gloss, diminished transparency, cracking, and yellowing.7 | ||
| Fig. 1 Polymer degradation process. Schematical representation of the polymer degradation process (blue) and the protective role of primary and secondary antioxidants (green). Figure adapted from ref. 4 and 5. | ||
To mitigate these effects, antioxidants are classified into two main categories: primary and secondary antioxidants (AOs, as shown in Fig. 1). Primary antioxidants, such as hindered phenols and secondary aromatic amines, function as hydrogen donors, neutralizing free radicals and stabilizing polymers against oxidation.7,8 These antioxidants inhibit oxidation through chain-terminating reactions, making them effective during both processing and long-term aging. Common examples include Irganox® 1076 (Irg 1076) and Irganox® 1010 (Irg 1010), which are favored for their high molecular weight (530.88 and 1176.79 g mol−1, respectively), ensuring low volatility and effectiveness.9 Spanning lower molecular weights, antioxidants like butylated hydroxytoluene (BHT, 220.18 g mol−1), butylated hydroxyanisole (BHA, 180.24 g mol−1), and tert-butyl hydroquinone (TBHQ, 166.22 g mol−1) are widely used across various applications today. Secondary antioxidants, such as phosphites and phosphonites, work by converting hydroperoxides into more stable alcohols, thereby preventing the formation of highly reactive alkoxy and hydroxy radicals.10 Irgafos® 168 (Irg 168) exemplifies a phosphite-type thioether antioxidant.11 These antioxidants are frequently combined with primary antioxidants to create a synergistic effect, improving overall effectiveness in preventing oxidation.3
Synthetic phenolic antioxidants (SPAs) are the most commonly used man-made antioxidants. However, despite their effectiveness, concerns have been raised about their potential toxicity.12 Some antioxidants, such as BHT and BHA, have been shown to readily migrate from the polymer matrix into the packaged materials or surrounding environment, raising environmental and health concerns.13–19 Table S1 in ESI,† adapted from Wiesinger et al.,20 gives an overview of the toxicity profile, regulations, and occurrence of lower molecular weight SPAs BHT, BHA, and higher molecular weight SPAs Irg 1010 and Irg 1076. Different health hazards can be related to these compounds, including mutagenic, carcinogenic, and endocrine-disrupting activities.12,21–24 In addition to the toxicity concerns associated with these commercial antioxidants, they are primarily derived from fossil resources. However, growing demand for more sustainable alternatives has spurred efforts to develop antioxidants from natural sources, in line with the EU's strategy for promoting safe and sustainable chemicals.25
Due to its natural polyphenolic structure, lignin stands out as an excellent candidate for a renewable and non-toxic antioxidant. In addition to its inherent antioxidant activity,26–32 it is the largest source of natural aromatics, important building blocks for the chemical industry.33,34 By employing lignin depolymerization techniques, such as Reductive Catalytic Fractionation (RCF), monomeric o-methoxyphenols like isoeugenol (IE) and isoallylsyringol (IAS) can be produced (Fig. 2).34–37 It has already been demonstrated that the additional o-methoxy groups present in lignin reduce the in vitro estrogenic activity (EA) of these molecules, making them promising candidates for developing safer and more sustainable alternatives.38–40 Furthermore, these electron-donating o-methoxy substituents improve the radical-scavenging capability of the phenolic moiety by providing conjugative and inductive effects that stabilize the phenoxy radical.41
 | ||
| Fig. 2 Primary synthetic phenolic antioxidants. Novel arylindane diols derived from isoeugenol (IE) and isoallylsyringol (IAS), both of which can be obtained from biomass. | ||
Diisoeugenol (DiE) is an o-methoxy substituted compound that can be synthesized from the lignin monomer IE. This dimer of IE contains two phenolic hydroxyl groups, each adjacent to an electron-donating methoxy group, and has been previously studied for its antioxidant properties.42–44 DiE can be classified as an arylindane diol, distinguished by its arylindane core structure. Compounds with this type of structure are known for their notable biological activities,45,46 as well as their ability to impart rigidity in polymers.47–49 A prominent example of an arylindane diol is 1,1,3-trimethyl-3-(p-hydroxyphenyl)-5-indanol (BPI), which has been extensively documented, primarily for its use as a building block in various polymers,48,50–65 but also as an antioxidant.66,67 However, BPI is a fossil-derived compound that does not contain o-methoxy groups and is included in Annex III of REACH68 (Registration, Evaluation, Authorisation and Restriction of Chemicals) due to its potential hazards, which include suspected carcinogenicity, environmental persistence, and reproductive toxicity. Additionally, it has been recognized for its endocrine activity.69,70 DiE has the potential to be a safer and more sustainable alternative.
An overview of the synthesis methods for DiE reported in the literature can be found in Table S2.† First identified in 1891,71 early syntheses primarily involved reacting IE with a homogeneous acid like HCl.42,72–75 Whereas these methods provided lower yields, more recent research has achieved DiE synthesis with yields as high as 99%.76–80 However, these modern methods typically involve complex, multi-step processes that require expensive specialized catalysts, as well as specific ligands and solvents. Recently, it was discovered that DiE can be produced as a side product in the reaction between IE and guaiacol, which yields bisguaiacol P, utilizing various types of acidic catalysts.39 DiE has four diastereoisomers,78 illustrated in Fig. 2, with the α-diastereoisomer predominantly formed. Despite this, the investigation into the specific synthesis and properties of different diastereoisomers has been relatively limited, and there is little to no information available regarding the impact of diastereoisomers on aspects such as toxicity.
Driven by the potential of DiE as a safe and renewable primary antioxidant, we investigated greener synthesis methods to obtain this compound in high yields. Its safety was assessed through cytotoxic evaluations and an EA screening to determine whether its structural similarity to estradiol, the female sex hormone (i.e., a hydrophobic structure with a phenolic group), poses an increased risk of EA. A comprehensive antioxidant evaluation was conducted using two antioxidant activity assays and a polymer application test. In addition to DiE, we also evaluated diisoallylsyringol (DiAS), which was synthesized from IAS.
Results and discussion
Catalytic synthesis of DiE
In the pursuit of a sustainable pathway to synthesize DiE, we investigated the potential of heterogeneous acidic catalysts, such as zeolites and ion exchange resins, and compared their effectiveness to that of homogeneous acidic catalysts. Selected commercial FAU zeolites (CBV712, CBV720, CBV760, and CBV780) also underwent a hierarchization treatment, as detailed in section M2 (ESI†), resulting in mesoporous hierarchized zeolites (CBV712-H, CBV720-H, CBV760-H, and CBV780-H). Fig. 3a illustrates the yield of DiE (in mol%) as a function of the conversion (in mol%) of IE in a batch setup, with 1 mmol of IE and 0.1 mmol of acid sites. Both yield and conversion were determined via GC. The methodology of the reaction analysis can be found in ESI M3.† Additional information and characterization of the heterogeneous catalysts used can be found in Tables M1 and M2 (ESI†), along with bar plots displaying product distribution, yield, conversion, and selectivity for each reaction (Fig. S1 and S2†). | ||
| Fig. 3 DiE synthesis with different catalysts. (a) total yield (mol%) of DiE in function of IE conversion (mol%) for different catalysts (with zoomed insert). Grey dotted line refers to 100% selectivity. (b) Isomer ratio [α-DiE/γ-DiE] in function of selectivity (mol%). Reaction conditions: 1 mmol IE, 0.1 mmol H+, 2.5 mL o-xyl, 80 °C, 4 h, 750 rpm. | ||
Interestingly, out of the more than 30 catalysts screened, 19 exhibited yields nearly equal to their conversion rates, resulting in a high selectivity of over 90 mol%, with data points clustering close to the grey dotted line. For comparison, a blank reaction conducted under the same conditions without a catalyst yielded no product. The reactions involving homogeneous acids achieved varied results with hydrochloric acid (HCl, 29) and nitric acid (HNO3, 30) solutions obtaining very low conversions (<4 mol%) and yields (<1 mol%). In contrast, using pure homogeneous acids, a complete conversion of 100 mol% was obtained, with para-toluene sulfonic acid (p-TSA, 28) achieving a maximum selectivity of 83 mol% and sulfuric acid (H2SO4, 27) showing a selectivity of 80 mol%. Among the zeolites tested, CBV780 (6), HSZ-980HOA (11), CBV28014 (15), and ZSM-22 (16) exhibited the lowest selectivity, with values of 68, 83, and 80 mol%, respectively. This was followed by CBV760 (5), Zeocat PB/65H (9), CBV720-H (4-H), and CBV780-H (6-H), which demonstrated slightly higher selectivities ranging from 85 to 90 mol%. In contrast, the zeolites CBV10A (1), CBV21A (2), CBV712 (3), CBV712-H (3-H), CBV720 (4), CBV760-H (5-H), CP814E* (7), CP814C* (8), CZB150 (10), CBV3024-E (12), CBV5524-G (13), CBV8014 (14), and FER18 (17) all achieved selectivities exceeding 90 mol%. The ion exchange resin Nafion™ NR-50 (23) demonstrated a selectivity of 83 mol%, while Dowex® 50Wx8 (100) (18) and Dowex® 50Wx2 (20–50) (19) had selectivities of 70% and 41%, respectively. Other ion exchange resins, including Amberlyst®-15 (20), Amberlyst®-36 (21), DIAION™ RCP160M (22), Aquivion® PW98 (24), Aquivion® PW79S (25), and Aquivion® PW87S (26), all displayed selectivities above 90 mol%. The high selectivity achieved with heterogeneous catalysts can be attributed to the more confined reaction environment they provide, which promotes site-specific catalysis, a characteristic often observed in zeolites and ion exchange resins.
Further evaluation of heterogeneous catalysts reveals that Dowex® 50Wx8 (100) (18), Dowex® 50Wx2 (20–50) (19), CBV10A (1), CBV3024E (12), Nafion™ NR-50 (23), CBV21-A (2), and CBV712 (3) exhibit low conversions and yields, below 35 mol%. It appears that smaller pore zeolites do not effectively promote the reaction. In contrast, CZB150 (10), Amberlyst®-15 (20), CBV720 (4), Diaion™ RCP 160M (22), CBV712-H (3-H), and CBV760-H (5-H) demonstrate high conversions and yields, exceeding 90 mol%, indicating their potential for efficient and selective DiE synthesis. This suggests that zeolites with higher mesopore surface areas (Smeso), which reflect the volume and distribution of mesopores crucial for enhancing the accessibility and diffusion of reactant molecules, and thus higher total surface areas (SBET), are more effective in catalyzing the reaction. This trend was confirmed for CBV712, where hierarchization significantly increased SBET (Smeso), leading to a 62% increase in yield. For CBV760, a moderate increase in SBET resulted in a smaller yield improvement of 14%. However, for other zeolites, hierarchization did not significantly impact SBET (Smeso), and consequently, it did not substantially improve the yield of DiE for CBV720 (−3%) or CBV780 (+6%). Furthermore, repeated experiments showed that the performance of CBV760-H, with the highest yield of 99%, did not significantly differ from CBV720, with a yield of 92 ± 7%. A comparison of parent and hierarchized zeolites in terms of yield, conversion, selectivity, Smeso, and SBET is given in Fig. S2.†
To identify the most suitable catalyst for the synthesis of DiE, we compared the production, costs, regeneration, toxicity, and performance of the catalyst categories, including ion exchange resins, homogeneous acids, zeolites, and hierarchized zeolites (Table S3†). Homogeneous catalysts present significant sustainability challenges due to their high environmental impact and toxicity. While ion exchange resins demonstrate high yield and selectivity in the DiE reaction, their production is energy-intensive, and they are more difficult to recycle or regenerate after use.81–84 Recycling experiments demonstrated superior regeneration of CBV720 compared to Amberlyst®-15. The results, presented in Fig. S3 and S4,† indicate that after two recycling steps, the zeolite CBV720 maintained its performance, showing only a slight 5 mol% yield reduction when washed with acetone, and a remarkable 7 mol% yield increase after calcination. In contrast, Amberlyst®-15 showed a significant 20 mol% decrease in yield after similar acetone washing. These results confirm that zeolites are more robust and regenerable under tested conditions, with calcination proving especially beneficial for their reusability.
Among the catalysts evaluated, non-hierarchical commercial zeolites are preferred over hierarchized zeolites, as they do not require the additional hierarchization treatment. Consequently, CBV720 was chosen as the optimal catalyst for DiE synthesis.
Formation of different diastereoisomers of DiE
As previously mentioned, DiE exists as four diastereoisomers, with α-DiE and γ-DiE being the two predominant forms. In Fig. 3b, the ratio of these main diastereoisomers (retroactively defined) is presented as a function of selectivity (in mol%). This ratio ranges from 1.5 to 5.9, with α-DiE consistently being the most abundant (α-DiE/γ-DiE > 1). Reactions with HCl (19) and HNO3 (20) yielded only α-DiE (low yields). A discernible trend emerges: the highest ratios are observed with homogeneous acids, followed by ion exchange resins, while zeolites yield the lowest ratios. This suggests that less confinement leads to a greater production of α-DiE. Among all the zeolites tested, FER (17) exhibits the highest ratio, indicating a greater formation of α-DiE. This observation may be attributed to FER having the lowest mesopore surface area (Smeso), whereas zeolites with higher Smeso tend to produce more γ-DiE, thereby confirming the confinement effect at the catalytic site. It can be inferred that catalysis occurs within the pores of zeolites with the lowest diastereoisomeric ratios, while for zeolites like FER, the reaction likely takes place more on the surface. For the selected catalyst, CBV720, the ratio is 1.95, indicating a 66%–34% split between the main isomers α and γ.To further investigate the greater preference for the α diastereomer in homogeneous acidic solutions compared to the reduced preference in zeolite confinement, we measured the ratio of [α]/[γ] as a function of temperature under both conditions. Assuming the rate-limiting step is the same for both scenarios, the change in ln([α]/[γ]) is related to (1/T), as depicted in Fig. 4. This relationship allows us to extract ΔΔH‡ (the difference in activation enthalpy) and ΔΔS‡ (the difference in activation entropy) using the equation derived in ESI D1:†

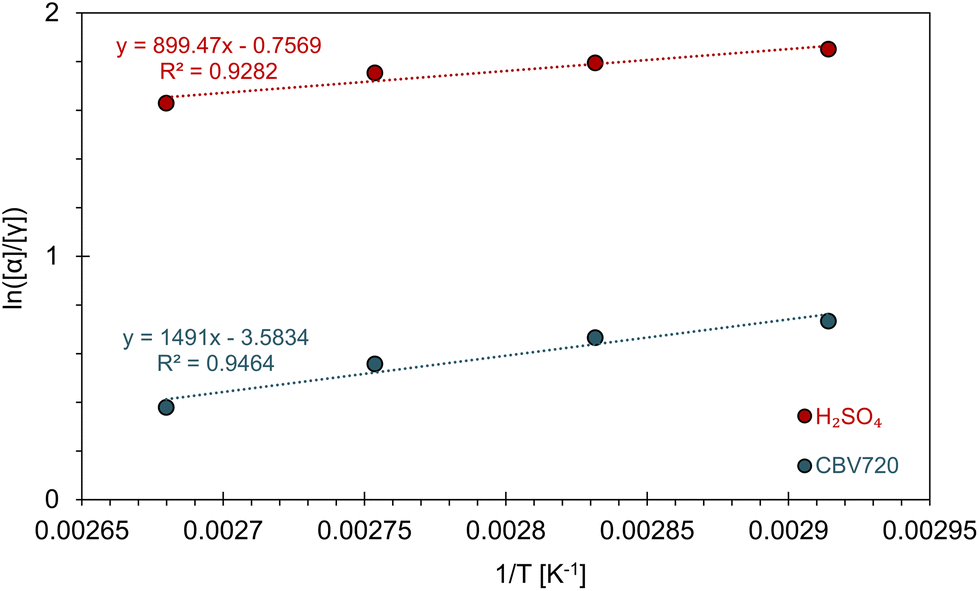 | ||
| Fig. 4 Plot of ln([α]/[γ]) as a function of 1/T for H2SO4 (red) and CBV720 (blue). Fits of the data (dashed lines) with parameters given. | ||
Under the H2SO4 condition, α-DiE has a lower enthalpic barrier by −7.48 kJ mol−1, accompanied by an entropic penalty of −6.29 J mol−1 K−1 (Table 1). In the temperature range of 70–100 °C, the enthalpic advantage prevails, resulting in a preference for the α isomer. When the reaction is conducted in CBV720, the enthalpic advantage for α increases slightly to −12.40 kJ mol−1; however, the entropic preference for γ becomes significantly larger, reaching −29.8 J mol−1 K−1, about five times greater than the previous condition (Table 1). In this case, while α remains the overall preferred isomer, the increased entropic preference for γ has a more pronounced effect, bringing the ratio closer to 1.
| H2SO4 | CBV720 | |
|---|---|---|
| ΔΔH‡ [kJ mol−1] | −7.48 | −12.40 |
| ΔΔS‡ [J mol−1 K−1] | −6.29 | −29.79 |
To gain a chemical understanding of these kinetic parameters, density functional theory (DFT) calculations were conducted for the α and γ isomers, revealing that the transition state preceding the coupling of two IE molecules (TS2, Fig. S12a†) is the determining factor for the diastereoisomeric ratio. The analysis and results are given in ESI D2 and D3.†
Solvent exploration
After identifying a suitable catalyst, the investigation progresses to explore a broader range of solvents, including less conventional options that have been proposed as greener alternatives to traditional solvents. More information on solvent selection is provided in M4, with Tables M3 and M4 (ESI).†Fig. 5 illustrates the yield of DiE as a function of the conversion of IE for reactions conducted with various solvents. The reaction using dimethyl isosorbide (DMI) did not produce any DiE, while reactions with Cyrene™, 2,5-dimethyl tetrahydrofuran (DMTHF), 1-butanol (BuOH), hexane, and heptane yielded only 25–35 mol% of DiE. Notably, the reactions in the aliphatic solvents hexane and heptane exhibited low selectivity, with values of 35% and 31%, respectively. In contrast, the reaction in Cyrene™ demonstrated higher selectivity at 87 mol%. Using methyl-tetrahydrofuran (Me-THF) and γ-valerolactone (GVL) resulted in yields of 50 and 64 mol% DiE, with selectivities of 82 and 92 mol%, respectively. Slightly higher yields were obtained for cumene (68 mol%) and acetonitrile (ACN, 70 mol%), with selectivities of 69 and 78 mol%, respectively. The best results were obtained with anisole, xylene (mixture of isomers), methyl isobutyl ketone (MIBK), cyclopentyl methyl ether (CPME), n-butyl acetate (n-BA), para-xylene (p-xyl), and ortho-xylene (o-xyl), which produced yields between 78 and 95 mol% DiE. However, when using anisole, the selectivity for DiE was limited to 81 mol% due to an undesired condensation reaction between the substrate IE and anisole. This issue was less pronounced with the xylenes, which lack an o-methoxy group that would promote such reactions through electron-donating effects. Notably, the pure isomers of xylene perform better than the mixture of isomers.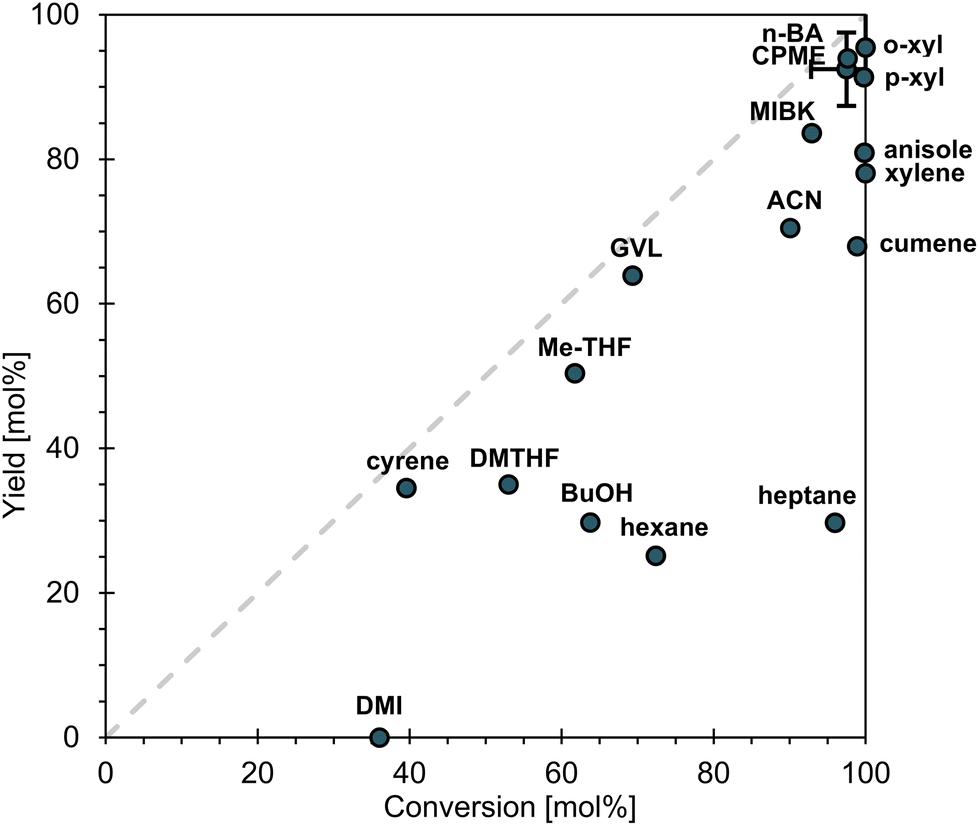 | ||
| Fig. 5 Total yield of DiE in function of IE conversion in different solvents. Grey dotted line refers to 100% selectivity. Reaction conditions: 1 mmol IE, 0.1 mmol H+ (CBV720), 2.5 mL solvent, 80 °C, 4 h, 750 rpm. | ||
Based on these findings, CPME, n-BA, and both xylene isomers emerged as the most suitable solvents for DiE synthesis, exhibiting the highest yield and selectivity. To select the optimal solvent for further investigation, a comparison is provided in Table S4.† Given their similar properties, o-xyl and p-xyl are grouped together as “xylene”. The comparison begins with the Health, Safety, and Environment (HSE) scores, as outlined in the CHEM-21 solvent selection guide.85 CPME, n-BA, and xylene received the same Health scores, and xylene and n-BA received the same Safety score. CPME had a higher Safety score, rated at 7 compared to 4 for xylene and n-BA. This higher score, indicating more safety concerns, is attributed to CPME's low auto-ignition point of 180 °C; however, it is noted that this could be a manageable hazard. n-BA has the lowest Environment score of 3, compared to 5 for xylene and CPME. Furthermore, all three solvents could potentially be sourced from natural materials.86–88 The boiling point and enthalpy of vaporization are higher for xylene, meaning more energy is needed if the solvent has to be evaporated, e.g., for its removal and recycling. However, it was found that DiE spontaneously precipitated fully as a mixture of diastereoisomers (66% α, and 34% γ) from the reaction mixture during cool down in o-xyl, which was not the case when using CPME or n-BA. This characteristic simplifies the work-up process by eliminating the need for solvent distillation. As a result, xylene, specifically the ortho-isomer, was chosen as the solvent to proceed with for DiE synthesis. However, CPME and especially n-BA, with its lower environmental score, present themselves as promising alternatives.
Productivity
To evaluate whether it is possible to reduce the reaction time or temperature without sacrificing productivity, experiments were conducted to vary both the temperature and the concentration of the substrate (IE). Bar plots with the yield, conversion, and selectivity for a varying reaction temperature and substrate concentration are given in Fig. 6a and b, respectively. Compared to the benchmark reaction temperature of 80 °C, both higher (90 °C and 100 °C) and lower (70 °C) temperatures resulted in slightly reduced DiE yields (decreasing by 6 mol% and 4 mol%, respectively) at 100% conversion. Consequently, 80 °C was determined to be the optimal reaction temperature, as it is the lowest temperature that does not compromise yield or selectivity. Additionally, it was found that the substrate concentration could be increased to 15 wt% while maintaining a high yield of 95 mol%. Under these optimal conditions, the reaction was monitored over time to evaluate the total DiE yield and isomer ratio, as shown in Fig. 6c. The results indicate that the DiE yield rapidly increases in the first hour, reaching 84 mol%, and then stabilizes to achieve a final yield of 95 mol% after 4 hours. | ||
| Fig. 6 (a) Yield, conversion, and selectivity (mol%) for reaction with CBV720 at different temperatures (4 h, 7.5 wt% IE). (b) Yield, conversion, and selectivity (mol%) for reaction with CBV720 at different substrate concentrations (4 h, 80 °C). (c) Time course plot giving the total DiE yield (mol%) and the α/γ diastereoisomer ratio at different points in time, employing the ideal reaction conditions (15 wt% IE, 80 °C). Reaction conditions: 1 mmol IE, 0.1 mmol H+ (CBV720), o-xyl, 750 rpm. | ||
A platform of arylindane diols to define structure–property relations
To investigate the effects of (i) the isomeric form and (ii) the lignin-related o-methoxy groups in arylindane diols on their physicochemical, toxicological properties, and antioxidant activity, a series of structurally related arylindanes was synthesized. The synthesis of DiE was initially scaled up to a 3-gram reaction. During the cooling phase, the spontaneous precipitation of the reaction product yielded a DiE composition of 63% α-diastereoisomer and 37% γ-diastereoisomer (α/γ = 1.70) with a total isolated mass yield of 86%. Following recrystallization, white crystals with an isomeric purity of 99% α-diastereoisomer were obtained, in a 53% isolated yield. Additionally, two other arylindane structures were synthesized: diisoallylphenol (DiAP) and DiAS. Alongside using BPI as a reference, this enables a comparative analysis of these four different molecular structures, investigating the influence of o-methoxy groups (none for DiAP, 2 for DiE, and 4 for DiAS) and varying core structures (DiAP vs. BPI). All molecular structures and their variations are given in Fig. 7. DiAP acts as a structural reference, while DiAS is examined alongside DiE for its potential as a renewable and safe antioxidant. | ||
| Fig. 7 Arylindane diols studied in this work. Chemical structures of 1,1,3-trimethyl-3-(p-hydroxyphenyl)-5-indanol (BPI), diisoallylphenol (DiAP), diisoeugenol (DiE) and diisoallylsyringol, (DiAS), showing the structure variations. DiE consists of a 63% α/37% γ diastereoisomeric ratio, α-DiE consist of 99% α. | ||
DiAP has been reported in literature as being formed by the demethylation of the dimer of trans-anethole,89,90 while DiAS has previously been formed by dimerization of syringylpropan-1-ol75,80,91,92 or 2,6-dimethoxy-4-(prop-1-en-1-yl)phenol (IAS).93 In this study, DiAP and DiAS were synthesized from allylphenol (AP) and allylsyringol (AS), respectively. Due to the unavailability of isoallylphenol (IAP) and IAS from commercial sources, an isomerization reaction was initially conducted. However, in future biorefinery applications, IAS should be as readily accessible as IE is.35,36 The synthesized compounds were purified through column chromatography, yielding 8% for DiAP (with 90% of the main isomer) and 53% for DiAS (with 75% of the main isomer). Further details on the synthesis, along with identification methods including GC-MS, 1H-NMR, HSQC, HMBC, 13C-NMR, and DEPT, are provided in ESI M5, M6 and Fig. S5–8,† which also includes data on α-DiE and γ-DiE.
Physicochemical and toxicological properties
| T m [°C] | T d,5% [°C] | T d,10% [°C] | T d,max [°C] | |
|---|---|---|---|---|
| BPI | 192 | 201 | 226 | 281 |
| DiAP | — | 191 | 209 | 260 |
| α-DiE | 186 | 260 | 277 | 342 |
| DiE | 158, 172 | 204 | 225 | 281 |
| DiAS | — | 210 | 222 | 265 |
The DSC results indicate that α-DiE and BPI have high and comparable melting points of 186 °C and 192 °C, respectively, while DiE had a double melting point of 158 °C and 172 °C. The melting points of DiAP and DiAS could not be determined. The TGA thermograms are illustrated in Fig. S9.† A significant difference in thermal stability was observed between the pure α-DiE diastereoisomer and its isomeric mixture, DiE. α-DiE displayed a high degradation onset temperature of 260 °C and a maximum degradation temperature (Td,max) of 342 °C, indicating greater stability than the mixed DiE, which began to degrade at 204 °C and reached a Td,max of 281 °C. Despite this, the DiE diastereoisomer mixture still demonstrated strong thermal stability, comparable to the reference compound BPI, which has a Td,5% of 201 °C and a Td,max of 281 °C. This finding suggests that purification to obtain pure diastereoisomers may not be necessary for applications requiring high thermal stability, as the mixtures still exhibit adequate stability. Furthermore, the presence of additional o-methoxy groups in DiAS and their absence in DiAP did not significantly affect thermal stability, highlighting the minimal impact of these substituents on the overall thermal properties.
The EA of various compounds was evaluated using the Chemically Activated Luciferase gene eXpression (CALUX) assay, which measures the activation of the human estrogen receptor alpha (hERα) in MCF-7 cells, transfected with an estrogen-responsive luciferase gene. All used concentrations were visually tested for cytotoxicity, and concentrations where any sign of cell death was observed were omitted. Key parameters, including the half-maximal effective concentration (EC50), log(EC50), and relative estrogenic efficacy (REE), were determined, with 17β-estradiol (E2) as a positive control and BPA as a reference. Commercial antioxidants BHA and BHT were also tested. Results are given in Fig. 8, accompanied by Table S5.†
 | ||
| Fig. 8 In vitro hERα activity of arylindane diols and references. Red, 17β-estradiol; blue, synthesized arylindane diols; black, references. (a) Concentration–response curves for the CALUX bioassay. Data are presented as mean ± s.d. of 3 replicate cellular exposures in 3 independent experiments (n = 3). (b) Scatterplot of potency and efficacy expressed in terms of REE and log(EC50), respectively. | ||
Fig. 8a illustrates the relative induction (RI) of luciferase activity as a function of compound concentration, while Fig. 8b displays the REE in relation to log(EC50). As anticipated, E2 exhibited the highest potency, with an EC50 of 7.9 × 10−12 M and an REE set to 100%. The steep induction curve for E2 acts as a benchmark for assessing the EA of other compounds. BPA demonstrated significant EA, with an EC50 of 6.4 × 10−7 M and an REE of 122.4%, indicating a higher relative efficacy compared to E2. This pronounced estrogenic effect of BPA is consistent with literature reports,94 underscoring its potency and potential health risks. In contrast, its dimeric analogue, BPI, exhibited reduced EA, with an EC50 of 6.2 × 10−6 M and an REE of 61.1%. This is likely due to its larger structure impeding its binding affinity to the estrogen receptor, resulting in reduced efficacy, even though both compounds possess free hydroxyl groups.95 Interestingly, DiAP, which also lacks o-methoxy groups, exhibited higher induction (REE of 92.8%) than BPI, but lower than BPA. However, with an EC50 of 9.4 × 10−9 M, a smaller concentration of this compound is needed to obtain an estrogenic effect. This suggests that even slight alterations in the core indane structure of these compounds can have a substantial effect on their EA.
DiE, with an o-methoxy group on each side, showed an EC50 of 5.9 × 10−6 M and an REE of 39.3%, while its pure isomer, α-DiE, demonstrated even lower activity with an EC50 of 2.5 × 10−6 M and an REE of 21.5%. This decrease in EA relative to DiAP implies that the presence of o-methoxy groups reduces estrogenic efficacy, likely due to steric hindrance or changes in receptor binding affinity. DiAS, which features two o-methoxy groups on each side, showed no detectable EA. The total absence of activity in DiAS, despite its structural similarity to DiE and α-DiE, further supports the trend that greater o-methoxy substitution results in diminished or non-existent EA. This finding aligns with previous research,39,40,96 reporting similar effects in the study of bisguaiacols, suggesting that o-methoxy groups may hinder the interaction with the estrogen receptor. Note that BHT and BHA did not exhibit significant EA in this assay, which further supports the beneficial effect of sterically ortho-substituents.
In summary, the CALUX assay results reveal significant variations in EA among the tested compounds, primarily influenced by structural differences such as o-methoxy substitution. Both DiE and DiAS demonstrated reduced EA, with DiAS showing no detectable activity, suggesting their potential as safe antioxidants with minimal endocrine-disrupting effects. Additionally, the core structure of the compounds significantly impacts EA, as evidenced by DiAP's lower EA compared to BPI. The comparison between DiE and α-DiE suggests that purification or crystallization to isolate the pure isomer may not be necessary for lowering EA, since both forms already exhibit low activity. These findings offer valuable insights for the development of safer antioxidant compounds for various applications.
 | ||
| Fig. 9 Cytotoxic activity of arylindane diols and references. Box plots representing the EC50 determined by the (a) XTT-assay and (b) LDH-assay. Blue, synthesized arylindane diols; black, references. | ||
The mean EC50 values for cell viability obtained from the XTT assay were as follows: 0.20 mM for BPI, 0.24 mM for α-DiE, 0.21 mM for DiAP, 0.29 mM for DiAS, 0.27 mM for BHT, and 0.81 mM for BHA. In the LDH assay, the mean EC50 values for LDH release were 0.13 mM for BPI, 0.19 mM for α-DiE, 0.16 mM for DiAP, 0.27 mM for DiAS, 0.16 mM for BHT, and 0.47 mM for BHA. Statistical analysis using the Tukey HSD test showed BHA to be significantly less cytotoxic than other compounds. No significant differences in cytotoxicity were found among BPI, DiAP, α-DiE, DiAS, and BHT in both assays, indicating the substitution pattern on the arylindane ring does not significantly influence cytotoxicity.
The consistent results from both the XTT and LDH assays highlight BHA as the least cytotoxic compound among those tested. The arylindane diols exhibited cytotoxicity levels comparable to BHT, represented by EC50 values in the range of 0.19 to 0.28 mM. These results indicate that these compounds could serve as potential alternatives to BHT, providing effective antioxidant properties while maintaining a similar safety profile. The concentrations required to elicit a cytotoxic effect are sufficiently high, posing minimal risk for their use as antioxidants, as they are well above typical leaching levels.12,99
Antioxidant activity
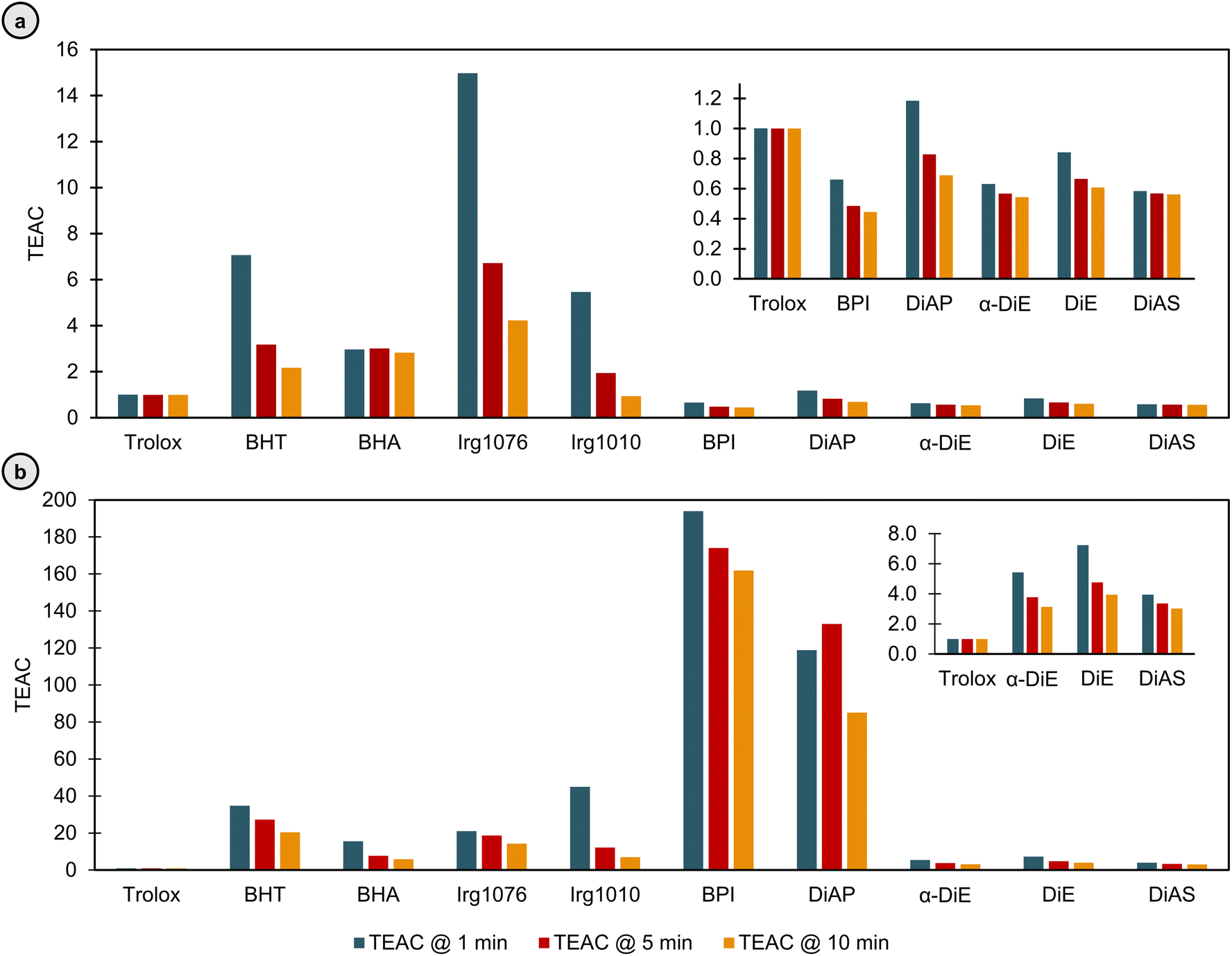 | ||
| Fig. 10 Trolox equivalent antioxidant capacity (TEAC) values after 1, 5, and 10 minutes, as determined by (a) the ABTS-assay, and (b) the DPPH assay. | ||
In the ABTS assay, the novel arylindane compounds demonstrated strong activity. BPI, without o-methoxy groups, exhibited a TEAC value of 0.66 at 1 minute (TEAC1 min), declining to 0.44 at 10 minutes (TEAC10 min), indicating effective radical scavenging. DiAP, also devoid of o-methoxy groups, had a TEAC1 min of 1.18, decreasing to 0.69 at 10 minutes. In contrast, DiE (two o-methoxy groups), showed a TEAC1 min of 0.84 and a TEAC10 min of 0.61. DiAS (four o-methoxy groups), recorded a TEAC1 min of 0.58, decreasing only slightly to 0.56 at 10 minutes. Additionally, the crystallized α-DiE isomer exhibited a TEAC1 min of 0.63, decreasing to a TEAC10 min of 0.54. While the presence of o-methoxy groups and the specific isomeric form seemed to enhance the antioxidant activity of these compounds, the differences among them were modest. Comparatively, BHT and BHA showed significantly higher TEAC values, but reduced over time, with BHT decreasing from 7.06 to 2.17, and BHA decreasing from 2.97 to 2.83. Irg 1076 and Irg 1010, primarily used in polymers, showed high initial TEAC values, with a TEAC1 min of 14.97 and 5.46, that significantly decreased to a TEAC10 min of 4.23 and 0.94, respectively, with Irg1010s lower value attributed to its four hydroxyl groups.
In the DPPH assay, significant antioxidant activity persisted. BPI recorded a TEAC1 min of 193.90, dropping to 161.88 at 10 minutes. DiAP, DiE, and DiAS also declined over time, with DiAP starting at a TEAC1 min of 118.85, ending at a TEAC10 min of 85.12, DiE reducing from a TEAC1 min of 7.24, to a TEAC10 min of 3.94, and DiAS recorded a TEAC1 min of 3.95, which further declined to 3.02. α-DiE displayed a TEAC1 min of 5.43, decreasing to a TEAC10 min of 3.14. Trolox, as a constant reference, maintained a TEAC of 1.00. BHT declined from a TEAC1 min of 34.78 to a TEAC10 min of 20.38, while BHA's TEAC dropped from 15.59 to 5.87. Irg 1076 and Irg 1010 displayed TEAC values decreasing from 21.10 and 45.03 at 1 minute to 14.26 and 6.95 at 10 minutes, respectively.
Whereas the ABTS and DPPH assay both evaluate antioxidants based on two different mechanisms, i.e., hydrogen atom transfer (HAT) and single electron transfer (SET),26 there is a notable difference, with the TEAC values for the DPPH assay being higher in general, and especially for BPI and DiAP. However, both assays show the considerable antioxidant activity of the arylindane diols, highlighting their potential as antioxidants.
Kinetic data, fitted according to two equations (ESI M9, Tables S8 and S9†),101–105 showed good parity between predicted and experimental OOTs, validating both equations to predict the oxidative behavior of PP. For k(Ti) = AkeDT (eqn (3) of ESI†), Fig. 11a illustrates the OOT vs. the heating rate with the fitted curve, while the parity plot (Fig. 11b) confirms the fit. Analogously, these figures are also given for eqn (4) of ESI (Fig. S11†). All OOT values are given in Table S8.†
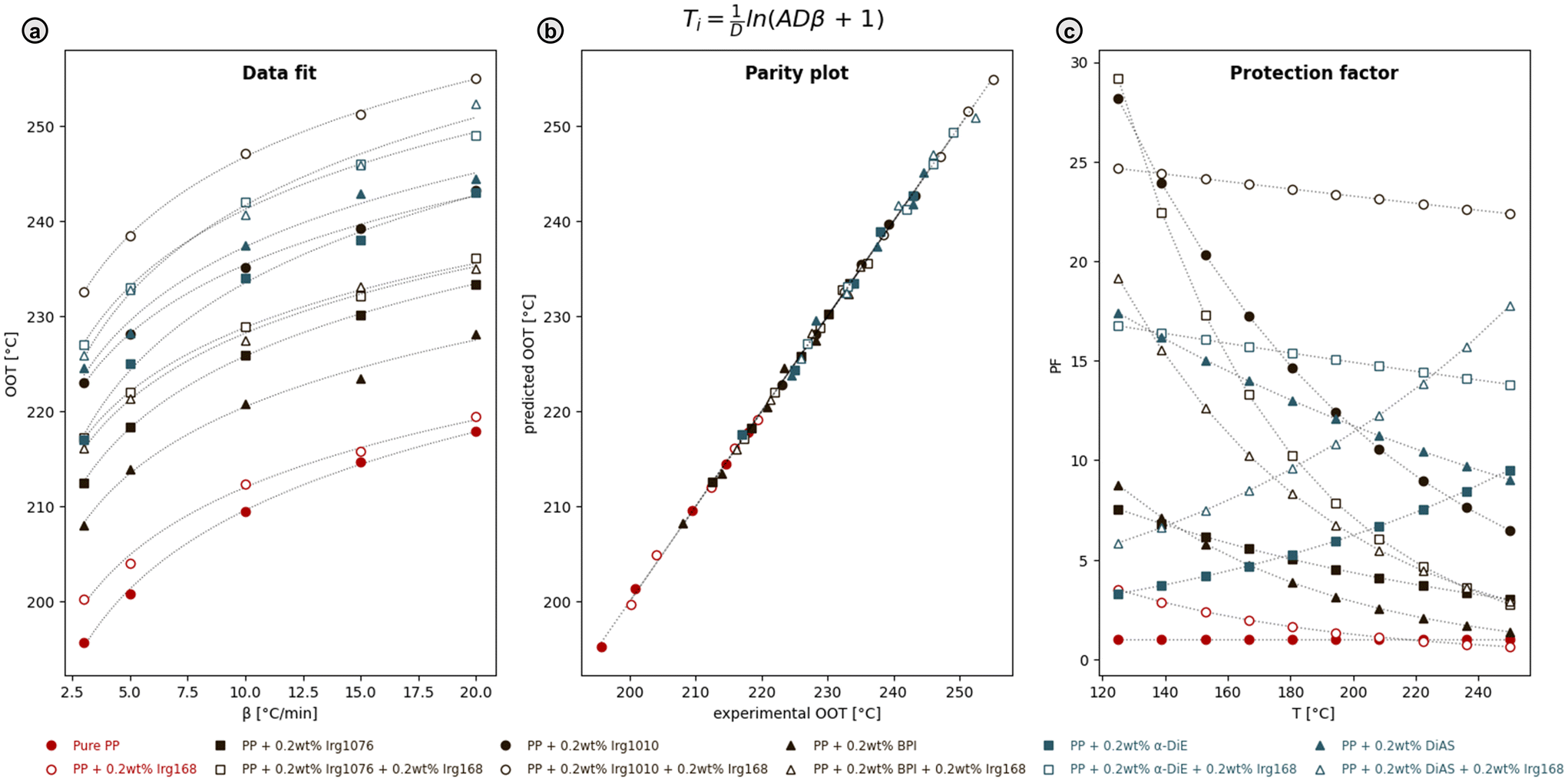 | ||
| Fig. 11 (a) Experimental and fitted dependencies of the onset oxidation temperatures (OOT) on the heating rates β, for pure PP and PP with added antioxidants, employing k(Ti) = AkeDT (eqn (3) from ESI M9†). Data points corresponding to samples with added heat stabilizer, Irg 168, are given as open symbols. (b) Parity plot of predicted and experimental OOT. (c) Dependence of PP protection factor (PF) on temperature. | ||
Addition of antioxidants significantly raises the OOT across all heating rates, compared to pure PP (196 °C at 3 °C min−1). With the inclusion of heat stabilizer Irg 168, the OOT increases to 200 °C, demonstrating a consistent trend where Irg 168 enhances the OOT by 1–10 °C for all samples, improving the polymer's stability. Furthermore, the incorporation of both commercial antioxidants and novel arylindane diols notably increases the OOT. At 3 °C min−1, the OOT is increased by 16 °C (Irg 1076), 27 °C (Irg 1010), 12 °C (BPI), 21 °C (α-DiE), and 29 °C (DiAS). DiAS, with four o-methoxy groups, showed superior performance, outperforming α-DiE (two o-methoxy groups), which in turn surpasses BPI (no o-methoxy groups), likely due to enhanced H-atom donation and chain stabilization. The results highlight that the electron-donating o-methoxy groups, abundant in biomass, can enhance antioxidant activity. Notably, the novel arylindane diols were comparable or superior to industrial standards, positioning them as suitable alternatives.
The length of the induction period (ti)—representing the stability of the polymer before oxidation begins—was calculated at 140 °C (before melting) and 180 °C (after melting), as shown in Table S9.† The results from both equations are highly comparable, ranging from a ti of 14.4 minutes for pure PP at 180 °C (the shortest lifespan) to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 228.5 minutes for PP with 0.2 wt% Irg 1010 and 0.2 wt% Irg168 at 140 °C (the longest lifespan). Protection factors (PFs)—comparing the ti of PP with added antioxidants to that of pure PP—are illustrated in Fig. 11c and S11c† across 120–250 °C. Numerical values for 140 °C and 180 °C are presented in Table S10.† All PFs, excluding the reference of pure PP, exceed one, confirming the stabilizing effect of the additives. The highest PFs—24.37 or 25.18 at 140 °C and 23.63 or 23.55 at 180 °C—were observed with the addition of Irg 1010 and Irg 168. Arylindane diols also achieved high PFs, with the highest recorded for α-DiE and Irg 168 (16.37 or 16.66 at 140 °C and 15.39 or 15.15 at 180 °C), outperforming BPI. Notably, DiAS alone yielded a higher PF at both temperatures (16.08 or 16.50 at 140 °C and 13.03 or 12.90 at 180 °C), but combining DiAS with Irg168 reduced performance (PF of 6.67 or 6.75 at 140 °C and 9.53 or 9.40 at 180 °C), indicating antagonistic cooperation. This observation is supported by a negative S-factor at 140 °C, as calculated and detailed in Table S11.† In contrast, all other combinations of the primary antioxidants with the additional heat stabilizer Irg 168 exhibited synergistic interactions. These calculations are based on comparing the antioxidant effectiveness (AEX)—represented by the PF, adjusted for the amount of antioxidant added—of the mixture against a calculated AEX based on the separate effects of the primary antioxidant and heat stabilizer. Formulas and derivation are based on Černá et al.101 and are given in ESI M9.†
228.5 minutes for PP with 0.2 wt% Irg 1010 and 0.2 wt% Irg168 at 140 °C (the longest lifespan). Protection factors (PFs)—comparing the ti of PP with added antioxidants to that of pure PP—are illustrated in Fig. 11c and S11c† across 120–250 °C. Numerical values for 140 °C and 180 °C are presented in Table S10.† All PFs, excluding the reference of pure PP, exceed one, confirming the stabilizing effect of the additives. The highest PFs—24.37 or 25.18 at 140 °C and 23.63 or 23.55 at 180 °C—were observed with the addition of Irg 1010 and Irg 168. Arylindane diols also achieved high PFs, with the highest recorded for α-DiE and Irg 168 (16.37 or 16.66 at 140 °C and 15.39 or 15.15 at 180 °C), outperforming BPI. Notably, DiAS alone yielded a higher PF at both temperatures (16.08 or 16.50 at 140 °C and 13.03 or 12.90 at 180 °C), but combining DiAS with Irg168 reduced performance (PF of 6.67 or 6.75 at 140 °C and 9.53 or 9.40 at 180 °C), indicating antagonistic cooperation. This observation is supported by a negative S-factor at 140 °C, as calculated and detailed in Table S11.† In contrast, all other combinations of the primary antioxidants with the additional heat stabilizer Irg 168 exhibited synergistic interactions. These calculations are based on comparing the antioxidant effectiveness (AEX)—represented by the PF, adjusted for the amount of antioxidant added—of the mixture against a calculated AEX based on the separate effects of the primary antioxidant and heat stabilizer. Formulas and derivation are based on Černá et al.101 and are given in ESI M9.†
Overall, antioxidants significantly enhanced the oxidative stability of PP. Combining antioxidants with Irg 168 further prolonged polymer lifespan and increased PF. The promising performance of DiE and DiAS, with DiAS showing the highest OOT and longest ti, indicates their potential as effective and more sustainable alternatives to traditional synthetic antioxidants.
Conclusions
In conclusion, this study successfully derived novel, safe, and sustainable primary antioxidants from lignin, specifically focusing on the synthesis and evaluation of diisoeugenol (DiE) and diisoallylsyringol (DiAS). By utilizing zeolite catalysis, we optimized a selective synthesis process for DiE, achieving a high yield of 95 mol% with the recyclable H-USY zeolite CBV720. DFT studies further revealed the formation of two main diastereoisomers (α and γ). This approach, using potentially lignin-derived compounds, aligns with several Green Chemistry principles, including catalysis, atom economy, waste prevention, and the absence of derivatization steps. Additionally, catalysts and solvents were assessed for their safety and sustainability to minimize environmental impact and health risks. While n-butyl acetate (n-BA) is preferred for its lower environmental score, o-xylene (o-xyl) was selected for scale-up due to easier work-up.Aligned with the EU's Safe and Sustainable by Design (SSbD) framework, we screened the in vitro estrogenic activity (EA) and cytotoxicity of these novel arylindane diols early in development. The results showed that DiE (EC50 of 5.9 × 10−6 M) and DiAS (no detectable EA) have significantly lower EA than bisphenol A (BPA) (EC50 of 6.4 × 10−7 M) and estradiol (EC50 of 7.9 × 10−12 M), with low cytotoxicity comparable to conventional antioxidants. Based on these promising findings, we plan further toxicity assessments, including tests on persistence, bioaccumulation, and ecotoxicity.
The antioxidant activities of DiE and DiAS were assessed using ABTS and DPPH assays, showing significant radical scavenging properties with Trolox equivalent antioxidant capacity (TEAC) values exceeding those of commercial antioxidants. Oxidation onset temperature (OOT) measurements for polypropylene (PP) containing these antioxidants demonstrated improved thermal stability, with DiE and DiAS outperforming conventional antioxidants like Irganox® 1010 and Irganox® 1076. Additionally, the DiE isomer mixture was found suitable, eliminating the need for crystallization to the α-isomer, as it exhibited low EA and high antioxidant activity. Our findings highlight the role of lignin-characterizing o-methoxy groups in reducing EA and enhancing antioxidant properties, suggesting the potential of lignin-based compounds as alternatives to synthetic phenolic antioxidants (SPAs). Future work should focus on using lignin fractions directly as substrates for these antioxidants.
This work contributes to the field of sustainable materials by presenting a selective, zeolite-catalyzed pathway for developing potentially lignin-derived arylindane diols as promising antioxidants, achieving both performance and safety goals.
Experimental
Chemicals and materials
A list of all chemicals and materials used can be found in ESI M1.†Reaction procedures
In a typical batch screening reaction, 0.1 mmol of catalytic H+ (ESI M2†) was added to 1 mmol isoeugenol (IE, 0.164 g) in a 10 ml glass reaction vial containing a magnetic stirring bar (13 × 3 mm), whereto 2.5 mL of solvent was added. The vial was capped and put into a pre-heated, magnetically stirred (750 rpm), copper heating block set at 80 °C for 4 h. Samples for GC were taken and filtered using a Millex-FH hydrophobic PTFE syringe filter (0.45 μm pore size, 13 mm), after which they were cooled down by submerging the vial in ice water. The temperature and amount of IE were varied for the increased productivity screening. For the time discourse measurement, samples were taken at certain points in time using a syringe, following the same procedure. In the multigram-scale experiments, the standard reaction procedure was performed on a larger scale in a round-bottom flask, employing a heating mantle.Reaction analysis and product characterization
Analysis by Gas Chromatography with Flame Ionization Detection (GC-FID) was conducted using an Agilent GC system (6890 series) equipped with an Agilent HP5 capillary column (30 m × 0.32 mm, film thickness 0.25 μm) and an FID detector, controlled by ChemStation software. Prior to GC-FID analysis, samples underwent derivatization via trimethylsilylation using N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA). In a standard sample preparation, 10 mg of internal standard (n-propyl guaiacol), 70 μl of anhydrous pyridine, 150 μl of MSTFA, and 150 μl of acetonitrile (ACN) were added to either 100 mg of the reaction mixture or 5–10 mg of pure compounds and heated for 20 minutes at 80 °C.For Gas Chromatography-Mass Spectrometry (GC-MS) analysis, an Agilent GC system (6890 series) equipped with an HP5 capillary column was utilized, coupled with an Agilent Mass Spectroscopy detector (5973 series). This setup facilitated the identification of unknown signals.
The analytical methodology used for GC-FID and quantification details are provided in ESI M3.†
Liquid-phase 1H, 13C, 13C DEPT-135° and 1H–13C HSQC and HMBC NMR spectra were acquired on a Bruker Avance spectrometer (400 MHz) with automated sampler. In a typical sample preparation, 5–30 mg of dried sample was dissolved in 500 μl of deuterated solvent (CDCl3 and acetone-d6).
TGA and DSC were used to determine the compounds’ thermal stability and melting point, respectively. Using a TGA/DSC 3+ instrument of Mettler Toledo equipped with an autosampler, 5–15 mg of sample was heated at a rate of 10 °C min−1 to 600 °C under a constant N2-flow of 90 mL min−1.
Density functional theory (DFT)
DFT calculations were performed with Gaussian 16106 using the B3LYP functional,107 the 6-311G(d) basis set, and the GD3 empirical dispersion correction of Grimme.108 A polarizable continuum model was used to test for electric field solvent effects.109 Transition state structures were validated through intrinsic reaction coordinate calculations. Reaction coordinates include zero-point/thermal corrections. Zeolite Y model used was generated from coordinates of the FAU lattice deposited in the IZA-SC Database of Zeolite Structures.110 Molecular structures were visualized using Chemcraft.111Product work-up
Following a synthesis at a multigram scale, the reaction mixture underwent Büchner filtration to remove the catalyst. The resulting mixture was allowed to stand in an Erlenmeyer and cooled to room temperature, resulting in crystals, composed of 63% α-diastereoisomer and 37% γ-diastereoisomer. Subsequently, to obtain compound α-DiE, further hot recrystallization to achieve an even higher purity was done in ACN, resulting in crystals with a purity of 99% of the α-diastereoisomer. The synthesis and purification procedures for DiAP and DiAS can be found in ESI M5 and M6.†In vitro estrogen receptor transactivation bioassay
The EA of the novel arylindane diols and chosen references was evaluated using the CALUX in vitro human ERα transactivation bioassay. This assay utilizes VM7Luc4E2 cells, defined as human breast cancer MCF-7 cells, which were stably transfected with an estrogen-responsive luciferase gene pGudLuc7.ERE. Activation of ERα transcriptional activity by estrogenically active compounds led to the induction of luciferase reporter gene expression, measured in relative light units. Results were presented as the ER activation induction relative to the positive control's (17β-E2) luciferase induction, set at 100%. The CALUX assay was conducted according to previous protocols.112 Further experimental details are provided in ESI M7.†Data from three replicate cellular exposures in three independent experiments (3 × 3) for each test condition were averaged (± s.d.). EC50 values were determined by fitting a four-parameter sigmoidal dose–response curve (i.e., Hill equation) using Python. For partial concentration-response curves, the fit was constrained at the top and/or bottom value. However, if the fit was deemed unreliable (R2 < 0.95), no EC50 concentration was defined (ND). The Relative Estrogenic Efficacy (REE) was calculated as the top value of the induction relative to the positive control (17β-E2).
Determination of cytotoxicity
Cytotoxicity was assessed using two methods: (i) impact on cell metabolism was measured by the 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) test and (ii) cell membrane damage was quantified by the lactate dehydrogenase (LDH) assay.Tests were performed on primary human gingival fibroblasts (HGFs), where to multiple concentrations of the compounds were dosed. Each concentration was tested in triplicate. For each test, the half-maximal effective concentration (EC50) was determined. A full experimental procedure is provided in ESI M8.†
Antioxidant activity
The antioxidant properties were determined by three methods: (i) the 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS) assay, (ii) the 2,2′-diphenyl-1-picrylhydrazyl (DPPH) assay and (iii) the determination of the Oxidation Onset Temperature (OOT) of polypropylene.Author contributions
T. H., L. T. and B. F. S. conceived and directed the project. T. H. carried out key experiments and wrote the manuscript. As an expert in DFT, A. J. H. contributed to the DFT analysis and section thereof. F. R. assisted with the isomerization reaction. I. B., Y. S, B. M., and M. E. performed the CALUX bioassay. M. T., D. P., K. L. V. L., and B. V. M. performed the XTT and LDH cytotoxicity assays. P. V. P. contributed to the extrusion of PP samples. All authors discussed the results and commented on the manuscript.Data availability
The data supporting this article are reported within the Article and its ESI,† and are available from the corresponding author upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. H. acknowledges LIBRA (Lignin-based flame retardants) – G0D9923N – FWO, Bioeconomy project. L. T. thanks ARD (Aromatic renewable diols for tomorrow's circular economy) – HBC.2023.0158 – Flanders Innovation & Entrepreneurship (VLAIO), for the Innovation Manager funding. T. H. and B. F. S. acknowledge the Flemish Government for the iBOF Next-BIOREF project (Next generation lignocellulose biorefinery concepts and implementation) – The Flemish Interuniversity Council, iBOF/21/105. Measurements were performed with the help of Bijzonder Onderzoeksfonds – Small research infrastructure project KA/20/038 – Advanced thermogravimetric analysis (TGA) tools for advanced study of materials and sustainable processes, and the AgriChemWhey project (BBI-JU, EU Horizon 2020, 744310). Funding for this work was provided by the Anne T. and Robert M. Bass Stanford Graduate Fellowship (to A. J. H.) and the Commission for Educational Exchange between the United States, Belgium, and Luxembourg Fulbright Fellowship (to A. J. H.). Some of the computing for this project was performed on the Sherlock cluster. We would like to thank Stanford University and the Stanford Research Computing Center for providing computational resources and support that contributed to these research results. Y. S. was supported by the China Scholarship Council, grant number 202107650006. B. M. was supported by EXCELLENT SCIENCE—Future and Emerging Technologies (FET), grant agreement ID, 829047. I. B. and M. E. were supported by Research Foundation Flanders project G089016N of the Free University of Brussels (VUB). M. T., D. P., L. T., K. V. L., and B. V. M. were supported by KU Leuven research project C24E/19/078and Research Foundation Flanders project G0C7320N. The authors kindly thank Wisse Hermans and Christophe Courtin (Laboratory of Food Chemistry and Biochemistry, KU Leuven) for the use of the cryomill and Matthew Hickson for his assistance with Python.References
- M. Rubens, P. Falireas, K. Vanbroekhoven and W. Van Hecke, Biomacromolecules, 2023, 24, 3498–3509 CrossRef PubMed.
- J. N. Hahladakis, C. A. Velis, R. Weber, E. Iacovidou and P. Purnell, J. Hazard. Mater., 2018, 344, 179–199 CrossRef PubMed.
- S. Ügdüler, K. M. Van Geem, M. Roosen, E. I. P. Delbeke and S. De Meester, Waste Manage., 2020, 104, 148–182 CrossRef.
- S. Dorey, F. Gaston, N. Girard-Perier, N. Dupuy, S. R. A. Marque, M. Barbaroux and G. Audran, J. Appl. Polym. Sci., 2020, 137, 49336 CrossRef CAS.
- J. Li, C. Zhou, S. Xu and L. Shen, Sci. Rep., 2020, 10, 10189 CrossRef CAS PubMed.
- J. D. Crounse, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard and P. O. Wennberg, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS.
- V. Ambrogi, Modification of Polymer Properties, in 4-Additives in Polymers, ed. C. F. Jasso-Gastinel and J. M. Kenny, William Andrew Publishing, 2017, pp. 87–108 Search PubMed.
- J. Pospíšil, Polym. Degrad. Stab., 1993, 40, 217–232 CrossRef.
- E. Reingruber and W. Buchberger, J. Sep. Sci., 2010, 33, 3463–3475 CrossRef CAS PubMed.
- R. Gensler, C. J. G. Plummer, H.-H. Kausch, E. Kramer, J.-R. Pauquet and H. Zweifel, Polym. Degrad. Stab., 2000, 67, 195–208 CrossRef CAS.
- L. Hermabessiere, J. Receveur, C. Himber, D. Mazurais, A. Huvet, F. Lagarde, C. Lambert, I. Paul-Pont, A. Dehaut, R. Jezequel, P. Soudant and G. Duflos, Sci. Total Environ., 2020, 749, 141651 CrossRef CAS PubMed.
- R. Liu and S. A. Mabury, Environ. Sci. Technol., 2020, 54, 11706–11719 CrossRef CAS.
- Y. Gao, Y. Gu and Y. Wei, J. Agric. Food Chem., 2011, 59, 12982–12989 CrossRef CAS PubMed.
- M. S. Dopico-García, J. M. López-Vilariño and M. V. González-Rodríguez, J. Chromatogr. A, 2003, 1018, 53–62 CrossRef PubMed.
- J. A. Garde, R. Catalá, R. Gavara and R. J. Hernandez, Food Addit. Contam., 2001, 18, 750–762 CrossRef CAS PubMed.
- J. Alin and M. Hakkarainen, J. Appl. Polym. Sci., 2010, 118, 1084–1093 CrossRef CAS.
- J. Alin and M. Hakkarainen, J. Agric. Food Chem., 2011, 59, 5418–5427 CrossRef CAS PubMed.
- G. Beldì, S. Pastorelli, F. Franchini and C. Simoneau, Food Addit. Contam.,:Part A, 2012, 29, 836–845 CrossRef PubMed.
- I. Reinas, J. Oliveira, J. Pereira, F. Machado and M. F. Poças, Food Control, 2012, 28, 333–337 CrossRef CAS.
- H. Wiesinger, Z. Wang and S. Hellweg, Environ. Sci. Technol., 2021, 55, 9339–9351 CrossRef CAS.
- S. P. den Braver-Sewradj, R. van Spronsen and E. V. S. Hessel, Crit. Rev. Toxicol., 2020, 50, 128–147 CrossRef CAS.
- D. Miller, B. B. Wheals, N. Beresford and J. P. Sumpter, Environ. Health Perspect., 2001, 109, 133–138 CAS.
- A. Pop, T. Drugan, F. Loghin, B. Kiss and D. Lupu, J. Appl. Toxicol., 2018, 1–14 Search PubMed.
- A. Pop, B. Kiss and F. Loghin, Clujul Med., 2013, 86, 16–20 Search PubMed.
- European Commission, Chemicals strategy – The EU's chemicals strategy for sustainability towards a toxic-free environment, https://environment.ec.europa.eu/strategy/chemicals-strategy_en#:~:text=The%20European%20Commission%20published%20a,of%20the%20European%20Green%20Deal, (accessed 25 July 2024).
- K. Li, W. Zhong, P. Li, J. Ren, K. Jiang and W. Wu, Int. J. Biol. Macromol., 2023, 251, 125992 CrossRef CAS.
- A. Gregorová, Z. Cibulková, B. Košíková and P. Šimon, Polym. Degrad. Stab., 2005, 89, 553–558 CrossRef.
- X. Lu, X. Gu and Y. Shi, Int. J. Biol. Macromol., 2022, 210, 716–741 CrossRef PubMed.
- T. Dizhbite, G. Telysheva, V. Jurkjane and U. Viesturs, Bioresour. Technol., 2004, 95, 309–317 CrossRef PubMed.
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC.
- D. Ruijten, T. Narmon, K. Van Aelst, H. De Weer, R. van der Zweep, T. Hendrickx, C. Poleunis, L. Li, K. M. Van Geem, D. P. Debecker and B. F. Sels, ACS Sustainable Chem. Eng., 2023, 11, 4776–4788 CrossRef.
- M. A. Jedrzejczyk, S. Van den Bosch, J. Van Aelst, K. Van Aelst, P. D. Kouris, M. Moalin, G. R. M. M. Haenen, M. D. Boot, E. J. M. Hensen, B. Lagrain, B. F. Sels and K. V. Bernaerts, ACS Sustainable Chem. Eng., 2021, 9, 12548–12559 CrossRef.
- Y. Liao, S.-F. Koelewijn, G. van den Bossche, J. van Aelst, S. van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef PubMed.
- T. Renders, S. Van Den Bosch, S.-F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- S. Van Den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, D. Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- W. Schutyser, T. Renders, S. Van Den Bosch, S.-F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- Y. Guo, L. Alvigini, M. Trajkovic, L. Alonso-Cotchico, E. Monza, S. Savino, I. Marić, A. Mattevi and M. W. Fraaije, Nat. Commun., 2022, 13, 7195 CrossRef PubMed.
- A. Amitrano, J. S. Mahajan, L. S. T. J. Korley and T. H. Epps, RSC Adv., 2021, 11, 22149–22158 RSC.
- L. Trullemans, S.-F. Koelewijn, I. Boonen, E. Cooreman, T. Hendrickx, G. Preegel, J. Van Aelst, H. Witters, M. Elskens, P. Van Puyvelde, M. Dusselier and B. F. Sels, Nat. Sustain., 2023, 6, 1693–1704 CrossRef.
- S.-F. Koelewijn, D. Ruijten, L. Trullemans, T. Renders, P. Van Puyvelde, H. Witters and B. F. Sels, Green Chem., 2019, 21, 6622–6633 RSC.
- L. Ross, C. Barclay and M. R. Vinqvist, The Chemistry of Phenols, in Phenols as antioxidants, ed. Z. Rappoport, John Wiley & Sons, 2003, pp. 840–902 Search PubMed.
- C. I. Jarowski, US Pat, 2455256, 1946 Search PubMed.
- T. Atsumi, Y. Murakami, K. Shibuya, K. Tonosaki and S. Fujisawa, Anticancer Res., 2005, 25, 4029–4036 CAS.
- D. R. Merchán Arenas, A. Muñoz Acevedo, L. Y. Vargas Méndez and V. V. Kouznetsov, Sci. Pharmacol., 2011, 79, 779–792 CrossRef PubMed.
- B. Lantaño, J. M. Aguirre, E. A. Ugliarolo, M. L. Benegas and G. Y. Moltrasio, Tetrahedron, 2008, 64, 4090–4102 CrossRef.
- E. Nomura and Y. Kakimoto, Res. Chem. Intermed., 2016, 42, 3567–3577 CrossRef CAS.
- Y. Eguchi and K. Kadowaki, Jpn. Pat, 2021028371A, 2021 Search PubMed.
- B. Dasgupta, S. K. Sen, S. Maji, S. Chatterjee and S. Banerjee, J. Appl. Polym. Sci., 2009, 112, 3640–3651 CrossRef CAS.
- G. Gascoyne and J. L. Gordon, US Pat, 6093785, 2000 Search PubMed.
- K. Yamaguchi, Y. Yoshikawa, K. Sugimoto, Y. Tanabe and T. Yamaguchi, GB Pat, 2153347A, 1985 Search PubMed.
- M. Tamai, S. Kawashima, Y. Sonobe, M. Ota, H. Oikawa and T. Yamaguchi, EP Pat, 0235294A1, 1987 Search PubMed.
- S. Tamai, S. Kawashima, Y. Sonobe, M. Ohta, H. Oikawa and A. Yamaguchi, US Pat, 4734482A, 1988 Search PubMed.
- C. W. Paul, K. M. Patel and S. C. Arnold, US Pat, 4988785A, 1991 Search PubMed.
- Y. Uetani, EP Pat, 0461388A1, 1991 Search PubMed.
- K. M. Patel and F. Mares, US Pat, 5145926A, 1992 Search PubMed.
- J. L. Gordon, K. R. Stewart and C. Park, US. Pat, 5777063, 1998 Search PubMed.
- K. P. Chan, US Pat, 5830988A, 1998 Search PubMed.
- G. C. Davis, A. J. Caruso, J. R. Wetzel, R. Hariharan and M. B. Wisnudel, US Pat, 6001953A, 1999 Search PubMed.
- G. C. Davis, US Pat, 6060577, 2000 Search PubMed.
- K. Takuma and K. Yokohama-shi, EP Pat, 0987567A1, 2000 Search PubMed.
- C. Bailly, B. Idage, S. Sivaran, N. Chavan, R. Faber, S. Menon, G. Chatterjee, G. Varadarajan, J. Day, P. Mcloskey, J. King and A. Jadhav, WO Pat, 0114453A, 2001 Search PubMed.
- C. T. Vijayakumar, S. S. Rishwana, R. Surender, N. D. Mathan and S. Alam, Des. Monomers Polym., 2013, 37–41 Search PubMed.
- S. S. Rishwana, A. Mahendran and C. T. Vijayakumar, High Perform. Polym., 2015, 27, 802–812 CrossRef CAS.
- J. P. Dhanalakshmi, M. A. Raj and C. T. Vijayakumar, Chin. J. Polym. Sci., 2016, 34, 253–267 CrossRef CAS.
- S. Malik, W. Reinerth, B. De, R. Sakamuri, O. Dimov and A. Naiini, WO Pat, 2018085099A1, 2018 Search PubMed.
- I. G. Hinton and R. F. Web, GB Pat, 903062A, 1962 Search PubMed.
- S. Dai, US Pat, 4334106A, 1982 Search PubMed.
- ECHA, Annex III inventory, https://echa.europa.eu/information-on-chemicals/annex-iii-inventory (accessed 8 October 2024).
- M. Terasaki, M. Nomachi, J. S. Edmonds and M. Morita, Chemosphere, 2004, 55, 927–931 CrossRef CAS PubMed.
- M. Terasaki, F. Shiraishi, T. Nishikawa, J. S. Edmonds, M. Morita and M. Makino, Environ. Sci. Technol., 2005, 39, 3703–3707 CrossRef CAS PubMed.
- F. Tiemann, Ber. Dtsch. Chem. Ges., 1891, 24, 2870–2877 CrossRef.
- E. Puxeddu, Gazz. Chim. Ital., 1909, 29, 131–137 Search PubMed.
- R. Haworth and C. Mavin, J. Chem. Soc., 1931, 1363–1366 RSC.
- H.-C. Chiang and S.-Y. Li, J. Chinese Chem. Soc., 1978, 25, 141–147 CrossRef CAS.
- L. S. Smirnova and K. A. Abduazimov, Chem. Nat. Compd., 1996, 32, 98–99 CrossRef.
- A. Gonzalez-De-Castro and J. Xiao, J. Am. Chem. Soc., 2015, 137, 8206–8218 CrossRef CAS PubMed.
- N. Morita, R. Mashiko, D. Hakuta, D. Eguchi, Y. Hashimoto, I. Okamoto and O. Tamura, Synthesis, 2016, 48, 1927–1933 CrossRef CAS.
- T. Yamamoto, B. Riehl, K. Naba, K. Nakahara, A. Wiebe, T. Saitoh, S. R. Waldvogel and Y. Einaga, Chem. Commun., 2018, 54, 2771–2773 RSC.
- T. Takubo, N. Kikuchi, H. Nishiwaki and S. Yamauchi, Org. Biomol. Chem., 2021, 19, 2168–2176 RSC.
- B. Yang, K. Dong, X. S. Li, L. Z. Wu and Q. Liu, Org. Lett., 2022, 24, 2040–2044 CrossRef CAS PubMed.
- J. Lange, W. D. Van De Graaf and R. J. Haan, ChemSusChem, 2009, 2, 437–441 CrossRef CAS PubMed.
- H. Hattori and Y. Ono, in Solid Acid Catalysis, Pan Stanford Publishing, Boca Raton, 2015 Search PubMed.
- F. de Dardel and T. V. Arden, Ion exchangers, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2008 Search PubMed.
- R. Broach, D.-Y. Jan, D. Lesch, S. Kulprathipanja, E. Roland and P. Kleinschmit, Zeolites, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2012 Search PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. Mcelroy, S. Abou-shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- Y. Wang, Z. Chen, M. Haefner, S. Guo, N. DiReda, Y. Ma, Y. Wang and C. T. Avedisian, Fuel, 2021, 304, 121324 CrossRef CAS.
- G. de Gonzalo, A. R. Alcántara and P. Domínguez de María, ChemSusChem, 2019, 12, 2083–2097 CrossRef CAS PubMed.
- Y. Hu, N. Li, G. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, ChemSusChem, 2017, 10, 2880–2885 CrossRef CAS PubMed.
- M. C. Davis, A. J. Guenthner, T. J. Groshens, J. T. Reams and J. M. Mabry, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4127–4136 CrossRef CAS.
- M. D. Garrison, P. J. Storch, W. S. Eck, V. H. Adams, P. W. Fedick and B. G. Harvey, Green Chem., 2021, 23, 8016–8029 RSC.
- L. S. Smirnova, S. Mukhamedova and K. A. Abduazimov, Chem. Nat. Compd., 1991, 27, 648–649 CrossRef.
- L. S. Smirnova and K. A. Abduazimov, Chem. Nat. Compd., 1993, 28, 493–495 CrossRef.
- A. F. A. Wallis, Aust. J. Chem., 1973, 26, 585–594 CrossRef.
- F. S. vom Saal and C. Hughes, Environ. Health Perspect., 2005, 113, 926–933 CrossRef PubMed.
- L. Trullemans, S.-F. Koelewijn, I. Scodeller, T. Hendrickx, P. Van Puyvelde and B. Sels, Polym. Chem., 2021, 12, 5870–5901 RSC.
- S.-F. Koelewijn, C. Cooreman, T. Renders, C. Andecochea Saiz, S. Van Den Bosch, W. Schutyser, W. De Leger, M. Smet, P. Van Puyvelde, H. Witters, B. Van Der Bruggen and B. F. Sels, Green Chem., 2018, 20, 1050–1058 RSC.
- K. Gordon, T. Clouaire, X. X. Bao, S. E. Kemp, M. Xenophontos, J. I. de Las Heras and I. Stancheva, Nucleic Acids Res., 2014, 42, 3529–3541 CrossRef PubMed.
- P. Hughes, D. Marshall, Y. Reid, H. Parkes and C. Gelber, BioTechniques, 2007, 43, 575–586 CrossRef PubMed.
- Y. Wu, M. Venier and R. A. Hites, Environ. Sci. Technol. Lett., 2019, 6, 443–447 CrossRef.
- M. Xin, Y. Ma, K. Xu and M. Chen, J. Appl. Polym. Sci., 2014, 39850, 1–7 Search PubMed.
- A. Černá, Z. Cibulková, P. Šimon, J. Uhlár and P. Lehocký, Polym. Degrad. Stab., 2012, 97, 1724–1729 CrossRef.
- P. Šimon, D. Hynek, M. Malíková and Z. Cibulková, J. Therm. Anal. Calorim., 2008, 93, 817–821 CrossRef.
- P. Šimon, T. Dubaj, Z. Cibulková and A. Vykydalová, presented in part at Central European Symposium on Thermophysics, Banska Bystrica, October, 2019.
- P. Šimon, J. Therm. Anal. Calorim., 2009, 97, 391–396 CrossRef.
- K. Vizárová, M. Reháková, S. Kirschnerová, A. Peller, P. Šimon and R. MikuláŠik, J. Cult. Herit., 2011, 12, 190–195 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- C. Baerlocher and L. B. McCusker, IZA Structure Database, https://www.iza-structure.org/databases.
- Chemcraft – graphical software for visualization of quantum chemistry computations. Version 1.8, build 682. https://www.chemcraftprog.com.
- T. Vandermarken, S. De Galan, K. Croes, K. Van Langenhove, J. Vercammen, H. Sanctorum, M. S. Denison, L. Goeyens, M. Elskens and W. Baeyens, J. Steroid Biochem. Mol. Biol., 2016, 155, 182–189 CrossRef CAS PubMed.
- I. R. Ilyasov, V. L. Beloborodov, I. A. Selivanova and R. P. Terekhov, Int. J. Mol. Sci., 2020, 21, 1131 CrossRef CAS PubMed.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radicals Biol. Med., 1999, 26, 1231–1237 CrossRef CAS PubMed.
- K. M. Schaich, X. Tian and J. Xie, J. Funct. Foods, 2015, 14, 111–125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05190d |
| This journal is © The Royal Society of Chemistry 2025 |