
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImportance of hydrogen oxidation reaction current in quantifying hydrogen crossover in PEM water electrolyzers at high differential pressure†
Ramchandra
Gawas
 ,
Douglas I.
Kushner
,
Xiong
Peng
* and
Rangachary
Mukundan
*
,
Douglas I.
Kushner
,
Xiong
Peng
* and
Rangachary
Mukundan
*
Energy Storage and Distributed Resources Division, Lawrence Berkeley National Laboratory, Berkeley, California, USA. E-mail: xiongp@lbl.gov; rmukundan@lbl.gov
First published on 15th April 2025
Abstract
Understanding hydrogen permeation in proton exchange membrane water electrolyzers (PEMWEs) operating at high differential pressures (>25 bar) is critical towards developing effective gas recombination strategies that enable safe operation and high efficiency. Developing this understanding relies on accurate quantification of hydrogen crossover rates in water electrolyzers operating under such conditions. In this work, we show that PEMWEs operating at high differential pressures exhibit noticeable hydrogen oxidation reaction (HOR) currents. As the HOR consumes part of the permeated hydrogen at the anode, neglecting HOR currents leads to severe underestimation of the hydrogen crossover rate. We implemented a new method combining hydrogen oxidation current with online gas chromatography measurements to accurately quantify hydrogen crossover rates as a function of operating current density in PEMWEs operating at high differential pressures (10–30 barg).
Broader contextThe proton exchange membrane water electrolyzer (PEMWE) remains the emergent technology for clean hydrogen production due to its ability to dynamically follow loads, enabling facile integration with renewable energy sources. Moreover, operation at high current densities and high differential pressure further make this technology very attractive for low-cost hydrogen production. While the use of thin membranes improves the efficiency of PEMWE systems by reducing ohmic losses, it also exacerbates safety concerns associated with hydrogen crossover. This study investigates the hydrogen crossover phenomenon in PEM water electrolyzers operating at high differential pressure using electrochemical techniques combined with online gas chromatography. Our results show that PEMWE cells exhibit hydrogen oxidation reaction (HOR) currents under differential pressure. This HOR current, only observed at differential pressure, is diffusion limited and consumes a significant portion of the permeated hydrogen. Overlooking these HOR currents could lead to severe underestimation of the hydrogen crossover rates in PEMWEs operating at high differential pressure. This work demonstrates the importance of HOR current and proposes a new method that integrates HOR current measurement with online gas chromatography for accurate quantification of hydrogen crossover rates at various operating current densities and differential pressures. |
Proton exchange membrane water electrolyzers (PEMWEs) have been considered as a frontrunner for realizing large-scale, clean hydrogen production owing to their ability to operate at high differential pressures and intermittent operation that allows facile integration with renewable energy sources.1,2 The high differential pressure operation improves the overall system efficiency by increasing the hydrogen feed pressure to the compressor with the help of electrochemical compression (>25 bar) on the cathode side, eliminating the need for an additional mechanical compressor required otherwise.3,4 The water-oxygen loop on the anode side is operated at ambient pressure as pressurizing oxygen above 10 bar can be extremely dangerous, and oxygen is not collected in water electrolyzer systems anyway. This differential pressure operation results in significant hydrogen crossover from cathode to anode across the proton exchange membrane (PEM).5–7 Apart from modest losses in the system efficiency, this hydrogen crossover results in a safety risk as hydrogen and oxygen at the anode form a flammable gas mixture above 4% hydrogen in oxygen. The risk of formation of a flammable gas mixture on the anode side is higher at lower current densities, as less oxygen is produced for diluting hydrogen while the rate of hydrogen diffusion remains high at differential pressures. Common strategies to ensure safe operation of PEMWEs at differential pressure include incorporating gas recombination catalysts (GRCs) in the membrane, PTL or downstream in the anode gas outlet.8–11 Gas recombination catalysts facilitate the combining of hydrogen and oxygen to produce water, thus reducing the hydrogen concentration at the anode. Manufacturing economically viable PEMWE systems to achieve the DOE Hydrogen ShotTM target of $1 per kg of hydrogen relies on the use of thinner membranes to reduce ohmic losses.12–14 Thinner membranes, however, severely exacerbate safety concerns associated with hydrogen crossover.15,16 Consequently, the successful deployment of PEMWEs at scale would require successful implementation of gas recombination catalysts to reduce the hydrogen concentration at the anode to enable safe operation at high efficiency.17,18 To this end, the fundamental understanding of the hydrogen permeation through PEMs in PEMWEs at differential pressure would immensely help in developing Multiphysics simulations to predict the hydrogen crossover phenomenon as well as design effective gas recombination strategies.
Existing studies to investigate hydrogen permeation in PEMWEs operating at differential pressure employ online gas chromatography or mass spectrometry measurements to quantify hydrogen concentration at the anode, reported as %H2 in O2.10,15,16,19,20 This hydrogen concentration is then converted to hydrogen permeation flux as a function of current and differential pressure. Trinke et al. used this methodology to measure the hydrogen permeation rate in PEMWEs utilizing a 250 μm PFSA membrane as a function of differential pressure, current density, and temperature.20 They found that the hydrogen permeation rate in PEMWEs increases linearly with current density and attributed this increase to supersaturation of hydrogen in the ionomer phase. The hydrogen permeation rate measured by Wrubel et al.15 as well as Martin et al.16 in 50 μm PEMs also exhibited a similar increase with current density. However, most of these measurements overlook the possibility of hydrogen oxidation at the anode catalyst layer. If some of the permeated hydrogen is oxidized at the anode, hydrogen concentration measurement at the anode outlet would be insufficient to quantify hydrogen permeation rates through the PEM. Although previous studies have shown facile hydrogen oxidation reaction (HOR) kinetics on metallic Ir surfaces,21–23 the hydrogen oxidation reaction on commercial oxygen evolution reaction (OER) electrocatalysts i.e., iridium oxide surfaces, remains unexplored. The existing PEMWE literature assumes that iridium (Ir) exists as iridium oxide (IrOx) under the operating potential of the anode in an electrolyzer, with negligible HOR occurring on this IrOx surface.18,19 However, the permeated hydrogen can potentially reduce the iridium oxide catalyst in the anode catalyst layer and form surface metallic Ir sites that can facilitate the HOR.24–26 In this work, we show conclusive evidence of the hydrogen oxidation reaction (HOR) at the anode catalyst layer in PEMWEs operating at high differential pressure (10–30 barg). Our careful investigation reveals that the HOR, only observed at differential pressures, is diffusion-limited and consumes a significant portion of permeated hydrogen. These hydrogen oxidation current measurements were combined with online gas chromatography measurements to accurately quantify hydrogen permeation rates in PEMWEs at various operating current densities and differential pressures (0–30 barg).
Measuring hydrogen crossover at high differential pressures is significantly more challenging than simply evaluating the electrolyzer cell performance.27 The presence of any small leak in the system can lead to the loss of hydrogen and severely underestimate hydrogen concentration measured at the anode. Taking this into consideration, we carefully modified the electrolyzer components and the testing setup to ensure leak-free and precise measurements at high differential pressures. The ESI,† contains a detailed description of the catalyst-coated membrane fabrication process, modified electrolyzer cell components (Fig. S1, ESI†), test station with a custom backpressure unit (Fig. S2, ESI†), and measurement protocols used for electrochemical testing as well as online gas chromatography measurements (Fig. S3, ESI†). Each membrane electrode assembly (MEA) was identically conditioned prior to performance and hydrogen crossover evaluation at ambient and high differential pressure (10–30 barg) to ensure consistency.
Fig. 1(a) and (b) show the PEMWE cell performance and the high-frequency resistance (HFR) at 0–30 barg cathode backpressures. The high-frequency resistance (HFR) was extracted from the galvanostatic electrochemical impedance spectra (GEIS) performed at each current density. The ambient cell performance of 1.88 V at 3 A cm−2 shows good agreement for MEAs at comparable Ir loadings (0.4 mgIr cm−2) and membrane thickness (127 μm).28,29 The reversible cell voltages of PEMWE operating at high cathode backpressures are expected to increase due to elevated hydrogen partial pressures (Table S1, ESI†). This effect has been discussed in detail in Section 1(d) of the ESI.† HFR-corrected polarization curves demonstrate this expected Nernstian shift at high differential pressures (Fig. 1(c)). However, cell voltages in Fig. 1(a) do not exhibit a notable difference under different cathode backpressures at currents above 1 A cm−2. The HFR-free overpotentials, i.e., polarization curves corrected for both the iR-drop and the Nernstian shift (Fig. 1(d)), demonstrate the impact of differential pressure on electrode kinetics, with ∼ 60 mV improvement at 4 A cm−2 and 30 barg. This improvement offsets the Nernstian penalty at high cathode pressures (62 mV at 30 barg), resulting in identical overall cell performance in Fig. 1(a). A full voltage breakdown analysis needs to be performed in order to attribute these voltage gains to specific electrochemical phenomena.
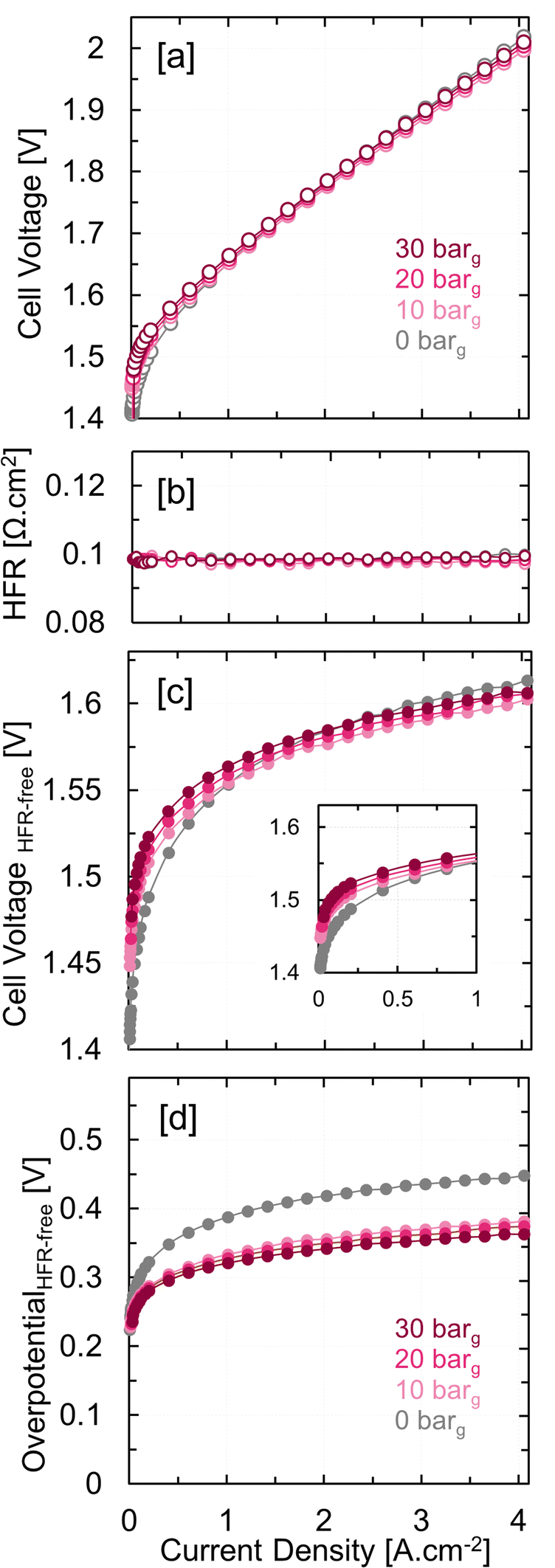 | ||
| Fig. 1 (a) PEM water electrolyzer cell voltage, (b) high frequency resistance (HFR), (c) HFR-free cell voltage, and (d) HFR-free overpotential as a function of current density at 0–30 barg cathode backpressures for a catalyst coated Nafion 115 membrane (127 μm dry thickness); 50 wt% Pt/C (0.1 mgPt cm−2) was used as the cathode catalyst with Toray 120 as the GDL and amorphous IrOx (0.4 mgIr cm−2) was used as the anode catalyst with platinized Mott sintered PTL; HFR was extracted from galvanostatic EIS at each current density. | ||
The HFR-free overpotentials can be further deconvoluted into kinetic and residual (transport) contributions.30,31 Kinetic contributions are extrapolated using the Tafel equation with kinetic parameters extracted from HFR-free overpotentials at low currents (<80 mA cm−2). Fig. S4 (ESI†) shows the voltage breakdown analysis at ambient backpressure showing ohmic, kinetic and residual overpotentials. However, this analysis wasn’t feasible at high differential pressures i.e., 10–30 barg, as we observed that the cell voltages dropped below 1.0 V at low currents (Fig. S5, ESI†). Furthermore, cyclic voltammograms (CVs) of the anode measured at 10–30 barg cathode backpressures showed only positive currents in both the forward and reverse directions. CVs of PEMFC cathodes also show such positive currents due to the background hydrogen oxidation current (Fig. S6, ESI†). These two observations pointed towards the oxidation of permeated H2 at the anode in electrolyzers operating at differential pressure even when utilizing IrOx catalysts. Although the hydrogen oxidation reaction (HOR) is relatively well characterized on metallic Ir surfaces,21–23 HOR on iridium oxide surfaces remains poorly understood. Fig. S7 (ESI†) shows rotating disk electrode (RDE) measurements in H2-saturated acidic electrolytes confirming the presence of HOR on the TKK amorphous IrOx surface. These measurements, discussed in detail in Section S2a of the ESI,† reveal that IrOx thin film electrodes exhibit similar HOR activity to metallic Ir or Pt/C.
We systematically analyzed the hydrogen oxidation reaction (HOR) current in PEMWE single cells at 10–30 barg cathode backpressures using linear sweep voltammetry at 1 mV s−1 from open circuit voltage (OCV) until the oxygen evolution reaction (OER) onset potential. The slow scan rate, 1 mV−1, was chosen to minimize contributions from the double layer capacitance and other faradaic reactions on the surface. Fig. 2(a) shows HOR currents for three commercial anode catalysts as a function of anode potential at 10, 20, and 30 barg cathode pressures. These three OER electrocatalysts were chosen to represent three kinds of iridium oxide catalysts – amorphous iridium oxyhydroxide (Tanaka Kikinzoku (TKK) amorphous IrOx), rutile iridium dioxide (TKK rutile IrO2), and iridium oxide with some metallic Ir (Alfa Aesar (AA) IrOx). The HOR polarization curves, irrespective of the catalyst and differential pressure, exhibited three distinct regions – (i) from OCV to 0.1 V showing very facile HOR kinetics, (ii) from 0.1 to 1.4 V showing diffusion limited HOR current and (iii) beyond 1.4 V showing OER current. The diffusion-limited HOR current is proportional to the differential pressure as H2 permeation without any water oxidation current is largely driven by diffusion. As the most ubiquitous PTL coating layer, Pt, is also an active HOR catalyst, we used Au-coated PTL at the anode with an IrOx anode catalyst layer to eliminate any HOR contributions from Pt present as the coating.32 The PEMWE single cells consisting of the HOR inactive Au-coated PTL showed similar HOR currents as Pt- and Ir-coated PTLs. Fig. 2(b) further shows that HOR current is independent of the PGM-coating material (Pt, Ir, Au) confirming that IrOx catalyzes hydrogen oxidation at the anode. We would like to clarify that HOR current was observed only at differential pressures (Fig. S8, ESI†); HOR current was negligible under ambient pressure due to the lack of hydrogen at the anode.
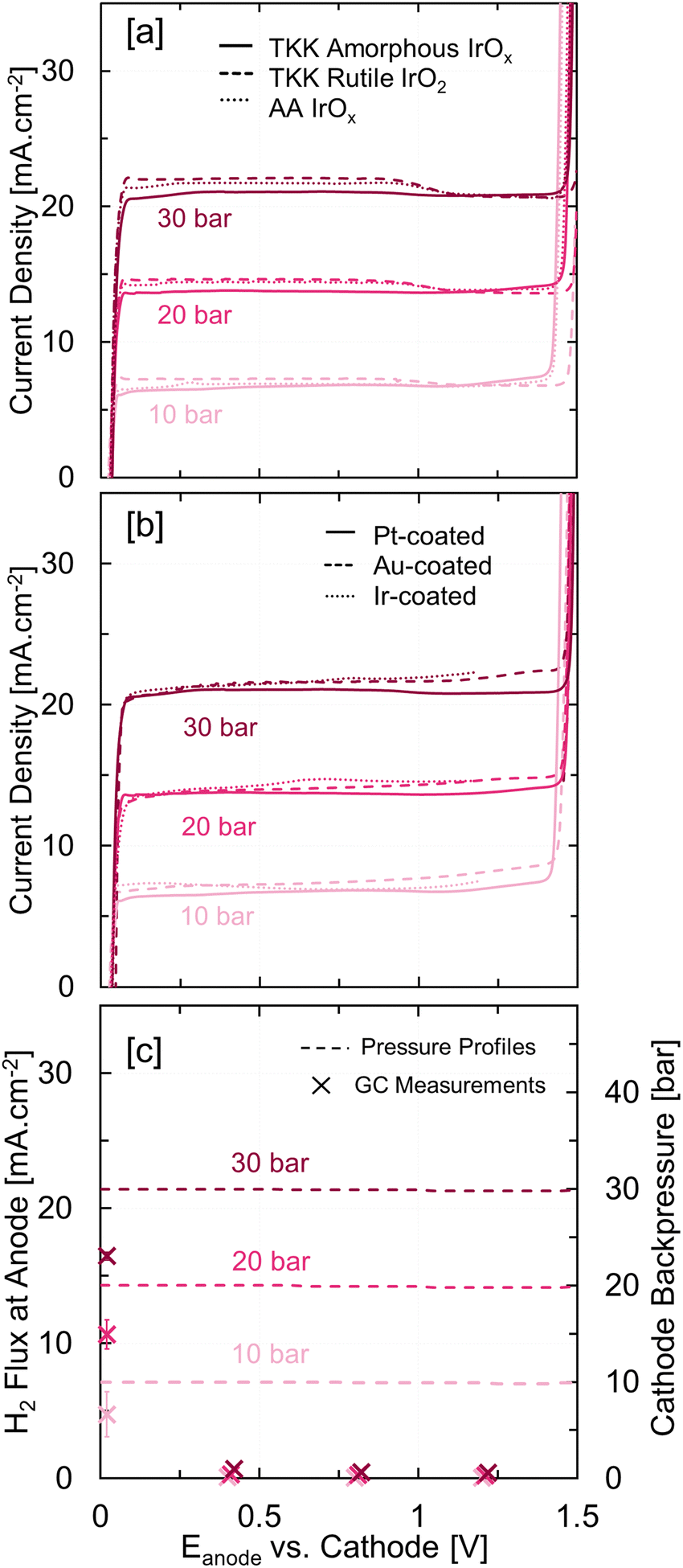 | ||
| Fig. 2 The hydrogen oxidation current measurements for (a) three different anode catalysts – TKK amorphous IrOx (solid), TKK rutile IrO2 (Dashed), and AA IrOx (dotted) and (b) three different PTL coatings – Pt (solid), Au (dashed), and Ir (dotted) recorded using linear sweep voltammetry (LSV) from OCV to 1.5 V vs. cathode at 1 mV s−1 at high differential pressure (10–30 barg); (c) H2 flux at the anode outlet (left ordinate) calculated using gas chromatography measurements with argon (Ar) as the sweep gas and cathode backpressure (right ordinate) recorded at various potentials during the HOR at 0–30 barg (initial) cathode backpressure; error bars represent measurements on three independent MEAs. | ||
Furthermore, we coupled HOR measurements with online gas chromatography (GC) to assess whether the mass transport-limited HOR consumes all permeated H2. These GC measurements were taken using Ar as the sweep gas, as shown in Fig. S2(b) (ESI†) at various operating potentials and cathode backpressures and converted to H2 flux at the anode or H2 permeation rate (the ESI† contains further details of this measurement in Section S1e). Fig. 2(c) shows zero H2 flux at the anode outlet in the diffusion limited region at all (10–30 barg) cathode backpressures, indicating that the HOR consumes all permeated hydrogen. Thus, the diffusion-limited HOR current represents an electrochemical H2 permeation measurement in PEMs at differential pressures. These H2 permeation rates or HOR currents concur with H2 permeation rates reported by Schalenbach et al. for a 127 μm PFSA membrane (N115) at the corresponding differential pressures.33 Note that the diffusion limited HOR current measures H2 permeation rate at zero operating current (no water oxidation or OER). Fig. 2(c) also shows cathode backpressures (dotted line) recorded during HOR measurements. Interestingly, we did not observe a decrease in the cathode backpressure despite H2 permeation from the cathode to the anode. The HOR at the anode generates protons that come back to the cathode and regenerate H2 by the hydrogen evolution reaction. These observations suggest that the PEMWE functions as a hydrogen pump at differential pressures and voltages below 1.4 V (Fig. S5, ESI†). Moreover, cathode backpressure retention during the hydrogen pump operation confirms that our system does not have any leaks. Hydrogen pump operation was also observed during low-current galvanostatic measurements at high differential pressure when the applied current was below the diffusion limited HOR current at the corresponding cathode backpressure (Fig. S5, ESI†). H2 flux at the anode at OCV, i.e., without any HOR current, depicting H2 permeation rate, showed reasonable agreement with the HOR currents at 10–30 barg cathode backpressures.
The existing PEMWE literature widely neglects HOR currents while analyzing polarization curves and reporting faradaic efficiencies, which is valid only for ambient pressure operation. However, our careful investigations reveal that PEMWEs operating at high differential pressure exhibit noticeable HOR currents. As more laboratories acquire the capability to operate under differential pressure, the procedures reported in this manuscript can be utilized to quantify the HOR currents. Since the HOR consumes some of the permeated H2, it needs to be accurately measured and accounted for while estimating H2 permeation rates through the PEM (Fig. 3). In the next section, we will discuss the implications of the HOR at the anode on H2 crossover rates in PEMWEs under practical operating currents and 0–30 barg cathode backpressures. We implemented online gas chromatography to quantify the hydrogen volume fraction at the anode. Gas chromatography offers the highest accuracy among techniques used for on-line quantification of hydrogen concentration at the anode outlet.
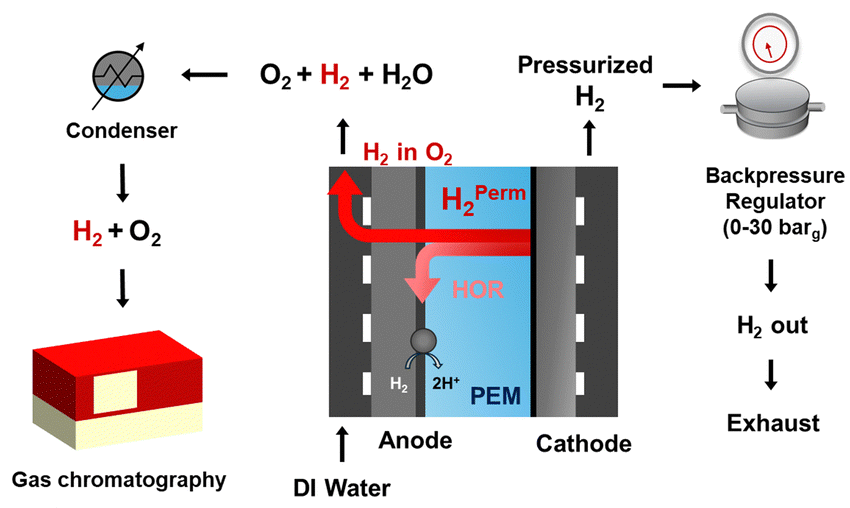 | ||
| Fig. 3 Schematic of the experimental setup showing PEMWE equipped with manual backpressure at the cathode and online gas chromatography to measure hydrogen content at the anode. | ||
Fig. 4(a) shows the hydrogen volume fraction at the anode (%H2 in O2, dry basis) as a function of current density (0.25 to 3.5 A cm−2) at 0 to 30 barg cathode backpressures with error bars obtained from on-line GC measurements on three independent MEAs. As expected, the hydrogen concentration at the anode increases with cathode backpressure due to an increase in H2 permeation rate and decreases with current density as oxygen from the OER dilutes the hydrogen concentration. The volume fraction of hydrogen in oxygen was converted to H2 flux at the anode using eqn (S1) (ESI†). Fig. 4(b) shows the H2 flux at the anode outlet as a function of current density in comparison with the H2 permeation rate measured at zero OER current, i.e., HOR current at 0–30 barg cathode backpressures. Previously, this H2 flux at the anode outlet measured using online gas chromatography was solely projected as the H2 permeation rate. However, Fig. 4(b) clearly shows that GC measurements alone severely underestimate the H2 permeation rate at high differential pressures. The H2 flux at the anode outlet at low current densities is significantly smaller than the ex situ H2 permeation rate at high differential pressures (indicated by crosses at zero current at 10–30 barg). This discrepancy cannot be explained by a recombination reaction on Pt in the PTL without considering the hydrogen oxidation reaction at the anode. The MEAs with an Ir-coated PTL also show significantly lower H2 flux at the anode outlet than ex situ permeation rate measurements (Fig. S9, ESI†). In fact, identical H2 fluxes at the anode outlet for MEAs with Pt and Ir-coated PTLs suggest that Pt present in the PTL is not an effective recombination catalyst. Since, some of the permeated H2 is oxidized at the anode catalyst layer, the HOR current needs to be considered when computing H2 crossover rates in PEMWEs. Assuming that the diffusion-limited HOR current would not change beyond 1.5 V, we computed the H2 permeation rate by adding the HOR current to the H2 flux at the anode calculated using in operando GC measurements. Fig. 4(c) shows H2 permeation rate as a function of current density for 0–30 barg cathode backpressures illustrating the effect of differential pressure as well as the current density on hydrogen crossover.
 | ||
| Fig. 4 (a) %H2 in O2 measured at the anode outlet using online gas chromatography measurements, (b) H2 flux at the anode calculated using online GC measurements and HOR current (crosses and faded solid lines) representing H2 permeation rate at zero OER current, (c) hydrogen permeation rate calculated by combining the H2 flux at the anode and HOR measurements for various operating current densities at 0–30 barg cathode backpressures; error bars represent measurements on three independent MEAs. | ||
Trends in H2 permeation rates at 0–30 barg cathode backpressure (Fig. 4(c)) point towards two distinct driving forces for H2 crossover through PFSA membranes in PEMWEs operating at high differential pressures. The hydrogen permeation under differential pressure without any operating current, measured as the mass transport-limited HOR current, is directly proportional to the cathode backpressure and represents diffusion or a ‘pressure-driven permeation’ through the PFSA membrane. The H2 flux at the anode i.e., hydrogen permeation rate excluding the HOR, increases significantly with operating current density, showing a weak dependence on differential pressure. This increase in H2 permeation rate with current density suggests H2 supersaturation in ionomer or water phases, representing a ‘concentration-driven permeation’ through PEMs.5,19,20,34 This analysis further emphasizes the importance of hydrogen oxidation current in understanding the hydrogen crossover phenomenon in PEMWE systems operating under high differential pressure. These findings also underscore the significance of detailed electroanalytical studies at the single-cell level to gain insights into complex phenomena occurring in large scale systems. Combining this methodology with H2 and O2 mole balance experiments could further advance our understanding of H2 crossover in these systems and aid in maximizing the efficiency of electrolyzer systems.35,36
Conclusions
In this work, we evaluated standard membrane electrode assemblies (MEAs) using electrochemical characterization and online gas chromatography to understand hydrogen crossover in PEMWEs at high differential pressure. Our detailed analysis showed that part of the permeated hydrogen is oxidized at the anode catalyst layer at high differential pressure. We characterized the HOR current at the anode catalyst layer with LSV at 1 mV s−1 at 0–30 barg differential pressure. The HOR current observed in PEMWEs is diffusion-limited, directly proportional to cathode backpressures, and independent of the type of iridiun oxide catalyst or PGM coating on PTL. Furthermore, we quantified hydrogen concentration at the anode outlet using online gas chromatography. As some of the permeated H2 is oxidized at the anode catalyst layer, GC measurements at the anode outlet severely underestimate the H2 permeation rates at high differential pressures. Therefore, we combined these measurements with HOR current measurements to accurately compute the hydrogen permeation rates in PEMWEs at 0–30 barg differential pressure at various operating current densities. The hydrogen crossover rate in PEMWEs increases as a function of increasing differential pressure as well as increasing operating current densities, pointing towards two distinct gas permeation mechanisms – a diffusion or pressure driven pathway and a supersaturation or concentration driven pathway.Experimental methods
5 cm2 single-cell PEMWEs consisting of spray-coated catalyst coated membranes (CCMs) with Pt as the cathode catalyst (Pt/C, TKK TEC10V50E, 0.1 mgPt cm−2) and iridium oxide as the anode catalyst (IrOx, TKK 77100, 0.4 mgIr cm−2), a Mott sintered PTL (254 μm, 37% porosity, 0.2 mg cm−2 PGM coating), Toray carbon paper as the GDL (370 μm, 5 wt% PTFE), 10 mil thick skived PTFE gaskets on both sides, custom-built titanium flow fields coated with platinum and aluminum end plates with Au-plated current collectors from Scribner were assembled using Belleville spring washers at 40 lb-in. compression. Each PEMWE single cell at 80 °C was fed deionized water at the anode and subjected to four different cathode backpressures (0 barg or ambient, 10 barg, 20 barg, and 30 barg) using a custom-built backpressure unit. Detailed electrochemical characterization (polarization curves, electrochemical impedance spectroscopy, cyclic voltammograms, and hydrogen oxidation reaction current measurements) was carried out using a Biologic Potentiostat with a 20 A current booster at ambient as well as 10 barg, 20 barg and 30 barg cathode pressures. Furthermore, % hydrogen in oxygen at the anode (%H2 in O2, dry basis) was quantified using online GC measurements (SRI Instruments, 8610C) at various operating current densities (0.25 to 3.5 A cm−2) and cathode backpressures (0 to 30 barg).Author contributions
Ramchandra Gawas: conceptualization, methodology, investigation, data curation, formal analysis, validation, visualization, writing – original draft, writing – review and editing. Douglas Kushner: methodology, software, resources, writing – review. Xiong Peng: conceptualization, funding acquisition, supervision, validation, writing – review and editing. Rangachary Mukundan: conceptualization, funding acquisition, supervision, writing – review and editing.Data availability
The original data used in this article is included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge the Department of Energy (DOE), Office of Energy Efficiency and Renewable Energy (EERE), Hydrogen and Fuel Cell Technologies Office (HFTO), and the Hydrogen from the Next-generation Water Electrolyzers (H2NEW) consortium for funding this research under contract no. DE-AC02-05CH11231. All opinions expressed in this article are those of the authors and do not necessarily reflect the policies and views of the DOE. The authors would like to express gratitude to Dr Jason Lee, Dr Arthur Dizon, Dr Paul Rudnicki, and Dr Guanzhi Wang for feedback and fruitful discussions.References
- K. Ayers, N. Danilovic, R. Ouimet, M. Carmo, B. Pivovar and M. Bornstein, Perspectives on Low-Temperature Electrolysis and Potential for Renewable Hydrogen at Scale, Annu. Rev. Chem. Biomol. Eng., 2019, 10(1), 219–239 CrossRef CAS PubMed.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, A Comprehensive Review on PEM Water Electrolysis, Int. J. Hydrogen Energy, 2013, 38(12), 4901–4934 CrossRef CAS.
- K. Ayers, N. Danilovic, K. Harrison and H. Xu, PEM Electrolysis, a Forerunner for Clean Hydrogen, Electrochem. Soc. Interface, 2021, 30(4), 67–72 CrossRef CAS.
- X. Wang, A. G. Star and R. K. Ahluwalia, Performance of Polymer Electrolyte Membrane Water Electrolysis Systems: Configuration, Stack Materials, Turndown and Efficiency, Energies, 2023, 16(13), 4964 CrossRef CAS.
- R. Omrani and B. Shabani, Hydrogen Crossover in Proton Exchange Membrane Electrolysers: The Effect of Current Density, Pressure, Temperature, and Compression, Electrochim. Acta, 2021, 377, 138085 CrossRef CAS.
- P. Trinke, P. Haug, J. Brauns, B. Bensmann, R. Hanke-Rauschenbach and T. Turek, Hydrogen Crossover in PEM and Alkaline Water Electrolysis: Mechanisms, Direct Comparison and Mitigation Strategies, J. Electrochem. Soc., 2018, 165(7), F502 CrossRef CAS.
- S. Fahr, F. K. Engel, S. Rehfeldt, A. Peschel and H. Klein, Overview and Evaluation of Crossover Phenomena and Mitigation Measures in Proton Exchange Membrane (PEM) Electrolysis, Int. J. Hydrogen Energy, 2024, 68, 705–721 CrossRef CAS.
- C. Klose, P. Trinke, T. Böhm, B. Bensmann, S. Vierrath, R. Hanke-Rauschenbach and S. Thiele, Membrane Interlayer with Pt Recombination Particles for Reduction of the Anodic Hydrogen Content in PEM Water Electrolysis, J. Electrochem. Soc., 2018, 165(16), F1271–F1277 CrossRef CAS.
- D. Abbas, A. Martin, P. Trinke, M. Bierling, B. Bensmann, S. Thiele, R. Hanke-Rauschenbach and T. Böhm, Effect of Recombination Catalyst Loading in PEMWE Membranes on Anodic Hydrogen Content Reduction, J. Electrochem. Soc., 2022, 169(12), 124514 CrossRef CAS.
- S. Garbe, U. Babic, E. Nilsson, T. J. Schmidt and L. Gubler, Communication—Pt-Doped Thin Membranes for Gas Crossover Suppression in Polymer Electrolyte Water Electrolysis, J. Electrochem. Soc., 2019, 166(13), F873–F875 CrossRef CAS.
- N. Briguglio, F. Pantò, S. Siracusano and A. S. Aricò, Enhanced Performance of a PtCo Recombination Catalyst for Reducing the H2 Concentration in the O2 Stream of a PEM Electrolysis Cell in the Presence of a Thin Membrane and a High Differential Pressure, Electrochim. Acta, 2020, 344(136153), 136153 CrossRef CAS.
- A. Badgett, M. Ruth and B. Pivovar, Economic Considerations for Hydrogen Production with a Focus on Polymer Electrolyte Membrane Electrolysis, in Electrochemical Power Sources: Fundamentals, Systems, and Applications, ed. T. Smolinka and J. Garche, Elsevier, 2022, ch. 10, pp. 327–364 Search PubMed.
- K. Ayers, High Efficiency PEM Water Electrolysis: Enabled by Advanced Catalysts, Membranes, and Processes, Curr. Opin. Chem. Eng., 2021, 33(100719), 100719 CrossRef.
- Department of Energy, Hydrogen Shot: Water Electrolysis Technology Assessment, 2024, https://www.energy.gov/sites/default/files/2024-12/hydrogen-shot-water-electrolysis-technology-assessment.pdf.
- J. A. Wrubel, C. Milleville, E. Klein, J. Zack, A. M. Park and G. Bender, Estimating the Energy Requirement for Hydrogen Production in Proton Exchange Membrane Electrolysis Cells Using Rapid Operando Hydrogen Crossover Analysis, Int. J. Hydrogen Energy, 2022, 47(66), 28244–28253 CrossRef CAS.
- A. Martin, P. Trinke, B. Bensmann and R. Hanke-Rauschenbach, Hydrogen Crossover in PEM Water Electrolysis at Current Densities up to 10 A Cm−2, J. Electrochem. Soc., 2022, 169(9), 094507 CrossRef CAS.
- A. Stähler, M. Stähler, F. Scheepers, W. Lehnert and M. Carmo, Scalable Implementation of Recombination Catalyst Layers to Mitigate Gas Crossover in PEM Water Electrolyzers, J. Electrochem. Soc., 2022, 169(3), 034522 CrossRef.
- M. Schalenbach, M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Pressurized PEM Water Electrolysis: Efficiency and Gas Crossover, Int. J. Hydrogen Energy, 2013, 38(35), 14921–14933 CrossRef CAS.
- M. Bernt, J. Schröter, M. Möckl and H. A. Gasteiger, Analysis of Gas Permeation Phenomena in a PEM Water Electrolyzer Operated at High Pressure and High Current Density, J. Electrochem. Soc., 2020, 167(12), 124502 CrossRef CAS.
- P. Trinke, B. Bensmann and R. Hanke-Rauschenbach, Current Density Effect on Hydrogen Permeation in PEM Water Electrolyzers, Int. J. Hydrogen Energy, 2017, 42(21), 14355–14366 CrossRef CAS.
- J. Durst, C. Simon, F. Hasché and H. A. Gasteiger, Hydrogen Oxidation and Evolution Reaction Kinetics on Carbon Supported Pt, Ir, Rh, and Pd Electrocatalysts in Acidic Media, J. Electrochem. Soc., 2015, 162(1), F190–F203 CrossRef CAS.
- C. G. Zoski, Scanning Electrochemical Microscopy: Investigation of Hydrogen Oxidation at Polycrystalline Noble Metal Electrodes, J. Phys. Chem. B, 2003, 107(26), 6401–6405 CrossRef CAS.
- M. A. Montero, J. L. Fernández, M. R. Gennero de Chialvo and A. C. Chialvo, Kinetic Study of the Hydrogen Oxidation Reaction on Nanostructured Iridium Electrodes in Acid Solutions, J. Phys. Chem. C, 2013, 117(48), 25269–25275 CrossRef CAS.
- S. M. Alia, K. S. Reeves, D. Cullen, H. Yu, K. Jeremy, N. N. Kariuki, J. H. Park and D. J. Myers, Simulated Start-Stop and the Impact of Catalyst Layer Redox on Degradation and Performance Loss in Low-Temperature Electrolysis, J. Electrochem. Soc., 2024, 171(4), 0 CrossRef.
- M. Milosevic, T. Böhm, A. Körner, M. Bierling, L. Winkelmann, K. Ehelebe, A. Hutzler, M. Suermann, S. Thiele and S. Cherevko, In Search of Lost Iridium: Quantification of Anode Catalyst Layer Dissolution in Proton Exchange Membrane Water Electrolyzers, ACS Energy Lett., 2023, 8(6), 2682–2688 CrossRef CAS.
- A. Weiß, A. Siebel, M. Bernt, T.-H. Shen, V. Tileli and H. A. Gasteiger, Impact of Intermittent Operation on Lifetime and Performance of a PEM Water Electrolyzer, J. Electrochem. Soc., 2019, 166(8), F487–F497 CrossRef.
- M. N. I. Salehmin, T. Husaini, J. Goh and A. B. Sulong, High-Pressure PEM Water Electrolyser: A Review on Challenges and Mitigation Strategies towards Green and Low-Cost Hydrogen Production, Energy Convers. Manage., 2022, 268, 115985 CrossRef CAS.
- E. Padgett, G. Bender, A. Haug, K. Lewinski, F. Sun, H. Yu, D. A. Cullen, A. J. Steinbach and S. M. Alia, Catalyst Layer Resistance and Utilization in PEM Electrolysis, J. Electrochem. Soc., 2023, 170(8), 084512 CrossRef CAS.
- S. M. Alia, K. S. Reeves, H. Yu, J. H. Park, N. N. Kariuki, A. J. Kropf, D. J. Myers and D. A. Cullen, Catalyst-Specific Accelerated Stress Tests in Proton Exchange Membrane Low-Temperature Electrolysis for Intermittent Operation, J. Electrochem. Soc., 2024, 171(2), 024505 CrossRef CAS.
- S. Chatterjee, X. Peng, S. Intikhab, G. Zeng, N. N. Kariuki, D. J. Myers, N. Danilovic and J. Snyder, Nanoporous Iridium Nanosheets for Polymer Electrolyte Membrane Electrolysis, Adv. Energy Mater., 2021, 11(34), 2101438 CrossRef CAS.
- J. K. Lee, G. Anderson, A. W. Tricker, F. Babbe, A. Madan, D. A. Cullen, J. D. Arregui-Mena, N. Danilovic, R. Mukundan, A. Z. Weber and X. Peng, Ionomer-Free and Recyclable Porous-Transport Electrode for High-Performing Proton-Exchange-Membrane Water Electrolysis, Nat. Commun., 2023, 14(1), 4592 CrossRef CAS PubMed.
- P. S. Ruvinsky, S. N. Pronkin, V. I. Zaikovskii, P. Bernhardt and E. R. Savinova, On the Enhanced Electrocatalytic Activity of Pd Overlayers on Carbon-Supported Gold Particles in Hydrogen Electrooxidation, Phys. Chem. Chem. Phys., 2008, 10(44), 6665–6676 RSC.
- M. Schalenbach, T. Hoefner, P. Paciok, M. Carmo, W. Lueke and D. Stolten, Gas Permeation through Nafion. Part 1: Measurements, J. Phys. Chem. C, 2015, 119(45), 25145–25155 CrossRef CAS.
- P. Trinke, G. P. Keeley, M. Carmo, B. Bensmann and R. Hanke-Rauschenbach, Elucidating the Effect of Mass Transport Resistances on Hydrogen Crossover and Cell Performance in PEM Water Electrolyzers by Varying the Cathode Ionomer Content, J. Electrochem. Soc., 2019, 166(8), F465 CrossRef CAS.
- A. T. S. Freiberg and S. Thiele, Closing the Hydrogen and Oxygen Mass Balance for PEM Water Electrolysis, J. Electrochem. Soc., 2025, 172, 034506 CrossRef CAS.
- J. A. Wrubel, R. F. P. M. Duarte, E. D. Pomeroy, A. Kowal and G. Bender, Advanced Hydrogen Crossover Characterization: Closing the Mole Balance through Coupled Anode and Cathode Exhaust Analysis, Meet. Abstr., 2024,(45), 3112 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed descriptions of experimental methods, supporting results and discussion, and supporting figures (Fig. S1–S9). See DOI: https://doi.org/10.1039/d5ee00048c |
| This journal is © The Royal Society of Chemistry 2025 |