
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anchoring ligand engineering enables highly stable MA-free perovskite solar cells with a minimal VOC deficit of 0.32 V†
Chaohui
Li
 *aj,
Andrej
Vincze
c,
Hyoungwon
Park
d,
Fabian
Streller
e,
Klaus
Götz
f,
Shudi
Qiu
a,
Jiwon
Byun
g,
Ying
Shang
h,
Zhangyu
Yuan
h,
Lirong
Dong
a,
Jingjing
Tian
aj,
Zijian
Peng
aj,
Chao
Liu
ab,
Fu
Yang
i,
Yanxue
Wang
ab,
Andreas
Späth
el,
Andres
Osvet
a,
Karen
Forberich
ab,
Thomas
Heumueller
ab,
Silke H.
Christiansen
dk,
Marcus
Halik
f,
Rainer H.
Fink
e,
Tobias
Unruh
f,
Ning
Li
h,
Larry
Lüer
*a and
Christoph J.
Brabec
*ab
*aj,
Andrej
Vincze
c,
Hyoungwon
Park
d,
Fabian
Streller
e,
Klaus
Götz
f,
Shudi
Qiu
a,
Jiwon
Byun
g,
Ying
Shang
h,
Zhangyu
Yuan
h,
Lirong
Dong
a,
Jingjing
Tian
aj,
Zijian
Peng
aj,
Chao
Liu
ab,
Fu
Yang
i,
Yanxue
Wang
ab,
Andreas
Späth
el,
Andres
Osvet
a,
Karen
Forberich
ab,
Thomas
Heumueller
ab,
Silke H.
Christiansen
dk,
Marcus
Halik
f,
Rainer H.
Fink
e,
Tobias
Unruh
f,
Ning
Li
h,
Larry
Lüer
*a and
Christoph J.
Brabec
*ab
aInstitute of Materials for Electronics and Energy Technology (i-MEET), Department of Materials Science and Engineering, Friedrich-Alexander-Universität Erlangen-Nürnberg, Martensstrasse 7, 91058 Erlangen, Germany. E-mail: chaohui.li@fau.de; larry.lueer@fau.de; christoph.brabec@fau.de
bHelmholtz-Institute Erlangen-Nürnberg for Renewable Energy (HI ERN), FAU Profile Center Solar, Immerwahrstrasse 2, 91058 Erlangen, Germany
cInternational Laser Centre SCSTI, Ilkovičova 3, 84104 Bratislava, Slovak Republic
dFraunhofer Institute for Ceramic Technologies and Systems IKTS, Äußere Nürnberger Strasse 62, 91301 Forchheim, Germany
ePhysical Chemistry II and Interdisciplinary Center for Molecular Materials, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstrasse 3, 91058 Erlangen, Germany
fInstitute for Crystallography and Structural Physics, Friedrich-Alexander-Universität Erlangen-Nürnberg, Staudtstrasse 3, 91058 Erlangen, Germany
gOrganic Materials and Devices, Department of Materials Science, Interdisciplinary Center for Nanostructured Films (IZNF), Friedrich-Alexander-Universität Erlangen-Nürnberg, 91058 Erlangen, Germany
hInstitute of Polymer Optoelectronic Materials and Device, Guangdong Basic Research Center of Excellence for Energy and Information Polymer Materials, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China
iLaboratory of Advanced Optoelectronic Materials, Suzhou Key Laboratory of Novel Semiconductor-Optoelectronics Materials and Devices, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, 215123, China
jErlangen Graduate School in Advanced Optical Technologies (SAOT) Paul-Gordan-Straße 6, 91052 Erlangen, Germany
kPhysics Department, Freie Universität Berlin, 14195 Berlin, Germany
lFAU Competence Center Engineering of Advanced Materials, Friedrich-Alexander-Universität Erlangen-Nürnberg, Cauerstrasse 3, 91058 Erlangen, Germany
First published on 26th June 2025
Abstract
Ligand engineering is an effective method to reduce defects in perovskite solar cells (PSCs) and to enhance efficiency. Likewise, enhancing device stability through ligand engineering is currently emerging as a key focus to suppress the bidirectional migration of halides and silver ions, which otherwise can cause irreversible chemical corrosion to the electrode and perovskite layer. Here, triphenylphosphine oxide (TPPO) is demonstrated to improve the long-term operational stability of PSCs when introduced at the interface between the perovskite and the electron transport layer (ETL). TPPO effectively eliminates uncoordinated Pb2+ and thus reduces surface defects. Accordingly, the target solar cell yields a hero power conversion efficiency (PCE) of 26.01% and a maximum open-circuit voltage (VOC) of 1.23 V, representing the minimum voltage deficit (0.32 V) reported for methylammonium-free (MA-free) PSCs. Moreover, long-term operational analysis reveals that the bidirectional migration of halides and silver ions is significantly suppressed, resulting in enhanced device stability. TPPO-modified PSCs retain 90% of the initial PCE after 1200 hours of operation in maximum power point tracking. Ligand engineering with TPPO marks a significant advancement in enhancing the stability of PSCs and is fully compatible to upscaling scenarios.
Broader contextPerovskite solar cells (PSCs) exhibit outstanding power conversion efficiency and low fabrication cost, yet their practical deployment is hindered by ion migration and interfacial degradation. Here, we introduce an anchoring ligand strategy employing triphenylphosphine oxide (TPPO) to passivate the buried interface and suppress ion transport. The resulting methylammonium-free PSCs achieve a champion efficiency of 26.01% with maximun open-circuit voltage of 1.23 V and retain 90% of their initial performance after 1200 hours of continuous operation. This approach effectively addresses both voltage loss and long-term operational stability, offering a scalable route toward durable, commercially viable perovskite photovoltaics. |
Introduction
Self-assembled monolayers (SAMs) have recently emerged as a prominent class of hole-selective layers for inverted perovskite solar cells (PSCs), achieving photoelectric conversion efficiencies (PCE) over 27%.1–4 However, despite the remarkable progress in performance, the stability of PSCs remains the major challenge and represents a risk scenario for product development. The instability of halide PSCs is mainly attributed to a rather low enthalpy for halide void formation when stimulated by the combination of heat and light or through decomposition under humidity or a corrosive chemical environment, followed by a strong tendency for ions to migrate and cause decomposition processes.5–7 That primary decomposition process cannot be mitigated with advanced encapsulation strategies but instead needs to be resolved by material design. The most promising concepts are the design of perovskites with a larger enthalpy for void formation, the suppression of ion migration by additives or binders or the mitigation of ion diffusion by the insertion of ion blocking layers to protect the electrode.8–10 Despite significant progress in mitigating the negative effects of ion migration, only a limited number of studies have reported stable perovskite cells operating at elevated temperatures (85 °C),11–13 particularly those employing the p–i–n architecture with PCBM as the electron transport layer.14A smart strategy to mutually improve both, efficiency and stability of PSCs, is the interface engineering between the perovskite layer and the charge transport layers.8,15,16 Effective passivation of interface defects is crucial to reduce non-radiative recombination and does improve device performance.17 Increasing endeavor has been devoted to also enhancing device stability through interface engineering. Commonly, molecular agents with tailored structures and properties have been employed to reduce defect densities in perovskite materials, proving to be an effective approach for improving the photovoltaic performance of PSCs.18–20 However, conventional interface passivation methods, such as depositing two-dimensional phases, can effectively suppress non-radiative recombination but often suffer from lattice mismatch and interface stress at high temperatures, limiting their long-term operability.21–25 In addition, intrinsic ion migration is also an important issue for perovskite instability. Among all possible mobile ions, halide ions are the most difficult to stabilize due to their weak bonding to the metal cation Pb2+.9,26,27 Under light or thermal activation, halogen species (I−) will diffuse from the perovskite absorber layer towards the charge transport layers and further on to the metal electrode. In the case of Ag as the top electrode, the reaction between iodine or iodide with Ag+ will form insulating AgI, leading to device degradation.28,29 Therefore, the development of strategies that inhibit halide migration is one smart strategy to minimize the trade-off between device performance and stability.
Although triphenylphosphine oxide (TPPO) has been extensively utilized in perovskite light-emitting diodes (PeLEDs) to enhance electron transport and minimize non-radiative recombination, its application in MA-free perovskite solar cells (PSCs), particularly for stability enhancement, remains largely unexplored (Table S1, ESI†).30,31 In this work, we employ the strong polarity of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in TPPO to effectively passivate uncoordinated Pb2+ and reduce interfacial nonradiative recombination, resulting in devices with a champion PCE of 26.01%. We further show that the TPPO successfully delays the interdiffusion of Ag+ and I− even under long-term thermal and light stress operation conditions. After 1200 hours of continuous maximum power point tracking and 1 sun illumination of continuous heating at 65 °C in N2 atmosphere, the PCE of the target device can still maintain 90% of the initial value. Therefore, TPPO surface anchoring engineering represents a promising strategy for enhancing both the efficiency and long-term stability of PSCs.
O in TPPO to effectively passivate uncoordinated Pb2+ and reduce interfacial nonradiative recombination, resulting in devices with a champion PCE of 26.01%. We further show that the TPPO successfully delays the interdiffusion of Ag+ and I− even under long-term thermal and light stress operation conditions. After 1200 hours of continuous maximum power point tracking and 1 sun illumination of continuous heating at 65 °C in N2 atmosphere, the PCE of the target device can still maintain 90% of the initial value. Therefore, TPPO surface anchoring engineering represents a promising strategy for enhancing both the efficiency and long-term stability of PSCs.
Results and discussion
We fabricated glass/ITO/NiOx/SAM/perovskite/TPPO (with and without)/PCBM/Ag layered solar cells, as illustrated in Fig. 1a. The schematic representation of the interaction between TPPO and the perovskite layer is also shown in Fig. 1a. Initially, to analyze the interaction between TPPO molecules and perovskite, X-ray photoelectron spectroscopy (XPS) was carried out. XPS analysis confirmed the formation of a strong interaction between TPPO and under-coordinated Pb2+ ions, as indicated by the shift in Pb 4f binding energy, providing direct evidence of enhanced passivation at the perovskite/electron transport layer interface (Fig. 1b). For control sample, Pb 4f peaks at 143.30 and 138.41 eV were observed, and with the introduction of TPPO, the Pb 4f peak shifted to lower binding energies at 142.84 and 137.75 eV, providing evidence for the formation of a complex between –PO and the under-coordinated [PbI6]4− cage.32,33 Similarly, the I 3d core levels in Fig. S1 (ESI†) move from 630.89 and 619.40 eV to lower binding energies of 630.17 eV and 618.71 eV. These changes indicate the strong passivation interaction between TPPO and perovskite. The X-ray Diffraction (XRD) results further show that new characteristic peaks different from those of the control and TPPO powder appear at positions 2θ = 4 and 8°, which confirms that a new complex has been formed (Fig. 1c). In summary, we report the surface interaction of TPPO with the uncoordinated [PbI6]4− octahedral. | ||
| Fig. 1 (a) The schematic diagram of interface modification in inverted perovskite solar cells via TPPO (left) and the chemical structure of TPPO molecule (right). (b) XPS spectra of Pb 4f (c) XRD patterns for control (PVK), target(w/TPPO), TPPO powder, a.u., arbitrary units. | ||
Influence of the TPPO molecule
The surface morphology of both perovskite films (control and target) was investigated using scanning electron microscopy (SEM) measurements. Fig. 2a and b present the SEM micrographs of the two films. To exclude the effect of solvent and annealing, the morphology of the perovskite film without molecular modification remained unchanged after post-annealing (Fig. S2, ESI†), indicating that TPPO molecules play a crucial role in promoting grain growth during annealing. Meanwhile, from the across section SEM image (Fig. S3, ESI†), the TPPO-modified perovskite film exhibits larger crystals that are vertically aligned, whereas the control sample contains smaller, fragmented crystals that are randomly distributed. This random distribution leads to poor interlayer contact and introduces defects. In addition, the effect of TPPO molecular passivation on the crystallinity of perovskite was directly studied by further X-ray diffraction (XRD) measurements. As shown in the XRD pattern of Fig. S4 (ESI†), the peak of the (100) plane is 24% stronger than that of control sample, confirming that TPPO induces enhanced crystallization of the perovskite film. Additionally, we performed roughness measurements on samples with and without TPPO treatment before and after PCBM deposition. To eliminate the influence of IPA solvent on film morphology, we performed IPA spin coating only on the film without TPPO modification. By comparing the roughness of Control and Control@IPA, the root mean square (RMS) roughness values were 21.12 and 24.21 nm, respectively. However, after TPPO deposition, the roughness was significantly reduced from 21.12 to 15.25 nm (Fig. S5, ESI†). This demonstrates that TPPO effectively reduces surface roughness by reacting with Pb2+, which enhances the interaction between the transport layer and reduces charge recombination at the interface. A similar trend in roughness changes was observed after PCBM deposition (Fig. S6, ESI†). Lower RMS roughness leads to smoother surfaces, which is more conducive to better interaction of the transport layer and helps to reduce charge recombination at the interface.34 | ||
| Fig. 2 Top-view SEM images of the (a) control and (b) TPPO-treated perovskite films, Sample structure: ITO/NiOx/SAM/PVK (w, w/o TPPO). KPFM images of (c) the control and (d) TPPO-treated perovskite films. The corresponding line profiles (e) for the control film and TPPO-treated perovskite films (f). Sample structure: silicon wafer (Si)/NiOx/SAM/PVK (w, w/o TPPO). | ||
We further analyzed the surface potential distribution of the samples using Kelvin probe force microscopy (KPFM) (Fig. 2c–f). Fig. 2c and d shows the corresponding surface potential images, it is worth noting that various surface defects will change the potential distribution on the perovskite surface. Fig. 2e compares the line scans of the surface potential changes in a typical perovskite area, and these potential fluctuations can reach up to about 200–250 mV in the length range of about 0–2 μm, indicating that the uneven plane causes strong potential fluctuations.34 The bright PbI2 clusters on the perovskite surface are the centers of non-radiative recombination (Fig. 2a and b), which can lead to changes in the local surface charge.35 In contrast, TPPO treatment significantly reduces and suppresses unpaired Pb2+, smoothing the surface potential changes (Fig. 2f). This reactive surface engineering approach enhanced the X-ray diffraction (Fig. S4, ESI†) and UV-vis absorption intensity (400–800 nm) (Fig. S7, ESI†), which is consistent with the TPPO treatment being primarily a surface modification. A smoother, less defective surface is expected to be beneficial for improving carrier transport across perovskite/ETL junction.36
Carrier dynamics of perovskite films and devices
To further evaluate the impact of TPPO on the charge recombination mechanism in perovskite devices, the carrier dynamics were first investigated by steady-state photoluminescence (PL) and time-resolved photoluminescence (TRPL) spectroscopy. As shown in Fig. 3a, the higher emission intensity of the TPPO-treated film, indicating a significant reduction in non-radiative recombination to directly contribute to the observed improvements in PCE and VOC.37,38 Synchronously, the introduction effect of TPPO passivation on the non-radiative recombination of the device was further quantified by photoluminescence quantum yield (PLQY) (Fig. 3b). The quasi-Fermi level splitting (QFLS) of the bare PSCs film was 1.21 V. After the deposition of PCBM, it dropped significantly. However, it was gratifying that after the modification with TPPO, the QFLS could be restored to 1.214 V. This shows that TPPO can indeed effectively reduce the non-radiative recombination between active layer and the electron transport layer. Subsequently, we used a 405 nm laser to study the recombination dynamics of the samples under three different excitation densities and recorded time-resolved photoluminescence (TRPL). Here, we used the perovskite carrier recombination simulator (PEARS) tool to fit the bimolecular capture-detrapping model.39 The TRPL spectra are shown in Fig. 3c. | ||
| Fig. 3 (a) The steady-state PL. (b) PLQY of ITO/PVK, ITO/PVK/PCBM, ITO/PVK/TPPO/PCBM, respectively. (c) TRPL spectra of control and TPPO-treated perovskite films on glass substrate at different laser intensities. (d) Dark J–V cure of control and target devices (e) Mott–Schottky plots of PSCs based on control, target. (f) VOCversus light intensity of control and target samples. | ||
The data were fitted at three different laser intensities (Tables S2 and S3, ESI†). Usually, the relationship between trap density and trapping rate can be simplified as:
| Dt = NT × d |
where d is the thickness of perovskite. From cross-sectional SEM analysis, the thickness of both films is about 650 nm (Fig. S3, ESI†). NT is corresponding trap state density. Dt is the first-order surface trap density. The TPPO surface treatment significantly reduces the first-order surface trap density in perovskite thin films. Considering the film thickness of approximately 650 nm, the Dt is reduced from 7.62 × 109 cm−2 (control sample) to 2.72 × 108 cm−2 This corresponds to a reduction by a factor of 28, highlighting the excellent passivation capability of TPPO in suppressing surface and interface traps. This reduction directly decreases surface recombination losses and improves the potential performance of the perovskite device. The results underline that reducing the first-order surface trap density is a critical strategy for enhancing the efficiency and stability of PSCs, providing valuable insights for future device optimization.40,41 Furthermore, we observed that the significant decrease in trap state concentration was consistent with the increase in PL intensity.
To elucidate the charge transfer and recombination mechanisms behind the enhanced photovoltaic performance of TPPO-treated PSCs, we explored the charge recombination dynamics of the device with dark J–V (Fig. 3d). Compared to the Control sample, the leakage current of the Target sample is significantly reduced under negative bias, demonstrating that TPPO modification effectively reduces the defect density at the interface, thereby minimizing current leakage. This result aligns with the 28-fold reduction in trap state density observed in PL analysis. Furthermore, the forward current of the Target sample increases sharply at lower bias levels, indicating enhanced charge injection and transport efficiency due to TPPO modification, resulting in a more ideal diode behavior. Overall, TPPO modification significantly enhances the device's electrical performance by reducing surface trap density and passivating interface defects, confirming its effectiveness as an interface modifier.42 Meanwhile, the charge recombination kinetics were evaluated at the device level, and the performance under varying light intensities (JSC mode) was analyzed using electrochemical impedance spectroscopy (EIS). The TPPO-modified samples exhibited lower charge transport resistance (Rct) values under 0.1–1 sun illumination (Fig. S8 and Table S4, ESI†), indicating significantly enhanced interfacial charge transport after TPPO surface modification. Notably, this improvement was particularly pronounced under low light intensity conditions. Furthermore, the Target sample demonstrated higher charge recombination resistance (Rrec) values, reflecting the effective passivation of trap states by TPPO, which reduced carrier recombination. This aligns with the previously observed 28-fold reduction in trap state density, further validating the effectiveness of TPPO modification. In summary, the Target sample exhibited more stable Rct and Rrec values across different light intensities, reinforcing the superiority of TPPO as an effective modifier in PSCs. In addition, according to the Mott–Schottky analysis, we further understand the reason for the enhanced VOC of the target device. As shown in Fig. 3e,The built-in potential (Vbi) of the target device (1.12 V) is greater than that of the control device (1.03 V), which is due to the reduced defect density in the perovskite film after TPPO treatment.43 Subsequently, to deepen the understanding of the mechanism of photovoltaic performance improvement, the dependence of VOC and JSC stability on light intensity (P) was studied to better understand the mechanism of TPPO's effect on charge recombination performance. According to:
VOC ∝ n(kBT/q)ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P P |
In summary, the results indicate that the trap density in TPPO-modified perovskite films is significantly reduced, which may be due to the effective passivation of the dangling Pb2+ defects by TPPO, forming new ligand complexes that are beneficial for suppressing non-radiative recombination in solar cells.
Photovoltaic performance of PSCs
The perovskite samples with a bandgap of 1.55 eV (Fig. S10, ESI†) were processed in a p–i–n device with the following architecture: ITO/NiOx/SAM/Perovskite (w, w/o TPPO)/PCBM/BCP/Ag. The optimal TPPO concentration was obtained by comparing the photovoltaic parameters of the devices (Fig. S11, ESI†). All target samples, whether thin films or devices, were modified with 0.25 mg ml−1 TPPO. The TPPO-treated perovskite device with an active area of 0.04 cm2 in the reverse scan obtained the best PCE of 26.01%, VOC value of 1.214 V, JSC value of 25.57 mA cm−2, and FF value of 83.8% (Fig. 4a). It is worth noting that this is one of the lowest VOC deficits (0.336 V) obtained so far (Fig. 4b). In comparison, the control perovskite solar cell obtained 23.79%. Individual target devices achieved the highest VOC of 1.230 V, corresponding to a VOC defect of only 0.32 V (Fig. S12 and Table S5, ESI†), which is the lowest voltage defect (0.32 V) reported for MA-free PSCs.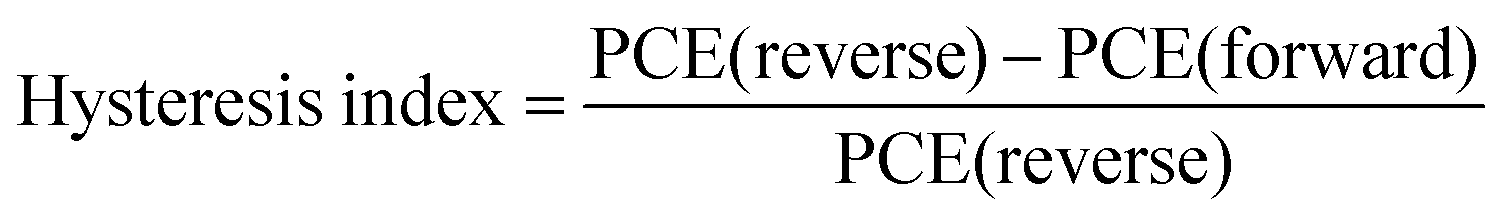
 | ||
| Fig. 4 (a) J–V cure of the control, target PSCs. (b) VOC and PCE statistics of devices with bandgaps close to approximately 1.55 eV. (c) EQE curves of champion-PSCs. Long-term operational stability under continuous output at MPP conditions (1 sun, LED lamps with ultraviolet filter); 65 °C (d) and 85 °C (e) in N2-filled Chamber. | ||
The external quantum efficiency (EQE) spectra, integrated JSC of the corresponding champion device is confirmed in Fig. 4c. The integrated JSC of the TPPO-treated devices calculated from the EQE spectra is 25.14 mA cm−2, in good agreement with the JSC values from J–V measurements. Additionally, the TPPO-modified device exhibited a lower hysteresis index (0.019) compared to the control device (0.027) (Table S6, ESI†).45 The potential for upscaling of the device is supported by fabricating a large-area (1.028 cm2) device (Fig. S13, ESI†), which yields high VOC of 1.201 (reverse scan). However, as expected from previous investigations, it was pleasing to see that after modification with TPPO, the high QFLS of the unmodified film was fully restored to 1.214 V after PCBM deposition. This shows that TPPO effectively reduces non-radiative recombination between the PVK and the electron transport layer (Fig. 3b).46,47 To further verify the versatility of TPPO across different perovskite compositions, we applied it to two wide-bandgap perovskites: Cs0.22FA0.78PbI1.8Br1.2 (bandgap ≈1.797 eV) and FA0.82MA0.13Cs0.05Pb(I0.87Br0.13)3 (bandgap ≈1.59 eV) (Fig. S14, ESI†). The TPPO-modified devices achieved PCEs of 19.01% with a VOC of 1.335 V, and 24.15% with a VOC of 1.204 V, respectively. In contrast, the corresponding control devices without TPPO exhibited lower PCEs of 17.43% with a VOC of 1.271 V, and 22.77% with a VOC of 1.175 V. These results demonstrate the broad applicability and effectiveness of TPPO as a surface passivation agent for perovskites with varying bandgaps in photovoltaic applications.
Evaluating stability is crucial for the commercialization of PSCs. Fig. 4d and e depict the maximum power point (MPP) tracking performance of unencapsulated control and TPPO-treated devices, tested at 65 °C, 85 °C, under 1 sun white LED illumination and in N2 atmosphere. After 1200 hours of aging at 65 °C, the control device retained only 68% of its initial PCE, whereas the TPPO-treated device retained over 85% of its original performance (Fig. 4d and Fig. S15, ESI†). To further accelerate the evaluation of operational stability, we performed tests at 85 °C and 1 sun. After 800 hours of continuous MPP tracking at 85 °C, the TPPO-treated device retained 80% of its initial efficiency, while the control device's efficiency dropped to 62% (Fig. 4e and Fig. S15, ESI†). For a preliminary assessment of stability, we calculate the acceleration factor (AF) between two temperatures based on the Arrhenius equation48

Analyzing long-term stability of PSCs and films
To further explore the reasons for the enhanced device stability, we first evaluated the stability of the perovskite film under the combined action of thermal stress and light stress. By comparing the PL and XRD spectra of the fresh film and the film after aging for 300 h (Fig. S16, ESI†), the effect of TPPO on the reduction of ion migration and defect formation under thermal-and photo-stress conditions plays a key role in maintaining the structural integrity of the perovskite film, thereby extending operational lifetimes under realistic conditions. The control sample exhibits significant degradation after aging, as evidenced by the pronounced redshift and broadening of the PL peak (Fig. S16a, ESI†). During the aging process, the control perovskite has a relatively low tolerance to heat or light, so more defects are generated. These defect states can form new energy levels and increase non-radiative recombination pathways, resulting in a red shift and broadening of the PL peak.49,50 In contrast, the PL position of the target sample modified by TPPO has almost no change and the peak width is relatively narrow, which indicates that TPPO effectively enhances the stability of the film. This indicates that TPPO modification effectively passivates surface defects, stabilizes the perovskite structure, and mitigates ion migration, thereby preserving the film's optoelectronic performance over time (Fig. S16b, ESI†). Subsequently, the XRD diffraction test was used to observe the changes in the film crystal structure in an ambient atmosphere with a relative humidity of 45% (Fig. S17, ESI†). In the film without encapsulation after 240 h, the peak representing PbI2 showed obvious differences over time. The results showed that obvious peaks for lead iodide appeared in the control film. However, no PbI2 was found in the TPPO-modified film, confirming the hydrophobicity of the TPPO-modified perovskite film (Fig. S18, ESI†)51,52 To gain insight into the differences in device operational stability after the introduction of TPPO, we performed time-of-flight secondary ion mass spectrometry (TOF-SIMS) tests on the device before and after stability testing (Fig. 5a and b). For the aged control device, a large amount of I− ions was found throughout the PCBM/BCP layer and at the Ag electrode. In parallel, Ag+ ions were found to penetrate the perovskite layer. However, for the target device, under the protection of TPPO, the diffusion of I− and Ag+ ions in the aged device was effectively suppressed. This result shows that the photothermal stability of the TPPO-based device comes from the surface passively built by complexes protecting the perovskite layer, effectively preventing the probability of bidirectional diffusion between I− and Ag+ under accelerated aging conditions of 85 °C and 1 sun. TPPO enhances the stability of perovskite solar cells through a multifaceted mechanism targeting key degradation pathways. Briefly, the TPPO protective layer delays the service life of the target device under aging conditions by minimizing the chance of encounter between I− and Ag+, effectively improving the stability of the device.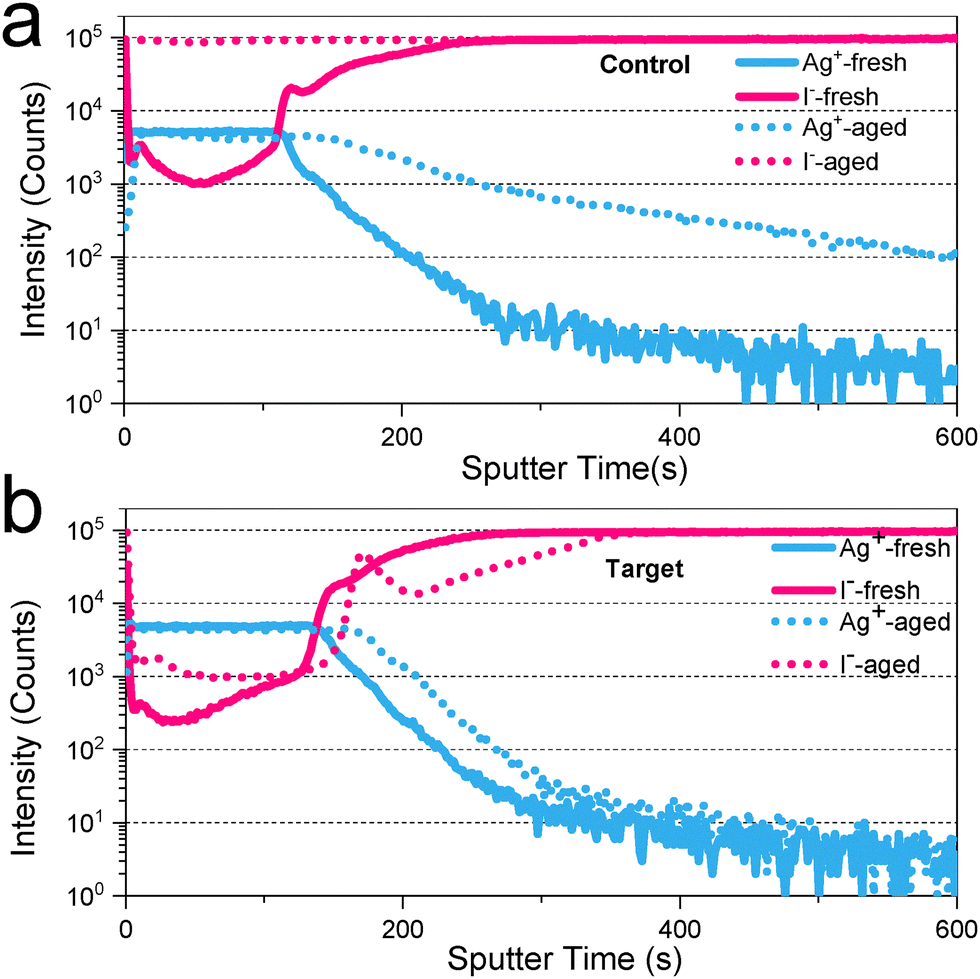 | ||
| Fig. 5 TOF-SIMS characterization of control (a) and target (b) devices for stability testing before and after aging. | ||
Conclusions
In summary, we have demonstrated that incorporating an oxide TPPO interlayer effectively prevents interlayer ion diffusion within PSCs, significantly enhancing the stability of p–i–n structured PSCs. This approach yielded a champion power conversion efficiency (PCE) exceeding 26%. Specifically, the TPPO interlayer inhibits ion migration from the perovskite layer to the ETL under thermal and light stress, thereby mitigating the degradation of the Ag electrode caused by ion-induced corrosion. Furthermore, unpaired Pb2+ ions are efficiently passivated, reducing interface recombination losses and resulting in a higher VOC. These findings highlight the potential of polymer charge-selective interlayers as a promising strategy to enhance the operational stability and efficiency of perovskite solar cells, paving the way for their commercial application.Author contributions
C. H. Li and C. J. B conceive the concept. A. V. performed the time-of-flight secondary ion mass spectrometry (ToF-SIMS) measurements. H. P., and S. H. C. tested and analyzed KPAFM data. J. B. and M. H. performed common AFM analysis. K. G. and T. U. performed the XRD measurement and discussed the results. F. S. A. S. and R. H. F. recorded and analyzed XPS data. S. Q tested the SEM image. Y. Shang. Z.Y, L.D., Z.P., J. T., C. Liu, Y.W., analyzed PL and PLQY data. F. Yang., A. O., K. F, and T. H. gave guidance on stabilization and the laser. N. Li., L. L, and C. J. B. supervised the work and contributed mainly to the revision of the manuscript. All authors discussed the results and revised the manuscript.Conflicts of interest
The authors declare no conflict of interest.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Acknowledgements
C. H. Li., S. Q., J. T., Z.P., C. Liu are grateful for the financial support from China Scholarship Council (CSC). C. J. B. gratefully acknowledges the financial support through the Bavarian Initiative “Solar Technologies go Hybrid” (SolTech), the DFG-SFB95367 (project no. 182849149), DFG-INST 90/917-1 FUGG. A. V. acknowledges funding from the Slovak Research and Development Agency under the Contract no. APVV 23-0462. H. P, and S. H. C, gratefully acknowledge funding by the ERC under the research project 4D+ nanoSCOPE with ID: ERC-SyG-810316. J. B. acknowledges the financial support RTG2861-Planar carbon lattice (RTG2861) of DFG. C. H. Li. gratefully acknowledges funding of the Erlangen Graduate School in Advanced Optical Technologies (SAOT) by the Bavarian State Ministry for Science and Art. N. Li acknowledges the financial support by the TCL Science and Technology Innovation Fund (20242065).References
- H. Chen, C. Liu, J. Xu, A. Maxwell, W. Zhou, Y. Yang, Q. Zhou, A. S. R. Bati, H. Wan, Z. Wang, L. Zeng, J. Wang, P. Serles, Y. Liu, S. Teale, Y. Liu, M. I. Saidaminov, M. Li, N. Rolston, S. Hoogland, T. Filleter, M. G. Kanatzidis, B. Chen, Z. Ning and E. H. Sargent, Science, 2024, 384, 189–193 CrossRef CAS.
- S. Liu, J. Li, W. Xiao, R. Chen, Z. Sun, Y. Zhang, X. Lei, S. Hu, M. Kober-Czerny, J. Wang, F. Ren, Q. Zhou, H. Raza, Y. Gao, Y. Ji, S. Li, H. Li, L. Qiu, W. Huang, Y. Zhao, B. Xu, Z. Liu, H. J. Snaith, N.-G. Park and W. Chen, Nature, 2024, 632, 536–542 CrossRef.
- G. Qu, S. Cai, Y. Qiao, D. Wang, S. Gong, D. Khan, Y. Wang, K. Jiang, Q. Chen, L. Zhang, Y.-G. Wang, X. Chen, A. K. Y. Jen and Z.-X. Xu, Joule, 2024, 8, 2123–2134 CrossRef CAS.
- J. Du, J. Chen, B. Ouyang, A. Sun, C. Tian, R. Zhuang, C. Chen, S. Liu, Q. Chen, Z. Li, X. Wu, J. Cai, Y. Zhao, R. Li, T. Xue, T. Cen, K. Zhao and C.-C. Chen, Energy Environ. Sci., 2025, 18, 3196–3210 RSC.
- S. Ma, G. Yuan, Y. Zhang, N. Yang, Y. Li and Q. Chen, Energy Environ. Sci., 2022, 15, 13–55 RSC.
- Q. C. Burlingame, Y.-L. Loo and E. A. Katz, Nat. Energy, 2023, 8, 1300–1302 CrossRef CAS.
- J. Luo, B. Liu, H. Yin, X. Zhou, M. Wu, H. Shi, J. Zhang, J. Elia, K. Zhang, J. Wu, Z. Xie, C. Liu, J. Yuan, Z. Wan, T. Heumueller, L. Lüer, E. Spiecker, N. Li, C. Jia, C. J. Brabec and Y. Zhao, Nat. Commun., 2024, 15, 2002 CrossRef CAS.
- L. Shen, P. Song, K. Jiang, L. Zheng, J. Qiu, F. Li, Y. Huang, J. Yang, C. Tian, A. K. Y. Jen, L. Xie and Z. Wei, Nat. Commun., 2024, 15, 10908 CrossRef.
- C. Gong, H. Li, H. Wang, C. Zhang, Q. Zhuang, A. Wang, Z. Xu, W. Cai, R. Li, X. Li and Z. Zang, Nat. Commun., 2024, 15, 4922 CrossRef CAS.
- F. Zhang and K. Zhu, Adv. Energy Mater., 2020, 10, 1902579 CrossRef CAS.
- C. Liu, Y. Yang, H. Chen, I. Spanopoulos, A. S. R. Bati, I. W. Gilley, J. Chen, A. Maxwell, B. Vishal, R. P. Reynolds, T. E. Wiggins, Z. Wang, C. Huang, J. Fletcher, Y. Liu, L. X. Chen, S. De Wolf, B. Chen, D. Zheng, T. J. Marks, A. Facchetti, E. H. Sargent and M. G. Kanatzidis, Nature, 2024, 633, 359–364 CrossRef CAS.
- H. Zai, P. Yang, J. Su, R. Yin, R. Fan, Y. Wu, X. Zhu, Y. Ma, T. Zhou, W. Zhou, Y. Zhang, Z. Huang, Y. Jiang, N. Li, Y. Bai, C. Zhu, Z. Huang, J. Chang, Q. Chen, Y. Zhang and H. Zhou, Science, 2025, 387, 186–192 CrossRef CAS.
- Y. Yang, H. Chen, C. Liu, J. Xu, C. Huang, C. D. Malliakas, H. Wan, A. S. R. Bati, Z. Wang, R. P. Reynolds, I. W. Gilley, S. Kitade, T. E. Wiggins, S. Zeiske, S. Suragtkhuu, M. Batmunkh, L. X. Chen, B. Chen, M. G. Kanatzidis and E. H. Sargent, Science, 2024, 386, 898–902 CrossRef CAS.
- Q. Li, Y. Zheng, H. Wang, X. Liu, M. Lin, X. Sui, X. Leng, D. Liu, Z. Wei, M. Song, D. Li, H. G. Yang, S. Yang and Y. Hou, Science, 2025, 387, 1069–1077 CrossRef CAS.
- M. Wang, Z. Shi, C. Fei, Z. J. D. Deng, G. Yang, S. P. Dunfield, D. P. Fenning and J. Huang, Nat. Energy, 2023, 8, 1229–1239 CrossRef CAS.
- Y. Yang, S. Cheng, X. Zhu, S. Li, Z. Zheng, K. Zhao, L. Ji, R. Li, Y. Liu, C. Liu, Q. Lin, N. Yan and Z. Wang, Nat. Energy, 2024, 9, 37–46 CrossRef CAS.
- T. Duan, S. You, M. Chen, W. Yu, Y. Li, P. Guo, J. J. Berry, J. M. Luther, K. Zhu and Y. Zhou, Science, 2024, 384, 878–884 CrossRef CAS.
- B. Li, D. Gao, S. A. Sheppard, W. D. J. Tremlett, Q. Liu, Z. Li, A. J. P. White, R. K. Brown, X. Sun, J. Gong, S. Li, S. Zhang, X. Wu, D. Zhao, C. Zhang, Y. Wang, X. C. Zeng, Z. Zhu and N. J. Long, J. Am. Chem. Soc., 2024, 146, 13391–13398 CrossRef CAS.
- C. Zhao, Z. Zhou, M. Almalki, M. A. Hope, J. Zhao, T. Gallet, A. Krishna, A. Mishra, F. T. Eickemeyer, J. Xu, Y. Yang, S. M. Zakeeruddin, A. Redinger, T. J. Savenije, L. Emsley, J. Yao, H. Zhang and M. Grätzel, Nat. Commun., 2024, 15, 7139 CrossRef CAS.
- Y. Wen, T. Zhang, X. Wang, T. Liu, Y. Wang, R. Zhang, M. Kan, L. Wan, W. Ning, Y. Wang and D. Yang, Nat. Commun., 2024, 15, 7085 CrossRef CAS.
- Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin and J. You, Nat. Photonics, 2019, 13, 460–466 CrossRef CAS.
- S. Teale, M. Degani, B. Chen, E. H. Sargent and G. Grancini, Nat. Energy, 2024, 9, 779–792 CrossRef CAS.
- H. Guo, X. Wang, C. Li, H. Hu, H. Zhang, L. Zhang, W. H. Zhu and Y. Wu, Adv. Mater., 2023, 35, e2301871 CrossRef.
- C. Zhu, X. Niu, Y. Fu, N. Li, C. Hu, Y. Chen, X. He, G. Na, P. Liu, H. Zai, Y. Ge, Y. Lu, X. Ke, Y. Bai, S. Yang, P. Chen, Y. Li, M. Sui, L. Zhang, H. Zhou and Q. Chen, Nat. Commun., 2019, 10, 815 CrossRef CAS.
- X. Wang, Z. Ying, J. Zheng, X. Li, Z. Zhang, C. Xiao, Y. Chen, M. Wu, Z. Yang, J. Sun, J.-R. Xu, J. Sheng, Y. Zeng, X. Yang, G. Xing and J. Ye, Nat. Commun., 2023, 14, 2166 CrossRef CAS.
- N. Li, S. Tao, Y. Chen, X. Niu, C. K. Onwudinanti, C. Hu, Z. Qiu, Z. Xu, G. Zheng, L. Wang, Y. Zhang, L. Li, H. Liu, Y. Lun, J. Hong, X. Wang, Y. Liu, H. Xie, Y. Gao, Y. Bai, S. Yang, G. Brocks, Q. Chen and H. Zhou, Nat. Energy, 2019, 4, 408–415 CrossRef CAS.
- N. Li, Z. Shi, C. Fei, H. Jiao, M. Li, H. Gu, S. P. Harvey, Y. Dong, M. C. Beard and J. Huang, Nat. Energy, 2024, 9, 1264–1274 CrossRef CAS.
- X. Ren, J. Wang, Y. Lin, Y. Wang, H. Xie, H. Huang, B. Yang, Y. Yan, Y. Gao, J. He, J. Huang and Y. Yuan, Nat. Mater., 2024, 23, 810–817 CrossRef CAS.
- Y. Kato, L. K. Ono, M. V. Lee, S. H. Wang, S. R. Raga and Y. B. Qi, Adv. Mater. Interfaces, 2015, 2, 1500195 CrossRef.
- D. Ma, K. Lin, Y. Dong, H. Choubisa, A. H. Proppe, D. Wu, Y.-K. Wang, B. Chen, P. Li, J. Z. Fan, F. Yuan, A. Johnston, Y. Liu, Y. Kang, Z.-H. Lu, Z. Wei and E. H. Sargent, Nature, 2021, 599, 594–598 CrossRef CAS.
- J. Han, J. M. Ha, S. H. Kweon, Y. W. Noh, D. Lee, M. H. Lee, N. Kim, J. K. Shin, S. Park, J. W. Min, W. B. Im, S. K. Kwak, M. H. Song and H. Y. Woo, ACS Nano, 2025, 19, 1044–1055 CrossRef CAS.
- R. A. Belisle, K. A. Bush, L. Bertoluzzi, A. Gold-Parker, M. F. Toney and M. D. McGehee, ACS Energy Lett., 2018, 3, 2694–2700 CrossRef CAS.
- C. Zhao, W. Wu, H. Zhan, W. Yuan, H. Li, D. Zhang, D. Wang, Y. Cheng, S. Shao, C. Qin and L. Wang, Angew Chem., Int. Ed., 2022, 61, e202117374 CrossRef CAS.
- Q. Jiang, J. Tong, Y. Xian, R. A. Kerner, S. P. Dunfield, C. Xiao, R. A. Scheidt, D. Kuciauskas, X. Wang, M. P. Hautzinger, R. Tirawat, M. C. Beard, D. P. Fenning, J. J. Berry, B. W. Larson, Y. Yan and K. Zhu, Nature, 2022, 611, 278–283 CrossRef CAS.
- Q. Jiang, Z. Chu, P. Wang, X. Yang, H. Liu, Y. Wang, Z. Yin, J. Wu, X. Zhang and J. You, Adv.Mater, 2017, 29, 1703852 CrossRef.
- S. Akin, N. Arora, S. M. Zakeeruddin, M. Grätzel, R. H. Friend and M. I. Dar, Adv. Energy Mater., 2019, 10, 1903090 CrossRef.
- Q. Ye, W. Hu, J. Zhu, Z. Cai, H. Zhang, T. Dong, B. Yu, F. Chen, X. Wei, B. Yao, W. Dou, Z. Fang, F. Ye, Z. Liu and T. Li, Energy Environ. Sci., 2024, 17, 5866–5875 RSC.
- C. Li, K. Zhang, S. Maiti, Z. Peng, J. Tian, H. Park, J. Byun, Z. Xie, L. Dong, S. Qiu, A. J. Bornschlegl, C. Liu, J. Zhang, A. Osvet, T. Heumueller, S. H. Christiansen, M. Halik, T. Unruh, N. Li, L. Lüer and C. J. Brabec, ACS Energy Lett., 2024, 9, 779–788 CrossRef CAS.
- E. V. Péan, S. Dimitrov, C. S. De Castro and M. L. Davies, Phys. Chem. Chem. Phys., 2020, 22, 28345–28358 RSC.
- M. Stolterfoht, C. M. Wolff, J. A. Márquez, S. Zhang, C. J. Hages, D. Rothhardt, S. Albrecht, P. L. Burn, P. Meredith, T. Unold and D. Neher, Nat. Energy, 2018, 3, 847–854 CrossRef CAS.
- Y. Li, Y. Duan, Z. Liu, L. Yang, H. Li, Q. Fan, H. Zhou, Y. Sun, M. Wu, X. Ren, N. Yuan, J. Ding, S. Yang and S. Liu, Adv. Mater., 2024, 36, e2310711 CrossRef.
- W. Tress, M. Yavari, K. Domanski, P. Yadav, B. Niesen, J. P. Correa Baena, A. Hagfeldt and M. Graetzel, Energy Environ. Sci., 2018, 11, 151–165 RSC.
- L. Ye, J. Wu, S. Catalán-Gómez, L. Yuan, R. Sun, R. Chen, Z. Liu, J. M. Ulloa, A. Hierro, P. Guo, Y. Zhou and H. Wang, Nat. Commun., 2024, 15, 7889 CrossRef CAS.
- M. Liu, M. L. Li, Y. X. Li, Y. D. An, Z. F. Yao, B. B. Fan, F. Qi, K. K. Liu, H. L. Yip, F. R. Lin and A. K. Y. Jen, Adv. Energy Mater., 2024, 14, 2303742 CrossRef CAS.
- S. N. Habisreutinger, N. K. Noel and H. J. Snaith, ACS Energy Lett., 2018, 3, 2472–2476 CrossRef CAS.
- D. Luo, R. Su, W. Zhang, Q. Gong and R. Zhu, Nat. Rev. Mater., 2020, 5, 44–60 CrossRef CAS.
- H. Zhu, Y. Liu, F. T. Eickemeyer, L. Pan, D. Ren, M. A. Ruiz-Preciado, B. Carlsen, B. Yang, X. Dong, Z. Wang, H. Liu, S. Wang, S. M. Zakeeruddin, A. Hagfeldt, M. I. Dar, X. Li and M. Gratzel, Adv. Mater., 2020, 32, e1907757 CrossRef.
- X. Zhao, T. Liu, Q. C. Burlingame, T. Liu, R. Holley, III, G. Cheng, N. Yao, F. Gao and Y. L. Loo, Science, 2022, 377, 307–310 CrossRef CAS.
- K. P. Goetz, A. D. Taylor, F. Paulus and Y. Vaynzof, Adv. Funct. Mater., 2020, 30, 1910004 CrossRef CAS.
- H. M. Jang, S. H. Lee, K. Y. Jang, J. Park and T.-W. Lee, Commun. Phys., 2023, 6, 372 CrossRef CAS.
- L. Shi, M. P. Bucknall, T. L. Young, M. Zhang, L. Hu, J. Bing, D. S. Lee, J. Kim, T. Wu, N. Takamure, D. R. McKenzie, S. Huang, M. A. Green and A. W. Y. Ho-Baillie, Science, 2020, 368, eaba2412 CrossRef CAS.
- G. Tumen-Ulzii, C. Qin, D. Klotz, M. R. Leyden, P. Wang, M. Auffray, T. Fujihara, T. Matsushima, J. W. Lee, S. J. Lee, Y. Yang and C. Adachi, Adv. Mater., 2020, 32, e1905035 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee03162a |
| This journal is © The Royal Society of Chemistry 2025 |