
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolar production of fuels from CO2 with high efficiency and stability via in situ transformation of Bi electrocatalysts†
Woo Seok
Cheon
a,
Su Geun
Ji
ab,
Jaehyun
Kim
a,
Sungkyun
Choi
a,
Jin Wook
Yang
a,
Sang Eon
Jun
a,
Changyeon
Kim
a,
Jeewon
Bu
a,
Sohyeon
Park
a,
Tae Hyung
Lee
a,
Jinghan
Wang
a,
Jae Young
Kim
a,
Sol A
Lee
ac,
Jin Young
Kim
 *a and
Ho Won
Jang
*ad
*a and
Ho Won
Jang
*ad
aDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: hwjang@snu.ac.kr; jykim.mse@snu.ac.kr
bChemistry and Nanoscience Center, National Renewable Energy Laboratory, Golden, Colorado 80401, USA
cLiquid Sunlight Alliance (LiSA), Department of Applied Physics and Materials Science, California Institute of Technology, Pasadena, CA 91106, USA
dAdvanced Institute of Convergence Technology, Seoul National University, Suwon 16229, Republic of Korea
First published on 19th November 2024
Abstract
The sustainable electrocatalytic reduction of carbon dioxide into solar fuels offers a potential pathway to mitigate the impact of greenhouse gas-induced climate change. Here, we successfully achieved a high solar-to-fuel (STF) efficiency of 11.5% by integrating a low-cost tandem solar cell with robust, high-performance, non-precious metal-based electrocatalysts. The bismuth-based cathode exhibited a high formic acid selectivity of 97.2% at a potential of −1.1 VRHE, along with an outstanding partial current density of 32.5 mA cm−2. Furthermore, upon undergoing more than 24 hours of electrolysis, we observed an enhancement in the catalytic activity. Through comprehensive analysis including in situ Raman spectroscopy and density functional theory (DFT) calculations, we elucidated that the in situ transformation of bismuth into bismuth subcarbonate (BOC) induces multiple effects: (i) the formation of grain boundaries between phases with distinct lattice parameters, (ii) electronic modulation due to defect formation, and (iii) changes in the binding modes of key reaction intermediates on active sites, resulting in the stabilization of *OCHO species. The cause of these phase transformations was attributed to the structural similarity between the cathode template and BOC. The sustainability of the STF efficiency sets a new benchmark for all cost-effective photovoltaic-coupled electrochemical systems.
Broader contextAs climate change continues to impact human survival negatively, interest in achieving net-zero emissions has increased. To reduce the atmospheric concentration of the predominant greenhouse gas, CO2, renewable energy sources must replace fossil fuels and drive electrochemical CO2 reduction reactions. In this study, we combine non-noble metal-based electrocatalysts with perovskite-Si tandem cells to investigate the feasibility of a sustainable electrolyzer. Additionally, we explore the in situ transformation of Bi-based electrocatalysts, which exhibit exceptional catalytic activity and selectivity. The partial transition of the cathode material during the reduction reaction induces modulation of the electronic structure and, thus, stabilization of key reaction intermediates. The insights gained from this work could significantly contribute to the design of catalytic materials and integrated systems for CO2 reduction. |
Introduction
Global warming has become more than a looming threat to the environment and human society; it is a current crisis with devastating consequences such as crop failure and climate refugees. These realities call for urgent actions to reduce emissions of greenhouse gases such as carbon dioxide (CO2). Various strategies have been proposed to mitigate CO2 emissions, among which the electrochemical CO2 reduction reaction (CO2RR) using renewable energy has attracted much attention.1 Although renewable energy has been extensively researched as an alternative to fossil fuels, its utilization is hindered by inherent limitations, such as the intermittent nature of power sources. In the case of solar energy harvesting, excessive energy generated during periods of abundant sunlight can misalign with the actual electricity demand. Storing surplus electricity as chemical energy appears to be an effective solution, and among various electrochemical reactions, the CO2RR stands out for its ability to mitigate greenhouse gas emissions directly and effectively.2,3 Photovoltaic-electrochemical CO2 reduction reaction (PV-EC CO2RR) systems offer multiple advantages. For instance, compact electrolyzers can be installed at emission sources such as the stacks of power plants, cement factories, and steel mills to directly convert gas containing high concentrations of CO2.4 Additionally, the chemical feedstock produced via the CO2RR enhances the economic viability of carbon dioxide reduction.5 Therefore, developing catalysts that can achieve high selectivity for commercially valuable products with low overpotentials and ensure long-term stability is crucial.According to Kuhl's research, there are 16 products obtainable from the CO2RR.6 Among these, formic acid is one of the most valuable chemicals due to its high market price and effectiveness in hydrogen storage and transportation.7 Moreover, the reaction requires only two electrons per molecule, making it kinetically favorable and cost-effective. While several metals, including Sn, In, Cd, and Pb, demonstrate selectivity for formate production, Bi outperforms them in terms of faradaic efficiency (FE) and energy efficiency. Furthermore, the low toxicity of bismuth compared to other metals makes it environmentally friendly. Several studies were conducted to maximize the catalytic activity of Bi-based electrocatalysts. Fan et al. designed Bi nanotubes with a large current density and high selectivity in a wide potential window.8 In Li's work, Bi nanoribbons with modified edge sites enhanced the robustness of the reaction.9 While exploring synergistic effects with other metals, Zeng's group developed Bi@Sn with a core–shell structure that exhibited a high HCOOH production rate of HCOOH, attributed to the compressive strain in the Sn shell.10
However, Bi is susceptible to oxidation and restructuring, and comprehensive studies regarding catalyst transformation during electrolysis are still lacking. In the case of Cu-based cathodes, extensive research has been conducted on in situ phase or morphological transitions. For instance, the electrochemical fragmentation of Cu oxide enhances hydrocarbon production selectivity, whereas potential-driven clustering of Cu nanocubes leads to diminished CO2RR performance.11 Therefore, a deeper understanding of in situ transformation mechanisms and their impacts is essential to improve the long-term stability of catalysts.
In this study, we integrated cost-effective, high-efficiency perovskite-Si tandem cells with non-precious metal electrocatalysts. The unassisted PV-EC CO2RR system exhibited unmatched solar-to-fuel (STF) efficiency and demonstrated the capability of the solar cells and the electrocatalysts and the effectiveness of their integration. BiOI-derived cathodes, featuring abundant edge sites and a nanostructure favorable for mass transfer, exhibited a high HCOOH production rate of 606.3 μmol cm−2 h−1 and 97.2% selectivity. We also examined the in situ transformation of Bi during long-term electrolysis and its effect on catalytic activity. Utilizing first-principles calculations, we elucidate how the phase transformation of a Bi-based catalyst influences the binding mode of key reaction intermediates on the surface. We believe that these findings will significantly advance catalyst and solar-driven system designs for effective greenhouse gas reduction.
Experimental
Materials
Bismuth nitrate pentahydrate (Bi(NO3)3·5H2O, 98.0%), nitric acid (60–61%), and 1,4-benzoquinone (98%) were purchased from Junsei Chemical. Lactic acid solution (85–90% in H2O) was purchased from Sigma Aldrich. Toray carbon paper and Cu foil were purchased from Thermofisher. Nickel chloride hexahydrate (NiCl2·6H2O, 96%), iron chloride hexahydrate (FeCl3·6H2O, 98%), potassium iodide (KCl, 99%), potassium iodide (KI, 99.5%), and ethyl alcohol (EtOH, 99.9%) were purchased from Daejung. Nickel foam (pore size 450 μm, 1.5 mm thick) was purchased from Alantum. All chemicals were used as received, without further purification.Synthesis of cathodes
Carbon paper was cut into pieces of regular size (25 mm × 10 mm) and cleaned with deionized water (DI) and EtOH. For the preparation of solution A, 16.6835 g of KI was added to 250 mL of DI. After 10 minutes of stirring, 0.15 mL of nitric acid was added to lower the pH of the solution. Bi(NO3)3·5H2O was added after 5 minutes, and after waiting until all precursors were dissolved. 0.658 mL of lactic acid was added and the solution was stirred. Solution B was prepared by mixing 0.5075 g of 1,4-benzoquinone with 100 mL of EtOH. 50 mL of solution A and 20 mL of solution B were mixed for 15 minutes before electrodeposition, and the temperature was kept at room temperature. Electrodeposition was carried out in a 5 cm × 5 cm × 5 cm quartz cell in a three-electrode configuration. A 3 M Ag/AgCl reference electrode and a Pt plate as the counter electrode were used. At first, a voltage of −0.35 VAg/AgCl was applied to the carbon paper for 20 s. And then, −0.1 VAg/AgCl was applied for 540 s. BiOI on carbon paper (BiOI-VNS) was then rinsed with DI and EtOH and dried. BiOI-VNS was reduced to Bi (Bi-VNS) in a 0.2 M KHCO3 solution (pH 8.5) by applying −1.2 VAg/AgCl in an H-cell and then purged with CO2 gas for FE measurement.For comparison, Bi film with a thickness of 100 nm was deposited onto electropolished Cu foil by an e-beam evaporator at a deposition rate of 0.3 Å s−1. The base pressure of the vacuum chamber was 1.5 × 10−6 torr.
Synthesis of NiFe-LDH and perovskite-Si tandem solar cells
Nickel foams were cleaned with DI and EtOH and dried. 0.1691 g of KCl was dissolved in 150 mL of DI. After 5 minutes, 0.7428 g of NiCl2·6H2O and 0.2069 g of FeCl3·6H2O were added to the solution. The solution was stirred for 10 minutes and kept at room temperature. Then, a constant current of −10 mA was applied for 600 s to the dried nickel foam with an exposed area of 1 cm2. The NiFe-LDH obtained from the electrodeposition was rinsed and dried under vacuum.For the fabrication of the perovskite-Si tandem solar cells, a-Si:H layers were deposited on both sides of n-type Si wafers using plasma-enhanced chemical vapor deposition (PE-CVD). ITO/poly(triarylamine) (PTAA)/perovskite/C60/PEIE/ITO/Ag grid/MgF2 layers were subsequently deposited, with their thickness varying from 15 nm to 500 nm. For more details, please refer to Ji et al.'s work.12
Characterization
Scanning electron microscopy (SEM, Carl Zeiss, SIGMA) was used to examine the sample morphologies with an acceleration voltage of 2 kV. Also, transmission electron microscopy (TEM, JEM-2100F, JEOL) was conducted to see the morphology and obtain high-resolution TEM (HRTEM) images of the catalyst powder, which was exfoliated from the sample by sonication. X-Ray diffraction (XRD, D8-Advance, BRUKER) patterns were obtained in the 2θ range of 20 to 70° with a step size of 0.02° and a scan speed of 4° min−1 using an X-ray diffractometer with Cu Kα radiation (λ = 1.54056 Å). X-ray photoelectron spectroscopy (XPS, Versaprobe III, UL-PHI) was conducted using an Al Kα X-ray source. Pass energies of 280 eV and 112 eV were employed for the survey and high-resolution spectra, respectively. All spectra were normalized by the integrated intensity, followed by baseline correction using a Shirley model, and the Gaussian–Lorentzian sum function was employed for fitting the spectra using the CasaXPS program. The contact angle was measured by referring to the method of Chen et al.13Electrochemical measurement and product analysis
All electrochemical measurements were carried out in an H-cell using a high-precision potentiostat from Ivium Technologies (Model: nStat). In a three-electrode configuration, the active area of the working electrode was kept at 0.5 cm2 and graphite rod and saturated Ag/AgCl reference electrodes were used. All electrodes were cleaned before their utilization in the experiment to ensure accurate measurements. A proton exchange membrane (Nafion 117, Dupont) separated the cathodic and anodic compartments. 15 mL of 0.2 M KHCO3 solution saturated with CO2 gas (pH 7.0) was used as the catholyte, and 1 M KOH as the anolyte. For calculation of the electrochemically active surface area (ECSA), the specific capacitance value of 0.22 mF cm−2 was used. ECSA was measured in 1 M KOH solution. All data were collected under standard temperature and pressure conditions. The linear sweep voltammetry (LSV) curves and cyclic voltammetry (CV) curves were recorded at a scan rate of 10 mV s−1. Electrochemical impedance spectroscopy (EIS) analysis was conducted by applying −0.7 VRHE without agitation and with frequencies ranging from 5 kHz to 0.1 Hz. EIS data were fitted to the corresponding equivalent circuits. All data were calibrated to a reversible hydrogen electrode (RHE) unless mentioned specifically. FEs of the gaseous products (CO, H2) were analyzed by using online gas chromatography (Agilent 7890B) equipped with a thermal conductivity detector and a flame ionization detector. The FEs were calculated using the following equation:| FEgas (%) = Qgas/Qtotal × 100 (%) = (z × ngas × F)/Qtotal × 100 (%) |
| = (z × P × V × Cgas × F)/R × T × Qtotal × 100 (%) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 325 Pa), volume, gas concentration, Faraday constant (96485 C mol−1), gas constant (8.314 J mol−1 K−1), and the temperature (298.15 K), respectively. The quantitative analysis of liquid products, mainly HCOOH, was conducted using a high-resolution nuclear magnetic resonance spectrometer (NMR, Avance 600, Bruker). 500 mM of dimethyl sulfoxide-d6 in D2O was used as the internal standard. The STF efficiency was calculated by using the following equation:
325 Pa), volume, gas concentration, Faraday constant (96485 C mol−1), gas constant (8.314 J mol−1 K−1), and the temperature (298.15 K), respectively. The quantitative analysis of liquid products, mainly HCOOH, was conducted using a high-resolution nuclear magnetic resonance spectrometer (NMR, Avance 600, Bruker). 500 mM of dimethyl sulfoxide-d6 in D2O was used as the internal standard. The STF efficiency was calculated by using the following equation:where E0product is the standard cell potential for each reaction product (E0CO = 1.34 V, E0HCOOH = 1.43 V), J is the current density, and Wsol is the solar irradiance.14

Theoretical calculations
In this study, all the spin-polarized density functional theory (DFT) calculations were performed using the Vienna Ab initio Simulation Package (VASP) with the projector augmented wave method for the core region and a plane-wave kinetic energy cutoff of 500 eV. The generalized gradient approximation (GGA) in the form of the revised Perdew–Burke–Ernzerhof (RPBE) for the exchange–correlation potentials was used. The DFT-D3 method of Grimme with the zero-damping function was employed to correct the van der Waals interactions of the system. The surfaces of Bi2CO5 (110) and Bi (111) were carried out using the slab models composed of p(2 × 2) supercells with four and six atomic layers, respectively. The large vacuum layers of these slab models were set at least 15 Å in the z-direction for the isolation of the surface to prevent the interaction between two periodic units. A 2 × 2 × 1 Monkhorst–pack sampled k-point grid was employed to sample the reciprocal space for the slab models. The bottom two atomic layers of Bi2CO5 (110) and three atomic monolayers of Bi (111) were fixed at their bulk positions, while the rest of the atomic layers and adsorbates were free to move in all directions until the convergence of energy and residual force on each atom were less than 1 × 10−4 eV and 0.05 eV Å−1, respectively. The Gibbs free energy was calculated using the following equation:in which EDFT is the electronic energy acquired from DFT calculations, Cp is the heat capacity, and S is the entropy, and ZPE is the zero-point energy from vibrational analysis calculations.

Results and discussion
Vertically grown bismuth nanosheets (Bi-VNS) prepared by electrodeposition
We synthesized BiOI with vertical nanosheet (BiOI-VNS) morphology, controlled height, and maximized specific surface area via a two-step electrodeposition process. A relatively higher potential was applied to form BiOI nuclei on the carbon paper, followed by the application of a lower potential to grow BiOI-VNS (Fig. S1, ESI†). By adding lactic acid to the aqueous solution, we maintained a consistent change in the local pH, enabling the formation of a BiOI layer with uniform height across the entire substrate (Fig. S2, ESI†).15,16 The common synthesis method for 2D structured BiOX (X = Cl, I, Br) is the solvothermal method, but electrodeposition offers several advantages. It allows for direct synthesis on a large-area substrate, making the process simple and scalable and facilitating charge transfer between the substrate and catalyst.17 The catalyst can be grown in a vertical direction on the substrate, creating pathways for the transport of the electrolyte. The absence of binders like Nafion or conductors like carbon black minimizes the obstruction of the catalyst's active sites.18 Additionally, the thickness of the BiOI layer can be controlled. By adjusting the growth time of BiOI through control experiments, we identified the optimized time for the second step of electrodeposition to be 540 s. When the growth time was too short, the carbon paper was not completely covered, or the layer's thickness was insufficiently thick. On the other hand, when the growth time was too long, an agglomerated structure formed (Fig. S3, ESI†). This aggregated shape was observed to detach after the electrochemical CO2 reduction reaction, suggesting that it may have poor bonding with the underlying structure and could negatively affect stability. By observing the cyclic voltammogram of BiOI in a 0.2 M KHCO3 solution, it was recognized that the reduction from Bi3+ to Bi0 occurs at approximately −0.5 VRHE (Fig. S4, ESI†). Applying the reductive potential for 5 minutes led to stabilization of the current, suggesting completion of the electrochemical reduction to the metallic state (Fig. S5, ESI†).The complete transformation from BiOI to Bi was verified through XRD patterns and Raman spectra (Fig. 1a and Fig. S6, ESI†). Comparison of the diffraction patterns before and after 5 minutes of electrochemical reduction reveals that the XRD peaks corresponding to BiOI (JCPDS No. 04-012-5693) completely disappear, and new peaks associated with rhombohedral Bi (JCPDS No. 00-005-0519) are newly formed. However, the nanosheet morphology of BiOI is overall maintained, and the surface becomes more porous, as evidenced by SEM images (Fig. 1b and c). An increase in the surface area leads to improved exposure of active sites and the mass transport of reactants. The thickness of a single vertical nanosheet was approximately 30 nm, and the height measured from the substrate was approximately 1 μm. Energy-dispersive X-ray spectroscopy (EDS) mapping on the bismuth vertical nanosheet (denoted as Bi-VNS) confirmed the uniform distribution of bismuth and oxygen, indicating the generation of native oxide on the Bi surface after exposure to the air (Fig. 1d).19 The lattice distances of 0.227 nm and 0.328 nm, corresponding to Bi (110) and (012) planes, were discerned from the high-resolution transmission electron microscopy (HRTEM) image and corresponding fast Fourier transform (FFT) pattern (Fig. 1e).
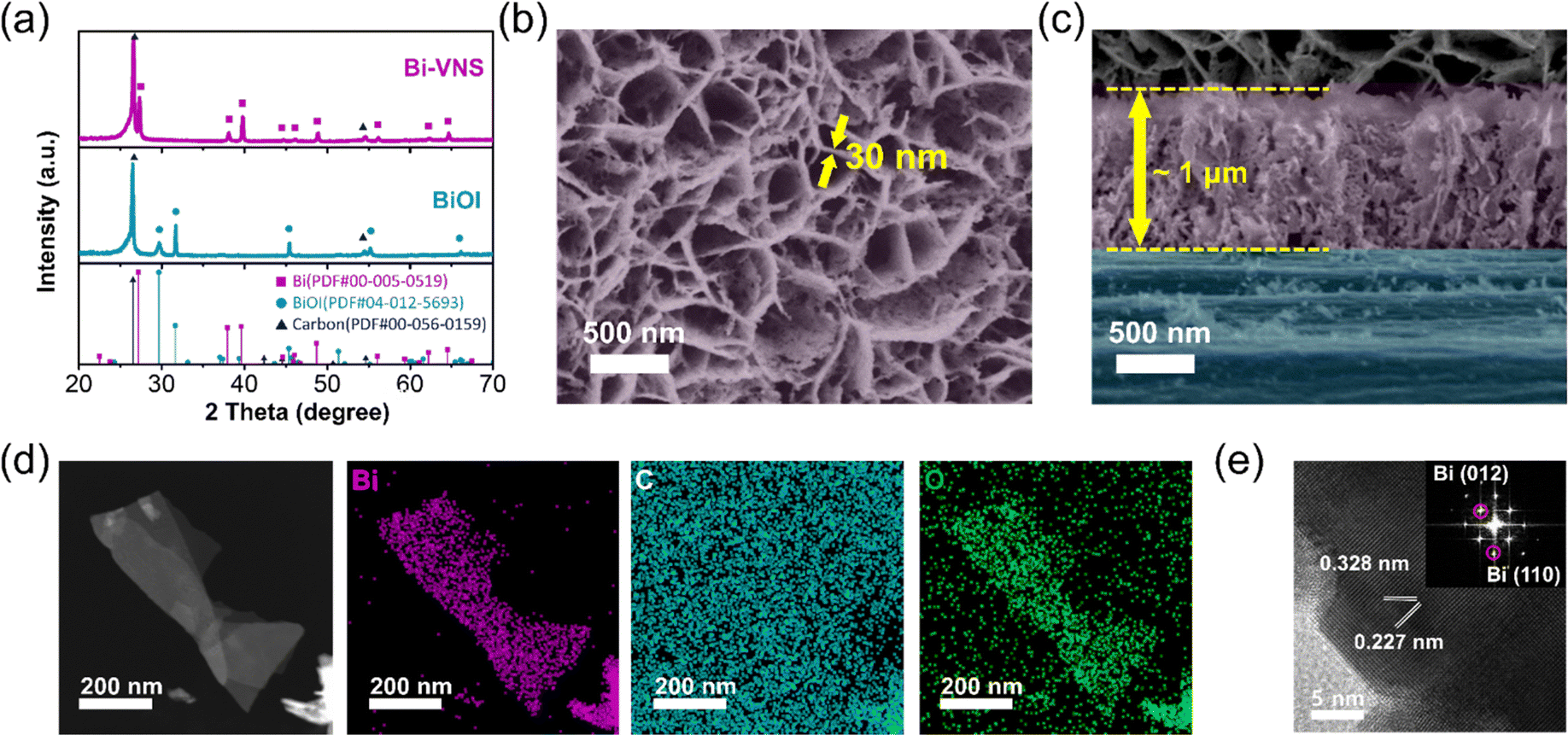 | ||
| Fig. 1 Characterization of Bi-VNS. (a) XRD patterns of BiOI and Bi-VNS (bismuth vertical nanosheet). SEM images of Bi-VNS in (b) top view, and (c) cross-sectional view. For visual clarity, Bi-VNS and carbon paper are colored purple and blue, respectively. (d) HAADF-STEM image and EDS elemental mapping of Bi-VNS. (e) HRTEM image and FFT pattern (inset) of Bi-VNS. | ||
EC CO2RR performance of Bi-VNS
The electrochemical CO2RR performance of the samples was evaluated in a Nafion membrane-separated H-type cell. LSV curves were recorded in Ar- and CO2-saturated 0.2 M KHCO3 electrolyte. Both the Bi film and Bi-VNS generated over two times more current density in the CO2-saturated electrolyte than in the Ar-saturated electrolyte, as shown in Fig. 2a. Still, Bi-VNS exhibited a much smaller onset potential and a substantially larger current density compared to the Bi film. One of the primary reasons for the superior catalytic activity of Bi-VNS is its greater ECSA, which is about three times that of the Bi film, as confirmed by CV measurements (Fig. S7, ESI†). The intrinsic activity is also enhanced, as seen in the ECSA-normalized LSV (Fig. S8, ESI†). This enhancement can be attributed to Bi-VNS having more edge sites exposed to the electrolyte, thus contributing to much higher activity. Edge sites display greater reactivity than basal planes due to features like the dangling bonds of undercoordinated atoms.20–22 According to a study by Su et al., the partial density of states (PDOS) near the Fermi level in the p band is higher in the Bi nanosheet than in bulk Bi. This leads to enhanced orbital hybridization between the catalyst and the adsorbed reactants and optimized adsorption of intermediate species.23 The thinner the nanosheet, the greater the number of unsaturated pits at the edge sites, which, upon calculating differential charge density distribution, can lower the reaction's activation barrier by reducing the formation energy of *OCHO due to charge enrichment at the defect-rich edge.24,25 Additionally, materials with large curvature in their nanostructures can reduce CO2 molecules more quickly at the same voltage due to the field-induced reagent concentration (FIRC) effect.8,26 The 2D nanosheet morphology of Bi-VNS is well-suited to maximizing the FIRC effect. | ||
| Fig. 2 Electrochemical measurements of Bi-VNS for the CO2RR. (a) LSV curves of Bi-VNS and Bi film in the Ar-saturated (dashed line) and CO2-saturated (solid line) 0.2 M KHCO3 electrolyte. (b) Faradaic efficiencies of Bi-VNS at different applied potentials. (c) Partial current densities of different products at applied potentials. (d) Nyquist plots for Bi-VNS and Bi-film with the equivalent circuit shown as an inset. Rs, Rct, and Qdl represent the system resistance, charge transfer resistance, and capacitance for a constant phase element (electrical double layer). (e) Current density and FEHCOOH during the long-term stability measurement of Bi-VNS at a potential of −0.9 VRHE. (f) Calculated partial current densities for HCOOH, CO, and H2. | ||
Nanostructures influence not only activity but also selectivity. In addition to the FIRC effect, the plastron effect may also occur due to the nanostructure. When contact angles were measured using a 10 μL water droplet, the angles were 80.4° for the Bi film and 135.0° for Bi-VNS (Fig. S9, ESI†). It is well known that greater hydrophobicity of the cathode leads to enhanced CO2RR selectivity. Gas chromatography (GC) and nuclear magnetic resonance (NMR) spectroscopy were used to quantitatively analyze the gas and liquid products formed during the reaction. Measurements of faradaic efficiency at various potentials revealed that CO2RR's selectivity was predominant over the hydrogen evolution reaction (HER) across a wide voltage window, recording a high formate/formic acid FE (FEHCOOH) of 97.2% at −1.1 VRHE (Fig. 2b). As formic acid (HCOOH) exists in the form of formate in the electrolyte with a neutral pH, these terms are used interchangeably in this paper. The FE value for CO remained almost constant at about 3%. The FE of H2 gas produced due to the competing HER gradually decreased as the voltage increased. While the control sample, Bi film, generally favored the CO2RR over the HER and exhibited an increase in FEHCOOH as the voltage rose, its liquid FE and partial current density were significantly lower than those of Bi-VNS (Fig. S10, ESI†). In the partial current density plot, the portions attributed to gaseous products were kept low, while the formate partial current density (jHCOOH) continued to increase with the applied potential, exceeding −32 mA cm−2 at −1.1 VRHE (Fig. 2c). This value is among the very superior performances measured in a gas-tight H-cell (Table S1, ESI†).
EIS analysis was conducted to evaluate the charge transfer capability at the interface. Upon analysis of the Nyquist plot, the charge transfer resistance (Rct) of Bi-VNS was significantly lower, thus facilitating the electron transfer to the adsorbed reactant (Fig. 2d).27,28 The simulated Rct values are listed in Table S2 (ESI†).
The commercialization of CO2 electrolyzers necessitates resolving the stability issue, a paramount concern.29–31 Many preceding studies on the CO2RR have observed a degradation in catalyst performance during measurements. To assess the stability of Bi-VNS, long-term electrolysis was conducted at −0.9 VRHE (Fig. 2e). Surprisingly, aside from an abrupt decrease in the initial three minutes, the current density continued to increase until saturation occurred around 24 h. The initial sharp decline in current density is observed in chronoamperometric electrolysis (CA) over a wide voltage range (Fig. S11, ESI†). The current density value after approximately 3 minutes followed the V–j relationship depicted in the LSV. As the CA at −0.9 VRHE proceeded, the current density increased while the FEHCOOH remained stable at 80%. Calculating the partial current density revealed that jHCOOH increased from −16.3 mA cm−2 to −24 mA cm−2. jCO was maintained below −1 mA cm−2 without significant changes, while jH2 increased from −1.8 mA cm−2 to −5.3 mA cm−2 (Fig. 2f). At first glance, the increased HER activity may seem to affect the selectivity of the CO2RR negatively. However, since the main product, formate, exists in liquid form and hydrogen is a gaseous product, their separation is facile. Moreover, the addition of the total quantity of profitable chemical substances contributes to the economic viability of the CO2 electrolyzer.14 The generation of CO, another gaseous product, remained suppressed, maintaining the purity of hydrogen gas. To the best of our knowledge, no reports have detailed the activation behaviour of Bi catalysts for both the CO2RR and HER. Additionally, such phenomena were not observed in the control sample of Bi film (Fig. S12, ESI†). We conducted ex situ characterization studies to gain a comprehensive understanding of this extraordinary phenomenon.
Partial transformation of Bi-VNS into BOC during the CO2RR
Through XRD analysis, we observed a partial phase change; the initial bismuth metal phase (JCPDS No. 00-005-0519) was supplemented by the formation of bismuth subcarbonate (BOC, JCPDS No. 00-041-1488) during the CA test in the KHCO3 solution (Fig. 3a). The 6 h sample revealed the presence of BOC through peaks at 30.3° and 32.7°. The two peaks’ intensity increased in the 12 h sample, indicating further BOC formation. After 24 h, when the current density saturation occurred, only the peak corresponding to the BOC's (110) plane remained, and its position shifted to a higher angle. Although most of the catalyst was still comprised of a metallic phase, the formation of BOC occurred near the surface through reactions described in eqn (1)–(5), leaving the stable plane (110) dominantly present.32Bi3+ (in BiOI) + 3e− ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) Bi Bi | (1) |
| CO2 + H2O + 2e− → HCOO− + OH− | (2) |
| 2H2O + 4e− → H2 + 2OH− | (3) |
| Bi + 3OH− Bi(OH)3 | (4) |
| Bi(OH)3 + CO2 Bi2O2CO3 + 3H2O | (5) |
 | ||
| Fig. 3 Phase transition of Bi-VNS. (a) XRD patterns of BOC@Bi-VNS formed after different CO2RR times. (b) HRTEM image and FFT pattern (inset), and (c) HAADF-STEM image and EDS elemental mapping of BOC@Bi-VNS after 24 h of the CO2RR. (d) High resolution XPS Bi 4f spectra of BOC@Bi-VNS and Bi-VNS. (e) O 1s, and (f) C 1s spectra of BOC@Bi-VNS. | ||
Also, strain-induced lattice contraction appeared to have occurred due to the creation of a heterojunction between Bi and BOC, which have different lattice parameters. The co-existence of Bi and BOC results in a higher current density, an increased faradaic efficiency, and reduced overpotential compared to a single phase.33,34
The existence of a heterojunction between Bi and BOC in BOC-incorporated Bi-VNS (BOC@Bi-VNS) has been confirmed through HRTEM. The inset in Fig. 3b demonstrates that the lattice fringe distances of 0.372 and 0.296 nm correspond to the (011) and (013) interplanar spacing of BOC.23 The elemental mapping reveals a uniform distribution of Bi, O, and C across the nanosheet (Fig. 3c). This observation further confirms the formation of BOC. It is well known that a heterojunction between metal and metal oxide or other metal compounds can enhance the catalytic activity.35 As a consequence of junction formation, surface wettability may be enhanced,36 and charge transfer across the interface could become more efficient. Furthermore, it should be emphasized that lattice strain-induced distortions or built-in electric fields can significantly alter the electronic structure of surface atoms.37–40 As observed in the diffraction pattern in Fig. 3a, lattice contraction in BOC occurred due to the lattice mismatch as the CA duration extended. Cho et al. elucidated through DFT calculations that interfacial strain lowered the thermodynamic barrier of formate production.37 By disrupting the linear scaling relationship, lattice strain can potentially improve both the activity and selectivity of electrochemical processes.38 After Bi's in situ partial transformation into BOC, the charge transfer resistance decreased, as can be seen in the EIS analysis (Fig. S13, ESI†). The enhancement in electron transfer might have benefited from the newly formed BOC/Bi junction.
The possibility of electronic modulation of Bi species due to the BOC formation reaction near the surface was verified through XPS analysis (Fig. 3d). The high-resolution spectrum of Bi 4f exhibits two pairs of doublets, corresponding to Bi0 and Bi3+. In BOC@Bi-VNS, the ratios of the Bi3+ peaks (158.66, 163.96 eV) to the Bi0 peaks are larger than in Bi-VNS, a result of an increased quantity of oxidized Bi due to BOC formation after 24 h of electrolysis. Comparing this with Fig. 3a, where the Bi metal phase is dominant, it can be inferred that BOC is concentrated near the surface. Bi-VNS also exhibits Bi3+ peaks (158.95, 164.25 eV), which is attributed to the native oxide formation by exposure to air.41 Unlike the position of metallic Bi, which remains at 156.90 eV for Bi 4f7/2 and 162.22 eV for Bi 4f5/2, the position of the Bi3+ peak in BOC@Bi-VNS shifted by 0.29 eV to a lower binding energy. The reduction in binding energy could indicate the formation of oxygen vacancies in BOC@Bi-VNS, and this may have redistributed the negative charge to Bi atoms, benefitting the stabilization of *OCHO.20,42 The presence of CO32− within BOC was also confirmed in the O 1s and C 1s spectra (Fig. 3e and f).
To provide evidence for the proposed reaction pathway and identify the state of surface adsorbates during the reaction, in situ Raman spectroscopy analysis was conducted in the same electrolyte, 0.2 M KHCO3 solution. The changes were closely monitored by incrementally adjusting the reductive potential in 0.1 V steps, with spectra recorded at the open circuit potential (OCP) both before and after the reaction. At OCP, ions from the electrolyte near the electrode surface, CO32− and HCO3−, were observed at 1000 and 1064 cm−1, respectively.37,43–45 Additionally, adsorbed species such as *OCOOH and *O2CO were detected at 1347 and 1583 cm−1,46 respectively. Given that the focus was on the catalyst surface, the intensities of these surface adsorbates were pronounced. Furthermore, peaks corresponding to the stretching and bending modes of water molecules were located at 3223/3403 cm−1 and 1648 cm−1, respectively.45,47,48 Upon applying the reductive potential, the peaks for adsorbates like HCO3− and *OCOOH, located near or on the surface, disappeared, and the presence of the key reaction intermediate *OCHO was confirmed at 1360 cm−1.42,45,49,50 The disappearance of these adsorbate peaks indicates that, under the influence of the applied negative potential, the adsorbates were consumed in the reduction reaction. The *OCHO peak first appeared at an applied potential of −0.2 VRHE, gradually decreased in intensity as the potential was increased to −0.7 VRHE, and ultimately disappeared at −0.8 VRHE. This behavior can be attributed to the conversion of *OCHO to *OCHOH via proton-coupled charge transfer on the catalyst surface, a process that requires overcoming an activation barrier. As the applied voltage increases, a greater number of molecules can overcome this barrier.50,51 The complete disappearance of the *OCHO peak at −0.8 VRHE aligns well with the sharp increase in the faradaic efficiency for formic acid at this potential. Moreover, the peak intensity of water, the reactant molecule required for the CO2 reduction reaction, also varied with increasing voltage. Similar to *OCHO, the water peaks sharply decreased at −0.8 VRHE, which can be understood in the same context as the disappearance of the *OCHO peak. However, unlike *OCHO, the water peak partially recovered at OCP. This reappearance occurs because, unlike *OCHO, which forms only under a reductive potential, water is always present in the electrolyte, allowing its concentration near the surface to recover after the potential is removed.
DFT calculations were conducted to simulate CO2RR and HER processes on Bi and BOC surfaces. The (110) plane was chosen as the model surface for BOC, corroborated by XRD observations, while the (111) plane was selected for Bi. Despite the thermodynamically preferential growth of the (012) plane,52 the (111) surface was preferred for comparison due to its recognized activity based on first-principles calculations and widespread utilization.35,53 The free energy diagrams for the CO2RR were derived and depicted along the reaction pathway, comprising 2 sequential proton-coupled electron transfer (PCET) steps followed by desorption from the catalyst surface (Fig. 5a). The generation of *OCHO, a key reaction intermediate in the proposed pathway, was previously confirmed by in situ Raman (Fig. 4). While the reaction Gibbs free energy for *OCHO formation on the BOC (110) surface was only −0.07 eV, it was as high as 0.95 eV on the Bi (110) surface, indicating a significant difference in the reaction energetics. On Bi (111), the considerable energy barrier for forming the key intermediate species emphasizes that the initial step of CO2 conversion was the rate-determining step (RDS). Conversely, on the BOC (110) surface, *OCHO was greatly stabilized. As a result, the second PCET step, which requires an uphill energy of 0.54 eV, was identified as the rate-determining step (RDS). In Fig. 5b and c, charge density redistribution and Bader charge analysis were used to compare *OCHO stabilization on BOC and Bi surfaces. During the CO2-to-formic acid reaction pathway, the key reaction intermediate, *OCHO, adheres to the catalyst surface via oxygen atoms in a bidentate mode.54 On the BOC surface, the oxygen atoms attach to individual Bi atoms, intensifying the interaction and stabilizing *OCHO.34 The cyan-colored depletion regions in the charge density map clearly illustrate this point. Comparing the Bader charge changes of C, O, and Bi in BOC, bismuth is the only element whose charge changes significantly on the catalyst surface before and after the adsorption of *OCHO, indicating that Bi atoms serve as the active site. The Bader charge values of Bi atoms with *OCHO bound on BOC (110) are +1.95, 1.88 e (Fig. 5b). Since electron depletion is more pronounced than in Bi (111), the nucleophilic attack of oxygen atoms is stronger, allowing the key reaction intermediate to remain stably bound on the surface. The high HCOOH FE of Bi-based cathodes is linked to the adsorption mode of CO2 molecules on the metal surface. For CO-selective metals like Au and Ag, a nucleophilic attack occurs at the carbon atom, leading to the formation of the *COOH intermediate. In contrast, metals such as Bi and Sn undergo a nucleophilic attack at the oxygen atom, producing *OCHO. *COOH can either become *CO via PCET, which is released as CO gas if its binding energy to the surface is weak or transform into the C2+ product via C–C coupling if the binding energy is appropriate. In the case of bismuth, the energy required for *COOH generation is significantly higher than for *OCHO, resulting in a low CO faradaic efficiency at all potentials. The DFT results are consistent with previous studies suggesting that BOC is a stable and efficient phase for formate production under CO2RR conditions.27,32,35,55,56 Similar to the CO2RR, the BOC (110) surface exhibited key intermediate stabilization in the free energy diagram for the HER. The increased partial current densities for both hydrogen and formate observed in Bi-VNS's CA tests were attributed to the incorporation of the BOC phase. As proposed by Chen et al., the primary limitation for formate production is sluggish water dissociation; hence, strategies to stabilize *H and enhance CO2RR activity are effective.42 Wang et al.'s research further supports the result, as the authors demonstrated experimentally and computationally that the increasing amount of Bi–O bonds promotes the electrochemical reduction of CO2.57
 | ||
| Fig. 4 In situ Raman spectroscopy (a) schematic of the electrochemical CO2RR (eCO2RR) to the formic acid pathway and (b) in situ Raman spectra in CO2-saturated 0.2 M KHCO3 electrolyte solution. The Raman spectra were recorded stepwise starting from OCP to −1.2 VRHE and back to OCP. | ||
 | ||
| Fig. 5 DFT calculations. (a) Gibbs free energy diagrams for the formation of formic acid from CO2 on the BOC (green) and Bi (yellow) surface. The differential charge density spatial distribution of (b) BOC and (c) Bi with *OCHO adsorption (the charge accumulation and depletion areas are represented in yellow and cyan, respectively). The calculated Bader charge values are also presented. (d) Gibbs free energy diagrams for the formation of hydrogen and carbon monoxide. | ||
To conclude, during the CO2RR, Bi-VNS underwent an in situ partial transformation, leading to the surface incorporation of BOC. The co-existence of BOC with Bi facilitated the stabilization of the key reaction intermediate, *OCHO, subsequently enhancing intrinsic activity and increasing the production rates of HCOOH on the cathode. The escalation in the catalytic performance of Bi-VNS also has roots in other factors, including increased roughness and active surface area, which led to improved active site exposure and mass transport (Fig. S14, ESI†). Additionally, ECSA analysis authenticated the growth in the specific surface area of BOC@Bi-VNS (Fig. S15, ESI†). However, this phenomenon was not observed in the control sample, Bi film. Fig. S12 (ESI†) shows the chronoamperometric result at 14 h. Other than small fluctuations, there was no increase in its catalytic activity, which is consistent with other literature in the field.8,19,23,35,43,58–61 XRD patterns indicate that the Bi film didn’t undergo phase transformation during the CO2 electrolysis (Fig. S16, ESI†). A plausible speculation related to this occurrence can be traced to the similarity found between layered structures of BiOX (X = Cl, Br, I) and BOC. BOC possesses a layered structure consisting of [Bi2O2]2+ and CO32− layers, a configuration strikingly analogous to that of BiOX, with the only difference being the type of interlayer anion.33 As the [Bi2O2]2+ slabs and iodide ion slabs in BiOI are stacked through weak van der Waals (vdW) force, the iodide ions are easily replaced with CO32− ions.62 Also, BiOX-derived Bi catalysts inherit the interlayer spacing of the precursor.63 Consequently, the easy formation of stable crystalline BOC through interlayer ion exchange in BiOI at the cathodic potential seems reasonable.34 The change of morphology and phase is well depicted in Fig. 6 and Fig. S14 (ESI†). While there have been studies starting from BOC to create phases that coexist with reduced Bi, there are only a few papers that have tried to create BOC incorporated Bi metal starting from BiOI and elucidate the conversion process and the mechanism of the high activity conversion process and the mechanism of high activity.
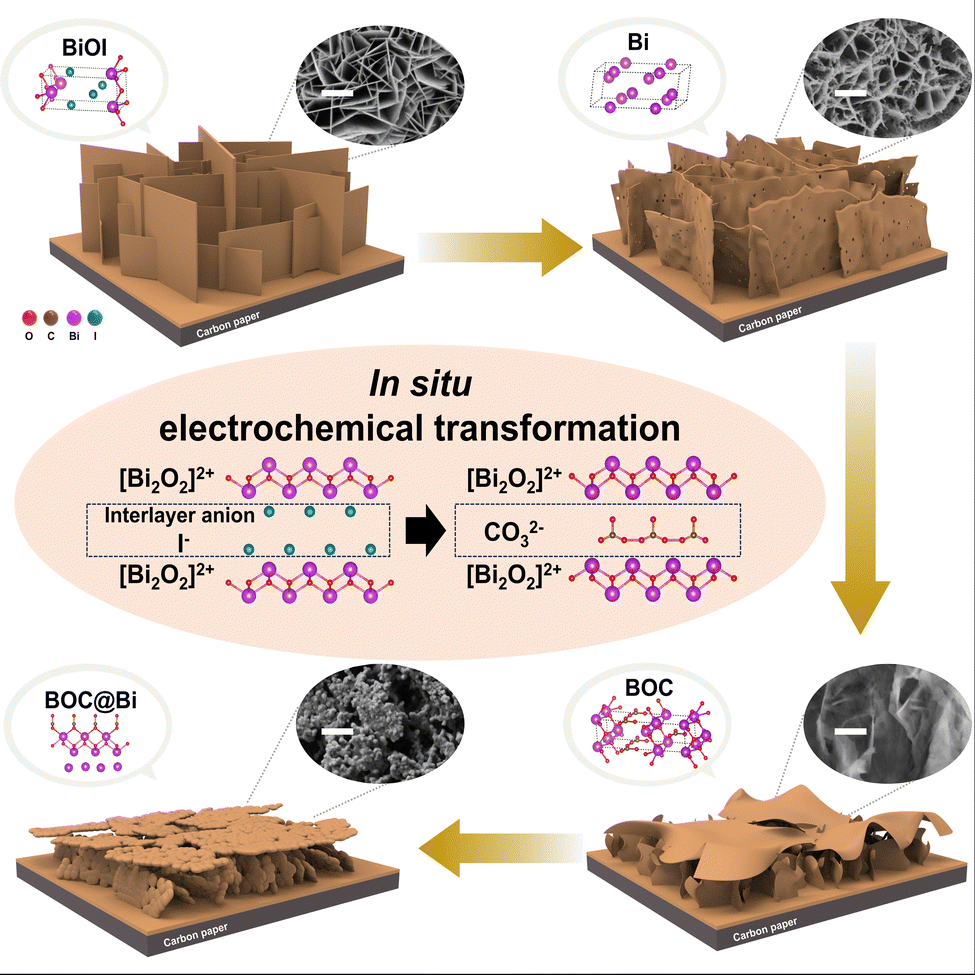 | ||
| Fig. 6 Schematic illustration of the in situ transformation of the BiOI-derived cathode. The morphology and phase of BiOI vertical nanosheets change significantly during the electrochemical reduction reaction. The similarity between the layered structures of BiOI and BOC enables the in situ transformation of BiOI into Bi, and then BOC. | ||
PV-EC measurement
Bi-VNS, activated through an in situ transformation at the cathodic potential, is exceedingly useful for a stable PV-EC CO2RR. Initially, to construct a practical PV-EC system, a perovskite-Si tandem cell was employed (Fig. 7a). Utilizing solar energy as the sole energy source, two series-connected solar cells with approximately 1.8 V of open circuit voltage (VOC) and 19 mA cm−2 of short-circuit current (JSC) for each cell were used to induce an efficient and spontaneous CO2RR. The I–V characteristic of the connected PV module is depicted in Fig. 7b. By calculating the power conversion efficiency of the PV module, the operating point for maximum efficiency was identified, with the voltage and current of the maximum point (VMP, IMP) being 3.03 V and 8.75 mA cm−2, respectively. | ||
| Fig. 7 PV-EC measurements. (a) Schematic illustration and (b) I–V and P–V characteristics of a perovskite-Si tandem cell. (c) Faradaic efficiencies at different applied potentials in the 2-electrode configuration of Bi-VNS (working electrode) and NiFe-LDH (counter electrode). (d) Current density and STF efficiency during the unassisted light-driven electrochemical reaction. (e) Chopped-light chronoamperometric measurements of the PV-EC system without external bias under simulated 1 sun illumination (AM1.5G 100 mW cm−2). (f) Solar cell characteristics and operating points at the start and end of PV-EC measurements. (g) Comparison of the STF efficiency and stability with the reported PV-EC systems utilizing cost-effective solar cells. | ||
For unassisted CO2 reduction, low-cost, high-efficiency transition metal-based catalysts were employed at the cathode and anode. Synthesized through one-step electrodeposition, NiFe layered double hydroxide (NiFe-LDH) exhibited a porous structure suitable for robust oxygen evolution reaction (OER) catalytic activity (Fig. S17, ESI†). With the superior performance of an overpotential of 220 mV at a current density of 10 mA cm−2 and a very low Rct value, water splitting occurred vigorously.
The ratio between the solar cell's irradiated area and the catalyst area was determined based on LSV in a 2-electrode configuration employing Bi-VNS and NiFe-LDH. It was anticipated that a device operating point at VMP and IMP could be achieved when the catalyst's active area was set to 1.5 times the irradiated area. The faradaic efficiencies at various voltages were also measured in the 2-electrode configuration (Fig. 7c). FEHCOOH increases with higher voltage, reaching over 80% at 3 V. The sum of the faradaic efficiencies does not reach 100% at low voltages due to the low current values, resulting in inaccurate data when the product concentration was near or below the detecting limit.
After connecting all solar cells and electrodes in series, the CO2RR and water splitting occurred solely through electromotive force provided by the solar cell for an extended period of approximately 13 h (Fig. 7d). The experiment measured the longest stability in PV-EC CO2 electrolysis using low-cost PV cells, with the fuel production rates gradually increasing, as anticipated from Bi-VNS's activation. The sharp peaks and decreasing tails are attributed to bubble formation on the electrodes. The bubbles formed on the NiFe-LDH surface hindered reactant transport, and when the bubbles reached the threshold size, they detached from the surface. The current density was recovered upon each bubble's escape, showing an instant increase. This phenomenon was more prominent on the anode. Overall, STF was maintained above 10% and steadily increased. Unlike in Bi-VNS's 3-electrode measurement, a significant increase in current density was not observed, limited by the current that could flow through the connected PV module. The whole system's operation was confirmed through chopped-light measurement (Fig. 7e). As seen in Fig. 7f, the initial STF was only 10.8%, but it increased to 11.5% after 13 h as performance improvement occurred through Bi's in situ transformation. After 13 h of CO2 electrolysis measurement, the PV cell exhibited slight degradation. To our knowledge, maintaining and enhancing STF over such an extended period in a PV-EC system was unprecedented. This study achieved the highest STF efficiency in experiments using cost-effective PV cells (Fig. 7g and Table S3, ESI†).
Conclusions
In this study, we successfully demonstrated the in situ transformation of a BiOI-derived electrocatalyst and its impact on stable and cost-effective solar fuel production. Highly selective and reactive bismuth nanosheets were fabricated, achieving FEHCOOH exceeding 97% for formate production and a current density of 32.5 mA cm−2 at −1.1 VRHE. Notably, unlike most electrocatalysts, the catalytic activity of Bi-VNS continuously increased, which was attributed to the in situ formation of BOC. The coexistence of BOC and bismuth in the cathode led to lattice strain and charge redistribution, thereby modulating the electrocatalyst's electronic structure and facilitating efficient charge transfer. DFT calculations revealed a change in the interaction between the *OCHO intermediate and Bi atoms, shedding light on the mechanism of reaction intermediate stabilization. The activation of Bi-VNS proved advantageous for long-term electrolysis, driven by an efficient, low-cost perovskite-silicon tandem solar cell. Through the optimization of the PV-EC system, we achieved a high STF conversion efficiency of 10.8%. Remarkably, the initial STF was further enhanced to 11.5%, even in the presence of slight PV module decay. It is inferred that the beneficial reconstruction of Bi-VNS originated from the structural similarities between BOC and BiOI. Our findings offer valuable insights into electrocatalyst and system design for practical and applicable CO2RR powered by renewable energies. A promising direction for future research would be to integrate advanced PV cells with flow cell-based electrolyzers, paving the way for the development of a commercially applicable PV-EC system.Author contributions
W. S. Cheon contributed to the research as a first author. He conducted experiments and wrote the manuscript under the supervision of H. W. Jang and J. Y. Kim. All authors contributed to the general discussion. S. G. Ji: resources, methodology. J. Kim: software, validation, formal analysis. S. Choi: formal analysis, methodology. J. W. Yang: methodology. S. E. Jun: conceptualization. C. Kim: methodology, conceptualization. J. Bu: visualization, formal analysis. S. Park: visualization, formal analysis. T. H. Lee: formal analysis. J. Wang: formal analysis. J. Y. Kim: formal analysis. S. A. Lee: investigation.Data availability
The data supporting the findings of this study are available within this article and its ESI,† or from the corresponding authors upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the KRISS (Korea Research Institute of Standards and Science) MPI Lab. Program. And this work was also supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT), South Korea (RS-2024-00405016, RS-2024-00421181). The Inter-University Semiconductor Research Center and Institute of Engineering Research at Seoul National University provided research facilities for this work.References
- J. He and C. Janáky, ACS Energy Lett., 2020, 5, 1996–2014 CrossRef CAS.
- S. Xu and E. A. Carter, Chem. Rev., 2019, 119, 6631–6669 CrossRef CAS.
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS.
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- Y. Cheng, P. Hou, X. Wang and P. Kang, Acc. Chem. Res., 2022, 55, 231–240 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- K. Fan, Y. Jia, Y. Ji, P. Kuang, B. Zhu, X. Liu and J. Yu, ACS Catal., 2020, 10, 358–364 CrossRef CAS.
- Y. Li, J. Chen, S. Chen, X. Liao, T. Zhao, F. Cheng and H. Wang, ACS Energy Lett., 2022, 7, 1454–1461 CrossRef CAS.
- Y. Xing, X. Kong, X. Guo, Y. Liu, Q. Li, Y. Zhang, Y. Sheng, X. Yang, Z. Geng and J. Zeng, Adv. Sci., 2020, 7, 1902989 CrossRef CAS.
- J. Huang, N. Hörmann, E. Oveisi, A. Loiudice, G. L. De Gregorio, O. Andreussi, N. Marzari and R. Buonsanti, Nat. Commun., 2018, 9, 3117 CrossRef.
- S. G. Ji, I. J. Park, H. Chang, J. H. Park, G. P. Hong, B. K. Choi, J. H. Jang, Y. J. Choi, H. W. Lim, Y. J. Ahn, S. J. Park, K. T. Nam, T. Hyeon, J. Park, D. H. Kim and J. Y. Kim, Joule, 2022, 6, 2390–2405 CrossRef CAS.
- H. Chen, J. L. Muros-Cobos and A. Amirfazli, Rev. Sci. Instrum., 2018, 89, 35117 CrossRef CAS.
- M. Schreier, L. Curvat, F. Giordano, L. Steier, A. Abate, S. M. Zakeeruddin, J. Luo, M. T. Mayer and M. Grätzel, Nat. Commun., 2015, 6, 7326 CrossRef CAS PubMed.
- J. W. Yang, I. J. Park, S. A. Lee, M. G. Lee, T. H. Lee, H. Park, C. Kim, J. Park, J. Moon, J. Y. Kim and H. W. Jang, Appl. Catal., B, 2021, 293, 120217 CrossRef CAS.
- D. K. Lee and K.-S. Choi, Nat. Energy, 2018, 3, 53–60 CrossRef CAS.
- S. A. Lee, J. W. Yang, S. Choi and H. W. Jang, Exploration, 2021, 1, 20210012 CrossRef PubMed.
- D. Wu, G. Huo, W. Chen, X.-Z. Fu and J.-L. Luo, Appl. Catal., B, 2020, 271, 118957 CrossRef CAS.
- X. Chen, H. Chen, W. Zhou, Q. Zhang, Z. Yang, Z. Li, F. Yang, D. Wang, J. Ye and L. Liu, Small, 2021, 17, 2101128 CrossRef CAS.
- J. Xu, S. Yang, L. Ji, J. Mao, W. Zhang, X. Zheng, H. Fu, M. Yuan, C. Yang, H. Chen and R. Li, Nano Res., 2023, 16, 53–61 CrossRef CAS.
- G. Liu, B. Wang, X. Zhu, P. Ding, J. Zhao, H. Li, Z. Chen, W. Zhu and J. Xia, Small, 2022, 18, 2105228 CrossRef CAS PubMed.
- S. E. Jun, S.-P. Hong, S. Choi, C. Kim, S. G. Ji, I. J. Park, S. A. Lee, J. W. Yang, T. H. Lee, W. Sohn, J. Y. Kim and H. W. Jang, Small, 2021, 17, 2103457 CrossRef CAS.
- P. Su, W. Xu, Y. Qiu, T. Zhang, X. Li and H. Zhang, ChemSusChem, 2018, 11, 848–853 CrossRef CAS.
- F. Yang, A. O. Elnabawy, R. Schimmenti, P. Song, J. Wang, Z. Peng, S. Yao, R. Deng, S. Song, Y. Lin, M. Mavrikakis and W. Xu, Nat. Commun., 2020, 11, 1088 CrossRef CAS PubMed.
- A. Xu, D. Wei, X. Chen, T. Yang, Y. Huang, H. He and J. Xu, Chem. Eng. J., 2023, 452, 139227 CrossRef CAS.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS.
- Z. Chen, K. Mou, X. Wang and L. Liu, Angew. Chem., Int. Ed., 2018, 57, 12790–12794 CrossRef CAS.
- A. H. Shah, Y. Wang, A. R. Woldu, L. Lin, M. Iqbal, D. Cahen and T. He, J. Phys. Chem. C, 2018, 122, 18528–18536 CrossRef CAS.
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen, X. Gu, R. Zhang, W. Zhou and Y. Gong, Nat. Commun., 2022, 13, 1877 CrossRef CAS PubMed.
- L. Li, A. Ozden, S. Guo, F. P. García de Arquer, C. Wang, M. Zhang, J. Zhang, H. Jiang, W. Wang, H. Dong, D. Sinton, E. H. Sargent and M. Zhong, Nat. Commun., 2021, 12, 5223 CrossRef CAS.
- W. Choi, D. H. Won and Y. J. Hwang, J. Mater. Chem. A, 2020, 8, 15341–15357 RSC.
- T. Fan, W. Ma, M. Xie, H. Liu, J. Zhang, S. Yang, P. Huang, Y. Dong, Z. Chen and X. Yi, Cell Rep. Phys. Sci., 2021, 2, 100353 CrossRef CAS.
- Y. Zhang, X. Zhang, Y. Ling, F. Li, A. M. Bond and J. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13283–13287 CrossRef CAS PubMed.
- P. F. Liu, M. Y. Zu, L. R. Zheng and H. G. Yang, Chem. Commun., 2019, 55, 12392–12395 RSC.
- L. Lin, X. He, X.-G. Zhang, W. Ma, B. Zhang, D. Wei, S. Xie, Q. Zhang, X. Yi and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202214959 CrossRef CAS PubMed.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li, S. Smart, Z. Zhu and Z. Yuan, Appl. Catal., B, 2021, 286, 119945 CrossRef CAS.
- W. S. Cho, D. M. Hong, W. J. Dong, T. H. Lee, C. J. Yoo, D. Lee, H. W. Jang and J.-L. Lee, Energy Environ. Mater., 2022, e12490 Search PubMed.
- R. P. Jansonius, L. M. Reid, C. N. Virca and C. P. Berlinguette, ACS Energy Lett., 2019, 4, 980–986 CrossRef CAS.
- X. Wei, Z. Li, H. Jang, M. G. Kim, Q. Qin and X. Liu, Sci. China Mater., 2023, 66, 1398–1406 CrossRef CAS.
- P.-F. Sui, C. Xu, M.-N. Zhu, S. Liu, Q. Liu and J.-L. Luo, Small, 2022, 18, 2105682 CrossRef CAS.
- G. Piao, S. H. Yoon, D. S. Han and H. Park, ChemSusChem, 2020, 13, 698–706 CrossRef CAS.
- X. Chen, J. Chen, H. Chen, Q. Zhang, J. Li, J. Cui, Y. Sun, D. Wang, J. Ye and L. Liu, Nat. Commun., 2023, 14, 751 CrossRef CAS PubMed.
- N. Li, P. Yan, Y. Tang, J. Wang, X.-Y. Yu and H. Bin Wu, Appl. Catal., B, 2021, 297, 120481 CrossRef CAS.
- Y. Zhao, X.-G. Zhang, N. Bodappa, W.-M. Yang, Q. Liang, P. M. Radjenovica, Y.-H. Wang, Y.-J. Zhang, J.-C. Dong, Z.-Q. Tian and J.-F. Li, Energy Environ. Sci., 2022, 15, 3968–3977 RSC.
- T. Shi, D. Liu, N. Liu, Y. Zhang, H. Feng and Q. Li, Adv. Sci., 2022, 9, 2204472 CrossRef CAS.
- R. Amirbeigiarab, J. Tian, A. Herzog, C. Qiu, A. Bergmann, B. Roldan Cuenya and O. M. Magnussen, Nat. Catal., 2023, 6, 837–846 CrossRef CAS.
- Q. Hu, H. Zhao and S. Ouyang, Phys. Chem. Chem. Phys., 2017, 19, 21540–21547 RSC.
- S. Burikov, T. Dolenko, S. Patsaeva, Y. Starokurov and V. Yuzhakov, Mol. Phys., 2010, 108, 2427–2436 CrossRef CAS.
- L. Huang, G. Gao, C. Yang, X.-Y. Li, R. K. Miao, Y. Xue, K. Xie, P. Ou, C. T. Yavuz, Y. Han, G. Magnotti, D. Sinton, E. H. Sargent and X. Lu, Nat. Commun., 2023, 14, 2958 CrossRef CAS PubMed.
- T. Zhonghao, Z. Jianling, Y. Yisen, Z. Jiajun, Z. Yingzhe, H. Jingyang, W. Yanyue and S. Zhuizhui, CCS Chem., 2023, 6, 100–109 Search PubMed.
- W. Shan, R. Liu, H. Zhao, Z. He, Y. Lai, S. Li, G. He and J. Liu, ACS Nano, 2020, 14, 11363–11372 CrossRef CAS PubMed.
- X. Fu, J. Wang, X. Hu, K. He, Q. Tu, Q. Yue and Y. Kang, Adv. Funct. Mater., 2022, 32, 2107182 CrossRef CAS.
- H. Yang, N. Han, J. Deng, J. Wu, Y. Wang, Y. Hu, P. Ding, Y. Li, Y. Li and J. Lu, Adv. Energy Mater., 2018, 8, 1801536 CrossRef.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- Y. Wang, B. Wang, W. Jiang, Z. Liu, J. Zhang, L. Gao and W. Yao, Nano Res., 2022, 15, 2919–2927 CrossRef CAS.
- X. An, S. Li, X. Hao, X. Du, T. Yu, Z. Wang, X. Hao, A. Abudula and G. Guan, Sustainable Energy Fuels, 2020, 4, 2831–2840 RSC.
- Y. Wang, L. Cheng, J. Liu, C. Xiao, B. Zhang, Q. Xiong, T. Zhang, Z. Jiang, H. Jiang, Y. Zhu, Y. Li and C. Li, ChemElectroChem, 2020, 7, 2864–2868 CrossRef CAS.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, P. Zhao, X. Xue, R. Chen, S. Yang, J. Ma, J. Liu and Z. Jin, Nano Energy, 2018, 53, 808–816 CrossRef CAS.
- H. Xie, T. Zhang, R. Xie, Z. Hou, X. Ji, Y. Pang, S. Chen, M.-M. Titirici, H. Weng and G. Chai, Adv. Mater., 2021, 33, 2008373 CrossRef CAS.
- X. Zhang, X. Sun, S.-X. Guo, A. M. Bond and J. Zhang, Energy Environ. Sci., 2019, 12, 1334–1340 RSC.
- W. Zhang, S. Yang, M. Jiang, Y. Hu, C. Hu, X. Zhang and Z. Jin, Nano Lett., 2021, 21, 2650–2657 CrossRef CAS.
- T. Zhou, H. Zhang, X. Zhang, W. Yang, Y. Cao and P. Yang, J. Phys. Chem. C, 2020, 124, 20294–20308 CrossRef CAS.
- J. Ma, J. Yan, J. Xu, J. Ni, H. Zhang and L. Lu, Chem. Eng. J., 2023, 455, 140926 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ey00209a |
| This journal is © The Royal Society of Chemistry 2025 |