
In vivo mechanism of the interaction between trimethylamine lyase expression and glycolytic pathways†
Qian
Li
 abc,
Di
Wu
ab,
Yu
Song
ab,
Lu
Zhang
ab,
Ting
Wang
ab,
Xiaoxu
Chen
abc and
Min
Zhang
*abcd
abc,
Di
Wu
ab,
Yu
Song
ab,
Lu
Zhang
ab,
Ting
Wang
ab,
Xiaoxu
Chen
abc and
Min
Zhang
*abcd
aTianjin Agricultural University, Tianjin 300392, PR China. E-mail: zhangmin @tjau.edu.cn
bKey Laboratory of Smart Breeding Ministry of Agriculture and Rural Affairs, Tianjin Agricultural University, Tianjin 300392, PR China
cTianjin Agricultural University Nutritious and Healthy Food Sino-Thailand Joint Research Center, Tianjin Agricultural University, Tianjin 300392, PR China
dState Key Laboratory of Nutrition and Safety, Tianjin University of Science & Technology, Tianjin 300457, PR China
First published on 31st October 2024
Abstract
Recent studies confirmed that host–gut microbiota interactions modulate disease-linked metabolite TMA production via TMA lyase. However, microbial enzyme production mechanisms remain unclear. In the present study, we investigated the impact of dietary and intervention factors on gut microbiota, microbial gene expression, and the interplay between TMA lyase and glycolytic pathways in mice. Using 16S rRNA gene sequencing, metagenomics, and metabolomics, the gut microbiota composition and microbial functional gene expression profiles related to TMA lyase and glycolytic enzymes were determined. The results revealed that distinct diets and intervention factors altered gut microbiota, gene expression, and metabolites linked to glycine metabolism and glycolysis. Notably, an arabinoxylan-rich diet suppressed genes linked to choline, glycine, glycolysis, and TMA lyase, favoring glycine utilization via pyruvate pathways. Glycolytic inhibitors amplified these effects, mainly inhibiting pyruvate kinase. Our findings underscored the crosstalk between TMA lyase and glycolytic pathways, regulating glycine levels, and suggested avenues for targeted interventions and personalized diets to curb choline TMA lyase production.
Introduction
The quality of dietary nutrition is a key indicator of the advancement of human civilization. With economic development and the continuous improvement in living standards, people have increasingly been adopting a dietary structure characterized by nutritional surplus, insufficient grain consumption, and significantly increased consumption of animal-derived foods, thus leading to rising incidence rates of metabolic syndrome (MS).1 Among such syndromes, atherosclerosis has been considered one of the primary causes of morbidity and mortality worldwide, increasing over the past few decades. According to the data from the World Health Organization, over 2 billion people worldwide live with atherosclerosis, having been the primary cause of death of approximately 18 million people in 2023, hence accounting for 32% of the global death toll.2Excessive or unbalanced nutrition has been shown to be a significant cause of MS.3 It has been described that foods rich in choline compounds, such as red meat (beef and mutton), egg products, dairy products, animal brain and liver, among others, can induce the production of trimethylamine (TMA) and its oxides (TMAO) under the action of gut microbiota and host flavin monooxygenase. In addition, it is known that elevated levels of TMA and TMAO can lead to lipid disorders, such as cholesterol deposition and plaque formation, thus increasing the risk of developing MS, including atherosclerosis and thrombosis.4,5 Moreover, findings of epidemiological and animal model studies strongly point to the interaction between gut microorganisms and the host playing a key role in the development of atherosclerotic cardiovascular disease.
The gut microbiota has a primary contribution to the conversion of choline into TMA compounds.6 In fact, it is involved in key physiological processes such as nutrient metabolism and absorption, energy supply, maintenance of intestinal homeostasis, host gut–liver axis, and immune regulation. Moreover, the ecological balance of the gut microbiota is crucial for conserving and promoting overall health, while dysbiosis may lead to metabolic diseases.7 Harmful products produced by the gut microbiota could adversely influence the host's internal stability, tissue structure, or gene expression by disrupting the gut microbiota, ultimately triggering modifications within the intricate gut–liver axis signaling mechanisms, with potentially detrimental consequences for the host's overall well-being.8,9 Research suggests that gut microbiota regulation through gut–liver axis signaling pathways may underlie the development of cardiovascular diseases.10 The development of cardiovascular disease could be influenced by a multitude of metabolic factors, with key causative roles played by metabolic disorders encompassing elevated blood glucose levels, dyslipidemia, obesity, etc. These disorders, in conjunction, contribute significantly to the onset and progression of cardiovascular disease. Therefore, exploring the interplay between the gut microbiota and host gut–liver axis signaling pathways might provide a solid theoretical foundation for effectively preventing and addressing metabolic disorders resulting from unhealthy lifestyle and dietary choices.
Several studies have revealed that inhibiting TMA formation at its source is a promising approach to reducing TMAO levels and potentially minimizing related side effects, such as inflammation.11 Previous studies have explored human-associated microorganisms in various sequence databases and identified 216 TMA-lyase-containing microbial species across 102 genera. These microbial species contained homologous sequences of the genes cntA/B, yeaW/X, and/or cutC/D, which have been found to be involved in TMA metabolism. Notably, 13 strains from 5 genera (with cntA sequences) and 30 strains from 14 genera (with cutC) have been detected in healthy individuals.12 CutC is a key TMA lyase involved in the initial step of choline metabolism leading to TMA production, thus playing a crucial role in the biological conversion of TMA.13 Consequently, TMA lyase is emerging as a molecular target for intervening in the metabolism of dietary choline components, which may potentially help to mitigate associated health risks. It has been demonstrated that a reduced intake of dietary precursors or a decreased abundance of TMA-producing bacteria in the gut leads to lower TMA and TMAO levels. Thus, the goal of modifying the gut microbiota structure, suppressing TMA lyase activity, and subsequently reducing circulating TMA and TMAO levels can be achieved by reducing precursor intake, improving the gut microbiota composition, or intervening in TMA and TMAO metabolic pathways.
Escherichia coli is among the most abundant cutC-harboring bacterial species, being found in significantly higher abundance in atherosclerotic patients compared to healthy individuals. In addition, it is known that E. coli can effectively convert choline into TMA. Moreover, in vivo studies showed that E. coli and choline treatment increased TMAO levels and promoted aortic plaque accumulation in mice.
Methylglyoxal is a highly active α-dicarbonyl compound primarily produced in the glycolytic pathway, and it can also be generated from glycine conversion.14 The pathway leading to methylglyoxal production can be activated by increasing the cellular uptake of carbon-containing molecules, such as glucose, glucose-6-phosphate, lactate, and glycerol. In a preliminary study conducted by our research group, we found a correlation between glycine formation, methylglyoxal levels, and the expression of TMA lyase and its activating enzyme at metagenomic and metabolomic levels.15 The methylglyoxal bypass pathway provides an alternative route for many bacteria to degrade glycerone phosphate (G3P) into pyruvic acid via the formation of an intermediate product, namely fructose-1,6-bisphosphate (FBP). Subsequently, FBP is converted into dihydroxyacetone phosphate (DHAP), methylglyoxal, and lactic acid. The physiological role of this bypass is to allow G3P to be converted into pyruvic acid under phosphate-limited conditions, thereby inhibiting the activity of glycerone phosphate dehydrogenase (GAPDH) due to the scarcity of its substrate or one of the inorganic phosphates.16,17 The potential harmful effects of methylglyoxal can be mitigated by regulating the reactions involving this substrate.18,19
The present study aimed to explore the intricate relationship between dietary factors, gut microbiota composition, and expression patterns of genes related to choline metabolism and glycolysis regulation in a mouse model. To achieve this, an induced system was conceived for the regulation of choline intestinal metabolism and glycolytic pathways in mice. Moreover, the formation of choline-derived intestinal metabolites and physiological indicators was evaluated. In addition, the impact of the diet and regulatory factors on the gut microbiota structure and the expression patterns of functional microbial genes was investigated.
Materials and methods
Chemicals
Choline chloride was purchased from Kangzhiyuan Food Ingredients Co., Ltd (Guangdong, China). Arabinoxylan (derived from wheat bran) was purchased from Weiyi Peptide Biotechnology Co., Ltd, China, and comprised 99.6% of carbohydrates, 0.1% of protein, 0.1% of lipids and 0.2% of ash (w/w, on a dry basis). Basic dietary formulated feed was purchased from Synergy Biotechnology Co., Ltd, Jiangsu Province, China, and comprised 18.0% of crude protein, 4.0% of crude fat, 5.0% of crude fiber, 1.0–1.8% of calcium, 0.6–1.2% of phosphorus, 10.0% of moisture, and 8.0% of crude ash.20 All the other chemicals used were of analytical grade, unless otherwise specified.Construction of the induction system for the regulation of choline intestinal metabolism and the glycolytic pathway
During a four-week experiment, 120 specific pathogen-free (SPF) clean-grade C57BL/6J male mice (SBF Biotechnology Co., Ltd, Beijing, China) (20 g of body weight, 6 weeks of age) were selected based on body weight block randomization and then divided into different groups based on dietary and metabolic regulatory factors.21To evaluate the impact of different dietary components on mouse metabolism, the animals were subjected to a dietary induction phase spanning 14 days. Three different feeding groups were constituted: the standard diet feeding (SF) group; choline diet feeding (CF) group; and standard diet + choline + arabinoxylan feeding (AX) group. Mice in the SF group were fed a basic formulated feed. Mice in the CF group received a diet rich in choline via drinking water with added 1% choline chloride. Mice in the AX group were fed a combination of the standard diet, choline diet, and arabinoxylan (0.2 mL, 0.613 mg mL−1) calculated based on the recommended daily dietary fiber intake of 25–30 g and body surface area for adults.
To preliminary understand the effects of inhibitor molecules on the glycolytic pathway and other metabolic processes, the first stage of glycolysis inhibitor induction phase (days 15–21 of the experiment) was performed. Mice in the AX group were divided into four groups: (i) AX group (n = 10), ten mice fed a standard diet + choline + arabinoxylan; (ii) AX.H group, mice fed a standard diet + choline + arabinoxylan + inhibitor I; (iii) AX.L group, mice fed a standard diet + choline + arabinoxylan + inhibitor II; (iv) AX.P group, mice fed a standard diet + choline + arabinoxylan + inhibitor III. Inhibitors I, II, and III (MedChemExpress China) specifically target key enzymes in the glycolytic pathway, namely, hexokinase (HK), lactate dehydrogenase (LDHA), and pyruvate kinase (pyk) respectively. Mice in the AX.H, AX.L, and AX.P groups were administered 0.2 mL of inhibitor I (3 mg mL−1, Lonidamrie), inhibitor II (1 mg mL−1, GNE-140), or inhibitor III (0.5 mg mL−1, Shikonin), respectively.
To further investigate the impact of inhibitor molecules of the glycolytic pathway and other metabolic processes at different concentrations, the second stage of glycolysis inhibitor induction (days 22–28 of the experiment) was conducted. Mice in each inhibitor group were distributed evenly into three cages (10 mice per cage) and administered a high, medium, or low dose of the inhibitor once daily by gavage. Firstly, mice received a standard diet + choline + arabinoxylan + inhibitor I low-dose (AX.HL group); inhibitor I medium-dose (AX.H.M group); and inhibitor I high-dose (AX.H.H group), and were administered 0.2 mL of inhibitor I solution at various concentrations (i.e., 1 mg mL−1, 3 mg mL−1, and 5 mg mL−1; dissolved in physiological saline). Secondly, mice received a standard diet + choline + arabinoxylan + inhibitor II low-dose (AX.L.L group); inhibitor II medium-dose (AX.L.M group); and inhibitor II high-dose (AX.L.H group), and were administered 0.2 mL of inhibitor II solution at various concentrations (i.e., 0.5 mg mL−1, 1 mg mL−1, and 5 mg mL−1; dissolved in physiological saline). Lastly, mice received a standard diet + choline + arabinoxylan + inhibitor III low-dose (AX.P.L group); inhibitor III medium-dose (AX.P.M group); or inhibitor III high-dose (AX.P.H group), and were administered 0.2 mL of inhibitor III solution at various concentrations (i.e., 0.1 mg mL−1, 0.5 mg mL−1, and 1 mg mL−1; dissolved in physiological saline).
All animal model studies were approved by the Experimental Animal Ethics Committee of the Institute of Radiology, Chinese Academy of Medical Sciences (2022LLSC20), and all experiments were conducted using the guidelines for the Care and Use of Laboratory Animals.
The daily choline intake per mouse was calculated by multiplying daily water intake by the choline concentration in drinking water of 1% (Table 1).22 Arabinoxylan and glycolytic inhibitor intake were calculated by determining the gavage dose (Table 1). Body weight of mice was recorded weekly, and the results are shown in Table 1.
| Group | Choline intake (mg d−1) | Arabinoxylan intake (mg d−1) | Inhibitor intake (mg d−1) | Body mass records of mice (g) | ||||
|---|---|---|---|---|---|---|---|---|
| Adaptation period | 7d | 14d | 21d | 28d | ||||
| Note: the results are expressed as mean ± SD values. n = 10. Different letter marks indicate significant differences between groups, p < 0.05. | ||||||||
| SF | 0.00b | 0.00b | 0.00c | 23.68 ± 0.38a | 25.18 ± 0.49b | 25.18 ± 0.91a | 25.36 ± 0.43a | 25.58 ± 0.70b |
| CF | 23.34 ± 0.00a | 0.00b | 0.00c | 23.62 ± 1.12a | 24.74 ± 0.40b | 25.06 ± 0.34a | 25.68 ± 0.43a | 25.96 ± 0.61a |
| AX | 23.34 ± 0.00a | 0.12 ± 0.00a | 0.00c | 23.84 ± 0.95a | 25.74 ± 0.66a | 25.20 ± 0.74a | 25.42 ± 0.80a | 25.52 ± 0.41b |
| AX.H | 23.35 ± 0.01a | 0.12 ± 0.00a | 0.60 ± 0.00a | — | — | — | 24.86 ± 0.61a | — |
| AX.L | 22.98 ± 0.01b | 0.12 ± 0.00a | 0.20 ± 0.00b | — | — | — | 25.34 ± 0.94a | — |
| AX.P | 23.33 ± 0.00a | 0.12 ± 0.00a | 0.10 ± 0.00b | — | — | — | 25.34 ± 0.48a | — |
| AX.H.L | 23.34 ± 0.01a | 0.12 ± 0.00a | 0.20 ± 0.00b | — | — | — | — | 25.56 ± 0.68b |
| AX.H.M | 23.34 ± 0.00a | 0.12 ± 0.00a | 0.60 ± 0.00a | — | — | — | — | 25.38 ± 0.53b |
| AX.H.H | 23.34 ± 0.01a | 0.12 ± 0.00a | 1.00 ± 0.00a | — | — | — | — | 25.44 ± 0.69b |
| AX.L.L | 23.36 ± 0.00a | 0.12 ± 0.00a | 0.10 ± 0.00b | — | — | — | — | 25.36 ± 0.78b |
| AX.L.M | 23.35 ± 0.01a | 0.12 ± 0.00a | 0.20 ± 0.00b | — | — | — | — | 25.42 ± 0.66b |
| AX.L.H | 23.36 ± 0.01a | 0.12 ± 0.00a | 1.00 ± 0.00a | — | — | — | — | 25.36 ± 0.80b |
| AX.P.L | 23.34 ± 0.00a | 0.12 ± 0.00a | 0.02 ± 0.00c | — | — | — | — | 25.36 ± 0.71b |
| AX.P.M | 23.34 ± 0.01a | 0.12 ± 0.00a | 0.10 ± 0.00b | — | — | — | — | 25.54 ± 0.74b |
| AX.P.H | 23.34 ± 0.02a | 0.12 ± 0.00a | 0.20 ± 0.00b | — | — | — | — | 25.44 ± 0.56b |
At the end of the experiment, mice were sacrificed, and cecal digesta samples (at least 2 g) were collected immediately by gently squeezing the cecal content into a pre-sterilized sealed tube after abdominal dissection of the mice at 25 °C within a nitrogen-filled chamber in an anaerobic glove box.23 The sealed tube was then stored at approximately −80 °C in liquid nitrogen until further use or immediate use (within 1 h) in subsequent experiments.
Microbiotal sequencing of digesta samples
Using an Illumina NovaSeq6000 platform (Illumina Inc, San Diego, USA), 16S rRNA gene sequencing was conducted to characterize microbial species in the gut microbiota of mice in different sample groups as previously described. Digesta samples obtained on day 28 were subjected to 16S rRNA gene sequencing following the procedures described in a previous study. Briefly, after blood sampling, mice were sacrificed, their abdomens carefully dissected, and a portion of the cecal tissue (30 cm) was obtained. The cecal content was then obtained by gently squeezing it using a sharp forceps, washed with 0.2 M PBS buffer (pH = 7.0, 5.0 mL), placed in 10 mL sterile cryopreservation tubes, and immediately frozen in liquid nitrogen, followed by storage at −80 °C.DNA (100 ng) was extracted from digesta samples using the cetyl trimethyl ammonium bromide (CTAB) method. The quality and purity of DNA samples were assessed by agarose gel electrophoresis. DNA samples were then subjected to PCR amplifications to obtain sufficient material for subsequent sequencing analysis using Phusion® high-fidelity PCR master mix with GC buffer and DNA polymerase (New England Biolabs Ltd, Beijing, China). PCR products were then purified using magnetic beads and quantified using enzymatic labeling. Samples were mixed in equal amounts based on PCR product concentration. After thorough mixing, PCR products were subjected to 2% agarose gel electrophoresis, and the desired bands were excised and purified using a gel extraction kit (Qiagen Company, CA, USA).
The sequencing library was constructed using a TruSeq® DNA PCR Free Sample Preparation Kit (Illumina, USA) according to the manufacturer's recommendations. Briefly, the DNA sample was normalized with a resuspension buffer to a final volume of 100 μL to reach a final concentration of 2 ng μL−1. The diluted sample was then transferred to a Covaris microTUBE (Covaris Inc.) to generate 350 bp fragments in a Covaris M220 focused-ultrasonicator. The obtained fragments were then evaluated for size distribution using Qubit2.0 (Thermo Fisher Scientific-CN, Shanghai, China) and the 2100 Bioanalyzer (Agilent, Santa Clara, USA) The library was then subjected to 250 bp paired-end sequencing on the Illumina NovaSeq6000 platform.
Sequencing data for each sample was split based on barcode sequence and PCR primer sequence and then removed using FLASH software v.1.2.11 (https://ccb.jhu.edu/software/FLASH/). Raw data underwent quality trimming by removing low-quality base sites with a default quality threshold of ≤19 and a default length value of 15 bp using Fastp software. Host read removal was conducted using Bowtie2 software v.2.3.4 (https://bowtie-bio.sourceforge.net/bowtie2/index.shtml) to obtain clean reads.
High-quality, clean DNA sequences for each sample were assembled using MEGAHIT v.1.2.9 (https://github.com/voutcn/megahit) with k-mers within the range of 35–95. Additionally, clean sequencing data for each sample after quality control were compared to contigs obtained for each sample after assembly using Bowtie2 to obtain PE reads which were not utilized, which were then combined for mixed assembly.
To identify open reading frames (ORF), gene prediction was conducted on the obtained unique contigs with a minimum length of 500 bp using MetaGeneMark software v.3.38 (https://exon.gatech.edu/GeneMark/meta_gmhmmp.cgi). Predicted genes whose length was below 100 nt as well as redundant genes were filtered out using CD-HIT software v.4.8.1 (https://www.bioinformatics.org/cd-hit/) to obtain a non-redundant initial gene catalogue. Gene sequences with a minimum sequence identity of 95% and coverage of 90% were clustered, and the longest sequence was selected as the representative sequence. Genes with less than two representative reads in each sample were filtered out to obtain a unigene catalogue for subsequent analysis. The abundance information of each gene in each sample was calculated by counting the number of reads and the gene length.
To investigate species composition in each sample, contigs were annotated against the NCBI NR database using DIAMOND software considering E-value ≤1e−5 and a sequence identity of ≥95%. To ensure biological significance, the final species annotation information of the sequence was obtained using the Least Common Ancestors (LCA) algorithm in MEGAN software. Based on the results of LCA annotation analysis and the gene abundance table, abundance information at different classification levels (i.e., phylum, family, genus, species) was obtained for each sample. Operational taxonomic unit (OTU) clustering was conducted on the effective tags of all samples, using a 95% similarity threshold. Representative sequences for these OTUs were then annotated with their taxonomic information.
To gain insights into the microbial diversity of the samples, principal component analysis (PCA) and weighted UniFrac beta diversity analysis based on OTUs were performed. Moreover, LEfSe multivariate statistical analysis was employed to test the significance of inter-group species differences. In addition, to explore the impact of different environmental factors on microbial adaptability, as well as determining the dominant species and closely interacting species groups in a certain environment, network analysis was conducted.24
Determination of the levels of TMA and related metabolites
To determine the impact of regulatory factors on the levels of TMA and related metabolites, the contents of choline, TMA, TMAO, betaine, glycine methylglyoxal and pyruvate in digesta samples of mice were analyzed in an QTRAP® 6500 + LC-MS/MS system equipped with an electrospray ionization (ESI) turbo interface, operating in both positive and negative ion modes, and data were collected using Analyst 1.6 software (LC, Waters Co., Milford, MA, USA; MS, AB Sciex LLC, Framingham, MA, USA).After thawing, cecal content samples were vortexed vigorously for 10 s to ensure homogeneity, and then 50 μL of the sample was carefully transferred to a centrifuge tube, mixed with 250 μL of methanol, and vortexed vigorously for 5 min to ensure the complete extraction of metabolites. After centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min at 4 °C, 150 μL of the obtained supernatant was transferred to a new centrifuge tube and placed at −20 °C for 30 min. After this, the samples were subjected to centrifugation at 12000 rpm for 20 min at 4 °C to precipitate any residual solids. Subsequently, 140 μL of the supernatant was filtered through a protein precipitation plate for further LC-MS analysis.25
000 rpm for 10 min at 4 °C, 150 μL of the obtained supernatant was transferred to a new centrifuge tube and placed at −20 °C for 30 min. After this, the samples were subjected to centrifugation at 12000 rpm for 20 min at 4 °C to precipitate any residual solids. Subsequently, 140 μL of the supernatant was filtered through a protein precipitation plate for further LC-MS analysis.25
Analytical conditions were as follows: column, ACQUITY UPLC BEH Amide (i.d. 2.1 × 100 mm, 1.7 μm); solvent system, MilliQ water (Millipore, Bradford, USA) with 10 mM ammonium acetate and 0.3% ammonium hydroxide (solvent A, v/v, Sigma-Aldrich, St Louis, MO, USA), and 95% acetonitrile/water (solvent B, v/v, Sigma-Aldrich, St Louis, MO, USA). The gradient started at 95% B (0–1.2 min), decreased to 70% B (8 min), 50% B (9–11 min), and finally increased to 95% B (11.1–15 min); flow rate, 0.4 mL min−1; temperature, 40 °C; and injection volume, 2 μL.
ESI source operation parameters were as follows: ion source, turbo spray; source temperature 550 °C; ion spray voltage (IS), 5500 V (positive); curtain gas (CUR), 35.0 psi. The declustering potential (DP) and collision energy (CE) for individual multiple reaction monitoring (MRM) transitions were optimized. A specific set of MRM transitions was monitored for each period based on the metabolites eluted, namely, the precursor to product ion transitions m/z 104 → m/z 60 for choline, m/z 60 → m/z 44 for TMA, m/z 76.0 → m/z 58.0 for TMAO; m/z 118.0 → m/z 58.0 for betaine; m/z 114.0→ m/z 86.0 for creatine; m/z 75.0 → m/z 30.0 for glycine; m/z 72.0 → m/z 44.0 for methylglyoxal.
MultiQuant 3.0.3 software was used to process MS data. Retention time and peak shape information of the reference standards were used to integrate and correct chromatographic peaks to ensure accurate quantitative and qualitative analysis.26,27 The integrated correction results of the quantitative analysis of substances in different sample groups were obtained, with the horizontal axis referring to the detected retention time (Time, min) and the vertical axis referring to the ion detection current (Intensity, cps). Peak intensity in MS data corresponding to the standard substance at different concentrations was caculated.28 Standard curves for different substances were drawn using concentration on the x-axis and area on the y-axis. The integrated peak area of all detected samples was incorporated into the linear equation of the standard curve for calculation and then into the calculation formula to determine the actual content of the substance in the sample. Metabolite data were processed using unit variance scaling, and a heatmap was drawn using the ComplexHeatmap R package to perform hierarchical cluster analysis (HCA) on the accumulation patterns of metabolites among different sample groups.29
Metabonomics of digesta samples
To identify differences in functional gene expression profiles among different sample groups, digesta samples on day 28 from mice of the SF, CF, AX, and groups showing significant differences based on 16S rRNA gene sequencing and metabolomics data, namely AXH, AXL, and AXP groups, were subjected to metagenomic analysis. Metagenomes of digesta samples of mice were analyzed using an Illumina NovaSeq6000 platform (Illumina Inc, San Diego, USA), as previously described.30Digesta samples and DNA samples were collected using the method described in the section “Microbiotal sequencing for digesta samples”, and extracted DNA samples were subjected to qualification, quantification, construction of a sequencing library, quality trimming, quality control, and gene prediction, as described in the same previous section.31
To understand the functions of assembled sequences, unigenes were compared with each functional database entry using DIAMOND software (Blastp parameter, E-value ≤1E−5). Based on the BLAST results for each sequence, the unique sequence was selected for subsequent analysis. The relative abundance of genes at each functional level was equal to the sum of the relative abundance of genes annotated at a given functional level. Functional levels included six levels in the KEGG database, and three levels in the CAZy database. Based on these results, the number of microbial functional genes with nonzero abundances was determined.
Based on the abundance of reads at each classification level (EC, KO, pathway analysis), the number of annotated genes, relative abundance overview, abundance clustering heatmap, PCA and NMDS dimensionality reduction analysis, ANOSIM inter-group (within) difference analysis based on functional abundance, and comparative metabolic pathway analysis were carried out. Metastat and LEfSe analyses of functional differences between sample groups were also performed.
Knockout of key glycolytic genes of E. coli and in vitro induction expression
To further verify the interaction between glycolysis and TMA metabolism, a key glycolytic gene was knocked out using CRISPR/Cas9 technology.32To obtain a CRISPR-Cas9 guide RNA (gRNA) that could accurately recognize and direct the Cas9 protein to the target DNA sequence, an online CRISPR design tool (https://en.rc-crispr.com/; Red CottonTM, Guangzhou, China) was used to design the sgRNA sequence, and the optimal cleavage target for CRISPR/Cas9 genome editing in the gene sequence was determined. Based on the specificity and efficiency scores of sgRNA, the online tool generated a ranked list of potential cleavage sites for each, and the two top-ranked sgRNA targets were selected. A pair of oligomers (gRNA1/gRNA2) was synthesized based on the sgRNA sequence of each target gene. Oligomers were annealed in vitro to form double-stranded DNA and ligated to the CB-001 vector (containing expression elements for the Cas9 protein and multiple cloning sites for gRNA insertion). The CB-001-rtcR[gRNA] plasmid containing the target gRNA sequence and donor was transformed into E. coli MG1655 strain by electroporation.
To prepare competent cells, E. coli MG1655 was inoculated on Luria–Bertani (LB) agar containing antibiotics (Kana, 50 μg mL−1; Str, 50 μg mL−1) (Ubigene Biosciences Co., Ltd, Guangzhou, China), followed by incubation overnight at 37 °C in an incubator (Shanghai Yiheng Scientific Instrument Co., Ltd, Shanghai, China). A freshly activated single colony of E. coli DH5α was selected and inoculated into 5 mL of LB liquid medium, followed by incubation at 37 °C under shaking at 220 rpm until late logarithmic growth phase. Then, 1 mL of the obtained bacterial suspension was transferred to 100 mL of LB, incubated at 37 °C under shaking at 220 rpm until reaching an OD600 of 0.6. The bacterial suspension was immediately aliquoted into a 50 mL centrifuge tube and placed on ice for 10 min, then centrifuged at 4 °C and 4000 rpm. The precipitate was collected, and 10 mL of sterilized and pre-chilled glycerol-CaCl2 solution (glycerol 10%, v/v, CaCl2 0.1 M; Shanghai Shenggong Biotechnology Co., Ltd, China) was added, followed by gentle mixing to resuspend the cells. The suspension was centrifuged and resuspended by repeating the above steps twice. The cell suspension was collected and immediately stored at −80 °C until future use.
An aliquot of 200 μL of the competent cell suspension was thawed at room temperature and immediately placed on ice. Subsequently, 10 μL of CRISPR-B plasmid solution (5 ng μL−1) was added to competent cells, and the mixture was gently agitated to ensure uniform distribution, followed by incubation on ice for 10 min. Subsequently, the suspension was transferred to a pre-cooled 1 mm electrode cuvette (Bio-Rad, California, USA) and incubated on ice for 10 min. Cells were electroporated at 1.8 kV for 5 ms with 1 pulse, followed by the addition of 1 mL of LB liquid medium and gentle resuspension. The suspension was then transferred to a 1.5 mL EP tube and incubated at 30 °C under shaking at 180 rpm for 2 h for recovery.
After the recovery period, the suspension was centrifuged, and the supernatant was partially aspirated; precipitated cells were gently mixed to ensure uniform distribution and then spread onto LB agar supplemented with antibiotics (Kana, 50 μg mL−1, Str, 50 μg mL−1), followed by incubation at 37 °C for 12 h.
Bacterial genomic material from clones was extracted using a DNA extraction kit (Shanghai Shenggong Biotechnology Co., Ltd, China) according to the manufacturer's instructions. PCR amplification was performed using primers gene-JD-F/gene-JD-R and gene-BWD-F/gene-JD-R (Suzhou Genomics Biotechnologies Co., Ltd, China). PCR products were separated using agarose gel electrophoresis, and the recovery of DNA fragments was made using a gel recovery kit (Shanghai Shenggong Biotechnology Co., Ltd, China) according to the manufacturer's instructions. Colonies showing band sizes consistent with the theoretically expected PCR product size were subjected to sequencing, and the results were compared with the wild-type gene sequence. The CRISPR-B plasmid in clones with the correct sequence was eliminated through serial passages on antibiotic-free selection culture medium, thus allowing obtaining knockout-positive clone strains. Successful positive clones were preserved and stored at −80 °C for the construction of the in vitro induction system described herein in a previous section.33,34 Additionally, the content of choline, TMA, TMAO, glycine, and methylglyoxal was determined using the UPLC-MS/MS method described earlier. Similarly, differential expression of functional genes was determined using the method described earlier.35,36
Statistical analysis
Phylogenetic trees were drawn using the ggtree package. Statistical analyses and graphs were carried out using R software v.3.3.2, OriginPro 2022 SR1 software (OriginLab Corporation, Northampton, USA), and/or SPSS 22.0 (IBM, NYS, USA). The ComplexHeatmap package was used to visualize heatmaps. Box plots and scatter plots were drawn using the ggplot2 package. Rarefaction analysis was performed to characterize gene richness. Our samples were randomly sampled 100 times with replacement, and the total number of identifiable genes was estimated using R software v.3.3.2. Differences were considered significant and extremely significant when P values were <0.05 and <0.01, respectively.Results and discussion
Impact of the diet and inhibitor intake on body weight of mice
As depicted in Table 1, no significant changes (P < 0.05) were observed in choline intake in mice in the CF and AX groups, as well as in the inhibitor groups (AX.H.L, AX.H.M, AX.H.H, AX.L.L, AX.L.M, AX.L.H, AX.P.L, AX.P.M, and AX.P.H), which was approximately 23 mg day−1, being significantly greater than that of mice in the SF group (0 mg day−1). Compared to mice in the SF and CF groups, mice in the AX and inhibitor groups exhibited a relatively higher arabinoxylan intake (approximately 0.12 mg day−1), although no significant differences were observed among these groups. Additionally, mice in the inhibitor groups individually ingested varying doses of inhibitors (I, II, or III), distinguishing them from those in the SF, CF, and AX groups.During the initial adaptation period, the body weight of mice in all experimental groups was 23–24 g. Following this period, the mice sustained daily eating patterns and maintained overall health, resulting in a body weight of 24–26 g. When comparing body weight of mice among the various experimental groups, no significant differences (P < 0.05) were observed, except for a minor reduction in the body weight of mice in the AX.H group during the third week of the experiment. This indicates that the addition of 1% choline chloride to drinking water and the administration of 0.12 mg day−1 of arabinoxylan solution or varying doses of inhibitors by gavage did not significantly influence the growth and development of mice.
Distinction in gut microbiota structure of mice
An in-depth analysis of the top 10 phyla and 35 most abundant genera of bacteria was conducted to elucidate the composition of the gut microbiota in mice receiving specific dietary components and metabolic inhibitors over a four-week period.The gut microbiota composition in the digesta samples of mice in various experimental groups exhibited distinct differences, as shown in Fig. 1. Compared to mice in the CF group (1.50), the gut microbiota in mice of the SF (2.73), AX (2.88), and medium-dose and high-dose inhibitor groups (AX.H.M, 6.51; AX.H.H, 2.86; AX.L.M, 2.51; AX.L.H, 2.91; AX.P.M, 2.53; AX.P.H, 2.93) showed varying degrees of increase in the Bacteroidetes to Firmicutes ratio (Table S1† and Fig. 1a). This has been suggested to reduce energy absorption and can be attributed to the food ingredient intake as shown in Table 1.37,38
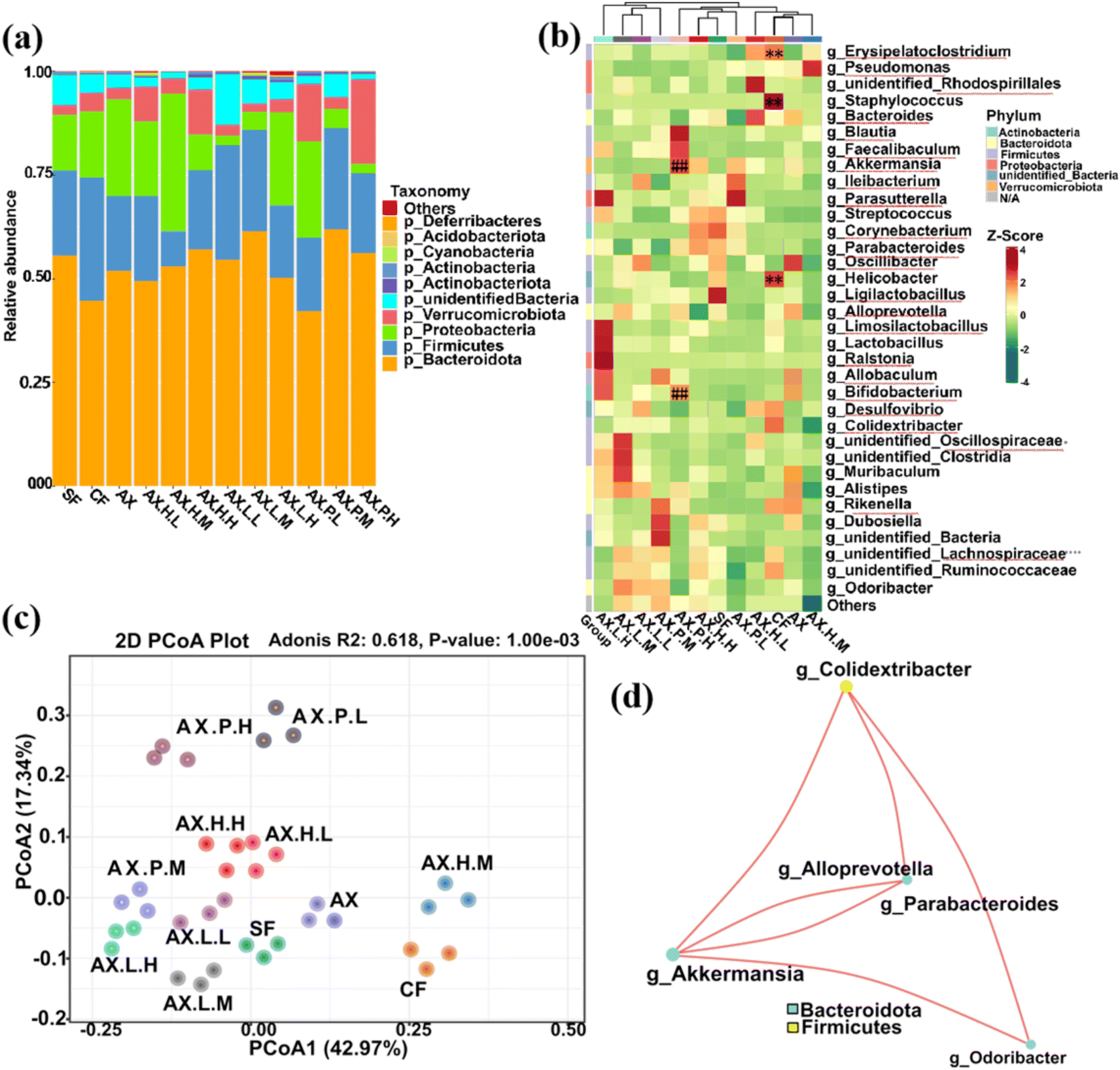 | ||
| Fig. 1 Differences in the structure of intestinal flora for mice with distinct induction groups. (a) The relative abundance of gut bacteria at the phylum level. (b) The quantitative data heatmap of species and their abundance based on OTUs identified in mice with distinct induction groups. The different colors indicate the differences in microbial abundance in the corresponding groups (red represents relatively high abundance, yellow represents intermediate abundance, and green represents relatively low abundance). ** indicates a significant positive correlation with the concentrations of TMA from intestinal digestion samples (P-value <0.01, Spearman correlation analysis). ## indicates a significant negative correlation with the concentrations of TMA from intestinal digestion samples (P-value <0.01, Spearman correlation analysis). (c) Principal component analysis (PCA) plot of gut microbiota at the genus level in mice. The horizontal and vertical axes represent principal components, and the percentages represent the contribution value of the principal component to the sample difference. (d) Boxplot of weighted UniFrac beta-diversity based on OTUs. SF group, the standard diet feeding group. CF group, the standard diet + choline diet feeding group. AX group, the standard diet + choline + arabinoxylan feeding group. AX.H, AX.L, and AX.P groups, respectively, referring to standard diet + choline + arabinoxylan + inhibitor I, inhibitor II and inhibitor III feeding groups; the suffixes .L, .M, .H, respectively, referring to high, medium, and low dose groups. | ||
The diversity in the clustering heatmap revealed that the relative abundance of Erysipelatoclostridium, Staphylococcus, and Helicobacter at the genus level positively correlated with TMA levels and was higher in the CF group (P < 0.01, Fig. 1b). In contrast, Akkermansia and Bifidobacterium were the most abundant genera in the AX groups, which were significantly negatively associated with TMA production (P < 0.01, Fig. 1b). Additionally, the relative abundances of the genera Lachnospiraceae, Ligilactobacillus, and Oscillibacter showed a significant downward trend in the CF group compared to the SF group. These genera have been reported to benefit gut microbiota balance and the environment.39 Comparing the AX group with the CF and SF groups showed that the relative abundances of the genera Lachnospiraceae, Ligilactobacillus, Oscillibacter, Muribaculum, and Alistipes were remarkably increased due to dietary fiber supplementation. These genera participate in the regulation of polysaccharide degradation and play a key role in the regulation of gene expression involved in glucose utilization, such as glycolysis/gluconeogenesis.40,41
Compared to the AX group, changes were observed in the gut microbial community structure of mice in the intervention groups with glycolytic inhibitors (i.e., AX.H., AX.L., AX.P.), which could be attributed to alterations in the host's homeostatic environment to a certain extent. For instance, compared with the AX group, the relative abundance of beneficial bacteria within the genus Bifidobacterium in the digesta samples of mice in the inhibitor groups was significantly reduced (P < 0.01, Fig. S2–S4†), except for the high-dose inhibitor II and III groups AX.L.H and AX.P.H, which showed increased abundances of Limosilactobacillus, Lactobacillus, Parabacteroides, Faecalibaculum and Blautia. This has attracted considerable attention due to the potential contribution to alleviating inflammatory and metabolic diseases, as well as the antibacterial activity against specific microorganisms.42 This could favor the inhibition of TMA lyase expression and TMA production. Moreover, compared to the AX group, a significant inhibition in Desulfovibrio, Pseudomonas, or Helicobacter genera was found in the gut microbiota of mice of the inhibitor III groups AX.P.L, AX.P.M, and AX.P.H (P < 0.01, Fig. S4†), thus indicating that inhibitor III is advantageous in reducing the abundance of harmful microorganisms.
Differences in group diversity observed in PCA analysis (Fig. 1c) further support the observations that choline and dietary fiber interventions, as well as inhibitor induction interventions, significantly impacted the gut microbiota of mice. Specifically, 42.97% and 17.34% of the variations in the gut microbiota (Fig. 1c) explained the main differences in gut bacterial patterns induced by the interventions conducted herein.43,44 The gut microbiota of the mice in the inhibitor III groups, AX.P.L, AX.P.M, and AX.P.H, exhibited notably greater relative distances from those of the CF and AX groups, implying that the inhibition of pyruvate kinase resulted in more pronounced alterations within the gut microbial community.
The co-occurrence network diagram allows for the study of the community structure and function of complex microbial environments. Given the vastly different co-occurring relationships among microorganisms in different environments, the species co-occurrence network diagram can intuitively illustrate the impact of different environmental factors on microbial adaptability, as well as identify the dominant species and closely interacting species groups in a certain environment.45 Such dominant species and species groups often play a unique and crucial role in maintaining the stability of the microbial community structure and function in which they are found.46 Thus, the co-occurrence network diagram of the gut microbiota of mice receiving different dietary interventions was analyzed based on correlation analysis. The results showed that the dominant species induced by the distinct intervention factors comprised the genera Colidextribacter, Akkermansia, Alloprevotella, Parabacteroides, and Odoribacter (Fig. 1d). This can be attributed to the distinct microbial composition influenced by the intake of different food ingredients and the diverse inhibitor interventions conducted herein.
Content of TMA and related metabolites in experimental groups
Subsequently, the ability of gut microbiota to convert choline into TMA and TMAO in mice fed different dietary and intervention factors was assessed by determining the content of their related metabolites, i.e., glycine and methylglyoxal.Higher TMA, TMAO, glycine, and methylglyoxal levels were found in the CF group compared to SF and AX groups (Fig. 2a). As expected, decreased TMA, TMAO, glycine, methylglyoxal and pyruvate levels were observed in mice of the AX group. Compared to the AX group, significantly decreased levels of choline, TMA, and TMAO were found in mice in the inhibitor induction groups AX.H., AX.L., and AX.P., especially in the high-dose inhibitor groups (AX.H.H, AX.L.H and AX.P.H). This suggests that glycolytic inhibitors may inhibit the activity of key glycolytic enzymes and reduce TMA lyase abundance, thereby lowering TMA and TMAO levels.47,48 Interestingly, relatively higher levels of glycine, methylglyoxal and pyruvate were found in mice in the low-dose inhibitor intervention groups, namely AX.H.L and AX.H.H groups. This could be unfavorable for inhibiting TMA lyase synthesis that is generated using glycine as a substrate.49
 | ||
| Fig. 2 Differential analysis of TMA and the relative metabolite content. (a) The quantitative data heatmap of TMA and the relative metabolite content identified in the intestinal digestion samples from mice with distinct induction groups. The horizontal and vertical axes represent the group names and metabolite information. Different colors represent different values obtained after standardization of relative content (red represents relatively high content, yellow represents intermediate content, and green represents relatively low content). (b) The quantitative data heatmap of TMA and the relative metabolite content identified in vitro from cultures of engineered bacteria and gene knockout engineered bacteria. (c) Abundance clustering heatmap of enzymes associated with glycolysis and glycine metabolism. The different colors indicate the differences of microobial abundance in the corresponding groups. SF group, the standard diet feeding group. CF group, the standard diet + choline diet feeding group. AX group, the standard diet + choline + arabinoxylan feeding group. AX.H, AX.L, and AX.P groups, respectively referring to standard diet + choline + arabinoxylan + inhibitor I, inhibitor II and inhibitor III feeding groups; the suffixes .L, .M, .H, respectively, referring to high, medium, and low dose groups. E. coli MG 1665-WT represents in vitro culture samples of wild-type Escherichia coli; E. coli MG 1665-GK represents in vitro culture samples of gene knockout Echerichia coli. | ||
Thus, considering the significant differences in the results of the 16S rRNA gene sequencing analysis and metabolomic analysis, the high-dose inhibitor groups (AX.H.H, AX.L.H and AX.P.H) were chosen for subsequent metagenomic analysis and were henceforth relabeled as AXH, AXL, and AXP groups.
Expression of functional genes in gut microbiota of mice in inhibitor induction groups
KEGG analysis of the functional metagenomic profiles of the gut microbiota of mice revealed that specific functional genes were distinctly enriched in different inhibitor induction groups (Fig. 3). As shown in Fig. 3a and based on PCA results, the CF and AXH groups showed similar functional gene expression profiles. Based on the Bray–Curtis distance method, the AX, AXL, and AXP groups were clustered into one category with similar functional gene structures, while the CF, SF, and AXH groups clustered into another category (Fig. 3b). This suggests that changes in functional metabolism were likely the result of the dietary interventions proposed herein with choline, arabinoxylan, and glycolytic inhibitors, likely due to changes in carbon cycle functions (glycolysis/sugar metabolism) among the experimental groups.50,51 | ||
| Fig. 3 Differences in KEGG analysis for the functional metagenomic profiles of gut microbiota. (a) PCA analysis results based on functional gene abundance. The horizontal axis, vertical axis and Z axis represent principal components, and the percentages represent the contribution value of the principal component to the sample differences. (b) Functional gene abundance clustering tree identified in intestinal digestion samples from mice with distinct induction groups. (c) Relative abundance (%) of CutC (choline trimethylamine-lyase) and CutD (choline trimethylamine-lyase activating enzyme). The different superscript letters in the same histogram represent significant differences (P < 0.05). (d) The relative abundance heatmap of genes responsible for glycolysis and glycine metabolism identified in intestinal digestion samples from mice with distinct induction groups. ** indicates a significant positive correlation with CutC and CutD expression from intestinal digestion samples (P-value <0.01, Spearman correlation analysis). ## indicates a significant negative correlation with the CutC and CutD expression from intestinal digestion samples (P-value <0.01, Spearman correlation analysis). (e) Functional gene abundance clustering heatmap identified in intestinal digestion samples from mice with distinct induction groups. The horizontal and vertical axes represent group names and annotated functional information (red represents relatively high abundance, yellow represents intermediate abundance, and green represents relatively low abundance). SF group, the standard diet feeding group. CF group, the standard diet + choline diet feeding group. AX group, the standard diet + choline + arabinoxylan feeding group. AXH, AXL, and AXP groups, respectively, referring to standard diet + choline + arabinoxylan + inhibitor I, inhibitor II and inhibitor III feeding groups, which showed significant differences in 16S rDNA and metabolomics detection results among the inhibitor groups. | ||
Interestingly, the functional gene structures of the mice in the AXH and CF groups were similar, indicating that the inhibition of hexokinase had a relatively weak inhibitory effect on choline metabolism. This could be attributed to the presence of alternative pathways that can generate glucose-6-phosphate in the glycolytic pathway. Therefore, the inhibition of hexokinase might not affect the generation of glucose-6-phosphate and subsequent steps.52,53
Based on the results of the differential expression analysis of genes coding for key enzymes shown in the KEGG database, the expression levels of genes encoding choline trimethylamine-lyase (cutC) and choline trimethylamine-lyase activating enzyme (cutD), Fig. 3c, were used to validate gene expression results.54 Compared to the SF group, the expression of cutC and cutD was found to be significantly upregulated in the CF group, which is consistent with previous findings.55 In contrast, when compared to the CF group, the AX group exhibited a significant downregulation of cutC and cutD (P < 0.05), which may be related to the reduced abundance of choline-TMA lyase activity in the gut microbiota of mice in the AX group, aligning with our previous findings.56 Conversely, compared to the digesta samples of mice in the AX group, mice in groups treated with inhibitors (i.e., AXH, AXL, AXP) showed a pronounced downregulation of cutC or cutD (P < 0.05). This suggested that alterations in the host environment (such as glycolysis inhibition) can modulate the expression of cutC or cutD in the gut microbiota, which can be associated with the glycine cleavage pathway leading to the production of methylglyoxal.57 Among the inhibitor induction groups, relatively lower expression levels of cutC and cutD were found in the AXP group treated with inhibitor III, which might be related to the inhibition of pyruvate kinase, leading to a reduced conversion of dihydroxyacetone phosphate into pyruvate.58 Consequently, decreased pyruvate levels directly promote the conversion of methylglyoxal to pyruvate, ultimately resulting in decreased glycine content and reduced expression of cutC and cutD. Nevertheless, compared to other inhibitor groups, relatively higher expression levels of cutC and cutD were found in the AXH group treated with the polyphosphate glucokinase inhibitor. This can be attributed to the insufficient impact of the polyphosphate glucokinase inhibitor on glycine, pyruvate, or methylglyoxal formation due to uncontrollable factors in lengthy metabolic pathways.59,60 For instance, D-glucose-6P can still be generated through the action of other enzymes (such as Pgm), even if polyphosphate glucokinase is suppressed, as illustrated in Fig. 4, which ensures that subsequent product generation remains unaffected.61
 | ||
| Fig. 4 The expression of key enzymes associated with glycolysis and glycine metabolism. | ||
KEGG analysis of the gut microbiota of mice showed that the genes specifically enriched in the metagenomes of mice in the CF group associated with glycine formation and utilization were distinct from those in the other groups (Fig. 3c). Notably, genes involved in the regulation of substrate and energy metabolism, such as glycolysis and glycine metabolism (Table S2† and Fig. 3), were more enriched in the CF group compared to the other groups. These genes included phosphoglucomutase (pgm), hexokinase (HK), glucose-6-phosphate isomerase (pgi), 6-phosphofructokinase (PFK9), diphosphate-dependent phosphofructokinase (pfp), fructose-bisphosphate aldolase class I (ALDO), triosephosphate isomerase (tpiA), glyceraldehyde 3-phosphate dehydrogenase (gapA), phosphoglycerate kinase (pgk), glyceraldehyde-3-phosphate dehydrogenase NADP+ (gapN), pyruvate kinase (pyk), choline dehydrogenase (gbsB), glycine betaine monooxygenase A (gbcA), glycine betaine-corrinoid protein co-methyltransferase (mtgB), N,N-dimethylglycine/sarcosine dehydrogenase (dgcA), glycine hydroxymethyltransferase (glyA), and L-serine/L-threonine ammonia-lyase (SDS), which are involved in choline dehydrogenation as well as in the formation of glyceraldehyde 3-phosphate, pyruvate, and glycine. This could result in increased glycine production in the CF group (Fig. 4). As expected, pyk genes were shown to be significantly and positively correlated (P < 0.01) with the expression of cutC and cutD. However, the relative abundance of these functional genes in the AX and inhibitor groups, especially in the AXP group (Table S2† and Fig. 3), was lower compared to the CF group, which is consistent with low glycine synthesis. This can be attributed to the increased level of choline intake and glucose utilization by the gut microbiota of mice in the CF group. Moreover, a relatively lower expression of 2,3-bisphosphoglycerate-independent phosphoglycerate mutase (gpmI), 2,3-bisphosphoglycerate-dependent phosphoglycerate mutase (gpmA), glycine C-acetyltransferase (kbl), threonine aldolase (ItaE), 3-hydroxy acid dehydrogenase/malonic semialdehyde reductase (ydfG), monoamine oxidase (aofH), threonine 3-dehydrogenase (TDH), and lactaldehyde dehydrogenase/glycolaldehyde dehydrogenase (aldA) was observed in the gut microbiota of mice in the CF group compared to those in the AX and inhibitor induction groups, which led to high pyruvate levels as well as to methylglyoxal and glycine accumulation in the CF group, thus improving TMA lyase synthesis. In contrast, gpmI, gpmA, kbl, ItaE, ydfG, aofH, TDH, and aldA were increased in the AX and inhibitor induction groups, which could circumvent the excessive increase in pyruvate levels as well as in methylglyoxal and glycine utilization induced by pyruvate accumulation (Fig. 4). This might be correlated with the weakened feedback inhibition induced by arabinoxylan or glycolysis inhibitors in the AX and inhibitor induction groups.
Moreover, correlation analysis between the relative abundance of these functional genes and TMA lyase activity showed that the abundance of kbl, aofH, and aldA was significantly and negatively associated (P < 0.01) with the expression of cutC and cutD. Among the inhibitor induction groups, the AXP group exhibited lower gene abundance positively correlated with cutC and cutD expression, and higher gene abundance negatively correlated with cutC and cutD expression. This observed phenomenon could be attributed to the more direct and effective effect of the pyruvate kinase inhibitor on pyruvate formation and glycine utilization.62
Differences in the abundance of a series of enzymes related to the utilization of substrates or ATP were observed among the groups (Fig. 3e). For instance, compared to the SF group, the abundance of peptidoglycan hydrolase (EC: 3.5.1.28) and ATP phosphohydrolase (EC: 7.4.2.8) in the CF group was increased. This indicates that the choline diet could be more conducive to the proliferation of microorganisms that utilize protein substances as growth substrates and ATP as energy sources, which may increase, to some extent, the number or diversity of microbial species in the gut microbiota.
Compared to the CF group, the expression of genes coding for enzymes including DNases, oxidoreductases, amino acid hydrolases, and other specific enzymes related to the carbon cycle, such as β-glucosidase (EC 3.2.1.21), β-galactosidase (EC 3.2.1.23), and α-amylase (EC 3.2.1.1) was enriched in the AX group. These enzymes have important functions such as antioxidation, blood sugar regulation, digestion promotion, and improvement of overall health indices. This indicates that an arabinoxylan diet improves gut microbiota structure and significantly activates the expression of starch-degrading enzymes, thus promoting gastrointestinal motility and regulating blood sugar.
Compared to the AX group, an upregulation of genes coding for certain enzymes including threonine protein kinase (EC 2.7.11.1) and alanine carboxypeptidase (EC 3.4.16.4) was observed in the gut microbiota of mice in the AXH group. However, a decreased abundance was observed for certain DNases and sugar metabolism enzymes such as α-L-glucosidase (EC 3.2.1.51), β-glucosidase (EC 3.2.1.21), and α-arabinosidase (EC 3.2.1.55), which might significantly inhibit key enzymes in the sugar metabolism pathway. This may be related to the action of the hexokinase inhibitor on sugar metabolism.
Moreover, the gut microbiota of mice in the AXL group showed a similar composition and abundance of carbon cycle-related enzymes, with a downregulation in the expression of genes coding for DNA- and certain protein-related enzymes. This indicates that the lactic dehydrogenase inhibitor could suppress other metabolism-related enzymes besides those involved in sugar metabolism.63
Compared to the AX group, a significant increase (P < 0.05) in the relative abundance of metabolic enzymes such as homeodomain interacting protein kinase (EC 2.7.11.1) and RNA-directed DNA polymerase (EC 2.7.7.49) was found in the gut microbiota of mice in the AXP group. This might be due to the increased expression of enzymes in other pathways such as protein cycling, induced by the pyruvate kinase inhibitor.
Changes in metagenomic and metabolomic profiles related to TMA and glycine metabolism in gene knockouts
To confirm whether the expression of functional genes related to glycolysis and glycine metabolism could regulate or control TMA lyase expression in the gut microbiota of mice, the metagenomic and metabolomic profiles of E. coli gene knockouts were analyzed.64,65Given the more significant differences observed in the AXP group based on metabolomics and metagenomics compared to the other experimental groups, as well as the feasibility of microbial gene knockout, the pyk gene was knocked out in E. coli. Quantitative heatmap analysis (Fig. 2b) revealed that, compared to wild-type E. coli cultured in vitro, the E. coli knockout exhibited reduced levels of glycine, choline, TMA, and TMAO, along with increased levels of methylglyoxal and pyruvate.66,67
A clustering heatmap analysis based on gene abundance (Fig. 2c) revealed that, compared to wild-type E. coli cultured in vitro, the E. coli knockout strain exhibited significantly reduced levels of pgm, ppgK, pgi, PFK9, pfp, ALDO, tpiA, gapA, pgk, gapN, eno, pyk, Idh, gbsB, gbcA, mtgB, dgcA, glyA, and SDS, which were observed in higher abundance in the digesta samples of mice in the CF group.68 This observation confirmed that the increased expression of these genes could be associated with choline utilization and the subsequent generation of TMA.69
Conversely, the levels of gpml, gpmA, kbI, ItaE, ydfG, aofH, TDH, and aldA were elevated, which were observed in higher abundance in the digesta samples of mice fed an arabinoxylan diet with the pyk inhibitor. This observation confirmed that the increased expression of these genes could weaken choline utilization and the subsequent generation of TMA.
Conclusions
The pioneering study comprehensively analyzes TMA lyase microbial biosynthesis in choline metabolism, emphasizing dynamic co-metabolic pathways and gene circuits in the gut microbiota of mice through multi-omics and gene knockout. High-choline, arabinoxylan diets, or glycolytic enzyme inhibitors could modulate gut microbiota–host interactions, impacting TMA lyase and its activating enzymes, as well as the levels of choline and its related metabolites in the gut based on the abundance of functional genes. Significant differences were made in metabolic pathways, altering choline-related metabolism. Arabinoxylan diets, with or without glycolytic inhibitors, significantly diminished gut TMA/TMAO levels and improved mouse physiology. The study underscored the influence of diet on gene expression profiles and suggested that the administration of glycolytic inhibitors could reduce the expression of cutC and cutD through non-specific pathways involving key enzymes (gbsB, ItaA, KbI, and TDH) in the choline–glycine–methylglyoxal pathway and the role of regulators on host homeostasis, advocating for a synergistic arabinoxylan-glycolytic inhibitor intervention. While validating glycolytic pathway–TMA lyase links, the host homeostasis–TMA lyase interplay remains complex, requiring deeper gut microbiota investigations.Author contributions
Qian Li and Di Wu contributed to the work equally and should be regarded as co-first authors, responsible for conceptualization, data curation, formal analysis, investigation, and writing the initial draft and revision. Yu Song, Lu Zhang, Ting Wang and Xiaoxu Chen contributed to the investigation, data curation and formal analysis. Min Zhang was responsible for project administration and supervision.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors have declared no conflict of interest.Acknowledgements
This work was supported by the project of the National Natural Science Foundation of China (No. 32101953); the 2024 open fund project from the Key Laboratory of Smart Breeding (Co-construction by Ministry and Province), Ministry of Agriculture and Rural Affairs (No. 2024-TJAULSBF-2309 and 2024-TJAULSBF-2303), 2024 Tianjin “the Belt and Road” Innovation Platform Project and the Key Research and Development Program of China (No. 2022YFF1100201). We thank the Instrument and Analysis Center at Tianjin University, Tianjin University of Science and Technology, and the College of Food Science and Bioengineering at Tianjin Agricultural University for their assistance in experiments.References
- J. L. M. Björkegren and A. J. Lusis, Atherosclerosis: Recent developments, Cell, 2022, 185, 1630–1645 CrossRef.
- P. Libby, The changing landscape of atherosclerosis, Nature, 2021, 592, 524–533 CrossRef CAS PubMed.
- L. Zhang, C. Xia, Y. Yang, F. Sun, Y. Zhang, H. Wang, R. Liu and M. Yuan, DNA methylation and histone post-translational modifications in atherosclerosis and a novel perspective for epigenetic therapy, Cell Commun. Signal, 2023, 21, 344 CrossRef CAS PubMed.
- Y. Y. Cai, F. Q. Huang, X. Lao, Y. Lu, X. Gao, R. N. Alolga, K. Yin, X. Zhou, Y. Wang, B. Liu, J. Shang, L. W. Qi and J. Li, Integrated metagenomics identifies a crucial role for trimethylamine-producing Lachnoclostridium in promoting atherosclerosis, npj Biofilms Microbiomes, 2022, 8, 11 CrossRef CAS.
- H. Lescinsky, A. Afshin, C. Ashbaugh, C. Bisignano, M. Brauer, G. Ferrara, S. I. Hay, J. He, V. Iannucci, L. B. Marczak, S. A. Mclaughlin, E. C. Mullany, M. C. Parent, A. L. Serfes, R. J. D. Sorensen, A. Y. Aravkin, P. Zheng and C. J. L. Murray, Health effects associated with consumption of unprocessed red meat: a Burden of Proof study, Nat. Med., 2022, 28, 2075–2082 CrossRef CAS.
- Z. Y. Bai, H. Zheng, Z. Luo, C. Hogstrand, L. J. Wang and Y. F. Song, Dietary Choline Mitigates High-Fat Diet-Impaired Chylomicrons Assembly via UPRer Modulated by perk DNA Methylation, Cells, 2022, 11, 38–48 Search PubMed.
- D. B. Morse, A. M. Michalowski, M. Ceribelli, J. D. Jonghe, M. Vias, D. Riley, T. D. Hill, T. Voss, S. Pittaluga, C. Muus, J. Liu, S. Boyle, D. A. Weitz, J. D. Brenton, J. D. Buenrostro, T. P. J. Knowles and C. J. Thomas, Positional influence on cellular transcriptional identity revealed through spatially segmented single-cell transcriptomics, Cell Syst., 2023, 14, 464–481 CrossRef CAS.
- O. Pabst, M. W. Hornef, F. G. Schaap, V. Cerovic, T. Clavel and T. Bruns, Gut-liver axis: barriers and functional circuits, Nat. Rev. Gastroenterol. Hepatol., 2023, 20, 447–461 CrossRef PubMed.
- H. Tilg, T. E. Adolph and M. Trauner, Gut-liver axis: Pathophysiological concepts and clinical implications, Cell Metab., 2022, 34, 1700–1718 CrossRef CAS PubMed.
- E. Barroso, M. M. Grau, W. Wahli, X. Palomer and M. V. Carrera, Striking a gut-liver balance for the antidiabetic effects of metformin, Trends Pharmacol. Sci., 2023, 44, 457–473 CrossRef CAS PubMed.
- K. Ri, T. H. Weng, A. C. Cabezudo, W. Jösting, Y. Zhang and A. Bazzone, Molecular mechanism of choline and ethanolamine transport in humans, Nature, 2024, 630, 501–508 CrossRef CAS PubMed.
- J. Kashyap, J. R. Ringiesn, N. Schwab and D. J. Ferguson Jr., Isolation and characterization of a novel choline degrading Citrobacter amalonaticus strain from the human gut, Curr. Res. Microb. Sci., 2022, 3, 100157 CAS.
- A. Y. M. Woo, M. A. A. Ramos, R. Narayan, K. C. R. Corke, M. L. Wang, W. J. S. Espinola and E. P. Balskus, Targeting the human gut microbiome with small-molecule inhibitors, Nat. Rev. Chem., 2023, 7, 319–339 CrossRef CAS.
- Y. Guan, W. Yao, H. Yu, Y. Feng, Y. Zhao, X. Zhan and Y. Wang, Chronic stress promotes colorectal cancer progression by enhancing glycolysis through β2-AR/CREB1 signal pathway, Int. J. Biol. Sci., 2023, 19, 2006–2019 CrossRef CAS PubMed.
- L. Liu and K. Shah, The Potential of the Gut Microbiome to Reshape the Cancer Therapy Paradigm: A Review, JAMA Oncol., 2022, 8, 1059–1067 CrossRef.
- X. Wang, S. Q. Fu, X. Yuan, F. Yu, Q. Ji, H. W. Tang, R. K. Li, S. Huang, P. Q. Huang, W. T. Qing, H. Zuo, C. Du, L. L. Yao, H. Li, J. Li, D. X. Li, Y. Yang, S. Y. Xiao, A. Tulamaiti, X. F. Wang, C. H. Dai, X. Zhuang, S. H. Jiang, L. P. Hu, X. L. Zhang and Z. G. Zhang, A GAPDH serotonylation system couples CD8(+) T cell glycolytic metabolism to antitumor immunity, Mol. Cell, 2024, 84, 760–775.e7 CrossRef CAS PubMed.
- W. Song, X. Zhai, J. Shi, X. Zou, Y. Xue and Y. Sun, A ratiometric fluorescence amine sensor based on carbon quantum dot-loaded electrospun polyvinylidene fluoride film for visual monitoring of food freshness, Food Chem., 2024, 434, 137423 CrossRef CAS.
- J. Song, X. Shi, X. Li and J. Zheng, Choline diet improves serum lipid parameters and alters egg composition in breeder ducks, Vet. Med. Sci., 2022, 8, 1553–1562 CrossRef CAS PubMed.
- A. Leone, A. Nicolò, I. Prevenzano, F. Zatterale, M. Longo, A. Desiderio, R. Spinelli, M. Campitelli, D. Conza, G. A. Raciti, F. Beguinot, C. Nigro and C. Miele, Methylglyoxal Impairs the Pro-Angiogenic Ability of Mouse Adipose-Derived Stem Cells (mADSCs) via a Senescence-Associated Mechanism, Cells, 2023, 12, 1741 CrossRef CAS PubMed.
- L. Hao, D. Kshatriya, X. Li, A. Badrinath, Z. Szmacinski, M. J. Goedken, M. Polunas and N. T. Bello, Acute feeding suppression and toxicity of raspberry ketone [4-(4-hydroxyphenyl)-2-butanone] in mice, Food Chem. Toxicol., 2020, 143, 111512 CrossRef CAS PubMed.
- X. Yu, Z. Zhao, X. Yan, J. Xie, Q. Yu and Y. Chen, Extraction optimization of tea saponins from Camellia oleifera seed meal with deep eutectic solvents: Composition identification and properties evaluation, Food Chem., 2023, 427, 136681 CrossRef CAS PubMed.
- G. B. V. S. Lakshmi, A. K. Yadav, N. Mehlawat, R. Jalandra, P. R. Solanki and A. Kumar, Gut microbiota derived trimethylamine N-oxide (TMAO) detection through molecularly imprinted polymer based sensor, Sci. Rep., 2021, 11, 1338 CrossRef CAS PubMed.
- J. Lu, J. Liao, M. Qin, H. Li, Q. Zhang, Y. Chen and J. Cheng, Single-Cell RNAseq Resolve the Potential Effects of LanCL1 Gene in the Mouse Testis, Cells, 2022, 11, 4135 CrossRef CAS PubMed.
- R. Sharma, A. Chischolm, M. Parikh, A. I. Qureshi, P. Sahota and M. M. Thakkar, Ischemic Stroke Disrupts Sleep Homeostasis in Middle-Aged Mice, Cells, 2022, 11, 2818 CrossRef CAS.
- J. Tang, M. Qin, L. Tang, D. Shan, C. Zhang, Y. Zhang, H. Wei, L. Qiu and J. Yu, Enterobacter aerogenes ZDY01 inhibits choline-induced atherosclerosis through CDCA-FXR-FGF15 axis, Food Funct., 2021, 12, 9932–9946 RSC.
- J. W. Blayney, H. Francis, A. Rampasekova, B. Camellato, L. Mitchell, R. Stolper, L. Cornell, C. Babbs, J. D. Boeke, D. R. Higgs and M. Kassouf, Super-enhancers include classical enhancers and facilitators to fully activate gene expression, Cell, 2023, 186, 5826–5839.e18 CrossRef CAS.
- L. Funk, K. C. Su, J. Ly, D. Feldman, A. Singh, B. Moodie, P. C. Blainey and I. M. Cheeseman, The phenotypic landscape of essential human genes, Cell, 2022, 185, 4634–4653.e22 CrossRef CAS.
- C. S. Cowan, M. Renner, M. D. Gennaro, B. G. Scherf, D. Goldblum and Y. Hou, Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution, Cell, 2020, 182, 1623–1640.e34 CrossRef CAS.
- H. H. Shi, L. P. Chen, C. C. Wang, Y. C. Zhao, Y. M. Wang, C. H. Xue and T. T. Zhang, Docosahexaenoic acid-acylated curcumin diester alleviates cisplatin-induced acute kidney injury by regulating the effect of gut microbiota on the lipopolysaccharide- and trimethylamine-N-oxide-mediated PI3K/Akt/NF-κB signaling pathway in mice, Food Funct., 2022, 13, 6103–6117 RSC.
- V. L. Zogopoulos, A. Malatras, K. Kyriakidis, C. Charalampous, E. A. Makrygianni, S. Duguez, M. A. Koutsi, M. Pouliou, C. Vasileiou, W. J. Duddy, M. Agelopoulos, G. P. Chrousos, V. A. Iconomidou and I. Michalopoulos, HGCA2.0: An RNA-Seq Based Webtool for Gene Coexpression Analysis in Homo sapiens, Cells, 2023, 12, 388 CrossRef CAS PubMed.
- M. Gong, H. Lu, L. Li, M. Feng and Z. Zou, Integration of transcriptomics and metabonomics revealed the protective effects of hemp seed oil against methionine-choline-deficient diet-induced non797 alcoholic steatohepatitis in mice, Food Funct., 2023, 14, 2096–2111 RSC.
- A. T. Lee, E. F. Chang, M. F. Paredes and T. J. Nowakowski, Large-scale neurophysiology and single-cell profiling in human neuroscience, Nature, 2024, 630, 587–595 CrossRef CAS.
- Y. Zou, E. Geuverink, L. W. Beukeboom, E. C. Verhulst and L. Zande, A chimeric gene paternally instructs female sex determination in the haplodiploid wasp Nasonia, Science, 2020, 370, 1115–1118 CrossRef CAS PubMed.
- S. Panicker, G. Chengizkhan, R. Gor, I. Ramachandran and S. Ramalingam, Exploring the Relationship between Fusion Genes and MicroRNAs in Cancer, Cells, 2023, 12, 2467 CrossRef CAS.
- C. C. Galouzis and B. Prud'homme, Transvection regulates the sex-biased expression of a fly X-linked gene, Science, 2021, 371, 396–400 CrossRef CAS PubMed.
- J. Li, P. Huang, W. Cheng and Q. Niu, Stilbene-based derivatives as potential inhibitors of trimethylamine (TMA)-lyase affect gut microbiota in coronary heart disease, Food Sci. Nutr., 2023, 11, 93–100 CrossRef CAS PubMed.
- H. Lee, X. Liu, J. P. An and Y. Wang, Identification of Polymethoxyflavones (PMFs) from Orange Peel and Their Inhibitory Effects on the Formation of Trimethylamine (TMA) and Trimethylamine-N-oxide (TMAO) Using cntA/B and cutC/D Enzymes and Molecular Docking, J. Agric. Food Chem., 2023, 71, 16114–16124 CrossRef CAS PubMed.
- L. I. Carres, E. S. Krueger, J. A. Herring, J. S. Tessem and A. P. Neilson, Potential of Phenolic Compounds and Their Gut Microbiota-Derived Metabolites to Reduce TMA Formation: Application of an In Vitro Fermentation High-Throughput Screening Model, J. Agric. Food Chem., 2022, 70, 3207–3218 CrossRef.
- J. Wang, N. Zhu, X. Su, Y. Gao and R. Yang, Gut-Microbiota-Derived Metabolites Maintain Gut and Systemic Immune Homeostasis, Cells, 2023, 12, 793 CrossRef CAS PubMed.
- H. Chen, Y. Li, H. Li, X. Chen, H. Fu, D. Mao, W. Chen, L. Lan, C. Wang, K. Hu, J. Li, C. Zhu, I. Evans, E. Cheung, D. Lu, Y. He, A. Behrens, D. Yin and C. Zhang, NBS1 lactylation is required for efficient DNA repair and chemotherapy resistance, Nature, 2024, 631, 663–669 CrossRef CAS PubMed.
- I. P. Torres, M. E. Soto, V. G. Lans, L. M. Pech and E. S. Castro, The Possible Role of Glucose-6-Phosphate Dehydrogenase in the SARS-CoV-2 Infection, Cells, 2022, 11, 1982 CrossRef.
- L. Ma, Y. Ma and Y. Liu, β-Sitosterol protects against food allergic response in BALB/c mice by regulating the intestinal barrier function and reconstructing the gut microbiota structure, Food Funct., 2023, 14, 4456–4469 RSC.
- Q. Nie, J. Hu, H. Chen, F. Geng and S. Nie, Arabinoxylan ameliorates type 2 diabetes by regulating the gut microbiota and metabolites, Food Chem., 2022, 371, 131106 CrossRef CAS PubMed.
- D. Zhang, R. C. Rudjito, S. Pietiäinen, S. C. Chang, A. Idström, L. Evenäs, F. Vilaplana and A. J. Quero, Arabinoxylan supplemented bread: From extraction of fibers to effect of baking, digestion, and fermentation, Food Chem., 2023, 413, 135660 CrossRef CAS PubMed.
- S. Luo, L. He, H. Zhang, Z. Li, C. Liu and T. Chen, Arabinoxylan from rice bran protects mice against high-fat diet-induced obesity and metabolic inflammation by modulating gut microbiota and short-chain fatty acids, Food Funct., 2022, 13, 7707–7719 RSC.
- Z. He, H. Zhu, J. Liu, E. Kwek, K. Y. Ma and Z. Y. Chen, Mangiferin alleviates trimethylamine-N-oxide (TMAO)-induced atherogenesis and modulates gut microbiota in mice, Food Funct., 2023, 14, 9212–9225 RSC.
- H. Zhuang, X. Ren, Y. Zhang, F. Jiang and P. Zhou, Trimethylamine-N-oxide sensitizes chondrocytes to mechanical loading through the upregulation of Piezo1, Food Chem. Toxicol., 2023, 175, 113726 CrossRef CAS.
- T. Đorđević, M. Milošević and M. Antov, Advance diversity of enzymatically modified arabinoxylan from wheat chaff, Food Chem., 2021, 339, 128093 CrossRef.
- C. Zhao, J. Shen, S. Xu, J. Wei, H. Liu, S. Xie, Y. Pan, Y. Zhao and Y. Zhu, Ultra-efficient trimethylamine gas sensor based on Au nanoparticles sensitized WO(3) nanosheets for rapid assessment of seafood freshness, Food Chem., 2022, 392, 133318 CrossRef CAS.
- N. Buawangpong, K. Pinyopornpanish, A. Phrommintikul, N. Chindapan, S. Devahastin, N. Chattipakorn and S. C. Chattipakorn, Increased plasma trimethylamine-N-oxide levels are associated with mild cognitive impairment in high cardiovascular risk elderly population, Food Funct., 2022, 13, 10013–10022 RSC.
- Y. Li, S. Song, Y. Li, F. Du, S. Li and J. Li, Novel insights into the inhibitory mechanism of (+)-catechin against trimethylamine-N-oxide demethylase, Food Chem., 2022, 373, 131559 CrossRef CAS.
- M. Zeidler, A. Hüttenhofer, M. Kress and K. K. Kummer, Intragenic MicroRNAs Autoregulate Their Host Genes in Both Direct and Indirect Ways-A Cross-Species Analysis, Cells, 2020, 9, 232 CrossRef PubMed.
- L. Zhang, J. Wei, X. Liu, D. Li, X. Pang, F. Chen, H. Cao and P. Lei, Gut microbiota-astrocyte axis: new insights into age-related cognitive decline, Neural Regener. Res., 2025, 20, 990–1008 CrossRef.
- A. N. Gorelick, F. J. S. Rivera, Y. Cai, C. M. Bielski, E. Biederstedt and P. Jonsson, Phase and context shape the function of composite oncogenic mutations, Nature, 2020, 582, 100–103 CrossRef CAS.
- L. Jing, H. Zhang, Q. Xiang, H. Hu, C. Zhai, S. Xu and H. Tian, Role of Trimethylamine N-Oxide in Heart Failure, Rev. Cardiovasc. Med., 2024, 25, 240 CrossRef.
- Q. Li, T. Wu, M. Zhang, H. Chen and R. Liu, Induction of the glycolysis product methylglyoxal on trimethylamine lyase synthesis in the intestinal microbiota from mice fed with choline and dietary fiber, Food Funct., 2021, 12, 9880–9893 RSC.
- W. Song, X. Zhai, J. Shi, X. Zou, Y. Xue, Y. Sun and M. Povey, A ratiometric fluorescence amine sensor based on carbon quantum dot-loaded electrospun polyvinylidene fluoride film for visual monitoring of food freshness, Food Chem., 2024, 434, 137423 CrossRef CAS PubMed.
- Y. Fang, Y. Yuan and J. Liu, Casting Light on the Adaptation Mechanisms and Evolutionary History of the Widespread Sumerlaeota, mBio, 2021, 12, e00350 CrossRef CAS.
- K. K. Oh, H. Gupta, B. H. Min, R. Ganesan, S. P. Sharma and S. M. Won, Elucidation of Prebiotics, Probiotics, Postbiotics, and Target from Gut Microbiota to Alleviate Obesity via Network Pharmacology Study, Cells, 2022, 11, 2903 CrossRef CAS PubMed.
- W. Yoo, J. K. Zieba, N. J. Foegeding, T. P. Torres, C. D. Shelton and N. G. Shealy, High-fat diet-induced colonocyte dysfunction escalates microbiota-derived trimethylamine N-oxide, Science, 2021, 373, 813–818 CrossRef CAS PubMed.
- L. Zhang, Q. Wu, N. Wang, L. Zhang, X. Yang and Y. Zhao, Quercetin inhibits hepatotoxic effects by reducing trimethylamine-N-oxide formation in C57BL/6J mice fed with a high L-carnitine diet, Food Funct., 2023, 14, 206–214 RSC.
- X. Gao, G. Sun, E. Randell, Y. Tian and H. Zhou, Systematic investigation of the relationships of trimethylamine N-oxide and L-carnitine with obesity in both humans and rodents, Food Funct., 2020, 11, 7707–7716 RSC.
- N. Krause and A. Wegner, Fructose Metabolism in Cancer, Cells, 2020, 9, 2635 CrossRef CAS.
- C. Simó, T. Fornari, M. R. G. Risco, A. P. Cearra, L. Abecia and J. Anguita, Resazurin-based high-throughput screening method for the discovery of dietary phytochemicals to target microbial transformation of L-carnitine into trimethylamine, a gut metabolite associated with cardiovascular disease, Food Funct., 2022, 13, 5640–5653 RSC.
- C. Simó and V. G. Cañas, Dietary bioactive ingredients to modulate the gut microbiota-derived metabolite TMAO, New opportunities for functional food development, Food Funct., 2020, 11, 6745–6776 RSC.
- C. M. Liao, V. C. Wulfmeyer, M. Swallow, C. S. Falk, H. Haller, R. Korstanje, A. Melk and R. Schmitt, Induction of Stress-Induced Renal Cellular Senescence In Vitro: Impact of Mouse Strain Genetic Diversity, Cells, 2021, 10, 1437 CrossRef CAS PubMed.
- Y. Hou, Y. Wang, Y. Zhang, Z. Lu, Z. Zhang, Z. Dong and Y. Qiu, Cotransport of nanoplastics with nZnO in saturated porous media: From brackish water to seawater, J. Environ. Sci., 2025, 148, 541–552 CrossRef.
- Y. Deng, J. Zou, Y. Hong, Q. Peng, X. Fu, R. Duan, J. Chen and X. Chen, Higher Circulating Trimethylamine N-Oxide Aggravates Cognitive Impairment Probably via Downregulating Hippocampal SIRT1 in Vascular Dementia Rats, Cells, 2022, 11, 3650 CrossRef CAS.
- Y. Zhang, L. Zhan, Q. Wen, Y. Feng, Y. Luo and T. Tan, Trapping Methylglyoxal by Taxifolin and Its Metabolites in Mice, J. Agric. Food Chem., 2022, 70, 5026–5038 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fo03809f |
| This journal is © The Royal Society of Chemistry 2025 |