
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Green phosphonate chemistry – Does it exist?
Konstantinos D.
Demadis
 *a,
Santosh Kumar
Adla
b,
Juri
Timonen
bc and
Petri A.
Turhanen
*b
*a,
Santosh Kumar
Adla
b,
Juri
Timonen
bc and
Petri A.
Turhanen
*b
aCrystal Engineering, Growth and Design Laboratory, Department of Chemistry, University of Crete, Heraklion, GR-71003, Greece. E-mail: demadis@uoc.gr
bUniversity of Eastern Finland, School of Pharmacy, Biocenter Kuopio, P.O. Box 1627, FIN-70211, Kuopio, Finland. E-mail: petri.turhanen@uef.fi
cDrug Research Program, Division of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, Viikinkaari 5E, P.O. Box 56, 00014 Helsinki, Finland
First published on 11th December 2024
Abstract
Organophosphorus chemistry plays a crucial role in several scientific disciplines, including chemistry, biology, medicine, and pharmacy. In particular, phosphonates have found important applications in diverse health- and technology-related areas, for example, drugs for calcium metabolic disorders, and corrosion and scale inhibitors. This review discusses the importance of phosphonate compounds from a green chemistry point of view, along the lines of synthesis, applications and recovery/recycling. It is the hope of the authors that readers will form their own answer to the question posed in the title.
Green foundation1. This review discusses phosphonate chemistry from the “green” perspective. It focuses on green methods in phosphonate synthesis, biodegradation issues, photo/chemical/catalytic degradation, recovery, and recycling.2. Phosphonates are used in several industrial, household, and medicinal/pharmaceutical applications. They are the most preferred additives for scale and corrosion control in industrial systems. They are used in nearly all detergents and several cleaning fluids. They constitute the first line of defense for pathologies such as osteoporosis, Paget's disease, tumor-induced hypercalcemia and others. 3. Due to continuous regulatory pressure for reducing phosphorus discharge to the environment, the “greening” of phosphonate chemistry will continue to thrive on four fronts: (a) novel synthetic methods, (b) enhanced biodegradability, (c) phosphorus recovery, and (d) phosphorus recycling. |
1. Introduction
Among the important fundamental worldwide issues to be tackled, the problem of exposure to plastic and plastic waste stands out. Micro- or nanoplastics (MNPs) are nowadays found everywhere, even in the most unlikely of places, such as the atmosphere.1 It is important to note that recently MNPs were found in human arterial plaques and even in the human brain. The relevant study indicated that patients with those artery plaques in which MNPs were detected had a higher risk of infarction, stroke, or death when compared with a control patient group.2 It is apparent that we need a more environmentally friendly and sustainable way of thinking in every facet of our daily life. From a chemist's point of view, the 12 green chemistry principles can greatly help to address such global problems. As a reminder, the 12 green chemistry principles are: (1) avoiding waste, (2) optimal atom economy, (3) preventing hazardous synthesis, (4) design of safer chemicals, (5) use of sustainable solvents, (6) energy efficiency, (7) use of renewable feedstocks, (8) reduced use of derivatives, (9) use of optimal catalysis (catalysts), (10) design for (bio)degradation, (11) real-time analysis for pollution prevention, and (12) safer chemistry to avoid accidents.3,4 Everyone can relate to the fact that re-using or recycling even a small plastic bottle can make a big difference if everyone follows this example. We just need to remember the motto: “mighty oaks from little acorns grow”!5,6Chemistry has its own share of responsibility for several global problems. Phosphonate chemistry deals with a certain category of organophosphorus compounds having a direct and very robust carbon phosphorus bond (C–P) instead of the C–O–P bond found in common phosphate esters. A phosphonate molecule displays one or more –PO3H2 moieties (in its acid, ester, or deprotonated form).7 Phosphonates embrace a great number of compounds utilized in a wide range of applications, such as drugs, imaging, pesticides, flame retardants, water treatment agents, metal organic framework (MOF) linkers, antioxidants, polymers, etc. The wide range of diverse applications is the driving force for the constant development of phosphonate chemistry, apart from the fact that phosphonate compounds are interesting molecules on their own.8 For example, etidronic acid (see Fig. 1) is one example of a phosphonate (called bisphosphonate) being used as a drug for the treatment of osteoporosis, as a scale and corrosion inhibitor in water treatment, and as an antioxidant in cosmetics. Hence, a single phosphonate compound may have many different uses and applications.9
 | ||
| Fig. 1 Some examples of antiviral and antiosteoporotic phosphonate drugs. | ||
Several years ago, natural compounds containing the C–P bond (natural phosphonates) were treated mostly as a curiosity and only scarcely studied. Nowadays there are several reports in the literature related to naturally occurring phosphonates, and their biological and environmental functions.10 It has been reported that the most common natural phosphonate is 2-aminoethylphosphonate (AEP) (see Fig. 2). Fosfomycin, the only phosphonate antibiotic available in the market (at least until 2019), was originally isolated and characterized from cultures of Streptomyces fradiae in 1969 and is now chemically synthesized. It is primarily used as a treatment of urinary infections.11 Several natural phosphonates are produced by many organisms like protozoa, bacteria, coelenterates, and mollusks. Rhizocticins, plumbemycins and bialaphos are examples of such phosphonate compounds having antibiotic properties (see structures in Fig. 2.).12,13
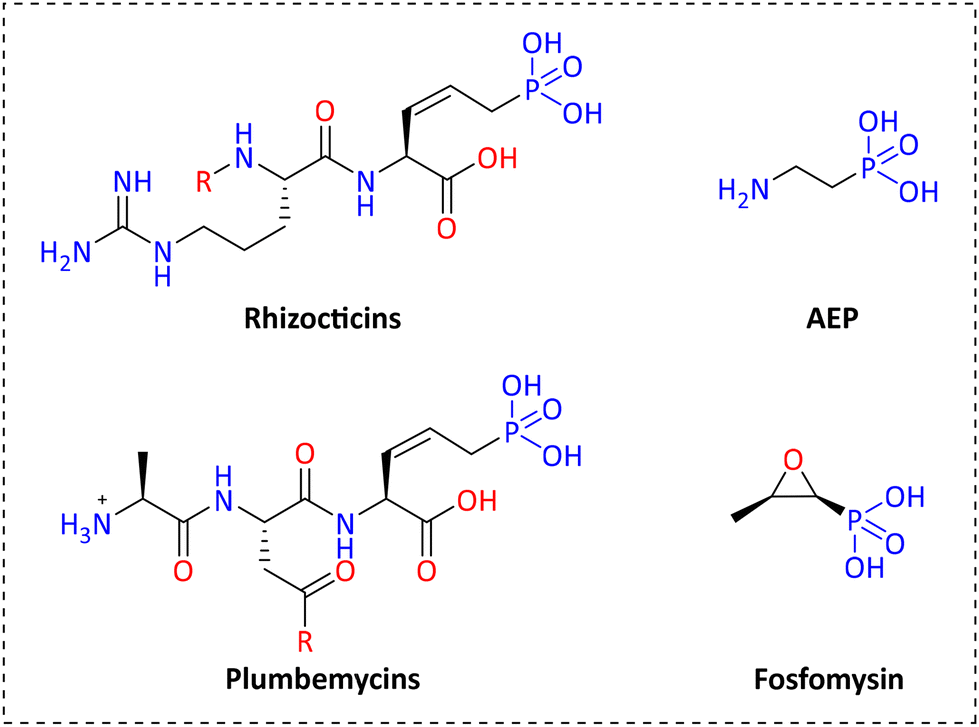 | ||
| Fig. 2 Examples of antibiotic phosphonate compounds. | ||
Cidofovir, adefovir, tenofovir, and foscarnet (and/or their prodrug forms) are among the most important antiviral phosphonate drugs in clinical use, for the treatment of a range of viral infections including HIV, hepatitis B, CMV retinitis, and herpes.14–17 Bisphosphonates like etidronate, clodronate, pamidronate, alendronate, ibandronate, risedronate, and zoledronate (and others of the same class of bisphosphonates) are well-known drugs in clinical use for the treatment of osteoporosis, Paget's disease, bone metastases, and other skeletal disorders.18,19Fig. 1 displays structures of some of the most important anti-viral and anti-osteoporotic drugs. Additionally, important phosphonate pesticides like Fosetyl-Al (fungicide) and potassium phosphite (for plant diseases), and other phosphonates for uses as insecticides, fungicides, and fertilizers have been reported.20 Phosphonates also play a significant role in water treatment processes. The importance of these compounds stems from their unique ability to prevent scale formation, a common challenge in water systems, by inhibiting the precipitation and aggregation of scale- and deposit-forming crystals.21
As stated above, phosphonate chemistry is evidently an important part of our life. In this review we present a comprehensive overview of the current state of the art of phosphonate chemistry, focusing on aspects of green synthesis methods, water treatment, possibility for recycling of phosphonates, and their degradation strategies. We attempt to address the question in the title in the light of the fact that the EU listed phosphorus as a critical raw material in 2017. Hence, is it realistic to use the terms “green” or “sustainable” when phosphorus is part of a molecule, such as a phosphonate?22,23
2. Green methods for the synthesis of phosphonates
According to recent literature studies, there are several methods for the synthesis of phosphonates that are considered as “green”, including e.g. solvent-free, ultrasound-assisted, microwave-promoted, electrochemical, and light-mediated strategies. Also, methods using water as solvent have been implemented. Additionally, green catalysts or catalyst-free syntheses and several combinations of all the above have been reported. To the best of our knowledge green methods for the synthesis of phosphonates have not been reviewed thus far, and only very few examples were presented in a recent review related to the topic.24 Herein, we review and present several sustainable methods related to the synthesis of phosphonates.2.1. Ultrasound-assisted methods
In general, ultrasound-assisted syntheses are considered eco-friendly, as the energy needed to agitate the reaction is transmitted via sonic waves travelling through the reaction medium. Sonication promotes cavitation inside the medium. The collapsing cavitands release energy for the molecules that are in close vicinity, thus promoting the formation of high-energy transition states.α-Aminophosphonates are compounds generally prepared by a three-component synthesis approach using aldehyde or ketone, amine and phosphite as starting materials. Correspondingly, α-hydroxyphosphonates are analogs of α-aminophosphonates prepared mostly from reactions between carbonyl compounds and phosphites. General methods for the synthesis of α-amino- and α-hydroxyphosphonates are presented in Scheme 1. Both groups of compounds are considered to have biological potential to act as enzyme inhibitors, herbicides, antioxidants, antibacterials and antifungals, antibiotics, antivirals, and antitumor agents.25,26
 | ||
| Scheme 1 General methods for the synthesis of α-amino- and α-hydroxyphosphonates. | ||
A catalyst-free Kabachnik–Fields reaction combining aromatic aldehydes, para-substituted anilines, and triethylphosphite was reported by Dar et al. in 2012. A model reaction of 4-chloroaniline (1 equivalent), 4-chlorobenzaldhyde (1 equivalent) and triethyl phosphite (1.3 equivalents) was completed with 99% yield in 20 seconds under solvent-free sonication conditions, while the use of a range of solvents resulted in lower yields [hexane (90%), ethyl acetate (89%), tetrahydrofuran (86%), dimethylformamide (85%), acetone (82%), acetonitrile (79%), methanol (79%), toluene (76%), dichloromethane (73%), ethanol (72%) and water (65%)]. The tested conditions were then used to carry out the reaction with 12 aromatic aldehydes and four para-substituted anilines to prepare α-aminophosphonates with high purity and yields ranging from 80% to 99% (see synthesis procedure in Scheme 2). However, the reaction yielded only a trace of the desired product or no product at all when ketones were used instead of aldehydes. Experimentation demonstrated that ultrasound radiation significantly influenced the direct production of α-aminophosphonates as the model reaction, giving 10% yield under reflux over 2 hours and only a trace amount during an 18 hour reaction at 30 °C.27
 | ||
| Scheme 2 Catalyst free synthesis of α-amino phosphonates by ultrasound. | ||
Study of ultrasound-assisted Kabachnik–Fields reaction of benzaldehyde, aniline and triethyl phosphate demonstrated, similarly to Dar's research, that several single-solvent systems resulted in low to moderate yields at ambient-temperature sonication even with 90 minutes of reaction time (see the Table 1, entries 1–13) The best result was obtained with ethyl lactate (75%), and when mixed in water (ethyl lactate/water 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), the yield increased to 95%. Using a series of 11 (hetero)aromatic aldehydes and cyclohexanecarboxaldehyde combined with seven substituted anilines, a series of 24 α-aminophosphonates was synthesized with 89–95% yield during 19–46 minutes, apart from the reaction of cyclohexanecarboxaldehyde that reached “only” 79% yield in 47 minutes.28
:2), the yield increased to 95%. Using a series of 11 (hetero)aromatic aldehydes and cyclohexanecarboxaldehyde combined with seven substituted anilines, a series of 24 α-aminophosphonates was synthesized with 89–95% yield during 19–46 minutes, apart from the reaction of cyclohexanecarboxaldehyde that reached “only” 79% yield in 47 minutes.28
| Entry | Phosphorus source | Catalyst (load) | Solvent | Time (min) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | P(O)(OEt)3 | — | — | 90 | Trace | 28 |
| 2 | P(O)(OEt)3 | — | Ethyl acetate | 90 | 35 | 28 |
| 3 | P(O)(OEt)3 | — | Dimethylformamide | 90 | 40 | 28 |
| 4 | P(O)(OEt)3 | — | Dichloromethane | 90 | 48 | 28 |
| 5 | P(O)(OEt)3 | — | Chloroform | 90 | 50 | 28 |
| 6 | P(O)(OEt)3 | — | PEG-400 | 90 | 51 | 28 |
| 7 | P(O)(OEt)3 | — | Tetrahydrofuran | 90 | 55 | 28 |
| 8 | P(O)(OEt)3 | — | Ethanol | 90 | 57 | 28 |
| 9 | P(O)(OEt)3 | — | Water | 90 | 42 | 28 |
| 10 | P(O)(OEt)3 | — | Methanol | 90 | 58 | 28 |
| 11 | P(O)(OEt)3 | — | Acetonitrile | 90 | 59 | 28 |
| 12 | P(O)(OEt)3 | — | Ethyl lactate | 90 | 75 | 28 |
| 13 | P(O)(OEt)3 | — | Ethyl lactate/water (3:2) |
90 | 95 | 28 |
| 14 | P(OEt)3 | CSA (10 mol%) | — | 30 | 91 | 35 |
| 15 | P(OEt)3 | CSA (10 mol%) | — | 8 | 93 | 35 |
Another procedure carried out in solution was developed using non-aromatic amines for the synthesis of a family of tertiary α-amino phosphonates (TAPs) through the Kabachnik–Fields reaction involving substituted salicylaldehydes or 2-hydroxy naphthaldehyde, di-n-alkylamine, and di-, or trialkylphosphites in water with the aid of ultrasound irradiation (see Scheme 3).
 | ||
| Scheme 3 Synthesis of tertiary α-amino phosphonates via the Kabachnik–Fields reaction. | ||
Additionally, the method was compared with conventional methods. The reaction worked with equimolar amounts of four substituted salicylaldehydes or 2-hydroxy naphtaldehyde and a series of amines including di-methylamine, di-n-butylamine, di-n-hexylamine, pyrrolidine and morpholine. Reactions were carried out at ambient temperature without catalyst in water, selected from a diverse set of solvents. The solvents (toluene (40%), dichloromethane (45%), tetrahydrofuran (50%), ethylene glycol (70%), ethanol (78%), PEG-400 (80%) and glycerol (85%)) were screened via a model reaction of salicylaldehyde, di-n-butylamine and dimethyl phospite. However, the highest yield of 87% was achieved when using water. Higher yields of TAPs were obtained (ranging from 84% to 94%) with sonication requiring shorter reaction times (6 minutes to 35 minutes) compared with conventional methods (yields 56%–79% and reaction times 80–180 min, respectively). The reaction was followed by a straightforward workup and purification process.29
Similarly to the Kabachnik–Fields reaction for the synthesis of α-aminophosphonates, ultrasound-assisted methods to prepare α-hydroxyphosphonates have been developed. A method of choice is a solvent-free and catalyst-free approach, involving the reaction of an aldehyde or a ketone (1 equivalent) with trialkylphosphite (1.2 equivalents) under ultrasound irradiation (as an example, see the reaction of ketones with trialkylphosphite in Scheme 4). The resulting α-hydroxyphosphonates were obtained in high yields (84–94%) and within a short reaction time (from 10 to 37 minutes). In contrast to three-component reactions to prepare α-aminophosphonates described above, this method was successful with aromatic, benzylic and aliphatic aldehydes as well as benzylic ketones and isatin.30
 | ||
| Scheme 4 Synthesis of α-hydroxyphosphonates from ketones. | ||
In the ultrasound-assisted Kabachnik–Fields reaction, heterogeneous catalysts, like nanoparticles (NPs), have been studied to accelerate the reaction. An example of mixed-material NPs is graphene nanosheets–silver nanoparticles (GNS-AgNPs) used in the synthesis of a range of novel α-aminophosphonate derivatives. While the use of 10 mg mmol−1 of this nanomaterial gave 98% yield of a model reaction (1 mmol of 4-chlorobenzaldehyde, 1 mmol of 5-aminoindan and 1.2 mmol of diethyl phosphite under ultrasonication at ambient temperature for 15 minutes) under solvent-free conditions (see in Scheme 5), the yields were decreased in different solvents, for example, water (45%), toluene (60%), ethanol (67%), acetonitrile (75%), and dichloromethane (80%).
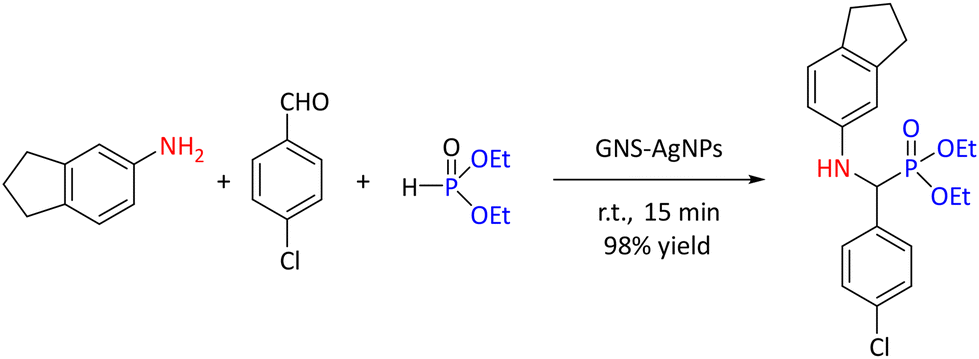 | ||
| Scheme 5 Model reaction for the synthesis of α-amino phosphonate from 5-aminoindan. | ||
Using the optimized conditions, a range of other catalysts was also screened, e.g., NPs (TiO2 NPs (69%), MgO NPs (70%), Ag NPs (76%), and ZnO NPs (84%)), AgNO3 (57%), graphite (65%), and graphene oxide (87%). Finally, a series of novel α-aminophosphonate derivatives was synthesized from 5-aminoindan or 3,4-(methylenedioxy)aniline, aromatic aldehydes, and diethyl phosphite with higher yields (84–97%) and shorter reaction times (10–15 minutes) compared with conventional methods (29–54% yields at 2 hours of reaction time). The recyclability of GNS-AgNPs for up to five runs with minimal loss of activity was noted. The GNS-AgNPs were prepared in situ by simultaneously reducing graphene oxide (GO) and silver nitrate (AgNO3) with sodium borohydride (NaBH4) as the reducing agent.31
NPs have also been utilized in the synthesis of more complex phosphonates, like 4H-chromen-4-yl phosphonates. Nasab and Kiasat developed a bifunctionalized acid–base mesoporous organosilica catalyst, SBA-Im/HSO3, for the preparation of these phosphonated heterocycles from a substituted salicylaldehyde, triethyl phosphite and a malonate derivative (see Scheme 6). The synthesis of SBA-Im/HSO3 was achieved by co-condensing trialkoxysilanes (3-chloropropyltrimethoxysilane and 3-mercaptopropyltrimetoxysilane) and tetraalkoxysilane in the presence of a triblock copolymer template as a structure-directing agent. The introduction of acidic and basic units in the nanocomposite involved oxidizing the thiol groups with H2O2 to provide sulfonate (–SO3H) groups, followed by the reaction of the chloro substituent with imidazole. The catalytic activity evaluation demonstrated that 5 mg mmol−1 of the nanocomposite exhibited high activity for a wide range of substrates for the synthesis of 4H-chromen-4-yl phosphonate derivatives under solvent-free ultrasonic conditions at ambient temperature, featuring yields from 55% up to 95% at 5–15 minutes.32
 | ||
| Scheme 6 Synthesis of 4H-chromen-4-yl phosphonate derivatives. | ||
As shown by the abovementioned examples, the solvent can have a drastic effect on the reaction yield. This effect was shown to be even stronger on the synthesis of α-hydroxyphosphonates from benzaldehyde and triethyl phosphite in the presence of potassium dihydrogen phosphate (KH2PO4) than on Kabachnik–Fields reactions. While utilizing 5 minutes of sonication or 45 minutes of stirring at ambient temperature, the solvents showed diverse results: while with both methods the lowest yields were achieved with water, ethanol and dimethylformamide (0/20%, 26/28%, and 27/45% for sonicated/stirred, respectively), the highest yields were obtained with methanol, acetonitrile and dichloromethane (32/53%, 38/56% and 44/60% for sonicated/stirred, respectively). The solvent-free system gave 86/80% yields. Optimization of the catalyst load showed that 10 mol% gave the same 83% yield as did the 5 mol%. With this optimized method, a series of 10 compounds was synthesized with yields ranging from 80% to 92%. However, the method was not successful with non-aromatic compounds, like cyclohexanone and propenone, and yielded only 48% and 56% for aliphatic aldehydes isobutyraldehyde and propionaldehyde, respectively.33
In addition to the above cases, the effect of sonication was described in the synthesis of α′-oxindole-α-hydroxyphosphonates using the ion-exchange resin Amberlyst-15 as catalyst under both conventional and sonication conditions (see Scheme 7.). The yields achieved under 15 minutes of sonication at 45 °C were comparable to those of a two-hour reaction at 100 °C. In the reactions of substituted isatin and dialkylphosphite, the product yields varied from 86% to 92%.34
 | ||
| Scheme 7 Synthesis of tertiary α-amino phosphonates via the Kabachnik–Fields reaction. | ||
The number of potential starting materials for the synthesis of α-aminophosphonates was increased by a consecutive reaction combining Kornblum and Kabachnik–Fields reactions. The process involves the formation of an aldehyde intermediate from benzyl halides through DMSO-promoted oxidative dehalogenation, followed by the combination with aryl amine and trimethyl phosphite under ultrasonic irradiation (see Scheme 8). This methodology is suitable for a variety of functional groups, (e.g. nitro, alkyls, nitriles, halides (Cl, I, F), hydroxyl) and heterocycles (e.g., furyl and pyridinyl) resulting in the formation of the desired products in high yields (ranging from 78% to 93%) under gentle conditions (ambient temperature and 40–55 minutes).36
 | ||
| Scheme 8 DMSO promoted synthesis of α-amino phosphonates. | ||
An even longer reaction path was utilized to prepare complex quinazolinone phosphonate derivatives in a one-pot multicomponent reaction of euparin, isatin (or their derivatives), primary amines, dialkyl acetylenedicarboxylates, trimethyl phosphite or triphenyl phosphite, and an acidic solution of hydrogen peroxide in an aqueous environment at ambient temperature. It is worth noting that the reaction could not progress without the use of ultrasonic irradiation, and achieving appropriate agitation of the reaction mixture was challenging. Despite the complexity of this reaction, the reaction time was less than one hour, yields were high (90% and above), and isolation was straightforward (see Scheme 9).37
 | ||
| Scheme 9 General synthesis of quinazolinone phosphonate derivatives. | ||
While most of the reactions to synthesize phosphonates utilizing sonication have focused on the formation of α-amino/hydroxyphosphonates, a few reactions to prepare phosphonates from isocyanates and isothiocyanates have also been described. As an example, a range of biologically interesting diethyl-substituted phenylcarbamoylphosphonates and phenylcarbamothioylphosphonates was synthesized with the addition of diethyl phosphite using a silica-supported Lewis acid catalyst, SiO2-CeCl3·7H2O. During the reaction optimization, 4-nitrophenyl isocyanate and diethyl phosphite were reacted under different temperatures, solvents and catalysts. When comparing catalysts, inorganic chlorides performed best (AlCl3 (56%), ZnCl2 (56%), CuCl2 (58%), FeCl3 (60%), CaCl2 (64%), CeCl3·7H2O (69%)) in tetrahydrofuran over 6–18 hour reaction times at 55 °C. Addition of an equimolar amount of an organic base e.g. triethylamine or 1,4-dimethylpiperazine afforded yields of only 49% and 51%, respectively, while the reaction without any catalyst or base resulted in 32% yield. However, when SiO-CeCl3·7H2O was used, the yield rose to 75% in tetrahydrofuran, and to 88% in a solvent-free 40 minute reaction at 50 °C. Other solvents resulted in lower yields, e.g., ethanol (56%), dichloromethane (64%), acetonitrile (72%), 1,4-dioxane (72%), and toluene (73%). The catalyst was also shown to perform well after five cycles: the yields decreased slightly from 89% to 81% in the ultrasound-assisted method and 85% to 78% in the conventional method. The actual load of CeCl3·7H2O on silica was not reported; however, the demand for SiO2-CeCl3·7H2O to promote the reaction was found to be high: the optimal loading was 200 mg for 1 mmol of starting material.38
2.2. Microwave-promoted synthesis
Microwaves (MWs) have been used in synthetic chemistry to transfer the energy needed in the reaction straight into the reaction mixture. This decreases the loss of power compared with hotplate and other conventional heating methods, as the energy is transmitted straight into the solvent and the reactant molecules. Additionally, MW-assisted syntheses can be conducted at higher temperatures when sealed vials are used.Although many MW-assisted methods are catalyzed, a few catalyst-free methods have been also developed. As an example, a Kabachnik–Fields reaction was used to synthesize two different sets of α-aminophosphonates (see Scheme 10). These reactions were conducted utilizing ethanol as solvent, without the need for a catalyst. The reaction times ranged from 20 to 40 minutes, resulting in a wide range of yields from 58% to 97% when ethyl 4-aminobenzoate was used as a starting amine. However, only 21% to 80% yields were obtained for the more complex amine (S)-4-amino-N-(2-hydroxy-1-phenylethyl)benzamide. The two lowest yields (21% and 34%) were obtained with aliphatic aldehydes, 2-ethylbutanal and 2-methylpropanal, respectively. Reactions were conducted at temperatures which were optimized for each reaction, ranging from 60 °C to 90 °C.39
 | ||
| Scheme 10 Synthesis of 4-aminobenzoic acid-derived α-amino phosphonates via the Kabachnik–Fields reaction. | ||
A catalyst-free synthesis of chiral bidentate aminophosphine ligands without the use of solvents was demonstrated. Two types of chiral bidentate aminophosphine ligands were synthesised, ligands with a chiral phosphorus, ethyl octyl α-aminophosphonates (see Scheme 11) and α-aminomethyl-phenylphosphinates, and ligands with a chiral carbon and an achiral phosphorus, (S)-α-phenylethylaminophosphonates and (S)-α-phenylethylaminophosphine oxides. The method utilized MW-assisted Kabachnik–Fields reaction of amines, paraformaldehyde, and diverse P-reagents. By starting with one equivalent of phosphorus compounds and paraformaldehyde the reaction resulted in 72%–92% yields of mono-products, while two equivalents resulted in a double Kabachnik–Fields reaction with similar yields of bis-products. Additionally, the structure of the phosphorus compound was found to affect the reaction time and product yield: with phosphites, the reaction times were 0.5 and 1–1.5 hours for mono- and bis-products, respectively, meanwhile the use of alkyl phenyl-H-phosphinates required 1 hour for both mono- and bis-products with yields of 42–91% and 59–97%, respectively. A MW-assisted double deoxygenation method to synthesize (bis(phosphinoylmethyl)-(S)-α-phenylethylamines) from C-chiral α-phenylethylaminophosphine derivatives was also developed. This chiral bidentate aminophosphine ligand was utilized in the synthesis of an optically active cyclic platinum complex. Furthermore, N-hydroxyethyl-α-aminophosphonates were also prepared with yield of 72% at 20 minutes as an extension of this approach. All the reactions were carried out at 80–100 °C, except the deoxylation, which needed stronger conditions, e.g. 5 hours at 140 °C.40
 | ||
| Scheme 11 MW-assisted synthesis of ethyl octyl a-aminohosphonates. | ||
In a recent study, a MW-assisted Pd-catalyzed Hirao reaction was used to prepare fifteen N-heterocycles. Three bromine-containing N-heterocycles, namely 9-(4-bromophenyl)-9H-carbazole, 3-bromo-9-phenyl-9H-carbazole, and 1-(5-bromoindolin-1-yl)ethan-1-one, were coupled with either dialkyl phosphite (e.g. (MeO)2P(O)H or (EtO)2P(O)H) or diarylphosphine oxide (e.g., Ph2P(O)H, 4-MePh2P(O)H, or 3,4-diMePh2P(O)H) using Pd(OAc)2 (10%) as the catalyst precursor and triethylamine as the base in ethanol (see example in Scheme 12). The need for a conventional phosphorus ligand in the catalysis was avoided by using excess of the phosphorus starting material Y2P(O)H as its trivalent tautomer (Y2POH) which served as the P-ligand in the formation of catalytic Pd-complex, thereby reducing cost and environmental impact. In comparison with a conventional method, the presented method yielded around 80% yields during one hour of reaction at 135 °C using MW, while conventional heating at 80 °C yielded somewhat lower yields from 59% to 79% with longer reaction times (from 5 to 8 hours). Shorter reaction times and avoiding the use of complex phosphorus-ligands in the synthesis improved the ecological safety of the protocol.44
 | ||
| Scheme 12 Example of Pd catalyzed Hirao reaction. | ||
An extensive optimization study of MW-assisted Michaelis–Arbuzov reaction both in batch and flow conditions resulted in a method for synthesizing dialkyl haloalkylphosphonates, crucial intermediates in the synthesis of Ethephon or acyclic nucleoside phosphonate drugs like adefovir, tenofovir, and cidofovir. The reaction of triisopropyl phosphite and 10-fold excess of 1,2-dichloroethane to produce diisopropyl 2-chloroethylphosphonate showed highest yield (89.2%) with optimal purity (190 °C reaction temperature and 160 minutes). Use of four-fold excess of dichloroethane resulted in 87.5% yield, and with equal ratio of starting materials the yield was still high, 63.1%. Comparison of solvent-free conditions with some solvents showed that the use of tetrahydrofuran and diglyme hindered the reaction remarkably (yields only 10.9% and 12.8%, respectively), while acetonitrile and dimethylformamide had only a minor effect (65.8% and 69.5%, respectively). Aromatic solvents, namely toluene, xylene and pyridine, showed the strongest effect on the reaction, with 0%, 8.8% and 2.4% yields, and even 1 equivalent of toluene blocked the reaction. Thus, batch reactions were carried out without any solvent. In order to optimize a large-scale flow reaction, acetonitrile was selected as the solvent. Process optimization to prepare diisopropyl 2-(2-chloroethoxy)ethylphosphonate, diidopropyl 2-bromoethylphosphonate, diisopropyl bromomethylphosphonate, and diisopropyl iodomethylphosphonate in preparative scale (tens or hundreds of grams) was also carried out using flow conditions. Depending on the starting materials, the method required only one equivalent of each starting compound to give high yields (63–88%). Impurities of the products were easily removed.45 One of the optimized reactions is shown in Scheme 13.
 | ||
| Scheme 13 Michaelis–Arbuzov reaction for the key intermediate for e.g. adefovir preparation. | ||
 | ||
| Scheme 14 Nano BF3·SiO2-catalyzed synthesis of phosphonates. | ||
Similarly, as with sonication, NPs and other nanomaterials can also be used in MW-assisted syntheses. In general, these nano catalysts are easy to remove from the reaction mixture and most of them are less sensitive to air, moisture, light and catalyst poisoning than other types of catalyst (e.g. transition metal catalysts used in cross-coupling reactions like the Hirao reaction described above). Additionally, several inorganic nanomaterials are considered as non-toxic.
Among the synthetic reactions for phosphorus compounds under MW irradiation utilizing nano catalysts, the Kabachnik–Fields reaction is the most common, as noted in the recent literature. Metal oxides are among the used nanocatalysts, for example nano ZnO, which was utilized in the synthesis of α-aminophosphonates from 2-hydroxy-4-methylaniline, various aromatic aldehydes and triethylphosphite. Products were obtained with 82–93% yields within 2–5 min reaction time using ethanol as solvent in an open reaction vessel.47
Another inorganic oxide, nano CaO, was proved to be an efficient catalyst for the synthesis of benzimidazolyl α-aminophosphonates from 1H-benzo[d]imidazol-2-amine, diverse aryl aldehydes, and diethyl phosphite (see Scheme 15). The CaO nanocatalyst was selected based on product yield of a model reaction of 4-ethoxybenzaldehyde under conventional heating over 24 hours. Thirteen catalysts were screened, including chlorides (MgCl2 (68%), CaCl2 (75%), FeCl3 (60%), VCl3 (68%), and CrCl3 (76%)), iodides (KI (60%) and CuI2 (79%)), acetates (Co(OAc)2·4H2O (60%) and Zn(OAc)2 (80%)), oxides (Al2O3 (55%) and nano CaO (80%)) and other inorganic salts (Mg(ClO4)2 (70%) and [NH4]2[Ce(NO3)6] (75%)). The optimal conditions were determined to be 10 mol% of catalyst under solvent-free conditions and with 5 minutes of reaction at 300 W irradiation. For comparison, optimal conditions for the conventional method were 30 minutes at ambient temperature under solvent-free conditions. The MW-assisted versus conventional method yielded 95% and 93% yields, respectively. Utilization of optimized MW conditions for the series of reactions resulted in higher yields than a conventional reaction at ambient temperature over 6 hours (89%–94% and 72%–85% for MW-assisted and conventional, respectively).48
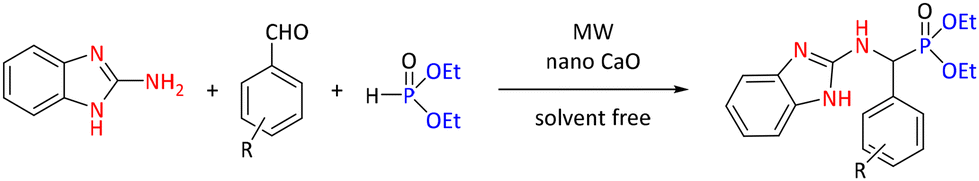 | ||
| Scheme 15 Synthesis of benzimidazolyl α-aminophosphonates using nano CaO as catalyst. | ||
Nanoparticles of stannic oxide (SnO2) were also shown to catalyze the Kabachnik–Fields reaction. The studied reaction involved conjugation of 2-amino-5-ethylthio-1,3,4-thiadiazole, various aromatic aldehydes, and diethylphosphite under solvent-free conditions yielding a series of 10 substituted diethyl (((5-(ethylthio)-1,3,4-thiadiazol-2-yl)amino)(phenyl)methyl)-phosphonates with high yields (89–97%).49
In Kabachnik–Fields reactions, “mixed” materials have also been used as catalysts. In contrast to single-type nanoparticles, these “mixed” catalysts can incorporate inorganic materials supported with organic compounds. Such an example is glutathione-supported nano ferrite (nano FGT) used in the synthesis of a range of diethyl-(((2,3-dihydrobenzo[b][1,4]dioxin-6-yl)amino)(substituted phenyl)methyl) phosphonates (see Scheme 16). In the study, a model reaction of 2,3-dihydrobenzo[b][1,4]dioxin-6-amine with 3-methoxy benzaldehyde was investigated to find the optimal conditions. The reaction was conducted at 80 °C and 8–10 hours with several metal chlorides/bromides (FeCl3 (32%), AlCl3 (35%), ZnCl2 (45%), and CrCl3 (46%), ZnBr2 (58%)) and p-toluenesulfonic acid (50%) and at room temperature for 30 minutes; with nano-FGT, the yield was 70%. Optimization of catalyst load, reaction time and solvent revealed that the reaction demonstrated optimal performance with 10 mol% catalyst load, under solvent-free conditions at ambient temperature for 15 min reaction time. From the studied solvents (toluene, acetonitrile, dichloromethane, tetrahydrofuran, isopropanol and ethanol), toluene gave the same yield of 93% as the solvent-free reaction. The same yield was reached within 5 minutes using MW at 400 W power. Reactions were carried out under these conditions using 2,3-dihydrobenzo[b] [1,4]dioxin-6-amine, eleven aromatic aldehydes and diethyl phosphite, yielding 85%–95% of the desired products during a 30 minute reaction time.50
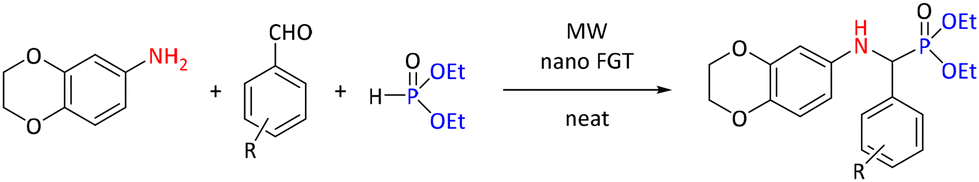 | ||
| Scheme 16 Nano FGT-catalyzed synthesis of α-aminophosphonates. | ||
Sulfonated reduced graphene oxide (rGO-SO3H) nanoparticles were used to produce aminomethylene bisphosphonates from fluorine-containing anilines, triethyl orthoformate, and dialkyl phosphite (see Scheme 17). For comparison, a set of other commonly used catalysts, i.e., metal chlorides/bromides (CuCl2 (40%), NiCl2 (40%), FeCl3 (45%), ZnCl2 (50%), AlCl3 (60%), and LaCl3 (60%)), TiO2 (30%), CuBr2 (35%), nickel bis-acetylacetonate (60%), and Rh(OAc)4 (60%) were screened in a model reaction at 40–60 °C for 6–10 hours of reaction time. The reaction using 10 mg mmol−1 rGO-SO3H gave 70% yield within 3 hours, which increased to 80% when 20 mol% of catalyst was used within 2 hours of reaction time. As observed in earlier studies, the use of solvents (toluene, tetrahydrofuran, acetonitrile, dichloromethane, dimethylformamide ethanol or methanol) compromised the reaction. Optimization of the MW power showed that the reaction reached 94% yield within 10 minutes when 300 W power was used. These conditions were used to conduct twenty reactions with ten different aromatic aldehydes, dimethyl or diethyl phosphite and triethyl ortoformate. Products were obtained within 10–20 minutes with yields ranging from 80% to 95%.51
 | ||
| Scheme 17 Synthesis of aminomethylene bisphosphonates. | ||
Porous materials have also been used as catalysts to increase the active surface area of the nanoparticles. A recent example of such a porous mixed material is mesoporous titania-ceria mixed oxide (MTCMO). It was utilized for synthesizing diethyl((substituted phenyl)((4-(N-(5-methyl-4,5-dihydroisoxazol-3-yl)sulfamoyl)phenyl) amino)methyl)-phosphonates from 4-amino-N-(5-methyl-4,5-dihydroisoxazol-3yl) benzenesulfonamide, different aldehydes and diethyl phosphite. Catalyst screening also included several known catalysts, including organic (super)bases (TMG (35%), DBU (45%), and DABCO (58%)), resins (PS-PTSA (32%) and Amberlyst-15 (46%)), p-toluenesulfonic acid (50%), CeCl3·7H2O (55%) and SiO2-BF3 (47%) at 10 mol% catalyst loading, at 60–80 °C, in 8–10 hours of reaction. However, MTCMO performed the best with 70% yield, in 30 minutes, and at ambient temperature. Optimization of conditions resulted in a 10 minute reaction time, under solvent-free conditions, at 400 W power, giving 93% yield. Broadening the scope of the reaction to ten different aromatic aldehydes gave products with yields in the range 89–93%.52
Silica-supported catalysts have also attracted interest in MW-assisted synthesis similarly to ultrasound-assisted methods described above. One example among these methods is the use of silica-supported TiO2 to catalyze the synthesis of a range of new bis(α-aminophosphonates) (bis(α-Aps)) under solvent-free conditions (see Scheme 18).
 | ||
| Scheme 18 Silica-supported TiO2-catalyzed synthesis of bis(α-aminophosphonates). | ||
Catalyst screening was carried out in tetrahydrofuran with conventional heating (70 °C for 4–8 hours) and 2.5 mol% catalyst loading, and included known catalysts, i.e., metal chlorides (ZnCl2 (62%), LaCl2 (65%), CuCl2 (66%), FeCl3 (66%), and AlCl3 (68%)), boron trifluorides (BF3 (62%) and BF3/Et2O (63%)), and silica-supported catalysts (SiO2-BF3 (70%), SiO2-ZnBr2 (76%)). The nano-SiO2-TiO2 catalyst shortened the reaction time to 3 hours giving 79% yield. Solvent-free conditions were found to increase the yield to 85% at 50 °C. On optimization of the catalytic system under solvent-free MW conditions, the reaction times were: 15 minutes of irradiation without any catalyst, 12 minutes with 2.5 mol% SiO2-ZnBr2 and either 5 minutes with 2.5 mol% or 7 minutes with 5 mol% of nano-SiO2-TiO2, and the yields were 86%, 89%, 90% and 97%, respectively.53
Other known catalysts, e.g., CeCl3 and LaCl3, have been used as silica-supported catalysts for MW-assisted one-pot Kabachnik–Fields reactions. In a solvent-free SiO-CeCl3·7H2O-catalyzed reaction of diethyl phosphite, aromatic aldehyde, and 4,4′-sulfonyldianiline, a total of 10 α-diaminophosphonates derived from 4,4′-sulfonyldianiline were synthesized with yields >90% (the same kinds of structures as presented in Scheme 18). The MW-assisted method was superior compared with conventional heating with or without solvent. Specifically, yields around 70% were reached within 15 hours in solution and within 5–8 hours under solvent-free conditions, while yields >80% were recorded within 6–7 minutes under MW-assisted and solvent-free conditions. The added value of SiO2-CeCl3·7H2O comes from the increased yield (91%) and short reaction time (5 minutes) with 10 mol% catalyst load. Additionally, the reaction time was lowered to 4 minutes and the yield rose to 96% when 12 mol% of catalyst was used. In a catalyst recycling study, the catalyst showed only 7% lower yield (82%) at the fifth run.54
The catalytic performance of SiO2-LaCl3·7H2O was evaluated based on the preparation of a diverse range of α-aminophosphonates from four substituted benzothiazole and thiadiazole amines, seven substituted aromatic aldehydes, and diethylphosphite under solvent-free conditions employing both MW and conventional heating (see reaction in Scheme 19). Reaction optimization showed that 10 mol% of catalyst with 5 min MW irradiation at 455 W performed best (97% yield) while 12.5 mol% was required to yield 95% under the conventional method (3 hours at 50 °C). Altogether, twenty compounds were synthesized with yields ranging from 85% to 97%. Studies also showed good reusability of the catalyst as the yield of the model reaction was 90% after eight cycles. Additionally, product isolation was easy through straightforward recrystallization without the need for column chromatography.55
 | ||
| Scheme 19 Synthesis of α-amino phosphonates via the Kabachnik–Fields reaction using SiO2–LaCl3 as catalyst. | ||
2.3. Solvent-free synthesis
As presented in the previous sections, several phosphonate compounds can be prepared under solvent-free conditions. Depending on the reaction under study, the nature of the solvent plays a pivotal role. For example, the formation of some α-hydroxyphophonates seems to be highly hindered in water,33 while some α-aminophophonates are produced in excellent yields in aqueous solutions.28 Similarly, the polarity, presence of exchangeable protons and acidity/basicity have shown diverse effects depending on the nature of the reactant compounds. However, avoiding a solvent in a reaction would reduce waste and increase the mass of reaction mixture, thus lowering the energy consumption due to heating. This is applicable to several phosphorus-containing starting materials because they are liquids and can be used neatly.An extensive study was carried out on the effects of diverse solvents and catalysts on the Kabachnik–Fields reaction. It was found that shorter reaction times were noted with higher amounts of diethylphosphite (see Table 2, entries 1–9). In this study, eighteen different α-aminophosphonates were synthesized with yields varying from 35% to 90% with reaction times from 3 to 24 hours. The study also showed that the formation of the imine intermediate is most likely the rate-limiting factor. The synthesis starting with the imine led to higher yields with compounds that resulted in only moderate yield in the Kabachink–Fields reaction (e.g. diethyl phosphinate yielded 40% when starting from p-methyl benzaldehyde and aniline vs. 95% for starting from the corresponding imine). The study also presented energy diagrams for the proposed reaction pathways calculated at B3LYP/6-31+G** level using density functional theory (DFT). Additionally, synthesis of N-deprotected α-aminophosphonates (similar to α-amino acids) from N-PMP-protected α-aminophosphonates in the presence of trichloroisocyanuric acid was demonstrated.56
| Entry | Phosphorus source | Catalyst (load) | Solvent | Temperature/method | Time (h) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | H(O)P(OEt)2 (1.1 eq.) | — | 1,4-Dioxane | Ambient | 24 | 15 | 56 |
| 2 | H(O)P(OEt)2 (1.1 eq.) | — | Toluene | Ambient | 24 | 25 | 56 |
| 3 | H(O)P(OEt)2 (1.1 eq.) | — | (1.1 eq.) | Ambient | 24 | 27 | 56 |
| 4 | H(O)P(OEt)2 (1.1 eq.) | — | Dichloromethane | Ambient | 24 | 32 | 56 |
| 5 | H(O)P(OEt)2 (1.1 eq.) | — | Tetrahydrofuran | Ambient | 24 | 52 | 56 |
| 6 | H(O)P(OEt)2 (1.1 eq.) | — | Acetonitrile | Ambient | 24 | 55 | 56 |
| 7 | H(O)P(OEt)2 (1.1 eq.) | — | — | Ambient | 24 | 78 | 56 |
| 8 | H(O)P(OEt)2 (1.5 eq.) | — | — | Ambient | 48 | 60 | 56 |
| 9 | H(O)P(OEt)2 (2.0 eq.) | — | — | Ambient | 6 | 90 | 56 |
| 10 | H(O)P(OEt)2 | — | — | 90 °C | 6 | 51 | 58 |
| 11 | H(O)P(OEt)2 | SPB (1 wt%) | 90 °C | 0.5 | 88 | 58 | |
| 12 | H(O)P(OEt)2 | SPB (5 wt%) | 90 °C | 5 min | 98 | 58 | |
| 13 | H(O)P(OEt)2 | SPB (5 wt%) | 70 °C | 0.25 | 97 | 58 | |
| 14 | H(O)P(OEt)2 | SPB (5 wt%) | Ethanol | Reflux | 0.5 | 91 | 58 |
| 15 | H(O)P(OEt)2 | SPB (5 wt%) | Acetonitrile | Reflux | 0.5 | 90 | 58 |
| 16 | H(O)P(OEt)2 | SPB (5 wt%) | Tetrahydrofuran | Reflux | 0.5 | 88 | 58 |
| 17 | H(O)P(OEt)2 | SPB (5 wt%) | Water | 90 °C | 0.5 | 30 | 58 |
| 18 | H(O)P(OEt)2 | SPB (5 wt%) | Dimethylformamide | 90 °C | 0.5 | 81 | 58 |
| 19 | H(O)P(OEt)2 | SPB (5 wt%) | Toluene | 90 °C | 0.5 | 30 | 58 |
| 20 | P(OMe)3 | ZnI2 (20 mol%) | — | Ambient | 0.25 | 96 | 58 |
| 21 | H(O)P(OEt)2 | Humic acid (50 mg mmol−1) | Water | Reflux | 6 | Trace | 64 |
| 22 | H(O)P(OEt)2 | Humic acid (50 mg mmol−1) | Methanol | Reflux | 6 | 42 | 64 |
| 23 | H(O)P(OEt)2 | Humic acid (50 mg mmol−1) | Ethanol | Reflux | 6 | 46 | 64 |
| 24 | H(O)P(OEt)2 | Humic acid (50 mg mmol−1) | Dimethylformamide | Reflux | 6 | 50 | 64 |
| 25 | H(O)P(OEt)2 | Humic acid (50 mg mmol−1) | — | 90 °C | 6 | 94 | 64 |
| 26 | H(O)P(OEt)2 | Humic acid (15 mg mmol−1) | — | 90 °C | 6 | 93 | 64 |
| 27 | H(O)P(OEt)2 | — | — | 90 °C | 6 | 0 | 64 |
| 28 | P(OMe)3 | — | — | Ambient | 24 | 41 | 65 |
| 29 | P(OMe)3 | Succinic acid (8.5 mol%) | — | Ambient | 5 min | 98 | 65 |
| 30 | P(OMe)3 | Succinic acid (8.5 mol%) | Ethyl acetate | Ambient | 0.3 | 79 | 65 |
| 31 | P(OMe)3 | Succinic acid (8.5 mol%) | Ethanol | Ambient | 0.5 | 65 | 65 |
| 32 | P(OMe)3 | Succinic acid (8.5 mol%) | Methanol | Ambient | 0.3 | 76 | 65 |
| 33 | P(OMe)3 | Succinic acid (8.5 mol%) | Tetrahydrofuran | Ambient | 25 min | 70 | 65 |
| 34 | P(OMe)3 | Succinic acid (8.5 mol%) | Chloroform | Ambient | 28 min | 67 | 65 |
| 35 | P(OMe)3 | Succinic acid (8.5 mol%) | Diethylether | Ambient | 35 min | 69 | 65 |
| 36 | P(OMe)3 | Succinic acid (8.5 mol%) | Acetone | Ambient | 25 min | 70 | 65 |
| 37 | P(OMe)3 | Succinic acid (8.5 mol%) | Dichloromethane | Ambient | 0.25 | 82 | 65 |
| 38 | P(OMe)3 | Succinic acid (8.5 mol%) | Acetonitrile | Ambient | 0.5 | 69% | 65 |
Also, phosphonylation of C(sp2) carbon was demonstrated in solvent- and catalyst-free synthesis of oxoindolin-3-ylphosphonates from oxoindolin-3-ylidenes using combinatiorial chemistry.57
One such class of catalysts comprises sulfated materials, like sulfated polyborates (SPB)58 and sulfated meglumine.59 These were proposed to promote a proton to the aldehyde to facilitate nucleophilic attack of the amine to the carbonyl carbon. Both sulfated materials showed lower yields if solvents were used. The impact on the yield was diverse: while sulfated meglumine gave 97% yield in a model reaction of 2-methoxy-5-trifluoromethylaniline, benzaldehyde and diethyl phosphite in optimized conditions without solvent, the yields were only 51% in toluene, 85% in ethanol, 86% in tetrahydrofuran, and 71% in acetonitrile (for sulfated polyborate, see entries 11–19 in Table 2).
Also, other compounds capable of activating the carbonyl C have been used, e.g., ZnI2 (see entry 20 in Table 2),60 which provided yields from 50% to 96% within 5–20 minute reaction times.
Similarly to MW- and ultrasound-assisted syntheses a variety of NPs and nanomaterials have been developed to catalyze synthesis of both α-aminophosphonates and α-hydroxyphosphonates. Nano Sb2O3 was used in a Kabachnik–Fields reaction of a series of dialkyl (aryl substituted) ((2-fluoro-4-(((2-(methylcarbamoyl)pyridine-4-yl)oxy)phenyl)amino)methyl)phosphonates from seven different aromatic aldehydes, 3-(4-amino-3-fluorobenzyl)-N-methylbenzamide and either dimethyl or diethyl phosphite (see the reaction in Scheme 20).
 | ||
| Scheme 20 Synthesis of α-amino phosphonate derivatives with anticancer properties. | ||
The yields were lower for other known catalysts, e.g., metal chlorides (BiCl3 (63%), AlCl3 (65%), ZnCl2 (65%)), CuO (54%), and SiO2-BF3 (54%). Additionally, solvent use was shown to compromise the yields (ethanol (50%), acetonitrile (60%), dichloromethane (65%), water (68%), toluene (70%), dimethylformamide (70%), and tetrahydrofuran (72%)). Notably, the solvent-free reaction gave 75% yield under the same conditions (40 °C and 40 minutes). Prepared compounds were evaluated to have anticancer properties.61
Another example of single-type NPs used in Kabachnik–Fields reactions was TiO2 (anatase) nanoparticles (see in Scheme 21).
 | ||
| Scheme 21 Anatase-catalyzed synthesis of α-aminophosphonates. | ||
Catalyst screening in the reaction of 2-iodo-4-trifluoromethyl aniline (1 eq.), 3,4-dimethoxy benzaldehyde (1 eq.) and diethylphosphite (1.2 eq.) included several metal halides [InCl3 (56%), NiBr2 (57%), CuI (60%), ZrCl4 (61%), FeCl3 (74%), and AlCl3 (79%), triflates (Ce(OTf)4, Al(OTf)3(89%)), oxides (ZnO (68%), and nano CuO (84%))]. The optimal results were achieved with the nano TiO2 (93%). Interestingly, the catalyst-free reaction outperformed several catalysts by yielding 83%. Similarly, as in other reactions above, most of the solvents decreased the yield (1,4-dioxane (74%), acetonitrile (76%), dimethylsufoxide (78%), dimethylformamide (79%), methanol (80%), chloroform (81%), ethanol (82%), and tetrahydrofuran (85%)). Temperature decrease from 50 °C to 45 °C decreased the yield to 87%. The optimized conditions were used to prepare a series of diethyl((2-iodo-4-(trifluoromethyl)phenyl)amino)(aryl)methyl)-phosphonates using aromatic aldehydes.62
Smaller particles, nano TiO2 in the form of anatase, were used to catalyze the synthesis of a series of α-aminophosphonates from 2,3-dihydrobenzo[b][1,4]dioxine-6-carbaldehyde, various (hetero)aromatic amines, and dimethyl phosphite at 50 °C. Fifteen α-aminophosphonates were produced with yields varying from 86% to 96%.63
Also, polymer-based NPs like the polyethyleneimine-grafted mesoporous nanomaterial MCM-41@PEI have been used to catalyze Kabachnik–Fields reactions. MCM-41@PEI improved the synthesis of quinoline α-aminophosphonate derivatives starting from aniline, 4-chloroaniline or 2-phenyl ethylamine, dimethyl or diethylphosphite and quinoline-4-carbaldehyde. The reaction was optimal under solvent-free conditions at temperatures ranging from 50 to 80 °C, to produce the desired α-aminophosphonates with 87%–91% yields within 5–6 hours.64
Amberlyst-15 has been used as a solid-phase catalyst to produce α-aminophosphonates. It showed the highest yield (93%) among the screened catalysts, i.e., H3PW12O40 (24%), KHSO4 (29%), BiCl2 (37%), InCl3 (52%), TiO2 (58%), FeCl3 (60%), Mg(ClO4) (64%). Amberlyst-15 performed best in solvent-free conditions, while with solvents only low yields were noted: methanol (27%), ethanol (30%), dichloromethane (35%), tetrahydrofuran (36%) and toluene (42%). Optimized reactions were used to couple ethyl-2-amino-α-(methoxy-imino)-4-thiazoleacetate, aromatic aldehydes, and diethylphosphite at ambient temperature with yields varying from 74% to 93% within 30–45 minutes.65
Humic acid is a non-toxic and environmentally friendly high molecular weight biopolymer. It was studied as a catalyst in the Kabachnik–Fields reaction. It showed the highest catalytic activity for the model reaction (see entries 21–27 in Table 2) with yield of 93% when 15 mg mmol−1 of humic acid was used. Using amines with various substitutions and aromatic aldehydes, thirty five diethyl(phenyl(phenylamino)methyl)phosphonates were prepared with yields ranging from trace amounts to 93%. The secondary amine pyrrolidine resulted only in a trace amount of product. The benzaldehydes with higher electron density on the ring (electron-donating groups attached) gave higher yields. The opposite was observed with electron-withdrawing groups. Steric hinderance at the ortho-position of aldehyde was shown to lower the yield. In addition to anilines, also butylamine and 2-aminopyridine resulted in high yields (85% and 88%, respectively).66
As discussed above, diverse materials and high molecular weight organic compounds have been used to catalyze phosphonylation. However, small molecules may act as organo-catalysts in Kabachnik–Fields reactions as well. Succinic acid has been shown to be an efficient catalyst under solvent-free conditions (see entries 28–38 in Table 2) and it was used to prepare thirty-two different α-aminophosphonates.67
Iron in combination with iodine was used in a novel and straightforward method for the preparation of α-aminophosphonates from aldehydes, diethyl phosphite, and azides at ambient temperature. The reaction times varied from 5 minutes to 12 hours depending on the structures of aldehyde and azide. Eighteen products were synthesized with 43–86% yields.68
 | ||
| Scheme 22 CaO-catalyzed synthesis of α-hydroxyphosphonates. | ||
Some common inorganic salts, like K2CO3 and KHSO4 were used to catalyze the formation of α-hydroxyphosphonates. K2CO3 was used to convert ketones into tertiary-α-hydroxyphosphonates with cyclic neopentylene phosphites reaching yields from 85% to 95% when using 1 equivalent of base. Instead, 20 mol% of KHSO4 was used to convert aromatic aldehydes and diethylphosphite into the corresponding α-hydroxyphosphonates with 82–91% yields for 2–4 hours reaction time.70,71
Triethylamine has also been used to facilitate the formation of tertiary α-hydroxyphosphonates. With one equivalent of triethylamine, unactivated ketones can undergo hydrophosphonylation, resulting in 63–89% yields.72
 | ||
| Scheme 23 Synthesis of potential candidates for antimicrobial, antileishmanial and anticancer drug discovery. | ||
Similarly to the study by Nasab and Kiasat,32 synthesis of various 2-amino-4H-chromen-4-yl phosphonates and β-phosphonomalonates was performed at ambient temperature using silica-bonded 2-hydroxyethylammonium acetate.76
Carbamoylphosphonates were synthesized under solvent-free conditions from alkyl- and aryl-substituted isocyanates and dialkyl phosphites using 0.1 mol% rare-earth (yttrium or erbium) monoalkyl or dialkyl complexes as catalysts (see Scheme 24). The solvent had only a slight effect on the reaction (96% to 99% yields in different solvents). Yttrium showed higher catalytic activity than erbium, by comparison. Optimized conditions were used to prepare twenty-one carbamoyl phosphonates with 87%–99% yields at ambient temperature within just 5 minutes.77
 | ||
| Scheme 24 Synthesis of carbamoylphosphonates catalyzed by rare-earth metal complexes. | ||
Several heteroaromatic aldehydes were shown to augment the formation of α-aminophosphonates from benzyl amines (see Scheme 25). The proposed mechanism proceeds through auto-oxidation of the aldehyde and transfer of a radical from the aldehyde to the benzylic sp3-carbon of the amine. In the next step, the amine is converted to an imine and subsequently to an aldehyde in a reaction involving in situ-formed H2O2. In the last step, the formed aldehyde reacts with a starting amine to form imine during auto-oxidation of the heteroaromatic aldehyde, which will react with phosphite. In the study, twelve reaction products were reported with yields ranging from 30–98%. However, the mechanism was supported by experiments carried out only under a few reaction conditions, and neither radical scavenging nor theoretical calculations were carried out to support what was proposed.78
 | ||
| Scheme 25 One-pot, metal- and solvent-free synthesis of α-amino phosphonates with auto-oxidation of heteroaromatic aldehydes. DEFINE “Het”. | ||
2.4. Water as solvent
Among the numerous reactions to prepare phosphonates, especially α-amino and α-hydroxyphosphonates (see Table 1), some reactions were carried out in solvent mixtures containing water or even in pure water. As an example, the abovementioned ethyl lactate/water mixture showed higher yields than the pure ethyl lactate.28 Also, a mixture of polyethylene glycol (PEG-600) and water as a solvent was suitable for the preparation of α-diaminophosphonates from 4,4-dioxyaniline, different aryl/heteroaryl aldehydes, and diphenyl H-phosphonate, giving high yields (74–93%). Reactions were carried out at 80 °C and the reaction time was 2 hours.79 Occasionally, instead of using a different solvent, a small amount of an organic compound might also enhance the synthesis outcome. This was demonstrated in the Kabachnik–Fields reaction, in which a range of solvents, additives and catalysts were screened (see Table 3). As a result, 1 mol% of ytterbium triflate reached maximum efficiency when supported with 2 wt% of polyoxyethanyl α-tocopheryl sebacate (PTS) in water (see in Scheme 26). The reaction was successful with a series of aromatic aldehydes and anilines (83–95%) and also gave a yield of 56% when 2-methylpropanal and aniline were used in the presence of 5 mol% of catalyst.80 | ||
| Scheme 26 Ytterbium triflate-catalyzed synthesis of α-aminophosphonates. | ||
| Entry | Catalyst (load) | Additive | Solvent | Yield (%) |
|---|---|---|---|---|
| 1 | Yb(OTf)3 (1 mol%) | — | — | 84 |
| 2 | Yb(OTf)3 (1 mol%) | — | Ethanol | 72 |
| 3 | Yb(OTf)3 (1 mol%) | — | Acetonitrile | 70 |
| 4 | Yb(OTf)3 (1 mol%) | — | Toluene | 38 |
| 5 | Yb(OTf)3 (1 mol%) | — | Ethyl acetate | 40 |
| 6 | Yb(OTf)3 (1 mol%) | — | Tetrahydrofuran | 52 |
| 7 | Yb(OTf)3 (1 mol%) | — | Dichloromethane | 50 |
| 8 | Yb(OTf)3 (1 mol%) | — | Water | 32 |
| 9 | Yb(OTf)3 (1 mol%) | 2 wt% p-toluenesufonic acid | Water | 95 |
| 10 | Yb(OTf)3 (1 mol%) | 2 wt% Triton-X 100 | Water | 78 |
| 11 | Yb(OTf)3 (1 mol%) | 2 wt% sodium dodecyl sulfate | Water | 70 |
| 12 | YbCl3 (1 mol%) | 2 wt% p-toluenesufonic acid | Water | 56 |
| 13 | Yb(PFO)3 (1 mol%) | 2 wt% tetrabutylammonium bromide | Water | 90 |
| 14 | Sc(OTf)3 (1 mol%) | 2 wt% tetrabutylammonium bromide | Water | 83 |
| 15 | Hf(OTf)4 (1 mol%) | 2 wt% tetrabutylammonium bromide | Water | 70 |
| 16 | Sn(OTf)4 (1 mol%) | 2 wt% tetrabutylammonium bromide | Water | 72 |
| 17 | Zn(OTf)2 (1 mol%) | 2 wt% tetrabutylammonium bromide | Water | 76 |
| 18 | Cu(OTf)3 (1 mol%) | 2 wt% tetrabutylammonium bromide | Water | 65 |
In addition to metal-catalyzed methods, organocatalytic methods have been developed for the synthesis of α-hydroxyphosphonates. As an example, pyridine 2,6-dicarboxylic acid was shown to perform best among a series of known (organo)catalysts (e.g. MgO nanoparticles (50%), cellulose sulfuric acid (60%), sulfamic acid (70%), silica sulfuric acid (70%), and H3PW12O40 (75%)) for converting 3-phenylpropanal into the corresponding α-hydroxyphosphonate. The optimized reaction conditions were used for reactions of sixteen (hetero)aromatic and aliphatic aldehydes and ketones with yields between 60% and 95% and reaction times from 1–4 hours.81 Molecular iodine is an inexpensive and green catalyst that has been extensively investigated. It has been studied as a catalyst for the synthesis of α-hydroxyphosphonates using (hetero)aromatic and aliphatic aldehydes and triethylphosphite. The reactions were of high yields (83–97%) in 15–120 minutes at 80 °C.82 Molecular iodine was also utilized by Rajasekhar et al. in the synthesis of 2-amino-4H-chromen-4-yl phosphonate derivatives by reaction of salicylaldehyde, malononitrile or ethylcyanoacetate, and triethyl phosphite or trimethyl phosphite (see Scheme 27). They found that the reaction gave low yields in several organic solvents but performed well in water. Yields ranged from 77% to 91%. Prepared compounds were reported to have antimicrobial and antioxidant properties.83
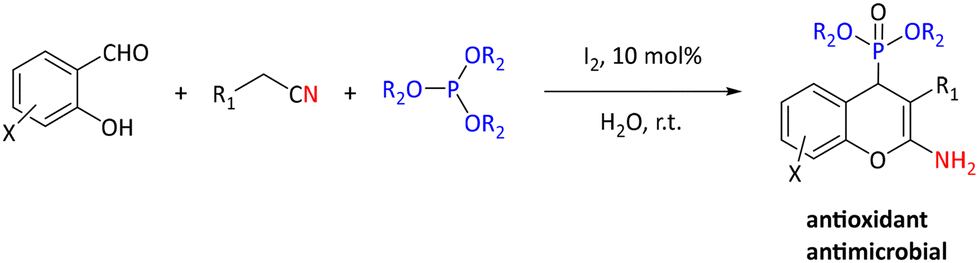 | ||
| Scheme 27 Synthesis of potential antimicrobial and antioxidant phosphonates. | ||
Water was also used as solvent in two other syntheses of 2-amino-4H-chromen-4-yl phosphonates. Both studies utilized a nature-derived substance in catalysis, namely β-cyclodextrin (β-CD) and chitosan. β-CD was used as a catalyst without modification, while chitosan was utilized as chitosan-coated Fe3O4 core–shell NPs (Fe3O4@CS NPs). In comparison with the methods described above,32,74,81 iodine and β-CD performed slower than the ultrasound-assisted method with SBA-im/SO3H NPs developed by Nasab and Kiasat32 and silica-bonded 2-hydroxyethylammonium acetate studied by Sobhani and Honarmand.74 However, the Fe3O4@CS NPs yielded comparative yields (88–97%) approximately within 30 minutes of reaction time. While NP-assisted methods yielded products in minutes or tens of minutes, reactions catalysed by iodine or β-CD took 3–5 hours to achieve 76–87% yields. An interesting advantage of Fe3O4@CS NPs was their magnetic properties that allowed catalyst separation by utilising a magnetic field.84,85
Similarly, the reaction for β-CD-catalyzed synthesis of α-hydroxyphosphonates from aromatic/heteroaromatic aldehydes and triethyl phosphite resulted in high yields (80–93%) but took a considerably long time, 8–14 hours (see the procedure in Scheme 28). However, no reaction was observed with acetaldehyde, phenylacetaldehyde and acetophenone. The catalytic activity of β-CD was ascribed to its ability to form an inclusion complex that dissolves the aldehyde more effectively and also interacts with aldehyde, thus enhancing the reaction.86
 | ||
| Scheme 28 β-Cyclodextrin-catalyzed synthesis of α-hydroxyphosphonates. | ||
Synthesis of complex (hetero)cyclic compounds through the formation of phosphonates has also been studied in aqueous media, as has been presented above in the syntheses of 2-amino-4H-chromen-4-yl phosphonates. Several cyclic compounds have been prepared and/or modified using phosphites, like (5H-furo[3,2-g]chromene-5-yl)phosphonates,87 coumarins88 and furanocoumarins89 derivatives. Although these reactions are specific for a certain type of scaffold, they nicely demonstrate the usability of water as a solvent in the synthesis of complex phosphonate-containing structures. In general, these reactions performed well under mild conditions with good to excellent yields: 68–85% at 70 °C for 5 hours for coumarin derivatization, 73–93% in 1 hour at ambient temperature for quinazolinyl-substituted furanocoumarins and 80–90% for 5 hours at ambient temperature for (5H-furo[3,2-g]chromene-5-yl)phosphonates. While the first reaction was catalyst-free, the last two were catalyzed by a 2D ZnO/Fe3O4 nanocomposite and piperidine, respectively.
In addition to direct formation of P–C bonds, coupling of phosphonate-containing building blocks with e.g. heterocyclic compounds is an important methodology for preparing new phosphonates. For example, S-alkylation of 5-aryl-1H-1,2,4-triazole-3-thiones with diethyl (3-bromopropyl)phosphonate was catalyzed by nano-Fe2O3. Interestingly, the reaction performed best in water (92%) for 1 hour of reaction at 80 °C, while in other solvents the yield varied from 57% (toluene) to 78% (PEG-400). The catalyst was easily retrieved using a magnet and reused without a significant loss in catalytic activity.90
A highly interesting type of reaction carried out in water is the cross-coupling reaction. Typically, these reactions need careful protocols as they utilize transition metals and their redox reactions. A recent example was of P–C bond formation using multimetallic Pd/Ni- and dual-ligand Pd-catalyzed C–P cross-coupling reactions in aqueous micelles under mild conditions (see Scheme 29). By utilizing micellar catalysis in water, a wide range of (hetero)aryl (thio)phosphonates, phosphinates, and phosphine oxides were prepared with good yields.91
 | ||
| Scheme 29 Synthesis of phosphonates in water medium to “examples”. | ||
2.5. Electrochemical synthesis
In recent years, electrochemistry has gained interest due to its sustainable nature. Many electrochemical reactions are very selective, and they can be carried out in very sophisticated ways. As an example, phosphonation of C3 and C4 of 2-phenyl quinazolines and quinoxalines was studied as part of a wide study of methods to activate C–H bonds in quinazoline moieties. The system was equipped with a graphite anode and a nickel foam cathode, while a constant current of 10 mA was used to carry out the reaction. Yields after 3 hours were in the range between 83% and 93%. The phosphite was used in excess (2 eq.) and 0.1 molar nBu4PF6 was applied as electrolyte in a mixture of acetonitrile and trifluoroethane (1:2) under ambient temperature in an undivided cell (see in Scheme 30).92
 | ||
| Scheme 30 Electrochemical synthesis of phosphonates. | ||
Similarly to quinazolines and quinoxalines, phosphonation of N-aryltetrahydroisoquinolines has been demonstrated by electrochemistry. The study showed a significant impact of the substitution of the aryl: mild electron-withdrawing groups (EWGs) (e.g., 4-F: 71%, 3-F: 91%. 2-F: 72%, 4-Cl:90%) provided higher yields than strong EWGs (e.g., 4-OMe: 43%, 3-OMe: 33%, 2-OMe: 26%) or strong electron-donating groups (EDGs) (e.g., 4-CN). As an anode, a graphite rod was used while a platinum plate was used as a cathode, and 0.1 M of nBu4NBr was applied in dichloromethane as electrolyte.93
Also, both C(sp2)–H and C(sp3)–H phosphonylations with other heterocycles, like 2-phenylimidazo[1,2-a]pyridine, 2-thiophenylimidazo-[1,2-a]pyridines, xanthene and N-methyl-9,10-dihydroacridine, N,N-dimethylaniline, and N-phenyl tetrahydroisoquinoline are reactive in electrochemical reactions, allowing access to the corresponding phosphonates. Similarly, as above, the yields were highly dependent on the electronic effects of the substituents in the heteroaromatic ring.94
A method for electrochemical domino-reaction of P–H iodination–phosphonation–oxygenation utilizes alcohols and phosphoramides to prepare carbamoylphosphonates with yields ranging from 75% to 92% (see Scheme 31). Reactions were performed at a constant current of 0.5 mA in the corresponding alcohol using platinum as both anode and cathode. Combination of 0.03 M (1.5 eq.) of LiClO4 and 0.002 M (10 mol%) of nBu4NI was applied as electrolyte/catalyst. The role played by n-Bu4NI was suggested to be rather that of a catalyst which provides iodine to catalyze the reaction.95
 | ||
| Scheme 31 Electrochemical synthesis of carbamoylphosphonates. | ||
Electrochemistry was also used to modify phosphonates. As an example, synthesis of α-ketiminophosphonates by oxidizing the corresponding α-amino phosphonates was carried out with sixteen diverse phosphonates with yields from 52 to 98%.96
In addition to carbamoyl phosphonate synthesis, another reaction proposed to occur via halide-catalyzed electrochemical addition to phosphorus is selenation of phosphites by electrochemical reaction of phosphonate and selenol. The reaction conditions were screened using benzeneselenol and di-n-butylphosphite. Optimum yields were reached using platinum anode and cathode, n-Bu4NBr as electrolyte and acetonitrile as solvent. The role played by n-Bu4NBr was proposed to be as a bromide donor to phosphonate, thus activating it towards selenol electrophilic attack onto the phosphorus. The optimized conditions were used to prepare twenty-two compounds with 57–96% yields.97
2.6. Light-mediated synthesis
In recent years, light-mediated synthetic chemistry has gained growing interest. Light-mediated reactions can be driven by different types of light, but the most common types are blue or white light produced by light-emitting diodes (LEDs) or white light produced by compact fluorescent lamps (CFLs) (a.k.a. energy-saving lamps). Light has even been used to prepare zinc oxide nanoparticles for the synthesis of phosphonates by utilizing focused sunlight.98Blue light LEDs are used for example in the synthesis of Arylazo Phosphine Oxides from anilines and phosphine oxides and phosphite derivatives in the presence of a NO source, like isoamylnitrite. As a solvent, dichloroethane (DCE) gave the highest yield. The reaction did not proceed in the solvents water, acetonitrile and methanol. The generality of the reaction was demonstrated with twenty-eight examples with yields in the 33–90% range (see example in Scheme 32).99 Another study achieved the blue light-mediated synthesis of phosphonates from α-diazoesters, cyclic ethers and diaryl phosphite oxides. This reaction was conducted under ambient aerobic conditions and yielded a range of phosphonates and phosphinates with good to excellent yields, without the requirement of photosensitizers.100
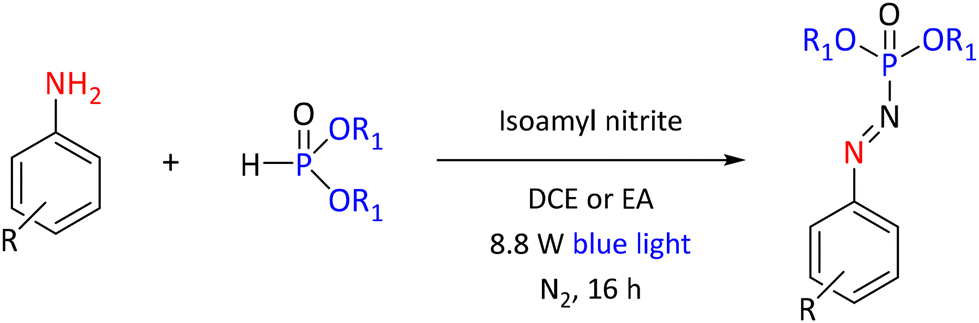 | ||
| Scheme 32 Blue light-mediated synthesis of phosphonate derivatives. | ||
In contrast to the abovementioned reactions, conversion of aryl iodides to the corresponding aryl phosphonates under blue light required 10 mol% of photoredox catalyst, 10H-phenothiazine (PTZ) and 2 eq. of (1,8-diazabicyclo(5.4.0)undec-7-ene) (DBU) as a base (see in Scheme 33).
 | ||
| Scheme 33 PTZ-catalyzed and blue light-mediated synthesis of phosphonates. | ||
The highest yield of a model reaction (triethyl phosphite with 4-methoxyiodobenzene) was achieved in acetonitrile (92%). An acetonitrile/water (1:3) mixture provided nearly the same result, 90%, and was preferred for its higher eco-friendliness. The reaction also worked with bromides and aryl trimethylammonium salts. However, changing from halides to trimethylammonium salts required the use of purple LED (390 nm) instead of blue (427 nm). Also, the radical mechanism of the reaction was supported by radical-trapping experiments. Under the optimized conditions, forty examples of a diverse range of starting materials were explored, allowing the conversion in the presence of ketone, amide, ester, amine, and alcohol functionalities.101
In addition to light with exact wavelength (or color), white light with a wide spectrum of wavelengths has been used. A white light-emitting LED lamp was used in an aerobic phosphorylation of 5-membered heteroarenes using dichromatic photoredox catalysis within a gel-based nanoreactor. A model reaction of 5-chloro-2-thiophenecarbonitrile and triethyl phosphite showed that the highest yield was reached with 5 eq. of phosphite, 1.2 eq. of di-isopropylethylamine, 10 mol% of 9,10-dicyanoanthracene as photocatalyst and 10 mg pf (N,N′-bis(octadecyl)-L-boc-glutamic diamide) as a gelator under 4 hours irradiation with cold white LED light (410–700 nm). The optimized reaction conditions were used to synthesize fifteen different thiophene derivatives and seventeen other five-membered O, N, S and Se-heterocycles and five other (hetero)aromatic ring systems (e.g. benzene, indole and quinoline).102
The efficiency of white light in comparison with blue light was demonstrated in the synthesis of alkynyl phosphonates from various alkynes in the presence of TiO2/Cu2O as photocatalyst. The white light produced by compact fluorescent lamp (CFL) was shown to provide similar yields as the blue light LED. During the optimization, organic solvents performed well (dimethylsulfoxide (80%), methanol (82%), ethyl acetate (83%), tetrahydrofuran (84%), and acetonitrile (87%)), but water hindered the reaction in mixtures of acetonitrile/water (40%) and provided only 20% as pure solvent. Pure TiO2 did not produce any product and Cu2O yielded only 75% in acetonitrile. Cu2O played the role of a visible-light absorber, while the conduction band of TiO2 facilitated electron acceptance, thus initiating the reaction. The optimized reaction conditions (dimethylformamide, 20 mg of catalyst/mmol and equimolar amounts of starting materials) were used to produce twelve examples of alkynyl phosphonates with 78–96% yields for 3–18 hours reaction time at ambient temperature (see reaction in Scheme 34).103
 | ||
| Scheme 34 Visible light-mediated synthesis of alkynylphosphonates. | ||
CFLs were also used in Ru-catalyzed cascade reactions involving photocatalyzed oxidation, [3 + 2] cycloaddition, and oxidative aromatization, producing pyrrolo[2,1-a]isoquinoline-substituted phosphonates. Reaction conditions were optimized using a model reaction of ethyl [(3,4-dihydroisoquinolin-2(1H)-yl)methyl]phosphonate and N-methylmaleimide. The optimized reaction conditions (1 eq. of phosphonate and 1.3 eq. of imide, dichloromethane as solvent with 5 mol% of Ru(bpy)3Cl2·6H2O as catalyst and sodium acetate as base) resulted in 71% yield within 15 hours of reaction at ambient temperature and using 24W CFL under an oxygen atmosphere. These conditions were used to demonstrate the generality of the reaction by synthesizing sixteen different products with 31–85% yields.104
2.7. Use of green solvents other than water
In addition to water, some other solvents are also considered as green, like 2-methyltetrahydrofuran, ethanol, and ethyl acetate. These solvents have also been used in phosphonate synthesis. As an example, Kabachnik–Fields reactions in ethanol have been used to prepare a variety of α-aminophosphonates. A reaction involving hydrazine derivatives performed well in ethanol when synthesizing α-aminophosphonate derivatives linked with indole-2,3-dione groups, specifically the diethyl(substituted phenyl/heteroaryl) (2-(2-oxoindolin-3-ylidene)hydrazinyl)methylphosphonates derivatives. These prepared compounds were reported to have anticancer properties. The reaction was conducted at ambient temperature in the presence of ceric ammonium nitrate (CAN) as catalyst (see reaction in Scheme 35).105 | ||
| Scheme 35 CAN-catalyzed synthesis of α-aminophosphonates with anticancer properties. | ||
Methyl sulphonic acid can catalyze the Kabachink–Fields reaction of 2,4-difluorobenzyl amine and substituted aromatic aldehydes with diethylphosphite in ethanol.106 The study demonstrated that the use of other solvents, e.g. acetonitrile, toluene or methanol, resulted in lower yields (60–75%) compared with ethanol (75–90%). Similar screening of solvents has also been reported for the abovementioned Fe3O4@CS-SO3H NPs83 catalyzed synthesis of 2-amino-4H-chromen-4-yl phosphonates and also confirmed ethanol as a potential solvent for this type of reaction. Specifically, the yield in ethanol was 80% after 45 minutes compared with that in water (93% after 30 minutes) while other organic solvents needed longer reaction times and resulted in lower yields, e.g., methanol (37% after 2 hours), toluene (40% after 2 hours), dichloromethane (48% after 2 hours), and acetonitrile (55% after 1.5 hours). In light of this, the selection of ethanol as solvent in dibutylamine-catalyzed synthesis of 2-amino-3-cyano-4H-chromen-4-ylphosphonates is reasonable (see reaction in Scheme 36). In the study, a range of other catalysts were also screened, including inorganic salts (CuCl2 (15%), ZrOCl2 (20%), ZnCl2(30%), and AlCl3 (35%)), organic amines (e.g. pyrrolidine (75%), morpholine (78%), piperidine (85%)) as well as some inactive compounds, such as Montmorillonite K-10, KHSO4, and Oxone which did not result in any product. The optimized conditions were used to prepare eleven 2-amino-4H-chromen-4-yl phosphonates with 85–95% yields.107
 | ||
| Scheme 36 Synthesis of 2-amino-3-cyano-4H-chromen-4-ylphosphonates in EtOH. | ||
In the case of graphene oxide as a catalyst, the polar solvents were more effective in dispersing the catalyst (see Table 4). The reaction showed slightly higher yields in methanol than in ethanol, and thus the former was selected for optimized conditions to prepare a series of α-aminophosphonates from trimethylphosphite, aromatic aldehydes and aliphatic amines or anilines.108 Graphene oxide was also shown to be a reusable catalyst, as the yield lowered slightly from 88% to 79% during five cycles. In another study, a methanol–water mixture was used in AgNO3-catalyzed hydration of alkynylphosphonates into β-ketophosphonates. Methanol was proposed to participate in the reaction and thus assist the reaction. The yields with other solvents as mixtures with water (10/1) were significantly lower than with methanol (97%), e.g. acetonitrile (9%), dimethylformamide (11%), dichloroethane (12%), tetrahydrofuran (23%), isopropanol (27%), 1,4-dioxane (34%), 1,3 propanediol (42%), ethanol (48%), and ethylene glycol (56%). The synergistic role played by Ag+ and NO3− ions was demonstrated, as no conversion was detected with HNO3, Fe(NO3)3 or Cu(NO3)2 and lower conversions were recorded with other Ag(I) salts, e.g., AgCl (83%), AgSO4 (83%), AgSbF6 (85%), AgOAc (86%), AgBF4 (87%), and AgOTf (92%). The optimized conditions (120 °C, 24 h, 10 mol% of catalyst) were used to provide 70–93% yields of twenty-two diverse (hetero)aromatic and aliphatic β-ketophosphonates.109 Another method for the synthesis of β-ketophosphonates demonstrated the synthesis of β-ketophosphonates from alkynes and dialkyl phosphites utilizing both AgNO3 and CuSO4·5H2O, as catalysts. The highest yield was reached using 10 mol% of CuSO4·5H2O and 5 mol% of AgNO3 as catalysts and 4 eq. of K2S2O8 as oxidant. The reaction was performed in a water/dichloromethane (1:1) solvent mixture. To show the generality of the reaction, a series of twenty-seven β-ketophosphonates was synthesized, with yields varying from 56% to 93%.110
| Entry | Phosphorus source | Catalyst (load) | Solvent | Temperature/method | Time (h) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | P(OMe)3 | Graphene oxide (3 mg mmol−1) | Methanol | Ambient | 3 | 88 | 108 |
| 2 | P(OMe)3 | Graphene oxide (3 mg mmol−1) | Water | Ambient | 3 | 25 | 108 |
| 3 | P(OMe)3 | Graphene oxide (3 mg mmol−1) | Dichloromethane | Ambient | 3 | 81 | 108 |
| 4 | P(OMe)3 | Graphene oxide (3 mg mmol−1) | Ethanol | Ambient | 3 | 85 | 108 |
| 5 | P(OMe)3 | Graphene oxide (3 mg mmol−1) | Toluene | Ambient | 3 | Trace | 108 |
| 6 | P(OEt)3 | T3P® (1eq.) | Ethyl acetate | Ambient | 5 min | 92 | 112 |
| 7 | P(OEt)3 | — | Ethyl acetate | Ambient | 24 | 50 | 112 |
| 8 | P(OEt)3 | — | — | Ambient | 48 | 80 | 112 |
Ethanol was also demonstrated to perform well in the synthesis of six α-aminophosphates using vortex agitation (50–95% in 5–25 minutes). Additionally, these compounds were monohydrolyzed in an ethanol–water mixture via an alkaline hydrolysis reaction (see Scheme 37), with 49–65% yields at 140 °C under MW for 20 minutes, and they were reported to have antifungal properties.111
 | ||
| Scheme 37 Vortex-mediated synthesis of α-aminophosphonates with antifungal properties. | ||
In addition to ethanol and methanol, non-alcohol solvents such as ethyl acetate, polyethylene glycol and 2-methyltetrahydrofuran have also been used in the synthesis of phosphonates. As an example, Kabachnik–Fields reaction has been carried out in ethyl acetate using 1 eq. of propylphosphonic anhydride (T3P®) as the condensing agent. Reactions of a diverse set of aromatic aldehydes and anilines resulted in 80–96% yields for 5–10 minutes of reaction time (for an example, see Table 4).112
Polyethyleneglycol (PEG) has been demonstrated to perform efficiently in cross-coupling reactions, e.g. KI/K2CO3-catalyzed and Pd(PPh3)4-catalyzed Michaelis–Becker type reactions (see example in Scheme 38). The first-mentioned reaction was carried out at ambient temperature. In the study, other solvents resulted in significantly lower yields compared with PEG-400 (60%): acetonitrile (23%), tetrahydrofuran (28%), and dimethylformamide (45%), and only 20% yield was reached in solvent-free conditions. No improved yields were obtained by using other bases, e.g. Li2CO3 (35%), Na2CO3 (48%) and KOH (60%), while the addition of 0.3 equivalents of KI with the base improved the yields significantly: Li2CO3 (52%), Na2CO3 (61%), Cs2CO3 (63%), and K2CO3 (97%). The solvent was proposed to take part in the reaction mechanism by interacting with potassium iodide and releasing the iodide ion to substitute the less reactive halide from the starting benzyl bromide or bromide. Reactions yielded from 82% to 96%.113
 | ||
| Scheme 38 PEG-400-mediated synthesis of phosphonates. | ||
In the latter reaction, triethylamine was used as a base and the screening of a series of solvents revealed that PEG-600 resulted in the highest yields (95%) at 130 °C for 15 hours compared with other solvents in reflux temperatures, e.g., 1,4-dioxane (25%/20 h), tetrahydrofuran (35%/20 h), ethanol (39%/23 h), toluene (45%/25 h), acetonitrile (56%/27 h), dichloromethane (68%/26 h), and dimethylformamide (74%/19 h). Similarly, other catalysts (e.g., CuI (15%/45h), Pd(OAc)2 (60%/17 h), Pd(PPh3)2Cl2 (62%/40 h)) and bases resulted in lower yields at 130 °C in PEG-600: Na2CO3 (60%/22 h), K2CO3 (15%/22 h), NaF (trace/21 h), NaOAc (23′5%/22 h), KOAc (15%/22 h), KOH (10%/23 h), NaOH (13%/23 h), NaHCO3 (16%/21 h), and 4-(dimethylamino)pyridine (5%/23 h). The optimized conditions were used to prepare twenty-six products with yields from 60% to 95%.114
The biomass-derived solvent 2-methyltetrahydrofuran has received growing attention, and a recent example is its use as solvent in an iodine-catalyzed Kabachnik–Fields reaction. The yield was higher in 2-methyltetrafydrofuran after 8 hours (91%) than in tetrahydrofuran after 24 hours (90%) when using 1.5 eq. of diethyl phosphite. The results with equimolar amounts of starting material were slightly different after 24 hours of reaction time: dichloromethane (31%), acetonitrile (60%), ethanol (71%), 2-methyltetrahydrofuran (74%) and tetrahydrofuran (84%). A series of twenty-five products was synthesized (see reaction in Scheme 39).115
 | ||
| Scheme 39 Iodine-catalyzed synthesis of α-aminophosphonates. | ||
2.8. Other methods
As an example of a MOF-catalyzed Kabachnik–Fields reaction, a nickel-based MOF (Ni-MOF) catalyzed α-aminophosphonate synthesis (see Scheme 40). The catalyst was shown to provide 90–98% yields for 30–60 minutes of reaction time under ambient temperature and without solvent.116
 | ||
| Scheme 40 Ni-MOF-catalyzed synthesis of α-aminophosphonates. | ||
Ionic liquids are efficient catalysts in several organic reactions as well as in diverse separation processes. They are often considered as green solvents or catalysts, as they can be reused several times. Various types of ionic liquid have been used in the preparation of phosphonates, e.g. organic and inorganic as well as acidic and basic ionic liquids. Based on the amount of ionic liquid used in synthetic reactions, they are considered as catalysts rather than solvents.
An example of an acidic ionic liquid is [Et3NH][HSO4], which was studied for its ability to promote the reaction of aromatic aldehydes, anilines and trimethyl phosphite. The reaction mechanism was proposed to be similar to other catalyzed Kabachnik–Fields reactions: the catalyst activated the carbonyl C, thus promoting formation of the imine intermediate, and later supported the elimination of methanol from the phosphonium intermediate. To compare the solvent-free method, yields were recorded in six solvents (see entries 1–10 in Table 5).117
| Entry | Phosphorus source | Catalyst (load) | Solvent | Time (min) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | P(OMe)3 | — | — | 60 | Trace | 117 |
| 2 | P(OMe)3 | [Et3NH][HSO4] (5 mol%) | 60 | 70 | 117 | |
| 3 | P(OMe)3 | [Et3NH][HSO4] (20 mol%) | — | 10 | 95 | 117 |
| 4 | P(OMe)3 | — | — | 60 | Trace | 117 |
| 5 | P(OMe)3 | [Et3NH][HSO4] (20 mol%) | Dichloromethane | 20 | 42 | 117 |
| 6 | P(OMe)3 | [Et3NH][HSO4] (20 mol%) | Tetrahydrofuran | 20 | 45 | 117 |
| 7 | P(OMe)3 | [Et3NH][HSO4] (20 mol%) | 1,4-Dioxane | 20 | 48 | 117 |
| 8 | P(OMe)3 | [Et3NH][HSO4] (20 mol%) | Toluene | 20 | 54 | 117 |
| 9 | P(OMe)3 | [Et3NH][HSO4] (20 mol%) | Acetonitrile | 20 | 56 | 117 |
| 10 | P(OMe)3 | [Et3NH][HSO4] (20 mol%) | Ethanol | 20 | 60 | 117 |
| 11 | H(O)P(OMe)2 | 2-Hydroxyethylammonium formate | — | 120 | 25 | 118 |
| 12 | H(O)P(OMe)2 | [SFHEA][NO3] (20) | — | 120 | 82 | 118 |
| 13 | H(O)P(OMe)2 | [SFHEA][CF3COO] (20) | — | 120 | 83 | 118 |
| 14 | H(O)P(OMe)2 | [SFHEA][CH3SO3] (10) | — | 120 | 86 | 118 |
| 15 | H(O)P(OMe)2 | [SFHEA][HSO4] (10) | Water | 120 | 84 | 118 |
| 16 | H(O)P(OMe)2 | [SFHEA][HSO4] (10) | Acetonitrile | 120 | 85 | 118 |
| 17 | H(O)P(OMe)2 | [SFHEA][HSO4] (10) | Ethanol | 120 | 91 | 118 |
| 18 | H(O)P(OMe)2 | [SFHEA][HSO4] (10) | Chloroform | 120 | 89 | 118 |
Another acidic ionic liquid, [SFHEA][HSO4] [SFHEA = cation of (2-(sulfoxy)ethyl)sulfamic acid] was reported as a potent and reusable ionic liquid. With 10 mol% it gave good yields with diverse aromatic and aliphatic aldehydes and anilines. The efficiency was marginally reduced from 91% to 87% after five cycles of use (see Scheme 41 and entries 11–18 in Table 5).118
 | ||
| Scheme 41 Synthesis of α-amino phosphonates using the ionic liquid [SFHEA][HSO4]. | ||
Also, diverse basic ionic liquids have been utilized in phosphonate synthesis. Two examples, choline hydroxide and a series of nitrogene heterocycles like 1-[3-(dimethylamino)propyl]-1,4-diazabicyclo[2.2.2]octan-1-ium and 3-(2-ethoxy-2-oxoethyl)-1-methyl-1H-imidazol-3-ium, 3-(2-amino-2-oxoethyl)-1-methyl-1H-imidazol-3-ium and 3-(2-hydroxyethyl)-1-methyl-1H-imidazol-3-ium hydroxides, chlorides, phosphates and prolinates were studied as ionic liquids catalyzing phosphonation.119,120
The choline hydroxide augments the reaction between aromatic aldehydes and diethylphosphite by lowering the reaction time and increasing the yields. The yield of a model reaction with benzaldehyde was 98% for 5 minutes with 10 mol% of catalyst. The generality of this reaction was demonstrated by the synthesis of twenty-two substituted α-hydroxyphosphonates with yields of 90–98% (see reaction in Scheme 42).
 | ||
| Scheme 42 Synthesis of α-hydroxyphosphonates using choline hydroxide. | ||
Several other basic catalysts were found to be less active, e.g. carbonates (CaCO3 (55%), K2CO3 (50%), CsCO3 (75%), hydroxides (LiOH·H2O (59%), KOH (60%))), organic bases (dimethylaminopyridine (81%), piperazine (82%), tetramethylethylenediamine (83%), dimethylamine (85%), di-n-hexylamine (86%), di-n-pentylamine (88%), pyrrolidine (88%), 4-ethyl morpholine (89%), di-n-butylamine (90%) and dimethyl ammonium carbonate (84%)). With other ionic liquids (ethylenediammonium diformate and ethylenediammonium diacetate) no reaction was observed. The effect of diverse solvents was also reported, and the results emphasised the solvent-free conditions, as the solvent-free protocol provided 90% yield within 5 minutes for the model reaction, while other solvents gave lower yields varying from benzene and toluene (50% and 55%, respectively) to dichloromethane, methanol and ethanol (85%, 85% and 83%, respectively). In contrast, the dual basic ionic liquids performed best in water/ethanol 1:1 mixture, giving 97% yields. However, the reaction gave no product in toluene and performed well in methanol (80%) and ethanol (97%). However, the importance of ethanol was clear, as the yield in water was only 40% and 55% in ethanol/water 1:2 mixture.
In addition to the use of ionic liquids as catalysts, a study of a nano-SiO2-BF3-catalyzed Michaelis–Arbuzov reaction was reported using ionic liquid 1,3-dibutylimidazolium bromide [BBim]Br as solvent at ambient temperature. The model reaction of 1-bromo-4-methyl-2-nitrobenzene and triethylphosphite was reported to yield 86% of product at 80–90 °C within 30 minutes without catalyst, and the addition of 0.25 g mmol−1 of nano-SiO2-BF3 gave 94% yield within 20 minutes under the same conditions. In this way fifteen different products were synthesized with 76–94% yields.121
In addition to ionic liquids, also DESs have been utilized to prepare pyrazolylphosphonate derivatives. The screening of a diverse set of catalysts and DESs showed that the highest yield (96%) of the model reaction with benzaldehyde, pyrazolone and triethyl phosphite was reached with 2 g mmol−1 of choline chloride:urea-DES, while with other catalysts, like inorganic salts (ZnCl2 (86%), InCl3 (87%), FeCl2 (87%)) lower yields were achieved. Series of peoducts were also achieved through a three-component reaction involving pyrazolone or phenylpyrazolone, various aldehydes, and triethyl or trimethyl phosphite in the presence of an ammonium-based deep eutectic solvent (DES) at 25 °C. The DES was easily prepared from choline chloride and urea (1:2) at 80 °C for 2 hours. This method is environmentally friendly, cost-effective, and allows for the reuse of the DES up to four times without losing its catalytic activity.122
MOFs constructed from phosphorus-containing organic precursors have also been studied. These materials have diverse properties, like the ability to produce highly reactive singlet oxygen (O2) species and acting as proton conductors, as well as catalysing chemical reactions, and thus they can find diverse uses in medicine, materials and battery technology. Preparation of these materials can be achieved for example though mechanochemical synthesis, or in an aqueous solution. Several tetradentate phosphonic acid ligands and bisphosphonates have been used, while copper, nickel, cobalt and yttrium were used as the metal ions.123–125
To widen the scope of α-aminophosphonate synthesis, methods to increase the range of starting materials have been studied. As an example, decarboxylative coupling of amino acids, aldehydes and diethyl phosphite opens up opportunities for α-amino acids as green starting materials. The reaction was proposed to proceed through a cyclic intermediate formed from the aldehyde and amino acid. The reactions using proline and diverse aldehydes produced yields from 43% to 75% within 1–5 hours refluxing in toluene.126
A different approach for forming C–P bonds with C(sp3), C(sp2) and C(sp) was implemented via desulfonative coupling with toluenesulfones and benzenesulfones. The method was used to prepare a series of alkynylphosphonates, alkenylphosphonates and alkylphosphonates. The optimization of reaction conditions resulted in conditions of acetonitrile as solvent and CsCO3 as base. The reaction was carried out using 1 equivalent of phosphite and 1.5 equivalents of sulfone and base. Using the optimized conditions, thirty alkynyl, alkenyl, and allyl phosphine oxides or phosphonates were prepared with 42–98% yields.127
Terminal alkynyl phosphonates (same kind of structures as presented in Scheme 34) were also synthesized using a Cu–MnO catalyst. The optimal conditions for these reactions were screened, and the best yield of a model reaction was reached by using 5 mol% of catalyst, 100 °C and dimetylsulfoxide as solvent. Toluene and dimethylformamide produced low yields (32% and 38%, respectively) while tetrahydrofuran and acetonitrile did not give any product. Using the optimized conditions, a diverse set of thirty-nine phosphonates was synthesized with yields ranging from 38% with hexynyl phosphonates to over 90% yields of several phenyl-substituted products. The catalyst was shown to be recyclable, and the reaction was also scaled to gram-scale.128
Triethylamine was used in the catalyzed Pudovic reaction for the preparation of bis-α-hydroxy phosphonates from aromatic and heteroaromatic dialdehydes. Ten reactions yielded the desired products, including biomass-based ones, with yields ranging from 52% to 95%, under solvent-free conditions or in a minimal amount of tetrahydrofuran at ambient temperature.129
BiCl3 serves as a cost-effective, environmentally friendly, and low-toxicity catalyst. The synthesis of substituted arylphosphonates/phosphinates in high yields was successfully demonstrated through the BiCl3-catalyzed Michaelis–Arbuzov reaction. The reaction used 5-iodovanillin/3,5-difluorobenzylbromide with various phosphites and dimethyl phenylphosphonate in the presence of the Lewis acid catalyst BiCl3, conducted under N2 atmosphere at 40 °C. The synthesized compounds were then evaluated for their antibacterial and antifungal activity.130
A reaction resembling the one reported by Shnigirev et al.69 was used to prepare α-aminophosphonates from cinnamaldehyde in catalyst-free Kabachnik–Fields reaction. The reactions provided 74–94% yields within 3–6 hours under reflux conditions using 1,4-dioxane as solvent.131
Phosphorus-containing ligands are important in catalysis, and chiral ligands especially are essential for stereoselective catalysts. Hence, efficient methods to prepare such ligands are needed. A green method to prepare axially chiral biaryl phosphonates using p-quinone phosphonates and 2-naphthols through chiral phosphoric acid catalysts was developed. The reaction yielded a series of chiral biaryl monophosphonates with excellent yields and enantioselectivities, reaching up to 99% yield and 95% ee in dichloroethane. Notably, this reaction could be conducted on a gram scale with a low catalyst loading of 0.5 mol%.132
3. Phosphonates in water-related fields – green aspects
In order to facilitate the reader in following this section smoothly, we refer to Fig. 3 and 4, where the schematic structures of all mentioned phosphonates and their abbreviations are presented. | ||
| Fig. 3 Schematic structures of various polyphosphonic acids. All of them are commercially available in bulk quantities. Color codes: arbon fragments, black; phosphonate groups, blue; protonated amine groups, green; carboxylate group, red. Abbreviations: HPAA = hydroxy-phosphonoacetic acid, EDTMP = ethylenediamine-tetrakis(methylenephosphonic acid), HEDP = hydroxyethylidene-1,1-diphosphonic acid (etidronic acid), NTMP = nitrilo-tris(methylenephosphonic acid), HDTMP = hexamethylene-tetrakis(methylenephosphonic acid), DTPMP = diethylenetriamine-pentakis(methylenephosphonic acid), PBTC = 2-phosphonobutane-1,2,4,-tricarboxylic acid, BHMTPAMP (bis-hexamethylenetriamine-pentakis(methylenephosphonic acid)). | ||
 | ||
| Fig. 4 Schematic structures of two families of phosphonic acids: diphosphonates (upper) and tetraphosphonates (lower). Color codes: carbon fragments, black; phosphonate groups, blue; protonated amine groups, green. | ||
3.1. (Bio)degradability of phosphonate additives used in water treatment applications
Naturally occurring phosphonic acids have unique features related to stability and mimicry. The C–P bond present in all these compounds is generally resistant to hydrolytic cleavage. On the other hand, the phosphoryl group is a versatile mimic of transition states, intermediates, and primary metabolites, a fact that explains why several organisms have extensively explored the use of phosphonates as bioactive secondary metabolites. Several phosphonates, e.g., fosfomycin (an antibiotic primarily used to treat lower urinary tract infections) and bialaphos (a natural herbicide produced by the bacteria Streptomyces hygroscopicus and Streptomyces viridochromogenes), have a prominent position in human health and agriculture, respectively. The enzymatic reactions that generate these molecules are a complex combination of chemical processes, adopted from primary metabolism. The phosphonate group is also a source of inorganic phosphate (via cleavage of the C–P bond) for microorganisms living in phosphate-poor environments. An excellent review has been published on the occurrence and function of phosphonate natural products and on the mechanisms by which enzymes synthesize and catabolize these molecules.133There is general agreement in the scientific community that phosphonates are not considered to be inherently biodegradable. Little or no biodegradation of phosphonates is noted in natural systems. Interestingly, microorganisms capable of degrading phosphonates have been isolated.134,135 Based on test data and the relevant literature on phosphonate degradation, these compounds are considered “not readily biodegradable” and are likely in the “red” category. However, there are degradation pathways that allow phosphonate molecules to be degraded to smaller fragments. This section contains a critical view of the relevant literature on phosphonate degradation.
Commonly, ready biodegradability of organic substances is assessed by OECD 301 standard tests. However, due to the chemical imbalance of carbon to phosphorus, synthetic phosphonates do not promote microbial growth and, thus, limit its biodegradation. Therefore, standard OECD test methods are not always reliable for predicting the real biodegradability of phosphonates. To overcome the drawbacks of the OECD 301 test a study was conducted via a standardized batch system suitable for synthetic phosphonates such as NTMP, EDTMP, DTPMP and others. The novel standard batch test is applicable with pure strains, activated sludge from different wastewater treatment plants (i.e., municipal, and industrial), and with tap water as inoculum.136
Phosphonates are utilized in a plethora of household and industrial applications. Some examples include boiler and cooling water systems, oil and gas production, the textile industry, detergents, shampoos, and even in the pharmaceutical and medicinal fields as drugs for calcium disorders (the most notable being osteoporosis). Phosphonates do not biodegrade during conventional wastewater treatment137 but are removed from the waste stream by adsorption onto biosolids (sludge)138 and onto ferric flocs used in tertiary wastewater treatment.139 Phosphonates are resistant to photodegradation in the absence of metal ions to coordinate to, but are readily photodegradable when they complex to Fe(III).140 For example, solutions that are able to generate radicals (e.g., hydrogen peroxide in the presence of Mn(II), or Fe(II), or Cu(II) and riboflavin, irradiated by 450 nm light) can also degrade phosphonates.141 It was reported that the polyphosphonates EDTMP and DTPMP suffered a 1% loss per day in non-illuminated, H2O2-free and metal-free solutions. However, solutions containing Mg(II), Ca(II), and Fe(II) increased EDTMP and DTPMP degradation rates mildly. Non-illuminated solutions of nitrilo-tris(methylenephosphonate) (NTMP) in the presence of metal ions such as Mg(II), Ca(II), Mn(II), Fe(III), Co(II), Cu(II), and Zn(II) demonstrated significant loss of the phosphonate in a variety of water qualities (river water, groundwater, reservoir water, tap water).142 In fact, in certain cases quantitative loss was observed within 48 hours, while there was a variety of degradation products, namely, hydroxymethylphosphonic acid (HMP), aminomethylphosphonic acid (AMP), imino(dimethylene)phosphonic acid (IDMP), and carbon dioxide, as detected by HPLC (Fig. 5).
 | ||
| Fig. 5 Schematic structures of HMP, AMP and IDMP, degradation products of EDTMP and DTPMP. | ||
Mn(II) as a “catalyst” for phosphonate degradation is a notable case that warrants further discussion. Nowack and Stone reported that both Mn(II) and molecular oxygen are required for effective NTMP degradation.143 Further studies were also carried out in a wide pH range for other phosphonates (EDTMP, DTPMP, and HEDP). It was found that formation of a Mn(II)–phosphonate intermediate complex is important for promotion of phosphonate degradation. The authors concluded that degradation arises from Mn(II)-catalyzed autoxidation. The proposed mechanism of Mn(II)-catalyzed NTMP degradation is shown in Fig. 6.
 | ||
| Fig. 6 Proposed mechanism for the degradation of NTMP in the presence of Mn(II) and molecular oxygen. Redrawn from reference 143. | ||
A carbon-centered radical has been proposed for the reaction of N-(phosphonomethyl)iminodiacetic acid (PMIDA, Fig. 7) oxidation catalyzed by V(IV,V)144 and Co(II,III).145 Results reported on the ability of MnOOH suspensions to degrade phosphonates showed that both C–N and C–P bonds of aminopolyphosphonates are susceptible to rapid oxidative cleavage in the presence of manganese. In oxygen-free MnOOH suspensions, C–N bond cleavage takes precedence over C–P bond cleavage, whereas in the presence of oxygen C–N and C–P bond cleavage occur at comparable rates.146
 | ||
| Fig. 7 Schematic structure of N-(phosphonomethyl)iminodiacetic acid (PMIDA). | ||
NTMP reacts rapidly with oxidizing biocides at room temperature and reaches a plateau after 1000 min. Even after 50 h of reaction time, only 20% of the NTMP has decomposed. Orthophosphate was identified as the inorganic by-product of the decomposition. NTMP degrades much more rapidly at 43 °C than at 25 °C due to faster kinetics of decomposition.147,148 PBTC, an important industrial phosphonate that is mainly used as a scale inhibitor, showed great resistance to oxidizing biocides.149
Phosphonates are also used in the oilfield sector as chemical additives for scale inhibition.150 The Norwegian Institute for Water Research (NIVA) compiled a large amount of available data on the biodegradation of selected offshore chemicals. This report compiled data for two phosphonates, namely, HEDP and DTPMP, that point to the conclusion that both are not readily biodegradable. The polyphosphonate DTPMP was shown to be biodegraded by cell-free extracts from cyanobacterial cells of the strain CCALA 007 of Anabaena variabilis grown in the absence of any phosphonate.151 Phosphonates adsorb very strongly onto almost all mineral surfaces, and adsorption of chelating agents by surfaces has been shown to decrease their biodegradability.152 Another study used isolated wild-type Streptomyces sp. strains that are able to utilise both naturally occurring and synthetic organophosphonates. The phosphonates included in the study were aminomethylphosphonate, 1-aminoethylphosphonate, 2-aminoethylphosphonate, 1-amino-1-methylethylphosphonate, 1-amino-3-methylbutylphosphonate, 1-amino-1-phenylmethylphosphonate, amino(p-carboxyphenyl)methylphosphonate, N-phosphonomethylglycine (glyphosate), 2-aminopropylphosphonate, phosphonoacetate, phosphonomycin (fosfomycin), methylphosphonate, ethylphosphonate, propylphosphonate, phenylphosphonate, and t-butylphosphonate (Fig. 8).
 | ||
| Fig. 8 Schematic structures of the phosphonic acids degraded by Streptomycete StC. | ||
Variable degradation results were reported. Streptomycete StC had a surprising ability to degrade glyphosate in a phosphate-independent manner via C–P bond cleavage accompanied by sarcosine formation.153 Various products of the degradation of organophosphonates by Escherichia coli were identified in an effort to unravel possible mechanisms of the biodegradation. The hypothesis that was put forth was consistent with radical-based dephosphorylation. The authors attempted chemical modeling of this process by the reaction of alkylphosphonic acids with Pb(IV) tetraacetate and electrochemical oxidation at a platinum anode.154
The degradation of phosphonates by common microbial pathogens belonging to 22 microbial species grown in phosphate-rich media was investigated employing nuclear magnetic resonance spectroscopy and bioinformatics searches.155 Fifteen bacterial and four fungal species were capable of cleaving phosphonate (C–P) bonds of α-aminomethylphosphonate, phosphonoacetate or phenylphosphonate, indicating that the enzymes responsible for these activities are expressed in the absence of P limitation. In silico analyses indicated that most of the microorganisms with phosphonate degradation activities did not have genes orthologous to those encoding C–P cleaving enzymes of the classical phosphonate catabolism pathways. The results suggested that phosphonate degradation in some bacteria and fungi, including human and animal pathogens, took place via novel pathways.
Advanced oxidation processes (AOPs) have been employed to remove several chelating agents from aqueous systems, including phosphonates. Photocatalysis with UV irradiation alone or coupled with TiO2, ozonation and Fenton's oxidation are frequently applied to mineralize target pollutants.156 Ozonation of solutions containing EDTMP generated glyphosate and NTMP.157 The performance and mechanism of degradation of phosphonates were investigated systematically via a Co(II)-triggered peroxymonosulfate (PMS) activation process. The phosphonates included in the study were HEDP, PBTC, EDTMP, and DTPMP. The primary reactive species involved in the Co(II)/PMS process was proposed to be a Co(II)–PMS complex, while free radicals, 1O2, and Co(III) play as the secondary reactive species that make a minor contribution in the oxidation of targeted phosphonate pollutant.158 A study investigated the methods of metal-catalyzed photolysis (UV/FeII), Fenton method (FeII/H2O2) and photoFenton method (UV/FeII/H2O2) for removal of phosphonates (HEDP, PBTC, NTMP, EDTMP, and DETMP) from a pure water matrix and industrial wastewaters.159 The degradability of phosphonates by UV/Fe(II) in pure water depended dramatically on pH, while it was found to be higher with fewer phosphonate moieties on the phosphonate backbone.
3.2. Non-polymeric phosphonates as scale inhibitors
The important feature of phosphonates for acting as scale inhibitors is ascribed to their high affinity for metal cations, either in a soluble form160 or on a surface of an inorganic salt.161 Scale deposits are formed by precipitation and crystal growth at a surface in contact with water. Precipitation occurs when solubilities are exceeded either in the bulk water or at the surface. The most common scale-forming salts that deposit on heat transfer surfaces are those that exhibit inverse solubility (higher tendency to form as temperature increases).162 Deposit control agents that inhibit precipitation are called threshold inhibitors and operate at dosages far lower than the stoichiometric level required for sequestration or chelation. These additives affect the kinetics of the nucleation and crystal growth of scale-forming salts, and permit supersaturation without scale formation. Mineral scale inhibitors can be “small” molecules or polymers. There are several examples of phosphonates belonging to the former category, but fewer to the second.The literature on the use of phosphonates as scale inhibitors is vast. The reader is referred to a number of reviews, which cover different aspects of mineral scale inhibition and crystal growth modification by phosphonate additives.163–166 This section will include examples of “greener” phosphonates and their application as scale inhibitors on some representative mineral scales.
The effect of phosphonate additives methylphosphonate, HEDP, NTMP, and DTPMP on calcite precipitation kinetics was studied using a Hastelloy autoclave fitted with sapphire windows for in situ light-scattering measurements.168 As the temperature increased from 25 °C to 200 °C the induction time for calcite nucleation following CO2 degassing decreased from more than an hour to a few minutes. The ability of phosphonate inhibitors to delay nucleation also decreased with increasing temperature. DTPMP was able to disperse the calcite precipitate, when used in concentrations sufficient to precipitate “calcium phosphonate”.
The phosphonates 2-(bis(phosphonomethyl)amino)alkane-1-sulfonic acid (alkane = methane, ethane, and propane labeled SI-1 to SI-3, respectively), N,N-bis(phosphonomethyl)cysteic acid (SI-4), and N,N-bis(phosphonomethyl) metanilic acid (SI-5) were subjected to seawater biodegradation over 28 days (BOD28) by the OECD 306 method (Fig. 9).169
 | ||
| Fig. 9 Schematic structures of phosphonate scale inhibitors subjected to seawater biodegradation over 28 days (BOD28) by the OECD 306 method. | ||
SI-1 and SI-4 gave BOD28 values of 79% and 84%, respectively, and can be classed as “readily biodegradable”, whereas the BOD28 value for SI-5 was 41%. All inhibitors demonstrated good calcite inhibition properties, but only SI-1 and SI-2 gave fairly good barite inhibition. SI-2 stood out as an excellent calcite inhibitor, suitable for use in very high calcium-containing brines. SI-2 could also provide protection against barite deposition under mild-to-medium scaling potentials. SI-5 showed the best thermal stability at 130 °C and could therefore be a useful downhole squeeze scale inhibitor for fairly high-temperature wells.
The adsorption of PBTC on calcite surfaces (1–10), (102), (104), (113), and (202) was studied by molecular simulations.170 The phosphonic and carboxylic acid functional groups energetically interacted with the faces and preferentially occupied the carbonate ion sites by chemisorption, which is in agreement with the critical pH experiments. The strength of adsorption on crystal planes followed the order of (1–10) > (113) > (102) > (202) > (104). The binding energy gradually decreased with increasing temperature. The relationship between the critical pH and the adsorbed bis-deprotonated PBTC2− state indicates that the adsorbed inhibitor configuration plays an important role in inhibitor efficiency.
The polyphosphonates EDTMP, DTPMP and the polymeric PPCA (phosphinopolycarboxylic acid) were evaluated as CaCO3 scale inhibitors based on continuous measurement of particle size distribution by a laser diffraction technique and simultaneous pH monitoring.171 The inhibitory performance was based on real-time monitoring of the homogeneous nucleation and growth of CaCO3 particles formed in the bulk phase after the addition of carbonate ions to synthetic formation water. The performance found followed the ranking EDTMP <DTPMP < PPCA. It was found that PPCA acts as a nucleation inhibitor for CaCO3, but also has the capacity for preventing further crystal growth. EDTMP and DTPMP, on the other hand, demonstrate a single function, as they act as crystal growth inhibitors.
The effect of three polyphosphonic acids, namely EDTMP, HDTMP, and DTPMP on the growth of CaCO3 was investigated by the pH curve method (pHCM).172 The result showed the inhibitor effectiveness DTPMP > EDTMP > HDTMP. Also, the interaction of the phosphonates with the calcite (104) surface was studied by means of molecular dynamics simulations under the periodic boundary condition in an industrial water environment. The results indicated that strong electrostatic interactions between the oxygen atoms of the phosphonate functional groups and the surface Ca2+ of the calcite (104) face played a dominant role in their adsorption. The least effective inhibition of CaCO3 was HDTMP because only one phosphonate group interacts strongly with the CaCO3 surface.
The effect of ultrasonic irradiation on the growth of calcite in the presence of the inhibitor NTMP was investigated at constant composition conditions.173 In seeded growth experiments, it was found that the inhibiting effect of NTMP on crystal growth could be seriously mitigated under the influence of ultrasonic irradiation. These results could be explained in part by the physical effect of ultrasound that causes breakage and attrition of poisoned crystals, which resulted in an increase in fresh surface area. Several NTMP breakdown products were identified, which showed that ultrasound caused the progressive loss of phosphonate groups from NTMP, probably by means of physicochemically generated free radicals.
In the field of reverse osmosis (RO) membrane desalination, the effects of phosphonate type, concentration, and pH on calcium precipitation were investigated through analysis of dissolved calcium, particle size distribution of precipitated calcium salts, scanning electron microscope imaging, and microfiltration flux during solid/liquid separation.174 The phosphonates studied were EDTMP, HDTMP, and DTPMP. The addition of phosphonate antiscalants to synthetic brackish water concentrate reduced the amount of precipitated CaCO3. At pH 8.0, addition of antiscalant caused 91% of the calcium to remain in solution, while in the “control” solution, only 13% of the calcium remained soluble. The antiscalant HDTMP was not effective in preventing CaCO3 precipitation, most likely due to poor adsorption of the antiscalant onto nucleating crystals. The presence of EDTMP, and DTPMP antiscalants during CaCO3 precipitation altered the particle size and shape of the precipitate and affected subsequent microfiltration. At high EDTMP or DTPMP dosages CaCO3 precipitation and crystal growth were completely prevented, and only nanoparticles of size 100–300 nm form.
The highly calcium-tolerant polyamino polyether methylene phosphonate (PAPEMP, Fig. 10) was very effective in preventing CaCO3 precipitation at high supersaturation and high pH.175
 | ||
| Fig. 10 Schematic structure of the highly calcium-tolerant polyamino polyether methylene phosphonate (PAPEMP). | ||
The inhibition of CaCO3 crystallization in the presence of PAPEMP at both low and high supersaturation was studied and then compared with the inhibitory ability of other phosphonates. It was found that the phosphonates exhibited a significant reduction in CaCO3 precipitation. A surface adsorption mechanism involving a Langmuir isotherm model, coupled with the inhibitor's ability to remain in solution, was proposed to account for the inhibition of CaCO3 growth in the presence of these phosphonates. The inhibitor molecules are preferentially adsorbed at active growth sites on the crystal surface, which are effectively blocked from further growth.
The interactions of two diphosphonic acid-based surfactants, namely alkylimino-bis-methylenediphosphonic acid (IMPA-8) and 1-hydroxy-alkylidene-1,1-diphosphonic acid (Flotol-8), with calcite surfaces were quantified on the basis of molecular modeling computations.176 Both force field (UFF) and semiempirical quantum mechanical (MNDO) methods were employed for this purpose. The findings suggested that to a first approximation one can use the molecular modeling calculations in a vacuum, so far as the relative strength of mineral–reagent interactions is concerned.
Polyoxyethylenes having a diphosphonate functional group at one end of their chain were found to strongly adsorb onto calcium carbonate particles in aqueous colloidal suspensions.177 An enhanced colloidal stability of such suspensions resulted in a concomitant drastic reduction of the viscosity of concentrated aqueous suspensions. The adsorption and viscosity reduction were studied as a function of polymer molecular weight. The diphosphonate group was found to be the most efficient anionic group when associated with a polyoxyethylene of polymerization degree larger than 20.
Adsorption and desorption experiments involving the DTPMP pentaphosphonate and barite were systematically carried out over a wide range of physiochemical conditions.179 Both adsorption and desorption of inhibitor to and from barite surfaces proceeded rapidly. At a low phosphonate concentration, the surface adsorption mechanism accounted for the interaction between phosphonate and barite. The findings provide fundamental information that can benefit the inhibition performance and efficiency for barite scale control in oilfield operations.
The effect of five phosphonates (HEDP, NTMP, methylene-diphosphonic acid MDP, NTMP, and PBTC) on the growth of the barite (001) face was investigated using atomic force microscopy.180 Both affinity constants (calculated from adsorption isotherms) and measurements of growth rates of barite monomolecular steps as a function of inhibitor concentration led to the ranking of inhibitor effectiveness PBTC > NTMP > MDP > HEDP ≫ NTMP. Molecular simulations of the interaction of the phosphonates with barite (001) surfaces indicated that only kink sites along monomolecular steps can be considered as possible inhibition sites. This agrees with the AFM observations and measurements.
Macrocyclic phosphonate additives, such as DOTP (1,4,7,10-tetraazacyclododecanetetrakis(methylenephosphonic acid)) and NOTP (1,4,7,-triazacyclononanetris(methylenephosphonic acid)) were found to inhibit precipitation of barium sulfate just as effectively as their non-cyclic counterparts (Fig. 11).181
 | ||
| Fig. 11 Schematic structures of two macrocyclic phosphonate additives, DOTP (1,4,7,10-tetraazacyclododecane-tetrakis(methylenephosphonic acid)) and NOTP (1,4,7,-triazacyclononane-tris(methylenephosphonic acid)). | ||
Overall, the inhibition could be explained by the number of deprotonated phosphonate groups and was not significantly impacted by the presence of the ring.
The inhibition effect of DTPMP against barite scale in the presence of cations, such as Mg2+, Ca2+, Al3+, Fe2+, and Fe3+, was evaluated in high-pressure dynamic tube blocking tests.182 In general, there were no adverse effects, and in some cases the inhibitory efficiency improved, except in the case of Mg2+ ions which caused a lowering in the performance of all the scale inhibitors.
Crystal growth modifiers based on 1,3,5-substituted benzene derivatives were tested for their effect on calcite and barite crystallization.183 The phosphonated derivative, as expected, was a much more efficient inhibitor than the analogous sulfonate in calcite precipitation. However, both additives demonstrated promotion of crystallization in the case of barite (with the phosphonate being a better promoter). These studies showed that the functional group (phosphonate vs. sulfonate) alone does not determine the impact of the additive on crystallization.
The phosphonates EDTMP, N-methyl-nitrilodi(methylenephosphonic acid) (MNDP) nitrilodi(methylenephosphonic acid) (NDP), NTMP, and DOTP were investigated in barite precipitation inhibition.184 The important outcome of this research was the conclusion that a relatively high number of phosphonate groups on the inhibitor molecule does not guarantee inhibition, while a relatively low number of phosphonate groups does not imply low inhibition.
A group of phosphonates, namely EDTMP, HDTMP, DTPMP, and HEDP, were tested as barite scale inhibitors based on solution conductivity measurements.185 The efficiency ranking obtained was DTPMP > HEDP > EDTMP > DTPMP. The retardation effect of these additives was attributed to the simultaneous chelation of an active-site metal ion and adsorption on the surface through a phosphonate/sulfate exchange.
The adsorption of a series of straight chain phosphonates (MNDP, NTMP, and EDTMP) onto barite was studied by empirical molecular mechanics and experimentally.186 It was found that inhibition could be predicted for this series of molecules, which differ by the number of phosphonate groups present as well as by the chain length. The modeling results could predict which barite faces are preferred for the interaction with the phosphonate, and this has been verified by scanning and transmission electron microscopy. The agreement between the experiment and the model confirms that the dominant mechanism of interaction of phosphonates with barite is that the deprotonated phosphonate groups interact with the barium ions on the barite surface.
The role played by phosphonate speciation on the inhibition of barite precipitation was investigated using the model phosphonates HEDP, NDP, NTMP, and EDTMP.187 Inhibition was pH dependent and was found to be compromised at very high and low pH values. Maximum inhibition for all phosphonate additives was observed at pH 8. Speciation curves showed that inhibition could be improved by the presence of two or more fully deprotonated phosphonate groups at pH ∼8, while at pH 12 inhibition was insensitive to the number of deprotonated phosphonate groups. Therefore, surface charge repulsion affects inhibition at very high pH.
The effect of a number of structurally related multidentate organic phosphonates on the rate of crystal growth of calcium oxalate was studied as a function of pH.189 Rate constants were obtained at various concentrations for the phosphonates HEDP, NTMP, EDTMP, and HDTMP at pH 5.00, 6.00, and 7.00. The effect of pH on the inhibitory activity of each of the phosphonates was considerable, with effective concentrations of inhibitor decreasing two orders of magnitude, in some cases, as the pH was increased. At a given pH the tetraphosphonates EDTMP and HDTMP were generally the most effective inhibitors. The results also suggested that HEDP, at currently administered doses in humans, provides only a moderate increase in the capacity of human urine to inhibit calcium oxalate crystal growth.
The kinetics of precipitation of calcium oxalate monohydrate was studied conductometrically at 298 K in both spontaneous and seeded growth systems. The rate of growth follows a quadratic dependence upon the relative supersaturation, which suggests a surface-controlled growth mechanism. The effect of phosphonate additives HEDP, EDTMP and HDTMP on the precipitation kinetics of calcium oxalate in the absence and presence of well-characterized seeds was investigated at various levels of additive concentration.190 The inhibiting activity of these additives was related to the surface characteristics of the inoculated seeds and structural features of the additive molecules. The fit of experimental data to the Langmuir adsorption isotherm supported a mechanism of inhibition through molecular adsorption of the foreign ions on the surface of the growing crystals.
The effects of DTPMP on the morphology and structure of precipitated calcium oxalate were reported.191 The results indicated that morphology and structure of the resulting calcium oxalate varied with the conditions. The architecture of the samples showed that DTPMP had a significant influence on the morphology and crystalline structure of calcium oxalate. Whewellite with a monoclinic structure was the most favored polymorph in the absence of DTPMP, whereas calcium oxalate hydrate having orthorhombic structure was favored in the presence of DTPMP.
Phosphorylated osteopontin peptides strongly inhibited COD crystal growth in solution as compared with the nonphosphorylated state, with increasing inhibitory potency correlating with the degree of peptide phosphorylation.192 Scanning electron microscopy revealed that the inhibition from the phosphopeptides resulted in distinctive, rosette-like crystal aggregates called spherulites. The phosphorylated peptides preferentially bound and specifically inhibited the {110} crystallographic faces of calcium oxalate dihydrate (COD), as identified by combining atomic force microscopy and computational simulation approaches. These {110} surfaces of COD have high lattice calcium occupancy, providing preferential binding sites for the highly acidic peptides. This binding and inhibition by the peptides at the {110} faces led to crystal aggregation and intergrowth.
 | ||
| Fig. 12 Schematic structure of the fluorescent-tagged HEDP derivative, 1-hydroxy-7-(6-methoxy-1,3-dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)heptane-1,1-diyl-di(phosphonic acid), named HEDP-F. | ||
Evaluation of phosphonates as gypsum scale inhibitors was reported as early as the mid-1970s with the commercial additives HEDP, NTMP, and HDTMP.199 A general overview of the scientific literature was recently published.200
The effect of four amino tetraphosphonate additives with systematically elongated polymethylene linker between the N atoms was investigated on the crystallization of calcium sulfate dihydrate (CaSO4·2H2O, gypsum).201 The additives were EDTMP HDTMP, ODTMP, and DDTMP. The inhibition efficiency was directly proportional to the number of methylene groups in the organic chain that connects the amino-bis(methylenephosphonate) moieties. The degree of inhibition of crystallization was measured as an increase in induction time and reduction in crystallization rate. Particle size and crystal morphology were determined with a particle sizer and SEM. According to the experimental results, phosphonate additives tested in this study are very effective retardants for the formation of calcium sulfate dihydrate scale and have an effect on crystal morphology.
The effects of HEDP, NTMP, PAPEMP, DTPMP, and BHMTPMP (all at 20 ppm) on the crystallization of gypsum were investigated by in situ UV-vis, X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), inductively coupled plasma optical emission spectrometry (ICP-OES) and scanning electron microscopy (SEM) techniques.202 BHMTPMP was the most efficient antiscalant by completely inhibiting crystallization. Due to the chain length of BHMTPMP, the crystallization kinetics decreased to a larger extent than DTPMP. The increase in pH of the solution from ∼4 to ∼7 enhanced the efficiency of the inhibitors. The results revealed that partially deprotonated phosphonate additives were strongly associated with gypsum crystals and/or potentially taken up into the crystal matrix, resulting in a sudden and sharp increase in turbidity plots. Furthermore, phosphonate additives altered the thin, twinned gypsum crystals into thick needles.
The retardation effect of NTMP on calcium sulfate dihydrate nucleation kinetics was evaluated by means of an optical technique for accurate measurement of the induction period.203 Sodium sulfate and calcium chloride were mixed to obtain a supersaturated calcium sulfate dihydrate solution. Experiments were carried out at various NTMP concentrations (5–50 ppm) in solution, while the saturation index was in the range of 4.04–4.82. The temperature was varied from 15 to 45 °C. It was found that NTMP is a very strong retardant for gypsum nucleation. The same technique was used to study PBTC.204 Comparative studies from the same group using NTMP and PBTC as inhibitors for gypsum nucleation by laser scattering showed that the former is a much more potent nucleation retardant that substantially extends the induction time of gypsum formation.205
The performance and gypsum inhibition of five scale inhibitor mixtures consisting of NTMP, DTPMP, and poly(acrylic acid) (PAA) were studied at three saturation indexes.206 The induction time effect was studied for 40 min for each mixture. The results revealed that the best inhibitive performance was achieved by the NTMP/DTPMP blend. This high performance was attributed to the synergistic effect of NTMP and DTPMP in the calcium sulfate inhibitive mechanism. This blend showed enhanced scale inhibition compared with polyacrylate inhibitors.
The effects of five phosphonic additives [EDTMP, NTMP, NDPA (nitrilodi(methylenephosphonic acid)), HEDP, and NPDA (nitrilomethylenephosphono-diacetic acid)] on the formation of calcium sulfate scale on a metal surface in a flow system were investigated thoroughly using a multiple pipe flow system.207 Their chemical structure, concentration and run time were the main factors that influenced the inhibitory capability of these antiscalants. The relationships between these factors and the scale rate were established. The Langmuir adsorption isotherm was applied to predict the critical concentrations above which complete inhibition of scaling would occur. The inhibition efficiency order of the investigated organic additives was found as follows: EDTMP > NTMP > NDPA > HEDP > NPDA, which corresponds to the number of phosphonate groups that the additives possess.
The “mixed” sulfonophosphonate 2-(N,N-di(methylenephosphonate))aminoethylsulfonate was used as a scale inhibitor for CaCO3 and gypsum scales.208 In comparison with the conventional NTMP, it was found to be a more efficient inhibitor. The incorporation of the sulfonate group into the aminoalkylenephosphonate backbone significantly improved the inhibition properties under harsh conditions and the crystal-modifying properties.
The dynamic light scattering (DLS) technique was used to study bulk gypsum precipitation in supersaturated solutions during the induction period.209 It was based on the standard Ag nanoparticles (ARGOVIT) injection into the supersaturated gypsum solution. These nanoparticles can act as an internal indifferent light scattering intensity reference and provide a semiquantitative measurement of a relative gypsum particles content in a blank solution and in the system treated with phosphonates NTMP, HEDP, and PBTC. It was found that NTMP sufficiently reduced the number of gypsum nuclei spontaneously formed in the supersaturated solutions. The less effective inhibitors of gypsum scaling HEDP and PBTC also reduced the gypsum nuclei number, but to a lesser extent.
The diphosphonate propane-2,2-phenylene-diylbisbis(4,1)-phenylene-bis(4,1)-bis (azanediyl)bis(methylene diphosphonic acid), see Fig. 13, was evaluated as a CaCO3 and gypsum scale inhibitor.210 The gypsum crystals changed in morphology, from thin needles with a smooth surface in the absence of the inhibitor, to thicker needles with several crystal defects in the presence of the inhibitor. The inhibitor presented high efficiency for common deposits of salts of carbonates (94%) and sulphates (97%) at a concentration of 200 ppm, as well as a high thermostability.
 | ||
| Fig. 13 Schematic structure of the propane-2,2-phenylene-diylbisbis(4,1)-phenylene-bis(4,1)-bis (azanediyl)bis(methylene diphosphonic acid) as CaCO3 and gypsum scale inhibitor. | ||
A limited number of relevant literature reports exist on the use of chemical additives in Mg-silicate scale control. Our group has reported that the well-known tetra-carboxylate EDTA can be effective in compromising the catalytic action of Mg2+ cations in enhancing amorphous silica formation, at pH regimes >9,212 where EDTA demonstrates its “infamous” high affinity for Mg2+ cations at high pH.213 Along the same lines, phosphonate-based additives were evaluated as chelators or as ligands for the Mg2+ ions, “turning off” their catalytic action on silicic acid autocondensation, by acting as stabilizers, again by complexing and deactivating them, thus preventing the formation of Mg-silicate.214 The phosphonic acids evaluated were PBTC, NTMP, HEDP, HDTMP, and BHMTPAMP. Their efficiency was correlated to their affinity for Mg2+.
Generally, phosphonates demonstrate high affinity for Al3+ (ref. 215) and this was exploited in aluminum silicate scale inhibition. Specifically, six different phosphonic acids were evaluated in our group for their ability to inhibit aluminum silicate formation, namely, HEDP, PBTC, AMP, HDTMP, DTPMP and BHMTPAMP.216 The phosphonates acted as chelators/ligands for Al3+ ions, and thus “turned off” their catalytic effect in silica polycondensation. This study concluded that phosphonate additives were able to function as chelators or ligands for Al3+ cations, thus “turning off” its catalytic action in silicic acid auto condensation. A similar mechanism is also valid for Mg-silicate control (see above). It was observed that the phosphonate additives were partially entrapped in the formed precipitates due to the formation of aluminum−phosphonate “complexes” that are sparingly soluble.
The formation, precipitation, and deposition of the so-called “iron silicate” in water systems have been among the problematic issues in specific systems, such as geothermal installations. The influence of phosphonate-based chemical additives on the formation of precipitates in the presence of both silicate and ferric (Fe3+) ions was systematically evaluated.217 For this purpose, various phosphonate additives that possess diverse chemical structures and variable numbers of functional groups were used. These additives were PBTC, HEDP, NTMP, HDTMP, DTPMP, and BHMTPAMP. Inhibition experiments were conducted in solutions containing silicate (150 ppm, expressed as SiO2) and iron (150 ppm, as Fe) at pH 7.0. The phosphonate additives used were found to act as stabilizing agents, most likely by complexing the Fe3+ cations, thereby preventing ferric silicate formation. The experimental design included a plethora of physicochemical parameters, such as the inhibitor concentration, salinity, and temperature. The optimal inhibitor was HEDP, which has three functional groups (one hydroxyl and two phosphonate). An increase in the number of functional groups did not enhance inhibitor effectiveness.
3.3. Polymeric phosphonates as scale inhibitors
Highly phosphonated double hydrophilic block copolymers (DHBCs, Fig. 14) were synthesized through chain transfer radical polymerization or polymerization from a macroinitiator.218 The DHBCs bearing phosphonate groups have a strong influence on CaCO3 crystallization. They can control the morphology of CaCO3 from smooth spherical particles to porous structures, even at a rather low concentrations due to the strong binding effect of the phosphonate groups on the surface Ca2+ ions of the salt. | ||
| Fig. 14 Schematic structure of the phosphonated double hydrophilic block copolymers as CaCO3 crystallization inhibitors. | ||
The effects of polyphosphinocarboxylic acid (PPCA, Fig. 15) scale inhibitor on CaCO3 nucleation and growth were studied using electrodeposition, coupled with microscopic observations.219 The study showed that PPCA is an efficient scale inhibitor based on the quantity of CaCO3 formed by electrodeposition. In addition, significant crystal distortions were observed with PPCA, pointing to a link between CaCO3 crystal morphology alterations and inhibition efficiency.
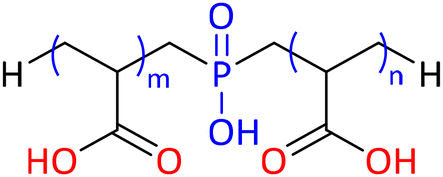 | ||
| Fig. 15 Schematic structure of polyphosphinocarboxylic acid (PPCA) as CaCO3 scale inhibitor. | ||
In a related study, the inhibiting effects of PPCA on CaCO3 were studied by in situ probing of crystal growth by synchrotron radiation wide-angle X-ray scattering (WAXS).220 It was found that PPCA lengthened the induction time of the surface deposition process. It also suppressed calcite formation and resulted in a vaterite-rich scale. Finally, it caused a change of lattice parameter for both calcite and vaterite crystals. The c-axis of unit cell increased, and the a-axis and b-axis decreased. PPCA (at 2.5 ppm) showed 100% inhibition gypsum inhibition efficiency at a saturation index of 0.31 at room temperature and without pH regulation after 24 h with practically no detectable gypsum crystallites even after two months.221
The polymer AA-APEM-H3PO3 (AA = acrylic acid, APEM = oxalic acid-allypolyethoxycarboxylate, see Fig. 16) demonstrated excellent scale inhibition performance for CaCO3, outstanding ability to disperse ferric oxide, and good corrosion inhibition properties.222 The study showed that AA-APEM-H3PO3 exhibited ∼90% CaCO3 inhibition. The proposed inhibition mechanism invoked surface complexation and chelation between the functional groups –P(O)(OH)2, –COOH and Ca2+ ions, with the polyethylene glycol segments increasing polymer solubility in water.
 | ||
| Fig. 16 Schematic structure of AA-APEM-H3PO3 polymer as CaCO3 scale inhibitor. | ||
A zwitterionic monomer 3-[diallyl{3-(diethoxyphosphoryl)propyl}ammonio]propane-1-sulfonate was synthesized and polymerized to yield the polymer PZA (Fig. 17), bearing phosphonate and sulfonate anionic moieties.223 PZA at a concentration of 20 ppm imparted excellent inhibition of the formation of calcium sulfate scale and as such it was proposed as a potential antiscalant in RO plants.
 | ||
| Fig. 17 Schematic structure of the PZA polymer as gypsum scale inhibitor. | ||
Amorphous silica is an “exotic” precipitate/deposit in silica-laden water systems. It does not conform to the known and well-recognized laws of crystal growth because it is not a mineral salt. Instead, it is a covalent, 3D random polymer that is formed by the polycondensation-induced formation of Si–O–Si bonds from silicic acid, see Fig. 18.224 Hence, its mitigation approaches differ from those used for mineral salts. Notably, non-polymeric phosphonates are ineffective silica scale inhibitors. Several polymers (cationic, uncharged, and zwitterionic) have been found to effectively control silica scale. Herein, only the ones derivatized from phosphonate groups will be mentioned.
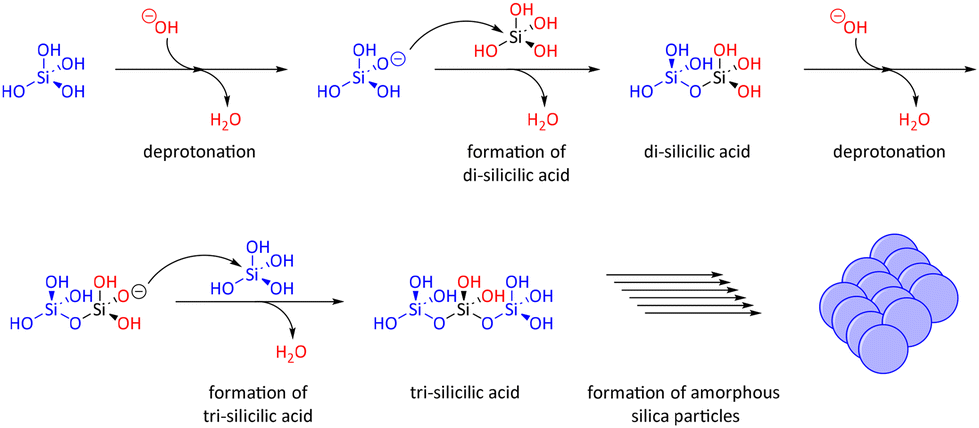 | ||
| Fig. 18 Schematic representation of the SN2-like mechanism of silicic acid polycondensation that eventually leads to amorphous silica particle formation. | ||
Phosphonate groups have been introduced to the backbone of polyethyleneimine (PEI) via the phospha-Mannich reaction resulting in the zwitterionic polymer phosphonomethylated polyethyleneimine (PPEI), see Fig. 19.225,226 By varying the reagent ratios, various phosphonomethylation degrees can be achieved, from as low as 8% (mole percent) to the fully substituted analog. PPEI has found numerous applications in diverse technical fields.227 PEI was used as a polymeric platform because of its acceptable low toxicity and environmental friendliness,228 which also make it a useful chemical for medicinal229 and pharmaceutical applications.230
 | ||
| Fig. 19 Derivatization of cationic polyethyleneimine (PEI, left) to zwitterionic phosphonated polyethyleneimine (PPEI, right). Cationic (amine) moieties are highlighted in red, whereas anionic (phosphonate) ones are in blue. | ||
PEI (non-phosphonated) has been used as a silica scale inhibitor, even at low dosages;231,232 PPEI was used as a silica inhibitor in silicate-loaded synthetic water.233 The results showed that phosphonate grafting on the initial cationic PEI polymeric chain causes enhancement of inhibitory activity by “relieving” the cationic charge of the parent polymer PEI. Hence, the polymeric inhibitor is not entrapped in the forming colloidal silica matrix, a problem severely exhibited by the parent PEI. Furthermore, it appears that the more extensive the phosphonate grafting is on PEI, the higher the inhibitory activity is of the PPEI modified polymer.
The phospha-Mannich reaction was also used to generate the zwitterionic polymer phosphonomethylated chitosan (PCH) from chitosan, a partial hydrolysis product of the natural polymer chitin,234 see Fig. 20. PCH was evaluated as a silica scale inhibitor, either as a stand-alone additive,235 or in combination with anionic (carboxymethyl inulin, CMI) or cationic (PEI) polyelectrolytes.236 PCH was found to be an inhibitor of silicic acid condensation at 40–200 ppm dosage levels in 8 h condensation reactions.
 | ||
| Fig. 20 Schematic structure of the zwitter-ionic phosphonomethylated chitosan backbone (m = 0.16, n = 0.37, p = 0.24, q = 0.14). | ||
The most frequently encountered inorganic scales in geothermal systems include amorphous silica, magnesium silicate, aluminum silicate, iron(III) silicate, zinc sulfide, lead sulfide, iron(II) sulfides, and calcium carbonate. Against these scales, four interrelated methacrylate-structured polymers were tested as inhibitors, grafted either with polyethylene glycol (PEG), or phosphonic acid (PHOS) side chains, or both, see Fig. 21. Based on several results obtained, the inhibitor PEGPHOS-LOW (containing 34 PEG grafts and 14 phosphonate grafts; LOW stands for “low molecular weight”) was selected as the most efficient and was subsequently tested in artificial geothermal brines of variable stress, containing all the scales together. This is why the name coined for this polymeric additive was “universal scale inhibitor”. The polymer showed variable inhibiting characteristics, but also demonstrated dispersion properties, particularly with the most challenging scales, such as PbS.237,238
 | ||
| Fig. 21 Schematic structures of the polymeric scale inhibitors tested. Neutral PEG groups are highlighted in red, anionic phosphonate groups, in blue. The lower image gives a perspective of the density of the two grafts on the polymeric backbones. | ||
3.4. Phosphonates and metal phosphonates as corrosion inhibitors
Phosphonic acids hold a prominent place among the corrosion inhibitors of choice in industrial water systems.239,240 At appropriate pH regions (depending on the particular application) phosphonic acids exist in their (partially or completely) deprotonated form. There are several reports on the deprotonation chemistry of phosphonic acids.241 Thus, in the presence of metal cations (commonly alkaline earth metals), they form sparingly soluble compounds, which eventually precipitate onto the metallic surface to form an ideally two-dimensional protective thin film. Phosphonates are introduced into the system to be protected in the acid form or as alkali metal soluble salts, but readily form more stable complexes with other metal cations found in the process stream (most commonly Ca, Mg, Sr or Ba), depending on the application. Research in this area has been stimulated by the need to develop inhibitor formulations that are free of chromates, nitrates, nitrites, inorganic phosphorus compounds, etc. Phosphonates, when blended with certain metal cations and polymers, reduce the optimal inhibitor concentration needed for inhibition due to synergistic effects. Synergism is one of the important effects in the inhibition process and serves as the basis for the development of all modern corrosion inhibitor formulations. This section will include applications of phosphonates as corrosion inhibitors for steel, a material that is very common in industry.Mixtures of 2-aminothiophenol and HEDP showed a cooperative effect of inhibition of stainless steel (SS 41).242 The actual role played by HEDP was explained in terms of its scale inhibition activity towards FeCO3.
Kuznetsov et al. have studied the inhibitor efficiency of various phosphonate inhibitors of the aminomethylenephosphonate type (R-N-CH2-PO3H2).243 The protective properties of aminophosphonic acids and their magnesium and calcium complexes were studied in soft water. 1,1-Hydroxycarboxypropane-3-amino-di(methylenephosphonic) acid (Fig. 22) and HDTMP could suppress the corrosion of steel in water completely.
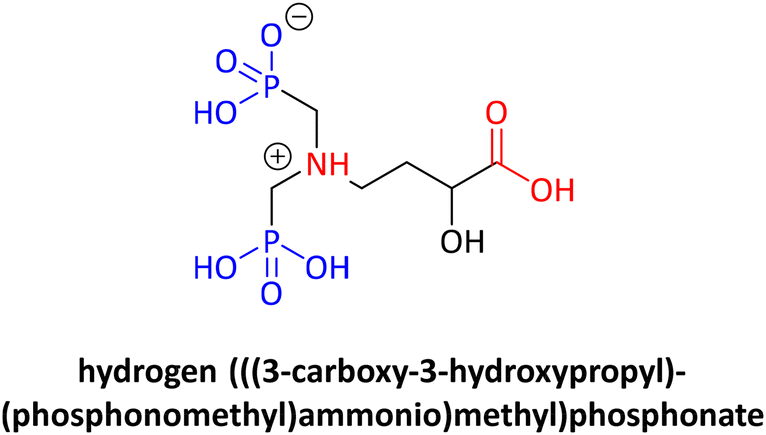 | ||
| Fig. 22 Schematic structure of the corrosion inhibitor 1,1-hydroxycarboxypropane-3-amino-di(methylenephosphonic) acid. | ||
Kouznetsov proposed that metal phosphonate complexes are much more effective than the corresponding acids; if the complexing agent is fixed, then the stability constants of the complexes become the major factor. For phosphonates of Mg2+ and Ca2+, which are usually less stable than the corresponding iron complexes, the dependence of the protective concentration on the stability constant passes through a maximum; the complexes of those acids whose own protective properties are weaker are more effective. Imino-N,N-diacetic-N-methylenephosphonic acid can serve as an example. By contrast, the relatively more stable complexes, for example Ca-NTMP, are much less effective than the acid itself.
Molybdate is a well-known corrosion inhibitor. Its combination with phosphonates enhances corrosion efficiency.244 A level of 300 ppm MoO42− had only 32% efficiency in inhibiting the corrosion of mild steel immersed in a neutral aqueous environment containing 60 ppm Cl−, whereas a formulation of NTMP (50 ppm)–MoO42− (300 ppm)–Zn2+ (50 ppm) exhibited 96% inhibition efficiency. The lower inhibition efficiency in the former case was due to the dissolution of the protective film formed on the metal surface and getting precipitated in the bulk of the solution; this system controlled the anodic reaction only. The latter system controlled both the anodic and cathodic reactions; the dissolution of the protective film formed on the metal surface was reduced to a greater extent.
Protective layer formation of α,ω-diphosphono-alkane compounds on an iron surface was studied.245 Layer formation proved to be a spontaneous process on iron, and can be accomplished by simple immersion into an aqueous solution of phosphonate additives, resulting in a thin dense multimolecular adsorption layer with a high corrosion protection effect. It was concluded that the mechanism of inhibition was anodic type, hindering active iron dissolution due to the blocking of the metal surface. The protective layer formation completes after a few days of immersion, resulting in a thin but dense multimolecular adsorption layer. A “self-healing” effect of diphosphonates was demonstrated. Based on the high corrosion protection effect, surface modification by diphosphonates may be regarded as a potential anticorrosive treatment.
Surface treatments have been carried out on carbon steel in solutions containing different phosphonates.246 The compounds were dissolved in an ethanol/water mixture (80:20). Corrosion protection afforded by laurylphosphonic acid (LPA), ethyllaurylphosphonate (ELP; also called lauryl phosphonic acid monoethylester) and diethyllaurylphosphonate (DELP) (Fig. 23) was studied by steady-state current–voltage curves and electrochemical impedance measurements with a rotating disc electrode.
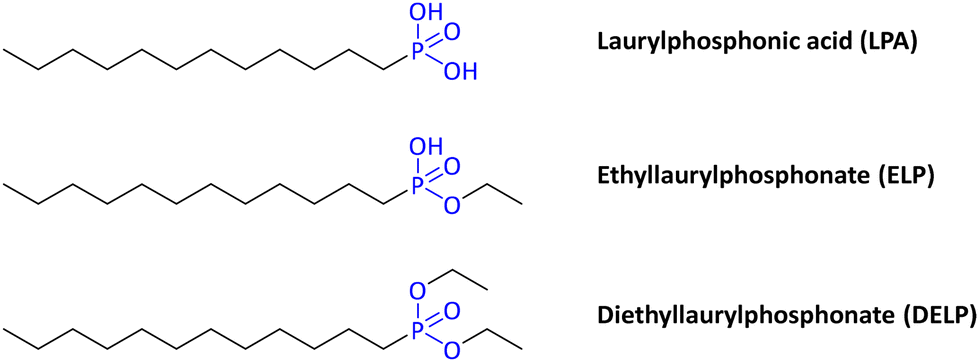 | ||
| Fig. 23 Schematic structure of the “fatty” corrosion inhibitors laurylphosphonic acid (LPA), ethyllaurylphosphonate (ELP) and diethyllaurylphosphonate (DELP). | ||
Corrosion protection was only obtained for ethyllaurylphosphonate (ELP), which was able to form a relatively thick film on the carbon steel surface. Electron probe microanalysis corroborated that the film is thick and porous. Infra-red spectroscopy indicated that the film was formed by reaction of the organic phosphonate with the steel surface to produce a metal salt.
The effect of phosphonates used in Russian heat-power engineering on the corrosion of carbon steel in deaerated delivery water at 90 °C was studied.247 It was demonstrated that the introduction of phosphonates reduced the susceptibility of steel to local corrosion. A Zn2+ complex of hydroxyethylidenediphosphonic acid (Zn-HEDP) was the most effective inhibitor of the anodic reaction.
For the protection of carbon steel from corrosion, NTMP was more effective than HEDP, N,N-dimethylidenephosphonoglycine (DMPG), 1-ethylphosphonoethylidenediphosphonic acid (EEDP) (Fig. 24), and EDTMP.248
 | ||
| Fig. 24 Schematic structures of the corrosion inhibitors N,N-dimethylidenephosphonoglycine (DMPG) and 1−ethylphosphonoethylidenediphosphonic acid (EEDP). | ||
A 20 min treatment in 1.0 M of AMP at pH 0.23 at 45 °C formed an anti-corrosive complex film, identified by XPS and Auger electron spectroscopy. From differences in binding energies of Fe, N, and O, in the shift of C–N and P–O vibration, in the FT-IR spectra, and in the change of P–OH and Fe–N vibration before and after film formation, it was deduced that N and phosphonate O in NTMP were coordinated with Fe2+ in the film.
The corrosion inhibition of iron in 0.5 M sulfuric acid by N,N-dipropynoxymethylamine-trimethylphosphonate was investigated by means of potentiodynamic polarization and electrochemical impedance spectroscopy (EIS) techniques.249 It was studied in concentrations 40–320 ppm at ambient temperature. The results revealed that the inhibition mechanism is a combination of anodic and cathodic type. It was also found that this inhibitor obeys the Frumkin adsorption isotherm and Flory–Huggins isotherm based on a substitutional adsorption process.
The role played by pH and Ca2+ in the adsorption of an alkyl N-aminodimethylphosphonate on mild steel (E24) surfaces was investigated by XPS.250 At pH 7, the adsorbed diphosphonate layer was continuous, and its equivalent thickness was ∼24 Å (monolayer). In the presence of Ca2+ ions, calcium phosphonate (and Ca(OH)2, in very small amounts) was formed on the steel surface. The adsorption of the diphosphonate molecules on the steel surface was promoted in alkaline solution (pH > 7.55). The measured values for the Ca:P intensity ratio were in the range 0.75–1.00, which suggested that the diphosphonate molecules were adsorbed on steel forming a coordination polymer cross-linked by Ca2+ through their phosphonate groups. In the presence of Ca2+ ions in alkaline solution, the adsorbed diphosphonate layer was discontinuous, and the surface coverage was ∼34%.
The effect of a new class of corrosion inhibitors, namely piperidin-1-yl-phosphonic acid (PPA) and (4-phosphono-piperazin-1-yl)phosphonic acid (PPPA) (Fig. 25) on the corrosion of iron in NaCl medium was investigated by electrochemical measurements.251
 | ||
| Fig. 25 Schematic structures of the corrosion inhibitors piperidin-1-yl-phosphonic acid (PPA) and (4-phosphono-piperazin-1-yl)phosphonic acid (PPPA). | ||
Potentiodynamic polarization studies clearly revealed the type of the inhibitor. The addition of increasing concentrations of phosphonic acids caused a shift of the pitting potential (Epit) in the positive direction, indicating the inhibitive effect of the added phosphonic acid on the pitting attack. The potential of corrosion was moved towards negative values and the corrosion current was reduced. The values of the current were lower in the presence of PPA and PPPA. This was explained by the fact that most of the surface of the electrode was covered by the adsorbed molecules. PPPA had a strongly inhibitive effect on chloride pitting corrosion. It was proposed that the addition of the –NCH2PO3H− group in the PPA para-position, giving PPPA, increased its inhibition efficiency.
Phosphonate layer formation on a passive iron surface was investigated by electrochemical and atomic force microscopy techniques.252 It was found that phosphonate groups bond more strongly to the oxide surface, while metallic iron surface is disadvantageous for phosphonate layer formation in aqueous solutions. The rate of anodic dissolution was continually decreasing due to the time-dependent formation of protective phosphonate layer. The kinetics of phosphonate layer formation on passive iron was determined by the potential applied for preceding passive film formation. The size and shape of iron oxide grains depended slightly on the potential of passivation. Changes in morphology due to the phosphonate layer formation were studied by AFM.
The passivation ability of the zinc complex with HEDP in a borate buffer solution was studied.253 An in situ ellipsometric method was used to study the mechanism of formation of a protective film on iron in the presence of HEDP, Zn-HEDP, and ZnSO4 in the course of the cathodic polarization of the electrode. The studies of Zn-HEDP adsorption on iron (at E = −0.65 V) in combination with X-ray photoelectron spectroscopy (XPS) showed that on the metal surface a multilayer protective film formed consisting of an internal layer of Zn(OH)2 and an outer layer consisting of HEDP complexes with Fe2+ and/or Zn2+. It was found that the thickness of the passivation film does not exceed 60 Å, of which 7–10 Å correspond to the sparingly soluble Zn(OH)2.
Electrochemical techniques were used for investigating the inhibitory activity on carbon steel of thiomorpholin-4-ylmethyl-phosphonic acid (TMPA) and morpholin-4-methyl-phosphonic acid (MPA) (Fig. 26) in natural seawater.254
 | ||
| Fig. 26 Schematic structures of the corrosion inhibitors thiomorpholin-4-ylmethyl-phosphonic acid (TMPA) and morpholin-4-methyl-phosphonic acid (MPA). | ||
Measurement of the free corrosion potential indicated that the phosphonic acids tested inhibit the corrosion of carbon steel in seawater. Potentiodynamic polarization curves proved that addition of these molecules was responsible for corrosion current density decrease and a corresponding reduction of the corrosion rate. The phosphonic acids tested as corrosion inhibitors of carbon steel in natural seawater were effective even at low concentrations. FT-IR spectroscopy was used to obtain information on the interaction between the metallic surface and the inhibitors.
The interaction of six homologous amino-diphosphonate additives shown in Fig. 4, MBMP (C1-D), EBMP (C2-D), BBMP (C4-D), HBMP (C6-D), OBMP (C8-D), and DBMP (C12-D), with carbon steel surfaces was studied by XPS at pH = 3.0.255 Structurally, they all possessed two methylenephosphonate moieties connected to a single N atom. The third substituent on N was a non-polar, variable-length alkyl chain, –(CH2)xCH3, where x = 0 (C1-D), 1 (C2-D), 3 (C4-D), 5 (C6-D), 7 (C8-D), and 11 (C12-D). The XPS studies showed significant deprotonation of the diphosphonic acid groups upon interaction with the metallic surface. This surface interaction resulted in adsorption on the surface via the deprotonated phosphonic acid moieties. The adsorption of inhibitors on the metal surface was investigated by potentiodynamic polarization and electrochemical impedance spectroscopy. The changes detected in the charge transfer resistance (Rct) and constant phase element (CPE) independently confirmed inhibitors’ adsorption on metal surface. Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR) spectroscopy and SEM was used to investigate the nature of the deposited film. Small alkyl chain diphosphonates (C1-D, C2-D and C4-D) exhibited lower corrosion resistance due to the thin, porous and/or incomplete layer formed on carbon steel surface. Longer alkyl chain molecules (C6-D, C8-D and C12-D) were found to adsorb more efficiently and form a more organized and denser layer. The best results were obtained in the case of C8-D (lower corrosion current, higher Rct and surface coverage). In the presence of C8-D the corrosion rate was reduced by a factor of 6.
The interaction of six systematically varied diamino-tetraphosphonate molecules, shown in Fig. 4, with carbon steel surfaces by XPS at pH 3 was reported.256 All the tetraphosphonates [EDTMP (C2), TDTMP (C4), HDTMP (C6), ODTMP (C8) and DDTMP (C12)] belong to the aminomethylene-phosphonate family, and they possess a systematically elongated backbone (from two to twelve methylene groups separating the N atoms). XPS studies were performed on powdered samples and also on immersed carbon steel specimens in aqueous solutions. The XPS results also suggest that the tetraphosphonic acid molecules become significantly deprotonated upon interaction with the carbon steel surface and that the surface interaction of the tetraphosphonic acid family leads to additive adsorption on the steel surface via the phosphonate groups. The mode of corrosion inhibition was studied by potentiodynamic polarization and electrochemical impedance spectroscopy. The changes observed in the impedance parameters, like charge transfer resistance (Rct) and constant phase element (CPE), confirmed the strong adsorption on the metal surface. The nature of the protective layer formed on the carbon steel surface was examined by Attenuated Total Reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy and optical microscopy.
The efficiency of phosphonate additives as corrosion inhibitors can often be enhanced by the presence of certain metal ions.257 Such synergistic systems have been focused on metal ions commonly found in natural waters (mostly alkaline-earth cations) or Zn2+ (which is purposely added to augment corrosion protection due to surface-formed Zn(OH)2),258 and a wide spectrum of phosphonate additives, such as tri-phosphonates,259 tetra-phosphonates,260 and “mixed” carboxy/phosphonates.261
3.5. Phosphonate additives as dissolvers of sparingly soluble solids and deposits
Apart from the precipitation of sparingly soluble salts, another topic of great interest for water system operators is their attachment to critical surfaces to form deposits.262,263 Such deposits compromise system integrity because they impede heat transfer. Ideally, they should be prevented by applying several practical strategies, such as water softening, low cycles of concentration, and use of scale inhibitors. However, prevention of precipitation is not always possible, and, as a result, deposition frequently occurs, with its severity varying with system stresses. Hence, water system operators are occasionally faced with the challenge of removing these deposits, either by mechanical or chemical means.264 This section will review scale removal strategies that are based on the utilization of chemical additives, specifically phosphonates. A prerequisite for effective scale dissolution is the strong interaction between the scale surface and the dissolver molecule.265,266During the last few years, there have been many improvements in the technology utilized to chemically clean industrial equipment. New corrosion inhibitors, organic acids and chelant solvents have been developed. Improved techniques for removing refinery/chemical plant deposits have also been produced. Most importantly, there have been significant improvements in our understanding of the chemical mechanisms underlying many of these processes. New developments have impacted important technology areas with the commercial use of chemicals to clean large industrial equipment.267
One of the deposit removal methods is the use of acid. Cleaning of gypsum scale deposits from the surface of a heat exchanger was performed with dilute acetic acid augmented with the presence of wood pulp fibers (from Eucalyptus).268 Generally, acid cleaning requires supplementing the cleaning program with a corrosion inhibitor, to avoid unwanted acid corrosion.269 The dissolution of CaCO3270 and barite with hydrochloric acid was also studied.271
The dissolution of (10–14) calcite surfaces was investigated in the presence of HEDP at the concentration range 0–10 mM at pH = 8 using in situ AFM.272 The presence of HEDP resulted in a change in the appearance of the dissolution features from the typical rhombohedral to elongated, tear shapes. Additionally, dissolution rates were drastically reduced, although they progressively increased with increasing additive concentration. Stabilization of polar steps and effects of HEDP on the structure and dynamics of the hydration shell of Ca2+ may explain such observations.
In a similar study the rates of dissolution of calcitic Carrara marble were reported to be significantly reduced in alkaline pH (8.25) at 25 °C in the presence of HEDP.273 The adsorption takes place at the calcite/water interface at the double layer through the interaction of charged surface species with the charged solution species of the adsorbate. The formation of surface complexes between the charged surface species of calcite and the species of HEDP (dominant at pH 8.25) were invoked to interpret the results.
The surface of precipitated calcite particles was modified by reaction with the monophosphonic acids phenylphosphonic and dodecylphosphonic in the organic solvents tetrahydrofuran and ethanol.274 Under appropriate conditions, dense phosphonate monolayers were deposited at the surface of the CaCO3 particles. The nature of the grafted phosphonates was investigated using 31P and 1H Magic Angle Spinning (MAS) solid state NMR spectroscopy, FTIR spectroscopy and N2 physisorption. These monolayers were found to increase the hydrophobic character of the CaCO3 surface, especially dodecylphosphonic acid.
Six organophosphonate molecules (tetraphosphonates and diphosphonates) with systematically varied length were evaluated in the dissolution process of the (10.4) surface of calcite using in situ dynamic atomic force microscopy.275 For each of the two groups of the organophosphonate derivatives, the same formation of etch pits (olive-shaped for the tetraphosphonate and triangular-shaped for the diphosphonate molecules), respectively, was observed. This finding indicates that the number of functional ends decisively determines the resulting calcite (10.4) surface morphology, whereas the size of the organophosphonate molecule within one group does not seem to be important.
The dissolution of silica (either in the crystalline or amorphous form) is of great importance in geology, chemistry, biology, and water technology.276
The effect of various environmentally friendly chemical additives (among which are PBTC and DTPMP) on the dissolution of colloidal silica has been systematically studied.277 These silica scale dissolvers are principally polycarboxylates with one to five –COOH groups, mixed polycarboxylates/phosphonates and amino acids. Based on the results, the dissolution performance of the additives was correlated to their structural features in a structure/function study. The presence of additional groups (e.g. –PO3H2, –NH2, or –OH) in the dissolver molecule augmented the dissolution process. Three follow-up papers extended the study to other additives to include NTMP, PMIDA, PPCA, and EDTMP (as well as a plethora of other additives).278,279
3.6. Phosphonate-containing materials for metal ion adsorption and removal
Phosphonates have been used in chelation solvent extraction for separation of metal ions.280–282 This is due to their impressive chelation capabilities towards dissolved metal ions.283 In this section a survey of two types of phosphonate-bearing materials will be presented. The first type includes purely organic polymeric materials with phosphonates covalently grafted onto the polymer backbone. The second type consists of inorganic (metal-containing) materials modified with phosphonates (showing surface adsorption features), and metal phosphonate materials (in which the phosphonate is an integral part of the material network). A recent review on polymeric supports for water treatment applications was published.284Different polymeric sorbents exhibiting various physical states and containing phosphonic acids were synthesized for targeted separation processes.285 Phosphonic acids are the most studied materials for metal ion removal. Compared with carboxylic acids, phosphonic acids are more potent chelators of metal ions. Based on the pKa constants of the –PO3H2 group, the sorption capacity of phosphonic acid is pH dependent. The –PO3H2 group is singly dissociated at pH values higher than ∼2 and up to ∼7, whereas it loses its second proton at pH > 7.
A diethyl[3-(methoxydimethylsilyl)propyl]phosphonate (DMPP, Fig. 27) polymer was synthesized by reacting diethylallylphosphonate with poly(methylhydro)siloxane.287
 | ||
| Fig. 27 Schematic structure of the polymeric corrosion inhibitor diethyl[3-(methoxydimethylsilyl)propyl]phosphonate (DMPP). | ||
The synthesized polymer was used for sequestering Sr(II) in aqueous solutions. The metal binding was evaluated by EDS and SEM. Batch adsorption studies were performed by varying initial pH, adsorbent dose and the contact time. The reaction kinetics was determined by the Langmuir, Freundlich, and pseudo-first and second-order models. Results of this study indicated that DMPP was effective at recovering Sr(II) from the aqueous solution.
Two bifunctional extractants, which possess one phosphonic group on each side of the backbone, were synthesized and targeted for the separation of Zn(II) and Cu(II),288 see Fig. 28. Their separation properties and extractability were investigated using the liquid–liquid extraction technique. These extractants demonstrated high selectivity towards Zn(II) over Cu(II) compared with their monophosphonate analog. The length of the spacer was an important factor for selectivity enhancement. The analog with n = 8 (ODPA) was found to be optimal based on computational studies and performance data.
 | ||
| Fig. 28 Schematic structures of the bifunctional phosphonate extractants used as Zn(II) and Cu(II) adsorbers. | ||
A library of bisphosphonate-based ligands was prepared using solution-phase parallel synthesis and tested for its uranium-binding properties, see Fig. 29.289 Based on a chromophoric complex displacement procedure, 23 dipodal and tripodal chelates bearing bisphosphonate chelating functions were found to display very high affinity for the uranyl ion and were selected for evaluation of their in vivo uranyl-removal efficacy. Among them, 11 ligands induced a significant modification of the uranyl biodistribution by deviating the metal from kidneys and bones to liver. Among the other ligands, the most potent was the dipodal bisphosphonate 3C, which reduced the retention of uranyl and increased its excretion by around 10% of the injected metal. In addition, preliminary experiments on tripods 5A, 5H and 5C highlighted a high level of metal specificity toward the uranyl ion over Ca(II), Mg(II), and Zn(II), but a very low specificity over Fe(III).
 | ||
| Fig. 29 Synthetic methodology and the structures of the most potent bisphosphonate-based ligands. | ||
An aminophosphonate-modified chitosan-supported adsorbent was prepared from the natural biopolymer chitosan.290 In the first step, modified chitosan with aminophosphonic acid groups was prepared using the “one-pot” Kabachnik–Fields reaction. Next, the polymer was impregnated with Ni(II) ions using a hydrothermal reaction at different values of pH (5, 6 and 7). This composite (named ChitPNi) was used to remove Pb(II) ions from aqueous solutions by studying the effect of pH, contact time, and Pb(II) ion concentration. The kinetic and equilibrium studies confirmed the fact that the Pb(II) adsorption is a complex process involving both physisorption of the metal ions in the pores of the adsorbent and also coordination-facilitated chemisorption. An analogous phosphorylated chitosan-based material containing Co(II) ions was used to adsorb Sr2+ ions from aqueous solutions.291
An interesting study described a novel method to separate Cr(III) from aqueous solutions based on the use of solid bisphosphonates with a P–C–P backbone. Five classes of bisphosphonates with different functional groups and alkyl chain lengths at the center carbon (16 compounds in total) were prepared and their suitability for metal ion complexing as chelating agents was investigated, see Fig. 30.292 Two of the studied compounds, which were almost insoluble in water, were effective at removing Cr(III) quantitatively from aqueous solutions and real wastewater samples from tanneries. Cr(III) could be absorbed rapidly, safely and with good capacity from aqueous solutions and tannery effluents in the pH range of 2–10 by compound 5d. It was also possible to separate Cr(III) and Cr(VI) from each other by 5d since it did not bind Cr(VI). Furthermore, 5d had low affinity for Fe(III), thus it could be utilized in the removal of Cr(III) and other metals from solutions containing high concentrations of Fe(III).
 | ||
| Fig. 30 Schematic structures of the bisphosphonates used as Cr(III) adsorbers. | ||
The above study was further expanded and focused on 11-amino-1-hydroxyundecylidene-1,1-bisphosphonic acid (named N10O), see Fig. 31.293 N10O was evaluated for its ability to adsorb metal cations [Mg(II), Ca(II), Sr(II), Ba(II), Cr(III), Mn(II), Fe(II), Fe(III), Co(II), Ni(II), Cu(II), Zn(II), Cd(II), and Al(III)] from different sources, such as ground water and mining process waters. Additionally, it could also be regenerated and reused over a number of adsorption/desorption cycles, which is crucial for environmental friendliness. Appropriate comparisons were made with the commercially available Diphonix® cation exchange resin.294
 | ||
| Fig. 31 Schematic structure of N10O used as multi-metal adsorber. | ||
Phenylphosphonic acid was imbedded into the matrix of the polyurethane foam during its fabrication process.295 The extraction of uranyl ions by phosphonic acid-imbedded polyurethane foam and blank polyurethane (i.e., foam without phosphonic acid functional groups) was investigated. Phosphonic acid-imbedded foam showed superior extractability of uranyl ions from solutions with pH ∼7 over a wide range of temperatures. The ability to sorb uranyl from solutions at a temperature as low as 4 °C would make this polymer material attractive for use with environmental water at any time of the year.
The extraction of Au(III), Pt(IV), and Pd(II) ions from aqueous hydrochloric acid solutions with solutions of bis(2-ethylhexyl)-N-butyl-N-octylaminomethylphosphonate in chloroform and xylene was studied (Fig. 32).296 The recovery of the noble metal ions is most efficient at low acidities of the aqueous solution, with a high selectivity of separation from the concomitant Fe(III), Cu(II), Ni(II), and Co(II) ions.
 | ||
| Fig. 32 Schematic structure of the Au(III), Pt(IV), and Pd(II) extractant bis(2-ethylhexyl)-N-butyl-N-octylaminomethylphosphonate. | ||
A crosslinked polyacrylamide-based polymer bearing phosphonate groups was synthesized, characterized and investigated for the removal of alkaline earth metal ions from water.297 Parameters studied involved pH, contact time, polymer dose, and initial concentration of the metal ion. The optimum pH was found to be ∼7 for all tested metal ions. The maximum sorption capacities of the prepared polymer were 667, 794, 769 and 709 (mg g−1) for Mg, Ca, Sr and Ba ions, respectively. The exothermic sorption process obeyed the Langmuir isotherm model and followed a pseudo-second order mechanism. The possibility of polymer reuse was also investigated.
Phosphonate-grafted polystyrene-divinylbenzene resins were prepared by an Arbuzov-type reaction between chloromethyl polystyrene-divinylbenzene copolymers and triethylphosphite, yielding the phosphonate ester copolymer (resin A) (Fig. 33).298 This was then hydrolyzed by HCl to yield the phosphonate/phosphonic acid copolymer (resin B). The total sorption capacity of the phosphonate ester-functionalized resin (A) and phosphonate/phosphonic acid-functionalized resin (B) for divalent metal ions such as Ca2+, Cu2+, and Ni2+ was studied in aqueous solutions. Resin A retained ∼3.25 mg of Ca2+ per g of copolymer and 2.75 mg of Cu2+ per g of copolymer but retained no Ni2+ at pH 1. On the other hand, resin B retained 8.46 mg of Ca2+ per g of copolymer, 7.17 mg of Cu2+ per g of copolymer, and no Ni2+ at pH 1. Efficient Ni2+ retention was observed at pH 7 only for the phosphonate/phosphonic acid-functionalized resin (B) at the level of 19 mg of Ni2+ per g of polymer B.
 | ||
| Fig. 33 Schematic structures of the phosphonate ester copolymer (resin A) and the phosphonate/phosphonic acid functionalized resin (B). | ||
PPEI was synthesized from commercially available polyethylenimine and P,P-dichlorophenylphosphine oxide.299 It was investigated in the liquid–solid extraction of uranium. The extraction strongly depended on the pH, initial concentration of uranium, extractant to analyte ratio (mol/mol), ionic strength of the liquid medium and their mutual interactions. The recovery of U(VI) was almost quantitative.
 | ||
| Fig. 34 Schematic structure of the linker 1,3,5-benzenetriphosphonic acid (BTP). | ||
Phosphonates EDTMP and DTPMP were anchored to the titania network homogeneously.301 The synthesized titania–phosphonate hybrids possessed irregular mesoporosity formed by the assembly of nanoparticles in a crystalline anatase phase. These porous titania–phosphonate materials exhibited high photocatalytic activity in photodecomposition of rhodamine B dye molecules whether under UV or visible-light irradiation, and a large capacity for selective adsorption of Cd(II) ions, making them promising adsorbents and photocatalysts for practical applications including wastewater cleanup.
A three-dimensionally ordered macroporous titanium phosphonate material was synthesized by an inverse opal method using HEDP.302 The inorganic–organic hybrid titanium phosphonate frameworks possessed an amorphous nature with intraframework organic functional groups that acted as binding sites for heavy metal ion adsorption. The macroporous material was efficient in the adsorption of Cu2+, Cd2+, and Pb2+ ions. The results showed specificity for the adsorption of Cu2+ over Cd2+ and Pb2+.
The adsorption of aqueous HEDP, Cu(II), and Zn(II) onto boehmite (γ-AlOOH) as single solutes was evaluated.303 The results indicated that the presence of HEDP significantly promoted metal adsorption at low pH in defects of surface sites. In conclusion, the presence of HEDP in natural waters could aid in the removal of Cu(II) and Zn(II) dissolved by aluminum oxides.
A family of hybrid surface-phosphonated titania, titania–phosphonate, and titanium phosphonate porous materials with different organic groups in the network was synthesized by utilizing a series of organophosphonic acids as the coupling molecules.304 The used phosphonates were HEDP, EDTMP, and DTPMP. As the molar ratio of the added phosphonate and titanate increased, the structural phase transformed from crystalline phosphonated titania to semicrystalline titania–phosphonated hybrid to amorphous titanium phosphonate, and simultaneously the nanoarchitecture also changed from disordered mesoporous to hierarchically meso-/macroporous structure. Mesoporous phosphonated titanias exhibited unique selective complexation affinity for Cd2+ with the sequence of Pb2+ < Cu2+ < Cd2+, and titania–phosphonate showed a selective complexation affinity for Cu2+ with the sequence of Cd2+ < Pb2+ < Cu2+, while hierarchical meso/macroporous titanium phosphonates exhibited large capacity for all the three kinds of ion adsorption with a preference sequence of Cd2+ < Cu2+ < Pb2+ and effective regeneration ability.
Apatite particles prepared from natural phosphate rock and grafted with NTMP were evaluated for Pb2+ and Zn2+ sorption from aqueous solutions.305 Sorption capacities as high as 640 mg g−1 and 300 mg g−1 were obtained for the highest NTMP content (10 wt%). Analysis of the sorption isotherms using Langmuir, Freundlich and Dubinin–Kaganer–Radushkevich models revealed that Pb2+ ions had a higher affinity for apatite than for NTMP, hence the phosphonate-modified surfaces led to a heterogeneous adsorption process. In contrast, Zn2+ interacted weakly and in a similar fashion with the apatite surface and with the phosphonate moieties of NTMP, indicating a homogeneous sorption process. Such an association of organic metal ligands with reactive apatite surfaces within porous materials appears as a promising strategy to obtain efficient adsorbents at low cost and with limited environmental impact.
A nanostructured mesoporous silicon adsorbent with grafted (1-hydroxyundec-10-ene-1,1-diyl)bis(phosphonic acid) ligand (Fig. 35) was evaluated for selectively removing uranium from a tailing obtained from the processing of a real ore sample by the Knelson concentration method.306
 | ||
| Fig. 35 Schematic structure of (1-hydroxyundec-10-ene-1,1-diyl)bis(phosphonic acid). | ||
The adsorbent consisted of bisphosphonate ligands grafted on highly stable carbonized surfaces of mesoporous silicon. The porous structure of the adsorbent enhanced its permeability, allowing it to be used in a column setup where metal solutions were flowed through the adsorbent. The adsorbent was capable of repeatedly adsorbing and desorbing uranium without significant reduction in performance. Importantly, the adsorbent showed essentially higher selectivity towards uranium than towards other less harmful metal ions, and the material could be regenerated with an acid. Desorption was carried out with sulfuric acid, resulting in 15-fold enrichment of uranium compared with the initial solution, while other metals did not concentrate efficiently. The adsorbent was capable of selectively capturing uranium from a solution with various other metals, and the adsorbed uranium was rapidly desorbed and quantified with a reasonable purity, indicating the adsorbent as a potential candidate for industrial applications.
Nanostructured silicon carbide produced from silica particles was functionalized via grafting with bisphosphonate ((undec-10-ene-1,1-diyl-bisphosphonate) with up to 8 wt% content) for utilization as a metal ion adsorbent and evaluated for the adsorption of various metal ions (Al, Cr, Mn, Fe, Co, Ni, Zn, Pb) from landfill water with pH 8.307 The functionalization of the silicon carbide with bisphosphonates increased the adsorption capacity by 32% and the material was able to withstand at least 5 adsorption/desorption cycles.
A Zr(IV)-based coordination porous material (Zr-EDTMP) was synthesized and employed for the fabrication of sandwich adsorptive membranes.308 The removal of Cu(II) from water was studied, and the static adsorption thermodynamics, dynamic adsorption/desorption and reusability were investigated. The results showed that the adsorption capacity of the sandwich adsorptive membranes was much higher than that of conventional mixed matrix membranes. From the breakthrough curves, the relationship between the removal rate and the filtrate volume was obtained. The adsorption capacity was not compromised after five cycles, exhibiting excellent application potential for the removal of Cu(II).
A series of lanthanide (Nd, Eu, Tb and Er) diphosphonates was synthesized with the semirigid diphosphonate ligand, (5-methyl-1,3-phenylene)bis(methylene)bisphosphonic acid.309 These lanthanide diphosphonates were investigated for the luminescence sensing of ferric (Fe3+) and dichromate (Cr2O72−) ions, with good selectivity and sensitivity.
Two Cd(II) phosphonates, [Cd(MPBP)(bipy)(H2O)]·H2O (1) and [Cd(H2BPDP)(bipy)2(H2O)]·H2O (2) [H2MPBP = 4,4-Bis(monoethylphosphonomethyl)biphenyl, H4BPDP = biphenyl-4,4′-diylbis(methylene)diphosphonic acid, bipy = 2,2′-bipyridine] (Fig. 36) were synthesized under hydrothermal conditions.310 Both compounds exhibited remarkable luminescence properties and could be applied as highly selective and sensitive luminescence sensors for dichromate (Cr2O72−) and chromate (CrO42−) anions through the luminescence quenching effect. Low detection limits were found of 3.5 × 10−7 M and 4.6 × 10−7 M (for compound 1) and 6.1 × 10−7 M and 4.3 × 10−7 M (for compound 2), respectively.
 | ||
| Fig. 36 Schematic structures of the ligands H2MPBP (4,4-bis(monoethylphosphonomethyl)biphenyl), H4BPDP (biphenyl-4,4′-diylbis(methylene)diphosphonic acid), and bipy (2,2′-bipyridine). | ||
Phosphonate MOFs were obtained with divalent metals Co and Cu and phosphonoacetic (Cp) or styrylphosphonic (Sp) acids under hydrothermal conditions.311 The phosphonate MOFs were used for the removal of Cs(I) and Tl(I) ions from aqueous solutions carried out in batch mode. The experimental data showed that the adsorption capacity of the studied materials increased in the order CuCp < CoCp < CoSp, for the removal of both Cs(I) and Tl(I) ions. The adsorption kinetics showed a fit with a pseudo-second-order kinetic model. The experimental data showed a good fit with the Langmuir isotherm.
An inorganic–organic hybrid material manganese phosphonate (Mn-HEDP, MnP) was synthesized under hydrothermal conditions, and was used to remove Pb(II) and Cu(II) ions from aqueous solutions.312 The adsorption results showed that MnP had excellent adsorption capacity for Pb(II) and Cu(II), with increased selectivity for Pb(II). The Langmuir model was better than the Freundlich model to fit the adsorption isotherms of MnP for Pb(II) and Cu(II), and the maximum adsorption capacities for Pb(II) and Cu(II) were 740.74 mg g−1 and 154.32 mg g−1, respectively. The kinetics data indicated that the adsorption processes were governed by the film diffusion and followed pseudo-second-order kinetics.
Nanoplates of a hybrid cobalt phosphonate material (Poly-CoAEDP), AEDP = aminoethylidenediphosphonic acid, (Fig. 37) possessing a two-dimensional structure were synthesized under hydrothermal conditions and applied in the removal of Pb(II) and Hg(II) ions from aqueous solutions.313 Poly-CoAEDP exhibited good performance for heavy metal removal, especially for Pb(II). The effects of different experimental parameters on the competitive adsorption capacities were investigated. The results suggested that the nanoplates of poly-CoAEDP have promising applications in environmental protection, having demonstrated good adsorption ability for lead removal.
 | ||
| Fig. 37 Schematic structure of the linker aminoethylidenediphosphonic acid (AEDP). | ||
The Cr(VI) adsorption potential of three types of Co(II)-based phosphonate metal organic frameworks was investigated, where the phosphonate was phosphonoacetic acid (CP), N,N-bis(phosphonomethyl)glycine (Gly), or vinyl phosphonic acid (VP).314 The study involved batch types of experiments evaluating the effects of solution pH, solid:liquid ratio, initial concentration of Cr(VI), and contact time upon the adsorption efficiency of the studied materials. Langmuir, Freundlich, Temkin, and Dubinin–Radushkevich isotherm models were applied to adsorption equilibrium data to find the best among these models. The kinetics of adsorption was found to follow the pseudo-second-order model. It was found that the adsorption efficiency of the studied materials in the removal process of Cr(VI) ions from aqueous solutions followed the order Co-CP < Co-Gly < Co-VP. In a similar study by the same authors, analogous Ni(II)-based phosphonate metal organic frameworks were used for removal of Cr(VI) ions from aqueous solutions.315 The efficiency followed the order: Ni-CP < Ni-Gly ≤ Ni-VP.
Two open-framework and microporous materials, Pb-EDTMP and Zn-EDTMP, were synthesized and structurally characterized.316 Both compounds exhibited high ion sorption and exchange capacities for millimolar concentrations of Fe(III). They were also found to adsorb ferric ions selectively over other metal ions, such as Ca(II), Cr(II), Mn(II), Cu(II), Zn(II), Cd(II). Their special ferric ion uptake capacities may be attributed to the cation exchange, coordination bonding, and electrostatic attraction between ferric ions and the metal phosphonate framework.
An anionic zirconium phosphonate framework material using methylene diphosphonic acid [CH2(PO3H2)2], named SZ-5, was synthesized by solvothermal methods, and the crystal structure was elucidated.317 SZ-5 exhibited efficient Sr(II) exchange capability with high uptake capacity and selectivity, as demonstrated by the radioactive Sr-90 removal from a real contaminated seawater sample with an extremely high ionic strength. The efficient ion exchange ability opens potential applications of SZ-5 for remediation of contaminated water.
Two functional SBA-15 mesoporous silicas were prepared by a post-grafting method using the phosphonates diethylethylphosphonate (DEP) and ethylphosphonic acid (PA).318 The effect on uranium(VI) sorption behavior of the functionalized SBA-15 materials was studied. Typical sorption isotherms (Langmuir and Freundlich) were determined for the sorption process, and the maximum sorption capacity was calculated. Outer-sphere complexation for SBA-15 and inner-sphere complexation for SBA-15-DEP and SBA-15-PA were proposed as the main sorption mechanisms of U(VI), respectively. Material SBA-15-PA possessed good sorption ability and a desirable selectivity for U(VI) over a range of competing metal ions but also excellent reusability, and had potential application in the separation of uranium(VI).
A number of phosphonate ligands were incorporated into a zirconium-based metal–organic framework (MOF-808, with trimesic acid as the linker), via ligand exchange.319 The derived MOFs were synthesized by replacing the trimesic acid ligands with methylphosphonic acid (MPA), ethanephosphonic acid (EPA), and vinylphosphonic acid (VPA). The phosphonate-substituted MOFs (808-MPAs) demonstrated high porosity, with only small changes in the pore diameter and specific surface area when compared with the parent MOF-808. An expansion of the lattice for all MOFs after decorating with phosphonate ligands was observed. The phosphonate-substituted MOFs showed ultrafast adsorption performance for uranyl ions using the ion-exchange properties of the P–OH sites in their cavity environment, with an equilibrium time of 10 min, much quicker than the previous adsorbents. The present study demonstrated a proof-of-concept example of pKa-directed Zr-MOFs with tunable phosphonate ligands.
Hierarchically meso-/macroporous titanium tetraphosphonates (Ti-EDTMP) were prepared.320 The synthesized materials showed high efficiency for heavy metal ion adsorption with a preference sequence of Cd2+ < Cu2+ < Pb2+ and good regeneration ability, which could be attributed to the remarkable interaction between organic functional groups of Ti-EDTMP framework and heavy metal ions.
Various phosphonate-based hybrid materials containing metal centers such as aluminum,321 manganese,322 titanium,323 zirconium,324 and nickel325 were investigated for the adsorption behavior for heavy metal ions. Featuring tunable porosity, stable skeleton, and enriched organic functional groups, these phosphonate-based hybrids were outstanding adsorbents with impressive adsorption capability for Cu2+ and good reusability, indicating their great potential in environmental remediation.
3.7. Phosphonate-based materials for organic dye adsorption and removal
Co-, Ni-, and Cu-phosphonate compounds were prepared with vinyl phosphonic acid (VP) under hydrothermal conditions.326 The synthesized materials were evaluated for their efficiency as adsorbents for diverse initial dye concentrations at different pH values and temperatures. They demonstrated good efficiency in the elimination of anionic as well as cationic types of dye from aqueous solutions. The highest adsorption capacities were obtained working at optimum solution pH 4.2 for Acid Orange 7 and 10.0 for Basic Fuchsine, using 1 g L−1 of adsorbent at ambient temperature. The adsorption capacities increased in the order CuVP < NiVP < CoVP.A hybrid ion exchange material Zr(IV)-HEDP was synthesized by the sol–gel method.327 It was used for wastewater treatment containing the anti-infective pharmaceutical products Acriflavin (AF) and Brilliant Green (BG). Breakthrough capacity indicated good affinity of dyes towards Zr-HEDP, found to be in the order BG > AF.
Periodic mesoporous titanium phosphonate (PMTP-1) materials were synthesized and used as an adsorbent for the removal of a cationic dye, methylene blue (MB), from aqueous solutions.328 The adsorption equilibrium was achieved after 30 minutes of contact time, and the adsorption of MB on PMTP-1 had the best fit with the Langmuir isotherm model with the maximum monolayer adsorption capacity of 617.28 mg g−1. The study results indicated that the PMTP-1 powder could be used as an efficient adsorbent for the removal of textile dyes from effluents.
Using surfactant-assistant solvo/hydrothermal methods, nanoscale calcium aminodiphosphonate species (CadP) with different morphologies, namely nanobelts, nanorods, and nano-networks were synthesized.329 (Cyclohexylazanediyl)bis(methylene)diphosphonic acid was used as the phosphonate source (Fig. 38). Removal of MB from aqueous solution was investigated using synthesized nano-networks (CadP-N) as adsorbents. The effects of process parameters such as adsorbent morphology, adsorbent dose, pH, contact time, and initial dye concentration on the extent of MB adsorption were investigated. Both the Langmuir and the Freundlich isotherms were fit to explain the MB adsorption on CadP-N. The maximum adsorption capacity of CadP-N was found to be 138.89 mg g−1.
 | ||
| Fig. 38 Schematic structure of the linker (cyclohexylazanediyl)bis(methylene)diphosphonic acid. | ||
A Mn(II) phosphonate has been shown to be an excellent adsorbent for the rapid separation of methyl orange (MO) dye from aqueous solution, having the advantages of rapid adsorption rate, high uptake capacity, and good recovery of raw materials.330 The phosphonate used was thiophene-2-phosphonic acid. Interestingly, it also displayed visible-light-induced photosensitive behavior, and a ligand-to-ligand electron-transfer mechanism was proposed to rationalize its photochromic phenomena.
A study reported on a nanomaterial containing a novel tri-cationic phosphonate ionic liquid (containing the phosphonate NTMP), copper ferrite magnetic nanoparticles and partially oxidized modified boron nitride nanosheets.331 This hybrid material was used as an efficient catalyst to reduce a series of nitro-anilines, and dyes. The catalytic reduction of a series of nitro-anilines such as 2-nitroaniline (2-NA), 3-nitroaniline (3-NA), 4-nitroaniline (4-NA) and 4-nitro-2-phenylenediamine (4-NPDA) and dyes methylene blue (MB) and allura red (AR) in aqueous solution at ambient temperature was effective in all compounds tested (Fig. 39). The order of reduction, based on reaction time, was 4-NPD > 4-NA > 3-NA > 2-NA. The nanocatalyst was easily recovered and re-used for more than seven times with negligible loss of its catalytic activity.
 | ||
| Fig. 39 Schematic structures of 2-nitroaniline (2-NA), 3-nitroaniline (3-NA), 4-nitroaniline (4-NA), 4-nitro-2-phenylenediamine (4-NPDA), methylene blue (MB) and allura red (AR). | ||
A series of phosphonate-based MOF materials, namely STA-12(M) (M = Mn, Fe, Co, Ni), demonstrated photocatalytic activity for the degradation of methylene blue and methyl orange from aqueous solution augmented by natural sunlight.332 Thus, photoFenton oxidative discoloration of dyes was carried out by H2O2 catalyzed by STA-12(M). This work demonstrated an example of facilitating photoFenton excitation of H2O2via phosphonate-based metal organic frameworks as photocatalysts and opened a new opportunity for sunlight-induced photocatalytic environmental remediation and protection. In a similar study, STA-12(Fe) exhibited synergistic photocatalytic activity for both degradation of dyes MO and rhodamine B and for Cr(VI) reduction.333
P,P-Bis(2-oxooxazolidin-3-yl)-N-(3-(triethoxysilyl)propyl)phosphinic amide (APTES-BOP)-modified SBA-15 (SBA-15-BOP) (Fig. 40) was prepared by a post-synthesis grafting method for the removal of anionic azo dyes from aqueous solutions.334 SBA-15-BOP showed high adsorption capacity for Congo red (CR) and Reactive Red 2 (RR2) dyes, with the maximum adsorption capacities of 518.1 mg g−1 and 253.8 mg g−1, respectively. The thermodynamic study indicated that the adsorption processes of CR and RR2 dyes on SBA-15-BOP were spontaneous and exothermic.
 | ||
| Fig. 40 Schematic structure of P,P-bis(2-oxooxazolidin-3-yl)-N-(3-(triethoxysilyl)propyl)phosphinic amide (APTES-BOP). | ||
A cadmium-phosphonate coordination polymer was prepared containing a tetrahedrally shaped tetraphosphonic acid linker (H8L = tetraphenylsilane tetrakis-4-phosphonic acid) and the auxiliary ligand 1,10-phenanthroline (phen) (Fig. 41).335 The resulting three-dimensional compound Cd4(H4L)2(phen)2(H2O)4 exhibited a rapid and efficient adsorption of the Congo Red dye, the adsorption capacity of which reached 684 m g−1. Furthermore, this adsorbent showed excellent structural stability and adsorptive recyclability after three cycles of adsorption and desorption. The absorption kinetics had a good fit with the pseudo-second-order kinetic model, revealing that the adsorption of the cadmium-phosphonate compound underwent a chemical process, where hydrogen bonding between the amide groups (from Congo Red) and the uncoordinated phosphonate oxygen atoms of the compound played a pivotal role.
 | ||
| Fig. 41 Schematic structures of the tetrahedrally-shaped tetraphosphonic acid linker tetraphenylsilane tetrakis-4-phosphonic acid and the auxiliary ligand 1,10-phenanthroline (phen). | ||
4. Possibilities for recycling of phosphonates/phosphorus
As we have seen in previous sections, phosphonates play a pivotal role in several water treatment applications and other sustainability related processes, and synthetic methods that obey the twelve rules of green chemistry have been developed to manufacture these compounds in more sustainable manner. In this fourth part, we will focus on the question: is it possible to recycle phosphonates or should they just be removed? To understand the importance and challenges of phosphonate recycling, we will take a short look at the general recycling of phosphorus.Phosphorus is crucial for all living organisms’ natural life cycles on Earth. As an elemental raw material, it is not renewable, and unlike fossil-derived compounds it cannot be substituted in industrial or biological systems. Excess phosphorus is a strong pollutant causing eutrophication in aquatic systems, which in turn leads to the proliferation of algae followed by anoxia and lethality to fish. The principal demand for removing phosphorus from wastewater and restricting its leakage into natural stems from this polluting effect.
The main consumer of phosphorus in the chemical industry is the fertilizer industry. Phosphorus-containing fertilizers are manufactured mainly from impure phosphoric acid, so called “green acid” prepared from phosphate rock using sulphuric acid. For fine-chemical production, this impure phosphoric acid is further purified and used to generate starting materials for e.g. phosphonates.336 As a raw material, phosphate rock sources are unequally distributed. There are insufficient phosphate rock sources in Europe to fulfil its needs. However, after the European Union defined phosphorus as a critical raw material, interest in recycling and recovery of phosphorus instead of just removing it has emerged.
Similarly, most of the phosphorus discharge into nature originates from agriculture, e.g. animal manure and leaking phosphorus from fertilizers. Additionally, municipal waste (wastewater, manure, solid waste, water treatment sludges etc.) and industrial waste (wastewater, waste sludges, side streams etc.) contain remarkable amounts of phosphorus. In these streams, phosphorus may exist in either organic or inorganic compounds, and most methods to remove, recycle and recover phosphorus are based on the removal of phosphate ions from solution. Numerous methods for wastewater treatment to remove phosphorus in a cost-efficient manner have been studied. Among these, methods to convert organic phosphorus and insoluble inorganic phosphorus into soluble inorganic phosphorus have been developed to enrich the phosphorus content. Further methods to recover phosphorus from enriched streams are usually based on binding and conversion of soluble phosphorus (mainly as phosphate, PO43−) to less soluble compounds.337
In wastewater treatment plants, approximately 90% of phosphorus binds onto sewage sludge. Because the use of pure sludge as fertilizer has met with criticism, recovery of its phosphorus has gained emerging interest. Methods to reduce the unnecessary volume and mass (water, organic compounds) include, for example, incineration and pyrolysis, and their effects on phosphorus content and recovery have been studied.338,339 Pyrolysis and incineration products of domestic sewage sludge as well as (dried) sludge and other phosphorus-containing wastes, e.g. slaughterhouse waste, have been evaluated as fertilizers without any further recovery of phosphorus.340–342 However, the risks of using pure sewage sludge as a fertilizer pose significant concerns.343,344
Extraction of phosphorus from water treatment sludge has been demonstrated using chemical leaching with either added sulphuric acid or by bioleaching using in situ biologically generated sulphuric acid.345 Recovery of phosphorus from the original sewage sludge was shown to be higher than from incinerated sewage sludge ashes. The authors proposed that the lower recovery of phosphorus is due to partial loss due to emission of phosphorus compounds during incineration. Another downside is chemical leaching, because acidic conditions also cause leaching of (heavy) metals including Ca, Fe, Al, Cd, Pb, Cu, and Cr, which consequently contaminate the final product. Although Fe and Al are much less toxic than heavy metals, they are strongly chelated by phosphorus-containing compounds, thus limiting the bioavailability of phosphorus from sludge-derived fertilizers.346
In addition to physicochemical methods for valorisation of sludges, biological methods for enriching phosphorus from sewage have also been developed. As an example, an increase in the phosphorus content from 10 mg mL−1 to 100 mg mL−1 of PO43− was achieved when a simultaneous nitrification, denitrification and phosphorus recovery (SNDPr) protocol was applied.347 Other methods used to recover phosphorus from solution include the use of adsorbents, nanofiltration, precipitation and crystallization of sparingly soluble phosphate salts, membrane filtration and ion exchange.
Among the aforementioned technologies, adsorption seems to be the most cost-efficient method. Thus, diverse adsorbents have been studied in the recent literature. Notable examples include metal-modified biochar particles treated with a single metal, e.g., Fe,348 Mg, or Ca,349 or a combination of metals, like Fe–Mn.350 In addition to biochar, other sustainable materials have been studied, e.g. recycled sponge iron,351 and pine needles,352 as well as other inorganic or synthesized nanoparticles, like metal-modified mesoporous silica353 or Zr-doped layered double hydroxides-based microspheres.354
After removing, recovering, and concentrating phosphorus, it can be used for the preparation of inorganic phosphorus salts, like struvite (MgNH4PO4·6H2O) or calcium phosphate (Ca3(PO4)2). Preparation of struvite has been studied at a modified pilot scale to demonstrate the recovery of phosphorus from tapioca starch anaerobic digestion effluent355 and its theoretical background was studied using ion exchange utilized for struvite synthesis.356 In a recent study, a 10 m3 day−1 hybrid anion exchanger (HAIX) demonstration plant was used to show that ion exchange can be efficiently used for removal of PO43− as calcium phosphate.357 The average phosphate ion concentration was 6 mg L−1 and 0.5 mg L−1 for inlet wastewater and outlet wastewater, respectively.
In the recent literature, several methods have been developed to lower the metals contaminating the end product of phosphorus recovery. Three organic extractants were tested in the reactive extraction from acidic sludge ashes leaching liquor. These were Alamine 336 (trioctylamine), Ketrul D80 (a medium petroleum fraction) and Exxal 10 (isodecanol) and were shown to reduce the metal ion contamination.358 Also, nanofiltration using a modified polyamide filter was shown to increase the ratio of phosphorus to heavy metal ions.359 Nanofiltration has also been generally used to concentrate the phosphate ions from solutions of sewage sludge ashes.360 Both the abovementioned nanofiltration studies emphasised the importance of careful tuning of process parameters like pH, pressure, and composition of feed solution to obtain the optimal result.
4.1. Targeted removal and recovery of phosphorus and phosphonates
In contrast to domestic wastewater, industrial wastewater and industrial side streams contain well-defined contaminants in specific concentration ranges. Usually, concentrations of these contaminants are higher in industrial than in domestic wastewater, thus making the waste treatment strategies challenging. Some examples are mentioned below. The first is related to the phosphorus-containing antibiotic, Fosfomycin, which is found in the effluent of certain pharmaceutical industries.361 The second refers to the antiscalant additive NTMP discussed above in section 3.2.362 Finally, phosphogypsum is formed as a solid side product from phosphoric acid production from phosphate rock.363 These examples from industry emphasize the challenges posed by the toxic or harmful features of the effluent due to either the high toxicity of certain contaminants (e.g. arsenic in phosphogypsum wastewater) or the high concentration of a contaminant (e.g. the case of fosfomycin). The recovery of phosphorus from organophosphorus compounds is often achieved by first converting the phosphorus-containing constituents into inorganic phosphate and then removing it. In their recent review article, Altaf et al. discussed adsorption methods for phosphonate removal and recovery.364 Therefore, in the present review we will focus on other strategies to remove and recover phosphonates.In the recent literature, NTMP has been oxidized and precipitated as Ca3(PO4)2 in an electrochemical system.365 Another example of oxidation and recovery of phosphonate is the removal of HEDP using ozone in combination with lava rock or expanded clay aggregates as packing material in the oxidation column. According to this method, HEDP was decomposed to PO43− and adsorbed onto iron-coated sand granules.366 The same authors also used non-thermal plasma treatment to improve their system.367 Other methods for degrading phosphonates are discussed above in section 3.1.
In a very recent study, nanofiltration and catalytic oxidation of another antiscalant and anticorrosive agent, EDTMP, were used.368 For the removal (but not recovery) of phosphonates, MOFs (also discussed above in this paper) have been utilized for selective removal of phenyl phosphonic acid.369 The MOF material was efficient, and reusable after regeneration. Another efficient method to remove and recover HEDP and NTMP from reverse osmosis concentrates was electrochemical flocculation. The phosphonates were also recovered from the formed floc. The authors claimed that electrochemical flocculation is an efficient and cost-effective method for water treatment and antiscalant recovery.370
Although several methods to remove phosphorus from water have been developed, only a few of these achieve recycling of the original phosphonate back into use. One such method is the use of membranes in organocatalytic reactions to separate the phosphorus-containing organocatalyst from the reaction medium and recycle/reuse the catalyst.371
5. Conclusions and future prospect of green phosphonate chemistry
It comes as no surprise that the answer to the title question cannot be a simple “yes” or “no”. Looking at the big picture, phosphorus is critical raw material (as already mentioned in the Introduction) and there are, thus far, no sufficient and satisfactory recycling solutions. Hence, if one considers that every compound/molecule that contains phosphorus (particularly the organic ones) can be tagged as “critical” material, then the answer is “no”. However, according to the literature data and examples presented in this review, there are several sustainable and green methods to prepare phosphonates, and several studies indicate the recyclability potential of phosphonates in water treatment processes.The major single source of phosphorus pollution is agriculture and related activities. Hence, historically, methods for phosphonate recovery have not gained particular attention. However, recently, intense interest has emerged to develop strategies for phosphonate recovery, instead of resorting to degradation processes first. Due to pressing needs for more efficient phosphorus recycling and widespread acceptance of the principles of green chemistry (especially by the general public), there is an urgent need for methods to produce starting materials for the chemical industry (both at the fine/specialty and bulk level) from recovered phosphorus.
The quest for further “greening” of all aspects of phosphonate chemistry (from synthesis, all the way to applications and the end-user) is ongoing. Importantly, phosphonate chemistry is a highly interdisciplinary area, and it can be viewed as a convergence point of several scientific and technical disciplines. Hence, making phosphonate chemistry greener can bring together scientists from all directions in a concerted and combined effort. Such activities are certainly welcome from the scientific community and the general public.
Data availability
Data availability is not applicable to this article as no new data were created or analyzed in this study.Conflicts of interest
There are no conflicts to declare.References
- S. Allen, D. Allen, S. Karbalaei, V. Maselli and T. R. Walker, J. Hazard. Mater. Adv., 2022, 6, 100057 Search PubMed.
- R. Marfella, F. Prattichizzo, C. Sardu, G. Fulgenzi, L. Graciotti, T. Spadoni, N. D'Onofrio, L. Scisciola, R. La Grotta, C. Frigé, V. Pellegrini, M. Municinò, M. Siniscalchi, F. Spinetti, G. Vigliotti, C. Vecchione, A. Carrizzo, G. Accarino, A. Squillante, G. Spaziano, D. Mirra, R. Esposito, S. Altieri, G. Falco, A. Fenti, S. Galoppo, S. Canzano, F. C. Sasso, G. Matacchione, F. Olivieri, F. Ferraraccio, I. Panarese, P. Paolisso, E. Barbato, C. Lubritto, M. L. Balestrieri, C. Mauro, A. E. Caballero, S. Rajagopalan, A. Ceriello, B. D'Agostino, P. Iovino and G. Paolisso, N. Engl. J. Med., 2024, 390, 900–910 CrossRef PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- A. D. Kreuder, T. House-Knight, J. Whitford, E. Ponnusamy, P. Miller, N. Jesse, R. Rodenborn, S. Sayag, M. Gebel, I. Aped, I. Sharfstein, E. Manaster, I. Ergaz, A. Harris and L. N. Grice, ACS Sustainable Chem. Eng., 2017, 5, 2927–2935 CrossRef.
- P. A. Turhanen, J. Leppänen and J. J. Vepsäläinen, ACS Omega, 2019, 4, 8974–8984 CrossRef CAS PubMed.
- T.-L. Chen, H. Kim, S.-Y. Pan, P.-C. Tseng, Y.-P. Lin and P.-C. Chiang, Sci. Total Environ., 2020, 716, 136998 CrossRef CAS PubMed.
- M. Krečmerová, P. Majer, R. Rais and B. S. Slusher, Front. Chem., 2022, 10, 889737 CrossRef PubMed.
- P. A. Turhanen, K. D. Demadis and P. Kafarski, Front. Chem., 2021, 9, 695128 CrossRef.
- https://en.wikipedia.org/wiki/Etidronic_acid .
- P. Kafarski, in Contemporary Topics about Phosphorus in Biology and Materials, ed. D. G. Churchill, M. Dutour Sikirić, B. Čolović and H. Füredi Milhofer, IntechOpen, 2020 Search PubMed.
- Y. Cao, Q. Peng, S. Li, Z. Deng and J. Gao, RSC Adv., 2019, 9, 42204–42218 RSC.
- E. Zangelmi, F. Ruffolo, T. Dinhof, M. Gerdol, M. Malatesta, J. P. Chin, C. Rivetti, A. Secchi, K. Pallitsch and A. Peracchi, iScience, 2023, 26, 108108 CrossRef PubMed.
- M. Gahungu, A. Arguelles-Arias, P. Fickers, A. Zervosen, B. Joris, C. Damblon and A. Luxen, Bioorg. Med. Chem., 2013, 21, 4958–4967 CrossRef CAS.
- M. Krečmerová, P. Majer, R. Rais and B. S. Slusher, Front. Chem., 2022, 10, 889737 CrossRef.
- E. de Clercq, Biochem. Pharmacol., 2011, 82, 99–109 CrossRef CAS PubMed.
- E. de Clercq and G. Li, Clin. Microbiol. Rev., 2016, 29, 695–747 CrossRef.
- G. Andrei, D. Topalis, T. de Schutter and R. Snoeck, Antiviral Res., 2015, 114, 21–46 CrossRef CAS PubMed.
- J. Park, V. R. Pandya, S. J. Ezekiel and A. M. Berghuis, Front. Chem., 2020, 8, 612728 CrossRef CAS.
- E. S. Rudge, A. H. Y. Chan and F. J. Leeper, RSC Med. Chem., 2022, 13, 375–391 RSC.
- M. C. Manghi, M. Masiol, R. Calzavara, P. L. Graziano, E. Peruzzi and B. Pavoni, Chemosphere, 2021, 283, 131187 CrossRef CAS.
- https://www.waterlinepublication.org.uk/articles/chelants-in-water-treatment-an-overview/ .
- https://en.wikipedia.org/wiki/Critical_Raw_Materials_Act .
- https://single-market-economy.ec.Europa.eu/sectors/raw-materials/areas-specific-interest/critical-raw-materials_en .
- S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Chem. Rev., 2022, 122, 3637–3710 CrossRef PubMed.
- T. Olszewski, Synthesis, 2014, 46, 403–429 Search PubMed.
- Z. Rádai and G. Keglevich, Molecules, 2018, 23, 1493 Search PubMed.
- B. Dar, A. Singh, A. Sahu, P. Patidar, A. Chakraborty, M. Sharma and B. Singh, Tetrahedron Lett., 2012, 53, 5497–5502 Search PubMed.
- G. Gao, M.-N. Chen, L.-P. Mo and Z.-H. Zhang, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 528–532 CrossRef.
- R. M. N. Kalla, J. Bae and I. Kim, New J. Chem., 2017, 41, 6653–6660 RSC.
- A. Bouzina, N. Aouf and M. Berredjem, Res. Chem. Intermed., 2016, 42, 5993–6002 CrossRef.
- S. A. Rasal, M. S. Tamore and N. G. Shimpi, ChemistrySelect, 2019, 4, 2293–2300 CrossRef.
- M. J. Nasab and A. R. Kiasat, Microporous Mesoporous Mater., 2016, 223, 10–17 CrossRef.
- P. G. Mandhane, R. S. Joshi, D. R. Nagargoje and C. H. Gill, Tetrahedron Lett., 2010, 51, 1490–1492 CrossRef.
- K. U. Maheswara Rao, G. D. Reddy and C.-M. Chung, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 1104–1109 CrossRef.
- P. V. Shinde, A. H. Kategaonkar, B. B. Shingate and M. S. Shingare, Tetrahedron Lett., 2011, 52, 2889–2892 CrossRef.
- R. Amin and T. Ara, Phosphorus, Sulfur Silicon Relat. Elem., 2023, 198, 417–423 CrossRef.
- S. Sharafian, Z. Hossaini, F. Rostami-Charati and M. A. Khalilzadeh, Chem. Heterocycl. Compd., 2020, 56, 1283–1291 CrossRef.
- D. S. Rao, G. Madhava, S. Rasheed, S. Thahir Basha, M. N. Lakshmi Devamma and C. Naga Raju, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 574–584 CrossRef CAS.
- E. L. Loredo-Calderón, C. A. Velázquez-Martínez, M. A. Ramírez-Cabrera, E. Hernández-Fernández, V. M. Rivas-Galindo, E. A. Espinoza and S. T. López-Cortina, Med. Chem. Res., 2019, 28, 2067–2078 CrossRef.
- Á. Tajti, E. Bálint and G. Keglevich, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 379–381 CrossRef.
- A. Hirao, S. Itsuno, S. Nakahama and N. Yamazaki, J. Chem. Soc., Chem. Commun., 1981, 315–317 RSC.
- R. Henyecz, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 372–376 CrossRef CAS.
- R. Henyecz and G. Keglevich, Curr. Org. Synth., 2019, 16, 523–545 CrossRef CAS.
- B. Huszár, R. Szolga, S. Bősze, R. O. Szabó, A. Simon, K. Karaghiosoff, M. Czugler, L. Drahos and G. Keglevich, Chemistry, 2023, 29, e202302465 CrossRef.
- P. Jansa, A. Holý, M. Dračinský, O. Baszczyňski, M. Česnek and Z. Janeba, Green Chem., 2011, 13, 882 RSC.
- D. Ravikumar, C. Subramanyam, S. Mohan, D. Chandra Sekhar and K. Prasada Rao, Mater. Today: Proc., 2018, 5, 25832–25842 Search PubMed.
- R. Bahadi, M. Berredjem, C. Benzaid, F. Bouchareb, A. Dekir, M. L. Djendi, M. Ibrahim-Ouali, M. Boussaker, S. Bouacida, A. R. Bhat, S. Ahmed, K. Bachari and R. Redjemia, J. Mol. Struct., 2023, 1289, 135849 CrossRef.
- D. Renuka, G. Mohan, S. Santhisudha and C. S. Reddy, Russ. J. Gen. Chem., 2023, 93, 136–147 CrossRef.
- S. Sarva, R. Dunnutala, S. Tellamekala, M. Gundluru and S. R. Cirandur, Synth. Commun., 2022, 52, 268–279 CrossRef.
- Y. H. Shaik, V. Chintha, M. Gundluru, S. Sarva and S. R. Cirandur, Synth. Commun., 2022, 52, 129–144 CrossRef.
- M. Sudileti, S. Nagaripati, M. Gundluru, V. Chintha, S. Aita, R. Wudayagiri, N. Chamarthi and S. R. Cirandur, ChemistrySelect, 2019, 4, 13006–13011 CrossRef.
- M. Gundluru, S. Sarva, S. Poreddy, B. Poojitha, S. Ethiraj and S. R. Cirandur, Synth. Commun., 2023, 53, 2117–2133 CrossRef.
- M. P. P. Kumar, A. Vejendla, C. Gladis Raja Malar, S. Chennamsetty, S. Talari and N. Vedula, Phosphorus, Sulfur Silicon Relat. Elem., 2023, 198, 808–821 CrossRef.
- S. R. Devineni, S. Doddaga, R. Donka and N. R. Chamarthi, Chin. Chem. Lett., 2013, 24, 759–763 CrossRef.
- S. K. T. Basha, R. M. N. Kalla, M. Varalakshmi, H. Sudhamani, R. M. Appa, S. C. Hong and C. N. Raju, Mol. Divers., 2022, 26, 2703–2715 CrossRef PubMed.
- B. Kaboudin, S. Faghih, S. Alavi, M. R. Naimi-Jamal and A. Fattahi, Synthesis, 2023, 55, 121–130 CrossRef.
- G. I. Shakibaei, S. Samadi, R. Ghahremanzadeh and A. Bazgir, J. Comb. Chem., 2010, 12, 295–297 CrossRef.
- C. K. Khatri, V. B. Satalkar and G. U. Chaturbhuj, Tetrahedron Lett., 2017, 58, 694–698 CrossRef.
- S. Tellamekala, M. Gundluru, S. Sarva, M. R. Nadiveedhi, M. Sudileti, R. Allagadda, A. R. Chippada and S. R. Cirandur, Synth. Commun., 2019, 49, 563–575 CrossRef.
- Z. Karimi-Jaberi, M. Mardani and M. Amiri, Green Process. Synth., 2012, 1, 191–193 Search PubMed.
- S. Poola, M. R. Nadiveedhi, S. Sarva, M. Gundluru, S. Nagaripati, M. S. Shaik, P. Kotha, N. Chamarthi and S. R. Cirandur, Med. Chem. Res., 2019, 28, 528–544 CrossRef.
- M. S. Shaik, M. R. Nadiveedhi, M. Gundluru, S. Poola, R. Allagadda, A. R. Chippada and S. R. Cirandur, Synth. Commun., 2020, 50, 587–601 CrossRef.
- C. S. Sundar, N. B. Reddy, S. S. Prasad, K. U. M. Rao, S. H. Jaya Prakash and C. S. Reddy, Phosphorus, Sulfur Silicon Relat. Elem., 2014, 189, 551–557 CrossRef CAS.
- J. Taran, A. Ramazani, H. Aghahosseini, F. Gouranlou, R. Tarasi, M. Khoobi and S. W. Joo, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 776–781 CrossRef.
- S. S. Sudha, C. S. Sundar, N. B. Reddy, K. U. M. Rao, S. H. Jaya Prakash and C. S. Reddy, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 1402–1411 CrossRef.
- K. Datta, B. Mitra and P. Ghosh, ChemistrySelect, 2023, 8, e202301255 CrossRef.
- N. Hazeri, M. T. Maghsoodlou, S. M. Habibi-Khorassani, J. Aboonajmi, M. Lashkari and S. S. Sajadikhah, Res. Chem. Intermed., 2014, 40, 1781–1788 CrossRef.
- Y.-Q. Yu, Synthesis, 2013, 45, 2545–2550 CrossRef.
- R. B. Shnigirev, E. V. Kondrashov, I. A. Ushakov and A. Y. Rulev, Tetrahedron Lett., 2021, 85, 153466 CrossRef.
- C. Wang, J. Zhou, J. Liu, L. Chu and H. He, Phosphorus, Sulfur Silicon Relat. Elem., 2014, 189, 1361–1366 CrossRef.
- K. U. M. Rao, C. S. Sundar, S. S. Prasad, C. R. Rani and C. S. Reddy, Bull. Korean Chem. Soc., 2011, 32, 3343–3347 CrossRef.
- C. Wang, J. Zhou, X. Lv, J. Wen and H. He, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 1334–1339 CrossRef.
- M. Varalakshmi, D. Srinivasulu and V. S. Kotakadi, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 1518–1524 CrossRef.
- U. R. Kunda, V. R. Mudumala, C. S. Reddy Gangireddy, B. R. Nemallapudi, K. N. Sandip and S. R. Cirandur, Chin. Chem. Lett., 2011, 22, 895–898 CrossRef.
- H. Jelali, I. S. Al Nasr, W. S. Koko, T. A. Khan, E. Deniau, M. Sauthier, F. Alresheedi and N. Hamdi, J. Heterocycl. Chem., 2022, 59, 493–506 CrossRef CAS.
- S. Sobhani and M. Honarmand, Catal. Lett., 2013, 143, 476–485 CrossRef CAS.
- X. Zhu, D. Guo, Y. Zhang, Y. Wei, S. Zhou, M. Xu, S. Wang, Y. Yang and Y. Qi, Organometallics, 2020, 39, 4584–4591 CrossRef CAS.
- H. Aghahosseini, A. Ramazani, J. Taran, K. Ślepokura and T. Lis, Asian J. Org. Chem., 2019, 8, 1519–1527 CrossRef.
- C. Sampath and P. Harika, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 1893–1900 CrossRef.
- M. Shen, S. Shang, Z. Song, D. Wang, X. Rao, H. Gao and J. Wang, Synth. Commun., 2014, 44, 361–367 CrossRef.
- F. Jahani, B. Zamenian, S. Khaksar and M. Tajbakhsh, Synthesis, 2010, 3315–3318 Search PubMed.
- H.-S. Wang and J.-E. Zeng, Phosphorus, Sulfur Silicon Relat. Elem., 2010, 185, 1425–1428 CrossRef.
- M. Rajasekhar, K. U. Maheswara Rao, C. S. Sundar, N. B. Reddy, S. K. Nayak and C. S. Reddy, Chem. Pharm. Bull., 2012, 60, 854–858 CrossRef PubMed.
- S. N. Murthy, B. Madhav, V. P. Reddy and Y. Nageswar, Tetrahedron Lett., 2010, 51, 3649–3653 CrossRef.
- R. Mohammadi and M. Z. Kassaee, J. Mol. Catal. A: Chem., 2013, 380, 152–158 CrossRef.
- K. Ramesh, B. Madhav, S. N. Murthy and Y. V. Durga Nageswar, Synth. Commun., 2012, 42, 258–265 CrossRef.
- Z. Hossaini, F. Rostami-Charati, M. Ghambarian and S. A. Siadati, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 1177–1182 CrossRef.
- Z. Hossaini, F. R. Charati and M. Ghasemian, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 555–560 CrossRef.
- E. Ezzatzadeh and Z. Hossaini, Appl. Organomet. Chem., 2020, 34, e5596 CrossRef.
- M. S. R. Murty, M. R. Katiki, B. R. Rao, J. B. Nanubolu, S. K. Buddana and R. S. Prakasham, Synth. Commun., 2014, 44, 2724–2737 CrossRef.
- R. Navrátil, K. Kellovská and O. Baszczyňski, Green Chem., 2023, 25, 9779–9794 RSC.
- R. D. Mandal, M. Saha, D. Das and A. R. Das, J. Org. Chem., 2023, 88, 6071–6095 CrossRef.
- W. Xie, N. Liu, B. Gong, S. Ning, X. Che, L. Cui and J. Xiang, Eur. J. Org. Chem., 2019, 2498–2501 CrossRef.
- Y. Yuan, J. Qiao, Y. Cao, J. Tang, M. Wang, G. Ke, Y. Lu, X. Liu and A. Lei, Chem. Commun., 2019, 55, 4230–4233 RSC.
- Q.-L. Wu, X.-G. Chen, C.-D. Huo, X.-C. Wang and Z.-J. Quan, New J. Chem., 2019, 43, 1531–1535 RSC.
- M. Kondo, A. Sugizaki, M. I. Khalid, H. D. P. Wathsala, K. Ishikawa, S. Hara, T. Takaai, T. Washio, S. Takizawa and H. Sasai, Green Chem., 2021, 23, 5825–5831 RSC.
- S. Guo, S. Li, Z. Zhang, W. Yan and H. Cai, Tetrahedron Lett., 2020, 61, 151566 CrossRef.
- A. B. Patil, D. S. Patil and B. M. Bhanage, Mater. Lett., 2012, 86, 50–53 CrossRef.
- B.-R. Shen, P. Annamalai, R. Bai, S. S. Badsara and C.-F. Lee, Org. Lett., 2022, 24, 5988–5993 CrossRef PubMed.
- C. Qu, J. Hao, H. Ding, Y. Lv, X.-E. Zhao, X. Zhao and W. Wei, J. Org. Chem., 2022, 87, 12921–12931 CrossRef PubMed.
- L. Pan, A. S. Kelley, M. V. Cooke, M. M. Deckert and S. Laulhé, ACS Sustainable Chem. Eng., 2022, 10, 691–695 CrossRef PubMed.
- J. C. Herrera-Luna, D. D. Díaz, M. C. Jiménez and R. Pérez-Ruiz, ACS Appl. Mater. Interfaces, 2021, 13, 48784–48794 CrossRef PubMed.
- M. Hosseini-Sarvari and F. Jafari, Dalton Trans., 2020, 49, 3001–3006 RSC.
- L. Wang, T. Ma, M. Qiao, Q. Wu, D. Shi and W. Xiao, Synthesis, 2019, 51, 522–529 CrossRef.
- S. V. Tiwari, N. S. Sharif, R. I. Gajare, J. A. S. Vazquez, J. N. Sangshetti, M. D. Damale and A. P. G. Nikalje, Molecules, 2018, 23, 1981 CrossRef.
- N. Ravi, M. Venkatanarayana, H. Sharathbabu and K. R. Babu, Phosphorus, Sulfur Silicon Relat. Elem., 2021, 196, 1018–1024 CrossRef.
- R. M. N. Kalla, J.-S. Choi, J.-W. Yoo, S. J. Byeon, M. S. Heo and I. Kim, Eur. J. Med. Chem., 2014, 76, 61–66 CrossRef.
- K. B. Dhopte, D. S. Raut, A. V. Patwardhan and P. R. Nemade, Synth. Commun., 2015, 45, 778–788 CrossRef.
- J. Xiang, N. Yi, R. Wang, L. Lu, H. Zou, Y. Pan and W. He, Tetrahedron, 2015, 71, 694–699 CrossRef.
- X. Chen, X. Li, X.-L. Chen, L.-B. Qu, J.-Y. Chen, K. Sun, Z.-D. Liu, W.-Z. Bi, Y.-Y. Xia, H.-T. Wu and Y.-F. Zhao, Chem. Commun., 2015, 51, 3846–3849 RSC.
- A. Cordero-Díaz, E. Robledo-Leal, E. Hernández-Fernández, E. Hernández-Núñez, M. Elizondo-Zertuche and S. T. López-Cortina, Molecules, 2022, 27, 3886 CrossRef.
- M. Milen, P. Ábrányi-Balogh, A. Dancsó, D. Frigyes, L. Pongó and G. Keglevich, Tetrahedron Lett., 2013, 54, 5430–5433 CrossRef.
- S. Disale, S. Kale, G. Abraham, S. Kahandal, A. N. Sawarkar and M. B. Gawande, Front. Chem., 2016, 4, 35 Search PubMed.
- P. Wang, J. Lu and Z.-H. Zhang, J. Chem. Res., 2013, 37, 359–361 CrossRef.
- W. Fan, Y. Queneau and F. Popowycz, RSC Adv., 2018, 8, 31496–31501 RSC.
- S. A. Rasal, P. P. Dhavan, B. L. Jadhav and N. G. Shimpi, Appl. Organomet. Chem., 2020, 34, e5317 CrossRef CAS.
- M. H. Shaikh, D. D. Subhedar, F. A. K. Khan, J. N. Sangshetti and B. B. Shingate, Res. Chem. Intermed., 2016, 42, 5115–5131 CrossRef CAS.
- A. Ying, S. Liu, J. Yang and H. Hu, Ind. Eng. Chem. Res., 2014, 53, 16143–16147 CrossRef CAS.
- R. M. N. Kalla, Y. Zhang and I. Kim, New J. Chem., 2017, 41, 5373–5379 RSC.
- D. S. Gaikwad, K. A. Undale, A. A. Patravale and P. B. Choudhari, Res. Chem. Intermed., 2020, 46, 621–637 CrossRef CAS.
- S. Rasheed, D. S. Rao, C. Subramanyam, S. T. Basha and C. N. Raju, Synth. Commun., 2014, 44, 2988–2998 CrossRef CAS.
- R. M. N. Kalla and I. Kim, Mol. Catal., 2019, 473, 110396 CrossRef CAS.
- D. Chakraborty, D. Musib, R. Saha, A. Das, M. K. Raza, V. Ramu, S. Chongdar, K. Sarkar and A. Bhaumik, Mater. Today Chem., 2022, 24, 100882 CrossRef CAS.
- I. Akhmetova, M. Rautenberg, C. Das, B. Bhattacharya and F. Emmerling, ACS Omega, 2023, 8, 16687–16693 CrossRef CAS PubMed.
- A. D. G. Firmino, R. F. Mendes, M. M. Antunes, P. C. Barbosa, S. M. F. Vilela, A. A. Valente, F. M. L. Figueiredo, J. P. C. Tomé and F. A. A. Paz, Inorg. Chem., 2017, 56, 1193–1208 CrossRef CAS.
- B. Kaboudin, L. Karami, J. Kato, H. Aoyama and T. Yokomatsu, Tetrahedron Lett., 2013, 54, 4872–4875 CrossRef CAS.
- H.-M. Guo, Q.-Q. Zhou, X. Jiang, D.-Q. Shi and W.-J. Xiao, Adv. Synth. Catal., 2017, 359, 4141–4146 CrossRef CAS.
- H. Singh, T. Sahoo, C. Sen, S. M. Galani and S. C. Ghosh, Catal. Sci. Technol., 2019, 9, 1691–1698 RSC.
- Z. Mou, Y. Wang and X. Man, Phosphorus, Sulfur Silicon Relat. Elem., 2021, 196, 195–199 CrossRef CAS.
- C. Subramanyam, G. Madhava, S. K. T. Basha, S. Rasheed, A. U. Sankar and C. N. Raju, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 1948–1957 CrossRef CAS.
- G. Brahmachari, S. Begam, I. Karmakar and V. K. Gupta, Phosphorus, Sulfur Silicon Relat. Elem., 2021, 196, 769–779 CrossRef CAS.
- G. Sun, Z. Deng, Z. Luo, Z. Wang and J. Zhang, Org. Lett., 2021, 23, 7630–7634 CrossRef.
- G. P. Horsman and D. L. Zechel, Chem. Rev., 2017, 117, 5704–5783 CrossRef.
- D. Drzyzga, G. Forlani, J. Vermander, P. Kafarski and J. Lipok, Environ. Microbiol., 2017, 19, 1065–1076 CrossRef PubMed.
- S. V. Kononova and M. A. Nesmeyanova, Biochemistry – Biokhimiia, 2002, 67, 184–195 CrossRef PubMed.
- R. Riedel, K. Krahl, K. Buder, J. Böllmann, B. Braun and M. Martienssen, J. Microbiol. Methods, 2023, 212, 106793 CrossRef PubMed.
- J. Steber and P. Wierich, Chemosphere, 1987, 16, 1323–1337 CrossRef.
- J. Steber and P. Wierich, Chemosphere, 1986, 15, 929–945 CrossRef.
- B. Nowack, Water Res., 1998, 32, 1271–1279 CrossRef.
- E. Matthus, N. T. de Oude, M. Bolte and J. Lemaire, Water Res., 1989, 23, 845–851 CrossRef.
- D. Schowanek and W. Verstraete, J. Environ. Qual., 1991, 20, 769–776 CrossRef.
- J. Steber and P. Wierich, Chemosphere, 1987, 16, 1323–1337 CrossRef.
- B. Nowack and A. T. Stone, Environ. Sci. Technol., 2000, 34, 4759–4765 CrossRef CAS.
- D. P. Riley, D. L. Fields and W. Rivers, J. Am. Chem. Soc., 1991, 113, 3371–3378 CrossRef CAS.
- D. P. Riley and D. L. Fields, J. Am. Chem. Soc., 1992, 114, 1881–1882 CrossRef CAS.
- B. Nowack and A. T. Stone, Environ. Chem. Lett., 2003, 1, 24–31 CrossRef CAS.
- K. D. Demadis, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 167–176 CrossRef CAS.
- K. D. Demadis and A. Ketsetzi, Sep. Sci. Technol., 2007, 42, 1639–1649 CrossRef CAS.
- K. D. Demadis, B. Yang, P. R. Young, D. L. Kouznetsov and D. G. Kelley, in Advances in Crystal Growth Inhibition Technologies, ed. Z. Amjad, Kluwer Academic Publishers, Boston, 2002, pp. 215–234 Search PubMed.
- M. F. Mady, A. Bagi and M. A. Kelland, Energy Fuels, 2016, 30, 9329–9338 CrossRef CAS.
- D. Drzyzga, G. Forlani, J. Vermander, P. Kafarski and J. Lipok, Environ. Microbiol., 2017, 19, 1065–1076 CrossRef CAS.
- B. Nowack, Water Res., 2003, 37, 2533–2546 CrossRef CAS PubMed.
- A. Obojska, B. Lejczak and M. Kubrak, Appl. Microbiol. Biotechnol., 1999, 51, 872–876 CrossRef CAS.
- J. W. Frost, S. Loo, M. L. Cordeiro and D. Li, J. Am. Chem. Soc., 1987, 109, 2166–2171 CrossRef CAS.
- E. M. Fox and G. L. Mendz, Enzyme Microb. Technol., 2006, 40, 145–150 CrossRef CAS.
- M. E. T. Sillanpää, T. A. Kurniawan and W. Lo, Chemosphere, 2011, 83, 1443–1460 CrossRef.
- J. Klinger, M. Lang, F. Sacher, H.-J. Brauch, D. Maier and E. Worch, Ozone Sci. Eng., 1998, 20, 99–110 CrossRef CAS.
- J. Zhu, S. Wang, H. Li, J. Qian, L. Lv and B. Pan, Water Res., 2021, 202, 117397 CrossRef CAS.
- E. Rott, R. Minke, U. Bali and H. Steinmetz, Water Res., 2017, 122, 345–354 CrossRef PubMed.
- P. Cardiano, R. M. Cigala, M. Cordaro, C. de Stefano, D. Milea and S. Sammartano, New J. Chem., 2017, 41, 4065–4075 RSC.
- M. A. Blesa, A. D. Weisz, P. J. Morando, J. A. Salfity, G. E. Magaz and A. E. Regazzoni, Coord. Chem. Rev., 2000, 196, 31–63 CrossRef.
- https://www.watertechnologies.com/handbook/handbook-industrial-water-treatment .
- F. Jones and M. I. Ogden, CrystEngComm, 2010, 12, 1016–1023 RSC.
- S. Dobberschütz, M. R. Nielsen, K. K. Sand, R. Civioc, N. Bovet, S. L. S. Stipp and M. P. Andersson, Nat. Commun., 2018, 9, 1578 CrossRef.
- T. A. Hoang, in Water-Formed Deposits, Elsevier, 2022, pp. 13–47 Search PubMed.
- M. Boon and F. Jones, in Water-Formed Deposits, Elsevier, 2022, pp. 697–722 Search PubMed.
- M. M. Reddy and G. H. Nancollas, Desalination, 1973, 12, 61–73 CrossRef.
- R. G. Jonasson, K. Rispler, B. Wiwchar and W. D. Gunter, Chem. Geol., 1996, 132, 215–225 CrossRef.
- M. F. Mady, H. Malmin and M. A. Kelland, Energy Fuels, 2019, 33, 6197–6204 CrossRef.
- J. Jiang, J. Zhao and Y. Xu, Desalin. Water Treat., 2016, 57, 2152–2158 CrossRef.
- M. F. B. Sousa and C. A. Bertran, J. Colloid Interface Sci., 2014, 420, 57–64 CrossRef PubMed.
- M. Xia and C. Chen, Int. J. Environ. Sci. Dev., 2015, 6, 300–304 CrossRef.
- L. Boels, R. M. Wagterveld and G. J. Witkamp, Ultrason. Sonochem., 2011, 18, 1225–1231 CrossRef PubMed.
- L. F. Greenlee, F. Testa, D. F. Lawler, B. D. Freeman and P. Moulin, Water Res., 2010, 44, 2957–2969 CrossRef PubMed.
- Y. Tang, W. Yang, X. Yin, Y. Liu, P. Yin and J. Wang, Desalination, 2008, 228, 55–60 CrossRef.
- Pradip, B. Rai, T. K. Rao, S. Krishnamurthy, R. Vetrivel, J. Mielczarski and J. M. Cases, Langmuir, 2002, 18, 932–940 CrossRef.
- Y. Chevalier, S. Brunel, P. Perchec, M. Mosquet and J.-P. Guicquero, in Trends in Colloid and Interface Science XI, ed. J. B. Rosenholm, B. Lindman and P. Stenius, Steinkopff, Darmstadt, 1997, vol. 105, pp. 6–10 Search PubMed.
- S. N. Black, L. A. Bromley, D. Cottier, R. J. Davey, B. Dobbs and J. E. Rout, Faraday Trans., 1991, 87, 3409 Search PubMed.
- Y. Liu, Z. Dai, A. T. Kan, M. B. Tomson and P. Zhang, J. Pet. Sci. Eng., 2022, 208, 109425 CrossRef.
- C. Pina, C. Putnis, U. Becker, S. Biswas, E. Carroll, D. Bosbach and A. Putnis, Surf. Sci., 2004, 553, 61–74 Search PubMed.
- T. Radomirovic, M. I. Ogden, A. L. Rohl and F. Jones, CrystEngComm, 2019, 21, 807–815 RSC.
- M. A. Kelland, Ind. Eng. Chem. Res., 2011, 50, 5852–5861 CrossRef.
- A. Baynton, B. D. Chandler, F. Jones, G. Nealon, M. I. Ogden, T. Radomirovic, G. K. H. Shimizu and J. M. Taylor, CrystEngComm, 2011, 13, 1090–1095 Search PubMed.
- F. Jones, A. Oliveira, A. L. Rohl, G. M. Parkinson, M. I. Ogden and M. M. Reyhani, J. Cryst. Growth, 2002, 237–239, 424–429 Search PubMed.
- E. N. Rizkalla, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 1857 Search PubMed.
- F. Jones, W. R. Richmond and A. L. Rohl, J. Phys. Chem. B, 2006, 110, 7414–7424 Search PubMed.
- F. Jones, A. Stanley, A. Oliveira, A. Rohl, M. Reyhani, G. Parkinson and M. Ogden, J. Cryst. Growth, 2003, 249, 584–593 CrossRef.
- M. D. Sikirić and H. Füredi-Milhofer, Adv. Colloid Interface Sci., 2006, 128–130, 135–158 Search PubMed.
- J. L. Meyer, K. E. Lee and J. H. Bergert, Calcif. Tissue Res., 1977, 23, 83–86 Search PubMed.
- E. N. Rizkalla and M. M. Moawad, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 1617 RSC.
- M. Nirmaladevi, K. S. Raj and V. K. Subramanian, Nephrol. Open J., 2017, 3, 1–8 CrossRef.
- Y.-C. Chien, A. Mansouri, W. Jiang, S. R. Khan, J. J. Gray and M. D. McKee, J. Struct. Biol., 2018, 204, 131–144 CrossRef PubMed.
- A. Lancia, D. Musmarra and M. Prisciandaro, in Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, 2001 Search PubMed.
- M. Weijnen and G. M. van Rosmalen, J. Cryst. Growth, 1986, 79, 157–168 CrossRef.
- T. A. Hoang, H. M. Ang and A. L. Rohl, Aust. J. Chem., 2009, 62, 927 CrossRef.
- Y. O. Rosenberg, I. J. Reznik, S. Zmora-Nahum and J. Ganor, Desalination, 2012, 284, 207–220 CrossRef.
- M. F. Mady, in Water-Formed Deposits, Elsevier, 2022, pp. 325–352 Search PubMed.
- M. Oshchepkov, S. Kamagurov, S. Tkachenko, A. Ryabova and K. Popov, ChemNanoMat, 2019, 5, 586–592 CrossRef.
- A. E. Austin, J. F. Miller, D. A. Vaughan and J. F. Kircher, Desalination, 1975, 16, 345–357 CrossRef.
- M. Prisciandaro, A. Lancia and D. Musmarra, in Water-Formed Deposits, Elsevier, 2022, 283–294 Search PubMed.
- E. Akyol, M. Öner, E. Barouda and K. D. Demadis, Cryst. Growth Des., 2009, 9, 5145–5154 CrossRef.
- T. Rabizadeh, C. L. Peacock and L. G. Benning, Ind. Eng. Chem. Res., 2020, 59, 14970–14980 CrossRef.
- M. Prisciandaro, E. Olivieri, A. Lancia and D. Musmarra, Ind. Eng. Chem. Res., 2009, 48, 10877–10883 CrossRef.
- M. Prisciandaro, E. Olivieri, A. Lancia and D. Musmarra, Ind. Eng. Chem. Res., 2012, 51, 12844–12851 CrossRef.
- M. Prisciandaro, E. Olivieri, A. Lancia and D. Musmarra, Chem. Eng. Trans., 2009, 17, 669–674 Search PubMed.
- A. Zeino, M. Albakri, M. Khaled and M. Zarzour, J. Water Proc. Eng., 2018, 21, 1–8 CrossRef.
- T. A. Hoang, H. M. Ang and A. L. Rohl, Desalin. Water Treat., 2011, 29, 294–301 CrossRef.
- M. Mpelwa, S. Tang and L. Jin, Int. J. Ind. Chem., 2020, 11, 133–145 CrossRef.
- K. Popov, M. Oshchepkov, E. Afanas'eva, E. Koltinova, Y. Dikareva and H. Rönkkömäki, Colloids Surf., A, 2019, 560, 122–129 CrossRef.
- Y. A. Issabayev, G. I. Boiko, N. P. Lyubchenko, Y. M. Shaikhutdinov, H. Muhr, L. Colombeau, P. Arnoux and C. Frochot, J. Water Proc. Eng., 2018, 22, 192–202 CrossRef.
- G. Topcu, A. Çelik, A. Kandemir, A. Baba, H. Sahin and M. M. Demir, Geothermics, 2019, 77, 106–114 CrossRef.
- K. D. Demadis, A. Ketsetzi and E.-M. Sarigiannidou, Ind. Eng. Chem. Res., 2012, 51, 9032–9040 CrossRef.
- J. J. Stezowski, R. Countryman and J. L. Hoard, Inorg. Chem., 1973, 12, 1749–1754 CrossRef.
- A. Spinthaki, J. Matheis, W. Hater and K. D. Demadis, Energy Fuels, 2018, 32, 11749–11760 CrossRef.
- S. Lacour, V. Deluchat, J. C. Bollinger and S. Bernard, Talanta, 1998, 46, 999–1009 CrossRef PubMed.
- A. Spinthaki, M. Kamaratou, D. Disci, W. Hater and K. D. Demadis, Geothermics, 2023, 111, 102690 CrossRef.
- M. Kamaratou, A. Spinthaki, D. Disci and K. D. Demadis, Geothermics, 2024, 123, 103118 CrossRef.
- T. Wang, G. Rother and H. Cölfen, Macromol. Chem. Phys., 2005, 206, 1619–1629 CrossRef.
- A. Martinod, A. Neville, M. Euvrad and K. Sorbie, Chem. Eng. Sci., 2009, 64, 2413–2421 CrossRef.
- T. Chen, A. Neville, K. Sorbie and Z. Zhong, Chem. Eng. Sci., 2009, 64, 912–918 CrossRef.
- K. Malaie, O. Shojaei, S. Iranpour and Z. Taherkhani, Heliyon, 2021, 7, e06064 CrossRef PubMed.
- Y. Chen, Y. Zhou, Q. Yao, Y. Bu, H. Wang, W. Wu and W. Sun, J. Appl. Polym. Sci., 2015, 132, 41447 CrossRef.
- S. A. Haladu and S. A. Ali, J. Polym. Sci., Part A:Polym. Chem., 2013, 51, 5130–5142 CrossRef.
- K. D. Demadis, M. Preari and I. Antonakaki, Pure Appl. Chem., 2014, 86, 1663–1674 Search PubMed.
- R. R. Navarro, S. Wada and K. Tatsumi, Sep. Sci. Technol., 2003, 38, 2327–2345 CrossRef.
- R. R. Navarro and K. Tatsumi, Sep. Sci. Technol., 2002, 37, 203–216 CrossRef CAS.
- D. Villemin, C. Monteil, N. Bar and M. A. Didi, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 879–890 CrossRef CAS.
- Y. Wen, S. Pan, X. Luo, X. Zhang, W. Zhang and M. Feng, Bioconjugate Chem., 2009, 20, 322–332 CrossRef CAS.
- P. Vicennati, A. Giuliano, G. Ortaggi and A. Masotti, Curr. Med. Chem., 2008, 15, 2826–2839 CrossRef CAS PubMed.
- K. D. Demadis, M. Paspalaki and J. Theodorou, Ind. Eng. Chem. Res., 2011, 50, 5873–5876 CrossRef CAS.
- K. D. Demadis and A. Stathoulopoulou, Ind. Eng. Chem. Res., 2006, 45, 4436–4440 CrossRef CAS.
- A. Spinthaki, A. Stathoulopoulou and K. D. Demadis, Int. J. Corros. Scale Inhib., 2015, 4, 125–138 CrossRef CAS.
- A. Spinthaki, G. Skordalou, A. Stathoulopoulou and K. D. Demadis, Pure Appl. Chem., 2016, 88, 1037–1047 CrossRef CAS.
- A. Heras, Carbohydr. Polym., 2001, 44, 1–8 CrossRef CAS.
- K. D. Demadis, A. Ketsetzi, K. Pachis and V. M. Ramos, Biomacromolecules, 2008, 9, 3288–3293 CrossRef CAS PubMed.
- K. D. Demadis, K. Pachis, A. Ketsetzi and A. Stathoulopoulou, Adv. Colloid Interface Sci., 2009, 151, 33–48 CrossRef CAS PubMed.
- A. Spinthaki, M. Kamaratou, G. Skordalou, G. Petratos, I. Petrou, A. Tramaux, G. David and K. D. Demadis, Geothermics, 2021, 89, 101954 CrossRef.
- A. Spinthaki, M. Kamaratou, G. Skordalou, G. Petratos, A. Tramaux, G. David and K. D. Demadis, Geothermics, 2021, 89, 101972 CrossRef.
- A. Chirkunov and Y. Kuznetsov, in Mineral Scales and Deposits, Elsevier, 2015, pp. 85–105 Search PubMed.
- J. Telegdi, in Water-Formed Deposits, Elsevier, 2022, pp. 49–68 Search PubMed.
- K. Popov, H. Rönkkömäki and L. H. J. Lajunen, Pure Appl. Chem., 2001, 73, 1641–1677 CrossRef.
- I. Sekine, T. Shimode, M. Yuasa and K. Takaoka, Ind. Eng. Chem. Res., 1992, 31, 434–439 CrossRef.
- Y. I. Kuznetsov, G. Y. Kazanskaya and N. V. Tsirulnikova, Prot. Met., 2003, 39, 120–123 CrossRef.
- S. Rajendran, B. V. Apparao and N. Palaniswamy, Anti-Corros. Methods Mater., 1998, 45, 158–161 CrossRef.
- Y. V. Balaban-Irmenin, A. M. Rubashov and N. G. Fokina, Prot. Met., 2006, 42, 133–136 CrossRef.
- J. L. Fang, Y. Li, X. R. Ye, Z. W. Wang and Q. Liu, Corrosion, 1993, 49, 266–271 CrossRef.
- T. Du, J. Chen and D. Cao, J. Mater. Sci., 2001, 36, 3903–3907 CrossRef.
- I. Frateur, A. Carnot, S. Zanna and P. Marcus, Appl. Surf. Sci., 2006, 252, 2757–2769 CrossRef.
- H. Amar, J. Benzakour, A. Derja, D. Villemin and B. Moreau, J. Electroanal. Chem., 2003, 558, 131–139 CrossRef.
- A. Paszternák, I. Felhősi, Z. Keresztes and E. Kálmán, Mater. Sci. Forum, 2007, 537–538, 239–246 Search PubMed.
- H. Amar, J. Benzakour, A. Derja, D. Villemin, B. Moreau and T. Braisaz, Appl. Surf. Sci., 2006, 252, 6162–6172 CrossRef.
- A. Pilbáth, I. Bertóti, I. Sajó, L. Nyikos and E. Kálmán, Appl. Surf. Sci., 2008, 255, 1841–1849 CrossRef.
- Y. I. Kuznetsov, G. V. Zinchenko, L. P. Kazanskii, N. P. Andreeva and Y. B. Makarychev, Prot. Met., 2007, 43, 648–655 CrossRef.
- H. Amar, T. Braisaz, D. Villemin and B. Moreau, Mater. Chem. Phys., 2008, 110, 1–6 CrossRef.
- A. Moschona, N. Plesu, R. M. Colodrero, A. Cabeza, A. G. Thomas and K. D. Demadis, Chem. Eng. J., 2021, 405, 126864 CrossRef.
- A. Moschona, N. Plesu, G. Mezei, A. G. Thomas and K. D. Demadis, Corros. Sci., 2018, 145, 135–150 CrossRef.
- M. Papadaki and K. D. Demadis, Comments Inorg. Chem., 2009, 30, 89–118 CrossRef.
- R. Sabzi and R. Arefinia, Corros. Sci., 2019, 153, 292–300 CrossRef.
- K. D. Demadis, S. D. Katarachia and M. Koutmos, Inorg. Chem. Commun., 2005, 8, 254–258 CrossRef.
- K. D. Demadis, C. Mantzaridis, R. G. Raptis and G. Mezei, Inorg. Chem., 2005, 44, 4469–4471 CrossRef PubMed.
- K. D. Demadis and M. Papadaki, ACS Appl. Mater. Interfaces, 2010, 2, 1814–1816 CrossRef.
- Z. Amjad and K. D. Demadis, Mineral Scales and Deposits, Elsevier, 2015 Search PubMed.
- Water-Formed Deposits, ed. Z. Amjad and K. D. Demadis, Elsevier, 2022 Search PubMed.
- J. W. Morse, R. S. Arvidson and A. Lüttge, Chem. Rev., 2007, 107, 342–381 CrossRef.
- S. M. Hamza and S. H. El-Hamouly, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 3725 RSC.
- H.-C. Chang, T. W. Healy and E. Matijević, J. Colloid Interface Sci., 1983, 92, 469–478 CrossRef.
- W. W. Frenier and M. Ziauddin, Formation, Removal, and Inhibition of Inorganic Scale in the Oilfield Environment, Society of Petroleum EngineersRichardson, Texas, USA, 2008 Search PubMed.
- S. N. Kazi, G. G. Duffy and X. D. Chen, Chem. Eng. Commun., 2012, 199, 1263–1278 CrossRef.
- T. Hodgkiess, K. H. Al-Omari, N. Bontems and B. Lesiak, Desalination, 2005, 183, 209–216 CrossRef.
- K. Lund, H. S. Fogler, C. C. McCune and J. W. Ault, Chem. Eng. Sci., 1975, 30, 825–835 CrossRef.
- K. S. Wang, R. Resch, B. E. Koel, P. J. Shuler, Y. Tang, H. Chen and T. F. Yen, J. Colloid Interface Sci., 1999, 219, 212–215 CrossRef PubMed.
- E. Ruiz-Agudo, D. Di Tommaso, C. V. Putnis, N. H. de Leeuw and A. Putnis, Cryst. Growth Des., 2010, 10, 3022–3035 CrossRef.
- N. Spanos, D. G. Kanellopoulou and P. G. Koutsoukos, Langmuir, 2006, 22, 2074–2081 CrossRef.
- W. El Malti, D. Laurencin, G. Guerrero, M. E. Smith and P. H. Mutin, J. Mater. Chem., 2012, 22, 1212–1218 RSC.
- M. Nalbach, A. Moschona, K. D. Demadis, S. Klassen, R. Bechstein and A. Kühnle, Cryst. Growth Des., 2017, 17, 5867–5874 CrossRef.
- H. Ehrlich, K. D. Demadis, O. S. Pokrovsky and P. G. Koutsoukos, Chem. Rev., 2010, 110, 4656–4689 CrossRef PubMed.
- K. D. Demadis and E. Mavredaki, Environ. Chem. Lett., 2005, 3, 127–131 CrossRef.
- K. D. Demadis, E. Mavredaki and M. Somara, Ind. Eng. Chem. Res., 2011, 50, 12587–12595 CrossRef.
- K. D. Demadis, E. Mavredaki and M. Somara, Ind. Eng. Chem. Res., 2011, 50, 13866–13876 CrossRef.
- H. Itabashi and T. Nakahara, in Sample Preparation for Trace Element Analysis, Elsevier, 2003, 41, 459–494 Search PubMed.
- X.-W. Lv, C.-C. Weng, Y.-P. Zhu and Z.-Y. Yuan, Small, 2021, 17, e2005304 Search PubMed.
- T.-Y. Ma and Z.-Y. Yuan, ChemSusChem, 2011, 4, 1407–1419 CrossRef.
- T. Knepper, Trends Anal. Chem., 2003, 22, 708–724 CrossRef CAS.
- N. Plesu, L. Macarie, A. Popa and G. Ilia, in Water-Formed Deposits, Elsevier, 2022, pp. 397–433 Search PubMed.
- V. Beaugeard, J. Muller, A. Graillot, X. Ding, J.-J. Robin and S. Monge, React. Funct. Polym., 2020, 152, 104599 CrossRef.
- X. Wang, L. Song, F. Yang, D. Yun, J. Wang and H. Lu, Desalin. Water Treat., 2017, 71, 343–358 CrossRef.
- S. M. Bhosle, S. Ponrathnam, S. S. Tambe and N. N. Chavan, Bull. Mater. Sci., 2016, 39, 1541–1556 CrossRef.
- K. Araki, K. Uezu, M. Goto and S. Furusaki, Anal. Sci., 1999, 15, 651–656 CrossRef.
- M. Sawicki, D. Lecerclé, G. Grillon, B. Le Gall, A.-L. Sérandour, J.-L. Poncy, T. Bailly, R. Burgada, M. Lecouvey, V. Challeix, A. Leydier, S. Pellet-Rostaing, E. Ansoborlo and F. Taran, Eur. J. Med. Chem., 2008, 43, 2768–2777 CrossRef.
- A. Popa, A. Visa, B. Maranescu, I. Hulka and L. Lupa, Materials, 2021, 14, 7894 CrossRef PubMed.
- B. Maranescu, A. Popa, L. Lupa, V. Maranescu and A. Visa, Sep. Sci. Technol., 2018, 53, 1058–1064 CrossRef.
- A.-L. Alanne, M. Tuikka, K. Tõnsuaadu, M. Ylisirniö, L. Hämäläinen, P. Turhanen, J. Vepsäläinen and S. Peräniemi, RSC Adv., 2013, 3, 14132 RSC.
- P. A. Turhanen, J. J. Vepsäläinen and S. Peräniemi, Sci. Rep., 2015, 5, 8992 CrossRef PubMed.
- R. Chiariza, E. P. Horwitz, S. D. Alexandrators and M. J. Gula, Sep. Sci. Technol., 1997, 32, 1–35 CrossRef.
- S. Katragadda, H. D. Gesser and A. Chow, Talanta, 1997, 44, 1865–1871 CrossRef PubMed.
- A. R. Garifzyanov, S. V. Zakharov, S. V. Kryukov, V. I. Galkin and R. A. Cherkasov, Russ. J. Gen. Chem., 2005, 75, 1208–1211 CrossRef CAS.
- A. A. Younes, H. H. El-Maghrabi and H. R. Ali, J. Hazard. Mater., 2017, 334, 1–9 CrossRef CAS PubMed.
- A. Popa, C.-M. Davidescu, P. Negrea, G. Ilia, A. Katsaros and K. D. Demadis, Ind. Eng. Chem. Res., 2008, 47, 2010–2017 CrossRef.
- O. Abderrahim, M. A. Didi and D. Villemin, J. Radioanal. Nucl. Chem., 2009, 279, 237–244 CrossRef.
- V. Luca, J. J. Tejada, D. Vega, G. Arrachart and C. Rey, Inorg. Chem., 2016, 55, 7928–7943 CrossRef.
- X.-J. Zhang, T.-Y. Ma and Z.-Y. Yuan, J. Mater. Chem., 2008, 18, 2003–2010 RSC.
- T.-Y. Ma, X.-J. Zhang, G.-S. Shao, J.-L. Cao and Z.-Y. Yuan, J. Phys. Chem. C, 2008, 112, 3090–3096 CrossRef.
- M. C. Zenobi, L. Hein and E. Rueda, J. Colloid Interface Sci., 2005, 284, 447–454 CrossRef.
- T.-Y. Ma, X.-J. Zhang and Z.-Y. Yuan, J. Mater. Sci., 2009, 44, 6775–6785 CrossRef.
- S. Saoiabi, S. El Asri, A. Laghzizil, A. Saoiabi, J. L. Ackerman and T. Coradin, Chem. Eng. J., 2012, 211–212, 233–239 CrossRef PubMed.
- R. Thapa, A. Rahmani, P. Turhanen, A. Taskinen, T. Nissinen, R. Neitola, J. Vepsäläinen, V.-P. Lehto and J. Riikonen, Sep. Purif. Technol., 2021, 272, 118913 CrossRef.
- O. Haluska, A. Rahmani, A. Salami, P. Turhanen, J. Vepsäläinen, R. Lappalainen, V.-P. Lehto and J. Riikonen, Microporous Mesoporous Mater., 2021, 324, 111294 CrossRef.
- Y. Guo and Z. Jia, Mater. Lett., 2018, 228, 239–241 CrossRef.
- X. Hou, C.-C. Yan, X. Xu, A.-Q. Liang, Z.-W. Song and S.-F. Tang, Dalton Trans., 2020, 49, 3809–3815 RSC.
- Y. Su, X. Cai, H. Sun, Z. Sun, D. Dong, Y. Zhu and T. Wang, ChemistrySelect, 2018, 3, 6845–6851 CrossRef CAS.
- B. Maranescu, L. Lupa and A. Visa, Appl. Surf. Sci., 2019, 481, 83–91 CrossRef CAS.
- Z. Wang, P. Yin, Z. Wang, Q. Xu, R. Qu and Q. Tang, Sep. Sci. Technol., 2012, 48, 281–287 CrossRef CAS.
- F. Cao, P. Yin, J. Zhang, H. Chen and R. Qu, J. Ind. Eng. Chem., 2014, 20, 2568–2573 CrossRef CAS.
- L. Lupa, B. Maranescu and A. Visa, Sep. Sci. Technol., 2018, 53, 1017–1026 CrossRef CAS.
- B. Maranescu, L. Lupa and A. Visa, Pure Appl. Chem., 2018, 90, 35–47 CrossRef CAS.
- J. Wu, H. Hou, H. Han and Y. Fan, Inorg. Chem., 2007, 46, 7960–7970 CrossRef CAS.
- J. Zhang, L. Chen, D. Gui, H. Zhang, D. Zhang, W. Liu, G. Huang, J. Diwu, Z. Chai and S. Wang, Dalton Trans., 2018, 47, 5161–5165 RSC.
- Y.-L. Wang, L. Zhu, B.-L. Guo, S.-W. Chen and W.-S. Wu, New J. Chem., 2014, 38, 3853–3861 RSC.
- W. Zhang, A. Bu, Q. Ji, L. Min, S. Zhao, Y. Wang and J. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 33931–33940 CrossRef CAS.
- T.-Y. Ma, X.-J. Zhang and Z.-Y. Yuan, Microporous Mesoporous Mater., 2009, 123, 234–242 CrossRef CAS.
- T.-Y. Ma, X.-J. Zhang and Z.-Y. Yuan, J. Phys. Chem. C, 2009, 113, 12854–12862 CrossRef CAS.
- Y.-P. Zhu, Y.-L. Liu, T.-Z. Ren and Z.-Y. Yuan, Nanoscale, 2014, 6, 6627–6636 RSC.
- T.-Y. Ma, X.-Z. Lin and Z.-Y. Yuan, J. Mater. Chem., 2010, 20, 7406 RSC.
- X.-Z. Lin and Z.-Y. Yuan, Eur. J. Inorg. Chem., 2012, 2012, 2661–2664 CrossRef.
- Y.-P. Zhu, Y.-L. Liu, T.-Z. Ren and Z.-Y. Yuan, RSC Adv., 2014, 4, 16018–16021 RSC.
- M. A. Nistor, S. G. Muntean, B. Maranescu and A. Visa, Appl. Organomet. Chem., 2020, 34, e5939 CrossRef.
- A. Jayswal and U. Chudasama, Acta Chim. Slov., 2007, 54, 654–660 Search PubMed.
- T.-Z. Ren, X.-H. Zhu, T.-Y. Ma and Z.-Y. Yuan, Adsorpt. Sci. Technol., 2013, 31, 535–548 CrossRef.
- S. Dehghanpour, A. Mahmoudi, F. Esbati and S. H. Rasanani, Mater. Res. Bull., 2012, 47, 2126–2134 CrossRef.
- C.-Y. Gao, Y. Yang, J. Ai, H.-R. Tian, L.-J. Li, W. Yang, S. Dang and Z.-M. Sun, Chemistry, 2016, 22, 11652–11659 CrossRef PubMed.
- V. Arumugam, P. Sriram, T.-J. Yen, G. G. Redhi and R. M. Gengan, Appl. Catal., B, 2018, 222, 99–114 CrossRef.
- A. Farrokhi, M. Jafarpour and M. Alipour, Polyhedron, 2019, 170, 325–333 CrossRef.
- A. Farrokhi, F. Bivareh, S. Dejbakhshpour and A. Z. Moghaddam, Appl. Organomet. Chem., 2020, 34, e5938 CrossRef.
- F. Zhang, C. Yang, Y. Li, M. Chen, S. Hu and H. Cheng, RSC Adv., 2019, 9, 13476–13485 RSC.
- J. Ai, H.-R. Tian, X. Min, Z.-C. Wang and Z.-M. Sun, Dalton Trans., 2020, 49, 3700–3705 RSC.
- G. Morse, S. Brett, J. Guy and J. Lester, Sci. Total Environ., 1998, 212, 69–81 CrossRef.
- L. Dai, H. Li, F. Tan, N. Zhu, M. He and G. Hu, GCB Bioenergy, 2016, 8, 852–858 CrossRef.
- Z. Fang, F. Liu, Y. Li, B. Li, T. Yang and R. Li, J. Clean Prod., 2021, 287, 125550 CrossRef.
- Y. Zhu, Q. Zhao, D. Li, J. Li and W. Guo, J. Clean Prod., 2022, 372, 133728 CrossRef.
- M. L. Bornø, A. Zervas, F. Bak, T. Merl, K. Koren, M. H. Nicolaisen, L. S. Jensen and D. S. Müller-Stöver, Sci. Total Environ., 2023, 905, 166888 CrossRef PubMed.
- M. Jastrzebska, M. Kostrzewska, K. Treder, P. Makowski, A. Saeid, W. Jastrzebski and A. Okorski, Plant, Soil Environ., 2018, 64, 504–511 CrossRef.
- M. Jastrzebska, M. K. Kostrzewska and A. Saeid, Molecules, 2022, 27, 2769 CrossRef.
- N. Nunes, C. Ragonezi, C. S. Gouveia and M. Â. Pinheiro de Carvalho, Sustainability, 2021, 13, 2317 CrossRef.
- S. Delibacak, L. Voronina and E. Morachevskaya, Eurasian J. Soil Sci., 2020, 9, 126–139 Search PubMed.
- Y. Lee, M. Sethurajan, J. van de Vossenberg, E. Meers and E. D. van Hullebusch, J. Environ. Manage., 2020, 270, 110818 CrossRef PubMed.
- Z. A. Shariff, L. Fraikin, A. Bogdan, A. Léonard, E. Meers and A. Pfennig, Environ. Technol., 2024, 45, 2820–2832 CrossRef PubMed.
- S. Salehi, K. Y. Cheng, A. Heitz and M. P. Ginige, J. Environ. Manage., 2019, 238, 41–48 CrossRef.
- A. L. Gao and Y. Wan, Chemosphere, 2023, 313, 137434 CrossRef PubMed.
- Z. Hu, R. Wu, X. Pang, C. Yu and X. Jian, Sustainable Chem. Pharm., 2023, 36, 101279 CrossRef.
- L. Meina, M. Qiao, Q. Zhang, S. Xu and D. Wang, Sci. Rep., 2024, 14, 1235 CrossRef PubMed.
- P. Cheng, B. Wang, X. Wang and W. Xiao, Minerals, 2022, 12, 730 CrossRef.
- S. Balbay, Desalin. Water Treat., 2019, 161, 283–292 CrossRef.
- E. Pellicer-Castell, C. Belenguer-Sapiña, P. Amorós, J. El Haskouri, J. M. Herrero-Martínez and A. Mauri-Aucejo, Talanta, 2018, 189, 560–567 CrossRef PubMed.
- F. Tang, H. Yang, H. Chen, M. Zhou, P. Huang, Y. He, P. Song and R. Wang, J. Environ. Chem. Eng., 2022, 10, 108484 CrossRef.
- K. Riewklang, C. Polprasert, K. Nakason, S. Polprasert, S. Kwonpongsagoon, S. Mahasandana and B. Panyapinyopol, Environ. Res., 2023, 231, 116277 CrossRef PubMed.
- F. Mijangos, M. Kamel, G. Lesmes and D. Muraviev, React. Funct. Polym., 2004, 60, 151–161 CrossRef.
- S. Guida, G. Rubertelli, B. Jefferson and A. Soares, Chem. Eng. J., 2021, 420, 129913 CrossRef.
- Z. A. Shariff, L. Fraikin, A. Bogdan, A. Léonard, E. Meers and A. Pfennig, Environ. Technol., 2024, 45, 2820–2832 CrossRef.
- Y. Zhao, G. S. Lai, C. Li and R. Wang, Chem. Eng. J., 2023, 453, 139825 CrossRef.
- C. Niewersch, K. Meier, T. Wintgens and T. Melin, Desalination, 2010, 250, 1021–1024 CrossRef.
- C. Xing, J. Shi, F. Cui, J. Shen and H. Li, Chemosphere, 2021, 277, 130343 CrossRef.
- Y. Lei, M. Saakes, R. D. van der Weijden and C. J. N. Buisman, Water Res., 2020, 169, 115206 CrossRef.
- H. Miroslav, H. Pavel, B. Josef and K. Jarmila, J. Taiwan Inst. Chem. Eng., 2021, 129, 91–96 CrossRef.
- R. Altaf, B. Sun, H. Lu, H. Zhao and D. Liu, Crit. Rev. Environ. Sci. Technol., 2023, 53, 1032–1058 CrossRef.
- Y. Lei, M. Saakes, R. D. van der Weijden and C. J. N. Buisman, Water Res., 2020, 169, 115206 CrossRef PubMed.
- X. Yang, D. Manhaeghe, R. Zhang, S. Song, K. Demeestere and S. W. van Hulle, ACS Sustainable Chem. Eng., 2021, 9, 16946–16955 CrossRef.
- C. Chen, C. Ma, X. Yang, K. Demeestere, A. Nikiforov and S. W. van Hulle, Chem. Eng. J., 2023, 464, 142753 CrossRef.
- W. Wu, Y. Wang, K. Du, Q. Liu, T. Zhou, N. Wei, G. Liu and J. Guo, Appl. Catal., B, 2024, 354, 124118 CrossRef.
- S. Li, Y. Zhang, S. Zhao, L. Zhang, S. Qiao and J. Zhou, Sci. Total Environ., 2024, 914, 169760 CrossRef CAS.
- V. M. T. Serrano, L. E. Eshun, A. Farinha, G.-J. Witkamp and S. Bucs, J. Environ. Chem. Eng., 2022, 10, 109031 CrossRef CAS.
- J. Großeheilmann, H. Büttner, C. Kohrt, U. Kragl and T. Werner, ACS Sustainable Chem. Eng., 2015, 3, 2817–2822 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |