
Polarity coupling in biphasic electrolytes enables iodine/polyiodide co-extraction for portable Zn–iodine batteries following a liquid–liquid conversion route†
Hai
Xu
ab,
Ruanye
Zhang
a,
Derong
Luo
a,
Kangsheng
Huang
a,
Jiuqing
Wang
a,
Gengzhi
Sun
 *b,
Hui
Dou
*ac and
Xiaogang
Zhang
*ac
*b,
Hui
Dou
*ac and
Xiaogang
Zhang
*ac
aJiangsu Key Laboratory of Electrochemical Energy Storage Technologies, College of Materials Science and Technology, Nanjing University of Aeronautics and Astronautics, Nanjing, 210016, China. E-mail: dh_msc@nuaa.edu.cn; azhangxg@nuaa.edu.cn
bInstitute of Advanced Materials, Nanjing Tech University, Nanjing, 211816, China. E-mail: iamgzsun@njtech.edu.cn
cNational Key Laboratory of Mechanics and Control for Aerospace Structures, Institute for Frontier Science, Nanjing University of Aeronautics and Astronautics, Nanjing, 210016, P. R. China
First published on 17th June 2025
Abstract
The shuttle effect of polyiodides and aggregation of solid iodine on the cathode surface in aqueous Zn–iodine batteries are considered the main issues for their unsatisfactory cycling stability and slow charge transfer kinetics, respectively. Herein, we develop a biphasic (BP) electrolyte composed of immiscible organic (ethyl acetate, EA) and aqueous solvents for the co-extraction of iodine/polyiodides. The underlying mechanism is clarified by the principle of polarity coupling between iodine species and solvent molecules. Notably, distinct from the formation of solid iodine in an aqueous electrolyte, the electrochemical redox reactions of iodine/polyiodides at the cathodic side (organic phase) investigated by rotating ring electrodes follow the liquid–liquid conversion route. Accordingly, the diffusion of polyiodides is effectively suppressed at the interface of the BP electrolyte and the absence of solid iodine deposition significantly enhances charge transfer kinetics. Moreover, the quasi-solid-state Zn–iodine batteries featuring a gravity-independent stratified architecture are demonstrated, enabled by a BP system consisting of microspace-confined EA and PAM-CMC hydrogel. The fabricated portable device exhibits an areal capacity of 1.40 mA h cm−2 at 1 mA cm−2, improved rate performance and stable cycling performance over 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 10 mA cm−2, indicating extraordinary reliability for wearable applications.
000 cycles at 10 mA cm−2, indicating extraordinary reliability for wearable applications.
Broader contextRecently, Zn–iodine batteries composed of a Zn metal anode and an iodine/host cathode have received worldwide attention because of their Earth-abundance, high theoretical capacities and stable potential plateau. However, the shuttle effect of polyiodides and aggregation of solid iodine on the cathode surface in aqueous Zn–iodine batteries are considered the main issues for their unsatisfactory cycling stability and slow charge transfer kinetics, respectively. Although many effort has been devoted to solving the above issues, there remains a great challenge for the state-of-the-art strategies to simultaneously inhibit polyiodide shuttle and reform the deposition of solid iodine. Herein, we developed a biphasic electrolyte composed of immiscible organic and aqueous solvents for stable Zn–iodine batteries. Specifically, the co-extraction of iodine and polyiodides is experimentally achieved and the mechanism is correlated to the principle of polarity coupling, effectively restricting I-species in the organic phase. Moreover, the electrochemical redox reactions of iodine/polyiodides at the cathodic side (organic phase) following a liquid–liquid conversion route are explored, which are distinct from the formation of a solid iodine passivation layer in an aqueous electrolyte and significantly enhance charge transfer kinetics. |
Introduction
The rapid development of portable/wearable electronics, driven by growing demands of wireless communication, smart human–machine interfaces, artificial intelligence, humanoid robotics and unmanned aerial vehicles, has led to the exploitation of advanced energy storage devices with the expected features of high effectiveness, low-cost and long-serving lifespan.1–4 Although lithium ion batteries (LIBs) have been commercialized for over 30 years and are considered the primary choice as the power supply in existing consumable electronics and electric vehicles, the critical challenge in terms of scarce resources of Li-salt undoubtedly hinders their sustainable availability.5–8 A great amount of effort has been devoted to pursuing alternative substitutions. Among all potential candidates, Zn–iodine batteries composed of a Zn metal anode and an iodine/host cathode receive worldwide attention because of their Earth-abundance, high theoretical capacities and stable potential plateau.9–12In principle, the overall reaction mechanism in Zn–iodine batteries involves the stripping/deposition of the Zn metal anode and the redox of I−/I0 following a solid–liquid conversion path.13–15 At the cathode side, I− is firstly oxidized to I2(s) via a two-electron transfer process, forming a layer of solid aggregation at the interface between the electrode and the electrolyte; subsequently, I− needs to cross the solid I2(s) layer obeying a Grotthuss-like hopping mechanism for further oxidation and unavoidably complex with I2(s) to produce dissolvable polyiodides (I3−, I5−, etc.). In this whole reaction process, there are two main issues that hinder the development of Zn–iodine batteries:
Firstly, the generated soluble polyiodides at the cathode side will diffuse across the separator to the Zn anode, causing severe corrosion and promoting side reactions, which leads to low Coulombic efficiency and unsatisfactory cycling performance.16–19 Such a shuttle effect of polyiodides has become the most attractive issue. To address this dilemma, the commonly used strategies are adopting porous carbon as the iodine host material via physical adsorption.20 Nevertheless, special attention to the pore size, structure and distribution is required because of the weak attraction between non-polar carbon and iodine species. By comparison, stronger iodine adsorption can be achieved by organic materials or functional substrates through the chemical bonding of H–I, N–I and O–I.21–23 However, the loss of active iodine is inevitable after numerous charging/discharging cycles owing to the irreversible formation of the iodine-containing complex.
Secondly, the accumulation of solid iodine with poor electron conductivity (10−6 to 10−9 S cm−1) generates a passivation layer on the electrode surface, limiting the charge transfer process and slowing down cathodic reactions.24,25 Although recent efforts have been made to develop highly active electrocatalysts for fast iodine conversion, such as single-atom catalysts,26 heteroatom-doping23 and metal–organic frameworks,27 the aggregation of solid iodine is unavoidable due to its limited solubility in aqueous electrolytes. Therefore, the state-of-the-art strategies face a great challenge to simultaneously inhibit the polyiodide shuttle and reform the deposition of solid iodine.
Alternatively, a biphasic electrolyte (BP) composed of two immiscible solutions, which was primarily applied in redox flow batteries (RFB) to substitute the combination of traditional single-phase electrolytes and ion-exchange membranes, naturally produces an artificial liquid–liquid interface for effectively separating dissolved active species based on their polarities,28–31 offering a clever tactic to address the abovementioned issues. Recently, the different BP electrolytes of CH2Cl2/H2O,49 acetonitrile (ACN)/ZnSO4,50 ACN-ionic liquid/ZnSO4,51 and ionic liquid/H2O52 for Zn–iodine batteries were reported to restrict polyiodides. Nevertheless, the effectiveness of these BP electrolytes in portable Zn–iodine batteries is limited and the detailed reaction mechanism involved in the organic phase remains unexplored. (i) Owing to the reversible and rapid conversion between I2 and polyiodides, the dissolved polyiodides inevitably contain I2 components, while it is hard to co-extract I2 and polyiodides via a single solvent owing to their different polarities, which results in inefficient confinement of soluble iodine species through BP systems and the fundamental design principles governing the compatibility between solvents and different iodine species remains unknown; (ii) the cathodic reaction of I−/I0 proceeds through a solid–liquid conversion path in aqueous electrolytes, while the unexplored reaction mechanism in the organic phase deserves a detailed study in the BP system; and (iii) the reported BP Zn–iodine/bromine batteries are still prototypes typically in stationary beaker cells, which is impractical for the design of portable/wearable energy storage devices.
Herein, the co-extraction of iodine and polyiodides is experimentally achieved by polarity coupling in a newly developed BP electrolyte for Zn–iodine batteries. In detail, the BP interface is formed between the immiscible organic ethyl acetate (EA) phase containing bis(trifluoromethanesulfonyl)-imide zinc (Zn(TFSI)2) and the aqueous phase of ZnSO4/KI solution, where the dissolved iodine and polyiodides are confined in the organic phase electrolyte (cathode side). The mechanism of co-extraction of iodine/polyiodides is explored and correlated to the principle of polarity coupling between iodine species and solvent molecules. Besides, a liquid–liquid conversion route of iodine at the cathodic side in the BP electrolyte was revealed using a rotating ring electrode (RRE), which is completely distinct from the traditional liquid–solid conversion in aqueous electrolytes, exhibiting much improved charge transfer kinetics. According to these understandings, quasi-solid-state (QSS) portable Zn–iodine batteries featuring a gravity-independent stratified interface based on the BP electrolyte system composed of a microspace-confined EA phase and a PAM-CMC hydrogel further demonstrate superior cycling stability to other reported BP Zn–I/Br batteries. For proof-of-concept applications, the assembled BP devices are successfully employed for health monitoring under multiple practical operation conditions (Fig. 1)
 | ||
| Fig. 1 Schematic diagram of the comparison between aqueous SP Zn–iodine batteries and portable BP Zn–iodine batteries. | ||
Results and discussion
Iodine (I2) is a diatomic molecule with a symmetric linear shape. Since both I atoms share an equal proportion of charge, the net dipole moment of I2 comes out to be zero, making it a nonpolar molecule. As a typical polyiodide, triiodide ion (I3−) has a similar linear configuration with no dipole moment in the vapor phase. Nevertheless, the experimental results suggest that I3− becomes polarizable and soluble in polar solvents due to the molecule interaction-induced symmetry breaking.32–35 The differences in polarity make it challenging to co-extract I2 and polyiodides according to the principle of “like dissolve like”. However, in the application of Zn–iodine batteries, co-extraction is highly necessary because the cathodic reactions involve the reversible electrochemical conversions between I2 and polyiodides, since dissolved polyiodide species inevitably contain the I2 components owing to the chemical reaction equilibrium. Unfortunately, the feasibility of constructing a BP electrolyte to restrict I2/polyiodides on the cathode side via a single solvent and prevent their shuttling remains an unknown question to date. In our experiment, an aqueous solution containing I2 and I3− (0.1 M KI/20 mM I2) was prepared to simulate dissolved iodine species in the aqueous phase and then respectively mixed with a number of solvents with different polarities including petroleum ether (PE), cyclohexane (CH), carbon tetrachloride (CTC), benzene (BZ), ethyl acetate (EA), methyl acetate (MA), pyrrole (Py) and acetonitrile (ACN) (Fig. 2a and b).36 After a few seconds, self-stratified phases are produced except for the high polar Py and ACN. Notably, the aqueous phase becomes the lightest yellow upon the addition of EA, indicating the nearly complete extraction of polyiodides/iodine. Direct evidence is provided by the ultraviolet-visible (UV-vis) spectra through quantifying the content of I-species respectively in the organic and aqueous phases. Owing to the chemical equilibrium between I2 and polyiodides, the R value via the absorbance changes of I3− (288 nm) in the aqueous phase (I3− after and I3− before) was defined to quantitatively evaluate the extraction ability of different organic solvents for iodine species according to the Lambert–Beer law.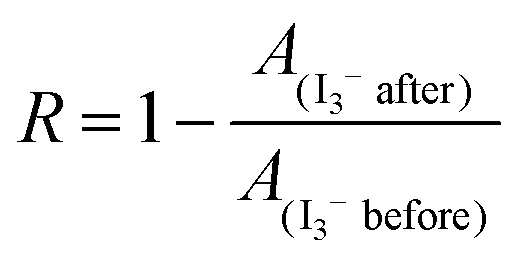
 | ||
| Fig. 2 Selection of organic solvents and extraction effects of iodine and polyiodides. (a) The photos of different BP extract systems composed of the organic phase (PE, CH, CTC, BZ, EA, MA, Py and ACN) and 0.1 M KI/20 mM I2 aqueous phase. (b) The dielectric constants of different organic solvents. The UV-vis spectra of (c) different organic phases and (d) the 0.1 M KI/20 mM I2 aqueous phase before and after extraction. (e) R value of I3− peaks of the H2O phase after extraction. | ||
As displayed in Fig. 2c, the obvious peaks at ∼518 nm of the PE, CH and CTC phases indicate the presence of I2, corresponding to the violet color of organic phases. The absorption of I2 blueshifts to ∼504 nm in the case of BZ, giving a color of red-brown due to the stronger molecular interaction with electron transfer from an aromatic hydrocarbon to I2, which is reflected by the newly emerged peak of the BZ–I2 complex at 288 nm.37 The corresponding intensity changes of I-species in the UV-vis spectra of the aqueous phase after extraction in Fig. 2d suggest that the non-polar solvents of PE, CH, CTC and BZ exhibit a high selectivity for I2, while I3− cannot be completely extracted into the organic phase with R values of 31%, 35%, 50% and 83% respectively (Fig. 2e). In comparison, both I3− (294 and 364 nm) and I2 (450 nm) are effectively extracted in EA,38,39 resulting in a calculated R-value of as high as 98%. In the case of MA with a similar structure but slightly increased polarity to EA, only I3− is detected in the MA phase with a comparatively decreased R-value of 88%. In addition, the corresponding absorbances of I3− (288 nm) in the aqueous phase after extraction by different organic solutions are exhibited in Fig. S1 (ESI†).
The distinct advantages of EA were validated by analyzing the charge distribution and molecular electrostatic potential (ESP). According to the comparison presented in Fig. 3a and Fig. S2 (ESI†), the nonpolar alkyl group and symmetric structure enable the uniform charge distribution in PE, CH, CTC and BZ, while the polar ester group induces the charge separation in EA and MA. Therefore, combined with the experimental results, the principle for the co-extraction of iodine/polyiodides enabled by polarity coupling is summarized in Fig. 3b. The nonpolar PE with hydrophobic alkyl chains ensures efficient extraction of iodine and facilitates the construction of the BP interface, but excludes polyiodides; Py and ACN with strong polarity can homogeneously mix with water, unlikely producing phase separation; EA containing both a nonpolar hydrophobic alkyl group and a polar ester group exhibits moderate molecule polarity and allows the formation of a self-stratified BP system with water and simultaneous extraction of iodine and polyiodides. As a control experiment, although MA possesses a similar structure to EA, the slightly increased polarity due to the shortened alkyl chain limits the ability to extract iodine, further validating the principle of polarity coupling.
 | ||
| Fig. 3 (a) Electrostatic potential distribution of different solvents. (b) The relationship between molecular polarity and iodine species extraction. (c) The photos of different BP extract systems composed of the organic phase (PE, EA, and MA) and I3− and I2 aqueous phase. (d) and (e) The UV-vis spectra of different aqueous phases before and after extraction. (f) and (g) R values of I3− peaks (288 nm) and I2 peaks (460 nm) of the aqueous phase after extraction. | ||
More compelling evidence was obtained through an experiment investigating the difference between the non-polarity and polarity groups in extracting the I2 aqueous solution and the I3− aqueous solution. The I3− aqueous solution was prepared by mixing 0.1 M KI and 1 mM I2; excess KI ensures the nearly complete conversion of I2 to I3−. The I2 aqueous solution was obtained by ultrasonication of solid iodine in water. As shown in Fig. 3c, PE, EA and MA were mixed with the I3− aqueous solution and I2 aqueous solution, respectively, spontaneously producing self-stratified phases. Taking solution color, R-value and the corresponding absorbance changes into consideration (Fig. 3d–g and Fig. S3, ESI†), it is reasonable to conclude that PE exhibits excellent extraction efficiency for I2 but weak extraction for I3−. Conversely, MA displays good ability for extracting I3− together with moderate extraction efficiency for I2. These results suggest that polar and non-polar functional groups in organic molecules function differently in their tendencies to extract I2 and I3−, Notably, EA demonstrates exceptional capability in extracting I2 and I3− from aqueous solutions because of its well-suited polarity coupling.
Molecular dynamics (MD) simulations were also conducted to investigate the co-extraction ability of the EA-H2O BP system for I3− and I2 (Fig. S4, ESI†). I3− and I2 distributed in the H2O phase in the initial state rapidly diffuse to and fully fill in the EA phase in the final state, suggesting the strong interaction between I2/I3− and EA. This was further proved by DFT calculations (Fig. S5, ESI†). According to molecular orbital theory, an empty antibonding (σ*) orbital is formed in I2 and I3− molecules due to the unique outermost electronic configuration of 5s25p5 in the I atom (Fig. S6, ESI†),40,41 which is prone to accept the donated lone-pair electrons from the negatively charged C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/C–O. Such a charge transfer behavior between EA and I3−/I2 is clearly evidenced in the optimized charge-density-difference pattern (Fig. S7, ESI†).
O/C–O. Such a charge transfer behavior between EA and I3−/I2 is clearly evidenced in the optimized charge-density-difference pattern (Fig. S7, ESI†).
In order to ensure that the ionic conductivity of the EA phase was high enough, Zn(TFSI)2 was selected as the Zn-salt because the other commonly used ones such as Zn(ClO4)2, ZnSO4 and Zn(OTf)2 cannot be dissolved in EA due to their relatively strong interaction between the anion and cation (Fig. S8, ESI†). Consequently, an immiscible BP electrolyte system was successfully prepared by simply mixing 1 M Zn(TFSI)2 in EA and an aqueous solution containing 1 M ZnSO4 + 0.5 M KI (Fig. S9, ESI†). The ion conductivity of the obtained upper EA (1 M Zn(TFSI)2) phase is found to be 14.34 mS cm−1 (Fig. S10, ESI†), better than most of the reported values for organic electrolytes even though still lower than that of the bottom aqueous solution (76.41 mS cm−1).
The redox behavior of iodine in the BP electrolyte system is revealed in a visualized two-electrode system with a glass carbon (GC) as the cathode inserted in the EA phase (1 M Zn(TFSI)2) and a Zn rod as the anode in the aqueous phase (1 M ZnSO4 + 0.5 M KI) (Fig. S11, ESI†). Chronoamperometry (CA) was adopted by respectively applying 1.4 V and 0.4 V for 250 s to oxidize and reduce I-species. In the corresponding current–time curve (Fig. 4a), four representative points of A, B, C and D are selected for the subsequent acquisition to characterize the chemical composition of the resultant products. As shown in Fig. 4b, yellow polyiodides emerge on the surface of the GC electrode and then gradually dissolve in the EA phase during the electrochemical oxidation from A to C. Upon switching the polarization voltage to 0.4 V, the color of the EA phase continuously fades from the C to the D state attributable to the reduction of polyiodides. It is noted that the bottom aqueous phase remains colorless during the whole process, suggesting the effective inhibition of polyiodide shuttling. Comparably, in the single phase (SP) aqueous electrolyte (1 M ZnSO4 + 0.5 M KI), once the polyiodides are generated, they freely diffuse to the anode side without any restriction (Fig. S12, ESI†). The UV-vis spectra provide direct evidence for the concentration variation of I− and I3− in the organic and aqueous phases (Fig. 4c and d). In the aqueous phase, the content of I− gradually decreases under oxidation and increases at reduction, without any discernible signal from I3− during the entire process. Oppositely, in the organic phase, the content of I3− gradually increases in the process of oxidation from state A to C and decreases in the process of reduction from state C to D. The UV-vis absorbance of I− in the H2O phase and that of I3− in the EA phase in the different states are summarized in Fig. 4e. Therefore, it could be concluded that I− can cross the BP interface and be oxidized at the GC surface, whereas the produced polyiodides are thoroughly restrained in the organic phase.
 | ||
| Fig. 4 Characterization of the ion transport behavior across the interface of the BP electrolyte. (a) The CA curves marked with four represented states (A, B, C and D). (b) The digital photos of the visualized two electrode system corresponding to the states of A–D. The UV-vis spectra in the states of A–D of (c) the EA phase and (d) the H2O phase. The summarized (e) UV-vis absorbance changes of I3− in the EA phase and I− in the H2O phase and (f) concentration changes of Zn2+ and K+ in the H2O phase from states A to D. (g) The Raman spectra of the EA phase and the H2O phase in the states of A–D, 1 M ZnSO4 in H2O and 1 M Zn(TFSI)2 in EA. (h) The schematic of the ion transport mechanism during the charging–discharging process. | ||
Additionally, inductively coupled plasma mass spectrometry (ICP-MS) and Raman spectra were further utilized to monitor the transport of ions involved in the charging–discharging process. In Fig. 4f, the concentrations of K+ and Zn2+ in the aqueous phase increase from states A to C and then decrease from C to D according to the ICP-MS results, indicating the migration of Zn2+ and K+ across the BP interface. Moreover, Raman spectra show typical vibration peaks of SO at 980 cm−1 and C–F at 1240 cm−1, respectively ascribing to SO42− and TFSI−.42–44 The results displayed in Fig. 4g indicate that SO42− in the aqueous phase and TFSI− in the organic phase are not able to cross the BP interface even under an electric field. Based on the above analysis, the ion transport mechanism can be concluded in Fig. 4h. From state A to C, Zn2+ and K+ migrate to the anode side, where Zn2+ accepts the electrons and is reduced to Zn0; I− moves to the cathode side and is oxidized to generate polyiodides and iodine. Once the polarization voltage is switched to 0.4 V, Zn metal is dissolved in the aqueous phase and I3− restrained in EA is reduced to I−, accompanied by the diffusion of Zn2+ and K+ to the cathode side and I− to the anode side. Besides, TFSI− and SO42− do not cross the BP interface during the charge–discharge process, although they diffuse under the electric field.
In principle, the cathodic reactions of Zn–iodine batteries involve the electrochemical conversion of I−, the complexation between I− and I2(s), and the formation of polyiodides in the charging process, which are difficult to detect by traditional spectroscopic techniques. A viable strategy to explore the reaction mechanism that occurred at the electrode/electrolyte interface is to use a rotating ring disk electrode (RRE: GC disk and Pt ring in this study) as the working electrode, coupled with Ag/AgCl as the reference electrode and Pt plate as the counter electrode (Fig. S13, ESI†). Experimentally, SP and BP were respectively utilized as electrolytes, a potential of 0–0.7 V was applied at the GC disk, and a constant potential of 0.2 V was held at the Pt ring to reduce the iodine species, with the rotation speed of the RRE maintained at 150 rpm.
Fig. 5a shows the linear sweep voltammetry (LSV) curves for the oxidation of I− in the SP electrolyte, which can be divided into four distinct regions: region-I 0–0.48 V, region-II 0.48–0.5 V, region-III 0.5–0.53 V and region-IV 0.53–0.7 V. In region-I, the oxidative current (denoted as io) at the GC disk corresponding to the conversion from I−(aq) to I3−(aq) through the soluble I2(aq) and reductive current (denoted as ir) of the conversion from I3−(aq) to I−(aq) at the Pt ring grow synchronously following the reactions (1) and (2).
| 3I−(aq) − 2e− ⇆ I2(aq) + I−(aq) ⇆ I3−(aq) | (1) |
| I3−(aq) + 2e− ⇆ 3I−(aq) | (2) |
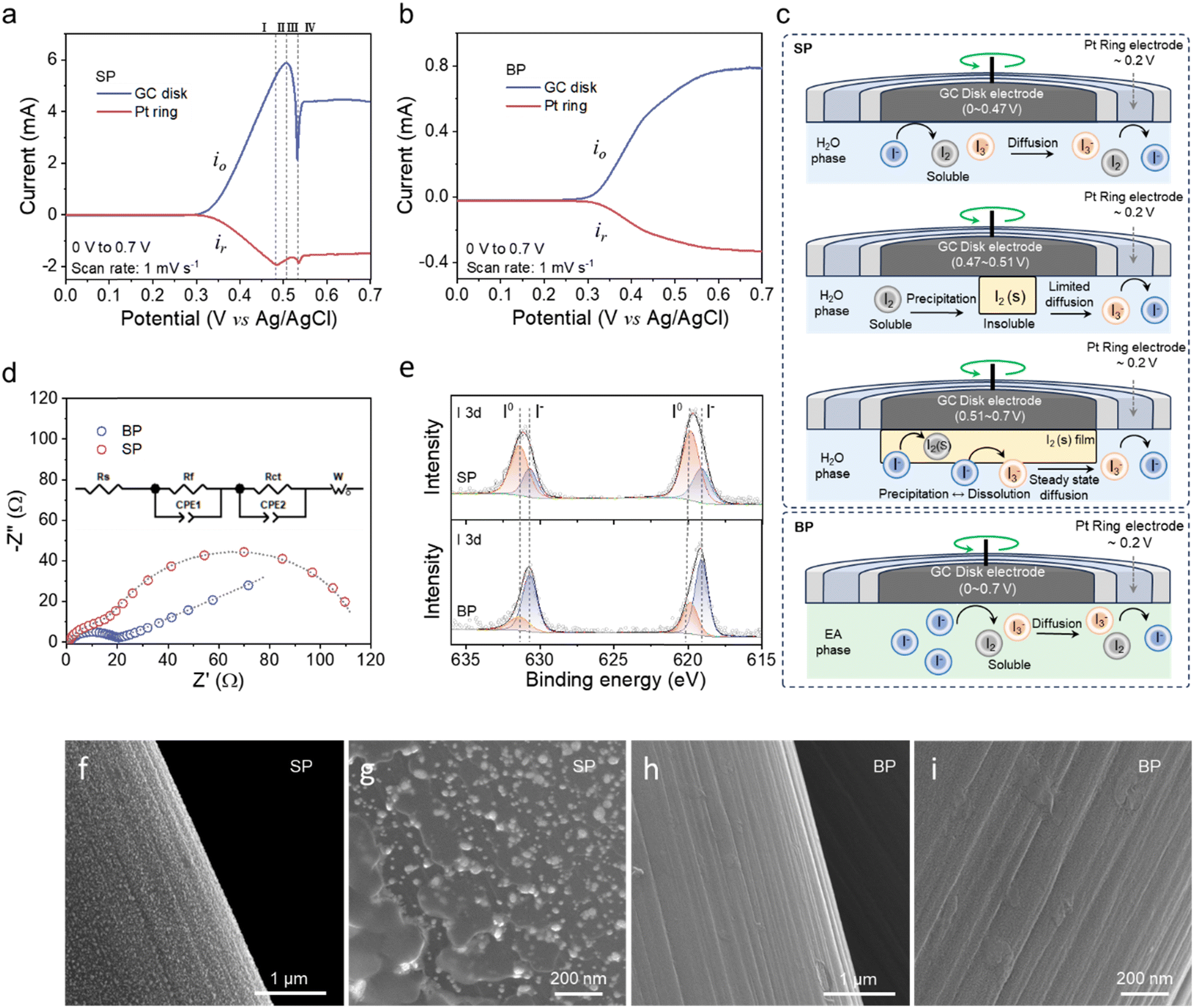 | ||
| Fig. 5 Characterization of the cathodic reactions in the BP and SP electrolytes. LSVs on RRE in the (a) SP and (b) BP electrolytes from 0 V to 0.7 V. (c) The schematic of redox reactions at the interface between the electrode and the electrolyte. (d) The EIS plots, (e) XPS spectra and (f)–(i) SEM images of the CC cathode for coin cells based on BP and SP after 15 s polarization under 1.5 V. | ||
The accumulation of I2(aq) in the vicinity of the GC disk leads to the precipitation of solid I2(s) upon further increase in the potential owing to its limited solubility in the aqueous electrolyte as the equilibrium shown in reaction (3).
| I2(aq) ⇆ I2(s) | (3) |
The precipitated I2(s) on the GC disk cannot transport to the Pt ring, thus ir gradually decreases in region-II. Subsequently, further aggregation of I2(s) on the GC disk slows down the oxidation reaction caused by the limited ion diffusion, even though the partial dissolution of I3−(aq) occurs as depicted in reaction (4), thus leading to a dramatically decreased io and thereafter an increased ir in region-III.
| I2(s) + I−(aq) ⇆ I3−(aq) | (4) |
When the formed I2(s) layer completely covers the surface of the GC disk, io reaches the lowest point and the manner of I−(aq) transport changes from direct diffusion to “carrier”-type diffusion according to the Grotthuss mechanism through the I2(s) film. Finally, both io and ir reach the steady-state in region-IV, indicating that the dissolution–precipitation of the I2(s) film attains the equilibrium state. The above analysis supports the solid–liquid reaction route which is in good accordance with our previous work.12 Comparably, in the case of BP electrolyte, io and ir monotonously increase and ultimately reach the equilibrium state with the applied potential (Fig. 5b), indicating a totally different reaction mechanism from that in the aqueous SP electrolyte without the presence of I2(s) owing to the high dissolubility of both I2 and polyiodides in EA following the so-called liquid–liquid conversion route. Such a special mechanism is confirmed by LSV from 0.7 V to 0 V in Fig. S14 (ESI†), where a peak attributable to the decomposition of the I2(s) film occurs in the SP system rather than in the BP system. The abovementioned reaction mechanisms in SP and BP electrolytes are summarized in Fig. 5c and Fig. S15 (ESI†) based on the principle that EA with moderate polarity possesses much higher solubility of both I2 and polyiodides than H2O, avoiding the aggregation of I2(s) and realizing the liquid–liquid conversion.
In order to further validate the above mechanism, the coin cells assembled with the Zn anode and carbon cloth (CC) cathode separately in BP and SP electrolytes were characterized by the CA technique at 1.5 V for 30 s and then their electrochemical impedance spectroscopy (EIS) results are displayed in Fig. 5d. Obviously, the wide semicircle in the intermediate/low frequency region for the batteries based on the SP electrolyte suggests an extra barrier resistance owing to I2(s) precipitation. Direct evidence can be found in the I 3d XPS spectra (Fig. 5e) and SEM images (Fig. 5f–i) of the CC cathode,45,46 where a stronger I0 signal is obtained and apparent I2(s) particles present on the cathode disassembled from the SP-based batteries but not with the BP-based batteries, verifying that the cathodic reactions in the BP electrolyte follows a different liquid–liquid reaction mechanism.
Although the feasibility of the BP electrolyte in prohibiting polyiodide shuttling and the reaction mechanism of I-species in the organic phase have been validated theoretically and experimentally, challenges still remain in the construction of portable Zn–iodine batteries. Firstly, the long diffusion distance of dissolved polyiodides in organic electrolytes leads to an irreversible charging/discharging process (Fig. 4b). Secondly, the gravity-driven stratification is inadequate for complex operational environments. We therefore proposed a QSS portable Zn–iodine battery based on a gravity-independent stratified interface realized by a synergistic effect between a microspace-confined EA phase in an activated carbon (AC) cathode and hydrogel as an aqueous phase to address the above issues (Fig. 6a). With AC serving as an effective cathode host, its ultra-high specific surface area (∼2198 cm2 g−1) and organophilic properties enable the rapid absorption of the EA electrolyte, and the abundant mesopores (10–20 Å) also could function as reaction micro-vessels, immensely shortening the diffusion distance of dissolved polyiodides and avoiding irreversible redox reactions (Fig. S16 and S17, ESI†). On the other hand, a hydrogel containing 1 M ZnSO4/0.5 M KI is prepared by chemically crosslinking polyacrylamide and carboxymethyl cellulose (PAM-CMC) (Fig. S18, ESI†), in which water molecules are constrained in the polymeric network, guaranteeing efficient ion diffusion and at the meantime further limiting the diffusion of iodine species. As displayed in Fig. 6b, after 24 h, the added I3−/I2 are effectively constrained in the EA phase, leaving hydrogel colorless, whereas severe shuttling of I3− was observed in the water//hydrogel SP system. When PAM-CMC and the EA phase were sequentially vialed in a bottom-up manner, forming a distinct interface, it can remain intact upon flipping over and keep stable even after 30 days (Fig. 6c and Fig. S19, ESI†). Combining with the advantages of the AC cathode and PAM-CMC hydrogel, the QSS devices were assembled in the configuration Zn//hydrogel//AC-EA based on the above BP electrolytes and then electrochemically evaluated at a constant current of 0.5 mA cm−2. In situ Raman spectra confirm the highly reversible reactions at the cathode side with the appearance and disappearance of I3− at 110 cm−1 and I5− at 160 cm−1 respectively in the charging and discharging processes (Fig. 6d). Moreover, almost the same GCD curves at 1 mA cm−2 with a high coulombic efficiency of 98.6% are obtained no matter how the batteries work in the way of anode upward or cathode upward, further ensuring the effectiveness of the design for gravity-independent BP-based energy storage systems (Fig. S20, ESI†).
 | ||
| Fig. 6 (a) The design of gravity-dependent and gravity-independent BP interfaces. (b) The photos for the shuttle experiment of I3−/I2 in EA/PAM-CMC (BP) and H2O/PAM-CMC (SP) systems. (c) The photos of gravity-independent test of EA/PAM-CMC. (d) The in situ Raman spectra of Zn–iodine based on the BP electrolyte during the charging/discharging process. | ||
In our previous study,13 it has been proved that the side reactions occurring at the Zn anode mainly originate from the corrosion by dissolvable polyiodides, resulting in a highly porous and loose surface which subsequently accelerates the hydrogen evolution reaction (HER), consumes protons in the aqueous electrolyte, and locally induces the precipitation of Zn4SO4(OH)6·xH2O (Fig. 7a). Herein, the anodes respectively disassembled from the batteries of Zn//hydrogel//AC (SP) and Zn//hydrogel//AC-EA (BP) after 10 cycles at 0.5 mA cm−2 were characterized to verify the suppression to the side reactions by the BP electrolyte via restricting the shuttle of polyiodides. The stronger XRD signals ascribed to Zn4SO4(OH)6·xH2O in the SP electrolyte reveal the more severe corrosion that occurred at the Zn anode in comparison with that in the BP electrolyte (Fig. 7b and Fig. S21a, ESI†). SEM images also confirm the severe corrosion of the Zn anode in the SP electrolyte with more platelets generated on the surface (Fig. 7c and d and Fig. S21b, ESI†). The composition of the cycled Zn anodes was analyzed in depth direction using X-ray photoelectron spectroscopy (XPS) equipped with an etching technique (Fig. 7e and 7f). The platelets can be identified as Zn4SO4(OH)6·xH2O with the characteristic peaks of Zn2+ at 1023.1 eV and SO42− at 169.8 eV at the outermost layer of Zn anodes.22,47,48 It is noteworthy that, according to the emergence of XPS signals of Zn0 and disappearance of SO42−, the corrosion depths of the Zn anode disassembled from the BP and SP batteries vary significantly with 100 nm and 500 nm respectively, demonstrating that the co-extraction of iodine and polyiodides in BP effectively inhibits the side reactions of corrosion and hydrolysis and thereby guarantees a relatively stable electrolyte/anode interface.
 | ||
| Fig. 7 Characterization of the anode in BP and SP electrolytes. (a) The schematic diagram of polyiodide-induced corrosion at the Zn anode surface. (b) The XRD patterns, (c) and (d) SEM images and the etching XPS results of (e) Zn 2p2/3 and (f) S 2p of Zn electrodes after 50 cycles in the Zn–iodine batteries based on the BP and SP electrolytes. | ||
The electrochemical performances of QSS Zn–iodine batteries respectively based on the BP and SP electrolytes were fully evaluated. The CV curves at 0.1 mV s−1 display similar shape and characteristic redox peaks of iodine, yet different voltage gaps between the anodic and cathodic peaks for the BP (0.20 V) and SP (0.25 V) electrolytes (Fig. 8a). Moreover, the anodic peak respectively shifts 80 and 110 mV for BP and SP devices when the scan rate increases from 0.2 mV s−1 to 1 mV s−1 (Fig. S22, ESI†). Both the large voltage gap and anodic peak shift in the SP electrolyte are ascribed to the slow electron transfer caused by the formation of solid iodine precipitation. As a consequence, the batteries based on the BP electrolyte deliver an areal capacity of 1.40 mA h cm−2 at 1 mA cm−2 and retain 0.75 mA h cm−2 at 10 mA cm−2 with a superior retention of 54% compared to 38% of the SP device (Fig. 8b). Besides, the relatively higher mid-discharge voltages at different current densities further demonstrate the fast redox reactions and stable electrode/electrolyte interface in the BP system (Fig. 8c and d and Fig. S23, ESI†). The optimized energy/power densities of 1.52 mW h cm−2 (at 1.1 mW cm−2) and 9.73 mW cm−2 (at 0.73 mW h cm−2) are achieved for Zn–iodine batteries based on the BP electrolyte (Fig. S24, ESI†).
 | ||
| Fig. 8 The electrochemical performances of QSS Zn–iodine batteries based on the BP and SP electrolytes. (a) The CV curves at 0.1 mV s−1. (b) The rate performance. (c) The GCD curves of the BP electrolyte at different current densities. (d) The mid-discharge voltages. (e) and (f) The in situ EIS spectra during the charging process. (g) The GITT curves. (h) The cycling performance at 5 mA cm−2. (i) The GCD curves of the BP electrolyte at 5 mA cm−2. (j) The cycling performance of the BP electrolyte at 10 mA cm−2. | ||
The in situ EIS tests monitor the resistance changes of QSS Zn–iodine batteries (Fig. 8e and f). Owing to the absence of a solid iodine layer, the device based on the BP electrolyte demonstrates a much lower charge transfer resistance than that based on the SP electrolyte during the charging process, realizing faster reaction kinetics. As displayed by the galvanostatic intermittent titration technique (GITT), the BP device shows a smaller voltage fluctuation due to the minimized polarization in comparison with the SP device (Fig. 8g), which agrees well with the results from EIS. Benefiting from the restriction of polyiodides and regulation of the cathode surface in the BP electrolyte, the assembled cell exhibits significantly enhanced stability with similar GCD curves at the 10th, 1000th and 2000th cycles, achieving a 100% areal capacity retention over 2500 cycles at a current density of 5 mA cm−2 (Fig. 8h and i). Even at a high current density of 10 mA cm−2, the batteries exhibit outstanding cycling stability over 22000 cycles (Fig. 8j), which is superior to other reported BP Zn–I/Br batteries (Tables S1 and S2, ESI†), indicating great potential to meet the requirements under practical conditions.
The QSS pouch cells (3 cm × 4 cm) of Zn anode//hydrogel//AC-EA cathode with CC current collector were also fabricated in an aluminum-plastic package. The QSS pouch cells output areal capacities of 1.45, 1.33, 1.26, 1.20 and 1.15 mA h cm−2 with a high coulomb efficiency of ∼100% at 10, 20, 30, 40, and 50 mA, respectively (Fig. 9a and b). The calculated maximum energy density reaches 1.59 mW h cm−2. Benefiting from the special design, the pouch cell exhibits excellent environmental adaptability with 100%, 96% and 98% capacity retentions as the device was inverted, bent (90°), and hammered (20 times) (Fig. 9c and d). When the cell was cycled in a 100% depth of discharge for 450 h (Fig. 9e), it still can sustain negligible capacity loss with an average coulomb efficiency of 99.5% (Fig. 9f). Besides, such a device maintains stability over 30 cycles of 90° bending–unbending, suggesting its excellent mechanical reliability (Fig. S25, ESI†). The portable QSS pouch cells were further integrated with a wearable sensing system for continuous health monitoring. In detail, three QSS pouch cells connected in series offer the capability to safely power a smart watch that can acquire the corresponding data of heart beat and blood pressure (Fig. 9g). The tandem device mounted on human limb maintains stable operation under various movements. These application measurements demonstrate the feasibility of our rational design for daily usage and potential in a broader context (Fig. 9h).
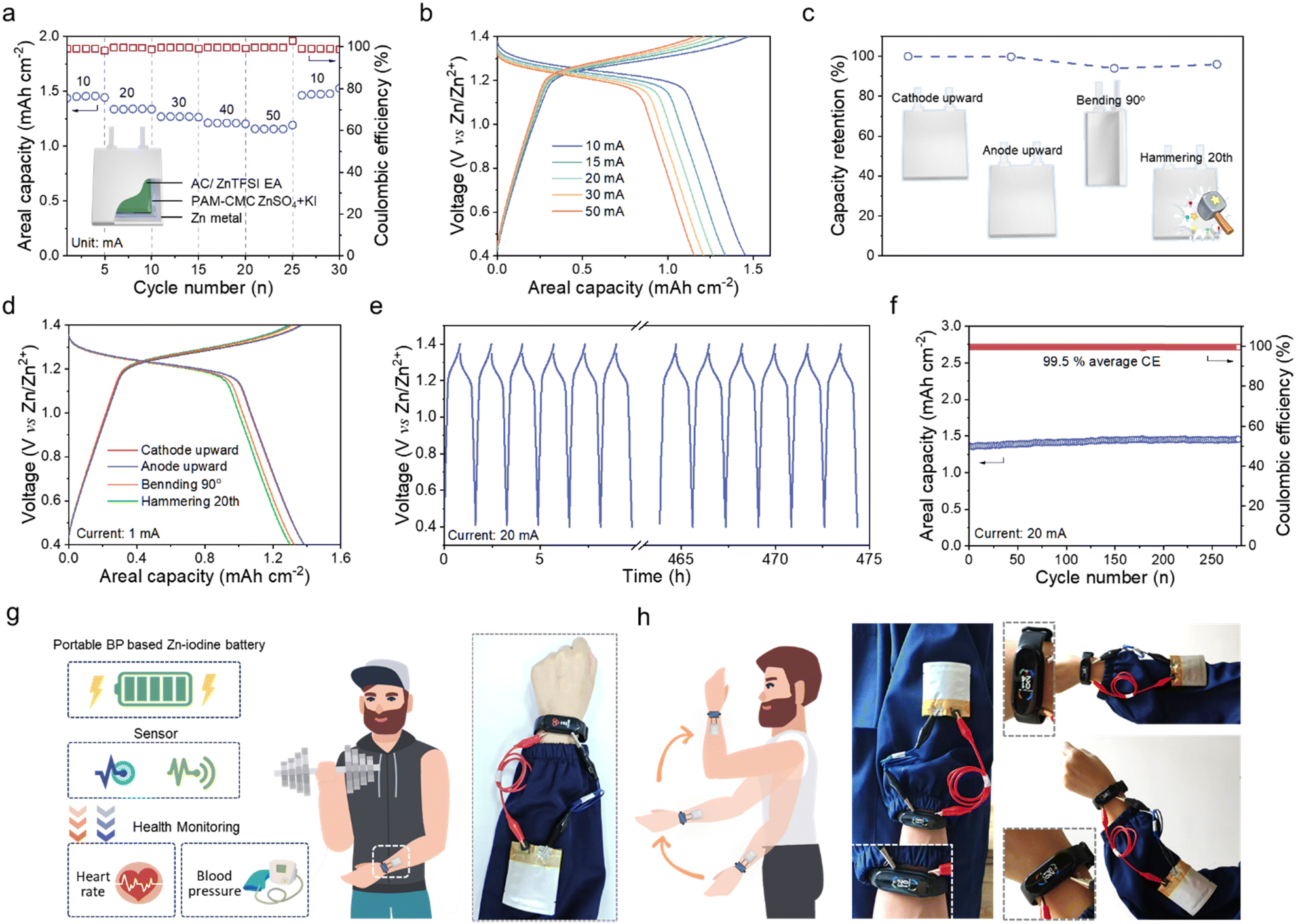 | ||
| Fig. 9 The performance of the QSS pouch cells based on the BP electrolyte. (a) The rate performance. (b) The GCD curves from 10 mA to 50 mA. (c) The capacity retention and (d) GCD curves under different working conditions. (e) The voltage–time curves and (f) cycling performance at 20 mA. (g, h) Schematic diagram of an integrated portable energy storage-sensing system and demonstration photos of health monitoring. | ||
Conclusions
In summary, we developed a BP electrolyte system composed of two immiscible solutions of Zn(TFSI)2 in EA and ZnSO4/KI in water to address the key issues of Zn–iodine batteries. Due to the proper nonpolar hydrophobic alkyl chain and polar ester groups, EA with moderate molecule polarity shows the polarity coupling with iodine species, and it allows the formation of a self-stratified BP system with water and simultaneous extraction of iodine and polyiodides, effectively preventing the shuttling effect of polyiodides. More importantly, multiple spectral characterization methods and electrochemical testing disclose that the cathodic reactions follow an unconventional liquid–liquid conversion route without the formation of a solid iodine passivating layer, achieving much faster charge transport kinetics. The designed QSS portable Zn–iodine battery with a gravity-independent stratified interface based on the BP electrolyte with microspace-confined EA and PAM-CMC hydrogel exhibits an enhanced areal capacity of 1.40 mA h cm−2 at 1 mA cm−2, excellent rate performance of 0.75 mA h cm−2 at 10 mA cm−2, 100% capacity retention after 2500 cycles at 5 mA cm−2 and cycling stability of over 22000 cycles at 10 mA cm−2. As proof-of-application, the assembled pouch cell based on the BP electrolyte can adapt to complex operation environments and realize energy storage supply for human health monitoring.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Data are available upon request from the authors.Acknowledgements
This work was supported by the National Key Research and Development Project Intergovernmental International Science and Technology Innovation Cooperation (2022YFE0109400), the National Key Research and Development Program of China (2023YFB2405800) and the Leading Edge Technology of Jiangsu Province (BK20232022 and BK20220009). This work was supported by the facilities in the Center for Microscopy and Analysis at Nanjing University of Aeronautics and Astronautics.References
- S. Ding, T. Saha, L. Yin, R. Liu, M. I. Khan, A.-Y. Chang, H. Lee, H. Zhao, Y. Liu and A. S. Nazemi, Nat. Electron., 2024, 7, 1–12 CrossRef.
- C. Lu, H. Jiang, X. Cheng, J. He, Y. Long, Y. Chang, X. Gong, K. Zhang, J. Li and Z. Zhu, Nature, 2024, 629, 1–6 CrossRef PubMed.
- S. Saifi, X. Xiao, S. Cheng, H. Guo, J. Zhang, P. Müller-Buschbaum, G. Zhou, X. Xu and H.-M. Cheng, Nat. Commun., 2024, 15, 6546 CrossRef CAS PubMed.
- K. Zhang, X. Shi, H. Jiang, K. Zeng, Z. Zhou, P. Zhai, L. Zhang and H. Peng, Nat. Protoc., 2024, 19, 1557–1589 CrossRef CAS PubMed.
- C. P. Grey and D. S. Hall, Nat. Commun., 2020, 11, 1–4 CrossRef PubMed.
- F. Duffner, N. Kronemeyer, J. Tübke, J. Leker, M. Winter and R. Schmuch, Nat. Energy, 2021, 6, 123–134 CrossRef CAS.
- J. T. Frith, M. J. Lacey and U. Ulissi, Nat. Commun., 2023, 14, 420 CrossRef CAS PubMed.
- J. L. Guelfo, P. L. Ferguson, J. Beck, M. Chernick, A. Doria-Manzur, P. W. Faught, T. Flug, E. P. Gray, N. Jayasundara and D. R. U. Knappe, Nat. Commun., 2024, 15, 5548 CrossRef CAS PubMed.
- D. Han, C. Cui, K. Zhang, Z. Wang, J. Gao, Y. Guo, Z. Zhang, S. Wu, L. Yin, Z. Weng, F. Kang and Q. H. Yang, Nat. Sustainability, 2021, 5, 205–213 CrossRef.
- Q. Cao, Y. Gao, J. Pu, X. Zhao, Y. Wang, J. Chen and C. Guan, Nat. Commun., 2023, 14, 641 CrossRef CAS PubMed.
- R. Zhang, H. Xu, H. Dou and X. Zhang, Adv. Funct. Mater., 2024, 15, 2416497 Search PubMed.
- H. Wang, L. Zhao, H. Zhang, Y. Liu, L. Yang, F. Li, W. Liu, X. Dong, X. Li and Z. Li, Energy Environ. Sci., 2022, 15, 311–319 RSC.
- H. Xu, R. Zhang, D. Luo, J. Wang, H. Dou, X. Zhang and G. Sun, ACS Nano, 2023, 17, 25291–25300 CrossRef CAS PubMed.
- Y. Zhao, Y. Li, J. Mao, Z. Yi, N. Mubarak, Y. Zheng, J.-K. Kim and Q. Chen, J. Mater. Chem. A, 2022, 10, 14090–14097 RSC.
- C. Prehal, H. Fitzek, G. Kothleitner, V. Presser, B. Gollas, S. A. Freunberger and Q. Abbas, Nat. Commun., 2020, 11, 4838 CrossRef CAS PubMed.
- H. Wu, J. Hao, S. Zhang, Y. Jiang, Y. Zhu, J. Liu, K. Davey and S. Z. Qiao, J. Am. Chem. Soc., 2024, 146, 16601–16608 CrossRef CAS PubMed.
- J. Hao, S. Zhang, H. Wu, L. Yuan, K. Davey and S. Z. Qiao, Chem. Soc. Rev., 2024, 53, 4312–4332 RSC.
- D. Lin and Y. Li, Adv. Mater., 2022, 34, 2108856 CrossRef CAS PubMed.
- R. Zhang, H. Xu, D. Luo, J. Wang, H. Dou and X. Zhang, Energy Storage Mater., 2024, 67, 103294 CrossRef.
- H. Yu, Z. Wang, R. Zheng, L. Yan, L. Zhang and J. Shu, Angew. Chem., Int. Ed., 2023, 62, e202308397 CrossRef CAS PubMed.
- L. Zhang, M. Zhang, H. Guo, Z. Tian, L. Ge, G. He, J. Huang, J. Wang, T. Liu, I. P. Parkin and F. Lai, Adv. Sci., 2022, 9, 2105598 CrossRef CAS PubMed.
- S. J. Zhang, J. Hao, H. Li, P. F. Zhang, Z. W. Yin, Y. Y. Li, B. Zhang, Z. Lin and S. Z. Qiao, Adv. Mater., 2022, 34, 2201716 CrossRef CAS PubMed.
- J. He, H. Hong, S. Hu, X. Zhao, G. Qu, L. Zeng and H. Li, Nano Energy, 2024, 119, 109096 CrossRef CAS.
- X. Yang, H. Fan, F. Hu, S. Chen, K. Yan and L. Ma, Nano-Micro Lett., 2023, 15, 126 CrossRef CAS PubMed.
- S. Yang, X. Guo, H. Lv, C. Han, A. Chen, Z. Tang, X. Li, C. Zhi and H. Li, ACS Nano, 2022, 16, 13554–13572 CrossRef CAS PubMed.
- M. Liu, Q. Chen, X. Cao, D. Tan, J. Ma and J. Zhang, J. Am. Chem. Soc., 2022, 144, 21683–21691 CrossRef CAS PubMed.
- L. Ma, Y. Ying, S. Chen, Z. Huang, X. Li, H. Huang and C. Zhi, Angew. Chem., Int. Ed., 2021, 60, 3791–3798 CrossRef CAS PubMed.
- P. Vanýsek and L. Basáez Ramírez, J. Chil. Chem. Soc., 2008, 53, 1455–1463 Search PubMed.
- P. Navalpotro, N. Sierra, C. Trujillo, I. Montes, J. Palma and R. Marcilla, ACS Appl. Mater. Interfaces, 2018, 10, 41246–41256 CrossRef CAS PubMed.
- J. Meng, Q. Tang, L. Zhou, C. Zhao, M. Chen, Y. Shen, J. Zhou, G. Feng, Y. Shen and Y. Huang, Joule, 2020, 4, 953–966 CrossRef CAS.
- A. F. Molina Osorio, A. Gamero Quijano, P. Peljo and M. D. Scanlon, Curr. Opin. Electrochem., 2020, 21, 100–108 CrossRef CAS.
- R. M. Lynden Bell, R. Kosloff, S. Ruhman, D. Danovich and J. Vala, J. Chem. Phys., 1998, 109, 9928–9937 CrossRef CAS.
- F. S. Zhang and R. M. Lynden Bell, Eur. Phys. J. D, 2005, 34, 129–132 CrossRef CAS.
- G. Manca, A. Ienco and C. Mealli, Cryst. Growth Des., 2012, 12, 1762–1771 CrossRef CAS.
- C. J. Margulis, D. F. Coker and R. M. Lynden Bell, Chem. Phys. Lett., 2001, 341, 557–560 CrossRef CAS.
- J. P. Cerón Carrasco, D. Jacquemin, C. Laurence, A. Planchat, C. Reichardt and K. Sraïdi, J. Phys. Org. Chem., 2014, 27, 512–518 CrossRef.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- M. Telfah, A. A. Ahmad, A. M. Alsaad, Q. M. Al-Bataineh and A. Telfah, Polym. Bull., 2022, 79, 10803–10822 CrossRef CAS.
- H. Naorem and S. D. Devi, Spectrochim. Acta, Part A, 2013, 101, 67–73 CrossRef CAS PubMed.
- J. Ma, M. Liu, Y. He and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 12636–12647 CrossRef CAS PubMed.
- D. Li, T. Xia, W. Feng and L. Cheng, RSC Adv., 2021, 11, 32852–32860 RSC.
- H. Wang, X. Liu, J. Zhong, L. Du, S. Yun, X. Zhang, Y. Gao and L. Kang, Small, 2024, 20, 2306947 CrossRef CAS PubMed.
- A. C. Hayes, P. Kruus and W. A. Adams, J. Solution Chem., 1984, 13, 61–75 CrossRef CAS.
- I. Rey, J. C. Lassègues, J. Grondin and L. Servant, Electrochim. Acta, 1998, 43, 1505–1510 CrossRef CAS.
- D. Zhao, Q. Zhu, Q. Zhou, W. Zhang, Y. Yu, S. Chen and Z. Ren, Energy Environ. Mater., 2024, 7, e12522 CrossRef CAS.
- Q. Deng, F. Liu, X. Wu, C. Li, W. Zhou and B. Long, J. Energy Chem., 2024, 89, 670–678 CrossRef CAS.
- M. Prabakaran, S. Ramesh and V. Periasamy, J. Adhes. Sci. Technol., 2016, 30, 24–44 CrossRef CAS.
- Z. Hou, M. Dong, Y. Xiong, X. Zhang, H. Ao, M. Liu, Y. Zhu and Y. Qian, Small, 2020, 16, 2001228 CrossRef CAS PubMed.
- J. Zou, G. Wang, S. Lin, H. Yang, X. Ma, H. Ji, S. Zhu, Y. Lyu, C. Wang and Y. Zhou, Adv. Funct. Mater., 2024, 34, 2415607 CrossRef CAS.
- X. Hu, Z. Zhao, Y. Yang, H. Zhang, G. Lai, B. Lu, P. Zhou, L. Chen and J. Zhou, InfoMat, 2024, 6, e12620 CrossRef CAS.
- L. Zhang, H. Ding, H. Gao, J. Gong, H. Guo, S. Zhang, Y. Yu, G. He, T. Deng and I. P. Parkin, Energy Environ. Sci., 2025, 18, 2462–2473 RSC.
- K. Zhang, Y. Ge, Q. Yu, P. Zhang, Y. Feng, Z. Tie, J. Ma and Z. Jin, Energy Storage Mater., 2024, 67, 103296 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee02593a |
| This journal is © The Royal Society of Chemistry 2025 |