
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElevated temperature and pressure driven ampere-level CO2 electroreduction to CO in a membrane electrode assembly electrolyzer†
Yang
Li
 ,
Huiyue
Liu
,
Jithu
Raj
,
Mohammad
Pishnamazi
and
Jingjie
Wu
*
,
Huiyue
Liu
,
Jithu
Raj
,
Mohammad
Pishnamazi
and
Jingjie
Wu
*
Department of Chemical and Environmental Engineering, University of Cincinnati, Cincinnati, OH 45221, USA. E-mail: jingjie.wu@uc.edu
First published on 22nd April 2025
Abstract
Achieving high selectivity for carbon monoxide (CO) in the electrochemical reduction of carbon dioxide (CO2) at industrially relevant current densities, particularly using dilute CO2 feedstocks, remains a significant challenge. Herein, we demonstrate that combining elevated temperature and CO2 pressure substantially enhances CO production in a membrane electrode assembly (MEA) electrolyzer using commercially available silver nanoparticles. Elevated CO2 pressures increase CO2 concentration and reduce the diffusion layer, counteracting the reduced CO2 solubility in water and enhanced wetting of catalyst layer caused by high temperature. The synergy of high pressure and temperature ensures high CO2 flux to the catalyst surface while leveraging elevated temperatures to accelerate reaction kinetics. Therefore, the pressurized and heated CO2 electrolyzer achieves an FECO of 92% at a high current density of 2 A cm−2 and a low cell voltage of 3.8 V under 10 bar and 80 °C when using 0.1 M KHCO3 as the anolyte. Even when using pure water as the anolyte, the system maintains a FECO value of 90% at 300 mA cm−2 and a cell voltage of 3.6 V. Furthermore, the system demonstrates exceptional performance with dilute 10 vol% CO2 feedstocks, achieving a FECO of 96% at 100 mA cm−2 and 2.4 V. These findings underscore the potential of combined temperature and pressure optimization to overcome mass transport limitations and enhance reaction kinetics, offering a viable pathway for scaling up CO2 electrolyzers for industrial applications.
Broader contextEfforts to mitigate the adverse effects of carbon dioxide emissions while meeting global energy demands have driven extensive research in carbon capture and utilization technologies. The electrochemical CO2 reduction reaction (CO2RR) has emerged as a promising pathway to convert CO2 into valuable products such as carbon monoxide, formate, and hydrocarbons using renewable electricity. Among these, CO is particularly attractive due to its versatility in producing chemicals and fuels with positive technoeconomic potential. Advancements in the CO2RR, particularly under industrially relevant conditions, hold the potential to revolutionize sustainable energy and environmental catalysis by reducing reliance on fossil fuels and lowering greenhouse gas emissions. The successful integration of optimized reaction parameters, such as high pressure and temperature, addresses mass transport and kinetic limitations, advancing scalable solutions for industrial CO2 conversion. As renewable-powered CO2 electrolyzers are developed, they could seamlessly integrate with CO2 capture systems, offering a circular carbon economy that aligns with decarbonization goals. |
1. Introduction
In response to the escalating carbon dioxide (CO2) emissions driven by increased fossil fuel consumption, CO2 capture and utilization has become a global priority with accelerated research efforts.1 The electrochemical CO2 reduction reaction (CO2RR) presents a dual function to mitigate greenhouse gas emissions and generate sustainable feedstocks by integrating with renewable electricity.2 By tailoring the catalyst, reaction environment, and operating potential, the CO2RR enables the production of a wide array of valuable products.3–6 Among these, carbon monoxide (CO) stands out as a versatile feedstock for downstream upgrading to various hydrocarbon chemicals and fuels with promising market potential. Technoeconomic assessments indicate that CO is among the few CO2RR products capable of achieving positive gross margins.7 Extensive research has identified silver (Ag) as an optimal catalyst for selective CO production, yet achieving high CO selectivity at a high current density (>1 A cm−2) remains a significant challenge due to the high energy barrier of CO2 activation and sluggish kinetics of multi-electron/proton transfer steps.8A significant advancement in CO2RR systems was achieved with the introduction of gas diffusion electrodes (GDEs), which effectively reduce the diffusion layer of gas phase CO2, thereby enabling operation at industrially relevant current densities.9–11 Among various cell configurations, the membrane electrode assembly (MEA) cell stands out as a promising approach, integrating GDEs to offer low ohmic resistance and scalability potential for multicell stacks.12,13 It is widely considered that,14 under operating conditions, the catalyst layer pores become saturated with liquid electrolyte, limiting the reaction primarily to the aqueous phase via dissolved CO2.15–17 However, high current densities often induce electrode flooding that thickens the diffusion layer of CO2, posing mass transfer limitation in MEA cells. Efforts to overcome these current density limitations have primarily focused on modifying the catalyst layer by incorporating with materials such as polytetrafluoroethylene (PTFE) to enhance hydrophobicity, silicon dioxide (SiO2) to consume the hydroxide ions and thereby reducing the local pH, and cesium (Cs+) with induced electric field to lower the barrier of CO2 activation at high current densities.18–20 Despite these advances, there has been comparatively little exploration of process intensification. To date, most CO2RR-MEA cell studies have been conducted under ambient conditions, with only limited reports on investigating pressurized MEA cells.21,22 In these studies, pressure was typically applied only to the cathode side, leading to gas crossover through the membrane due to pressure imbalances when the differential exceeded 6 bar, ultimately resulting in decreased performance.23
On the other hand, increasing the reaction temperature enhances CO2RR kinetics, as the rate generally increases exponentially with temperature.24,25 Industrial CO2 electrolyzers are expected to operate under elevated temperatures due to heat generated by overpotentials, resistive losses, as well as the high temperatures of flue gas streams, often exceeding 100 °C.26,27 However, as temperature rises, CO2 solubility in aqueous electrolytes decreases, where the hydrogen evolution reaction (HER) tends to accelerate, complicating the optimization of CO2RR selectivity.28 Several studies have examined the effects of temperature on GDE-based CO2RR systems with varying results depending on catalysts and cell configurations. For instance, in MEA cells using Ag catalysts, rising temperatures have been associated with reduced jCO and FECO at reported cell voltages of 2.2–3.4 V, largely attributed to diminished CO2 adsorption, lower solubility, and increased water presence.21,23 Conversely, under constant current conditions (100–500 mA cm−2), elevated temperatures have been shown to enhance FECO.19 In flow cell systems, peak FECO occurred at moderate temperatures under certain current, with performance declining at higher temperatures due to CO2 solubility constraints.29 For Au catalysts, FECO generally decreased with increasing temperature under both constant potential (−0.7 VRHE in the flow cell) and constant current conditions (100 mA cm−2 in the MEA cell), consistent with CO2 solubility limitations.30,31 Similarly, Sn-based catalysts exhibited a decline in formate selectivity at higher temperatures in both flow cell and MEA cell systems under the same cell voltage of 2.2 V, although partial current densities of formate plateau at elevated temperatures in MEA systems.25 Notably, these previous studies often focused on a single cell voltage or a narrow temperature range at ambient pressure, leaving a gap in the understanding of how combined temperature and pressure impacts catalytic performance across varying cell voltages. Given that practical CO2 electrolyzers are expected to operate at elevated temperatures and pressures for seamless integration with upstream and downstream processes,32,33 a systematic investigation into the interplay of these parameters on CO2RR performance is essential to advance catalyst and electrode design as well as intensifying process operation.
In this work, by systematically varying reaction temperature and pressure using a commercial Ag catalyst in a MEA cell, we demonstrate the synergy of high temperature and pressure operation to drive the CO2-to-CO conversion at simultaneously high current density and selectivity. Our results reveal that (i) high-pressure operation effectively enhances CO2 availability and promotes selective CO2 adsorption, thus facilitating the CO2RR at high current density while suppressing the parasitic HER; (ii) the effect of temperature on jCO is strongly influenced by the cell voltage and CO2 partial pressure. At lower cell voltages and higher CO2 pressures, elevated temperatures positively improve the CO formation rate. The combined effects of high temperature and pressure achieve an impressive FECO exceeding 92% at a current density of 2 A cm−2 at a cell voltage of 3.8 V when using 0.1 M KHCO3 as the anolyte, a stark improvement over that under ambient conditions, where FECO drops from 95% at 100 mA cm−2 to 73% at 200 mA cm−2. Additionally, pressurized and high-temperature operation presents a compelling strategy to substantially enhance CO2RR performance when using pure water as the anodic feedstock or processing under dilute CO2 concentrations.
2. Experimental section
2.1. Chemicals
Potassium bicarbonate (KHCO3, 99.7%) and potassium hydroxide (KOH, 99.99%) were purchased from Sigma-Aldrich and used as received without further purification. Silver nanoparticles (Ag, 20–40 nm) were purchased from Thermo Scientific. All solutions were prepared using Milli-Q water (17.8 MΩ cm).2.2. Preparation of the Ag electrode
The Ag electrodes were fabricated using a standard air-brush technique. Initially, the Ag catalyst ink was prepared by dispersing Ag nanoparticles (40 mg) in iso-propanol (4 mL), followed by sonication for 30 minutes. The resulting ink was uniformly air-brushed onto carbon paper (Sigracet GDL 34BC, Fuel Cell Store) to achieve a catalyst loading of approximately 0.8 mg cm−2. The geometric area of the GDE cathode was 1.0 cm × 1.0 cm.2.3. Electrochemical measurements
The CO2RR performance under varying temperatures and pressures was evaluated in a MEA cell with 0.1 M KHCO3 as the anolyte. The GDE cathode and an IrO2/Ti felt anode were separated by a PiperION anion exchange membrane (AEM, 20 μm, Fuel Cell Store). For the pure CO2RR, dry CO2 gas was supplied to the cathode at a flow rate of 250 sccm via a mass flow controller (Alicat Scientific) without external humidification. For the diluted CO2RR, a CO2/N2 gas mixture was used, with the total mass flow controlled at 250 sccm. For instance, for 10 vol% CO2RR, 25 sccm of CO2 was mixed with 225 sccm of N2, whereas for 50 vol% CO2RR, 125 sccm of CO2 was mixed with 125 sccm of N2. A potentiostat (Gamry Interface 5000E) was used to apply a constant current to the MEA cell and record the corresponding cell voltage without iR correction. The cell temperature was controlled by electrical heating rods directly connected to both the cathode and anode flow fields, with a thermocouple inserted into the cell to maintain the desired temperature (Fig. S1, ESI†), which was regulated by a PID temperature controller (Cole-Parmer TC5000).A schematic and photograph of the pressure setup is shown in Fig. S2 and S3 (ESI†). In all pressurized MEA setups, the pressures on the cathode and anode sides were balanced to ensure consistent conditions. The anode side pressure was controlled using a back-pressure regulator (BPR, Equilibar model LF2 with PEEK non-reinforced diaphragm) downstream of the cell, equipped with a high-pressure electronic pilot controller (Equilibar). The anolyte was fed into the anode using a high-pressure syringe pump (Fusion 6000X, Chemyx) at a flow rate of 0.5 mL min−1. As for the cathode side, gas pressure was maintained using stacked back-pressure regulators (Swagelok, KBP1J0A4A5A20000). A cold trap was positioned downstream of the cathode effluent to separate gaseous and liquid products. Due to liquid product crossover, the FEs of the liquid products were calculated based on the total amount collected from both the anode and cathode sides during the same time period. Gas samples were collected downstream of the BPR, ensuring that the gas was at atmospheric pressure.
2.4. Product detection
During the electrochemical reaction, an in-line gas chromatograph (GC, Agilent 8860) was employed to monitor gaseous products. To calibrate the outlet gas flow rate of CO2, a mass flow meter (MFM, Alicat Scientific) was used to measure the outlet gas stream from the cathode prior to sampling to the GC loop.34 The FE for gaseous products was calculated using the following equation:where z is the number of electrons transferred for producing a target product; F is the Faraday constant; x is the molar fraction of a target product determined by GC; V is the molar flow rate of gas; and jtotal is the total current density.

The liquid products after electrolysis were collected and quantified via1H nuclear magnetic resonance (NMR) spectroscopy using a Bruker NEO 400 MHz spectrometer. The electrolyte (500 μL) was mixed with an internal standard (100 μL of 5 mM 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt in D2O). The partial current densities of CO and H2 (jCO and jH2) at different cell voltages were determined by multiplying the overall current density by the corresponding FE. The single-pass CO2 conversion efficiency (SPCE) is calculated as follows:

3. Results and discussion
3.1. Pressurized electrolysis of CO2 to CO
We systematically examined the effects of CO2 partial pressure on the performance of an Ag GDE for the CO2RR over a pressure range of 1 to 10 bar. Fig. 1 illustrates the influence of pressure on FECO and cell voltage under galvanostatic conditions at various temperatures of 20 °C, 40 °C, 60 °C, and 80 °C. We note that only CO and H2 were detected across all experiments, with no liquid products that were observed or under the detection limit. Under ambient pressure and temperature (Fig. 1a), FECO reached 95% at 100 mA cm−2, demonstrating the superior capability of Ag catalyst in converting CO2 to CO. However, FECO sharply declined to below 40% as the current density increased to 600 mA cm−2. This trend highlights a key challenge in MEA cells with AEM: high current densities drive substantial electroosmotic water flow accompanied by cation migration from the anode, resulting in electrode flooding and thickened CO2 diffusion layer. The reduced flux of CO2 near the catalyst surface leads to HER dominance. | ||
| Fig. 1 FECO and cell voltage as a function of current density for the CO2RR at various pressures (1 bar, 3 bar, 6 bar, and 10 bar) and temperatures: (a) 20 °C, (b) 40 °C, (c) 60 °C, and (d) 80 °C. A consistent input flow of 250 sccm CO2 was employed in all experiments. The cathode was Ag GDE and the anode was Ir/Ti felt. 0.1 M KHCO3 was used as an anolyte. The error bars represent standard deviations of three independent measurements. | ||
Meanwhile, the elevated CO2 consumption rate at higher current densities exacerbates mass transport limitations, hindering conversion efficiency. To substantiate this claim, we evaluated the CO2 single pass conversion efficiency (SPCE) under varying current densities at 1 bar and 10 bar CO2 pressures. As shown in Fig. S4 (ESI†), under 1 bar CO2, the SPCE initially increases with current density but plateaus at ∼400 mA cm−2, indicating mass transport constraints. In contrast, at 10 bar CO2, the SPCE continues to rise, reaching a maximum at ∼800 mA cm−2. These observations confirm that rapid CO2 consumption at high current densities intensifies mass transport limitations, particularly under low CO2 partial pressures.
By increasing the CO2 pressure up to 10 bar, we effectively mitigate these limitations, resulting in higher FECO at elevated current densities. Specifically, under 10 bar and 20 °C, FECO remained above 95% even at 600 mA cm−2. This trend was also observed at higher temperature conditions (Fig. 1b–d). FECO consistently increased with pressure under the current density, signaling the effectiveness of pressurized conditions for the CO2RR to CO production.
The enhancement of FECO at high j with increasing pressure is associated with multiple factors: (i) Henry's law predicts that elevated CO2 pressure increases the dissolved CO2 concentration,35 boosting CO2 availability in the wetted catalyst layer as well as reducing proton adsorption, thereby effectively suppressing the HER; (ii) elevated pressure reduces the density difference between gas and liquid phases, thereby mitigating water flooding under high current densities.36
Encouragingly, elevated pressures across all temperatures consistently led to reductions in cell voltage, as shown in Fig. 1. At relatively lower current density, the drop is insignificant, as shown in Fig. 1(a), from 100 mA cm−2 to 300 mA cm−2 and the cell voltage variation is within 0.1 V from 1 bar to 10 bar. However, at higher current densities, the decrease of cell voltage with increasing pressure becomes particularly evident. For instance, at 80 °C under a current density of 1.8 A cm−2, increasing the pressure from 3 bar to 10 bar lowers the cell voltage from 5 V to 3.6 V. Calculations of thermodynamic potential across the studied range of pressure and temperature indicate minimal variation (∼0.1 V; see Fig. S5 and S6, ESI†) for both the CO2RR and OER. Considering that under high current density, the fast CO2 consumption rate leads to severe mass transfer limitation, we assume that the cell voltage reductions with increasing pressure are primarily due to decreased mass transfer resistance.
3.2. Effect of elevated temperature on CO2-to-CO conversion
Increasing the temperature also effectively reduces the overall cell voltage across all CO2 pressures under a current density range of 0.1 to 2 A cm−2 (Fig. S7, ESI†), consistent with previous high temperature MEA studies. The AEM shows negligible increase of ionic conductivity by 10 mS cm−1 from 20 to 80 °C at 1 bar,30,37,38 corresponding to an ohmic potential drop of around 0.20 V at 1 A cm−2, much lower than the cell voltage drop by 2.3 V. Considering the minimal thermodynamic potential variations for the pressure and temperature range under investigation (Fig. S5 and S6, ESI†), we posited that the reduction in cell voltage mainly arises from diminished kinetic overpotentials.Fig. 2(a–c) illustrates the trend of FECO and jCO as temperature increases under different CO2 pressures (0.1 bar to 10 bar) and different applied cell voltages of 3 V, 3.4 V, and 3.8 V. At ambient CO2 pressure (1 bar), FECO and jCO exhibit a distinct temperature-dependent response related to cell voltage. Specifically, at a lower cell voltage of 3 V, FECO initially increased slightly as temperature rose from 20 to 40 °C before decreasing beyond 60 °C. Conversely, at higher cell voltages (3.4 V and 3.8 V), a progressive decline in FECO was observed with increasing temperature from 20 to 80 °C, with the rate of decrease becoming more pronounced at higher cell voltage. Regarding jCO, at 3 V, a positive correlation with temperature was observed from 20 °C to 80 °C. However, at elevated cell voltages, jCO followed a volcano-shaped trend, peaking at 60 °C for 3.4 V and at 40 °C for 3.8 V, indicating that excessive heating suppresses CO production at higher cell voltages.
 | ||
| Fig. 2 Effect of reaction temperature on the CO2RR performance. (a–c) FECO and jCO as a function of temperature at various CO2 pressures for applied cell voltages of (a) 3 V, (b) 3.4 V, and (c) 3.8 V, (d) temperature for peak jCO as a function of CO2 pressure under different cell voltages. For the diluted CO2RR (0.1 bar to 0.75 bar) in (a), a CO2/N2 gas mixture was fed with a total mass flow rate of 250 sccm. For the CO2RR under 1 bar and above in (b) and (c), dry pure CO2 gas was supplied to the cathode at a flow rate of 250 sccm. | ||
Temperature influences not only the intrinsic reaction kinetics of the CO2RR but also other critical parameters, such as CO2 solubility and diffusion coefficients.39 With increasing temperature, the diffusion coefficient of CO2 in water rises (Fig. S8, ESI†),40 potentially enhancing mass transport. However, CO2 solubility within the wet catalyst layer decreases (Fig. S9, ESI†), which could limit CO2 availability at the catalyst surface. The interplay of these factors can be described by the diffusion-limited current density equation:

As demonstrated in Fig. S8 and S9 (ESI†), increasing the temperature from 20 °C to 80 °C results in a 3-fold decrease in CO2 solubility, but a concurrent ∼3-fold increase in its diffusivity. Intuitively, this would suggest a neutral net effect on jCO if only D and C were to be considered. The decrease of jCO with increasing temperature is linked to the improved surface wettability. Elevated temperatures also reduce the contact angle of water on the cathode surface, from 137° at 20 °C to 122° at 80 °C (Fig. S10, ESI†). Enhanced wettability increases the effective diffusion layer (δ), imposes an additional mass transport barrier for CO2.
At lower cell voltages (e.g., 3.0 V), CO2 consumption rates are modest, and the available CO2 concentration remains in excess across the studied temperature range. Under these conditions, mass transport limitations are minimal, and jCO benefits from enhanced intrinsic reaction kinetics as temperature rises under 1 bar CO2 (Fig. 2a). In contrast, at higher cell voltages (e.g., 3.4 V and 3.8 V), CO2 consumption rates rise substantially, and mass transport limitations become a dominant factor. As temperature increases, the combined effects of reduced CO2 solubility and increased diffusion layer limit CO2 flux to the catalyst surface. This results in a decline in jCO with increasing temperature under ambient pressure (Fig. 2b and c).
While δ, D, and CO2 solubility are all sensitive to temperature, CO2 solubility is also tunable by pressure. Elevating the CO2 pressure above 1 bar significantly increases solubility, thereby improving CO2 flux and enabling a linear increase in jCO as temperature rises (Fig. 2b and c). This synergy between high pressure and high temperature effectively overcomes the limitations imposed by mass transport and enhances overall CO2RR performance. Conversely, reducing the operating pressure below 1 bar shifts the temperature for peak jCO to lower values (Fig. 2d), as the system becomes increasingly constrained by limited CO2 solubility.
The temperature-dependent performance of CO2 reduction at varying cell voltages is closely linked to the shift in the reaction order of CO2. As shown in Fig. 3a, at 3 V, the reaction order approaches zero from a pressure range of 0.75 bar to 10 bar, indicating sufficient CO2 availability to drive the reduction process. In contrast, at 3.4 V and 3.8 V, the reaction order increases to 0.15 and 0.3, respectively. At higher cell voltages, increased adsorption free energy of CO2, as well as the rising surface coverage of adsorbed hydrogen (θHad), which introduces repulsive effects on the adsorbed carboxyl intermediate (θCOOHad), improve the reliance on CO2 availability.41,42
 | ||
| Fig. 3 (a) Reaction order for CO2 derived from the logarithmic dependence of jCO on CO2 pressure at various applied cell voltages (3 V, 3.4 V, and 3.8 V) from a pressure range of 0.75 bar to 10 bar. (b) Ed for the CO2RR to CO at 2.8 V, 3 V, and 3.2 V at 1 bar. (c) Ed for the HER at 2.8 V, 3 V, and 3.2 V at 1 bar. The error bars represent the standard deviations of three independent measurements. | ||
To elucidate the temperature dependence of product selectivity in the CO2RR, jCO and jH2 were analyzed as a function of reciprocal temperature. Here we define the electrochemical driving energy (Ed) using the following relationship:43
 | (1) |
| Ed = Ea − αFη | (2) |
3.3. Synergy of pressure and temperature effects
Temperature and pressure were found to have synergistic effects on CO2RR performance. The CO2 availability plays a crucial role in modulating the temperature effect on CO2RR performance, including the FECO and jCO. Under a constant current density for the CO2RR, at a pressure of 1 bar, increasing the temperature from 20 to 40 °C slightly enhances the FECO ranging from 100 to 1000 mA cm−2 (Fig. S13, ESI†). Specifically, at 500 mA cm−2, increasing the temperature from 20 to 40 °C leads to the FECO increase from 48% to 64%. However, further increasing the temperature did not result in substantial improvements in FECO, with only a 10% variation (60 °C > 80 °C > 40 °C) in FECO observed. In contrast, at pressures exceeding 3 bar, the FECO value shows a gradual increase with rising temperatures from 20 to 80 °C, signaling that the impact of temperature on FECO under constant current density is more evident at higher pressures, particularly under high current densities. This trend underscores the critical interplay between pressure and temperature in enhancing FECO under constant current density. At higher pressures, the increased CO2 concentration around the catalyst layer counteracts the solubility limitation imposed by elevated temperatures, overcoming mass transfer limitations of reactants to sustain high reaction rates of the CO2RR. Remarkably, as illustrated in Fig. 1d, at 80 °C, the FECO value increases from 19% at 1 bar to nearly 100% at 10 bar under a current density of 1 A cm−2, further maintaining a high FECO close to 95% from 1 A cm−2 to 2 A cm−2.Under constant cell voltage conditions for the CO2RR, the intrinsic temperature-dependent increase in reaction rates drives simultaneous increases in jCO and jH2 at lower cell voltages where CO2 availability remains sufficient to sustain the CO2RR despite diminished solubility at elevated temperatures (e.g., 80 °C) even under ambient pressure, demonstrating the positive kinetic effects of temperature. However, at higher cell voltages, where CO2 reliance and mass transfer limitations increases, the reduced CO2 solubility at elevated temperatures results in a decline in jCO. Increasing CO2 availability through elevated CO2 pressure mitigates these limitations and allows for the full utilization of the temperature-dependent enhancement of jCO. As shown in Fig. 2(c), at an applied cell voltage of 3.8 V and a pressure of 3 bar, jCO increases steadily with temperature up to 60 °C, although the rate of improvement diminishes at 80 °C. However, as long as the CO2 pressure is sufficiently high (above 6 bar), the positive temperature effects on jCO still consistently outweigh the adverse impact of reduced CO2 solubility, enabling sustained increases in jCO with temperature. Additionally, we observed that elevated pressure effectively constrains the increase in jH2 with temperature (Fig. S11, ESI†), further underscoring the efficacy of high-pressure operations at high temperatures to constrain the HER.
To further validate the critical role of CO2 availability in shaping the temperature dependence of CO2RR performance, we explored CO2RR performance under diluted CO2 conditions at varying temperatures. CO2 was mixed with N2 to get controlled concentrations of 10 vol%, 50 vol%, and 75 vol% (with a total flow rate of 250 sccm). As shown in Fig. 2a, at 3 V, both FECO and jCO declined sharply with increasing temperatures under extremely low CO2 partial pressure (0.1 bar), where the positive kinetic effect of temperature on the CO2RR is entirely offset by reduced CO2 solubility. In contrast, when CO2 pressure exceeded 0.5 bar, FECO shows a slight increase from 20 to 40 °C before declining significantly above 60 °C. Interestingly, with increasing CO2 pressure, the peak temperature for jCO gradually shifted to a higher position: 20 °C, 40 °C, 60 °C, and 80 °C for 0.1 bar, 0.5 bar, 0.75 bar, and 1 bar, respectively (Fig. 2d). It highlights that the temperature effect on jCO is highly reliable on CO2 pressure: as CO2 partial pressure increases, the CO2RR could benefit more from temperature elevation. In addition, we investigated the effect of CO2 supply rate on CO2RR performance by varying flow rates (10–250 sccm) under pure CO2 conditions. The nearly constant FECO across all flow rates at 20–80 °C suggests that reduced performance at low CO2 partial pressure is due to decreased CO2 concentration rather than the absolute CO2 supply (Fig. S14, ESI†).
Although previous studies have explored the individual effects of elevated pressure and temperature on Ag-based catalysts for CO2 reduction to CO, comprehensive investigations that systematically examine the combined influence of temperature, pressure, and cell voltage within MEA systems remain limited. In contrast, our study presents a holistic optimization strategy, integrating temperature, pressure, and cell voltage control within an MEA system. Notably, we demonstrate a remarkable jCO value of 1840 mA cm−2 with an FECO value of 92% at 10 bar and 80 °C. To the best of our knowledge, this represents one of the highest performances reported for the CO2RR to CO under industrially relevant conditions in an MEA configuration. This performance significantly surpasses previous reports that explored either elevated temperature or pressure independently (Fig. S16, ESI†).
Furthermore, while earlier studies have investigated pressurization in MEA cells, they typically applied pressure solely to the cathode compartment. Such asymmetric pressurization can result in significant pressure differentials across the membrane, leading to gas crossover, increased membrane degradation, and ultimately compromised system stability and performance. Our study, by contrast, adopts a balanced pressure strategy, applying equalized pressures to both the anode and cathode compartments. This approach mitigates pressure-driven membrane stress, eliminates crossover issues, and enables stable operation at high pressures, thereby advancing MEA durability and practical scalability.
To evaluate long-term operational stability, the pressurized CO2 electrolyzer employing an Ag-based cathode was tested at 40 °C and 10 bar for over 100 hours at a constant current density of 300 mA cm−2. During earlier experiments, we found that the 20 μm AEM was prone to mechanical failure under combined high-temperature (>60 °C) and high-pressure (>6 bar) conditions. To address this, a thicker 40 μm AEM was employed for the stability test. While this modification improved mechanical robustness, it also led to higher cell voltages due to increased ohmic resistance.
As shown in Fig. 4, the FECO remained above 95% for the first 83 hours, indicating highly stable performance. Beyond this point, a gradual decline in FECO was observed. Notably, this drop in selectivity was not accompanied by a significant increase in the HER, and the total FE remained around 85% after 95 hours. We hypothesize that the decline in FECO arises from increased gas permeability of the membrane. This is supported by the observed 10% reduction in the measured flow rate at the cathode outlet and the presence of gas bubbles on the anode side when the electrochemical reaction was paused. These signs suggest that prolonged exposure to elevated pressure and temperature may compromise membrane integrity. In additional tests conducted at 60 °C and 10 bar, membrane degradation occurred more rapidly, with total FE decreasing from nearly 100% to 60% within 30 hours, further highlighting the challenge of maintaining membrane stability under desired temperature and pressure conditions.
 | ||
| Fig. 4 Long-term stability test of the pressurized CO2 electrolyzer using a Ag cathode at 40 °C and 10 bar with a constant current density of 300 mA cm−2. The cell voltage (left axis, grey) and FECO (pink) and H2 (green) (right axis) are shown over 100 hours of continuous operation. A 40 μm AEM was employed. | ||
3.4. Pure-water-fed electrolysis under high pressure and temperature
To reduce the cell voltage, we explored the use of 1 M KOH as the anolyte. Alkaline electrolytes like 1 M KOH offer reduced ohmic losses compared to 0.1 M KHCO3 due to their higher conductivity. As shown in Fig. 5a, substituting 1 M KOH for 0.1 M KHCO3 did not affect the FECO, which still reached 90% at 2 A cm−2 under 10 bar and 80 °C. Importantly, the cell voltage was further reduced to 3.2 V for 2 A cm−2 (Fig. S17, ESI†).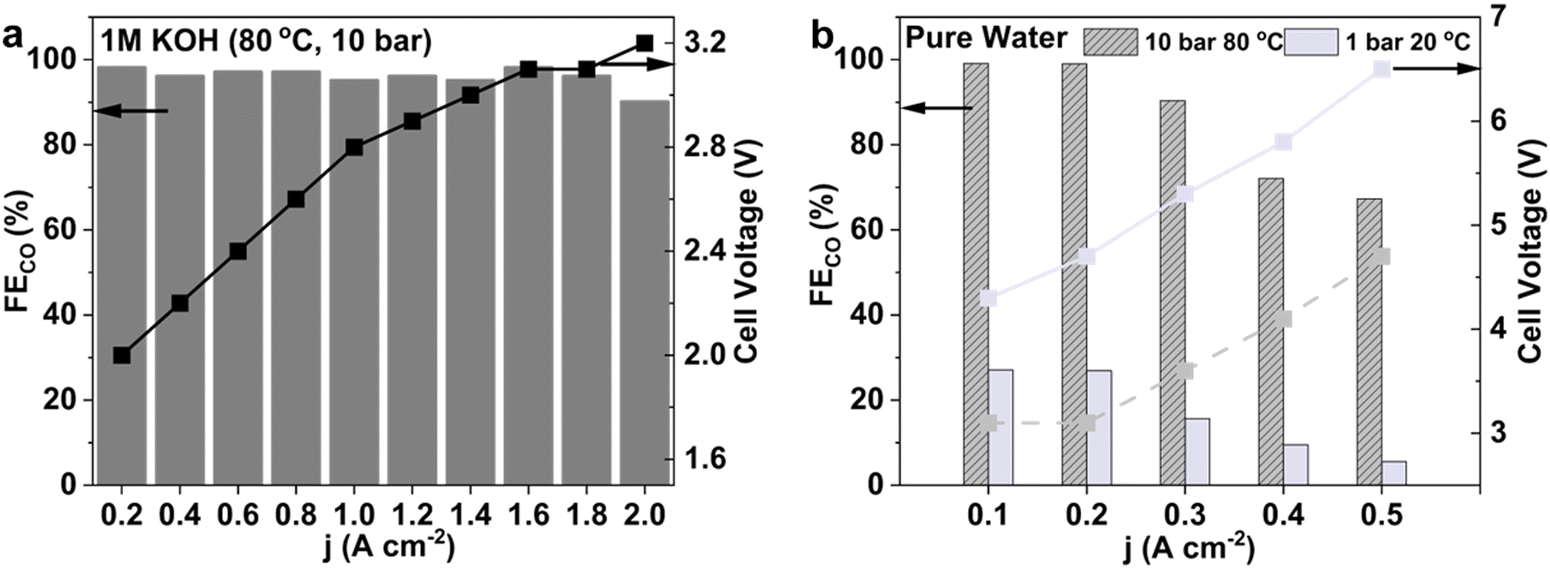 | ||
| Fig. 5 (a) FECO and cell voltage as a function of current density for the CO2RR in 1 M KOH anolyte at 80 °C and 10 bar. (b) Comparison of FECO and cell voltage at varying current densities for the pure-water-fed CO2RR at 10 bar and 80 °C versus ambient conditions (1 bar, 20 °C). | ||
During experimentation, however, we observed that using 1 M KOH under 10 bar and 20 °C led to blockage of the cathode flow field due to salt precipitation within five minutes, causing the CO2 inlet pressure to rise over 1.4 bar. This effect is likely due to the increased CO2 concentration, which enhances the reaction between CO2 and cathodically generated OH−, resulting in intense salt formation. The resulting salt precipitation obstructs the flow field and GDE, limiting CO2 mass transfer. Interestingly, when operating at an elevated temperature of 80 °C under the same 10 bar pressure, salt precipitation is significantly mitigated. Blockage only occurred after two hours of continuous operation, as indicated by a similar rise in CO2 inlet pressure beyond 1.4 bar. While some studies suggest that K2CO3 is the dominant salt precipitating at the cathode and that CO2 crossover occurs primarily via carbonate ions rather than bicarbonate,23,44,45 others observed the exclusive formation of KHCO3 at the cathode which has lower solubility than K2CO3.46 Regardless of the specific salt species, elevated temperatures effectively increase the solubility of both KHCO3 and K2CO3 (Fig. S18 and S19, ESI†), thereby reducing the extent of salt buildup and mitigating flow field obstruction.
Although elevated temperatures can mitigate salt accumulation, eventual blockage remains inevitable due to the crossover of K+ ions, resulting in system instability over time.47 Several strategies have been explored to address this issue. Acidic electrolytes, for example, allow bulk protons to react with carbonate, regenerating CO2 locally. However, acidic media require high concentrations of alkali metal cations to suppress the HER in the proton-rich environment.48–51 Their continuous accumulation in the Helmholtz layer can eventually cause alkali metal salt crystallization on the catalyst and GDL.52 Bipolar membrane (BPM) systems present an alternative strategy by creating an acidic cathode environment that eliminates carbonate formation. These systems regenerate CO2 through the reaction of carbonate or bicarbonate with protons, effectively addressing salt precipitation.53,54 However, the acidic cathode environment promotes the HER, thus reducing CO2RR selectivity, while BPM systems suffer from intrinsic drawbacks, including high resistance and long-term instability.
We employed a pure water feed (deionized water, 17.8 MΩ cm) at the anode of the AEM-based MEA cell. No anion exchange ionomer was incorporated into the cathodic catalyst layer. However, the PiperION AEM used in this study is functionalized with highly stable piperidinium cations, which are embedded within a rigid, hydrophobic, ether-bond-free aryl backbone.55 Recent studies suggest that organic cations, such as tetraalkylammonium species, could efficiently catalyze the CO2RR by modulating the interfacial electric field, facilitating the activation of CO2 and stabilizing the transition state, improving both the rate and selectivity of the CO2RR.56 Similarly, the piperidinium cations in the PiperION AEM have been proposed to enhance CO2RR performance via a comparable mechanism, despite the absence of alkali metal cations in the cathode compartment.
Under ambient conditions, the HER was the dominant reaction. FECO was below 30% at 100 mA cm−2, further dropping to less than 10% at 500 mA cm−2 (Fig. 5b). In stark contrast, under 10 bar and 80 °C, FECO reached nearly 100% at 100 mA cm−2 and 200 mA cm−2, with a slight reduction to 90% at 300 mA cm−2. In addition, the cell voltage decreased dramatically under high-pressure and high-temperature conditions. For example, at 300 mA cm−2, the cell voltage dropped from 5.3 V at ambient conditions to 3.6 V under 10 bar and 80 °C.
Compared to previous MEA studies employing pure water feeds to anode, our system exhibits comparatively lower performance.21,30 Notably, Zhuang et al.30 reported an impressive FECO value exceeding 85% at 60 °C and 1 bar, achieving current densities as high as 500 mA cm−2. Our system achieved 65% FECO at 500 mA cm−2 at 10 bar and 80 °C. This performance difference likely arises from variations in the membrane composition and the use of ionomers with smaller organic cations, known to strengthen interfacial electric fields and enhance CO2 activation kinetics.57 Nevertheless, our findings uniquely highlight that increasing CO2 pressure markedly improves CO2RR efficiency beyond ambient limitations, thus presenting a promising strategy for optimizing electrochemical CO2 conversion under H2O feedstock.
3.5. CO2RR using dilute CO2 feedstock
Currently, CO2 capture and reduction are typically conducted as separate processes, with the purification of CO2 from flue gas contributing substantially to the overall cost of the CO2 electrolysis system.58 Industrial CO2 capture technologies, such as those using monoethanolamine (MEOA), are estimated to cost at least $44 per ton of CO2 captured.59 Direct utilization of low-concentration CO2 (10 vol% balanced by N2), similar to real flue gas compositions,60 as a feedstock for the CO2RR could drastically lower costs and enhance the overall efficiency of the process.61,62 However, the low volume fraction of CO2 in such streams limits the electroreduction process and exacerbates the HER, particularly at high current densities where CO2 consumption increases rapidly.63,64We establish that our pressurized MEA configuration could significantly enhance the conversion of dilute CO2 feedstocks. Electrolysis experiments were conducted using gas feeds where CO2 was diluted with N2, with pressurization employed to elevate the partial pressure of CO2. Using a dilute CO2 feed (10 vol%) at 1 bar resulted in consistently low FECO values across 100–500 mA cm−2, with nearly zero FECO values beyond 400 mA−2 (Fig. 6a). However, when 10 vol% CO2 feed was pressurized to 10 bar, there was a notable enhancement in FECO, similar to that of pure CO2 feeds, demonstrating that elevated pressure effectively increases CO2 availability at the catalyst surface and overcomes the mass transfer limitations under the diluted CO2RR. Notably, pressurized (10 bar) CO2RR with 10 vol% CO2 demonstrated performance comparable to, or slightly exceeding, that of 1 bar CO2RR with 100% CO2 (Fig. 6a).
 | ||
| Fig. 6 (a) FECO as a function of current density under different CO2 concentrations (10 vol% and 100 vol%) and reaction pressures (1 bar and 10 bar) at 20 °C. (b) FECO and cell voltage as functions of temperature for the CO2RR at 100 mA cm−2 for 10 vol% CO2 and reaction pressure of 1 bar and 10 bar. (c) and (d) FECO at different temperatures (20 °C, 40 °C, 60 °C, and 80 °C) for 10 vol% CO2 and reaction pressure of (c) 1 bar and (d) 10 bar. | ||
Additionally, we observed that for the 10 vol% CO2RR at 1 bar, elevated temperatures adversely affected FECO (Fig. 6b and c). However, for the 10 vol% CO2RR at 10 bar, increasing temperature had a minimal effect on FECO at a current density of 100 mA cm2, while dramatically reducing the cell voltage by 0.7 V from 20 to 80 °C (Fig. 6b and Fig. S20, ESI†). At higher current densities (200–500 mA cm−2), an operational temperature of 60 °C yielded the maximum FECO. Further increasing the temperature to 80 °C resulted in a decline of performance, as the decreased CO2 solubility at this elevated temperature offsets the kinetic benefits gained from temperature increase.
Techno-economic analysis (see Supplementary Notes, ESI†) reveals that operating under elevated pressure introduces only a modest increase in dedicated capital and operating costs. In contrast, the enhanced reaction rate at higher current densities substantially reduces the required electrolyzer area, resulting in a net reduction in system-scale cost. For pure CO2 feedstock, this corresponds to a ∼90% reduction in electrolyzer capital cost compared to operation under ambient conditions. Furthermore, for low-purity feedstocks (e.g., 10% CO2 in simulated flue gas), the cost of direct pressurization to 10 bar is estimated at US$ 23 per ton of CO2 equivalent, which is nearly 80% lower than the cost of conventional CO2 capture technologies (∼US$ 100 per ton CO2).65
4. Conclusions
In summary, this study demonstrates that the combined effects of elevated temperature and pressure significantly enhance CO2 electrolysis to CO at industrially relevant current densities in a MEA electrolyzer employing commercial Ag nanoparticles. Pressurized CO2 not only increases the CO2 concentration at the catalyst surface, thereby suppressing the competing HER at ambient temperatures, but also sustains high CO2 reduction rates at elevated temperatures by counteracting the reduced CO2 solubility and enhanced wetting of catalyst layer caused by rising temperatures. This unique synergy of high temperature and pressure boosts CO current density to 2 A cm−2 with exceptional FECO (>90%) values under 80 °C and 10 bar. This is because increased thermal energy accelerates reaction kinetics while sufficient CO2 availability by high pressure mitigates mass transport limitations. Meanwhile, elevated temperature and pressure effectively lowers the cell voltage by reducing the mass transfer and kinetic overpotentials. Additionally, the pressurized MEA cell exhibits stable CO production, achieving FECO > 90% at 300 mA cm−2 and a cell voltage of 3.6 V under 80 °C and 10 bar, even using pure water as the anolyte. Moreover, under dilute CO2 feed conditions (10 vol% CO2), the system achieves an FECO of 96% at 100 mA cm−2, under 10 bar and ambient temperature. Increasing the temperature to 80 °C maintains the FECO while dramatically reducing the cell voltage by 0.7 V. Given that the operational conditions of commercial CO2 electroreduction systems are expected to function at elevated temperatures and pressures, these findings present a scalable pathway for CO2 electrolyzers to meet the demands of industrial applications.Author contributions
J. W. conceptualized and led the project, supervised the research activities, and provided critical revisions to the manuscript. Y. L. performed the experiments, analyzed the data, and drafted the manuscript. H. L. and R. J. contributed to the construction of the experimental setup. M. P. helped with characterization.Data availability
All data supporting the findings of this study are included within the article and its ESI.† Any additional information related to the data is available upon request from the corresponding author.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was financially supported by the Office of Research at the University of Cincinnati.References
- A. Rode, T. Carleton, M. Delgado, M. Greenstone, T. Houser, S. Hsiang, A. Hultgren, A. Jina, R. E. Kopp, K. E. McCusker, I. Nath, J. Rising and J. Yuan, Estimating a Social Cost of Carbon for Global Energy Consumption, Nature, 2021, 598(7880), 308–314 CrossRef CAS PubMed.
- B. Tang and F. Xiao, An Overview of Solar-Driven Photoelectrochemical CO2 Conversion to Chemical Fuels, ACS Catal., 2022, 12(15), 9023–9057 CrossRef CAS.
- Q. Cheng, M. Huang, L. Xiao, S. Mou, X. Zhao, Y. Xie, G. Jiang, X. Jiang and F. Dong, Unraveling the Influence of Oxygen Vacancy Concentration on Electrocatalytic CO2 Reduction to Formate over Indium Oxide Catalysts, ACS Catal., 2023, 13(6), 4021–4029 CrossRef CAS.
- P. Li, J. Liu, Y. Wang, X. Zhang, Y. Hou, Y. Zhang, X. Sun, X. Kang, Q. Zhu and B. Han, Manipulation of Oxygen Species on an Antimony-Modified Copper Surface to Tune the Product Selectivity in CO2 Electroreduction, J. Am. Chem. Soc., 2024, 146(38), 26525–26533 CrossRef CAS PubMed.
- J. Jin, J. Wicks, Q. Min, J. Li, Y. Hu, J. Ma, Y. Wang, Z. Jiang, Y. Xu, R. Lu, G. Si, P. Papangelakis, M. Shakouri, Q. Xiao, P. Ou, X. Wang, Z. Chen, W. Zhang, K. Yu, J. Song, J. Jiang, P. Qiu, Y. Lou, D. Wu, Y. Mao, A. Ozden, C. Wang, B. Y. Xia, X. Hu, V. P. Dravid, Y. M. Yiu, T. K. Sham, Z. Wang, D. Sinton, L. Mai, E. H. Sargent and Y. Pang, Constrained C2 Adsorbate Orientation Enables CO-to-Acetate Electroreduction, Nature, 2023, 617(7962), 724–729 CrossRef CAS PubMed.
- H. Deng, T. Liu, W. Zhao, J. Wang, Y. Zhang, S. Zhang, Y. Yang, C. Yang, W. Teng, Z. Chen, G. Zheng, F. Li, Y. Su, J. Hui and Y. Wang, Substituent Tuning of Cu Coordination Polymers Enables Carbon-Efficient CO2 Electroreduction to Multi-Carbon Products, Nat. Commun., 2024, 15(1), 1–12 Search PubMed.
- S. Verma, B. Kim, H.-R. Jhong, S. Ma and P. J. A. Kenis, A Gross-Margin Model for Defining Technoeconomic Benchmarks in the Electroreduction of CO2, ChemSusChem, 2016, 9(15), 1972–1979 CrossRef CAS PubMed.
- A. Reyes, R. P. Jansonius, B. A. W. Mowbray, Y. Cao, D. G. Wheeler, J. Chau, D. J. Dvorak and C. P. Berlinguette, Managing Hydration at the Cathode Enables Efficient CO2 Electrolysis at Commercially Relevant Current Densities, ACS Energy Lett., 2020, 5(5), 1612–1618 CrossRef CAS.
- D. Kopljar, A. Inan, P. Vindayer, N. Wagner and E. Klemm, Electrochemical Reduction of CO2 to Formate at High Current Density Using Gas Diffusion Electrodes, J. Appl. Electrochem., 2014, 44(10), 1107–1116 CrossRef CAS.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. Kenis, One-Step Electrosynthesis of Ethylene and Ethanol from CO2 in an Alkaline Electrolyzer, J. Power Sources, 2015, 301, 219–228 CrossRef.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li and Z. Yuan, Gas Diffusion Electrodes (GDEs) for Electrochemical Reduction of Carbon Dioxide, Carbon Monoxide, and Dinitrogen to Value-Added Products: A Review, Energy Environ. Sci., 2021, 14(4), 1959–2008 RSC.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Gas Diffusion Electrodes and Membranes for CO2 Reduction Electrolysers, Nat. Rev. Mater., 2021, 7(1), 55–64 CrossRef.
- D. Wakerley, S. Lamaison, J. Wicks, A. Clemens, J. Feaster, D. Corral, S. A. Jaffer, A. Sarkar, M. Fontecave, E. B. Duoss, S. Baker, E. H. Sargent, T. F. Jaramillo and C. Hahn, Gas Diffusion Electrodes, Reactor Designs, and Key Metrics of Low-Temperature CO2 Electrolysers, Nat. Energy, 2022, 7(2), 130–143 CrossRef CAS.
- D. A. Salvatore, C. M. Gabardo, A. Reyes, C. P. O’Brien, S. Holdcroft, P. Pintauro, B. Bahar, M. Hickner, C. Bae, D. Sinton, E. H. Sargent and C. P. Berlinguette, Designing Anion Exchange Membranes for CO2 Electrolysers, Nat. Energy, 2021, 6(4), 339–348 CrossRef CAS.
- Z. Xing, L. Hu, D. S. Ripatti and X. Feng, Enhancing Carbon Dioxide Gas-Diffusion Electrolysis by Creating a Hydrophobic Catalyst Microenvironment, Nat. Commun., 2021, 12(1), 136 CrossRef CAS PubMed.
- L. Weng, A. T. Bell and A. Z. Weber, Modeling Gas-Diffusion Electrodes for CO2 Reduction, Phys. Chem. Chem. Phys., 2018, 20(25), 16973–16984 RSC.
- T. Burdyny and W. A. Smith, CO2 Reduction on Gas-Diffusion Electrodes and Why Catalytic Performance Must Be Assessed at Commercially Relevant Conditions, Energy Environ. Sci., 2019, 12(5), 1442–1453 RSC.
- Z. Xing, X. Hu and X. Feng, Tuning the Microenvironment in Gas-Diffusion Electrodes Enables High-Rate CO2 Electrolysis to Formate, ACS Energy Lett., 2021, 6(5), 1694–1702 CrossRef CAS.
- Y. Sun, J. Chen, X. Du, J. Cui, X. Chen, C. Wu, X. Yang, L. Liu and J. Ye, Anchoring Cs+ Ions on Carbon Vacancies for Selective CO2 Electroreduction to CO at High Current Densities in Membrane Electrode Assembly Electrolyzers, Angew. Chem., Int. Ed., 2024, 63, e202410802 CrossRef CAS PubMed.
- C. Lim, S. Kim, J. H. Song, M. H. Han, Y. Ko, K. Lee, J. Choi, W. H. Lee and H. Oh, Breaking the Current Limitation of Electrochemical CO2 Reduction via a Silica-Hydroxide Cycle, Energy Environ. Sci., 2024, 17(17), 6215–6224 RSC.
- M. Heßelmann, J. K. Lee, S. Chae, A. Tricker, R. G. Keller, M. Wessling, J. Su, D. Kushner, A. Z. Weber and X. Peng, Pure-Water-Fed Forward-Bias Bipolar Membrane CO2 Electrolyzer, ACS Appl. Mater. Interfaces, 2024, 16(19), 24649–24659 CrossRef PubMed.
- J. Park, E. Kim, S. Kim, C. Lim, H. Kim, Y. Ko, J. Choi, H. Oh and W. H. Lee, Deriving an Efficient and Stable Microenvironment for a CO2 MEA Electrolyzer by Reverse Osmosis, ACS Energy Lett., 2024, 9(7), 3342–3350 CrossRef CAS.
- B. Endrődi, E. Kecsenovity, A. Samu, F. Darvas, R. V. Jones, V. Török, A. Danyi and C. Janáky, Multilayer Electrolyzer Stack Converts Carbon Dioxide to Gas Products at High Pressure with High Efficiency, ACS Energy Lett., 2019, 4(7), 1770–1777 CrossRef PubMed.
- B. Yang, K. Liu, H. J. W. Li, C. Liu, J. Fu, H. Li, E. J. Huang, P. Ou, T. Alkayyali, C. Cai, Y. Duan, H. Liu, P. An, N. Zhang, W. Li, X. Qiu, C. Jia, J. Hu, L. Chai, Z. Lin, Y. Gao, M. Miyauchi, E. Cortés, S. A. Maier and M. Liu, Accelerating CO2 Electroreduction to Multicarbon Products via Synergistic Electric-Thermal Field on Copper Nanoneedles, J. Am. Chem. Soc., 2024, 144(7), 3039–3049 CrossRef PubMed.
- W. Lee, Y. E. Kim, M. H. Youn, S. K. Jeong and K. T. Park, Catholyte-Free Electrocatalytic CO2 Reduction to Formate, Angew. Chem., Int. Ed., 2018, 57(23), 6883–6887 CrossRef CAS PubMed.
- R. Küngas, Review—Electrochemical CO2 Reduction for CO Production: Comparison of Low- and High-Temperature Electrolysis Technologies, J. Electrochem. Soc., 2020, 167(4), 044508 CrossRef.
- D. Aaron and C. Tsouris, Separation of CO2 from Flue Gas: A Review, Sep. Sci. Technol., 2005, 40(1–3), 321–348 CrossRef CAS.
- S. Piontek, C. Andronescu, A. Zaichenko, B. Konkena, K. J. Puring, B. Marler, H. Antoni, I. Sinev, M. Muhler, D. Mollenhauer, B. R. Cuenya, W. Schuhmann and U. Apfel, Influence of the Fe:Ni Ratio and Reaction Temperature on the Efficiency of (FexNi1−x)9S8 Electrocatalysts Applied in the Hydrogen Evolution Reaction, ACS Catal., 2017, 8(2), 987–996 CrossRef.
- E. J. Dufek, T. E. Lister and M. E. McIlwain, Bench-Scale Electrochemical System for Generation of CO and Syn-Gas, J. Appl. Electrochem., 2011, 41(6), 623–631 CrossRef CAS.
- Z. Yin, H. Peng, X. Wei, H. Zhou, J. Gong, M. Huai, L. Xiao, G. Wang, J. Lu and L. Zhuang, An Alkaline Polymer Electrolyte CO2 Electrolyzer Operated with Pure Water, Energy Environ. Sci., 2019, 12(8), 2455–2462 RSC.
- K. Li, S. Zou, J. Zhang, Y. Huang, L. He and X. Feng, Superhydrophobicity-Enabled Efficient Electrocatalytic CO2 Reduction at High Temperature, ACS Catal., 2023, 13(14), 9346–9351 CrossRef CAS.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah and P. S. Fennell, Carbon Capture and Storage Update, Energy Environ. Sci., 2013, 7(1), 130–189 RSC.
- D. Corral, J. T. Feaster, S. Sobhani, J. R. DeOtte, D. U. Lee, A. A. Wong, J. Hamilton, V. A. Beck, A. Sarkar, C. Hahn, T. F. Jaramillo, S. E. Baker and E. B. Duoss, Advanced Manufacturing for Electrosynthesis of Fuels and Chemicals from CO2, Energy Environ. Sci., 2021, 14(5), 3064–3074 RSC.
- H. Simonson, W. E. Klein, D. Henckel, S. Verma, K. C. Neyerlin and W. A. Smith, Direct Measurement of Electrochemical Selectivity Gradients over a 25 cm2 Copper Gas Diffusion Electrode, ACS Energy Lett., 2023, 8(9), 3811–3819 CrossRef CAS.
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang, C. M. Gabardo, C. T. Dinh, T.-T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Constraining CO Coverage on Copper Promotes High-Efficiency Ethylene Electroproduction, Nat. Catal., 2019, 2(11), 1124–1131 CrossRef CAS.
- H. Wu, B. Tian, W. Xu, K. K. Abdalla, Y. Kuang, J. Li and X. Sun, Pressure-Dependent CO2 Electroreduction to Methane over Asymmetric Cu–N2 Single-Atom Sites, J. Am. Chem. Soc., 2024, 146(32), 22266–22275 CrossRef CAS PubMed.
- C. A. G. Rodriguez, N. C. Kani, A. B. Moss, B. O. Joensen, S. Garg, W. Deng, T. Wilson, J. R. Varcoe, I. Chorkendorff and B. Seger, Insights into Zero-Gap CO2 Electrolysis at Elevated Temperatures, EES Catal., 2024, 2(3), 850–861 RSC.
- X. She, L. Zhai, Y. Wang, P. Xiong, M. M. Li, T. Wu, M. C. Wong, X. Guo, Z. Xu, H. Li, H. Xu, Y. Zhu, S. C. E. Tsang and S. P. Lau, Pure-Water-Fed Electrocatalytic CO2 Reduction to Ethylene Beyond 1000 h Stability at 10 A, Nat. Energy, 2024, 9(1), 81–91 CrossRef CAS.
- R. E. Vos, J. P. Smaak and M. T. M. Koper, The Temperature Dependence of Electrochemical CO2 Reduction on Ag and CuAg Alloys, J. Catal., 2024, 436(1), 115613 CrossRef CAS.
- A. Löwe, C. Rieg, T. Hierlemann, N. Salas, D. Kopljar, N. Wagner and E. Klemm, Influence of Temperature on the Performance of Gas Diffusion Electrodes in the CO2 Reduction Reaction, ChemElectroChem, 2019, 6(17), 4497–4506 CrossRef.
- M. R. Singh, J. D. Goodpaster, A. Z. Weber, M. Head-Gordon and A. T. Bell, Mechanistic Insights into Electrochemical Reduction of CO2 over Ag Using Density Functional Theory and Transport Models, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(42), E8812–E8821 CrossRef CAS PubMed.
- X. Zhu, J. Huang and M. Eikerling, Electrochemical CO2 Reduction at Silver from a Local Perspective, ACS Catal., 2021, 11(23), 14521–14532 CrossRef CAS.
- D. M. Koshy, S. S. Nathan, A. S. Asundi, A. M. Abdellah, S. M. Dull, D. A. Cullen, D. Higgins, Z. Bao, S. F. Bent and T. F. Jaramillo, Bridging Thermal Catalysis and Electrocatalysis: Catalyzing CO2 Conversion with Carbon-Based Materials, Angew. Chem., Int. Ed., 2021, 60(32), 17472–17480 CrossRef CAS PubMed.
- P. Mardle, S. Cassegrain, F. Habibzadeh, Z. Shi and S. Holdcroft, Carbonate Ion Crossover in Zero-Gap, KOH Anolyte CO2 Electrolysis, J. Phys. Chem. C, 2021, 125(46), 25446–25454 CrossRef CAS.
- J. Y. Kim, P. Zhu, F. Chen, Z. Wu, D. A. Cullen and H. Wang, Recovering Carbon Losses in CO2 Electrolysis Using a Solid Electrolyte Reactor, Nat. Catal., 2021, 5(4), 288–299 CrossRef.
- A. B. Moss, S. Garg, M. Mirolo, C. A. G. Rodriguez, R. Ilvonen, I. Chorkendorff, J. Drnec and B. Seger, In Operando Investigations of Oscillatory Water and Carbonate Effects in MEA-Based CO2 Electrolysis Devices, Joule, 2023, 7(2), 350–365 CrossRef CAS.
- L. Weng, A. T. Bell and A. Z. Weber, Towards Membrane-Electrode Assembly Systems for CO2 Reduction: A Modeling Study, Energy Environ. Sci., 2019, 12(6), 1950–1968 RSC.
- M. C. O. Monteiro, F. Dattila, N. López and M. T. M. Koper, The Role of Cation Acidity on the Competition between Hydrogen Evolution and CO2 Reduction on Gold Electrodes, J. Am. Chem. Soc., 2021, 144(4), 1589–1602 CrossRef PubMed.
- J. E. Huang, F. Li, A. Ozden, A. S. Rasouli, F. P. G. De Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C. Dinh, D. Sinton and E. H. Sargent, CO2 Electrolysis to Multicarbon Products in Strong Acid, Science, 2021, 372(6546), 1074–1078 CrossRef CAS PubMed.
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Modulating Electric Field Distribution by Alkali Cations for CO2 Electroreduction in Strongly Acidic Medium, Nat. Catal., 2022, 5(4), 268–276 CrossRef CAS.
- J. Hou, B. Xu and Q. Lu, Influence of Electric Double Layer Rigidity on CO Adsorption and Electroreduction Rate, Nat. Commun., 2024, 15, 1926 CrossRef CAS PubMed.
- Y. Xu, J. P. Edwards, S. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O’Brien, J. Li, E. H. Sargent and D. Sinton, Self-Cleaning CO2 Reduction Systems: Unsteady Electrochemical Forcing Enables Stability, ACS Energy Lett., 2021, 6(2), 809–815 CrossRef CAS.
- B. Siritanaratkul, M. Förster, F. Greenwell, P. K. Sharma, E. H. Yu and A. J. Cowan, Zero-GAP Bipolar Membrane Electrolyzer for Carbon Dioxide Reduction Using Acid-Tolerant Molecular Electrocatalysts, J. Am. Chem. Soc., 2022, 144(17), 7551–7556 CrossRef CAS PubMed.
- K. Yang, M. Li, S. Subramanian, M. A. Blommaert, W. A. Smith and T. Burdyny, Cation-Driven Increases of CO2 Utilization in a Bipolar Membrane Electrode Assembly for CO2 Electrolysis, ACS Energy Lett., 2021, 6(12), 4291–4298 CrossRef CAS PubMed.
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. B. Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Poly(aryl piperidinium) membranes and ionomers for hydroxide exchange membrane fuel cells, Nat. Energy, 2019, 4(5), 392–398 CrossRef CAS.
- S. Weng, W. L. Toh and Y. Surendranath, Weakly coordinating organic cations are intrinsically capable of supporting CO2 reduction catalysis, J. Am. Chem. Soc., 2023, 145(30), 16787–16795 CrossRef CAS PubMed.
- J. McGregor, J. T. Bender, A. S. Petersen, L. Cañada, J. Rossmeisl, J. F. Brennecke and J. Resasco, Organic electrolyte cations promote non-aqueous CO2 reduction by mediating interfacial electric fields, Nat. Catal., 2025, 8(1), 79–91 CrossRef CAS.
- S. Chen, J. Liu, Q. Zhang, F. Teng and B. C. McLellan, A Critical Review on Deployment Planning and Risk Analysis of Carbon Capture, Utilization, and Storage (CCUS) Toward Carbon Neutrality, Renewable Sustainable Energy Rev., 2022, 167, 112537 CrossRef CAS.
- A. Raksajati, M. T. Ho and D. E. Wiley, Reducing the Cost of CO2 Capture from Flue Gases Using Aqueous Chemical Absorption, Ind. Eng. Chem. Res., 2013, 52(47), 16887–16901 CrossRef CAS.
- G. V. Last and M. T. Schmick, A Review of Major Non-Power-Related Carbon Dioxide Stream Compositions, Environ. Earth Sci., 2015, 74(2), 1189–1198 CrossRef CAS.
- Y. Liu, J. Huang, H. Zhu, P. Liao and X. Chen, Simultaneous Capture of CO2 Boosting Its Electroreduction in the Micropores of a Metal–Organic Framework, Angew. Chem., Int. Ed., 2023, 62, e202311265 CrossRef CAS PubMed.
- Z. Wang, Y. Zhou, D. Liu, R. Qi, C. Xia, M. Li, B. You and B. Y. Xia, Carbon-Confined Indium Oxides for Efficient Carbon Dioxide Reduction in a Solid-State Electrolyte Flow Cell, Angew. Chem., Int. Ed., 2022, 61, e202200552 CrossRef CAS PubMed.
- Z. Zhao, J. Huang, D. Huang, H. Zhu, P. Liao and X. Chen, Efficient Capture and Electroreduction of Dilute CO2 into Highly Pure and Concentrated Formic Acid Aqueous Solution, J. Am. Chem. Soc., 2024, 146(20), 14349–14356 CrossRef CAS PubMed.
- D. Kim, W. Choi, H. W. Lee, S. Y. Lee, Y. Choi, D. K. Lee, W. Kim, J. Na, U. Lee, Y. J. Hwang and D. H. Won, Electrocatalytic Reduction of Low Concentrations of CO2 Gas in a Membrane Electrode Assembly Electrolyzer, ACS Energy Lett., 2021, 6(10), 3488–3495 CrossRef CAS.
- R. F. Service, Cost of carbon capture drops, but does anyone want it?, Science, 2016, 354(6318), 1362–1363 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information of the schematic of setup; calculated thermodynamic potential; temperature effect on diffusivity of CO2 in water, solubility of CO2, K2CO3 and KHCO3; and electrochemical results. See DOI: https://doi.org/10.1039/d5ey00034c |
| This journal is © The Royal Society of Chemistry 2025 |