
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identifying the role of Zn self-dissolution in the anode corrosion process in Zn-ion batteries†
Yi Yuan*ab,
Zixuan Lib,
Rongyu Dengac,
Shengda D. Pub,
Marc Walker a,
Mingzhi Caib,
Feixiang Wuc,
Peter G. Brucebd and
Alex W. Robertson*a
a,
Mingzhi Caib,
Feixiang Wuc,
Peter G. Brucebd and
Alex W. Robertson*a
aDepartment of Physics, University of Warwick, Coventry, CV4 7AL, UK. E-mail: yi.yuan.1@warwick.ac.uk; alex.w.robertson@warwick.ac.uk
bDepartment of Materials, University of Oxford, Parks Road, Oxford, OX1 3PH, UK
cNational Engineering Research Center of Advanced Energy Storage Materials, School of Metallurgy and Environment, Central South University, Changsha, 410083, China
dDepartment of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QZ, UK
First published on 24th April 2025
Abstract
Zinc-ion (Zn-ion) batteries for practical applications face several challenges, some of which arise from the inevitable degradation of the Zn metal anode. The intrinsic thermodynamic instability of Zn metal anodes in mildly acidic Zn-ion batteries can trigger spontaneous interfacial corrosion, which leads to hydrogen evolution, the formation of byproducts, and the irreversible loss of active species during both storage and operation. Here, we delve into the intricate corrosion processes of the Zn metal anode in mildly acidic electrolytes. With the help of operando electrochemical liquid cell transmission electron microscopy, the self-dissolution of Zn is observed, and the capacity loss due to such corrosion behaviour during the cell rest period is quantified. This dissolution of Zn is found to be closely related to the initial pH value of the electrolyte and can be mitigated by pH adjustment through the slight addition of a pH buffer additive. The self-dissolution of Zn, which causes an increase in the local pH, is a prelude to the formation of corrosion byproducts that continues throughout the entire storage and cycling period. These corrosion issues are exacerbated by the presence of excess Zn metal in the system, suggesting that the feasibility of using excess Zn metal in Zn-ion batteries should be carefully evaluated. These findings further emphasise the importance of considering electrolyte pH in future electrolyte modification research, as well as its potential impacts on the stability of both the anode and cathode, and the shelf life of the entire battery.
Broader contextThe recent wave of interest in rechargeable aqueous Zn-ion battery chemistries has led to a consolidated view as to what some of the major outstanding limitations are. One of the most persistent, and unique to aqueous Zn-ion batteries due to their mildly acidic chemistry, is the occurrence of corrosion and the ensuing formation of inactive byproducts. This locks away the active Zn ions, reducing capacity, and leads to hydrogen evolution, which is a danger for cell pressure and generates gas bubbles that occlude the electrode interfaces. This occurs even when the cell is not being cycled, with such calendar or rest aging posing a serious obstacle to any commercial application of aqueous Zn-ion batteries. Understanding the mechanistic underpinnings behind corrosion is therefore of fundamental importance. We use operando electron microscopy imaging to identify self-dissolution of metal zinc under aging conditions, which leads to the local pH changes that culminate in the formation of corrosion products. The pivotal role of these local pH changes is confirmed by the inclusion of a small quantity of pH-mediating buffer additive, that stops the dissolution and thus limits corrosion reactions. |
Introduction
Zinc-ion (Zn-ion) batteries are emerging as promising candidates for grid-scale energy storage due to their safety, low cost, and relatively high capacity.1,2 Metallic Zn has been widely considered to be the gold standard for anode materials due to offering the highest capacity (820 mA h g−1 or 5854 mA h cm−3), low cost, high abundancy, non-toxicity, as well as good stability in water.3,4 Despite these advantages, Zn metal anodes still face many challenges and remain far from satisfactory.5–7 For instance, dendrite formation due to uneven electrodeposition, and the hydrogen evolution reaction (HER) due to the decomposition of water, are inevitable issues during cycling that eventually result in poor reversibility and low coulombic efficiency. Various strategies have been proposed to alleviate these metal anode issues in mildly acidic/neutral aqueous Zn-ion batteries,8–10 including controlling the composition and crystal structure of the Zn metal anode, which can curb the growth of Zn dendrites;11–14 or isolating the Zn metal anode from the electrolyte by applying artificial surface coatings, which can suppress side reactions as well as promote homogeneous deposition.15–17 Beyond modifying the anode, many methods have instead sought to enhance the properties of the electrolyte, including optimising the types of salts and solvents,18,19 adjusting salt concentrations,20 incorporating additives,21–23 and developing innovative electrolyte formulations such as ionic liquids,24 deep eutectic electrolytes,25 gel electrolytes,26 solid–liquid hybrid electrolytes,27 and all-solid-state electrolytes.28While considerable efforts have been devoted to understanding and addressing Zn dendrite growth and hydrogen evolution, the issue of corrosion has received less attention.29 The water stability of Zn metal is only relative rather than absolute.30 In fact, the Zn metal anode does suffer from various degradation processes over prolonged storage and cycling in aqueous solutions. The corrosion of the Zn metal anode in Zn-ion batteries is largely due to the protic nature of aqueous electrolytes.31 When Zn salts are dissolved in water, the solution becomes acidic due to the spontaneous hydrolysis of Zn ions. The generated protons continue to react with Zn metal and lead to the hydrogen evolution, which induces a local pH increase, consequently generating the precipitation of basic insoluble corrosion byproducts.30–32 Taking the most commonly used zinc sulphate (ZnSO4) aqueous electrolyte as an example, the main corrosion byproduct is zinc hydroxide sulphate Zn4(OH)6SO4·xH2O (ZHS), which typically exhibits a porous and loose structure. This corrosion-induced species, unlike the dense ZnO/Zn(OH)2 layer formed in alkaline Zn battery systems, lacks the ability to shield the electrode surface from the electrolyte, leading to continuing consumption of active Zn, water and Zn salts.6 As a result, the corrosion reactions are not naturally self-limiting and will continue to proceed throughout the lifetime of the cell. This leads to the continuous consumption of the metallic Zn anode and electrolytes during both storage and operation, which is unsustainable for a practical cell.33 To compensate for this loss of active species and ensure a good cycling performance of full cells, a common practice is to increase the amount of both the electrolyte and Zn metal anode. However, an immediate downside of this approach is the low utilisation rate of Zn, which inevitably reduces the gravimetric energy density of Zn-ion batteries.34–36
It is worth noting that the corrosion of the Zn metal anode not only consumes active species but also contributes to the broader degradation of the entire battery system. Parasitic reactions between the Zn metal and the electrolyte will introduce uneven interfaces, triggering heterogeneous Zn nucleation and formation of dendrites.7,37 Such surface irregularities further increase the active surface area of the electrode accessible to the electrolyte, aggravating corrosion of the electrode and other side reactions.38 Prolonged rest in sealed Zn-ion batteries can lead to volume expansion, which poses safety risks.11,30 Additionally, hydrogen gas produced from corrosion can deactivate the zinc anode; the bubbles isolate Zn by physically displacing the electrolyte away, causing capacity loss.39 Furthermore, studies have shown that the limited lifespan and degradation of the cathode materials partially stem from pH fluctuations caused by Zn anode corrosion.40 These challenges arising from Zn metal anode corrosion significantly reduce not only the cycling life but also the shelf life after battery assembly even before use, thereby considerably constraining the practical application of Zn-ion batteries.5,41 The complexity of these interconnected degradation processes also complicates the quantification of coulombic efficiency losses due to the anode corrosion reactions. As a result of this complexity, there remains a gap in understanding as to the relationship between the different aspects of corrosion losses, how they evolve with time, and how these may be altered to limit corrosion. Diagnosing complex relationships typically benefit from in situ observation, allowing the decoupling of various processes and uncovering any causal links that develop with time.39
Here, we delve into the complex Zn anode corrosion processes that occur during battery rest (i.e., when not being cycled). We show that the corrosion of Zn can be considered a two-stage process, as revealed by direct in situ liquid cell transmission electron microscopy (TEM) imaging of the anode. This technique has been used in previous studies to investigate Zn dendrite growth and to analyse micro/nanostructures that cannot be observed with optical microscopy.42–44 Using in situ liquid cell TEM imaging, we observed the self-dissolution of Zn into the electrolyte during rest, which we identify as being the start of the zinc corrosion process that eventually leads to the generation of ZHS products. Performing these experiments with in situ TEM imaging permits us to verify that the source of the measured coulombic efficiency loss during cell rest must originate from Zn dissolution, instead of dendrite detachment or irreversible byproduct formation. We show that this self-dissolution step precedes ZHS formation during corrosion; the formation of H2 that accompanies aqueous Zn dissolution yields an excess of OH− ions at Zn dissolution sites, thus causing an increase in the local pH at the electrode. This can cause the interfacial pH to rise sufficiently to allow ZHS to form. Using surplus Zn metal at the anode, which is a common practice, was found to promote the anode corrosion process, as the local pH fluctuations were exacerbated by increased dissolution and byproduct formation. This model is supported by correlative studies with an electrolyte additive, zinc acetate [Zn(OAc)2], which regulates the pH to ensure that the initial Zn dissolution is limited, and also acts to buffer against any local pH changes. Coin cell studies corroborate that the coulombic efficiency loss following extended periods of rest were significantly curtailed by inclusion of Zn(OAc)2. This new picture for Zn corrosion in resting cells illustrates the critical role that excess zinc at the anode can play in facilitating corrosion byproducts, as the self-dissolution of this Zn facilitates a local pH environment necessary for ZHS formation.
Results and discussion
The poor reversibility and the stability of the Zn metal anode are the main issues that contribute to its degradation during battery operation and rest, and are correlated with each other.38 The loose structure of Zn dendrites forming on the surface increases the active area of the electrode, which exacerbates the corrosion process, while the corrosion byproduct layer and morphological changes generated by parasitic side reactions lead to an uneven distribution of the electric field and heterogeneous nucleation, triggering the formation of further dendrites. However, the complexity in their interactions makes it challenging to decouple their contributions to the performance degradation of the Zn metal anode. Previous studies have attempted to decouple and quantify the capacity loss of the Zn metal anode after aging, attributing it to various causes through electrochemical tests and acidic titration.33,39 However, the complexity of sample preparation and the challenges in disentangling interactions among different factors can lead to potential misinterpretations in the analysis. Therefore, operando liquid cell TEM is used here to reveal the mechanism for the capacity loss, which ensures detailed observation of dynamic processes within a confined microenvironment.The liquid cell for the operando experiments is described in the ESI.† A diagram of the Pt working electrode before and after plating is illustrated in Fig. 1a. We performed two experiments. In both, Zn was deposited from 1 M ZnSO4 electrolyte onto the Pt working electrode at a current density of 20 mA cm−2 to a capacity of 0.4 mA h cm−2, then the deposited Zn was either stripped immediately (Fig. 1a) or after a resting period of 5 minutes (Fig. 1b). For the sample with a rest period, the viewing field was moved outside of the chip window to avoid electron beam damage during the five-minute rest period. The galvanostatic voltage profiles recorded while plating and stripping for the two cycles are shown in Fig. 1c and d, with points in time corresponding to the shown sequential TEM images highlighted.
 | ||
| Fig. 1 (a) Schematic illustration of the Pt electrode before and after plating, and in situ liquid cell TEM imaging of the plating and the immediate stripping of Zn in the ZnSO4 electrolyte (20 mA cm−2 to a capacity of 0.4 mA h cm−2). Scale bars are 1 μm. (b) In situ liquid cell TEM imaging after plating (20 mA cm−2 to a capacity of 0.4 mA h cm−2) and resting for 5 minutes, followed by stripping. Scale bars are 1 μm. Corresponding galvanostatic voltage profiles of in situ liquid cell TEM experiments for (c) plating and immediate stripping, and (d) plating and stripping after 5 minutes. | ||
The coulombic efficiency can be calculated based on the plating duration and the time taken for complete stripping of the plated Zn (tplat and tstrip) according to:

The coulombic efficiency can be influenced by various cycling parameters, such as the current density and depth of discharge.45,46 To compare the coulombic efficiency under other conditions, Zn was also deposited at different current densities with the same capacity (0.1 mA h cm−2). Sequential liquid cell TEM images of Zn plating and stripping at current densities of 10 mA cm−2 and 20 mA cm−2 are shown in Fig. S1 and S2 (ESI†), respectively. Zn deposits under these two current densities exhibited similar morphologies, with no obvious residues left at the end of stripping. A higher coulombic efficiency was achieved using a higher current density of 20 mA cm−2 (Fig. S3, ESI†), which is consistent with previous studies in coin cells;47 indeed, ultra-high current densities are frequently used in order to avoid longer cycling durations and thus minimise side reactions.45
However, when comparing the results of using 20 mA cm−2 for different deposition capacities, the coulombic efficiency increased from 83.1% to 96.1% by decreasing the capacity to a quarter (Fig. 1c and Fig. S3, ESI†). Considering the concomitantly longer cycling time required for higher capacities, this suggests that the side reactions occurring simultaneously during plating/stripping periods should also be taken into account as an important factor affecting the coulombic efficiency.
To further investigate the decrease in the coulombic efficiency and examine the influence of side reactions during rest, Zn was deposited at a smaller current density of 8 mA cm−2 for 72 seconds to a capacity of 0.16 mA h cm−2, and was left for varying times before stripping. The images of Zn deposits before and after rest are compared in Fig. 2a. There was no significant change in the shape and density of Zn deposits after resting for a short period of 3 minutes. However, it was found that the contrast of the edge of deposits started to fade after 10 minutes, and a partial disappearance of the deposition was clearly observed after 20 minutes (shown in the orange dashed frame). Moreover, the coulombic efficiency was also found to decrease from 77.2% to 49.6% after resting for 20 minutes (Fig. 2b). The TEM images of electrodes after stripping are shown in Fig. S4 (ESI†), where all electrodes were found to be clean with no residual deposits left. Therefore, it is likely that the corrosion of the Zn deposits accounts for the capacity loss during rest, rather than the formation of dendrites and dead Zn.
 | ||
| Fig. 2 (a) In situ liquid cell TEM images of the end of plating before and after rest for different periods of time. Scale bars are 1 μm. (b) The corresponding coulombic efficiency after rest for different periods of time. | ||
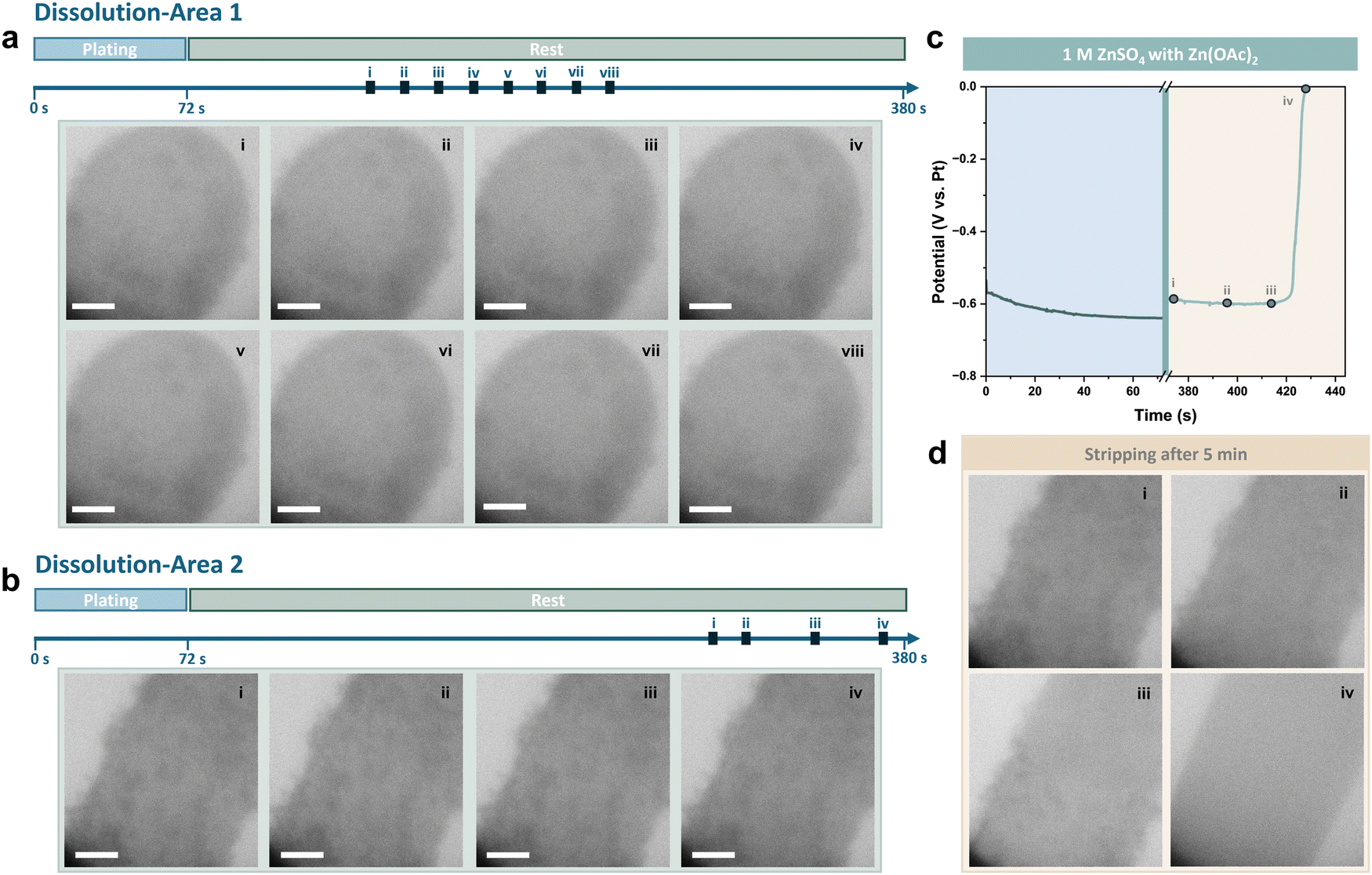
The current density and deposition capacity were further reduced to 5 mA cm−2 and 0.05 mA h cm−2 to highlight the decrease in the coulombic efficiency. Sequential images of plating and the corresponding voltage profile are shown in Fig. S5 (ESI†). To reveal the corrosion behaviour of Zn during rest, the changes to the Zn deposits on the Pt electrode were continuously recorded after deposition (see also Movie S1, ESI†). The viewing field was initially moved to the other side of the Pt electrode, which had a relatively smaller number of deposits. As shown in Fig. 3a, it was observed that the contrast of deposits began to fade within 20 seconds of the rest period. The deposits continued dissolving as the rest period extended, and distinct gaps were observed from stage iii. Zn deposits on this side of the Pt electrode almost completely disappeared after resting for approximately 200 seconds.
 | ||
| Fig. 3 In situ liquid cell TEM imaging of the dissolution of Zn deposits during cell rest in the ZnSO4 electrolyte for (a) area 1 and (b) area 2. Scale bars are 1 μm. (c) Corresponding galvanostatic voltage profile. (d) In situ liquid cell TEM imaging of the stripping process after rest for 5 minutes. Scale bars are 1 μm. | ||
The viewing field was then moved back to the initial side of the Pt electrode. As is shown in Fig. 3b, compared with straight after the end of plating, the majority of Zn deposits had been dissolved. Besides, the shape of the residues remained similar, indicating no new byproducts had formed on the electrode. The starting point of the dissolution appears to be random when comparing images recorded from the two dissolution areas on the Pt electrode.
Stripping was conducted after resting for 5 minutes, with the voltage profile and sequential TEM images shown in Fig. 3c and d. It was found that nearly all Zn deposits were dissolved and the coulombic efficiency, calculated based on the stripping time, was less than 10%. The Pt electrode surface after stripping remained clean, with the remaining thin deposit removed at the beginning of stripping, further demonstrating the lack of byproducts.
These results indicate that the dissolution of Zn precedes the formation of ZHS byproducts during the rest period. The self-dissolution of the Zn metal electrode is a crucial process in alkaline Zn batteries, where Zn continuously forms soluble zincate ions according to the Pourbaix diagram.6,30 However, the nature and impact of this dissolution in mild acidic systems can be quite different from that in alkaline batteries, and has been overlooked.
To better investigate the role of self-dissolution in directing the corrosion process of Zn, and to explore its relationship to the formation of ZHS byproducts, additional ex situ experiments with extended rest time were conducted using the same liquid cell as used for the in situ TEM experiments (see the methods section). Zn was electroplated onto the working electrode at 50 mA cm−2 to a capacity of 1 mA h cm−2. Afterward, the cell was dismantled and the chip with the Pt electrode was immersed in a vial of 1 M electrolyte for 40 hours. The morphologies of Zn deposits on the Pt electrode were characterised by scanning electron microscopy (SEM) before and after resting, as shown in Fig. 4a and b. Zn flakes with a loose structure covered the Pt electrode surface after the electrodeposition. However, after resting in 1 M ZnSO4 electrolyte for 40 hours, instead of forming ZHS byproducts on the Zn surface, most deposits had disappeared from the Pt electrode, as observed in our in situ experiments. The failure of ZHS to form suggests that the corrosion conditions were not met simply by immersing the plated Zn in 1 M ZnSO4 electrolyte, and that the dissolution of the plated Zn happened prior to any ZHS being able to form.
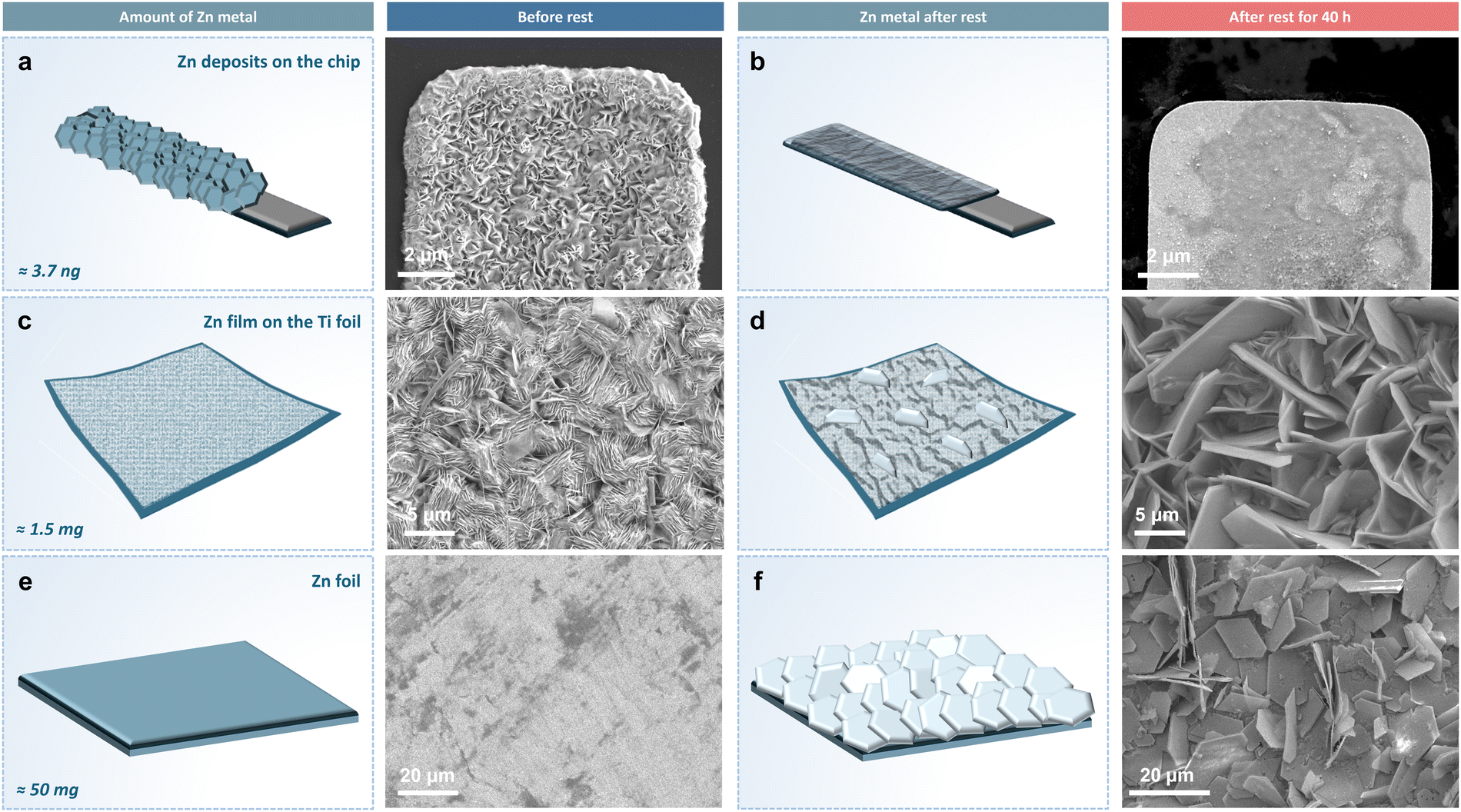 | ||
| Fig. 4 Schematic illustrations and SEM images of electrodes with increasing quantity of Zn metal before and after resting in a 1 M ZnSO4 electrolyte vial for 40 hours. (a) and (b) Zn deposited to 1 mA h cm−2 on a liquid cell TEM chip Pt electrode (approximately 3.7 ng of Zn). (c) and (d) A thin Zn film deposited on a Ti foil electrode (approximately 1.5 mg of Zn). (e) and (f) Zn foil electrode (approximately 50 mg of Zn). | ||
The formation of ZHS corrosion byproducts on the electrode/electrolyte interface should be a spontaneous process as long as the chemical equilibrium condition is satisfied with an adequate supply of Zn2+, OH−, SO42− and H2O in the electrolyte environment, regardless of the electrode substrate material used.48 Despite the mildly acidic condition of ZnSO4 electrolytes, OH− ions may also be generated due to a series of side reactions, such as water decomposition reaction during cycling, which can lead to a local alkaline environment that is conducive to ZHS formation.6 However, during battery rest, we suggest that the presence of sufficient Zn metal is essential to provide this OH− ion source for the formation of ZHS; the dissolution of the Zn metal is accompanied by hydrogen gas evolving from the electrolyte, and thereby a concomitant increase of additional OH− ions in the electrolyte.
To confirm this, Zn foil, Cu foil, and Ti foil were soaked in vials of the electrolyte for 18 hours. As is shown in Fig. S6 (ESI†), flaky byproducts formed on the Zn foil, a few formed on Cu, and the Ti foil remained clean. However, after soaking the Cu or Ti foil in ZnSO4 electrolyte together with Zn foil, their surfaces were found to be covered by Zn byproducts instead (Fig. S7a and b, ESI†), as confirmed by their X-ray diffraction (XRD) patterns (Fig. S7c, ESI†). We attribute this to the pH increase provided by the self-dissolution of the Zn foil, thus promoting the chemical equilibrium necessary for ZHS precipitation on these surfaces. These findings highlight the self-corrosion property of Zn metal in Zn-ion batteries, which is a process where the metal reacts with its environment without the influence of external galvanic action from a more noble metal.49 This self-corrosion property of the Zn metal anode reduces the stability of the anode side and also contributes to the degradation of other parts within the battery, such as the current collector and the cathode material, during battery rest periods.33,40,41
It should be noted that excess Zn metal is often used in Zn-ion batteries to offset Zn depletion. This practice is likely to increase the amount of dissolution and promote the formation of byproducts, thus exacerbating corrosion during battery storage. Given the above results showing the importance of the presence of Zn metal in facilitating corrosion, the influence of the amount of Zn metal used was investigated and compared with the ex situ liquid cell TEM results in Fig. 4a and b, where only a small quantity of metal Zn is plated onto the small electrode. Zn was deposited onto 0.5 cm2 inert Ti foil in a pouch cell at a current density of 100 mA cm−2 to 1.25 mA h (approximately 1.5 mg), and then soaked in 2 ml 1 M ZnSO4 electrolyte in a vial for 40 hours. SEM images of the Zn thin film on the Ti foil surface before and after soaking are presented in Fig. 4c and d. The thin film of Zn deposits showed a relatively compact morphology, while numerous larger hexagonal flakes were observed growing from the deposition layer. The amount of Zn metal was further increased to study the effect on the corrosion process. Zn foil of an area of 0.5 cm2 and a thickness of 0.14 mm (approximately 50 mg) was soaked under the same conditions. Initially, the Zn foil exhibited a relatively smooth surface (Fig. 4e). After soaking in the electrolyte for 40 hours, extensive large hexagonal flakes with a loose structure were observed forming on the surface (Fig. 4f). In both cases, the large hexagonal flakes are likely ZHS byproducts; however, as they share a similar morphology to the Zn metal deposits, it is challenging to identify ZHS flakes from only SEM imaging. Its presence on the electrode can be identified by a stronger oxygen signal in the energy dispersive X-ray spectroscopy (EDX) spectrum (Fig. S8, ESI†). From the EDX elemental mapping in Fig. S9 (ESI†), the larger ZHS flakes mixed with the smaller Zn deposits showed more oxygen signals. The presence of ZHS was further confirmed by XRD of the Ti foil, with a pronounced peak characteristic of ZHS emerging after soaking.
What these comparisons have shown is that the formation of ZHS byproducts occurs only after sufficient Zn is dissolved into the electrolyte during the self-corrosion process. This observation is also supported by the liquid cell TEM experiments, where only Zn dissolution, instead of byproducts, was observed during the self-corrosion process in both in situ and ex situ liquid cell experiments. This is because only a miniscule amount of Zn was deposited on the Pt electrode in these liquid cell experiments, as the electrode area is small, and so the interfacial pH did not reach the threshold for ZHS formation even after the complete dissolution of Zn deposits. Therefore, no byproducts were observed even after extended periods of rest (40 hours) in the liquid cell setup. Moreover, by comparing the XRD patterns in Fig. S10 (ESI†), it is evident that the peak intensity ratio of ZHS to Zn was significantly higher when soaking Zn foil compared to soaking Zn-coated Ti foil in the electrolyte for the same rest duration. The size of ZHS particles forming on the Zn foil was found to significantly increase as well. With more Zn present in the electrolyte, the self-dissolution was intensified, leading to a rapid accumulation of OH− ions in the local environment, thus promoting the uncontrolled growth of ZHS. This indicates that excess available Zn facilitates the corrosion process.
The relatively low initial pH of the electrolyte is possibly one of the main factors contributing to the Zn dissolution issue discussed above. The pH of ZnSO4 electrolyte can be mildly adjusted by adding small amounts of weak-basic and weak-acidic salts, which has been reported to result in varying levels of corrosion on Zn foils after soaking.50,51 Consequently, 0.025 M Zn(OAc)2 was introduced into the 1 M ZnSO4 electrolyte as a regulator of the pH value. To uncover the underlying mechanism of Zn dissolution and ZHS formation during the self-corrosion process of Zn, operando observations of the plating/stripping behaviour and the dissolution process of Zn deposits were performed in the electrolyte with Zn(OAc)2 to compare with that in pure ZnSO4, which has a relatively lower pH value. Zn was deposited at a low current density of 5 mA cm−2, but with a capacity that was doubled to 0.1 mA h cm−2 compared to the results in Fig. 3. The deposits were stripped after resting for 5 minutes, and the dissolution process was recorded during rest (see also Movie S2, ESI†). Sequential in situ liquid cell TEM images acquired during the plating process are shown in Fig. S11 (ESI†). The morphology of Zn deposits with Zn(OAc)2 was found to be slightly less compact compared to those in the pure ZnSO4 electrolytes, with more nucleation sites evident on the Pt electrode surface.
The dissolution of Zn deposits was recorded at areas around both the tip and the bottom of the Pt electrode. As shown in Fig. 5a, the shape of Zn deposits remained unchanged after 180 seconds of rest. This indicates that neither Zn dissolution nor the formation of byproducts occurred during the rest period. The viewing field was then shifted to the bottom of the Pt electrode, where Zn deposits with a looser structure were found. As previously demonstrated in Fig. 3a and b, the dissolution of Zn tends to be more visible at the edge of the electrode and in areas where a thinner amount of Zn was deposited, due to both a more sufficient supply of electrolytes and a more distinct change in the contrast of the images. However, it was observed that the small Zn deposits at the edge of the electrode were preserved well even after resting for 5 minutes (Fig. 5b). The coulombic efficiency is calculated to be 77.7%, compared to less than 10% after rest in the pure ZnSO4 electrolyte, based on the voltage profile given in Fig. 5c, and sequential in situ liquid cell TEM images of the stripping process after the rest period are shown in Fig. 5d. The Pt electrode surface was clean, with no residues left after stripping, thus no dead Zn or ZHS was formed.
 | ||
| Fig. 5 In situ liquid cell TEM imaging of the dissolution of plated Zn deposits in ZnSO4 electrolyte with Zn(OAc)2: (a) area 1 and (b) area 2. Scale bars are 1 μm. (c) Corresponding galvanostatic voltage profile of plating and stripping after rest for 5 minutes. (d) In situ liquid cell TEM imaging of the stripping process after rest for 5 minutes. Scale bars are 1 μm. | ||
These in situ liquid cell TEM experiments have confirmed that the self-dissolution of Zn must be related to the pH value of electrolytes, with more protons in the electrolyte from lower pH triggering more severe Zn dissolution. To further explore the relationship between the pH of electrolytes and the self-corrosion property of Zn metal, and given that the pH at the interface can be distinct to the electrolyte bulk, we measured in situ the pH change near the Zn surface using a home-made setup (Fig. S12, ESI†). The results of in situ pH monitoring in the 1 M ZnSO4 electrolyte and the 1 M ZnSO4 electrolyte with Zn(OAc)2 were compared (Fig. 6a and b). The initial pH values of two electrolytes were determined to be 4.46 and 5.43 respectively. After immersion of the Zn foil into the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M ZnSO4 electrolyte, a quick increase in pH was observed from 4.46 to 4.73 within two minutes. The pH value continued to grow over the 20 hours of resting time. In contrast, with Zn(OAc)2 added to the electrolyte, the pH value increased slightly to 5.5 after 10 minutes and then remained steady for the whole rest period. It is worth noting that after resting for 20 hours, the pH near the Zn surface in the 1 M ZnSO4 electrolyte was even higher than that in the 1 M ZnSO4 electrolyte with Zn(OAc)2. Additionally, the pH of the 1 M ZnSO4 electrolyte exhibited a continuous rising tendency, whereas with Zn(OAc)2 added, slight fluctuations of increasing and decreasing pH were observed after resting for 1 hour until the end of recording. This could be due to the role of acetate ions as a buffering agent, which helps to stabilise pH changes by neutralising extra OH− ions generated during the self-corrosion process of Zn.50
M ZnSO4 electrolyte, a quick increase in pH was observed from 4.46 to 4.73 within two minutes. The pH value continued to grow over the 20 hours of resting time. In contrast, with Zn(OAc)2 added to the electrolyte, the pH value increased slightly to 5.5 after 10 minutes and then remained steady for the whole rest period. It is worth noting that after resting for 20 hours, the pH near the Zn surface in the 1 M ZnSO4 electrolyte was even higher than that in the 1 M ZnSO4 electrolyte with Zn(OAc)2. Additionally, the pH of the 1 M ZnSO4 electrolyte exhibited a continuous rising tendency, whereas with Zn(OAc)2 added, slight fluctuations of increasing and decreasing pH were observed after resting for 1 hour until the end of recording. This could be due to the role of acetate ions as a buffering agent, which helps to stabilise pH changes by neutralising extra OH− ions generated during the self-corrosion process of Zn.50
 | ||
| Fig. 6 In situ pH recorded near the Zn foil surface soaked (a) in the 1 M ZnSO4 electrolyte and (b) in the 1 M ZnSO4 electrolyte with Zn(OAc)2. SEM images of Zn foils soaked in vials of electrolyte (c) without and (d) with Zn(OAc)2 for 40 hours, and soaked in pouch cells (PC) for 18 hours. (e) XRD patterns after soaking in vials of electrolyte without and with Zn(OAc)2 additive for 40 hours. (f) The voltage profiles of Zn‖Ti coin cells after rest for different times before stripping. (g) The voltage profiles of Zn‖Zn symmetric coin cells after rest for 35 days using the ZnSO4 electrolyte without and with Zn(OAc)2 addition. | ||
The self-corrosion property of Zn was further investigated by ex situ SEM and XRD analysis of Zn foils after resting under different conditions. Zn foils were immersed in these two electrolytes in vials for an extended rest period of 40 hours. Also, Zn foils were sealed in pouch cells containing 140 μl of 1 M ZnSO4 electrolyte without and with Zn(OAc)2 added, respectively. The pouch cells were then left to rest for 18 hours. SEM images of Zn foils after rest and the corresponding XRD patterns are displayed in Fig. 6c–e and Fig. S13 (ESI†). The formation of ZHS byproducts was observed on both Zn foils after resting. However, as demonstrated in the SEM images in Fig. 6c and d, both the size and the thickness of the ZHS flakes were reduced with the addition of Zn(OAc)2. Also, the byproduct layer on the Zn foils generated in the ZnSO4 electrolyte with Zn(OAc)2 was found to be more compact. This was also confirmed in the ZHS size distribution and cross-sectional imaging on the Zn foils after rest (Fig. S14 and S15, ESI†). These results indicate the constrained and gradual growth of byproducts, which could be attributed to the reduced accumulation of OH− ions due to alleviated Zn dissolution. The XRD peaks of ZHS exhibited lower intensity with the addition of Zn(OAc)2, indicating suppression of ZHS growth. In addition, the hydrogen evolution during rest in the ZnSO4 electrolyte without and with Zn(OAc)2 was quantified and compared via online mass spectrometry (MS), as detailed in Fig. S16 and S17 (ESI†). The amount of H2 in the system without Zn(OAc)2 (9.70 μmol) was approximately three times that in the system with Zn(OAc)2 (3.38 μmol) after 18 hours of rest.
Linear sweep voltammetry (LSV) measurements (Fig. S18a, ESI†) indicate a suppression of the hydrogen evolution reaction in the electrolyte containing Zn(OAc)2, with a lower current response at −0.15 V (vs. Zn2+/Zn). Tafel plots were constructed to determine the corrosion rates in the different electrolytes (Fig. S18b, ESI†). The exchange current density of the electrolyte with Zn(OAc)2 was found to be lower than that of the pure 1 M ZnSO4 electrolyte. Chronoamperometry tests were conducted with Zn‖Zn coin cells using two electrolytes at a fixed potential of −150 mV (Fig. S18c, ESI†). A slightly higher current density in the electrolyte with Zn(OAc)2 was observed, suggesting a larger active surface area, which is consistent with the greater number of nucleation sites observed in the liquid cell TEM images.
The self-dissolution of Zn in different electrolytes was further quantified in Zn‖Ti coin cells. Zn was deposited on the Ti electrode at a current density of 10 mA cm−2 with a capacity of 2 mA h cm−2. The Zn deposits were then stripped after resting for 6, 12 and 24 hours, respectively. According to the voltage profiles shown in Fig. 6f, the capacity loss in the cell with the addition of Zn(OAc)2 was significantly reduced.
As previously discussed, the addition of Zn(OAc)2 can alleviate the self-corrosion of the Zn metal electrode, which is supposed to be beneficial for improving the shelf life of Zn-ion batteries. To gain a deeper insight into the influence of pH adjustment on the electrochemical properties of the Zn anode after long-term rest, symmetric Zn‖Zn coin cells were prepared and left to rest for different durations, and then cycled at a current density of 10 mA cm−2 with a capacity of 1 mA h cm−2. As is shown in Fig. S19 (ESI†), after resting for 5 days, Zn‖Zn coin cells using both electrolytes exhibited steady curves with similar voltage polarisation, despite slight fluctuations using the pure 1 M ZnSO4 electrolyte (denoted by red arrows). After an extended rest period of 35 days, as shown in Fig. 6g, the Zn‖Zn coin cell using the 1 M ZnSO4 electrolyte with Zn(OAc)2 continued to exhibit stable cycling performances, while the cell without the addition of Zn(OAc)2 displayed significant voltage fluctuations from the beginning of cycling, indicating poor performance after long-term rest.
A schematic of the proposed corrosion process of the Zn metal anode in Zn-ion batteries during rest is shown in Fig. 7. In an acidic electrolyte environment, Zn metal undergoes the self-dissolution process first, which consumes H+ ions and leaves excess OH− ions, leading to hydrogen evolution and a local pH increase. The accumulation of the OH− ions fosters the formation of a ZHS corrosion byproduct layer and morphological changes to the anode surface, which will induce an uneven distribution of the electric field during cycling, causing heterogeneous nucleation and the ensuing formation of dendrites. These parasitic corrosion reactions persist throughout the entire storage period and are exacerbated by the use of excess Zn within the battery system. To mitigate corrosion during battery rest and extend the shelf life of Zn-ion batteries, it is essential to control the Zn dissolution process to enhance the stability of the Zn metal anode. One viable strategy to eliminate the Zn dissolution is to control the acidity of the electrolyte. By adding Zn(OAc)2 to the ZnSO4 electrolyte, the interfacial pH value is adjusted and stabilised, lowing the number of protons and thereby decreasing the activity of the Zn metal anode during the self-corrosion process. The Zn(OAc)2 functions as a pH buffer, helping to balance fluctuations in the chemical microenvironment, and suppressing the accumulation of OH− ions and the development of corrosion byproducts. Consequently, a more stable pH value can be maintained, leading to improved overall battery performance, especially after extended periods of rest.
 | ||
| Fig. 7 Schematic illustrations of (a) the underlying mechanism of the Zn metal anode corrosion process during rest in mildly acidic electrolytes, and (b) the mechanism for mitigating corrosion through pH adjustment with addition of the buffer additive. | ||
Some previous studies have also reported the elimination of ZHS corrosion byproducts by the addition of strong acidic additives,52 but this can result in more severe Zn self-dissolution under such circumstances, and may also trigger issues for the cathode materials, such as various manganese and vanadium based materials.53,54 This again underscores the importance of considering the potential influence of various electrolyte engineering strategies on the pH of electrolytes. Furthermore, most of the Zn-ion batteries have been evaluated with excess electrolytes and Zn metal to ensure a sufficient supply of active species, thereby prolonging the cycling life of the entire battery.31,35,55 Previous studies have reported that a large electrolyte dosage can help mitigate changes in the microenvironment, whereas with limited electrolytes, the local pH value is likely to become perturbed due to side reactions such as HER and corrosion.56,57 However, it should be noted that excess electrolyte does not prevent the dissolution of the Zn metal anode, which will consequently lead to the formation of corrosion byproducts after long-term storage. Moreover, although Zn depletion can be compensated by these additions, the excessive use of both electrolyte and Zn metal may exacerbate the corrosion issues, resulting in an increased formation of byproducts that pierce the separator, which will ultimately contribute to battery failure.58 A high utilisation rate of Zn in Zn-ion batteries is therefore crucial not only for achieving higher energy density but also for reducing corrosion, and optimising the Zn loading to balance corrosion and Zn depletion should be taken into account.
Conclusion
In this work, the self-corrosion process of Zn during battery resting was explored via in situ liquid cell TEM. The self-dissolution of Zn was observed, and this dissolution was believed to contribute to the majority of capacity loss during the initial phase of the corrosion process. The relationship between this self-dissolution and the formation of ZHS products was also investigated, which has highlighted the crucial role of the electrolyte pH in affecting corrosion during rest, and the two-stage nature of the corrosion process, with ZHS formation following after Zn self-dissolution. The addition of Zn(OAc)2 as a pH regulator in the 1 M ZnSO4 electrolyte was found to mitigate the self-dissolution behaviour significantly. In situ pH monitoring near the Zn surface in different electrolytes confirmed the distinct pH changes attributable to Zn dissolution during self-corrosion. SEM and XRD analysis of Zn foils soaked in electrolyte with Zn(OAc)2 demonstrated the additive alleviated the formation of ZHS byproducts. Moreover, the cycling performance of Zn‖Zn and Zn‖Ti coin cells after extended rest was significantly improved with Zn(OAc)2. These experimental results have elucidated the underlying mechanisms for the self-corrosion of Zn in a mild acidic environment, emphasising the importance of considering the electrolyte pH in future electrolyte modification research. As the formation of ZHS products and hydrogen evolution during battery rest are believed to result from Zn dissolution, the feasibility of using excess Zn metal in Zn-ion batteries should also be carefully evaluated.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AWR thanks the support of the Royal Society (UF221018) and UKRI (EP/Z000483/1). The authors acknowledge financial support provided by the Henry Royce Institute (grant ref EP/R010145/1), and the Henry Royce Institute for Advanced Materials (EP/R00661X/1, EP/S019367/1 and EP/R010145/1). FW gratefully acknowledges the National Natural Science Foundation of China (52172264), and the Science and Technology Innovation Program of Hunan Province (2023RC1016). The authors gratefully acknowledge the David Cockayne Centre for Electron Microscopy at the University of Oxford (Phani Karamched, Graham Wyatt, Neil Young, Gareth Hughes), and the Photoemission Research Technology Platform at the University of Warwick (Marc Walker), for their access and support.References
- M. Song, H. Tan, D. Chao and H. J. Fan, Recent Advances in Zn-Ion Batteries, Adv. Funct. Mater., 2018, 28, 1802564 CrossRef.
- X. Zeng, J. Hao, Z. Wang, J. Mao and Z. Guo, Recent progress and perspectives on aqueous Zn-based rechargeable batteries with mild aqueous electrolytes, Energy Storage Mater., 2019, 20, 410–437 CrossRef.
- W. Du, et al., Challenges in the material and structural design of zinc anode towards high-performance aqueous zinc-ion batteries, Energy Environ. Sci., 2020, 13, 3330–3360 RSC.
- T. Wang, et al., Anode Materials for Aqueous Zinc Ion Batteries: Mechanisms, Properties, and Perspectives, ACS Nano, 2020, 14, 16321–16347 CrossRef CAS PubMed.
- N. Dong, F. Zhang and H. Pan, Towards the practical application of Zn metal anodes for mild aqueous rechargeable Zn batteries, Chem. Sci., 2022, 13, 8243–8252 RSC.
- J. Hao, et al., Deeply understanding the Zn anode behaviour and corresponding improvement strategies in different aqueous Zn-based batteries, Energy Environ. Sci., 2020, 13, 3917–3949 RSC.
- Q. Li, et al., Dendrites issues and advances in Zn anode for aqueous rechargeable Zn-based batteries, EcoMat, 2020, 2, e12035 CrossRef CAS.
- Z. Li and A. W. Robertson, Electrolyte engineering strategies for regulation of the Zn metal anode in aqueous Zn-ion batteries, Battery Energy, 2023, 2, 20220029 CrossRef CAS.
- J. Cao, et al., Strategies of regulating Zn2+ solvation structures for dendrite-free and side reaction-suppressed zinc-ion batteries, Energy Environ. Sci., 2022, 15, 499–528 RSC.
- Z. Hou and B. Zhang, Boosting Zn metal anode stability: from fundamental science to design principles, EcoMat, 2022, 4, e12265 CrossRef CAS.
- Z. Cai, et al., Chemically resistant Cu–Zn/Zn composite anode for long cycling aqueous batteries, Energy Storage Mater., 2020, 27, 205–211 CrossRef.
- S. D. Pu, et al., Achieving Ultrahigh-Rate Planar and Dendrite-Free Zinc Electroplating for Aqueous Zinc Battery Anodes, Adv. Mater., 2022, 34, 2202552 CrossRef CAS.
- Y. Zhang, et al., Manipulating the Zinc Deposition Behavior in Hexagonal Patterns at the Preferential Zn (100) Crystal Plane to Construct Surficial Dendrite-Free Zinc Metal Anode, Small, 2022, 18, 2105978 CrossRef CAS.
- M. Zhou, et al., Surface-Preferred Crystal Plane for a Stable and Reversible Zinc Anode, Adv. Mater., 2021, 33, 2100187 CrossRef CAS PubMed.
- B. Li, et al., Interfacial Engineering Strategy for High-Performance Zn Metal Anodes, Nano-Micro Lett., 2021, 14, 6 CrossRef.
- L. Kang, et al., Nanoporous CaCO3 Coatings Enabled Uniform Zn Stripping/Plating for Long-Life Zinc Rechargeable Aqueous Batteries, Adv. Energy Mater., 2018, 8, 1801090 CrossRef.
- T. Foroozan, et al., Non-Dendritic Zn Electrodeposition Enabled by Zincophilic Graphene Substrates, ACS Appl. Mater. Interfaces, 2019, 11, 44077–44089 CrossRef CAS.
- P. Samanta, et al., Recent Progress on the Performance of Zn-Ion Battery Using Various Electrolyte Salt and Solvent Concentrations, ACS Appl. Electron. Mater., 2023, 5, 100–116 CrossRef CAS.
- L. Miao, et al., Three-functional ether-based co-solvents for suppressing water-induced parasitic reactions in aqueous Zn-ion batteries, Energy Storage Mater., 2022, 49, 445–453 CrossRef.
- C. Zhang, et al., A ZnCl2 water-in-salt electrolyte for a reversible Zn metal anode, Chem. Commun., 2018, 54, 14097–14099 RSC.
- S. Bai, et al., Electrolyte Additives for Stable Zn Anodes, Adv. Sci., 2024, 11, 2304549 CrossRef CAS.
- L. Hong, et al., An effective descriptor for the screening of electrolyte additives toward the stabilization of Zn metal anodes, Energy Environ. Sci., 2024, 17, 3157–3167 RSC.
- Z. Hu, et al., Screening metal cation additives driven by differential capacitance for Zn batteries, Energy Environ. Sci., 2024, 17, 4794–4802 RSC.
- L. Yu, et al., Ionic Liquid “Water Pocket” for Stable and Environment-Adaptable Aqueous Zinc Metal Batteries, Adv. Mater., 2023, 35, 2210789 CrossRef CAS.
- L. Geng, et al., Eutectic Electrolyte with Unique Solvation Structure for High-Performance Zinc-Ion Batteries, Angew. Chem., Int. Ed., 2022, 61, e202206717 CrossRef CAS PubMed.
- Y. Hao, et al., Gel Electrolyte Constructing Zn (002) Deposition Crystal Plane Toward Highly Stable Zn Anode, Adv. Sci., 2022, 9, 2104832 CrossRef CAS.
- R. Deng, Y. Yuan, Z. Li, A. W. Robertson and F. Wu, A liquid-infiltrated Al2 O3 framework electrolyte enables aqueous zinc batteries, Chem. Commun., 2024, 60, 14423–14426 RSC.
- L. Ma, et al., Hydrogen-Free and Dendrite-Free All-Solid-State Zn-Ion Batteries, Adv. Mater., 2020, 32, 1908121 CrossRef CAS PubMed.
- J. Hao, L. Yuan, Y. Zhu, M. Jaroniec and S.-Z. Qiao, Triple-Function Electrolyte Regulation toward Advanced Aqueous Zn-Ion Batteries, Adv. Mater., 2022, 34, 2206963 CrossRef CAS.
- Z. Cai, J. Wang and Y. Sun, Anode corrosion in aqueous Zn metal batteries, eScience, 2023, 3, 100093 CrossRef.
- R. Yao, et al., A corrosion-free zinc metal battery with an ultra-thin zinc anode and high depth of discharge, Energy Environ. Sci., 2024, 17, 3112–3122 RSC.
- W.-G. Lim, X. Li and D. Reed, Understanding the Role of Zinc Hydroxide Sulfate and its Analogues in Mildly Acidic Aqueous Zinc Batteries: A Review, Small Methods, 2024, 8, 2300965 CrossRef CAS PubMed.
- B. Zhang and H. J. Fan, Overlooked calendar issues of aqueous zinc metal batteries, Joule, 2025, 9, 101802 CrossRef CAS.
- M. Tribbia, G. Zampardi and F. La Mantia, Towards the commercialization of rechargeable aqueous zinc ion batteries: The challenge of the zinc electrodeposition at the anode, Curr. Opin. Electrochem., 2023, 38, 101230 CrossRef CAS.
- C. Li, S. Jin, L. A. Archer and L. F. Nazar, Toward practical aqueous zinc-ion batteries for electrochemical energy storage, Joule, 2022, 6, 1733–1738 CrossRef.
- S. W. D. Gourley, R. Brown, B. D. Adams and D. Higgins, Zinc-ion batteries for stationary energy storage, Joule, 2023, 7, 1415–1436 CrossRef CAS.
- B. Tang, L. Shan, S. Liang and J. Zhou, Issues and opportunities facing aqueous zinc-ion batteries, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- W. He, et al., Challenges and strategies of zinc anode for aqueous zinc-ion batteries, Mater. Chem. Front., 2021, 5, 2201–2217 RSC.
- S. D. Pu, et al., Decoupling, quantifying, and restoring aging-induced Zn-anode losses in rechargeable aqueous zinc batteries, Joule, 2023, 7, 366–379 CrossRef CAS.
- Y. Kim, et al., Corrosion as the origin of limited lifetime of vanadium oxide-based aqueous zinc ion batteries, Nat. Commun., 2022, 13, 2371 CrossRef CAS PubMed.
- Q. Li, et al., Calendar Life of Zn Batteries Based on Zn Anode with Zn Powder/Current Collector Structure, Adv. Energy Mater., 2021, 11, 2003931 CrossRef CAS.
- Y. Yuan, et al., Diagnosing the Electrostatic Shielding Mechanism for Dendrite Suppression in Aqueous Zinc Batteries, Adv. Mater., 2024, 36, 2307708 CrossRef CAS PubMed.
- Y. Huang, et al., Unraveling dynamical behaviors of zinc metal electrodes in aqueous electrolytes through an operando study, Energy Storage Mater., 2022, 46, 243–251 CrossRef.
- Y. Sasaki, et al., In situ electron microscopy analysis of electrochemical Zn deposition onto an electrode, J. Power Sources, 2021, 481, 228831 CrossRef CAS.
- Y. Yang, H. Yang, R. Zhu and H. Zhou, High reversibility at high current density: the zinc electrodeposition principle behind the “trick”, Energy Environ. Sci., 2023, 16, 2723–2731 RSC.
- X. Shi, et al., Metallic Zinc Anode Working at 50 and 50 mA h cm−2 with High Depth of Discharge via Electrical Double Layer Reconstruction, Adv. Funct. Mater., 2023, 33, 2211917 CrossRef CAS.
- Z. Liu, et al., Unraveling Paradoxical Effects of Large Current Density on Zn Deposition, Adv. Mater., 2024, 36, 2404140 CrossRef CAS PubMed.
- X. Yu, et al., Ten concerns of Zn metal anode for rechargeable aqueous zinc batteries, Joule, 2023, 7, 1145–1175 CrossRef CAS.
- A. Nazarov, T. Yurasova and A. Marshakov, Hydrogen Absorption and Self-Corrosion of Mg Anode: Influence of Aqueous Electrolyte Species, Corros. Mater. Degrad., 2024, 5, 350–369 CrossRef CAS.
- D. Han, et al., A Self-Regulated Interface toward Highly Reversible Aqueous Zinc Batteries, Adv. Energy Mater., 2022, 12, 2102982 CrossRef CAS.
- L. Miao, et al., A Universal Descriptor in Determining H2 Evolution Activity for Dilute Aqueous Zn Batteries, Adv. Funct. Mater., 2023, 33, 2306952 CrossRef CAS.
- Q. Nian, et al., Highly reversible zinc metal anode enabled by strong Brønsted acid and hydrophobic interfacial chemistry, Nat. Commun., 2024, 15, 4303 CrossRef CAS.
- Y. Zhao, Y. Zhu and X. Zhang, Challenges and perspectives for manganese-based oxides for advanced aqueous zinc-ion batteries, InfoMat, 2020, 2, 237–260 CrossRef CAS.
- X. Chen, et al., Vanadium-based cathodes for aqueous zinc-ion batteries: Mechanism, design strategies and challenges, Energy Storage Mater., 2022, 50, 21–46 CrossRef CAS.
- L. Yuan, et al., Regulation methods for the Zn/electrolyte interphase and the effectiveness evaluation in aqueous Zn-ion batteries, Energy Environ. Sci., 2021, 14, 5669–5689 RSC.
- M. Chen, et al., Suppressing Rampant and Vertical Deposition of Cathode Intermediate Product via PH Regulation Toward Large-Capacity and High-Durability Zn//MnO2 Batteries, Adv. Mater., 2024, 36, 2304997 CrossRef CAS.
- H. Yang, et al., Protocol in Evaluating Capacity of Zn–Mn Aqueous Batteries: A Clue of pH, Adv. Mater., 2023, 35, 2300053 CrossRef CAS PubMed.
- Z. Wu, Y. Wang and C. Zhi, Zinc-anode reversibility and capacity inflection as an evaluation criterion, Joule, 2024, 8, 2442–2448 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00485c |
| This journal is © The Royal Society of Chemistry 2025 |