
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning for degradation: the importance of considering biotic and abiotic polymer degradation†
Omar
Tantawi
 a,
Wontae
Joo
b,
Elijah E.
Martin
a,
Sarah H. M.
Av-Ron
b,
K'yal R.
Bannister
b,
Kristala L. J.
Prather
b,
Bradley D.
Olsen
b and
Desiree L.
Plata
*a
a,
Wontae
Joo
b,
Elijah E.
Martin
a,
Sarah H. M.
Av-Ron
b,
K'yal R.
Bannister
b,
Kristala L. J.
Prather
b,
Bradley D.
Olsen
b and
Desiree L.
Plata
*a
aDepartment of Civil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA. E-mail: dplata@mit.edu
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA, USA
First published on 10th April 2025
Abstract
Considering the increasing global plastic demand, there is a critical need to gain insight into environmental processes that govern plastic degradation in order to inform novel design of sustainable polymers. Current biological degradation testing standards focus on formation of CO2 (i.e., mineralization) alone as a diagnostic, ultimately limiting identification of structure–degradation relationships in a timely fashion. This work developed a sequential abiotic (i.e., photodegradation and hydrolysis) and biotic degradation test and applied it to a suite of 18 polymers, including ten lab produced, novel polyhydroxyalkanoate polyesters, and eight commercially available, bio-based (i.e., polylactic acid and poly-3-hydroxybutyrate) and fossil-derived (i.e., polystyrene, polypropylene, low density polyethylene, poly(ethylene terephthalate) and tire rubber) polymers. Biomineralization alone following standard methods (i.e., ASTM 6691-17, ISO 23977-1 2020) underestimated polymer degradation up to two-fold over 28 days. Simulated sunlight enhanced the overall polymer degradation by mobilizing dissolved organic carbon (DOC). After photoirradiation, up to 100% of released dissolved organic carbon was bioavailable for marine microbes over 14 days. Photodegradation and hydrolysis could be explained by structural drivers in the commodity polymers, and the lab-synthesized polymers illustrated a limit to total degradation beyond which no enhancements in degradation were achieved. Taken together, this workflow allows for relatively fast experimental determination of environmentally relevant stimuli to help support eventual elucidation of structure–property relationships for enhanced a priori design of degradable polymers.
Environmental significanceGrowing plastic consumption demands an improved fundamental understanding of the environmental processes that degrade polymers. Standard biodegradation tests focus mainly on mineralization to CO2 and can underestimate plastic fate in natural systems. This study illustrates that combined abiotic (photodegradation and hydrolysis) and biotic (biodegradation and mineralization) processes substantially contribute to degradation, mobilizing dissolved organic carbon that marine microbial organisms can consume. Adopting an expanded definition of “environmental degradability”—tracking CO2, biomass, and dissolved carbon release—will enable more comprehensive test frameworks for elucidation of the mechanisms by which the polymer structure imparts degradability to a material. Such holistic approaches can guide new design strategies, reduce pollution, and better align emerging materials with the Earth's ecosystems, helping to meet society's growing material demands sustainably. |
Introduction
Due to the design of waste management infrastructure or complete lack thereof in some regions of the world, the United Nations estimates that over 50% of all plastics produced since 1950 have accumulated in the environment, including landfills and oceans.1 Indeed, all industrial chemicals and materials have some release to the environment, and without implicit degradation that exceeds the rate of input, accumulation will occur.2 Despite the importance of the challenge, current understanding of the fate of polymeric materials in the marine environment is not sufficient to inform design-for-environment guidelines.3 This is due to a limited capacity to accurately simulate natural systems in lab experiments,4 the complexity of natural environments, and undisclosed additives in commercial polymers, and a lack of systematic data collection to relate chemical structural properties to environmental degradation rates.Environmental degradation mechanisms for polymers can be classified as abiotic or biotic, where abiotic degradation refers to transformations that occur due to non-living stimuli, such as light, physical stress, or chemical modifications. Two examples include hydrolysis and oxidation, which can be accelerated by light, heat, or some combination thereof.4,5 Biotic degradation refers to the ability of organisms, often microbial organisms, to consume carbon-rich polymers and convert those solid materials to biomass, carbon dioxide (CO2), water (H2O) and/or dissolved organic carbon (DOC).6 Despite these known myriad endpoints, biodegradation is often measured as CO2 generation alone (or corresponding oxygen (O2) consumption), which provides a limited view of what is truly bioavailable and correspondingly limits the development of structure–degradation relationships by obfuscating the distribution in degradation rates that may arise with modest structural modifications. While seldom considered together in standard tests, in the natural world, abiotic and biotic degradation mechanisms likely work in tandem to enhance mineralization of polymer-derived carbon.7 Investigating both processes may result in a more comprehensive representation of environmental weathering pathways and yield degradation rate results that can be related to chemical structures of the reactive substrates, ultimately serving to inform the design of more environmentally compatible polymers.
Currently, there are several standards available to measure polymer degradability, each of which simulates distinct environments (e.g., non-floating,8,9 buried in sandy marine sediment,10 sediment–seawater interface11,12 and pelagic11,13,14) (Table S1†). A few recently published standards attempt to provide better simulation of environmental conditions by accounting for biotic and abiotic degradation implicitly (i.e., physical and biological weathering), but typically only via visual assessment or dry mass change.9,12 These approaches have several limitations, including (1) poor-reproducibility due to the diversity of inocula, leading to a lack of intercomparable results, (2) slow degradation kinetics as a result of operating in the dark and often with biologically dilute inocula, translating to testing that takes between 28 days and up to 2 years, and (3) providing low temporal and chemical resolution (e.g., monitoring CO2 evolution alone), limiting understanding of kinetic processes that can be related to the material structure. Here, it is worth noting that these test methods are often used as regulatory assays rather than to guide innovation. Nevertheless, these factors limit the utility of the results to inform design of new materials with engineered degradation rates, particularly by limiting the size of the intercomparable data set, the pace of discovery, and the resolution of the data (i.e., the distribution between kinetic rates or total degradability as a function of polymer structure). That is, when many polymers return “non-degradable” results (due to the use of CO2 alone as an endpoint, with a dilute inoculum, in the dark), there is insufficient variation in degradation rates as a function of material type to relate the structure to environmental performance.
Indeed, CO2 is often a minor product of polymer degradation in standard tests, and one of the primary products of abiotic degradation can be DOC.15 Previous work to characterize the impact of photo-transformations on DOC fate provides somewhat conflicting results. For instance, Romera-Castillo et al.16 first reported the rate of microbial consumption of photo-weathered DOC as slower compared to that of non-photoweathered DOC generated from low density polyethylene (LDPE), high density polyethylene (HDPE), polyethylene (PE) and polypropylene (PP). This finding was further echoed by Zhu et al.,17 who hypothesized that photo-weathered DOC released from PE could have an inhibitory effect on marine microbial community growth. However, Romera-Castillo et al.18 later reported higher derived DOC bioavailability in irradiated samples, contradicting their earlier findings,16 and this could have indicated a sensitivity to either the experimental photoweathering, the polymer variability (e.g., due to additives), or known heterogeneities in marine inocula. These factors could be a cause for the great distribution of results present in the literature.19 Others have shown widely variable influences of polymer-derived DOC on marine microorganism growth.18,20–36 For example, plastic leachate was found to impair growth and oxygen production in Prochlorococcus (a photosynthetic bacterium),23,37 but enhance photosynthesis in four other marine microalgal species.31 Overall, while it is important to consider potential impairments in the microbial community, these prior results support the inclusion of mobilized DOC in the quantification of bioavailable, polymer-derived carbon, and that DOC formation is a critical step in polymer breakdown.
This work considered DOC generation from abiotic degradation (i.e., photodegradation and hydrolysis), giving a distributed set of total degradation and providing a better understanding of polymer breakdown mechanisms. Applying this approach to a series of lab-synthesized materials, this study illustrated the utility of such data to guide more informed decisions during the material development phase. Note that commercial polymers, with a variety of additives, may exhibit distinct behaviors compared to the relatively pure materials used here, which are preferable for a systematic study of structure–function relationships.38 The methods described provide an experimental assay of biotic and abiotic degradation together, allowing for a more representative assessment of degradation mechanisms of new or proposed materials, with the ultimate goal of informing sustainable polymer invention practices.
Materials and methods
Materials and the overarching approach
Test materials included a suite of petroleum-based polymers, commercially available biopolymers, and lab-synthesized polymers of systematically varied composition (Table 1). All polymers acquired were sold as the highest purity commercially available materials, which was desired to isolate the impact of the molecular structure of the polymers on degradation, except for tire rubber and polyhydroxyalkanoate straws (Phade®, which were colored light blue). While all industrial materials should be characterized independently for purity, comprehensive identification of additives in a non-target fashion was not possible; crystallinity and molecular weight distribution were investigated using methods described in the ESI.† The authors acknowledge here that off-the-shelf products contain many diverse additives that substantively impact environmental lifetime, often by design (e.g., UV stabilizers), and caution that the lifetimes reported here may be distinct from that of commercial products.15 Lab-synthesized materials were prepared via catalyst-free polycondensation using different starting ratios of 3-hydroxypivalic acid (3HP) and 6-hydroxyhexanoic acid (6HA).39 The shorthand P(3HPxx-6HAyy) indicates the relative proportions (by mass) of 3HP (given by xx) and 6HA (given by yy) in a series of poly[(hydroxypivalic acid)-r-(hexanoic acid)]. The polymer crystallinity dropped on increasing 6HA loading from 74% to less than 1% for P(3HP100-co-6HA00) and P(3HP65-co-6HA35), respectively. The melting and glass transition temperatures ranged from 154 ± 13 °C and 48 ± 6 °C to 50 ± 4 °C and −39 ± 1 °C for P(3HP100-co-6HA00) and P(3HP65-co-6HA35), respectively (Table S2†).| Polymer (material) name | Label | Structure | Supplier/acquisition date |
|---|---|---|---|
| a Poly[(hydroxypivalic acid)-r-(hexanoic acid)] is a polymer formed by the copolymerization of hydroxypivalic acid and hydrodxyhexanoic acid. “r” is a qualifier used to denote a polymer made of more than one monomer (i.e., a copolymer). b Commercial polyhydroxyalkanoate phade straws were purchased and cut into small square shaped pieces using a new, acetone washed razor blade. c Micronized Tire Rubber40 was obtained from MRH (Mülsener Rohstoff-und Handelsgesellschaft mbH, Mülsen, Germany). | |||
| 3-Hydroxy-2,2-dimethylpropanoic acid (3-hydroxypivalic acid) | 3 HP |
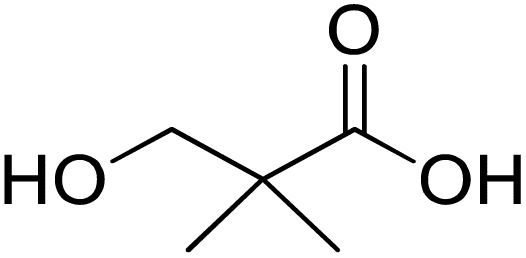
|
Sigma Aldrich |
| 6-Hydroxyhexanoic acid | 6 HA |

|
Sigma Aldrich |
| Poly(hydroxypivalic acid) | P(3HP100-co-6HA00) |

|
— |
| Poly(hydroxyhexanoic acid) | P(3HP00-co-6HA100) |

|
— |
| Poly[(hydroxypivalic acid)-r-(hexanoic acid)]a | P(3HPxx-co-6HAyy) |

|
— |
| Low density polyethylene | LDPE |

|
Sigma Aldrich (427772-250 G) 10/18/2020 |
| Poly[(R)-3-hydroxybutyric acid] | P3HB |

|
Sigma Aldrich (363502-10 G) 10/18/2020 |
| Commercial polylactic acid | NPLA |

|
NatureWorks (INEGO biopolymer 2003D) 10/18/2020 |
| Polypropylene | PP |

|
Sigma Aldrich (428116-250 G) 10/18/2020 |
| Poly(ethylene terephthalate) | PET |
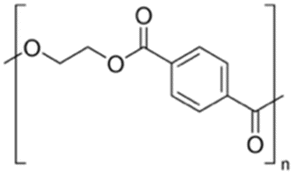
|
Sigma Aldrich (429252-250 G) 10/18/2020 |
| Polystyrene | PS |
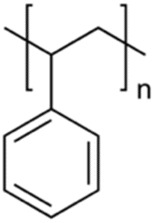
|
Sigma Aldrich (331651-500 G) 11/20/2021 |
| Polyhydroxyalkanoates (commercial phade straws)b | PHA | N/A | Phade 7.75′′ Jumbo straws (10/20/2021) |
| Tire rubberc | TR | N/A | MRH |
Pre-combusted (450 °C in air for 8 h) sample quartz cuvettes (Technical Glass Products; 3 cm dia. × 10 cm length) were filled with 5 mg of test polymer and 30 mL of synthetic sea water (prepared according to ASTM D1141-98 (41)), then closed with Teflon-lined polypropylene caps, and held horizontally in a solar simulator (described below and in the ESI†) on orbital shakers for 14 days. For each polymer material, six total samples (three irradiated and three covered with aluminum foil as dark controls) were prepared. On day 14, the samples were filtered using 0.45 μm, pre-combusted glass fiber filters (GFF; Whatman) to separate particles from colloids and dissolved compounds. (No ultracentrifugation or nanofiltration was conducted to remove colloidal materials or those below 0.45 μm; little is known about the mobilization rates from larger particles of plastics versus nano-sized particles of the same materials, although this is the subject of subsequent study from the Plata research group). Aliquots of the filtered liquid were split, where 10 mL was transferred to 60 mL serum borosilicate bottles (Wheaton) for further experimentation (see the Biodegradation testing section below) and the remainder were stored in pre-combusted, 40 mL EPA VOA vials at 4 °C for further experimentation and TOC analysis (Scheme 1). Collected filters, along with their particulate content, were transferred into 60 mL serum bottles for further experimentation. A traditional biodegradation test of the polymer was conducted in parallel according to the modified ASTM 6691-17 standard (28 days).
 | ||
| Scheme 1 Experimental schematic of sequential polymer photodegradation and biodegradation. (A) Polymers were irradiated for 14 days under simulated sunlight, and then the dissolved and particulate (over 0.45 μm) fractions were separated and incubated with natural sea water (NSW) for 14 additional days (I and II, respectively). (B) A traditional biodegradation test of the polymer was conducted in parallel according to the modified ASTM 6691-17 standard (28 days). DOCp, DOC0 and DOCf represent the amount of polymer-derived DOC, total DOC at time 0 and total DOC at the end of the experiment, respectively. DOC measurements were conducted to evaluate carbon mobilization into synthetic sea water and DOC assimilated by microorganisms after incubation (I). CO2 measurements were conducted in all biodegradation tests. The natural sea water (NSW) inoculum contributed no more than 20% of the total organic carbon after the addition of the test polymer, following the recommended standard. The illustration was produced using BioRender.42 | ||
Photoweathering of polymers
All photodegradation experiments were conducted in a custom-built solar simulator, which is described in detail in the ESI.† Briefly, this low-cost (less than $3000) simulator emulated the UV and visible ranges of the solar output spectrum, had a wide operating area (5000 cm2), and was evaluated using an Ocean Optics FLAME-S-XR1-ES to have less than 10% variability over the irradiated surface. The solar simulator irradiance dose across the UV and visible light range is 0.91 kW h m−2 h−1, comparable to the ASTM G173 standard total irradiance of 0.9 and 1 kW h m−2 h−1 for direct and hemispherical tilted spectra, respectively.43 The power delivered by the solar simulator is 21.9 kW h m−2 day−1. The average daily solar power in Boston, Massachusetts, ranges from 4.93 to 7.46 kW h m−2 day−1 in December and September, respectively.44 The full solar simulator description and characterization spectrum are provided in the ESI (Fig. S1, S2 and Tables S3–S5†). Here, the authors note that higher fluency is generally accepted for accelerated photoweathering protocols, and because the wavelengths of light are not changed, the energies of the associated transitions and subsequent reactions hold.15,45–49 After 14 days of irradiation, DOC released from studied polymers was quantified (see Carbon analysis, below) and assessed for bioavailability (see Biodegradation testing, below) and this was related to traditional biodegradation testing (Scheme 1). The temperature during the photo-degradation and leaching (dark controls) was 25 ± 5 °C; all water was equilibrated with air, and had a pH of 8.04. We prepared and analyzed triplicate samples and triplicate blanks that included synthetic seawater without added polymers.Biodegradation testing
Aerobic biodegradation experiments were conducted on photoweathered polymers, released DOC, and unweathered polymers using seawater collected from Boston, MA near Castle Island (42°19′49.9′′N 71°00′54.8′′W). Note that two distinct inocula were used: one collected on December 10, 2021 for commercial polymers and one collected on March 30, 2022 for all laboratory-synthesized polymers. Hence, the individual sets were self-consistent, but intercomparisons between the two are not valid due to potential variations in the marine community at the time of sampling. A probe (YSI 556 handheld Multiparameter Instrument) was submerged in a 500 mL beaker to determine pH, temperature, and specific conductance (Table S6†). Then, seawater was collected in two pre-combusted, 3 L Duran glass media bottles and returned to the laboratory. Within an hour of collection, the seawater was filtered into clean, pre-combusted 3 L glass media bottles using a paper filter (Whatman, England; Cat. No. 1440 090; pore size 2.7 μm) to remove coarse particles. The bottles were left slightly ajar and placed on a shaker table (80 rpm) for 7 days to reduce background dissolved organic matter (DOM) and/or allow enrichment of microbial culture, as recommended by the ASTM standard. On day 7, 0.1 g L−1 of monopotassium phosphate (KH2(PO4)) and 0.05 g L−1 ammonium chloride (NH4Cl) were added as inorganic nutrients. All blanks and experimental samples were tested in triplicate in 60 mL serum bottles sealed with bromobutyl rubber septa (Bellco Glass, NJ) secured with aluminum crimp caps (Supetco) for 14 days (following photoirradiation) or 28 days (traditional biodegradation testing) as described below.Following 14 days of photoweathering (Scheme 1), the samples were filtered using 0.45 μm GFFs. The filters, along with any remaining particles, were transferred into clean, 60-mL serum bottles, and 10 mL aliquots of post-filtration liquid with DOC were transferred to additional clean, 60-mL serum bottles. To preserve a low available-carbon-to-native seawater DOC ratio of less than 20% (recommended by aerobic biodegradation ASTM 6691-17), 18 mL of natural seawater were added to the filter-and-particle containing serum bottles, whereas 0.5 mL of natural seawater brought to a final volume of 18 mL using artificial seawater was added to the DOC samples. CO2 measurements were carried out at 5, 7 and 14 days. On day 14, after the CO2 measurement, serum bottles were opened, and the solution was filtered using a 0.45 μm GFF to measure total DOC loss and pH during the biodegradation, where the net change in DOC concentration was attributed to mineralization and biomass formation. Particulate organic carbon (POC) was not measured because the particulate matter was carried forward in the degradation experiments.
As a point of comparison, unweathered polymers were subjected to traditional biodegradation testing over 28 days (modified ASTM 6691-17); these samples combined 5 mg of native polymer materials (Table 1) and 18 mL of natural seawater inoculum. The temperature during biodegradation experiments was 20 ± 5 °C.
Carbon analysis
DOC was measured via an Elementar Vario-EL analyzer (TOC analyzer) with a liquid interface. Calibration was conducted using a potassium hydrogen phthalate standard solution (LabCem, USA). The limit of detection (LOD) and limit of quantification (LOQ) were determined to be 0.13 μg L−1 and 0.27 μg L−1, respectively (computed according to Harris et al., 2020 (ref. 50)). Briefly, the standard deviation (SD) was calculated from seven replicate measurements of the lowest calibration point (0.5 mg L−1). This standard deviation was then used to compute the method's LOD and LOQ. The LOD was calculated as 3 times the standard deviation divided by the slope of the calibration curve, while the LOQ was calculated as 7 times the standard deviation divided by the slope of the calibration curve. The samples were first filtered through 0.45 μm GFFs (Kinesis KX, Canada) and diluted 5 times with Milli-Q water to reduce the salt content. The samples were then acidified with 3 drops of 12.1 M (37% w/w) hydrochloric acid and analyzed via a TOC analyzer. The samples (0.5 mL aliquots) were analyzed in triplicate directly after collection, and a flush sequence was conducted between samples. Blanks and control experiments were conducted for all experimental conditions. We prepared blanks that did not include polymer derived carbon (polymer particles or DOC) with a marine inoculum and controls that had polymer derived carbon (polymer particles or DOC) without a marine inoculum. Controls of a marine inoculum alone and polymer carbon alone were also tested.Mineralization to CO2 was measured via an SRI 8610C gas chromatograph (GC) equipped with a methanizer connected to a flame ionization detector calibrated with authentic gas standards using standard sample loops. Measurements were conducted on day 5, 7, and 14 by extracting 1 mL of headspace (through the butyl rubber stoppers) using a gas-tight syringe (Hamilton Company) and replacing the displaced volume with lab air. After the samples were opened, we measured the pH to account for the dissolved inorganic carbon (DIC). All samples' pH ranged from 7 to 8 at the end of the biodegradation experiments.
Results and discussion
Consideration of abiotic and biotic degradation processes
For ease of access and to reduce cost, the majority of standard biodegradation assays rely on the measurement of CO2 production or O2 consumption alone as an indicator of polymer remineralization (Table S1†).13,14,51 Applying one such standard approach with a natural sea water inoculum, commodity polymers poly(ethylene terephthalate) (PET), polystyrene (PS), LDPE, PP, and tire rubber (TR) exhibited less than 7 ± 2 (by mass) mineralization to CO2 over 28 days (Fig. 1; exact values were 0.01 ± 1 (insignificant), 0.01 ± 1 (insignificant), 2 ± 1, 0.4 ± 0.3 (insignificant), and 7 ± 2%, respectively). Similarly, one commercially available biopolymer, NatureWorks™ poly(lactic acid) (NPLA) was recalcitrant in this marine inoculum, showing no additional mineralization compared to LDPE (less than 0.6 ± 1%). In contrast, the polymers polyhydroxyalkanoate (PHA, a biopolymer) straw fragment and poly(R-3-hydroxybutyric acid) (P3HB, a biopolymer) showed 17 ± 6 and 80 ± 7% mineralization to CO2, respectively. Note that the straws contain a light blue dye, and the effect of this dye is unknown but certainly would be expected to be important to photo transformation rates.15 Efforts to obtain dye-free PHA were unsuccessful due to supply chain limitations, which may be relieved in the future and enable further study. While the observed results were consistent with previous understanding of broad categories of degradation52 (i.e., more or less biodegradable), CO2 generation/mineralization (headspace CO2 and DIC; see ESI Section 3†) represents only a single possible product of polymer-derived carbon and excludes the possibility that carbon mobilized from the polymer as dissolved organic matter (DOC) will be bioavailable as well. Zakem et al. recently argued that nearly 84% of marine dissolved organic matter is bioavailable over some timescale,53 but it remains to be demonstrated that polymer-derived DOC can be similarly utilized and for which polymer resins specifically. To account for the carbon mobilization pathway via DOC, we subjected test polymers to 14 days of photoweathering and quantified the possible transfer of solid carbon to DOC via photolysis, hydrolysis, or desorption (i.e., leaching) (Table S7†). Accounting for this pathway, LDPE, TR, and PHA all showed enhanced carbon release, where LDPE and TR underwent a more than 2-fold increase (from 2 ± 1 to 12 ± 2% and 7 ± 2 to 12 ± 4%, respectively) and PHA mobilization increased modestly (from 17 ± 6 to 19 ± 4%). TR was not purchased as a high-purity polymer and certainly contains additives,54 and the release rates may include both polymer-derived and additive-derived materials. Note that LDPE is commonly a negative control for such releases, and other tests of LDPE in the same photoreactor showed differential release rates (2 ± 1% for 28 days) in the same photoreactor. High-purity polymers were used in all cases, so additive leachate should have been minimized. Differences in light scattering associated with polymer crystallinity (subject of subsequent study by the authors), additives and morphology would feasibly impact photochemical degradation and could potentially account for the LDPE-derived DOC mobilization.15 In contrast, there was no measurable photochemical, hydrolytic, or desorptive enhancement of carbon transfer from the particles to the dissolved phase for PET, PS, PP, or NPLA above the 0.001% of the added polymer detection limit. This is unsurprising for PP, but PET and PS should show some degree of photolability (i.e., light absorbing aromatic rings in the monomers terephthalic acid and styrene), and NPLA should be hydrolysable in principle (i.e., hydrolysable ester bonds).45,55 Indeed, previous studies of commercial PS56 and PET46 illustrated degradation in the presence of light. This apparent disparity could have resulted from previous work using higher light irradiance, strong UV wavelengths (i.e., UVC), a higher polymer ratio of DOC and/or polymers with photoactive additives.47,48 | ||
Fig. 1 Polymer degradation measured as mineralization to CO2 and dissolved organic carbon (DOC) versus mineralization to CO2 alone for (a) commercially available polymers and (b) laboratory-synthesized polymers. In panel (a), PET is poly(ethylene terephthalate); PP is polypropylene; PS is polystyrene; LDPE is low density polyethylene; PHA is polyhydroxyalkanoate Phade® straw fragment; TR is tire rubber; P3HB is poly(3-hydroxybutyrate); NPLA is Nature Works® polylactic acid (PLA). In panel (b), polymers with variable hydroxypivalic acid (3HP) and hexanoic acid (6HA) loading were prepared, and the shorthand P(3HPxx-6HAyy) indicates the relative proportions (by mass) of 3HP (given by xx) and 6HA (given by yy) in a series of poly[(hydroxypivalic acid)-r-(hexanoic acid)]. Circles symbolize these polymers, where the blue-to-red gradient corresponds to increasing proportions of 6HA. Squares indicate pure 3HP (blue) and pure 6HA (red). A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 relationship is shown by the solid dark line. Error bars represent standard deviation on triplicate measurements (data in Table S8†). :1 relationship is shown by the solid dark line. Error bars represent standard deviation on triplicate measurements (data in Table S8†). | ||
There are two noteworthy implications of the findings: first, accounting for mineralization alone may give a false indication of what is truly degradable in the environment, as DOC formation is an important step on the path to remineralization of carbon-derived plastics15,57 (see subsequent discussion of DOC bioavailability and fate). It is important to recognize that some mobilized carbon, over long timescales, will become CO2 whereas other materials may crosslink with other particles or form larger, non-degradable DOC structures.58 Second, accounting for carbon mobilization to the dissolved phase may produce a wider distribution of degradation rates, which is necessary to construct structure–degradation relationships to inform design. Specifically, elucidating the fundamental mechanisms of environmental degradation that can be related to the material structure will ultimately require a broad spectrum of polymers with variable structural or physicochemical properties (e.g., light absorption or molecular composition) and measurable distinction between corresponding transformation rates. Third, and documented elsewhere, the role of additives is clear in photochemical weathering, which must be considered in any efforts to produce intercomparable datasets from the literature or cross-laboratory comparisons.47,48,59,60 The study of pure polymers adds utility for first principles understanding to inform design, whereas the study of commercial materials may help constrain relevant environmental degradation timelines and/or the impact of additives.
To explore the possibility that systematic variation in the chemical structure could lead to measurable, and someday predictable, modifications in the degradation rate, we produced a series of polymers of variable 3-hydroxypivalic acid (3HP) and 6-hydroxyhexanoic acid (6HA) composition and subjected them to paired abiotic–biotic degradation testing. The shorthand P(3HPxx-6HAyy) indicates the relative proportions (by mass) of 3HP (given by xx) and 6HA (given by yy) used to produce the series of poly[(hydroxypivalic acid)-r-(hexanoic acid)] materials, where r indicates a co-polymer (Table 1). Increasing the fraction of hexanoic acid (6HA) led to systematically increasing rates of bioavailability, as measured by CO2 generation in the ranges of 12 ± 7% and 50 ± 3% mineralization for P(3HP100-6HA00) and P(3HP00-6HA100), respectively (Fig. 1). The impact of 6HA on increasing mineralization may be a result of the structure of bioavailable carbon; 6HA contains a linear alkane chain, whereas 3HP includes a branched structure at the alpha carbon position. Linear alkanes are known to be more bioavailable to marine organisms than branched structures,61 which tend to be more persistent.53 This is presumed to be due to steric limitations on enzymatic processing of branched structures.61 These structural differences could account for the higher CO2 generation in polymers with greater 6HA content. Accounting for the formation of DOC produced from a similar trend as CO2 generation, where total carbon released from the polymers increased with 6HA content, varying from 15 ± 8% and 54 ± 11% for P(3HP950-6HA5) and P(3HP40-6HA60), respectively. Although 3HP and 6HA introduce ester linkages throughout the polymer, no particular moiety enhanced DOC formation more than another; for example, P(3HP95-6HA05), P(3HP90-6HA10), P(3HP85-6HA15) and P(3HP80-6HA20) showed approximately equivalent enhancements of DOC release ranging from 1.2- to 2.1-fold higher carbon mobilization (DOC and CO2, as compared to CO2 alone), respectively. Following this release, it is important to understand what fraction of this polymer-sourced dissolved organic carbon is labile for consumption and/or mineralization.
Fate of polymer released DOC
To develop a quantitative understanding of the bioavailability and lability of DOC derived from polymers, the DOC pool was quantified before and after incubation with a marine inoculum to develop a mass balance. The contribution of biologically generated DOC was approximated via a control experiment, in which only a natural sea water inoculum was added to synthetic sea water. Here, bioavailability is defined as uptake into the marine biomass plus CO2 mineralization, and this was reported as a percentage of the original DOC (a detailed explanation of the calculation is available in ESI Section 3†). Among eight tested commercially available polymers, bioavailability and lability ranged from 2 ± 8% (i.e., insignificant) to 84 ± 18% (Fig. 2a). DOC derived from PP showed insignificant bioavailability (p-value > 0.05) compared to the control. DOC derived from LDPE, PET, TR, NPLA, PS, PHA and P3HB exhibited progressively increasing lability of 25 ± 17, 44 ± 19, 29 ± 10, 17 ± 7, 63 ± 19, 79 ± 12, and 84 ± 18% bioavailability, respectively (mean and standard deviation on triplicate measurements). Residual fractions ranged from less than 16% for P3HB-derived DOC to insignificant changes for PP-derived DOC. Considering the total fraction of carbon taken up as biomass or mineralized to CO2 illustrated that relatively large fractions of DOC were labile for many of the tested polymers.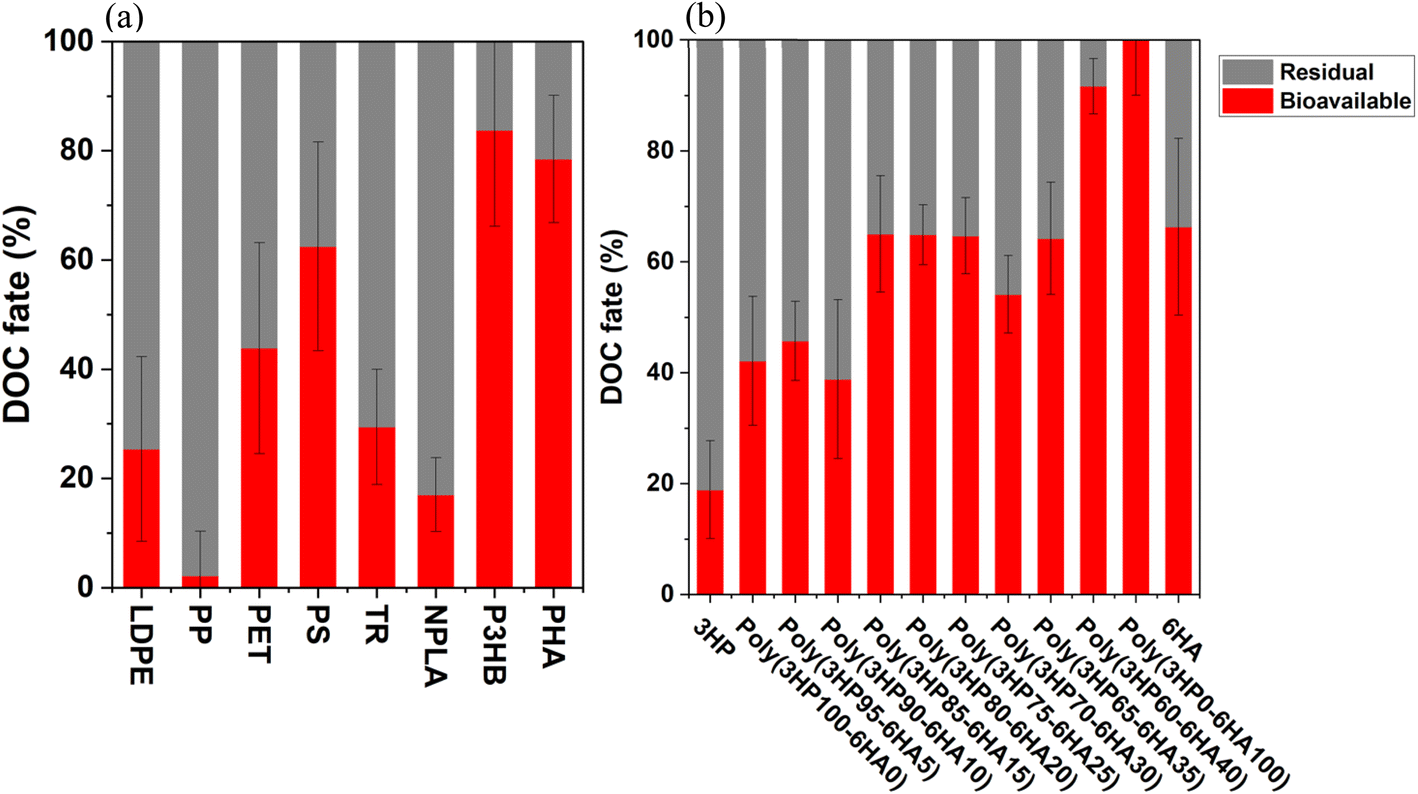 | ||
| Fig. 2 Biological fate of DOC derived from polymer photo-weathering for 14-days. DOC fate is measured as bioavailable (red) and residual (gray). Bioavailable carbon reflects the amount of DOC that is consumed by microorganisms (i.e., mineralization + biomass). The residual is the difference between the initial concentration of DOC and remaining (see ESI Section 3†). (a) Commercial polymers: PET, PP, PS, LDPE, PHA, TR, P3HB, and NPLA. (b) Lab-synthesized poly[(hydroxypivalic acid)-r-(hexanoic acid)] co-polymer with varying mass fractions of hydroxypivalic acid (3HP) and hexanoic acid (6HA). Error bars represent standard deviation on triplicate measurements. | ||
These results are challenging to compare to prior work due to variable approaches in experimentation. Nevertheless, there are some prior studies that sought to account for both photo and biological transformations and should be noted. First, Zhu et al.62 found that up to 76 ± 8% of DOC derived from photo-weathered expanded PS was bioavailable in 96 days, whereas this 14 day experiment gave 63 ± 20%. Furthermore, Romera-Castillo et al.18 found that DOC derived from PLA lacked elevated bioavailability relative to LDPE, PS and EPS, contrasting our results that NPLA, PHA and P(3HPxx-6HAyy) were more bioavailable. The broad heterogeneity among biodegradation studies could result from many influences: differences in polymer formulations (e.g., inclusion of photoactive additives), and known variability in marine microbial consortia in space, time, and competing carbon substrates. Thus, it is important that all biodegradation studies benchmark material environmental performance with well-known standards (e.g., both recalcitrant and labile polymers) prior to declaring materials as stable or bioavailable. Furthermore, intercomparison studies that inform material design principles or machine learning models relating the structure to the absolute transformation rate should draw from datasets where there is some consistency in the experimental approaches. Note that any studies that rely on natural inocula could be subject to poor interoperability, and efforts to build degradation datasets may ultimately require reliance on abiotic approaches to probe biological degradation potential (e.g., enzyme–substrate activity tests) or well-controlled cultures.
Using the same inoculum to compare bioavailability amongst systematically varied polymer structures, DOC lability varied but in a non-linear fashion (Fig. 2b). Specifically, the addition of bioavailable copolymer (e.g., 6HA) did not enhance the bioavailability of DOC derived from polymers in a systematic way quantitatively. For instance, DOC produced from P(3HP85-6HA15) and P(3HP65-6HA35) showed a similar bioavailability (uptake of DOC into bacterial biomass plus remineralization to CO2) of 65 ± 11% and 64 ± 10%, respectively (i.e., no change in bioavailability in spite of a 20% increase in 6HA loading). Similarly, a 60% increase in 6HA content between P(3HP60-6HA40) to P(3HP00-6HA100) resulted in only a modest increase in total lability of DOC (92 ± 5 to 100 ± 10%). In other cases, a minor (5%) addition of 6HA from P(3HP65-6HA35) to P(3HP60-6HA40) enhanced DOC bioavailability dramatically, from 64 ± 10 to 92 ± 5%, respectively. Interestingly, all polymerized co-polymers generated DOC with higher bioavailability than the respective monomers. For example, 3HP (a fully soluble monomer) showed 19 ± 9% bioavailability, whereas 42 ± 12% total C was bioavailable for consumption from P(3HP100-6HA00)-released DOC. Similarly, while 6HA showed 66 ± 16% lability, P(3HP00-6HA100) showed 100 ± 10% DOC mobilization into biomass uptake and CO2. Finally, the authors acknowledge that the self-consistency within each of these sets of experiments (Fig. 2a and b) does not necessarily imply that the order of degradation would be the same in every circumstance. It is well documented that several factors, including the composition of available DOC, influence the overall bacterial growth and associated degradation. Nevertheless, this is true for all biodegradation studies, and a strong effort to standardize degradation tests38 is needed alongside the development of deeper understanding of the fate of polymers in natural systems.
Comparison of dark and light-mediated processes on polymer bioavailability
One argument for processing standard biological tests in the dark is to avoid light-induced effects on the microbial species (e.g., death) and the test materials. To investigate the impact of photo-weathering on the overall carbon mobilized from the test materials and the impact on the growth of the surrounding marine microbes, we compared dark controls subjected to standard biodegradation tests to photo-irradiated samples treated with subsequent biological testing. First, DOC liberated by photo-processing was as or more bioavailable than that in dark biodegradation experiments alone (i.e., sunlight contributed to a net enhancement of carbon mobilization, Fig. 3a and b). Two commercial polymers (PHA and TR at 0.8 ± 0.6% and 2 ± 1%, respectively) and six of the lab-synthesized co-polymers (P(3HP85-6HA15), P(3HP75-6HA25), P(3HP70-6HA30), P(3HP65-6HA35), P(3HP60-6HA40) and P(3HP00-6HA100) at 7 ± 3, 4 ± 2, 3 ± 2, 5 ± 3, 14 ± 3 and 1.3% ± 0.9%, respectively), showed enhanced DOC bioavailability, whereas all others showed no significant difference between photo-bio processing testing and standard dark biodegradation approaches. This suggests that the light mediated release was not significant for those polymers. Second, CO2 generated from photo-weathered polymer particles was not significantly different from CO2 generated in standard dark biodegradation studies except in the cases of LDPE, P3HB and PHA. LDPE showed a minor reduction in CO2 generation (−0.9 ± 0.6% (light–dark)) while P3HB and PHA showed an increase in CO2 mobilization due to photo-irradiation at 6 ± 2% and 4.6 ± 0.7%, respectively (Fig. 3c and d). For the polymers tested here, a minor mobilization enhancement was expected, because the majority of these solids are not known to have a high degree of photo activity (Table S7†). Nevertheless, the results are consistent with previous evidence that photo-degradation accelerates the rate of DOC formation.21,23,31,49,57,63,64 For the 18 polymers tested, there was no measurable negative impact of photo-weathered DOC on microbial activity. Furthermore, ten out of the 18 tested polymers showed that irradiation for only 14 days (with elevated fluence relative to the Boston sun) caused the mobilization of up to 14 ± 3% more carbon (Fig. 3), which eventually was more bioavailable for marine microbial degradation.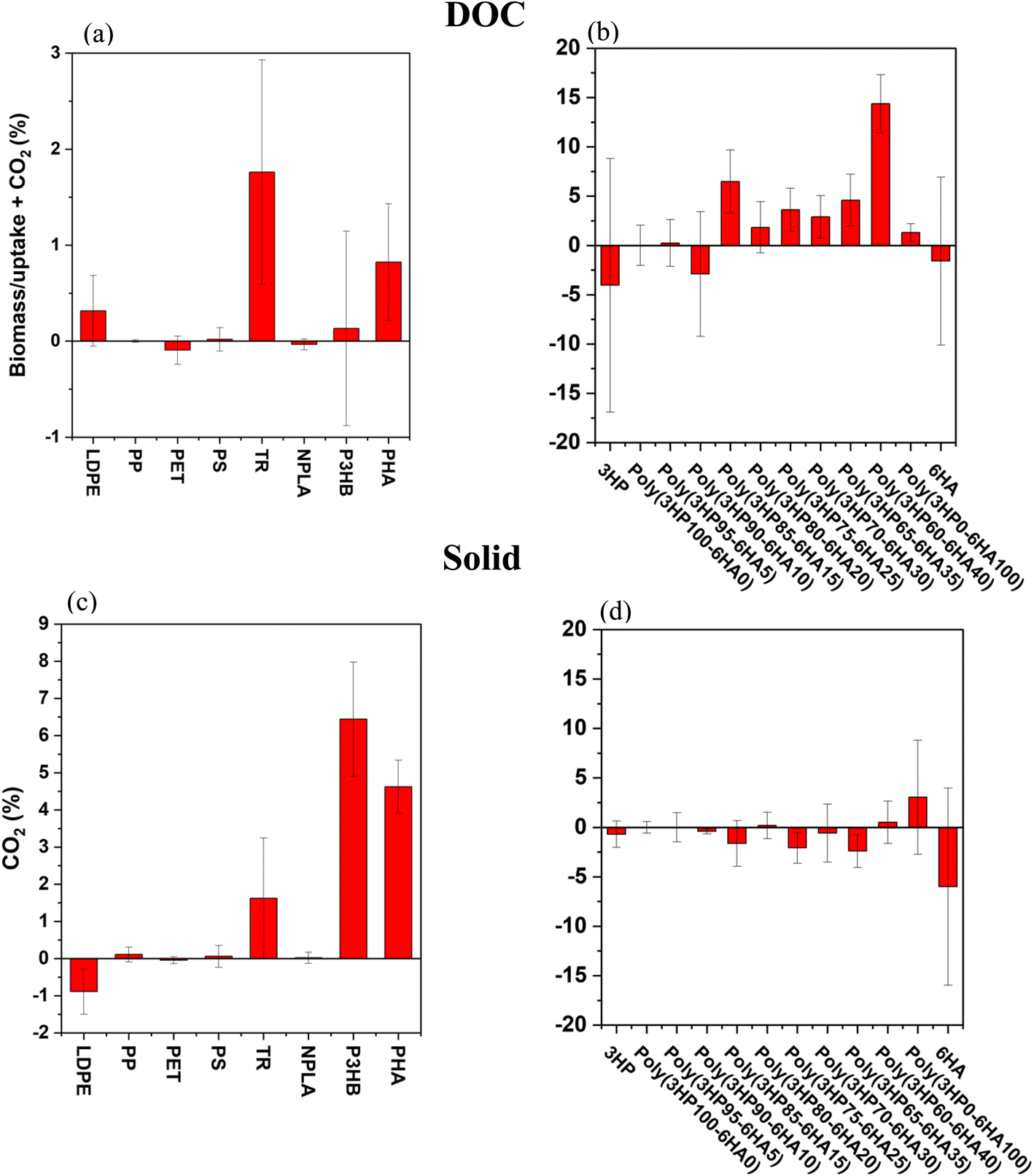 | ||
| Fig. 3 Comparison between photoweathered polymers and dark controls (light–dark). DOC produced from abiotic testing was taken up by biomass or converted to CO2 (combined) for photo-weathered polymers minus the dark control in (a) commercial polymers and (b) laboratory-synthesized co-polymers. Residual solids from abiotic testing were converted to CO2 for (c) commercial polymers and (d) laboratory-synthesized co-polymers. Error bars represent the standard deviation on triplicate measurements. | ||
Pathways of polymer degradation
 | ||
| Fig. 4 Intercomparison of biotic and abiotic degradation. Abiotic degradation is defined as the percent of carbon mobilized from fresh polymers as DOC after 14 days of photoweathering under simulated sunlight. Biotic degradation is defined as the percent of carbon consumed as CO2 from fresh polymers by natural marine microorganisms after 14 days of incubation (i.e., without photostimulation). The authors underscore that these are operational definitions highly dependent on the experimental system. Polymer degradation (a) presented in a framework to simultaneously show abiotic and biotic degradability of polymers, (b) commercial polymers and (c) lab-synthesized co-polymers of poly[(hydroxypivalic acid)-r-(hexanoic acid)]. Circles symbolize these polymers, where the blue-to-red gradient corresponds to increasing proportions of 6HA. Squares indicate pure 3HP (blue) and pure 6HA (red). Error bars represent standard deviation on triplicate measurements. | ||
Finally, readers are encouraged to recognize that the results reported here are a function of the experimental conditions (e.g., light fluence, innocula, and polymer types) and duration (e.g., time), where further degradation is likely possible over longer timescales (Tantawi et al., unpublished work). The ability to track polymer dynamics in high throughput and with chemical specificity would be a great asset for a more complete understanding of polymer fate in environmental and experimentally simulated systems.
Implications
These results illustrate that polymer-derived DOC is available for consumption by marine organisms and represents a non-trivial allocation of carbon mobilized from polymers, in addition to fully remineralized CO2. Efforts to trace polymer-derived byproducts could be aided by isotopic labeling studies (i.e., 13C-enriched polymers to enable explicit tracking of DOC in complex mixtures), as demonstrated by Zumstein et al.,67 and natural abundance isotope experiments may be useful in restricted circumstances where the starting polymer's isotopic signature is unique compared to the environmental background (Foster et al., unpublished work). Furthermore, experimentation leveraging advanced high-throughput experimentation and data analysis58,68 to understand polymer fate more comprehensively will rapidly augment current knowledge of polymer chemical fate. Specifically, the identity of polymer-derived DOC products will be critical to anticipate their degradability, partitioning, or ecological effects, and ongoing efforts to accelerate polymer-derived DOC classification and identification are needed.The work presented here underscores that the definition of biodegradable should be expanded in standard tests to include the production of DOC as well as CO2 and biomass.15,16,57,63,64,69,70 Furthermore, inclusion of abiotic transformation processes is critical to capture a broader range of environmentally relevant removal mechanisms and a more encompassing term (i.e., “environmental degradability”) may provide a more accurate description of material fate than biodegradation alone. Such a description would inform sustainable design practices by virtue of providing a range of degradation rates (i.e., rather than simple assignment as biodegradable or not) and mechanisms that can be related to underlying chemical or physical structures of the materials.38,71,72 Hence, intercomparable databases could be constructed to both inform new fundamental understanding of what gives rise to environmental degradability and enable prediction of degradation rates from novel structures alone. In this way, novel materials necessary to meet the growing demands of society can be reconciled with the requirement to build materials that are fundamentally compatible with the earth system.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
OT designed and executed the experiments, interpreted the results, and wrote the paper, WJ fabricated novel polymer chemistries, EEM and SHMA helped execute the experiments, KRB helped in experimental design, KLJP and BDO designed and supported the experiments, and DLP designed and supported the experiments, interpreted the results, and wrote the paper. All authors edited the report.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the DIC Corporation (Tokyo, Japan).References
- United Nations Environment Program, From Pollution to Solution: A Global Assessment of Marine Litter and Plastic Pollution, 2021 Search PubMed.
- D. M. I. René P. Schwarzenbach and P. M. Gschwend, Environmental Organic Chemistry, Environmental Organic Chemistry, 2016, p. 7 Search PubMed.
- K. Min, J. D. Cuiffi and R. T. Mathers, Ranking environmental degradation trends of plastic marine debris based on physical properties and molecular structure, Nat. Commun., 2020, 11(1), 1–11, DOI:10.1038/s41467-020-14538-z.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, Degradation Rates of Plastics in the Environment, ACS Sustain. Chem. Eng., 2020, 8, 3494–3511, DOI:10.1021/acssuschemeng.9b06635.
- A. L. Andrady, Microplastics in the marine environment, Mar. Pollut. Bull., 2011, 62, 1596–1605, DOI:10.1016/J.MARPOLBUL.2011.05.030.
- N. Mohanan, Z. Montazer, P. K. Sharma and D. B. Levin, Microbial and Enzymatic Degradation of Synthetic Plastics, Front. Microbiol., 2020, 11, 2837, DOI:10.3389/FMICB.2020.580709/BIBTEX.
- A. C. Albertsson and S. Karlsson, The influence of biotic and abiotic environments on the degradation of polyethylene, Prog. Polym. Sci., 1990, 15, 177–192, DOI:10.1016/0079-6700(90)90027-X.
- International Organization for Standardization (ISO), ISO 22404:2019 Plastics — Determination of the aerobic biodegradation of non-floating materials exposed to marine sediment — Method by analysis of evolved carbon dioxide.
- ASTM D7473/D7473M-21, Standard Test Method for Weight Attrition of Non-floating Plastic Materials by Open System Aquarium Incubations, 2021 Search PubMed.
- ASTM D7991-15, Standard Test Method for Determining Aerobic Biodegradation of Plastics Buried in Sandy Marine Sediment under Controlled Laboratory Conditions, 2016 Search PubMed.
- ISO 23832:2021, Plastics — Test Methods for Determination of Degradation Rate and Disintegration Degree of Plastic Materials Exposed to Marine Environmental Matrices under Laboratory Conditions, 2021 Search PubMed.
- ISO 22766:2020, Plastics — Determination of the Degree of Disintegration of Plastic Materials in Marine Habitats under Real Field Conditions, 2020 Search PubMed.
- ASTM D6691-17, Standard Test Method for Determining Aerobic Biodegradation of Plastic Materials in the Marine Environment by a Defined Microbial Consortium or Natural Sea Water Inoculum, 2017 Search PubMed.
- ISO 23977-1:2020, Plastics — Determination of the Aerobic Biodegradation of Plastic Materials Exposed to Seawater — Part 1: Method by Analysis of Evolved Carbon Dioxide Search PubMed.
- A. N. Walsh, C. M. Reddy, S. F. Niles, A. M. McKenna, C. M. Hansel and C. P. Ward, Plastic Formulation is an Emerging Control of Its Photochemical Fate in the Ocean, Environ. Sci. Technol., 2021, 55, 12383–12392, DOI:10.1021/acs.est.1c02272.
- C. Romera-Castillo, M. Pinto, T. M. Langer, X. Antón Álvarez-Salgado and G. J. Herndl, Dissolved organic carbon leaching from plastics stimulates microbial activity in the ocean, Nat. Commun., 2018, 9, 1430, DOI:10.1038/s41467-018-03798-5.
- L. Zhu, S. Zhao, T. B. Bittar, A. Stubbins and D. Li, Photochemical dissolution of buoyant microplastics to dissolved organic carbon: rates and microbial impacts, J. Hazard. Mater., 2020, 383, 121065, DOI:10.1016/j.jhazmat.2019.121065.
- C. Romera-Castillo, R. Mallenco-Fornies, M. Saa-Yanes and X. Á. Alvarez-Salgado, Leaching and bioavailability of dissolved organic matter from petrol-based and biodegradable plastic. Marine Environmental Research, Mar. Environ. Res., 2022, 176, 105607, DOI:10.1016/j.marenvres.2022.105607.
- C. P. Ward and C. M. Reddy, We need better data about the environmental persistence of plastic goods, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 14618–14621, DOI:10.1073/pnas.2008009117.
- R. Mallenco Fornies, Characterization of dissolved organic matter leached from plastics and its effects on marine microbes, CSIC - Instituto de Ciencias del Mar (ICM), http://hdl.handle.net/10261/212214 Search PubMed.
- S. Birnstiel, M. Sebastián and C. Romera-Castillo, Structure and activity of marine bacterial communities responding to plastic leachates, Sci. Total Environ., 2022, 834, 155264, DOI:10.1016/j.scitotenv.2022.155264.
- K. L. Rogers, J. A. Carreres-Calabuig, E. Gorokhova and N. R. Posth, Micro-by-micro interactions: how microorganisms influence the fate of marine microplastics, Limnol. Oceanog.r Lett., 2020, 5, 18–36, DOI:10.1002/LOL2.10136.
- S. G. Tetu, I. Sarker, V. Schrameyer, R. Pickford, L. D. H. Elbourne, L. R. Moore and I. T. Paulsen, Plastic leachates impair growth and oxygen production in Prochlorococcus, the ocean’s most abundant photosynthetic bacteria, Commun. Biol., 2019, 2, 184, DOI:10.1038/S42003-019-0410-X.
- Y. Su, Z. Cheng, Y. Hou, S. Lin, L. Gao, Z. Wang, R. Bao and L. Peng, Biodegradable and conventional microplastics posed similar toxicity to marine algae Chlorella vulgaris, Aquat. Toxicol., 2022, 244, 106097, DOI:10.1016/j.aquatox.2022.106097.
- X. Chen, Y. Wang, S. Chen, Y. Sun, Q. Tan, Z. Ding, Y. Lu and Y. Yu, Microplastics as carbon-nutrient sources and shaper for microbial communities in stagnant water, J. Hazard. Mater., 2021, 420, 126662, DOI:10.1016/j.jhazmat.2021.126662.
- A. Agostino, N. R. H. Rao, S. Paul, Z. Zhang, G. Leslie, P. Le-Clech and R. Henderson, Polymer leachates emulate naturally derived fluorescent dissolved organic matter: understanding and managing sample container interferences, Water Res., 2021, 204 DOI:10.1016/j.watres.2021.117614.
- A. N. Walsh, C. M. Reddy, S. F. Niles, A. M. McKenna, C. M. Hansel and C. P. Ward, Plastic Formulation is an Emerging Control of Its Photochemical Fate in the Ocean, Environ. Sci. Technol., 2021, 55, 12383–12392, DOI:10.1021/acs.est.1c02272.
- Y. K. Lee, C. Romera-Castillo, S. Hong and J. Hur, Characteristics of microplastic polymer-derived dissolved organic matter and its potential as a disinfection byproduct precursor, Water Res., 2020, 175, 115678, DOI:10.1016/j.watres.2020.115678.
- J. Duan, N. Bolan, Y. Li, S. Ding, T. Atugoda, M. Vithanage, B. Sarkar, D. C. W. Tsang and M. B. Kirkham, Weathering of microplastics and interaction with other coexisting constituents in terrestrial and aquatic environments, Water Res., 2021, 196, 117011, DOI:10.1016/j.watres.2021.117011.
- X. Wang, H. Zheng, J. Zhao, X. Luo, Z. Wang and B. Xing, Photodegradation Elevated the Toxicity of Polystyrene Microplastics to Grouper (Epinephelus moara) through Disrupting Hepatic Lipid Homeostasis, Environ. Sci. Technol., 2020, 54, 6202–6212 CrossRef CAS PubMed.
- Y. Chae, S. H. Hong and Y. J. An, Photosynthesis enhancement in four marine microalgal species exposed to expanded polystyrene leachate, Ecotoxicol. Environ. Saf., 2020, 189, 109936, DOI:10.1016/j.ecoenv.2019.109936.
- A. Vaksmaa, K. Knittel, A. A. Asbun, M. Goudriaan, A. Ellrott, H. J. Witte, I. Vollmer, F. Meirer, C. Lott, M. Weber, J. C. Engelmann and H. Niemann, Microbial Communities on Plastic Polymers in the Mediterranean Sea, Front. Microbiol., 2021, 12, 673553, DOI:10.3389/fmicb.2021.673553.
- Y. Su, Z. Cheng, Y. Hou, S. Lin, L. Gao, Z. Wang, R. Bao and L. Peng, Biodegradable and conventional microplastics posed similar toxicity to marine algae Chlorella vulgaris, Aquat. Toxicol., 2022, 244, 106907, DOI:10.1016/j.aquatox.2022.106907.
- H. Luo, Y. Li, Y. Zhao, Y. Xiang, D. He and X. Pan, Effects of accelerated aging on characteristics, leaching, and toxicity of commercial lead chromate pigmented microplastics, Environ. Pollut., 2020, 257, 113475, DOI:10.1016/j.envpol.2019.113475.
- K. Gunaalan, E. Fabbri and M. Capolupo, The hidden threat of plastic leachates: a critical review on their impacts on aquatic organisms, Water Res., 2020, 184, 116170, DOI:10.1016/j.watres.2020.116170.
- Z. Li, Y. Xie, Y. Zeng, Z. Zhang, Y. Song, Z. Hong, L. Ma, M. He, H. Ma and F. Cui, Plastic leachates lead to long-term toxicity in fungi and promote biodegradation of heterocyclic dye, Sci. Total Environ., 2021, 806(Pt 1), 150538, DOI:10.1016/j.scitotenv.2021.150538.
- M. T. Zumstein, R. Narayan, H. P. E. Kohler, K. McNeill and M. Sander, Dos and Do Nots When Assessing the Biodegradation of Plastics, Environ. Sci. Technol., 2019, 53, 9967–9969, DOI:10.1021/ACS.EST.9B04513.
- K. A. Fransen, S. H. M. Av-Ron, T. R. Buchanan, D. J. Walsh, D. T. Rota, L. Van Note and B. D. Olsen, High-throughput experimentation for discovery of biodegradable polyesters, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2220021120 CrossRef CAS PubMed.
- K. L. Jones Prather, D. Plata, B. D. Olsen, W. Joo, S. Av-Ron, K. R. Bannister, O. Tantawi, Biodegradable sustainable polyesters, US Pat., US11787900B2, 2023 Search PubMed.
- E. E. Emecheta, D. B. Borda, P. M. Pfohl, W. Wohlleben, C. Hutzler, A. Haase and A. Roloff, A comparative investigation of the sorption of polycyclic aromatic hydrocarbons to various polydisperse micro- and nanoplastics using a novel third-phase partition method, Microplast. Nanoplast., 2022, 2(1), 1–17 Search PubMed.
- ASTM D1141-98, Standard Practice for Preparation of Substitute Ocean Water, 2021.
- Scientific Image and Illustration Software|BioRender, https://www.biorender.com/, accessed 7 April 2023 Search PubMed.
- ASTM, ASTM G173 - 03: Standard Tables for Reference Solar Spectral Irradiances: Direct Normal and Hemispherical on 37° Tilted Surface, 2003 Search PubMed.
- National Renewable Energy Laboratory (NREL), NSRDB: National Solar Radiation Database, 2022 Search PubMed.
- E. Yousif and R. Haddad, Photodegradation and photostabilization of polymers, especially polystyrene: review, Springerplus, 2013, 2, 1–32 CrossRef CAS PubMed.
- C. R. Hurley and G. J. Leggett, Quantitative investigation of the photodegradation of polyethylene terephthalate film by friction force microscopy, contact-angle goniometry, and X-ray photoelectron spectroscopy, ACS Appl. Mater. Interfaces, 2009, 1, 1688–1697 CrossRef CAS.
- L. V. Bora, M. Bhatt, A. Patel and N. V. Bora, Plastic Degradation by Photocatalysis: Basic Concepts and General Mechanisms, ACS Symp. Ser., 2024, 1–22 CAS.
- A. M. Díez, N. Licciardello and Y. V. Kolenko, Photocatalytic processes as a potential solution for plastic waste management, Polym. Degrad. Stab., 2023, 215, 110459 CrossRef.
- L. Zhu, S. Zhao, T. B. Bittar, A. Stubbins and D. Li, Photochemical dissolution of buoyant microplastics to dissolved organic carbon: Rates and microbial impacts, J. Hazard. Mater., 2020, 383, 121065 CrossRef CAS.
- D. C. Harris and C. A. Lucy, Quantitative Chemical Analysis, ed. W. H. Freeman Search PubMed.
- International Organization for Standardization (ISO), ISO 14851:2019(en), Determination of the ultimate aerobic biodegradability of plastic materials in an aqueous medium — Method by measuring the oxygen demand in a closed respirometer, 2019 Search PubMed.
- A. Künkel, J. Becker, L. Börger, J. Hamprecht, S. Koltzenburg, R. Loos, M. B. Schick, K. Schlegel, C. Sinkel, G. Skupin and M. Yamamoto, Polymers, Biodegradable, Ullmann’s Encyclopedia of Industrial Chemistry, 2016, pp. 1–29 Search PubMed.
- E. J. Zakem, B. B. Cael and N. M. Levine, A unified theory for organic matter accumulation, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(6), e2016896118, DOI:10.1073/pnas.2016896118.
- Z. Tian, H. Zhao, K. T. Peter, M. Gonzalez, J. Wetzel, C. Wu, X. Hu, J. Prat, E. Mudrock, R. Hettinger, A. E. Cortina, R. G. Biswas, F. V. C. Kock, R. Soong, A. Jenne, B. Du, F. Hou, H. He, R. Lundeen, A. Gilbreath, R. Sutton, N. L. Scholz, J. W. Davis, M. C. Dodd, A. Simpson, J. K. McIntyre and E. P. Kolodziej, A ubiquitous tire rubber–derived chemical induces acute mortality in coho salmon, Science, 1979, 2021(371), 185–189 Search PubMed.
- H. Inderthal, S. L. Tai and S. T. L. Harrison, Non-Hydrolyzable Plastics – An Interdisciplinary Look at Plastic Bio-Oxidation, Trends Biotechnol., 2021, 39, 12–23 CrossRef CAS.
- X. Wang, H. Zheng, J. Zhao, X. Luo, Z. Wang and B. Xing, Photodegradation Elevated the Toxicity of Polystyrene Microplastics to Grouper (Epinephelus moara) through Disrupting Hepatic Lipid Homeostasis, Environ. Sci. Technol., 2020, 54, 6202–6212 CrossRef CAS.
- R. C. Cristina, M. F. Rebeca, S. Y. Marola and Á. S. Xosé Antón, Leaching and bioavailability of dissolved organic matter from petrol-based and biodegradable plastics, Mar. Environ. Res., 2022, 176, 105607 CrossRef CAS.
- V. Albergamo, W. Wohlleben and D. L. Plata, Tracking Dynamic Chemical Reactivity Networks with High-Resolution Mass Spectrometry: A Case of Microplastic-Derived Dissolved Organic Carbon, Environ. Sci. Technol., 2024, 58, 4314–4325 CAS.
- W. Shi, K. Xue, E. R. Meshot and D. L. Plata, The carbon nanotube formation parameter space: data mining and mechanistic understanding for efficient resource use, Green Chem., 2017, 19, 3787–3800 RSC.
- E. Kim, K. Huang, A. Saunders, A. McCallum, G. Ceder and E. Olivetti, Materials Synthesis Insights from Scientific Literature via Text Extraction and Machine Learning, Chem. Mater., 2017, 29, 9436–9444 CrossRef CAS.
- F. Rojo, Degradation of alkanes by bacteria, Environ. Microbiol., 2009, 11, 2477–2490 CrossRef CAS PubMed.
- L. Zhu, S. Zhao, T. B. Bittar, A. Stubbins and D. Li, Photochemical dissolution of buoyant microplastics to dissolved organic carbon: rates and microbial impacts, J. Hazard. Mater., 2020, 383, 121065, DOI:10.1016/j.jhazmat.2019.121065.
- B. Gewert, M. M. Plassmann and M. Macleod, Pathways for degradation of plastic polymers floating in the marine environment, Environ. Sci. Process. Impacts, 2015, 17, 1513–1521 RSC.
- C. Romera-Castillo, S. Birnstiel, X. A. Álvarez-Salgado and M. Sebastián, Aged Plastic Leaching of Dissolved Organic Matter Is Two Orders of Magnitude Higher Than Virgin Plastic Leading to a Strong Uplift in Marine Microbial Activity, Front. Mar. Sci., 2022, 9, 329 Search PubMed.
- P. Shieh, W. Zhang, K. E. L. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun, K. A. Nelson, D. L. Plata and J. A. Johnson, Cleavable comonomers enable degradable, recyclable thermoset plastics, Nature, 2020, 583(7817), 542–547 CrossRef CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3, e1700782, DOI:10.1126/sciadv.1700782.
- M. T. Zumstein, A. Schintlmeister, T. F. Nelson, R. Baumgartner, D. Woebken, M. Wagner, H. P. E. Kohler, K. McNeill and M. Sander, Biodegradation of synthetic polymers in soils: tracking carbon into CO2 and microbial biomass, Sci. Adv., 2018, 4, eaas9024, DOI:10.1126/sciadv.aas9024.
- V. Albergamo, W. Wohlleben and D. L. Plata, Photochemical weathering of polyurethane microplastics produced complex and dynamic mixtures of dissolved organic chemicals, Environ. Sci. Process. Impacts, 2023, 25, 432–444 RSC.
- B. Singh and N. Sharma, Mechanistic implications of plastic degradation, Polym. Degrad. Stab., 2008, 93, 561–584 CrossRef CAS.
- R. Li, Espi, C. Wayman and H. Niemann, The fate of plastic in the ocean environment – a minireview, Environ. Sci. Process. Impacts, 2021, 23, 198–212 RSC.
- A. Jayaraman and B. Olsen, Convergence of Artificial Intelligence, Machine Learning, Cheminformatics, and Polymer Science in Macromolecules, Macromolecules, 2024, 57(16), 7685–7688, DOI:10.1021/acs.macromol.4C01704.
- C. M. Hartquist, S. Wang, B. Deng, H. K. Beech, S. L. Craig, B. D. Olsen, M. Rubinstein and X. Zhao, Fracture of polymer-like networks with hybrid bond strengths, J. Mech. Phys. Solid., 2025, 195, 105931 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Eight tables, two figures and a detailed description of the carbon analysis calculations, which include standard marine biodegradation tests, polymer crystallinity, molecular weight, morphology characterization, custom-built solar simulator parts, comparative analysis of the custom-built solar simulator vs. similar commercial systems, collected seawater conditions, dissolved organic carbon (DOC) release after 14 days of photo-weathering, polymer degradation measured as mineralization to CO2 and DOC versus mineralization to CO2 alone for commercial and laboratory-synthesized polymers, solar simulator diagrams and characterization, and additional experimental details and discussion. See DOI: https://doi.org/10.1039/d5em00079c |
| This journal is © The Royal Society of Chemistry 2025 |