
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrosynthesis of benzyl-tert-butylamine via nickel-catalyzed oxidation of benzyl alcohol†
P. J. L.
Broersen‡
,
V.
Paschalidou‡
and
A. C.
Garcia
 *
*
Van't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH, Amsterdam, The Netherlands. E-mail: a.c.garcia@uva.nl
First published on 3rd January 2025
Abstract
The development of sustainable synthetic methods for converting alcohols to amines is of great interest due to their widespread use in pharmaceuticals and fine chemicals. In this work, we present an electrochemical approach by using green electrons for the selective oxidation of benzyl alcohol to benzaldehyde using a NiOOH catalyst, followed by its reductive amination to form benzyl-tert-butylamine. The number of Ni monolayer equivalents on the catalyst was found to significantly influence selectivity, with 2 monolayers achieving up to 90% faradaic efficiency (FE) for benzaldehyde in NaOH, while 10 monolayers performed best in a tert-butylamine solution (pH 11), yielding 100% FE for benzaldehyde. Reductive amination of benzaldehyde was optimized on Ag and Pb electrodes, with Ag achieving 39% FE towards the amine product, though hydrogen evolution remained a competing reaction. In situ FTIR spectroscopy confirmed the formation of benzaldehyde and its corresponding imine intermediate during oxidation, while reduction spectra supported the formation of the amine product. These results demonstrate the potential of paired electrolysis for alcohol-to-amine conversion, achieving an overall 35% FE for the synthesis of benzyl-tert-butylamine. This work paves the way for more efficient and sustainable electrochemical routes to amine synthesis.
Green foundation1. This work advances green chemistry by developing an electrochemical method for synthesizing benzyl-tert-butylamine, replacing traditional methods that rely on stoichiometric reagents, high pressures, and noble metals. By utilizing nickel oxyhydroxide (NiOOH) and silver electrodes, the approach integrates oxidation and reductive amination steps into a single sustainable process, minimizing waste and improving energy efficiency.2. The electrochemical system achieves up to 90% faradaic efficiency (FE) for benzaldehyde and 39% FE for the final amine, resulting in an overall 35% FE for benzyl-tert-butylamine synthesis. This paired electrolysis strategy significantly reduces the environmental impact compared to conventional routes, avoiding harmful oxidants, reducing catalyst dependence, and operating under ambient conditions. 3. To further improve sustainability, research could optimize catalyst stability, enhance faradaic efficiency, and integrate renewable electricity sources. Scaling up the process and exploring alternative electrode materials with higher selectivity and durability could make this method more efficient and commercially viable. |
Introduction
Organic amines are essential intermediates and final products in industries such as pharmaceuticals, agriculture, and specialty chemicals.1,2 Traditionally, strong reducing (LiAlH4) or alkylation agents are needed in amine synthesis to drive reductive alkylation or SN2 type reactivity.3 This leads to harsh reaction conditions (e.g., strong stoichiometric redox reagents), salt production or overalkylation byproducts.1,4,5 Additionally, industrial production of amines rely on fossil fuel and ammonia (NH3) as feedstock.6 These methods often lead to significant challenges, including the generation of stoichiometric waste, high energy requirements, and the production of undesired byproducts. Additionally, industrial-scale amine production typically relies on fossil-based feedstocks and ammonia (NH3), further contributing to environmental concerns. Among the various pathways to amine synthesis, the conversion of alcohols to amines has garnered significant attention due to the wide availability and functional diversity of alcohols. Alcohols, easily accessible through oxidation of biomass or petrochemical precursors, provide a versatile starting point for sustainable amine synthesis.7Traditional chemical routes for alcohol-to-amine conversion, such as the Mitsunobu reaction or hydrogen borrowing catalysis, have demonstrated considerable utility but are often accompanied by limitations. The Mitsunobu reaction requires toxic reagents and generates significant waste, while hydrogen borrowing catalysis typically involves noble metal catalysts under high temperatures and pressures, limiting its scalability.8,9 Enzymatic pathways, although milder, are constrained by the availability and stability of biocatalysts, which complicates their industrial implementation (Scheme 1).10,11
 | ||
| Scheme 1 Prior work on alcohol to amine conversion reactions and the electrochemical alternative presented here. | ||
Whilst formally a redox neutral process, all mentioned strategies to perform the alcohol to amine conversion rely on a two-step reaction: one oxidation and one reduction step to perform the amination reaction.8–11 Oxidation of a suitable alcohol to the aldehyde can be superseded by imine formation in the presence of an amine.12 This imine can be further reduced to form the amine, completing the catalytic circuit. Since oxidation and reduction steps are involved, it should be possible to perform the reaction in an electrochemical fashion.
Compared to traditional methods, electrochemical approach demonstrates significant improvements in sustainability and efficiency. Unlike chemical oxidation processes that rely on oxidants like chromium-based reagents, the NiOOH catalyst enables selective oxidation under mild conditions, minimizing waste and energy consumption. Similarly, the reductive amination avoids the use of high-pressure hydrogen gas or precious metal catalysts, which are standard in conventional methods. By integrating these reactions into a single electrochemical system, our approach highlights a pathway towards more sustainable and scalable amine synthesis, addressing key limitations of existing technologies.13,14
In this study, we explore benzylamines due to their superior thermodynamic stability compared to aliphatic amines. This enhanced stability makes benzylamines particularly attractive from a synthetic perspective. Moreover, their broad applicability, especially in the pharmaceutical and coatings industries, underscores their importance as a subject of study.15–18
Most literature on electrochemical amination reactions to benzylamine has focused on the electrochemical reductive amination of benzaldehyde using inner or outer sphere electrode materials such as Ag, Au, and carbon, respectively.19–21 Benzaldehyde itself can be directly produced in situ via the electrochemical oxidation of benzyl alcohol, a process involving a two-electron transfer reaction. However, a significant challenge in alcohol oxidation is maintaining selectivity towards the aldehyde and to prevent its further oxidation to the corresponding carboxylic acid, in this case benzoic acid (Scheme 2).22
 | ||
| Scheme 2 Overoxidation of benzaldehyde to benzoic acid. | ||
Nickel-based catalysts have shown great selectivity for the chemical oxidation of benzyl alcohol to benzaldehyde, reaching 95% selectivity in organic solvent and H2O.23 Electrochemically, Ni-based catalysts were used to explore potential dependence for electrochemical oxidation of alcohols to their corresponding aldehydes and carboxylic acids.1,24–27 For instance, Bender et al. showed that benzyl alcohol is oxidized to benzaldehyde by following a potential-dependent oxidation pathway (Scheme 3, top), with benzaldehyde being further oxidized to benzoic acid when the indirect oxidation pathway is favored (Scheme 3, bottom).22 This research hints that the selectivity for the reaction can be tuned, even in an electrochemical setup. Whilst there are many examples available that show full electrochemical alcohol oxidation to carboxylic acids using nickel, we believe it is possible to steer the reactivity towards the corresponding aldehyde intermediate.
 | ||
| Scheme 3 Mechanism for the alcohol oxidation on NiOOH through both potential dependent and indirect pathways as suggested by Bender et al.28 | ||
For the second step, which is the reductive amination of benzaldehyde, many catalysts have been employed, like Ag and Pb electrodes.19–21,29 In aqueous solvent, a Ag electrode demonstrated better selectivity to the amine product compared to other transition metal electrodes, however its overall efficiency is limited by the competing hydrogen evolution reaction (HER). Pb is a less efficient catalyst for hydrogen evolution due to its high overpotential for HER, therefore it can also be a good catalyst for the reaction.
Besides the catalyst and the applied potential, the pH also plays an important role in the reaction, since the amine needs to be unprotonated to form the aldimine.21 When the pH is lower than the pKa of the amine (ca. 10.5–11.0 for linear amines), the aldimine is either not formed at all or it is hydrolyzed, which inhibits the imine reduction reaction.
Since both an oxidation and reduction step are involved in the alcohol to amine conversion reaction in a so-called paired electrolysis,30 optimization of both the anodic (oxidation) and cathodic (reduction) processes are needed. Therefore, we optimized the two-step electrochemical amination reaction using (i) a Ni oxyhydroxide (NiOOH) catalyst for the selective oxidation of benzyl alcohol to benzaldehyde and (ii) Ag and Pb catalysts for the electrochemical reduction of aldehyde to amine. We tested an aqueous NaOH solution (pH 14) for the oxidation reaction (i) and aqueous tert-butylamine (pH 11) for the reductive process (ii). To streamline the process into a single reaction, we found that 0.7 M tert-butylamine was the optimal solvent condition for both steps.
Ni oxyhydroxide was prepared by electrodeposition of 2, 3, 5 and 10 nickel monolayer equivalents on a glassy carbon electrode. We found that the FE to benzaldehyde increases as the Ni monolayers decrease, reaching almost 90% FE for 2 Ni monolayer equivalents. Whilst for the reductive amination (step ii), in agreement with the literature,19,21 the Ag electrode showed superior catalytic activity and selectivity to the final amine product (39% FE) in comparison to a Pb electrode (26% FE). Therefore, by multiplying the FE for both processes we estimated that the theoretical FE for the full conversion from benzyl alcohol to benzyl-tert-butylamine is 35%, which is double that of previous research.31
Experimental section
All water used in this research was purified using a Milli-Q Millipore system with a total organic carbon content lower than 3 ppb and a resistivity higher than 18 MΩ cm at room temperature. Prior to electrochemical experiments, all glassware was stored overnight in a large beaker containing an aqueous 1 g L−1 KMnO4 (VWR chemicals, GPR RECTAPUR) and 0.5 M H2SO4 (Sigma-Aldrich, ACS reagent) solution. Before use, the KMnO4 solution was removed from the beaker and the glassware in the beaker was rinsed three times with Milli-Q water. The glassware was then immersed in a dilute aqueous H2SO4/H2O2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.3 M) solution (so-called ‘piranha solution’) to further oxidize the leftover organic and KMnO4 residue. After removing the piranha solution and rinsing the glassware, the glassware was boiled in fresh Milli-Q water three times before use.
:0.3 M) solution (so-called ‘piranha solution’) to further oxidize the leftover organic and KMnO4 residue. After removing the piranha solution and rinsing the glassware, the glassware was boiled in fresh Milli-Q water three times before use.
All electrochemical experiments were performed at room temperature. Unless stated otherwise, chemicals were purchased from commercial companies and used as received.
Prior to all procedures, the solutions were purged with N2 gas (Linde Gas, 99.999%) for 15 min and subsequently kept under inert atmosphere by passing a constant N2 stream over them.
For the nickel electrodeposition, the cyclic voltammetry (CV), and the bulk electrolysis a circular glassy carbon (GC, MaTecK, ∅ = 7 mm) electrode was used. Prior to each measurement, the glassy carbon was mechanically polished with a 1.0 μm diamond suspension (Buehler) on a MicroCloth polishing cloth (Buehler). For both CV and bulk electrolysis graphite (MaTecK, ∅ = 6 mm, l = 90 mm) and a Hydroflex reverse hydrogen electrode (RHE, Gaskatel) were used as counter and reference electrodes (CE and RE), respectively.
For the electrochemical impedance spectroscopy (EIS), a circular gold (MaTeck, ∅ = 5 mm) electrode was used as the substrate for Ni electrodeposition. Prior to the electrodeposition, the electrode was polished with a 1.0 μm, a 0.025 μm and a 0.005 μm diamond suspension on a MicroCloth polishing cloth. Additionally, it was electropolished by cycling for 200 cycles from 0.05 V to 1.75 V vs. RHE at 1 V s−1 in 0.1 M H2SO4 using a gold CE and RHE as RE.17
For the reductive amination process, a circular Ag (∅ = 10 mm, MaTecK, 99.99+%) and a Pb rod (Asurface = 3.10 cm2, MaTecK, 99.99+%) were used as electrodes. The Ag and the Pb electrode were mechanically polished with 3.0 μm, 1.0 μm diamond suspension on a MicroCloth polishing cloth. The Pb electrode was subsequently electropolished in an aqueous 0.5 M H2SO4 solution at −1.8 V vs. a Pt counter electrode for 500 seconds.
For the NaOH purification a process was adapted from literature.9,32 First, 2.0 M NaOH (MaTecK, 99+%) was mixed with Ni(NO3)2·6H2O (Sigma-Aldrich, >97.0%) to precipitate high-purity Ni(OH)2 based on the following equation:
| 2Ni(NO3)2 + 4NaOH → 2Ni(OH)2(s) + 4Na(NO3)aq |
After that, the Ni(OH)2 was washed 3 times with 20 mL Milli-Q H2O and 2 mL of 2.0 M NaOH by redispersing the solid and subsequently centrifuging for 10 minutes. Finally, each centrifugation tube was filled with 50 mL of 2.0 M NaOH. After the final centrifugation, the purified NaOH supernatant was collected in plastic bottles.
Procedure for NiOOH synthesis and electrochemical characterization
The electrodeposition of Ni was performed by applying a cathodic current of 10 μA on the GC working electrode which was submerged in a 5 × 10−3 M Ni(NO3)2·6H2O (Sigma-Aldrich, 99.999%) solution using 0.1 M NaClO4 (Sigma Aldrich, ACS reagent, >98.0%) as the supporting electrolyte. The duration of the electrodeposition was determined by assuming an imperfect sphere stacking of a Ni atom with a diameter of 0.124 nm, leading to a Ni loading of 2.07 × 1015 cm−1. Therefore, to deposit 1 monolayer equivalent on a ∅ = 7 mm (0.385 cm2) electrode, a total of 255.3 μC charge was required, so at a cathodic current of 10 μA one monolayer equivalent of Ni would be deposited in 25.5 seconds. Electrodeposition was performed until the desired amount of monolayer equivalents were deposited.Subsequently, the GC electrode with the electrodeposited Ni0 was transferred to a plastic cell with 20 mL of purified 1.0 M NaOH. The graphite CE and the RHE RE were added. The NiOOH formation and stabilization was performed by cycling 20 times from 1.01 V to 1.81 V at 50 mV s−1. Subsequently, for the characterization the electrode was cycled 3 times from 1.01 V to 1.56 V starting from 200 mV s−1 down to 25 mV s−1, decreasing the scan rate by 25 mV s−1 each time. The peak anodic currents from the characterization CVs. were plotted against the scan rate (V s−1). Using eqn (1) the surface coverage (τ) can be calculated, since the slope of the linear trendline (ipvs. υ) is equal to n2F2Aτ/4RT:
 | (1) |
485.33 C mol−1), A is the geometric surface area of the electrode (cm2), τ is the surface coverage (mol Ni), υ is the scan rate, R is the universal gas constant (8.314 J mol−1 K−1), T is the temperature (K) and ip is the anodic peak potential.
Bulk electrolysis
The preparative scale reactions were performed in a two-compartment electrochemical glass cell configuration divided by an anion-exchange membrane (Fumatech, Fumasep FAA-3-PK-130). Each compartment was filled with 30 mL 1.0 M purified NaOH. The catholyte compartment contained a graphite CE. The anolyte compartment contained a RHE RE and the NiOOH-deposited GC WE. The electrolysis was performed at the selected potential for 3 hours, while stirring at ∼500 rpm in the anolyte compartment and with an unstirred catholyte compartment, using an Autolab Potentiostat (Metrohm M204).Electrochemical impedance spectroscopy (EIS)
The NiOOH catalyst was first synthesized and characterized via the aforementioned procedures on a gold (MaTeck, ∅ = 5 mm) electrode. Subsequently, the WE was transferred to a plastic cell containing 10 mL 0.1 M purified-NaOH. The EIS was performed at 1.6 V vs. RHE.Reductive amination of benzaldehyde
High-performance liquid chromatography (HPLC)
After performing the benzyl alcohol oxidation a 400 μL aliquot was take from the working and counter electrode compartments. The sample was diluted to 1 mL with acetonitrile and water at known concentrations. For the reductive amination process, after the bulk electrolysis was completed, 1 mL of the WE and the CE compartment was collected in HPLC vials.Analysis was done on an Agilent 1260 Infinity II system equipped with an Agilent Technologies Inc. Poroshell 120 EC-C18 column (150 × 3 mm, 2.7 μm) and an RID and VWD detector. The column temperature was maintained at 35 °C. The mobile phase was acetonitrile/5 mM H2SO4, which was run in a gradient from 10/90 v/v to 80/20 v/v after 25 min at a flow rate of 0.5 mL min−1. The benzaldehyde derivative was detected via UV at 200 nm and the final concentration was determined by fitting the results to a predetermined regression line (Fig. S1 and S2†).
In situ Fourier transform infrared spectroscopy
To investigate the mechanistic details of our reaction we performed in situ Fourier Transform Infra-Red spectroscopy (FTIR) experiments for the oxidation and the reduction processes. FTIR spectroscopic measurements were carried out in a Bruker Vertex 80-V IR spectrometer equipped with a liquid nitrogen cooled MCT detector. A Veemax III (PIKE Technologies) was positioned in the spectrometer, wherein a home-made three electrode spectroelectrochemical cell with a CaF2 prism attached to the bottom was placed. We performed the FTIR in both solvents used in bulk electrolysis, namely NaOH (1 M) and tert-butylamine (0.7 M). The FTIR cell was filled with approximately 5 mL of each solvent containing 0.1 M benzyl alcohol. Glassy carbon or Ag (MaTeck, ∅ = 5 mm) was used as WE, RHE and Pt wire were used as reference and counter electrodes respectively. On the GC electrode 10 monolayers of NiOOH were electrodeposited following the process described earlier. The cell was purged with argon for 15 minutes and subsequently kept under inert atmosphere. The WE was pressed against the CaF2 prism to obtain a thin film configuration. For the oxidation, spectra were collected over a potential range in 0.05 V increments from 1.0 V to 1.9 V vs. RHE, or until oxygen evolution deteriorated the quality of the data. For the reduction, spectra were recorded on the Ag electrode from 0.0 to −0.65 V vs. RHE. FTIR spectra were collected in a range of 4000–1000 cm−1 at a resolution of 8 cm−1 for 100 scans. The spectra are presented as absorbance, according to A = −log(R/R0), where R and R0 are the reflectance corresponding to the single beam spectra obtained at the sample and reference potentials, respectively. In these difference spectra, negative bands (pointing down) correspond to the species that were present on or near the electrode surface at the reference potential and that are “consumed” at the sample potential. Positive bands (pointing up) correspond to the formation of species at the sample potential. All the spectro-electrochemical experiments were performed at room temperature.
Results and discussion
Electrochemical oxidation of benzyl alcohol
The NiOOH catalyst was synthesized by electrodepositing a precise amount of nickel onto a glassy carbon electrode, according to the experimental procedure, followed by its electrochemical cycling in a Fe-free purified 1.0 M NaOH solution to ensure the formation of the active catalyst (Fig. S3†). Subsequent cyclic voltammetry at various scan rates (Fig. 1A) allowed us to quantify the catalyst weight loading, expressed in monolayer equivalents. From the resulting voltammograms, the monolayer equivalents can be determined by plotting the peak anodic currents against the scan rate and adding the slope to eqn (1) as further described in the Experimental section. During these experiments, conditions were maintained that mitigate the influence of Fe trace impurities from the alkaline electrolyte, which can otherwise favor the oxygen evolution reaction over alcohol oxidation33 and therefore inhibit alcohol oxidation.24,27 | ||
| Fig. 1 (A) Cyclic voltammogram profile of the 5 monolayer equivalents NiOOH catalyst in 1.0 M purified NaOH at room temperature (scan rates ranging from 200 mV s−1 decreasing to 25 mV s−1; arrow indicates the scan direction) and (B) cyclic voltammogram of the 5 monolayer equivalents NiOOH catalyst in 0.1 M benzyl alcohol and 1.0 M NaOH (scan rate = 50 mV s−1; arrow indicates the scan direction). | ||
The CV profile in Fig. 1A shows the well-defined peaks characteristic for the Ni(OH)2/NiOOH (Ni2+/Ni3+) redox couple at approximately 1.38 V and 1.3 V vs. RHE, which increase as a function of the scan rate. The reductive currents are lower than the oxidative currents due to the poor electrochemical reversibility of NiOOH,34 as evidenced by the increased peak-to-peak separation with higher scan rates, indicating an electrochemically quasi-reversible process.35 The electrochemical activity of the NiOOH catalysts towards benzyl alcohol oxidation was also investigated by cyclic voltammetry in a Fe-free purified 1.0 M NaOH solution containing 0.1 M benzyl alcohol (Fig. 1B). These initial investigations were performed at 5 monolayer equivalents of Ni coverage. We chose 5 monolayer equivalents as a baseline to provide enough weight loading of Ni to get adequate catalytic activity for easier product quantification.
We observed three distinct regions in the CV profile. A small peak at around 1.4 V vs. RHE related to the (Ni2+/Ni3+) redox couple, which then overlaps with the benzyl alcohol oxidation (A), resulting in a significant increase of the current density up to 9.0 mA cm−2 (peak potential B (Ep)) at ca. 1.48 V. This shows the high catalytic activity of NiOOH. Subsequently, the current drops (C), likely due to carbonyl species blocking the catalyst active sites. During the reverse scan, the current increases again due to the reactivation of the electrode resulting in a slightly higher current density at peak B (10 mA cm−2), attributed to an increased amount of nickel oxyhydroxide active phase and the regeneration of active sites from the removal of adsorbed intermediates and products formed during the forward scan.36
The small cathodic peak at 1.3 V vs. RHE indicates the consumption of NiOOH during benzyl alcohol oxidation, leading to the formation of Ni(OH)2. These results support both the direct and indirect electron transfer mechanism proposed by Bender et al. for alcohol oxidation on a Ni electrode in an alkaline medium.28
In Fig. 1B, points A, B and C refer to the half-peak potential Ep1/2 (ca. 1.45 V), the peak potential Ep (ca. 1.48 V) and the potential after the peak (ca. 1.58 V), respectively. Therefore, we selected those potentials to perform chronoamperometric analysis of alcohol oxidation.
Fig. S4† shows the liquid products from the oxidation of benzyl alcohol at three different applied potentials after 3 hours of bulk electrolysis using a 5 Ni monolayer equivalent electrode. The electrolysis at 1.58 V vs. RHE (point C) achieved an 81% faradaic efficiency (FE) for benzaldehyde, compared to 58% and 73% at points A and B, respectively. However, the catalytic activity at this potential was five times lower than at point B, which also maintained a higher carbon balance due to the absence of oxygen evolution.
To explore the influence of Ni monolayers (2, 3, 5, and 10 monolayer equivalents) on benzyl alcohol oxidation, we conducted electrolysis at 1.48 V vs. RHE, which provided an optimal balance of activity and selectivity. Fig. S5† shows a typical profile for the current over time for the electrolysis reactions, showing a slight decrease due to catalyst leaching and deactivation. Fig. 2 shows that benzaldehyde is the main product of benzyl alcohol oxidation across all NiOOH monolayer equivalents, with benzoic acid forming as a secondary product from its further oxidation. Interestingly, the FE for benzaldehyde decreases as the number of monolayer equivalents increase, thereby favoring the production of benzoic acid.
 | ||
| Fig. 2 Faradaic efficiency towards benzaldehyde and benzoic acid in relation to the number of NiOOH monolayer equivalents. All electrolysis reactions were performed in a 1.0 M purified NaOH solution at E = 1.48 V vs. RHE for 3 hours at room temperature. | ||
We propose that this effect is due to the three-dimensional structure of the electrode material.37–39 NiOOH catalysts are characterized by a highly porous, leaf-like geometry, where a greater catalyst loading results in deeper pores, while lower loading yields shallower pores. Benzyl alcohol trapped in deep pores is more likely to undergo overoxidation to benzoic acid, whereas shallower pores promote the diffusion of benzaldehyde into the bulk solution. The critical role of mass transfer in Ni-catalyzed alcohol oxidation was previously observed on a macroscopic scale, where Ni foam showed to better mass transport due to its larger pore structure in comparison to flat Ni electrodes.22 In our study, we extend this understanding by demonstrating that similar principles apply at the nanoscale, where diffusion likely occurs within the confined nanopores of NiOOH rather than through the larger macropores characteristic of Ni foam. The relationship between the FE ratio of benzaldehyde to benzoic acid and the number of Ni monolayer equivalents follows an exponential decay trend (Fig. 3A). The intercept of this trend with the y-axis represents the theoretical behaviour of an “ideal” single-atom catalyst40,41 which lacks porosity and facilitates immediate mass transfer of the reaction products to the bulk solution. Such a catalyst would minimize overoxidation of benzyl alcohol, favoring benzaldehyde formation. Based on eqn (2), which is the exponential decay fit along the different NiOOH monolayer equivalents, we calculated that for an ideal catalyst (where x = 0 and diffusion of the product to the bulk is assumed to be immediate), the selectivity ratio of benzaldehyde to benzoic acid would be 12.7:1, corresponding to a maximum achievable FE ratio of 93:7%. This highlights the potential upper limit of selectivity for catalysts optimized to minimize porosity and maximum product diffusion.
| y = 4.14 × e(−(x−2)/1.995) + 1.453 | (2) |
 | ||
| Fig. 3 (A) Ratio of selectivity towards benzaldehyde and benzoic acid related to the NiOOH monolayer equivalents (equation = 4.14 × e(−(x−2)/1.995) + 1.453, R2 = 0.9334) (B) representation of the deposited 2 (left) and 10 (right) monolayer NiOOH equivalents. For the 10 monolayer equivalents, two deposition possibilities are shown, a close packed and a porous structure. | ||
To support our hypothesis regarding the relationship between catalyst porosity and selectivity, we calculated the roughness factor of our synthesized nickel catalysts using Electrochemical Impedance Spectroscopy (EIS) at 1.6 V vs. RHE in a 0.1 M purified NaOH electrolyte, following the Armstrong–Henderson equivalent circuit model (Fig. S6†).24 The roughness factor is proportional to the electrochemical active surface area (ECSA), which provides insight into the catalyst's surface characteristics.
Our measurements indicated capacitances of 11 μF cm−2 for 2 monolayer equivalents and 296 μF cm−2 for 10 monolayer equivalents, suggesting that the 10 monolayer equivalents are approximately 27 times rougher than the 2 monolayer equivalents. Fig. 3B illustrates the possible deposition structures for 2 (left, green box) and 10 (right, blue box) monolayer equivalents. For the 10 monolayer equivalents, the catalyst crystals can adopt either a close-packed structure (I) with a lower ECSA and roughness factor, or a more open, porous structure (II) with a higher ECSA due to a greater number of exposed nickel atoms acting as active sites.
Structure (II) is characterized by longer, more porous ‘canals’ formed by the nickel atoms, which is consistent with the significantly higher ECSA observed for the 10 monolayer equivalents. These results support the case for structure (II), suggesting that the nickel atoms create porous pathways in which benzaldehyde can become trapped and potentially undergo overoxidized to benzoic acid, in agreement with the results showed in Fig. 2. These findings align with the work by Deng et al., who demonstrated that NiOOH material reconstructs into a stacked configuration, creating longer pores when the applied potential exceeds 1.41 V vs. RHE.42
To obtain more information on the structure of the NiOOH catalyst, we attempted XRD spectroscopy. However, because of the low weight loading that was used for these experiments, the amount of NiOOH present was too low to obtain an XRD spectrum. However, we can compare our electrode material structure with previous study by Laan et al. who performed benzyl alcohol oxidation using NiOOH chemically synthesized.23 From their XRD and XPS spectra, it is clear that upon reaction with benzyl alcohol, NiOOH (in both β- and γ-phase) are converted to β-Ni(OH)2. Despite their electrode contained higher quantities of NiOOH we therefore expect similar behavior of our catalytic material after electrochemical reaction with benzyl alcohol.
After optimization of the anodic benzyl alcohol oxidation reaction using the NiOOH electrocatalyst in 1.0 M NaOH solution, we investigated its behavior in the same conditions required for the reductive amination reaction (0.7 M tert-butylamine, pH = 11). tert-Butylamine was chosen for its lack of β-hydrogens, to avoid major byproduct formation from its oxidation.43
The blank CV profile (red line) in Fig. 4, shows significantly lower current densities in pH 11 than in pH 14 (Fig. 1A), in accordance with the literature.44 The peak related to the Ni(OH)2/NiOOH couple shifts to higher potentials, which is expected, since the pH was decreased by 3 compared to the normal oxidation conditions (1.0 M NaOH). Additionally, we observe an increase in current density at approximately 1.63 V vs. RHE, which is not observed at the same potential in pH 14 (Fig. 1A). While this increase could be attributed to the oxygen evolution reaction (OER), Diaz-Morales et al. have demonstrated that the OER on NiOOH catalyst is less prominent at pH 11 compared to pH 14.45 Therefore, we propose that this current increase is likely caused by oxidation of the amine in solution, which may also account for the small cathodic peak observed at 1.4 V.
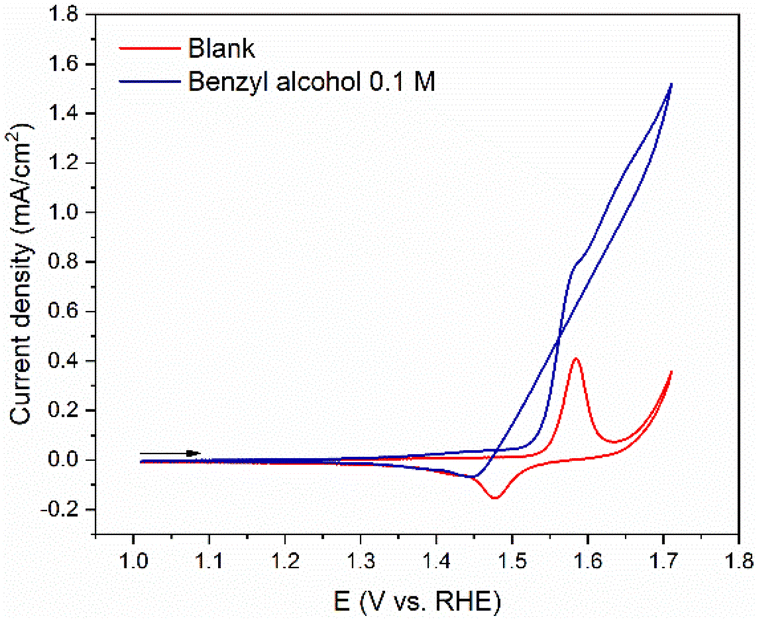 | ||
| Fig. 4 Cyclic voltammograms of 2 NiOOH monolayer equivalents in 0.7 M tert-butylamine before (red line) and after (blue line) the addition of 0.1 M benzyl alcohol (pH = 11; scan rate = 50 mV s−1; arrow indicates the scan direction). | ||
The addition of benzyl alcohol to the tert-butylamine solution resulted in significant changes in the CV profile (blue line – Fig. 4) compared to both its blank profile (red line – Fig. 4) and the CV profile at pH 14 (Fig. 1B). At 1.53 V, the current increases, and a small peak appears at approximately 1.55 V, which is associated with the Ni(OH)2/NiOOH redox couple. As the potential becomes more positive, the current further increases, resulting in a large wave due to the oxidation of the alcohol. The absence of reactivation of the electrode in the reverse scan (blue line) suggests that in the tert-butylamine solution, regeneration of NiOOH active sites is less pronounced than at pH 14. This means that either the removal of adsorbed species happens less readily in amine solution, or that less adsorbed species are present on the electrode, possibly through sequestration by the amine to form imine species.
The selectivity of the reaction towards benzaldehyde at pH 11 was investigated by long-term electrolysis at 1.63 V vs. RHE for 20 hours. In comparison to the previous condition (pH 14 – Fig. 2), in Fig. 5 we observe that the FE% for 2 Ni monolayers remains almost the same, while for 10 Ni monolayers, there is an increase to 100% FE of benzaldehyde. For these experiments, the final concentration of benzaldehyde was approximately 2 mM. The activity of the catalyst was substantially lower than in 1.0 M NaOH.
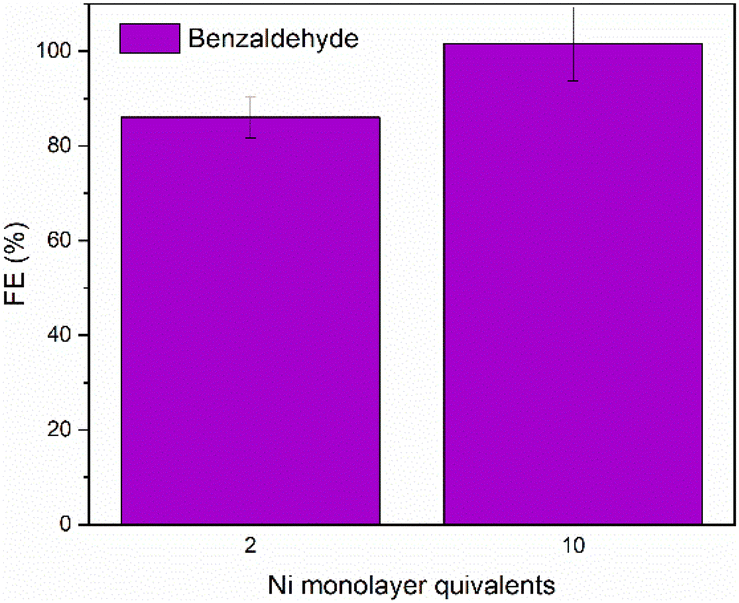 | ||
| Fig. 5 Faradaic efficiency towards benzaldehyde when the oxidation was performed in 0.7 M tert-butylamine (pH = 11) for 2 and 10 Ni monolayer equivalents. E = 1.63 V vs. RHE for 20 hours at room temperature. The resulting solution contains approximately 2 mM benzaldehyde. | ||
Both the increased product selectivity and lower catalyst activity in these experiments can largely be attributed to the change in pH. At lower pH, the activity of NiOOH is significantly lower. When it comes to selectivity, previous studies on NiOOH-catalyzed oxidation reactions indicate that for the oxidation of aldehydes to benzoic acid, the aldehyde must first be converted to its geminal diol form.23 However, this diol form is only significantly present at higher pH levels. At pH 11, most of the benzaldehyde remains in its carbonyl form, which protects it from further oxidation. Furthermore, due to the excess of amine in the system, the benzaldehyde can quickly react with the amine to form an imine, thereby providing additional protection against overoxidation.8 Because of the lower pH and presence of the amine provide an inherent protection against overoxidation of the benzyl alcohol, the effect of the amount of monolayers that was present in the NaOH electrolyte is no longer seen.
Reductive amination of benzaldehyde
Once the anodic reaction has been optimized, we performed optimization of the reductive amination reaction on Ag and Pb electrodes. Fig. 6A compares the CV profiles of a Ag electrode in 0.7 M tert-butylamine solution (pH = 11) in absence and presence of 0.02 M benzaldehyde. The blank CV (red line) shows an increase in current density at approximately −0.4 V vs. RHE which is indicative of HER on a Ag catalyst.46 When benzaldehyde is added into the solution (blue line), a broad irreversible redox peak gives rise between −0.25 V and −0.6 vs. RHE, which is likely related to the reduction of the imine formed from benzaldehyde and tert-butylamine, although HER is a competing reaction. At potentials more negative than −0.6 V vs. RHE, HER prevails over imine reduction.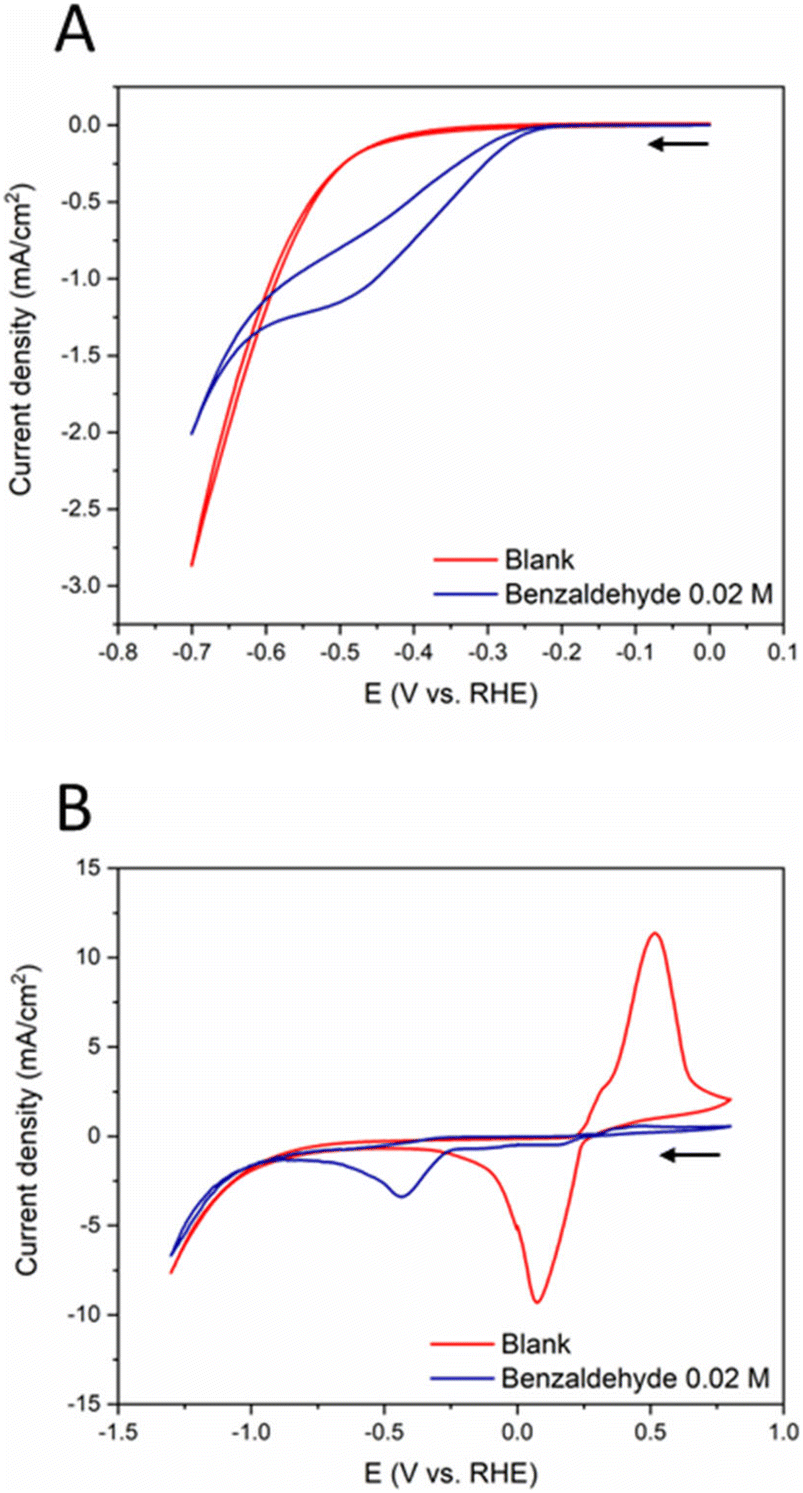 | ||
| Fig. 6 Cyclic voltammograms for (A) a Ag electrode and (B) a Pb electrode in 0.7 M tert-butylamine after acidification to pH = 11 before (red line) and after (blue line) adding 0.02 M benzaldehyde (scan rate = 10 mV s−1; arrow indicates the scan direction). | ||
Different profiles are obtained for a Pb electrode (Fig. 6B). In the blank solution (red line), a well-defined irreversible redox process is observed at approximately 0.1 V and 0.45 vs. RHE. The anodic peak is associated with the oxidation of Pb to form PbSO4, while the cathodic peak is associated with the desorption of the PbSO4 layer.47–49 An increase in current density is observed from −0.75 V vs. RHE due to HER. Upon addition of benzaldehyde (blue line), the peaks associated to the Pb/PbSO4 redox process significantly decreases because of interaction of the organic substrates with the metal surface, similar to previous work reported in our group.50 In addition, a new cathodic peak at −0.44 V, with a small shoulder at approximately −0.58 V vs. RHE is observed, which are associated with the reduction of imine and benzaldehyde, respectively. Afterwards, bulk electrolysis on both Ag and Pb electrodes was carried out for 20 hours to investigate their catalytic activity towards benzyl-tert-butylamine.
On the Ag electrode, electrolysis was performed at −0.37 V vs. RHE and not at the peak potential (−0.5 V vs. RHE), to avoid HER as much as possible, whereas on the Pb electrode, electrolysis was performed at both −0.44 V and −0.58 V vs. RHE.
Fig. 7 shows the FE for the two electrodes. On the Pb electrode, we found that the optimal potential is −0.44 V vs. RHE. This resulted in 26% FE towards benzyl-tert-butylamine and only trace amounts of benzyl alcohol from the reduction of benzaldehyde. In contrast, on the Ag electrode the FE% to the amine product is higher than on Pb, reaching almost 40%, although benzyl alcohol is also detected in higher amounts. These results suggest that even though the Ag electrode consumes part of the benzaldehyde via a hydrogenation reaction to the corresponding alcohol, it simultaneously promotes the amination reaction, with the last one being favorable. Fig. S7† shows a current profile of the electrolysis reaction. After an initial current decrease, a steady state is reached with only minor deactivation of the catalyst for the 20 hour reaction time, showing that the Ag electrode is stable for these reaction times. The remaining FE for this electrode is likely due to HER. On the Pb electrode, HER is unlikely, since the onset for HER is at a much later potential as discussed previously. We searched for typical side products of benzaldehyde reduction on a Pb electrode, such as hydrobenzoin, but could only detect them in trace amounts. Possibly, corrosion processes of the electrode could partially explain the remaining FE.
 | ||
| Fig. 7 Faradaic efficiency towards benzyl-tert-butylamine and benzyl alcohol for the two tested electrodes. Electrolysis was performed in 0.7 M tert-butylamine (pH 11) with 0.02 M benzaldehyde at E = −0.37 V vs. RHE (Ag) and −0.44 V vs. RHE (Pb) for 20 hours at room temperature. | ||
In situ Fourier transform infrared spectroscopy
We further studied the mechanism for the oxidation and reduction reactions through in situ FTIR spectroscopy. Fig. 8 shows the spectra obtained as function of the potential for the oxidation of benzyl alcohol on a 10-monolayer equivalent Ni catalyst. We can identify several negative bands, which grow in intensity as the potential becomes more positive, related to species being consumed near the electrode surface. | ||
| Fig. 8 In situ FTIR spectra of benzyl alcohol oxidation on a 10-monolayer equivalent NiOOH catalyst. Aqueous 0.7 M tert-butylamine (pH = 11) was used as solvent and electrolyte. Reference spectra were taken at 1.0 V vs. RHE. | ||
The bands at 1225 and 1252 cm−1 are attributed to the C3C–N antisymmetric stretching and the CC4 skeletal stretching vibrations of the tert-butylamine,51 respectively, whereas the band at 1370 cm−1 is assigned to the O–H bending of benzyl alcohol.52 This band could also be associated with the CH3 symmetric deformation of tert-butylamine.51 At higher wavenumbers, two more consumption bands associated with benzylic C–H stretching frequencies of benzyl alcohol at 2875 and 2969 cm−1 are identified.53,54
The spectra also present a series of positive bands related to the product formation as a function of the potential. The bands at 2740 and 2830 cm−1 are associated with the C–H bond of the carbonyl group of benzaldehyde, confirming the oxidation of alcohol to the aldehyde, in agreement with our results showed in Fig. 5.55 The formation of protonated imine is evidenced by the positive band at 2525 cm−1,56 which appears at potentials where both tert-butylamine and benzaldehyde are consumed. The presence of protonated imine can be the result of the added H2SO4.
The spectra for the reductive step, which involves that reduction of the imine on Ag electrode is shown in Fig. 9. At less negative potentials, we observe a small positive band at 1383 cm−1 and 2630 cm−1, which are associated with benzaldehyde (Fig. S8†).57 At more negative potentials, these peaks shift from positive to negative, showing that the surface has affinity for benzaldehyde, until an appropriate potential is reached at which it can be reduced, at approximately −0.35 V vs. RHE. At this potential, a small positive band becomes visible at 1370 cm−1, assigned to the O–H bending frequency of benzyl alcohol.52 At slightly less negative potentials (−0.20 V vs. RHE), another positive peak appears at 2850 cm−1. When looking at the IR spectrum of the final product, benzyl-tert-butylamine, the benzylic C–H stretching peaks are found at 2964 cm−1 and 2861 cm−1.58 Whilst these positive bands are present in the IR spectra, they are highly similar to the C–H stretching vibrations of benzyl alcohol. Therefore, these peaks can be assigned to both alcohol and amine species. However, since benzyl-tert-butylamine is a base and benzyl alcohol is not, the presence of the H2SO4 can protonate the amine product. This causes a redshift of the C–H stretching vibration, so that it is now visible at 2850 cm−1, confirming the formation of the desired amine product under these conditions (Fig. S9†). Additional peaks at 2969 cm−1, 2916 cm−1 and 2875 cm−1 are associated with the production of benzyl alcohol or production of the unprotonated amine product, both of which are synthesized during the reaction.52,58 At lower wavenumbers a new positive peak at 1252 cm−1 is associated with the C–N stretching frequency of benzyl-tert-butylamine, also starting at approximately −0.20 V vs. RHE. Consumption of the protonated imine can only be seen at potentials lower than −0.55 V vs. RHE by a consumption band at 2525 cm−1, although the presence of the product bands at 2850 cm−1 and 1252 cm−1 shows imine consumption must be happening earlier than this.
 | ||
| Fig. 9 In situ FTIR spectra of benzyl-tert-butylimine reduction on a Ag catalyst. Aqueous 0.7 M tert-butylamine (pH = 11) was used as solvent and electrolyte. Reference spectra were taken at 0.0 V vs. RHE. | ||
The results of the FTIR experiments show that during the oxidation reaction, benzyl alcohol is oxidized to form benzaldehyde and benzyl-tert-butylimine, whilst overoxidation to benzoic acid is not observed in the presence of tert-butylamine. These results are in accordance with the results obtained from the bulk electrolysis, where high selectivity for benzyl alcohol oxidation was reached in the presence of tert-butylamine. The reduction reaction tells a slightly different story. At approximately −0.20 V vs. RHE, the benzyl-tert-butylamine product can be identified. Only at −0.35 V vs. RHE benzaldehyde reduction to benzyl alcohol is observed. Combined with the results obtained from the electrolysis, it is clear that imine reduction is preferred on the Ag catalyst, but benzaldehyde reduction to benzyl alcohol still occurs. The imine reduction is therefore less selective than the benzyl alcohol oxidation, and a point of future improvement.
The research provided here shows that there is a possibility to improve traditional alcohol to amine transformation reactions through electrochemistry. This avoids the use of stoichiometric redox reactions, high pressures or temperatures. In a direct comparison with the state of the art in hydrogen borrowing catalysis (see Table S1†), it is apparent that the reaction conditions are significantly milder. When compared to the work by Appiagyei et al.,30 the only other example of an electrochemical alcohol to amine conversion reaction, the lower temperature, avoidance of platinum group metals and higher FE are clear improvements on the sustainability of the process.
Conclusion
This study highlights the successful electrochemical oxidation of benzyl alcohol to benzaldehyde using a NiOOH catalyst, where the number of Ni monolayer equivalents plays a crucial role in tuning the selectivity. A thinner layer of 2 Ni monolayers achieves up to 90% faradaic efficiency (FE) in NaOH, favoring benzaldehyde formation, while thicker layers lead to overoxidation to benzoic acid due to increased catalyst porosity. However, when the conditions were shifted to a solution of tert-butylamine (pH 11), 10 Ni monolayers demonstrated the best performance, achieving 100% FE towards benzaldehyde. This result highlights the protective role of tert-butylamine against overoxidation and underscores the influence of solution conditions on catalyst performance.For the reductive amination step, Ag electrodes outperformed Pb, with a 39% FE towards benzyl-tert-butylamine, though hydrogen evolution remained a competing reaction. The overall electrochemical process, optimized at pH 11 with tert-butylamine as the solvent, yielded a 35% FE for the conversion of benzyl alcohol to benzyl-tert-butylamine.
In situ FTIR measurements further supported these findings, revealing the sequential formation of benzaldehyde and its corresponding imine during oxidation on the NiOOH catalyst. The data showed no overoxidation to benzoic acid in the presence of tert-butylamine, which correlates with the bulk electrolysis results. During the reductive step on Ag electrodes, the FTIR spectra confirmed the formation of the desired amine product at potentials that favored imine reduction over benzaldehyde reduction. These spectroscopic results align with the electrolysis outcomes, reinforcing the mechanistic understanding of both oxidative and reductive processes.
This work presents a promising pathway for electrochemical amine synthesis, with potential for further catalyst optimization to enhance the overall process efficiency.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. C. G. and P. B. acknowledges the project number ECCM.TT.ECCM.008, which was granted in the framework of the Electrochemical Conversion and Materials (ECCM) program and received funding from the Dutch Research Council (NWO). V. P. would like to thank Hellenic Energy for the scholarship.References
- M. K. K. Goetz, M. T. Bender and K. S. Choi, Nat. Commun., 2022, 13, 1–10 CrossRef PubMed.
- R. Kumar, N. J. Flodén, W. G. Whitehurst and M. J. Gaunt, Nature, 2020, 581, 415–420 CrossRef CAS PubMed.
- T. C. Nugent and M. El-Shazly, Adv. Synth. Catal., 2010, 352, 753–819 CrossRef CAS.
- J. Zhang, M. Jiang, C. S. Wang, K. Guo, Q. X. Li, C. Ma, S. F. Ni, G. Q. Chen, Y. Zong, H. Lu, L. W. Xu and X. Shao, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- Y. Zhong, H. Xiong, J. Low, R. Long and Y. Xiong, eScience, 2023, 3, 100086 CrossRef.
- B. Zhang, T. Guo, Y. Liu, F. E. Kühn, C. Wang, Z. K. Zhao, J. Xiao, C. Li and T. Zhang, Angew. Chem., Int. Ed., 2021, 60, 20666–20671 CrossRef CAS PubMed.
- A. L. Marshall and P. J. Alaimo, Chem. – Eur. J., 2010, 16, 4970–4980 CrossRef CAS PubMed.
- K. Wisniewski, A. S. Koldziejczyk and B. Falkiewicz, J. Pept. Sci., 1998, 4, 1–14 CrossRef CAS PubMed.
- H. Huang and J. Y. Kang, J. Org. Chem., 2017, 82, 6604–6614 CrossRef CAS PubMed.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed.
- R. Labes, C. Mateos, C. Battilocchio, Y. Chen, P. Dingwall, G. R. Cumming, J. A. Rincón, M. J. Nieves-Remacha and S. V. Ley, Green Chem., 2019, 21, 59–63 RSC.
- J. T. Reeves, M. D. Visco, M. A. Marsini, N. Grinberg, C. A. Busacca, A. E. Mattson and C. H. Senanayake, Org. Lett., 2015, 17, 2442–2445 CrossRef CAS PubMed.
- D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed.
- Y. Holade, K. Servat, S. Tingry, T. W. Napporn, H. Remita, D. Cornu and K. B. Kokoh, ChemPhysChem, 2017, 18, 2573–2605 CrossRef CAS PubMed.
- L. Heuer, Benzylamine, in Ullmann's Encyclopedia of Industrial Chemistry, 2006 Search PubMed.
- G. Van Lommen, M. De Bruyn, M. Schroven, W. Verschueren, W. Janssens, J. Verrelst and J. Leysen, Bioorg. Med. Chem. Lett., 1995, 5, 2649–2654 CrossRef CAS.
- D. Choi, J. P. Stables and H. Kohn, J. Med. Chem., 1996, 39, 1907–1916 CrossRef CAS PubMed.
- J. Li, Z. Weng, Q. Cao, Y. Qi, B. Lu, S. Zhang, J. Wang and X. Jian, Chem. Eng. J., 2022, 433, 134512 CrossRef CAS.
- Z. J. Schiffer, M. Chung, K. Steinberg and K. Manthiram, Chem. Catal., 2023, 3, 1–12 Search PubMed.
- N. Wang, J. Liu, X. Li and L. Ma, Catal. Commun., 2023, 175, 106620 CrossRef CAS.
- J. J. Roylance and K. S. Choi, Green Chem., 2016, 18, 5412–5417 RSC.
- M. T. Bender and K. S. Choi, ChemSusChem, 2022, 15, e202200675 CrossRef CAS PubMed.
- P. C. M. Laan, F. J. de Zwart, E. M. Wilson, A. Troglia, O. C. M. Lugier, N. J. Geels, R. Bliem, J. N. H. Reek, B. de Bruin, G. Rothenberg and N. Yan, ACS Catal., 2023, 13, 8467–8476 CrossRef CAS PubMed.
- A. C. Garcia, T. Touzalin, C. Nieuwland, N. Perini and M. T. M. Koper, Angew. Chem., 2019, 131, 12999–13003 CrossRef PubMed.
- Z. Li, Y. Yan, S. M. Xu, H. Zhou, M. Xu, L. Ma, M. Shao, X. Kong, B. Wang, L. Zheng and H. Duan, Nat. Commun., 2022, 13, 1–14 Search PubMed.
- W. Jud, C. A. Salazar, J. Imbrogno, J. Verghese, S. M. Guinness, J. N. Desrosiers, C. O. Kappe and D. Cantillo, Org. Process Res. Dev., 2022, 26, 1486–1495 CrossRef CAS.
- C. S. Santana, E. Gjonaj and A. C. Garcia, ChemElectroChem, 2024, 11, e202300570 CrossRef CAS.
- M. T. Bender, Y. C. Lam, S. Hammes-Schiffer and K. S. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef CAS PubMed.
- H. Hong, Z. Zou, G. Liang, S. Pu, J. Hu, L. Chen, Z. Zhu, Y. Li and Y. Huang, Org. Biomol. Chem., 2020, 18, 5832–5837 RSC.
- B. Appiagyei, S. Bhatia, G. L. Keeney, T. Dolmetsch and J. E. Jackson, Green Chem., 2020, 22, 860–869 RSC.
- G. Hilt, Curr. Opin. Electrochem., 2024, 43, 101425 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- L. Wei, M. D. Hossain, M. J. Boyd, J. Aviles-Acosta, M. E. Kreider, A. C. Nielander, M. B. Stevens, T. F. Jaramillo, M. Bajdich and C. Hahn, ACS Catal., 2023, 13, 4272–4282 CrossRef CAS.
- H. Angerstein-Kozlowska and B. E. Conway, J. Electroanal. Chem. Interfacial Electrochem., 1979, 95, 1–28 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- M. S. E. Houache, E. Cossar, S. Ntais and E. A. Baranova, J. Power Sources, 2018, 375, 310–319 CrossRef CAS.
- J. L. Gunjakar, A. I. Inamdar, B. Hou, S. Cha, S. M. Pawar, A. A. Abu Talha, H. S. Chavan, J. Kim, S. Cho, S. Lee, Y. Jo, H. Kim and H. Im, Nanoscale, 2018, 10, 8953–8961 RSC.
- L. Yu, X. Ji, X. Zhao, Y. Zhang and H. Zhu, Appl. Phys. A, 2023, 129, 1–8 CrossRef.
- X. Wang, X. Liu, C. J. Tong, X. Yuan, W. Dong, T. Lin, L. M. Liu and F. Huang, J. Mater. Chem. A, 2016, 4, 7762–7771 RSC.
- L. Li, X. Chang, X. Lin, Z. J. Zhao and J. Gong, Chem. Soc. Rev., 2020, 49, 8156–8178 RSC.
- N. Cheng, L. Zhang, K. Doyle-Davis and X. Sun, Electrochem. Energy Rev., 2019, 2, 539–573 CrossRef.
- J. Deng, M. R. Nellist, M. B. Stevens, C. Dette, Y. Wang and S. W. Boettcher, Nano Lett., 2017, 17, 6922–6926 CrossRef CAS PubMed.
- P. L. Theofanis and W. A. Goddard, Organometallics, 2011, 30, 4941–4948 CrossRef CAS.
- A. Govind Rajan and E. A. Carter, Energy Environ. Sci., 2020, 13, 4962–4976 RSC.
- O. Diaz-Morales, D. Ferrus-Suspedra and M. T. M. Koper, Chem. Sci., 2016, 7, 2639–2645 RSC.
- T. Hatsukade, K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Phys. Chem. Chem. Phys., 2014, 16, 13814–13819 RSC.
- E. N. Codaro and J. R. Vilche, Electrochim. Acta, 1997, 42, 549–555 CrossRef CAS.
- C. Martin, H. Huser, K. Servat and K. B. Kokoh, Electrochim. Acta, 2005, 51, 111–117 CrossRef CAS.
- M. Gençten, K. B. Dönmez, Y. Şahin, K. Pekmez and E. Suvacı, J. Solid State Electrochem., 2014, 18, 2469–2479 CrossRef.
- P. J. L. Broersen, T. de Groot, D. F. Bruggeman, E. S. Caarls, J. A. Trindell, D. Anastasiadou, M. C. Figueiredo, G. Rothenberg and A. C. Garcia, ChemCatChem, 2024, 16, e202301370 CrossRef CAS.
- P. K. Kipkemboi, P. C. Kiprono and J. J. Sanga, Bull. Chem. Soc. Ethiop., 2003, 17, 211–218 CAS.
- C. Keresszegi, D. Ferri, T. Mallat and A. Baiker, J. Phys. Chem. B, 2005, 109, 958–967 CrossRef CAS PubMed.
- NIST Chemistry Webbook, SRD 69 (2023). Benzyl Alcohol. https://webbook.nist.gov/cgi/cbook.cgi?Name=benzyl+alcohol&Units=SI (accessed 17 September 2024).
- Y. C. Ning, Interpretation of Organic Spectra, Wiley, 2011 Search PubMed.
- D. L. Pavia, G. S. Kriz, G. M. Lampman and R. G. Engel, A Small Scale Approach to Organic Laboratory Techniques, Cencage Learning, 2015 Search PubMed.
- L. S. Lussier, C. Sandorfy, H. O. L. E. Thanh and D. Vocelle, Photochem. Photobiol., 1987, 45, 801–808 CrossRef CAS.
- NIST Chemistry Webbook, SRD 69 (2023). Benzaldehyde. https://webbook.nist.gov/cgi/cbook.cgi?ID=C100527&Type=IR-SPEC&Index=2# (accessed 17 September 2024).
- P. Q. Huang, Q. W. Lang and Y. R. Wang, J. Org. Chem., 2016, 81, 4235–4243 CrossRef CAS PubMed.
- A. Afanasenko, S. Elangovan, M. C. A. Stuart, G. Bonura, F. Frusteri and K. Barta, Catal. Sci. Technol., 2018, 8, 5498–5505 RSC.
- D. Pingen, O. Diebolt and D. Vogt, ChemCatChem, 2013, 5, 2905–2912 CrossRef CAS.
- Y. Kita, M. Kuwabara, S. Yamadera, K. Kamata and M. Hara, Chem. Sci., 2020, 11, 9884–9890 RSC.
- Y. Liu, P. Ji, G. Zou, Y. Liu, B.-M. Yang and Y. Zhao, Angew. Chem., Int. Ed., 2024, e202410351 CAS.
- S. Jia, T. Tong, X. Liu, Y. Guo, L. Dong, Z. Chen and Y. Wang, J. Catal., 2024, 432, 115407 CrossRef CAS.
- X. Wu, M. De Bruyn and K. Barta, Catal. Sci. Technol., 2022, 12, 5908–5916 RSC.
- R. Labes, C. Mateos, C. Battilocchio, Y. Chen, P. Dingwall, G. R. Cumming, J. A. Rincón, M. J. Nieves-Remacha and S. V. Ley, Green Chem., 2019, 21, 59–63 RSC.
- X. Bai, F. Aiolfi, M. Cettolin, U. Piarulli, A. Dal Corso, L. Pignataro and C. Gennari, Synthesis, 2019, 51, 3545–3555 CrossRef CAS.
- X. Xi, Y. Li, G. Wang, G. Xu, L. Shang, Y. Zhang and L. Xia, Org. Biomol. Chem., 2019, 17, 7651–7654 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: The authors have cited additional references within the ESI59–67. See DOI: https://doi.org/10.1039/d4gc05171h |
| ‡ Both authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2025 |