
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Singlet oxygen is not the source of ethylene carbonate degradation in nickel-rich Li-ion cells†
Rory C.
McNulty‡
 abcd,
Kieran D.
Jones‡
abcd,
Benjamin M. G.
Denison
abcd,
Elizabeth
Hampson
abc,
Israel
Temprano
ef,
Darren A.
Walsh
abcd,
Hon Wai
Lam
bc,
Graham N.
Newton
abcd,
Wesley M.
Dose
dg,
Clare P.
Grey
de and
Lee R.
Johnson
*abcd
abcd,
Kieran D.
Jones‡
abcd,
Benjamin M. G.
Denison
abcd,
Elizabeth
Hampson
abc,
Israel
Temprano
ef,
Darren A.
Walsh
abcd,
Hon Wai
Lam
bc,
Graham N.
Newton
abcd,
Wesley M.
Dose
dg,
Clare P.
Grey
de and
Lee R.
Johnson
*abcd
aNottingham Applied Materials and Interfaces Group, School of Chemistry, University of Nottingham, NG7 2TU, UK. E-mail: lee.johnson@nottingham.ac.uk
bThe GSK Carbon Neutral Laboratories for Sustainable Chemistry, University of Nottingham, Jubilee Campus, Triumph Road, Nottingham, NG7 2TU, UK
cSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
dThe Faraday Institution, Quad One, Harwell Science and Innovation Campus, Didcot, OX11 0RA, UK
eYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
fCICA – Interdisciplinary Center for Chemistry and Biology, University of A Coruña, 15071, A Coruña, Spain
gSchool of Chemistry, University of Sydney, Camperdown, NSW 2006, Australia
First published on 25th June 2025
Abstract
Nickel-rich intercalation electrodes (i.e. Ni0.8Mn0.1Co0.1O2) are seeing widespread adoption in high-performance lithium-ion batteries due to their high energy density and reduced need for cobalt. However, as nickel content increases, so too does the rate of cell capacity fade, which in part has been assigned to reactions between ethylene carbonate (EC) and reactive oxygenic species formed at the surface of nickel-rich electrodes. In particular, singlet oxygen (1O2) has long been suspected as a primary source of ethylene carbonate degradation and has been proposed to drive its conversion to either vinylene carbonate, a graphite stabilising additive, or to complete oxidation products such as CO2, and protic species (i.e. H2O, H2O2) that accelerate cell failure. Contrary to this understanding, we show using online mass spectrometry and quantitative 1H NMR spectroscopic analysis that ethylene carbonate is stable in the presence of photocatalytically generated 1O2. Furthermore, this study indicates the use of rose bengal as a photocatalyst to study ethylene carbonate reactivity with 1O2 may lead to unexpected side-reactions under operationally-relevant conditions, producing misleading results. We conclude that the choice of photocatalyst is critical when assessing degradation with 1O2 for battery applications. Despite eliminating the direct reaction of 1O2 with EC as a source of degradation, ethylene carbonate to vinylene carbonate conversion is still found to occur in cells. We demonstrate that vinylene carbonate production begins before gas release from the positive electrode. These findings show that degradation driven by 1O2 reaction with EC is unlikely to be an important factor at nickel-rich intercalation electrodes highlighting the need for the community to explore alternative degradation pathways in nickel-rich lithium-ion batteries.
Broader contextTo meet the high volumetric energy densities and sustainable supply chains needed for long-range electric vehicles, industry is moving towards nickel-rich materials for the positive electrodes of Li-ion batteries. Increasing the nickel content can potentially increase the energy stored, but it also increases their degradation rate, decreasing the battery lifetime. Consequently, stabilising these materials is a critical challenge in the field. Degradation of the battery electrolyte at nickel-rich electrodes has been identified as a leading cause of cell failure. One of the more prominent theories is by reaction of singlet oxygen, released from the electrode during charging, with the electrolyte component ethylene carbonate forming side-products that trigger a cascade of failure mechanisms. Here we demonstrate for the first time that ethylene carbonate is in fact stable when in contact with singlet oxygen, even after prolonged exposure, hence does not contribute to the failure of the cell. We show that degradation occurs in the absence of singlet oxygen, suggesting that alternative reactions, such as at the surface of the nickel-rich electrode, are the origin of degradation. These findings will refocus academic and industrial efforts towards the study of interface-based degradation mechanisms and mitigating strategies, accelerating the development of high-energy nickel-rich electrodes. |
Introduction
As demand for electrification of global infrastructure increases, the availability and versatility of our battery systems need to advance.1–3 The positive electrode limits the energy density of conventional lithium-ion (Li-ion) cells; the ubiquitous LiCoO2 (LCO) has an effective capacity of ∼150 mAh g−1, which is significantly smaller than that of graphite (Gr) at 372 mAh g−1.4,5 While the practical capacity of Li-ion batteries can be improved by increasing upper cut-off voltage to facilitate the extraction of more Li+ ions, a balance between increasing the capacity and maintaining structural stability must be struck to avoid rapid and irreversible capacity fade. Nickel-rich layered oxide materials, such as LiNi0.6Mn0.2Co0.2O2 (NMC622) and LiNi0.8Mn0.1Co0.1O2 (NMC811), nominally offer higher capacities than LiCoO2.6–8 However, these nickel-rich materials suffer from lattice instability, and can yield reactive oxygenic species that are proposed to react with the electrolyte. These complications, paired with interphase degradation at the negative electrode caused by transition-metal ion migration, account for active ion loss9–11 and, ultimately, premature cell failure.Recently, it has been speculated that many of the degradation modes in nickel-rich electrodes originate from decomposition of electrolyte components, generating antagonistic intermediates that are often protic species such as HF or H2O.12–16 State-of-the-art electrolytes used in nickel-rich cells currently comprise of LiPF6 in mixtures of ethylene carbonate (EC) with linear carbonates such as ethyl methyl carbonate (EMC). However, EC is considered to be particularly prone to degradation reactions in the presence of an oxygen-releasing electrode, promoting gas release and metal oxide surface layer reconstruction, negatively impacting cycling stability.13,17–21 The exact degradation routes of EC within the nickel-rich cells remain contentious. Online mass spectrometry has been shown by multiple reports to correlate lattice oxygen release of nickel-rich electrodes to EC degradation to CO2,13,17,18,22 while small organics, specifically vinylene carbonate (VC), have been detected to form within the nickel-rich cells prior to O2 release by Raman,23 FTIR,24 and NMR22 spectroscopies. Moreover, the trigger for EC degradation (Fig. 1a) in Gr-NMC811 full-cells has been suggested to be (electro)chemical oxidation at the metal oxide surface (<4.3 V),17,22–24 or by direct reaction with singlet oxygen (1O2) released at higher upper cut-off voltages (≥4.3 V).17,25 The latter suggestion involves the release of 1O2 as the nickel-rich layered oxide surface degrades to a rock-salt surface structure, which may be driven by reaction between the surface oxygen atoms and solvent,26–28 or may occur spontaneously, as seen, for example, in Gasteiger and co-worker's study at elevated temperatures when heating delithiated NMC811 under an inert atmosphere (Ar).26 One thing is clear, namely that at high voltages, CO2 is the dominant gas that is released, not O2. Since oxygen/CO2 evolution is correlated with a specific state of charge (amongst different NMCs), Jung et al. proposed that electrolyte oxidation is mainly a chemical reaction, rather than a direct electrochemical oxidation, as is commonly assumed.17 A similar mechanism had also been proposed in lithium-rich materials by Jiang et al. in earlier work.29 Despite these discussions, the role of 1O2 in electrolyte degradation remains unclear.
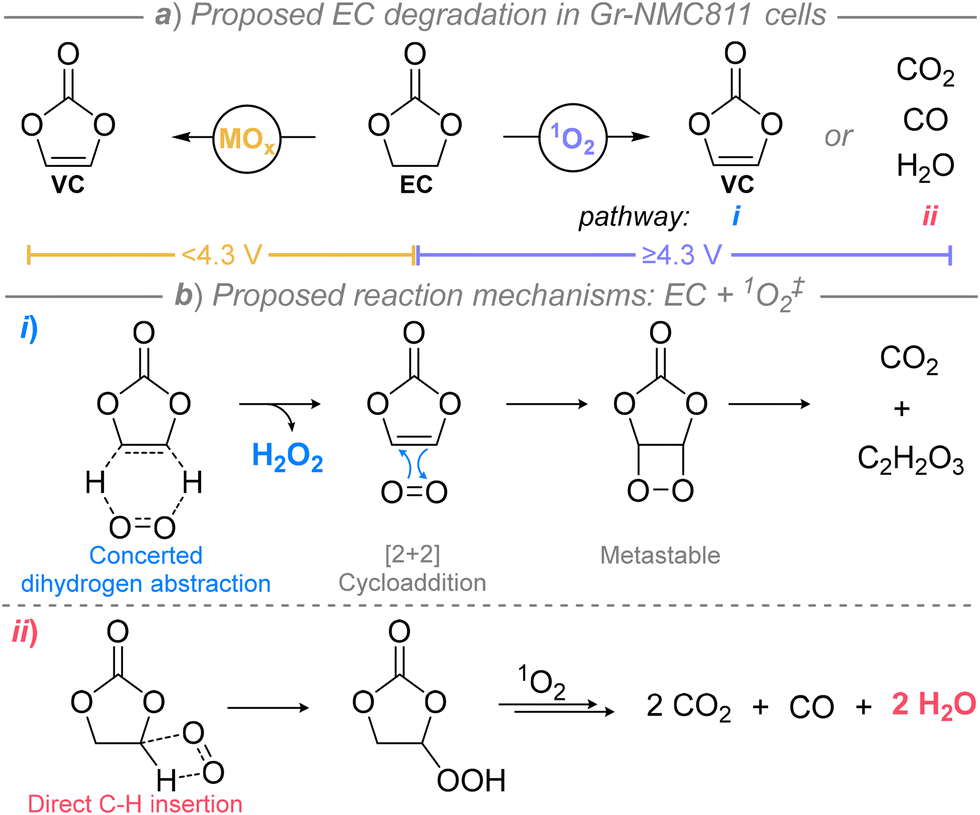 | ||
| Fig. 1 Overview of ethylene carbonate degradation pathways to vinylene carbonate25 or small molecular products22 in NMC811 cells (a) with proposed reaction mechanisms between 1O2 and EC summarised (b). ‡Studies were reported using 1O2 generated by the rose bengal photosensitiser. | ||
Reports to decouple electrolyte degradation by chemical oxidation at the electrode surface or by 1O2 reactivity with EC have been conducted by ex situ methods using rose bengal (RB, disodium salt form),22,25 a well-known photosensitiser for generating 1O2 from ground state 3O2.28 Gasteiger et al. proposed that the reaction between EC and 1O2 yields VC and H2O2 by a concerted dihydrogen abstraction mechanism (Fig. 1b, pathway i).25 While VC was not detected, CO2 release was observed by online mass spectrometry which was assigned to a subsequent reaction of 1O2 with VC. Alternatively, Rinkel et al. proposed 1O2 reacts with EC by direct C–H insertion (Fig. 1b, pathway ii),22 with the resulting hydroperoxide intermediate reacting further with 1O2 to form CO, CO2, and H2O, the latter supported by NMR spectroscopic analysis of reaction mixtures. In this case, H217O, rather than H217O2, was detected when 17O-enriched O2 gas was used, with no evidence of VC formation. Although, in the same report, VC was detected by analysis of extracted electrolyte from nickel-rich cells cycled at voltages below 1O2 release, leading the authors to suggest dehydrogenation of EC by the oxidised electrode surface may be a route to VC.22 Critically, these degradation pathways (i and ii) of EC by 1O2 are likely to be accompanied by the formation of antagonistic protic species (hydroperoxides, H2O2, H2O, etc.) which can trigger loss of lithium inventory and cell failure in nickel-rich cells. Recently, a DFT study has suggested that reactions involving 1O2 and EC are kinetically limited,31 drawing into question the impact of 1O2 in electrolyte degradation, hence, understanding this degradation mode is critical to enhancing long-term cycle stability of cells.
Herein, we seek to clarify the role of 1O2 within nickel-rich cells and to understand its reactivity with ethylene carbonate. Gr-NMC811 full-cells containing 1 M LiPF6 in EC:EMC 3/7 v/v (LP57) electrolyte have been cycled, the electrolyte analysed ex situ to determine at what state-of-charge vinylene carbonate forms and if this correlates with the release of 1O2. Our findings indicate that VC is formed within operational voltages (i.e. ≤4.2 V) with a notable increase in VC concentration being observed at ≥4.3 V, coinciding with lattice oxygen release, supporting previous 1O2-driven EC degradation mechanisms. However, our investigation revealed that RB may not be a suitable photocatalyst for the study of EC degradation reactions and appears to mediate side reactions thereby impacting the reliability of analytical data. By developing alternative methods to study 1O2 reaction routes with EC, we show that no degradation of EC occurs through this route and that direct reaction with 1O2 is not the source of EC degradation within Gr-NMC811 full-cells.
Results & discussion
We first sought to evaluate the link between cell potential, 1O2 release, and the formation of the organic by-product VC from electrolyte degradation within Gr-NMC811 full-cells.22–24 This was achieved by quantitatively assessing the amount of VC formed within VC-free LP57 electrolyte of cycled Gr-NMC811 full-cells using a gas chromatography-mass spectrometry (GC-MS) method (see ESI† for details). The concentrations of VC in the extracted electrolyte solutions were measured following three C/20 formation cycles to different upper cut-off voltages (UCV) between 3.9–4.5 V (Fig. 2a). The chromatograms showed no deviation from the baseline for electrolyte solution extracted from cells cycled to an UCV of 3.9 V, but small peaks corresponding to the retention time of VC were observed in the measurements of samples with UCVs of 4.0 V and greater. GC-MS analysis of electrolyte solutions extracted from cells after 10 cycles at C/2 (including three formation cycles) indicated formation of VC in cells cycled with an UCV as low as 3.9 V (Fig. 2b and c), however, we note this is below the limit of quantification.32 A clear relationship between UCV and VC concentration is observed in both cycling regimes, with a notable increase in VC concentration in electrolytes extracted from cells cycled to UCVs of ≥4.3 V (Fig. 2c). Online electrochemical mass spectrometry analysis of a Li-NMC811 half-cell indicated the onset of O2 release on charge to be at 4.22 V (Fig. 2d), which coincides with the sharp increase in electrolyte degradation, indicated by an increase in VC concentration, at UCVs greater than 4.2 V (Fig. 2c). Considering a fraction of this O2 has been shown to be in the 1O2 spin state,26 these findings are consistent with previously proposed hypothesis that EC degradation and VC formation may be a 1O2-driven process.25 However, in lower-nickel-content Gr-Ni0.33Co0.33Mn0.33O2 (Gr-NMC111) full-cells, where O2 release is not reported until ∼4.7 V,17 we also detect significant levels of VC formation at an UCV of 4.3 V (Fig. S4, ESI†), further indicating that EC-degradation can occur independently from O2 release as previously observed.22–24 | ||
| Fig. 2 Detection of vinylene carbonate formation within Gr-NMC811 full-cells showing increased concentration at higher UCVs which coincides with oxygen release from the nickel-rich electrode. Ex situ GC-MS analysis of VC from electrolyte solutions extracted from Gr-NMC811 full-cells after (a) 3 formation cycles and (b) with an additional 10 cycles at the UCVs indicated in the figure. (c) Plot of VC concentration within electrolyte extracted from cycled Gr-NMC811 cells (LOQ: limit of quantification). (d) Online electrochemical mass spectrometry (OEMS) of O2 evolution from a Li-NMC811 half-cell. The electrolyte solution was LP57 for OEMS measurements, cell potential corrected in relation to graphite full-cell (see ESI† for details). | ||
Typically, VC is added to nickel-rich cells as a sacrificial additive which participates in the formation of a robust and protective solid electrolyte interphase (SEI) on the negative electrode, mitigating reductive decomposition of other electrolyte components, and prolonging cell life.33,34 To explore the necessity of VC when using nickel-rich electrodes, Gr-NMC811 full-cells containing an LP57 electrolyte with (LP57-VC) and without 2 wt% VC additive were cycled at C/2 for 500 cycles. An UCV of 4.2 V was employed due to the reduced oxidative stability of the VC containing electrolyte (see ESI,† Note S1). A hysteresis voltage plot of the cell containing LP57-VC (Fig. 3a) shows an initial charge capacity and coulombic efficiency (CE) of 217.2 mAh gNMC−1 and 91.2%, respectively. These metrics are consistent with the reversible capacity expected for the first formation cycle of Gr-NMC811 cells containing a SEI optimising additive, where some loss of lithium inventory due to SEI formation is expected. Cycling of cells containing only LP57 (Fig. 3a) demonstrated a marginally higher initial charge capacity of 226.1 mAh gNMC−1 but an expectedly lower first cycle CE of 86.0%. The drop in CE can be attributed to an inefficient SEI formation stage. Conversely, long-term cycling data at C/2 (Fig. 3b) demonstrates that the cell without VC outperforms that with the additive over the first 400 cycles, with an average CE of 99.92%. By the 400th cycle, the capacity of the cells containing LP57-VC and VC-free electrolyte have reached 148.2 and 149.4 mAh gNMC−1, respectively, marking ca. 80% capacity retention. These data indicate that LP57 Gr-NMC811 full-cells can perform comparable to, or even exceed, the performance of Gr-NMC811 cells doped with 2 wt% VC, which is at least in part ascribed to the in situ VC formation, which coincides with prior reports supported by operando Raman spectroscopy.23 However, following 400 cycles, the greater VC concentration shows slight improvement, emphasising its importance in electrolyte formulation.
 | ||
| Fig. 3 Cycling data for Gr-NMC811 full-cells showing cells with and without vinylene carbonate achieve similar performance. (a) Hysteresis plots of Gr-NMC811 full-cells with and without 2 wt% VC. (b) Capacity performance and coulombic efficiency vs. cycle number for every fifth cycle over 500 cycles. Cells contained an LP57 electrolyte solution with or without 2% VC, and were cycled for three C/20 formation cycles (not shown) followed by C/2 cycling between 2.5–4.2 V. | ||
Having confirmed the formation of vinylene carbonate within the cell, we now consider the reaction between ethylene carbonate and 1O2. By use of an online mass spectrometry (OMS) method we were able to monitor the reaction between 1O2 and EC.35 Ground state oxygen (3O2) is flowed through a solution that contains a photocatalyst and the O2 flux is measured using mass spectrometry. Upon illumination, 1O2 is generated and any decrease in O2 signal can be related to reactions between 1O2 and the analyte. In the absence of a degradation reaction between 1O2 and EC, O2 flux should remain constant. In addition, release of CO2 may suggest reaction between 1O2 and EC, as it is a by-product common to the two proposed degradation pathways (Fig. 1)22,25 and a product of complete oxidation. To demonstrate this method, a positive control using 10 mM of the 1O2 trap 9,10-dimethylanthracene (DMA) in EC with 10 μM of the photosensitiser RB at ca. 40 °C with constant flow of 20% O2 in Ar gas was used (Fig. 4a). Upon irradiation with a green LED lamp (5 W), an on–off response was observed, seen as the decrease in O2 flux as photogenerated 1O2 reacts with DMA by a hetero-Diels–Alder reaction.36,37 The Diels–Alder reaction forms the peroxy-bridged product, 9,10-dimethyl-9,10-dihydro-9,10-epidioxyanthracene (DMA–O2), as supported by 1H NMR spectroscopy (Fig. S8, ESI†), albeit under these reaction conditions moderate selectivity was achieved (55%) as minor by-products were also observed. When 10 μM of RB was used to generate 1O2 in EC without DMA under similar reaction conditions (Fig. 4b and c), no decrease in O2 flux was detected indicating no reaction between 1O2 and EC; however, CO2 release was still recorded with peak flux at 0.0062 nmol s−1, which aligns with prior reports.25 In the absence of a photocatalyst or light there were no significant change in O2 or CO2 flux (Fig. S11, ESI†). The RB concentration was increased 50-fold (500 μM) which increased peak CO2 flux to 0.093 nmol s−1, and still no O2 consumption was observed (Fig. S10, ESI†). Bleaching of the RB photocatalyst was noted following 110 min of illumination in EC (Fig. S12, ESI†). This observation, paired with the release of CO2, and absence of O2 consumption, indicates that RB takes part in unexpected side reactions that generate CO2 but are not associated with EC degradation in the cell. We note that a weak NMR signal from 17O-enriched H2O was detected in prior studies with RB and 17O2,22 and thus minor reactions with 1O2 including isotopic scrambling reactions cannot be excluded. We conclude, therefore, that RB is not a suitable photocatalyst for these studies.
 | ||
| Fig. 4 OMS analysis demonstrating that ethylene carbonate itself is stable to 1O2 and that CO2 release is coupled to the photocatalyst instability. (a) Change in O2 (32 m/z) flux for solutions of 10 mM DMA in EC showing 1O2 is generated and reacting with DMA when illuminated; (b) change in O2 (32 m/z) flux for EC solutions showing no notable reactivity of 1O2; (c) change in CO2 (44 m/z) flux for EC solutions indicating CO2 release for reactions containing RB but not TPP. All EC solutions contained 10 μM of photocatalyst (RB: red line; TPP: purple line) and a constant flow of 20% O2 in Ar gas was used during OMS measurements. | ||
To decouple CO2 release from RB instability, control experiments were performed using of 5,10,15,20-tetraphenyl-21H,23H-porphine (TPP), as an alternative photosensitiser to RB.30,38 10 μM TPP was added to EC containing 10 mM DMA and O2 flux was monitored by OMS (Fig. 4a). Illumination by a red LED lamp (40 W) at ca. 40 °C under a constant flow of 20% O2 in Ar gas showed O2 consumption responded in an on–off manner to the irradiation, similar to that observed when using RB, but with greater 1O2 production (Fig. S13, ESI†). 1H NMR spectroscopic analysis indicated that conversion of DMA to DMA–O2 progressed with greater selectivity (77% selectivity, Fig. S9, ESI†) than that observed when using RB, with minimal bleaching of TPP, suggesting that TPP has greater stability under the reaction conditions. The experiment was then performed in the absence of DMA, with O2 and CO2 flux monitored using OMS (Fig. 4b and c). The data indicated no consumption of O2 or CO2 release upon generation of 1O2 suggesting that 1O2 does not readily react with EC and confirms that CO2 release is a RB-coupled phenomenon, thereby highlighting the importance of considered catalyst selection (see ESI† for details).
To account for a slow reaction between 1O2 and ethylene carbonate, which may impact long term cycling of a Li-ion cell, a solution of 0.1 M EC and 100 μM TPP in CDCl3 was irradiated for 22 hours under an atmosphere of O2. The use of deuterated chloroform allowed for an increase in photocatalyst concentration and an extended 1O2 half-life,39–41 thereby increasing the probability for EC to react with 1O2 (see ESI,†Note S2). Despite these allowances, analysis by quantitative 1H NMR spectroscopy showed no consumption of EC (Fig. 5) or formation of related products, VC or otherwise, under these reaction conditions. Ultimately, EC appears stable to 1O2 which importantly is in agreement with recent computational reports,31,42 and highlights the need for careful consideration of 1O2 reactivity within electrochemical cells.43 This suggests that VC formation occurs through alternative pathways in Gr-NMC811 full-cells, which should be the focus of further study.
 | ||
| Fig. 5 1H NMR spectra of ethylene carbonate after long-term exposure to 1O2 showing no detectable reaction between EC and 1O2. 0.1 M EC with 100 μM TPP in CDCl3 under an atmosphere of O2 was illuminated by a red LED lamp (40 W) for 22 hours. Intensities corrected to the internal standard (dimethyl sulfone). (inset) A plot showing no relevant percentage change in moles of EC over the course of this reaction. Aliquots were taken for analysis by quantitative 1H NMR spectroscopy. This reaction was conducted in duplicate and results from the two reactions averaged. | ||
Conclusion
Lattice oxygen release in the singlet spin state from NMC811 has long been suspected to be a major degradation mode for ethylene carbonate and a route to vinylene carbonate, both common components in commercial Li-ion electrolytes. Here we have shown that this reaction pathway is not operative. While we have demonstrated that EC degradation to VC in Gr-NMC811 full-cells does occur, this reaction is not due to singlet oxygen and future studies should focus on the reactivity of the nickel-rich surface or alternative oxygenic species. Interestingly, we have shown that this adventitious VC formation within NMC811 cells may preclude the need for the direct addition of VC, as cells containing VC-free LP57 perform comparably to cells with VC-containing LP57 up to 400 cycles. Finally, our studies show that particular care must be taken when studying 1O2 under battery conditions, as the introduction of battery components and solvents add additional complexity.Author contributions
RCM and KDJ contributed equally to the study. All authors contributed to the conception and design of the study. RCM, KDJ, BMGD and EH performed the experiments and processed the data. IT performed OEMS measurements. All authors contributed to manuscript writing. DAW, HWL, GNN, WMD, CPG and LRJ supervised the project.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
Special thanks to Dr Kevin S. Butler (University of Nottingham) for assistance with quantitative 1H NMR spectroscopy. L. J. thanks the Engineering and Physical Sciences Research Council (EPSRC) Fellowship scheme (EP/S001611/1), and the University of Nottingham's Propulsion Futures Beacon of Excellence. This work was supported by the Faraday Institution grant number FIRG017 and FIRG065. I. T. acknowledges support from a Beatriz Galindo senior fellowship (BG22/00148) from the Spanish Ministry of Science and Innovation. W. M. D. acknowledges support from an Australian Research Council Discovery Early Career Award (DE220100350) and a University of Sydney Horizon Fellowship.References
- S. G. Booth, A. J. Nedoma, N. N. Anthonisamy, P. J. Baker, R. Boston, H. Bronstein, S. J. Clarke, E. J. Cussen, V. Daramalla, M. De Volder, S. E. Dutton, V. Falkowski, N. A. Fleck, H. S. Geddes, N. Gollapally, A. L. Goodwin, J. M. Griffin, A. R. Haworth, M. A. Hayward, S. Hull, B. J. Inkson, B. J. Johnston, Z. Lu, J. L. MacManus-Driscoll, X. Martínez De Irujo Labalde, I. McClelland, K. McCombie, B. Murdock, D. Nayak, S. Park, G. E. Pérez, C. J. Pickard, L. F. J. Piper, H. Y. Playford, S. Price, D. O. Scanlon, J. C. Stallard, N. Tapia-Ruiz, A. R. West, L. Wheatcroft, M. Wilson, L. Zhang, X. Zhi, B. Zhu and S. A. Cussen, APL Mater., 2021, 9, 109201 CrossRef CAS.
- E. A. Olivetti, G. Ceder, G. G. Gaustad and X. Fu, Joule, 2017, 1, 229–243 CrossRef.
- B. E. Murdock, K. E. Toghill and N. Tapia-Ruiz, Adv. Energy Mater., 2021, 11, 2102028 CrossRef CAS.
- Q. Liu, X. Su, D. Lei, Y. Qin, J. Wen, F. Guo, Y. A. Wu, Y. Rong, R. Kou, X. Xiao, F. Aguesse, J. Bareño, Y. Ren, W. Lu and Y. Li, Nat. Energy, 2018, 3, 936–943 CrossRef CAS.
- W. Zhao, C. Zhao, H. Wu, L. Li and C. Zhang, J. Energy Storage, 2024, 81, 110409 CrossRef.
- G.-L. Xu, X. Liu, A. Daali, R. Amine, Z. Chen and K. Amine, Adv. Funct. Mater., 2020, 30, 2004748 CrossRef CAS.
- W. Li, E. M. Erickson and A. Manthiram, Nat. Energy, 2020, 5, 26–34 CrossRef CAS.
- M. D. Radin, S. Hy, M. Sina, C. Fang, H. Liu, J. Vinckeviciute, M. Zhang, M. S. Whittingham, Y. S. Meng and A. Van der Ven, Adv. Energy Mater., 2017, 7, 1602888 CrossRef.
- W. M. Dose, J. K. Morzy, A. Mahadevegowda, C. Ducati, C. P. Grey and M. F. L. D. Volder, J. Mater. Chem. A, 2021, 9, 23582–23596 RSC.
- Z. Ruff, C. Xu and C. P. Grey, J. Electrochem. Soc., 2021, 168, 060518 CrossRef CAS.
- W. M. Dose, C. Xu, C. P. Grey and M. F. L. De Volder, Cell Rep. Phys. Sci., 2020, 1, 100253 CrossRef.
- Z. Cui and A. Manthiram, Angew. Chem., Int. Ed., 2023, 62, e202307243 CrossRef CAS PubMed.
- W. M. Dose, I. Temprano, J. P. Allen, E. Björklund, C. A. O’Keefe, W. Li, B. L. Mehdi, R. S. Weatherup, M. F. L. De Volder and C. P. Grey, ACS Appl. Mater. Interfaces, 2022, 14, 13206–13222 CrossRef CAS PubMed.
- S. Wiemers-Meyer, M. Winter and S. Nowak, Phys. Chem. Chem. Phys., 2016, 18, 26595–26601 RSC.
- B. Salomez, S. Grugeon, M. Armand, P. Tran-Van and S. Laruelle, J. Electrochem. Soc., 2023, 170, 050537 CrossRef.
- Y. Liu, Z. Li, Y. Gao, C. Wang, X. Wang, X. Wang, X. Xue, K. Wang, W. Cui, F. Gao, S. He, Z. Wu, F. Qi, J. Gan, Y. Wang, W. Zheng, Y. Yang, J. Chen and H. Pan, Small, 2024, 20, 2311500 CrossRef CAS PubMed.
- R. Jung, M. Metzger, F. Maglia, C. Stinner and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, A1361 CrossRef CAS.
- W. M. Dose, W. Li, I. Temprano, C. A. O’Keefe, B. L. Mehdi, M. F. L. De Volder and C. P. Grey, ACS Energy Lett., 2022, 7, 3524–3530 CrossRef CAS PubMed.
- C. Xu, Y. Liang, R. Zhang, J. Cheng and H. Yu, Energy Fuels, 2024, 38, 18208–18226 CrossRef CAS.
- C.-Y. Li, Y. Yu, C. Wang, Y. Zhang, S.-Y. Zheng, J.-F. Li, F. Maglia, R. Jung, Z.-Q. Tian and Y. Shao-Horn, J. Phys. Chem. C, 2020, 124, 4024–4031 CrossRef CAS.
- L. Giordano, P. Karayaylali, Y. Yu, Y. Katayama, F. Maglia, S. Lux and Y. Shao-Horn, J. Phys. Chem. Lett., 2017, 8, 3881–3887 CrossRef CAS PubMed.
- B. L. D. Rinkel, J. P. Vivek, N. Garcia-Araez and C. P. Grey, Energy Environ. Sci., 2022, 15, 3416–3438 RSC.
- E. Miele, W. M. Dose, I. Manyakin, M. H. Frosz, Z. Ruff, M. F. L. De Volder, C. P. Grey, J. J. Baumberg and T. G. Euser, Nat. Commun., 2022, 13, 1651 CrossRef CAS PubMed.
- Y. Zhang, Y. Katayama, R. Tatara, L. Giordano, Y. Yu, D. Fraggedakis, J. G. Sun, F. Maglia, R. Jung, M. Z. Bazant and Y. Shao-Horn, Energy Environ. Sci., 2020, 13, 183–199 RSC.
- A. T. S. Freiberg, M. K. Roos, J. Wandt, R. de Vivie-Riedle and H. A. Gasteiger, J. Phys. Chem. A, 2018, 122, 8828–8839 CrossRef CAS PubMed.
- J. Wandt, A. T. S. Freiberg, A. Ogrodnik and H. A. Gasteiger, Mater. Today, 2018, 21, 825–833 CrossRef CAS.
- G. J. Páez Fajardo, E. Fiamegkou, J. A. Gott, H. Wang, I. Temprano, I. D. Seymour, M. J. W. Ogley, A. S. Menon, I. E. L. Stephens, M. Ans, T.-L. Lee, P. K. Thakur, W. M. Dose, M. F. L. De Volder, C. P. Grey and L. F. J. Piper, ACS Energy Lett., 2023, 8, 5025–5031 CrossRef.
- A. R. Genreith-Schriever, H. Banerjee, A. S. Menon, E. N. Bassey, L. F. J. Piper, C. P. Grey and A. J. Morris, Joule, 2023, 7, 1623–1640 CrossRef CAS.
- M. Jiang, B. Key, Y. S. Meng and C. P. Grey, Chem. Mater., 2009, 21, 2733–2745 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- E. W. C. Spotte-Smith, S. Vijay, T. B. Petrocelli, B. L. D. Rinkel, B. D. McCloskey and K. A. Persson, J. Phys. Chem. Lett., 2024, 15, 391–400 CrossRef CAS PubMed.
- D. A. Armbruster, M. D. Tillman and L. M. Hubbs, Clin. Chem., 1994, 40, 1233–1238 CrossRef CAS.
- A. L. Michan, B. S. Parimalam, M. Leskes, R. N. Kerber, T. Yoon, C. P. Grey and B. L. Lucht, Chem. Mater., 2016, 28, 8149–8159 CrossRef CAS.
- H. Ota, Y. Sakata, A. Inoue and S. Yamaguchi, J. Electrochem. Soc., 2004, 151, A1659 CrossRef CAS.
- C. Zor, K. D. Jones, G. J. Rees, S. Yang, A. Pateman, X. Gao, L. R. Johnson and P. G. Bruce, Energy Environ. Sci., 2024, 17, 7355–7361 RSC.
- D. Zehm, W. Fudickar and T. Linker, Angew. Chem., Int. Ed., 2007, 46, 7689–7692 CrossRef CAS PubMed.
- Y. Yang, T. K. Ronson, D. Hou, J. Zheng, I. Jahović, K. H. Luo and J. R. Nitschke, J. Am. Chem. Soc., 2023, 145, 19164–19170 CrossRef CAS PubMed.
- R. Dědic, V. Vyklický, A. Svoboda and J. Hála, J. Lumin., 2011, 131, 442–444 CrossRef.
- M. Bregnhøj, M. Westberg, F. Jensen and P. R. Ogilby, Phys. Chem. Chem. Phys., 2016, 18, 22946–22961 RSC.
- C. A. Long and D. R. Kearns, J. Am. Chem. Soc., 1975, 97, 2018–2020 CrossRef CAS.
- Ł. Ożog and D. Aebisher, Eur. J. Clin. Exp. Med., 2018, 16, 123–126 CrossRef.
- K. Leung and M. Zhang, J. Phys. Chem. Lett., 2024, 15, 4686–4693 CrossRef CAS PubMed.
- A. Schürmann, B. Luerßen, D. Mollenhauer, J. Janek and D. Schröder, Chem. Rev., 2021, 121, 12445–12464 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00956a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |