
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalysts for selective CO2/CO electroreduction to C3+ compounds
Ngoc Huan
Tran
a,
Moritz W.
Schreiber
b and
Marc
Fontecave
 *a
*a
aLaboratoire de Chimie des Processus Biologiques, CNRS UMR 8229, Collège de France, Sorbonne Université, 11 Place Marcelin Berthelot, 75231 Cedex 05, Paris, France. E-mail: marc.fontecave@college-de-france.fr
bTotalEnergies OneTech Belgium, B-7181 Seneffe, Belgium
First published on 15th May 2025
Abstract
Electroreduction of carbon dioxide and carbon monoxide to organic compounds is considered a promising way for (i) exploring a source of carbon alternative to fossil carbon; (ii) storing electrical energy as stable chemical energy; and (iii) producing useful e-chemicals and e-fuels for the chemical industry. While it is generally considered that only Cu-based catalysts facilitate the formation of multicarbon compounds, which are mainly limited to ethylene and ethanol, recent studies have challenged this assumption. In this review, we provide exhaustive, structural and mechanistic analyses of the solid materials that have been reported as catalysts for electroreduction of CO2 and CO to more complex molecules. This review elucidates that besides copper, metals such as nickel, iron and molybdenum have the potential to favor C–C coupling reactions to form important molecules in the chemical industry, such as propane, propanol, and butanol, along with offering substantial faradaic efficiencies. Thus, this review offers fresh perspectives on CO2R and COR.
Broader contextThe valorization of captured CO2, as a source of carbon, via electroconversion into organic compounds useful for the chemical industry, such as hydrocarbons and alcohols, is one of the rare alternatives to the current petrochemistry. Because the reactions involve multiple electron- and proton-transfers, the development of this technology depends on the discovery of cheap, selective and efficient catalysts. However, research has mainly focused on Cu-based electrocatalysts that are appropriate for the production of methane, ethylene and ethanol, with less efforts devoted to catalysts for the more challenging electroconversion of CO2 into C3+ molecules, containing three carbon atoms or more. This is unfortunate given that important feedstocks for the chemical industry belong to this category, such as propanol, propane, propylene, butane and butanol. This review provides an overview of the current state-of-the-art electrocatalysts used for CO2 to C3+ product formation and provides directions for their further development. |
Introduction
Because of anthropogenic CO2 emissions and the resulting global warming, current energy policy trajectories aim at drastically and quickly reducing the combustion of fossil carbon sources, which currently represent more than 80% of the global energy consumption. Although this implies the massive electrification of transport, residential heating and industries via the development of low-carbon renewable (wind, solar and hydroelectric) and nuclear energy, some sectors will continue to rely on carbon-based compounds with high-energy densities, which are presently derived from fossil resources.1,2 These sectors include functional organic chemical (solvents, polymers, and drugs) production, aviation and shipping. The development of potential new sectors such as long-term energy storage and transport may also depend on these fuels. Accordingly, sustainable alternatives to fossil carbon, such as agricultural biomass and domestic and industrial waste can be used to produce (bio)methane, (bio)ethanol and (bio)diesel. However, the limited availability of these feedstocks allows only to replace a fraction of fossil resources that are currently used.3CO2 is a promising source of carbon as enough CO2 is available in the atmosphere to fully replace the fossil-derived chemicals consumed by our society with e-fuels and e-chemicals.4–7 CO2 conversion has several advantages. Firstly, as it relies on CO2 captured from point sources (power plants, cemeteries, steel factories, and digesters), with the possibility of direct capture from the air (DAC) in the future, the process is carbon neutral. Secondly, it provides a mechanism to convert renewable intermittent electricity into stable chemical energy (energy in chemical bonds) for long-term storage. Thirdly, it can convert CO2, as a carbon source, into a variety of useful organic chemicals for the chemical industry and for applications such as transport and heat production. However, currently, this approach is not industrially mature. The development of economically viable e-fuels and e-chemicals on a large scale will depend on successfully addressing the challenges encountered in the (i) massive production of cheap low-carbon electricity; (ii) cost-effective capture of massive amounts of CO2; (iii) development of efficient electrolyzers (with high energy efficiency, high current density, high selectivity and long lifetime).
CO2 valorization requires the initial conversion of electrical energy to chemical energy. The most mature pathways to convert CO2 to organic molecules rely on H2O electrolysis to H2 and subsequent thermochemical reduction steps. Alternatively, CO2 electroreduction (CO2R) allows the synthesis of organic molecules in a single step. This has the potential to greatly increase the energy efficiency and reduce the capital expenditure required for this transformation compared with the electrolytic H2 pathway.8,9
The CO2R performance can be optimized via the appropriate development of electrolyzers and catalysts. Electrolyzers suffer from various sources of ohmic losses, which need to be minimized via specific cell designs and optimization of the electrolyte composition to maximize their energy efficiencies.10 An important recent breakthrough in the development of flow cells is that the catalyst is deposited onto a gas diffusion layer (GDL) and the resulting gas diffusion electrode (GDE) is fed with CO2 gas, resulting in high CO2 coverage on the catalyst and industrially relevant current densities (several hundred mA cm−2 up to A cm−2). In the classical H-cells, the electrolyte is saturated with CO2 and the low CO2 concentration in water (30 mM) results in mass transport limitations and much lower current densities (below 50 mA cm−2). The technological issues related to the electrolyzer have been extensively presented in several recent review articles, and thus will not be discussed here.11–13
Catalysis is a critical component of this technology for optimizing the reaction kinetics and selectivity. CO2R involves multi-electron and multi-proton reactions, with electrons coming from the cathode and protons from the aqueous electrolyte. These reactions are associated with large overpotentials and low kinetics, deteriorating the energy efficiency. Thus, catalysts, which facilitate the transfer of electrons and protons, are required to minimize these barriers.14,15 Furthermore, a variety of products can be obtained by the addition of electrons and protons to CO2, within a limited potential range (Table 1). The selectivity can be controlled by tailoring the electronic and structural properties of catalysts. It should be noted that the hydrogen evolution reaction (HER) via proton reduction always competes with CO2R, and thus catalysts have also been developed to minimize HER. Obviously, the carbon number increase in more complex product molecules, requiring more electrons and protons to be transferred, which results in larger overpotentials, and hence slower reaction kinetics and low selectivity. This explains why CO2R to C1 products, such as carbon monoxide and formic acid,16,17 is more industrially mature than CO2R to C2 products, such as ethylene and ethanol, and the formation of C3+ products (see some examples in Table 1) is more challenging, and thus rarely observed at low current densities and with low faradaic efficiencies. Previous articles reviewed the field of CO2R to C2 compounds (namely ethylene, acetate and ethanol).8,18,19 Thus, the focus of this review article is on the recently discovered catalysts allowing production of complex C3+ organic molecules from CO2R, and also from COR.
| Products | Half reaction | E 0 (V vs. RHE) | |
|---|---|---|---|
| Hydrogen | 2e− + 2H+ → H2 | 0.00 | |
| C1 | Formic acid | CO2 + 2e− + 2H+ → HCOOH(L) | −0.12 |
| Carbon monoxide | CO2 + 2e− + 2H+ → CO(g) + H2O | −0.1 | |
| Methane | CO2 + 8e− + 8H+ → CH4(g) + H2O | 0.17 | |
| C2 | Ethylene | 2CO2 + 12e− + 12H+ → C2H4(g) + 4H2O | 0.08 |
| Ethanol | 2CO2 + 12e− + 12H+ → C2H5OH(L) + 3H2O | 0.09 | |
| Ethane | 2CO2 + 14e− + 14H+ → C2H6(g) + 4H2O | 0.14 | |
| Ethylene glycol | 2CO2 + 10e− + 10H+ → C2H6O2(L) + 2H2O | 0.2 | |
| Acetic acid | 2CO2 + 8e− + 8H+ → C2H4O2(L) + 2H2O | 0.11 | |
| C3 | Propanol | 3CO2 + 18e− + 18H+ → C3H7OH(L) + 5H2O | 0.1 |
| Acetone | 3CO2 + 16e− + 16H+ → C3H6O(L) + (H2O | −0.14 | |
| Propylene | 3CO2 + 18e− + 18H+ → C3H6(g) + 6H2O | 0.13 | |
| Propane | 3CO2 + 20e− + 20H+ → C3H8(g) + 6H2O | 0.14 | |
| C4 | Methyl glyoxal | 3CO2 + 12e− + 12H+ → C4H4O2(L) + 4H2O | 0.02 |
| Butane | 4CO2 + 26e− + 26H+ → C4H10(g) + 8H2O | 0.14 | |
| Butanol | 4CO2 + 24e− + 24H+ → C4H10O(L) + 7H2O | 0.14 | |
| 2,3-Furandiol | 4CO2 + 14e− + 14H+ → C4H4O3(L) + 5H2O | 0.01 |
The selectivity can be controlled based on the choice of the metal used at the cathode.20 The majority of metals (Pt, Pd, Rh, Fe, Ni, Co, etc. favor HER over CO2R. Alternatively, Ag, Au and Zn, to a low extent, are well known to favor CO production, which is a C1 product obtained via the 2-electron reduction of CO2. Sn, Bi and In are selective for formic acid production, which is also a C1 product obtained via 2-electron reduction. Although methanol is rarely observed, methane, another C1 product, can be obtained with a very high faradaic efficiency using Cu-derived catalysts.21 Cu is considered a unique catalyst, given that it is frequently claimed to be the only metal capable of promoting C–C coupling reactions. As demonstrated herein, this is not correct given that other metals can also promote these reactions. It should be noted that the Cu-dependent CO2R to multi-carbon compounds is generally limited to the generation of C2 compounds such as ethylene and ethanol, whereas propanol is also formed in substantial yield in only a few circumstances, discussed in detail in a dedicated chapter. Because these reactions rely on a large number of electrons and protons, their mechanism is very complex, implying the involvement of a large number of surface adsorbed intermediates, and thus has been a matter of intense experimental and computational research.22 Although this capacity makes Cu very attractive, generally Cu-based catalysts suffer from a lack of selectivity, and resulting in a complex mixture of a variety of C1 (CO, HCOOH, and CH4) and C2 products (ethylene and ethanol) together with H2. Accordingly, numerous strategies have been developed to better control the selectivity of Cu-based catalysts including tuning the morphology and size of nanoparticles, introducing defects, alloying and metal-doping, surface functionalization with molecules and polymers, and electrolyte engineering.23,24
CO2R suffers from an additional drawback, namely the formation of carbonate/bicarbonate salts via the reaction between OH− formed during the reaction and CO2 dissolved in the electrolyte or at the gas-electrolyte interphase.25,26 This results in unproductive CO2 consumption, changes in the membranes and electrolyte, instability of the whole electrochemical system and extra costs associated with the recovery and recycling of the electrolyte as well as the recovery of CO2. Thus, a strategy to limit the problem of CO2 loss and carbonate formation is shifting from single-step to tandem CO2R, involving the first step of CO2R to CO, followed by COR to C2+ compounds using Cu-based catalysts.27 The rational is based on the fact that CO does not react with OH−, and thus highly alkaline electrolytes, favoring C–C coupling and suppressing HER, can be used in CO electrolyzers. Furthermore, given that high CO surface coverage favors C–C coupling, COR competes favorably with HER, and generally is more selective towards C2+ products than CO2R, despite their lower water solubility.28 Because of this very low solubility of CO in water, flow electrolyzers and GDEs for COR have been developed recently, showing remarkable achievements with respect to ethylene and ethanol production, with high selectivity at high current densities.29,30 However, the tandem scenario requires that the CO2-to-CO electrochemical step does not degrade CO2 into carbonate too extensively. This is the case if a high-temperature solid oxide electrolysis cell (SOEC) is used for highly efficient and selective CO2R to CO, given that it avoids carbonate formation.27 A recent techno-economic benchmarking analysis indeed showed that this tandem system is the most economically promising for the production of ethylene due to the very high energy efficiency and excellent selectivity of already available systems for CO2R to CO under conditions of limited CO2 loss to carbonate.31 Therefore, here we also discuss interesting catalysts for CO electroreduction (COR) to C3+ products.
An alternative strategy for limiting carbonate formation and carbon loss is the utilization of acidic electrolytes for CO2R, although specific catalysts have to be designed for limiting HER.32,33 However, the specific conditions will not be discussed here given that, to our knowledge, there is no report of significant C3+ product formation during acidic CO2R.
Thus, the aim of this review is to summarize the current knowledge regarding solid catalysts specifically promoting the formation of C3+ products containing three carbon atoms or more during both CO2R and COR. Remarkably, as expected, most reports focused on Cu-based catalysts; however, a significant number of studies showed the potential of other transition metals for catalyzing the formation of these complex molecules, although with low faradaic efficiencies (FEs) and low current densities. It is important to appreciate the development of highly sensitive analytic methods (NMR, gas and liquid chromatography), which has allowed the detection of very low amounts of products (with faradaic efficiencies as low as 0.1%), although in some cases, the detection of the products was only possible via long-term electrolysis and the use of large cathodes.
Considering the industrial importance, with increasing demand and high potential growth rate, of some C3+ compounds, such as propanol, propylene, propane, butane and butanol, it is worth focusing on these reactions as a sustainable alternative way to produce them. These compounds are indeed key precursors of polymers, fuels and rubber, making them attractive targets for CO2R and COR. The most important C3 and C4 organic chemicals according to their market volume are shown in Table 2.
| Products | Volume (2023) millions of tons | USD billion (2023) | Expected growth rate (%) 2023–2032 | Data source |
|---|---|---|---|---|
| a The data are obtained from: https://www.chemanalyst.com/industry-report/propylene-market-633. b The data are obtained from: https://www.chemanalyst.com/industry-report/isopropyl-alcohol-ipa-187. c The data are obtained from: https://www.mordorintelligence.com/industry-reports/propane-market. d The data are obtained from: https://www.statista.com/statistics/1245205/acetone-market-volume-worldwide/. e The data are obtained from: https://www.statista.com/statistics/1245211/n-butanol-market-volume-worldwide/. f The data are obtained from: https://www.globalinsightservices.com/reports/butane-market/. g The data are obtained from: https://prismaneconsulting.com/report-details/butene-1-market-insights-trends-analysis-and-forecast#:~:text=Butene-1%20Capacity%20Overview,number%20of%20on%20purpose%20capacity. h The data are obtained from: https://eippcb.jrc.ec.europa.eu/sites/default/files/2019-11/JRC109279_LVOC_Bref.pdf. i The data are obtained from: https://www.statista.com/statistics/1067436/global-butadiene-production-capacity/#:~:text=The%20production%20capacity%<?pdb_no 20of?>20of<?pdb END?>%20the,additions%<?pdb_no 20in?>20in<?pdb END?>%20the%20next%20years. | ||||
| Propylene | 100 | 121 | 5.4 | |
| Propanol | 3.2 | 4.2 | 7.0 | |
| Propane | 189 | 98 | 12.5 | |
| Acetone | 8.1 | 6.5 | 3.8 | |
| Butanol | 5.5 | 9 | 6.2 | |
| Butane | 200 | 112 | 4.1 | |
| Butene-1 | 3 | 3.2 | 3.7 | |
| Propylene oxide | 10 | 23.5 | 5.8 | |
| Butadiene | 12 | 12.1 | 4.7 | |
Based on the data analyzed here, we present a full view of the C3+ products accessible via CO2R and COR and a comparison with the list of industrially relevant compounds, providing a rational perspective regarding the reactions that should be further studied and developed. We also provide a benchmarking of the different catalysts favoring the formation of these products in terms of their performances and catalytic mechanisms, opening new research directions with respect to the elaboration of catalysts for CO2 and CO valorization. During the preparation and finalization of this article, a review article was published on a similar topic.34 However, our approach is from a different perspective, and thus we believe that these two articles are nicely complementary.
1. Non-Cu-based catalysts
To our knowledge, only a few metals, the most interesting one being Mo, Ni and Fe, were reported as catalysts for C–C coupling reactions leading to multi-carbon compounds from CO2R. Metal-free catalysts have also been reported to catalyze these reactions, and one example will be described at the end of this chapter. The few available reports are summarized in Table 3, presenting a list of the products and most relevant performances reflecting the activity of non-Cu catalysts, namely, Faraday efficiencies, applied potentials and geometric current densities (total or partial), under specific reaction conditions. In a few cases, the reaction mechanisms are proposed to explain the formation of a given product, but one should be aware that computational and experimental mechanistic validations are missing generally. It should also be noted that non-Cu catalysts have not been studied for COR to date, with the exception of silver under high CO pressure conditions (see Section 1.6).35| Catalyst | Conditions | Applied total current densitya | Products | FE (%) | Ref. |
|---|---|---|---|---|---|
| Potential vs. RHE | |||||
| a Partial current density can be derived from the total current density (column 3) with the FE for a given product (column 5). b The information regarding current density is not available. | |||||
| PdAu | H-cell | −1.4 Vb | C2 (ethylene + ethane) | 0.7 | 36 |
| C3 (propene + propane) | 0.3 | ||||
| 0.1 M KPO4 | C4 (butene + butane + isobutene) | 0.16 | |||
| C5 (pentene + pentane + 2-methyl-butane) | 0.07 | ||||
| MoS2 | H-cell | 0.6 mA cm−2 | 1-Propanol | 4 | 37 |
| 0.1 M KPO4, pH 6.8 | −0.6 V | Ethylene glycol | 0.65 | ||
| Tert-Butanol | 0.2 | ||||
| Mo3P-Im | Flow cell | 390 mA cm−2 | Propane | 91 | 38 |
| 1.0 M KOH | |||||
| Ni3Al | H-cell | 2.1 mA cm−2 | 1-Propanol | 2 | 39 |
| 0.1 M K2SO4, pH 4.5 | |||||
| Ni2P | H-cell | <0.5 mA cm−2 | Methyl glyoxal | 25 | 40 |
| 0.5 M KHCO3, pH 6.8 | 2,3-Furandiol | 71 | |||
| NiP2 | H-cell | <0.5 mA cm−2 | Methyl glyoxal | 84 | 40 |
| 0.5 M KHCO3, pH 6.8 | 2,3-Furandiol | 15 | |||
| Ni2P–CTAB | H-cell | 0.8 mA cm−2 | Methyl glyoxal | 97 | 41 |
| 0.5 M KHCO3, pH 6.8 | 2,3-Furandiol | 1 | |||
| Flow cell | 50 mA cm−2 | Methyl glyoxal | 50 | ||
| 0.4 M K2SO4 | 200 mA cm−2 | Methyl glyoxal | 40 | ||
| Ni2P-Ho2O3 | H-cell | 0.95 mA cm−2 | Acetone | 25.4 | 42 |
| 0.1 M KHCO3, pH 6.8 | |||||
| Ni phosphate | Flow cell | 12 mA cm−2 | Propene | 2.2 | 43 |
| Propane | 1.2 | ||||
| 1-Propanol | 0.5 | ||||
| 1-Butene | 0.8 | ||||
| Iso-butane | 0.3 | ||||
| 0.1 M KHCO3, pH 6.8 | n-Butane | 0.3 | |||
| 1-Butanol | 0.3 | ||||
| 1-Pentene | 0.5 | ||||
| n-Pentane | 0.2 | ||||
| n-Hexane | 0.16 | ||||
| Ni-doped (Cr2O3)3Ga2O3 | H-cell | −0.88 V | 1-Butanol | 42 | 44 |
| 0.1 M KH2PO4/0.1 M K2HPO4, pH 6.7 | 0.2 mA cm−2 | 3-Hydroxy butanal | 5 | ||
| AuNi | H-cell | 2 mA cm−2 | CnH2n | 0.12 | 45 |
| 0.1 M KHCO3, pH 6.8 | CnH2n+2 (n > 2) | ||||
| Fe2P | H-cell | <1.0 mA cm−2 | Methyl glyoxal | 20 | 46 |
| 0.5 M KHCO3, pH 6.8 | 2,3-Furandiol | 8 | |||
| Co3O4 | Flow cell | 29.8 mA cm−2 | C2–C7 hydrocarbons | 0.56 | 47 |
| 0.1 M KHCO3 | −0.9 V vs. RHE | ||||
1.1. PdAu-based catalysts
The bimetallic PdAu material, obtained via the electrodeposition of Pd on an Au substrate, was shown to not only catalyze the electroreduction of CO2 to formic acid as the main product but also a variety of multi-carbon products, essentially hydrocarbons, albeit with very low faradaic efficiencies (FEs < 1%) including C2 (ethylene and ethane), C3 (propane and propylene), C4 (butane, 1-butene and isobutene), and C5 (pentane, pentene and 2-methyl-butane) (Table 3).36 To the best of our knowledge, this was the first non-Cu-based material that showed the ability to catalyze C–C coupling reactions. Although no mechanistic study was provided in this report, it was proposed without experimental and theoretical support that the reaction proceeds via CO2R to *CO, and then *CH2 groups are adsorbed on the catalyst surface, followed by polymerization (via *CH2–*CH2 coupling), in agreement with the absence of oxygenates in the product mixture. In the following, an asterisk associated with a molecular formula, such as *CO, indicates that the molecule, here CO, is adsorbed on the surface of the solid catalyst. This putative mechanism is similar to the polymerization mechanism occurring in the Fischer–Tropsch (F–T) process.48,49 In this case, the reaction of CO with H2 (syngas) over a thermal catalyst at high temperature and pressure involves the adsorption of *CO and dissociative adsorption of H2, leading to the adsorption of *H. These two primary intermediates react via C hydrogenation and C–Cn coupling steps, leading to the formation of long-chain hydrocarbon products. During the electroreduction of CO2, the same intermediates, *CO, coming from CO2R, and *H, coming from H+/H2O reduction, are formed and can potentially proceed along comparable pathways. However, thus far, C–Cn coupling is limited to low n values and the production of long-chain hydrocarbons is not considered in general competitive with respect to H2 formation and to CO desorption owing to its very low FEs (<1%).1.2. Mo-based catalysts
Mo-based materials have been rarely used for CO2R/COR. In contrast, MoS2 is one of the most studied catalyst for proton electroreduction given that it has been proven to proceed with very low energy barriers and overpotentials.50 Actually, when implemented in an H-cell containing a CO2-saturated potassium phosphate electrolyte, H2 was the major product. However, interesting organic products were observed, albeit in very low amounts and at very low current density (0.6 mA cm−2) including 1-propanol (FE = 4%), ethylene glycol (FE: 0.65%) and t-butanol (FE = 0.2%) (Table 3).37 These reactions were not studied further and no experimental and computational insight was provided into the understanding of the C–C coupling pathways on the surface of MoS2.In 2023, M. Asadi and coworkers reported a study showing that molybdenum phosphide, Mo3P, has the potential to catalyze CO2R with remarkable selectivity towards propane, achieving a very high FE of 91% using a flow cell with the catalyst deposited on a GDL at a high applied current density of 390 mA cm−2 during 100 h electrolysis (Table 3).38 The observed selectivity and stability were attributed to the combination of the following factors: (i) surface functionalization with a monolayer of imidazolium molecules via electrodeposition and (ii) coating the GDE with an anion-exchange ionomer, which suppressed HER and helped maintain the molecular layer during long-term electrolysis. Alternatively, the bare Mo3P material produced a mixture of CO (FE = 75%) and CH4 (FE = 24%) under the same electrolytic conditions. The experimental and computational studies suggested that the active sites were the Mo atoms and that the presence of an imidazolium layer on Mo3P decreased the charge-transfer resistance, favored *CO2 adsorption via electrostatic interactions/H-bonds, stabilized *CO adsorption and promoted *CO coverage, favoring C–C coupling. These results are in agreement with the in situ electrochemical Raman spectroscopy results, showing high *CO coverage. However, this work did not provide any clue regarding the high selectivity for propane formation. DFT calculations showed a favorable mechanistic pathway from CO2 to propane, implying an intriguing trimerization step, *CO + *CH + *CO → *CO − CH −CO, with a quite low energy barrier. They also suggested that the surface was carbophilic enough to stabilize *C intermediates on the surface and favor dehydration, in agreement with the lack of oxygenates.51 This class of materials clearly deserves further investigation.
1.3. Ni-based materials
Up to recently, Ni was not expected to promote C–C coupling from CO2R, where Hori and collaborators earlier described CO poisoning of Ni surfaces, leading to their generally high catalytic activity for HER.52 Nevertheless, the theoretical study by Norskov and collaborators predicted that Ni–Ga alloys can catalyze CO2R.53 Subsequently, N. Lewis and collaborators demonstrated CO2R to methane, ethylene and ethane using different phases of Ni–Ga as catalysts; however quite inefficiently, with very low FEs (below 2%) and low current densities.54 A. B. Bocarlsy extended this study to Ni–Al materials and reported that Ni3Al thin films supported on glassy carbon could catalyze the formation of a variety of C1, C2 and also C3 products from CO2R, providing, to our knowledge, the first illustration of an Ni-based material capable of promoting C–C coupling from CO2 up to a C3 product.39 Although the major products of the reaction carried out under the conditions of 0.1 M CO2-saturated K2SO4 and pH 4.5 were H2 (FE > 60%) and CO (FE = 33%) at the optimal applied potential, giving a current density of 2.1 mA cm−2, 1-propanol was the major liquid product (FE = 2%), together with minor amounts of methanol, formate, ethanol (FE < 1%) and traces of acetone (Table 3). This system proved remarkably stable during several days of electrolysis. However, although preliminary mechanistic studies indicated that CO is a key intermediate, no further chemical study was provided regarding how Ni3Al facilitates the formation of C3 products (propanol and acetone).Despite the fact that nickel phosphides were reported as highly active HER catalysts, G.C. Dismukes and collaborators discovered that they could also be used as electrocatalysts for CO2R.40 In their study, different nickel phosphide compounds (Ni3P, Ni2P, Ni2P5, Ni5P4 and NiP2) were synthesized and deposited on an aluminium die as a working electrode support within an H-cell electrolyzer. Remarkably, CO2 electrolysis led to the production of multi-carbon oxygenates, such as methylglyoxal, a C3 product, and 2,3-furandiol, a C4 product; however, only when a very low potential of −0.1 V vs. reversible hydrogen electrode (RHE) (thus with a very low current density, <−0.5 mA cm−2) was applied. The most selective catalyst for methylglyoxal was NiP2, with an FE of 84% at −0.10 V vs. RHE, while the maximum FE for 2,3-furandiol was 71% observed at 0 V vs. RHE on Ni2P (Table 3). At slightly more cathodic potentials (<−0.2 V vs. RHE), the reaction selectivity shifted to HER. Although formic acid was produced at all potentials, FEformate never exceeded 5% for any of the Ni phosphides. With these low cell potentials, high energy efficiency values were obtained, namely 99% and 92%, for Ni2P and NiP2, respectively. The authors observed a preference for P-rich Ni phosphides in forming C3/C4 products, suggesting that the nucleophilic surface P sites are the potential binding sites for hydridic *H and *CO2.
Regarding the mechanisms, the low potentials, close to equilibrium potentials, at which product formation takes place exclude CO2 adsorption, followed by activation via proton-coupled electron transfer, which requires much more cathodic potentials (−0.7 to −1.0 V vs. RHE). This suggests a hydride transfer mechanism (Fig. 1) in the initial rate-determining step during CO2 conversion to formic acid. Regarding the formation of multi-carbon products, the system is quite intriguing, considering the nature of the products, methylglyoxal and 2,3-furandiol, highly oxygenated compounds, and the absence of CO formation, which excludes *CO–*Cn coupling pathways for chain elongation as in the case of Cu-based catalysts (see Section 2). Based on thermodynamic considerations, it has been proposed that the reaction proceeds via a series of aldehyde self-condensation steps (Fig. 1). The reaction would start from surface adsorbed formaldehyde, which is derived from formic acid reduction by a hydride species, giving rise to adsorbed glycoaldehyde, then adsorbed glyceraldehyde (from the condensation of formaldehyde with glycoaldehyde), and finally methylglyoxal (from the condensation of formaldehyde with glyceraldehyde) (Fig. 1). Thus, methylglyoxal is proposed to be derived from the condensation of three formaldehyde molecules. 2,3-Furandiol is formed by the last condensation between methylglyoxal and formaldehyde, followed by furan five-membered cycle formation and hydride abstraction for ring aromatization. There is precedent in the literature for hydride abstraction by nickel phosphides.55 It was proposed that these aldehyde condensation reactions are catalyzed by Ni phosphides, which display favorable Lewis acid character (given that carbonyl binding to a Lewis acid surface lowers the barrier for proton abstraction from the C–H of formaldehyde), and furthermore, as mentioned, Ni phosphides also favor the last hydride abstraction.55 Thus, the proposed mechanism is dependent on two initial successive hydride transfers to produce formate, and then formaldehyde and with formic acid/formaldehyde, leading to multi-carbon products, which greatly differs from the CO2R mechanism on Cu-based catalysts (see Section 2). This was supported by experiments using the intermediates (formate, formaldehyde, and methylglyoxal) as substrates in the absence of CO2. A computational study established the key role of Ni-bound surface-adsorbed hydride H* species (bound at an Ni3 hollow site) in the formation of formic acid, and then formaldehyde.56 However, the strong affinity for surface H* is responsible for the large kinetic barrier for these two first steps (>1 eV), limiting the catalytic activity of Ni2P, which is consistent with the very low turnover frequency observed. It also shown that H2CO* self-coupling giving glycoaldehyde is thermodynamically downhill by 0.41 eV, and thus more favorable than the further reduction of adsorbed formaldehyde to C1 products (CH3OH and CH4) displaying high kinetic barriers. Glyceraldehyde formation via coupling glycoaldehyde and a third molecule of H2CO* and the subsequent water elimination giving 2-hydroxy-2-propenal are also downhill by 0.19 and 0.96 eV, respectively. Finally, the formation of methylglyoxal (enol–keto tautomerization) is downhill by 0.31 eV, while the formation of the C4 molecule, furandiol, is nearly thermoneutral. Thus, the overall energetics of the proposed mechanism is strongly exergonic, with the formation of surface hydrides being essential, and with the first step of hydride transfer to CO2 being the rate-limiting step. Thus, this is the key target to study and optimize to improve the catalytic activity via active site engineering.
 | ||
| Fig. 1 Proposed mechanism for methyl glyoxal and 2,3-furandiol formation catalyzed by nickel phosphides.40 | ||
Dismukes and collaborators achieved a remarkable improvement in the system performances by combining the following: (i) a high surface area Ni2P material (Ni2P–CTAB) with a much larger surface density of catalytic sites, owing to a specific soft-templating synthesis procedure using a surfactant, cetyltrimethylammonium bromide (CTAB), to control the material morphology; (ii) surface modification of the catalyst with a hydrophobic anionic polymer (PFAEM) as a co-catalyst binder to increase the CO2R vs. HER selectivity; and (iii) a flow cell with a GDE to improve CO2 mass transport to the catalyst and limit HER under larger current densities.41 Indeed, the use of an H-cell system allowed remarkably high FE for methylglyoxal (97%) at applied potentials close to the RHE, at very low current densities (Table 3). Instead, with a flow cell system based on a bipolar membrane, high total current densities in the range of 50 to 200 mA cm−2 could be applied, while achieving remarkably high FE for CO2-derived products, almost exclusively methylglyoxal, accounting for a total FE of up to 40–50%. The control experiments showed that Ni2P, in contrast to Ni2P–CTAB, produced only H2 at the applied current densities, under the same electrolytic conditions.
Ni2P was also studied as a part of pure monodisperse core/shell nanoparticles (CSNPs), in which the inner core was made of crystalline Ni2P and the 1.3 nm-thick outer shell was made of amorphous Ho2O3.42 The amorphous shell was designed to provide a high density of active sites, defects, and undercoordinated sites, while the crystalline conductive core facilitated charge transfer between the core and shell. This material behaved as a catalyst; however, it was not stable for CO2R in an H-cell using CO2-saturated 0.1 M KHCO3 as the electrolyte, leading to the large production of acetone (FE = 25.4%) at an applied potential of −0.98 V vs. RHE (total current density 0.95 mA cm−2), together with H2 as the main product (FE > 60%), HCOOH and CH3OH (Table 3). Coupling between the *C1 and *C2 intermediates was proposed for the formation of the C3 product acetone, but owing to the lack of mechanistic studies, it is difficult to understand why only acetone is formed as a C2+ product given that no C2 product and no propanol were observed.
Inorganic nickel oxygenates (nickel phosphate, nickel carbonate, nickel bicarbonate, nickel hydroxide and nickel oxide), deposited on a GDL, were also shown to catalyze CO2R in a flow cell electrolyzer, using a 0.1 M KHCO3 electrolyte and an applied potential of −1.0 V vs. RHE.43 Remarkably, while H2 was the major product (FE = 65%), a great variety of CO2-derived carbon compounds was detected. These compounds not only included C1 (CO, formate, and methane), with a total FE of 13.5%, and C2 (ethylene, ethanol, and acetaldehyde), with a total FE of 6%, products usually found in Cu-catalyzed CO2R, but also a long list of C3, C4, C5 and C6, linear and branched, products. In the case of the most productive catalyst, namely Ni phosphate, C3+ hydrocarbon products accounted for a total FE of 6.5% with a partial current density of 0.91 mA cm−2, while lower amounts of oxygenates (FE = 3.8%), including alcohols up to C4 (1-butanol), were detected. Nevertheless, each product accounted for FE < 2%, with the C3+ products being propene (2.2%), propane (1.2%), 1-propanol (0.5%), 1-butene (0.8%), isobutene (0.3%), n-butane (0.3%), 1-butanol (0.3%), 1-pentene (0.5%), and n-pentane (0.2%) (Table 3). The heaviest compound and unique C6 compound was n-hexane (FE = 0.16%).
Using operando X-ray absorption near edge structure (XANES), it was observed that unlike Cu systems, the inorganic Ni oxygenates do not undergo full reduction to metallic Ni, where the absorption of the Ni K-edge indicated the existence of stable Niδ+ sites. The presence of Ni2+ was proposed to be due to the retention of (near)-surface oxygenated species and stable Ni–O bonds, as shown by operando EXAFS. This might explain why CO is moderately bound at the catalyst surface compared to Ni0, freeing it from CO poisoning, a characteristic of metallic Ni, and allowing further COR and C–C coupling. This was confirmed by density functional theory (DFT) calculations using surface models with a wide degree of polarization owing to O or OH doping, which showed that the CO binding strength was weakened and C–C coupling favored on surfaces with increased positively charged Ni sites. Considering the extreme complexity of the reactions, implying a huge number of electrons and protons for the formation of each of the C4–C6 products, and thus a huge number of possible intermediates (hundreds), the mechanism is almost impossible to decipher. Nevertheless, it was proposed that *CH and *CH2, derived from CO or CH2O, respectively, were the key species in C–C coupling reactions, leading to an extension of the carbon chains during the formation of long-chain hydrocarbons. In this work, according to the proposed mechanism, the first C–C bond formation on the polarized Ni surface proceeds via the *CH/*CH2 + *COOH coupling pathway, followed by *CH and *CH2 insertions to form C3+ hydrocarbons. Recently, M. T. M. Koper's group reported further information regarding this Ni phosphate material.57 They showed that while the effect of temperature and pressure on selectivity towards long-chain hydrocarbons was minor, the catalyst was more sensitive to variations in the electrolyte composition, with K+ cations and better proton-donating anions, such as phosphate, favoring long-chain hydrocarbons.
A study in 2023 confirmed the ability of Ni to promote C3+ product formation.44 Ni-doped (Cr2O3)3Ga2O3 was indeed shown to catalyze the electroreduction of CO2 to 1-butanol, with a remarkable FE of 42%, during 20 h electrolysis at an applied potential of −1.48 V vs. Ag/AgCl; however, at low current density of <1 mA cm−2 using an H-cell containing KCl/NaHCO3 as the electrolyte at pH 4 (Table 3). 3-Hydroxybutanal, a C4 product, was also obtained with an FE of 5% (FE of 25% was obtained at pH 5 with an applied potential of −1.4 V). Acetic acid was also detected (FE = 9%). In contrast, dopant-free (Cr2O3)3Ga2O3 produced mostly hydrogen (FEH2 = 92%), with very minor amounts of CO2-derived products, such as acetic acid, acetaldehyde and acetone. As a control experiment, all the products previously identified with the Ni-doped (Cr2O3)3Ga2O3 catalyst were detected after 20 h electrolysis of formic acid used as the substrate in place of CO2. This supported the hypothesis that formate could serve as the primary intermediate towards multi-carbon compounds, a mechanism similar to that proposed for Ni phosphides.40 Acetaldehyde was also proposed to be a secondary intermediate given that the electrolysis of acetaldehyde using Ni-doped (Cr2O3)3Ga2O3 resulted in the production of 1-butanol. (Cr2O3)3Ga2O3 was proposed as the source of acetaldehyde and nickel atoms as the critical component for acetaldehyde conversion to butanol given that the electrolysis of acetaldehyde using a coiled Ni wire or a planar glassy electrode electroplated with Ni resulted in the formation of significant amounts of 1-butanol. In conclusion, according to the proposed mechanism, the catalyst activates surface hydrides to allow the conversion of CO2 to formic acid and its reduction to formaldehyde, which couples with a second molecule of formaldehyde to form acetaldehyde. Thus, this step is thus facilitated by (Cr2O3)3Ga2O3. Then, the Ni sites promote the coupling of two molecules of acetaldehyde to generate 3-hydroxybutanal, which is then further reduced to 1-butanol (Fig. 2).
 | ||
| Fig. 2 Proposed mechanism for 1-butanol formation catalyzed by Ni-doped (Cr2O3)3Ga2O3.44 | ||
Finally, the recent report on an Au–Ni catalyst leading to the formation of long-chain hydrocarbons should be mentioned.45 Following the concept that the F–T mechanism could operate during CO2R with an appropriate combination of metals, providing *CO and *H intermediates (see Section 1.1), the Au–Ni material, synthesized via Au deposition on an Ni sheet, was chosen for the following reasons. Ni served to enhance CO adsorption and favor surface *H coverage, while Au was selected for generating a high surface density of *CO. Under these favorable conditions, *CO and *H could combine to form *CHx and promote *C1–*Cn polymerization steps. Actually, besides the large production of H2 and CO, a series of CnH2n+2 and CnH2n products, up to n = 7, was detected, including isomers of butane (isobutane and 2-methylpropane) and pentane (isopentane and 2-methylbutane) during CO2R in an H-cell at a potential of −0.977 V vs. RHE using 0.1 M KHCO3 as the electrolyte. No liquid products were detected. Nevertheless, the total FEs for these hydrocarbons were very low (FE = 0.23%, with small variations depending on the applied potential and the Au thickness) at a total current density of about 2 mA cm−2 and with the C2 products C2H4 and C2H6 accounting for about half of total FE (FEC2 = 0.11%, FEC2H6 = 0.07%, and FEC2H4 = 0.04%). In the absence of the Au deposit, hydrocarbons were also detected but with even lower total FEs (<0.02%). The production of alkanes was much higher than alkenes. The same products were obtained during COR; however, with lower FEs. The product distribution was interpreted as an indication for a C–C coupling polymerization reaction mechanism, leading to long-chain hydrocarbons, similar to the conventional F–T synthesis.48,49 The same products, also with a low FE of about 0.35% for C2–C5 hydrocarbons, were obtained using a Cd electrode but the FE increased to 0.45% upon modification by sputter deposition of Ni (FE also increased with Pt or Ag deposition).58,59
Besides the findings described above, it is interesting to mention a recent work confirming the ability of Ni-based materials to catalyze CO2R to multi-carbon compounds.60 In this case, a stable material composed of Ni particles encapsulated in N-rich carbon nanotubes was proven to be remarkable for converting CO2 selectively to ethanol, a unique liquid product, and CO with very little production of H2 (FE < 10%) using both H-cells and flow cells equipped with GDEs. Thus, it was possible to achieve FE values for ethanol in the range of 30–40%, within a wide voltage range of −0.6 to −1.2 V vs. RHE, enabling high current densities to be obtained (from 12 mA cm−2 in an H-cell to 127 mA cm−2 in a flow cell).
1.4. Fe-based catalysts
Based on a previous study on nickel phosphides, it was reasoned and confirmed computationally that iron phosphides, displaying weaker binding, and thus greater reactivity of surface hydrides, should catalyze CO2R at greater rates.46 Two initial reports indeed showed the ability of iron phosphides to catalyze C–C coupling specifically towards the formation of ethanol. Firstly, an FeP nanoarray on Ti mesh was shown to be able to catalyze CO2R to methanol (FE = 80%) and ethanol (FE = 14%); however, at very low applied potentials (close to the onset potentials) and low current densities (∼1 mA cm−2) in 0.5 M KHCO3.61 Secondly, an Fe2P2S6 sheet was shown to favor ethanol formation with a maximum FE of 23.1%; however, at even lower current densities (<0.5 mA cm−2).62 Later, another class of Fe-based catalysts with a core–shell architecture, with a nitrogen-doped γ-Fe2O3 material as the core and carbonitride as the shell, was also shown to catalyze CO2R to a C2 product, namely ethane, reaching an FE value of 42% at a significant current density of 32 mA cm−2 in an H-cell using a mixture of an ionic liquid, organic solvent (acetonitrile) and water as the electrolyte.63Interestingly, a study confirmed the ability of Fe2P to catalyze C–C coupling reactions, allowing CO2 conversion to C3 and C4 products.46 In contrast, Fe2P could not catalyze COR, where CO essentially behaved as a poison. In 0.5 M KHCO3, CO2 electrolysis, at a very low applied potential (0.00 V) and low current density (<0.1 mA cm−2), resulted in the production of formic acid (FE = 15%), methylglyoxal (FE = 20%), ethylene glycol (FE = 10%) and 2,3-furandiol (FE = 8%), the major product being H2 (Table 3). The total FE for CO2R products decreased upon applying more cathodic potentials (FEmax = 53% at 0.00 V), with H2 accounting for more than 95% at −0.2 V, but the maximum FE for ethylene glycol (22%) was obtained at −0.05 V. The proposed mechanism leading to methylglyoxal and 2,3-furandiol for Ni2P was also applied in the case of Fe2P, consisting of CO2R to formate, and then to formaldehyde, from which C–C coupling occurs giving glycoaldehyde, then glyceraldehyde, and then methylglyoxal and furandiol via consecutive couplings with formaldehyde (Fig. 1). Ethylene glycol formation was proposed to proceed via the reduction of glycoaldehyde. The presence of this C2 product in the case of Fe2P and not Ni2P is likely related to the greater reactivity of surface hydride on Fe2P, favoring the C2 pathway, which requires the addition of a hydride, while the C3/C4 pathway requires formaldehyde coupling. These experimental and computational studies indicate that surface hydrides and their binding affinities are potentially critical for promoting multi-carbon formation from CO2.
1.5. Co-based catalysts
Based on the performances of inorganic nickel oxygenates43 as well as the activity of Co-based catalysts for thermocatalytic CO hydrogenation and Fischer–Tropsch synthesis (FTS) reaction to produce long-chain hydrocarbons, a Co3O4 material was recently studied.47 Co3O4 was deposited on carbon black, and then on a GDL of a three-compartment flow cell, and using a bipolar membrane, it could indeed catalyze the electroreduction of CO2 into C2–C7 hydrocarbons (saturated and unsaturated including branched and linear isomers) during short term electrolysis, with the product distribution closely following the Anderson–Schulz–Flory distribution observed for thermocatalytic FTS. At an applied potential of −0.9 V vs. RHE in CO2-saturated 0.1 M KHCO3 as the electrolyte, the total FE for C2+ products was 0.56% and the total partial current density was 0.12 mA cm−2, with the major product being H2 (Table 3). Co3O4 was the most active catalyst compared to CoO and Co oxygenates (phosphate, carbonate, and bicarbonate), while metallic Co only produced H2. However, with prolonged electrolysis, the hydrocarbon production rate decayed as a consequence of a reduction-induced deactivation mechanism, following the accumulation of inactive metallic cobalt, as shown by in situ analysis of the catalyst under the operating conditions. Actually, upon reoxidation at an anodic potential of +0.6 V for 5 min, Co3O4 could be regained, and further CO2 electrolysis showed that the catalytic capabilities to form long-chain hydrocarbons were recovered. CO was shown to be the primary coupling component and interfacial Co–Co3O4 centers were proposed to be the catalytically active sites. Indeed, DFT calculations showed that the *CO adsorption energy on these sites was comparable to metallic copper, and thus compatible with *CO activation for C–C coupling, in contrast to pure metallic Co (too strong adsorption energy leading to CO poisoning) or pure Co3O4 (too weak adsorption energy). DFT calculations also showed that chain elongation was more favorable via *CO–CH2*, and then *CO–CH3(CH2)nCH* coupling followed by termination via hydrogenation (reaction with H*), and furthermore that preference for chain growth over hydrogenation well explains the propensity of the interfacial sites to form long-chain hydrocarbons. This work demonstrated the possibility of using metal–metal oxide interfaces as an effective pathway towards CO2R to long chain hydrocarbons, although the appropriate balanced population of each component may be difficult to control under the highly cathodic potentials used during CO2R.1.6. Ag-based catalysts
Ag is well-established as a catalyst for the selective formation of CO from CO2R due to its low surface CO adsorption energy, and thus surface *CO desorbs faster than it reacts via C–C coupling. However, an interesting theoretical study suggested that Ag could have a lower onset potential for ethanol production from COR than that of Cu, suggesting that Ag can catalyze ethanol formation from COR if the surface CO coverage is sufficiently high.64 This was confirmed a few years later using a pressure cell allowing alkaline COR to run under a high pressure of CO (from 10 to 60 bar) as a way to increase the CO coverage on the surface of a silver GDE.35 Interestingly, very low amounts of C2+ products were detected, predominantly oxygenates, in agreement with the DFT predictions, including C2 products (ethanol, acetic acid and ethylene glycol), and also a C3 product, namely propanol. The largest amount of propanol was obtained at 60 bar (the total FE of the C2+ products was below 2% with a partial current density of <8 mA cm−2) and was proposed to be derived from the coupling reaction between CO and a surface-bound *C2 oxygenated intermediate, likely to be one hydrogen short of acetaldehyde and precursor of ethanol as well. Thus, the product spectrum of Ag can resemble that of Cu under a high pressure of CO; however, at orders of magnitude lower formation rates.Very recently, two other examples of Ag-based catalysts were shown to allow CO electroreduction to C2+ products. The first one was a PdAg alloy containing isolated Pd atoms.65 This configuration allowed an increase in the *CO coverage owing to the presence of Pd atoms and a balance in the *CO adsorption energy, enabling C–C coupling to occur as in the case of Cu surfaces. Only C2 products could be obtained (no C3 product), such as ethylene, acetate and ethanol, with a total FEC2 of 37% at −0.83 V vs. RHE and a partial current density of about 25 mA cm−2 in 1 M KOH. In the second example, it was found that the chirality-induced spin polarization of chiral nanostructured Ag films could promote *CO–*CO coupling during CO2R in a pressure H-cell (P = 12.5 atm) in KHCO3, leading, in addition to CO as the major product, to minor amounts of C1 products (methane and methanol) and C2 products (ethylene, ethane, acetate and ethanol) with only one C3 product, propane, accounting for the maximum FE of about 1%.66 The total FEC2+ was 4.7% for a partial current density of 22 mA cm−2.
1.7. Metal-free catalyst
A completely different approach was proposed recently, which is based on the ability of molecular metal-free frustrated Lewis acid–base pairs (FLPs) to activate CO2, owing to electron donation from the lone pair of each O atom in CO2 to the Lewis acid (LA) center and from the Lewis base (LB) to the C atom of CO2.67 A heterogeneous version of this concept, consisting of highly conductive graphene powder doped with boron atoms (6.8 at%) as LA and nitrogen atoms (4.2 at%) as LB, via the reaction of graphene with B and N dopants under high-frequency ultrasound, proved highly selective for propanol formation during CO2R.68 A record FEpropanol of 50%, together with a high FEethanol, was obtained; however, in an H-cell system using 0.5 M Na2SO4 as the electrolyte at potentials more positive that −0.7 V vs. RHE, and thus at a very low total current density (2–6 mA cm−2). A higher applied cathodic potential resulted in a large drop in FEpropanol. The control graphene doped with only one heteroatom, B or N, gave only a mixture of formic acid, as the major product, and small amounts of CO and H2, demonstrating that C–C coupling occurred at the B/N-co-doping sites. DFT calculations showed a thermodynamically favorable pathway (favorable reaction free energy and activation energy barrier) involving the following steps: (i) binding CO2 in a bidentate mode with N–C and B–O bonds favoring CO formation remaining attached to the B,N-site and (ii) C–C coupling between the CO intermediate and a second molecule of CO2. This coupling reaction is more favorable than CO–CO coupling, owing to the cooperative effect of the electron enrichment on C in N–CO–B and the electron deficiency on C in CO2. Thus, here, the mechanism is proposed to be different from that occurring in Cu-based materials, where the coupling of *C1 intermediates with CO2 is excluded in general. Furthermore, the absence of hydrocarbons as products and the high selectivity for alcohols (ethanol and propanol) was explained by the suppressed deoxygenation steps given that DFT calculations demonstrated the high stabilization of the C–O bonds in the *C2 and *C3 intermediates by B/N-sites.2. Cu-based catalysts
Presently, it is well-established that Cu-based materials have the greatest potential to catalyze the reduction of CO2 to multi-carbon molecules with significant faradaic efficiencies and partial current densities. Although C2 molecules such as ethylene and ethanol are the major multi-carbon products, propanol, a C3 product, is also generally formed to a large extent, as discussed below. In 2012, employing highly sensitive analytical methods, T. Jaramillo and collaborators reported that the product mixture derived from Cu-dependent CO2R is in fact much more complex than earlier anticipated, containing 16 different CO2 conversion products, notably with very low amounts of C3 products. The heaviest compounds, such as propionaldehyde, allyl alcohol, glycoaldehyde, ethylene glycol, acetone and hydroxyacetone, accounted individually for FE < 1%.69 In a more recent study by the M. Koper group, glyoxal and 1,2-propanediol, with FEs between 0.5% and 1%, were added to the list.70 In 2015, the formation of low amounts of other C3 compounds (propane and propene) as well as a C4 compound, butane, was reported.71 The latter study reported the highest FE for propanol (8.7%) at that time but larger values were obtained later, as discussed in a specific chapter on propanol formation. To our knowledge, the first report of C4 oxygenate formation during CO2R catalyzed by a Cu-based catalyst was published in 2020.72 These studies clearly established the general complexity of the reaction product mixtures from Cu-based CO2R, and thus confirmed the lack of selectivity of Cu-based systems. In the following, we only discuss Cu-based systems of interest, specifically producing C3, notably propanol, propane and propylene, and C4, notably butanol, compounds with relatively large total FEs.2.1. Mechanistic theoretical considerations
In the case of Cu, the mechanism of CO2 conversion to multi-carbon compounds has been extensively investigated theoretically.73–75 Specifically, several excellent review articles have appropriately discussed our current knowledge, regarding the mechanism for the formation of C2 compounds, ethylene, ethanol and acetate.8,76,77DFT calculations are a major component of most studies reported on CO2R and COR as a way to identify the most relevant intermediates, understand reaction mechanisms and provide some rational with respect to the effect of the structure, morphology and composition of catalysts on their activity and selectivity. This is an attractive approach given that tremendous progress has been achieved in the development of DFT methods for describing periodic solids. However, this field suffers from the excessively systematic use of DFT calculations of reaction free energies and activation barriers, as a type of requested exercise, which is not always accessible and relevant. The frequent lack of relevance is attributed to two reasons. Firstly, the reactions leading to C2 products and C3+ products are greatly complicated, involving a large diversity of potential intermediates, most of them not observable experimentally, despite efforts to use, for example, in situ vibrational spectroscopy, which is the most appropriate for the detection and identification of CO-derived intermediates.78–81 For example CO2R to the C2 products ethylene and ethanol, the most studied pathway, requires 12 electrons and 12 protons, respectively. Thus, combining C–C coupling steps, coupled H+/e− transfer steps and deoxygenation steps, and considering the multiple configurations in which the same intermediate binds to the catalyst surface, involve more than 30 possible intermediates.8,77,82 Reduction to propanol, a pathway much less studied thus far, requires 18 electrons and 18 protons, and thus potentially involves much more intermediates.83,84 In general, under these circumstances, for simplification, a very small population of these intermediates is considered for calculating the energetics of the critical C–C coupling steps leading to C2 or C3 products. The second reason for the irrelevance is due to the growing complexity of the structure and the composition of the catalysts, which are no longer a pure single-facet metal, making it difficult to model their surface appropriately, and thus a large gap exists between the real catalyst and the model, given that it is necessarily oversimplified to make it accessible to current computing tools and time. The complexity is even more challenging if one considers that the dynamic reconstruction of the catalyst surface during electrolysis, which is almost always systematically overlooked, due to the harsh reaction conditions (polarization at very high cathodic potentials, extreme pH sometimes and large concentration of salts). Consequently, the theoretical structural models used to calculate the reaction energies and characterize the reaction mechanism are often too far from the real structure of the experimentally studied solid catalyst, and thus the conclusions from these studies should be considered with great care, which is not always the case. It is even more problematic in studies where the DFT calculations are performed first as a way to rationally identify a potential catalyst target for a given reaction, given that there is little chance that the synthesized material will possess a structure similar to the anticipated simple model. Nevertheless, if one uses DFT calculations with sufficient modesty for these complex catalysts and reactions, they are useful in providing some rational for understanding some observed experimental trends.
The theoretical efforts have made it possible to propose the mechanism and intermediates for CO2R and COR to C2 products catalyzed by Cu-based catalysts. Briefly, as shown in the simplified version in Fig. 3, the first key intermediate is *CO, adsorbed on the surface, which can dimerize into *OCCO, forming a C–C bond. Then, a series of coupled electron/proton transfer steps generate a variety of *C2 intermediates up to a few ones at which the ethylene and ethanol pathways bifurcate. Regarding C3 product formation specifically, the proposed mechanisms in general involve C–C coupling steps between the *CO and *C2 intermediates given that the trimerization of *C1 intermediates is excluded because of its too high energy barrier.83,84 DFT calculations of the reaction energies and barriers either simply consider coupling between *CO and *OCCO85,86 or, in some cases, between *CO or *CHO and a variety of the most likely *C2 intermediates, such as *HCCH, *CCO, *CHCO, *CHCHO and *CH3CO, for example. This is consistent with these reactions being the most feasible steps towards the formation of C3 products on Cu(100).83,84,87,88 However, it is still unknown which pathways are the dominant ones.
 | ||
| Fig. 3 Simplified mechanism for CO2R showing the involvement of adsorbed *CO, *C2 and *C3 intermediates.83 | ||
Recently, another important issue computationally addressed regards the control of the selectivity among C2+ products towards oxygenates, specifically alcohols, ethanol and propanol, with respect to hydrocarbons, in particular ethylene.51,75 F. Abild-Pedersen et al. found that carbophilicity of the surface was a simple and primary guide to interpret the oxygenate/hydrocarbon selectivity, where more carbophilic surfaces stabilize the intermediates, favoring dehydration towards ethylene, while a carbophobic surface disfavors C-bonded reaction intermediates in the ethylene pathway. DFT calculations were performed to determine the adsorption energy of C*, as the descriptor of surface carbophilicity, confirming, for example, that Cu(111) was less carbophilic than Cu(100), thus disfavoring ethylene. Oxophilicity can also be considered, as probed based on the calculated adsorption free energy of OH*, given that an oxophilic surface will have good affinity for the surface O-bonded intermediates, species. These species are widely accepted as intermediates towards the formation of alcohols, but this parameter is less instrumental in discriminating between oxygenates and hydrocarbons.51 A recent study using in situ surface-enhanced Raman spectroscopy (SERS) presented new insights into the specific Cu sites and surface intermediates that favor ethylene (intermediate *COCO) or ethanol (intermediate *OCHCH2).81
In agreement with the importance of *CO for C–C and C–C2 coupling, efforts have been devoted to elaborating Cu-based catalysts that favor CO coverage. This can be achieved in particular by tailoring the surface of the material by introducing grain boundaries, defects and undercoordinated sites.8,89–97
2.2. C3 oxygenate formation via CO2R and COR
Considering its high energy-mass density (30.94 kJ g−1) and high octane number (118), the fuel efficiency of n-propanol is close to that of gasoline. Furthermore, it can be blended with gasoline to form a cleaner fuel. Finally, it is a precursor for the polymer industry, and thus it has a high market value. It is currently produced in the industry from fossil-derived ethylene via hydroformylation to propionaldehyde, followed by reduction. Propanol is almost systematically formed during CO2R catalyzed by a variety of Cu-based materials, including polycrystalline Cu, albeit with low selectivity, namely with FEs lower than about 10%.98–102 This suggests that Cu surfaces partially stabilize *C2 intermediates enough to allow coupling with *C1 intermediates, before the C2 products desorb (Fig. 3). The recent study by M Koper and collaborators indeed nicely showed that a *CO trimerization mechanism is unlikely and that n-propanol formation can only be optimized via the fine balance of the relative surface coverage of CH3–CO* methyl carbonyl (dehydrogenated acetaldehyde), a common possible *C2 intermediate towards ethanol and propanol, and *CO, given that these two intermediates compete for the same active sites on the catalyst surface.103 This explains why the selective formation of propanol is challenging given that HER is favored at low *CO coverage, while at high *CO coverage, CH3–CO* coverage and C1–C2 coupling are disfavored. Nevertheless, some interesting studies led to the development of selective, efficient and cost-effective catalysts for propanol formation using CH3–CO* or CO electrolysis fueled by low-carbon electricity. Very few studies reported FEpropanol exceeding 15%. Here, we exclusively discuss these systems.Although CuS is a good catalyst for CO2R to C1 products, notably HCOOH and CH4, a recent study showed that the introduction of double sulfur vacancies, generated by an electrochemical lithium tuning strategy (CuS + Li+ + e− → CuSx + Li2S), led to a large FEpropanol of up to 15.4%, using an H-cell with 0.1 M KHCO3 as the electrolyte at an applied potential of −1.05 V vs. RHE, giving a partial current density of about 3.0 mA cm−2, which was 10-times larger than that obtained with a CuS catalyst without sulfur vacancies.109 A much lower FEpropanol was obtained using a flow cell and 1.0 M KOH as the electrolyte. Furthermore DFT calculations showed that owing to the presence of these vacancies, both *CO and *C2 (*OCCO) intermediates were stabilized and the *CO–OCCO* coupling was favored.109
It has been discovered that Cu-phtalocyanines and N,Cu-doped carbon materials, containing single Cu sites consisting of Cu2+ ions in N-coordination, are precursors of the small Cu clusters transiently formed during CO2R, as shown by operando characterization techniques. It has been well established that the formation of C2 products, notably ethanol, is due to the activity of these clusters in both cases.110,111 This led B. Yang and coworkers to explore a series of molecular dinuclear Cu2+ complexes using N-based macrocyclic ligands, including expanded porphyrins such as hexaphyrins and octaphyrins.112 In one case, a high Fepropanol of 18% (together with FEethanol of 32%) was obtained at an applied potential of −1.2 V vs. RHE (total current density of 9.4 mA cm−2) using an H-cell with the complex loaded onto a Ketjen black cathode and 0.1 M CO2-saturated KHCO3 as the electrolyte. Post-electrolysis characterization by mass spectrometry, UV-visible spectroscopy, X-ray photoelectron spectroscopy (XPS), microscopy and X-ray absorption spectroscopy (XAS) consistently showed that the complex was partially reduced and Cu ions partly converted into small Cu0 clusters, leading to the formation of inorganic/organic hybrids. Although the production of multi-carbon products was possible owing to C–C coupling reactions at the surface of the under-coordinated Cu clusters, as confirmed computationally, it was experimentally well established that the presence of both the partially reduced molecular complex and metallic clusters was necessary for the cathode to be selective for alcohols. However, the same catalyst tested in a flow cell using 1.0 M KOH as the electrolyte gave very low yields of propanol (FEpropanol < 10%).
A high FEpropanol of 17.9% was obtained using oxide-derived copper.94 In this interesting study, metallic Cu derived from CuO (CuOD–Cu) was compared to metallic Cu derived from Cu2O (Cu2OD–Cu). The two materials proved distinct in terms of both structure and activity. Upon reduction under CO2R conditions, all the Cu ions converted rapidly into metallic Cu0 in both cases, as shown by XAS. However, CuOD–Cu had a richer population of undercoordinated Cu sites and a rougher surface, with a higher surface Cu atom density. Furthermore, in situ surface-enhanced Raman measurements clearly showed that CuOD–Cu was much better at promoting the generation of surface-adsorbed *CO and *(H)OCCOH, a key C2 intermediate, likely favoring C–C2 coupling and the formation of C3 products. Although both catalytic materials resulted in relatively high yields of H2 (FEH2 > 40%), CO2R carried out in an H-cell using 0.1 M KHCO3 as the electrolyte led to the much higher production of propanol in the case of CuOD–Cu (FEpropanol = 17.9% at −0.94 V vs. RHE and a partial current density of 4.0 mA cm−2).
Manipulating the oxidation states to combine Cu+ and Cu0 sites seems to favor *CO to *OCCO coupling and propanol formation. The first example concerns an R–Cu–C material, a composite of CuCl and CuO, in which the Cl− anions serve to stabilize the Cu+ species during electrocatalysis due to their strong affinity for Cu surfaces.113 Actually, during electroreduction at large cathodic potentials, Cu+ and Cu0 coexist on the surface, as shown by XPS, (X-ray diffraction) XRD and XAS. Furthermore, the presence of abundant defect sites, favoring multi-carbon product formation, was shown from low coordination numbers. In an H-cell, this material efficiently in catalyzed the formation of alcohols. The maximum FEpropanol of 17.3% was obtained at an applied potential of −1.05 V vs. RHE (with a partial current density of 8.2 mA cm−2), together with an FEethanol of 32.5%. A slightly lower FEpropanol of 14% was obtained when CO2R was carried out in a flow cell using 1 M KOH as the electrolyte at an applied potential of −1 V vs. RHE (270 mA cm−2). In 2024, the same strategy was explored with the synthesis of a Cu-based material possessing a bicontinuous structure, assembling ultra small domains of Cu2O and Cu, with numerous grain boundaries between the Cu2O and Cu phases and a high roughness factor.114 For unknown reasons, Cu2O resisted electroreduction and the valence state of Cu fluctuated between 0.42 and 0.55 during 100 min electrolysis. Using an H-cell and 0.1 M KHCO3 electrolyte, the maximum FEpropanol of 16.2% was obtained at −1.4 V vs. RHE (6.8 mA cm−2 partial current density). A slightly lower FEpropanol of 12.1% was obtained in a flow cell with 1 M KOH as the electrolyte and an applied current density of 0.84 A cm−2 (thus a partial current density of 101.6 mA cm−2). In situ Raman spectroscopy, in situ attenuated total reflection surface enhanced infrared absorption spectroscopy (ATR-SEIRAS), differential electrochemical mass spectroscopy (DEMS) and DFT calculations supported the mechanism of propanol formation via *CO–*OCCO coupling, followed by propionaldehyde formation, and then reduction to propanol.
As discussed below, high FEs for propanol were obtained during COR with bimetallic catalysts, specifically Ag- or Au-doped Cu. Recently, this has been observed in very few cases during CO2R as well. Generally, bimetallic systems have been developed based on the working hypothesis that a tandem catalysis mechanism operates, in which Ag or Au, catalyzing CO2 to COR, contributes increased surface CO coverage, followed by CO spillover from Ag or Au onto Cu favoring C–C coupling.115 The addition of foreign heavy metals can modulate the atomic ensembles for adsorbate binding and induce lattice strain and charge transfer, altogether playing a role in enhancing catalytic activity and selectivity.116 Specifically, DFT calculations of CO2R on Ag- or Au-doped Cu nicely showed that doping decreased the activation energy barriers for the model reactions used as indicators for C–C and C–C2 coupling reactions, producing C3 compounds.85,88,117 Finally, it was shown that doping Cu with Ag or Au increased the carbophobicity of the catalyst surface, an effect disfavoring surface C-bonded reaction intermediates in the ethylene pathway, explaining why the formation of oxygenates is promoted.51
A record, while still limited, FEpropanol of 18% was obtained during CO2R with colloidal Au-doped Cu nanorods (NRs), Au0.02Cu0.98-NR, using a flow cell and 1 M KOH as the electrolyte at an applied current of 70 mA cm−2, corresponding to a moderate cathodic potential of −0.41 V vs. RHE, given that larger current densities gave lower FEs.117 This performance could be related to the increased capacity of the catalyst to maintain high CO coverage, favoring the formation of C3 products during CO2R, as shown by Raman studies monitoring the *CO-characteristic peaks. Furthermore, DFT calculations showed that Au doping lowers the energy barriers for *CO coupling to two *C2 intermediates, namely *HCCH and *HCCH3. These intermediates were chosen based on a previous DFT study showing that *CO–*HCCH coupling displayed the smallest kinetic barrier, among many other scenarios involving other *C2 intermediates.83
Finally, a recent study showed an interesting effect of CO2 pressure on the formation of propanol.118 There are only a few examples of CO2R investigations at elevated pressure,119–123 but none before the case described here showed the formation of high amounts of C3+ products, although increased surface CO coverage, which can be potentially obtained by increasing the CO2 pressure, was shown to increase C3 product formation, including propanol.124 The catalyst, developed by D. Voiry et al. was a dendritic Cu94Ag6 alloy with highly dispersed Ag atoms and preferential CuAg(100) facets on its surface, which was obtained via co-electrodeposition under conditions that prevented galvanic replacement.118 They used a supersaturation strategy to prepare a 1 M CsHCO3 electrolyte containing dissolved CO2 at a concentration above the saturation limit via several steps of bubbling the solution with CO2 at a pressure of 10 bar. This led to the maximum content of 0.3 M CO2 in the supersaturated electrolyte at atmospheric pressure compared to 0.05 M by simply bubbling CO2 at 1 bar. Using an H-cell functioning at atmospheric pressure, it led to the remarkable and intriguing formation of 2-propanol with a record FE of 39.6% at an applied potential of −0.73 V vs. RHE (partial current density 12 mA cm−2). Interestingly, no C3 product could be detected using an H-cell and electrolyte CO2-saturated at 1 bar or using a flow cell or MEA electrolyzer, confirming the importance of elevated CO2 concentration for the formation of 2-propanol. A Cs-based electrolyte was also required. The selectivity increased further with an FE for 2-propanol of 56.7% at a specific current density of 59 mA cm−2 when the H-cell was functioning with a supersaturated solution and a CO2 pressure of 10 bar, a system proven to be stable for 200 h operation. Thus, the data established that the high FE for 2-propanol was derived from the combination of a supersaturated electrolyte, which favored CO coverage and C–C coupling reactions, and the presence of Ag, favoring the formation of isopropanol.
Operando Raman spectroscopy showed an increased CO coverage upon CO2 supersaturation, with an increased ratio between *CObridge (defect-like) and *COatop (terrace-like) sites. Operando FTIR also showed an increase in the *CO and *OCH2CH3 signals, in parallel to an increase in FEisopropanol, suggesting that a high density of these two intermediates triggered the formation of 2-propanol under CO2 supersaturation conditions. Finally, DFT calculations of the energies of the pathways derived from coupling the *OCH2CH3 intermediate to *CO showed that the selective formation of 2-propanol in the case of the CuAg alloy was due to the effect of adjacent Ag on increasing the C–O bond dissociation energy of the *O–CH2CH3 intermediate, resulting in the selective formation of 2-propanol.
Fig. 4, showing the partial current density as a function of applied potential derived from the data in Table 4, nicely shows that the doping strategy, specifically with Ag-doped Cu materials, currently offers the best impact with respect to the formation of propanol.
 | ||
| Fig. 4 Catalysts for CO2R to propanol in H-cells; partial current density as a function of applied cathode cell voltage. Data are given in Table 4. | ||
| Catalysts | Conditions | Partial current density | FE (%) | Ref. |
|---|---|---|---|---|
| Potential vs. RHE | ||||
| a In all other cases, the product is 1-propanol, and the Cu94Ag6 alloy produces only 2-propanol. | ||||
| Cu(100)-rich | H-cell | 9.3 mA cm−2 | 15 | 108 |
| Cu nanocubes | 0.25 M KHCO3 | −0.96 V | ||
| CuSx-DSV | H-cell | 3 mA cm−2 | 15.4 | 109 |
| 0.1 M KHCO3 | −1.05 V | |||
| Hex-2Cu–O | H-cell | 1.7 mA cm−2 | 18 | 112 |
| 0.1 M KHCO3 | −1.2 V | |||
| CuOD–Cu | H-cell | 4 mA cm−2 | 17.9 | 94 |
| 0.1 M KHCO3 | −0.94 V | |||
| R–Cu–C | H-cell | 8.2 mA cm−2 | 17.3 | 113 |
| 0.1 M KHCO3 | −1.05 V | |||
| Cu2O–Cu | H-cell | 6.8 mA cm−2 | 16.2 | 114 |
| 0.1 M KHCO3 | −1.4 V | |||
| Au0.02Cu0.98-NR (Au-doped Cu nanorods) | Flow cell | 12.6 mA cm−2 | 18 | 117 |
| 1.0 M KOH | −0.41 V | |||
| Cu94Ag6 alloya | H-cell (CO2 supersaturated) | 12 mA cm−2 | 39.6 (2-Propanol) | 118 |
| 1 M CsHCO3 | −0.73 V | |||
| P = 1 bar | 59 mA cm−2 | 56.7 (2-Propanol) | ||
| P = 10 bar | −0.7 V | |||
| Catalysts | Conditions | Partial current density | FE (%) | Ref. |
|---|---|---|---|---|
| Potential vs. RHE | ||||
| OD–Cu nanocavities | Flow cell | 7.77 mA cm−2 | 21 | 124 |
| 1.0 M KOH | −0.56 V | |||
| Cu2O NPs (multi hollow) | Flow cell | 12.84 mA cm−2 | 30.2 | 127 |
| 1.0 M KOH | −0.7 V | |||
| CuO adparticles | Flow cell | 11 mA cm−2 | 23 | 89 |
| 1.0 M KOH | −0.47 V | |||
| Fragmented Cu | Flow cell | 8.6 mA cm−2 | 20 | 128 |
| 1.0 M KOH | −0.45 V | |||
| Ag-doped Cu | Flow cell | 4.5 mA cm−2 | 33 | 85 |
| 1.0 M KOH | −0.46 V | |||
| AgRu-doped Cu | MEA | 111 mA cm−2 | 37 | 86 |
| 1.0 M KOH | ||||
| Ru-doped Cu NW | Flow cell | 10 mA cm−2 | 35.9 | 129 |
| 1.0 M KOH | −0.5 V | |||
| CuAg5%N | Flow cell | 67.5 mA cm−2 | 45 | 88 |
| 1.0 M CsOH | −1 V | |||
| Au-doped Cu nanosheets | Flow cell | 23.3 mA cm−2 | 46.6 | 95 |
| 1.0 M KOH | −0.58 V | |||
| Pb-doped Cu NPs | Flow cell | 17.86 mA cm−2 | 47 | 90 |
| 1.0 M KOH | −0.68 V | |||
| SnCu | MEA | 70.5 mA cm−2 | 47 | 130 |
| 3.0 M KOH |
Various strategies have been explored to stabilize and concentrate *C2 intermediates, favoring coupling to *CO or other *C1 adsorbed species and the formation of C3 compounds. However, in general, high selectivity for propanol is achieved at a low overpotential, and thus at a low current density. The first example involved the introduction of OD–Cu via the gentle acidic etching of Cu2O in nanocavities, allowing a confinement effect.124 In this case, using a flow cell with 1 M KOH electrolyte, a peak FEpropanol of 21% was obtained at −0.56 V vs. RHE (total current density of 37 mA cm−2), with decreased values at more cathodic potentials and larger current densities. This catalyst was unstable, undergoing reconstruction into aggregates during electrolysis, and thus becoming much less selective for propanol. In 2022, this strategy was further developed.127 Multi-hollow Cu2O nanoparticles containing nanocavities were synthesized via the reduction of copper acetate with hydrazine hydrate, followed by etching with HCl, and tested for COR in a flow cell using 1 M KOH as the electrolyte. The maximum FEpropanol of 30.2% was obtained at an applied current density of 42.5 mA cm−2. Based on this interesting finding, G. Wu and collaborators used a two-step tandem catalytic system (consisting of a first flow electrolyzer converting CO2 selectively into CO and a second flow cell using multi-hollow Cu2O nanoparticles at the cathode for electrolyzing the CO gas derived from the former) and achieved propanol formation from CO2 with an FE of 15.9%.125
The second example involved the introduction of adparticles, which are small clusters with a size of a few nm and possess a high population of low-coordinated sites, on the surface of metallic Cu.89 Adparticle growth on Cu could be achieved via the electroreduction of a Cu oxide precursor under a flow of CO. This rough surface was expected to increase the CO coverage and stabilize the *C2 intermediates, and DFT calculations established that it also allows lower energy barriers for *CO–*C2 (specifically *OCCOH and *CCH2 intermediates) coupling. Actually, this catalyst deposited on a GDL resulted in a peak FEpropanol of 23% at −0.47 V vs. RHE (partial current density of 11 mA cm−2), using a flow cell and 1 M KOH as the electrolyte. The role of adparticles in this selectivity was confirmed by the significant decrease in FEpropanol upon erasing the adparticle texture via thermal-annealing under N2 gas. The third example involved using fragmented Cu, exhibiting a high degree of distinct facet fragments, based on the fact that the *C1 intermediates are preferentially stabilized on Cu(111), while the *C2 intermediates are stabilized on Cu(100).131,132 Based on a Cu surface model with interfaces between the Cu(100) and Cu(111) domains, DFT calculations showed that this combination lowers the barriers of both *CO–*CO and *CO–*OCCO coupling reactions.128 This fragmented catalyst, with a large density of fragments of Cu(100) and Cu(111) that are adjacent to each other, and thus with an abundance of sites, where the two facets conjoin, could be obtained via CuO synthesis from cuprous iodide, a salt allowing slow nucleation and the generation of a variety of crystalline phases. Employing this catalyst in a flow cell and using 1 M KOH as the electrolyte, CuO was reduced to metallic Cu and an FEpropanol of 20% was obtained at −0.45 V vs. RHE (with a total current density of 43 mA cm−2).128 Less fragmented control samples, with less interfaces between the two types of facets, were less selective for propanol. Finally, it was shown that increasing the catalyst loading on a GDL potentially provides a way to stabilize and accumulate the *C2 intermediates before they diffuse out as C2 products, favoring their coupling to *CO and the formation of C3 products.133 Using commercial Cu nanoparticles, propanol production increased dramatically upon increasing the catalyst loading during COR in a flow cell with 1 M KOH electrolyte from 2% at 1.0 mg cm−2 to 20% at 10 mg cm−2 (partial current density 31 mA cm−2 at 4.0 V cell potential). A simulation confirmed an increase in the *C2 intermediate retention time in the catalyst layer as a function of catalyst loading.
Another recently developed strategy favoring C3 product formation from COR, specifically propanol, is doping Cu-based materials with one or two heavy metals. There are several examples of bi- and tri-metallic M- and M,M′-doped Cu catalysts reported in the literature exhibiting quite high FEpropanol. The stimulation of C3 product formation was ascribed to various effects of doping with main group metals possessing a large radius, such as Ag, Au and Pb. Due to the larger radius of these metals compared to that of Cu, doping may produce surface compressive strain and increase the number of low-coordinated sites within a defect-rich Cu structure.85,134 These sites are known to favour *CO surface coverage, and thus *CO–*CO and *CO–*C2 coupling.89,93,96 It was shown by DFT calculations that the diversity of Cu atoms environments is responsible for the asymmetric C–C coupling active sites, which decrease the energy barriers for *CO dimerization and the *CO–*OCCO coupling reaction.85 The calculations were performed for a series of dopants, Pd, Ru, Rh, Ag and Au, among which the largest effects on the reaction barriers were obtained for Ag doping. Other computational studies, based on a larger scope of the most stable computed C2 intermediates on Cu (100), confirmed that the presence of Ag or Au atoms on Cu results in a large decrease in the activation barriers of *C2–*CO coupling reactions.88,95 Furthermore, CuAg materials are prone to favor alcohols vs. hydrocarbons, which can be rationalized through the principle of lowering the *C affinity, given that Ag addition renders the surface more carbophobic.51
The first reported example of an Ag-doped Cu (atomic Ag percentage of 4%) COR catalyst, prepared by the galvanic exchange reaction between Cu nanoparticles and silver nitrate, allowed the formation of propanol with high selectivity (FE = 33%, partial current density of 4.5 mA cm−2) at −0.46 V vs. RHE in a flow cell reactor with 1 M KOH electrolyte.85 The selectivity decreased upon applying a more cathodic potential, indicating that C–C2 coupling becomes slower at high potential, favoring C2 protonation and the formation of C2 products such as ethylene. Later, the same authors slightly increased the FEpropanol to 37% using an Ru/Ag-doped Cu material, which was prepared via a two-step galvanic exchange between Cu and RuCl3, followed by AgNO3.86 This performance was obtained with an MEA electrolyzer using 1 M KOH as the anolyte, with an applied current density of 300 mA cm−2 associated with a full cell voltage of 2.75 V. Interestingly, this selectivity was maintained during long-term electrolysis (100 h). This result was supported by DFT calculations, showing that the addition of Ru to Ag-doped Cu was the most effective dopant in decreasing the energy barriers of *CO dimerization and *CO coupling to *OCCO compared to Au, Pd, Ni, Fe and Pt.
Following the same strategy, more recently, Ru–CuNW, CuO-derived Cu0 nanowires doped with Ru (1 at%) without Ru phase segregation, showed high selectivity for propanol with an FE of 35.9% at −0.5 V vs. RHE from COR (corresponding to a low partial density of about 10 mA cm−2) using a flow cell and 1 M KOH as the electrolyte.129 This selectivity was proposed notably by DFT calculations to derive from the combination of a low-coordinated Cu step surface favoring alcohol formation vs. ethylene,97 as shown by XAS analysis on the Cu edge of the activated catalyst, with the presence of a doped heavy metal displaying high CO affinity, thus favoring *C2 intermediate coupling to *CO for propanol formation. This agreed with the much lower FE for propanol with the undoped CuNW catalyst (FE = 22%). However, electrolysis at a more cathodic potential and higher current densities led to a decrease in the FE for propanol.
Although metal doping was mainly achieved on Cu NPs or OD–Cu (oxide-derived Cu), a recent study showed that silver or gold doping on copper nitride, Cu3N, resulted in significantly higher selectivity in COR for propanol.88 The CuAg5%N catalyst, consisting of Cu3N nanoparticles and nanorods doped with metallic Ag, was prepared via a galvanic replacement reaction using CuNPs and AgNO3, followed by calcination, and then a nitridation step, consisting of pyrolysis in the presence of NaNH2. Subsequently, it was used at the cathode of a flow cell using 1 M CsOH as the electrolyte, allowing COR reduction with a remarkable FEpropanol of 45% at a high applied current density of 150 mA cm−2 (−1 V vs. RHE), which was found to be stable over 9 h electrolysis. In situ XAS and XRD showed that under catalytic conditions, Cu3N was totally reduced to metallic Cu0, indicating that the catalytic species are nitride-derived copper (ND–Cu) sites. Although the pristine Cu NPs, OD–Cu and ND–Cu all possessed catalytic Cu0 sites, intriguingly ND–Cu was different from that derived from the two other materials, given that different product distributions in COR were obtained. Thus, further investigation is needed to better understand the specific local structure, surface coordination and electronic properties of the ND–Cu sites that make them unique in favoring propanol formation.
Gold is indeed another metal used for doping Cu. A recent study reported the propanol-selective activity of a Cu material doped with Au NPs.95 Au NPs (6.1 wt%) were homogeneously deposited on the surface of CuO nanosheets before electroreduction under CO. This led to the reconstruction of the material, during which Cu species migrated to the surface of Au NPs, leading to a disordered layer of Cu atoms around Au NPs. The detailed EXAFS analysis indicated a lower Cu–Cu coordination number on RCu/Au (R for reconstructed) compared to the control undoped RCu sample, indicating that Au doping is responsible for the presence of rich undercoordinated sites. Employing this catalyst and a flow cell with 1 M KOH as the electrolyte, an FEpropanol of 46.6% was obtained at −0.58 V vs. RHE with a total current density of 50 mA cm−2. The FE dropped to 25% at more cathodic potentials; however, leading to a partial current density for propanol formation at −0.78 V vs. RHE of 124 mA cm−2.
Finally, Pb-doped Cu NPs, obtained by the electrodeposition of Pb atoms onto oxide-derived Cu surfaces in the presence of CO, proved to be excellent catalysts for COR to propanol.90 SEM and TEM analysis showed that Pb deposition led to the formation of small grains and a surface rich in grain boundaries (GB), which are not observed on the control undoped Cu catalyst. A higher density of Pb atoms was present in the GB zones, suggesting that the Pb doping is the origin of the formation of GBs and the increased density of undercoordinated Cu sites, as confirmed by XAS. Furthermore, operando Raman spectroscopy measurements during COR showed stronger *CO binding on the Pb–Cu sample compared to the undoped Cu. Employing this Pb–Cu material, containing 8% Pb atoms, and using a flow cell and 1 M KOH electrolyte, a record FEpropanol of 47% was obtained at −0.68 V vs. RHE (current density 38 mA cm−2), while the highest value was 28% at a more cathodic potential in the case of the undoped OD–Cu. Variations in the Pb loading and CO partial pressure, as well as operando ATR-SEIRAS, clearly indicated that the improvement in propanol formation was well related to Pb favoring CO binding and enhancing the *CO coverage, thus favoring C–C coupling up to C3 products. A stable FEpropanol of 30% (current density of 76 mA cm−2) was obtained using an MEA electrolyzer during 110 h electrolysis.
During the revision of this article, an interesting study was reported, showing that metallic Cu doped with dispersed Sn atoms (Sn–Cu) was highly selective for propanol formation during COR using an MEA electrolyzer and 3.0 M KOH electrolyte (Table 5).130 An FEpropanol of 47% was obtained at an applied current density of 150 mA cm−2 and the system was proven to be stable for 120 h. This was consistent with DFT calculations establishing that the combination of Cu with Sn atoms favors C1–C2 coupling better than other combinations (Zn–Cu, Ga–Cu, In–Cu, Sb–Cu, Pb–Cu and Bi–Cu). Furthermore, owing to the construction of an Sn–Cu/carbon/ionomer heterojunction, propanol crossover through the membrane was greatly limited, achieving a high concentration of propanol (30 wt%) at the cathode after 120 h electrolysis, a concentration that minimizes the downstream separation cost.
Fig. 5 summarizes the best catalysts and partial current density for propanol from COR in flow cells as a function of applied potential, illustrating the superiority of metal-doped Cu materials, as is the case for CO2R to propanol (Fig. 4).
 | ||
| Fig. 5 Catalysts for COR to propanol in flow cells; partial current density as a function of applied cathode cell voltage. Data are given in Table 5. | ||
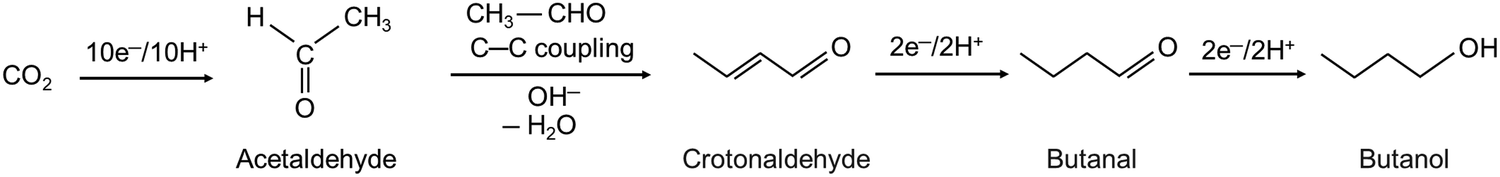 | ||
| Fig. 6 Proposed mechanism for butanol formation.72 | ||
However, recently, butanol formation was reported to be increased using copper phosphide, CuP2, as the catalyst.137 Using a flow cell with a 9.0 cm−2 electrode and 0.5 M KHCO3 as the electrolyte, the maximum FE of 3.9% was obtained at a very low applied potential of −0.6 V vs. RHE (resulting in a very low current density of <3.0 mA cm−2). XPS and HAADF-STEM-EDS (high-angle annular dark field scanning transmission electron microscopy energy-dispersive X-ray spectroscopy) indicated that copper phosphide was partially covered with phosphate and that phosphorous was partially leached, forming copper oxide after CO2R. Although no mechanistic studies were carried out, a mechanism was proposed for butanol production, which was very similar to that proposed for Ni- and Fe-phosphide-based CO2R to C3 and C4 products (Fig. 1). In contrast to the usual Cu-based catalysts, the vibrational bands associated with adsorbed *CO could not be observed, thus excluding the involvement of CO–CO coupling during the formation of butanol. CuP2, with highly oxophilic surfaces due to the presence of P atoms, is likely to favor the conversion of CO2 to formate rather than CO as the first reaction step. The reduction of formate to formaldehyde and condensation of two molecules of formaldehyde, followed by water elimination, can generate acetaldehyde. Then, the reaction proceeds via the mechanism shown in Fig. 6, i.e., the aldol condensation of two molecules of acetaldehyde, generating crotonaldehyde, which is finally reduced to butanol. Accordingly, the electroreduction of formate under identical conditions was shown to produce acetaldehyde, while the electroreduction of acetaldehyde produced 1-butanol.
Following these studies, several reports showed the potential of bimetallic M–Cu materials to catalyze the electroreduction of CO2R to butanol with larger FEs (Table 6). The first one described an amorphous CuTi alloy, in which Ti is expected to transfer electrons to and increase the electron density of the coordinatively unsaturated Cu active sites.138 Given that Ti is prone to favor HER over CO2R, the surface Ti is further dealloyed in a dilute HF solution, forming the a-CuTi@Cu catalyst. Employing this catalyst, the highest FE value for butanol (FE = 6.85%) was obtained in an H-cell with 0.1 M KHCO3 as the electrolyte at an applied potential of −0.8 V vs. RHE, the major products being H2 and ethanol (FE = 24%), together with a C3 product, namely acetone (FE = 11,14%) and small amounts of C1 products (mainly methanol). Although DFT calculations showed that subsurface Ti atoms favored C–C coupling on the Cu active sites, no insight was presenting into why this catalyst allowed the relatively high formation of a C4 compound.
| Catalysts | Conditions | Partial current density | Products | FE (%) | Ref. |
|---|---|---|---|---|---|
| Potential vs. RHE | |||||
| OD–Cu | Flow cell | 0.08 mA cm−2 | 1-Butanol | 0.056% | 72 |
| 1.0 M KOH | −0.48 V | ||||
| CuP2 | Flow cell | <0.12 mA cm−2 | 1-Butanol | 3.9% | 137 |
| 0.5 M KHCO3 | −0.6 V | ||||
| CuTi | H-cell | 2 mA cm−2 | 1-Butanol (+acetone) | 6.85% (11.14%) | 138 |
| 0.1 M KHCO3 | −0.8 V | ||||
| Cu-Ir | H-cell | 0.21 mA cm−2 | Tert-Butanol (+acetone) | 14.8% (5.0%) | 139 |
| 0.1 M KHCO3 | −0.57 V |
The second study used a Cu0.48Ir0.52 alloy, Cu–Ir, as shown in Table 6.139 This catalyst, in an H-cell with 0.1 M KHCO3 electrolyte mainly generated H2 (FE = 67%), together with C1 products (methanol and formic acid accounting for FE = 14%), but most remarkably acetone and tert-butanol. Under the optimized applied potential of 0.57 V vs. RHE, with a total current density of −1.4 mA cm−2, tert-butanol was obtained with an FE of 14.8%, a current record, and acetone with FE = 5%. The formation of acetone and tert-butanol requires 16- and 24-electron transfer, respectively. It was proposed that acetone is an intermediate towards tert-butanol via CO insertion into adsorbed acetone, given that the addition of acetone to the electrolyte during electrolysis increased the production of tert-butanol. DFT calculations showed that this reaction is thermodynamically favorable. Thus, the reaction was proposed to proceed via *CO–*CO coupling, followed by a series of proton-coupled electron transfers, generating the CH3CH2O* intermediate. The presence of an oxophilic metal (Ir) favors binding of the intermediates via the O atom and stabilizes the C–O bond. The oxygen atom is retained during CO insertion and further reduction leads to adsorbed acetone. At this stage, either acetone is released in solution or it further reacts with a fourth molecule of CO to generate tert-butanol after the elimination of water and reduction (see Fig. 7).
 | ||
| Fig. 7 Proposed mechanism for the formation of tert-butanol catalyzed by Cu0.48Ir0.52 alloy.139 | ||
2.3. C3+ hydrocarbon formation via CO2R (Table 7)
Propylene is an important chemical feedstock, in particular for the polymer industry, reaching an annual global capacity of about 150 Mt; however, with a parallel emission of about 100 Mt of CO2, given that its current production is entirely derived from crude oil. Propane is used as a fuel source for ovens and furnaces in the production of glass, ceramics, and other materials as well as for boilers in the manufacturing of paper products, textiles, and plastics. CO2RR and COR using renewable electricity may be an interesting alternative for the production of these C3 hydrocarbons. However, propylene and propane are rarely observed during CO2R and COR using Cu-based catalysts. Here, we describe the most important systems showing this capability.Using Cu nanocubes and an MEA-type electrolyzer, CO2R at an applied current density of 200 mA cm−2 resulted in the production of H2 and ethylene as the major products together with very small amounts of propylene (jC3H4 = 1.5 mA cm−2 and FE < 1%).140
Using a biphasic Cu2O–Cu catalyst and KCl electrolyte during 1 h electrolysis in an H-cell, in addition to the usual C2 products ethylene and ethanol (total FE 45%), not only a relatively high FE of 8.7% for propanol was obtained but also low amounts of propane and propylene (FE = 1%), and for the first time, butane, a C4 product, with a FE of 0.9%.71 However, during longer electrolysis, productivity of the catalyst towards C3–C4 products declined with time. This was correlated with the decline in the surface Cu+/Cu0 ratio, as shown by XPS analysis and operando Cu K-edge XANES, suggesting the importance of the Cu+ sites for promoting the formation of hydrocarbons with long chains. Furthermore, it was shown that the KCl electrolyte played a critical role in slowing down the reduction of the oxide to the metallic phase. It was proposed that Cl adsorption stabilized the Cu2O phase, allowing higher binding energy for CO adsorption and increasing the CO coverage, which is known to favor C–C coupling reactions. The stronger CO adsorption on a surface containing Cu+ sites has been studied theoretically and experimentally, as well as the increase in CO binding due to the presence of ad-atoms such as Cl.141–143
More recently, the significant production of ethylene was reported during CO2R using Cu nanocrystals (CuNanoCs) as catalysts and an alkaline flow cell.144 The surface of CuNanoCs predominantly consisted of Cu(100) and Cu(111) facets, with a distribution that remained unchanged during long-term electrolysis, despite the considerable reconstruction of their morphology. As mentioned earlier, the presence of both types of facets is an interesting property given that Cu(100) was shown to favour the propagation of carbon chains,74 while its coexistence with Cu(111) provides sites stabilizing the key intermediates for multi-carbon products.128 CO2R at −0.6 V vs. RHE led to the formation of ethylene (FE = 50%) and ethanol (FE = 15%) as the major CO2-derived products, together with propanol (FE = 4%) and propylene accounting for an FE of 1.4%. A partial current density of 5.5 mA cm−2 was obtained at a slightly lower applied potential (−0.65 V vs. RHE). Surprisingly, only trace amounts of propylene (FE = 0.06%) could be detected during COR under similar electrolysis conditions, while ethylene was still produced with a high FE. This led to the intriguing consideration that propylene was unlikely to be derived from surface *CO coupling to *C2 intermediates and that the active *C1 intermediate was missing in CO electrolysis. This led to the hypothesis that the key *C1 intermediate involved in the propylene pathway was either adsorbed CO2 or *COOH. This is in contrast with the propanol pathway in which the C3 backbone is formed via *CO–*C2 coupling,84 in agreement with the observation that the formation of propanol was less affected by the change in the feed gas. The working mechanistic hypothesis for propylene formation was nicely substantiated by isotopic labeling experiments using various mixtures of 12CO2/13CO2 and 12CO/13CO. Notably, using the 13CO2/12CO = 20%/80% mixture, the majority of the formed ethylene had two 12C atoms, while the majority of formed propylene had two 12C atoms and one 13C atom, in agreement with the hypothesis that propylene arises from the coupling of 13CO2 or *13COOH intermediates with the *C2 species that are produced from 12COR and are precursors of ethylene (and ethanol). The reason why the *C3 intermediate, derived from the coupling of three *CO molecules, involved in the propanol pathway does not produce propylene may be kinetic. Interestingly, the same study evaluated a broad library of monometallic, bimetallic and trimetallic Cu-based catalysts (a total of 20 different materials) and none gave FE larger than 1.8% for propylene formation.128
An interesting report showed that bismuth-doped copper nanowires were active for the formation of propane.145 Cu foam was converted into Cu nanowires (NWs), on which Bi was electrodeposited using different deposition times. Characterization of the material showed that the Cu foam was covered with Cu2O NWs and Bi particles. Using a filter-press cell working at constant current density of −45 mA cm−2 with 0.1 M KHCO3 or 0.45 M KHCO3 + 0.5 M KCl as the catholyte, different products were obtained. In the second case, the major product was formate (FE = 86%) and HER (FEH2 = 10%) was limited. In the first case, HER increased slightly and formate decreased to 5–30%, depending on the Bi electrodeposition time. However, the most interesting and intriguing observation was the production of a C3 hydrocarbon, specifically propane, with very high selectivity, where the highest amount (FE = 85%) was obtained with the sample derived from the longest Bi electrodeposition time. This gave a record partial current density for propane of 38 mA cm−2. Unfortunately, the origin of this selectivity was not studied, and thus this material clearly deserves further investigation.
Finally, propane has also been observed during CO2R with a substantial FE of 3.3% using a Cu2O/MXene catalyst, in which Cu2O nanoparticles were deposited on titanium carbide (Ti3C2Tx), a 2D material known as MXene.146 These results were obtained using an H-cell with a CO2-saturated 0.1 M KHCO3 catholyte, at a cathodic potential of −1.3 V vs. RHE (current density of about 35 mA cm−2). A much lower FE of 0.1% was obtained using the Cu/MXene catalyst, whereas no propane could be detected using Cu-free MXene. In all cases, the major products were CO and H2. The importance of combining Cu2O and MXene for propane formation was rationalized by DFT calculations, where Cu2O and MXene stabilize the *C2 and *CO intermediates, respectively, thus favoring *C2–*CO coupling at the interface of the two components.
3. Conclusion
The deep analysis of the current knowledge regarding CO2R and COR to C3+ compounds provided in this review leads to the following notable observations and perspectives.(1) Further investigation of the potential of CO2R/COR to generate C3+ products
Indeed, it is possible to generate a variety of C3+ compounds via CO2R and COR, with quite high selectivity, the most common being propanol (Tables 4 and 5). For example, C4 molecules such as butanol, methylglyoxal and furandiol are sometimes formed with reasonable selectivity (Tables 3 and 6). Although rare, C5 and C6 compounds have also been observed; however, with very low current densities and faradaic efficiencies. Except in a few cases, as discussed below, oxygenated compounds (alcohols and acetone) are generally more abundant than hydrocarbons, although the remarkable formation of propane, with quite high faradaic efficiency, has been reported; however, without being reproduced in the literature thus far (Tables 3 and 7).38,145| Catalysts | Conditions | Applied total current | Products | FE (%) | Ref. |
|---|---|---|---|---|---|
| Potential vs. RHE | |||||
| Cu nanocubes | MEA | 200 mA cm−2 | Propylene | <1 | 140 |
| Cu2O–Cu | H-cell | 7 mA cm−2 | Propanol | 8.7 | 71 |
| Propane | 0.9 | ||||
| 0.1 M KCl | −1.6 V | Propylene | 1 | ||
| Butane | 0.9 | ||||
| CuNanoCs | Flow cell | 0.6 A cm−2 | Propanol | 4 | 144 |
| 1.0 M KOH | −0.6 V | Propylene | 1.4 | ||
| 0.2 M CsI | Allyl alcohol | 0.5 | |||
| Bi–Cu2O nanowire | Filter-press cell | 45 mA cm−2 | Propane | 85 | 145 |
| 0.1 M KHCO3 | |||||
| Cu2O/Ti3C2TxMXene | H-cell | 35 mA cm−2 | Propane | 3.3 | 146 |
| 0.1 M KHCO3 | −1.3 V |
These results are summarized in Table 2, showing the most industrially relevant C3 and C4 products and highlighting the potential of CO2R and COR for alcohols and alkanes synthesis. Presently, alcohols are important commodity chemicals. Beyond their current uses, new high-volume applications such as precursors for the production of synthetic aviation fuels may arise in the future. Alkanes, such as propane and butane, have the highest market volume to date due to their use as fuels. However, this might be reduced in the future because of the electrification of the applications using propane and butane as fuel today. In the case of olefins, although they can be synthesized from CO2R or COR derived alkanes, such as propylene from propane, via thermal dehydrogenation, direct CO2R or COR to propylene would be much preferred. This will result in lower capital expenditures compared to pathways relying on CO2 hydrogenation using electrolytic H2. It should be noted that in some cases, small amounts of propylene were observed (Tables 3 and 7), justifying further studies aimed at improving the corresponding catalysts. Table 2 also shows that butadiene is the C4 olefin with the highest market volume, larger than that of butene. However, thus far, there is no study reporting butadiene formation from CO2R or COR. These considerations indicate the potential for further optimizing catalysts for C3+ alcohols, propane and propylene formation.
(2) Re-evaluating non-Cu materials as catalysts for CO2R/COR
It is timely and worth reinvestigating non-Cu metals, in particular Mo, Fe and Ni, as catalysts for these reactions. However, these metals suffer from strong limitations, the most important one being their low productivity in general. As shown in Table 1, C3+ compounds are mostly formed at quite low overpotentials, and thus with very low current densities in most cases. Indeed, increased polarization results in the increased production of H2, which is greatly favored on the surface of these metals because they are prone to adsorbing H atoms from protons and providing hydridic surfaces, where the reactivity of the latter with protons exceeds that with CO2. Furthermore, they display too strong *CO binding, which favors HER.52 Thus, efforts should be devoted to weakening the *CO adsorption energy on the surface of these non-Cu metals. One interesting illustration of the possibility to allow Fe- and Ni-based materials to efficiently catalyze CO2 activation is the case of Fe- and Ni-based single-atom catalysts (SACs).147–149 They exhibit much lower *CO binding strength compared to their bulk metal counterparts; however, too low that they can catalyze CO2R to CO exclusively with almost FECO = 100%, with no further conversion to C2+ products. Further research on Fe- and Ni-based catalysts should focus on tuning their *CO binding energy to achieve mild strength comparable to that on Cu. Presently, there is no clear trend regarding the correlation between structure/morphology and selectivity/productivity with this class of catalysts, and thus no rationale pointing to obvious targets. Therefore, a large variety of potential catalysts should be explored. Whether artificial intelligence and machine learning can play a role in this respect is an open question.150–152(3) Exploring surface modification of non-Cu catalysts
Mo3P-Im is a unique catalyst for CO2R to propane with very high selectivity (FE = 91%) and high current density (390 mA cm−2).38 It is interesting to note that this is the only example of a non-Cu material whose activity and selectivity has been tuned by surface molecular modification, a strategy that has been largely and successfully used for improving the performances of Cu-based catalysts.153,154 This strategy can be used more systematically in the case of non-Cu metals for improving the selectivity, in particular with modifications preventing HER, and thus allowing the higher productivity of CO2-derived compounds at larger current densities.(4) Gaining deeper insight into the reaction mechanisms in the case of non-Cu catalysts
Reactivity studies coupled with spectroscopic and microscopic characterization of catalysts under in situ and operando conditions and DFT calculations, in the case of simple catalysts whose modelling is accessible, and despite the multitude of possible intermediates, may give deeper insight into the mechanisms, which are still far from understood. Interestingly, the few studies available seem to suggest mechanisms significantly different from that proposed and partly established for Cu-based catalysts, thus involving different reactive intermediates such as formaldehyde and acetaldehyde in the case of Ni- and Fe-based materials.40,46,56 Indeed, given that C–C coupling occurs at potentials excluding CO2 activation, it is very likely, as generally proposed, that the reaction does not proceed via *CO–*CO coupling. Instead, it might in some cases involve formaldehyde intermediate formation, followed by a series of thermal aldehyde condensation steps, as in the case of Ni- and Fe-phosphides. Based on this, one should consider future research on engineering (multi)metallic materials that combine low kinetic barriers for CO2R to formic acid, and then to formaldehyde with catalytic activity for thermal aldehyde self-condensation. Thus far, although Ru-, Co-, Ni- and Fe-based materials show some catalytic potential for the latter, there are exceedingly few good systems for producing formaldehyde from CO2R or COR.155–159(5) Further investigating multi-metallic Cu-based catalysts
Cu is still the metal of choice and has been extensively studied during the last 20 years. Tuning its morphology and surface as well as the reaction conditions (pressure, electrolyte, and flow rate) has recently led to improved selectivity in particular for propanol formation via both CO2R and COR. Propanol is the most important C3+ product in Cu-based CO2R and COR, where in the case of CO2R, there are still very few Cu-based systems achieving FEpropanol exceeding 15%, while the record FEpropanol of almost 50% has been obtained via COR. Improvements have been obtained through strategies aimed at stabilizing the *C2 intermediates to allow *CO–*C2 coupling including confinement effects owing to the introduction of nanocavities, facet engineering, or tuning the catalyst layer and the CO2 pressure. The most effective way to favor propanol formation, both in CO2R and COR, is consistently via the introduction of dopants (Ag, Au, and Pb), which leads to *C2 stabilization and favors *CO–*C2 coupling, as supported by DFT calculations, owing to the surface compressive strain and increase in low-coordination sites and grain boundaries. These initial studies should encourage the further extensive exploration of combinations of metal dopants on Cu-based materials towards not only bimetallic but also polymetallic materials for propanol formation via CO2R and COR.(6) Further studies on the selectivity for oxygenates/hydrocarbons
Cu-based materials promoting the formation of C3+ hydrocarbons are still lacking, with only a few exceptions, such as the Bi-doped Cu catalyst described above with a unique FE value of 85% for propane.145 Also, it is still necessary to better understand how to control the selectivity for oxygenates/hydrocarbons to favour the formation of propylene and propane, which have been observed only rarely and with low selectivity in general. The other limitation of Cu-based catalysts is the general absence of C4 compounds, with the exception of tert-butanol formation via CO2R, with the maximum FE of 14.8% in the case of a Cu–Ir alloy material,139 again suggesting the prevalence of alcohols over hydrocarbons. It is very likely that this is due to the impossibility thus far to engineer Cu surfaces to stabilize the *C3 intermediates and allow them to couple with *CO towards C4 products before the C3 products desorb from their surface. It should be noted that no C4+ products have been reported during COR.(7) Investigating tandem pathways
Although the formation of C3+ compounds via CO2R and COR provides an opportunity to valorize CO2 into industrially relevant products, the scope of compounds is still limited to C3 and C4 derivatives. It is not unlikely that producing more complex molecules with a larger carbon number will be significantly more difficult. Indeed, an increase in the number of electrons and protons to be transferred results in lower reaction kinetics and an increased number of possible products, making it difficult to achieve high partial current densities and faradaic efficiencies for a target product, and thus limiting the industrial interest towards the direct synthetic approach. Under these conditions, CO2-derived complex molecules should preferentially be synthesized via a tandem scenario, converting CO2R/COR-derived C2/C3 products via mature and inexpensive thermochemical processes for reactions such as oligomerization and aromatization.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by TotalEnergies SE.References
- Y. Li, F. Zhu, E. Liu, H. Ouyang, W. Lu, H. Gu, J. Ren, W. Peng, H. Hou and Y. He, Adv. Compos. Hybrid Mater., 2024, 7, 147 CrossRef.
- M. N. Anwar, A. Fayyaz, N. F. Sohail, M. F. Khokhar, M. Baqar, A. Yasar, K. Rasool, A. Nazir, M. U. F. Raja, M. Rehan, M. Aghbashlo, M. Tabatabaei and A. S. Nizami, J. Environ. Manage., 2020, 260, 110059 CrossRef CAS PubMed.
- S. C. Peter, ACS Energy Lett., 2018, 3, 1557–1561 CrossRef CAS.
- E. T. C. Vogt and B. M. Weckhuysen, Nature, 2024, 629, 295–306 CrossRef CAS PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- R. G. Grim, J. R. Ferrell Iii, Z. Huang, L. Tao and M. G. Resch, Joule, 2023, 7, 1684–1699 CrossRef CAS.
- X. Tan, C. Yu, Y. Ren, S. Cui, W. Li and J. Qiu, Energy Environ. Sci., 2021, 14, 765–780 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- G. Leonzio, A. Hankin and N. Shah, Chem. Eng. Res. Des., 2024, 208, 934–955 CrossRef CAS.
- W. Lai, Y. Qiao, J. Zhang, Z. Lin and H. Huang, Energy Environ. Sci., 2022, 15, 3603–3629 RSC.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- D. Wu, F. Jiao and Q. Lu, ACS Catal., 2022, 12, 12993–13020 CrossRef CAS.
- T. N. Nguyen and C.-T. Dinh, Chem. Soc. Rev., 2020, 49, 7488–7504 RSC.
- J. Han, X. Bai, X. Xu, X. Bai, A. Husile, S. Zhang, L. Qi and J. Guan, Chem. Sci., 2024, 15, 7870–7907 RSC.
- X. Zhang, S.-X. Guo, K. A. Gandionco, A. M. Bond and J. Zhang, Mater. Today Adv., 2020, 7, 100074 CrossRef.
- D. Ewis, M. Arsalan, M. Khaled, D. Pant, M. M. Ba-Abbad, A. Amhamed and M. H. El-Naas, Sep. Purif. Technol., 2023, 316, 123811 CrossRef CAS.
- Z. Liu, J. Qian, G. Zhang, B. Zhang and Y. He, Sep. Purif. Technol., 2024, 330, 125177 CrossRef CAS.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2023, 6, eaay3111 CrossRef PubMed.
- B. Chang, H. Pang, F. Raziq, S. Wang, K.-W. Huang, J. Ye and H. Zhang, Energy Environ. Sci., 2023, 16, 4714–4758 RSC.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- B. Li, L. Liu, M. Yue, Q. Niu, M. Li, T. Zhang, W. Xie and Q. Wang, Green Chem., 2024, 26, 103–121 RSC.
- Z. Sun, Y. Hu, D. Zhou, M. Sun, S. Wang and W. Chen, ACS Energy Lett., 2021, 6, 3992–4022 CrossRef CAS.
- Y. Yan, L. Ke, Y. Ding, Y. Zhang, K. Rui, H. Lin and J. Zhu, Mater. Chem. Front., 2021, 5, 2668–2683 RSC.
- L. Xie, Y. Jiang, W. Zhu, S. Ding, Y. Zhou and J.-J. Zhu, Chem. Sci., 2023, 14, 13629–13660 RSC.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS PubMed.
- C. Chen, Y. Li and P. Yang, Joule, 2021, 5, 737–742 CrossRef.
- A. Ozden, Y. Wang, F. Li, M. Luo, J. Sisler, A. Thevenon, A. Rosas-Hernández, T. Burdyny, Y. Lum, H. Yadegari, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, Joule, 2021, 5, 706–719 CrossRef CAS.
- N. S. Romero Cuellar, K. Wiesner-Fleischer, M. Fleischer, A. Rucki and O. Hinrichsen, Electrochim. Acta, 2019, 307, 164–175 CrossRef CAS.
- M. Jouny, G. S. Hutchings and F. Jiao, Nat. Catal., 2019, 2, 1062–1070 CrossRef CAS.
- Y. Ji, A. Guan and G. Zheng, Cell Rep. Phys. Sci., 2022, 3, 101072 CrossRef CAS.
- J. Sisler, S. Khan, A. H. Ip, M. W. Schreiber, S. A. Jaffer, E. R. Bobicki, C.-T. Dinh and E. H. Sargent, ACS Energy Lett., 2021, 6, 997–1002 CrossRef CAS.
- A. Perazio, C. E. Creissen, J. G. Rivera de la Cruz, M. W. Schreiber and M. Fontecave, ACS Energy Lett., 2023, 8, 2979–2985 CrossRef CAS.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- X. Zhong, H.-J. Peng, C. Xia and X. Liu, J. Mater. Chem. A, 2024, 12, 19663–19684 RSC.
- S. J. Raaijman, M. P. Schellekens, P. J. Corbett and M. T. M. Koper, Angew. Chem., Int. Ed., 2021, 60, 21732–21736 CrossRef CAS PubMed.
- R. Kortlever, I. Peters, C. Balemans, R. Kas, Y. Kwon, G. Mul and M. T. M. Koper, Chem. Commun., 2016, 52, 10229–10232 RSC.
- S. A. Francis, J. M. Velazquez, I. M. Ferrer, D. A. Torelli, D. Guevarra, M. T. McDowell, K. Sun, X. Zhou, F. H. Saadi, J. John, M. H. Richter, F. P. Hyler, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, Chem. Mater., 2018, 30, 4902–4908 CrossRef CAS.
- M. Esmaeilirad, Z. Jiang, A. M. Harzandi, A. Kondori, M. Tamadoni Saray, C. U. Segre, R. Shahbazian-Yassar, A. M. Rappe and M. Asadi, Catalysts, 2021, 11, 330 CrossRef.
- A. R. Paris and A. B. Bocarsly, ACS Catal., 2017, 7, 6815–6820 CrossRef CAS.
- K. U. D. Calvinho, A. B. Laursen, K. M. K. Yap, T. A. Goetjen, S. Hwang, N. Murali, B. Mejia-Sosa, A. Lubarski, K. M. Teeluck, E. S. Hall, E. Garfunkel, M. Greenblatt and G. C. Dismukes, Energy Environ. Sci., 2018, 11, 2550–2559 RSC.
- M. Dhiman, Y. Chen, Y. Li, A. B. Laursen, K. U. D. Calvinho, T. G. Deutsch and G. C. Dismukes, J. Mater. Chem. A, 2023, 11, 717–725 RSC.
- M.-G. Kim, Y. Choi, E. Park, C.-H. Cheon, N.-K. Kim, B. K. Min and W. Kim, ACS Appl. Energy Mater., 2020, 3, 11516–11522 CrossRef CAS.
- Y. Zhou, A. J. Martín, F. Dattila, S. Xi, N. López, J. Pérez-Ramírez and B. S. Yeo, Nat. Catal., 2022, 5, 545–554 CrossRef CAS.
- S. P. Cronin, S. Dulovic, J. A. Lawrence, K. A. Filsinger, A. P. Hernandez-Gonzalez, R. Evans, J. W. Stiles, J. Morris, I. Pelczer and A. B. Bocarsly, J. Am. Chem. Soc., 2023, 145, 6762–6772 CrossRef CAS PubMed.
- Y. J. Kim, J. Y. Maeng, S. Y. Hwang, J. H. Yang, I. Yoon, C. W. Myung, C. K. Rhee and Y. Sohn, Nano Energy, 2023, 118, 108995 CrossRef CAS.
- K. U. D. Calvinho, A. W. Alherz, K. M. K. Yap, A. B. Laursen, S. Hwang, Z. J. L. Bare, Z. Clifford, C. B. Musgrave and G. C. Dismukes, J. Am. Chem. Soc., 2021, 143, 21275–21285 CrossRef CAS PubMed.
- P. Preikschas, J. Zhang, R. R. Seemakurthi, Z. Lian, A. J. Martín, S. Xi, F. Krumeich, H. Ma, Y. Zhou, N. López, B. S. Yeo and J. Pérez-Ramírez, Adv. Energy Mater., 2024, 14, 2401447 CrossRef CAS.
- H. Mahmoudi, M. Mahmoudi, O. Doustdar, H. Jahangiri, A. Tsolakis, S. Gu and M. LechWyszynski, Biofuels Eng., 2017, 2, 11–31 CrossRef.
- Z. Teimouri, N. Abatzoglou and A. K. Dalai, 2021, 11.
- X. Zhang, S. Hua, L. Lai, Z. Wang, T. Liao, L. He, H. Tang and X. Wan, RSC Adv., 2022, 12, 17959–17983 RSC.
- H.-J. Peng, M. T. Tang, J. Halldin Stenlid, X. Liu and F. Abild-Pedersen, Nat. Commun., 2022, 13, 1399 CrossRef CAS PubMed.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2014, 6, 320–324 CrossRef CAS PubMed.
- D. A. Torelli, S. A. Francis, J. C. Crompton, A. Javier, J. R. Thompson, B. S. Brunschwig, M. P. Soriaga and N. S. Lewis, ACS Catal., 2016, 6, 2100–2104 CrossRef CAS.
- P. Bui, J. A. Cecilia, S. T. Oyama, A. Takagaki, A. Infantes-Molina, H. Zhao, D. Li, E. Rodríguez-Castellón and A. Jiménez López, J. Catal., 2012, 294, 184–198 CrossRef CAS.
- S. Banerjee, A. Kakekhani, R. B. Wexler and A. M. Rappe, ACS Catal., 2021, 11, 11706–11715 CrossRef CAS.
- R. E. Vos and M. T. M. Koper, ACS Catal., 2024, 14, 4432–4440 CrossRef CAS PubMed.
- J. Y. Maeng, S. Y. Hwang, Y. J. Kim, I. Yoon, C. W. Myung, C. K. Rhee and Y. Sohn, J. Phys. Chem. C, 2023, 127, 11448–11461 CrossRef CAS.
- J. Y. Maeng, S. Y. Hwang, C. K. Rhee and Y. Sohn, Appl. Surf. Sci., 2023, 631, 157576 CrossRef CAS.
- X. Liu, Y. Hou, F. Yang, Y. Liu, H. Yu, X. Han, J. Chen, S. Chen, S. Zhou, S. Deng and J. Wang, Carbon, 2023, 201, 460–466 CrossRef CAS.
- L. Ji, L. Li, X. Ji, Y. Zhang, S. Mou, T. Wu, Q. Liu, B. Li, X. Zhu, Y. Luo, X. Shi, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2020, 59, 758–762 CrossRef CAS PubMed.
- L. Ji, L. Chang, Y. Zhang, S. Mou, T. Wang, Y. Luo, Z. Wang and X. Sun, ACS Catal., 2019, 9, 9721–9725 CrossRef CAS.
- P. Chen, P. Zhang, X. Kang, L. Zheng, G. Mo, R. Wu, J. Tai and B. Han, J. Am. Chem. Soc., 2022, 144, 14769–14777 CrossRef CAS PubMed.
- S. Hanselman, M. T. M. Koper and F. Calle-Vallejo, ACS Energy Lett., 2018, 3, 1062–1067 CrossRef CAS.
- Y. Ji, X. Lv, R. Wei, A. Guan, C. Yang, Y. Yan, M. Kuang and G. Zheng, Angew. Chem., Int. Ed., 2024, 63, e202411194 Search PubMed.
- W. Zhang, J. Ai, T. Ouyang, L. Yu, A. Liu, L. Han, Y. Duan, C. Tian, C. Chu, Y. Ma, S. Che and Y. Fang, J. Am. Chem. Soc., 2024, 146, 28214–28221 CAS.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- H.-T. Kim, J. Park, J. Mun, H. Shin, D.-H. Roh, J. Kwon, S. Kim, S.-J. Kim, G. Lee, S. J. Kang and T.-H. Kwon, ACS Catal., 2024, 14, 10392–10402 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- A. H. M. da Silva, G. Karaiskakis, R. E. Vos and M. T. M. Koper, J. Am. Chem. Soc., 2023, 145, 15343–15352 CrossRef CAS PubMed.
- S. Lee, D. Kim and J. Lee, Angew. Chem., Int. Ed., 2015, 54, 14701–14705 Search PubMed.
- L. R. L. Ting, R. García-Muelas, A. J. Martín, F. L. P. Veenstra, S. T.-J. Chen, Y. Peng, E. Y. X. Per, S. Pablo-García, N. López, J. Pérez-Ramírez and B. S. Yeo, Angew. Chem., Int. Ed., 2020, 59, 21072–21079 CrossRef CAS PubMed.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS PubMed.
- H. Peng, M. T. Tang, X. Liu, P. Schlexer Lamoureux, M. Bajdich and F. Abild-Pedersen, Energy Environ. Sci., 2021, 14, 473–482 RSC.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS.
- I. V. Chernyshova, P. Somasundaran and S. Ponnurangam, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9261–E9270 CrossRef CAS PubMed.
- S. Jiang, K. Klingan, C. Pasquini and H. Dau, J. Chem. Phys., 2018, 150, 041718 CrossRef PubMed.
- X. Li, S. Wang, L. Li, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2020, 142, 9567–9581 CAS.
- C. Zhan, F. Dattila, C. Rettenmaier, A. Herzog, M. Herran, T. Wagner, F. Scholten, A. Bergmann, N. López and B. Roldan Cuenya, Nat. Energy, 2024, 9, 1485–1496 CrossRef CAS PubMed.
- J. Santatiwongchai, K. Faungnawakij and P. Hirunsit, ACS Catal., 2021, 11, 9688–9701 CrossRef CAS.
- M. T. Tang, H.-J. Peng, J. H. Stenlid and F. Abild-Pedersen, J. Phys. Chem. C, 2021, 125, 26437–26447 CrossRef CAS.
- S. Pablo-García, F. L. P. Veenstra, L. R. L. Ting, R. García-Muelas, F. Dattila, A. J. Martín, B. S. Yeo, J. Pérez-Ramírez and N. López, Catal. Sci. Technol., 2022, 12, 409–417 RSC.
- X. Wang, Z. Wang, T.-T. Zhuang, C.-T. Dinh, J. Li, D.-H. Nam, F. Li, C.-W. Huang, C.-S. Tan, Z. Chen, M. Chi, C. M. Gabardo, A. Seifitokaldani, P. Todorović, A. Proppe, Y. Pang, A. R. Kirmani, Y. Wang, A. H. Ip, L. J. Richter, B. Scheffel, A. Xu, S.-C. Lo, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 5186 CrossRef PubMed.
- X. Wang, P. Ou, A. Ozden, S.-F. Hung, J. Tam, C. M. Gabardo, J. Y. Howe, J. Sisler, K. Bertens, F. P. García de Arquer, R. K. Miao, C. P. O’Brien, Z. Wang, J. Abed, A. S. Rasouli, M. Sun, A. H. Ip, D. Sinton and E. H. Sargent, Nat. Energy, 2022, 7, 170–176 CrossRef CAS.
- X. Chang, A. Malkani, X. Yang and B. Xu, J. Am. Chem. Soc., 2020, 142, 2975–2983 CrossRef CAS PubMed.
- H. Phong Duong, J. G. Rivera de la Cruz, N.-H. Tran, J. Louis, S. Zanna, D. Portehault, A. Zitolo, M. Walls, D. V. Peron, M. W. Schreiber, N. Menguy and M. Fontecave, Angew. Chem., Int. Ed., 2023, 62, e202310788 CrossRef CAS PubMed.
- J. Li, F. Che, Y. Pang, C. Zou, J. Y. Howe, T. Burdyny, J. P. Edwards, Y. Wang, F. Li, Z. Wang, P. De Luna, C.-T. Dinh, T.-T. Zhuang, M. I. Saidaminov, S. Cheng, T. Wu, Y. Z. Finfrock, L. Ma, S.-H. Hsieh, Y.-S. Liu, G. A. Botton, W.-F. Pong, X. Du, J. Guo, T.-K. Sham, E. H. Sargent and D. Sinton, Nat. Commun., 2018, 9, 4614 CrossRef PubMed.
- W. Niu, Z. Chen, W. Guo, W. Mao, Y. Liu, Y. Guo, J. Chen, R. Huang, L. Kang, Y. Ma, Q. Yan, J. Ye, C. Cui, L. Zhang, P. Wang, X. Xu and B. Zhang, Nat. Commun., 2023, 14, 4882 CrossRef CAS PubMed.
- H. Li, P. Wei, T. Liu, M. Li, C. Wang, R. Li, J. Ye, Z.-Y. Zhou, S.-G. Sun, Q. Fu, D. Gao, G. Wang and X. Bao, Nat. Commun., 2024, 15, 4603 CrossRef CAS PubMed.
- X. K. Lu, B. Lu, H. Li, K. Lim and L. C. Seitz, ACS Catal., 2022, 12, 6663–6671 CrossRef CAS.
- K. Jiang, Y. Huang, G. Zeng, F. M. Toma, W. A. Goddard, III and A. T. Bell, ACS Energy Lett., 2020, 5, 1206–1214 CrossRef CAS.
- C. Long, X. Liu, K. Wan, Y. Jiang, P. An, C. Yang, G. Wu, W. Wang, J. Guo, L. Li, K. Pang, Q. Li, C. Cui, S. Liu, T. Tan and Z. Tang, Sci. Adv., 2023, 9, eadi6119 CrossRef CAS PubMed.
- C. Long, K. Wan, Y. Chen, L. Li, Y. Jiang, C. Yang, Q. Wu, G. Wu, P. Xu, J. Li, X. Shi, Z. Tang and C. Cui, J. Am. Chem. Soc., 2024, 146, 4632–4641 CrossRef CAS PubMed.
- T. Cheng, H. Xiao and W. A. Goddard, J. Am. Chem. Soc., 2017, 139, 11642–11645 CrossRef CAS PubMed.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Joule, 2021, 5, 429–440 CrossRef CAS.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 Search PubMed.
- D. Ren, N. T. Wong, A. D. Handoko, Y. Huang and B. S. Yeo, J. Phys. Chem. Lett., 2016, 7, 20–24 Search PubMed.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. L. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS PubMed.
- D. Zhong, Z.-J. Zhao, Q. Zhao, D. Cheng, B. Liu, G. Zhang, W. Deng, H. Dong, L. Zhang, J. Li, J. Li and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 4879–4885 Search PubMed.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- A. H. M. da Silva, Q. Lenne, R. E. Vos and M. T. M. Koper, ACS Catal., 2023, 13, 4339–4347 CrossRef CAS PubMed.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, ACS Catal., 2017, 7, 1749–1756 CrossRef CAS.
- K. J. P. Schouten, Z. Qin, E. Pérez Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Phys. Chem. B, 2002, 106, 15–17 Search PubMed.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179–5182 CrossRef CAS PubMed.
- K. Jiang, R. B. Sandberg, A. J. Akey, X. Liu, D. C. Bell, J. K. Nørskov, K. Chan and H. Wang, Nat. Catal., 2018, 1, 111–119 Search PubMed.
- C. Peng, G. Luo, J. Zhang, M. Chen, Z. Wang, T.-K. Sham, L. Zhang, Y. Li and G. Zheng, Nat. Commun., 2021, 12, 1580 CrossRef CAS PubMed.
- Z. Weng, Y. Wu, M. Wang, J. Jiang, K. Yang, S. Huo, X.-F. Wang, Q. Ma, G. W. Brudvig, V. S. Batista, Y. Liang, Z. Feng and H. Wang, Nat. Commun., 2018, 9, 415 Search PubMed.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- B. Yang, L. Chen, S. Xue, H. Sun, K. Feng, Y. Chen, X. Zhang, L. Xiao, Y. Qin, J. Zhong, Z. Deng, Y. Jiao and Y. Peng, Nat. Commun., 2022, 13, 5122 CrossRef CAS PubMed.
- D. Tan, B. Wulan, J. Ma, X. Cao and J. Zhang, Chem. Catal., 2023, 3, 100512 CrossRef CAS.
- R. Zhang, J. Zhang, S. Wang, Z. Tan, Y. Yang, Y. Song, M. Li, Y. Zhao, H. Wang, B. Han and R. Duan, Angew. Chem., Int. Ed., 2024, 63, e202405733 CrossRef CAS PubMed.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- J. D. Lee, J. B. Miller, A. V. Shneidman, L. Sun, J. F. Weaver, J. Aizenberg, J. Biener, J. A. Boscoboinik, A. C. Foucher, A. I. Frenkel, J. E. S. van der Hoeven, B. Kozinsky, N. Marcella, M. M. Montemore, H. T. Ngan, C. R. O’Connor, C. J. Owen, D. J. Stacchiola, E. A. Stach, R. J. Madix, P. Sautet and C. M. Friend, Chem. Rev., 2022, 122, 8758–8808 CrossRef CAS PubMed.
- S. Jeong, C. Huang, Z. Levell, R. X. Skalla, W. Hong, N. J. Escorcia, Y. Losovyj, B. Zhu, A. N. Butrum-Griffith, Y. Liu, C. W. Li, D. Reifsnyder Hickey, Y. Liu and X. Ye, J. Am. Chem. Soc., 2024, 146, 4508–4520 Search PubMed.
- K. Qi, Y. Zhang, N. Onofrio, E. Petit, X. Cui, J. Ma, J. Fan, H. Wu, W. Wang, J. Li, J. Liu, Y. Zhang, Y. Wang, G. Jia, J. Wu, L. Lajaunie, C. Salameh and D. Voiry, Nat. Catal., 2023, 6, 319–331 CrossRef CAS.
- M. Ramdin, A. R. T. Morrison, M. de Groen, R. van Haperen, R. de Kler, L. J. P. van den Broeke, J. P. M. Trusler, W. de Jong and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2019, 58, 1834–1847 CrossRef CAS PubMed.
- C. I. Shaughnessy, D. J. Sconyers, T. A. Kerr, H.-J. Lee, B. Subramaniam, K. C. Leonard and J. D. Blakemore, ChemSusChem, 2019, 12, 3761–3768 CrossRef CAS PubMed.
- C. M. Gabardo, A. Seifitokaldani, J. P. Edwards, C.-T. Dinh, T. Burdyny, M. G. Kibria, C. P. O’Brien, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2018, 11, 2531–2539 RSC.
- J. P. Edwards, Y. Xu, C. M. Gabardo, C.-T. Dinh, J. Li, Z. Qi, A. Ozden, E. H. Sargent and D. Sinton, Appl. Energy, 2020, 261, 114305 CrossRef CAS.
- S. Lamaison, D. Wakerley, J. Blanchard, D. Montero, G. Rousse, D. Mercier, P. Marcus, D. Taverna, D. Giaume, V. Mougel and M. Fontecave, Joule, 2020, 4, 395–406 CrossRef CAS.
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 Search PubMed.
- J. M. Spurgeon and B. Kumar, Energy Environ. Sci., 2018, 11, 1536–1551 RSC.
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang, C. M. Gabardo, C.-T. Dinh, T.-T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
- G. Wu, Y. Song, Q. Zheng, C. Long, T. Fan, Z. Yang, X. Huang, Q. Li, Y. Sun, L. Zuo, S. Lei and Z. Tang, Adv. Energy Mater., 2022, 12, 2202054 CrossRef CAS.
- Y. Pang, J. Li, Z. Wang, C.-S. Tan, P.-L. Hsieh, T.-T. Zhuang, Z.-Q. Liang, C. Zou, X. Wang, P. De Luna, J. P. Edwards, Y. Xu, F. Li, C.-T. Dinh, M. Zhong, Y. Lou, D. Wu, L.-J. Chen, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 251–258 CrossRef CAS.
- G. Zhou, B. Li, G. Cheng, C. J. Breckner, D. P. Dean, M. Yang, N. Yao, J. T. Miller, J. B. M. Klok, N. Tsesmetzis, G. Wang and Z. J. Ren, J. Am. Chem. Soc., 2024, 146, 31788–31798 CrossRef CAS PubMed.
- Y. Chen, X. Wang, X.-Y. Li, R. K. Miao, J. Dong, Z. Zhao, C. Liu, J. E. Huang, J. Wu, S. Chu, W. Ni, Z. Guo, Y. Xu, P. Ou, B. Xu, Y. Hou, D. Sinton and E. H. Sargent, Nat. Catal., 2025, 8, 239–247 Search PubMed.
- H. Xiao, T. Cheng and W. A. Goddard, III, J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS PubMed.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed.
- S. Guo, Y. Liu, Y. Huang, H. Wang, E. Murphy, L. Delafontaine, J. L. Chen, I. V. Zenyuk and P. Atanassov, ACS Energy Lett., 2023, 8, 935–942 CrossRef CAS.
- H. Xiao, W. A. Goddard, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS PubMed.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- C. E. Creissen and M. Fontecave, Nat. Commun., 2022, 13, 2280 CrossRef CAS PubMed.
- M. Choi, S. Bong, J. W. Kim and J. Lee, ACS Energy Lett., 2021, 6, 2090–2095 CrossRef CAS.
- F. Hu, L. Yang, Y. Jiang, C. Duan, X. Wang, L. Zeng, X. Lv, D. Duan, Q. Liu, T. Kong, J. Jiang, R. Long and Y. Xiong, Angew. Chem., Int. Ed., 2021, 60, 26122–26127 CrossRef CAS PubMed.
- M.-G. Kim, J. Park, Y. Choi, H. C. Song, S.-H. Kim, K.-M. Bang, H. C. Ham, N.-K. Kim, D. H. Won, B. K. Min, S. J. Yoo and W. Kim, Adv. Energy Mater., 2023, 13, 2300749 CrossRef CAS.
- G. O. Larrazábal, V. Okatenko, I. Chorkendorff, R. Buonsanti and B. Seger, ACS Appl. Mater. Interfaces, 2022, 14, 7779–7787 CrossRef PubMed.
- D. F. Cox and K. H. Schulz, Surf. Sci., 1991, 249, 138–148 CrossRef CAS.
- L.-Y. Gan and Y.-J. Zhao, J. Chem. Phys., 2010, 133, 094703 CrossRef PubMed.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- J. Gao, A. Bahmanpour, O. Kröcher, S. M. Zakeeruddin, D. Ren and M. Grätzel, Nat. Chem., 2023, 15, 705–713 CrossRef CAS PubMed.
- C. Azenha, C. Mateos-Pedrero, M. Alvarez-Guerra, A. Irabien and A. Mendes, Chem. Eng. J., 2022, 445, 136575 CrossRef CAS.
- J. Y. Kim, W. T. Hong, T. K. C. Phu, S. C. Cho, B. Kim, U. Baeck, H.-S. Oh, J. H. Koh, X. Yu, C. H. Choi, J. Park, S. U. Lee, C.-H. Chung and J. K. Kim, Adv. Sci., 2024, 11, 2405154 Search PubMed.
- W. Huang, J. Zhu, S. Liu, W. Zhang, L. Zhou and L. Mai, EES Catal., 2023, 1, 434–458 Search PubMed.
- X. Yan, C. Duan, S. Yu, B. Dai, C. Sun and H. Chu, Renewable Sustainable Energy Rev., 2024, 190, 114086 CrossRef CAS.
- Q. Sun, C. Jia, Y. Zhao and C. Zhao, Chin. J. Catal., 2022, 43, 1547–1597 Search PubMed.
- D. H. Mok, H. Li, G. Zhang, C. Lee, K. Jiang and S. Back, Nat. Commun., 2023, 14, 7303 CrossRef CAS PubMed.
- X. Ma, Z. Li, L. E. K. Achenie and H. Xin, J. Phys. Chem. Lett., 2015, 6, 3528–3533 CrossRef CAS PubMed.
- Z. Gariepy, G. Chen, A. Xu, Z. Lu, Z. W. Chen and C. V. Singh, Energy Adv., 2023, 2, 410–419 RSC.
- O. Christensen, S. Zhao, Z. Sun, A. Bagger, J. V. Lauritsen, S. U. Pedersen, K. Daasbjerg and J. Rossmeisl, ACS Catal., 2022, 12, 15737–15749 Search PubMed.
- Q. Zhu, C. J. Murphy and L. R. Baker, J. Am. Chem. Soc., 2022, 144, 2829–2840 CrossRef CAS PubMed.
- C. Dong, M. Ji, X. Yang, J. Yao and H. Chen, Catalysts, 2017, 7, 5 CrossRef.
- A. Singh, A. Zamader, R. Khakpour, K. Laasonen, M. Busch and M. Robert, J. Am. Chem. Soc., 2024, 146, 22129–22133 CrossRef CAS PubMed.
- S. Zhao, H.-Q. Liang, X.-M. Hu, S. Li and K. Daasbjerg, Angew. Chem., Int. Ed., 2022, 61, e202204008 CrossRef CAS PubMed.
- S. Desmons, J. Bonin, M. Robert and S. Bontemps, Chem. Sci., 2024, 15, 15023–15086 RSC.
- L. Deng, Z. Wang, X. Jiang, J. Xu, Z. Zhou, X. Li, Z. You, M. Ding, T. Shishido, X. Liu and M. Xu, Appl. Catal., B, 2023, 322, 122124 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |