
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePorous organic material-based atomically dispersed metal electrocatalysts
Hao
Zhang†
 *ab,
Suwen
Wang†
c,
Enmin
Lv†
*d,
Menghui
Qi
c,
Chengchao
He
e,
Xinglong
Dong
d,
Jieshan
Qiu
f,
Yong
Wang
*c and
Zhenhai
Wen
*e
*ab,
Suwen
Wang†
c,
Enmin
Lv†
*d,
Menghui
Qi
c,
Chengchao
He
e,
Xinglong
Dong
d,
Jieshan
Qiu
f,
Yong
Wang
*c and
Zhenhai
Wen
*e
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: hzhchem@mit.edu
bDepartment of Chemistry, University of Oxford, Oxford OX1 3TA, UK
cAdvanced Materials and Catalysis Group, Institute of Catalysis, Department of Chemistry, Zhejiang University, Hangzhou 310058, P. R. China. E-mail: chemwy@zju.edu.cn
dSchool of Materials Science and Engineering, Dalian University of Technology, Dalian 116024, P. R. China. E-mail: lvenmin456@163.com
eCAS Key Laboratory of Design and Assembly of Functional Nanostructures, and Fujian Provincial Key Laboratory of Materials and Techniques toward Hydrogen Energy, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, P. R. China. E-mail: wen@fjirsm.ac.cn
fCollege of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China
First published on 26th March 2025
Abstract
The transition to renewable energy sources and the need for efficient energy conversion technologies have led to the development of various types of catalysts, among which atomically dispersed metal catalysts (ADMCs) supported by porous organic materials (POMs) have attracted attention for their high catalytic efficiency and stability. This review focuses on the development and application of ADMCs supported by POMs, such as MOFs, COFs, and HOFs, which offer catalytic performance due to their high atomic utilization, stability, and selectivity. This paper systematically explores various strategies for synthesizing ADMCs, including the use of organic linkers, metal nodes, and pore spaces within POMs to stabilize metal atoms and prevent aggregation. Key applications highlighted include energy conversion and storage technologies, such as fuel cells, water splitting, CO2 reduction and nitrogen reduction, where ADMCs demonstrate the potential to replace noble metals. Despite the progress, challenges remain in achieving high metal loading, long-term stability, and cost-effective large-scale production. This study underscores the importance of advanced characterization techniques and computational models to deepen the understanding of ADMCs’ catalytic mechanisms and guide future material design, paving the way for their broader application in sustainable energy technologies.
Broader contextThe development of atomically dispersed metal catalysts (ADMCs) supported by porous organic materials (POMs) marks a significant leap in the field of energy and environmental science. By leveraging the unique properties of POMs such as MOFs, COFs, and HOFs, this research addresses critical challenges in renewable energy conversion and storage. These materials offer unparalleled catalytic activity, stability, and selectivity, enabling applications across fuel cells, water splitting, CO2 reduction and nitrogen reduction. Notably, ADMCs achieve high atomic utilization, thus presenting a sustainable alternative to traditional noble metal-based catalysts. However, the work transcends catalyst development, emphasizing the role of advanced synthesis techniques, characterization methods, and computational models in elucidating the structure–performance relationships of ADMCs. This understanding is crucial for designing next-generation materials capable of accelerating the global shift towards low-carbon energy systems. Despite challenges such as achieving high metal loading and scalability, this research provides a blueprint for overcoming these barriers, setting the stage for breakthroughs in clean energy technologies and environmental remediation. The integration of ADMCs into sustainable practices could transform energy industries, fostering a cleaner and more efficient future. |
1. Introduction
The transition from traditional fossil fuels to renewable energy sources is becoming increasingly urgent due to rising energy demands and the push for a low-carbon society. The global energy crisis has been exacerbated by the depletion of fossil fuels such as coal and oil and their uneven distribution (Fig. 1a).1 Additionally, the consumption of fossil fuels has led to severe environmental and climate issues. As renewable energy technologies (e.g., hydropower, wind, nuclear, solar, tidal, and geothermal) continue to advance, the reliance on fossil fuels is expected to decrease. New energy is approaching a golden age of development (Fig. 1b), potentially ushering in the “new energy era” and accelerating the “new energy revolution” sooner than anticipated. | ||
| Fig. 1 Global energy landscape: fossil fuel distribution and consumption trends. (a) Layout of global fossil energy distribution and (b) trends and forecasts of global energy consumption.1 Copyright 2016, Elsevier. | ||
Therefore, in order to seize the technological commanding heights of the next generation of energy revolution, exploring environmentally friendly and low-pollution renewable clean energy, and developing technologies for the efficient and clean utilization of traditional energy resources have become hot spots and key points in various countries. Currently, a significant amount of research is focused on developing advanced energy conversion technologies, such as fuel cells,2 metal–air batteries,3 CO2 conversion,4 water splitting,5 supercapacitors6 and NH3 production,7 which are essential for the efficient use and storage of renewable energy.8 The key new energy technologies, such as fuel cells (FCs), batteries, and solar cells, are compared in Table 1.2 Fuel cells, which directly convert the chemical energy in fuel into electrical energy, are becoming a new direction of technological development because of their high efficiency and low emissions.9 Combining the design features of conventional batteries and fuel cells, metal–air batteries have much higher theoretical energy densities than lithium-ion batteries, 3–30 times higher than Li-ion batteries, and are often advocated as the next-generation electrochemical energy storage solution for applications such as electric vehicles or grid energy storage (Fig. 2b).10 Moreover, supercapacitors are devices that store and release electrical energy when needed. Compared to rechargeable batteries such as lithium-ion batteries, supercapacitors are high capacitance/capacity capacitors with higher capacitance values than ordinary capacitors, but with lower potential limits. Their high energy and power densities compared to conventional capacitors and batteries have led to their great popularity in the past few years.11 In addition, H2 comes from 48% of natural gas, 30% of oil, 18% of coal, and 4% of renewable energy.2 The production of H2 from fossil energy is key to the widespread deployment of H2, which requires further reduction in its production costs. Therefore, the cost competitiveness of H2 extraction can be ensured by combining the high utilization rate of the electrolyzer reactor and the low price of renewable electricity.12
| Device | Energy density | Life time | Advantage | Disadvantage |
|---|---|---|---|---|
| Fuel Cell | Very high | 5000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (hours) 000 (hours) |
Modular and compact | Slow cold start |
| High efficiency | Expensive | |||
| Smooth power output | Hazards of H2 | |||
| Rapid H2 refuelling | Fuel price is high | |||
| Minimal emission | ||||
| Battery | High | 4–6 (years) | Portable and rechargeable | Recharging slowly |
| Low cost | Lifetime is short and preparing and recycling batteries lead to environmental pollution | |||
| Established technology | Electrolyte flammable | |||
| Supercapacitor | Very low | 10–20 (years) | Recharging and quick reaction | Short time energy storage |
| High cost | ||||
| Photovoltaic panel | Medium | 15–20 (years) | Eco-friendly | Power output is intermittent |
| Huge for light transport | ||||
| Flywheels | High | 5–10 (years) | High power output and rating | Charging slowly |
| Eco-friendly | Heavy weight | |||
| Superconducting magnetic energy storage system | Low | 25–30 (years) | High power output and rating | Short-term energy storage |
| High efficiency | High cost | |||
| Eco-friendly | ||||
| Quick response |
 | ||
| Fig. 2 Comparison of fuel cells and metal–air batteries: performance and energy density. (a) The relationship between the operating temperature and output power for several fuel cells.2 Copyright 2023, Elsevier. (b) Theoretical energy densities for different types of metal–air batteries.10 Copyright 2017, American Chemical Society. | ||
However, the efficiency of these technologies is greatly limited by the sluggish kinetics of key reactions such as the hydrogen precipitation reaction (HER), hydrogen oxidation reaction (HOR), oxygen reduction reaction (ORR), oxygen precipitation reaction (OER), carbon dioxide reduction reaction (CO2RR), nitrogen reduction reaction (NRR), nitrate reduction reaction (NO3−RR) and alcohol oxidation reaction (AOR).8,13
The development of high performance and durable electrocatalysts is essential for accelerating these reactions and thereby increasing the energy conversion efficiency, helping to lower the energy barrier and facilitating the kinetics of electrochemical processes. Currently, noble metals and noble metal-based materials are considered as state-of-the-art catalysts for various electrochemical reactions, but their high cost, low natural abundance and scarcity limit their large-scale practical applications.14
Generally, metal nanoparticles have inactive metal atoms that do not actively participate in the reaction. In addition, the active sites of metal nanoparticles are usually located in the corner or/and edge positions, leading to the dissipation of the main precious metals. In order to address this bottleneck, the miniaturization strategy of metal particles has been used in the preparation of catalysts.15 It has been shown that as the size of nanoparticles decreases, their surface free energy increases, the number of active sites and the exposed active surface area increase, and the intrinsic properties of the metal sites can be enhanced by tuning their geometrical configuration and electronic structure (Fig. 3).16–21
 | ||
| Fig. 3 Atomically dispersed metal catalysts: structure–activity relationships and size effects. (a) Scheme of atomically dispersed metal catalysts or single-atom catalysts, and the relationship between surface free energy and metal size. (b) Specific activity as a function of metal loadings/sizes.21 Copyright 2013 American Chemical Society. (c) Ratio of surface atoms at different particle sizes.22 Copyright 2020 American Chemical Society. (d) Geometric and electronic structures of single atom, clusters, and nanoparticles.20 Copyright 2018 American Chemical Society. | ||
In this context, porous organic materials (POMs) have emerged as an ideal carrier for atomically dispersed metal catalysts (ADMCs) due to their unique structural characteristics. POMs, such as metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and hydrogen-bonded organic frameworks (HOFs), possess a high specific surface area, tunable pore size distribution, and abundant coordination sites, enabling precise anchoring of metal atoms through organic ligands, metal nodes, or pore spaces, effectively suppressing the migration and aggregation of metal atoms. For instance, the periodic pore structure of MOFs can stabilize single metal atoms through coordination interactions, while the π-conjugated system of COFs can optimize electron transfer pathways, enhancing catalytic activity. Research has shown that POMs not only provide highly dispersed active sites for ADMCs but also regulate the adsorption energy barriers of reaction intermediates through electronic interactions between the carrier and the metal, significantly improving catalytic efficiency. This synergistic effect between POMs and ADMCs offers new insights for the design of materials in key energy conversion technologies such as fuel cells, water splitting, and CO2 reduction, while also indicating directions to overcome the cost and stability bottlenecks of traditional precious metal catalysts.
2. Atomically dispersed metal catalysts (ADMCs)
In recent years, the well-defined ADMCs are particularly attractive for reducing the cost of noble metal-based catalytic materials due to their highest atomic utilization (100%), obvious active sites, high catalytic activity, good stability, and good selectivity. It is expected to become the main catalyst for many catalytic reactions.22 Depending on the number of metal atoms in the centre, ADMCs can be classified into several different types, such as single-atom catalysts (SACs), dual-atom catalysts (DACs) and triatomic catalysts (TACs).23 Among them, SACs are one of the fastest developing branches in recent years in terms of design and fabrication. In 2011, Zhang's team first outlined the theory of ADMCs,24 which have received widespread attention in the field of electrocatalysis and have shown good catalytic performance in photochemical and thermal catalysis reactions.25–28 The important progress of ADMCs in several typical catalytic reactions is summarized in Fig. 4. In Fig. 5, the elements of reported ADMCs are summarized and marked.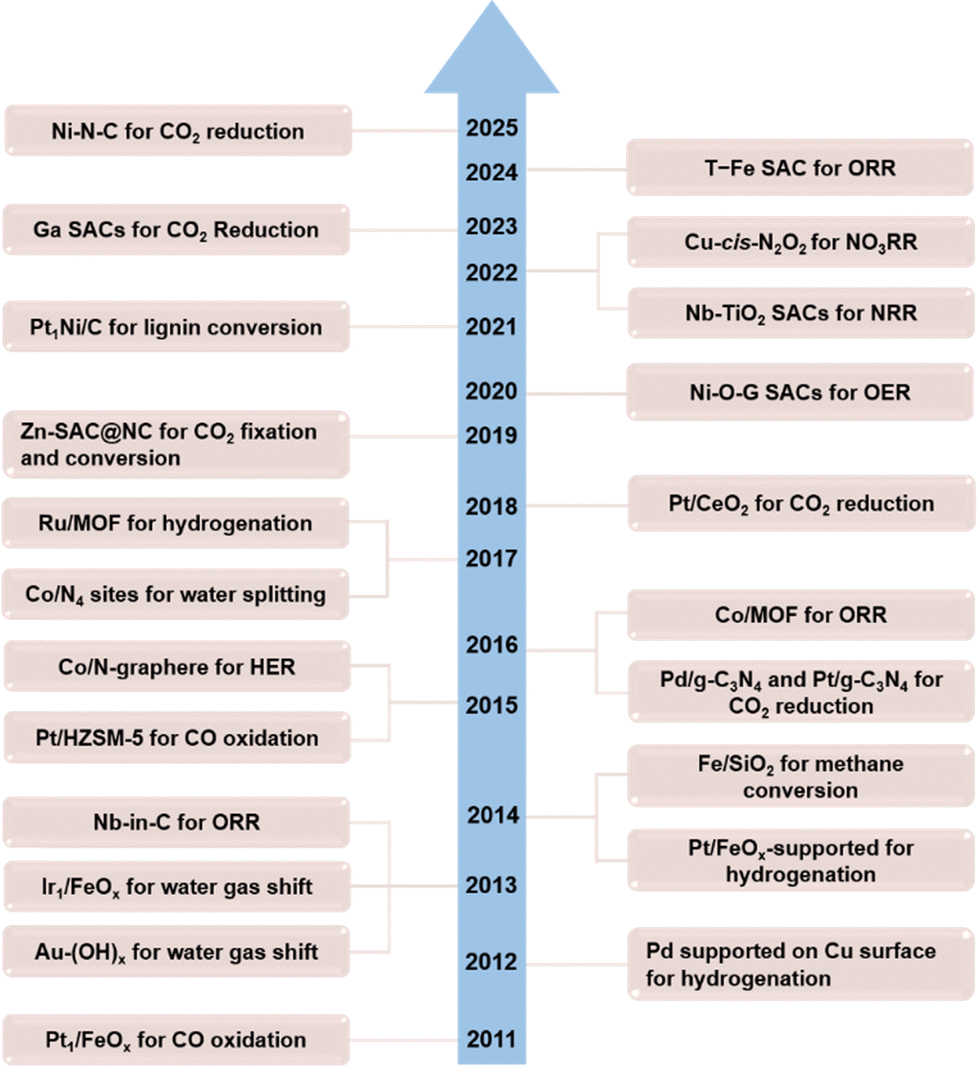 | ||
| Fig. 4 Timeline of important developments of ADMCs. | ||
 | ||
| Fig. 5 Schematic illustration of kinds of metal elements for the reported ADMCs. | ||
Compared with homogeneous catalysts, the anchoring of uniformly distributed metal atoms on a carrier makes them have heterogeneous properties, such as easy separation, high selectivity reusability and good stability, so it provides an ideal bridge for connecting heterogeneous and homogeneous catalysts (Fig. 6a).29–32 However, due to the dispersion of the catalyst at the atomic level, the surface free energy increases dramatically and tends to aggregate. Although great efforts have been made to bypass this problem, ADMCs still suffer from low metal loading and easy aggregation, partly due to the low surface area of the substrate and the weak interactions between the metal atoms and the substrate, which hinders the large-scale synthesis and practical application of ADMCs. The main idea to address this problem is centered on loading of active metals onto a variety of substrates with high surface area and desired anchoring positions.
 | ||
| Fig. 6 Advancements in ADMCs: catalyst integration and stabilization strategies. (a) Relationship among ADMCs, homogeneous catalysts, and heterogeneous catalysts. (b) Potential advantages of MOFs for stabilizing ADMCs as compared with conventional porous materials. | ||
3. Porous organic materials (POMs)
POMs represent an innovative category of porous substances characterized by a diverse array of organic molecular components linked together through delicate supramolecular forces, including hydrogen bonding, π–π stacking, van der Waals interactions, and electrostatic interactions. According to the definition of the International Union of Pure and Applied Chemistry (IUPAC), the pores in POMs can be classified into micropores (<2 nm), mesopores (2–50 nm) and macropores (>50 nm).33 Compared to conventional inorganic skeletal materials such as zeolites, silica, activated carbon, POMs stand out for their unique composition of lightweight elements, functionality for organic reactions, high stability in air and conventional humidity conditions, and resistance to acidic and alkaline environments.In all application scenarios of POMs, having a rich pore structure, i.e., having a high specific surface area, is an indispensable property. In recent years, with the development of new types of building blocks and advances in efficient linking strategies, it has become possible to precisely regulate the pore properties of POMs and their surface area. The emergence of various types of POMs such as hyper-cross-linked polymers (HCPs), conjugated microporous polymers (CMPs), polymers of intrinsic microporosity (PIMs), covalent triazine frameworks (CTFs), COFs, porous aromatic frameworks (PAFs), HOFs, conjugated microporous polymers, hyper crosslinked polymers, MOFs, porous polymer networks (PPNs), and porous aromatic backbones (PAFs) over the past decade has signaled significant progress in this field.33 These new materials not only broaden the application scope of POMs, but also provide new impetus for the development of related technologies.
3.1. MOFs
MOFs are porous crystalline materials consisting of metal ions or cluster nodes with organic ligands, and have a wide range of potential applications, especially excelling in the field of catalysis.33 The specific surface area of common MOF compounds can generally exceed 2000 m2 g−1, and some of them can reach 7000 m2 g−1. The structure, morphology, and porosity of MOFs can be tailored by adjusting the metal nodes, organic ligands, or preparation conditions. Almost all transition and main group metal elements can be assembled with suitable organic ligands to prepare functionalized MOFs. In addition, MOFs can be controllably integrated with other materials (e.g., molecules, quantum dots, polymetallic oxides, etc.) to prepare MOF-composites with specific morphologies and functions. These materials can be used as precursors and/or ADMCs' sacrificial templates for further transformation into micro/nanomaterials with specific catalytic functions.The unique properties of MOFs, including abundant internal molecular metal sites, ultra-high surface area, flexible customization, ordered porous structures, accurate design ability and chemical stability, fully meet the requirements of a stable ADMC substrate compared to conventional porous materials such as zeolites and carbon nanotubes (Fig. 6b). Therefore, the MOF is expected to be one of the most promising and versatile platforms for preparing ADMCs with high metal loading and good stability. Meanwhile, MOF-derived materials that have partially inherited the properties or structures of the original MOF have been found to be ideal candidates for the construction of MOF-derived ADMCs.34
In 1995, Yaghi et al. put forward the concept of “metal–organic framework” and created a new era of MOFs.35 In the course of their development in recent years, various series of MOFs have continuously appeared. Fig. 7 shows the classification of MOFs and the reported structural schematic diagram of MOFs.36
 | ||
| Fig. 7 Classification of MOFs and the reported structural schematic diagram of MOFs.36 Copyright 2016 Elsevier. | ||
MIL was discovered by the Ferry research group in France. They named the MOF after their own scientific research institution (Materials of Institute Lavoisier).36 The MIL uses trivalent metals such as iron, aluminum, lead, and cadmium as the coordination center, and phthalic acid as the ligand, and they are prepared by a solvothermal method. The most representative MOFs of this series are MIL-53(Al), MIL-101(Fe), MIL-88(Fe) and MIL-100(Al).37 The ZIF series was also researched and developed by Yaghi and his collaborators. It is based on the coordination of metal ions with nitrogen elements to form three-dimensional porous materials, which have the advantages of structural diversity and adjustable pore size.38 UiO was discovered by researchers at the University of Oslo and named after the school abbreviation. There are not many types of UiO, with UiO-66 the most representative one.39 It is formed by coordination of zirconium oxygen clusters and twelve p-phthalic acid molecules, which is the highest coordination number among known MOFs.40,41 UiO-66 has very good chemical and thermal stability in a variety of solvents and is widely used in various catalytic fields. In addition, by modifying the organic ligands, UiO-66 with different group substituents such as –NH2, –CH3, –SO3H can be obtained.42
Despite the existence of over 2000 MOFs, only a limited number of them demonstrate stable existence and practical applicability, posing a hindrance to their advancement. Consequently, the development and investigation of novel MOFs with enhanced chemical and thermal stability remain crucial endeavors within the academic community.
3.2. MOF derivatives
MOFs are distinguished by their complex coordination between organic ligands and inorganic entities. By precisely manipulating the metal ions and organic ligands, researchers can tailor the particle dimensions, pore distribution, active surface area, and crystalline structure of MOFs to meet diverse and specific needs. These frameworks are characterized by their extensive skeletal structures and unique pore architectures, which offer high pore volumes and substantial specific surface areas. Since the pioneering use of MOF-5 for synthesizing porous carbon in 2008,43 there has been increase in the development of nanomaterials derived from MOFs, including porous carbon, metal oxides, metal phosphides, and metal sulfides (Fig. 8a). On account of the co-existence of both organic and inorganic substances in MOFs, they can be used as templates and precursors under certain circumstances, which brings new ideas for their research and development.44 For example, when the precursor MOF is heat-treated in an inert atmosphere, commonly used organic ligands (such as trimesic acid, p-phthalic acid, etc.) can be converted into carbon materials in high yield, and the metal nodes uniformly distributed in MOFs are reduced by the carbothermal process to metal-particles supported on the porous carbon. After etchings by acid, the generated metal particles and other components are further removed, and finally a porous carbon material can be obtained, as shown in Fig. 8b.45,46 | ||
| Fig. 8 Transformation and thermal conversion of MOFs into functional nanomaterials. (a) Transformation of MOFs/coordination polymers into functional nanostructured materials.47 Copyright 2017 American Chemical Society. (b) Scheme and TEM images of the thermal conversion of [Ni2(EDTA)] into 3D mesoG.45 Copyright 2015 Royal Society of Chemistry. | ||
Furthermore, as a template, the MOF has also excelled in the synthesis of metal oxides, diversifying their forms from zero-dimensional quantum dots to three-dimensional complex porous structures through self-template and external template strategies during the heat treatment process. This imparts enhanced thermal stability, catalytic activity, and unique electrical and magnetic properties to these oxide materials. The synthesis of metal sulfides and metal phosphides further expands the application scope of MOFs, as the precursor MOF is transformed into metal sulfides/phosphides with high conductivity, good mechanical properties, and thermal stability through a one-step sulfidation or phosphorization process, enhancing the performance of the materials in areas such as batteries, supercapacitors, and electrochemical catalysis.
It is worth noting that the successful preparation and application of the aforementioned MOF-derived materials provide new insights into the design and development of single-atom catalysts. As a cutting-edge research direction in the field of catalysis, single-atom catalysts have active centers in the form of isolated individual atoms, enabling atomic-level utilization efficiency and maximization of catalytic activity.48 Given the unique advantages of MOFs in constructing complex nanostructures and achieving precise element doping, it is foreseeable that through further exploration and optimization of MOF-derived strategies, particularly in combination with single-atom loading and anchoring techniques, the creation of high-performance single-atom catalysts derived from MOFs is promising. These catalysts not only have the potential to address the bottleneck issues of traditional catalysts in terms of activity, selectivity, and stability, but also hold broad application prospects in areas such as energy conversion and storage, environmental purification, and fine chemical synthesis.
3.3. COFs/HOFs
COFs are a class of porous polymers with highly ordered two- or three-dimensional structures, possessing high surface area, structural regularity, designability and synthetic controllability.49 COFs containing π-conjugated systems enhance electron transport and stability through carbon–carbon double bonds.50 By introducing functionalized ligands, such as bipyridine, and combining them with metal ions, COF@ADMC composites can be prepared, in which the metal atoms are uniformly distributed and firmly attached to the COF framework.51 In addition, the bulk COFs can be exfoliated into two-dimensional nanosheets to increase the contact area of the active sites, thus optimizing their performance in catalytic applications.52 It is expected that COFs will show great potential for application as platforms with external coordination capabilities. All of these benefits of COFs meet the substrate requirements for stabilization of ADMCs (Fig. 9a). COFs have shown great potential for application as a platform with external coordination capabilities (Fig. 9b).53 | ||
| Fig. 9 COF-based materials for ADMCs and electrocatalysis: structural advantages and publication trends. (a) The structural merits of COF-based materials over traditional substrates for the production of ADMCs.54 Copyright 2022 Elsevier. (b) Statistics of publications on COF-based single-sites for electrocatalysis from 2014 to 2024.53 Copyright 2024 RSC. | ||
COFs can be modified by post-metallization methods by introducing functional groups of organic linkers into the COFs and using interlayer interactions. Metal atoms can be uniformly coordinated to free functional groups in the COF skeleton, such as –OH, –CN, –C@C–, etc.55 The unpaired electrons of the low-density metal atoms facilitate the formation of coordination bonds with other atoms, resulting in the formation of ADMCs.
COF-based ADMCs combine the advantages of homo- and heterogeneous catalysis to provide innovative pathways for catalytic reactions. The interaction between metal atoms and COF carriers is key in this field, as refining metal particles to the single atom level exposes more d-orbitals, which in turn optimizes their electronic properties and catalytic activity.56 This COF-based ADMC combines the properties of intrinsic COFs with the unique activity of individual metal atoms in a synergistic behavior.
HOF materials are a class of porous solids with high porosity and/or flexibility.57 HOFs are self-assembled from functional building blocks and form porous two-dimensional (2D) or three-dimensional (3D) frameworks through hydrogen-bonding interactions, but stabilization of their structures and prediction of their frameworks remain challenging.58 Weaker hydrogen bonding compared to POMs and COFs makes it more challenging to fabricate robust and porous HOFs. Nevertheless, HOFs have been widely used in gas separation and storage, optics and catalysis due to their high crystallinity, large surface area and abundant porosity. HOFs exhibit good processability, reparability and profitability on top of high porosity and tunable pore size due to the reversible nature of hydrogen bonding. Four key advances have driven the development of HOF chemistry: (1) the use of rigid backbones connected with oriented hydrogen bonding modules to achieve geometrical architectures;59 (2) the tuning of the network structure by varying the branching length of the organic nuclei to prepare HOFs with the same topology but different pore structures;59 (3) the structural flexibility of HOFs that allows for adaptive dynamics in the presence of encapsulation, which aids molecular recognition studies; and (4) the solution-processability of HOFs becomes an effective means of remodeling the morphology, and the reversibility of hydrogen bonding permits the repair and regeneration of HOFs by reconnecting broken units.60 By carefully selecting coordinated hydrogen bonding units and rigid organic cores, robust porous HOFs with interconnected multidimensional structures and diverse shapes can be designed and synthesized to create stable materials with complex pore networks (Fig. 10).57
 | ||
| Fig. 10 Hydrogen-bonding units (inner region) employed in the construction of HOFs (outer region).57 Copyright 2021 Elsevier. | ||
4. Synthesis of POM-derived ADMCs
To systematically outline current methodologies for the synthesis of POM-derived ADMCs, we present a detailed evaluation of the advantages and limitations of representative synthetic approaches, accompanied by a comparative analysis table (Table 2). It is intended to provide a foundational reference for subsequent discussions on systems stabilized by POMs and ADMCs derived from POMs.| Scheme | Synthetic method | Advantage | Disadvantage |
|---|---|---|---|
| ADMCs stabilized by POMs | Organic linkers | Strong coordination capability | Constrained choice of organic linkers |
| Coordination flexibility | |||
| Metal nodes | Abundant binding sites | Limited carrier availability | |
| High stability | Demanding reaction conditions | ||
| Precise localization | |||
| Pore space | Abundant interaction sites | High compatibility requirements | |
| Synergistically enhanced activity | |||
| POM-derived ADMCs | Metal nodes | Effectively inhibits agglomeration | Limited metal selection |
| High activity | Metal sites require concurrent functionality | ||
| Pore structure | Precise control of atomic distribution | Limited applicability | |
| High activity | Complex preparation with high requirements | ||
| High stability |
4.1. ADMCs Stabilized by POMs
Recent advancements have demonstrated the tremendous potential of functionalizing organic linkers, metal nodes, and pore spaces in POMs, establishing them as an ideal platform for immobilizing desired single atom species.61 By integrating effective active sites with organic components, MOF-stabilized ADMCs can be synthesized to exhibit synergistic effects as catalysts.62 Typically, three primary techniques are utilized for the production of POM-based ADMCs. These involve the dispersion of metal atoms within MOF metal nodes, the attachment of desired metal atoms onto organic linkers featuring appropriate chelating sites, and the adaptation of the pore space to enable the decoration of metal atoms using a range of building compounds like MOF-derived carbon materials, metal complexes, inorganic oxides, and polyoxometalates (Fig. 11).32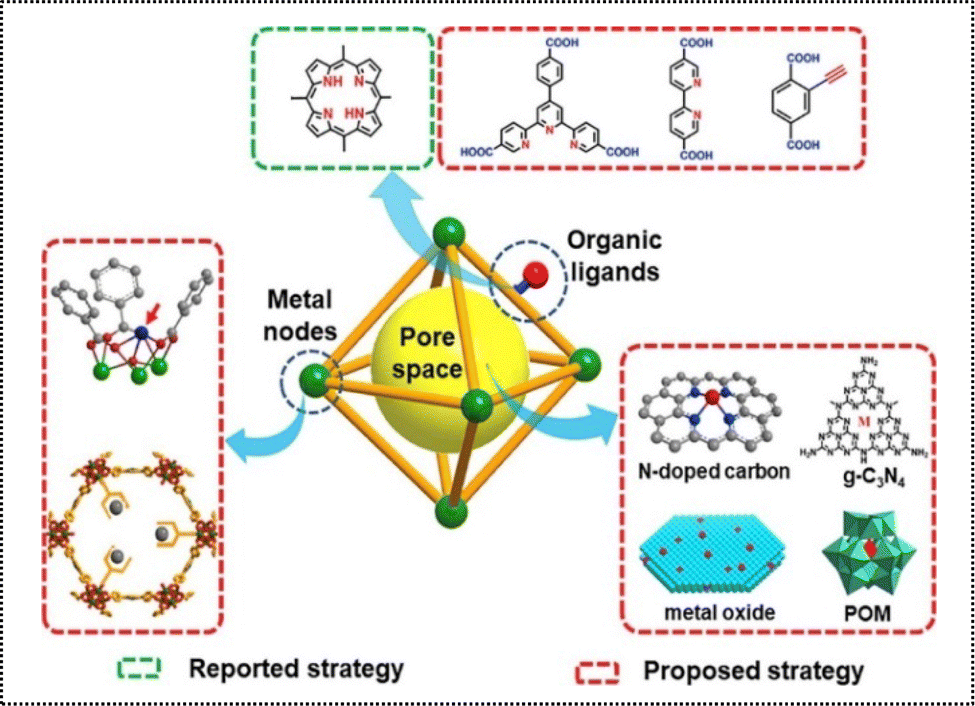 | ||
| Fig. 11 Strategies for the production of ADMCs based on POMs.32 Copyright 2019 Elsevier. | ||
 | ||
| Fig. 12 Schematic approaches for the preparation of ADMCs and MOF-derived catalysts. (a) Schematic diagram of the preparation of Fe ADMC-MIL101-T using organic ligands.65 Copyright 2021 Wiley. (b) Schematic synthesis procedure of BMOF-derived ZnxCoy.67 Copyright 2024 American Chemical Society. (c) Schematic diagram illustrating the preparation of a POM-supported Pt catalyst utilizing the pore space within POMs.68 Copyright 2021 Springer Nature. | ||
Similarly, AIM can be utilized in the gas phase for the synthesis of POMs. This method enables the synthesis of metal single atom sites on the POM nodes. By altering the coordination sites, additional binding sites are generated, allowing for the attachment of metal species and the formation of catalysts with isolated atom active sites.69 Through the use of continuous pulse precursors and either H2O or H2S, a diverse range of metal ions, such as AlxOy, In2O3, CoOxHy, ReOx, Pt, ZnO, and CoSx can be successfully deposited on NU-1000 metal nodes, offering the opportunity for various chemical reactions. However, it is worth noting that the AIM method's stringent standards limit the availability of POM supports, primarily NU-1000. Therefore, future research should focus on identifying alternative suitable POMs.70
4.2. POM-derived ADMCs

5. Identification of POM-based ADMCs
In the realm of catalytic science, advanced characterization techniques hold paramount significance in constructing the structure–reactivity correlations of catalysts, particularly in the context of ADMCs. Recent technological advancements have ushered in a multitude of methods for probing the active sites of ADMCs. Foremost among these are the Aberration corrected scanning transmission electron microscopy (AC-STEM) and high-energy synchrotron radiation X-ray absorption spectroscopy (XAS). AC-STEM, with its sub-atomic resolution capabilities, enables direct visualization of the electronic imaging information of individually dispersed atoms.94 By harnessing Rutherford scattered electrons, it discriminates between elements of varying atomic numbers, offering unequivocal evidence for the existence of single atoms. This technique revolutionizes the understanding of catalyst fundamentals by resolving the spatial arrangement and distribution of metal atoms on the support. As depicted in Fig. 14a, the transmission electron microscopy (TEM) image of Pt SAC/PCN reveals an absence of conspicuous Pt particles, indicating the absence of metal aggregation. Conversely, the densely packed bright spots in the AC HAADF-STEM image in Fig. 14b demonstrate that the isolated Pt atoms are uniformly distributed on the PCN surface, confirming the atomic-level dispersion of Pt sites.95 Electron energy loss spectroscopy (EELS) is a high spatial resolution technique that combines TEM to reveal the elemental composition, chemical states, and local electronic structure of materials by analyzing the characteristic energy loss of incoming electrons interacting with the sample.96 With advancements in AC-STEM, EELS can now achieve atomic-level resolution, effectively characterizing single metal atoms anchored on the surface of a support.97 By comparing the spatial distribution of EELS signals from the support (such as the C-K edge, N-K edge) and the metal single atoms (such as the Pt–M edge), one can verify the phenomena of charge transfer or bonding at the metal–support interface.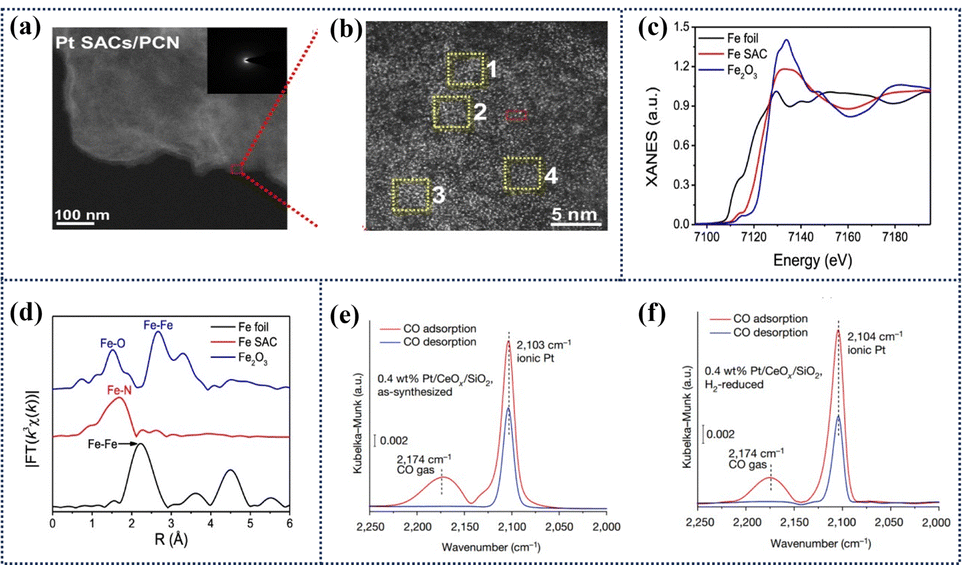 | ||
| Fig. 14 Characterization of ADMCs: TEM, XANES, EXAFS, and IR Spectra. (a) TEM image of Pt ADMCs/PCN and (b) aberration-corrected HAADF-STEM image.95 Copyright 2024 Springer Nature. (c) XANES spectra at the Fe K-edge of the Fe ADMC, referenced with Fe foil and Fe2O3. (d) FT k3-weighted χ(k)-function of the EXAFS spectra at Fe K-edge.98 Copyright 2021 Springer Nature. (e) IRAS spectra of 0.4 wt% Pt/CeOx/SiO2 after oxidation in air and (f) after reduction with hydrogen.99 Copyright 2022 Springer Nature. | ||
Complementing AC-STEM, XAS, comprising EXAFS and XANES, delves into the atomic-scale intricacies of ADMCs.100 By measuring the X-ray absorption coefficients, XAS unveils the atomic valence states, coordination environments, and neighboring elements of ADMCs, thereby furnishing detailed insights into their local structures and electronic configurations. Using Fe ADMC as an example, the Fe K-edge XANES exhibits an absorption energy between Fe metal foil and Fe2O3 references, suggesting an oxidation state of Fe single atoms between Fe0 and Fe3+. The corresponding EXAFS spectrum displays a prominent Fe–N peak near 1.6 Å, with no observable Fe–Fe peak, demonstrating the evolution pathway of Fe ADMC (Fig. 14c and d).98 This information is vital for formulating accurate structure–reactivity correlations.
Electrochemical surface-enhanced Raman spectroscopy (EC-SERS) serves as a pivotal characterization technique in the field of spectroelectrochemistry, boasting dual advantages of high-sensitivity surface detection and in situ monitoring of electrochemical responses.101 It enables the dynamic tracking of structural evolution of active sites on electrocatalysts and reaction pathways. For instance, Tang et al. utilized EC-SERS to real-time capture the dynamic stability characteristics of FeCo–N6–O active sites in the FeCo-NCNS bimetallic catalyst during the ORR, unveiling the correlation mechanism between O2 accumulation behavior at bimetallic sites and the rate-determining steps of the reaction.102 The team led by Chernyshova utilized EC-SERS to reveal the formation of key intermediates (*COOH) during the proton-coupled electron transfer process in the early stages of CO2 electroreduction, while simultaneously observing the dynamic restructuring of the electrode/electrolyte interface double layer structure.103 This provides direct experimental evidence for establishing a multi-scale structure–activity relationship of “active sites-interface microenvironment-reaction kinetics.”
Furthermore, infrared absorption spectroscopy (IRAS) stands as a pivotal tool in the characterization of ADMCs due to its high sensitivity and broad applicability.104 Leveraging the distinct interaction strengths of probe molecules (e.g., CO) at specific adsorption sites, IRAS meticulously scrutinizes spectral bandwidths, frequencies, and symmetries, unraveling the local structures, oxidation states, and coordination environments of the materials. For example, after CO adsorption, the IRAS spectra of 0.4 wt% Pt/CeOx/SiO2 in both oxidized and reduced states exhibit a sharp single peak near 2103 cm−1 with a half-peak width of 12.8 cm−1 (Fig. 14e and f),99 which is attributed to CO adsorbed on isolated cationic Pt, confirming the existence of Pt in a single-atom state.
Solid-state nuclear magnetic resonance (SSNMR) has the capability to elucidate the local coordination structure of metal centers in catalysts, particularly suitable for systems that are amorphous or challenging to characterize with X-ray diffraction.105 For instance, the group led by Venkatesh utilized 195Pt SSNMR to quantitatively analyze the coordination bond lengths between Pt single atoms and the hydroxyl groups of the carrier.106 Simultaneously, Mössbauer spectroscopy can achieve atomic-level quantitative analysis of the chemical states of specific isotopes (such as 57Fe and 119Sn). Li et al. through operando 57Fe Mössbauer spectroscopy confirmed that the oxygen adsorption step is the rate-determining step for the ORR.107 These two techniques, operating in the dimensions of nuclear spin interactions and nuclear hyperfine fields, respectively, provide complementary quantitative experimental evidence for the electronic structure and dynamic evolution of catalytic active sites.
The synergistic application of these advanced characterization techniques profoundly enhances our comprehension of the geometric and electronic structures of ADMCs' active sites. Additionally, it fosters research into the intricate relationships between the structure and performance during catalytic processes. As three-dimensional imaging technologies continue to evolve, the future holds immense promise for a deeper understanding of atomic-level catalyst structures and properties, thereby propelling the advancement of catalytic science.
6. Applications
As quintessential POMs, MOFs, HOFs, and COFs have emerged as ideal precursors for constructing ADMCs due to their unique electronic structural characteristics and high controllability. By precisely controlling the coordination connection modes of POMs (such as MOFs’ coordination bonds, COFs’ covalent bonds, and HOFs’ hydrogen bond networks) and coupling with ADMC active sites, it is possible to effectively overcome the traditional dilemma of “activity–stability” balance in catalytic materials, thereby constructing electrocatalytic materials with both high intrinsic activity and excellent durability in various electrocatalytic reactions (Fig. 15 and Table 3). This chapter will focus on the directed synthesis strategies of POM-based ADMCs, elucidating the mechanism of electronic structure regulation of catalysts and the synergistic enhancement principle of active sites, providing a scientific basis for the rational design of the next generation of highly efficient energy conversion materials. | ||
| Fig. 15 Applications of POM-derived ADMCs in electrocatalytic reactions. | ||
| Reaction | Reaction equation | E (vs. RHE) | Catalysts | Performance |
|---|---|---|---|---|
| ORR | O2 + 4H+ + 4e− → 2H2O | 1.23 | Fe1/d-CN108 | E 1/2 = 0.95 V |
| Fe/I-N-CR109 | E 1/2 = 0.92 V | |||
| Fe SAC-MIL101-T65 | E 1/2 = 0.94 V | |||
| Zn/Co–N–C67 | E 1/2 = 0.94 V | |||
| OER | 2H2O → O2 + 4H+ + 4e− | 1.23 | FeCo3(DDA)2110 | 260 mV@10 mA cm−2 |
| Ni BTC111 | 279 mV@10 mA cm−2 | |||
| FeCo-SAs112 | 270 mV@10 mA cm−2 | |||
| CoxZn3−x(HITP)2113 | 210 mV@10 mA cm−2 | |||
| HER | 2H+ + 2e− → H2 | 0 | NiRu0.13-BDC114 | 36 mV@10 mA cm−2 |
| W-SAC115 | 85 mV@10 mA cm−2 | |||
| MOF-MoSAWSA116 | 57 mV@10 mA cm−2 | |||
| W1Mo1-NG117 | 24 mV@10 mA cm−2 | |||
| CO2RR | CO2 + 2H+ + 2e− → CO + H2O | −0.1 | Ni1–N–C118 | FE = 96%, j = 20 mA cm−2 |
| Ni/Cu–N–C119 | FE = 97.7%, j = 95.2 mA cm−2 | |||
| NiSA–N2–C120 | FE = 98%, j = 27 mA cm−2 | |||
| In SAC121 | FE = 97.2%, j = 40 mA cm−2 | |||
| CO2 + 6H+ + 6e− → CH3OH + H2O | 0.03 | Cu3(HHTQ)2122 | FE = 53.6%, j = 0.5 mA cm−2 | |
| CuSAs/TCNFs123 | FE = 44%, j = 62 mA cm−2 | |||
| CO2 + 2H+ + 2e− → HCOOH | −0.12 | Zn1Sn1/SNC124 | FE = 94.6%, j = 32.8 mA cm−2 | |
| In-BDC125 | FE = 88%, j = 6.49 mA cm−2 | |||
| CO2 + 4H+ + 4e− → CH4 + 2H2O | 0.17 | Cu SAC126 | FE = 40%, j = 15 mA cm−2 | |
| Cu SAC127 | FE = 75.3%, j = 47.8 mA cm−2 | |||
| NRR | N2 + 6H+ + 6e− → 2NH3 | 0.09 | W-NO/NC128 | FE = 8.35%, yield = 12.6 μg h−1 mg−1cat |
| Fe–Co SAC129 | FE = 73.2%, yield = 574.8 μg h−1 mg−1 | |||
| Ru SAs/N-C130 | FE = 29.6%, yield = 120.9 μg h−1 mg−1 | |||
| NOxRR | NO3− + 9H+ + 8e− → NH3 + 3H2O | 0.69 | Fe SAC131 | FE = 73.2%, yield = 9.2 mg h−1 cm−2 |
| Cu(I)–N3C1132 | FE = 94.8%, yield = 5466 mmol gCu−1 h−1 | |||
| Cu12-NND-H133 | FE = 98.7%, yield = 35.1 mg h−1 mgcat−1 | |||
| In-MOF In8134 | FE = 90.1%, yield = 256.9 μg h−1 mg−1cat |
6.1 Fuel cells and electrolysers
Hydrogen has long been studied as an alternative to fossil fuels due to its low carbon footprint and high gravimetric energy density. While direct hydrogen combustion offers short-term benefits,135 the electrochemical harnessing of hydrogen using fuel cells is ā more popular field of research. This is due to their versatility. The use of hydrogen in engines is largely limited to applications in automotive transport. Conversely, fuel cells can be applied to support renewable energy use in homes,136 decarbonisation of the energy grid,137 and are becoming increasingly feasible for widespread production, allowing sustainable energy storage on a global scale.138Cost effective and practical storage methods are therefore of key importance. Further to those methods discussed, nanoporous materials such as activated carbon, porous carbons, covalent organic frameworks, and POMs are being researched as an alternate means of ambient temperature and pressure gas storage.139 In each case, the primary mechanism of storage is reversible adsorption of gaseous molecules onto active sites, with surface area increasing the number of available active sites. While able to operate under ambient conditions, POMs currently suffer from issues regarding incomplete desorption or fuel at lower pressures and a trade-off between volumetric and gravimetric hydrogen densities due to each being enhanced by total surface area and pore volume, respectively.140,141 Considering the additional issues with energy intensive mass-production, POMs are not currently a viable wide-scale option for gas storage.142
While not directly relevant to POMs use in energy storage, the full context of the demands of hydrogen fuel systems should be established. Following development of effective storage, infrastructure for their refuelling of fuel cells, and costs associated with the technology need to decrease before hydrogen fuel cells can be successfully integrated and accepted by consumers as replacements to conventional fossil fuel transport.143
Hydrogen fuel cells produce energy via the electrochemical conversion of hydrogen and oxygen into water. Furthermore, provided the energy supplied to the system is from a renewable source, the hydrogen required for its operation can be sustainably produced via the electrolysis of water. Despite these properties, both fuel cells and water electrolysers are slowed through the unfavourable multi-step kinetics of the ORR and OER at the cathode and anode, respectively. Therefore, electrolysers with efficient anodes and cathodes remain a key challenge. The current optimum catalysts for these reactions are noble metals Pt and IrO2 for the ORR and OER, respectively – which are unsustainable for wide-scale fuel cell application and hydrogen production given the estimated global production levels.140,141 Hence, current research efforts are focused on the synthesis of non-noble metal catalysts with comparable performance to noble metal catalysts. To date, a number of atomically dispersed non-noble metal-based catalysts have been developed to enhance the performance of the OER and ORR. Gong et al. successfully prepared a class of Cu, Fe/NC bimetallic SACs for pH universal ORR using hydrothermal and ball milling assisted secondary pyrolysis methods.144 This unique bimetallic single atom catalyst exhibits ORR performance and stability in pH universal electrolytes (half potentials: 0.895 V (alkaline), 0.757 V (acidic), 0.815 V (neutral), superior to Pt/C catalyst) (Fig. 16a). With its unique monatomic iron active sites, hierarchical pore structure, oriented mesoporous channels, and good electrical conductivity, this material exhibits oxygen reduction activity and stability under both alkaline and acidic conditions, and outperforms most of the nonprecious metal catalysts and advanced Pt/C. More importantly, this MOF-based hybrid ligand strategy provides a new pathway for the precise construction of highly efficient monatomic catalysts. Wang et al. designed a core–shell structure of Fe-doped bilayer ZIFs, and successfully synthesized a composite structured catalyst (named Fe3C–Co–NC) containing Fe3C, Co nanoparticles, and M–Nx monoatoms, which can be used as efficient bifunctional catalysts for both the ORR and OER (Fig. 16b).145 Fe3C–Co–NC excels in performance with a half-wave potential of 0.89 V and an overpotential of 1.67 V, both of which outperform commercially available Pt/C and RuO2 catalysts.
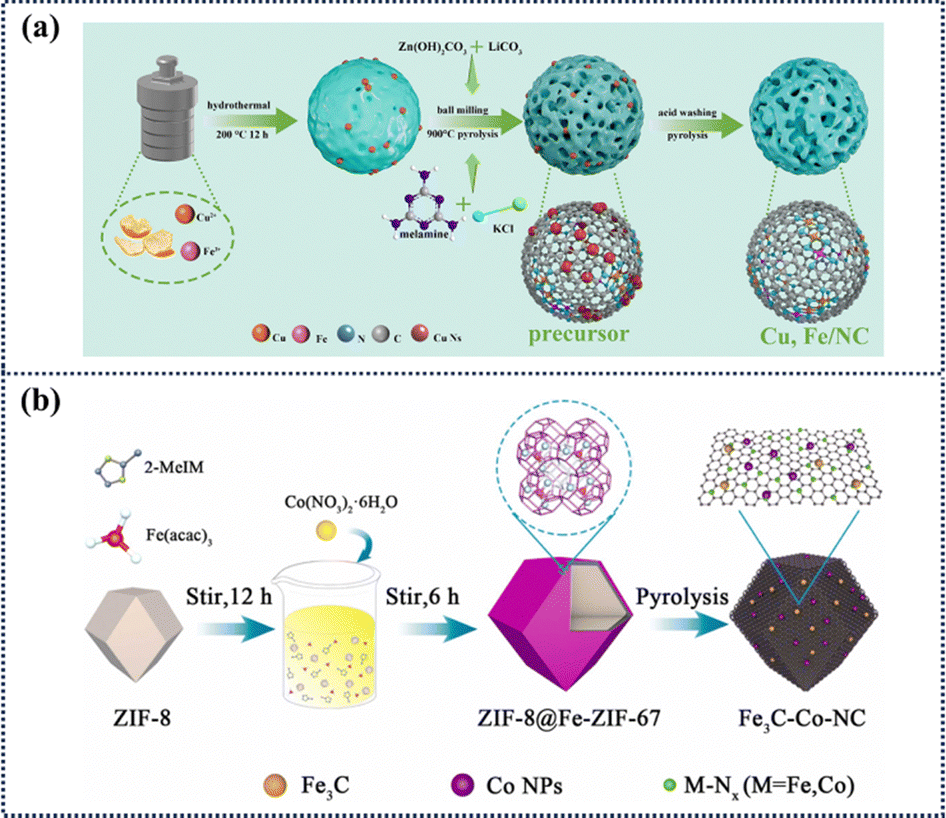 | ||
| Fig. 16 Synthesis routes for Cu, Fe/NC and Fe3C–Co–NC catalysts. (a) Schematic synthesis route of Cu, Fe/NC.144 Copyright 2025 Elsevier. (b) Schematic diagram of the Fe3C–Co–NC synthesis.145 Copyright 2023 Elsevier. | ||
6.2. POM-derived catalysts for the ORR and OER
In recent years, global energy demand has risen sharply, and the extensive use of fossil fuels such as coal and oil has exacerbated carbon dioxide emissions and environmental problems. To cope with the energy shortage and environmental challenges, solutions are being actively sought, especially the development of clean and renewable energy sources to replace traditional petroleum fuels. Although Pt catalysts are widely used in electrochemical applications, non-Pt catalysts (e.g., POMs-derived materials) are increasingly favored because of their affordability due to their cheap cost. These materials show potential in electrocatalytic processes such as the OER and ORR.The ORR is essential for environmentally friendly and promising energy conversion technologies such as fuel cells and metal–air batteries. The process involves multi-electron reactions, including proton-coupled electron transfer. Oxygen can be produced from water via a direct four-electron pathway or an indirect pathway that converts it to hydrogen peroxide and then further reduces it to water.146 The ORR in acidic media proceeds via the following steps:
| O2 + H+ + e− → *OOH |
| *OOH + H+ + e− → *O + H2O |
| *O + H+ + e− → *OH |
| *OH + H+ + e− → H2O |
| O2 + H2O + e− → *OOH + OH− |
| *OOH + e− → *O + OH− |
| *O + H2O + e− → *OH + OH− |
| *OH− + e− → OH− |
The ORR mechanism can be optimized through synergistic structural engineering of catalytic systems. Atomically dispersed metal centers, exemplified by ADMCs embedded in COFs/MOFs (e.g., Fe–N4 and Co–N4 configurations), mimic enzymatic active sites such as heme-like structures, thereby achieving exceptional ORR activity through precise electronic modulation. Graphitic and pyridinic nitrogen sites further enhance reaction kinetics by strengthening O2 adsorption/activation and stabilizing critical intermediates (*O2 and *OOH), effectively promoting the energetically favorable 4e− transfer pathway. Concurrently, π-conjugated organic networks serve as electron highways, substantially improving bulk electrical conductivity and enabling rapid charge transfer to sustain continuous catalytic cycles. Complementarily, tuning the hydrophobicity/hydrophilicity in MOF-derived carbon frameworks optimizes interfacial pore-wettability, ensuring efficient O2 diffusion to active sites while mitigating mass transport limitations. These multidimensional strategies collectively address thermodynamic, kinetic, and mass transfer challenges in ORR electrocatalysis.
MOF-based ADMCs are of interest for their enhanced ORR efficiency and durability, and the dispersed metal centers in POMs provide abundant active sites for efficient oxygen adsorption and reduction. In addition, the controlled environment and organic ligands of the MOF help to tune the electronic structure, promote favorable reaction pathways and reduce the energy barrier for the ORR. Liu et al. developed a bifunctional OER/ORR electrocatalyst composed of Ni single atom sites and FeN0.0324 nanoclusters, with a unique core–shell structure of FeN0.0324@NiN4/C.147 Benefiting from the efficient synergistic electronic effect of single atom Ni and FeN0.0324, FeN0.0324@NiN4/C exhibits electrocatalytic activity for the OER, with an overpotential of 258 mV at 10 mA cm−2 and a half-wave potential of 0.89 V for the ORR. The liquid zinc-air battery assembled with FeN0.0324@NiN4/C achieved a maximum peak power density of 180.9 mW cm−2 and a cycle durability stability of more than 150 h (Fig. 17a). Chen et al. successfully prepared a ZIF-derived two-site electrocatalyst (Fe/CoSx-SNC), which is composed of iron single atoms and ultrafine cobalt sulfide nanoparticles supported on S and N co-doped porous carbon (Fig. 17b).148 The Fe/CoSx-SNC electrocatalyst exhibits large specific surface area, high porosity, high metal center density, and a dual-site synergistic effect, showing electrocatalytic ORR activity. The half-wave potential is 0.885 V, and the dynamic current density at 0.80 V is 27.00 mA cm−2, and the stability is better than that of the commercial 20% Pt/C catalyst. Quirós-Díez et al. elegantly employed a thiol-functionalization method to covalently assemble POMs onto the surface of gold nanoparticles (AuNPs), resulting in AuNP@POM.149 This nanostructure effectively reduces unwanted intermediate products during the ORR, such as hydroperoxyl radicals (HO2−), thereby enhancing selectivity. This study represents the first demonstration of utilizing a POM-based electrochemically active coating to enhance the selectivity of gold nanoparticles with remarkable success. This innovative design strategy holds promise for extending to other metal surfaces with varying activities and POM clusters.
 | ||
| Fig. 17 Synthesis and characterization of various metal-based catalysts. (a) Schematic illustration of the synthesis process of FeN0.0324@NiN4/C.147 Copyright 2025 Wiley. (b) Schematic diagram of the synthesis of the Fe/CoSx-SNC electrocatalyst.148 Copyright 2024 Elsevier. (c) Synthesis and morphology characterization of Fe ADMC NCs.150 Copyright 2023 Wiley. (d) Preparation process of Co/Ce@NC.151 Copyright 2024 American Chemical Society. | ||
The OER plays a vital role in the conversion of various renewable energy sources, especially in rechargeable metal–air batteries.152,153 Four-electron reactions usually require high overpotentials, leading to slow reaction rates and high energy consumption, which affects electrocatalytic efficiency. Generally, the OER process involves the sequential formation of OH*, O*, and OOH* intermediates. In alkaline media, the process proceeds as follows:
| 4OH− → OH* + 3OH− + e− |
| OH* + 3OH− + e− → O* + H2O + 2OH− + 2e− |
| O* + H2O + 2OH− + 2e− → *OOH + H2O + OH− + 3e− |
| OOH* + H2O + OH− + 3e− → O2 + 2H2O + 4e− |
In acidic media, a similar process occurs:
| 2H2O → OH* + H2O + H+ + 2e− |
| OH* + H2O + H+ + e− → O* + H2O + 2H+ + 2e− |
| O* + H2O + 2H+ + 2e− → OOH* + 3H+ + 3e− |
| OOH* + 3H+ + 3e− → O2 + 4H+ + 4e− |
The OER mechanism can be strategically enhanced through multifunctional engineering of electrocatalytic architectures. Metal node functionalization in MOFs/COFs involves transition metal centers (Co, Ni, Fe, and Mn) coordinated to organic linkers as catalytically active sites, where high-valency metal species critically stabilize key intermediates (*O and *OOH) to accelerate the O–O bond formation kinetics. Heteroatom doping (e.g., N, S, P) in covalent materials systematically modulates electron density distribution at active centers, particularly through pyridinic-N and graphitic-N sites that optimize metal–oxygen orbital interactions, thereby reducing the energy barrier for intermediate stabilization and lowering overpotential. Concurrently, redox-active linkers such as π-conjugated porphyrins and phthalocyanines enable efficient electron delocalization across the framework, synergistically promoting charge transfer dynamics and improving overall OER activity. Complementing these strategies, defect engineering introduces oxygen vacancies in MOF/COF matrices to strengthen oxygen adsorption capacity and create electron-rich regions, further facilitating charge transfer while maintaining structural stability. These coordinated modifications collectively address the inherent limitations of sluggish kinetics and high energy barriers in OER electrocatalysis.
ADMCs derived from POMs are ideal for enhancing the efficiency and stability of the OER due to their unique structure and electronic properties.154 The metal atoms modulate the band structure of the catalyst, providing a favorable electronic environment for OER intermediates such as OOH* and O*, facilitating their adsorption and activation.155,156 Li et al. developed atomically dispersed Fe atoms loaded on mesoporous nitrogen-doped carbon with accessible metal sites and optimized electronic structures (Fig. 17c).150 This material possesses a high specific surface area and mesoporosity, which facilitates mass and electron transport and improves the utilization of metal sites. The catalyst exhibits good OER activity under alkaline conditions due to optimized metal–carrier electronic interactions. Importantly, the zinc–air battery based on this catalyst exhibits a peak power density of 306.1 mW cm−2, a specific capacity of 746.9 mA h g−1, and a long cycling stability of more than 315 h, which is superior to previously reported non-precious-metal catalysts and standard Pt/C + RuO2 catalysts. Theoretical calculations suggest that electronic structure tuning can optimize the adsorption of oxygen-containing intermediates and thus improve the catalytic performance. Wu et al. prepared catalysts with atomically dispersed Ce and Co on nitrogen-doped carbon substrates by doping Ce and Co into metal–organic skeleton precursors (Fig. 17d).151 Taking advantage of the unique structure and high oxygen affinity of Ce in order to modulate the catalytic activity, the catalyst exhibited electrocatalytic performance. At a current density of 10 mA cm−2, the overpotential of the OER was 340 mV, which was lower than that of commercial IrO2 (370 mV); the half-wave potential of the ORR reached 0.79 V, which was close to that of commercial Pt/C (0.8 V). The porous structure, three-dimensional interconnected carbon network and large specific surface area of the catalyst together contributed to its catalytic performance. In the realm of Janus materials, Tang et al. embarked on a pioneering journey by utilizing the 2D MOF as a precursor to fabricate FeN3@C, followed by employing acetylacetone cobalt as a precursor to introduce Co–O3 motifs into the defects of d-FeN3@C through low-temperature pyrolysis at 330 °C.157 The low-temperature annealing process serves to govern the thermal motion of ligands, thereby preventing the aggregation of metal atoms. Meanwhile, the synergistic interplay between the adsorption forces of metal atoms and the metastable states of defect sites propels the preferential anchoring of Co–O3 motifs at defect sites. The synthesized FeCo–N3O3@C exhibits a low overpotential at a current density of 10 mA cm−2 (OER), while achieving a high half-wave potential of 0.936 V in the ORR. These exceptional performances endow it with immense potential for applications in sustainable energy conversion and storage technologies.
6.3. POM-derived catalysts for the HER
Hydrogen has been widely used in transport fuels, grid energy storage, chemical feedstocks, etc., which has driven the development of efficient and economical hydrogen production and conversion technologies. The HER is a multi-step electrochemical process. According to the pH value of the electrolyte, it may follow two different ways. The first step is electrochemical hydrogenadsorption (Volmer reaction):in acid solution| H+ + * + e− → H* |
| H2O + * + e− → H* + OH− |
In the next step, there are two different ways to produce the final product of hydrogen. If the H* coverage is low, the adsorbed hydrogen atom prefers to couple with a new electron and another proton in the electrolyte to form hydrogen. This is known as the Heyrovsky reaction,in acid solution
| H+ + e− + H* → H2 + * |
| H2O + e− + H* → * + OH− + H2 |
However, hydrogen is produced by chemical desorption (Tafel reaction) when the H* coverage is relatively high and the combination between the neighbouring adsorbed hydrogen atoms is favourable, resulting in the formation of hydrogen, in both acid and alkaline solution:
| 2H* → H2 + * |
The HER mechanism can be comprehensively optimized through hierarchical engineering of catalytic systems. Metal–organic hybrid catalysts leverage transition metals (Ni, Mo, Co) embedded in MOF/COF frameworks as proton adsorption centers, where precise coordination environments enable efficient H+ capture and activation. pH-responsive active sites, achieved by incorporating Lewis acid/base functional groups into the catalytic architecture, dynamically tune the electronic environment of metal centers to maintain high HER activity across varying pH conditions. Simultaneously, synergistic π-conjugation within the organic matrix enhances charge delocalization, significantly accelerating electron transfer kinetics and stabilizing reaction intermediates. To further optimize adsorption energetics, defect engineering and heteroatom doping introduce sulfur-doped porous carbon networks that lower the Gibbs free energy barrier for hydrogen adsorption (*H), while oxygen vacancies in MOF-derived materials promote water dissociation by weakening O–H bonds. These multiscale strategies collectively address critical challenges in the HER, including sluggish proton-coupled electron transfer kinetics and unfavorable hydrogen adsorption/desorption equilibrium, ultimately achieving efficient hydrogen production.
Hydrogen production by electrochemical water electrolysis and low-temperature membrane hydrogen fuel cells using renewable energy sources is the current focus of hydrogen energy research.158 These technologies rely on high-performance electrocatalysts to accelerate the reactions, especially in proton exchange membrane (PEM) fuel cells and water electrolysers. While proton exchange membrane technologies have made progress due to their compactness and high efficiency for the HER, their commercialization faces obstacles due to the high dependence on platinum group metals for the OER/ORR reaction. As an alternative, anion exchange membrane (AEM) technology has gained attention due to its low cost and materials suitable for oxygen reactions. However, the slower rate of hydrogen reaction in alkaline environments leads to an increased demand for platinum group metals. Therefore, there is an urgent need to develop lower-cost electrocatalysts suitable for both acidic and alkaline conditions to reduce costs and promote technology diffusion.159 ADMCs derived from POMs show great potential for enhancing the efficiency of the HER due to their unique structural and electronic properties. Tuning the electronic structure is a key means to optimize the interaction between ADMC and reactant molecules. By finely tuning the electronic properties, an optimal balance of binding strength, neither too strong nor too weak, can be achieved, thus providing ideal conditions for efficient HER. Mishra et al. prepared a Ru single-atom and Ni nanoparticle co-modified electrocatalyst (Ru1/Ni-NPGC), which was immobilised on porous graphitic carbon by controlled pyrolysis of Ni-MOF (Fig. 18a).160 The catalyst combines the unique coordination feature of Ru-N4 single-atom sites with the strong metal–carrier interactions of Ni nanoparticles, exhibiting electrocatalytic activity and long-term stability over a wide pH range. Under alkaline conditions, the overpotentials of the OER and HER were ∼195 mV and 54 mV, respectively, for achieving a current density of 10 mA cm−2. The constructed electrolytic cell could achieve a current density of 10 mA cm−2 in acidic, neutral and alkaline media with only a cell voltage of 1.5 V, 1.55 V and 1.48 V, respectively, and exhibit long-term stability at low to high current densities. This work provides a new idea for the development of highly efficient bifunctional electrocatalysts for hydrogen production from water electrolysis over the full pH range. Li et al. have devised a dual-metal MOF approach to encapsulate phosphomolybdic acid for the synthesis of P, Mo co-doped Ru ultra-small nano clusters (P,Mo-Ru@PC).161 The heterostructure coupling of the doped phosphorus porous carbon matrix with ultra-small nano clusters effectively exposes active sites and provides rapid electron transfer channels, thus facilitating a favorable HER process. Liang et al. developed a method to homogeneously immobilise Pt nanoparticles on porous nitrogen-doped carbon substrates derived from ZnCo-ZIF by exploiting the segregation of Pt by cobalt single-atom sites and the strong interactions between the two (Fig. 18b).162 This catalyst (Pt@CoSAs ZIF NC) with ultra-low Pt loading and optimized particle size not only increases the number of active centres, but also accelerates the catalytic kinetics and enhances the catalytic performance of the ORR and HER. It achieved a half-wave potential of 0.917 V in an acidic environment, which was better than that of commercial Pt/C (0.868 V), and a mass activity of 0.48 A mgPt−1 at 0.9 V, which was three times higher than that of commercial Pt/C, exceeding the target set by the U.S. Department of Energy. In addition, the mass activity was 4.5 and 13.6 times higher than that of Pt/C at 20 and 30 mV overpotentials, respectively. When applied to the seawater HER, its mass activity is about 4 times that of commercial Pt/C, showing a promising application.
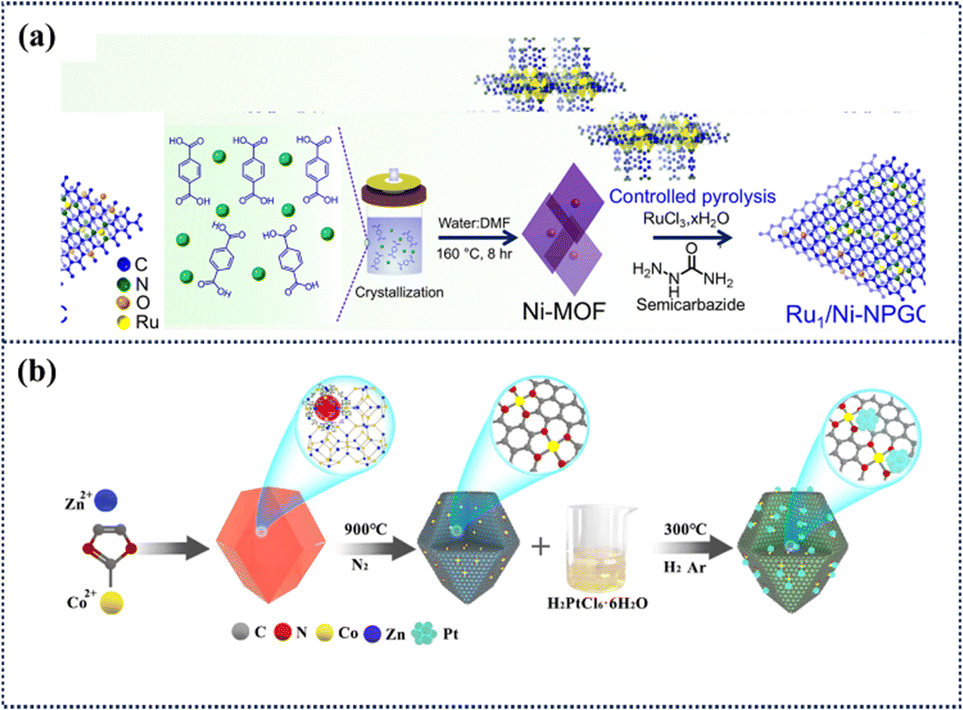 | ||
| Fig. 18 Synthesis routes for Ru1/Ni-NPGC and Pt@Co SAs-ZIF-NC catalysts. (a) Schematic representation of the synthesis procedure of Ru1/Ni-NPGC.160 Copyright 2025 RSC. (b) Schematic diagram of the synthesis for Pt@Co SAs-ZIF-NC.162 Copyright 2021 Elsevier. | ||
6.4. POM-derived catalysts for the CO2RR
In addressing the challenges of global climate change, the conversion of carbon dioxide (CO2) into valuable chemicals and fuels has become a prominent research focus. The CO2RR is a crucial technology in this field, with its efficiency and selectivity directly determining the feasibility of its commercial applications. ADMCs, derived from the meticulous design of POMs, exhibit outstanding performance in the CO2RR, providing robust catalytic support for the green conversion of CO2. The earliest application of POM-related materials in the CO2RR can be traced back to 2012 when Hinogami and colleagues creatively applied copper–yttrium salt metal–organic frameworks (CR-MOFs) to the reduction of CO2, achieving high selectivity in catalyzing the conversion of CO2 into formic acid (98%) at lower applied potentials.163 In CO2RR research, achieving the efficient and selective generation of different C1, C2, and C2+ products is a goal for scientists. However, the high chemical stability of CO2 molecules and the possibility of various competing reactions during the process present certain challenges in realizing this objective. Compared to other types of catalysts, MOF-derived materials exhibit lower overpotentials and relatively simple reaction mechanisms.164 Therefore, most MOF-derived materials tend to generate C1 products in the electrocatalytic CO2RR, mainly including CO, HCOOH, CH4, and methanol. Among these products, HCOOH and CO are the most common electrocatalytic reduction products, as they involve simple two-electron transfer processes.146 In order to enhance the selectivity and yield of CO, Lim et al. designed a stable Ni ADMC prepared from thermally activated Ni ZIF-8, which features low-coordinated Ni–Nx sites.165 By controlling the pyrolysis temperature, the catalyst's structure and composition can be optimized. Theoretical calculations and experimental results indicate that increasing the proportion of pyrrole N and reducing the coordination number of Ni help to lower the energy barrier for CO desorption. Ni ADMC-1000 exhibited a CO faradaic efficiency (FE CO) of 98.24% at −0.8 V RHE and showed enhanced current density. Combined with a flow cell system, Ni ADMC-1000 demonstrated good catalytic performance and industrial application potential. In promoting the conversion of CO2 to formic acid, Zhao et al. first reported using dilute CO2 (15% volume concentration) as a reactant for the eCO2RR (electrochemical CO2 reduction), achieving the generation of high-purity and high-concentration formic acid aqueous solution.65 The catalytic reaction was carried out on a Bi-HHTP conductive MOF (Fig. 19a), with an FE HCOOH of 93.6% in high-purity CO2 at 2.6 V and a current density of 88 mA cm−2; while in dilute CO2, the FE HCOOH was 91%, with a current density of 80 mA cm−2 (Fig. 19b and c). Specifically, the pore structure of Bi-HHTP provides abundant adsorption sites for CO2, while Bi active sites are responsible for catalyzing the conversion of CO2 to formic acid. This dual functionality enables the catalyst to perform well under dilute CO2 conditions. Huang et al. elegantly prepared a Bi-MOF using bismuth acetate and tannic acid as precursors, followed by the encapsulation of nickel ions within the MOF cavities through an ion exchange process.166 Subsequently, by subjecting the material to thermal treatment with dicyandiamide (DCDA), they successfully synthesized a bimetallic Ni/Bi–N–C catalyst. This catalyst exhibited a remarkable CO selectivity close to 100% during the process of CO2 reduction to CO, with a maximum CO partial current density reaching 312 mA cm−2, surpassing industrial standards of 200 mA cm−2. Moreover, it demonstrated exceptional stability and enduring catalytic performance. | ||
| Fig. 19 Electrocatalytic pathways and performance of MOF-based catalysts. (a) Schematic illustration of the Bi-HHTP structure. (b) LSV curves of Bi-HHTP in high-purity CO2 and dilute CO2 atmospheres, and (c) corresponding formic acid FEs and current densities under varying cell voltages in both high-purity and dilute CO2 conditions.65 Copyright 2021 Wiley. (d) Diagram of the Zn–N2S2-MOF NRR reaction pathway. (e) Schematic illustration of enhanced Zn–N2S2-MOF NRR activity achieved through increased symmetry breaking.167 Copyright 2024 Wiley. (f) Schematic illustration of the Fe2Co-MOF structure.168 (g) Comparison of the electrocatalytic performance of Fe2Co-MOF for the NO3RR with widely reported electrocatalysts. Copyright 2023 Wiley. (h) Schematic diagram of Ni-BDC@Co-HHTP heterogeneous MOF-on-MOF electrocatalyst for NO3−RR.169 Copyright 2024 Wiley. | ||
In common ADMCs, copper is considered to be the most effective catalyst for forming C2 and C2+ compounds. This is because copper exhibits moderate adsorption abilities towards *CO and *H, allowing the generation of C1 products (such as CO and formic acid) or multi-carbon compounds by adjusting the adsorption of key intermediates.170 The latter, due to their high value-added and energy density, become more desirable products. However, copper catalysts suffer from high overpotentials and low selectivity, limiting their practical application.171 Therefore, maintaining catalytic activity while enhancing the selectivity of target products is a worthwhile issue to explore. To address this challenge, Shao et al. electrochemically reduced a three-dimensional porous copper-based MOF, synthesizing low-nuclearity copper clusters-based catalysts (LNCCs) derived from POMs.172 In these catalysts, isolated copper single atoms and low-nuclearity copper clusters formed within the MOF framework retain the porosity of the MOF and introduce highly active catalytic sites, achieving high selectivity for the CO2RR to produce ethanol. Under optimal conditions, the FE of ethanol reached 82.5%, which is relatively high among similar catalysts currently available. Furthermore, copper-based materials can also modulate the distribution of CO2RR products by adjusting the coordination number of copper metal sites. Building upon Cu-Zn/MOF-74 precursors, Song et al. designed and synthesized copper single-atom catalysts (CuN3O/C and CuCO3/C) with different asymmetric atomic interfaces.173 This design enhances the catalytic efficiency of the CO2RR by precisely controlling the coordination environment of copper atoms. It is evident that well-designed atomic-scale ADMC catalysts represent a rational approach. In the future, further optimizing the CO2 capture capability of catalysts and the catalytic performance of active sites holds promise for achieving a wider range of carbon dioxide conversion applications.
6.5. POM-derived catalysts for the NRR and NOxRR
By utilizing the clean N2 and water present in the natural environment, the NRR can directly produce NH3 through photochemical and electrochemical reduction strategies. In this regard, the electrochemical NRR is more effective due to its lack of involvement in various wavelengths and rapid charge carrier recombination.174 However, due to the high stability of nitrogen molecules, efficient catalysts are usually required to lower the reaction energy barrier in NRR reactions, with most studies facing the issue of low ammonia yield.175 The unique large surface area and high porosity of POMs provide a number of exposed active sites for electron transfer, N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond cleavage, and N2 adsorption, demonstrating distinctive advantages in the field of NRR reduction.176 To enhance the intrinsic activity of active sites, Sun et al. synthesized different coordination mode Zn-MOF materials (Zn–N4-MOF, Zn–N3S-MOF, and Zn–N2S2-MOF) for electrocatalytic NRR through symmetry breaking strategies.167 Among these, Zn–N2S2-MOF exhibited an NH3 production rate of 25.07 ± 1.57 μg h−1 cm−2, with a FE of 44.57 ± 2.79%. The source of the catalyst's good activity lies in the unpaired low-valent Znδ+ in Zn–N2S2-MOF, which lowers the PDS energy barrier, suppresses the HER, promotes kinetics, and enhances NRR activity (Fig. 19d and e). This approach of breaking symmetrical POMs paves a new path for designing efficient catalysts. In terms of enhancing the catalytic activity for nitrogen fixation to ammonia, Ren et al. utilized defect-induced deposition of Ru single atoms on the UiO-66(Zr) framework, resulting in Ru1/d-UiO-66 with a catalytic yield of 53.28 μmol g−1 h−1.177 DFT results indicate the establishment of electron-metal–support interactions (EMSI) through covalent bonds between the ruthenium single atoms and the carrier, optimizing the local electronic structure of the catalyst, facilitating electron transfer, and activating N2 adsorption. Similarly, based on the Zr-oxo clusters in UiO-66, Wen et al. introduced PdCu single atoms into the MOF, simultaneously modifying it with polydimethylsiloxane (PDMS) to synthesize PdCu@UiO-S@PDMS.178 The electron transfer within PdCu NPs promotes N2 conversion, while the hydrophobic PDMS coating inhibits water interaction with PdCu, creating an ideal microenvironment in PdCu@UiO-S@PDMS that provides protons but suppresses competitive HER. Further calculations and theories suggest that the presence of copper atoms increases the electron density of palladium atoms, causing an upward shift in the d-band center of palladium atoms, thereby facilitating the reduction of the energy barrier in the NRR reaction and promoting catalytic progress. In summary, these works demonstrate that synergistically enhancing the NRR through electron transfer and modulation of POMs’ microenvironments is achievable, and rational tailoring and engineering of POMs themselves can advance nitrogen photocatalytic fixation.
N bond cleavage, and N2 adsorption, demonstrating distinctive advantages in the field of NRR reduction.176 To enhance the intrinsic activity of active sites, Sun et al. synthesized different coordination mode Zn-MOF materials (Zn–N4-MOF, Zn–N3S-MOF, and Zn–N2S2-MOF) for electrocatalytic NRR through symmetry breaking strategies.167 Among these, Zn–N2S2-MOF exhibited an NH3 production rate of 25.07 ± 1.57 μg h−1 cm−2, with a FE of 44.57 ± 2.79%. The source of the catalyst's good activity lies in the unpaired low-valent Znδ+ in Zn–N2S2-MOF, which lowers the PDS energy barrier, suppresses the HER, promotes kinetics, and enhances NRR activity (Fig. 19d and e). This approach of breaking symmetrical POMs paves a new path for designing efficient catalysts. In terms of enhancing the catalytic activity for nitrogen fixation to ammonia, Ren et al. utilized defect-induced deposition of Ru single atoms on the UiO-66(Zr) framework, resulting in Ru1/d-UiO-66 with a catalytic yield of 53.28 μmol g−1 h−1.177 DFT results indicate the establishment of electron-metal–support interactions (EMSI) through covalent bonds between the ruthenium single atoms and the carrier, optimizing the local electronic structure of the catalyst, facilitating electron transfer, and activating N2 adsorption. Similarly, based on the Zr-oxo clusters in UiO-66, Wen et al. introduced PdCu single atoms into the MOF, simultaneously modifying it with polydimethylsiloxane (PDMS) to synthesize PdCu@UiO-S@PDMS.178 The electron transfer within PdCu NPs promotes N2 conversion, while the hydrophobic PDMS coating inhibits water interaction with PdCu, creating an ideal microenvironment in PdCu@UiO-S@PDMS that provides protons but suppresses competitive HER. Further calculations and theories suggest that the presence of copper atoms increases the electron density of palladium atoms, causing an upward shift in the d-band center of palladium atoms, thereby facilitating the reduction of the energy barrier in the NRR reaction and promoting catalytic progress. In summary, these works demonstrate that synergistically enhancing the NRR through electron transfer and modulation of POMs’ microenvironments is achievable, and rational tailoring and engineering of POMs themselves can advance nitrogen photocatalytic fixation.
Compared to the slow kinetics and very low NH3 yield hindering the NRR process, NOxRR bypasses the activation of NN and complex gas–liquid–solid interface reactions, offering a more promising route for NH3 production.179,180 The NOxRR mainly involves the electrocatalytic reduction of NO3−, NO2−, and NO, but the reduction of the former often includes the latter two, so we mainly discuss the NO3−RR. Since the conversion of the NO3−RR to NH3 is a complex 9-proton-coupled 8-electron transfer reaction that involves not only the adsorption/activation of NO3− but also the hydrogenation of active hydrogen (Hads) in the process of water decomposition/reduction, the advantages of MOF-related materials with dispersed active sites, adjustable structure, and composition are highlighted.181 As a porous substrate anchoring catalytic active nanostructures, MOF structures often appear together with Cu-based materials in the field of the NO3−RR.182 As a typical common non-precious metal material, Cu-based catalysts can quickly adsorb nitrate and reduce it to nitrite, attributed to the presence of unpaired electrons in their electronic structure and the energy of the d-band being close to the LUMO (lowest unoccupied molecular orbital) π* energy level of nitrate, promoting effective electron transfer and stable adsorption of nitrate molecules.183 Combined with the UiO-66 framework, Wang et al. confined CuZn nano-clusters within its molecular selective skeleton, forming highly immobilized and intelligent channels (UiO-66-CuZn).184 Experimental findings showed that the dangling Brønsted acidic groups (COOH) in the UiO-66 framework interacted with the CuZn nano-clusters through a secondary coordination effect, which not only effectively stabilized the catalytic intermediates and promoted the overall reaction process, but also increased the proton activation energy barrier and inhibited hydrogen evolution side reactions. At a potential of −1.0 V (vs. RHE), the nitrate conversion rate, ammonia selectivity, and FE reached 97.6%, 95.2%, and 91.4%, respectively. Similarly, Liu et al. designed a dual-metal C-MOF containing Cu nanoparticles for ammonia production via NO3− electroreduction. Cu, as the main active site, plays a core role in NO3RR reactions, while Co atom doping effectively modulates the electronic structure of Cu active sites, enhancing the selectivity for the reduction of *NO2 to *NO and reducing the generation of NO2−.185 Moreover, Lv et al. developed a series of MOF structures coupling iron-based trinuclear clusters (Fe2M-MOF, M = Fe, Co, Ni, Zn) with dinitrogen ligands (Fig. 19f).168 The trinuclear clusters ensured high adsorption and reduction of nitrate, while the dinitrogen ligand (H4TPBD) promoted proton conduction and electron transfer within the framework, further enhancing catalytic efficiency. The synthesized Fe2Co could efficiently reduce nitrate to ammonia under strongly acidic conditions (pH = 1) (Fig. 19g). This overcomes the dependence of traditional biological denitrification and existing inorganic electrocatalysts under neutral/alkaline conditions, avoiding pretreatment steps and competition with hydrogen evolution side reactions. Recently, an interesting nanostructured MOF-on-MOF design has been applied in the field of NO3−RR. This special structure creates nanostructures with abundant surface metal nodes and diverse ligands, inducing interface charge redistribution and embedded electric fields by selecting different Fermi level POMs to optimize the electronic structure of active sites and accelerate charge transfer. For example, Zou et al. achieved the construction of interface dual active sites (Ni and Co) and an embedded electric field by growing Co-HHTP nanorods on Ni-BDC nanosheets (Ni-BDC@Co-HHTP) (Fig. 19h), forming a highly efficient synergistic catalytic mechanism, with an ammonia production rate of 11.46 mg h−1 cm−2 and a FE of 98.4%.169
The synergistic mechanism between POM-based ADMCs and POMs can be attributed to the dynamic interactions between the two at the atomic/electronic scale. This interaction is orchestrated through a triple-core pathway that regulates the catalytic process: firstly, the strong coordination ability of POMs can induce significant electronic structural reorganization in the metal active sites. For example, the downward shift of the d-band center of the Fe1N4O1 site leads to an increase in the occupancy of antibonding orbitals, weakening the adsorption strength of oxygen intermediates at the active site and optimizing the ORR pathway from a thermodynamic perspective.150 Secondly, the charge redistribution between atomically dispersed metal sites and the POM framework can create novel synergistic active domains. For instance, bimetallic Co/Ni sites form electron complementary structures through orbital hybridization, significantly enhancing the activation efficiency of oxygen species.186 Furthermore, the nanoscale confinement effect of POMs can stabilize the transition state configuration of crucial reaction intermediates. For instance, the Cu(I)–N3C1 site selectively stabilizes *H adsorption states through a dynamic proton-coupling mechanism,132 suppressing side reaction pathways while regulating the rate-determining steps of nitrate hydrogenation. This electron-space synergistic regulation strategy can also trigger a transition in reaction mechanisms, as exemplified by the Ru-UiO system activating the lattice oxygen participation mechanism (LOM) through strong metal–support interactions, shifting the oxygen evolution pathway from an adsorption-evolution mechanism (AEM) to a more efficient four-electron transfer channel.187 Essentially, the aforementioned interactions involve the dynamic coupling between the POM framework and the metal active centers, precisely controlling the coordination microenvironment of active sites, intermediate adsorption free energy, and reaction energy barriers to ultimately achieve a synergistic enhancement of catalytic activity and selectivity. This unique synergistic effect provides a new paradigm for key catalytic processes such as the NRR and NOxRR, breaking through the efficiency and selectivity bottlenecks of traditional catalysts through the synergistic optimization of electronic structure and microenvironment. A comprehensive analysis of the structure–function relationships of POMs in the future will propel their precise design and efficient application in fields such as sustainable ammonia synthesis.
7. SAC electrocatalysis: ML-based prediction and sieving
With the flourishing development of computational materials science, machine learning (ML) driven full-scale materials libraries are gradually becoming a powerful tool for accelerating material design, as they are less time-consuming compared to traditional experimental methods. As a potent computational tool, machine learning is a subset of artificial intelligence (AI) and is commonly used to predict various material properties such as thermal conductivity, atomic electron interactions, free energy, grain boundary energy, and crystal structure.188 Furthermore, when combined with DFT calculations, it can also be utilized to design high-performance materials by selecting material synthesis parameters, which is a crucial step in material commercialization.The fundamental process of constructing ML models, as illustrated in Fig. 20, encompasses the following key steps: the primary task is to collect data to build a training dataset; subsequently, a series of relevant mathematical descriptors encoding material properties need to be generated and refined; based on this, appropriate algorithms are chosen to construct the model; finally, a comprehensive evaluation of the model's quality and predictive ability is conducted.189 Specifically, the three main sources of material data are: structure and property databases, material property data obtained through experimental or computational means, and literature sources. It is noteworthy that regardless of whether the data come from databases or the literature, the reliability of the data needs to undergo rigorous validation. After obtaining the data, the material information needs to be transformed into mathematical forms suitable for training machine learning models and presented in the form of descriptors. Once the optimal feature subset is determined, a variety of linear and nonlinear methods can be employed to train machine learning models.190
 | ||
| Fig. 20 The development of machine learning techniques catalyzes the workflow of materials research. Copyright 2022 American Chemical Society.191 | ||
Taking the advancements in machine learning in the ORR and CO2RR as examples, efficient catalysts are required to reduce the reaction overpotential and promote reaction activity. The ORR is commonly used in clean energy production, and the active electrocatalysts involved in this process need to have a near-zero ΔGH*, as well as moderate binding energies for reaction intermediates (H*, OH*, and O*). Recently, Zhang et al. collected the O* adsorption free energy of 149 single-atom catalytic materials in the ORR process,192 calculated the theoretical limiting potentials (UL) of 31 SACs using DFT, and established five machine learning models to obtain accurate descriptors of the adsorption and catalytic performance of active carbons. It was found that the UL values of dual-metal single-atom Zn@Pc-N3C1, Au@Pd-N4, Au@Pd-N1C3, and Au@Py-N3C1 were all higher than the standard values, demonstrating the effectiveness of the ML model. Furthermore, Lin et al. developed a descriptor (ARSC) that can be used to unify various electrocatalytic reactions.189 Unlike current methods that require extensive DFT calculations, this descriptor can quickly pinpoint the optimal catalysts for various reactions. The synthesized dual-single-atom Co-Co/Ir-Qv3 was successfully verified as a high-performance bifunctional ORR/OER catalyst. Yu et al. comprehensively explored the potential of DAC catalyzing the CO2RR to C1 products on defective graphene. The ML model obtained can further be used to predict 154 potential electrocatalysts among 784 DACs with similar reverse sandwich structures.193 Similarly, Sun et al. conducted a comprehensive theoretical exploration of the complete C2 reaction path on GDY-supported ACs.194 A series of novel GDY-SACs based on transition metals, GDY-Pr, and GDY-Pm SACs, were proven to be efficient electrocatalysts for producing CH3CH2OH, CH3COOH, CH3CHO, and CH2OHCH2OH.
In conclusion, machine learning is profoundly impacting the theoretical and methodological design of materials science, particularly demonstrating immense potential in the field of electrocatalysis. However, its deep application still heavily relies on a large number of DFT calculations, and the accuracy of predictions needs to be improved. Balancing the accuracy of machine learning models with computational efficiency to accelerate the collaborative development of materials science theory and experimental techniques poses a key issue that urgently needs to be addressed. In the future, the development of more efficient algorithms is essential to drive the innovative application of machine learning in materials science.
8. Conclusions and outlooks
In conclusion, the development of efficient and sustainable energy conversion and storage technologies is of paramount importance for addressing the global energy crisis and environmental concerns. ADMC, as the most advanced atomic-level catalysts, exhibit catalytic performance due to their high density of active sites, unsaturated coordination environment, quantum-size effect, and enhanced metal–carrier interaction. The utilization of POMs as precursors and/or templates has further facilitated the controlled synthesis of ADMCs with high metal loading and excellent stability. This paper reviews various synthesis strategies for preparing ADMCs from POMs, the latest advances in characterization techniques, and the applications of derived ADMCs in multiple fields such as fuel cells and electrolyzers, exploring the structure–performance relationship of ADMCs at the atomic scale (Fig. 21). However, despite these significant advancements, challenges still exist in achieving high metal loading, preventing aggregation, and enhancing the long-term stability of ADMCs.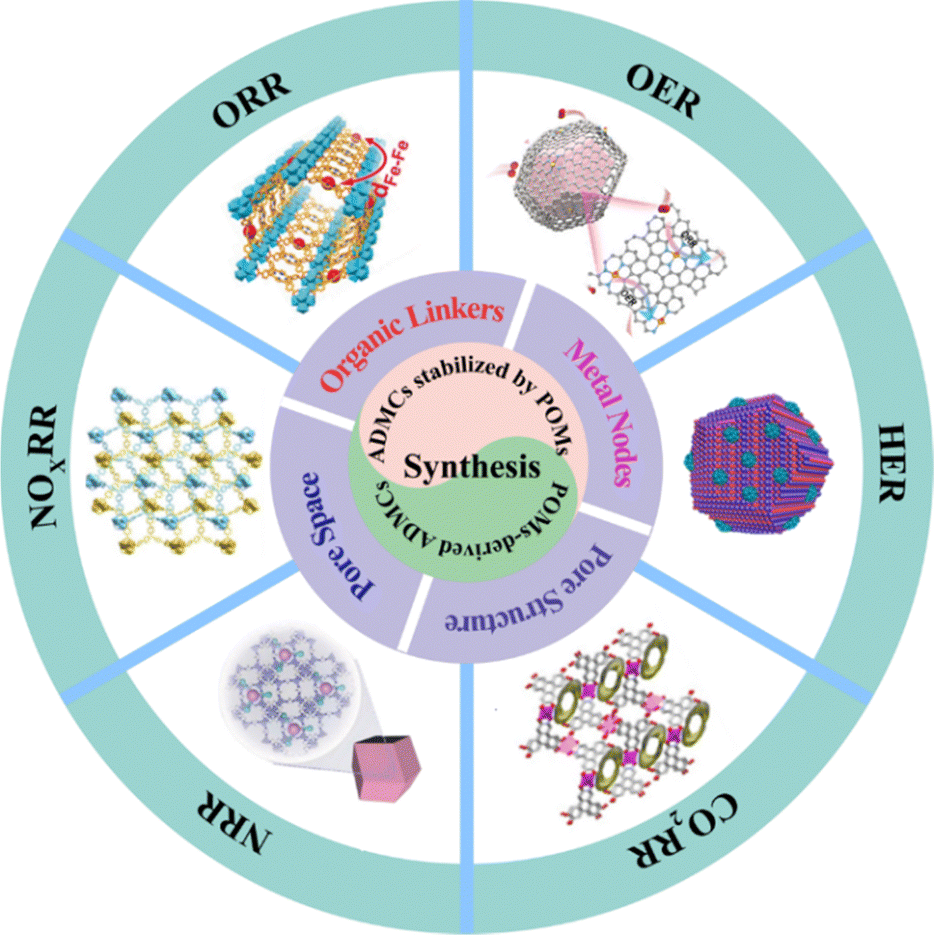 | ||
| Fig. 21 Overview of the porous organic material-based atomically dispersed metal catalysts. | ||
(1) The synthesis of MOF-derived materials typically involves intricate procedures, especially when it comes to the creation of ADMCs derived from POMs. This process necessitates precise control over the loading amount and distribution of metal atoms to prevent aggregation and maintain their high activity. The synthesis process often requires specialized equipment and conditions, resulting in high production costs. Thus, there is a need to develop more efficient and controllable synthesis methods.
(2) MOF-derived materials are susceptible to corrosion and degradation during electrochemical processes, leading to structural changes and loss of active sites. ADMCs, due to their atomic-level dispersion, are more sensitive to environmental conditions such as temperature, pH, and electrolytes. Further research is needed to enhance their stability, such as through heteroatom doping, defect engineering, and optimized preparation techniques.
(3) The electrochemical removal mechanisms of MOF-derived materials and ADMCs remain inadequately explored from a theoretical perspective, particularly concerning the electronic structure of active sites and reaction pathways. This gap in understanding constrains the optimization of their performance and the development of novel materials. Consequently, there is a critical need to intensify theoretical research and to leverage existing in situ techniques and characterization methods to advance the design and synthesis of catalysts.
(4) Future research should focus on developing innovative synthesis strategies that can precisely control the structure and composition of ADMCs at the atomic level. The integration of computational models and advanced characterization techniques will play a key role in elucidating the fundamental mechanisms of ADMCs' catalytic behavior.
Although there are still numerous issues awaiting resolution, we firmly believe that through the adjustment of process parameters and the optimization of material design, single-atom catalysts derived from POMs will garner a broader array of applications. In the future, the comprehensive exploration of the preparation, characterization, performance modulation, and mechanistic studies of POM-derived ADMCs catalysts will hold paramount practical significance. This is due to its potential to reduce costs, expand the industrial utilization of ADMCs, and facilitate the transition towards a future powered by more sustainable and renewable energy sources.
Author contributions
H. Zhang conceived the idea. H. Zhang, S. Wang and E. Lv co-wrote the paper. H. Zhang, M. Qi, C. He, X. Dong, J. Qiu, Y. Wang and Z. Wen discussed the results and revised the manuscript.Data availability
The authors declare that the data supporting the findings of this study are available in the article.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
H. Z. acknowledges the University of Oxford for the Mathematical, Physical and Life Sciences Division (MPLS) Enterprise and Innovation Fellowship and the support from the Massachusetts Institute of Technology. Y. W. acknowledges the support from the National Key R&D Program of China (2021YFB3801600) and the National Natural Science Foundation of China (22325204). X. L. D. acknowledges the support from the National Natural Science Foundation of China (U1908220). Z. H. W. acknowledges the support from the National Natural Science Foundation of China (22225902 and U22A20436) and the National Key Research & Development Program of China (2022YFE0115900 and 2021YFA1501500).References
- C. Zou, Q. Zhao, G. Zhang and B. Xiong, Nat. Gas Ind. B, 2016, 3, 1–11 CrossRef.
- M. Aminudin, S. Kamarudin, B. Lim, E. Majilan, M. Masdar and N. Shaari, Int. J. Hydrogen Energy, 2023, 48, 4371–4388 CrossRef CAS.
- N. Javed, T. Noor, N. Iqbal and S. R. Naqvi, RSC Adv., 2023, 13, 1137–1161 RSC.
- L. Lin, X. He, S. Xie and Y. Wang, Chin. J. Catal., 2023, 53, 1–7 CrossRef CAS.
- Y. Zhang, Y. Yang, B. Lu, D. Wang, X. Guo, X. Zhou and Z. Lei, Int. J. Hydrogen Energy, 2023, 66, 540–547 Search PubMed.
- E. Kianfar, Case Stud. Chem. Environ. Eng., 2023, 9, 100584 Search PubMed.
- J. Kong, H. Kim and H. S. Park, Appl. Catal., B, 2023, 338, 123019 CrossRef CAS.
- J.-T. Ren, L. Chen, H.-Y. Wang and Z.-Y. Yuan, Chem. Soc. Rev., 2023, 52, 8319–8373 RSC.
- Y. Wang, B. Seo, B. Wang, N. Zamel, K. Jiao and X. C. Adroher, Energy AI, 2020, 1, 100014 CrossRef.
- Y. Li and J. Lu, ACS Energy Lett., 2017, 2, 1370–1377 CrossRef CAS.
- S. Samantaray, D. Mohanty, I.-M. Hung, M. Moniruzzaman and S. K. Satpathy, J. Energy Storage, 2023, 72, 108352 CrossRef.
- S. Jiao, X. Fu, S. Wang and Y. Zhao, Energy Environ. Sci., 2021, 14, 1722–1770 RSC.
- T. Sun, L. Xu, D. Wang and Y. Li, Nano Res., 2019, 12, 2067–2080 CrossRef CAS.
- Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30, 2000768 CAS.
- J. Su, L. Zhuang, S. Zhang, Q. Liu, L. Zhang and G. Hu, Chin. Chem. Lett., 2021, 32, 2947–2962 CAS.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, Y. Wang, L. Jiang, Z. Wu, D.-J. Liu and L. Zhuang, Nat. Commun., 2017, 8, 15938 CAS.
- T. Sun, L. Xu, Y. Yan, A. A. Zakhidov, R. H. Baughman and J. Chen, ACS Catal., 2016, 6, 1446–1450 CAS.
- T. Tang, W.-J. Jiang, S. Niu, N. Liu, H. Luo, Y.-Y. Chen, S.-F. Jin, F. Gao, L.-J. Wan and J.-S. Hu, J. Am. Chem. Soc., 2017, 139, 8320–8328 CAS.
- C. Panda, P. W. Menezes, C. Walter, S. Yao, M. E. Miehlich, V. Gutkin, K. Meyer and M. Driess, Angew. Chem., Int. Ed., 2017, 129, 10642–10646 Search PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CAS.
- Y. Wang, H. Su, Y. He, L. Li, S. Zhu, H. Shen, P. Xie, X. Fu, G. Zhou and C. Feng, Chem. Rev., 2020, 120, 12217–12314 CAS.
- J. Zhong, Z. Liang, N. Liu, Y. Xiang, B. Yan, F. Zhu, X. Xie, X. Gui, L. Gan and H. B. Yang, ACS Nano, 2024, 18, 5258–5269 CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CAS.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- K. Du, M. Sun, J. Peng, S. Zhou, G. Sheng, R. Shen, L. Deng, C. Hu, Y. Sun and P. Zhang, Appl. Surf. Sci., 2023, 625, 157115 CAS.
- M. Humayun, M. Israr, Z. Li, W. Luo and C. Wang, Coord. Chem. Rev., 2023, 488, 215189 CAS.
- H. Li, Y. Wen, M. Jiang, Y. Yao, H. Zhou, Z. Huang, J. Li, S. Jiao, Y. Kuang and S. Luo, Adv. Funct. Mater., 2021, 31, 2011289 CAS.
- X. Wu, K. Rigby, D. Huang, T. Hedtke, X. Wang, M. W. Chung, S. Weon, E. Stavitski and J.-H. Kim, Environ. Sci. Technol., 2021, 56, 1341–1351 Search PubMed.
- C. Chen, T. Ma, Y. Shang, B. Gao, B. Jin, H. Dan, Q. Li, Q. Yue, Y. Li and Y. Wang, Appl. Catal., B, 2019, 250, 382–395 CAS.
- L. Jiao and H.-L. Jiang, Chem, 2019, 5, 786–804 CAS.
- D.-D. Ma and Q.-L. Zhu, Coord. Chem. Rev., 2020, 422, 213483 CrossRef CAS.
- H. Huang, K. Shen, F. Chen and Y. Li, ACS Catal., 2020, 10, 6579–6586 CrossRef CAS.
- O. M. Yaghi, Mol. Front. J., 2019, 3, 66–83 CrossRef CAS.
- Y. Zhao, Z. Song, X. Li, Q. Sun, N. Cheng, S. Lawes and X. Sun, Energy Storage Mater., 2016, 2, 35–62 Search PubMed.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CAS.
- S. Y. Tee, K. Y. Win, W. S. Teo, L. D. Koh, S. Liu, C. P. Teng and M. Y. Han, Adv. Sci., 2017, 4, 1600337 Search PubMed.
- O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703–706 CAS.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CAS.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 Search PubMed.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CAS.
- D. Venkataraman, G. B. Gardner, S. Lee and J. S. Moore, J. Am. Chem. Soc., 1995, 117, 11600–11601 CAS.
- K. J. Lee, Y. J. Sa, H. Y. Jeong, C. W. Bielawski, S. H. Joo and H. R. Moon, Chem. Commun., 2015, 51, 6773–6776 CAS.
- J. Duan, M. Higuchi, J. Zheng, S.-I. Noro, I.-Y. Chang, K. Hyeon-Deuk, S. Mathew, S. Kusaka, E. Sivaniah and R. Matsuda, J. Am. Chem. Soc., 2017, 139, 11576–11583 CrossRef CAS.
- K. J. Lee, J. H. Lee, S. Jeoung and H. R. Moon, Acc. Chem. Res., 2017, 50, 2684–2692 CrossRef CAS PubMed.
- W. Guo, Z. Wang, X. Wang and Y. Wu, Adv. Mater., 2021, 33, 2004287 CrossRef CAS PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- S. Xu, M. Richter and X. Feng, Acc. Mater. Res., 2021, 2, 252–265 CAS.
- K. Chi, Y. Wu, X. Wang, Q. Zhang, W. Gao, L. Yang, X. Chen, D. Chang, Y. Zhang and T. Shen, Small, 2022, 18, 2203966 CAS.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 CAS.
- X. Yan, N. Liu, W. Liu, J. Zeng, C. Liu, S. Chen, Y. Yang, X. Gui, G. Yang and D. Yu, Chem. Commun., 2024, 60, 11890–11898 Search PubMed.
- V. Hasija, S. Patial, P. Raizada, A. A. P. Khan, A. M. Asiri, Q. Van Le, V.-H. Nguyen and P. Singh, Coord. Chem. Rev., 2022, 452, 214298 CAS.
- Y. Shao, Z. Zha and H. Wang, J. Energy Chem., 2021, 63, 54–73 CAS.
- A. Kuc, M. A. Springer, K. Batra, R. Juarez-Mosqueda, C. Wöll and T. Heine, Adv. Funct. Mater., 2020, 30, 1908004 CAS.
- R.-B. Lin and B. Chen, Chem, 2022, 8, 2114–2135 CAS.
- X. Zhao, Q. Yin, X. Mao, C. Cheng, L. Zhang, L. Wang, T.-F. Liu, Y. Li and Y. Li, Nat. Commun., 2022, 13, 2721 CAS.
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 CAS.
- F. Hu, C. Liu, M. Wu, J. Pang, F. Jiang, D. Yuan and M. Hong, Angew. Chem., Int. Ed., 2017, 56, 2101–2104 CrossRef CAS PubMed.
- Z. Chen, K. O. Kirlikovali, P. Li and O. K. Farha, Acc. Chem. Res., 2022, 55, 579–591 CrossRef CAS PubMed.
- H. Dong, H. Zhu, Q. Li, M. Zhou, X. Ren, T. Ma, J. Liu, Z. Zeng, X. Luo and S. Li, Adv. Funct. Mater., 2023, 33, 2300294 CrossRef CAS.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang and X. Meng, Angew. Chem., Int. Ed., 2016, 128, 14522–14526 CrossRef.
- X. Fang, Q. Shang, Y. Wang, L. Jiao, T. Yao, Y. Li, Q. Zhang, Y. Luo and H. L. Jiang, Adv. Mater., 2018, 30, 1705112 CrossRef PubMed.
- X. Xie, L. Peng, H. Yang, G. I. Waterhouse, L. Shang and T. Zhang, Adv. Mater., 2021, 33, 2101038 CrossRef CAS PubMed.
- J. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu and Y. Pan, Nat. Catal., 2021, 4, 719–729 CrossRef CAS.
- W. Xu, R. Zeng, M. Rebarchik, A. Posada-Borbón, H. Li, C. J. Pollock, M. Mavrikakis and H. D. Abruña, J. Am. Chem. Soc., 2024, 146, 2593–2603 CrossRef CAS.
- Y. Liu, X. Wu, Z. Li, J. Zhang, S.-X. Liu, S. Liu, L. Gu, L. R. Zheng, J. Li and D. Wang, Nat. Commun., 2021, 12, 4205 CrossRef CAS.
- Y. Peng, H. Huang, Y. Zhang, C. Kang, S. Chen, L. Song, D. Liu and C. Zhong, Nat. Commun., 2018, 9, 187 CrossRef.
- K. Tan, S. Jensen, L. Feng, H. Wang, S. Yuan, M. Ferreri, J. P. Klesko, R. Rahman, J. Cure and J. Li, Chem. Mater., 2019, 31, 2286–2295 CrossRef CAS.
- P. Jiang, S. Chen, C. Wang, D. Wang, J. Diao, Z. Cao, Z. Lin, Q. Luo, J. Lu and H. Huang, Mater. Today Sustainability, 2020, 9, 100039 CrossRef.
- Y. Zhu, W. Wang, J. Cheng, Y. Qu, Y. Dai, M. Liu, J. Yu, C. Wang, H. Wang and S. Wang, Angew. Chem., Int. Ed., 2021, 133, 9566–9574 CrossRef.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265–11269 CrossRef PubMed.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang and H. Sun, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo and D. Zang, Science, 2016, 352, 797–800 CAS.
- B. Zhang, H. Asakura, J. Zhang, J. Zhang, S. De and N. Yan, Angew. Chem., Int. Ed., 2016, 128, 8459–8463 CrossRef.
- J. Sui, H. Liu, S. Hu, K. Sun, G. Wan, H. Zhou, X. Zheng and H. L. Jiang, Adv. Mater., 2022, 34, 2109203 CrossRef CAS PubMed.
- T. Rasheed and M. T. Anwar, Coord. Chem. Rev., 2023, 480, 215011 CrossRef CAS.
- S. Ding, Y. Guo, M. J. Hülsey, B. Zhang, H. Asakura, L. Liu, Y. Han, M. Gao, J.-Y. Hasegawa and B. Qiao, Chem, 2019, 5, 3207–3219 CAS.
- K. Xue, Y. Mo, B. Long, W. Wei, C. Shan, S. Guo and L. Niu, InfoMat, 2022, 4, e12296 CAS.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong and Z. Deng, Angew. Chem., Int. Ed., 2016, 128, 10958–10963 Search PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao and S. Wei, J. Am. Chem. Soc., 2017, 139, 17281–17284 CAS.
- X. Han, X. Ling, Y. Wang, T. Ma, C. Zhong, W. Hu and Y. Deng, Angew. Chem., Int. Ed., 2019, 131, 5413–5418 CrossRef.
- C. Lv, B. Li, Y. Ren, G. Zhang, Z. Lu, L. Li, X. Zhang, X. Yang and X. Yu, Chem. Eng. J., 2024, 495, 153670 CAS.
- Y. Gu, Z. Zhou, Z. Lin, X. Liu, T. Xu, W. Chen and W. Lu, Chem. Eng. J., 2023, 455, 140759 CAS.
- H. Dai, Z. Zhao, K. Wang, F. Meng, D. Lin, W. Zhou, D. Chen, M. Zhang and D. Yang, J. Hazard. Mater., 2024, 465, 133399 CAS.
- L. Liu and A. Corma, Nat. Rev. Mater., 2021, 6, 244–263 CrossRef CAS.
- S. M. Wu, X. Y. Yang and C. Janiak, Angew. Chem., Int. Ed., 2019, 131, 12468–12482 CrossRef.
- X. Wang, W. Chen, L. Zhang, T. Yao, W. Liu, Y. Lin, H. Ju, J. Dong, L. Zheng and W. Yan, J. Am. Chem. Soc., 2017, 139, 9419–9422 CrossRef CAS PubMed.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang and D. Wang, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed.
- Y. Koizumi, K. Yonesato, S. Kikkawa, S. Yamazoe, K. Yamaguchi and K. Suzuki, J. Am. Chem. Soc., 2024, 146, 14610–14619 CrossRef CAS.
- H. Li, Z. Qin, X. Yang, X. Chen, Y. Li and K. Shen, ACS Cent. Sci., 2022, 8, 718–728 CrossRef CAS PubMed.
- S. Mitchell, F. Parés, D. Faust Akl, S. M. Collins, D. M. Kepaptsoglou, Q. M. Ramasse, D. Garcia-Gasulla, J. Pérez-Ramírez and N. López, J. Am. Chem. Soc., 2022, 144, 8018–8029 CrossRef CAS PubMed.
- Y. Wang, C. Li, X. Han, J. Bai, X. Wang, L. Zheng, C. Hong, Z. Li, J. Bai and K. Leng, Nat. Commun., 2024, 15, 5675 CrossRef CAS PubMed.
- Z. W. Yin, W. Zhao, J. Li, X. X. Peng, C. Lin, M. Zhang, Z. Zeng, H. G. Liao, H. Chen and H. Lin, Adv. Funct. Mater., 2022, 32, 2107190 CrossRef CAS.
- L. Wang, J. Li, S. Ji, Y. Xiong and D. Wang, Energy Environ. Sci., 2024, 17, 8482–8528 CAS.
- Z.-Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri and F.-Y. Chen, Nat. Commun., 2021, 12, 2870 CAS.
- X. Li, X. I. Pereira-Hernández, Y. Chen, J. Xu, J. Zhao, C.-W. Pao, C.-Y. Fang, J. Zeng, Y. Wang and B. C. Gates, Nature, 2022, 611, 284–288 CAS.
- M. K. Wong, J. J. Foo, J. Y. Loh and W. J. Ong, Adv. Energy Mater., 2024, 14, 2303281 CAS.
- A. Stefancu, J. Aizpurua, I. Alessandri, I. Bald, J. J. Baumberg, L. V. Besteiro, P. Christopher, M. Correa-Duarte, B. De Nijs and A. Demetriadou, ACS Nano, 2024, 18, 29337–29379 CAS.
- X. Tang, Y. Zhang, S. Tang, D. Lützenkirchen-Hecht, K. Yuan and Y. Chen, ACS Catal., 2024, 14, 13065–13080 CAS.
- W. Shan, R. Liu, H. Zhao, Z. He, Y. Lai, S. Li, G. He and J. Liu, ACS Nano, 2020, 14, 11363–11372 CAS.
- L. Piccolo, Catal. Today, 2021, 373, 80–97 CAS.
- L. Liu, J. Liu, G. Li, X. Shi, J. Yin, S. Zheng, K. F. Yung, H. B. Yang and T. W. B. Lo, Angew. Chem., Int. Ed., 2025, e202422744 Search PubMed.
- A. Venkatesh, A. Lund, L. Rochlitz, R. Jabbour, C. P. Gordon, G. Menzildjian, J. Viger-Gravel, P. Berruyer, D. Gajan and C. Copéret, J. Am. Chem. Soc., 2020, 142, 18936–18945 CrossRef CAS PubMed.
- X. Li, C.-S. Cao, S.-F. Hung, Y.-R. Lu, W. Cai, A. I. Rykov, S. Miao, S. Xi, H. Yang and Z. Hu, Chem, 2020, 6, 3440–3454 CAS.
- M. Zhao, H. Liu, H. Zhang, W. Chen, H. Sun, Z. Wang, B. Zhang, L. Song, Y. Yang and C. Ma, Energy Environ. Sci., 2021, 14, 6455–6463 RSC.
- M. Du, B. Chu, Q. Wang, C. Li, Y. Lu, Z. Zhang, X. Xiao, C. Q. Xu, M. Gu and J. Li, Adv. Mater., 2024, 36, 2412978 CrossRef CAS.
- L. Lin, C. Zhang, C. Liang, H. Zhang, Z. Wang, P. Wang, Z. Zheng, H. Cheng, D. Xing and Y. Dai, Adv. Mater., 2024, 36, 2402388 CrossRef CAS PubMed.
- J. Zhu, J.-W. Zhao, D. Luan and X.-W. Lou, Angew. Chem., Int. Ed., 2025, e202500261 Search PubMed.
- Z. Li, X. Zhong, L. Gao, J. Hu, W. Peng, X. Wang, G. Zhou and B. Xu, ACS Nano, 2024, 18, 13006–13018 CrossRef CAS PubMed.
- J. Liu, S. Zhao, C. Wang, B. Hu, L. He, M. Wang, Z. Zhang and M. Du, Adv. Mater. Interfaces, 2022, 9, 2200913 Search PubMed.
- Y. Sun, Z. Xue, Q. Liu, Y. Jia, Y. Li, K. Liu, Y. Lin, M. Liu, G. Li and C.-Y. Su, Nat. Commun., 2021, 12, 1369 Search PubMed.
- W. Chen, J. Pei, C. T. He, J. Wan, H. Ren, Y. Wang, J. Dong, K. Wu, W. C. Cheong and J. Mao, Adv. Mater., 2018, 30, 1800396 Search PubMed.
- C.-C. Cheng, T.-Y. Lin, Y.-C. Ting, S.-H. Lin, Y. Choi and S.-Y. Lu, Nano Energy, 2023, 112, 108450 Search PubMed.
- Y. Yang, Y. Qian, H. Li, Z. Zhang, Y. Mu, D. Do, B. Zhou, J. Dong, W. Yan and Y. Qin, Sci. Adv., 2020, 6, eaba6586 CAS.
- M. Wen, N. Sun, L. Jiao, S. Q. Zang and H. L. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202318338 CAS.
- J. Zhu, M. Xiao, D. Ren, R. Gao, X. Liu, Z. Zhang, D. Luo, W. Xing, D. Su and A. Yu, J. Am. Chem. Soc., 2022, 144, 9661–9671 CAS.
- Y. N. Gong, L. Jiao, Y. Qian, C. Y. Pan, L. Zheng, X. Cai, B. Liu, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2020, 132, 2727–2731 Search PubMed.
- W. Guo, X. Tan, J. Bi, L. Xu, D. Yang, C. Chen, Q. Zhu, J. Ma, A. Tayal and J. Ma, J. Am. Chem. Soc., 2021, 143, 6877–6885 CAS.
- J. Liu, D. Yang, Y. Zhou, G. Zhang, G. Xing, Y. Liu, Y. Ma, O. Terasaki, S. Yang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 14473–14479 CAS.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CAS.
- B. Peng, H. She, Z. Wei, Z. Sun, Z. Deng, Z. Sun and W. Chen, Nat. Commun., 2025, 16, 2217 CAS.
- S.-Z. Hou, X.-D. Zhang, W.-W. Yuan, Y.-X. Li and Z.-Y. Gu, Inorg. Chem., 2020, 59, 11298–11304 CAS.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian and X. Xu, ACS Energy Lett., 2020, 5, 1044–1053 CAS.
- S. Wei, X. Jiang, C. He, S. Wang, Q. Hu, X. Chai, X. Ren, H. Yang and C. He, J. Mater. Chem. A, 2022, 10, 6187–6192 CAS.
- Y. Gu, B. Xi, W. Tian, H. Zhang, Q. Fu and S. Xiong, Adv. Mater., 2021, 33, 2100429 CAS.
- S. Zhang, M. Han, T. Shi, H. Zhang, Y. Lin, X. Zheng, L. R. Zheng, H. Zhou, C. Chen and Y. Zhang, Nat. Sustainability, 2023, 6, 169–179 Search PubMed.
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Adv. Mater., 2018, 30, 1803498 CrossRef PubMed.
- W.-D. Zhang, H. Dong, L. Zhou, H. Xu, H.-R. Wang, X. Yan, Y. Jiang, J. Zhang and Z.-G. Gu, Appl. Catal., B, 2022, 317, 121750 Search PubMed.
- Y. Xue, Q. Yu, Q. Ma, Y. Chen, C. Zhang, W. Teng, J. Fan and W.-X. Zhang, Environ. Sci. Technol., 2022, 56, 14797–14807 Search PubMed.
- Q. Li, L. Luo, X. Guo, R. Wang, J. Liu, W. Fan, Z. Feng and F. Zhang, J. Am. Chem. Soc., 2024, 147, 1884–1892 Search PubMed.
- Y. Lv, J. Su, Y. Gu, B. Tian, J. Ma, J.-L. Zuo and M. Ding, JACS Au, 2022, 2, 2765–2777 CrossRef CAS PubMed.
- C. Gong, Z. Li, K. Huang and F. Liu, Fuel, 2020, 260, 116403 CrossRef CAS.
- H. Mehrjerdi, Renewable Energy, 2020, 156, 183–192 CrossRef CAS.
- L. Valverde-Isorna, D. Ali, D. Hogg and M. Abdel-Wahab, Renewable Sustainable Energy Rev., 2016, 53, 1313–1332 CrossRef.
- I. Staffell, D. Scamman, A. V. Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- Z. Chen, P. Li, R. Anderson, X. Wang, X. Zhang, L. Robison, L. R. Redfern, S. Moribe, T. Islamoglu and D. A. Gómez-Gualdrón, Science, 2020, 368, 297–303 CrossRef CAS PubMed.
- M. D. Allendorf, Z. Hulvey, T. Gennett, A. Ahmed, T. Autrey, J. Camp, E. S. Cho, H. Furukawa, M. Haranczyk and M. Head-Gordon, Energy Environ. Sci., 2018, 11, 2784–2812 RSC.
- D. A. Gómez-Gualdrón, T. C. Wang, P. García-Holley, R. M. Sawelewa, E. Argueta, R. Q. Snurr, J. T. Hupp, T. Yildirim and O. K. Farha, ACS Appl. Mater. Interfaces, 2017, 9, 33419–33428 Search PubMed.
- B. M. Connolly, D. G. Madden, A. E. Wheatley and D. Fairen-Jimenez, J. Am. Chem. Soc., 2020, 142, 8541–8549 CrossRef CAS PubMed.
- A. Miele, J. Axsen, M. Wolinetz, E. Maine and Z. Long, Transp. Res D Transp. Environ., 2020, 81, 102275 Search PubMed.
- W. Gong, P. Guo, L. Zhang, R. Fu, M. Yan, C. Wu, M. Sun, G. Su, Y. Wang and J. Ye, Chem. Eng. J., 2025, 507, 160400 Search PubMed.
- H. Wang, C. Sun, E. Zhu, C. Shi, J. Yu and M. Xu, J. Alloys Compd., 2023, 948, 169728 CrossRef CAS.
- M. Cui, B. Xu, X. Shi, Q. Zhai, Y. Dou, G. Li, Z. Bai, Y. Ding, W. Sun and H. Liu, J. Mater. Chem. A, 2024, 12, 18921–18947 RSC.
- Q. Liu, J. Chen, L. Cao, Y. Wang, Y. Qi, Y. Wei, Q. Ma, J. Huang, X. Fan and Y. Feng, Adv. Funct. Mater., 2025, 2424597 CrossRef.
- J. Rong, J. Wu, Y. Zhang, H. Ao, X. Zheng, C. Jing, R. Yang, Z. Zhou and Z. Li, Chem. Eng. J., 2024, 502, 158080 CrossRef.
- E. P. Quirós-Díez, C. Herreros-Lucas, J. M. Vila-Fungueiriño, L. Vizcaíno-Anaya, Y. Sabater-Algarra and M. D. C. Giménez-López, Small Methods, 2024, 8, 2301805 CrossRef PubMed.
- Z. Li, S. Ji, C. Xu, L. Leng, H. Liu, J. H. Horton, L. Du, J. Gao, C. He and X. Qi, Adv. Mater., 2023, 35, 2209644 CrossRef CAS PubMed.
- K. Wu, D. Wang, Q. Fu, T. Xu, Q. Xiong, S. G. Peera and C. Liu, Inorg. Chem., 2024, 63, 11135–11145 CrossRef CAS PubMed.
- R. Cao, J. S. Lee, M. Liu and J. Cho, Adv. Energy Mater., 2012, 2, 816–829 CrossRef CAS.
- M. Mechili, C. Vaitsis, N. Argirusis, P. K. Pandis, G. Sourkouni and C. Argirusis, Renewable Sustainable Energy Rev., 2022, 156, 111970 CrossRef CAS.
- H. Hu, J. Wang, P. Tao, C. Song, W. Shang, T. Deng and J. Wu, J. Mater. Chem. A, 2022, 10, 5835–5849 RSC.
- W. H. Lai, Z. Miao, Y. X. Wang, J. Z. Wang and S. L. Chou, Adv. Energy Mater., 2019, 9, 1900722 CrossRef CAS.
- Y. Li, Y. Sun, Y. Qin, W. Zhang, L. Wang, M. Luo, H. Yang and S. Guo, Adv. Energy Mater., 2020, 10, 1903120 CrossRef CAS.
- B. Tang, Y. Zhou, Q. Ji, Z. Zhuang, L. Zhang, C. Wang, H. Hu, H. Wang, B. Mei and F. Song, Nat. Synth., 2024, 3, 1–13 CrossRef.
- N. Kakati, L. Anderson, G. Li, D. M. Sua-An, A. Karmakar, J. D. Ocon and P.-Y. A. Chuang, ACS Appl. Mater. Interfaces, 2023, 15, 55559–55569 CrossRef CAS PubMed.
- T. Tang, L. Ding, Z. C. Yao, H. R. Pan, J. S. Hu and L. J. Wan, Adv. Funct. Mater., 2022, 32, 2107479 CrossRef CAS.
- B. Mishra, S. Biswal and B. P. Tripathi, J. Mater. Chem. A, 2025, 13, 6792–6803 RSC.
- C. Li, H. Jang, S. Liu, M. G. Kim, L. Hou, X. Liu and J. Cho, Adv. Energy Mater., 2022, 12, 2200029 CrossRef CAS.
- L. Liang, H. Jin, H. Zhou, B. Liu, C. Hu, D. Chen, Z. Wang, Z. Hu, Y. Zhao and H.-W. Li, Nano Energy, 2021, 88, 106221 CrossRef CAS.
- R. Hinogami, S. Yotsuhashi, M. Deguchi, Y. Zenitani, H. Hashiba and Y. Yamada, ECS Electrochem. Lett., 2012, 1, H17 CrossRef CAS.
- J. Liu, D. Zhu, C. Guo, A. Vasileff and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700518 CrossRef.
- J. W. Lim, D. H. Choo, J. H. Cho, J. Kim, W. S. Cho, O. F. N. Okello, K. Kim, S. Lee, J. Son and S.-Y. Choi, J. Mater. Chem. A, 2024, 12, 11090–11100 RSC.
- X. Huang, S. Wu, Z. Xiao, L. Zhi and B. Wang, Nano Today, 2024, 59, 102477 CrossRef CAS.
- Y. Sun, W. Fan, Y. Li, N. L. Sui, Z. Zhu, Y. Zhou and J. M. Lee, Adv. Mater., 2024, 36, 2306687 CrossRef CAS PubMed.
- Y. Lv, S. W. Ke, Y. Gu, B. Tian, L. Tang, P. Ran, Y. Zhao, J. Ma, J. L. Zuo and M. Ding, Angew. Chem., Int. Ed., 2023, 135, e202305246 CrossRef.
- Y. Zou, Y. Yan, Q. Xue, C. Zhang, T. Bao, X. Zhang, L. Yuan, S. Qiao, L. Song and J. Zou, Angew. Chem., Int. Ed., 2024, 63, e202409799 CrossRef CAS PubMed.
- K. Xiang, F. Shen, Y. Fu, L. Wu, Z. Wang, H. Yi, X. Liu, P. Wang, M. Liu and Z. Lin, Environ. Sci.: Nano, 2022, 9, 911–953 CAS.
- J. Yu, J. Wang, Y. Ma, J. Zhou, Y. Wang, P. Lu, J. Yin, R. Ye, Z. Zhu and Z. Fan, Adv. Funct. Mater., 2021, 31, 2102151 CAS.
- B. Shao, D. Huang, R. K. Huang, X. L. He, Y. Luo, Y. L. Xiang, L. B. Jiang, M. Dong, S. Li and Z. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202409270 CAS.
- P. Song, B. Hu, D. Zhao, J. Fu, X. Su, W. Feng, K. Yu, S. Liu, J. Zhang and C. Chen, ACS Nano, 2023, 17, 4619–4628 CAS.
- X. Zhao, G. Hu, G. F. Chen, H. Zhang, S. Zhang and H. Wang, Adv. Mater., 2021, 33, 2007650 CAS.
- S. Zhao, X. Lu, L. Wang, J. Gale and R. Amal, Adv. Mater., 2019, 31, 1805367 Search PubMed.
- I. E. Khalil, C. Xue, W. Liu, X. Li, Y. Shen, S. Li, W. Zhang and F. Huo, Adv. Funct. Mater., 2021, 31, 2010052 CAS.
- G. Ren, J. Zhao, Z. Zhao, Z. Li, L. Wang, Z. Zhang, C. Li and X. Meng, Angew. Chem., Int. Ed., 2024, 63, e202314408 CAS.
- L. Wen, K. Sun, X. Liu, W. Yang, L. Li and H. L. Jiang, Adv. Mater., 2023, 35, 2210669 CAS.
- S. Wang, C. Song, Y. Cai, Y. Li, P. Jiang, H. Li, B. Yu and T. Ma, Adv. Energy Mater., 2023, 13, 2301136 CAS.
- S. Wang, Z. Zhuang, J. Xu, C. Fu, Z. Qiu, H. Feng, H. Xiang, Z. Chen, H. Li and L. Zhang, Nano Energy, 2024, 130, 110088 CAS.
- J. Xia, J. Xu, B. Yu, X. Liang, Z. Qiu, H. Li, H. Feng, Y. Li, Y. Cai and H. Wei, Angew. Chem., Int. Ed., 2024, 136, e202412740 Search PubMed.
- T. Ren, Y. Sheng, M. Wang, K. Ren, L. Wang and Y. Xu, Chin. J. Struct. Chem., 2022, 41, 2212089–2212106 CAS.
- G.-F. Chen, Y. Yuan, H. Jiang, S.-Y. Ren, L.-X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- Z. Wang, S. Liu, M. Wang, L. Zhang, Y. Jiang, T. Qian, J. Xiong, C. Yang and C. Yan, ACS Catal., 2023, 13, 9125–9135 CrossRef CAS.
- P. Liu, J. Yan, H. Huang and W. Song, Chem. Eng. J., 2023, 466, 143134 CrossRef CAS.
- Z. Li, H. He, H. Cao, S. Sun, W. Diao, D. Gao, P. Lu, S. Zhang, Z. Guo and M. Li, Appl. Catal., B, 2019, 240, 112–121 CrossRef CAS.
- N. Yao, H. Jia, J. Zhu, Z. Shi, H. Cong, J. Ge and W. Luo, Chem, 2023, 9, 1882–1896 CAS.
- C. Lv, X. Zhou, L. Zhong, C. Yan, M. Srinivasan, Z. W. Seh, C. Liu, H. Pan, S. Li and Y. Wen, Adv. Mater., 2022, 34, 2101474 CrossRef CAS.
- X. Lin, X. Du, S. Wu, S. Zhen, W. Liu, C. Pei, P. Zhang, Z.-J. Zhao and J. Gong, Nat. Commun., 2024, 15, 8169 CrossRef CAS.
- J. Liu, W. Luo, L. Wang, J. Zhang, X. Z. Fu and J. L. Luo, Adv. Funct. Mater., 2022, 32, 2110748 CrossRef CAS.
- H. Mai, T. C. Le, D. Chen, D. A. Winkler and R. A. Caruso, Chem. Rev., 2022, 122, 13478–13515 CrossRef CAS PubMed.
- X. Zhang, J. Liu, R. Li, X. Jian, X. Gao, Z. Lu and X. Yue, J. Colloid Interface Sci., 2023, 645, 956–963 CrossRef CAS.
- L. Yu, F. Li, J. Huang, B. G. Sumpter, W. E. Mustain and Z. Chen, ACS Catal., 2023, 13, 9616–9628 CrossRef CAS.
- M. Sun, H. H. Wong, T. Wu, Q. Lu, L. Lu, C. H. Chan, B. Chen, A. W. Dougherty and B. Huang, Adv. Energy Mater., 2023, 13, 2203858 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |