
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonitoring chemical processes in redox flow batteries employing in situ and in operando analyses†
Ahmad
Alem
a,
Pooria
Poormehrabi
b,
Jonas
Lins
c,
Lukas
Pachernegg-Mair
 de,
Christine
Bandl
a,
Virginia
Ruiz
fg,
Edgar
Ventosa
fg,
Stefan
Spirk
*de and
Torsten
Gutmann
*c
de,
Christine
Bandl
a,
Virginia
Ruiz
fg,
Edgar
Ventosa
fg,
Stefan
Spirk
*de and
Torsten
Gutmann
*c
aInstitute of Chemistry of Polymeric Materials, Technical University of Leoben, Otto Glöckel-Strasse 2, A-8700 Leoben, Austria
bFlow-Nano S.r.l, Milano, Italy
cInstitute of Inorganic and Physical Chemistry, Technical University of Darmstadt, Peter-Grünberg-Straße 8, D-64287 Darmstadt, Germany. E-mail: gutmann@chemie.tu-darmstadt.de
dInstitute of Bioproducts and Paper Technology (BPTI), Graz University of Technology, Inffeldgasse 23, A-8010 Graz, Austria. E-mail: stefan.spirk@tugraz.at
eEcolyte GmbH Puchstraße 17, A-8020 Graz, Austria
fInternational Research Centre in Critical Raw Materials-ICCRAM, University of Burgos, Plaza Misael Bañuelos s/n, E-09001 Burgos, Spain
gDepartment of Chemistry, University of Burgos, Plaza Misael Bañuelos s/n, E-09001 Burgos, Spain
First published on 7th July 2025
Abstract
Redox flow batteries (RFBs) are promising solutions for large-scale stationary energy storage due to their scalability and long cycle life. The efficient operation of RFBs requires a thorough understanding of the complex electrochemical processes occurring during charging and discharging. This review provides an overview and perspective of in situ and in operando analytical techniques to monitor RFBs. In more detail, these advanced techniques allow for real-time observation of redox reactions, ion transport, and electrode–electrolyte interactions under working conditions, offering insights into formation of intermediate species and mechanisms of electrolyte degradation, State-of-Charge (SoC), and ion crossover. By discussing the principles, capabilities, and limitations of techniques such as nuclear magnetic resonance (NMR), electron paramagnetic resonance (EPR), ultraviolet-visible (UV-vis) spectroscopy, Raman spectroscopy, Fourier transform infrared spectroscopy (FTIR), X-ray absorption spectroscopy (XAS), electrochemical impedance spectroscopy (EIS), tomography and radiography, mass spectrometry (MS), and fluorescence microscopy this review highlights the essential role of in situ and in operando approaches in advancing RFB technology.
Broader contextThe review addresses one of the most critical issues in the development of redox flow batteries (RFBs), namely the need to understand the complex processes occurring in the solution, at the interface, and within the bulk of flow battery components. Its goal is to identify and summarize the ongoing challenges in the field that hinder the widespread adoption of RFBs in large-scale commercial energy storage applications. The review primarily focuses on in operando and in situ studies of RFBs, placing them in a broader context that includes other battery systems, such as lithium batteries and beyond. To the best of our knowledge, there are currently no such comprehensive overviews available on in situ and in operando techniques. Therefore, we believe that our review makes a significant contribution to the development of methodologies for in situ and in operando approaches in electrochemical storage and conversion systems, extending even to areas not explicitly covered in this work such as fuel cell technology. |
1. Introduction
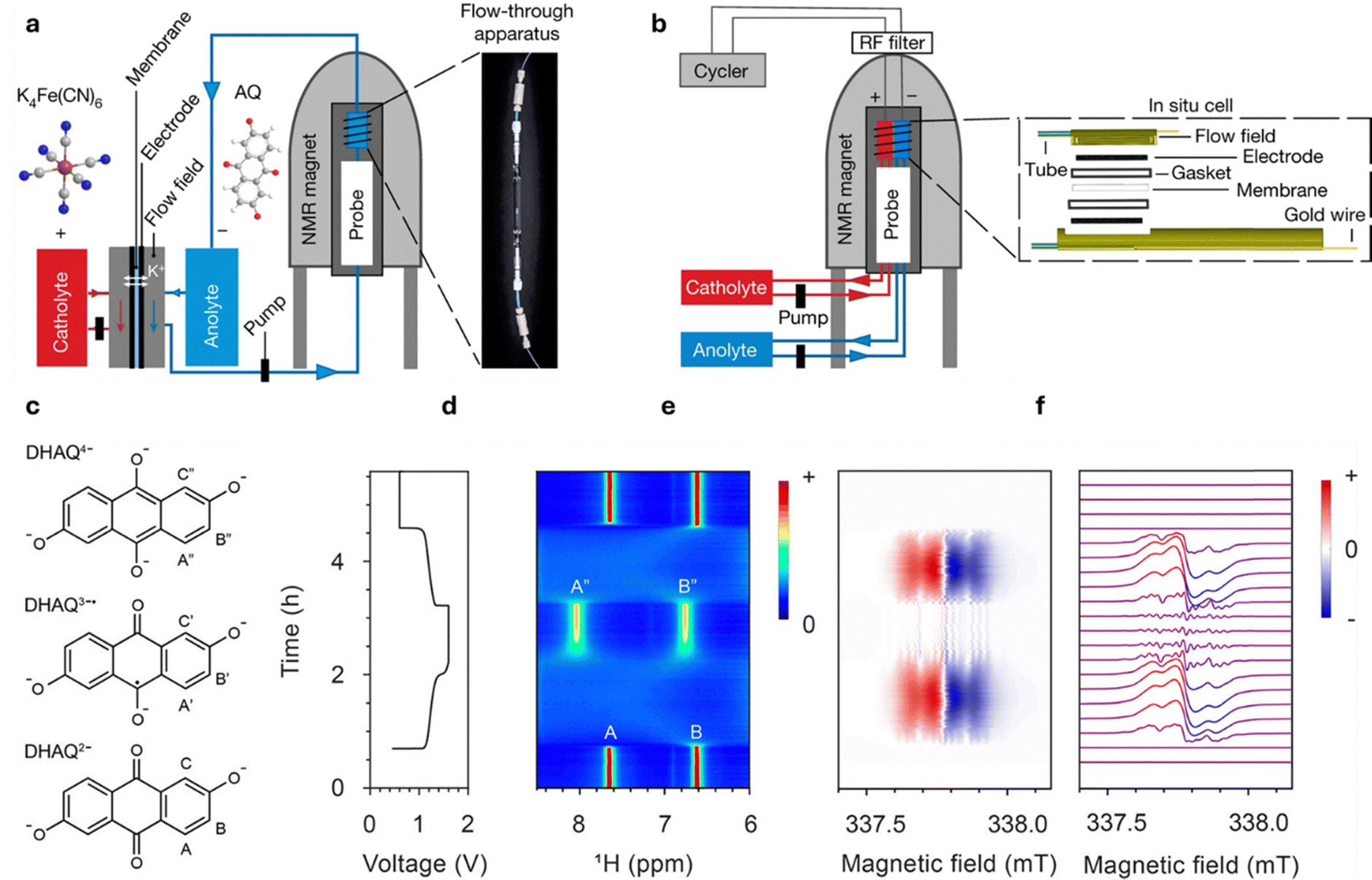
RFBs represent an energy storage technology that utilizes liquid electrolytes in large tanks to store electricity generated from intermittent renewable energy sources like solar, wind, and tidal power as well as providing backup energy for industrial applications.1–3 In contrast to traditional lithium-ion batteries (LIBs), storage capacity and power output are independent in RFBs, making them ideal for medium to large-scale stationary energy storage applications, which demand scalability and flexibility.4,5 Moreover, RFBs are supposed to have a longer lifespan than traditional batteries and feature long discharge times (12 hours and more), making them suitable for long-duration energy storage applications in microgrids and large-scale energy storage.6Fig. 1 (middle) illustrates the working principle of RFBs, in which dissolved electro-active materials stored in external electrolyte reservoirs, referred to as catholyte (or posolyte) on the positive side and anolyte (or negolyte) on the negative side. During charge and discharge the liquid electrolyte is circulated into the electrochemical cell compartment, where redox reactions take place at the electrode surfaces, allowing for the direct conversion of chemical and electrical energy.7 General cell charging/discharging reactions consisting of two half-cell reactions can be written as follows: | ||
| Fig. 1 Overview on challenges in progressing flow battery technology, which can be tackled by in operando monitoring. | ||
Cathode:
 | (1) |
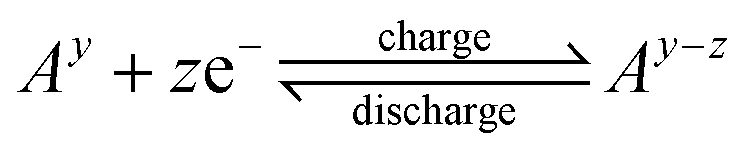 | (2) |
 | (3) |
1.1. State of the art of in situ and in operando analyses of batteries
A variety of advanced diagnostic tools have been developed and extensively used for LIBs, enhancing our understanding of their behavior.11–13 In general, one can distinguish between five types of in situ and in operando analytics, namely electrochemical techniques (cyclic voltammetry (CV), EIS), spectroscopy (e.g. Raman, UV-vis, NMR and EPR), scattering techniques (e.g. X-ray scattering, neutron scattering) as well as imaging and microscopic approaches (e.g. electron microscopy, computer tomography, radiography).14 As an example, imaging techniques for observing reaction front movement and reaction kinetics are increasingly applied, with in situ high-resolution transmission electron microscopy providing deep insights into interfacial processes.15 Recent advancements in synchrotron methodologies have facilitated a variety of spectroscopic techniques, two-dimensional transmission X-ray microscopy, three-dimensional tomographic reconstructions, and diffraction-based imaging methods.16,17 EPR imaging has been employed to monitor lithiation front displacement within paramagnetic electrodes during battery cycling, aiding in a better understanding of electrode behavior.18 Furthermore, in operando spectroscopy techniques such as Raman spectroscopy, UV-vis spectroscopy, FTIR spectroscopy, and MS have been utilized to study processes and phenomena occurring in electrolytes and electrodes.19–22 Finally, also techniques such as XPS, neutron diffraction, NMR, optical microscopy, scanning electron microscopy, atomic force microscopy, XAS, and magnetic resonance imaging (MRI) have been employed.23–29 While these techniques were mostly developed for LIBs, they are now being adapted to study RFBs,30–34 which face similar challenges related to electrochemical processes and degradation.1.2. Problem statement, motivation and scope
All-vanadium redox flow batteries (VRFBs) are the most developed RFBs and currently used in large scale energy storage.33,35 There are some detailed investigations concerning the effects of temperature on the stability of vanadium electrolytes, particularly on the formation of insoluble products such as VSO4, V2(SO4)3·xH2SO4·yH2O, VOSO4 and V2O5.36,37 However, there is still a lack of data that address processes at the interfaces and of suitable models describing the degradation of electrodes, electrolytes and capacity fade due to factors such as unwanted side reactions and pH change, especially at high current densities and high states of charge, as well as the crossover of active species through the membrane, leading to reactions with species on the opposite side.46 As crossover is dependent on the operation of the RFB e.g., current density, and other factors such as the state of charge (SoC), it is important to monitor and optimize the operation to minimize crossover.38 However, getting experimental data is non-trivial and requires in situ and in operando measurements as well as deep understanding of the chemical reactions and side-reactions.Information considering the whole RFB system is even more scarce when it comes to less investigated complex chemistries such as in Fe, Mn, Cu, and organic RFBs.47–49 For organic RFBs, the challenges are even harder as the involved chemistries and reactions vary to a large extent, which are additionally influenced by complex inter-species reactions triggered by crossover. However, the main decomposition mechanisms involve geminal diol formation & subsequent Michael addition reactions, nucleophilic substitutions, hydrolysis, disproportionation, dimerization, and tautomerization (Fig. 2), which have been extensively reviewed in the literature.39
 | ||
| Fig. 2 Decomposition mechanisms of main organic redox active materials.39 Reproduced with permission. Copyright 2020, ACS Publication. | ||
The sheer diversity of reactions leads to a larger need for understanding on the one hand the complex chemistries itself but also how they are affected by the components (felts, membranes, electrode) and the variation in operation conditions (e.g. current density, state of charge). As organic flow batteries have attracted a significant amount of interest in the past years, we summarize the main chemistries in Section 1.2.1.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds including benzene, naphthalene, anthracene, phenanthrene, and tetracene-based quinone-hydroquinone couples.41 Nucleophilic reactions are also responsible for the accelerated degradation of N-substituted viologen derivatives in the presence of traces of oxygen in the anolyte. Nucleophilic attack by hydroxide anions generated by the oxygen reduction leads to cleavage of the N-substituent and dealkylation of the viologen derivative.42 Dealkylation through a nucleophilic attack is the main degradation pathway of viologen derivatives in the presence of oxygen, leading to a significant decrease in the capacity of the RFB. Thus, it has been demonstrated that stability can be improved in molecularly engineered viologen derivatives by hindering nucleophilic attack.43
000 compounds including benzene, naphthalene, anthracene, phenanthrene, and tetracene-based quinone-hydroquinone couples.41 Nucleophilic reactions are also responsible for the accelerated degradation of N-substituted viologen derivatives in the presence of traces of oxygen in the anolyte. Nucleophilic attack by hydroxide anions generated by the oxygen reduction leads to cleavage of the N-substituent and dealkylation of the viologen derivative.42 Dealkylation through a nucleophilic attack is the main degradation pathway of viologen derivatives in the presence of oxygen, leading to a significant decrease in the capacity of the RFB. Thus, it has been demonstrated that stability can be improved in molecularly engineered viologen derivatives by hindering nucleophilic attack.43
Nucleophilic reactions also occur in high-potential substrates beyond quinones, including nitroxide radicals and organometallic complexes such as ferrocene (Fc) and ferrocyanide. Nitroxide radicals, for instance, possess reduction potentials around 0.8 V vs. standard hydrogen electrode (SHE), while ferrocene and ferrocyanide have reduction potentials ranging from approximately 0.4 to 0.6 V. A specific example involves the electrochemical oxidation of 4-hydroxy-(2,2,6,6-tetramethylpiperidin-1-yl)oxyl (4-HO-TEMPO), which produces 4-HO-TEMPO+. In alkaline electrolyte, this species is highly susceptible to nucleophilic addition by hydroxide ions (OH−), which leads to progressive acidification of the electrolyte. Under acidic conditions, 4-HO-TEMPO undergoes disproportionation to form 4-HO-TEMPO+ and 1,4-dihydroxy-2,2,6,6-tetramethylpiperidine, with the latter being redox-inactive.44
Furthermore, the electrochemical oxidation of ferrocene and ferrocyanide yields Fc+ and ferricyanide, respectively, both of which are also vulnerable to nucleophilic attack by OH−.45 For Fc+, nucleophilic substitution can still occur despite the presence of protective alkyl substituents, especially when such groups are in proximity to the Fc redox center. An example of this is seen with (ferrocenylmethyl)-trimethylammonium salts, where nucleophilic substitution of the quaternary ammonium group results in the loss of trimethylamine as a leaving group.44
Disproportionation of certain redox couples can trigger chemical decomposition in redox flow batteries (RFBs). This process occurs when two identical species with the same oxidation state undergo a redox reaction, resulting in one being reduced and the other oxidized, producing two products with different oxidation states. Capacity fade in RFBs can arise if either product is redox-inactive or has low solubility, leading to precipitation. Additionally, if one product has a significant different redox potential or similar potential but slower kinetics, this may also contribute to capacity fading during specific cycling conditions. Quinones are especially prone to disproportionation; for instance, anthrahydroquinone has been shown to disproportionate to anthrone and anthraquinone during the electrochemical reduction of unsubstituted anthraquinone in highly acidic media. The reduced form of viologen derivatives (the radical cation Vi+˙) can also undergo disproportionation (2Vi+˙→ Vi2+ + Vi0) yielding inactive neutral species that lead to capacity fading. Moreover, the radical cation of viologens can also dimerize to form inactive π-dimers, which reduces the effective concentration of active species.46 Dimerization can be prevented by structural modification of viologens with charged and bulky substituents to induce electrostatic repulsion and steric interactions. Tautomerization involves the formal migration of a hydrogen atom alongside the exchange between a single bond and an adjacent double bond, all while maintaining the same carbon skeleton. Tautomers typically exhibit different thermochemical properties compared to their original molecules. If these tautomers possess significantly higher reduction potentials or lower solubilities, their formation during electrochemical cycling can lead to capacity fading and a decrease in cell energy density. Aza-substituted conjugated π-systems, such as those based on quinones, viologens, and phenazines, are particularly prone to tautomerization during electrochemical cycling.47 This susceptibility arises because the redox reactions of these organic molecules involve the reorganization of conjugated double bonds, where a tautomer may act as a metastable intermediate. In contrast, simpler aromatic molecules, like phenols, primarily exist in their enolic form, as the energy gained from rearranging to the tautomeric keto structure is outweighed by a significant reduction in resonance energy. Ketonic tautomeric structures become energetically favorable in compounds such as naphthoquinones, polyhydric phenols, and hydroxyl derivatives of polycyclic aromatic hydrocarbons. Understanding the susceptibility of different classes of aqueous organic species to these decomposition mechanisms is essential for developing molecular engineering strategies aimed at enhancing the stability of RFB reactants. It is also crucial to recognize that decay rates can increase with reactant concentration for certain chemistries, particularly those that are vulnerable to bimolecular decomposition pathways like disproportionation and dimerization. Since practical RFBs ideally operate at high reactant concentrations to maximize energy densities, it is vital to comprehensively understand the trade-off between energy density and stability.
However, there are still many unresolved mechanistic questions on the behavior of even popular redox active materials such as AQDS. In particular, there is some uncertainty regarding whether AQDS can fully access its theoretical two-electron Coulombic capacity.48 Based on nuclear magnetic resonance (NMR), cyclic voltammetry (CV), and bulk electrolysis (BE) measurements, it has been suggested that reversible intermolecular dimerization of AQDS occurs at solution concentrations exceeding 10 mM, thereby inhibiting complete two-electron electrochemical reduction or reduction via chemical oxidants.49,50 On the basis of these works, it has been speculated that CO2 binds in a 1:1 anthraquinone–CO2 adduct within bulk AQDS powder. When dissolved in acidic electrolyte, this AQDS-CO2 adduct would undergo hydrolysis, leading to the formation of semianthraquinone, which could subsequently dimerize into semianthraquinone–anthraquinone units, yielding an overall Coulombic capacity of 1.5 electrons per AQDS molecule. In neutral to basic pH conditions, they anticipate the formation of hydroxyanthraquinone–anthraquinone dimers, resulting in a net capacity of one electron per AQDS molecule. However, the universality of this capacity-limiting mechanism is questionable, as Chen et al.51 demonstrated that more than 95% of the theoretical capacity of AQDS was accessible in an acidic AQDS/bromine cell. This high capacity is supported by multiple reports of AQDS or anthraquinone-based chemistries at different conditions, where electrochemical access to 90% or more of the full two-electron Coulombic capacity has been observed.52–54 Several studies have investigated the use of AQDS and its derivatives in full cells to analyze the current–voltage relationships in RFBs, partition voltage losses within the full cell stack, and assess the feasibility of integrating RFBs with solar cell charging systems. However, aside from contributions from bromine crossover, the origins of capacity fade in AQDS/bromine cells remain inadequately explored. Hypotheses regarding this issue include the hydroxylation and disproportionation of the reduced anthrahydroquinone into anthraquinone and redox-inactive anthrone. Another illustrative example involves TEMPO related chemistry. As mentioned above, TEMPO and its derivatives can undergo a wide range of reactions by reaction with e.g. Lewis acids to the oxonium derivatives, even without applying a potential. The decomposition of TEMPO derivatives typically involves ring opening and rearrangement reactions, which occur also simultaneously. Often the main degradation products are identified or even only proposed as shown for the example of TMA–TEMPO (TMA: trimethylammonium). This molecule can decompose by a variety of pathways, including ring opening and rearrangements, finally leading to the formation of CO2 (Fig. 3).55
 | ||
| Fig. 3 Proposed decomposition pathways of TMA–TEMPO towards carbon dioxide.56 Reproduced with permission. Copyright 2022, Elsevier Publication. | ||
Although the nitroxide can adopt many different mesomeric structures, for decomposition typically it is assumed that decomposition proceeds via the oxidized state.
| Technique | Working principle | Application | Limitations | Ref. |
|---|---|---|---|---|
| NMR spectroscopy | Detection of nuclear spins and identification of their local environment; tracking of dia-magnetic species | Monitoring the electrolyte decomposition and crossover of molecules; estimation of the SoC | Complexity of integrating the cell into the probe; no tracking of paramagnetic species feasible, limited sensitivity (typically 1017 nuclear spins are required) | 58 and 59 |
| EPR spectroscopy | Detection of unpaired electrons and their local environment; tracking of para-magnetic species | Monitoring radical species and electron transfer reactions; complementary technique to NMR | Complexity of integrating the cell into the probe; only tracking of paramagnetic species feasible | 60 and 61 |
| UV-vis spectroscopy | Measuring absorbance of UV-vis light absorbing active species; tracking changes in oxidation states during charge and discharge cycles | Real-time, non-invasive monitoring of SoC and total redox-active species concentration in half-cells; monitoring of redox stability in flow and static cells | Challenges for highly concentrated systems and light absorbing solvents; calibration complexity for industrial use | 62 and 63 |
| Raman spectroscopy | Probing bond-vibrations by Raman scattering | Monitoring of electrolyte composition/electrode surface/catalytic surface reactions; exploring origin of capacity fading, mechanisms of RFB failure | Sample heating; background noise by fluorescence; enhancement techniques (SERS/TERS) need calibration | 64 and 65 |
| FTIR spectroscopy | Probing bond-vibrations by infrared adsorption | Exploring mechanisms of electrochemical reactions in RFB and RMFB | Limited to IR-active species; complexity of the ATR-FTIR spectroelectrochemical setup | 66 and 67 |
| XAS | Probing element specific electron transitions through X-ray absorption/scattering | Monitoring structure and oxidation states of electrolytes and electrodes at surfaces/bulk | Requires special X-ray source (i.e. synchrotron); beam strongly dampened by liquid layers; characterization of surfaces is challenging, requires advanced setups; limited to heavy atoms and thin sample layers; requires uniform, homogenous samples | 68 and 69 |
| EIS | Application of a sinusoidal signal over a wide range of frequencies; monitoring of the sinusoidal response of the system | Monitoring of RFB performance | Limited frequency range by equipment; low resolution at low frequencies | 70 and 71 |
| Tomography and radiography | X-rays or neutrons are utilized to generate high-resolution 3D images; monitoring internal structures and dynamic processes | Visualization of dendrite growth on electrode surface and quantification of dendrite characteristics; mapping concentration profiles across electrodes; studying electrodes under varying compression to optimize porosity and wetting behavior | Requiring advanced imaging setups (e.g., synchrotron CT, neutron sources); difficulty in distinguishing phases (e.g., electrolyte vs. felt) due to similar attenuation coefficients | 72 and 73 |
| MS | Measure mass-to-charge ratios of molecules | Characterize reaction products of parasitic reactions at electrodes; explore reaction products and intermediates during cycling in electrolytes | Costly and hard-to-implement online instrumentation (electrochemical MS system); custom-made sampling systems connected to the gas carrier flow; time delay between sampling and detection; compounds with identical molecular weights cannot be distinguished. | 57 and 74 |
| Fluorescence microscopy | Visualization of redox-dependent fluorescence to track redox reactions | Mapping redox-active species concentration and distribution; monitoring mass transport phenomena within porous electrodes | Requires fluorescent redox-active species; potential optical interference and fluorescence quenching by electrolyte salts. | 75 and 76 |
A challenge in the design of in operando measurement setups lies also in the design of the flow cells. In some applications, there are restrictions in size and/or how to place or connect the flow battery cells to the respective instruments. In addition, challenges in material selection may be overcome, as some of the conventional cells are made of metal which is e.g. impeding any investigation using X-rays. The time scales of the electrochemical processes and the sensitivity of the methods need to match the ability of some measurements to capture transient phenomena accurately. For imaging techniques, the required resolution must be kept in mind: While maintaining the same environmental conditions is trivial in lab scale single cells, for in operando setups, control over temperature or other parameters can be tricky. As experimental in operando setups may have some peculiarities on cell design and operating conditions (e.g. flow rate, flow channels, tubing), the results may not be always completely comparable to standard cells. In the next section, we discuss the different techniques and setups.
1.3. In situ and in operando NMR spectroscopy
NMR spectroscopy has been widely used for several decades to identify the structure of organic and inorganic molecules. The introduction of the concept of flow-NMR spectroscopy has enabled various novel applications in recent years.77–79 As for example, it has been successfully established in reaction monitoring,80–82 in quantitative analysis,83–85 in combinatorial chemistry,86 liquid chromatography,87 and in hyperpolarization (i.e. PHIP or DNP).88,89 Moreover, it has also been applied in heterogeneous (electro)catalysis research.90In the past few years, it has been established as a powerful tool for analyzing electrochemical processes in RFBs on a molecular level. It allows to identify redox-active species and molecular structures of electrolytes and provides information on their concentrations. By tracking changes in the molecules/species involved in the electrochemical processes the kinetics and mechanisms of these reactions can be understood, which is one key step to optimize the performance of RFBs in the future. The adaptation needed for the RFB systems is to attach the RFB single cell to a flow through system (see Fig. 4a, inset). A potential difference to normal cells is that the tubing diameters are smaller, which may lead to different flow profiles.
 | ||
| Fig. 4 (a) In situ and (b) in operando NMR setup designed for RFBs according to Zhao et al.59 In the on-line setup (a), the battery comprises 5.0 cm2 carbon felt electrodes, with a catholyte and an anolyte of potassium ferrocyanide(II) and AQ, respectively, dissolved in 1 M KOH in D2O. The volume of the flow path through the magnet including the sampling apparatus (7.3 cm3) and excluding the reservoir is 15.0 cm3. At a flow rate of 13.6 cm3 min−1, the time of flight of the electrolyte out of and back into the reservoir is 1.1 min. In the operando setup (b), the miniaturized flow cell (shown on the right) consists of flow fields, tubes to flow electrolyte in and out, carbon electrodes, a cation-transport membrane and current collectors. The volume of the flow path including the cell cavity (0.032 cm3) is 7.8 cm3. At a flow rate of 2.5 cm3 min−1, the time of flight of the electrolyte out of and back into the reservoir is 3.1 min. Reproduced with permission. Copyright 2020, Nature Publishing Group. RF, radiofrequency (c)–(f) In situ1H-NMR and EPR investigations performed during electrochemical cycling by the same group.61 (c) Structures of the DHAQ2−, DHAQ3−˙, and DHAQ4− anions showing the labeling of the proton. (d) Voltage of a 10 mM DHAQ versus 15 mM K4[Fe(CN)6] and 3.75 mM K3[Fe(CN)6] full cell as a function of time. During charge, a constant current of 10 mA was applied, followed by a potential hold at 1.6 V. During discharge, a constant current of −10 mA (where the sign indicates the polarity of the electrodes) was applied, followed by a potential hold at 0.6 V. (e) NMR spectra of the anolyte in the aromatic region. The color bar (right) indicates the intensity of resonances in positive arbitrary units. The acquisition time per NMR spectrum is 95 s. (f) EPR spectra of the anolyte. The stack plot on the right shows every 10th spectrum. The acquisition time per EPR spectrum is 95 s, with a scanning time of 60 s, a coupling time of 30 s (time for automatic tuning), and a delay time of 5 s. Note that a typical continuous-wave EPR spectrum is detected and displayed as the first derivative of the absorption, and hence has negative and positive values. The color bar indicates the intensity of the resonance in arbitrary units. A different color scale is applied here because of the presence of negative peak intensities. Reproduced with permission. Copyright 2021, ACS Publication. | ||
As an early example in that field, Stimming and co-workers58 studied asymmetric polyoxometalate electrolytes in RFBs, focusing on [PV14O42]9− (PV14) as catholyte. To assess the stability of this diamagnetic anion upon reduction and its role as an electron carrier, they conducted in situ51V NMR studies. During the reduction a decrease in intensity of the three signals assigned to [PV14O42]9− with time was obtained. This was interpreted as conversion of diamagnetic PV14 to paramagnetic V(IV) species, which are not visible in NMR. During oxidation, the signals were partially recovered, suggesting the formation of PV14. However, the process was not quantitative since part of the species formed during reduction have diffused from the electrode, limiting the re-oxidation process.58
Next to this study, Zhao et al.59 conducted a thorough NMR analysis to monitor the electrolyte properties in organic RFBs employing a combined in situ and in operando approach. In the in situ setup (Fig. 4a), the RFB was externally connected to an NMR probe with a flow cell, allowing high-resolution time-resolved spectra without interference from cell components. For in operando studies (Fig. 4b), a tiny flow battery system was positioned inside the NMR probe. Using this operando mode, catholyte and anolyte can be investigated simultaneously, though with lower resolution due to signal interference from cell components.
In their in situ study Zhao et al.59 used a full cell model containing 2,6-dihydroxyanthraquinone (DHAQ) as anolyte and potassium hexacyanoferrate(II) as catholyte and the 1H NMR spectra of the anolyte were monitored as function of applied voltage to quantify the SoC. Upon charging, two signals attributed to the protons close to the carbonyl redox center of DHAQ vanished instantly, while a signal attributed to the farthest proton from the redox center was broadened and shifted towards higher chemical shifts. This observation was interpreted by electron delocalization over the semiquinone radical anion, resulting in significant line broadening. By charging to higher voltages, the signal further shifted to higher ppm values and then moved back after reaching a maximum. At the cut-off voltage (1.7 V), signals were observed, assigned to DHAQ4− which is a diamagnetic product. Moreover, the in situ NMR was employed to monitor the electrolyte decomposition in the RFB. By holding the voltage at 1.7 V to ensure full reduction of DHAQ, several new peaks appeared, which were attributed to the decomposition products anthrone (DHA3−) and anthrol (DHAL3−). The quantity of these species was much smaller when the voltage was kept at 1.2 V. This clearly pointed out that the reduction potential influences the degradation of the electrolyte. In addition to electrolyte degradation, in situ NMR enabled exploring battery self-discharge in real time.59 Similarly, in another research, in situ NMR was used to track the transformation of the precursor compound 2,4,6,8-tetramethylaminemethylene-1,5-naphthalenediol (TAND) to the active redox species 2,6,8-trismethylaminemethylene-3,5-dihydroxy-1,4-naphthoquinone (TANQ) during the electrochemical process.91
In a recent work, Zhao and co-workers92 investigated DHAQ as anolyte and potassium hexacyanoferrate(II) as catholyte under in situ conditions with a low-cost benchtop NMR spectrometer (43 MHz, 1 T). Although this spectrometer has a much lower field compared to the formerly used high-field spectrometer (300 MHz, 7 T), 1H NMR enabled them to identify and quantify intermediates with sufficient sensitivity, spectral and temporal resolution. Furthermore, they demonstrated the feasibility of their benchtop approach to investigate the crossover of DHAQ and impurity molecules through the Nafion separator membrane. The crossover of molecules in RFBs may significantly impact battery performance, resulting in a potential capacity loss of up to 50% and should be thus excluded.92
Finally, significant progress in development of special probes for in situ NMR and MRI investigations of RFBs has been made by Berthault and co-workers.93 By 3D printing, they designed a mini-RFB that was connected to a high field NMR probe and allowed to perform in situ NMR and imaging studies. As a proof of concept, they tested their equipment to study in real-time the redox cycle of the disodium salt of 9,10-anthraquinone-2,7-disulfonic acid (2,7 AQDS), revealing a crucial role of dimerization in electrolytes of organic RFBs.93
1.4. NMR combined with in situ and in operando EPR spectroscopy
In some cases, redox processes in RFBs produce paramagnetic species, having unpaired electrons. When present in high concentrations, such radicals can limit the detection of NMR active species in electrolyte solutions. Therefore, it is necessary to employ a complementary analytical technique which provides time-resolved information on electron spins. In situ and in operando EPR spectroscopy is such a powerful tool to monitor paramagnetic species and has been widely used in catalysis, electrocatalysis as well as biochemical research as shown by several reviews.69–74Recently, this technique has been increasingly employed to research energy storage systems, revealing information on charge transfer and storage processes.94–96 Simultaneous electrochemical-EPR measurements provide insights into reaction intermediates, products, and reaction kinetics. For instance, Zhao et al.61 reported on the development of a coupled NMR/EPR setup which was applied to investigate DHAQ in a DHAQ/K4Fe(CN)6 full cell in a time window of 5 h at voltages of 0.6 to 1.6 V (Fig. 4d). They demonstrated that in situ EPR can be employed for quantifying the concentration of radical species formed as reaction intermediates during electrochemical cycling, while in situ NMR enabled to quantify concentration changes of diamagnetic species in parallel. As shown in Fig. 4e, when signals A and B disappear in the 1H in situ NMR spectra because of the electron delocalization and intermolecular electron transfer, the in situ EPR spectra show a resonance at 337.78 mT, corresponding to the DHAQ3−˙ radical anion (Fig. 4f). As charging progresses, the EPR signal intensifies and broadens, reaching its maximum at 50% SoC. Thus, when NMR peaks disappear and provide limited information, EPR enables analyzing the radical species and the SoC.61 In that paper, Zhao et al.97 introduced an approach based on direct magnetic susceptibility measurements that allows the online in situ determination of the SoC which they applied on the DHAQ/K4Fe(CN)6 full cell system.
In a follow-up work, Jing et al.60 applied this NMR/EPR approach complemented with electrochemical analysis to investigate the mechanism of the recovery of DHAQ from its decomposition products 2,6-dihydroxy-anthranol (DHAL) and 2,6-dihydroxy-anthrone (DHA). This follows a two-step process, namely (i) the oxidation of DHA(L)2− to the dimer (DHA)24−, and (ii) the oxidation of (DHA)24− to DHAQ2−.
Moreover, Jethwa et al.98 used this NMR/EPR approach in combination with in situ UV-vis spectroscopy to study nitrogen-rich fused heteroaromatic quinones as promising candidates to be used in aqueous organic RFBs. As an interesting example, they investigated the electrochemical stability of the charged species during operation of a RFB containing 4,8-dioxo-4,8-dihydrobenzo[1,2-d:4,5-d′]bis([1,2,3]triazole)-1,5-diide as electrolyte, employing NMR/EPR to quantify formed species during electrochemical cycling.
Pyridinium derivatives represent another family of organic redox materials whose redox behavior has been probed by coupled in operando NMR and EPR spectroscopies under representative flow battery conditions.99 The powerful combination of both operando techniques to track the performance of an extensive library of bipyridinium compounds over a range of potentials provided insights into the air tolerance of batteries containing these electrolytes. The techniques allowed identifying a correlation between the singlet–triplet free energy gap (intramolecular electron pairing) and the onset of capacity fade mechanisms. Capacity fade was linked to the reactivity of the free radical species, and π-dimerization is the key to suppress reactivity between these radicals and trace impurities such as dissolved oxygen.99 The important findings allowed to propose strategies to mitigate capacity fade due to oxygen and stable pyridinium-based electrolytes for prolonged (days) cycling operation in air.
Combined ex situ NMR and in situ EPR characterization was also applied to investigate redox process intermediates and redox mechanisms in an aqueous organic RFB with fluorenone derivatives as anolytes.100 By identifying the fluorenone radical anion intermediates during charging process, the authors proposed a possible reaction mechanism that entailed electrochemical reduction–oxidation reactions and accompanying dis- and comproportionation reactions in the charge–discharge process for the reversible ketone hydrogenation and alcohol dehydrogenation of molecularly-engineered fluorenone-based anolytes.
In a recent work, Zhang et al.101 investigated methylene blue (MB) as catholyte in aqueous solution in a full cell. They utilized 1H in situ NMR spectroscopy and ex situ EPR spectroscopy to explore the redox chemistry of MB. They observed high oxidation-stability of MB radicals generated by comproportionation as well as for reduced MB in the electrolyte solution, and identified the important role of this stability for the high reversibility of the redox chemistry of MB. All these examples clearly show the power of combined in situ NMR/EPR techniques to investigate the redox chemistry of organic molecules in RFBs under working conditions.
Another application of simple in situ EPR experiments, which are not combined with NMR spectroscopy, is the evaluation of vanadium crossover in real-time in a VRFB.102 In that work, VO2+ crossover through Nafion in a VRFB was monitored as a function of sulfuric acid concentration and VO2+ concentration. In the experimental setup, a vanadium rich solution flowed through one side of the membrane of a permeation cell while a blank or vanadium deficit solution flowed on the other side. The blank flow solution was analyzed in an EPR cavity and the VO2+ concentration was determined from the intensity of the EPR signal. Permeability values of VO2+ ions across the membrane were analyzed at different temperatures to determine the effect of the viscosity of the flow solution on the permeability of the ion.102
1.5. In situ and in operando UV-vis spectroscopy
One significant limitation of RFBs is the loss of capacity over cycles, primarily due to ion and water crossover, hydrogen evolution, and inadequate mixing of electrolytes in the storage tanks. This leads to asymmetry in the electrolyte concentration and SoC between the positive and negative sides, resulting in charge imbalances. To address these issues effectively, real-time monitoring of both the total vanadium concentration and SoC in each half-cell is crucial. One approach to do this is to employ H-cells, as depicted in Fig. 5a. In that case, an optical fiber is inserted into the electrolyte solutions and the color change can be tracked. The advantage is that this setup is rather easy to assemble; a separate UV-vis apparatus is not needed. The disadvantage is that the kinetics and the efficiency of the battery may be compromised. The other option is to connect a flow battery to an UV-vis system and to pump the electrolyte solution through a flow-through cuvette (Fig. 5b), while continuously acquiring spectra, providing potentially a more precise determination of the SoC in a real battery. | ||
| Fig. 5 Comparison of (a) an H-cell and (b) a setup where the battery is connected to a flow-through cuvette of an UV-vis spectrometer.62 Reproduced with permission. Copyright 2019, IOP Science. | ||
In the cases where the color of electrolyte solutions significantly changes with the SoC as illustrated in Fig. 6a for vanadium species, UV-vis spectroscopy provides a direct and non-invasive method for analyzing the SoC.103 This method serves as an advantageous alternative to the traditional open circuit voltage (OCV) measurements. Unlike OCV, which only offers a general SoC status for the VRFB system, UV-vis spectroscopy can determine the SoC of each individual electrolyte and provides information on the e.g., total vanadium concentration. Optical sensing thus enhance VRFB performance monitoring by offering detailed insights into the processes governing efficiency and capacity decay, facilitating optimal real-time readings and enabling advanced control strategies.103 However, it is crucial to ensure the lack of electrolyte interference with the optical response of the electroactive species to deploy the full potential of UV-vis for quantitative analysis. In case of interference, contribution of the electrolyte solution to the total absorbance needs to be measured in a control experiment and subtracted.
 | ||
| Fig. 6 (a) H-cell experiments. During electrolytic reduction of a vanadium solution with a concentration of 0.04 M, the solution undergoes a series of color changes. Initially, the solution appears yellow due to the presence of V5+ ions. As the reduction process progresses, the color transitions from yellow to blue (V4+), then to green (V3+), and finally to purple (V2+).104 Reproduced with permission. Copyright 2019, IOP Science. Flow-cell spectro-electrochemistry showing the decrease in the features associated with VBH in vanadium(IV) oxidation state and growth of the feature associated with VBH in vanadium(V) oxidation state, (b) measured during the discharge to 40% SoC, and (c) plot of the change of the number of moles of reduced, oxidized and total VBH (blue, red and gray data points, respectively) present during oxidation, obtained using UV-vis data at 485 and 825 nm, respectively, and for comparison the number of moles of electrons removed electrochemically (black line).62 Reproduced with permission. Copyright 2019, IOP Science. (d) UV-vis spectra of 5 mM DMeQUIC after 0 min, 5 min, 2 h, and 16 h of potentiostatic reduction in a DMeQUIC–Fe(CN)6 flow cell at 1.6 V. (e) UV-vis spectra of rDMQ, tDMQ, and fDMQ estimated using Bayesian inference, displayed alongside the authentic spectrum of DMeQUIC.105 Reproduced with permission. Copyright 2024, ACS Publication. (f) Voltage profiles (top) and absorbance changes at 765 nm of the catholyte (bottom) in the ZMRFB with VOSO4 as function of time. (g) In situ UV-vis spectrum of the catholyte at the charge–discharge process of ZMRFB with 50 mM VOSO4.106 Reproduced with permission. Copyright 2024, ACS Publication. | ||
In research conducted by Gokoglan et al.,62in operando UV-vis spectroscopy was used to monitor redox events and to quantify the concentration of redox species during battery cycling, demonstrating strong redox stability of the bioinspired redox-active material vanadium(IV/V) bis-hydroxyiminodiacetate (VBH), highlighting smooth transitions between vanadium(IV) and vanadium(V) species in non-aqueous VRFBs. In operando UV-vis spectroscopy also allowed for quantitative tracking of each species in real-time, offering a simple method of SoC monitoring in VRFBs.84 Notably, Fig. 6b and c shows no signs of decomposition or irreversible side reactions in an operational flow cell at 10 mA cm−2. Furthermore, the data reveals a clear relationship between the change of the number of moles of electrons and of vanadium species, with their respective slopes showing a clear correlation.
In UV-vis spectroscopy, high vanadium concentration in the electrolyte often fails to follow Lambert–Beer's law, resulting in a low signal-to-noise ratio. Kyung-Hee et al.63 developed a straightforward method for real-time SoC monitoring of VRFB systems by UV-vis spectroscopy in operando mode that consisted in using cuvette windows with thin path lengths (below 0.1 mm) and a concept of difference absorbance. This approach is practical for large-scale engineering applications as well in which high vanadium concentrations (e.g., 2 M in 3 M sulfuric acid) without dilution are used.63
Despite extensive research, a critical question remains: How can UV-vis spectroscopy independently measure both the total vanadium concentration and SoC of both electrolytes in a VRFB, and with what accuracy? Existing literature often focused on specific electrolytes and lacked comprehensive analysis. Calibrations only provided SoC, ignoring the total vanadium concentration. Maurice et al.103 addressed this gap by presenting calibration methods capable of accurately quantifying both total vanadium concentration and SoC across all vanadium electrolyte mixtures, covering concentrations from 0.91 to 1.83 M. Their study addressed three types of vanadium electrolyte mixtures used in VRFBs: (i) anolyte V2+/V3+, (ii) V3+/VO2+, and (iii) catholyte VO2+/VO2+. Two calibration methods were provided for each electrolyte: (i) a quick empirical method based on linear regression requiring only one or two hand-picked absorbances, and (ii) a more detailed spectral deconvolution method using the entire spectrum, both based on the generalized Lambert–Beer law.107 The empirical methods are suited for industrial applications where low-cost optical sensors measure SoC in real-time with slightly less accuracy. The spectral deconvolution method, which is more powerful and computationally intensive, is better for laboratory use where spectrometers are available.
Operando UV-vis spectroscopy has also been utilized in organic RFBs. Among these, quinone-based aqueous flow batteries have the potential for large-scale, cost-effective energy storage, thanks to their reliance on earth-abundant elements, high aqueous solubility, reversible redox kinetics, and tunable chemical properties like reduction potential. In a specific quinone derivative, 9,10-anthraquinone-2,7-disulfonic acid, the formation of a quinhydrone dimer from an oxidized quinone and a reduced hydroquinone leads to deviations from ideal solution behavior and affects optical absorption according to the Lambert–Beer law.108 To address this, in situ UV-vis spectroscopy was used to determine the molar attenuation profiles of quinone, hydroquinone, and quinhydrone, along with an equilibrium constant (KQHQ) of about 80 M−1 for the quinhydrone dimer formation.108 These profiles helped identifying total molecular concentration and state of charge across various quinone and hydroquinone mixtures. Density functional theory (DFT) calculations supported UV-vis findings and offered insights into the dimerization conformations. A quinone–bromine flow battery was equipped with a Pd–H reference electrode to illustrate how complexation in both negative (quinone) and positive (bromine) electrolytes influences half-cell and full-cell voltage measurements. This study highlighted the importance of considering electrolyte complexation to enhance the accuracy of electrochemical modeling in flow battery systems.108
Modak et al.105 investigated the link between capacity fading and molecule decomposition in aqueous quinoxaline-based RFBs by combining flow cell cycling with various chemical analysis techniques, including UV-vis spectroscopy. Using Bayesian statistical inference, they calculated the rate constant of tautomerization, explaining the fade rates observed in cycled cells. They studied the cycling stability of 2,3-dimethylquinoxaline-6-carboxylic acid (DMeQUIC), pointing out that the tautomerization of its reduced form is the primary cause of capacity fading. Bayesian inference revealed that the time-varying absorbance profiles in Fig. 6d are driven by the evolution of three distinct species from DMeQUIC including its reduced form (rDMQ), a transient redox-active decay product (tDMQ), and a final redox-inactive species (fDMQ). The spectra of these pure species are shown in Fig. 6e. This method demonstrated the unique capability of combining Bayesian inference and in operando UV-vis spectroscopy to connect molecular decomposition with capacity fading.105
Additionally, UV-vis spectroscopy was applied for investigation of aqueous manganese RFBs, which rely on the Mn2+/MnO2 two-electron transfer reaction.106 These batteries are attractive due to their cost-effectiveness, high voltage, and excellent safety features. However, MnO2 tends to partially dissolve during discharge, hindering sustained operation. It dissolves and deposits even more easily when VO2+/VO2+ is added to the catholyte as a redox mediator. The chemical process between MnO2 and VO2+ was clarified by in situ UV-vis spectroscopy and kinetic analysis, which encouraged the recovery of “dead” MnO2 with little mediator that resulted in a reversible MnO2 cathode. By examining the VOSO4-mediated MnO2 reduction process in the Zn–Mn2+/MnO2 RFB (ZMRFB) during charging, it was observed that the characteristic peak at 765 nm decreased with the electrochemical oxidation of VO2+ to VO22+ at stage I (Fig. 6f).106 Thereafter, the intensity of wavelengths ranging from 1000 to 500 nm increased to varying extents (Fig. 6g), indicated the appearance of MnO2 in the flow cuvette. During discharging, decreased UV absorption suggested some MnO2 particles returned to the electrode, though some “dead” MnO2 remained.109 The redox mediator facilitated the swift reduction of residual MnO2 by VO2+, doubling the discharging time for the VO2+/VO22+ reaction, enhancing discharge capacity, and reducing MnO2 aggregation, thus significantly improving the ZMRFB cycle life.106 These findings underline the crucial role of UV-vis spectroscopy in enhancing the understanding and performance of RFBs, paving the way for more efficient energy storage solutions. Moreover, other metal battery systems have been explored in operando such as the alkaline zinc–iron/nickel hybrid flow battery with similar motivations.110
Next to these examples, in operando UV-vis spectroscopy has been applied for the analysis of kinetic processes in solution, more specific of the effect of redox boosters. Lu et al.111 investigated polysulfide-based flow batteries, whose main disadvantage is its rather sluggish kinetics. They designed a catalyst based on riboflavin, which improved the kinetics and finally the performance of the polysulfide-ferrocyanide battery to a large extent. Using in operando UV-vis spectroscopy, they were able to identify the kinetic processes and to finally design an efficient catalyst.
In the field of RMFBs, also known as redox-targeting flow batteries, in operando UV-vis spectroscopy has been applied in several works to investigate the charge transfer reaction between different pairs of solid energy boosters and soluble redox mediators. In an early report, M. Zhou et al.112 monitored concentration changes of the catholyte redox mediator, a ferrocene-grafted ionic liquid (FcIL), during cycling of a LiFePO4-based Lithium RFB. By using a spectro-electrochemical flow cell connected to the outlet cathodic flow channel, the concentration of FcIL during the discharging process was determined from the characteristic absorbance of the redox mediator at 630 nm. As the electrolyte was concentrated, UV-vis spectra were recorded as differential absorbance by using the fully charged catholyte as the reference. Comparison of the time-dependent concentration of the charged form of FcIL (FcIL+) in a cell without and loaded with the LiFePO4 solid booster showed extended time series spectra for the cell loaded with LiFePO4 during a galvanostatic measurement. In the presence of solid active material, the concentration changes of FcIL+ over the discharging process become stabilized while reaching the steady state, implying that the redox mediated process is the limiting step to sustain a constant flux of FcIL for electrode reaction under the conditions applied.112
The Prussian Blue(PB)-[Fe(CN)6]4−/3− is another example of solid booster/redox mediator pair where the redox mediated reaction mechanism has been investigated by in operando UV-vis spectroscopy, initially for an aqueous hybrid PB–Fe/Zn RFB113 and more recently, for a high energy density and low cost neutral aqueous PB–Fe/K2S RFB.114 In both studies, a custom-designed spectro-electrochemical cell was connected to the outlet cathodic flow channel of a symmetric flow cell and in operando UV-vis spectra of the catholyte ([Fe(CN)6]4−/3− redox pair) with or without PB granules were monitored during charging/discharging (Fig. 7). While [Fe(CN)6]4− shows nearly no absorption at wavelengths larger than 400 nm, [Fe(CN)6]3− has a high absorption band at 460 nm, allowing to probe the concentration of [Fe(CN)6]3− at different stages of charge/discharge. In absence of PB booster, a linear relationship between the absorbance at 460 nm and [Fe(CN)6]3− concentration was observed during the whole charge/discharge process, indicating the charge/discharge capacity solely arises from the redox reactions of the [Fe(CN)6]4−/3− redox pair. In presence of PB, the [Fe(CN)6]3− concentration changed linearly only initially at low SoC, indicating the electrochemical reduction of [Fe(CN)6]3− at the electrode, and started to deviate from linearity, reaching a steady state, at approximately 50% SoC of [Fe(CN)6]4/3− electrolyte, indicating that redox mediated reaction with Prussian White (PW, the reduced form of PB) occurred. At this stage, the charge/discharge capacity arises from the redox mediated reaction between [Fe(CN)6]4−/3− and PB/PW.
 | ||
| Fig. 7 Configuration of the setup for in operando UV-vis spectroscopy measurements at the catholyte of a RMFB.113 Reproduced with permission. Copyright 2019, Cell Press Publication. | ||
1.6. In situ and in operando Raman spectroscopy
During cycling of RFBs, redox reactions as well as absorption and desorption processes on the electrodes lead to changes in the redox states and the formation and/or breaking of chemical bonds in the components of the electrolytes and/or the electrode materials. Vibrational spectroscopic techniques, such as Raman scattering can probe these processes. Raman spectroscopy has been employed successfully in electrochemistry to monitor chemical reactions and probe intermediates as well as pH values in the electrolytes, monitor changes on catalytic electrode surfaces and investigate surface adsorbates.115,116Unlike FTIR spectroscopy, Raman scattering is less prone to absorption in aqueous media. However, care must be taken as the focusing of an intense laser beam on a spot of typically 1 μm in diameter causes significant heating of the sample. This may impact the reaction to be monitored and may lead to phase transitions in electrode materials or even damage the electrode material. To minimize the light-induced damage, the laser light needs to be attenuated, e.g., by using a line focus if available or fast rotation of electrodes. As such, extensive control experiments must be undertaken for each system to find the optimal laser power. Furthermore, control experiments are also needed to explore possible interfering bands from the electrolyte salt in the same spectral region as the charge-storing species using identical laser radiation conditions.
To achieve good resolution of the spectra, the weak Raman scattering needs to be acquired through confocal microscope optics with large numerical aperture while air gaps, as well as long passages of light through the medium should be avoided to minimize losses. A schematic representation of an operando Raman spectroscopy setup is shown in Fig. 8a–c. A typical standard Raman spectrum may require several minutes to be acquired. To increase the related time resolution, enhancement techniques such as surface-enhanced Raman spectroscopy (SERS)117 and tip-enhanced Raman spectroscopy (TERS)118 may be employed to collect spectra within seconds. As the enhancement factors can vary strongly depending on the electrical potential as well as the sample history a suitable normalization procedure is required when using these techniques.
 | ||
| Fig. 8 (a) Scheme of an in operando Raman spectroscopy setup for the investigation of redox flow cells.64 The laser can be focused onto the electrolyte by confocal microscope optics that will also pick up the Raman scattering and focus it on the detector. (b) Besides the electrolyte composition it is also possible to detect bound species on the surface of the electrodes.64 (c) Measuring the Raman spectra in operando allows to correlate changes in the relative composition of the redox active species with the electrochemical potential of the cell.64 Reproduced with permission. Copyright 2023, ACS Publication. | ||
Because of the small focus area of the laser, Raman spectroscopy also allows for a highly localized investigation of electrodes with spatial resolutions of 1–2 μm. In a recent example, Lan and Li et al.64 investigated an electrode material made from ordered hierarchical porous Co and N co-doped carbon (OHP-Co/NC) for usage in a polysulfide/ferrocyanide RFB. They monitored the composition of the electrolyte by in operando Raman spectroscopy, identifying different polysulfide species, i.e., S22−, S3−* and S42− through their specific S–S bond stretching modes. Following the composition of the electrolyte during the charge and discharge cycle revealed a high reversibility of the electrochemical processes in this system. Owing to the high spatial resolution of Raman spectroscopy they were also able to monitor the reaction on a single OHP-Co/NC particle of approximately 2 μm that revealed a high selectivity of this material towards S42− and S22−, whereas no S3−* was found. While in operando Raman spectroscopy of the electrolyte solution revealed more S22− than S42−, the in situ Raman spectroscopy performed on the surface of a single OHP-Co/NC particle showed a different distribution with more S42− than S22−. This reflects the complex interplay of the electrochemical reactions on the electrode surface and additional chemical reactions in the bulk phase of the electrolyte in this polysulfide system.119
Further applications for exploring reaction mechanisms were proposed by Zhang et al.,65 who investigated an atomic Bi–O–Mn catalytic interface on a mesoporous Mn3O4 sub-scaffold grafted onto carbon felts (Bi1–sMn3O4–CF) as electrodes for VRFBs. The atomic Bi–O–Mn interface catalyzes the V3+/V2+ conversion while the mesoporous Mn3O4 scaffold facilitates fast shuttling of redox active species from the surface to the pores leading to high performance RFBs with 77% energy efficiency up to a current density of 400 mA cm−2 and peak power density of 1.503 W cm−2 while maintaining durability over 1500 cycles. Using Raman spectroscopy, they confirmed the incorporation of bismuth onto Bi1–sMn3O4–CF by a downshift of 6 cm−1 in the strong phonon Mn–O deformation vibration compared to sMn3O4–CF without Bi. By in situ Raman spectroscopy, they identified bridging intermediates of the first solvation shell of vanadium ions dissolved in H2SO4 that are involved in the redox reaction after adsorption. For this, they compared S–O and S–OH stretch vibrations in the pure electrolyte and during oxidation and reduction at the electrode. During the oxidation they observed an increased intensity of the S–O vibration and a decreased intensity of the S–OH vibration as well as additional V–O–S modes. This allowed them to propose a feasible mechanism in which first H2O dissociates from [V(H2O)6]2+ upon binding to *Bi, and subsequently SO42− is added leading to a *Bi[V(H2O)5(SO42−)] intermediate. The intermediate promotes the charge transfer process due to its higher polarizability leading to the product [V(H2O)5(SO42−)]+. During the reduction process no changes in the signals of S-O and S-OH bond stretching vibrations were observed leading to the assumption of a direct adsorption and desorption process of [V(H2O)6]3+ without an intermediate species.
In another report by Nambafu et al.,120in operando Raman measurements, supported by DFT studies, provided compelling evidence for a specific molecular structure in a novel Fe anolyte for all-iron RFB (Fe-RFBs), specifically an Fe complex with a nitrogenous phosphonate/carboxylate mixed ligand, N,N-bis(phosphonomethyl)glycine (BMPG). The findings suggest that in BPMG, mixed phosphonate/carboxylate groups ensure that the Fe ion center maintains octahedral coordination exclusively with phosphonates, leaving the carboxylate unbound for both Fe(II) and Fe(III) complexes. This structural consistency between BPMG-based Fe(II) and Fe(III) complexes helps to alleviate the slow redox reaction kinetics typically seen in Fe(NTMPA)2 ((iron(II) nitrilotri(methylphosphonate acid)) anolyte, where significant ligand reorientation between the complexes occurs. The strategically modified ligand in the Fe anolyte allows enhanced performance in the Fe-RFB.120
In operando Raman spectroscopy was also employed by Radinger et al.121 to monitor the reactions that occurred at the interface between graphite-based model electrodes and vanadium-containing sulfuric acid electrolyte. The growth of various vibrational modes as the potential changed was linked to the electrocatalytic activity of the sample, allowing for the tracking of electrolyte species oxidation. Additionally, the findings revealed that vanadium reaction intermediates of dimeric origin appeared exclusively on the edge-exposed surface of graphite, which displayed notably higher electrochemical activity. Contrary to previously proposed interactions with surface oxygen, the Raman measurements revealed direct electronic interactions between vanadium ions and carbon atoms during charge transfer. More recently, Lubian et al.122 applied time-resolved Raman spectroscopy for operando investigation of reaction mechanisms responsible for charge storage capacity decay in a 2,6-dihydroxyanthraquinone–ferrocyanide alkaline flow battery. In their work, the faradaic imbalance process was explored, showing that the presence of oxygen in the anolyte leads to progressive loss of available ferrocyanide. Self-discharge of ferricyanide when left at an open circuit was also detected by Raman spectroscopy. Moreover, the operando technique allowed tracking the crossover of 2,6-dihydroxyanthraquinone.122 The report illustrated the power of Raman spectroscopy to monitor in real time chemical processes in flow battery electrolytes.
In addition to exploring reaction mechanisms, in situ Raman spectroscopy has been applied quantitatively for the real-time estimation of the SoC of a zinc-bromine RFB upon cycling.123 Accurate estimation of the SoC in zinc-bromine RFB is very challenging. The inhomogeneous mixing of the aqueous zinc bromide phase and non-aqueous polybromide phase in the positive electrolyte, together with the simultaneous occurrence of various equilibrium reactions in the battery, e.g., charge-transfer reactions, polybromide formation and complexation complicate SoC determination. To avoid errors arising from the inhomogeneous mixing of the aqueous and non-aqueous phases in the positive electrolyte, in situ Raman analysis was performed on the negative electrolyte flowing from the cell to the reservoir during the charge–discharge. In their study, Lee et al.123 observed a strong linear relation between the Raman band intensity and total zinc bromide concentration, proving that in situ Raman spectroscopy is very effective for the real-time SoC estimation under operando conditions.
1.7. In situ and in operando FTIR spectroscopy
FTIR is another type of vibrational spectroscopy which has been used to explore in operando electrolyte solutions for RFB. Kautek et al.66 conducted in situ absorption-reflection FTIR spectroscopy in the formation of bromine-complexes in a zinc/bromine flow battery at gold electrodes. In this RFB, storage reactions are the cathodic deposition of zinc and the anodic formation of a nonaqueous polybromide phase. In situ FTIR spectroscopy measurements revealed formation of ion pairs between electrode adsorbed polybromide anions and quaternary ammonium cations, which store the bromine as polybromide complexes. Studying the complex formation capability of different cations, they were able to identify electrolyte compositions for enhanced efficiency of zinc/bromine flow batteries by improving the capability to store bromine.Huang et al.67 performed attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) to monitor changes of bonding structures of 2,6-DHAQ and 1,5-DHAQ at different stages of reduction during CV scans using a commercialized spectro-electrochemical cell (Fig. 9a and b). The evolution of vibrations of C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of 2,6-DHAQ and 1,5-DHAQ in the range of 1300–1600 cm−1 were recorded during CV scans. The signal of CC gradually diminished in the cathodic scan and re-emerged in the anodic scan while C–O showed a reversed trend. Variations of the characteristic vibrations of water molecules were attributed to changes of the solvent environment associated with disruption of the water molecule network with quinone molecules through hydrogen bonding. New peaks emerging at around 1105 and 1242 cm−1 upon reduction followed by a resting process were attributed to the presence of 2,6-dihydroxyanthrone (2,6-DHA), suggesting a disproportionation reaction producing 2,6-DHAQ and 2,6-DHA (hydrogen bond-mediated degradation process). This was not the case for 1,5-DHAQ, whose reduced state (1,5-DHAQ2−) preferably forms 1,5-DHAQ2−·H2O, for which the neighboring oxygen sites are bonded with the same water molecule preventing it from joining another DHAQ molecule (hydrogen bond-mediated protection process). Their work highlights the power of in operando ATR-FTIR characterization to elucidate electrochemical reaction mechanisms.67
C bonds of 2,6-DHAQ and 1,5-DHAQ in the range of 1300–1600 cm−1 were recorded during CV scans. The signal of CC gradually diminished in the cathodic scan and re-emerged in the anodic scan while C–O showed a reversed trend. Variations of the characteristic vibrations of water molecules were attributed to changes of the solvent environment associated with disruption of the water molecule network with quinone molecules through hydrogen bonding. New peaks emerging at around 1105 and 1242 cm−1 upon reduction followed by a resting process were attributed to the presence of 2,6-dihydroxyanthrone (2,6-DHA), suggesting a disproportionation reaction producing 2,6-DHAQ and 2,6-DHA (hydrogen bond-mediated degradation process). This was not the case for 1,5-DHAQ, whose reduced state (1,5-DHAQ2−) preferably forms 1,5-DHAQ2−·H2O, for which the neighboring oxygen sites are bonded with the same water molecule preventing it from joining another DHAQ molecule (hydrogen bond-mediated protection process). Their work highlights the power of in operando ATR-FTIR characterization to elucidate electrochemical reaction mechanisms.67
 | ||
| Fig. 9 (a) Schematic representation of an in operando ATR-FTIR setup and (b) photograph of the cell for DHAQ electrolyte. The working electrode (WE) consisted of a Pt disk (diameter: 3mm with a Pt plate (4 × 2.5mm2) as counter electrode and a Ag wire (diameter: 0.5mm) as reference electrode to detect the structural evolution of 2,6-DHAQ and 1,5-DHAQ and PAQS/CB composites from 4000 to 900cm−1 with a resolution of 4cm−1. The CV scan rate was 2mV s−1, and the working distance was 0.05mm to the substrate. The blank background was 1M KOH solution. 0.1M 2,6-DHAQ and 1,5-DHAQ were dissolved into 1M KOH to obtain the operando FTIR spectra during CV scans.67 Reproduced with permission. Copyright 2022, Nature Publishing Group. (c)–(e) In situ ATR-FTIR setup for (c) 2,7-AQDS only, (d) PI only, (e) PI under reaction with 2,7-AQDS. (f) In situ FTIR spectra of PI during reaction with 2,7-AQDS, the color bar indicates transmittance. (g) FTIR spectra evolutions during reduction of 2,7-AQDS without PI substrate (upper), reduction of PI coated on carbon cloth without 2,7-AQDS (middle) and reduction of PI during redox-mediated reaction with 2,7-AQDS (lower).124 Adapted and reproduced with permission. Copyright 2020, Wiley-VCH. | ||
Redox mediated processes of organic solid booster/redox mediator pairs have also been investigated by in operando FTIR spectroscopy for different RMFB chemistries. In a first report, the redox reaction between the quinone derivative 9,10-anthraquinone-2,7-disulphonic acid (2,7-AQDS) and the 1,4,5,8-naphthalenetetracarboxylic dianhydride polyimide (PI) in neutral electrolyte was explored during a CV scan in an ATR-FTIR spectro-electrochemical setup (Fig. 9c–e).124 The PI solid booster was deposited on the ATR-FTIR window and the 2,7-AQDS solution was polarized in a three-electrode configuration. Spectral changes of 2,7-AQDS and PI alone and those of the 2,7-AQDS/PI system were monitored in separate CV experiments (Fig. 9f). The reversible vibrational changes in the region 1400–1800 cm−1 of the FTIR spectra (imide carbonyl CO bands) of PI measured for the 2,7-AQDS/PI system corroborated the reversible redox-mediated reactions between 2,7-AQDS and PI in both cathodic and anodic directions and the regeneration of 2,7-AQDS on the PI substrate (Fig. 9g). FTIR investigations allowed proposing a mechanism of hydrogen-bonding mediated charge transfer between 2,7-AQDS and PI in pH-neutral solutions that was corroborated by DFT calculations.124
Similarly, mechanistic studies were conducted by Huang et al.67 using in operando ATR-FTIR for the solid booster/redox mediator pair 1,5-DHAQ/poly(anthraquinonyl sulfide) (PAQS). Time-dependent evolution of the FTIR spectra of PAQS was monitored during repeated CV scans. During reduction of 1,5-DHAQ, a stretching vibration of C–O bond of reduced PAQS at 1371 cm−1 gradually appeared that vanished during oxidation of 1,5-DHAQ2−. Additional reversible changes of vibrations attributed to the aromatic rings also corroborated the reversibility of the redox mediated process.
1.8. In situ and in operando XAS
An important technique to probe the element specific oxidation states and structures of electrolytes and electrodes is the XAS. It allows the observation of electronic transitions of inner shell electrons, and selection of the X-ray energy regime allows for different penetration depths to probe the bulk phase or superficial regions.125 The penetration depth is also affected by the method of detection. Both, transmission and fluorescence mode mainly contain information about the bulk phase as the surface components only make a small contribution to the resulting signal. Samples for transmission also need to be thin enough for the radiation to pass through, which is especially challenging at low X-ray energies. Measurements at very small angles under total external reflection as well as measurements of Auger electrons can provide information on the sample surface. Samples need to be homogenous, and one must consider the energy dependent absorption lengths in the material of interest as well as of substrate materials at the utilized energy for transmission or fluorescence measurements. Heat buildup and radiation damage to the sample might occur which limit the energy of X-rays used and may require cooling of the sample, albeit without freezing the electrolyte solution which would impede the flow inside a flow-cell.126As tunable source for monochromatic X-rays a synchrotron is commonly applied and detection takes place at the absorption edge, that is the energy at which an electron is removed from an inner orbital by X-ray absorption. X-ray absorption near edge structure (XANES) yields information about the electronic configuration and coordination states while analyzing the extended X-ray absorption fine structure (EXAFS) yields information about the structure and environment of the atom.
Most XAS investigations of RFB materials have not been conducted in situ during operation. Instead ex situ investigations after cycling have been conducted to explore (i) changes undergone by electrocatalysis,127 (ii) the influence of functional groups in carbon felts on the kinetics of the VO2+/VO2+ reaction,128 and (iii) the chemical and morphological origins of improved ion conductivity in ion-conducting polymer membranes,129 among other applications.130 A first application of in situ XAS for redox flow cells was reported by Jia et al.68 who studied the oxidation states of vanadium ions during the first charge/discharge cycle of a VRFB. They observed the K-edge of vanadium in the anolyte and catholyte using XANES in a flow cell equipped with sections of a Kapton tube as observation windows with a time resolution of 6 min per spectrum. They found that during a first charging step the oxidation of the precursor VOSO4 to VO2+ on the cathode and the reduction of VOSO4 on the anode site to V2+ only reached about 82% of the theoretical maximum affecting all subsequent cycles as well. Based on their findings, they proposed an improved first charging cycle to utilize 100% of the available capacity in this system.
Probing the interface between electrode and electrolyte has proven a challenging task with XAS as the intensity of the X-ray beam is strongly attenuated already by liquid films of a few μm in thickness especially at low X-ray energies. Using higher energies can improve penetration into liquid layers but limits the observation to heavier nuclei. On the contrary, using thin liquid layers of a few nm leads to fast expiration of the electrolyte during operation while formation of gas bubbles in some reactions leads to strong perturbations in the measurements. Paparoni et al.69 presented a novel flow-cell design for in operando XAS with an X-ray opaque electrode surface and liquid layers down to 17 μm that allowed them to study the influence of Ni doping of Fe2O3 films grown on Pt substrates on the catalysis of the oxygen evolution reaction. Their cell design (Fig. 10) facilitated the adjustment of the liquid layer thickness above the electrode sample for gradient free reactions at stable cell potentials and allowed observation of in operando XAS at low energy absorption edges such as iron and titanium with total transmittances of 43% for Ti and 75% for Fe.
 | ||
| Fig. 10 Exploded view of the cell: (a) top PEEK plate, (b) mask, (c) Kapton window and (d) spacer, (e) O-ring, (f) counter electrode, (g) cell body, (h) reference electrode, (i) electrolyte outlet, (m) sample, (n) sample holder column, (o) bottom aluminum plate. (P) (inset) detail of the inlet/outlet channels passing through the counter electrode compartment. The top plate (a) is designed for fluorescence measurements with a 120° view angle, which extends to 150° through two channels, allowing near 15° incidence and emergence angles. The sample is positioned close to window (c) and can be adjusted using a retractable column (n). It is secured with PELCO carbon conductive glue, and sides are coated with nitrocellulose varnish, leaving the front uncoated. The cell body (g) has a circular aperture for the sample holder, surrounded by a Pt wire counter electrode (f). Electrolyte flows through apertures (i) and channels (inset P) for circulation before and after contact with the sample. A leak-free Ag/AgCl reference electrode (h) completes the three-electrode setup, ensuring compatibility and preventing contamination. The window assembly consists of 25 μm Kapton foil and a 50 μm Kapton spacer (c) and (d) bonded to a 1.5 mm thick Teflon mask (b) with a 3 × 14 mm hole for beam access. Top and bottom plates (a) and (o) press the window against the cell body (g), allowing free volume above the sample. The electrolyte fills the counter electrode compartment (f) and (P), with all components in contact with the electrolyte made from PEEK and sealed with FFKM o-rings for leak-tightness. A peristaltic pump circulates the solution continuously refreshing the electrolyte to maintain stable cell potential.69 Reproduced with permission. Copyright 2024, RSC Publication. | ||
Moreover, Lutz et al.131,132 also conducted synchrotron experiments with spatiotemporal resolution to unravel the vanadium species gradient variation during diffusion through the membrane at different points in the flow field. Their experiments confirmed the chemical formation of V(IV) inside the membrane after exposing to V(III) on one side and V(V) on the other one. XAS was also applied by Huang et al.133 in a Zn–Fe RFB to investigate in operando the Cr-doped NiFe(oxy)hydroxide (CrNiFeOOH) catalyst used to compensate the coulombic inefficiency caused by irreversible anodic parasitic reactions that lead to unbalanced charge state of catholyte/anolyte and subsequently, to poor cycling performance. The proposed coulombic efficiency compensating strategy aimed to equalize the charge state of the [Fe(CN)6]3−/4−-based catholyte by promoting the oxygen evolution reaction (OER) in the cathodic side aided by a CrNiFeOOH catalyst placed in an external catalytic reactor column via a redox-mediated process.133In operando XAS was employed to investigate the valence states of metal species in CrNiFeOOH catalyst and analyze the reaction mechanism during the redox-mediated OER process. The normalized Cr, Fe, and Ni K-edge XANES of CrNiFeOOH catalyst collected during the charge process revealed that the oxidation states were Fe3+, Ni2+, and Cr3+ in pristine CrNiFeOOH. At increased SoC using [Fe(CN)6]3−/4−-based electrolyte, observed energy shifts for the main edge and pre-edge features in Cr and Ni XANES indicated that the oxidation process of CrNiFeOOH upon charging involved the oxidation of Cr3+ to Cr6+ initially, followed by the oxidation of Ni2+ whereas the main edge of Fe in the CrNiFeOOH catalyst remained at the same peak position, suggesting that the valence of Fe3+ was unchanged. The whole redox-mediated process was similar to direct electrochemical OER with the same electrocatalyst grafted on electrodes.133
Similarly, Y. Cai et al.134 conducted operando synchrotron XAS measurements to explore interface reaction mechanism of the redox-mediated process between the LiMnxFe1−xPO4 solid energy booster and [Fe(CN)6]3−/4− redox mediator in a Zn–Fe RMFB. Operando synchrotron XAS unveiled the reversible changes of the Fe–O and Fe–Fe bonds in the [Fe(CN)6]3−/4−-LiMnxFe1−xPO4 system during real-time monitoring.
1.9. In situ and in operando electrochemical techniques
EIS is one of the most used techniques for characterizing RFBs. It offers insights into the charge transfer resistance, ohmic losses, and mass transport limitations within the battery. Usually, interpreting EIS data relies on fitting equivalent circuit models and requires prior knowledge of the system. However, Schilling and co-authors135 combined EIS with distribution of relaxation times (DRT) analysis, which is a mathematical approach, offering an alternative by transforming the impedance data from the frequency domain into a distribution of time constants without the need for a priori assumptions. This transformation reveals distinct peaks corresponding to different physicochemical processes, such as the electrochemical redox reaction, transport through the porous electrode structure, and ion transport in the electrolyte. They applied DRT analysis to investigate the V(IV)/V(V) redox reaction in VRFB half-cells, producing a spectrum that plots the distribution function of relaxation times γ(f) against frequency, where high-frequency peaks (above 50 Hz) correspond to redox reactions, mid-frequency peaks (10 mHz–50 Hz) to transport through the porous electrode, and low-frequency peaks (below 10 mHz) to ion diffusion in the electrolyte (Fig. 11a).135 Similarly work done by Schneider et al.136 deconvoluted the whole DRT spectra into the individual contributions from the RFB system utilizing full- and half-cell tests. Enabling the tracking of changes in the systems, such as changes in liquid flow rate, kinetics and mass transport. Both works showcase how the use of DRT can help to better understand the measured EIS spectra and monitor changes in the system. In addition, the method serves as a fingerprint for electrode materials based on their morphology and wettability, helping in the optimization of electrodes for VRFBs. Calculation of the DRT spectra from the measured EIS spectra can readily be accomplished with the software tools from Tichter et al.137,138 and Ciucci and co-workers.139–142 | ||
| Fig. 11 (a) Schematic display of the proposed peak assignments to the half-cell processes using an exemplary DRT spectrum.135 Reproduced with permission. Copyright 2022, Elsevier Publication. (b) Open-circuit current density and the obtained voltage–current density polarization curve of the Zn–Cr battery with its three distinct regions where I activation, II ohmic, and III residual effects (mass transfer) influence its voltage loss. (c) Resolved factors causing overpotentials in the battery systems leading to voltage loss: Charge transfer, ohmic, and residual factors. (d) Resolved polarization curve of the RFB to identify the side of major losses contributing to the system.70 Reproduced with permission. Copyright 2020, Elsevier Publication. | ||
In systems such as the zinc–cerium RFB (Zn–Ce RFB), in situ polarization measurements coupled with EIS have been utilized by Amini et al.68 to pinpoint performance limitations (Fig. 11b). By inserting reference electrodes in the flow paths, it was possible to decouple the contributions of the positive and negative electrodes to overall voltage losses (Fig. 11d). For example, most of the kinetic overpotential in Zn–Ce RFBs originates from the Zn/Zn2+ half-cell, particularly at low current densities, where kinetic limitations dominate (Fig. 11b and c). This allows for targeted improvements in electrolyte formulation or electrode material. Further, they were able to show how the current density influences the detrimental factors of the voltage loss. Another study employed EIS while using a pulse dampener in the hydraulic circuit to collect stable low-frequency impedance spectra at operationally relevant flow rate of 25 mL min−1 in a VRFB.143 This allowed for the quantification of charge transfer, finite diffusion, and ohmic resistances and assessing critical physical parameters such as rate constants and effective surface areas of various electrode materials, offering a deeper understanding of how electrode structure and morphology affect performance. For example, GFD3 felt was found to be mainly limited by charge transfer resistance, whereas 10AA carbon paper displayed considerable contributions from all three types of resistance.143
Addressing battery operation at elevated temperatures is, however, a key challenge for stationary storage applications. Thus, the maximum operating temperature for vanadium solutions in flow batteries is generally capped at 40 °C to avoid the thermal precipitation of V2O. In this regard, a comprehensive study was conducted to assess the high-temperature stability of redox solutions in VRFBs. The study compared batch (solution in flasks under temperature control) and in operando (RFB operation) precipitation experiments, revealing that relying solely on capacity fade to assess precipitation can be misleading due to significant influences from external oxidation and cycling parameters. In operando electrochemical experiments consistently showed a precipitation temperature 10–20 °C higher than batch tests at a 100% SoC for the same duration.144
A comparable method detailed the use of carbon fiber-based potential probes embedded in the membrane of a VRFB to monitor transport processes and redox reactions occurring within the membrane. By strategically placing these probes at various points within a prepared multilayer membrane stack, distinct redox potentials (Φ) were identified, which were linked to specific vanadium ion species (Φ < −0.083 V: V2+ and V3+; 0.183 V < Φ < 0.537 V: V3+ and VO2+; Φ > 0.823 V: VO2+ and VO2). This allowed the real-time observation of processes inside the membrane both in situ and in operando. Furthermore, it was determined that reactions occur in the through-plane area of the membrane, with the position of the reaction front being influenced by the SoC in the bulk electrolytes.145 Another approach in electrode design is a simple yet effective silver–silver sulfate reference electrode for use in VRFBs for evaluation of their electrochemical performance under in operando conditions. By inserting this reference electrode between two membrane pieces, Ventosa et al.71 were able to gain valuable insights, as demonstrated in the study of ammoxidized graphite felts. Findings from this study revealed that the kinetics of the VO2+/VO2+ reaction are slower compared to the V2+/V3+ reaction at the electrode, contributing to limitations in voltage efficiency.
The through-plane voltage-loss profile of a VRFB and the changes in electrolyte conditions during practical charge–discharge cycles have also been explored in several studies.146,147 Results indicated that the anode half-cell reactions and the conditions of the anolyte (SoC, vanadium ion concentrations) significantly influenced overall VRFB performance. Traditional methods for calculating efficiencies (coulombic, voltage, and energy), based on total electron input and output, have limitations, as they do not account for energy storage efficiency and charge capacity decay during cycles. A new methodology was introduced to analyze capacity decay and coulombic efficiency by focusing on electrolytes and internal cell resistance. Correlating through-plane voltage losses, electrolyte conditions, and charge storage capacity evolution helped to quantify the effects of coulombic round-trip loss, vanadium ion crossover loss, concentration loss of the limiting electrolyte, and charge imbalance loss on the coulombic storage efficiency. This comprehensive analysis method offered a valuable tool for quantifying capacity losses and optimizing high-performance VRFB systems.146
A novel response time-based approach for VRFB was developed by Wang et al.148 to decouple the various types of polarization—ohmic, activation, and concentration—during RFB operation. It leverages the different time constants of each polarization type to identify the contributions of each type of polarization under different working conditions (e.g., flow rate, compression ratio, and current density). This allowed a more detailed analysis of voltage losses during charge and discharge cycles. Results by Wang et al.148 showed that ohmic polarization is the dominant factor of voltage loss in VRFB. The contribution of the ohmic polarization ranges from 49.6% to 78.8% depending on the type of membrane used in the study. Furthermore, the contribution of the activation polarization was determined to be 32.4%, but can be reduced to about 2.5% by chemically modifying the electrode.
Steady-state amperometry using potential step experiments with microelectrodes is highly accurate for monitoring the state of charge in RFBs. Traditionally, only part of the current response has been used for SoC and SoH assessment. A study by Schubert and co-workers149 showed that analyzing the transient chronoamperometric signal provides detailed physicochemical information, allowing the use of macroelectrodes instead of microelectrodes for both in situ and ex situ SoC measurements. The use of these macroelectrodes also enables temperature- and concentration-independent SoH assessments. The technique offers fast and cost-effective in operando SoC measurement without the need for restricting electrolyte flow, unlike previous methods. Additionally, the developed ex situ microelectrode chronoamperometric method allows the investigation of electrolyte lifetime, promising a high-throughput SoH assessment.149 The same group introduced also the redox targetivity as a new parameter to study organic redox mediators in TEMPO based flow batteries. In operando characterization of the redox target (or solid booster) via electrolyte SoC monitoring provided estimates for the redox target's SoC changes during cycling. In addition, targetivity was proposed to provide insights into the efficiency of the targeting reaction and the future optimization of materials, cell designs, and operational parameters for redox targeting RFBs (RTFBs).150
Another sensitive in operando technique for monitoring the SoC and average oxidation state of vanadium in VRFBs is coulometric analysis.151 This technique provides real-time data on electrolyte imbalance, which is crucial for understanding degradation processes such as ion crossover and side reactions. The sensors are integrated into the battery management system and can operate without calibration, making them ideal for long-time monitoring in industrial-scale applications. Additionally, potentiometric sensors are commonly used to track changes in the ratio of oxidized and reduced vanadium species, though they are sensitive to changes in the supporting electrolyte.152,153 A microelectrode based sensor in a symmetric redox flow cell was shown to effectively measure in operando concentrations, highlighting its utility in such applications.13 Key factors for successful implementation include attention to measurement protocols, careful material selection, and thoughtful flow cell design. Constructed from commercially available off-the-shelf components, the sensor can be easily adopted by research laboratories and integrated into existing experimental workflows, making it a valuable tool for investigating flow battery materials.
1.10. In situ and in operando tomography and radiography
X-ray computed tomography (CT) has proven instrumental in characterizing felt electrodes in operando. Jervis et al.73 designed a miniature flow cell enabling lab-based and synchrotron CT imaging to investigate carbon felt materials (Fig. 12a–d). This study revealed incomplete wetting of certain felt regions, resulting in areas of empty pores and electrolyte-wetted fibers. Advanced image processing techniques facilitated calculations of wetting amounts, pore size distribution, and contact angles. Moreover, X-ray CT enables the study of felts under varying in situ compression, providing insights into porosity, pore size distribution, and tortuosity. For instance, it was observed that significant fiber-fiber contact occurs only at 90% compression, while fiber contact with the current collector increases proportionally with compression-induced porosity reduction.154 Additionally, tomography and radiography are useful tools as they enable the visualization of the interfacial processes taking place during formation of dendrites which has a detrimental effect on RFBs performance. Different tomographic methods were employed by Yufit et al.72 to visualize dendritic growth in a zinc-air RFB in real time and to quantify various dendrite characteristics at submicron and nanoscale levels. Accordingly, they visualized the deposition of zinc dendrites onto a cone-shaped zinc anode when a negative current of 30 mA cm−2 was applied between the zinc anode and a carbon air cathode. Zinc dendrites are formed after reaching a critical overpotential. The sequence of dendrite formation is shown in Fig. 12e–i, where small dendrites appear on the tip of the electrode after 214 s of deposition. As the deposition continued, dendrites at various locations on the electrode (Fig. 12f and g) are obtained. After sufficient deposition, secondary dendrites are formed on the already grown dendrites (Fig. 12h and i). This growth pattern is influenced by local crystallography and current distribution. Thereafter, ternary dendrites are grown from the secondary dendrites' trunks (Fig. 12i). In addition, Gebhard et al.155 employed computer tomography to analyze bismuth doping of the carbon felts to improve the electrode kinetics. These studies focused on deposition and dissolution processes at the carbon felt surface during charging and discharging and helped to better understand deposition patterns in operando. | ||
| Fig. 12 Design of the miniature flow cell (a), with transparent view showing internal workings of the cell (b). The path of electrolyte and working part of the cell (membrane, felt electrodes, and current collection) is shown in (c), and details of the imaging region, including the sealing that separates the two electrodes achieved by clamping the internal body (blue) onto a shelf created in the external body of the cell (red) are shown in (d). O-rings prevent electrolyte from flowing through the gap between the internal body and the adjustable piston.73 Reproduced with permission. Copyright 2016, IOP Science. (e)–(i) In operando tomography study of zinc dendrite growth, dissolution, and regrowth in the cell without separator at a current density of 30 mA cm−2. Zinc dendrite growth after (e) 214 s, (f) 388 s, (g) 561 s, (h) 734 s, and (i) 918 s.72 Reproduced with permission. Copyright 2019, Elsevier Publication. | ||
Neutron imaging was also employed to measure spatial and temporal concentration variations in an operating redox flow cell.156 By using the properties of redox-active organic materials and boron-containing electrolytes, subtractive neutron imaging was conducted to map concentration profiles across electrodes. This allowed linking concentration to cell performance through polarization experiments with changing conditions. Time-of-flight neutron imaging further clarified species and electrolyte concentrations during operation. This method provided insights into how cell polarity, voltage, and flow rate affect concentration distributions and correlated these with overall performance, offering new understanding in reactive mass transport. This technique can be applied to various (electro)chemical systems, aiding in the development of new materials and reactor designs. Boz et al.157 also employed in operando neutron radiography to monitor how electrolyte distributes within porous carbon electrodes in RFBs. The high sensitivity of neutrons to hydrogen enables this technique to distinguish between the aqueous electrolyte and the carbon electrode. This provided valuable insight into how wetting behavior changes with variations in flow rate, flow field geometry, and electrode surface treatment. However, because of the limited spatial resolution of neutron radiography, it is unable to capture interfacial phenomena like pore-level wetting dynamics or electrochemical reactions at the electrode–electrolyte interface. To overcome this limitation, neutron imaging was combined with electrochemical techniques such as EIS and CV. These methods were used to measure the double-layer capacitance, which serves as a proxy for the electrochemical surface area and reflects how well the electrolyte contacts the internal surfaces of the electrode.157 This novel combination enabled the researchers to obtain a detailed picture of how wetting behavior impacts overall battery performance.
To directly observe microporous mass transport in vanadium-based electrolytes, Tariq et al.158 employed real-time in situ X-ray tomography at high resolution. Their work identified mechanisms of electrolyte flow behavior and interactions with porous carbon electrodes during initial wetting. They confirmed that non-uniform infiltration arises from the evolving surface chemistry of carbon fibers, which become more hydrophilic over time. Controlling surface functional groups may enable optimized fluid flow into electrodes, presenting opportunities for tailored electrode designs. These advances in tomographic and radiographic techniques not only enhance the understanding of structural and transport phenomena in redox flow batteries but also provide actionable insights for optimizing material properties and operational parameters, thereby paving the way for more efficient and durable energy storage systems.
1.11. In situ and in operando MS
MS allows for in situ analysis of gases released from the RFB cells while being operated. Typically, the gases evolving from the working electrode are collected in an upside-down funnel above the electrode with the rim of the funnel just below the surface of the electrolyte. Using helium or argon as carrier gases the gaseous products can be transported into the mass spectrometer. The partial pressure of the produced gases can be monitored as a function of time and subsequently quantified. A scheme showing the elements is depicted in Fig. 13.159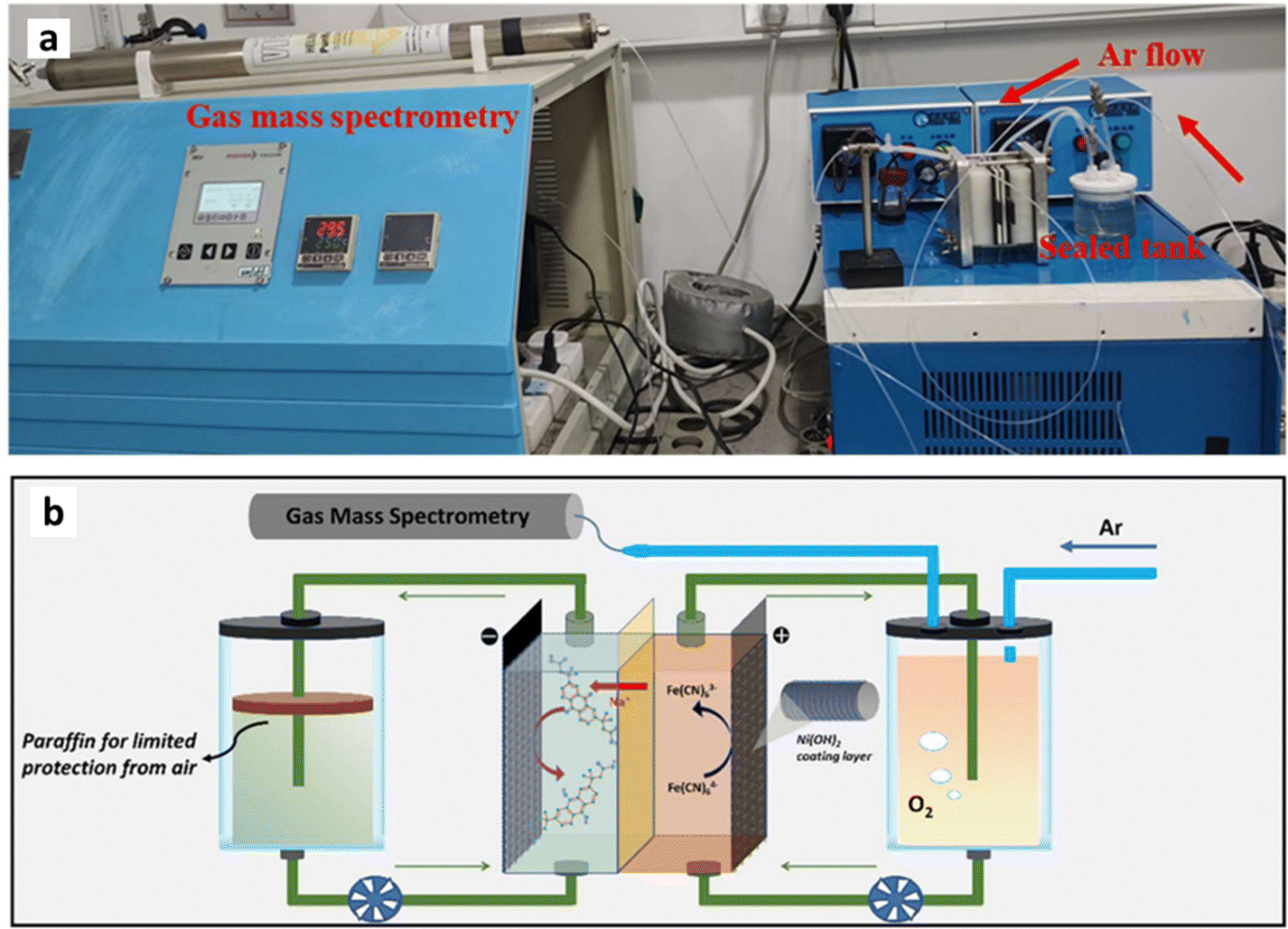 | ||
| Fig. 13 (a) Device and (b) schematic of a flow battery single cell linked to a mass spectrometer.159 Reproduced with permission from Wiley-VCH, Copyright 2022. | ||
While selectivity and sensitivity are major advantages of MS for analysis of mixtures of products from chemical processes in RFBs, time delay between sampling and detection due to sample transfer to the mass spectrometer represents a limitation for real-time monitoring applications or detection of time-evolving products. Gas evolution typically occurs on the electrode surface due to undesired secondary reactions such as decomposition of the electrolyte or degradation of the electrodes during operation of the RFB and hence it is important to be evaluated. Thus, Liu et al.57 conducted an on-line MS study of the electrochemical corrosion of the graphite electrode for VRFB. By monitoring and analyzing the composition of the gases evolved from the graphite anode during anodic polarization in 2 M H2SO4 + 2 M VOSO4, it was possible to gain insight into the electrochemical corrosion of the graphite electrode for VRB. Evolved gases were collected in an upside-down funnel placed above the electrode with the rim of the funnel just below the electrolyte surface and carried by helium gas to the mass spectrometer. The results showed preferred formation of CO2 and CO over O2 on the graphite anode due to electrochemical corrosion of the graphite electrode. The investigation also revealed that the O2 evolution rate is the highest one among all evolved gases if the polarization potential becomes > 1.8 V. Oxidation of VO2+ to VO2+ on the graphite electrode in the electrolyte with high concentration of VO2+ hindered carbon oxidation reaction, retarding the electrochemical corrosion of the graphite electrode. Overall, the results highlight the power of on-line MS to characterize reaction products of parasitic reactions at the electrodes.
Later on, Sun et al.74 demonstrated that MS can be a quantitative technique to determine the hydrogen evolution rate occurring at the negative carbon electrode of a VRFB. Samples were collected using a gas tight syringe flushed with argon gas prior to sampling and immediately injected into the mass spectrometer through a septum into the helium gas carrier. MS measurements confirmed that the evolved gas was primarily hydrogen and allowed quantifying the evolution rate. The performance of two types of carbon papers as electrodes was compared, concluding that the higher surface area electrode led to a higher hydrogen evolution rate.
More recently, operando MS experiments were performed in situ using a custom-made two-compartment electrochemical cell (H-cell) connected to an online electrochemical MS system to explore reaction products and intermediates during cycling of different organic electrolytes, such as anthraquinone derivatives59 and bispyridinium compounds.99 To sample the headspace of the anolyte in both works, the top plunger of the anolyte compartment of the designed cell was fitted with two stainless steel tubes connected to a gas line through double-shut-off quick connects. The flow of the argon carrier gas was controlled by a mass-flow controller and a pressure controller. After passing through the headspace of the anolyte, the sample gas was fed to the mass spectrometer through a heated capillary to prevent condensation. In the former report, evolution of D2 during a stepped-potential experiment in DHAQ anolyte was monitored, which was attributed to water reduction by reaction with reduced DHAQ, according to the reaction.59
| DHAQ4− + 2D2O → DHAQ2− + D2 + 2OD−. |
1.12. In situ and in operando fluorescence microscopy
Optical fluorescence microscopy has been demonstrated to provide both high spatial and temporal resolution in monitoring redox-dependent fluorescence within flowing electrolytes.76 By coupling fluorescence microscopy with electrochemistry, a direct observation of redox-active quinones' reactions and transport within porous carbon electrodes in real-time was performed.75 The cell design needs to accomplish optical transparence, which is realized by using transparent polymeric materials such as polyacrylics. A scheme of a microfluidic setup is depicted in Fig. 14.160 | ||
| Fig. 14 Microfluidic flow cell compatible with electrochemistry and confocal microscopy. (a) Scheme of the individual components. (b) Photographs of the assembled cell from above (top) and below (bottom). The working electrode (3 × 6 mm2) is visible in the center of the bottom image. For the bottom picture a working electrode made of gold was used to ensure visibility against the counter electrode in the background.160 Reproduced with permission. Copyright 2024, RSC Publication. | ||
Surprisingly, several porous electrodes exhibited electrolyte channeling, creating distinct regions dominated by advection and diffusion. For instance, Fig. 15 illustrates fluid flow dynamics of fully reduced H2AQDS (red) and fully oxidized AQDS (blue) electrolytes within 4 types of electrodes. Non-uniform flow is observed, with stagnant regions (red) where H2AQDS persists due to diffusion-dominated transport. These variations in flow patterns across different electrode designs highlight significant differences in mass transport properties, even among electrodes with similar physical characteristics. This finding challenges the typical assumption that transport in porous electrodes can be modeled with homogeneous Darcy-like permeability, especially at length scales critical to systems like redox flow batteries. This research introduced a new methodology for gaining detailed spatial and temporal insights into electrolyte reactions and transport behaviors within porous electrodes.75
 | ||
| Fig. 15 Time-based advection profile of 4 carbon paper electrodes across the middle land (a)–(p). The AQDS electrolyte is fully reduced (red) within the cell at t = 0. Fully oxidized electrolyte (blue) is then forced through the cell with a syringe pump at 100 mL h−1. Scale bars, 1 mm.75 Reproduced with permission. Copyright 2021, Cell Press. | ||
In operando quantitative electrochemical fluorescence state of charge mapping (QEFSM), a non-invasive method for studying active electrochemical systems has also been reported. This approach was complemented by a novel design of optically transparent microfluidic redox flow cells. QEFSM enabled quantitative mapping of the concentration of specific oxidation states of redox-active species within a porous electrode during its operation. In particular, confocal microscopy to map the fluorescence signal of the reduced form of 2,7-anthraquinone disulfonate (AQDS) in a series of multistep chronoamperometry experiments was employed.76,160 A similar concept was exploited for electrokinetic investigations of the 5,10-bis(2-methoxyethyl)-5,10-dihydrophenazine (BMEPZ) catholyte, recently proposed as a high-performance multiredox organic molecule. By leveraging BMEPZ's inherent colorimetric properties, the study revealed the intrinsic electrochemical properties in terms of charge and mass transfer kinetics during the multiredox reaction. This was achieved through in operando visualization, allowing for a theoretical examination of physicochemical hydrodynamics in electrochemical systems. Based on insights into electrokinetic limitations in RFBs, the study validated an electrode geometry design that minimized the depletion region, leading to enhanced cell performance.161
1.13. Other analytics
Next to the more common analytics, in operando techniques also include the measurement of the acoustic attenuation coefficient with an ultrasonic probing system to determine the SoC.162 The acoustic attenuation coefficient is only little affected by temperature variations, making it reliable for applications where temperature control is challenging. Hydrogen evolution is a significant side reaction in VRFBs affecting long-term cycling, which has also been addressed using a noninvasive ultrasonic probing method. This online technique monitors the SoC, predicts hydrogen generation, and detects hydrogen gas bubbles in anolyte solutions through a pulse-echo method that measures sound speed and the acoustic attenuation coefficient.163 In both static offline and online operando experiments, hydrogen gas presence led to notable changes in these measurements. Variations in the acoustic attenuation coefficient were highly correlated with gas flow rates. This method provides insights into the SoC and health of flow battery systems during operation.By employing a platform that combines dark-field light microscopy (DFLM) with a transparent electrochemical cell (Fig. 16a), Wu et al.164 investigated a zinc-bromine RFB and were able to observe the dynamics of polybromide formation in their native environment, capturing key phenomena such as the liquid nature of the charge products, their nucleation and growth behaviors, and interactions with the Pt electrode surface.164 Using a small current density (0.066 mA cm−1), sparsely distributed polybromide products were generated for easy tracing (Fig. 16b). A surface defect (circled in Fig. 16b(i)) ensured consistent focus during the experiment. At 5 minutes, yellow spherical microstructures (2–3 μm in size) are formed on the Pt surface and grew to approximately 10 μm by the end of the charging process (Fig. 16b(iv)). Neighboring droplets merged into larger ones to minimize surface energy (Fig. 16c(i, ii) and Fig. 16c(iii, iv)), showcasing their liquid nature, as evidenced by rapid merging (<1 s) and shape relaxation. During discharge (Fig. 16d(i–iv)), the droplets were reduced to Br− ions, shrinking anisotropically by maintaining lateral dimensions while decreasing in thickness. They gradually became transparent (Fig. 9d(iii)) before disappearing (Fig. 16d(iii)).
 | ||
| Fig. 16 (a) Schematic representation of the cell and microscope for the in operando visualization of bromine electrochemistry. Inset: Photograph of the setup. (b) Time-lapse DFLM images showing the formation of polybromide droplets upon charging. (c) Two sets of images recording the rapid merging of polybromide droplets, indicating their liquid nature. (d) Time-lapse DFLM images during discharging of the same cell as in (b).164 Reproduced with permission. Copyright 2019, Wiley-VCH. | ||
This DFLM approach also provided insights into critical issues like self-discharge and overcharging-induced side reactions, enhancing the understanding of complexing agents' roles and guiding future zinc-bromine RFB design improvements. Thus, it enables a deeper mechanistic understanding of electrochemical systems that are otherwise difficult to analyze due to sensitivity to environmental conditions.146
A novel imaging-based diagnostic approach for VRFBs was introduced by Ma et al.165 who developed a total internal reflection imaging (TIRi) sensor capable of mapping local electrochemical activity and reversibility on graphite felt electrodes with single-fiber resolution (∼16 μm). The TIRi system detects changes in refractive index at the electrolyte-electrode interface caused by redox reactions during CV, allowing quantification of peak oxidation and reduction currents, the reversibility ratio, and peak potential separation (ΔE). Their method successfully distinguished the electrochemical performance of pristine, thermally treated, and porous graphite felts, showing that higher surface area and activation improved both activity and uniformity. Beyond static mapping, the system captured dynamic bubble formation from oxygen evolution reaction (OER) in real-time, correlating bubble growth kinetics with cycle number and electrode surface characteristics. Importantly, the technique enabled spatial correlation between bubble generation zones and regions with high local activity and large ΔE, linking overpotential-induced side reactions (like OER) directly to inhomogeneous electrode behavior. This visualization capability supports the identification of degradation hotspots and facilitates design improvements in electrode architecture and flow field management.165 The TIRi method represents a significant advancement in non-invasive, high-resolution, in situ characterization tools for understanding spatially resolved kinetics in VRFBs.
Recent progress in in situ electrolyte diagnostics has introduced refractive index (RI) monitoring as a powerful tool for real-time assessment of VRFB health. Vlasov et al.166 proposed a Fresnel-based optical RI sensor integrated with an OCV cell to determine the electrolyte's SoH without relying on cycling history. By measuring RI shifts at fixed OCV points (particularly around 1.4 V), and applying a linear calibration model, they achieved a mean prediction error as low as 1.83%, with a maximum root mean square error of 4.45%. The sensor, based on reflection coefficient measurements at the fiber-electrolyte interface, is compact, calibration-friendly, and does not require bypass channels or spectral deconvolution, making it suitable for industrial VRFB monitoring systems. Notably, their method proved sensitive to the effects of vanadium ion crossover, preferential water transfer, and imbalance processes that typically distort coulomb-based SoH estimation. A major practical outcome of this method is its ability to predict capacity fading or trigger rebalance operations mid-cycle, using instantaneous OCV and RI values, thus enabling predictive maintenance and dynamic adjustment of operational protocols. This innovation adds a valuable dimension to in operando flow battery diagnostics and has strong implications for electrolyte rebalancing strategies and smart battery management systems.
2. Perspectives and future challenges
In the past ten years, exciting developments have been made in the field of in situ and in operando techniques for RFB analytics. Much of that progress has been inspired by the rapid development of lithium battery technology concomitant with the need to understand degradation processes. Still, the main challenge in RFBs is to ensure long term stability (20 years+) without substantial degradation of any of the battery components, which is hardly achieved for any flow battery chemistry so far. This point is important for various reasons but the major factor is that flow batteries feature high capital costs, requiring trust of the consumers to invest in the future. Therefore, longevity of the battery system is a must to mass-commercialize this technology. While in operando technologies just emerged in the RFB field about ten years ago, they mostly have been employed in lab scale single cell systems. While there has been a lot of progress here, there are still multiple challenges and further developments necessary, particularly with respect to novel battery chemistries and the need for improved analysis methods. Novel chemistries may open new ways of analysis for in operando studies. For example, novel redox active molecules carrying NMR active nuclei such as 31P have been very recently used as probes to study decomposition pathways ex situ and it is obvious that in operando setups can be employed for analysis of these systems as well.45,120,167,168 Like that, also redox active molecules carrying NMR active 19F may be feasible in future RFB technology. Future improvements in imaging techniques or integrating composite absorption with phase contrast methodologies may overcome limitations concerning the distinction between electrolyte and carbon felts due to their similar attenuation coefficients.169 Again, the introduction of markers with high atomic number (e.g., iodine) may be a strategy to allow the study of deposits or reactions taking place at the felt and to investigate the effect of compression on electrolyte flow. While it is highly important to understand the underlying processes and phenomena on a single cell level, in the operation of a real battery, there are additional effects contributing to the complexity of the system (e.g., shunt currents).170 A next logical step is therefore to explore multi-cell setups, employing the toolbox developed by researchers in the past years. It is obvious that such studies are more challenging to perform and will require even more engineering efforts and further adaptation of setups and experiments than needed already for single cell setups. In addition, parameters such as pressure drop and flow factor can play, depending on the used chemistry, a more important role in multicell setups.171 While these approaches are following engineering logics, the use of artificial intelligence (AI) will probably allow to make progress in the analysis and the design of in operando techniques.172,173 So far designs have been based on classic engineering approaches. The adaptation of AI based methods in the design of novel in operando setups is not a question of if, but when. AI derived inputs could comprise the design of novel sensor geometries, as well as experiments to monitor the battery. In addition, AI based methods may be used in data analysis to unravel non-linear correlations difficult for humans to recognize. AI applications in that field, however, must also overcome challenges related to experimental validation and computational costs. Overall, the use of AI based methods will not be the holy grail to solve the challenges of RFB technology development, but it will make a substantial contribution to advance the field in the years to come.Future research should prioritize the development of instrumentation such as customized flow cell setups specifically designed for easier integration/coupling into RFBs with minimal interference with battery components. Addressing issues of sensitivity, calibration, and real-time adaptability will be crucial to advance these in situ and in operando methods and realize their full potential as tools for improving RFB performance at industrial scales.
3. Conclusion
To accelerate the large-scale industrial adaptation of RFBs, in situ and in operando analyses are crucial for unraveling complex electrochemical and chemical processes, which enable improvements in efficiency and battery lifespan. Although real-time analytical methods have been widely applied to LIBs, they remain moderately explored in RFBs. Notably, the majority of in situ and in operando studies on RFBs have emerged only since 2013. By reviewing the literature, we have tracked advancements of these techniques and their impact on RFB optimization.In situ and in operando NMR have emerged as powerful tools, enabling researchers to monitor the formation of diamagnetic intermediate species in the electrolyte during battery operation, and to elucidate electrolyte composition changes in response to voltage variations. One significant challenge with NMR, however, is the complexity of integrating the RFB cell into the probe, making real-time monitoring difficult. EPR serves as an excellent complementary technique to NMR for tracking paramagnetic species. Like NMR, it also faces challenges in integrating operational cells. The use of UV-vis spectroscopy has proven to be a powerful tool for real-time monitoring of both SoC and total concentration of active electrolyte species, both in VRFBs and ORFBs. However, highly concentrated systems and light-absorbing solvents may introduce challenges. Raman spectroscopy, despite its power for monitoring electrode surfaces, electrolyte composition and catalytic reactions, faces shortcomings due to limited sensitivity, resolution, and background noise, often requiring enhancement techniques for broader application. Similarly, FTIR spectroscopy is useful for probing electrochemical mechanisms but often requires complex ATR-FTIR electrochemical setups. X-ray-based techniques, such as XAS, can provide valuable insights into the structure of both the electrolyte and electrode surfaces, though high-cost infrastructures and limited sensitivity to light atoms constrain their widespread use. EIS offers details on the SoH of the battery as well as helps to analyze voltage losses in the battery systems to determine limiting factors, though it is restricted by the frequency range and low resolution at low frequencies. Advanced imaging techniques such as tomography and radiography allow for high-resolution 3D imaging of electrode structures, dendrite formation, and concentration gradients. MS enables the characterization of reaction products and intermediates in the electrolyte but is costly and requires customized sampling systems. Finally, fluorescence microscopy provides a unique approach for tracking redox-active species concentration and mass transport phenomena but requires fluorescent redox-active species.
Each of the presented techniques provides important insights into different aspects of RFB working mechanisms. However, challenges such as instrumentation complexity, cost, and integration into in situ and in operando setups remain. Future developments to address integration and sensitivity challenges are thus crucial for optimizing RFB performance and enabling large scale industrial implementation.
Abbreviations
| AI | Artificial intelligence |
| ATR-FTIR | Attenuated total reflection Fourier transform infrared spectroscopy |
| BE | Bulk electrolysis |
| CE | Counter electrode |
| CT | Computed tomography |
| CV | Cyclic voltammetry |
| DFLM | Dark-field light microscopy |
| DFT | Density functional theory |
| DRT | Distribution of relaxation times |
| EIS | Electrochemical impedance spectroscopy |
| EPR | Electron paramagnetic resonance |
| EXAFS | X-ray absorption fine structure |
| FTIR | Fourier transform infrared spectroscopy |
| LIBs | Lithium-ion batteries |
| MB | Methylene blue |
| MRI | Magnetic resonance imaging |
| MS | Mass spectrometry |
| NMR | Nuclear magnetic resonance |
| OCV | Open circuit voltage |
| OER | Oxygen evolution reaction |
| QEFSM | Fluorescence state of charge mapping |
| RE | Reference electrode |
| RFBs | Redox flow batteries |
| RI | Refractive Index |
| RMFBs | Redox-mediated flow batteries |
| SERS | Surface-enhanced Raman spectroscopy |
| SHE | Standard hydrogen electrode |
| SoC | State-of-charge |
| SoH | State-of-health |
| TERS | Tip-enhanced Raman spectroscopy |
| TIRi | Total internal reflection imaging |
| UV-vis | Ultraviolet-visible |
| VRFBs | Vanadium redox flow batteries |
| XANES | X-ray absorption near edge structure |
| XAS | X-ray absorption spectroscopy |
| XPS | X-ray photoelectron spectroscopy |
| WE | Working electrode |
| ZMRFB | Zn–Mn2+/MnO2 RFB |
Data availability
This review paper does not contain original research data. Figures and schemes are adapted with permission from research papers accessible via DOI which may be requested by the corresponding authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding from the European Innovation Council (EIC) under grant agreements No. 101115293 (VanillaFlow) and No. 101046742 (MeBattery), and by the Spanish Government (Ministerio de Ciencia e Innovacion, Grant PID2021-124974OB-C22).References
- M. Pan, M. Shao and Z. Jin, SmartMat, 2023, 4, e1198 CrossRef CAS.
- X. Wei, W. Pan, W. Duan, A. Hollas, Z. Yang, B. Li, Z. Nie, J. Liu, D. Reed, W. Wang and V. Sprenkle, ACS Energy Lett., 2017, 2, 2187–2204 CrossRef CAS.
- A. Khataee, J. Azevedo, P. Dias, D. Ivanou, E. Dražević, A. Bentien and A. Mendes, Nano Energy, 2019, 62, 832–843 CrossRef CAS.
- W. Schlemmer, P. Nothdurft, A. Petzold, G. Riess, P. Frühwirt, M. Schmallegger, G. Gescheidt-Demner, R. Fischer, S. A. Freunberger, W. Kern and S. Spirk, Angew. Chem., Int. Ed., 2020, 59, 22943–22946 CrossRef CAS PubMed.
- K. Teenakul, S. A. Ahmad Alem, R. Gond, A. Thakur, B. Anasori and A. Khataee, RSC Adv., 2024, 14, 12807–12816 RSC.
- M. Wu, M. Bahari, Y. Jing, K. Amini, E. M. Fell, T. Y. George, R. G. Gordon and M. J. Aziz, Batteries Supercaps, 2022, 5, e202200009 CrossRef CAS.
- M. Shoaib, P. Vallayil, N. Jaiswal, P. Iyapazham Vaigunda Suba, S. Sankararaman, K. Ramanujam and V. Thangadurai, Adv. Energy Mater., 2024, 14, 2400721 CrossRef CAS.
- Z. Hou, X. Chen, J. Liu, Z. Huang, Y. Chen, M. Zhou, W. Liu and H. Zhou, J. Power Sources, 2024, 601, 234242 CrossRef CAS.
- E. Ventosa, Curr. Opin. Chem. Eng., 2022, 37, 100834 CrossRef.
- C. Santana Santos, T. Quast, E. Ventosa and W. Schuhmann, ChemElectroChem, 2024, 11, e202400283 CrossRef CAS.
- Y. Shen, S. Wang, H. Li, K. Wang and K. Jiang, J. Energy Storage, 2023, 64, 107164 CrossRef.
- Y. Yang, J. Feijóo, V. Briega-Martos, Q. Li, M. Krumov, S. Merkens, G. Salvo, A. Chuvilin, J. Jin, H. Huang, C. J. Pollock, M. B. Salmeron, C. Wang, D. A. Muller, H. D. Abruña and P. Yang, Curr. Opin. Electrochem., 2023, 42, 101403 CrossRef.
- B. J. Neyhouse, K. M. Tenny, Y.-M. Chiang and F. R. Brushett, ACS Appl. Energy Mater., 2021, 4, 13830–13840 CrossRef CAS.
- R. Zeis, in Flow Batteries: From Fundamentals to Applications, ed. C. Roth, J. Noack and M. Skyllas-Kazacos, Wiley-VCH GmbH, 2023, pp. 263–280 Search PubMed.
- Z. Sun, J. Pan, W. Chen, H. Chen, S. Zhou, X. Wu, Y. Wang, K. Kim, J. Li, H. Liu, Y. Yuan, J. Wang, D. Su, D.-L. Peng and Q. Zhang, Adv. Energy Mater., 2024, 14, 2303165 CrossRef CAS.
- M. T. Shuja, S. Thatipamula, M. W. Khan, M. Haris, R. Babarao and N. Mahmood, Battery Energy, 2024, 3, 20230043 CrossRef CAS.
- R. A. Vicente, I. T. Neckel, S. K. R. S. Sankaranarayanan, J. Solla-Gullon and P. S. Fernández, ACS Nano, 2021, 15, 6129–6146 CrossRef CAS PubMed.
- M. Sathiya, J.-B. Leriche, E. Salager, D. Gourier, J.-M. Tarascon and H. Vezin, Nat. Commun., 2015, 6, 6276 CrossRef CAS PubMed.
- L. Xue, Y. Li, A. Hu, M. Zhou, W. Chen, T. Lei, Y. Yan, J. Huang, C. Yang, X. Wang, Y. Hu and J. Xiong, Small Struct., 2022, 3, 2100170 CrossRef.
- M. M. Amaral, C. G. Real, V. Y. Yukuhiro, G. Doubek, P. S. Fernandez, G. Singh and H. Zanin, J. Energy Chem., 2023, 81, 472–491 CrossRef CAS.
- J.-H. Tian, T. Jiang, M. Wang, Z. Hu, X. Zhu, L. Zhang, T. Qian and C. Yan, Small Methods, 2020, 4, 1900467 CrossRef CAS.
- L. Meyer, N. Saqib and J. Porter, J. Electrochem. Soc., 2021, 168, 90561 CrossRef.
- M. B. Dixit, J.-S. Park, P. Kenesei, J. Almer and K. B. Hatzell, Energy Environ. Sci., 2021, 14, 4672–4711 RSC.
- H. Li, S. Guo and H. Zhou, J. Energy Chem., 2021, 59, 191–211 CrossRef CAS.
- D. Liu, Z. Shadike, R. Lin, K. Qian, H. Li, K. Li, S. Wang, Q. Yu, M. Liu, S. Ganapathy, X. Qin, Q.-H. Yang, M. Wagemaker, F. Kang, X.-Q. Yang and B. Li, Adv. Mater., 2019, 31, e1806620 CrossRef PubMed.
- F. Strauss, D. Kitsche, Y. Ma, J. H. Teo, D. Goonetilleke, J. Janek, M. Bianchini and T. Brezesinski, Adv. Energy Sustainability Res., 2021, 2, 2100004 CrossRef CAS.
- J. Tan, D. Liu, X. Xu and L. Mai, Nanoscale, 2017, 9, 19001–19016 RSC.
- E. Šić, D. Fredericks, O. Pecher, S. Wegner, H. Breitzke, V. Singh, G. Buntkowsky and T. Gutmann, Appl. Magn. Reson., 2024, 55, 575–583 CrossRef.
- O. Pecher, J. Carretero-González, K. J. Griffith and C. P. Grey, Chem. Mater., 2017, 29, 213–242 CrossRef CAS.
- O. Nolte, I. A. Volodin, C. Stolze, M. D. Hager and U. S. Schubert, Mater. Horiz., 2021, 8, 1866–1925 RSC.
- Y. A. Gandomi, D. S. Aaron, J. R. Houser, M. C. Daugherty, J. T. Clement, A. M. Pezeshki, T. Y. Ertugrul, D. P. Moseley and M. M. Mench, J. Electrochem. Soc., 2018, 165, A970 CrossRef CAS.
- L. Zhang, R. Feng, W. Wang and G. Yu, Nat. Rev. Chem., 2022, 6, 524–543 CrossRef PubMed.
- P. Arévalo-Cid, P. Dias, A. Mendes and J. Azevedo, Sustainable Energy Fuels, 2021, 5, 5366–5419 RSC.
- P. Leung, X. Li, C. Ponce de León, L. Berlouis, C. T. J. Low and F. C. Walsh, RSC Adv., 2012, 2, 10125–10156 RSC.
- E. Sánchez-Díez, E. Ventosa, M. Guarnieri, A. Trovò, C. Flox, R. Marcilla, F. Soavi, P. Mazur, E. Aranzabe and R. Ferret, J. Power Sources, 2021, 481, 228804 CrossRef.
- M. Skyllas-Kazacos, L. Cao, M. Kazacos, N. Kausar and A. Mousa, ChemSusChem, 2016, 9, 1521–1543 CrossRef CAS PubMed.
- J. Ren, Y. Li, Z. Wang, J. Sun, Q. Yue, X. Fan and T. Zhao, Int. J. Heat Mass Transfer, 2023, 203, 123818 CrossRef CAS.
- D. I. Kushner, A. R. Crothers, A. Kusoglu and A. Z. Weber, Curr. Opin. Electrochem., 2020, 21, 132–139 CrossRef CAS.
- D. G. Kwabi, Y. Ji and M. J. Aziz, Chem. Rev., 2020, 120, 6467–6489 CrossRef CAS PubMed.
- T. A. Hamlin, M. Swart and F. M. Bickelhaupt, ChemPhysChem, 2018, 19, 1315–1330 CrossRef CAS PubMed.
- D. P. Tabor, R. Gómez-Bombarelli, L. Tong, R. G. Gordon, M. J. Aziz and A. Aspuru-Guzik, J. Mater. Chem. A, 2019, 7, 12833–12841 RSC.
- C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49–82 RSC.
- R. Rubio-Presa, L. Lubián, M. Borlaf, E. Ventosa and R. Sanz, ACS Mater. Lett., 2023, 5, 798–802 CrossRef CAS PubMed.
- A. Orita, M. G. Verde, M. Sakai and Y. S. Meng, J. Power Sources, 2016, 321, 126–134 CrossRef CAS.
- M. Egger, I. Koehne, D. Wickenhauser, W. Schlemmer, S. Spirk and R. Pietschnig, ACS Omega, 2023, 8, 10899–10905 CrossRef CAS PubMed.
- B. Ambrose, R. Naresh, S. Deshmukh, M. Kathiresan and P. Ragupathy, Energy Fuels, 2023, 37, 18226–18242 CrossRef CAS.
- J. Maier, R. Yagmur, D. Wickenhauser, A. Torvisco, A.-M. Kelterer and S. Spirk, J. Electrochem. Soc., 2024, 171, 116504 CrossRef CAS.
- A. Pasadakis-Kavounis, V. Baj and J. Hjelm, J. Electrochem. Soc., 2024, 171, 20501 CrossRef CAS.
- C. Wiberg, T. J. Carney, F. Brushett, E. Ahlberg and E. Wang, Electrochim. Acta, 2019, 317, 478–485 CrossRef CAS.
- T. J. Carney, S. J. Collins, J. S. Moore and F. R. Brushett, Chem. Mater., 2017, 29, 4801–4810 CrossRef CAS.
- Q. Chen, L. Eisenach and M. J. Aziz, J. Electrochem. Soc., 2016, 163, A5057 CrossRef CAS.
- M. R. Gerhardt, E. S. Beh, L. Tong, R. G. Gordon and M. J. Aziz, MRS Adv., 2017, 2, 431–438 CrossRef CAS.
- S. Jin, Y. Jing, D. G. Kwabi, Y. Ji, L. Tong, D. Porcellinis, M.-A. Goulet, D. A. Pollack, R. G. Gordon and M. J. Aziz, ACS Energy Lett., 2019, 4, 1342–1348 CrossRef CAS.
- M.-A. Goulet and M. J. Aziz, J. Electrochem. Soc., 2018, 165, A1466 CrossRef CAS.
- S. Stefan, F. Belaj, T. Madl and R. Pietschnig, Eur. J. Inorg. Chem., 2010, 289–297 CrossRef CAS.
- O. Nolte, P. Rohland, N. Ueberschaar, M. D. Hager and U. S. Schubert, J. Power Sources, 2022, 525, 230996 CrossRef CAS.
- H. Liu, Q. Xu and C. Yan, Electrochem. Commun., 2013, 28, 58–62 CrossRef.
- J. Friedl, M. V. Holland-Cunz, F. Cording, F. L. Pfanschilling, C. Wills, W. McFarlane, B. Schricker, R. Fleck, H. Wolfschmidt and U. Stimming, Energy Environ. Sci., 2018, 11, 3010–3018 RSC.
- E. W. Zhao, T. Liu, E. Jónsson, J. Lee, I. Temprano, R. B. Jethwa, A. Wang, H. Smith, J. Carretero-González, Q. Song and C. P. Grey, Nature, 2020, 579, 224–228 CrossRef CAS PubMed.
- Y. Jing, E. W. Zhao, M.-A. Goulet, M. Bahari, E. M. Fell, S. Jin, A. Davoodi, E. Jónsson, M. Wu, C. P. Grey, R. G. Gordon and M. J. Aziz, Nat. Chem., 2022, 14, 1103–1109 CrossRef CAS PubMed.
- E. W. Zhao, E. Jónsson, R. B. Jethwa, D. Hey, D. Lyu, A. Brookfield, P. A. A. Klusener, D. Collison and C. P. Grey, J. Am. Chem. Soc., 2021, 143, 1885–1895 CrossRef CAS PubMed.
- T. C. Gokoglan, S. K. Pahari, A. Hamel, R. Howland, P. J. Cappillino and E. Agar, J. Electrochem. Soc., 2019, 166, A1745 CrossRef CAS.
- K.-H. Shin, C.-S. Jin, J.-Y. So, S.-K. Park, D.-H. Kim and S.-H. Yeon, J. Energy Storage, 2020, 27, 101066 CrossRef.
- J. Lan, K. Li, L. Yang, Q. Lin, J. Duan, S. Zhang, X. Wang and J. Chen, ACS Nano, 2023, 17, 20492–20501 CrossRef CAS PubMed.
- X. Zhang, A. Valencia, W. Li, K. Ao, J. Shi, X. Yue, R. Zhang and W. A. Daoud, Adv. Mater., 2024, 36, 2305415 CrossRef CAS PubMed.
- W. Kautek, A. Conradi, M. Sahre, C. Fabjan, J. Drobits, G. Bauer and P. Schuster, J. Electrochem. Soc., 1999, 146, 3211 CrossRef CAS.
- S. Huang, H. Zhang, M. Salla, J. Zhuang, Y. Zhi, X. Wang and Q. Wang, Nat. Commun., 2022, 13, 4746 CrossRef CAS PubMed.
- C. Jia, Q. Liu, C.-J. Sun, F. Yang, Y. Ren, S. M. Heald, Y. Liu, Z.-F. Li, W. Lu and J. Xie, ACS Appl. Mater. Interfaces, 2014, 6, 17920–17925 CrossRef CAS PubMed.
- F. Paparoni, G. Alizon, A. Zitolo, S. J. Rezvani, A. Di Cicco, H. Magnan and E. Fonda, Phys. Chem. Chem. Phys., 2024, 26, 3897–3906 RSC.
- K. Amini and M. D. Pritzker, J. Power Sources, 2020, 471, 228463 CrossRef CAS.
- E. Ventosa, M. Skoumal, F. J. Vázquez, C. Flox and J. R. Morante, J. Power Sources, 2014, 271, 556–560 CrossRef CAS.
- V. Yufit, F. Tariq, D. S. Eastwood, M. Biton, B. Wu, P. D. Lee and N. P. Brandon, Joule, 2019, 3, 485–502 CrossRef.
- R. Jervis, L. D. Brown, T. P. Neville, J. Millichamp, D. P. Finegan, T. M. M. Heenan, D. J. L. Brett and P. R. Shearing, J. Phys. D: Appl. Phys., 2016, 49, 434002 CrossRef.
- C.-N. Sun, F. M. Delnick, L. Baggetto, G. M. Veith and T. A. Zawodzinski, J. Power Sources, 2014, 248, 560–564 CrossRef CAS.
- A. A. Wong, S. M. Rubinstein and M. J. Aziz, Cell Rep. Phys. Sci., 2021, 2, 100388 CrossRef CAS.
- A. A. Wong, M. J. Aziz and S. Rubinstein, ECS Trans., 2017, 77, 153 CrossRef CAS.
- P. A. Keifer, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Academic Press, England, 2007, pp. 1–47 Search PubMed.
- F. Dalitz, M. Cudaj, M. Maiwald and G. Guthausen, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 60, 52–70 CrossRef CAS PubMed.
- Z. Zhu, R. Luo and E. W. Zhao, Magn. Reson. Lett., 2024, 4, 100096 CrossRef CAS.
- M. V. Gomez and A. La Hoz, Beilstein J. Org. Chem., 2017, 13, 285–300 CrossRef CAS PubMed.
- A. Friebel, E. Harbou, K. Münnemann and H. Hasse, Ind. Eng. Chem. Res., 2019, 58, 18125–18133 CrossRef CAS.
- P. Giraudeau and F.-X. Felpin, React. Chem. Eng., 2018, 3, 399–413 RSC.
- R. Kircher, S. Mross, H. Hasse and K. Münnemann, Appl. Magn. Reson., 2023, 54, 1555–1569 CrossRef CAS.
- A. Friebel, T. Specht, E. Harbou, K. Münnemann and H. Hasse, J. Magn. Reson., 2020, 312, 106683 CrossRef CAS PubMed.
- N. Zientek, K. Meyer, S. Kern and M. Maiwald, Chem. Ing. Tech., 2016, 88, 698–709 CrossRef CAS.
- P. A. Keifer, Curr. Opin. Chem. Biol., 2003, 7, 388–394 CrossRef CAS PubMed.
- A. Salgado, in Liquid Chromatography (Third Edition): Handbooks in Separation Science, ed. S. Fanali, B. Chankvetadze, P. R. Haddad, C. F. Poole and M.-L. Riekkola, Elsevier, 2023, pp. 743–794 Search PubMed.
- R. Kircher, H. Hasse and K. Münnemann, Anal. Chem., 2021, 93, 8897–8905 CrossRef CAS PubMed.
- D. Graafen, S. Ebert, O. Neudert, L. Buljubasich, M. B. Franzoni, J. F. Dechent and K. Münnemann, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Academic Press, England, 2014, pp. 167–215 Search PubMed.
- M. Hunger, Prog. Nucl. Magn. Reson. Spectrosc., 2008, 53, 105–127 CrossRef CAS.
- Z. Zhao, T. Li, C. Zhang, M. Zhang, S. Li and X. Li, Nat. Sustainability, 2024, 7, 1273–1282 CrossRef.
- B. Wu, R. L. E. G. Aspers, A. P. M. Kentgens and E. W. Zhao, J. Magn. Reson., 2023, 351, 107448 CrossRef CAS PubMed.
- B. Caja-Munoz, K. Chighine, J.-P. Dognon, L. Dubois and P. Berthault, Anal. Chem., 2023, 95, 6020–6028 CrossRef CAS PubMed.
- H. Nguyen and R. J. Clément, ACS Energy Lett., 2020, 5, 3848–3859 CrossRef CAS.
- X. Sang, X. Xu, Z. Bu, S. Zhai, Y. Sun, M. Ruan and Q. Li, Magnetochemistry, 2023, 9, 63 CrossRef CAS.
- B. Wang, W. Wang, K. Sun, Y. Xu, Y. Sun, Q. Li, H. Hu and M. Wu, Nano Res., 2023, 16, 11992–12012 CrossRef CAS.
- E. W. Zhao, E. J. K. Shellard, P. A. A. Klusener and C. P. Grey, Chem. Commun., 2022, 58, 1342–1345 RSC.
- R. B. Jethwa, D. Hey, R. N. Kerber, A. D. Bond, D. S. Wright and C. P. Grey, ACS Appl. Energy Mater., 2024, 7, 414–426 CrossRef CAS PubMed.
- M. E. Carrington, K. Sokołowski, E. Jónsson, E. W. Zhao, A. M. Graf, I. Temprano, J. A. McCune, C. P. Grey and O. A. Scherman, Nature, 2023, 623, 949–955 CrossRef CAS PubMed.
- R. Feng, X. Zhang, V. Murugesan, A. Hollas, Y. Chen, Y. Shao, E. Walter, N. P. N. Wellala, L. Yan, K. M. Rosso and W. Wang, Science, 2021, 372, 836–840 CrossRef CAS PubMed.
- Y. Zhang, F. Li, T. Li, M. Zhang, Z. Yuan, G. Hou, J. Fu, C. Zhang and X. Li, Energy Environ. Sci., 2023, 16, 231–240 RSC.
- J. S. Lawton, A. Jones and T. Zawodzinski, J. Electrochem. Soc., 2013, 160, A697 CrossRef CAS.
- A. A. Maurice, A. E. Quintero and M. Vera, Electrochim. Acta, 2024, 482, 144003 CrossRef CAS.
- H. Sun, H. Takahashi, N. Nishiumi, Y. Kamada, K. Sato, K. Nedu, Y. Matsushima, A. Khosla, M. Kawakami, H. Furukawa, P. Stadler and T. Yoshida, J. Electrochem. Soc., 2019, 166, B3125 CrossRef.
- S. V. Modak, D. Pert, J. L. Tami, W. Shen, I. Abdullahi, X. Huan, A. J. McNeil, B. R. Goldsmith and D. G. Kwabi, J. Am. Chem. Soc., 2024, 146, 5173–5185 CrossRef CAS PubMed.
- J. Cao, K. Yu, J. Zhang, B. Lu, J. Yu, S. Huang and F. Zhang, ACS Sustainable Chem. Eng., 2024, 12, 6320–6329 CrossRef CAS.
- P. Loktionov, R. Pichugov, D. Konev, M. Petrov, A. Pustovalova and A. Antipov, J. Electroanal. Chem., 2022, 925, 116912 CrossRef CAS.
- L. Tong, Q. Chen, A. A. Wong, R. Gómez-Bombarelli, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Phys. Chem. Chem. Phys., 2017, 19, 31684–31691 RSC.
- S. Park, H. Lee, H. J. Lee and H. Kim, J. Power Sources, 2020, 451, 227746 CrossRef CAS.
- D. Yu, L. Zhi, F. Zhang, Y. Song, Q. Wang, Z. Yuan and X. Li, Adv. Mater., 2023, 35, 2209390 CrossRef CAS PubMed.
- J. Lei, Y. Zhang, Y. Yao, Y. Shi, K. L. Leung, J. Fan and Y.-C. Lu, Nat. Energy, 2023, 8, 1355–1364 CrossRef CAS.
- M. Zhou, Q. Huang, T. N. Pham Truong, J. Ghilane, Y. G. Zhu, C. Jia, R. Yan, L. Fan, H. Randriamahazaka and Q. Wang, Chem, 2017, 3, 1036–1049 CAS.
- Y. Chen, M. Zhou, Y. Xia, X. Wang, Y. Liu, Y. Yao, H. Zhang, Y. Li, S. Lu, W. Qin, X. Wu and Q. Wang, Joule, 2019, 3, 2255–2267 CrossRef CAS.
- S. Yan, S. Huang, H. Xu, L. Li, H. Zou, M. Ding, C. Jia and Q. Wang, ChemSusChem, 2023, 16, e202300710 CrossRef CAS PubMed.
- S. Liu, L. D'Amario, S. Jiang and H. Dau, Curr. Opin. Electrochem., 2022, 35, 101042 CrossRef CAS.
- X. Jia, H. Kang, G. Hou, W. Wu, S. Lu, Y. Li, Q. Wang, W. Qin and X. Wu, Angew. Chem., Int. Ed., 2024, 63, e202318248 CrossRef CAS PubMed.
- S. E. J. Bell, G. Charron, E. Cortés, J. Kneipp, M. L. La Chapelle, J. Langer, M. Procházka, V. Tran and S. Schlücker, Angew. Chem., Int. Ed., 2020, 59, 5454–5462 CrossRef CAS PubMed.
- M. D. Sonntag, E. A. Pozzi, N. Jiang, M. C. Hersam and R. P. van Duyne, J. Phys. Chem. Lett., 2014, 5, 3125–3130 CrossRef CAS PubMed.
- Z. Li, G. Weng, Q. Zou, G. Cong and Y.-C. Lu, Nano Energy, 2016, 30, 283–292 CrossRef CAS.
- G. S. Nambafu, A. M. Hollas, P. S. Rice, J. M. Weller, D. Boglaienko, D. M. Reed, V. L. Sprenkle and G. Li, Adv. Energy Mater., 2025, 15, 2403149 CrossRef CAS.
- H. Radinger, F. Bauer and F. Scheiba, Batteries Supercaps, 2023, 6, e202200440 CrossRef CAS.
- L. Lubian, R. Rubio-Presa, V. Ruiz, A. Colina and E. Ventosa, J. Power Sources, 2025, 646, 237272 CrossRef CAS.
- H. J. Lee, D.-W. Kim and J. H. Yang, J. Electrochem. Soc., 2017, 164, A754 CrossRef CAS.
- M. Zhou, Y. Chen, M. Salla, H. Zhang, X. Wang, S. R. Mothe and Q. Wang, Angew. Chem., Int. Ed., 2020, 59, 14286–14291 CrossRef CAS PubMed.
- F. Paparoni, E. Mijit, H. Darjazi, F. Nobili, A. Zitolo, A. Di Cicco, R. Parmar, R. Gunnella and S. J. Rezvani, J. Phys. Chem. C, 2023, 127, 8649–8656 CrossRef CAS PubMed.
- Introduction to XAFS: A Practical Guide to X-ray Absorption Fine Structure Spectroscopy, ed. G. Bunker, Cambridge University Press, Cambridge, 2010 Search PubMed.
- S. Mehboob, G. Ali, H.-J. Shin, J. Hwang, S. Abbas, K. Y. Chung and H. Y. Ha, Appl. Energy, 2018, 229, 910–921 CrossRef CAS.
- J. Melke, J. Martin, M. Bruns, P. Hügenell, A. Schökel, S. Montoya Isaza, F. Fink, P. Elsässer and A. Fischer, ACS Appl. Energy Mater., 2020, 3, 11627–11640 CrossRef CAS.
- G. M. Su, I. A. Cordova, M. A. Yandrasits, M. Lindell, J. Feng, C. Wang and A. Kusoglu, J. Am. Chem. Soc., 2019, 141, 13547–13561 CrossRef CAS PubMed.
- Y. Wen and R. Jervis, Curr. Opin. Chem. Eng., 2022, 37, 100836 CrossRef.
- C. Lutz, S. Hampel, S. Beuermann, T. Turek, U. Kunz, J. Garrevoet, G. Falkenberg and U. Fittschen, J. Synchrotron Radiat., 2021, 28, 1865–1873 CrossRef CAS PubMed.
- C. Lutz, S. Hampel, X. Ke, S. Beuermann, T. Turek, U. Kunz, A. Guilherme Buzanich, M. Radtke and U. E. Fittschen, J. Power Sources, 2021, 483, 229176 CrossRef CAS.
- S. Huang, M. Li, Y. Song, S. Xi, C. Wu, Z. W. J. Ang and Q. Wang, Adv. Mater., 2024, 36, 2406366 CrossRef CAS PubMed.
- Y. Cai, H. Zhang, T. Wang, S. Xi, Y. Song, S. Rong, J. Ma, Z. Han, C. T. J. Low, Q. Wang and Y. Ji, Joule, 2025, 9, 101768 CrossRef CAS.
- M. Schilling, M. Braig, K. Köble and R. Zeis, Electrochim. Acta, 2022, 430, 141058 CrossRef CAS.
- J. Schneider, T. Tichter, P. Khadke, R. Zeis and C. Roth, Electrochim. Acta, 2020, 336, 135510 CrossRef CAS.
- T. Tichter, M. Gernhard and P. Vesborg, Electrochim. Acta, 2023, 469, 143119 CrossRef CAS.
- T. Tichter and A. T. Marshall, Curr. Opin. Electrochem., 2022, 34, 101027 CrossRef CAS.
- F. Ciucci and C. Chen, Electrochim. Acta, 2015, 167, 439–454 CrossRef CAS.
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- M. B. Effat and F. Ciucci, Electrochim. Acta, 2017, 247, 1117–1129 CrossRef CAS.
- J. Liu, T. H. Wan and F. Ciucci, Electrochim. Acta, 2020, 357, 136864 CrossRef CAS.
- A. M. Pezeshki, R. L. Sacci, F. M. Delnick, D. S. Aaron and M. M. Mench, Electrochim. Acta, 2017, 229, 261–270 CrossRef CAS.
- E. H. Kirk, F. Fenini, S. N. Oreiro and A. Bentien, Batteries, 2021, 7, 87 CrossRef CAS.
- T. Lemmermann, M. Becker, M. Stehle, M. Drache, S. Beuermann, M. S. Bogar, U. Gohs, U. E. Fittschen, T. Turek and U. Kunz, J. Power Sources, 2022, 533, 231343 CrossRef CAS.
- H. Lim, J. S. Yi and D. Lee, ChemSusChem, 2019, 12, 1459–1468 CrossRef CAS PubMed.
- H. Lim, J. S. Yi and D. Lee, J. Power Sources, 2019, 422, 65–72 CrossRef CAS.
- Z. Wang, Y. Li, J. Ren, J. Sun, T. Wang, B. Liu, X. Fan and T. Zhao, Cell Rep. Phys. Sci., 2023, 4, 101395 CrossRef CAS.
- I. A. Volodin, C. Stolze, C. Casas Mesa, U. Haagen, C. Terechin, M. D. Hager and U. S. Schubert, Sens. Actuators, B, 2024, 403, 135101 CrossRef CAS.
- E. Schröter, C. Stolze, J. Meyer, M. D. Hager and U. S. Schubert, ChemSusChem, 2023, 16, e202300296 CrossRef PubMed.
- P. Loktionov, D. Konev, R. Pichugov, M. Petrov and A. Antipov, J. Power Sources, 2023, 553, 232242 CrossRef CAS.
- T. Haisch, H. Ji and C. Weidlich, Electrochim. Acta, 2020, 336, 135573 CrossRef CAS.
- M. R. Mohamed, H. Ahmad and M. N. Abu Seman, Elektron. Elektrotech., 2013, 19, 37–42 Search PubMed.
- R. Jervis, M. D. Kok, T. P. Neville, Q. Meyer, L. D. Brown, F. Iacoviello, J. T. Gostick, D. J. Brett and P. R. Shearing, J. Energy Chem., 2018, 27, 1353–1361 CrossRef.
- M. Gebhard, T. Tichter, J. Schneider, J. Mayer, A. Hilger, M. Osenberg, M. Rahn, I. Manke and C. Roth, J. Power Sources, 2020, 478, 228695 CrossRef CAS.
- R. R. Jacquemond, M. van der Heijden, E. B. Boz, E. R. Carreón Ruiz, K. V. Greco, J. A. Kowalski, V. Muñoz Perales, F. R. Brushett, K. Nijmeijer, P. Boillat and A. Forner-Cuenca, Nat. Commun., 2024, 15, 7434 CrossRef CAS PubMed.
- E. B. Boz, M. van der Heijden, R. R. Jacquemond, P. Boillat, J. Hjelm and A. Forner-Cuenca, J. Electrochem. Soc., 2024, 171, 53509 CrossRef CAS.
- F. Tariq, J. Rubio-Garcia, V. Yufit, A. Bertei, B. K. Chakrabarti, A. Kucernak and N. Brandon, Sustainable Energy Fuels, 2018, 2, 2068–2080 RSC.
- T. Kong, J. Liu, X. Zhou, J. Xu, Y. Xie, J. Chen, X. Li and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202214819 CrossRef CAS PubMed.
- A. M. Graf, T. Cochard, K. Amini, M. S. Emanuel, S. M. Rubinstein and M. J. Aziz, Energy Adv., 2024, 3, 2468–2478 RSC.
- H. Park, G. Kwon, H. Lee, K. Lee, S. Y. Park, J. E. Kwon, K. Kang and S. J. Kim, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2114947119 CrossRef CAS PubMed.
- X. Zang, L. Yan, Y. Yang, H. Pan, Z. Nie, K. W. Jung, Z. D. Deng and W. Wang, Small Methods, 2019, 3, 1900494 CrossRef CAS.
- L. Yan, X. Zang, Z. Nie, L. Zhong, Z. D. Deng and W. Wang, J. Power Sources, 2023, 580, 233417 CrossRef CAS.
- Y. Wu, P.-W. Huang, J. D. Howe, Y. Yan, J. Martinez, A. Marianchuk, Y. Zhang, H. Chen and N. Liu, Angew. Chem., Int. Ed., 2019, 58, 15228–15234 Search PubMed.
- K. Ma, Y. Zhang, L. Liu, J. Xi, X. Qiu, T. Guan and Y. He, Nat. Commun., 2019, 10, 5286 CrossRef PubMed.
- V. I. Vlasov, M. A. Pugach, D. S. Kopylova, A. V. Novikov, N. A. Gvozdik, A. A. Mkrtchyan, A. I. Davletkhanov, Y. Gladush, F. M. Ibanez, D. A. Gorin and K. J. Stevenson, J. Power Sources, 2023, 584, 233600 CrossRef CAS.
- G. S. Nambafu, A. M. Hollas, S. Zhang, P. S. Rice, D. Boglaienko, J. L. Fulton, M. Li, Q. Huang, Y. Zhu, D. M. Reed, V. L. Sprenkle and G. Li, Nat. Commun., 2024, 15, 2566 CrossRef CAS PubMed.
- Y. Ji, M.-A. Goulet, D. A. Pollack, D. G. Kwabi, S. Jin, D. Porcellinis, E. F. Kerr, R. G. Gordon and M. J. Aziz, Adv. Energy Mater., 2019, 9, 1900039 CrossRef.
- J. T. Lang, D. Kulkarni, C. W. Foster, Y. Huang, M. A. Sepe, S. Shimpalee, D. Y. Parkinson and I. V. Zenyuk, Chem. Rev., 2023, 123, 9880–9914 Search PubMed.
- S. Prakash Yadav, M. K. Ravikumar, S. Patil and A. Shukla, ChemElectroChem, 2024, 11, e202400267 Search PubMed.
- H. Chen, S. Wang, H. Gao, X. Feng, C. Yan and A. Tang, J. Power Sources, 2019, 427, 154–164 CrossRef CAS.
- S. Wan, H. Jiang, Z. Guo, C. He, X. Liang, N. Djilali and T. Zhao, Energy Environ. Sci., 2022, 15, 2874–2888 RSC.
- T. Li, C. Zhang and X. Li, Chem. Sci., 2022, 13, 4740–4752 RSC.
Footnote |
| † This work is dedicated to Prof. Wolfgang Kern, who passed away during the preparation of this work. |
| This journal is © The Royal Society of Chemistry 2025 |