
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in hexaazatriphenylene-based COFs for rechargeable batteries: from structural design to electrochemical performance
Zhonghui
Sun
a,
Zhongping
Li
*bc,
Jinsong
Peng
a,
Xiaomeng
Yan
a,
Hang
Shang
a,
Yucheng
Jin
c,
Qiannan
Zhao
c,
Changqing
Li
*c,
Siliu
Lyu
c,
Chunxia
Chen
*a and
Jong-Beom
Baek
 *c
*c
aCollege of Chemistry, Chemical Engineering and Resource Utilization, and Center for Innovative Research in Synthetic Chemistry and Resource Utilization, Northeast Forestry University, Harbin 150040, P. R. China. E-mail: ccx0109@nefu.edu.cn
bKey Laboratory of Automobile Materials of MOE and School of Materials Science and Engineering, Jilin University, Changchun 130012, P. R. China. E-mail: lizhongping2025@jlu.edu.cn
cDepartment of Energy and Chemical Engineering/Center for Dimension-Controllable Organic Frameworks, Ulsan Nation Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: lizhongping2023@unist.ac.kr; changqingli@unist.ac.kr; jbbaek@unist.ac.kr
First published on 1st May 2025
Abstract
As commercial batteries reach capacity and energy density limits, especially with graphite anodes and transition metal cathodes, the need for advanced alternatives grows. Organic electrodes offer the promise of high capacity, sustainability, and tunable structures. Among them, hexaazatriphenylene (HATP)-based covalent organic frameworks (COFs) have gained considerable attention because of their distinctive characteristics. HATP-based COFs are formed with an electronegative skeleton within one-dimensional channels, and exhibit a strong affinity for metal ions (Li+, Na+, K+, Zn2+). Their distinct structure significantly enhances both ion transport and reaction kinetics. Moreover, HATP-based COFs exhibit highly ordered, permanent porosity and large surface areas, while their dense active sites and tunable conductivity facilitate rapid redox processes and enhanced capacity, leading to improved electrochemical performance. Additionally, their conjugated nature ensures robust physical and chemical stability, minimizing side reactions and maintaining structural integrity and cycling stability. As a result, HATP-based COFs are particularly well-suited for various rechargeable batteries, including lithium-ion, sodium-ion, potassium-ion, and aqueous zinc-ion batteries. This review explores the development and design principles of HATP-based COFs, analyzes their electrochemical performance and redox mechanisms, and addresses the challenges and future directions for their application in energy storage technologies.
Broader contextIn the current era of ubiquitous interconnectivity, demand for high-energy-density rechargeable batteries with superior electrochemical reliability continues to rise. Organic electrode materials have emerged as compelling alternatives to conventional inorganic counterparts, in part because they offer tunable molecular structures, environmental sustainability, and high theoretical capacity. Among them, hexaazatriphenylene-based covalent organic frameworks (HATP-based COFs) have been increasingly explored because of their reversible electrochemical redox activity, high theoretical specific capacities, and tunable electronic conductivity enabled by molecular design. Their electronegative skeletons confined within one-dimensional channels, coupled with exceptional structural stability, further enhance their potential for next-generation energy storage applications. This review provides a comprehensive analysis of HATP-based COFs as electrode materials for rechargeable batteries, including lithium-ion, sodium-ion, potassium-ion, and aqueous zinc-ion systems. It delves into their structural design principles, synthetic methodologies, and strategies for optimizing redox-active sites and morphological characteristics. The review further elucidates the underlying electrochemical mechanisms, emphasizing the correlation between molecular structure, charge transport dynamics, and ion diffusion kinetics. Additionally, it highlights recent advancements, persistent challenges, and key approaches for enhancing the stability, conductivity, and overall energy efficiency of HATP-based COFs. Finally, it presents prospective modification strategies and outlines future research directions for their development for high-performance rechargeable battery technologies. |
1. Introduction
The growing demand for efficient and sustainable energy storage solutions has accelerated the development of high-performance rechargeable secondary batteries capable of storing and releasing energy multiple times.1–3 Rechargeable batteries are increasingly utilized in applications such as electric vehicles, smart grids, and large-scale power systems.4–6 However, the conventional electrode materials used in rechargeable batteries, such as graphite for the anode and transition metal-based compounds for the cathode, are reaching their limits in terms of capacity and energy density (150–250 W h kg−1).7,8 The size and valence of the counterions need to be matched to the crystal inorganic structure due to lattice and structural stability limitations, inherently reducing the versatility as high-performance electrodes in lithium-ion batteries (LIBs) that often prove incompatible with other metal-ion systems.9,10 Additionally, the scarcity and toxicity of transition metal resources not only constrain the mass production to meet rapidly growing demands, but also complicate recycling processes and exacerbate environmental pollution.11,12 These factors have driven ongoing efforts to develop alternative rechargeable battery technologies with improved cost-effectiveness, safety, and sustainability, especially for large-scale applications.13Organic materials capable of reversible electrochemical redox reactions offer several advantages over traditional inorganic materials. They include higher theoretical specific capacities, adjustable voltage platforms through molecular design, and environmental benefits because of the absence of heavy metals.14–16 Organic materials composed of light elements such as like carbon (C), hydrogen (H), oxygen (O), nitrogen (N), and sulfur (S), and especially those containing electronegative elements, can effectively interact with a wide range of metal-ions (e.g., Li+, Na+, K+, Mg2+, Al3+, Zn2+), making them suitable for various secondary batteries.17–19 Additionally, organic materials are cost-effective, readily available, and possess soft mechanical properties, making them ideal for flexible batteries.20,21 However, traditional organic electrode materials have challenges, such as the dissolution of active materials during cycling, low conductivity, and the limited accessibility of redox-active sites.22 To overcome these limitations, a deeper understanding of fundamental properties, mechanisms, and structure–performance relationships is urgent and essential.
Covalent organic frameworks (COFs) have emerged as a promising class of crystalline materials linked by robust covalent bonds, resulting in highly stable conjugated π-skeletons.23–35 COFs can be synthesized with diverse topologies, skeletons, and pore structures due to a diverse range of topological design diagrams, the availability of building units, and the accessibility of linkages.36–40 One of the most significant features of COFs is the ability to pre-design their functional structures via topological diagrams, which enables materials to be created with well-defined wall interfaces.41–45 The unique properties of COFs—including permanent and adjustable porosity, large surface area, excellent thermal and chemical stability, and tailored structural designability—make them highly attractive for a wide range of applications, including gas adsorption/separation, photo/electro-catalysis, chemical detection, and energy storage.46–58
Redox-active anchored COFs were first employed in 2015 as electrode materials in lithium-ion batteries (LIBs), demonstrating their ability to undergo reversible redox reactions.59 Since then, COFs with various structural motifs including benzoquinone, imide, azo, pyrazine, imidazole, phenazine, anthraquinone, and so on, have been explored for use in LIBs, sodium-ion batteries (SIBs),60,61 potassium-ion batteries (KIBs),62,63 and aqueous zinc-ion batteries (AZIBs),64–66 these COFs offer highly ordered skeletons, large surface area, adjustable conductivity, and excellent solvent resistance.
Among them, hexaazatriphenylene (HATP)-based COFs have attracted significant attention due to their unique structural and functional properties (Fig. 1(a)). These COFs feature an electronegative skeleton within well-defined one-dimensional (1D) channels, and exhibit strong metal cation affinity (Li+, Na+, K+, and Zn2+), which enhances both ion transport and transport kinetics.67,68 Moreover, the exceptionally designable structure permits HATP-based COFs with precisely ordered permanent porosity, large surface areas, and adjustable conductivity, synergistically enabling accelerated redox reactions. Their highly π-conjugated structure not only ensures excellent physical and chemical stability but also facilitates the electron transfer processes, contributing to the longevity and fast charge efficiency of electrodes.69 Furthermore, the planar structure enables efficient π–π stacking interactions, further strengthening both solvent resistance and chemical degradation tolerance, even under prolonged cycling.70 The dense C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds serve as redox-active sites capable of undergoing a reversible 6-electron transfer process, giving the COFs an impressive theoretical capacity.71 The combination of these characteristics—excellent stability, tunable conductivity, high capacity, and fast cation transfer—make the HATP-based COFs highly promising as materials for next-generation electrode applications.
N bonds serve as redox-active sites capable of undergoing a reversible 6-electron transfer process, giving the COFs an impressive theoretical capacity.71 The combination of these characteristics—excellent stability, tunable conductivity, high capacity, and fast cation transfer—make the HATP-based COFs highly promising as materials for next-generation electrode applications.
 | ||
| Fig. 1 (a) Schematic representation of the key features of HATP-based COFs in rechargeable batteries. (b) Timeline of key developments in HATP-based COF electrodes for rechargeable batteries. | ||
Not surprisingly, increasing demand for sustainable, high-performance electrode materials has sparked significant interest in HATP-based COFs for rechargeable batteries over the past decade (Fig. 1). This review provides a comprehensive overview of their key research development, identify ongoing challenges, and explores innovative strategies for structural optimization and performance enhancement. It delves into various synthesis strategies and highlights how structural and functional modifications influence their electrochemical properties. Special attention has been given to the unique advantages and engineering of the redox mechanisms of HATP-based COF electrodes in LIBs, specifically addressing challenges such as insufficient and buried redox-active sites and limited electron transport, while also exploring potential solutions to overcome these limitations. Additionally, the review covers the applications of HATP-based COFs in SIBs, PIBs, and AZIBs, demonstrating the versatility of these materials across different types of energy storage systems. Finally, future research directions are outlined, focusing on the structural diversification of COFs, enhancement of their conductivity, regulation of morphology, development of scalable production methods, and optimization of advanced characterization methods. These insights are intended to guide the continued progress of HATP-based COFs, contributing to next-generation, sustainable energy storage technologies.
2. Synthesis strategy of HATP-based COFs
HATP-based COFs are synthesized through two primary strategies: direct one-step synthesis and indirect multi-step approaches (Fig. 2). The direct one-step synthesis involves the straightforward polymerization of cyclohexanehexone octahydrate (CHHO) with polyamine compounds,72–74 which is highly efficient. However, this method has difficultly precisely controlling connectivity and incorporating functional groups. In contrast, the indirect synthesis approach begins with the preparation of small molecules with functional groups capable of forming various dynamic covalent linkages, such as imine,75,76 imide,77 phenylimino,78 and triazine.79 This strategy enables superior control over the molecular structure, stability, and functionality of the COFs, offering greater flexibility and versatility when designing COFs with tailored properties for specific applications.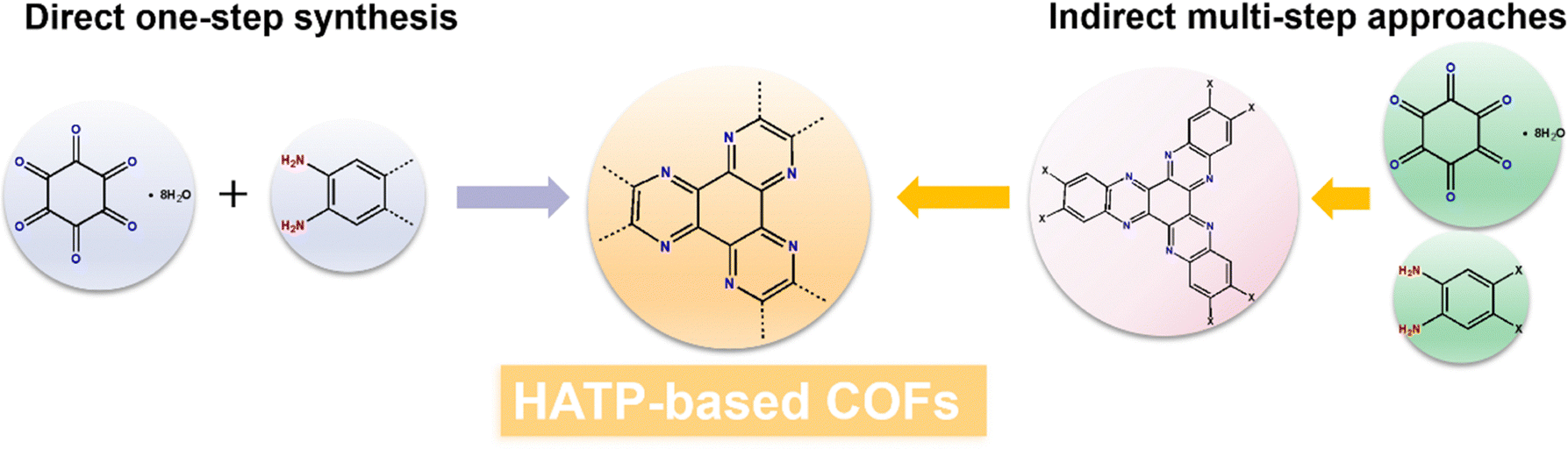 | ||
| Fig. 2 Schematic diagram of the synthesis method for HATP-based COFs. | ||
2.1 The direct one-step synthesis method
The direct one-step solvothermal synthesis of HATP-based COFs relies on the condensation reaction between CHHO and organic amines (tetramines, hexamines, and octamines), an efficient and straightforward approach for constructing highly symmetrical and rigid COF structures. This method is advantageous due to its simplicity, requiring only appropriate solvent selection and temperature control to promote imine bond formation without the need for expensive catalysts. The reaction mixture underwent flash frozen in a liquid nitrogen bath and evacuated under vacuum for three pump–thaw cycles, ensuring complete degassing and oxygen removal prior to hydrothermal reaction. Furthermore, the crystallinity and electrochemical performance of HATP-based COFs could be significantly enhanced through optimizing the catalyst systems (potassium hydroxide (KOH) or various acid systems) and reaction conditions. These optimizations achieved exceptional crystallinity, structural regularity, high surface areas and tunable conductivity, thereby providing efficient ion transport and electron transfer for high-performance electrodes.The selection of amines precursors also plays a pivotal role to determine the electrochemical performance of HATP-based COFs (Fig. 3). Redox-active amines, including 2,3,5,6-tetraminobenzoquinone (TABQ) and 2,3,7,8-tetraaminophenazine-1,4,6,9-tetraone (TAPT), incorporate additional redox-active CN and CO groups to HATP-based COFs that enable multi-electron redox reactions, elevating the theoretical capacities. Three-dimensional (3D) amines endowed HATP-based COFs with hierarchical porosity and enhanced surface areas, which synergistically optimized ion diffusion kinetics while ensuring accessibility of redox-active sites, collectively enabling high reversible capacity. Alternatively, non-redox-active two-dimensional (2D) amines promoted the construction of extended π-conjugation skeletons that exhibited enhanced charge transport properties and cycling stability, attributing to their improved electronic delocalization and dissolution resistance to electrolyte dissolution.
 | ||
| Fig. 3 (a) Schematic illustration of the direct one-step method. (b) Various reported amines precursors utilized in the synthesis of HATP-based COFs. | ||
In 2015, Baek's group pioneered this approach by synthesizing the first HATP-based COF, C2N-h2D crystal, through the condensation of hexaaminobenzene trihydrochloride and CHHO in N-methyl-2-pyrrolidone (NMP) with a few drops of sulphuric acid (H2SO4) or trifluoromethanesulphonic acid at 175 °C for 8 h (Fig. 4(a)).80 The scanning tunnelling microscopy (STM) results confirmed formation of a well-defined layered network featuring uniformly distributed nitrogen atoms within its porous structure (Fig. 4(b)). The measured inter-hole distance was 8.24 ± 0.96 Å obtained from the height profiles, agrees well with the theoretical value (Fig. 4(c)). Additionally, to fabricate large-area films, the as-synthesized C2N-h2D crystals were dispersed in trifluoromethanesulfonic acid and drop-cast onto the preheated (140 °C) SiO2/Si substrate, followed by high-temperature annealing at 700 °C under argon for 2 h. The solution-casted films were then transferred onto flexible polyethylene terephthalate substrates via poly(methylmethaacrylate)-mediated processes. Atomic force microscopy (AFM) characterization of the C2N-h2D crystal flakes demonstrated an average thickness of 8.0 ± 3.5 nm, confirming the formation of multilayer stacks (Fig. 4(d)). Furthermore, the shiny metallic reflection observed in the C2N-h2D film indicated a high degree of crystallinity (Fig. 4(e) and (f)). Compared to graphite, C2N-h2D exhibited a reduced interlayer distance and stronger interlayer interactions, leading to improved semiconducting behavior and excellent electrical properties.
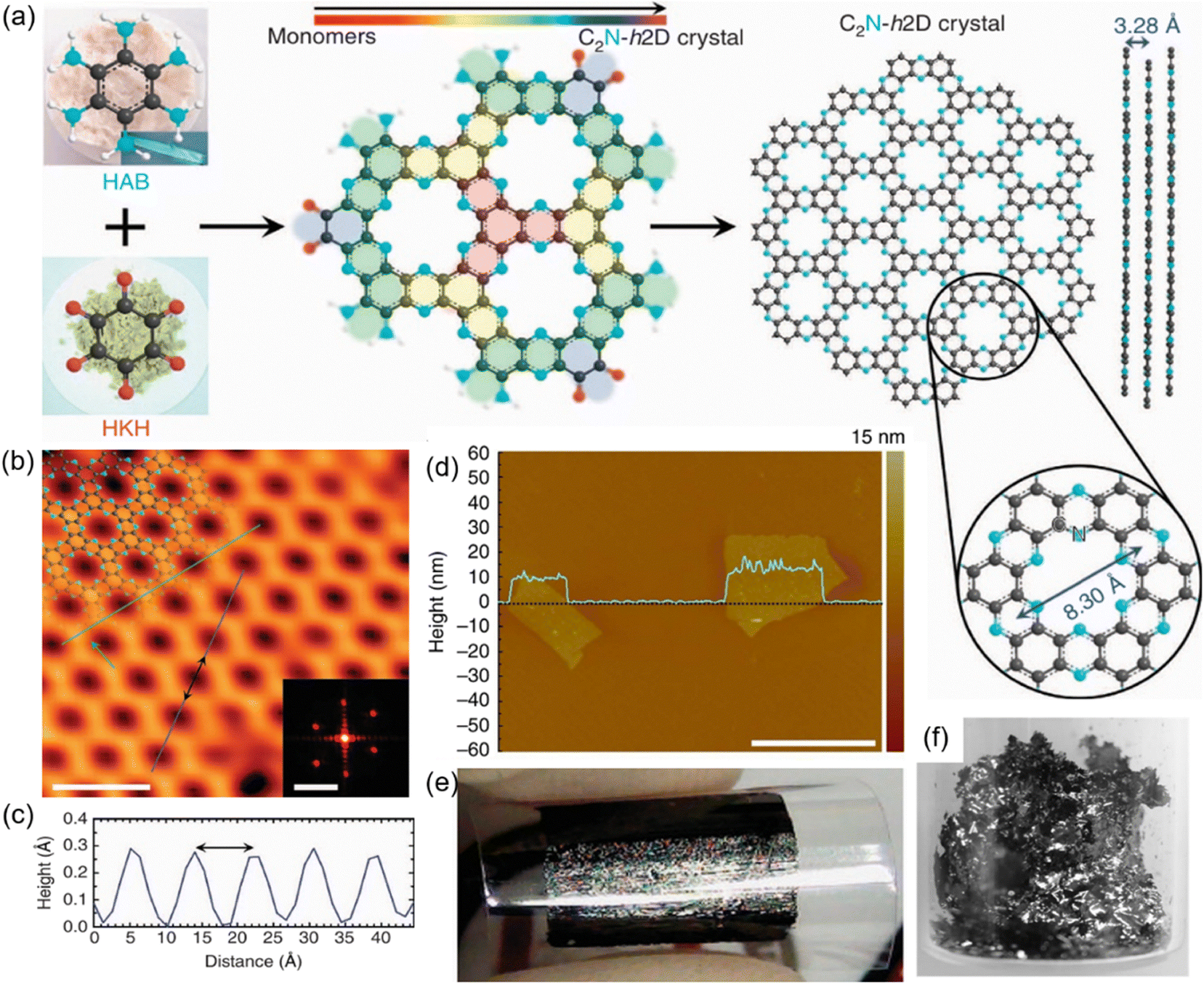 | ||
| Fig. 4 (a) Synthetic route for the preparation of C2N-h2D. (b) Atomic-resolution STM topography image of C2N-h2D. (c) The topographic height profile along deep-blue line. (d) AFM image of the C2N-h2D crystal flakes. (e) Crystal film of C2N-h2D transferred onto a PET substrate. (f) Digital photograph of solution-cast C2N-h2D on a SiO2 surface after heat-treatment at 700 °C. Reproduced with permission.80 Copyright 2015, Springer Nature. | ||
The designed amine precursors played a crucial role in tailoring pore structure properties, facilitating the creation of COFs for diverse applications. Inspired by this breakthrough, a uniformly microporous, robust 3D cage-like organic network (CON) structure was synthesized by condensing triptycene-based hexamine with CHHO in an ethylene glycol/acetic acid mixture at 130 °C for 80 h.74 The resulting 3D-CON exhibited a remarkably high Brunauer–Emmett–Teller (BET) surface area of 2247 m2 g−1 and a uniform pore size distribution of 0.55 nm, attributed to its periodic microporous structure and multi-dimensional connectivity. This work confirmed that HATP-based 3D network structures constructed with steric hexamines and octaamines were superior to traditional 2D frameworks in terms of surface area, porosity, and functional versatility. The combination of high surface area and rigid skeleton enabled fast and stable electrolyte infiltration and cation transport, making this material a promising candidate for high-performance energy storage applications.
Since these pioneering developments, HATP-based COFs have been continuously improved, with ongoing research focused on fine-tuning the reaction conditions to enhance crystallinity and functional properties. Mirica et al. synthesized fused π-conjugated aza-COF-1 and aza-COF-2 by condensing CHHO and 2,3,6,7,10,11-hexaaminotriphenylene hexahydrochloride and 1,2,4,5-benzenetetramine tetrahydrochloride (BTA·4HCl), respectively.81 After 5 days of reaction at 185 °C, well-ordered crystalline frameworks were achieved in the presence of concentrated H2SO4 as a catalyst. Powder X-ray diffraction (PXRD) patterns revealed distinct crystalline features: aza-COF-1 exhibited sharp peaks at 7.78, 13.44, and 26.67° corresponding to the (100), (110), and (001) planes of a layered structure, whereas aza-COF-2 showed broader peaks at 6.51 and 26.51° assigned to the (100) and (001) planes, respectively. Both COFs exhibited superior proton conductivity values at 10−3 S m−1, leading to the development of stable and high proton-conductive materials. Chou et al. synthesized CPT by the condensation reaction of CHHO with redox-active 2,3,7,8-phenazine-tetramine (PT) under an acetic acid-catalyzed condensation reaction at 150 °C for 4 days, which exhibited rich electron delocalization, needle-like morphology and thinner sheet thickness.82 The high intrinsic conductivity (1.58 × 10−1 S m−1) coupled with abundant redox-active sites enabled it a proper electrode material for high-performance energy storage systems. The optimized reaction conditions led to better crystalline COFs, which was important, to ensure uniform pore size distribution and improve the electrochemical performance of the electrode materials and supercapacitors.
The choice of catalyst has a crucial effect on the crystallinity and structural regularity of HATP-based COFs, which in turn significantly affects their electrochemical performance. Liu et al. focused on the condensation between BTA·4HCl and CHHO to synthesize highly crystalline PGF-1, which was obtained in 4 M aqueous KOH solution at 120 °C in a sealed vessel after 3 days (Fig. 5(a)).83 The PXRD pattern of the PGF-1 revealed two prominent peaks at 6.1° and 26.4°, which corresponded to the (100) and (001) planes, respectively (Fig. 5(b)). These PXRD features were matched well with the simulated AA eclipse stacking structure and the corresponding predicted pore size was 1.2 nm, consistent with the experimental values derived from the adsorption isotherm (Fig. 5(c)). The high-resolution transmission electron microscopy (HR-TEM) of PGF-1 further strongly supported its high crystallinity and ordered hexagonal micropores (Fig. 5(d) and (e)). The nitrogen-rich and fully fused π-conjugated aromatic framework of PGF-1 demonstrated remarkable electrical conductivity of 3 × 10−3 S m−1. In contrast, the amorphous polymer AP-1, synthesized utilizing the acid catalyst, only exhibited a broad and weak peak within the (100) region in its PXRD pattern. PGF-1, with an improved structural regularity and electronic conductivity, enabled more accessible redox-active sites and faster charge transport, resulting in higher capacity and excellent rate performance when used as the electrode material.
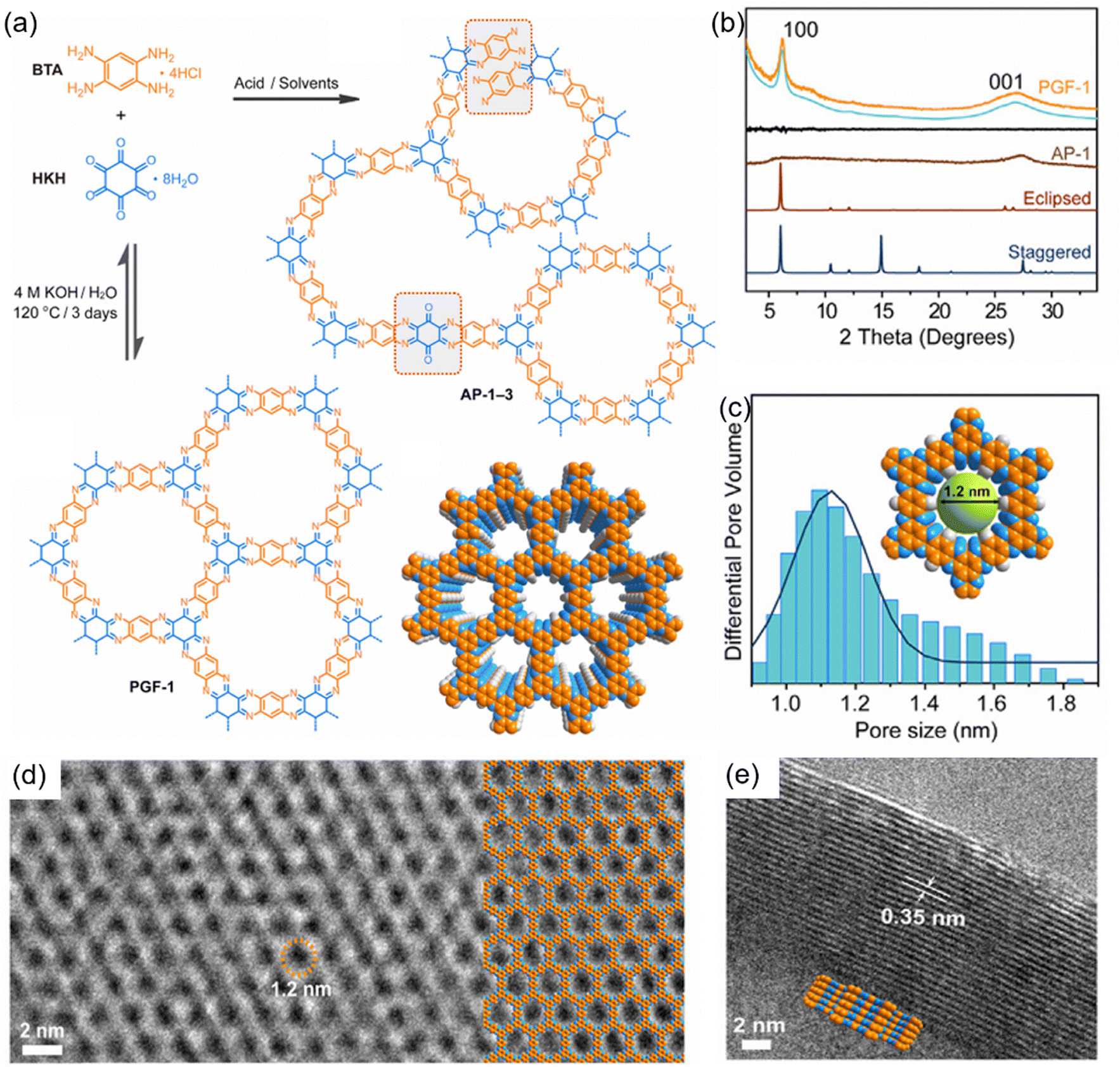 | ||
| Fig. 5 (a) Synthetic route for the preparation of PGF-1 and AP-1. (b) PXRD patterns of PGF-1 and AP-1. (c) Pore size distribution profile of PGF-1. (d) and (e) HR-TEM images of PGF-1. Reproduced with permission.83 Copyright 2020, Elsevier. | ||
2.2 The indirect synthesis methods
The indirect synthesis of HATP-based COFs typically involves a more intricate multi-step approach, which also provides enhanced control over the molecular architecture and functionality of the final COFs. Unlike the one-step direct synthesis, which generally relies on condensation reactions between CHHO and di-amino building blocks, the indirect synthesis methods involve additional steps to prepare the precursor materials before polymerization. This method typically begins with the functionalization of HATP and hexaazatrinaphthalene (HATN)-based building blocks, which can be modified by introducing different functional groups such as amino groups (–NH2),84 cyano groups (–CN),85,86 anhydride.87 After functionalizing the HATP derivatives, covalent bonds are used to assemble them into the designable COF structures, utilizing different reaction types based on the intended topology, functionality, and properties of the final COFs. Prevalent polymerization strategies employed in the construction of HATP-based COFs encompass Schiff base reactions, Knoevenagel reactions, and polyimide formation. These approaches enable the synthesis of the corresponding dynamic covalent structures (Fig. 6).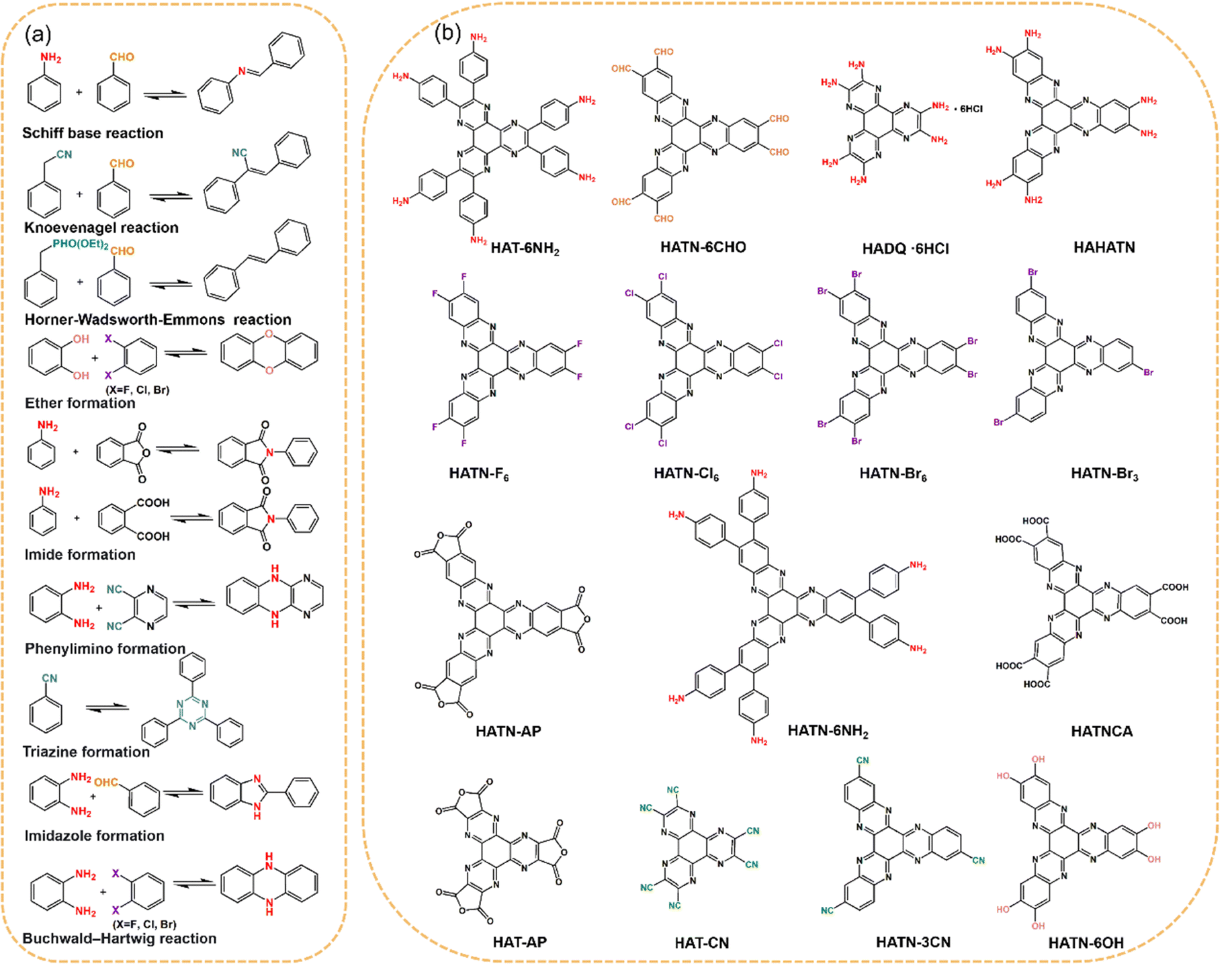 | ||
| Fig. 6 (a) Representative reactions involved in the indirect synthesis methods. (b) Structure of the precursor monomer for HATP-based COFs. | ||
N) bonds. This reaction not only underpins the creation of these materials but also lays the foundation for a wide array of structural and functional variations, making imine-linked HATP-based COFs an indispensable platform for the design of advanced electrode materials in next-generation rechargeable batteries. A significant contribution to the development of imine-linked HATP-based COFs was made by Zhao et al. through their indirect synthesis approach.75 By condensing HAT-6NH2 with terephthalaldehyde (TA) in a mixture of N,N-dimethylacetamide (DMAc), 1,3,5-trimethylbenzene and acetic acid at 120 °C for 3 days, HAT-COF was constructed with an eclipsed packing model with two triangular micropores of distinct sizes (11.3 and 15.2 Å). This unique topology provides the COF pores with distinct chemical environments: the larger pores contain aromatic CN groups from the HATP cores, while the smaller centers share only peripheral CN groups.
However, the BET surface area of HAT-COF was relatively low (486.15 m2 g−1) due to the twisted conformation of the phenyl units in HAT-6NH2, which distorts the framework and disrupts the ideal stacking of the COF layers. Building on this pioneering work, Zhao's group further extended their research by polymerizing HAT-6NH2 with two other aldehyde compounds, 4,4′,4′′-nitrilotribenzaldehyde (NTBA) and 4′,4′′′,4′′′′′-nitrilotris(([1,1′-biphenyl]-4-carbaldehyde)) (NTBCA), to synthesize two new HATP-based COFs, HAT-NTBA-COF and HAT-NTBCA-COF, which both exhibited an AA stacking model (Fig. 7).88 A distinctive feature of these COFs is the heterodromous orientation of the imine bonds, which alternate between clockwise and counterclockwise, and promote consistent bond formation between the building blocks. The specific surface areas of HAT-NTBA-COF and HAT-NTBCA-COF were 628.0 m2 g−1 and 439.9 m2 g−1, respectively. The pore sizes of HAT-NTBA-COF and HAT-NTBCA-COF were primarily distributed around 9.5 Å and 12.7 Å, respectively, which were consistent with the theoretical simulations (Fig. 7(c)–(g)). This variable orientation was crucial to achieving high crystallinity, and laid the ground work for the future development of multifunctional COF materials.
 | ||
| Fig. 7 (a) Construction of HATP-NTBA-COF using the variable orientation of imine linkages. (b) PXRD patterns of HATP-NTBA-COF. (c) N2 adsorption–desorption isotherms of HATP-NTBA-COF. (d) Pore size distribution profiles of HATP-NTBA-COF. (e) PXRD patterns of HATP-NTBCA-COF. (f) N2 adsorption–desorption isotherms of HATP-NTBCA-COF. (g) Pore size distribution profiles of HATP-NTBCA-COF. Reproduced with permission.88 Copyright 2017, Royal Society of Chemistry. | ||
In addition to constructing imine-linked COFs using HATP-based amino compounds, Feng et al. explored a different strategy for building 2D dual-porous COFs utilizing 2,3,8,9,14,15-hexa(4-formylphenyl)diquinoxalino[2,3-a:2′,3′-c]phenazine (HATN-6CHO) and 1,4-diaminobenzene (PDA)89 and benzidine (BZD),90 to form 2D imine-based HATN and HATN-BZD, respectively. The reactions were carried out in a mixture of DMAc, mesitylene and acetic acid (aq. 6 M) at 120 °C for 3 days, which contributed to the high crystallinity. The larger BZD molecule, in contrast to PDA, led to larger pore size distributions of 1.0–1.3 nm and 1.8–2.0 nm. Jiang et al. synthesized crystalline USTB-6 by the condensation reaction of HATN-6CHO with redox-active carbonyl-rich 2,7-diaminopyrene-4,5,9,10-tetraone (PTO-NH2), utilizing a mixed system containing DMAc, mesitylene, acetic acid (aq. 6 M) and p-toluidine.76 USTB-6 exhibited a specific surface area of 328 m2 g−1 with an AA stacking mode and a dual-pore structure comprising 0.7 and 1.3 nm pores. The well-defined porous structures and a high theoretical capacity of 272 mA h g−1 made it a suitable material for energy storage electrodes.
The above content describes the polymerization of monoamine or monoaldehyde groups with a HATP center; additionally, imine bonds and pyrazine rings can be generated between ortho-diamine and ortho-dicarbonyl groups. Specifically, Saleem et al. synthesized HADQ-COF by reacting 2,3,6,7,10,11-hexaamine dipyrazino quinoxaline hexahydrochloride (HADQ·6HCl) hexahydrochloride with CHHO in the mixture of NMP and H2SO4 at 150 °C for 7 days, followed by a heat treatment process at 250 °C to further enhance the material's properties.91 The thermal treatment removed edge groups and facilitated the formation of larger crystalline segments, in as confirmed by PXRD and HR-TEM observations. The enhanced crystallinity, surface area, and stability of 250-HADQ-COF made it a promising candidate for double-layer supercapacitors. Similarly, Zhu et al. utilized polymerization between hexaiminohexaazatrinaphthalene (HAHATN) and pyrene-4,5,9,10-tetrone (PTO) to create a 2D porous honeycomb-shaped material, which effectively improved utilization of the redox-active sites and facilitated ion transport.69 The extended π-electron delocalization within the framework reduced both the lowest unoccupied molecular orbital (LUMO) and bandgap, demonstrating enhanced redox potential and superior electronic conductivity. The structural diversity of imine-linked HATP-based COFs offers extensive potential for the creation of highly efficient, tailored electrode materials with enhanced electrochemical performance, which are poised to drive advances in energy storage technologies.
Based on these distinct features, Feng et al. initially investigated olefin-linked conjugated polymers to enhance electrochemical performance while constructing electron donor–acceptor structures (Fig. 8(a)). By condensing the precursor HATN-6CHO with 1,4-phenylenediacetonitrile, 2,2′-(biphenyl-4,4′-diyl)diacetonitrile (BDAN), and 2,2′-([2,2′-bithiophene]-5,5′-diyl)diacetonitrile (ThDAN) via Knoevenagel condensation in a mixture of DMAc, ortho-dichlorobenzene (o-DCB) and NEt4OH at 150 °C for 3 days, a series of olefin-linked HATP-based COFs—2D CCP-HATN, 2D CCP-BD, and 2D CCP-Th—were synthesized.89,90 All of these frameworks exhibited an AA stacking model, with layer stabilization energies of −97, −113.1, and −117.4 kcal mol−1 for 2D CCP-HATN, 2D CCP-BD, and 2D CCP-Th, respectively (Fig. 8(b)–(d)). In addition, the larger BDAN and ThDAN molecules resulted in larger pore size distributions, specifically ranging from 1.0–1.3 nm and 1.8–2.0 nm (Fig. 8(e)–(g)). Comparative studies between imine-based HATN and olefin-based CCP-HATN revealed that the latter exhibited sharper and more well-defined redox bands, underscoring its superior redox activity as a cathode material in LIBs. Further investigation into its electronic and structural properties highlighted the role of thiophene incorporation in tuning charge transport characteristics. The bithiophene-based donor–acceptor framework (2D CCP-Th) possessed an extended conjugated skeleton. Ultraviolet-visible analysis indicated a reduced energy bandgap attributed to the efficient intramolecular charge transfer (ICT) between the electron-donating bithiophene unit and the electron-accepting HATN core. This charge delocalization substantially enhanced the electronic conductivity, which was crucial to achieve excellent rate performance as an electrode material.
 | ||
| Fig. 8 (a) Synthetic route for the preparation of olefin-linked HATP-based COFs. (b) XRD patterns of 2D CCP-HATN. (c) XRD patterns of 2D CCP-Th. (d) XRD patterns of 2D CCP-BD. (e) N2 adsorption–desorption isotherms and pore size distributions of 2D CCP-HATN. (f) N2 adsorption–desorption isotherms and pore size distributions of 2D CCP-Th. (g) N2 adsorption–desorption isotherms and pore size distributions of 2D CCP-BD. Reproduced with permission.89 Copyright 2019, Wiley-VCH GmbH. Reproduced with permission.90 Copyright 2021, Wiley-VCH GmbH. | ||
The presence of additional cyano groups in olefin-linked HATP-based COFs, constructed via Knoevenagel polycondensation, induced significant structural twisting due to the steric hindrance effects. Therefore, the same group pioneered the first synthesis of unsubstituted olefin-linked HATP-based COFs—2D-PPQV1 and 2D-PPQV2—via the Horner–Wadsworth–Emmons reaction, utilizing Cs2CO3-mediated condensation of 1,4-bis(diethylphosphonomethyl)benzene and 4,4′-bis(diethylphosphonomethyl)biphenyl with HATN-6CHO in DMAc and mesitylene at 120 °C for 3 days, respectively.94 PXRD patterns confirmed the formation of highly crystalline and dual-pore frameworks. These COFs exhibited exceptional thermal and chemical stability, combined with enhanced π-conjugation and a narrow bandgap, rendering them outstanding candidates for electrode materials with superior long-term cycling stability.
Building on this, Xu et al. developed a high-yield, metal-free method for fabricating HOTT–HATN COF by combining 2,3,6,7,10,11-hexahydroxytriphenylene (HOTT) and 2,3,8,9,14,15-hexachloro-5,6,11,12,17,18-hexaaza-trinaphthylene (HATN-Cl6) as building blocks. The reaction was carried out in DMAc, with K2CO3 serving as the base catalyst at 170 °C for 5 days.98 Additionally, the incorporation of both oxygen (O) and nitrogen (N) heteroatoms in the HOTT–HATN COF played a crucial role in establishing strong electron donor–acceptor (D–A) interactions, which significantly enhanced its redox properties.
Baek et al. made significant progress in constructing a highly crystalline HD-COF by refining the synthetic approach. They replaced HATN-Cl6 with 2,3,8,9,14,15-hexafluoro-5,6,11,12,17,18-hexaazatrinaphthylene and optimized the reaction conditions using a K2CO3-mediated system in a mixed solvent of NMP and o-dichlorobenzene (o-DCB) at 180 °C for 3 days (Fig. 9(a)).99 This adjustment led to enhanced crystallinity and improved charge transport properties. The PXRD analysis of HD-COF revealed well-defined diffraction peaks at 4.9, 8.6, 10.1, and 13.1°, corresponding to the (100), (110), (200), and (210) facets of the eclipsed 2D trigonal unit cell, respectively (Fig. 9(b)). Notably, the interlayer spacing (∼3.3 Å), attributed to π–π stacking interactions, facilitated efficient out-of-plane charge transport, a key factor for high-performance electrode materials. Further structural characterization through HR-TEM confirmed the presence of well-defined 1.7 nm micropore channels, consistent with fast Fourier transform (FFT) analysis results (Fig. 9(c) and (d)). Capitalizing on the superior structural properties of the bulk HD-COF material, they extended investigation to thin-film fabrication. A SiO2 substrate was secured in a home-made Π-shaped polytetrafluoroethylene holder, which was placed in a Teflon-lined autoclave for solvothermal synthesis under standard conditions (Fig. 9(e)). Optical microscopy revealed uniform coloration and defect-free morphology at the macroscopic scale (Fig. 9(f)). AFM indicated HD-COF film thicknesses of 90–100 nm with a surface roughness below 10 nm, confirming smooth surface morphology (Fig. 9(g)). Additionally, grazing-incidence wide-angle X-ray scattering (GIWAXS) patterns (Fig. 9(h)) and z-axis line-cut profiles (Fig. 9(i)) validated the highly crystalline of HD-COF film. Combined its high electrical conductivity (1.58 × 10−1 S m−1) and superior structural properties, HD-COF film showed promising potential for application as a binder-free electrode material in rechargeable batteries.
 | ||
| Fig. 9 (a) Synthetic route for the preparation of HD-COF. (b) PXRD patterns of HD-COF. (c) HR-TEM image of HD-COF. Inset: FFT of the image in the cyan square. (d) Line intensity profile of the lattice planes from the yellow line in (c), corresponding to the (010) plane of HD-COF. (e) Schematic illustration of the in situ fabrication of the HD-COF film. (f) Optical microscope image of the HD-COF film. Scale bar: 5 μm. (g) AFM image of HD-COF film. (h) GIWAXS of HD-COF film. (i) z-Axis linecut plot of HD-COF film.99 Copyright 2023, Wiley-VCH GmbH. | ||
In a related development, Li et al. synthesized HATN-HHTP COF via an anhydrous Cs2CO3-catalyzed reaction in an NMP/mesitylene solvent system.100 This approach capitalized on the structural benefits of its highly ordered porosity and extended conjugation, both critical for optimizing electronic conductivity and redox activity. The strategic integration of robust ether linkages, high conductivity, and tunable porosity in these COFs established their significant potential for high-performance energy storage applications.
A recent study by Huang et al. introduced a novel COF, HAHATN-PMDA-COF, synthesized through the condensation reaction between hexa(p-anilinyl) hexaazatrinaphthalene and pyromellitic dianhydride (PMDA) in a mixture of NMP, mesitylene and water with isoquinoline as a catalyst at 180 °C after 5 days (Fig. 10(a)).103 The strong PXRD peaks observed at 3.52°, 7.03°, 9.31°, 10.56°, and 28.14° corresponded to (100), (200), (210), (300), and (001) facets, confirming the AA stacking mode (Fig. 10(b)). Additionally, HAHATN-PMDA-COF featured a 2D trigonal structure with dual well-defined pores of 1.58 and 0.95 nm, which were the smallest pore sizes among the reported polyimide COFs (Fig. 10(c)–(e)). The high density lithophilic quinoxaline and phthalimide units within the uniform 1D channel of the COF make it a promising candidate for solid electrolyte interfacial in lithium metal anodes, facilitating the homogeneous deposition of Li+ ions and suppressing the growth of lithium dendrites.
 | ||
| Fig. 10 (a) Synthetic route for the preparation of HAHATN-PMDA-COF. (b) PXRD patterns of HAHATN-PMDA-COF. (c) and (d) TEM images of HAHATN-PMDA-COF. (e) N2 sorption isotherm profile (inset: pore size distribution) of HAHATN-PMDA-COF. Reproduced with permission.103 Copyright 2024, Wiley-VCH GmbH. | ||
Jiang's group developed 2D HATN-AQ-COF by condensing 2,3,8,9,14,15-hexacarboxyl hexaazatrinaphthalene trianhydride (HATN-AP) with a redox-active linker, 2,6-diaminoanthraquinone (DAAQ), demonstrating a high theoretical capacity of 406 mA h g−1.104 The reaction was performed in the mixture of NMP, mesitylene and isoquinoline at 180 °C for 5 days. The PXRD patterns exhibited a sequence of diffraction peaks at 2.22°, 3.86°, 5.94° and 26.1°corresponding to (100), (101), (200), and (001) facets, respectively, which indicated a highly ordered and crystalline framework. The observed AA stacking mode was consistent with the pore size distribution of 3.8 nm calculated using density functional theory. HATN-AQ-COF exhibited an abundant pore structure and high specific surface area of up to 725 m2 g−1, facilitating rapid ion transport (10−13 to 10−8 cm2 s−1). These characteristics rendered it highly advantageous for use as a fast-charging electrode material.
The same group also developed redox-active 3D HATP-based COFs with cross-linked channels.105 They synthesized two COFs, 3D-TP-HATN-COF and 3D-TAM-HATN-COF, which were synthesized from triphenylene-2,3,6,7,10,11-hexacarboxylic acid (HATNCA) and N,N,N′,N′-tetraphenyl-1,4-phenylenediamine and tetrakis(4-aminophenyl)methane, respectively (Fig. 11(a)). The diffraction peaks in PXRD patterns observed at 3.78, 4.89, 11.35, and 27.17° for 3D-TP-HATN-COF corresponded to the (211), (310), (716), and (12,9,1) facets, respectively (Fig. 11(b)). Similarly, 3D-TAM-HATN-COF exhibits distinct diffraction peaks at 4.24, 12.25, and 26.07°, which were indexed to the (211), (541), and (12,9,1) facets (Fig. 11(c)). Both materials showed high porosity with uniform pore sizes of 1.51 nm for 3D-TP-HATN-COF and 1.26 nm for 3D-TAM-HATN-COF, as well as interconnected 3D nanochannel networks (Fig. 11(d) and (e)). These structural features enable exceptional ion diffusion performance, achieving ion diffusion coefficients of 7.4 × 10−1 and 4.5 × 10−1 cm2 s−1 for 3D-TP-HATN-COF and 3D-TAM HATN-COF, respectively. The strategy highlighted the critical role of ordered nanopores and tunable channel dimensions in optimizing ion transport kinetics, providing a robust platform for advanced high-energy density batteries.
 | ||
| Fig. 11 (a) Synthetic route for the preparation of 3D-TP-HATN-COF and 3D-TAM-HATN-COF. (b) PXRD patterns of 3D-TP-HATN-COF. (c) PXRD patterns of 3D-TAM-HATN-COF. (d) N2 adsorption–desorption isotherms of 3D-TP-HATN-COF. (e) N2 adsorption–desorption isotherms of 3D-TAM-HATN-COF. Reproduced with permission.105 Copyright 2025, Chinese Chemical Society. | ||
For instance, the introduction of phenylimino into HATP-based COFs introduces intriguing redox-active properties. In a recent study by Lv et al., a novel HPP-COF was synthesized in N,N-dimethylformamide by condensing BTA·4HCl and 1,4,5,8,9,11-hexaazatriphenylenehexacarbonitrile (HAT-CN) with Na2CO3 as a deacid reagent at 130 °C for 6 hours (Fig. 12(a)).107 HPP-COF featured both pyrazine and phenylimino functional groups. Structural characterization by PXRD analyses revealed two broad peaks at 7.0° and 26.6°, corresponding to the (100) and (001) facets, respectively (Fig. 12(b)). The broad peak at 26.6° was attributed to the π–π stacking interactions between aromatic units in the framework, confirming a multi-layered structure of HPP-COF. HPP-COF retained its crystallinity even after 7 days of treatment with 6 M H2SO4 and 6 M NaOH as verified by the characteristic peaks in the PXRD patterns (Fig. 12(c)), which was advantageous as an electrode material in aqueous acidic and alkaline batteries.
 | ||
| Fig. 12 (a) Synthetic route for the preparation of HPP-COF. (b) PXRD pattern of HPP-COF. (c) PXRD patterns of HPP-COF after treatments under 6 M NaOH and H2SO4 for 7 days. Reproduced with permission.107 Copyright 2023, Wiley-VCH GmbH. | ||
Building on their previous work with HPP-COF, Lv et al. also synthesized a novel COF, PTHATP-COF, by replacing BTA·4HCl with 2,3,5,6-tetraamine trihydrochloride in the design of PTHATP-COF, following the same approach.108 PTHATP-COF exhibited excellent crystallinity, further demonstrating that the regulation of amino building blocks could lead to high crystallinity while allowing for the introduction of functionalized groups. Featuring pyrazine and phenylimino units, PTHATP-COF showed remarkable performance as an electrode material in calcium-ion batteries, highlighting its potential for tuning the properties of these COFs for diverse applications.
Strategic incorporation of CO groups into phenylimino-linked HATP-based COFs (HTCOF) was pioneered by Yan et al. to amplify aromaticity and promote long-range electron delocalization.109 This innovative design was realized through the condensation of TABQ and HAT-CN. HTCOF exhibited extended π-conjugation with fully delocalized electronic states and optimized band structures, achieving an intrinsic electrical conductivity of 3.9 × 10−1 S m−1. Such exceptional charge transport properties enhanced redox activity and electron affinity, which enabled ultrafast ion storage kinetics, positioning it as a promising electrode material for next-generation energy storage systems.
Triazine, a six-membered aromatic ring with electron-deficient characteristics, has been explored as a linkage in HATP-based COFs to enhance redox activity and stability. Voort et al. synthesized HATN-CTFs by using diquinoxalino[2,3-a:2′,3′-c]phenazine-2,8,14-tricarbonitrile (HATN-3CN) as the starting material and ZnCl2 as a molten salt at 400 °C for 48 h.110 Two distinct covalent triazine frameworks were obtained by varying the ZnCl2/monomer ratio, specifically, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 10:1, which controlled the final structural characteristics. The higher ZnCl2 concentration led to the formation of HATN-CTF-2, which exhibited a larger BET surface area and improved pore volume, suggesting better charge transport in energy storage devices. The research group further extended their work by post-synthetically metalating the HATN framework with copper(II) acetate, which significantly enhanced its catalytic activity, particularly in the Henry reaction of aromatic aldehydes and nitromethane under mild conditions.111
:1 and 10:1, which controlled the final structural characteristics. The higher ZnCl2 concentration led to the formation of HATN-CTF-2, which exhibited a larger BET surface area and improved pore volume, suggesting better charge transport in energy storage devices. The research group further extended their work by post-synthetically metalating the HATN framework with copper(II) acetate, which significantly enhanced its catalytic activity, particularly in the Henry reaction of aromatic aldehydes and nitromethane under mild conditions.111
Additionally, imidazole, a heterocyclic aromatic compound with two nitrogen atoms in its five-membered ring, has proven to be an ideal linking unit in COFs.112 Its coordination ability to cations and redox-active properties make it particularly suitable for use in energy storage applications. El-Kaderi et al. synthesized imidazole-linked HATP-based COFs, known as BCOF-1, by condensing HAHATN and TA, adopting a solvothermal synthesis in 1,4-dioxane and mesitylene with acetic acid catalysis at 120 °C for 5 days (Fig. 13).106 The PXRD pattern of BCOF-1 closely matched the simulated diffraction pattern of an eclipsed AA model, confirming its well-ordered crystallinity. The honeycomb-like structure observed in the HR-TEM images was a hallmark of 2D materials. This ordered porous architecture and π-conjugated structure synergistically facilitated rapid ion diffusion coefficient (1.09 × 10−12 cm2 s−1) and substantial electronic conductivity (5.13 × 10−3 S m−1), demonstrating its great potential as a high-performance electrode material.
 | ||
| Fig. 13 (a) Synthetic route for the preparation of BCOF-1. (b) PXRD patterns of BCOF-1. (c) and (d) TEM images of BCOF-1. (e) Solid-state NMR of BCOF-1. Reproduced with permission.106 Copyright 2024, American Chemical Society. | ||
Although the synthesis methods for HATP-based COFs mentioned above represent significant progress in terms of mild reaction conditions and simplicity, challenges remain in further expanding the diversity of linkage and topologies. Addressing these challenges will enable the development of designable and functional materials with tailored properties, broadening their applicability across various fields. This progress holds great promise for advancing future research and applications in energy storage.
3. Applications of HATP-based COFs in rechargeable batteries
The unique chemical structures of HATP-based COFs endow them with superior electrochemical performance. Their electronegative skeletons are situated within well-defined 1D channels, which are ideally suited for accommodating and rapidly transporting metal cations (Li+, Na+, K+, and Zn2+). The delocalized electrons within the fused extended π-system significantly enhance the framework's conductivity and stability, which is critical for achieving fast charge efficiency and long cycling stability in rechargeable batteries.113 During the discharge process, the n-type CN bonds of these materials undergo reduction, causing cleavage of the π bond and the formation of negatively charged nitrogen atoms due to their higher electronegativity.114 Simultaneously, Li+ are intercalated and transported through the cathode, balancing the negative charge. In the reverse charging process, Li+ are released back into the electrolyte, and the electrode materials undergo oxidation, restoring the neutral states of the CN groups. This reversible mechanism allows these HATP-based electrodes to perform efficiently across multiple cycles.
In 2017, Loh's team made a significant advance, synthesizing π-conjugated HATN through a simple condensation process, paving the way further exploration of their Li storage mechanisms.113 A key aspect of their work involved the use of 15N solid-state nuclear magnetic resonance (NMR) to probe the different lithiation states of the HATN. In the discharged state, the NMR spectra revealed the N1 peak disappeared, and the appearance of new peaks (N2, N3, N4), indicating that the HATN molecule coordinated with one Li+ (forming 3Q-Li). With further discharge to 1.12 V, the final lithiated state (3Q-6Li) was confirmed by the increased intensity of the N5 resonance at −302 ppm. The DFT simulation revealed two discharge plateaus in the voltage range of 2.6–2.15 V (stage 1) and 1.68–1.38 V (stage 2). These results demonstrated that the HATN compound underwent a two-step lithiation process, with each step involving the transfer of three electrons, resulting in a high theoretical capacity of 418 mA h g−1. This work laid the foundation for the design and optimization of HATP-based cathodes which had the potential to surpass traditional inorganic cathodes in specific capacity and energy density.
HATP-based COFs show great promise for various energy storage applications, especially as electrode materials in rechargeable batteries. Their high-dense redox-active sites, electronegative channels, π-conjugated structure, excellent stability, and tunable electronic properties make them ideal candidates for use in LIBs, SIBs, PIBs as well as AZIBs. The modular and tunable nature of HATP-based COFs allows for precise control over their structural properties. Ongoing research aimed at enhancing performance and optimizing synthesis methods will undoubtedly unlock the full potential of HATP-based COFs in next-generation energy storage devices.
3.1 HATP-based COFs electrodes for LIBs
The application of HATP-based COFs as electrode materials for LIBs began to gain attention in 2019. Table 1 summarizes the overall electrochemical performance of various HATP-based COFs. To achieve HATP-based COFs electrodes with excellent electrochemical performance, particularly in terms of reversible capacity, extended cycle stability, and superior rate capability, advanced synthetic methodologies and modification strategies have been systematically proposed. These include the incorporation of a conductive carbon matrix (morphology optimization and conductivity enhancement) and rational chemical structure engineering.| Materials | Electronic conductivity (S m−1) | Metal-ion battery | Electrode type | Voltage range (V) | Capacity (mA h g−1), current density (A g−1) | High-rate capacity (mA h g−1), current density (A g−1) | Capacity retention (%), current density (A g−1), cycles | Ref. |
|---|---|---|---|---|---|---|---|---|
| NTCDI-COF | 1.7 × 10−6 | LIBs | Cathode | 1.5–3.5 | 210, 0.1 | 157, 5 | 86%, 2, 1500 | 67 |
| PGF-1 | 3 × 10−3 | LIBs | Cathode | 1–3.6 | 842, 0.1 | 189, 5 | 78.3%, 0.5, 1400 | 83 |
| USTB-6@G | Not mentioned | LIBs | Cathode | 1.2–3.9 | 285, 0.0544 | 188, 2.72 | 70%, 6000, 1.36 | 76 |
| CCP-HATN@CNT | 40 | LIBs | Cathode | 1.2–3.9 | 116, 0.1 | 94, 1 | 91%, 0.5, 1000 | 89 |
| HATN-HHTP@CNT | 5 | LIBs | Cathode | 1.2–3.8 | 230, 0.05 | 130, 2 | 100%, 0.5, 6900 | 100 |
| HATN-AQ-COF | 2.2 × 10−2 | LIBs | Cathode | 1.2–3.9 | 319, 0.179 | 226, 3.58 | 80%, 3.58, 3000 | 104 |
| HAPT-COF@rGO | Not mentioned | LIBs | Cathode | 1.2–3.6 | 558, 0.0788 | 318, 7.88 | 92%, 7.88, 1000 | 115 |
| BQ1-COF | 3.1 × 10−6 | LIBs | Cathode | 1.2–3.5 | 502.4, 0.0385 | 170.7, 7.73 | 81%, 1.54, 1000 | 116 |
| cHATN-CTF | Not mentioned | LIBs | Cathode | 1–4.5 | 310, 0.1 | 77, 2 | Not mentioned | 117 |
| aHATN-CTF | Not mentioned | LIBs | Cathode | 1–4.5 | 64, 0.1 | 10, 2 | Not mentioned | 117 |
| aHATN-CTF | Not mentioned | LIBs | Anode | 0.01–3 | 1171, 0.1 | 320, 10 | 82.9%, 1, 2000 | 117 |
| HATN-HHTP@CNT | 5 | SIBs | Cathode | 1–3.4 | 225, 0.05 | 128, 2 | 100%, 1, 6200 | 100 |
| 3D-TP-HATN-COF | Not mentioned | SIBs | Cathode | 1–4.1 | 250, 0.2 | 206, 10 | 93%, 10, 8000 | 105 |
| 3D-TAM-HATN-COF | Not mentioned | SIBs | Cathode | 1–4.1 | 202, 0.2 | 164, 10 | 82%, 10, 8000 | 105 |
| HTCOF | 3.9 × 10−1 | SIBs | Cathode | 2.8–3.9 | 157.1, 0.16 | 53.5, 10 | 94.8%, 10, 10000 |
109 |
| TQBQ-COF | 1.973 × 10−7 | SIBs | Cathode | 0.8–3.7 | 452, 0.02 | 134.3, 10 | 96.4%, 1, 1000 | 118 |
| HATN-PD-COF | 3.69 × 10−4 | SIBs | Cathode | 1–3.6 | 210, 0.2 | 195, 10 | 91%, 10, 7000 | 119 |
| HATN-TAB-COF | 2.96 × 10−4 | SIBs | Cathode | 1–3.6 | 150, 0.2 | 140, 10 | 100%, 10, 3000 | 119 |
| BCOF-1 | 5.13 × 10−3 | SIBs | Anode | 0.01–3 | 370, 0.0393 | 50, 5.895 | 77%, 1.179, 400 | 106 |
| HATN-HHTP@CNT | 5 | KIBs | Cathode | 1.2–3.8 | 218, 0.05 | 120, 0.5 | 86.5%, 0.5, 2400 | 100 |
| CPT | 1.58 × 10−1 | KIBs | Anode | 0.05–3 | 351, 0.1 | 186, 2 | 87%, 0.1, 200 | 82 |
| TQBQ-COF | 3.45 × 10−6 | KIBs | Anode | 0.01–3 | 423, 0.03 | 185, 3 | 90.9%, 0.9, 600 | 120 |
| PA-COF | Not mentioned | AZIBs | Cathode | 0.2–1.6 | 265, 0.05 | 68, 10 | 74%, 1, 10000 |
68 |
| HA-COF | Not mentioned | AZIBs | Cathode | 0.2–1.6 | 164, 0.1 | 35.4, 10 | 75.3%, 5, 10000 |
121 |
| HAQ-COF | Not mentioned | AZIBs | Cathode | 0.2–1.6 | 339, 0.1 | 95.6, 10 | 88%, 5, 10000 |
121 |
| D-HATN | Not mentioned | AZIBs | Cathode | 0.1–1.3 | 166, 1 | 101, 20 | 42.2%, 5, 10000 |
122 |
| GDAQ | Not mentioned | AZIBs | Cathode | 0.1–1.6 | 331, 0.1 | 207, 15 | 90%, 10, 20000 |
123 |
| GDA | Not mentioned | AZIBs | Cathode | 0.1–1.6 | 255, 0.1 | 100, 10 | 80%, 10, 20000 |
123 |
Electrochemical Li-storage properties of the HATP-based COFs were evaluated using CR2032-type coin cells assembled in argon-filled glove box (the oxygen and water concentration maintained below 1 ppm). The working electrode was fabricated by mixing redox-active materials (HATP-based COFs), conductive carbon and binder at an optimized ratio in NMP to form a slurry, which was subsequently casted on the current collector and dried under vacuum. Li foil was used as the counter electrode, while the electrolyte was selected based on the operating voltage windows (1 M lithium bis((trifluoromethyl)sulfonyl)azanide (LiTFSI) in 1,3-dioxane (DOL) and dimethoxyethane (DME) (1:1, v/v) for cathode and 1 M lithium hexafluorophosphate (LiPF6) in ethylene carbonate (EC):diethyl carbonate (DEC) (1:1, v/v) for anode).124 A Celgard polyethylene porous membrane was utilized as the separator.
 | ||
| Fig. 14 (a) Synthetic route for the preparation of 2D CCP-HATN. (b) Rate performance of 2D CCP-HATN. (c) Cycling performance at 0.5 A g−1 of 2D CCP-HATN. Reproduced with permission.89 Copyright 2019, Wiley-VCH GmbH. | ||
To address these challenges, the team explored a novel approach by in situ growing 2D CCP-HATN on the surface of carbon nanotubes (CNTs) during a solvothermal reaction, resulting in the formation of the 2D CCP-HATN@CNT hybrid material. This hybrid was intended to improve both the exposure of redox-active sites and the electrical conductivity. PXRD and SEM results indicated that when the amount of CNTs was optimized to 50%, the 2D CCP-HATN could be uniformly coated onto the CNTs. This optimization of morphology and structure led to a significant enhancement of the hybrid material's electrical conductivity (4 × 10−1 S cm−1), which in turn improved the rate capability (94 mA h g−1 at 1 A g−1). As a result, the electrochemical performance of the 2D CCP-HATN@CNT was significantly better than that of the pure 2D CCP-HATN. 2D CCP-HATN@CNT showed a stable capacity (116 mA h g−1 at 0.1 A g−1), reflecting 73% utilization of redox-active sites after accounting for the contribution from the CNTs. In addition, the hybrid material also retained 91% of its initial capacity after 1000 cycles at a current rate of 0.5 A g−1, demonstrating stable performance under fast charging/discharging conditions. This approach effectively addressed the issues of low electronic conductivity, slow Li+ diffusion and buried redox-active sites, significantly improving COF-based electrode performance. However, the presence of large non-redox portions in the 2D CCP-HATN structure still limited both the theoretical and practical capacity.
To further enhance specific capacity, it was crucial to reduce the proportion of redox-inactive sites, or introduce external redox-active sites in the frameworks. Li et al. in situ incorporated CNTs onto the outer surface of HATN–HHTP to regulate morphology, giving rise to a core–shell structure (HATN–HHTP@CNT).100 The electrical conductivity of the HATN–HHTP@CNT was enhanced (0.05 S cm−1) to seven orders of magnitude higher than that of HATN–HHTP (4.8 × 10−9 S cm−1). The discharge capacity was 230 mA h g−1 for the HATN–HHTP@CNT, closely approaching the theoretical capacity for a six-electron transfer mechanism and indicating a nearly ideal electrochemical performance. Remarkably, it still retained 100% capacity after 6900 cycles, a duration of over 4100 hours cycling, which was attributed to the stable chemical bond, π–π interaction, and pseudocapacitive effect.
Liu et al. developed an imide-linked HATP-based COF (HCBHAT-PH) with high-density redox-active CN and CO groups and incorporated carboxyl-functionalized carbon nanotubes in situ into the COF structure.125 This strategy achieved enhanced conductivity (1.18 × 10−1 S m−1) and Li+ diffusion kinetics (10−14 to 10−8 cm2 s−1), coupled with a uniform morphology of HCBHAT-PH on carboxyl-functionalized carbon nanotubes, resulting in high reversible capacity (303 mA h g−1 at 0.1 A g−1) and 73% utilization of redox-active sites.
The subsequent introduction of conductive carbon materials, such as graphene and (rGO), to optimize the morphology and exfoliate stacked COFs into nanosheets has proven to be an extremely promising approach to enhance their electrochemical properties. Jiang et al. described the fabrication of conductive graphene during the synthesis of USTB-6 to fabricate a USTB-6@G composite, which was employed as a cathode material (Fig. 15(a)).76 The HR-TEM analysis of USTB-6@G revealed lattice stripes similar to those of USTB-6, confirming that the USTB-6@G retained a crystalline structure after being dispersed onto graphene (Fig. 15(b)–(e)). The USTB-6 nanosheets had an average thickness of approximately 8.3 nm, which supported a high surface area, improving Li+ diffusion during electrochemical reactions (Fig. 15(f)–(i)). These enhancements allowed the USTB-6@G composite to achieve a high capacity (285 mA h g−1 at 0.2C) (Fig. 15(j)), excellent rate performance (188 mA h g−1 at 10C) and superior stability (170 mA h g−1 at 5C after 6000 cycles) (Fig. 15(k)).
 | ||
| Fig. 15 (a) Synthetic route for the preparation of USTB-6@G. (b) and (c) TEM images of USTB-6@G. (d) and (e) TEM images of USTB-6. (f) and (g) AFM topography images and AFM height profiles of graphene. (h) and (i) AFM topography images and AFM height profiles of USTB-6@G. (j) Cycling performance of USTB-6@G at 0.2C. (k) Cycling performance of USTB-6@G at 5C. Reproduced with permission.76 Copyright 2022, Wiley-VCH GmbH. | ||
In a more recent study, HAPT-COF was synthesized through a one-step condensation method between CHHO and TAPT.115 The resultant HAPT-COF exhibited a 2D π-conjugated, robust framework with abundant CO and CN functional groups, providing an ultrahigh theoretical capacity of 788 mA h g−1. In the subsequent step, a post-hydrothermal reaction was employed between HAPT-COF and graphene oxide (GO), resulting in the formation of HAPT-COF@rGO, where GO was reduced to rGO (Fig. 16(a)). The efficient intercalation of HAPT-COF with rGO ensured that HAPT-COF were uniformly distributed and well-dispersed on rGO, forming a sheet morphology of HAPT-COF@rGO, which led to the exposure of redox-active centers, and enhanced electron transport and Li+ diffusion (Fig. 16(b)–(i)). The strong π–π interactions between HAPT-COF and rGO not only improved conductivity but also helped maintain structural integrity. Theoretical simulations and practical characterizations revealed that up to 18 Li+ could be stored reversibly per repeating unit of the framework in three distinct steps, thus achieving a substantial capacity (558 mA h g−1 at 0.1C) (Fig. 16(j)). In addition, the HAPT-COF@rGO composite cathodes also demonstrated excellent rate capability (318 mA h g−1 at 10C), along with 92% capacity retention after 1000 cycles (Fig. 16(k)). These in situ polymerization and exfoliation-related investigations provided feasible strategies for the effective utilization of the redox-active sites of COFs, as well as an improvement in conductivity, thus facilitating application as electrode materials.
 | ||
| Fig. 16 (a) Synthetic route for the preparation of HAPT-COF@rGO. (b) Raman spectra and PXRD patterns of HAPT-COF and HAPT-COF@rGO. (c) TEM images of HAPT-COF. (d) HR-TEM images of HAPT-COF. (e) HR-TEM images of HAPT-COF@rGO. (f)–(i) The corresponding mapping images of C, N and O elements for HAPT-COF@rGO. (j) The galvanostatic charge/discharge curves at 0.05C. (k) Cycling performance at 10C. Reproduced with permission.115 Copyright 2024, Elsevier. | ||
Chen et al. made significant advancements in developing a novel redox-active COF cathode material, BQ1-COF, which was synthesized via a direct condensation reaction between TABQ and CHHO (Fig. 17(a)).116 The unique structure incorporated CO groups with minimal redox-inactive groups, resulting in an exceptionally high reversible capacity (502.4 mA h g−1 at 39 mA g−1), among the highest specific capacities ever reported for LIBs (Fig. 17(b)). However, BQ1-COF exhibited a capacity retention of 81% at 1.54 A g−1 after 1000 cycles with fluctuating Coulombic efficiency (Fig. 17(c)). The electrochemical behavior of BQ1-COF was governed by the reversible reaction of Li+ with the CO and CN groups in the framework. During the discharge process, up to 12 Li+ participated reversibly in the redox reaction in four distinct steps (Fig. 17(d)). The reaction involved the reduction of the carbonyl (CO) and imine (CN) groups, followed by re-oxidization during the subsequent charge process. However, the stability of the lithiated structure tended to decrease as more Li+ became involved in the redox process. The upper limitation appeared to be 12 Li+, beyond which the structural integrity and electrochemical performance might deteriorate.
 | ||
| Fig. 17 (a) Synthetic route for the preparation of BQ1-COF. (b) The galvanostatic charge/discharge curves of BQ1-COF at 39 mA g−1. (c) Cycling performance of BQ1-COF at 1.54 A g−1. (d) Structure evolution during the discharge procedure of BQ1-COF. Reproduced with permission.116 Copyright 2020, Elsevier. | ||
Moreover, Jiang et al. enhanced the stability and electrochemical performance of HATP-based COFs by constructing a redox-active imide-based structure, resulting in the development of HATN-AQ-COF. This material, with fully conjugated nature, well-defined porous network and densely redox-active sites, synergistically demonstrated exceptional charge transfer characteristics, achieving rapid ion diffusion coefficient (10−13 to 10−8 cm2 s−1) and high electrical conductivity (2.2 × 10−4 S cm−1), all of which contributed to its outstanding electrochemical properties (Fig. 18).104 As a result, HATN-AQ-COF achieved a high capacity (319 mA h g−1 at 0.5C). Even at a high-rate of 10C, it retained 80% of its initial capacity after 3000 cycles, demonstrating exceptional cycling stability and rate performance.
 | ||
| Fig. 18 (a) Synthetic route for the preparation of HATN-AQ-COF. (b) Cycling performance of HATN-AQ-COF at 10C. (c) Rate performance of HATN-AQ-COF. (d) Adsorption energy per Li+ at different sites. (e) Structure revolution during the discharge procedure of HATN-AQ-COF. Reproduced with permission.104 Copyright 2022, Wiley-VCH GmbH. | ||
To elucidate the underlying charge storage mechanism, an analysis of Li+ absorption energies was conducted, revealing a sequential multi-site coordination process. Initially, Li+ preferentially binds to the CN groups in the HATN units (site A), followed by interactions with the CO groups in quinone moieties (site B). Subsequently, Li+ coordinates with the CN groups in the HATN units (site A) and the CO groups in imide moieties (site C), enabling an overall 12-electron transfer process (Fig. 18(d) and (e)). This study underscores the importance of integrating multiple redox-active sites to enhance the utilization of existing redox-active sites. By strategically tailoring the spatial distribution and electronic environment of these sites within the framework, researchers can further enhance energy storage capacity, rate capability, and long-term cycling stability. Such structural refinements pave the way for the development of next-generation COF-based electrode materials with superior electrochemical performance, catering to the growing demands of high-performance energy storage technologies.
Because of their electronegative nature, HATP-based COFs predominantly function as cathode materials, where n-type CN groups facilitate reduction reactions during discharge (Li+ insertion) and oxidation reactions during charge (Li+ extraction). However, recent studies have shown that under certain conditions, these materials can also exhibit bipolar behavior, enabling them to operate at higher voltages and achieve greater energy density.
Zhu et al. investigated the impact of ZnCl2 concentration and reaction time on the crystallinity of the resulting HATN-CTF, which was developed by building on the ionothermal trimerization of HATN-3CN.117 When ZnCl2 was used in lesser amounts and the reaction time was short, the resulting cHATN-CTF exhibited a crystalline structure, which was consistent with the simulated eclipsed AA stacking model. However, due to its low specific surface area of 41.52 m2 g−1 and strong π–π interactions, cHATN-CTF provided limited accessible redox-active sites for Li+ interaction, resulting in a relatively low discharge capacity of 70 mA h g−1 within a voltage range of 1.0–4.5 V. In contrast, increasing ZnCl2 concentration and extending the reaction time, (monomer/ZnCl2 of 1:10 and a reaction time of 48 hours), yielded an aHATN-CTF with higher surface area and a porous structure. These effects, significantly improved the ability of aHATN-CTF to store and release cations, leading to a substantial capacity (251 mA h g−1 at 50 mA g−1). Notably, unlike other HATP-based COFs, the presence of electron-deficient triazine rings at higher voltages enabled HATN-CTF to bind with PF6− ions, further supporting its bipolar characteristics. This demonstrated that incorporating redox-active units with high potential, alongside precise polymerization control, could provide an effective strategy for tuning electrochemical properties, particularly in terms of capacity and operating voltage. Furthermore, the superlithiation of aHATN-CTF underscored its potential as an anode material. It achieved a high reversible capacity of 655 mA h g−1 at 1 A g−1 with an impressive capacity retention of 82.9% over 2000 cycles, positioning it as a promising candidate for next-generation organic anode materials.
3.2 HATP-based COFs electrodes for other alkali metal-ions batteries
SIBs and KIBs have emerged as a more sustainable alternative to LIBs due to the abundance and low cost of sodium and potassium.126–128 The standard potential (vs. standard hydrogen electrode) of K+/K and Na+/Na is −2.936 V and −2.714 V, respectively, giving PIBs a higher voltage and energy density compared to SIBs.19 Given the current challenges of accommodating larger sized sodium-ions (Na+) and potassium-ions (K+) in traditional inorganic electrode materials, HATP-based COFs present a promising solution. The interconnected porous structures in COFs provide abundant ion transport channels, facilitating the rapid transport and accommodation of Na+ and K+.62 Additionally, the designable structure of COFs enables the precise tuning of functionality in their building blocks, which enhances their metal-ion affinity (Na+ and K+), ultimately improving charge storage performance and stability.Another key feature of HATP-based COFs is their high-density redox-active sites, specifically CN groups within the HATP frameworks. These sites undergo reversible conversion reactions, particularly similar to those observed in LIBs during repetitive charge and discharge cycles. However, it is important to emphasize that the mechanisms for cation storage in SIBs and KIBs differ from those in LIBs. In PIBs, the π-electrons present in the aromatic rings of the COF structure can interact with K+, creating additional binding sites that enhance cation storage and the overall capacity of the electrode materials. This unique synergy between structural and chemical features positions HATP-based COFs as transformative materials for advanced SIBs and KIBs, contributing significantly to the development of high-performance, sustainable energy storage materials.
The assembly processes of HATP-based COFs for SIBs and PIBs followed a similar protocol to LIBs, with the primary differences lying in the employment of separator and electrolyte. Typically, a Whatman glass microfiber filter was utilized as the separator, and the electrolyte systems were formulated using the corresponding alkali metal salts.129
Building on the success of the HATP-based COFs in LIBs, Li et al. extended the application of in situ polymerization to SIBs, achieving remarkable electrochemical performance in HATN-HHTP@CNT composite cathodes through the incorporation of CNTs.100 The in situ polymerization process facilitated the formation of a well-integrated conductive polymer network, which improved electronic conductivity and promoted the rapid Na+ transport process during charge/discharge cycles. A particularly notable feature of HATN-HHTP@CNT in SIBs was its exceptional cycling stability, as it sustained 6200 charge/discharge cycles at 1 A g−1 with 100% capacity retention. This indicated the absence of significant capacity degradation over more than 1700 hours, marking a remarkable achievement for SIBs. The design strategy of HATP-based COFs with porous structures, high conductivity, and multiple redox-active sites remains a highly effective approach for enhancing SIBs performance.
While 2D COFs have demonstrated significant potential as cathode materials for SIBs, their intrinsic microporous structure presents a major challenge. With pore sizes typically below 2 nm, these materials impose considerable limitations on the diffusion of larger ions like Na+, thereby restricting high-rate charge/discharge performance.
Jiang et al. synthesized two imide-linked HATP-based COFs containing a mesoporous structure—HATN-PD-COF and HATN-TAB-COF—via hydrothermal reactions using HATNCA with PDA and 1,3,5-tris(4-aminophenyl) benzene, respectively (Fig. 19(a)).119 These COFs exhibited high crystallinity with an AA stacking configuration and featured mesopores of approximately 3.1 nm (HATN-PD-COF) and 2.0 nm (HATN-TAB-COF). The larger pore size of HATN-PD-COF facilitated faster Na+ transport, as evidenced by its higher diffusion coefficient of 10−9 cm2 s−1 compared to the HATN-TAB-COF (10−9.5 cm2 s−1). As a result, they demonstrated superior rate performance (195 mA h g−1 for HATN-PD-COF and 140 mA h g−1 for HATN-TAB-COF at 10 A g−1), outperforming most reported organic cathodes (Fig. 19(b) and (c)). The higher capacity of HATN-PD-COF was primarily attributed to the denser distribution of redox-active sites within its structure. Moreover, the HATN-PD-COF cathode exhibited excellent capacity retention of 91% after extensive 7000 cycles at 10 A g−1 (Fig. 19(d)). The binding strength analysis suggested that the N atoms in the HATN unit (site I) provided a more energetically favorable environment for Na+ coordination compared to the O atoms in the imide groups (site II) (Fig. 19(e)). Consequently, HATN-PD-COF achieved a more balanced and stable Na+ storage process by initially occupying the stronger-binding site I, followed by the utilization of slightly weaker site II.
 | ||
| Fig. 19 (a) Synthetic route for the preparation of HATN-PD-COF and HATN-TAB-COF. (b) Rate performance of HATN-PD-COF. (c) Rate performance of HATN-TAB-COF. (d) Cycling performance of HATN-PD-COF at 10 A g−1. (e) The adsorption energy per Na+ at different sites of HATN-PD-COF. Reproduced with permission.119 Copyright 2024, Springer. | ||
The same groups also developed 3D HATP-based COFs cathodes, 3D-TP-HATN-COF and 3D-TAM-HATN-COF, featuring cross-linked nanochannels and uniform pore sizes, which was also determined to be an effective strategy to enhance the ion transport in rechargeable batteries.105 Benefiting from the rapid ion diffusion and co-storage of Na+ and PF6−, the 3D-TP-HATN-COF cathode showed an ideal reversible capacity (250 mA h g−1 at 0.2 A g−1), which was higher than that of 3D-TAM HATN-COF (202 mA h g−1 at 0.2 A g−1), thanks to its high-density redox-active sites and larger channels. Additionally, the 3D-TP-HATN-COF cathode achieved an outstanding rate performance (206 mA h g−1 at 10 A g−1) and high capacity retention of 93% after 8000 cycles at 10 A g−1. Furthermore, the full battery, composed of 3D-TP-HATN-COF and Na3Bi, exhibited the exceptional capacity of 195 mA h g−1 at 10 A g−1 after 1000 cycles.
HATP-based COFs demonstrated remarkable potential not only as cathodes but also anode applications for SIBs. Notably, El-Kaderi et al. investigated BCOF-1 as an anode material for Na+ storage.106 Apart from the inherent six redox-active sites in HATP units, the imidazole linkages generated during polymerization provided additional Na+ coordination sites, significantly enhancing charge storage capacity. This synergistic combination of abundant redox-active sites, well-ordered pore structure, and extended π-conjugated frameworks contributed to a high reversible capacity (370 mA h g−1 at 0.1C), rate capability (50 mA h g−1 at 15C), and cycling stability (77% capacity retention after 400 cycles at 3C).
O and CN groups in TQBQ-COF serve as redox-active sites capable of reversibility with K+. The distinguishing factor of PIBs compared to other alkali metal-ion batteries is the additional involvement of the CC bonds in the framework. Fourier transform infrared (FT-IR) spectra revealed that the CC groups of the TQBQ-COF reversibly disappeared and reappeared during the discharging and charging cycles, indicating that K+ interacted directly with the π-electrons of the aromatic rings. This interaction enabled reversible modulation of the π-conjugation, providing additional capacity (423 mA h g−1 at 0.03 A g−1) beyond the simple interaction towards K+ with the CO and CN groups. Such a phenomenon has also been observed in multivalent ion batteries.130 Guided by the π–cation interaction, this behavior highlights the versatility of π-conjugated materials in facilitating ion storage. The additional capacity arising from these interactions may be a key factor driving the success of π-conjugated HATP-based COFs in energy storage applications.
The HATN-HHTP@CNT composite, recognized for exceptional cycling stability in LIBs and SIBs, also demonstrated remarkable K+ storage capabilities.100 HATN-HHTP@CNT cathode delivered a high discharge capacity (218 mA h g−1 at 0.05 A g−1) with characteristic voltage plateaus at 2.9–2.35 V and 2.0–1.5 V. These clear redox features revealed multi-step charge transfer processes, demonstrating a charge storage mechanism similar to that observed in LIBs and SIBs. Furthermore, the HATN-HHTP@CNT cathode exhibited ultralong cyclability with a capacity retention of 86.5% over 2400 cycles at 0.5 mA g−1, highlighting its robust coordination and structural integrity.
3.3 HATP-based COFs electrodes for aqueous zinc-ion batteries
The low cost and high abundance of zinc has contributed to a growing interest in AZIBs, which offer a cost-effective and environmentally friendly alternative for large-scale energy storage applications.131 Zinc is highly stable in aqueous electrolytes with a satisfactory theoretical specific capacity of 820 mA h g−1 and a suitable standard potential (vs. standard hydrogen electrode) of −0.76 V for Zn2+/Zn.132,133 Additionally, AZIBs combine high safety, cost-effectivity, non-toxicity, and higher ionic conductivity, making them promising alternatives to conventional LIBs.The use of HATP-based COFs as electrode materials for AZIBs represents an emerging area of research with great potential. A notable advantage of HATP-based COFs is their poor solubility in aqueous electrolytes, which contributes to structural robustness and exceptional long-term cycling stability. Certain HATP-based COFs exhibit the ability to cycle up to ten thousand cycles or more without significant degradation.
Determining the charge storage mechanism of organic electrode materials is a crucial factor in the context of AZIBs. The mechanism is primarily governed by the coordination interactions between active functional groups in the electrode material and the cations in the electrolyte. A key factor in this process is the competition between different cations, notably H+ and Zn2+, which significantly impacts both capacity and rate performance.
For the fabrication of AZIBs, the electrode slurry—containing HATP-based COF, conductive additive and binder—was typically coated onto stainless steel or carbon paper current collectors. The electrochemical system employed ZnSO4 electrolyte and the Whatman glass fiber separator.134
000 cycles at 1.0 A g−1 (Fig. 20(b)). Distinctive variations at 16.2° and 24.3° observed in the in situ XRD patterns during cycling confirmed the formation of Zn4(OH)6SO4·5H2O (Fig. 20(c)), which was further supported by the SEM results and elemental mapping images. To explore capacity contribution, they measured capacity in pure 1.0 M ZnSO4 and H2SO4 (pH = 5) electrolytes, revealing that the intercalation of Zn2+ accounted for 59.3%, 63.3%, and 66% in the first three cycles, respectively. These findings were consistent with the inductively coupled plasma atomic emission spectroscopy (ICP-AES) results (Fig. 20(d) and (e)), validating the dominant role of Zn2+ intercalation in the electrochemical process.
 | ||
| Fig. 20 (a) Synthetic route for the preparation of PA-COF. (b) Cycling performance of PA-COF at 1.0 A g−1. (c) In situ XRD patterns of PA-COF during the first three cycles. (d) Zinc percentage calculated from Zn2+ and Zn4(OH)6SO4·5H2O. (e) Quantitative capacity contribution of Zn2+ and H+. (f) Simulated binding energies with different quantities of Zn2+ in PA-COF. Reproduced with permission.68 Copyright 2020, American Chemical Society. | ||
The calculated binding energies of PA-COF bonded with different quantities of Zn2+—specifically 6, 12, and 18 Zn2+—were all negative, which signified that the interactions between the PA-COF and Zn2+ were thermodynamically favorable, and reflected the strong affinity of the PA-COF for Zn2+ (Fig. 20(f)). Building on this work, the same group extended the research by investigating the role of different organic building blocks of HATP-based COFs—specifically benzene rings and benzoquinone units—to understand their influence on the electrochemical performance and storage mechanism of AZIBs.121 The introduction of quinone-functional groups into the COF structure significantly enhanced the uptake capacity of Zn2+ and H+, outperforming the non-quinone-functionalized cathodes. Additionally, the presence of redox-active quinone groups served as favorable binding sites for Zn2+, promoting the formation of O⋯Zn⋯N coordination. This interaction not only stabilized Zn2+ within the framework but also favored the competitive intercalation of Zn2+ over H+, achieving high reversible capacity (339 mA h g−1 at 0.1 A g−1) and long cycling stability (128 mA h g−1 after 10000 cycles at 5 A g−1).
The linkage of HATP units played a pivotal role to modulate the accessibility and activity of redox-active sites. Yu et al. investigated this structure–activity relationship by synthesizing two distinct HATP-based COFs through polycondensation of CHHO with PT or dibenzo[b,e][1,4]dioxine-2,3,7,8-tetraamine (Dio-4NH2), constructing pyrazine-bridged P-HATN and dioxin-bridged D-HATN, respectively.122 Mulliken population analysis of orbital compositions revealed a remarkable enhanced redox activity for N atoms in D-HATN, with each active N atom contributing 10.0% to the total LUMO—nearly triple the 3.8% contribution observed in P-HATN. Furthermore, D-HATN exhibited significantly attenuated π–π stacking interactions compared to P-HATN as evidenced by reduced density gradient analyses. P-HATN maintained near-planar geometry (dihedral angle of 179.75°) due to its aromaticity, while D-HATN adopted a slightly distorted configuration (dihedral angle of 174.87°) enabled by the flexible dioxin linkage. The synergistic combination of enhanced π-electron density localization at N active sites, optimized LUMO distribution, and molecular flexibility to prevent interlayer stacking collectively endowed D-HATN with higher redox-active site utilization and reversible capacity (166 mA h g−1 at 1 A g−1) compared to P-HATN. D-HATN also achieved good cyclability, maintaining 42.2% capacity retention after 10000 cycles at 5 A g−1.
The use of in situ polymerization as a strategy to enhance the overall electrochemical performance of AZIBs is still considered to be effective. Zhu et al. initially proposed the in situ polymerization method in AZIBs, involving the polymerization of HATN units with dual thioether bonds on CNT substrates.135 Notably, this approach led to a remarkable increase in capacity, from 168 mA h g−1 to 328 mA h g−1, which was attributed to the creation of larger pore sizes and improved conductivity. The material exhibited exceptional cycling stability, retaining over 76% of its initial capacity after the extensive number of 10000 cycles at a high current density of 20 A g−1, underscoring its structural integrity and electrochemical robustness. It was evident from the CV and GCD measurements that the contribution of H+ to the overall capacity was quantified at 10.07% in a 2 M ZnSO4 electrolyte. These findings highlight the potential of in situ polymerization as a powerful strategy for optimizing the performance of AZIBs.
Shen et al.'s work on HATP-based COFs represents a notable example of efforts to enhance redox activity and H+ storage mechanisms.123 The synthesis began with bromine-functionalized HATN-3Br; these electronegative Br atoms are highly susceptible to nucleophilic attack, making them ideal for substitution reactions. Two amino precursors, 2,6-diaminoanthracene (DA) and DAAQ, were employed as nucleophiles to two distinct COFs: DA-HATN COF and DAQ-HATN COF, respectively (Fig. 21(a)). These materials exhibited distinct electronic properties, redox activity, and structural characteristics, primarily influenced by the incorporated CO groups. However, both DA-HATN COF and DAQ-HATN COF were relatively dense, with small micropores which could impede efficient ion diffusion and limit the accessibility of redox-active sites.
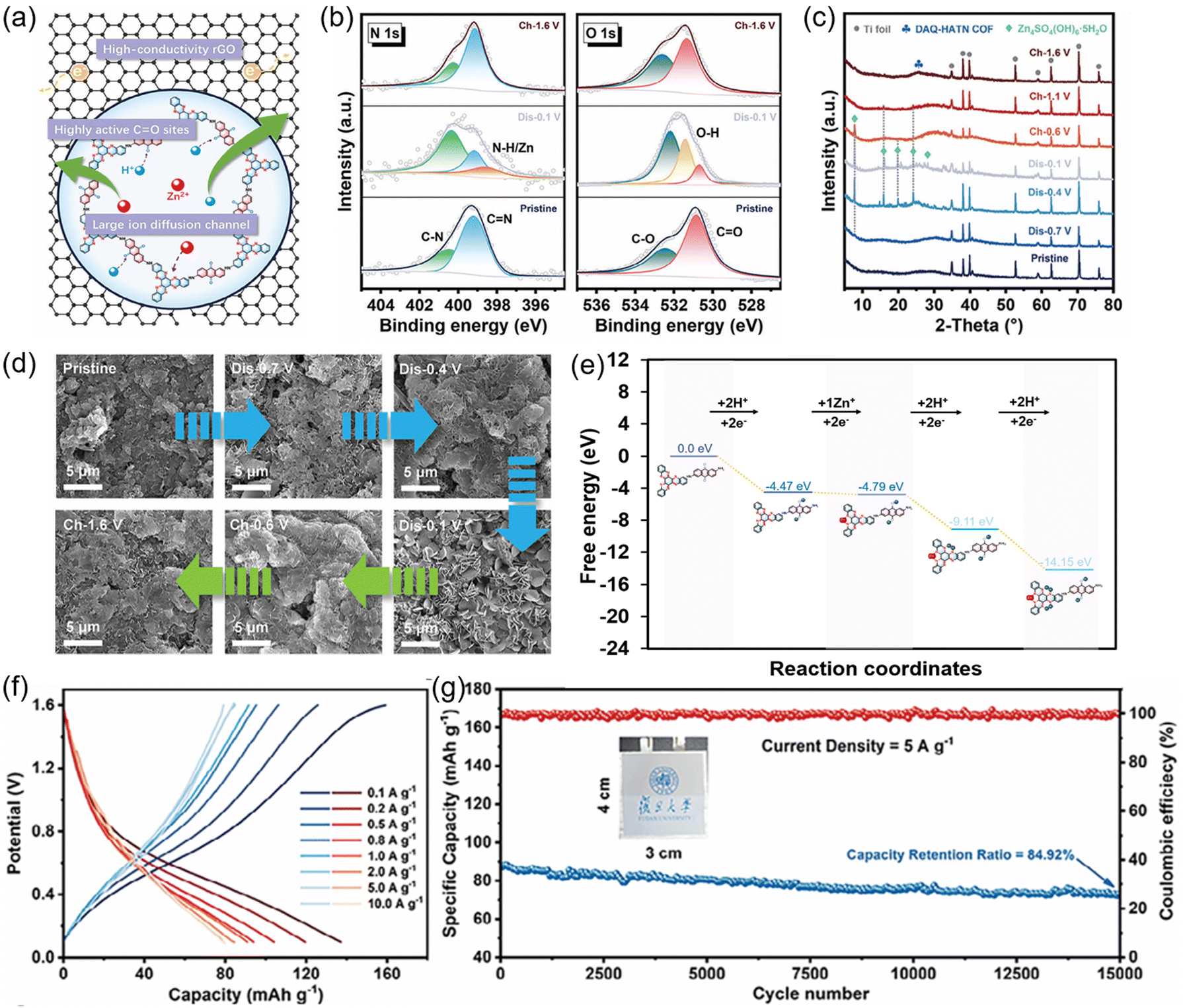 | ||
| Fig. 21 (a) Structure and mechanism of GDAQ. (b) Ex situ XPS spectra. (c) Ex situ XRD patterns. (d) Ex situ SEM images. (e) Calculated Gibbs free energy of DAQ-HATN COF during stepped H+/Zn2+ coordination process. (f) Rate performance of GDAQ//GDAQ-R pouch cells. (g) Cycling stability at 5 A g−1 of GDAQ//GDAQ-R pouch cells. Reproduced with permission.123 Copyright 2024, Wiley-VCH GmbH. | ||
To address these limitations, rGO was introduced using a post-hydrothermal method to create larger porous networks and improve conductivity, yielding materials donated as GDA and GDAQ, respectively. The incorporation of redox-active carbonyl groups with electron-withdrawing properties into GDAQ significantly improved its electrochemical properties compared to GDA. This modification enabled GDAQ cathode to achieve a higher capacity (331 mA h g−1 at 0.1 A g−1) with a higher plateau voltage of 0.66 V and excellent rate capability (207 mA h g−1 at 15 A g−1). The use of X-ray photoelectron spectra (XPS) to investigate the evolution of active groups in the GDAQ cathode during the charge/discharge cycles provided valuable insight into the electrochemical processes. The XPS spectra of the discharged state of the DAQ-HATN COF revealed a noticeable reduction in the intensity of peaks assigned to the CN and CO bonds, which were transformed into C–N and C–O bonds, respectively (Fig. 21(b)). Upon recharging, these changes reversed, confirming the involvement of the CO and CN groups in electron transfer during charge storage. The emergence of N–H and O–H bonds indicated that H+ were anchoring to the redox-active CN and CO sites during the discharge cycle. This was consistent with the reversible formation and disappearance of characteristic peaks for Zn4(OH)6SO4·nH2O (ZOHS) in ex situ XRD spectra (Fig. 21(c)) and the appearance of 2D flakes in SEM images (Fig. 21(d)).
By integrating the DFT calculations based on thermodynamic laws with the ICP-OES results, a detailed step-by-step mechanism for the coordination reaction involving Zn2+ and H+ ions was developed. This stepwise mechanism begins with the CO group coordinating with 2 H+, followed by the coordination of Zn2+ with CN groups, and finally, the protonation of the CN group by 4 H+ (Fig. 21(e)). The above comprehensive characterization demonstrated that the incorporation of anthraquinone units in the DAQ-HATN COF significantly enhanced the H+ storage capability over Zn2+ and elevated the average (dis-)charge potential. Building on these findings, they developed symmetric all-COF/graphene AZIBs (GDAQ//GDAQ-R) based on GDAQ as a cathode material and the reduced product of GDAQ (GDAQ-R) as an anode material. The symmetric pouch cells also demonstrated impressive rate performance (80 mA h g−1 at 10 A g−1) and cycling stability, retaining 84.92% capacity after 2000 cycles at a high current density of 5 A g−1 (Fig. 21(f) and (g)).
4. Conclusions and outlook
Organic electrode materials, particularly π-conjugated HATP-based COFs, have emerged as promising alternatives to conventional inorganic options. Their highly designable structures, electronegative channels, high-density redox-active sites, excellent stability, conjugated structure, and light elements make them ideal for use in rechargeable batteries such as LIBs, SIBs, PIBs, and AZIBs (Fig. 22). In this review, we provided a summary of key research developments, ongoing challenges, and innovative strategies for structural optimization and performance enhancement. We systematically summarized the synthetic strategies for HATP-based COFs, which can be categorized into direct one-step synthesis and indirect multi-step approaches. The former enables efficient fabrication of HATP-based COFs, while the latter facilitates the construction of more diversified architectures with versatile dynamic covalent linkages, such as imine, imide, phenylimino, and triazine. Special attention was given to the unique advantages and the engineering of redox mechanisms of HATP-based COF electrodes in LIBs, specifically addressing challenges, such as insufficient and buried redox-active sites and limited electronic conductivity, while exploring potential solutions to overcome these limitations, including the incorporation of a conductive carbon matrix and the optimization of chemical structure. Furthermore, the review covered the applications of HATP-based COFs in SIBs, PIBs, and AZIBs, highlighting the exceptional adaptability and versatility of these materials across distinct electrochemical energy storage systems. | ||
| Fig. 22 Advantages and outlook of HATP-based COFs electrodes. | ||
While considerable progress has been made, substantial ongoing research efforts are still imperative to simultaneously achieve high energy density and power density, including expanding structural diversity, optimizing molecular engineering and synthesis, enhancing conductivity, precisely controlling morphology, developing large-scale production, and refining electrode preparation and characterization (Fig. 23). Overcoming these challenges is crucial to unlock the full potential of HATP-based COFs in next-generation sustainable and high-performance rechargeable batteries.
 | ||
| Fig. 23 Effective approaches applied in HATP-based COFs electrode materials for enhanced electrochemical performance. | ||
4.1 Enrichment of functional building blocks
Currently, two primary methods are employed to fabricate HATP-based COFs: the direct one-step method and the indirect step-by-step method. While both have distinct advantages, expanding the range of functional building blocks and organic linkers is crucial to diversifying the structures and properties of HATP-based COFs. Although the one-step method is straightforward and efficient, improving its versatility for producing diverse HATP-based COFs remains a challenge, particularly due to the limited range of amino-functionalized organic linkers. Expanding these linkers, such as incorporating heteroatoms like S, P, or B, can enhance the electronic properties and stability of the COFs. The indirect method, by introducing new functional groups, offers additional diversity by incorporating various organic reactions, such as Debus Radziszewski, Suzuki coupling, and Yamamoto coupling reactions. Post-modification strategies could further enhance COF structure, properties, and electrochemical performance, allowing for fine-tuned control over functionality and stability.4.2 Incorporation of high-voltage redox-active groups
Most of the HATP-based COFs used as cathode materials are primarily materials involving n-type CN groups with (dis-)charge voltages below 3 V. To further enhance the energy density of these materials, strategic molecular engineering approaches can be developed, focusing on the incorporation of high-voltage redox-active units with operating potentials exceeding 3 V. Particularly, phenazine, phenothiazine, phenoxazine and thianthrene units with higher redox voltages and strong redox activity can be incorporated into the COFs skeleton, leading to not only higher redox potentials but also excellent electrochemical stability, thereby improving energy density.
4.3 Enhancement of conductivity
A major challenge for HATP-based COF electrodes is their poor intrinsic electrical conductivity, which limits reversible capacity and rate performance. Traditional methods, such as adding conductive agents, can increase conductivity but often reduce the content of active materials in the electrodes, negatively affecting the energy density and overall battery performance. To address this challenge, constructing HATP-based COFs with low bandgap is an effective strategy. The strategic alternation of electron-rich units and electron-deficient moieties within a donor–acceptor (D–A) configuration induces substantial bandgap narrowing, facilitating intramolecular charge transfer. Moreover, the extension of π-conjugated systems through molecular engineering facilitates electron delocalization across the framework via enhanced π–π orbital interactions, driven by resonance stabilization effects, acquiring high conductivity and fast charge/discharge states while preserving redox-active sites. These approaches improve charge separation, delocalization, and electron transport, essential for enhancing battery performance.4.4 Regulation of morphology
The dense stacking of 2D HATP-based COFs into bulk structures can hinder electrochemical performance by restricting the accessibility of redox-active sites and electrolyte penetration. To maximize the exposure of redox-active sites, designing open, porous structures with well-defined channels is crucial, to facilitate ion diffusion and electrolyte interaction. Controlling COF morphology at the microscale or nanoscale—such as creating nanoparticles or nanofibers—can improve surface area and shorten ion transport paths. The integration of stereoscopic building blocks to construct 3D COFs enables the formation of interconnected hierarchical pore structures combining microporosity and mesoporosity, which offer a balance between charge storage and fast ion diffusion, enhancing both specific capacity and rate performance. These synergistic effects simultaneously enhance the accessibility of redox-active sites and charge carrier dynamics, addressing the traditional trade-off between structural stability and accessibility in 2D HATP-based COFs.Furthermore, the development of HATP-based COF films also represents a significant advancement, offering ordered pore channels, uniform surface morphology, and distinct interfacial properties that can enhance metal-ion diffusion. HATP-based COF films can be prepared using a home-made Π-shaped PTFE holder with a SiO2 substrate to prevent powder contamination, with the entire reaction vessel sealed in a stainless steel autoclave under identical reaction conditions to powder preparation. Notably, the optimized prepare methods enable precise control over free-standing COF film electrodes, achieving tunable thickness, reduced surface roughness, and uniform morphology. Binder-free HATP-based COF films can also effectively minimize the grain boundary effect of bulk COFs while maintaining robust structure, potentially achieving simultaneous optimization of both ionic and electronic transport.
4.5 Developing large-scale production
Despite promising laboratory-scale performance, the commercialization of HATP-based COFs still involves significant challenges, primarily the high cost of raw materials. Many organic linkers and functional groups used in COF synthesis are expensive, and their availability can be limited. Developing alternative, cheaper precursors—such as renewable bio-based or waste-derived materials—could lower production costs and improve sustainability. Additionally, parallel efforts are focused on developing more efficient synthesis methodologies. Microwave-assisted synthesis, ultrasonic-assisted synthesis, and mechanical ball milling under ambient conditions can reduce energy consumption, shorten production times, and enhance COF synthesis efficiency. However, solvothermal methods remain the most commonly used approach, and diversifying synthesis techniques could enhance electrochemical properties and enable large-scale production. These combined advancements in precursor selection and synthesis optimization are essential to bridging the gap between laboratory-scale innovation and the industrial-scale implementation of HATP-based COFs electrodes.4.6 Optimizing electrode preparation and characterization methods
In conventional electrode preparation processes, current methods typically use HATP-based COF powders as active materials, which can result in structural heterogeneity and inconsistent particle morphologies. The development of single-crystal COF electrodes represents a transformative approach, which could offer higher crystallinity, uniform structures, and consistent molecular sizes, improving ion transport and overall electrode performance. Additionally, traditional slurry-casting methods often introduce non-conductive polymeric binders, such as polyvinylidene difluoride and polytetrafluoroethylene, hindering electron conduction and limiting electrochemical performance. The transition from non-conductive binders to conductive alternatives, such as PEDOT:PSS, can not only provide the necessary binding properties but also facilitate electron transport within the electrode, thereby enhancing overall electrochemical performance.For characterizing electrode behavior during discharge/charge cycles, advanced techniques such as in situ FT-IR, PXRD, XPS, and DFT calculations are valuable, but methods like in situ AFM could provide real-time insights into the electrode/electrolyte interface, mechanical properties, and morphological changes, enabling more targeted improvements in electrode design. Additionally, ex situ solid-state NMR spectroscopy can be exploited to detect specific redox reactions involving electrolytes in HATP-based COF electrodes, particularly by visualizing structural changes intuitively through colour-mapped profiles of solid-state NMR spectra.
In short, the development of high-performance HATP-based COF electrode materials necessitates a multifaceted approach combining precision synthesis protocols, molecular structural engineering, and advanced characterization methodologies. This synergistic integration enables the rational design of optimized electrodes that simultaneously achieve enhanced energy density, excellent cycling stability, environmental friendliness and cost-effectiveness.
The exceptional electrochemical properties of HATP-based COFs that enable their outstanding electrode performance also afford them uniquely suitable for addressing various challenges in rechargeable batteries. Their characteristic periodic arrangement of HATP units and well-defined nanochannels proves equally valuable for metal anode protection, where the high-density and electronegative sites within uniform 1D channels simultaneously guide homogeneous metal deposition, suppress dendrite growth, and promote the formation of solid electrolyte interphase during repeated plating/stripping cycles. The rational molecular design of HATP-based COFs allows optimization of these protective functions across various metal anodes, including Li, Zn, Na, and Mg, while maintaining exceptional structural stability during the prolonged cycling. Furthermore, this multifunctional adaptability of HATP-based COFs can also extend to sulfur host materials in lithium sulfur batteries (LSBs). The matched redox potential between HATP-based COFs and sulfur reveals their potential as Li2Sx reactive-type hosts, enabling efficient transformation into nanostructured Li2S and S within the porous frameworks.
This review highlights HATP-based COFs as advanced and highly promising candidates for electrode materials in various rechargeable batteries, based on the precise tuning of their structural, electronic, and chemical properties. By leveraging this design flexibility, researchers can exploit structure–property relationships to develop electrode materials with high energy densities, excellent cycling stability, and superior safety—critical factors for the commercialization of next-generation rechargeable batteries. While HATP-based COF electrode materials face challenges across diverse metal-ion batteries, the general molecular and structural-modification strategies are valid to achieve excellent electrochemical performance. With ongoing advancements and interdisciplinary research, the application of HATP-based COFs in batteries is poised to make a significant contribution to addressing the global energy crisis, positioning them as key materials for sustainable energy solutions in the future. The accelerating pace of research in this field is sure to lead to breakthroughs that will help meet the growing demand for high-performance, safe, and cost-effective energy storage technologies.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation (NRF) of Korea (RS-2023-00221668, RS-2024-00435493, RS-2024-00466616).References
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120, 6358–6466 CrossRef CAS PubMed.
- S. Haldar, A. Schneemann and S. Kaskel, J. Am. Chem. Soc., 2023, 145, 13494–13513 CrossRef CAS PubMed.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef.
- Z. Li, Y.-X. Yao, S. Sun, C.-B. Jin, N. Yao, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202303888 CrossRef CAS PubMed.
- J. Wang, Y.-F. Zhu, Y. Su, J.-X. Guo, S. Chen, H.-K. Liu, S.-X. Dou, S.-L. Chou and Y. Xiao, Chem. Soc. Rev., 2024, 53, 4230–4301 RSC.
- L. Wang, J. Wang, Y. Lu, S. Fang, C. Yang, X. Wu, Y. Xiao, Y. Wang, S. Chou and S. Chen, Chem. Soc. Rev., 2025 10.1039/d3cs00911d.
- C. Zhao, Z. Yang, X. Zhou, Z. Hao, J. Chen, Z. Wang, X. Chen, X. Wu, L. Li, L. Li, L. Jiao and S. Chou, Adv. Funct. Mater., 2024, 34, 2303457 CrossRef CAS.
- C.-H. Jo, N. Voronina, Y.-K. Sun and S.-T. Myung, Adv. Mater., 2021, 33, 2006019 CrossRef CAS PubMed.
- L. Zhu, G. Ding, L. Xie, X. Cao, J. Liu, X. Lei and J. Ma, Chem. Mater., 2019, 31, 8582–8612 CrossRef CAS.
- T. Sun, J. Xie, W. Guo, D.-S. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS.
- Y. Xu, Y. Du, H. Chen, J. Chen, T. Ding, D. Sun, D. H. Kim, Z. Lin and X. Zhou, Chem. Soc. Rev., 2024, 53, 7202–7298 RSC.
- H. Dong, N. Kang, L. Li, L. Li, Y. Yu and S. Chou, Adv. Mater., 2024, 36, 2311401 CrossRef CAS PubMed.
- H. Gao, A. R. Neale, Q. Zhu, M. Bahri, X. Wang, H. Yang, Y. Xu, R. Clowes, N. D. Browning, M. A. Little, L. J. Hardwick and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9434–9442 CrossRef CAS PubMed.
- H. Dong, N. Kang, L. Li, L. Li, Y. Yu and S. Chou, Adv. Mater., 2024, 36, 2311401 CrossRef CAS PubMed.
- C. Guo, Y. Gao, S.-Q. Li, Y. Wang, X.-J. Yang, C. Zhi, H. Zhang, Y.-F. Zhu, S. Chen, S.-L. Chou, S.-X. Dou, Y. Xiao and X. Luo, Adv. Funct. Mater., 2024, 34, 2314851 CrossRef CAS.
- L. Cao, C. Wang, H. Wang, X. Xu, X. Tao, H. Tan and G. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202402095 CrossRef CAS PubMed.
- J. Chu, Z. Liu, J. Yu, L. Cheng, H.-G. Wang, F. Cui and G. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202314411 CrossRef CAS PubMed.
- L. Zhong, C. Wang, J. He, Z. lin, X. Yang, R. Li, S. Zhan, L. Zhao, D. Wu, H. Chen, Z. Tang, C. Zhi and H. Lv, Adv. Mater., 2024, 36, 2314050 CrossRef CAS PubMed.
- Z. Sun, C. Yang, Y. Zhang, J. Zhang, Z. Chen, J. Peng, C. Chen, H. Yao and S. Guan, J. Colloid Interface Sci., 2025, 680, 456–463 CrossRef CAS.
- M. Cheng, H. Wang, L. Cao, J. Shao, Y. He, X. Tao and G. Zhu, ChemCatChem, 2025, 17, e202401141 CrossRef CAS.
- H. Wang, L. Cao, X. Tao and G. Zhu, Angew. Chem., Int. Ed., 2025, e202502943 Search PubMed.
- Z. Sun, M. Shu, J. Li, B. Liu, H. Yao, S. Guan and Z. Sun, J. Energy Chem., 2023, 78, 30–36 CrossRef CAS.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- H. Li, Z. Zhou, T. Ma, K. Wang, H. Zhang, A. H. Alawadhi and O. M. Yaghi, J. Am. Chem. Soc., 2024, 146, 35486–35492 CrossRef CAS PubMed.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef CAS.
- S. E. Neumann, J. Kwon, C. Gropp, L. Ma, R. Giovine, T. Ma, N. Hanikel, K. Wang, T. Chen, S. Jagani, R. O. Ritchie, T. Xu and O. M. Yaghi, Science, 2024, 383, 1337–1343 CrossRef CAS.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS.
- Y. Li, W. Chen, G. Xing, D. Jiang and L. Chen, Chem. Soc. Rev., 2020, 49, 2852–2868 RSC.
- Q. Yan, S. Tan, R. Liu, Y. Zhi and D. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202316092 CrossRef CAS PubMed.
- R. Liu, K. T. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yang and D. Jiang, Chem. Soc. Rev., 2021, 50, 120–242 RSC.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- W. Wang, W. Zhao, H. Xu, S. Liu, W. Huang and Q. Zhao, Coord. Chem. Rev., 2021, 429, 213616 CrossRef CAS.
- C. Niu, S. Zhao and Y. Xu, J. Am. Chem. Soc., 2024, 146, 3114–3124 CrossRef CAS.
- A. R. Camargo, K. Endo and B. V. Lotsch, Angew. Chem., Int. Ed., 2024, 63, e202413096 CrossRef.
- T. He and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202303086 CrossRef CAS.
- Q. Guan, L.-L. Zhou and Y.-B. Dong, J. Am. Chem. Soc., 2023, 145, 1475–1496 CrossRef.
- D. W. Burke, Z. Jiang, A. G. Livingston and W. R. Dichtel, Adv. Mater., 2024, 36, 2300525 CrossRef CAS.
- F. Hu, Z. Hu, Y. Liu, K. C. Tam, R. Liang, Q. Xie, Z. Fan, C. Pan, J. Tang, G. Yu and W. Zhang, J. Am. Chem. Soc., 2023, 145, 27718–27727 CrossRef CAS PubMed.
- L. Liu, Y. Gong, Y. Tong, H. Tian, X. Wang, Y. Hu, S. Huang, W. Huang, S. Sharma, J. Cui, Y. Jin, W. Gong and W. Zhang, CCS Chem., 2024, 6, 1255–1263 CrossRef.
- J. Cheng, Y. Wu, W. Zhang, J. Zhang, L. Wang, M. Zhou, F. Fan, X. Wu and H. Xu, Adv. Mater., 2024, 36, 2305313 CrossRef CAS PubMed.
- L. Qin, C. Ma, J. Zhang and T. Zhou, Adv. Funct. Mater., 2024, 34, 2401562 CrossRef CAS.
- Q. Zheng, A. Ren, A. Zagalskaya, H. Mao, D. Lee, C. Yang, K. C. Bustillo, L. F. Wan, T. A. Pham, J. A. Reimer, J. Zhang, Y. Liu and H. Zheng, J. Am. Chem. Soc., 2024, 146, 34167–34175 CrossRef CAS PubMed.
- F. Jin, H. L. Nguyen, Z. Zhong, X. Han, C. Zhu, X. Pei, Y. Ma and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144, 1539–1544 CrossRef CAS PubMed.
- X. Wang, T. Fellowes, M. Bahri, H. Qu, B. Li, H. Niu, N. D. Browning, W. Zhang, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2024, 146, 14128–14135 CrossRef CAS PubMed.
- L. Deng, W. Chen, G. Zhou, Y. Liu, L. Liu, Y. Han, Z. Huang and D. Jiang, J. Am. Chem. Soc., 2024, 146, 35427–35437 CrossRef PubMed.
- S. Ge, K. Wei, W. Peng, R. Huang, E. Akinlabi, H. Xia, M. W. Shahzad, X. Zhang, B. B. Xu and J. Jiang, Chem. Soc. Rev., 2024, 53, 11259–11302 RSC.
- C. Sun, D. Sheng, B. Wang and X. Feng, Angew. Chem., Int. Ed., 2023, 62, e202303378 CrossRef CAS PubMed.
- C. Wang, Z. Lv, W. Yang, X. Feng and B. Wang, Chem. Soc. Rev., 2023, 52, 1382–1427 RSC.
- Z. Xiong, L. Gu, Y. Liu, H. Wang, L. Shi, X. Wu, L. Liu and Z. Chen, CCS Chem., 2024, 6, 2835–2844 CrossRef CAS.
- H. Zhang, Z. Lin, P. Kidkhunthod and J. Guo, Angew. Chem., Int. Ed., 2023, 62, e202217527 CrossRef CAS PubMed.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- Z. Chen, K. Wang, Y. Tang, L. Li, X. Hu, M. Han, Z. Guo, H. Zhan and B. Chen, Angew. Chem., Int. Ed., 2023, 62, e202213268 CrossRef CAS.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed.
- F. Chen, H. Zheng, Y. Yusran, H. Li, S. Qiu and Q. Fang, Chem. Soc. Rev., 2025, 54, 484–514 RSC.
- M. Yuan, F. Ma, X. Dai, L. Chen, F. Zhai, L. He, M. Zhang, J. Chen, J. Shu, X. Wang, X. Wang, Y. Zhang, X. Fu, Z. Li, C. Guo, L. Chen, Z. Chai and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 21250–21255 CrossRef CAS PubMed.
- Y. Wang, J. Wang, J. Peng, Y. Jiang, Y. Zhu and Y. Yang, ACS Nano, 2024, 18, 23958–23967 CrossRef CAS.
- F. Xu, S. Jin, H. Zhong, D. Wu, X. Yang, X. Chen, H. Wei, R. Fu and D. Jiang, Sci. Rep., 2015, 5, 8225 CrossRef CAS.
- B. Sun, Z. Sun, Y. Yang, X. L. Huang, S. C. Jun, C. Zhao, J. Xue, S. Liu, H. K. Liu and S. X. Dou, ACS Nano, 2024, 18, 28–66 CrossRef CAS.
- L. Cheng, X. Yan, J. Yu, X. Zhang, H.-G. Wang, F. Cui and Y. Wang, Adv. Mater., 2025, 37, 2411625 CrossRef CAS.
- X.-X. Luo, W.-H. Li, H.-J. Liang, H.-X. Zhang, K.-D. Du, X.-T. Wang, X.-F. Liu, J.-P. Zhang and X.-L. Wu, Angew. Chem., Int. Ed., 2022, 61, e202117661 CrossRef CAS PubMed.
- J. Lee, H. Lim, J. Park, M.-S. Kim, J.-W. Jung, J. Kim and I.-D. Kim, Adv. Energy Mater., 2023, 13, 2300442 CrossRef CAS.
- H. Li, M. Cao, Z. Fu, Q. Ma, L. Zhang, R. Wang, F. Liang, T. Zhou and C. Zhang, Chem. Sci., 2024, 15, 4341–4348 RSC.
- S. Zheng, D. Shi, D. Yan, Q. Wang, T. Sun, T. Ma, L. Li, D. He, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202117511 CrossRef CAS PubMed.
- D. Ma, H. Zhao, F. Cao, H. Zhao, J. Li, L. Wang and K. Liu, Chem. Sci., 2022, 13, 2385–2390 RSC.
- D. Luo, J. Zhang, H. Zhao, H. Xu, X. Dong, L. Wu, B. Ding, H. Dou and X. Zhang, Chem. Commun., 2023, 59, 6853–6856 RSC.
- W. Wang, V. S. Kale, Z. Cao, S. Kandambeth, W. Zhang, J. Ming, P. T. Parvatkar, E. A. Hamad, O. Shekhah, L. Cavallo, M. Eddaoudi and H. N. Alshareef, ACS Energy Lett., 2020, 5, 2256–2264 CrossRef CAS.
- J. Wang, X. Zhang, Z. Liu, J. Yu, H.-G. Wang, X.-L. Wu, F. Cui and G. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202401559 CrossRef CAS PubMed.
- Z. Sun, H. Yao, J. Li, B. Liu, Z. Lin, M. Shu, H. Liu, S. Zhu and S. Guan, ACS Appl. Mater. Interfaces, 2023, 15, 42603–42610 CrossRef CAS.
- Z. Tian, V. S. Kale, Z. Shi, J. Yin, S. Kandambeth, Y. Wang, A.-H. Emwas, Y. Lei, X. Guo, J. Ming, W. Wang, N. Alsadun, O. Shekhah, M. Eddaoudi and H. N. Alshareef, ACS Nano, 2023, 17, 13961–13973 CrossRef CAS.
- J. L. Segura, R. Juárez, M. Ramos and C. Seoane, Chem. Soc. Rev., 2015, 44, 6850–6885 RSC.
- S. Kandambeth, J. Jia, H. Wu, V. S. Kale, P. T. Parvatkar, J. Czaban-Jóźwiak, S. Zhou, X. Xu, Z. O. Ameur, E. Abou-Hamad, A.-H. Emwas, O. Shekhah, H. N. Alshareef and M. Eddaoudi, Adv. Energy Mater., 2020, 10, 2001673 CrossRef CAS.
- J. Mahmood, S.-J. Kim, H.-J. Noh, S.-M. Jung, I. Ahmad, F. Li, J.-M. Seo and J.-B. Baek, Angew. Chem., Int. Ed., 2018, 57, 3415–3420 CrossRef CAS.
- S.-Q. Xu, T.-G. Zhan, Q. Wen, Z.-F. Pang and X. Zhao, ACS Macro Lett., 2016, 5, 99–102 CrossRef CAS.
- X. Liu, Y. Jin, H. Wang, X. Yang, P. Zhang, K. Wang and J. Jiang, Adv. Mater., 2022, 34, 2203605 CrossRef CAS.
- B. Yao, G. Li, X. Wu, H. Sun, X. Liu, F. Li and T. Guo, Chem. Commun., 2024, 60, 793–803 RSC.
- L. Xu, Y. Liu, Y. Li, X. Xuan, X. Xu, Z. Gong and L. Pan, J. Mater. Chem. A, 2024, 12, 29814–29825 RSC.
- N. Tahir, G. Wang, I. Onyshchenko, N. D. Geyter, K. Leus, R. Morent and P. V. D. Voort, J. Catal., 2019, 375, 242–248 CrossRef CAS.
- J. Mahmood, E. K. Lee, M. Jung, D. Shin, I.-Y. Jeon, S.-M. Jung, H.-J. Choi, J.-M. Seo, S.-Y. Bae, S.-D. Sohn, N. Park, J. H. Oh, H.-J. Shin and J.-B. Baek, Nat. Commun., 2015, 6, 6486 CrossRef CAS PubMed.
- Z. Meng, A. Aykanat and K. A. Mirica, Chem. Mater., 2019, 31, 819–825 CrossRef CAS.
- Q. Peng, H. Dong, H. Yan, Y. Xiao, Y. Wang, S. Chou and S. Chen, Energy Storage Mater., 2024, 73, 103849 CrossRef.
- X. Li, H. Wang, H. Chen, Q. Zheng, Q. Zhang, H. Mao, Y. Liu, S. Cai, B. Sun, C. Dun, M. P. Gordon, H. Zheng, J. A. Reimer, J. J. Urban, J. Ciston, T. Tan, E. M. Chan, J. Zhang and Y. Liu, Chem, 2020, 6, 933–944 CAS.
- H. Huang, Y. Zhao, Y. Bai, F. Li, Y. Zhang and Y. Chen, Adv. Sci., 2020, 7, 2000012 CrossRef CAS.
- M. Shi, R. Wang, J. He, L. Zhao, K. Dai and C. Yan, Chem. Eng. J., 2022, 450, 138238 CrossRef CAS.
- D. Zhao, Z. Li, D. Xu and Z. Yang, Adv. Funct. Mater., 2024, 34, 2316182 CrossRef CAS.
- L. M. Klivansky, D. Hanifi, G. Koshkakaryan, D. R. Holycross, E. K. Gorski, Q. Wu, M. Chai and Y. Liu, Chem. Sci., 2012, 3, 2009 RSC.
- S.-Q. Xu, R.-R. Liang, T.-G. Zhan, Q.-Y. Qi and X. Zhao, Chem. Commun., 2017, 53, 2431–2434 RSC.
- S. Xu, G. Wang, B. P. Biswal, M. Addicoat, S. Paasch, W. Sheng, X. Zhuang, E. Brunner, T. Heine, R. Berger and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 849–853 CrossRef CAS.
- S. Xu, H. Sun, M. Addicoat, B. P. Biswal, F. He, S. W. Park, S. Paasch, T. Zhang, W. Sheng, E. Brunner, Y. Hou, M. Richter and X. Feng, Adv. Mater., 2021, 33, 2006274 CrossRef CAS.
- R. Iqbal, M. K. Majeed, A. Hussain, A. Ahmad, M. Ahmad, B. Jabar, A. R. Akbar, S. Ali, S. Rauf and A. Saleem, Mater. Chem. Front., 2023, 7, 2464–2474 RSC.
- S. Xu, Z. Liao, A. Dianat, S.-W. Park, M. A. Addicoat, Y. Fu, D. L. Pastoetter, F. G. Fabozzi, Y. Liu, G. Cuniberti, M. Richter, S. Hecht and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202202492 CrossRef CAS PubMed.
- S. Ma, T. Deng, Z. Li, Z. Zhang, J. Jia, Q. Li, G. Wu, H. Xia, S.-W. Yang and X. Liu, Angew. Chem., Int. Ed., 2022, 61, e202208919 CrossRef CAS.
- D. L. Pastoetter, S. Xu, M. Borrelli, M. Addicoat, B. P. Biswal, S. Paasch, A. Dianat, H. Thomas, R. Berger, S. Reineke, E. Brunner, G. Cuniberti, M. Richter and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 23620–23625 CrossRef CAS PubMed.
- B. Zhang, M. Wei, H. Mao, X. Pei, S. A. Alshmimri, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 12715–12719 CrossRef CAS.
- X. Guan, H. Li, Y. Ma, M. Xue, Q. Fang, V. Valtchev and S. Qiu, Nat. Chem., 2019, 11, 587–594 CrossRef CAS.
- Y. Yusran, Q. Fang and S. Qi, Isr. J. Chem., 2018, 58, 971–984 CrossRef CAS.
- R. Xiao, J. M. Tobin, M. Zha, Y.-L. Hou, J. He, F. Vilela and Z. Xu, J. Mater. Chem. A, 2017, 5, 20180–20187 RSC.
- S.-W. Kim, H. Jung, M. S. Okyay, H.-J. Noh, S. Chung, Y. H. Kim, J.-P. Jeon, B. M. Wong, K. Cho, J.-M. Seo, J.-W. Yoo and J.-B. Baek, Angew. Chem., Int. Ed., 2023, 62, e202310560 CrossRef CAS.
- S. Li, Y. Liu, L. Dai, S. Li, B. Wang, J. Xie and P. Li, Energy Storage Mater., 2022, 48, 439–446 CrossRef.
- G. Zhao, H. Li, Z. Gao, L. Xu, Z. Mei, S. Cai, T. Liu, X. Yang, H. Guo and X. Sun, Adv. Funct. Mater., 2021, 31, 2101019 CrossRef CAS.
- H. Duan, K. Li, M. Xie, J.-M. Chen, H.-G. Zhou, X. Wu, G.-H. Ning, A. I. Cooper and D. Li, J. Am. Chem. Soc., 2021, 143, 19446–19453 CrossRef CAS PubMed.
- X. Wu, S. Zhang, X. Xu, F. Wen, H. Wang, H. Chen, X. Fan and N. Huang, Angew. Chem., Int. Ed., 2024, 63, e202319355 CrossRef CAS PubMed.
- X. Yang, L. Gong, X. Liu, P. Zhang, B. Li, D. Qi, K. Wang, F. He and J. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202207043 CrossRef CAS.
- X. Yang, L. Gong, Y. Jin, T. Zheng, X. Wang, Q. Zhi, B. Yu, K. Wang and J. Jiang, CCS Chem., 2025 DOI:10.31635/ccschem.024.202405020.
- M. K. Shehab and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2024, 16, 14750–14758 CrossRef CAS.
- Y. Lin, H. Cui, C. Liu, R. Li, S. Wang, G. Qu, Z. Wei, Y. Yang, Y. Wang, Z. Tang, H. Li, H. Zhang, C. Zhi and H. Lv, Angew. Chem., Int. Ed., 2023, 62, e202218745 CrossRef CAS.
- C. Wang, R. Li, Y. Zhu, Y. Wang, Y. Lin, L. Zhong, H. Chen, Z. Tang, H. Li, F. Liu, C. Zhi and H. Lv, Adv. Energy Mater., 2024, 14, 2302495 CrossRef CAS.
- D. Geng, H. Zhang, Z. Fu, Z. Liu, J. Yang and C. Yan, Energy Storage Mater., 2025, 75, 103996 CrossRef.
- G. Wang, N. Tahir, I. Onyshchenko, N. D. Geyter, R. Morent, K. Leus and P. V. D. Voort, Microporous Mesoporous Mater., 2019, 290, 109650 CrossRef CAS.
- X. Chen, D. Liu, C. Yang, L. Shi and F. Li, Inorg. Chem., 2023, 62, 9360–9368 CrossRef CAS.
- Z. He, T. Luan, S. Zhang, Q. Wei, D. Huang, L. Wang, Y. Wang, P. Li and W. W. Yu, Adv. Mater., 2024, 36, 2410363 CrossRef CAS PubMed.
- C. Peng, G.-H. Ning, J. Su, G. Zhong, W. Tang, B. Tian, C. Su, D. Yu, L. Zu, J. Yang, M.-F. Ng, Y.-S. Hu, Y. Yang, M. Armand and K. P. Loh, Nat. Energy, 2017, 2, 17074 CrossRef CAS.
- Q. Bai, J. Huang, K. Tang, Y. Zhu and D. Wu, Adv. Mater., 2025, 37, 2416661 CrossRef CAS PubMed.
- Z. Sun, J.-M. Seo, H. Liu, Y. Wei, Y. Zhang, Z. Li, H. Yao, S. Guan and J.-B. Baek, Nano Energy, 2024, 129, 110073 CrossRef CAS.
- M. Wu, Y. Zhao, B. Sun, Z. Sun, C. Li, Y. Han, L. Xu, Z. Ge, Y. Ren, M. Zhang, Q. Zhang, Y. Lu, W. Wang, Y. Ma and Y. Chen, Nano Energy, 2020, 70, 104498 CrossRef CAS.
- J. Chu, L. Cheng, L. Chen, H.-G. Wang, F. Cui and G. Zhu, Chem. Eng. J., 2023, 451, 139016 CrossRef CAS.
- R. Shi, L. Liu, Y. Lu, C. Wang, Y. Li, L. Li, Z. Yan and J. Chen, Nat. Commun., 2020, 11, 178 CrossRef CAS.
- X. Yang, L. Gong, Z. Liu, Q. Zhi, B. Yu, X. Chen, K. Wang, X. Li, D. Qi and J. Jiang, Sci. China: Chem., 2024, 67, 1300–1310 CrossRef CAS.
- X.-L. Chen, M. Xie, Z.-L. Zheng, X. Luo, H. Jin, Y.-F. Chen, G.-Z. Yang, D.-S. Bin and D. Li, J. Am. Chem. Soc., 2023, 145, 5105–5113 CrossRef CAS.
- W. Wang, V. S. Kale, Z. Cao, Y. Lei, S. Kandambeth, G. Zou, Y. Zhu, E. Abouhamad, O. Shekhah, L. Cavallo, M. Eddaoudi and H. N. Alshareef, Adv. Mater., 2021, 33, 2103617 CrossRef CAS.
- L. Zhong, C. Liu, Y. Zhang, J. Li, F. Yang, Z. Zhang and D. Yu, Angew. Chem., Int. Ed., 2025, 64, e202413971 CrossRef CAS PubMed.
- P. Yi, Z. Li, L. Ma, B. Feng, Z. Liu, Y. Liu, W. Lu, S. Cao, H. Fang, M. Ye and J. Shen, Adv. Mater., 2024, 36, 2414379 CrossRef CAS PubMed.
- J. Xie, Z. Wang, Z. J. Xu and Q. Zhang, Adv. Energy Mater., 2018, 8, 1703509 CrossRef.
- P. Xu, F. Gao and D. Liu, Adv. Mater. Interfaces, 2023, 10, 2300464 CrossRef CAS.
- M. Mao, C. Luo, T. P. Pollard, S. Hou, T. Gao, X. Fan, C. Cui, J. Yue, Y. Tong, G. Yang, T. Deng, M. Zhang, J. Ma, L. Suo, O. Borodin and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 17820–17826 CrossRef CAS PubMed.
- J. Park, M. Lee, D. Feng, Z. Huang, A. C. Hinckley, A. Yakovenko, X. Zou, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2018, 140, 10315–10323 CrossRef CAS.
- Y. Hu, W. Tang, Q. Yu, X. Wang, W. Liu, J. Hu and C. Fan, Adv. Funct. Mater., 2020, 30, 2000675 CrossRef CAS.
- J. Zoua, K. Fana, X. Wang, Y. Chen, Y. Cao, H. Dai, C. Zhang, M. Fu, Y. Gao, H. Liu and C. Wang, Chem. Eng. J., 2023, 460, 141703 CrossRef.
- S. Zhang, Y.-L. Zhu, S. Ren, C. Li, X.-B. Chen, Z. Li, Y. Han, Z. Shi and S. Feng, J. Am. Chem. Soc., 2023, 145, 17309–17320 CrossRef CAS.
- J. Wei, P. Zhang, J. Sun, Y. Liu, F. Li, H. Xu, R. Ye, Z. Tie, L. Sun and Z. Jin, Chem. Soc. Rev., 2024, 53, 10335–10369 RSC.
- S. Li, J. Shang, M. Li, M. Xu, F. Zeng, H. Yin, Y. Tang, C. Han and H.-M. Cheng, Adv. Mater., 2023, 35, 2207115 CrossRef CAS PubMed.
- Y. Chen, J. Li, Q. Zhu, K. Fan, Y. Cao, G. Zhang, C. Zhang, Y. Gao, J. Zou, T. Zhai and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202116289 CrossRef CAS PubMed.
- J. Ning, X. Zhang, D. Xie, Q. He, J. Hu, J. Tang, R. Li, H. Meng and K. X. Yao, Angew. Chem., Int. Ed., 2024, 63, e202319796 CrossRef CAS PubMed.
- J. Wang, Z. Liu, H.-G. Wang, F. Cui and G. Zhu, Chem. Eng. J., 2022, 450, 138051 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |