
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stability of multifunctional lignin-g-PLGA films as a function of lignin type and lignin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :PLGA ratio
:PLGA ratio
Omar
Mendez
 a,
Carlos E.
Astete
a,
Rafael
Cueto
b,
Alvaro
Garcia
b,
Jessica R.
Eberhard
c,
Fannyuy V.
Kewir
a,
Kevin
Hoffseth
a and
Cristina M.
Sabliov
*a
a,
Carlos E.
Astete
a,
Rafael
Cueto
b,
Alvaro
Garcia
b,
Jessica R.
Eberhard
c,
Fannyuy V.
Kewir
a,
Kevin
Hoffseth
a and
Cristina M.
Sabliov
*a
aDepartment of Biological & Agricultural Engineering, Louisiana State University, LSU Ag Center, Baton Rouge, Louisiana 70803, USA. E-mail: csabliov@lsu.edu
bDepartment of Chemistry, Louisiana State University, Baton Rouge, Louisiana 70803, USA
cDepartment of Biological Sciences, Louisiana State University, Baton Rouge, Louisiana 70803, USA
First published on 11th April 2025
Abstract
Biodegradable films were synthesized from lignin(LN)-grafted-PLGA polymers, and their stability was tracked over 12 months. The impact of the type of lignin, alkaline LN (ALN) and sodium lignosulfonate (SLN), and the LN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PLGA w/w ratio (1:4 and 1:6 w/w) on the mechanical, chemical, thermal and surface properties of the films was assessed. Films were made using an interphase formation process and were stored at two relative humidities (RH; 30% or 70%) for one year. Mechanical characterization revealed that ALN-g-PLGA films were stiffer than SLN-g-PLGA and control (PLGA) films. LN-grafted films had a glass transition temperature (Tg) of approximately 50.9 ± 4.6 °C, which remained consistent over 12 months at both RHs. For the LN-grafted films, the contact angles (CAs) and roughness coefficients (Rc) of the aqueous and organic sides differed. The aqueous side showed a lower CA and higher Rc, while the organic side had a higher CA and lower Rc, suggesting a direct correlation between wettability and roughness. The CA and Rc of most films showed no significant changes over time. UV (UV-A/B/C) shielding was above 95% for ALN-g-PLGA, and above 75% for SLN-g-PLGA films; in comparison, control PLGA films only blocked 67% of UV radiation at time zero. The UV-blocking by LN-g-PLGA films did not change over time, but for PLGA films it decreased from 67% to 23%. Our study shows that, compared with PLGA films, LN-g-PLGA films maintained their integrity for a longer period at both high and low RHs. Because of their higher durability, UV absorption properties and potential for tunability of mechanical and surface properties of the films, it is concluded that LN-g-PLGA films have a high versatility for applications ranging from packaging to coating materials.
:PLGA w/w ratio (1:4 and 1:6 w/w) on the mechanical, chemical, thermal and surface properties of the films was assessed. Films were made using an interphase formation process and were stored at two relative humidities (RH; 30% or 70%) for one year. Mechanical characterization revealed that ALN-g-PLGA films were stiffer than SLN-g-PLGA and control (PLGA) films. LN-grafted films had a glass transition temperature (Tg) of approximately 50.9 ± 4.6 °C, which remained consistent over 12 months at both RHs. For the LN-grafted films, the contact angles (CAs) and roughness coefficients (Rc) of the aqueous and organic sides differed. The aqueous side showed a lower CA and higher Rc, while the organic side had a higher CA and lower Rc, suggesting a direct correlation between wettability and roughness. The CA and Rc of most films showed no significant changes over time. UV (UV-A/B/C) shielding was above 95% for ALN-g-PLGA, and above 75% for SLN-g-PLGA films; in comparison, control PLGA films only blocked 67% of UV radiation at time zero. The UV-blocking by LN-g-PLGA films did not change over time, but for PLGA films it decreased from 67% to 23%. Our study shows that, compared with PLGA films, LN-g-PLGA films maintained their integrity for a longer period at both high and low RHs. Because of their higher durability, UV absorption properties and potential for tunability of mechanical and surface properties of the films, it is concluded that LN-g-PLGA films have a high versatility for applications ranging from packaging to coating materials.
Sustainability spotlightLignin is the second most abundant renewable polymer. A product of the pulping industry, most lignin is burned for energy production. This study investigates the development of value-added, lignin-based materials with adjustable biodegradability aimed at providing sustainable alternatives to conventional plastics. This work aligns well with the global effort of transitioning toward more sustainable and renewable materials. |
Introduction
Plastic pollution has become a critical environmental problem. In 2019, 446.2 million tons of plastics were produced,1 of which 50% were destined for single use products.2 The main types of plastics produced are petroleum based, such as low- and high-density polyethylene, polystyrene, polypropylene, polyester, polyvinylchloride, polyamide, and polyurethane.3 Because plastics have a low production cost, are long-lasting materials, and are used for disposable objects, they accumulate in the environment or in landfills or are burned.4 Microplastics and nanoplastics, formed due to fragmentation and weathering effects, are of great concern due to their low degradability, and potential health impact after ingestion. Around 13 million tons of plastics produced end up in the ocean every year.2 Polymer traces have been found in many aquatic organisms, such as fish, crustaceans, and zooplankton.5 Additionally, microplastics have been found to cause desiccation and cracking in soil surfaces.6Between 2014 and 2019, approximately 31% of plastic was produced by the packaging sector.1 We propose the production of biodegradable polymers as a solution to reduce the environmental impact of petroleum-based plastics used in packaging, particularly for single use products.
After cellulose, lignin (LN) is the second most plentiful biological polymer and is a good candidate for synthesis of biodegradable films used in packaging. It is a complex macromolecule composed of aromatic phenolic rings, namely coniferyl alcohol, sinapyl alcohol, and p-coumaryl alcohol.7 LN is a highly branched 3D molecule that contains several functional groups, such as hydroxyl (OH), carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and carboxyl (COOH) which permits easy chemical modification of the polymer.8 LN can be extracted in many forms depending on the process used and has different properties. For example, alkaline LN (ALN) has a molecular weight of 1000–15,000 g mol−1 and is more soluble in organic solvents, while sodium lignosulfonate (SLN) has a molecular weight of 1000–50,000 g mol−1, is more soluble in water, and contains a higher concentration of –SO3 groups.9 LN is mainly extracted from the pulping industry; nevertheless it is mainly used for production of energy and considered a biowaste making it a valuable resource for value-added products.10
O) and carboxyl (COOH) which permits easy chemical modification of the polymer.8 LN can be extracted in many forms depending on the process used and has different properties. For example, alkaline LN (ALN) has a molecular weight of 1000–15,000 g mol−1 and is more soluble in organic solvents, while sodium lignosulfonate (SLN) has a molecular weight of 1000–50,000 g mol−1, is more soluble in water, and contains a higher concentration of –SO3 groups.9 LN is mainly extracted from the pulping industry; nevertheless it is mainly used for production of energy and considered a biowaste making it a valuable resource for value-added products.10
LN has several desirable properties for packaging applications, such as antioxidant and antimicrobial activity, biodegradability, and UV absorbance.11 During storage, the degradation of kraft LN into phenolic components was found to be associated with increased antimicrobial activity.12 The UV-blocking properties of LN have been investigated by adding LN nanoparticles to poly-vinyl alcohol (PVA) to create films with UV shielding capabilities. The addition of 1% w/w LN nanoparticles increased the UV-B absorbance of films to >90%, which is much greater than the 30% absorbance of neat PVA.13 In addition, LN has been used to enhance the mechanical properties of biopolymeric films. When added to soy protein isolate, both alkaline LN and lignosulfonates have been shown to increase the tensile strength and decrease the elongation of the resulting films.14 LN has also been incorporated in PVA and PLA polymer films to reinforce their physical properties.8,15 Another biodegradable but synthetic polymer used to form soft films is poly(lactic-co-glycolic) acid (PLGA) with good gas barrier properties.16 Biodegradable PLGA films showed low CO2 and oxygen permeability,17 making them good candidates for packaging perishable foods susceptible to oxidation.
To take advantage of the degradability and other desirable properties of LN and PLGA, we synthesized films from LN (ALN and SLN) grafted with poly(lactic-co-glycolic) acid (PLGA) using an interphase formation process between an organic and an aqueous phase as described in a previous study.18 The amphiphilic nature of the grafted polymer and the synthesis method led to the formation of films with different characteristics on each side, as shown in this study. The side facing the organic solvent showed a higher contact angle compared to the side facing the aqueous solvent, and X-ray photoelectron analysis showed higher amounts of polar groups on the aqueous side of the films. The addition of LN to the polymer was shown to increase the stiffness of the films and reduce their ductility.
Building on these previous findings, we hypothesized that the type of LN and the ratio of LN:PLGA (w/w) would affect the stability of the films, and that incorporation of LN would extend the use-life of PLGA films. The novelty of the study consists of incorporating lignin in the film by covalently attaching it to PLGA prior to film formation. To test these hypotheses, we synthesized films using two types of LN and two different LN:PLGA ratios. The LN-g-PLGA films were then stored for 12 months at different relative humidities (RH; 30% and 70%) to emulate a wide range of humidity conditions relevant for many packaging applications. Their mechanical, thermal, surface, and chemical properties were measured over time.
Experimental
Materials
Alkaline lignin (ALN) and sodium lignosulfonate (SLN) were purchased from TCI (Portland, OR). Oxalyl chloride, dimethylformamide (DMF), ethyl acetate, dimethyl sulfoxide (DMSO), magnesium chloride hexahydrate, sodium chloride, and glycerol were obtained from Fisher Scientific (Fair Lawn, NJ). Dimethyl sulfoxide 99.9% for NMR was obtained from Thermo Fisher Scientific (Plaquemine, LA). Dichloromethane (DCM) was purchased from Supelco (Burlington, MA), and poly lactic-co-glycolic acid (1:1) (35000 to 45000 Da) (PLGA) was acquired from Sigma Aldrich (St. Luis, MO). Deionized (DI) water was obtained from a Barnstead Smart2Pure water purification system (Barnstead International, Dubuque, IA).
Methods
:PLGA ratios, 1:4 and 1:6 (w/w) in accordance with previous work,18 following an optimization protocol. 4 g of PLGA were dissolved in 100 mL dry dichloromethane (DCM). Argon gas was circulated during the reaction to remove humidity within the flask, preventing inactivation of oxalyl chloride, and to remove the hydrochloric acid generated during the reaction.20 Oxalyl chloride (110 μL) and 4 mL dimethylformamide (DMF) were added to PLGA and mixed for five hours. An R-300 Rotavapor (Buchi Corporation, New Castle, DE) was used to concentrate the PLGA-Cl solution, which was then dissolved in 30 mL dimethyl sulfoxide (DMSO). In a separate flask, A/S (LN) was dissolved in 30 mL dry DMSO; 1 g or 0.66 g of LN was used for the 1:4, and 1:6 LN:PLGA w/w ratios, respectively. After the solutions were mixed, 4 mL of DMF was added and the mixture was stirred for 24 hours. The A/S(LN)-g-PLGA polymer was precipitated in 200 mL ethyl ether for 20 min. Excess ethyl ether was removed, and new ethyl ether was added five times, after which the polymer was left to air dry for 10–15 min. DCM (40 mL) was added to the polymer and stirred until dissolved. The solution was added to a decantation flask and washed with water three times to remove unreacted LN. The A/S (LN)-g-PLGA polymer was concentrated using an R-300 Rotavapor (Buchi Corporation, New Castle, DE) and freeze-dried in a Labconco FreeZone 2.5 plus (Labconco, Kansas City, MO).
150 mg of (A/S) LN-g-PLGA were dissolved in 10 mL ethyl acetate at a concentration of 1.5% (w/v), stirred for 30 min at approximately 300 rpm, and sonicated for 5 min in a Branson 3510 sonicator bath (Branson Ultrasonics Corporation, Danbury, CT). After solubilization, glycerol was added as a plasticizer (0, 5, 20% w/w) and stirred for 30 min. The (A/S) LN-g-PLGA solution was poured into 50 mL beakers with 40 mL of DI water and saturated with 8 mL of ethyl acetate. The solutions were dried at room temperature under a Supreme Air fume hood (Kewaunee Scientific, Statesville, NC) with an airflow of 167 fpm for 3 days to evaporate all of the organic solvent. The resulting films on the water surface were collected and stored in a desiccator at room temperature at 30% RH (with a saturated magnesium chloride solution) or 70% RH (with saturated sodium chloride).
Composition analysis of the polymer films showed three main components: water fraction, bulk, and the ash remaining after decomposition. The LN% of the films was calculated using the ash% of ALN and SLN considering the water content in the films. Eqn (1) and (2) were used to calculate the ash and LN%:
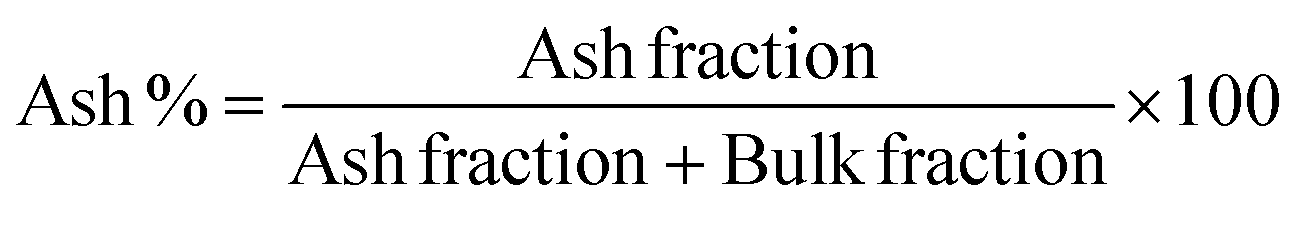 | (1) |
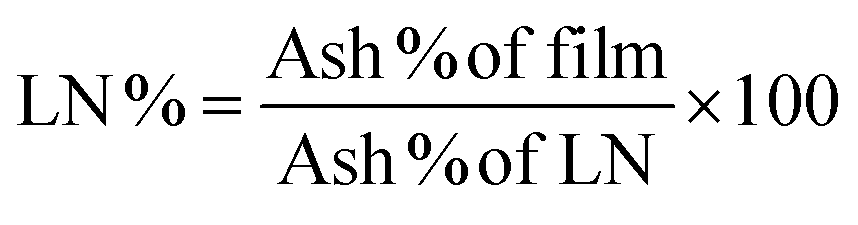 | (2) |
 | (3) |
Results
1H-NMR
1H-NMR spectra of the freshly synthesized copolymer films, and those after storage for 12 months at 30% and 70% RH are shown in Fig. 1. Both DMSO and H2O peaks were present in all polymers analysed and for all-time points (Fig. 1A–F). DMSO was used as a solvent for 1H-NMR analysis and corresponds to the peak at 2.5 ppm. At 3.3–3.5 ppm H2O was present in every film. Similar results were observed in ref. 24 in analyses of LN-g-PLGA polymers where DMSO was also used as an 1H-NMR solvent. The peak observed at 1.5 ppm indicated the presence of methyl groups in PLA (poly lactic acid),25 and this peak can be seen in the PLGA spectrum (Fig. 1B) as well as in those of all LN-g-PLGA polymers, but it was absent in the ALN and SLN spectra, as expected. The acetoxy group (–OCOCH3, 1.7–2.2 ppm), formed between PLGA and (A/S) LN during the grafting reaction was observed in all polymers at time 0, as well as after 6 and 12 months. | ||
| Fig. 1 (A) 1H-NMR spectra of newly synthesized (A/S) LN-g-PLGA polymers, and SLN-g-PLGA 1:4 w/w LN:PLGA ratio and ALN-g-PLGA 1:4 w/w LN:PLGA ratio films. Also shown are the spectra of the LN-grafted polymer components analysed separately: PLGA, sodium lignosulfonate (SLN), and alkaline lignin (ALN). (B) 1H-NMR spectra of PLGA films stored at two different relative humidities (30%, and 70%) and sampled at three time points: 0 months, 6 months, and 12 months. (C) 1H-NMR spectra of ALN-g-PLGA films (1:4 LN:PLGA ratio) stored at two different relative humidities (30% and 70%) and sampled at three time points: 0 months, 6 months, and 12 months. (D) 1H-NMR spectra of ALN-g-PLGA (1:6 LN:PLGA ratio) films stored at two different relative humidities (30%, and 70%) and sampled at three time points: 0 months, 6 months, and 12 months. (F) 1H-NMR spectra of SLN-g-PLGA (1:6 LN:PLGA ratio) films stored at two different relative humidities (30%, and 70%) and sampled at three time points: 0 months, 6 months, and 12 months. (E) 1H-NMR spectra of SLN-g-PLGA (1:4 LN:PLGA ratio) films stored at two different relative humidities (30%, and 70%) and sampled at three time points: 0 months, 6 months, and 12 months. | ||
Glyceride groups were found at 4.2 ppm in all polymer samples at the 6- and 12-month time points, but not at time zero. Glyceride groups are present in PGA molecules,26 which would demonstrate the degradation of PLGA into its constituents PGA and PLA. Peaks at 4.8 and 5.2 ppm which correspond to (CH2) of PGA and (CH–CH3) of PLA, respectively, were found, indicating a consistent presence of PLGA in the co-polymers over time.25 Similarly, Wang et al.27 showed that PLGA with different PLA:PGA ratios (12/88, and 6/94) degraded over 45 weeks and singlets found at 4.2 ppm corresponding to CH2 were related to the presence of glycolic acid. PLGA is a biodegradable polyester and the non-enzymatic hydrolysis of the ester bonds between PLA and PGA are indicators of its degradation.28 This correlates with the observed increase in the peak sizes at 4.2 ppm over time seen in Fig. 1B–F. These results indicate that even after the acylation reaction of LN with PLGA, PLGA was still susceptible to hydrolytic degradation.
Thermal analysis
Thermogravimetric (TGA) analysis showed that unmodified ALN and SLN had an ash content of 70.4 ± 0.8 and 57.1 ± 2.7% respectively; meanwhile, PLGA showed an almost complete degradation with only 0.8 ± 0.3% ash at the same temperature (600 °C). Using the known ash content of PLGA and LN, the LN% of the films was calculated (Fig. 2). LN% of the LN-g-PLGA films was 8.2 ± 0.4 and 7.8 ± 2% for the 1:4 and 1:6 LN:PLGA w/w ratios, respectively; meanwhile, SLN-g-PLGA films showed a lower LN% of 7.6 ± 0.5 and 6.1 ± 0.7% for the 1:4 and 1:6 LN:PLGA w/w ratios respectively.
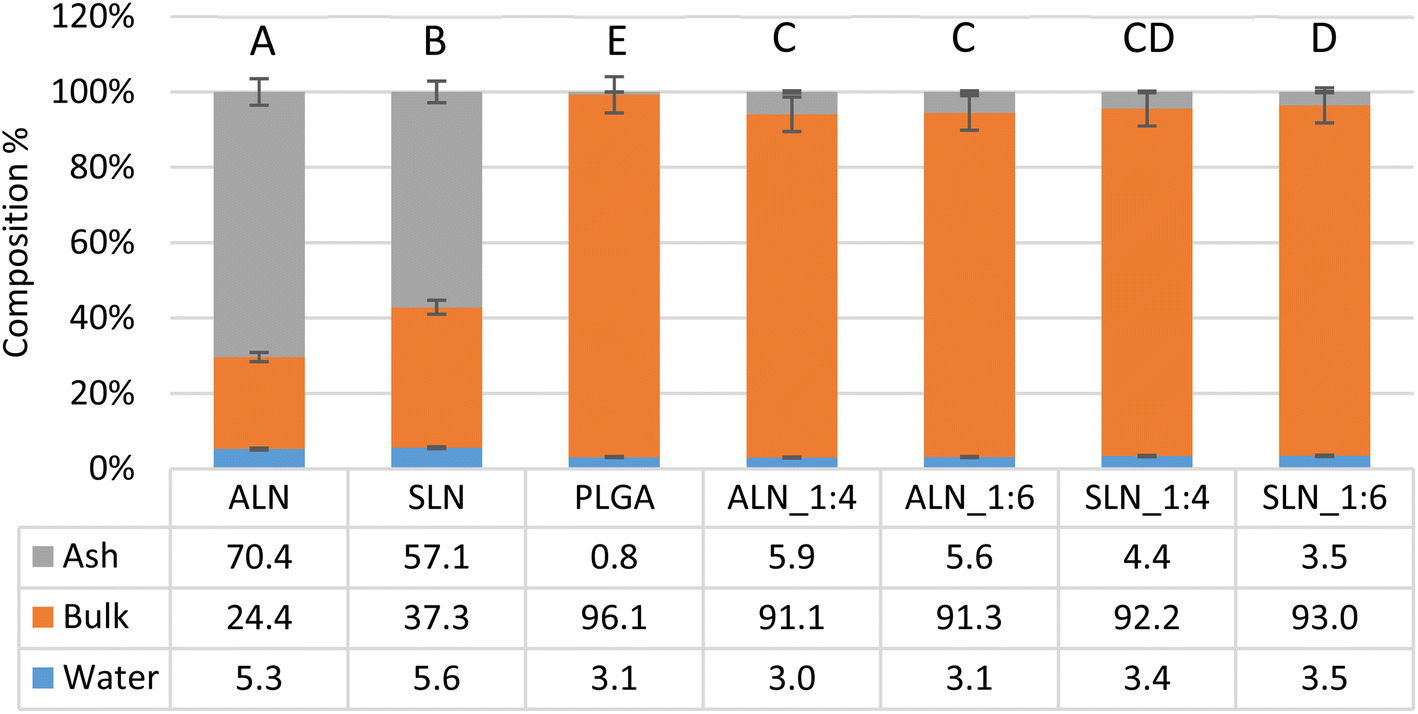 | ||
| Fig. 2 PLGA and LN-g-PLGA film composition as determined by thermogravimetric analysis. The results are reported as percent composition of three components: ash, bulk, and water. For each film type, three samples were analysed (n = 3). Multiple comparison Tukey's test results: different letters represent a significant difference (P < 0.05). | ||
Differential scanning calorimetry was performed on PLGA and LN-g-PLGA films. Polymers were confirmed to have an amorphous structure at room temperature, and the films had a glass transition temperature (Tg) between 38.5 °C and 50.1 °C, with PLGA having the highest Tg (Table 1). This corresponds to similar Tg reported for PLGA in the literature (45 °C).29 After 12 months, the Tg of the polymers increased to values ranging between 50 and 55 °C (Table 1). After storage, the polymers showed smaller endothermic curves, especially at 30% RH. This indicated that the polymers degraded over time and the amount of energy necessary for transition was reduced. PLGA showed no Tg after 12 months at both RHs, similar to what was reported by others.30
| Glass transition temperature (°C) | |||
|---|---|---|---|
| Polymers | 0 M | 12M_30% | 12M_70% |
| PLGA | 50.1 | N/A | Degraded |
| ALN-g-PLGA_1:4 |
48.8 | 52.6 | 53.8 |
| ALN-g-PLGA_1:6 |
47.9 | 51.3 | 53.5 |
| SLN-g-PLGA_1:4 |
40.1 | N/A | N/A |
| SLN-g-PLGA_1:6 |
38.5 | 52.8 | 52.2 |
Mechanical analysis
The mechanical properties of the films were measured at multiple time points (0, 1, 2, 3, 6, and 12 months) and reported in terms of yield strain (%), yield tensile strength (MPa), and Young's modulus (MPa). Two-way ANOVA analysis indicated a decrease in yield strain over time for the ALN-g-PLGA 1:4 (P < 0.001), ALN-g-PLGA 1:6 (P = 0.0141), and SLN-g-PLGA 1:6 (P = 0.0035) films, due to degradation. The yield strain of the SLN-g-PLGA 1:4 film did not change significantly over time; nevertheless, the low P-value (P = 0.0606) for this comparison suggests a trend that is consistent with observations for the other film types. For nearly all films, the humidity did not impact the strain of the films; the only exception was the SLN-g-PLGA 1:6 films. These films had a higher overall yield strain at 70% RH, similar to PLGA, suggesting that the higher amount of PLGA led to a higher ductility but also faster degradation. At time zero, PLGA films showed the highest ductility (8.9 ± 4.1%), which decreased to 2.1 ± 0.2% after 12 months at 30% RH; after 12 months at 70% RH, PLGA films depolymerized completely (Fig. 3).
 | ||
| Fig. 3 Yield strain of (A/S) LN:PLGA films over time stored at different relative humidities: (A) 30% and (B) 70%. (n = 3). Note: PLGA depolymerized fully after 12 months of storage, and hence no data are available for that time point. | ||
The yield strength of the films was affected by both the time and type of polymer at both RHs, with PLGA showing the most marked changes. At time zero, PLGA films had an initial strength of 1.7 ± 0.2 MPa, increasing to 18.7 ± 0.5 MPa after 12 months at 30% RH, and they depolymerized into a liquid state after 12 months at 70% RH. LN stabilized the change in the yield strength of the films over time at both RHs. At time zero, ALN-g-PLGA films had a higher yield strength than SLN-g-PLGA films (4.3 ± 2.4 and 4.5 ± 0.6 MPa for the ALN-g-PLGA 1:4 and 1:6 films, respectively; 1.6 ± 0.1 and 2.9 ± 0.2 MPa for the SLN-g-PLGA 1:4 and 1:6 films, respectively). After 12 months, films increased in yield strength when stored at 30% RH (17.7 ± 0.4, and 10.1 ± 7.2 MPa for the ALN-g-PLGA 1:4 and 1:6 films; and 13.2 ± 4, and 4 ± 1.9 MPa for the SLN-g-PLGA 1:4 and 1:6 films). Meanwhile at 70% RH films showed a yield strength of 3.9 ± 0.9, 10 ± 1.1, 4.9 ± 2.7, and 3.5 ± 0.9 MPa for the ALN-g-PLGA 1:4 and 1:6 films, and SLN-g-PLGA 1:4 and 1:6 films, respectively (Fig. 4).
 | ||
| Fig. 4 Stress values at yield (yield strength) of (A/S) LN:PLGA films over time stored at different relative humidities: (A) 30% and (B) 70%. (n = 3). Note: PLGA depolymerized fully after 12 months of storage, and hence no data are available for that time point. | ||
Storage time strongly affected the stiffness of the films (P < 0.0001) at both RHs. Young's modulus of the films generally increased over time; larger changes were observed for films stored at 30% RH. ALN-g-PLGA films showed the highest Young's modulus at time zero: 250.4 ± 60.4 MPa and 248.5 ± 39.4 MPa for the 1:4 and 1:6 LN:PLGA w/w ratios. After 12 months, the Young's modulus of these films increased to 624.8 ± 194.3 and 640.3 ± 194.3 MPa for the 1:4 and 1:6 (w/w) ratios at 30% RH; and 149.6 ± 16.8 and 389.9 ± 82.9 MPa for 1:4 and 1:6 LN:PLGA ratios respectively at 70% showing the effects of humidity on the stiffness of the films. SLN-g-PLGA films had an initial Young's modulus of 37.9 ± 7.6 and 132.6 ± 31.5 MPa. At 30% RH, the 1:4 w/w ratio films showed an increase to 617.6 ± 56 MPa, and the 1:6 w/w ratio films to 264.7 ± 67.3 MPa. At 70% RH, Young's modulus of the SLN-g-PLGA films increased to 198 ± 65.6 and 242.3 ± 84.9 MPa for the 1:4, and 1:6 w/w films after 12 months, respectively. PLGA showed the most changes in its stiffness. PLGA films went from the lowest to the highest Young's modulus when stored at 30% RH and depolymerized at 70% RH. The initial stiffness of PLGA was 26.7 ± 16.4 MPa, and after 12 months it increased to 1012.9 ± 270.6 MPa at 30% RH (Fig. 5).
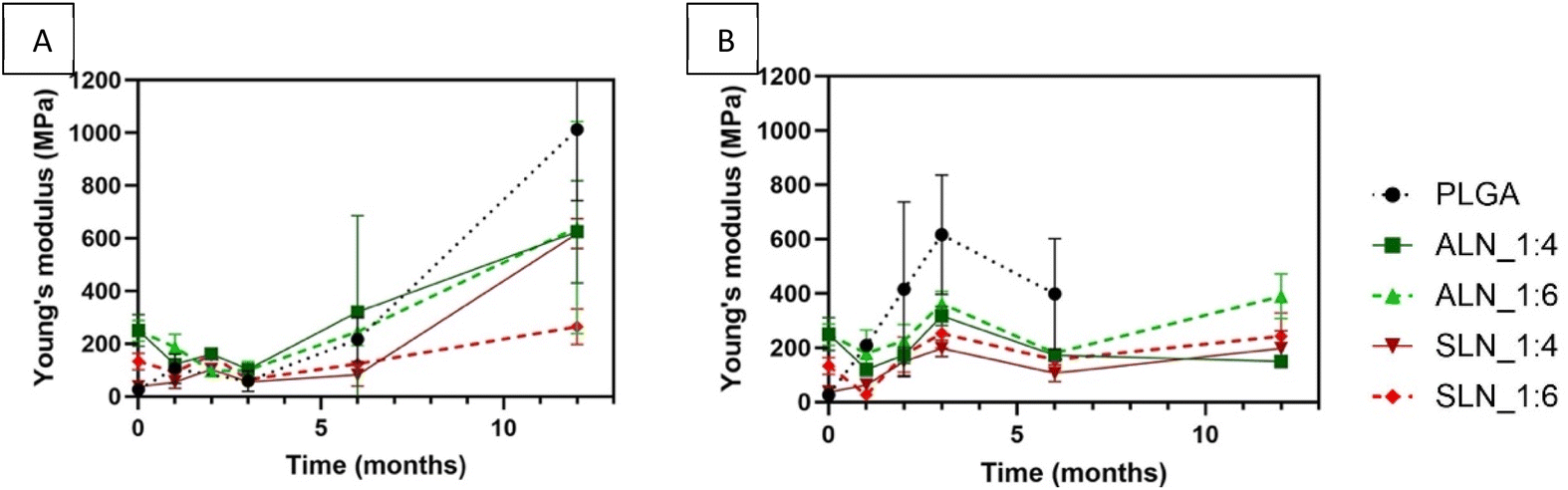 | ||
| Fig. 5 Young's modulus of (A/S) LN:PLGA films over time stored at different relative humidities: (A) 30% and (B) 70%. (n = 3). Note: PLGA depolymerized fully after 12 months of storage, and hence no data are available for that time point. | ||
Results similar to ours have been observed in other studies with different polymer films. Poly-lactic acid (PLA) films synthesized with an addition of 3% LN showed a 15% increase in yield strength.15 Pinecone LN changed the mechanical properties of faba bean protein (FBP) films, increasing their yield tensile strength from 4.9 ± 0.2 MPa (neat FBP films) to 9.3 ± 0.3 MPa (FBP) with an addition of 10% LN.31 Another study showed that the addition of 6% of either alkaline LN or lignosulfonate increased the yield tensile strength of soy-isolated protein films by 114% and 63%, respectively.14
These results suggest that the addition of LN can reinforce polymeric films; nevertheless it should be noted that PLGA chemistry is different compared to FBP or PLA, and that LN is covalently attached to PLGA in our study.
Crosslinking as well as possible polar–polar interactions between the LN and the PLGA polymers32 can also explain the decrease in the ductility of the films. The greater stiffness and yield tensile strength observed for ALN-g-PLGA films compared to SLN-g-PLGA could also be explained by differences in crosslinking of the polymers. SLN has a higher amount of –SO3,9 which could result in fewer polar interactions and consequently weaker intermolecular interactions with PLGA.
Our results also show that humidity plays an important role in the mechanical integrity of the films. When stored at 70% RH, the ductility of PLGA films decreased over time, while their yield strength and Young's modulus increased, and the films eventually depolymerized into the liquid form by 12 months. LN-grafted films, however, maintained a similar ductility at both RHs and only increased in yield strength at 30% RH. The increase in the yield strength would suggest that at low RH, films became more rigid, which correlates with the observed increase in Young's modulus over time. Overall, these results showed that LN stabilizes the mechanical properties of the films over a longer period, extending the life expectancy of the PLGA films.
Atomic force microscopy
AFM measurements of the films' surfaces showed that the sides facing the organic solvent had surface characteristics that were different from those of the side in contact with water, with the organic side generally being smoother than the aqueous side. Two-way ANOVA statistical analysis confirmed that at time zero, the roughness did not vary significantly across polymer types (P = 0.3186). However, films were significantly different between the organic and water sides (P = 0.0003). At time zero, the aqueous side of the PLGA films had a roughness of 51.7 ± 29.2 nm, compared to 3.8 ± 1.6 nm for the organic face. Comparatively, the aqueous face of the ALN-g-PLGA films was much rougher, with a roughness of 276.7 ± 126.7 and 238.7 ± 6.1 nm for the 1:4 and 1:6 w/w ratios, than the organic side with a roughness of 19.7 ± 5.6 nm and 14.4 ± 4.2 nm. Similarly, SLN-g-PLGA films presented a roughness of the aqueous face of 340.4 ± 81.1 and 202.9 ± 103.6 nm for the 1:4 and 1:6 w/w ratios, while the organic side had a roughness of 20.7 ± 10.5 and 106.4 ± 65.7 nm at time zero. During the interphase formation process, the polymer was positioned between an organic phase (ethyl acetate) and an aqueous phase (water). Due to its lower density and boiling point, ethyl acetate evaporated, leaving a smoother surface facing upwards. Meanwhile, more lignin will face the aqueous phase, positioned below, resulting in a rough texture on the water-facing side. These results were also observed in a previous study published by our group.18 In summary, our results indicate that the interphase formation process implemented for film synthesis differentially affected the two sides of the film, resulting in the aqueous side being rougher than the organic side (Fig. 6).
 | ||
| Fig. 6 Surface roughness measured using atomic force microscopy of PLGA and (A/S) LN:PLGA films. Films were stored at two relative humidities (30% and 70%) and measurements were made at time zero and after 12 months; both the aqueous side and organic side were analyzed. (A) Aqueous face, 30% RH; (B) aqueous face, 70% RH; (C) organic face, 30% RH; (D) organic face, 70% RH (n = 3). Multiple comparison Tukey's test results: ns = (P > 0.05), **** = (P < 0.00005). Note:PLGA depolymerized into a liquid form after 12 months of storage, and hence those samples were not analyzed at 12 months. | ||
For all films, the roughness of the aqueous side did not change after 12 months of storage, except for the PLGA films which depolymerized after 12 months at 70% RH. Similarly, roughness of the organic side did not change over time for most films; the only exceptions were the PLGA at 30% RH and ALN-g-PLGA 1:4 films at 70% RH, whose roughness increased over time (PLGA increased from 3.7 ± 1.6 to 77.1 ± 26.8 nm at 30% RH; ALN-g-PLGA 1:4 LN:PLGA increased from 19.7 ± 5.6 to 137.6 ± 52.8 nm at 70% RH).
Contact angle
Surfaces can be characterized as hydrophilic when their contact angle is below 90°.33 Two-way ANOVA analysis showed that on average the aqueous side of films was more hydrophilic than the organic side (P < 0.0001). In addition, we found that hydrophobicity varied significantly across polymers on both sides (P = 0.0037). The contact angle of the organic side of films did not differ between LN types or LN:PLGA ratios at time zero. The organic sides of PLGA films showed a contact angle of 79 ± 3°; meanwhile, ALN-g-PLGA 1:4 and 1:6 w/w films showed contact angles of 71 ± 21° and 78 ± 3° respectively, and SLN-g-PLGA films had contact angles of 68 ± 2° and 75 ± 6° for 1:4 and 1:6 w/w respectively. The contact angle of the aqueous side differed significantly between polymers (Table 2). The aqueous side of PLGA films showed a contact angle of 73 ± 6°; meanwhile, ALN-g-PLGA 1:4 and 1:6 w/w films showed contact angles of 27 ± 12° and 29 ± 5° respectively, and SLN-g-PLGA films had contact angles of 17 ± 4° and 57 ± 23° for 1:4 and 1:6 w/w respectively (Fig. 7C and D). PLGA's aqueous side had the highest contact angle among the polymers, and it was similar to that of its organic side (73 ± 6°). The aqueous sides of ALN-g-PLGA 1:4 and 1:6 films were significantly more hydrophilic, with contact angles of 27 ± 12° and 28 ± 6° respectively. SLN-g-PLGA 1:4 films showed the lowest contact angle (17 ± 4°) but were not significantly different from the ALN-g-PLGA films. Meanwhile, SLN-g-PLGA 1:6 films had a higher contact angle (57 ± 24°) (Table 2) (Fig. 7A and B).
| Polymer | Contact angle (organic) | Tukey's test | Contact angle (aqueous) | Tukey's test |
|---|---|---|---|---|
| PLGA | 78.7 ± 2.8 | A | 73 ± 6.2 | A |
| ALN-g-PLGA_1:4 |
71 ± 21.4 | A | 27.2 ± 12.4 | B |
| ALN-g-PLGA_1:6 |
78.4 ± 3.4 | A | 27.8 ± 5.5 | B |
| SLN-g-PLGA_1:4 |
67.8 ± 1.8 | A | 17.1 ± 4.4 | BC |
| SLN-g-PLGA_1:6 |
75.1 ± 6.4 | A | 57.3 ± 23.7 | AB |
 | ||
| Fig. 7 Contact angle measurements of the aqueous side and organic side of PLGA and (A/S) LN:PLGA films. Films were stored at two relative humidities (30% and 70%) and measurements were made at time zero and after 12 months. (A) Aqueous face, 30%; (B) aqueous face, 70%; (C) organic face, 30%; and (D) organic face, 70%. (n = 3). Multiple comparison Tukey's test results: ns = (P > 0.05), ** = (P < 0.005), and *** = (P < 0.0005). Note: PLGA depolymerized after 12 months of storage, and hence no data were collected for that time point. | ||
Humidity did not have a significant effect on the contact angle of the organic side of the films over time, except for the control PLGA films which depolymerized at 70% humidity. For the aqueous side of the films, neither the 1:4 nor 1:6 ALN-g-PLGA showed significant differences over time at either RH (Fig. 7A and B). SLN-g-PLGA films with a 1:4 LN:PLGA w/w ratio were the only type along with PLGA to show differences after 12 months. These films had an initial contact angle of 17 ± 4° at time zero. After 12 months, the contact angle increased to 71 ± 3° at 30% RH, and 67 ± 12° at 70% RH (Fig. 7A and B). On the other hand, neither did the hydrophobicity of SLN-g-PLGA 1:6 change over time, nor was there a difference between the aqueous and organic sides at either RH.
All films overall had a contact angle below 90°. LN-containing films had a lower contact angle and were thus more hydrophilic than the PLGA films. It is expected that roughness enhances the intrinsic properties of the films; in the case of hydrophilic surfaces, a higher roughness would reduce the contact angle.34 We observed a correlation between the roughness of films and their contact angle. This can be observed with the roughness and contact angle of SLN-g-PLGA 1:4 w/w films (340.4 ± 81.1 nm/17 ± 4°) which showed a decrease in the roughness in addition to an increase in the contact angle after 12 months at both 30% and 70% RH (220 ± 129.5 nm/61 ± 7° and 129.4 ± 186.7 nm/64 ± 11°). This suggests that the changes in hydrophilicity observed for the SLN-g-PLGA 1:4 films are caused by changes in the roughness of the films over the 12-month period. This phenomenon can also be observed between the aqueous and organic sides of the films and between polymers. ALN-g-PLGA films had lower contact angles compared with SLN-g-PLGA films, and they were also rougher. The differences in temporal changes in the roughness of the films between SLN-g-PLGA and ALN-g-PLGA films could also be explained by the differences in the stiffness of the films. Roughness can be correlated with Young's modulus due to frictional responses.35 This can also be a factor in the changes we observed, as SLN-g-PLGA films also showed lower Young's modulus than ALN-g-PLGA films and would thus be more susceptible to changes in roughness due to friction over time.
UV absorption
Ultraviolet radiation can be a determining factor in the degradation of products, so UV absorption can be a desirable property of plastic films.Both PLGA and PCL are susceptible to photodegradation by UV light. Irradiation of PLGA with UV light has been shown to cause chemical bond dissociation leading to a decrease in molecular weight and faster hydrolytic degradation.36 UV irradiation of PCL increases the degree of crystallinity leading to a reduction in the ductility of the polymer and bulk erosion.37 Therefore, an increase in the UV absorption caused by the presence of LN can be advantageous to the shelf life of the materials. All grafted films had a thickness between 60 and 120 μm and absorbed over 50% of both UV-AB and UV-C light at time 0 (Fig. 8). ALN-g-PLGA films showed the highest UV-A/B/C absorption (98 ± 0.0% for UV-A/B/C) which showed no meaningful change after 12 months of storage at both RHs. UV absorbance for SLN-g-PLGA 1:4 w/w (94 ± 1 and 87 ± 5% for UV-A/B and UV-C) was higher than that for SLN-g-PLGA 1:6 w/w (84 ± 0 and 79 ± 0% for UV-A/B and UV-C). Nevertheless, neither showed a decrease in absorbance over time at either RH (Fig. 8).
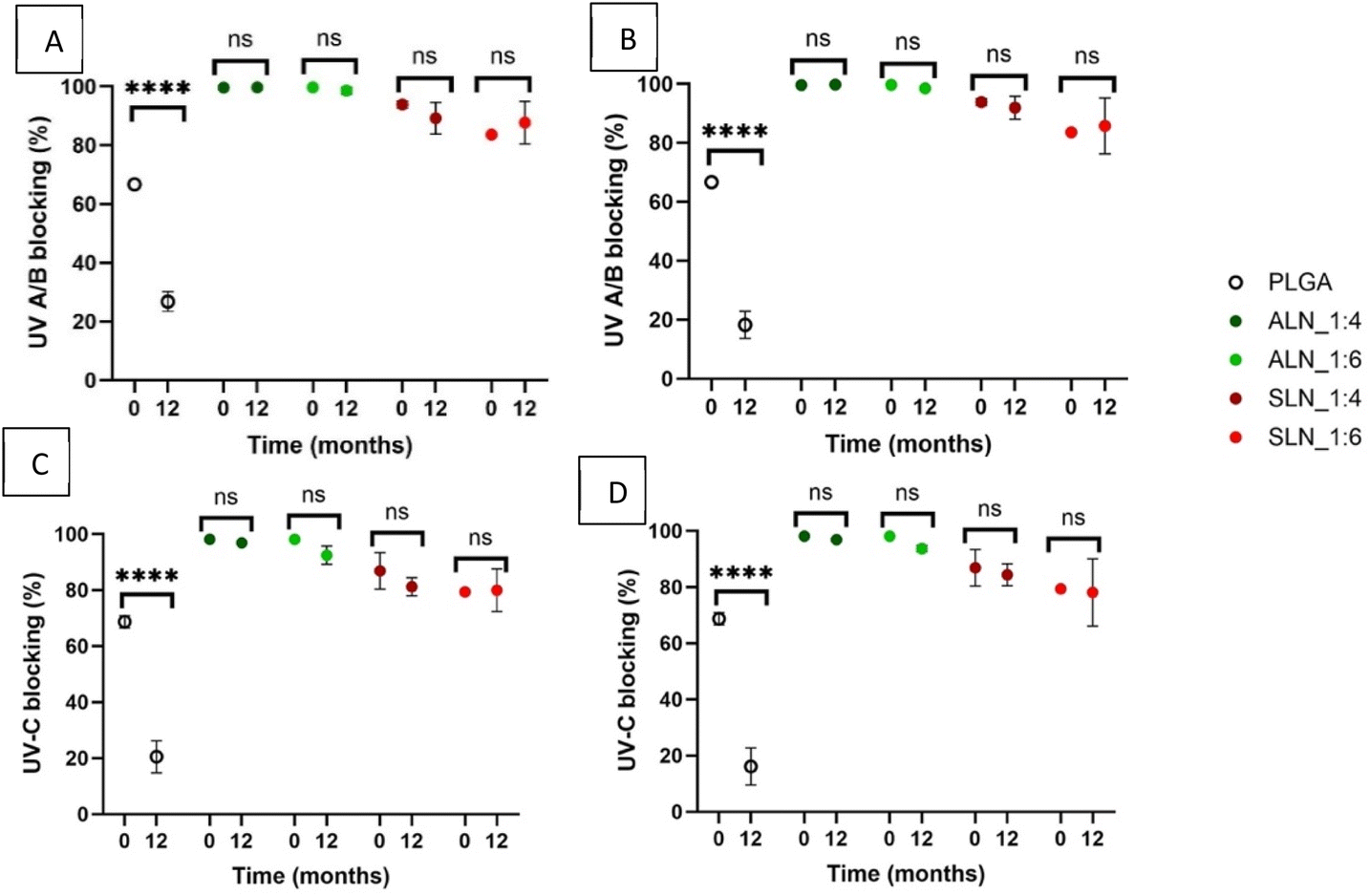 | ||
| Fig. 8 UV absorbance/blockage for UV-A/B and UV-C light by LN:PLGA films stored at two different relative humidities (30 and 70%); measurements were conducted at time zero and after 12 months. (A) UV-A/B light on films at 30% RH, (B) UV-A/B light on films at 70% RH, (C) UV-C light on films at 30% RH, and (D) UV-C light on films at 70% RH. (n = 3). Multiple paired “t” test results: ns = (P > 0.05), **** = (P < 0.00005). | ||
PLGA films were translucid and had the lowest UV absorbance among all the polymer types (67 ± 1%, and 69 ± 2% for UV-A/B and UV-C). PLGA films showed a very significant decrease (P < 0.0001) in their UV-A/B absorption (27 ± 3 and 18 ± 4% at 30 and 70% RH respectively) after 12 months, as well as UV-C absorption (P < 0.0001) (21 ± 5 and 16 ± 5% at 30 and 70% RH) after 12 months of storage (Fig. 8). This is caused by the decomposition of the PLGA polymer as UV degradation can cause the fracture of organic bond chains reducing the number of chromogenic groups in the films.38 Similar results have been observed in PLA films after accelerated weathering with a reduction of approximately 10% after 240 hours of weathering.39
In contrast, LN films were opaque, and our results showed that the addition of LN increased the UV absorbance of PLGA films. LN had different UV absorptions depending on the type, and both were higher than that of PLGA. This suggests that LN was able to increase the UV protection with the same efficacy even in the ratio with the lowest LN content (1:6) (Table 3).
| Polymer | UV-A/B absorption (%) | Tukey's test | UV-C absorption (%) | Tukey's test |
|---|---|---|---|---|
| ALN-g-PLGA_1:4 |
100 ± 0 | A | 98 ± 0 | A |
| ALN-g-PLGA_1:6 |
100 ± 0 | A | 98 ± 0 | A |
| SLN-g-PLGA_1:4 |
94 ± 1 | B | 87 ± 5 | B |
| SLN-g-PLGA_1:6 |
84 ± 0 | C | 79 ± 0 | B |
| PLGA | 67 ± 1 | D | 69 ± 2 | C |
Due to the chemical structure of LN, it provides UV protection to the films. LN possesses chromophores, ketones, and phenolic units, which are functional groups that can absorb UV radiation.40 Equivalent results have been observed in maleic anhydride (MAH)-g-LN films. A UV absorbance of 100% was obtained with 1:1 LN:MAH ratio polymer films with a thickness of 20 μm.41 LN nanoparticles have also been mixed into polylactic acid (PLA) films increasing the UV absorbance from 10% to 30% with the addition of 1% LN nanoparticles.39
Nevertheless, the addition of LN was shown to decrease the transparency of the films. Photodegradation is a cause of ageing and deterioration of polymers and LN can be used as a sustainable alternative for UV shielding. Even though LN addition decreases transparency, it can be an addition for UV protection in opaque materials.
Conclusions
After 12 months, DSC analysis showed that LN-g-PLGA films stored at 70% RH showed a reduction in the enthalpy required for glass transition suggesting increased degradation with humidity. The mechanical characteristics of the films were affected by both the polymer type used and the storage time. After being stored, films showed an increase in the yield strength and Young's modulus, but showed a decrease in the yield strain, particularly at 30% RH. LN-g-PLGA films also demonstrated better mechanical stability at higher RHs, as PLGA films depolymerized after 12 months when stored at 70% RH. Films showed two distinct sides, organic and aqueous. These sides showed different roughness and contact angle characteristics. The organic sides exhibited a lower roughness as well as higher contact angles, and the aqueous sides showed a higher roughness coefficient and lower contact angles. All films overall had a contact angle below 90°. LN-g-PLGA films showed a lower contact angle compared to PLGA films. The roughness and contact angle of the films showed no changes after storage at both RHs. UV absorbance was significantly higher for LN-g-PLGA films (>80%) compared to PLGA (67%), showing that LN addition considerably increases the shielding of UV-A/B and UV-C light. Contrary to PLGA which decreased its UV-blocking capability from 68.7 to 20.5%, LN-g-PLGA was able to retain its UV-blocking capability after 12 months at both RHs. The UV blocking properties of the films are promising for packaging applications as they can help reduce photodegradation of perishable products. Suitability of LN-g-PLGA films for long term storage under a wide range of conditions was demonstrated by their ability to maintain their chemical and mechanical properties over time, indicating that they are a good alternative for biodegradable, multifunctional packaging materials.Future work
Further research on different casting techniques such as solvent casting and compression molding may improve uniformity, mechanical strength and scalability. Barrier properties including oxygen and water permeability can provide more information for packaging applications. Additionally exploring electrospinning techniques could enable applications for biomedical and controlled release systems. These studies will advance the development of different applications for lignin-based films.Data availability
Data for this article, including figures and tables are available at the Science Data Bank at https://www.scidb.cn/en/s/FvUvEb.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation under NSF EPSCoR Track 2 RII, OIA 1632854, the USDA National Institute of Food and Agriculture, AFRI project # 2018-07406, and the USDA-NIFA Hatch Project #1008750.References
- OECD, Global Plastics Outlook – Plastics use by application, ourworldindata.org2022.
- N. Laskar and U. Kumar, Plastics and microplastics: A threat to environment, Environ. Technol. Innovation, 2019, 14, 100352 CrossRef.
- K. Karimi and A. Faghri, The Issues of Roadside Litter: A Review Paper, Curr. Urban Stud., 2021, 09, 779–803 CrossRef.
- J. P. da Costa, P. S. M. Santos, A. C. Duarte and T. Rocha-Santos, (Nano)plastics in the environment – Sources, fates and effects, Sci. Total Environ., 2016, 566–567, 15–26 CrossRef PubMed.
- L. C. de Sá, M. Oliveira, F. Ribeiro, T. L. Rocha and M. N. Futter, Studies of the effects of microplastics on aquatic organisms: What do we know and where should we focus our efforts in the future?, Sci. Total Environ., 2018, 645, 1029–1039 Search PubMed.
- Y. Wan, C. Wu, Q. Xue and X. Hui, Effects of plastic contamination on water evaporation and desiccation cracking in soil, Sci. Total Environ., 2019, 654, 576–582 CrossRef CAS PubMed.
- A. Duval and M. Lawoko, A review on lignin-based polymeric, micro- and nano-structured materials, React. Funct. Polym., 2014, 85, 78–96 CrossRef CAS.
- A. Naseem, S. Tabasum, K. M. Zia, M. Zuber, M. Ali and A. Noreen, Lignin-derivatives based polymers, blends and composites: A review, Int. J. Biol. Macromol., 2016, 93, 296–313 CrossRef CAS PubMed.
- M. S. Ganewatta, H. N. Lokupitiya and C. Tang, Lignin Biopolymers in the Age of Controlled Polymerization, Polymers, 2019, 11(7), 1176 CrossRef PubMed.
- V. Yadav, A. Kumar, M. Bilal, T. A. Nguyen and H. M. N. Iqbal, Chapter 12 - Lignin removal from pulp and paper industry waste streams and its application, in Nanotechnology in Paper and Wood Engineering, ed. R. Bhat, A. Kumar, T. A. Nguyen and S. Sharma, Elsevier, 2022, pp. 265–283 Search PubMed.
- A. Boarino and H.-A. Klok, Opportunities and Challenges for Lignin Valorization in Food Packaging, Antimicrobial, and Agricultural Applications, Biomacromolecules, 2023, 24(3), 1065–1077 CrossRef CAS PubMed.
- J. Yun, L. Wei, W. Li, D. Gong, H. Qin and X. Feng, et al., Isolating High Antimicrobial Ability Lignin From Bamboo Kraft Lignin by Organosolv Fractionation, Front. Bioeng. Biotechnol., 2021, 9, 683796 CrossRef PubMed.
- P. Posoknistakul, C. Tangkrakul, P. Chaosuanphae, S. Deepentham, W. Techasawong and N. Phonphirunrot, et al., Fabrication and Characterization of Lignin Particles and Their Ultraviolet Protection Ability in PVA Composite Film, ACS Omega, 2020, 5(33), 20976–20982 CrossRef CAS PubMed.
- E. M. Zadeh, S. F. O'Keefe and Y.-T. Kim, Utilization of Lignin in Biopolymeric Packaging Films, ACS Omega, 2018, 3(7), 7388–7398 CrossRef CAS PubMed.
- S. A. Shakoor, N. Wang, T. Chen, X. Zhao and Y. Weng, Development of PLA/Lignin Bio-Composites Compatibilized by Ethylene Glycol Diglycidyl Ether and Poly (ethylene glycol) Diglycidyl Ether, Polymers, 2023, 15(20), 4049 CrossRef PubMed.
- Y. Lu, D. Cheng, B. Niu, X. Wang, X. Wu and A. Wang, Properties of Poly (Lactic-co-Glycolic Acid) and Progress of Poly (Lactic-co-Glycolic Acid)-Based Biodegradable Materials in Biomedical Research, Pharmaceuticals, 2023, 16(3), 454 CrossRef CAS PubMed.
- T. Sun, J. Bian, Y. Wang, J. Hu, X. Yun and E. Chen, et al., One-Step Synthesis of Poly(L-Lactic Acid)-Based Soft Films with Gas Permselectivity for White Mushrooms (Agaricus bisporus) Preservation, Foods, 2023, 12(3), 586 CrossRef CAS PubMed.
- O. E. Mendez, C. E. Astete, S. Hermanová, D. Boldor, W. J. Orts and C. M. Sabliov, Biobased films from amphiphilic lignin-graft-PLGA copolymer, BioResources, 2023, 5887–5907 CAS.
- C. E. Astete, J. U. De Mel, S. Gupta, Y. Noh, M. Bleuel and G. J. Schneider, et al., Lignin-Graft-Poly(lactic-co-glycolic) Acid Biopolymers for Polymeric Nanoparticle Synthesis, ACS Omega, 2020, 5(17), 9892–9902 CrossRef CAS PubMed.
- L. Cruz, X. Yang, A. Menshikh, M. Brewer, W. Lu and M. Wang, et al., Adapting decarbonylation chemistry for the development of prodrugs capable of in-vivo delivery of carbon monoxide utilizing sweeteners as carrier molecules, Chem. Sci., 2021, 12, 10649–10654 RSC.
- A. Zirahi, H. Sadeghi Yamchi, A. Haddadnia, M. Zirrahi, H. Hassanzadeh and J. Abedi, Ethyl acetate as a bio-based solvent to reduce energy intensity and CO2 emissions of in situ bitumen recovery, AIChE J., 2020, 66(2), e16828 CrossRef CAS.
- D. Nečas and P. Klapetek, Gwyddion: An open-source software for SPM data analysis, Cent. Eur. J. Phys., 2011, 10, 181–188 Search PubMed.
- A. Marmur, C. Della Volpe, S. Siboni, A. Amirfazli and J. W. Drelich, Contact angles and wettability: towards common and accurate terminology, Surf. Innovations, 2017, 5(1), 3–8 CrossRef.
- A. Garcia, C. E. Astete, R. Cueto and C. M. Sabliov, Modulation of Methoxyfenozide Release from Lignin Nanoparticles Made of Lignin Grafted with PCL by ROP and Acylation Grafting Methods, Langmuir, 2024, 40(10), 5433–5443 CrossRef CAS PubMed.
- E. D. Pereira, R. Cerruti, E. Fernandes, L. Peña, V. Saez and J. C. Pinto, et al., Influence of PLGA and PLGA-PEG on the dissolution profile of oxaliplatin, Polímeros, 2016, 26, 137–143 CrossRef CAS.
- V. Taresco, J. Suksiriworapong, R. Creasey, J. C. Burley, G. Mantovani and C. Alexander, et al., Properties of acyl modified poly(glycerol-adipate) comb-like polymers and their self-assembly into nanoparticles, J. Polym. Sci., Part A: Polym. Chem., 2016, 54(20), 3267–3278 CrossRef CAS PubMed.
- Y. Wang, M. A. Murcia Valderrama, R. J. van Putten, C. J. E. Davey, A. Tietema and J. R. Parsons, et al., Biodegradation and Non-Enzymatic Hydrolysis of Poly(Lactic-co-Glycolic Acid) (PLGA12/88 and PLGA6/94), Polymers, 2021, 14(1), 15 CrossRef PubMed.
- T. G. Park, Degradation of poly(lactic-co-glycolic acid) microspheres: effect of copolymer composition, Biomaterials, 1995, 16(15), 1123–1130 CrossRef CAS PubMed.
- A. T. C. R. Silva, B. C. O. Cardoso, M. E. S. Ribeiro e Silva, R. F. S. Freitas and R. G. Sousa, Synthesis, Characterization, and Study of PLGA Copolymer in Vitro Degradation, J. Biomater. Nanobiotechnol., 2015, 06(01), 12 Search PubMed.
- M. L. Houchin and E. M. Topp, Physical properties of PLGA films during polymer degradation, J. Appl. Polym. Sci., 2009, 114(5), 2848–2854 CrossRef CAS.
- S. Rojas-Lema, K. Nilsson, M. Langton, J. Trifol, J. Gomez-Caturla and R. Balart, et al., The effect of pine cone lignin on mechanical, thermal and barrier properties of faba bean protein films for packaging applications, J. Food Eng., 2023, 339, 111282 CrossRef CAS.
- S. Sahoo, M. Misra and A. K. Mohanty, Enhanced properties of lignin-based biodegradable polymer composites using injection moulding process, Composites, Part A, 2011, 42(11), 1710–1718 CrossRef.
- M. Abraham, D. V. Claudio, S. Stefano, A. Alidad and J. Drelich, Contact angles and wettability: towards common and accurate terminology, Surf. Innovations, 2017, 5(1), 3–8 CrossRef.
- F. Sotoudeh, S. M. Mousavi, N. Karimi, B. J. Lee, J. Abolfazli-Esfahani and M. K. D. Manshadi, Natural and synthetic superhydrophobic surfaces: A review of the fundamentals, structures, and applications, Alexandria Eng. J., 2023, 68, 587–609 CrossRef.
- C. S. Sandeep and K. Senetakis, Effect of Young’s Modulus and Surface Roughness on the Inter-Particle Friction of Granular Materials, Materials, 2018, 11(2), 217 CrossRef PubMed.
- A. Shibata, S. Yada and M. Terakawa, Biodegradability of poly(lactic-co-glycolic acid) after femtosecond laser irradiation, Sci. Rep., 2016, 6(1), 27884 CrossRef CAS PubMed.
- D. C. França, D. D. Morais, E. B. Bezerra, E. M. Araújo and R. M. R. Wellen, Photodegradation Mechanisms on Poly(ε-caprolactone) (PCL), Mater. Res., 2018, 21(5) DOI:10.1590/1980-5373-mr-2017-0837.
- R.-M. Qiao, C.-P. Zhao, J.-L. Liu, M.-L. Zhang and W.-Q. He, Synthesis of Novel Ultraviolet Absorbers and Preparation and Field Application of Anti-Ultraviolet Aging PBAT/UVA Films, Polymers, 2022, 14(7), 1434 CrossRef CAS PubMed.
- W. Yang, F. Dominici, E. Fortunati, J. M. Kenny and D. Puglia, Effect of lignin nanoparticles and masterbatch procedures on the final properties of glycidyl methacrylate-g-poly (lactic acid) films before and after accelerated UV weathering, Ind. Crops Prod., 2015, 77, 833–844 CrossRef CAS.
- G. K. K. Anushikha, Lignin as a UV blocking, antioxidant, and antimicrobial agent for food packaging applications, Biomass Convers. Biorefin., 2023, 16755–16767 Search PubMed.
- Y. Zhang, S. Zhou, X. Fang, X. Zhou, J. Wang and F. Bai, et al., Renewable and flexible UV-blocking film from poly(butylene succinate) and lignin, Eur. Polym. J., 2019, 116, 265–274 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |