
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimulating solid electrolyte interphase formation spanning 108 time scales with an atomically informed phase-field model†
Kena
Zhang‡
 a,
Yanzhou
Ji‡
a,
Qisheng
Wu
b,
Seyed Amin
Nabavizadeh
a,
Yue
Qi
*b and
Long-Qing
Chen
*a
a,
Yanzhou
Ji‡
a,
Qisheng
Wu
b,
Seyed Amin
Nabavizadeh
a,
Yue
Qi
*b and
Long-Qing
Chen
*a
aDepartment of Materials Science and Engineering, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: lqc3@psu.edu
bSchool of Engineering, Brown University, Providence, RI 02912, USA. E-mail: yueqi@brown.edu
First published on 20th June 2025
Abstract
The solid electrolyte interphase (SEI) governs the reversibility of advanced electrochemical devices such as batteries, but the role of cations in its formation remains poorly understood. Here, the thickness and compositional evolution of the SEI are tracked over time scales from nanoseconds to seconds with a newly developed atomically informed phase-field multiscale model. We deconvolve the complex interplay among electron tunneling, species diffusion, and chemical/electrochemical reactions by probing different controlling factors separately and jointly to determine the rate-limiting steps. We show that the SEI growth begins with the formation of organic products, followed by the conversion of these organic products into inorganic ones, and in the end the inorganic products fully cover the lithium metal surface to form a passivation layer. While electron tunneling determines the thickness of these layers, the growth rates of the organic and inorganic SEI layers are controlled by the rates of Li-ion diffusion and electrochemical reactions, respectively. This predictive model is universally applicable to multiphase and multicomponent electrochemical systems and represents a significant advancement in simulating complex reaction processes.
Broader contextThe solid-electrolyte interphase (SEI) plays a critical role in battery performance and longevity, yet its formation process remains one of the most ambiguous issues in battery science due to the complex, spatially and temporally dynamic nature of this interfacial layer. To capture the rapid SEI formation process across various scales, an atomically informed phase-field model (AI-PFM), capable of handling complex reaction networks with multiple species, is developed. This model enables the investigation of SEI formation and initial growth from nanoseconds to seconds in time and angstroms to 100 nm in length. By tracking the evolution of SEI products and electrolyte species up to surface passivation, the interplay among reaction kinetics, species transport, and electron tunneling during SEI formation is successfully deconvoluted with the identification of the governing factors. For the first time, this study reveals that the competition between Li-ion diffusion and reaction kinetics is a key determinant of the growth rates of different SEI products. Such deconvolution is difficult to achieve with current modelling and experimental techniques, which underscores the major advancement and benefits of this AI-PFM framework. It offers a unique approach to evaluate the competing complex mechanistic pathways and understand the formation mechanism of the SEI. |
Introduction
The solid electrolyte interphase (SEI) plays a vital role in enabling advanced batteries where electrode materials operate beyond the electrochemical stability limits of electrolytes. Its chemical building blocks come from the sacrificial decomposition of electrolytes, which remains conductive to the working ions but prevents electron tunneling that drives the parasitic reactions.1–5 The SEI governs the reversibility and power density of the battery system. Given such importance, extensive efforts have been devoted to understanding its formation process, but a thorough mechanistic knowledge at a fine timescale is still absent. Recently, it was found that the SEI in Li-ion batteries formed at high charging current during the first cycle extends the battery cycle life by an average of 50%,6 revealing that the properties and chemistry of the SEI also rely on the rate of formation. Experiments indicated that the initial SEI formation on a bare Li metal surface completes in less than 1 second.7 This rate is comparable to the rate of lithium metal deposition, where a typical 1 mA cm−2 rate means depositing 8 Li (001) layers or 1.4 nm thick Li atoms in 1 second. Therefore, it is logical to infer that SEI formation and lithium growth compete at the same timescale, resulting in various deposition morphologies.8,9 However, experimental limitations in temporal and spatial resolutions often make it challenging to characterize fast in situ SEI formation processes and deconvolute the intricate physical and chemical processes across multiple length and time scales.At microscopic and mesoscopic timescales, atomic-scale theoretical approaches, such as density functional theory (DFT) calculations and ab initio molecular dynamics (AIMD) simulations, offer insights into the reaction energy profiles,10–12 species transport properties13,14 and electrolyte reduction pathway within a system11 that are otherwise unavailable from experiments. However, their simulation time is typically limited to scales of 10–100 picoseconds (10−11–10−10 s) for a system of hundreds of atoms,15,16 thus offering only limited information on SEI formation, which typically occurs over timescales approaching 1 s. A Monte Carlo-molecular dynamics (MC-MD) method can predict the time evolution of SEI species over 10 ns (10−8 s).15 Using classical reactive force fields, MD simulations can extend the process further up to 100 nanoseconds (10−7 s).17
At a macroscopic scale, continuum models have been developed to study the long-term SEI growth over hours and even months.18–23 Despite the widespread acceptance and observation of a two-layer structured SEI in many experimental works, the reaction networks and SEI compositions are simplified so that only a single SEI product is included in most continuum-level models.20,24,25 For example, Christensen and Newman20 proposed a mathematical model to estimate the growth rate of inorganic Li2CO3 and determined that the SEI grows around 20 nm in 15 h on graphite, which is limited by the electron transport via a Li interstitial diffusion mechanism. The continuum model in the study by Horstmann et al.23 predicted that the capacity fade shifts from a square-root-of-time dependence to a linear time dependence as the charging current density increased, suggesting a shift from diffusion-controlled to electron-migration-controlled SEI growth. Their simulations concluded that it would take several months for an SEI layer a few nanometers in thickness to form. While these models have been effective in predicting the battery lifetime25,26 at a macroscale, they overlooked several critical kinetic processes involved in SEI growth, which prevents them from accurately predicting the precise composition and morphology of the SEI. For instance, the detailed electrolyte reduction reaction networks and the competitive interactions between the various reaction products are neglected. Furthermore, these continuum models fail to account for the electron transport mechanisms, which is crucial for understanding the SEI formation and growth processes.
Since single-scale atomistic simulations are insufficient to capture the complex interfacial reactions and phase transformations involved in SEI formation, multiscale computational frameworks have therefore become indispensable for describing the growth, composition, and dynamic evolution of the SEI and its impact on battery performance. Among these, DFT and MD integrated kinetic Monte Carlo (kMC) methods have been used to track the stochastic reaction nature of electrolyte decomposition. For instance, Gerasimov et al.27 tracked EC decomposition and SEI formation on the Li metal for 100 ns, revealing an inorganic-rich inner layer (LiF/Li2CO3) and a porous organic-rich outer layer (Li2EDC) together forming a structure ∼11 nm thick. U. Krewer et al.28 constructed a DFT-kMC-continuum electroneutrality model that predicted a 7-nm-thick inorganic SEI layer within 1 μs (10−6 s), while the resulting bilayer architecture (porous Li2CO3 beneath dense LiF) deviates from experimental observations.29,30 Recently, chemical reaction networks (CRNs) have been developed to automatically identify the reaction pathways for over 80 million reactions among over 5000 species, in which DFT calculations are combined with kMC simulations to simulate the competition between SEI products within 10 μs (10−5 s), revealing the formation of distinct inorganic and organic layers in the SEI.31 While these kMC-based models have demonstrated success in capturing reaction mechanisms and compositional diversity at the molecular level, they remain limited in temporal and spatial scalability. Their inherently discrete spatial nature restricts the ability to simulate mesoscale structural evolution, such as growth, coarsening, polycrystallinity, and crack formation. Additionally, coupling external physical fields (e.g., mechanical stress relaxation, thermal transport, etc.) with kMC frameworks is generally indirect through modifications of reaction rates.
The phase-field (PF) method offers a continuum framework well-suited for modeling multicomponent, multiphase systems with intrinsic flexibility to incorporate physical fields. By introducing multiple concentrations or order parameter fields governed by free energy functionals, PF models can capture the spatiotemporal evolution of competing SEI phases, while naturally incorporating additional physical phenomena such as ion/electron transport, electrochemical reactions, elastic deformation, thermal transport, and other physical effects. Previously, phase-field modeling has been successfully applied to the investigation of Li electrodeposition32–38 and the interaction between Li dendrites and artificial SEIs.39,40 However, only a limited number of phase field investigations have been applied to the study of SEI formation and growth,29,41–43 with several important thermodynamic/kinetic parameters associated with SEI formation still absent. Compared with kMC-based models, previous PF models have simplified the SEI as a single homogeneous phase and thus failed to account for the chemical diversity observed experimentally.40,41 This is because PF simulations face numerical challenges in simulating multiple moving interfaces with distinct kinetics when tracking the evolution of reaction intermediates, which requires careful parameter calibration and efficient algorithms to ensure numerical stability and convergence.
In this work, we demonstrate that an atomically informed phase-field model (AI-PFM), which incorporates multiple electrochemical reactions, species transport and electron tunneling process, can track the temporal and spatial evolution of SEI formation from nanoseconds to seconds until it passivates the Li metal surface. The AI-PFM here incorporates these parameters obtained from DFT and MD calculations through simplification and parameterization. We apply this model to a 1-D prototypical battery system with a Li metal anode and a liquid electrolyte consisting of 1 M LiPF6 in ethylene carbonate (EC) and simulate the evolution of two common SEI products, i.e., organic component dilithium butylene dicarbonate (Li2BDC) and inorganic component lithium carbonate (Li2CO3). By tracking the temporal evolution of the spatial distribution of these products and electrolyte species, we analyze the effect of electron tunneling on SEI thickness, examine the competition between the reactive and diffusive processes during the growth of different SEI products, and identify the governing mechanism behind the formation of different SEI products. Our findings pinpoint the Li+ diffusion as the key limiting factor of the formation of organic Li2BDC during the initial 10−5 s scale, whereas Li2CO3 directly formed by two-electron reduction of EC is limited by its slow reaction kinetics within around 10−2 s. This multiscale approach, for the first time, provides profound insights into the SEI formation across time scales spanning 8 orders of magnitudes (from nanoseconds to seconds) and length scales spanning 3 orders of magnitudes (from angstroms to 100 nm). Furthermore, the ability of AI-PFM to simulate complex reaction networks that encompass multiphases and multicomponent systems has been demonstrated.
Results and discussion
Fig. 1 shows the overall framework for modeling the SEI growth integrating multiple electrochemical reactions, species transport, and electron tunneling. DFT calculations are utilized to determine the reaction pathways and their corresponding energy profiles, while MD simulations are employed to obtain the diffusivity of species. Next, phase-field simulations are performed to study the temporal and spatial evolution of SEI products, incorporating parameters derived from atomic-scale calculations. Details of all sub-models can be found in the Experimental section. | ||
| Fig. 1 Schematic modeling framework of SEI formation on a lithium metal. (A) Reaction networks and corresponding energy profiles (Gibbs free energy change ΔG0, reduction potential ψ0, and kinetic barrier ΔG* obtained from DFT calculations) of the considered reactions. MD simulations provide the diffusivity of species Di. (B) Phase-field model and boundary conditions (BC) for three coupled processes: electrochemical reactions, ion diffusion and electron tunneling. The simulation domain ranges from the surface of the Li metal electrode at x = 0 nm to the bulk liquid electrolyte region (the other boundary is at x = 100 nm). The initial concentrations of EC and LiPF6 in the electrolyte are 15 M and 1 M, respectively. Unless otherwise specified, the initial SEI nucleus on the Li metal surface consists of two layers: a 0.5 nm dense Li2CO3 layer adjacent to the Li metal and a 6 nm Li2BDC layer with a porosity of 50% adjacent to the electrolyte. A set of evolving non-conserved order parameters (ϕE, ϕS1, and ϕS2) represent the electrolyte, inorganic Li2CO3, and organic Li2BDC phases, respectively. | ||
To obtain the atomic-scale input parameters, we perform an extensive DFT study to establish the predefined reaction network that contains both electrochemical reactions (magenta arrows) and purely chemical reactions (green arrows) (Fig. 2). For each reaction, we calculate its standard Gibbs free energy change ΔG0 (for purely chemical reactions) or reduction potential ψ0 (for electrochemical reactions), as well as the electron transfer kinetic barrier ΔG* according to Marcus theory.44,45Fig. 2A shows all the reactions considered in our atomistic simulations, involving multiple reactants (Li+, c-EC, and e−), intermediate species (o-EC−, Li+/c-EC, Li+/o-EC−, Li+/CO32−, and 2Li+/o-EC−), and SEI products (Li2BDC, Li2CO3, and C2H4). Here, c-EC represents the neutral cyclic EC molecule, and o-EC− represents the reduced ring-opened EC.
 | ||
| Fig. 2 Illustration of the reaction pathways. (A) All the reaction products and pathways considered in atomistic simulations. The reduction potentials ψ0 (vs. SHE), the electron transfer kinetic barrier ΔG* for electrochemical reactions (green arrows), and Gibbs free energy change (ΔG0) for chemical reactions (blue arrows) are included. (B) and (C) Simplified reaction pathways and their corresponding thermodynamic and kinetic parameters. The reaction steps, R1, R2, and R3, along with the parameters in (C), are adopted for the phase-field simulations. | ||
While it is possible to develop a comprehensive phase-field model that incorporates all these reactions, such simulations would not efficiently bridge the length and time scales. Thus, we simplify the reaction paths and focus on the primary SEI products: the organic Li2BDC and inorganic Li2CO3 (Fig. 2B and C), based on DFT computed thermodynamics driving forces, consistent with extensive theoretical and experimental studies.15,28,46 We note that Li2BDC is thermodynamically more favorable than Li2EDC, in agreement with other computational studies,3,47 and both products have been experimentally observed to coexist within the SEI.46 Therefore, we consider Li2BDC as the representative organic product in our model. The impact of Li ions on EC reduction is considered, but the anions are not, as recent molecular dynamics simulations showed that the typical PF6− anions do not enter the electric double layer (EDL) in strong carbonate-based solvents.48 The simplification treatment involves two procedures: (1) for two or more parallel reactions, we select the smallest reaction barrier as the simplified reaction barrier and record the Gibbs free energy change (parallel reactions have the same Gibbs free energy change); (2) for series reactions, we select the largest reaction barrier as the simplified reaction barrier and record the sum of Gibbs free energies for these series reactions.
In the phase-field simulations, we focus on the formation kinetics of organic Li2BDC and inorganic Li2CO3via simplified reactions (R1, R2, and R3 in Fig. 2C) at a constant voltage of −3.04 V with respect to the standard hydrogen electrode potential, SHE (or 0 V versus Li+/Li0). As illustrated in Fig. 1, we employ a 1-D system representing a half-cell, and the simulation domain spans from the Li metal electrode surface at x = 0 nm into the bulk liquid electrolyte comprising EC and 1 M LiPF6 at x = 100 nm. A set of non-conserved order parameters (ϕE, ϕS1, and ϕS2) represent the electrolyte (E), inorganic Li2CO3 (S1), and organic Li2BDC (S2) phases, respectively. The phase evolution is governed by the Allen–Cahn equations (eqn (3)–(5) in the Experimental section). The total Gibbs free energy change ΔGrm and the linearized reaction rate Rm for each reaction (m is the reaction index for R1, R2, and R3) are related to its standard Gibbs free energy change ΔG0, reduction potential ψ0, and the activation energy ΔG* from DFT, as well as the local activities of species including electrons, Li+ and EC molecules, as shown in eqn (6) and (7) in the Experimental section. Electron tunneling is a key short-term electron transport mechanism for SEI formation, which is governed by the tunneling barrier of SEIs. Therefore, we numerically solve the steady-state Schrödinger electron tunneling equation by formulating a phase-dependent tunneling barrier to calculate the probability of electrons in the SEI, so that the SEI/electrolyte interface positions do not need to be explicitly tracked, taking advantage of phase-field modeling. The local electron activity can then be defined as the probability of electrons in the SEI, i.e., ae− = |Ψ*Ψ|, where Ψ is the electron wave function (see “Electron tunneling” in the Experimental section for details). At the Li metal surface, ae− is presumed to be 1 and decays exponentially through the SEI and the electrolyte. The time-dependent evolution of the concentration distribution of Li+ and EC is dominated by the reaction-diffusion equation (eqn (9) in the Experimental section), and the diffusivities of species are obtained from MD calculations (see the “Species transport” section in the Experimental section for details). We assume that the concentration of both Li+ and EC at the Li/SEI interface is 0 M. At the right electrolyte boundary, their concentrations are fixed at 1 M for Li+ and 15 M for EC, corresponding to their initial bulk concentration. The activities of species are given by ai = xi/x0i, where xi is the concentration of species Li+ and EC, and the standard concentrations of Li+ and EC (x0EC) are 1 M and 15 M, respectively. Therefore, the initial activity of both aLi+ and aEC is 1.
and EC (x0EC) are 1 M and 15 M, respectively. Therefore, the initial activity of both aLi+ and aEC is 1.
Electron tunneling effect on SEI thickness
SEI formation is initiated with the electrolyte reduction at the electrode surface, where electrons are transferred from the electrode via tunneling through the growing SEI layer. The electrolyte reduction products are precipitated on the electrode surface, serving as a protective layer against further electrolyte decomposition, and the thickness of the SEI is determined by the electron tunneling range. Therefore, in this section, we first investigate the effect of electron tunneling on the growth of both organic Li2BDC and inorganic Li2CO3via R1 and R3, respectively. We consider a 1-D system with different single SEI nuclei representing the initial dense organic Li2BDC and inorganic Li2CO3 seeds, as illustrated in Fig. 3A and B. To focus on investigating the electron tunneling behavior, we temporally disregarded the transport of Li+ and EC during SEI growth by assuming that the activities of Li+ and EC remain constant at their initial value (aLi+ = aEC = 1), mimicking a semi-infinite system with sufficient supplies of these species from the electrolyte. Thus, the Gibbs free energy changes of organic Li2BDC (ΔGrR1) and inorganic Li2CO3 formation (ΔGrR3) from eqn (6) in the Experimental section are modified as follows:ΔGrR1 = ΔG0R1 + F(ψe − ψsol − ψ0R1) − RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnae− lnae− | (1) |
| ΔGrR3 = ΔG0R3 + F(ψe − ψsol − ψ0R3) − 2RTlnae− | (2) |
 | ||
| Fig. 3 The effect of electron tunneling on the growth of the organic Li2BDC based on R1 and inorganic Li2CO3 based on R3. (A) and (B) Illustrations of reactions R1 and R3 considered and the 1D system, respectively, (C) comparison of organic Li2BDC growth behaviors between assuming ae− = 1 and electron tunneling, and (D) comparison of inorganic Li2CO3 growth behaviors assuming ae− = 1 and electron tunneling. | ||
To highlight the electron tunneling effect, we compare two cases: one assuming the electron activity ae− = 1 throughout the system (i.e., assuming that the SEI behaves like a metal) and the other with electron activity ae− obtained from the steady-state Schrödinger electron tunneling equation. As shown in Fig. 3C and D, starting with an initial thickness of 6 nm, the SEI will continuously grow when ae− = 1 until the electrolyte is fully consumed. However, when considering the electron tunneling effect, both organic and inorganic SEIs exhibit self-limiting growth behavior. They stop the initial quick growth after reaching a specific thickness. This occurs because the Gibbs free energy changes of reactions R1 and R3 (from eqn (1) and (2)) remain consistently negative, allowing the reactions to proceed indefinitely; but with electron tunneling, the electron activity decays exponentially as the SEI grows (Fig. S1, ESI†). Once the electron activity reduces to a certain level (i.e., when ΔGrR1 = 0 and ΔGrR3 = 0 in eqn (1) and (2)), reactions R1 and R3 reach equilibrium, where the SEI reaches a tunneling-limited thickness and stops further growth. In our model, the tunneling barrier for Li2CO3 (ΔELi2CO3 = 1.78 eV) is derived from the DFT calculation.49 The tunneling barrier for Li2BDC is estimated to be a lower bound of ΔELi2BDC = 0.24 eV due to porosity50 and the fact that the organic Li2DEC, structurally close to Li2BDC, has been experimentally measured to exhibit a tunneling barrier of ∼1 eV lower than that of inorganic SEI components.51 It aligns with the general trend that the inorganic component in the SEI blocks electron tunneling more effectively than the organic species. Using these values, our model predicts tunneling-limited SEI thicknesses of ∼29.4 nm and ∼11 nm for Li2BDC and Li2CO3, respectively, which are close to the experimentally reported values29,30 during SEI formation. These can be referred to as the “tunneling-limited thickness”, which leads to a good estimation of the first cycle capacity loss, corresponding to the Li consumed to form the SEI up to the tunneling-limited thickness, agreeing well with experiments.49,52
Fig. 3 also shows the time scales to grow the Li2BDC and Li2CO3 layers to reach a stable thickness. They are 66 ps and 20 ms for Li2BDC and Li2CO3 layers, respectively, under the assumption of no concentration variation of Li+ and EC during SEI growth. The electron tunneling generally occurs within a few attoseconds.53 Consequently, this discrepancy in timescales is primarily attributed to the kinetic barrier of the single-electron reduction reaction (R1), substantially lower than that of the two-electron reduction reaction (R3), despite the overall Gibbs free energy of R3 being much greater than that of R1.
Effect of Li+ and EC molecules on SEI formation rates
In addition to the electronic tunneling effect on the SEI thickness, the evolution of the concentration of Li+ and EC in the system is also critical to the growth dynamics of SEI products. To further investigate the governing factors for both organic and inorganic SEI growth kinetics, we performed a series of simulations by turning on/off the diffusivities of Li+ and EC. By assuming the activity of species, we can compare four different cases: (1) not evolving both Li+ and EC (aLi+ = 1 and aEC = 1) means that the concentrations of Li+ and EC remain constant at their initial values during SEI growth. Under this idealized condition, the reaction rate is governed mainly by charge-transfer kinetics. (2) Only evolving EC (aLi+ = 1, and aEC ≠ 1 calculated using eqn (9)) represents the scenario where only the consumption of EC is considered while the Li+ concentration remains at its initial value. The reaction rate is affected by both charge-transfer kinetics and the local activity of EC. (3) Only evolving Li+ (aLi+ ≠ 1 calculated using eqn (9), and aEC = 1) represents the scenario where only the consumption of Li+ is considered while the EC concentration remains at its initial value. The rate is thus governed by charge-transfer kinetics and the local activity of Li+. (4) Evolving both Li+ and EC (aLi+ ≠ 1 and aEC ≠ 1 calculated using eqn (9)) indicates that both Li+ and EC are consumed according to their stoichiometric ratio during SEI growth, and their concentration distributions over time are determined using the diffusion equation. Reaction rates in this case reflect a coupled control by charge-transfer kinetics and the activities of all species. The simulation system is the same as those presented in Fig. 3A and B.In case (1) shown in Fig. 4A, the organic SEI layer growth via R1 will reach their tunneling-limited thickness within around 66 ps (solid blue line), and its rate is purely governed by the reaction kinetics. Furthermore, we find that the EC diffusion has no significant effect on the Li2BDC growth (the solid and dotted blue lines overlap) by comparing cases (1) and (2), as the EC concentration in the electrolyte closely matches that in Li2BDC, suggesting that EC molecules could be reduced on-site without requiring additional EC supplied by the electrolyte. Considering the Li+ consumption and diffusion by comparing cases (3) and (4), it is found that the predicted Li2BDC growth time in Fig. 4A increased from 36 ns to ∼57 μs (solid and dotted purple lines). This is consistent with the time scale (∼29 μs) for Li+ to diffuse from the right boundary of the electrolyte region to the Li2BDC surface. It is estimated by using 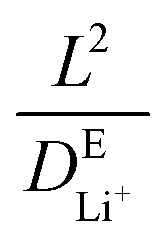 , where L = 100 nm is the diffusion length and
, where L = 100 nm is the diffusion length and  is the diffusivity of Li+ in the electrolyte obtained from MD simulations. The simulated Li2BDC growth time (∼57 μs) is close to the
is the diffusivity of Li+ in the electrolyte obtained from MD simulations. The simulated Li2BDC growth time (∼57 μs) is close to the  estimation, indicating the Li+ diffusion-controlled growth nature. This is because, in contrast to EC, the Li site density inside Li2BDC (∼13.5 M) is significantly higher than the initial concentration of Li+ in the electrolyte (1 M) and the reaction rate of R1 is much faster than that of the Li+ diffusion, which means that a large amount of Li+ needs to be consumed to grow the SEI, necessitating Li+ diffusion from the electrolyte to the SEI/electrolyte interface to sustain Li2BDC growth, as shown in Fig. S2A (ESI†).
estimation, indicating the Li+ diffusion-controlled growth nature. This is because, in contrast to EC, the Li site density inside Li2BDC (∼13.5 M) is significantly higher than the initial concentration of Li+ in the electrolyte (1 M) and the reaction rate of R1 is much faster than that of the Li+ diffusion, which means that a large amount of Li+ needs to be consumed to grow the SEI, necessitating Li+ diffusion from the electrolyte to the SEI/electrolyte interface to sustain Li2BDC growth, as shown in Fig. S2A (ESI†).
 | ||
| Fig. 4 1-D phase-field simulation of the growth of the organic and inorganic SEI products, considering reactions R1 and R3 in Fig. 2C. Parameter study for the governing kinetic factors for organic Li2BDC growth (A) and inorganic Li2CO3 growth (B). Four cases: (1) not evolving both Li+ and EC (aLi+ = 1 and aEC = 1): the concentrations of Li+ and EC remain constant at their initial values during SEI growth; (2) only evolving EC (aLi+ = 1, and aEC ≠ 1 calculated using eqn (9)): only the consumption of EC is considered while the Li+ concentration remains at its initial value; (3) only evolving Li+ (aLi+ ≠ 1 calculated using eqn (9), and aEC = 1): only the consumption of Li+ is considered, while the EC concentration remains at its initial value; (4) evolving both Li+ and EC (aLi+ ≠ 1 and aEC ≠ 1 calculated using eqn (9)): both Li+ and EC are consumed according to their stoichiometric ratio during SEI growth. | ||
Regarding the inorganic Li2CO3 growth via R3, the Li2CO3 layer grows to its tunneling-limited thickness of about 11 nm under ∼20 ms, which is much slower than the diffusion time of both Li+ (∼29 μs) and EC, and thus their diffusion does not influence the inorganic Li2CO3 SEI layer growth behavior (Fig. 4B and Fig. S2B, ESI†). In contrast to the organic Li2BDC, the growth rate of Li2CO3 is much (∼103 times) slower due to the significantly larger kinetic barrier  of R3 than
of R3 than 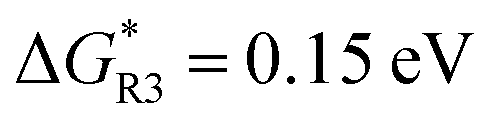 for R1. We further show that by increasing the Li+ concentration from 1 M to 4 M, the time required for Li2BDC to reach its stable thickness decreases from 57 μs to 1 μs, while the growth rate of dense Li2CO3 remains unchanged (Fig. S3, ESI†). Consequently, the growth rate of Li2BDC is determined by Li+ diffusion, whereas the growth of Li2CO3 is controlled by the reaction rate.
for R1. We further show that by increasing the Li+ concentration from 1 M to 4 M, the time required for Li2BDC to reach its stable thickness decreases from 57 μs to 1 μs, while the growth rate of dense Li2CO3 remains unchanged (Fig. S3, ESI†). Consequently, the growth rate of Li2BDC is determined by Li+ diffusion, whereas the growth of Li2CO3 is controlled by the reaction rate.
Spatial, chemical, and temporal evolution of the SEI
In the previous section, we separately investigate the effects of electron tunneling and species diffusion on the growth kinetics of single-layered dense organic Li2BDC or inorganic Li2CO3 products. However, experimental studies indicate that the formed SEI often consists of a two-layer structure, with an inner inorganic layer and an outer organic layer that usually exhibits a porous structure.29,30 Therefore, it is crucial to study the spatial, chemical, and temporal evolution of the SEI layer, as well as the competition between products, to gain an in-depth understanding of the limiting processes, their interactions, and the final SEI composition and thickness. To this end, we analyze the temporal SEI formation via R1 to R3 based on Fig. 2C. Initially, we assume that there is a two-layer structured SEI nucleus within the simulation system, comprising a 0.5 nm dense Li2CO3 layer adjacent to the Li metal and a 6 nm thick layer of Li2BDC at the outer layer with a constant porosity of 50%, as shown in Fig. S4 (ESI†). Subsequently, we calculate the SEI formation from nanoseconds to seconds. Fig. 5A illustrates the general trends of SEI thickness over time. Initially, from nanoseconds to microseconds, the porous Li2BDC will grow to its stable thickness (∼27.2 nm) via R1. After 21.7 μs, Li2BDC near the Li metal side transforms into porous Li2CO3via R2, growing to its stable thickness (∼11 nm). After 1 ms, the pores in the porous Li2CO3 are filled via R3. The eventual SEI consists of a porous outer organic layer (∼16.2 nm thick) and a thinner dense inner inorganic layer (∼11 nm) covering the anode surface. This aligns well with experimental finding of a two-layered structure.2,5,29 | ||
| Fig. 5 Temporal evolution of SEI growth in the Li/(EC + 1 M LiPF6) model system. (A) Temporal evolution of the SEI thickness. The dashed and solid lines represent the porous and dense products, respectively. Temporal evolution of the order parameters (B) indicating the products and the concentration distribution of Li+ (C) at 6 selected times. The position (0–100 nm) signifies the distance from the Li anode surface to the electrolyte region. (D) Schematic depiction of SEI growth in the Li/(EC + 1 M LiPF6) model system from nanoseconds to seconds. Color scheme: gray for the Li anode, blue for Li2BDC, orange for Li2CO3, light yellow for EC, and purple for Li+. | ||
To understand the underlying processes governing SEI formation, we conduct a detailed analysis of the temporal evolution in the distribution of chemical reaction species and SEI products, as shown in Fig. 5B and C. The corresponding SEI morphologies at 6 selected time frames are displayed in Fig. 5D. The diffusion rate of Li+ is slower than that of EC decomposition via R1 as the porous Li2BDC products gradually grow. As a result, Li+ ions are immediately consumed when they arrive at the SEI/electrolyte interface, leading to a concentration gradient in the electrolyte zone from 1 μs to 21.7 μs (stages I to II). This indicates that Li2BDC production is a Li+ diffusion-limited process. Despite the charge-transfer rate for the R2 pathway being slightly slower than that of R1, the formation of Li2CO3via R2 does not occur in this stage due to limited Li+ availability as Li+ is preferentially consumed by the faster-growing R1 process. After 21.7 μs, when Li2BDC reaches its tunneling-limited thickness, additional Li+ begins to diffuse into this porous layer (stages II to III), enabling Li2BDC to react with Li+ and electrons to form Li2CO3via R2, and the overall process still remains diffusion-limited. After ∼0.02 s, Li2CO3 can be generated inside the pores. Since direct two-electron reduction of EC to Li2CO3via R3 has a much slower charge-transfer rate, the Li+ diffusion from the electrolyte rapidly compensates for the consumption of Li+ during the two-electron reduction of EC, which is a reaction-controlled process (stages IV to VI), as shown in Fig. 5C. In contrast to the Li+ concentration profiles, no EC-concentration gradient develops during the entire process, as shown in Fig. S5 (ESI†). From this, we conclude that the first two steps of one-electron reduction process including the electrolyte degradation to organic Li2BDC and transformation to Li2CO3 are Li+ diffusion-limited processes (stages I to III); then, the two-electron reduction reaction that directly formed dense Li2CO3 is a reaction-controlled process (stages IV to VI).
Furthermore, after 0.02 s, the Li metal surface is fully covered with SEI products and the EC can no longer be reduced due to the blocking of electron tunneling. This indicates that the initial SEI formation is completed within this short time frame, which agrees with the experimental results.7,54 Direct in situ measurements of the initial SEI formation are rather challenging, as these reactions occur very fast,55 as pointed out by Odziemkowski and Irish,7 who tracked the corrosion potential time transients of the Li-metal electrode in various electrolyte systems and indicated that the passivating reactions, which lead to SEI formation, are often completed in less than 1 second. The two-layered SEI model, with the inorganic species (LiF, Li2CO3, and Li2O) close to the electrode surface and the porous organic layer (e.g. LEDC) closer to the electrolyte, obtained from postmortem analysis, does not reveal the formation sequence and timelines of these species.56 Notably, the absence of operando tools with nanosecond-to-second temporal resolution and nanoscale chemical specificity is a well-recognized gap.3 Recently, the formation sequence obtained from gas evolution combined with other spectroscopy analysis revealed LEDC as the major product with little Li2CO3 during initial SEI formation, and LEDC eventually evolved into Li2CO3.57–60 Using isotope exchange along with in situ time-of-flight secondary ion mass spectroscopy (TOF-SIM) measurements, a bottom-up SEI growth mechanism was proposed, suggesting that the SEI components formed in the early stage (organic species) are on the outer side (electrolyte side), while those formed in the latter stage (inorganic species) are on the inner side (electrode side).61 These experimental results on the temporal and spatial evolution of SEI formation collectively support the prediction from our atomically informed phase-field model (AI-PFM). This sequence, LEDC forms first and converts into Li2CO3 near the electrode surface, is different from the CRN-kMC, which predicted that LEDC continues to grow even after Li2CO3 stops growing.31
Influence of key parameters on the SEI growth dynamic
Having elucidated the growth dynamics and governing mechanisms of SEI formation on the Li metal in EC with LiPF6, we further employ our model to explore how key parameters, including electron tunneling barriers (ΔE), species diffusivities, and reaction kinetic barriers (ΔG*), affect SEI evolution.The relative tunneling barriers play an important role in determining the SEI morphology. Higher electron-tunneling barriers led to a thinner Li2BDC layer via R1 and a Li2CO3 layer via R3 and a shorter time to reach the tunneling-limited thickness (Fig. S6A–C, ESI†). Since the tunneling barrier of the organic phase (Li2BDC) is more susceptible to factors such as porosity and electrolyte composition,50 while only varying ΔELi2BDC from 0.24 to 1.8 eV, the overall SEI formation sequence remained the same: rapid porous Li2BDC deposition via R1, partial conversion to Li2CO3via R2, and final pore filling by Li2CO3via two-electron EC reduction in R3. However, the bilayer morphology was only formed when ΔELi2BDC < ΔELi2CO3. Conversely, when ΔELi2BDC ≥ ΔELi2CO3, a thinner initial Li2BDC layer is ultimately fully consumed and converted to a thicker dense Li2CO3 layer, yielding a predominantly single inorganic SEI layer (Fig. S8, ESI†). These results underscore the critical role of relative tunneling barriers in determining the SEI morphology.
The effect of Li+ diffusivity on SEI growth has also been systematically examined. Increasing Li+ diffusivity in the liquid electrolyte from 10−11 m2 s−1 to 10−8 m2 s−1 significantly accelerates Li2BDC growth via R1, reducing the growth timescale from ∼10−4 s to ∼10−7 s (Fig. S9A, ESI†), with no impact on the Li2CO3 growth via R3, as it is governed by the reaction kinetics rather than diffusion (Fig. S9C, ESI†). Notably, variations in Li+ diffusivity within both solid phases have a negligible influence on both Li2BDC and Li2CO3 growth (Fig. S9B and D, ESI†), further underscoring that liquid-phase Li+ transport is the rate-limiting step for initial organic SEI formation. Additionally, Li+ diffusivity has a minimal influence on the SEI growth sequence and the resulting bilayer architecture (Fig. S10, ESI†).
We further evaluated the impact of the charge-transfer kinetic barrier (ΔG*) on SEI growth by sweeping ΔG* from 0.15 to 0.75 eV for both Li2BDC (R1) and Li2CO3 (R3). This range corresponds to a decrease in the intrinsic electron-transfer rate constant k0m from 2.08 × 1010 s−1 to 1.736 s−1. As shown in Fig. S11 (ESI†), two kinetic regimes emerge: (i) Li+-diffusion-limited region: for lower ΔG*, both SEI products reach their tunneling-limited thickness around the same time and remain largely insensitive to ΔG* and (ii) charge-transfer kinetic-limited: for higher ΔG*, the growth rates of both Li2BDC and Li2CO3 decrease exponentially with increasing ΔG*. The transition occurs at ΔG* ≈ 0.49 eV, where the calculated charge-transfer reaction rate equals the Li+ diffusion rate, delineating the shift between Li+-diffusion and electron-transfer-controlled regimes (Fig. S11C, ESI†). These results highlight variations in kinetic barriers for each reaction, and their relative relationship to Li+ diffusion rates can lead to multiple rate hierarchies among R1–R3, ultimately altering the overall SEI growth sequence and one-layer or two-layer SEI structures.
It should be clarified that our simulations focus on the initial formation of the SEI, occurring on the timescale of seconds. Although our phase-field model can simulate and demonstrate the SEI evolution from nanoseconds to seconds, this self-limiting behavior is featured only during the initial short-term growth of SEIs. Ideally, the SEI would stabilize after initial formation, preventing further Li+ consumption and electrolyte degradation, since it is electronic insulating. However, the SEI layer thickness can change during cycling and calendar aging.3 Electron leakage mechanisms through hole polaron migration,62 the formation and transport of Li-atom interstitials,63 radical species shuttling,50 as well as grain boundaries,64,65 may dominate the SEI evolution over longer timescales. Shi et al.66 reported that Li atoms can diffuse through the SEI via interstitial mechanisms, forming positive-charged Li interstitials and electrons. A continuum model20 predicted that the SEI can continue to grow to 1400 nm in 1000 days, where the neutral Li atoms carrying electrons facilitate this long-term SEI growth. Moreover, defects such as grain boundaries and heterogeneous interfaces,64 cracks, and pores67 can serve as short-circuit transport paths for electrons and other reacting species. These mechanisms lead to continuous “growth” of the SEI as the battery degrades. We intend to thoroughly investigate these possibilities in our forthcoming study with a 2-D model.
The major strength demonstrated by this model framework is its ability to resolve the spatial and temporal evolution of the SEI composition and thickness that span from nanoseconds to seconds and deconvolute the interplay among multiple physical phenomena (reactions, species transport, and electron tunneling), as well as reveal the governing factors for each electrolyte degradation process. Although this deconvolution provides insights into the governing mechanisms of electrolyte decomposition that are currently inaccessible to experimental microscopy or spectroscopy techniques, the predictions from our simulations, such as the initial SEI formation and thickness of the SEI within milliseconds and the preferential formation of inorganic vs. organic products near the Li surface, serve as testable hypotheses for future experimental studies and can guide experimentalists by identifying key mechanistic signatures to be probed indirectly through ex situ quenching or rapid-interruption experiments followed by surface analysis.
The modeling framework developed in this study is broadly extensible to other electrochemical systems beyond lithium-based batteries, owing to two central features. First, it is explicitly informed by DFT calculations, which capture the chemical changes at the electrode/electrolyte interface. Second, the model is formulated as a general multiphase, multicomponent phase-field framework capable of capturing complex couplings among interfacial reactions, species transport, and microstructural evolution. To adapt this framework to other electrochemical systems, such as sodium-ion, lithium–sulfur, or solid-state batteries, the key steps involve determining the reaction networks and corresponding parameters via DFT calculations, redefining the phase-field variables to reflect the relevant phases, and modifying the free energy functional to include new phases and reaction intermediates. Transport parameters (e.g., diffusivities and electron tunneling barriers) can be similarly updated to reflect system-specific physical properties. For instance, the same model structure can simulate Na SEI formation by introducing Na-containing decomposition reactions and products (e.g., Na2CO3 and Na2O68) and recalibrating the thermodynamic driving force and reduction kinetics accordingly. In solid-state systems, the framework can be further extended to include coupled electrochemical-mechanical effects by incorporating additional mechanical energy terms or coupling coefficients in the free energy functional, thereby enabling the simulation of multiplephase evolution at the solid–solid interface. By extending to higher dimensional simulations, it can potentially capture the detailed SEI morphology and its competition with Li stripping/plating. Combining this model with high-throughput calculations and virtual screening for materials discovery would provide data-based design guidance.
Conclusions
In summary, we present an atomically informed phase-field framework that reveals SEI evolution across unprecedented time scales (from nanoseconds to seconds) and lengths (from angstroms to 100 nm). Initially, porous Li2BDC grows to ∼27.2 nm via one-electron reduction within microseconds. After 21.7 μs, part of the Li2BDC near the Li metal converts into porous Li2CO3 (∼11 nm). By 1 ms, the pores are filled with Li2CO3 directly via two-electron reduction, forming a final SEI with a porous organic outer layer (∼16.2 nm) and a dense inorganic inner layer (∼11 nm). Li2BDC formation and its transformation into Li2CO3 are limited by Li+ diffusion, while the final two-electron reduction of EC to form Li2CO3 is reaction-controlled. Electron tunneling determines the thickness of both layers. This study enhances the understanding of SEI formation and demonstrates potential for simulating complex reaction networks across broad time and length scales.Experimental
Phase-field model
We formulate the phase-field model by considering the simplified reaction networks as well as the reacting species Li+, e− and EC, based on our recently developed phase-field model of stoichiometric compounds and solution phases.69 With this approach, the phase-field governing equations can be directly derived via the variational derivatives of the free energies without any approximation or arbitrary treatments. The thermodynamic and kinetic parameters of the simplified reactions are shown in Fig. 2C and Table S2 (ESI†). We also consider the following assumptions/simplifications:• The SEI products are considered stoichiometric compounds whose free energy only exists at their stoichiometric composition points rather than being a continuous function of composition, as shown in Fig. S12 (ESI†). Meanwhile, the electrolyte is assumed to be an ideal solution, whose realistic interaction behavior will be investigated in our future work using MD simulations or Debye–Hückel approximations. The Li metal anode is assumed to be at the left boundary of the simulation region and not explicitly simulated.
• The electric potential is assumed to be uniform within the electrolyte and the SEI. Therefore, the applied voltage is imposed, and the Poisson equation is not solved, which significantly improves numerical efficiency and stability. The electron transport within the system is assumed to be dominated by tunneling.
• To improve numerical convergence, we use linear kinetics for most of the simulations involving SEI products in this work.
• The organic Li2BDC is inherently micro-porous due to molecular disorder and low packing density. In our 1D model, porosity is represented by a fixed value rather than explicitly evolving pore structures, which is a common simplification in 1-D SEI simulations.18,22 Thus, the porous SEI layer is considered as a mixture of electrolyte and solid products. While the current framework does not incorporate dynamic porosity, it retains the ability to resolve the competitive formation, spatial distribution, and temporal evolution of major inorganic and organic SEI species.
• The evolution of the gas phase (e.g., C2H4) is neglected because it escapes from the SEI or system and does not contribute to further reactions and the SEI film.31 Their formation energies and reaction barriers are still accounted for when selecting the dominant solid-phase pathways, influencing the free energy landscape and kinetics of the phase-field model. Thus, neglecting explicit gas-phase evolution has a minimal impact on the predicted solid-state SEI growth kinetics.
• The decomposition of both lithium hexafluorophosphate (LiPF6) and the formed SEI products (i.e., Li2O) is not considered in this model. We assume that the diffusion and dissolution of primary electrolyte reduction products (Li2CO3 and Li2BDC) formed during SEI formation are ignored due to relatively low solubility.70,71
Three phases are distinguished by a set of non-conserved order parameters, in which (ϕE,ϕS1,ϕS2) = (1,0,0), (0,1,0), and (0,0,1) represent the liquid electrolyte (E), Li2CO3 (S1) and Li2BDC (S2) phases, respectively. The kinetic evolution of the primary order parameters ξ is governed by the following Allen–Cahn equations:69
 | (3) |
 | (4) |
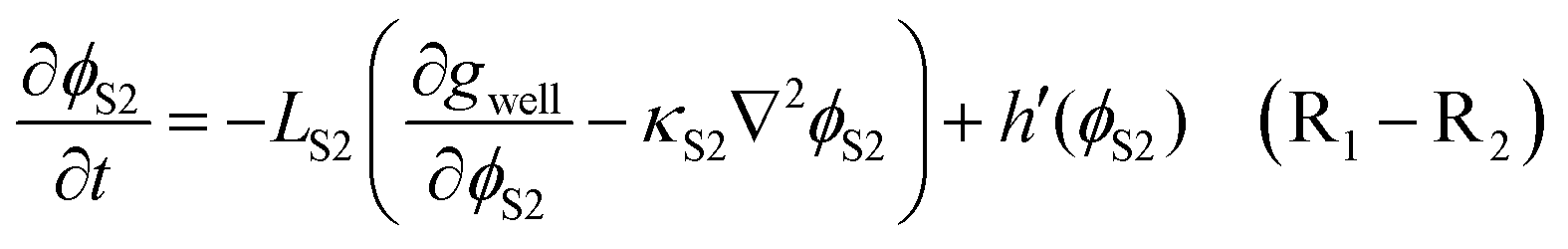 | (5) |
The total Gibbs free energy change ΔGrm and the linearized reaction rate Rm of reactions R1 to R3 can be written as follows:
 | (6) |
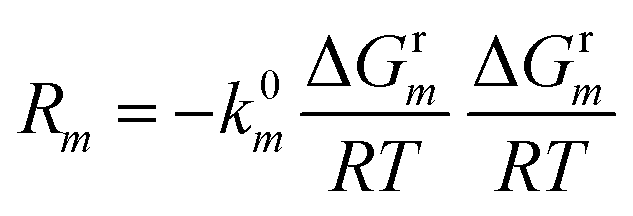 | (7) |
 is the electron transfer kinetic with the unit of (s−1),
is the electron transfer kinetic with the unit of (s−1), 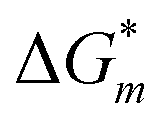 is the kinetic barrier of the electron transfer, m is the reaction index from R1 to R3, F is the Faraday constant, ψe is the electric potential in the electrode, ψsol is the electric potential in the electrolyte, and ψ0m is the reduction potential vs. SHE; ΔG0m is the standard Gibbs free energy change, R is the ideal gas constant, T = 298 K is the temperature, ak and al are the activities of the reactants and products of a given reaction, and νmk is the stoichiometric coefficient for species k in reaction m, which is positive for the reactants and negative for the products. In this work, we simulate the SEI formation at a constant voltage of −3.04 V vs. SHE (i.e., 0 V versus Li+/Li0). At this voltage, the overpotential of reaction Li+ + e− = Li is 0, which means ψe − ψsol − ψLi+/Li0 (vs. SHE) = 0 V. The interfacial potential difference at the Li metal and electrolyte interface is thus ψe − ψsol = −3.04 V.
is the kinetic barrier of the electron transfer, m is the reaction index from R1 to R3, F is the Faraday constant, ψe is the electric potential in the electrode, ψsol is the electric potential in the electrolyte, and ψ0m is the reduction potential vs. SHE; ΔG0m is the standard Gibbs free energy change, R is the ideal gas constant, T = 298 K is the temperature, ak and al are the activities of the reactants and products of a given reaction, and νmk is the stoichiometric coefficient for species k in reaction m, which is positive for the reactants and negative for the products. In this work, we simulate the SEI formation at a constant voltage of −3.04 V vs. SHE (i.e., 0 V versus Li+/Li0). At this voltage, the overpotential of reaction Li+ + e− = Li is 0, which means ψe − ψsol − ψLi+/Li0 (vs. SHE) = 0 V. The interfacial potential difference at the Li metal and electrolyte interface is thus ψe − ψsol = −3.04 V.
Electron transport
To describe the electron tunneling from the Li metal through the SEIs and electrolyte behavior, we develop a diffuse-interface description of the steady-state Schrödinger equation with a phase-dependent tunneling barrier, | (8) |
 is the phase-dependent tunneling barrier, with ΔE0tj being the electron tunneling barrier for each phase. The electron tunneling barrier for the porous structure is assumed to be a constant, without explicitly accounting for the local variations introduced by solvent-filled pores.50,72 As the 1D model does not resolve the pore geometry, the uniform electron tunneling barrier is a reasonable assumption.73 As such, the Schrödinger electron tunneling equation is numerically solved for the entire system without the need to distinguish the different phase regions and phase interfaces. The local electron activity can then be defined as the probability of electrons in the SEI, i.e., ae− = |Ψ*Ψ|. This numerical solution to the Schrödinger equation indicates an exponential decay of the electron concentration at the SEI/electrolyte interface with the increase of the SEI thickness, which would lead to extremely low electron concentrations when the SEI becomes thicker than its tunneling-limited thickness dlim and cause the SEI to shrink. However, the electron concentration is lower bounded by the intrinsic electron concentration of the SEI and electrolyte. Therefore, in the phase-field simulations, we use ae− = |Ψ*Ψ| with a lower cut-off of
is the phase-dependent tunneling barrier, with ΔE0tj being the electron tunneling barrier for each phase. The electron tunneling barrier for the porous structure is assumed to be a constant, without explicitly accounting for the local variations introduced by solvent-filled pores.50,72 As the 1D model does not resolve the pore geometry, the uniform electron tunneling barrier is a reasonable assumption.73 As such, the Schrödinger electron tunneling equation is numerically solved for the entire system without the need to distinguish the different phase regions and phase interfaces. The local electron activity can then be defined as the probability of electrons in the SEI, i.e., ae− = |Ψ*Ψ|. This numerical solution to the Schrödinger equation indicates an exponential decay of the electron concentration at the SEI/electrolyte interface with the increase of the SEI thickness, which would lead to extremely low electron concentrations when the SEI becomes thicker than its tunneling-limited thickness dlim and cause the SEI to shrink. However, the electron concentration is lower bounded by the intrinsic electron concentration of the SEI and electrolyte. Therefore, in the phase-field simulations, we use ae− = |Ψ*Ψ| with a lower cut-off of 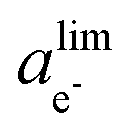 that ensures ΔGrm = 0 and an equilibrium SEI thickness of dlim to avoid the SEI shrinkage.
that ensures ΔGrm = 0 and an equilibrium SEI thickness of dlim to avoid the SEI shrinkage.
Species transport
The time-dependent evolution of the concentration distribution of Li+ and EC is dominated by diffusion. We directly evaluate the reduced concentration xi = ci/c0 in the electrolyte rather than the total concentration, which follows the reaction–diffusion equation below: | (9) |
Atomistic simulations
Density functional theory (DFT) calculations for ΔG0 and ψ0
All DFT calculations were conducted using the Gaussian 09 code.77 The double hybrid functional M06-2X78–81 and the basis set 6-31+G(d,p) together with the D3 dispersion correction82 were used. The SMD solvation model83 was used to account for the solvation environment with the dielectric constant set to ε = 20.5 for the carbonate-based electrolyte.84,85 The standard Gibbs free energy change (ΔG0) of the Li+ coordination reaction (the only chemical reaction considered in this work) is calculated as the Gibbs free energy difference between the product and the reactants. The Li+ coordination reaction was considered barrierless.31 The reduction potential of an electrochemical reaction with respect to the standard hydrogen electrode (SHE) was calculated via ψ0 = −ΔG0/F − 4.44. The calculation results of all reactions in Fig. 2A are presented in Table S1 (ESI†).Kinetic barrier ΔG* for electron transfer
Marcus theory44,45 was used to calculate the kinetics of reduction reactions. Specifically, it is assumed that all reduction reactions occur heterogeneously, with electrons transferred from the electrode.86 The energy barrier ΔG* for a reduction reaction is | (10) |
 | (11) |
Molecular dynamics simulations of diffusion coefficients
MD simulations were conducted to calculate the diffusion coefficients of Li+ and EC in the bulk electrolyte following our recent work.48 To be more specific, MD simulations were carried out using the Forcite module as implemented in the Materials Studio (MS) 202088 with the COMPASS III force field89 and a charge scale of 0.7 applied to the salt ions.48 The atomistic model for the carbonate-based electrolyte was constructed to be ∼1.0 M LiPF6 salt dissolved in the mixed solvent composed of 30 vol% EC and 70 vol % EMC. The MD simulations were first conducted under the constant particle number, volume, and temperature (NPT) ensemble for 2.0 ns at room temperature (20 °C). Then production runs under the NPT ensemble were conducted for 4.0 ns for statistical analyses to obtain the diffusion coefficients of Li+ and EC following the approach proposed in a recent work.90 The calculation results are presented in Table S2 (ESI†).Model implementation
The simulations are performed using COMSOL Multiphysics based on the finite element method. A one-dimensional model with a size of 100 nm is built in this work, and the simulation domain is discretized by a grid spacing of 0.5 nm. Zero-flux boundary conditions are applied for order parameters (ϕE, ϕS1, and ϕS2) at the left and right boundaries. The Li+ concentration in the electrolyte drops during SEI formation due to the consumption of large amounts of Li+ ions, and a new equilibrium needs to be established by stripping some Li+ into the electrolyte. The equilibrium Li-plating/stripping potential will shift due to the Li+ activity. However, the stripping of Li+ during initial SEI formation at potentiostatic voltages can be safely ignored, as the reaction rate of lithium deposition and stripping is much lower than that of SEI formation.35 Hence, we assume that the concentration of both Li+ and EC at the Li metal surface is 0 M. The concentrations of Li+ and EC at the right electrolyte boundary are 1 M and 15 M, respectively, corresponding to their initial bulk concentrations. The activity coefficients of Li+ and EC are 1 and 15, respectively. The boundary condition for solving the steady-state Schrödinger equation is maintained at Ψ = 1 (to mimic the pure Li metal) at the left boundary of our 1-D simulation region and at the right boundary. These boundary conditions are shown in Fig. 1. The phase-field parameters are summarized in Table S3 in the ESI.†
at the right boundary. These boundary conditions are shown in Fig. 1. The phase-field parameters are summarized in Table S3 in the ESI.†
Author contributions
K. N. Z.: methodology, investigation, data curation, writing – original draft, and visualization; Y. Z. J.: conceptualization, methodology, investigation, writing – review & editing, and supervision; Q. S. W.: methodology, investigation, data curation, and writing – review & editing; S. A. N.: methodology; L. Q. C. and Y. Q.: conceptualization, methodology, resources, writing – review & editing, supervision, project administration, and funding acquisition.Conflicts of interest
The work was performed when Dr Yanzhou Ji was at Penn State. His current affiliation is Ohio State University, Columbus, OH, USA. The work was performed when Dr Seyed Amin Nabavizadeh was at Penn State. His current affiliation is GE Power, Greenville, SC, USA. The work was performed when Dr Qisheng Wu was at Brown. His current affiliation is Suzhou Laboratory, Jiangsu, China.Data availability
The first-principles computational results on the structure of reaction products and the COMSOL input file for this model are available in the NOMAD repository at https://nomad-lab.eu/prod/v1/gui/user/uploads/upload/id/I6rPnwjtTAOafwYpwN_qPQ.Acknowledgements
We thank Dr John Lawson, Dr Mehta, and Dr Joakim H. Stenlid from NASA, as well as Dr Kang Xu from the SES AI Corp for stimulating discussions and reviewing the manuscript. The authors acknowledge financial support from NASA under grant number 80NSSC21M0107. K. N. Z. and L. Q. C. also appreciate the generous support from the Donald W. Hamer Foundation through a Hamer Professorship at Penn State. The computations were performed on the Roar supercomputers at Penn State and the Center for Computation and Visualization (CCV) at Brown.References
- X. B. Cheng, R. Zhang, C. Z. Zhao, F. Wei, J. G. Zhang and Q. Zhang, Adv. Sci., 2016, 3, 1500213 CrossRef PubMed.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- H. Wu, H. Jia, C. Wang, J. G. Zhang and W. Xu, Adv. Energy Mater., 2021, 11, 2003092 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- X. Cui, S. D. Kang, S. Wang, J. A. Rose, H. Lian, A. Geslin, S. B. Torrisi, M. Z. Bazant, S. Sun and W. C. Chueh, Joule, 2024, 8(11), 3072–3087 CrossRef CAS.
- M. Odziemkowski and D. E. Irish, J. Electrochem. Soc., 1992, 139, 3063–3074 Search PubMed.
- A. Yulaev, V. Oleshko, P. Haney, J. Liu, Y. Qi, A. A. Talin, M. S. Leite and A. Kolmakov, Nano Lett., 2018, 18, 1644–1650 Search PubMed.
- D. T. Boyle, Y. Li, A. Pei, R. A. Vila, Z. Zhang, P. Sayavong, M. S. Kim, W. Huang, H. Wang, Y. Liu, R. Xu, R. Sinclair, J. Qin, Z. Bao and Y. Cui, Nano Lett., 2022, 22, 8224–8232 Search PubMed.
- T. Li and P. B. Balbuena, Chem. Phys. Lett., 2000, 317, 421–429 CrossRef CAS.
- Y. Wang, S. Nakamura, M. Ue and P. B. Balbuena, J. Am. Chem. Soc., 2001, 123, 11708–11718 CrossRef CAS PubMed.
- J. Halldin Stenlid, P. Žguns, D. Vivona, A. Aggarwal, K. Gordiz, Y. Zhang, S. Pathak, M. Z. Bazant, Y. Shao-Horn, A. Baskin and J. W. Lawson, ACS Energy Lett., 2024, 9, 3608–3617 CrossRef CAS.
- O. Borodin and G. D. Smith, J. Phys. Chem. B, 2009, 113, 1763–1776 CrossRef CAS PubMed.
- K. Tasaki, J. Phys. Chem. B, 2005, 109, 2920–2933 CrossRef CAS PubMed.
- J. W. Abbott and F. Hanke, J. Chem. Theory Comput., 2022, 18, 925–934 CrossRef CAS PubMed.
- Y. Liu, P. Yu, Y. Wu, H. Yang, M. Xie, L. Huai, W. A. Goddard, 3rd and T. Cheng, J. Phys. Chem. Lett., 2021, 12, 1300–1306 CrossRef CAS PubMed.
- F. Ospina-Acevedo, N. Guo and P. B. Balbuena, J. Mater. Chem. A, 2020, 8, 17036–17055 RSC.
- P. J. Weddle, E. W. C. Spotte-Smith, A. Verma, H. D. Patel, K. Fink, B. J. Tremolet de Villers, M. C. Schulze, S. M. Blau, K. A. Smith, K. A. Persson and A. M. Colclasure, Electrochim. Acta, 2023, 468, 143121 CrossRef CAS.
- D. Li, D. Danilov, Z. Zhang, H. Chen, Y. Yang and P. H. L. Notten, J. Electrochem. Soc., 2015, 162, A858–A869 CrossRef CAS.
- J. Christensen and J. Newman, J. Electrochem. Soc., 2004, 151, A1977–A1988 CrossRef CAS.
- B. Horstmann, F. Single and A. Latz, Curr. Opin. Electrochem., 2019, 13, 61–69 CrossRef CAS.
- F. Single, B. Horstmann and A. Latz, J. Electrochem. Soc., 2017, 164, E3132–E3145 CrossRef CAS.
- L. von Kolzenberg, A. Latz and B. Horstmann, ChemSusChem, 2020, 13, 3901–3910 CrossRef CAS PubMed.
- M. Uppaluri, K. Shah, V. Viswanathan and V. R. Subramanian, J. Electrochem. Soc., 2022, 169, 040548 CrossRef CAS.
- M. Safari, M. Morcrette, A. Teyssot and C. Delacourt, J. Electrochem. Soc., 2009, 156, A145–A153 CrossRef CAS.
- R. Li, S. O’Kane, M. Marinescu and G. J. Offer, J. Electrochem. Soc., 2022, 169, 060516 CrossRef CAS.
- M. Gerasimov, F. A. Soto, J. Wagner, F. Baakes, N. Guo, F. Ospina-Acevedo, F. Röder, P. B. Balbuena and U. Krewer, J. Phys. Chem. C, 2023, 127, 4872–4886 CrossRef.
- J. Wagner-Henke, D. Kuai, M. Gerasimov, F. Roder, P. B. Balbuena and U. Krewer, Nat. Commun., 2023, 14, 6823 Search PubMed.
- P. Guan, L. Liu and X. Lin, J. Electrochem. Soc., 2015, 162, A1798–A1808 CrossRef CAS.
- S.-H. Lee, H.-G. You, K.-S. Han, J. Kim, I.-H. Jung and J.-H. Song, J. Power Sources, 2014, 247, 307–313 Search PubMed.
- E. W. C. Spotte-Smith, R. L. Kam, D. Barter, X. Xie, T. Hou, S. Dwaraknath, S. M. Blau and K. A. Persson, ACS Energy Lett., 2022, 7, 1446–1453 CrossRef CAS.
- L. Chen, H. W. Zhang, L. Y. Liang, Z. Liu, Y. Qi, P. Lu, J. Chen and L.-Q. Chen, J. Power Sources, 2015, 300, 376–385 CrossRef CAS.
- L. Liang and L.-Q. Chen, Appl. Phys. Lett., 2014,(105), 263903 CrossRef.
- L. Liang, Y. Qi, F. Xue, S. Bhattacharya, S. J. Harris and L.-Q. Chen, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 86, 051609 CrossRef PubMed.
- Z. Liu, Y. Li, Y. Ji, Q. Zhang, X. Xiao, Y. Yao, L.-Q. Chen and Y. Qi, Cell Rep. Phys. Sci., 2021, 2, 100294 CrossRef CAS.
- H. K. Tian, Z. Liu, Y. Ji, L.-Q. Chen and Y. Qi, Chem. Mater., 2019, 31, 7351–7359 CrossRef CAS.
- Q. Wang, G. Zhang, Y. Li, Z. Hong, D. Wang and S. Shi, npj Comput. Mater., 2020, 6, 176 CrossRef CAS.
- D. Cao, K. Zhang, W. Li, Y. Zhang, T. Ji, X. Zhao, E. Cakmak, J. Zhu, Y. Cao and H. Zhu, Adv. Funct. Mater., 2023, 33, 2307998 CrossRef CAS.
- W. Mu, X. Liu, Z. Wen and L. Liu, J. Energy Storage, 2019, 26, 100921 CrossRef.
- V. Yurkiv, T. Foroozan, A. Ramasubramanian, R. Shahbazian-Yassar and F. Mashayek, Electrochim. Acta, 2018, 265, 609–619 CrossRef CAS.
- J. Deng, G. J. Wagner and R. P. Muller, J. Electrochem. Soc., 2013, 160, A487–A496 CrossRef CAS.
- P. Guan, X. Lin and L. Liu, ECS Trans., 2014, 61, 29–41 CrossRef CAS.
- Z. Gao, Y. Bai, H. Fu, J. Yang, T. Ferber, J. Feng, W. Jaegermann and Y. Huang, Adv. Funct. Mater., 2022, 32, 2112113 CrossRef CAS.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- T. P. Silverstein, J. Chem. Educ., 2012, 89, 1159–1167 CrossRef CAS.
- A. L. Michan, M. Leskes and C. P. Grey, Chem. Mater., 2016, 28, 385–398 CrossRef CAS.
- D. Bedrov, O. Borodin and J. B. Hooper, J. Phys. Chem. C, 2017, 121, 16098–16109 CrossRef CAS.
- Q. Wu, M. T. McDowell and Y. Qi, J. Am. Chem. Soc., 2023, 145, 2473–2484 CrossRef CAS PubMed.
- Y. X. Lin, Z. Liu, K. Leung, L.-Q. Chen, P. Lu and Y. Qi, J. Power Sources, 2016, 309, 221–230 CrossRef CAS.
- F. A. Soto, Y. Ma, J. M. Martinez de la Hoz, J. M. Seminario and P. B. Balbuena, Chem. Mater., 2015, 27, 7990–8000 CrossRef CAS.
- L. Dong, H. J. Yan, Q. X. Liu, J. Y. Liang, J. Yue, M. Niu, X. Chen, E. Wang, S. Xin, X. Zhang, C. Yang and Y. G. Guo, Angew. Chem., Int. Ed., 2024, 63, e202411029 CAS.
- F. Joho, B. Rykart, A. Blome, P. Novák, H. Wilhelm and M. E. Spahr, J. Power Sources, 2001, 97–98, 78–82 CrossRef CAS.
- M. Yu, K. Liu, M. Li, J. Yan, C. Cao, J. Tan, J. Liang, K. Guo, W. Cao, P. Lan, Q. Zhang, Y. Zhou and P. Lu, Light: Sci. Appl., 2022, 11, 215 CrossRef CAS PubMed.
- S. Menkin, C. A. O'Keefe, A. B. Gunnarsdottir, S. Dey, F. M. Pesci, Z. Shen, A. Aguadero and C. P. Grey, J. Phys. Chem. C, 2021, 125, 16719–16732 CrossRef CAS PubMed.
- X. He, D. Bresser, S. Passerini, F. Baakes, U. Krewer, J. Lopez, C. T. Mallia, Y. Shao-Horn, I. Cekic-Laskovic, S. Wiemers-Meyer, F. A. Soto, V. Ponce, J. M. Seminario, P. B. Balbuena, H. Jia, W. Xu, Y. Xu, C. Wang, B. Horstmann, R. Amine, C.-C. Su, J. Shi, K. Amine, M. Winter, A. Latz and R. Kostecki, Nat. Rev. Mater., 2021, 6, 1036–1052 CrossRef CAS.
- D. Aurbach, J. Power Sources, 2000, 89, 206–218 CrossRef CAS.
- D. M. Seo, D. Chalasani, B. S. Parimalam, R. Kadam, M. Nie and B. L. Lucht, ECS Electrochem. Lett., 2014, 3, A91–A93 CrossRef CAS.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- G. M. Hobold, A. Khurram and B. M. Gallant, Chem. Mater., 2020, 32, 2341–2352 CrossRef CAS.
- B. von Holtum, C. Peschel, U. Rodehorst, D. Wang, Y. Shao-Horn, M. Winter, S. Nowak and S. Wiemers-Meyer, J. Electrochem. Soc., 2025, 172, 050511 CrossRef CAS.
- Z. Liu, P. Lu, Q. Zhang, X. Xiao, Y. Qi and L.-Q. Chen, J. Phys. Chem. Lett., 2018, 9, 5508–5514 CrossRef CAS PubMed.
- J. M. Garcia-Lastra, J. S. G. Myrdal, R. Christensen, K. S. Thygesen and T. Vegge, J. Phys. Chem. C, 2013, 117, 5568–5577 CrossRef CAS.
- S. Shi, Y. Qi, H. Li and L. G. Hector, J. Phys. Chem. C, 2013, 117, 8579–8593 CrossRef CAS.
- M. Feng, J. Pan and Y. Qi, J. Phys. Chem. C, 2021, 125, 15821–15829 CrossRef CAS.
- M. Smeu and K. Leung, Phys. Chem. Chem. Phys., 2021, 23, 3214–3218 RSC.
- S. Shi, P. Lu, Z. Liu, Y. Qi, L. G. Hector, H. Li and S. J. Harris, J. Am. Chem. Soc., 2012, 134, 15476–15487 CrossRef CAS PubMed.
- J. Heine, P. Hilbig, X. Qi, P. Niehoff, M. Winter and P. Bieker, J. Electrochem. Soc., 2015, 162, A1094–A1101 CrossRef CAS.
- S. Singsen, F. Ospina-Acevedo, S. Suthirakun, P. Hirunsit and P. B. Balbuena, Phys. Chem. Chem. Phys., 2023, 25, 26316–26326 RSC.
- Y. Ji and L. Q. Chen, Acta Mater., 2022, 234, 118007 CrossRef CAS.
- K. Tasaki, Z. A. Goldberg, J.-J. Lian, M. Walker, A. Timmons and S. J. Harrisc, J. Electrochem. Soc., 2009, 156, A1019–A1027 CrossRef CAS.
- K. Tasaki and S. J. Harris, J. Phys. Chem. C, 2010, 114, 8076–8083 CrossRef CAS.
- H. B. Akkerman, R. C. Naber, B. Jongbloed, P. A. van Hal, P. W. Blom, D. M. de Leeuw and B. de Boer, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11161–11166 CrossRef CAS PubMed.
- Z. Wang, D. L. Danilov, R. A. Eichel and P. H. L. Notten, J. Power Sources Adv., 2024, 29, 100157 CrossRef CAS.
- B. Tjaden, D. J. L. Brett and P. R. Shearing, Int. Mater. Rev., 2018, 63, 47–67 CrossRef CAS.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood, Carbon, 2016, 105, 52–76 CrossRef CAS.
- M. Esmaeilpour, S. Jana, H. Li, M. Soleymanibrojeni and W. Wenzel, Adv. Energy Mater., 2023, 13, 2203966 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ort and D. J. Fox, Gaussian 09, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. Debnath, V. A. Neufeld, L. D. Jacobson, B. Rudshteyn, J. L. Weber, T. C. Berkelbach and R. A. Friesner, J. Phys. Chem. A, 2023, 127, 9178–9184 CrossRef CAS PubMed.
- H. Neugebauer, F. Bohle, M. Bursch, A. Hansen and S. Grimme, J. Phys. Chem. A, 2020, 124, 7166–7176 CrossRef CAS PubMed.
- D. Coskun, S. V. Jerome and R. A. Friesner, J. Chem. Theory Comput., 2016, 12, 1121–1128 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- O. Borodin, W. Behl and T. R. Jow, J. Phys. Chem. C, 2013, 117, 8661–8682 CrossRef CAS.
- D. S. Hall, J. Self and J. R. Dahn, J. Phys. Chem. C, 2015, 119, 22322–22330 CrossRef CAS.
- R. A. Marcus, J. Chem. Phys., 1965, 43, 679–701 CrossRef CAS.
- S. F. Nelsen, S. C. Blackstock and Y. Kim, J. Am. Chem. Soc., 1987, 109, 677–682 CrossRef CAS.
- M. Nie, D. Chalasani, D. P. Abraham, Y. Chen, A. Bose and B. L. Lucht, J. Phys. Chem. C, 2013, 117, 1257–1267 CrossRef CAS.
- R. L. C. Akkermans, N. A. Spenley and S. H. Robertson, Mol. Simul., 2021, 47, 540–551 Search PubMed.
- X. He, Y. Zhu, A. Epstein and Y. Mo, npj Comput. Mater., 2018, 4, 18 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee01030f |
| ‡ Kena Zhang and Yanzhou Ji contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |