
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeveloping low-resistance ion migration pathways using perfluorinated chain-decorated COFs for enhanced performance in zinc batteries†
Kun
Zhang‡
 a,
Yijia
Yuan‡
a,
Gang
Wang‡
b,
Fangzheng
Chen
a,
Li
Ma
c,
Chao
Wu
d,
Jia
Liu
a,
Bao
Zhang
e,
Chenglin
Li
a,
Hongtian
Liu
a,
Changan
Lu
a,
Xing
Li
f,
Shibo
Xi
d,
Keyu
Xie
*c,
Junhao
Lin
*bg and
Kian Ping
Loh
*a
a,
Yijia
Yuan‡
a,
Gang
Wang‡
b,
Fangzheng
Chen
a,
Li
Ma
c,
Chao
Wu
d,
Jia
Liu
a,
Bao
Zhang
e,
Chenglin
Li
a,
Hongtian
Liu
a,
Changan
Lu
a,
Xing
Li
f,
Shibo
Xi
d,
Keyu
Xie
*c,
Junhao
Lin
*bg and
Kian Ping
Loh
*a
aDepartment of Chemistry, National University of Singapore, Singapore, Singapore. E-mail: chmlohkp@nus.edu.sg
bDepartment of Physics and Shenzhen Key Laboratory of Advanced Quantum Functional Materials and Devices, Southern University of Science and Technology, Shenzhen, China. E-mail: linjh@sustech.edu.cn
cState Key Laboratory of Solidification Processing, Center for Nano Energy Materials, Northwestern Polytechnical University and Shaanxi Joint Laboratory of Graphene (NPU), Xi’an, China. E-mail: kyxie@nwpu.edu.cn
dInstitute of Chemical and Engineering Sciences, Agency of Science Technology and Research, 1 Pesek Road, Jurong Island, Singapore, Singapore
eSchool of Materials and Energy, University of Electronic Science and Technology of China, Chengdu, Sichuan, China
fDepartment of Chemistry, State Key Laboratory of Marine Pollution, City University of Hong Kong, Hong Kong, China
gQuantum Science Center of Guangdong-Hong Kong-Macao Greater Bay Area (Guangdong), Shenzhen, 518045, People's Republic of China
First published on 20th March 2025
Abstract
Rechargeable aqueous zinc metal-based batteries present a promising alternative to conventional lithium-ion batteries due to their lower operating potentials, higher capacities, intrinsic safety, cost-effectiveness, and environmental sustainability. However, the use of aqueous electrolyte in zinc metal-based batteries presents its own unique set of challenges, which include the tendency for side reactions during discharge that encourages dendritic growth on Zn anodes, as well as sluggish kinetics caused by the large solvation shell of divalent Zn ions. Nanoporous materials can be deployed as coating on Zn anodes for enhancing both their performance and stability, particularly in addressing challenges associated with water reactivity and ion migration kinetics. In our study, we incorporated superhydrophobic fluorine chains into covalent organic frameworks (SPCOFs) to engineer nanochannels that facilitate efficient ion migration pathways. Molecular dynamics simulations demonstrate that these superhydrophobic fluorine chains significantly reduce interactions between the electrolyte and nanochannel walls, altering the confined electrolyte distribution. This modification enables rapid dehydration, reduces ion migration resistance, and promotes dense Zn deposition. The use of SPCOFs enable Zn batteries with exceptional stability, achieving over 5000 hours of runtime at high current densities and stable cycling across 800 cycles in full-cell configurations. This approach highlights the critical role of tailored nanochannel environments in advancing the functionality and durability of zinc metal-based batteries, offering a scalable and environmentally friendly alternative to traditional battery technologies.
Broader contextAs the global push for renewable energy gains momentum, there is a critical need for advancements in energy storage technologies that are both efficient and environmentally sustainable. Zinc-based batteries are stepping up as a formidable alternative to traditional lithium-ion batteries, lauded for their safety and cost-effectiveness. Yet, their broader application has been curtailed by persistent challenges such as dendritic growth and inefficient ion transport. Our research introduces a pioneering solution involving the use of superhydrophobic perfluoro chain-decorated covalent organic frameworks (SPCOFs). These frameworks are meticulously engineered to optimize ion migration pathways within zinc batteries, promoting rapid ion transport and consistent zinc deposition. This approach dramatically diminishes the resistance to ion migration, significantly boosting the battery's stability and longevity. Moreover, our study delves into the intricacies of ion interaction and distribution within sub-nanochannels, marking a notable first in this area of research. These enhancements could revolutionize zinc battery technology, positioning it as a cornerstone for future, more sustainable, and widely accessible energy storage systems. |
Introduction
Rechargeable aqueous zinc metal-based batteries (AZBs) have emerged as a promising alternative to traditional lithium batteries, offering advantages such as low operating potential (−0.762 V vs. the standard hydrogen electrode, SHE), high volumetric and specific capacities (5855 A h L−1 and 820 mA h g−1, respectively), enhanced safety, cost-effectiveness, and environmental sustainability.1,2 However, unstable anode surfaces and uncontrolled dendrite growth lead to rapid battery failure, posing significant challenges to practical application.3,4 Key issues include water decomposition-induced side reactions—such as the hydrogen evolution reaction (HER) and pH fluctuations5,6—as well as sluggish charge carrier diffusion caused by large hydrated ions, as illustrated in Fig. 1a. To address these limitations, strategies involving advanced electrolytes, robust solid electrolyte interphases (SEIs), and separator modifications have demonstrated effectiveness in suppressing water reactivity and improving ion transport. These approaches include tailoring solvation structures to facilitate desolvation, enhancing electrolyte–electrode wettability, and optimizing interfacial kinetics.7–11 Recent research has focused on utilizing nanoporous materials with highly ordered sub-nanochannels, such as covalent organic frameworks, to regulate ion and water migration behaviors and enhance the performance of AZBs.8,12–15 However, the intricate relationship between nanochannel structural properties, ion/water migration, and AZB performance remains poorly understood, necessitating further exploration and understanding. | ||
| Fig. 1 Schematic illustrations showing Zn interface with different interface. Schematic illustrations depicting various ion migration mechanisms and the Zn deposition process on different surfaces, along with the corresponding ion concentration profiles near the electrode surface simulated using COMSOL. (a) Bare Zn. (b) Zn with nanochannel interface (d < 5 nm). (c) Low surface energy nanochannel interface (d < 5 nm). | ||
Generally, the migration of Zn ions in the electrolyte across the nanochannel involves a dehydration and migration process. The predominant ion migration mechanism in sub-nanochannels is characterized by a ‘sites hopping’ pattern,8,13 as shown in Fig. 1b. In this process, partially dehydrated ions and water molecules adsorb on specific zincophilic sites and traverse the one-dimensional channels. However, such a migration mode faces considerable flow frictions due to the strong interactions between the electrolyte and the channel, resulting in increased energy barriers and resistance experienced by the electrolyte ions.16–18 Moreover, the functional groups used in sub-nanochannels, such as low-density doping atoms or small groups like –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –C–F, –COOH, and –SO3, were low-density and small-functional-area, decrease efficiency in improving ion transport or suppressing of dendritic growth.8,12,13 Therefore, there is a need to explore effective channel functionalization approaches. Inspired by the ultrafast solution permeation observed in aquaporins,19 which is facilitated by their hydrophobic interior surface, recent studies have focused on enhancing rapid water permeation by modifying water–wall interactions within nanochannels. These modifications include creating superhydrophobic regions within nanotubes or constructing fluorous oligoamide nanorings.20,21 By reducing interfacial frictions between water and the channel through a dewetting effect, the transport of solution within the nanochannels can be accelerated.21 Although this approach holds promise for addressing the slow ion migration kinetics in AZBs, it remains unexplored.
O, –C–F, –COOH, and –SO3, were low-density and small-functional-area, decrease efficiency in improving ion transport or suppressing of dendritic growth.8,12,13 Therefore, there is a need to explore effective channel functionalization approaches. Inspired by the ultrafast solution permeation observed in aquaporins,19 which is facilitated by their hydrophobic interior surface, recent studies have focused on enhancing rapid water permeation by modifying water–wall interactions within nanochannels. These modifications include creating superhydrophobic regions within nanotubes or constructing fluorous oligoamide nanorings.20,21 By reducing interfacial frictions between water and the channel through a dewetting effect, the transport of solution within the nanochannels can be accelerated.21 Although this approach holds promise for addressing the slow ion migration kinetics in AZBs, it remains unexplored.
In this study, we modified the nanochannels within COFs by incorporating superhydrophobic fluorine chains to construct SPCOFs. The introduction of fluorine effectively reduced the surface energy of the nanochannels, tailoring the interactions between the electrolyte solution and the channel walls to facilitate rapid dehydration. Through molecular dynamics (MD) simulations, we further observed a unique ion migration behavior in SPCOFs: the dehydrated Zn solvent sheath detached from the channel walls and occupied larger central areas within the unwetted nanochannels. This structural adjustment is crucial for reducing resistance to ion migration. SPCOF-modified Zn anode (SPCOF@Zn) exhibited exceptional stability and kinetics, achieving an impressive runtime of 5000 hours at a high current density of 10 mA cm−2. Furthermore, when the SPCOF@Zn anode was incorporated into a Zn||ZVO full cell, stable cycling was maintained for over 500 cycles and up to 800 cycles at low N/P ratios of approximately 2 and 4, respectively. These results highlight the significant potential of this innovative approach for practical battery applications.
Design principle for interface nanochannel
The primary objective of developing a protective layer for the zinc anode is to achieve the dual purpose of enhancing the mobility of zinc ions and also to prevent water infiltration, the latter is needed to ensure corrosion-resistant zinc deposition. To improve the mobility of Zn ions, it is essential to reduce the size of the solvation shell. Desolvation can be induced in a narrow channel with a diameter comparable to the Debye length of zinc ions in an aqueous electrolyte.22–24The Debye length of zinc ions in a 2 M ZnSO4 electrolyte can be calculated as follows:
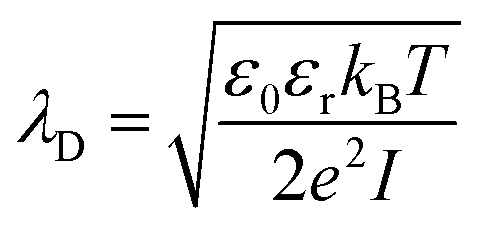 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) F m−1), εr is the relative permittivity of water (approximately 78.5 at room temperature), kB is the Boltzmann constant (1.381 × 10−23J K−1), T is the temperature in Kelvin (typically 298 K for room temperature), e is the elementary charge (1.602 × 10−19), I is the ionic strength in mol m−3. The Debye length of Zn ions in electrolyte (2 M ZnSO4) can be calculated as ∼2.14 nm.23
F m−1), εr is the relative permittivity of water (approximately 78.5 at room temperature), kB is the Boltzmann constant (1.381 × 10−23J K−1), T is the temperature in Kelvin (typically 298 K for room temperature), e is the elementary charge (1.602 × 10−19), I is the ionic strength in mol m−3. The Debye length of Zn ions in electrolyte (2 M ZnSO4) can be calculated as ∼2.14 nm.23
Fig. 1 illustrates three zinc anode scenarios: (a) a bare Zn anode, (b) a Zn anode coated with nanochanneled porous layers under 5 nm in diameter (∼2 times Debye length), and (c) a similar setup with low-surface-energy channels. In Fig. 1a, migration of Zn ions in the bare Zn anode is influenced by electromigration, this usually contributes to a multitude of issues such as uncontrolled deposition, electrode corrosion and HER, which promotes zinc dendrite formation. Fig. 1b demonstrates the dehydration of Zn ions using sub-5 nm nanochannels in the porous layers, where 5 nm is close to the Debye screening length in 2 M ZnSO4 solution. The migration of zinc ions occurs via a 'site hopping' mechanism, in which the interaction between electrolyte and partially wetted nanochannel imparts resistance to the flow.16–18Fig. 1c illustrates the case where a low-surface-energy wall (via inner fluorinated organic chains) reducing the electrolyte-channel interaction, thus promoting fast ion migration and dense zinc deposition.
Synthesis and characterization of SPCOF
Here, we designed a tritopic aldehyde COF precursor integrated with a perfluorohexyloxy chain, which enables us to make a highly crystalline, SPCOF. The synthesis process of these SPCOFs is illustrated in Fig. 2a, with further details provided in the experimental sections (Fig. S1 and S2, ESI†). As a control study, COF without the perfluorohexyloxy side chains, named here as SHCOF, were also synthesized. Nitrogen sorption experiments conducted at 77 K were utilized to evaluate the porosity of these COFs. The results are shown in Fig. S3 (ESI†), and the pore sizes were verified by non-local density functional theory calculations (Fig. 2b and d). The results reveal that SPCOF, modified with bulky perfluorohexyloxy side chains, exhibited a reduced pore size of approximately 1.40 nm, in contrast to the larger pore size of SHCOF at 1.74 nm. Adsorption–desorption isotherms using Argon at 87 K also demonstrate pore size distribution of SPCOF (Fig. S4a and b, ESI†). The porosity of SPCOF is further demonstrated by CO2 uptake tests. As is shown in Fig. S4c (ESI†), SPCOF demonstrate CO2 uptake capacities of 1.31 mmol g−1 at 273 K. The Brunauer–Emmett–Teller (BET)-derived pore diameter of SPCOF agrees well with that determined by HRTEM images. The smaller pore size in SPCOF is attributable to the perfluorohexyloxy chains integrated within the COF matrix. Moreover, this incorporation led to pore filling in SPCOF, resulting in a diminished BET surface area relative to SHCOF. Both COFs manifested pore sizes below the Debye length of Zn ions in the electrolyte.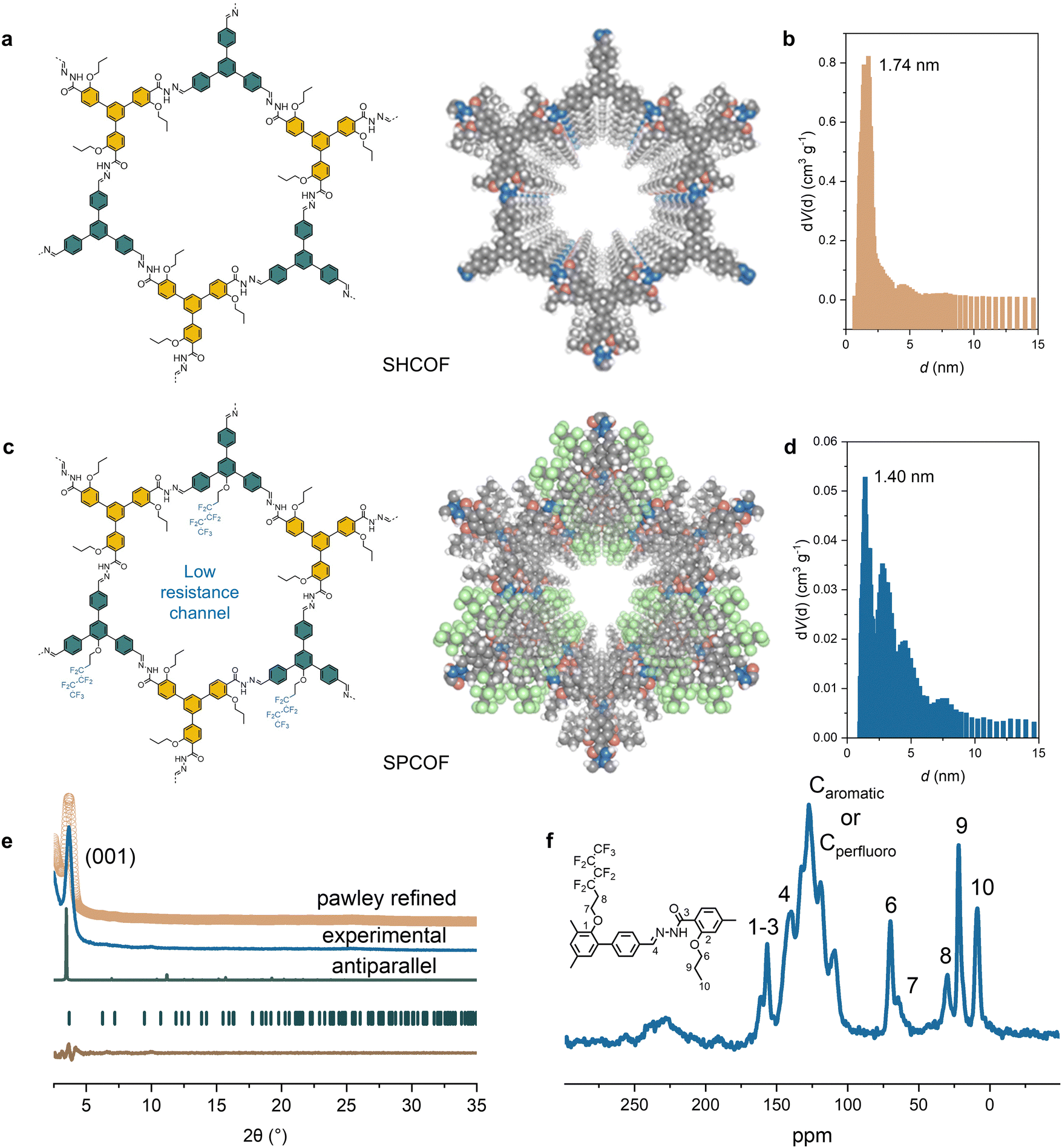 | ||
| Fig. 2 Synthesis and characterization of SPCOF and SHCOF. (a and b) Structure of SHCOF and corresponding pore size distribution. (c and d) Structure of SHCOF and corresponding pore size distribution. (e) XRD pattern and structure determination of SPCOF. (f) 13C CP-MAS solid-state NMR spectrum of SPCOF, with 13C chemical shift assignments indicated in the chemical structure. | ||
Both SPCOF and SHCOF displayed excellent crystallinity and an antiparallel-stacked architecture, as confirmed by powder X-ray diffraction (PXRD) analyses (Fig. 2e and Fig. S5 and S6, ESI†). The PXRD patterns for SPCOF revealed peaks at 3.66°, 6.35°, and 7.20°, correlating to the (100), (110), and (200) planes, respectively. Similarly, SHCOF exhibited peaks at 3.52°, 6.12°, and 7.08°, corresponding to these planes. Theoretical simulations of eclipsed, staggered, and antiparallel stacking configurations were conducted, with the antiparallel structures showing greater congruence with the experimental PXRD result. SPCOF is characterized by antiparallel stacking within a P3/C1 space group and refined cell parameters of a = 29.40 Å, b = 29.40 Å, c = 9.37 Å, α = γ = 90°, and β = 120°. In comparison, SHCOF, also exhibiting antiparallel stacking, possesses a slightly tilted unit cell within the same space group, with parameters of a = 30.14 Å, b = 30.14 Å, c = 7.37 Å, α = γ = 90°, and β = 120°.
The structural integrity and successful synthesis of SPCOFs were rigorously validated through Fourier-transform infrared spectroscopy (FT-IR) and solid-state nuclear magnetic resonance (NMR) spectroscopy (Fig. 2f and Fig. S7 and S8, ESI†). The FT-IR spectra featured a distinct peak at approximately 1155 cm−1, characteristic of C–F bond vibrations,25 affirming the successful integration of perfluorohexyloxy chains into the COF structure. Further confirmation was provided by solid-state cross-polarization/magic-angle-spinning (CP/MAS) 13C NMR spectroscopy, where peaks around 140 ppm indicated the carbon atoms involved in forming hydrazone linkages. Additional peaks ranging from 108.4 to 145.9 ppm were ascribed to the carbons in the perfluoro-chains and aromatic carbons within the COF framework. Notably, compared to SHCOF, SPCOF exhibited new peaks at higher fields, corresponding to the carbons on the perfluoro-chains.
The high crystallinity of SPCOF and SHCOF were validated using low-dose cryogenic transmission electron microscopy (cryo-TEM). Specifically, SPCOF and SHCOF were exfoliated using ethanol-assisted liquid sonication, and the thin flakes obtained were dispersed onto lacey carbon films. These samples were subsequently frozen rapidly at liquid nitrogen temperatures (∼77 K) for TEM analysis. The low-magnification cryo-TEM images shown in Fig. S9a and S10a (ESI†) display the characteristic morphology of large SPCOF and SHCOF nanosheets obtained by ultrasonic exfoliation. The high-resolution TEM results in Fig. S7b and S8b (ESI†) indicate that SPCOF and SHCOF share similar structural characteristics, with their nanosheets typically stacking in random orientations. This is reflected in the FFT patterns, which show sharp, concentric ring patterns, suggesting that these nanosheets have the same crystal structure but stack with isotropic orientations. The measured first-order reflection ring spacings for SPCOF and SHCOF are 0.39 nm−1 and 0.38 nm−1, respectively, consistent with the (100) series crystal planes determined by PXRD.
Although SPCOF and SHCOF show similar structure skeleton in the FFT analysis, the slight structural characteristic caused by the superhydrophobic perfluorohexyl chains can be distinguished in high-resolution TEM (HRTEM) images in real space. As shown in the selected area HRTEM image in Fig. 3a, well-aligned single-crystal regions were observed in certain small areas of SPCOF, captured at approximately 80 K with a cumulative electron dose below 100 e− Å−2. The high-resolution micrograph along the [001] axis indicate that the nanopores in SPCOF are arranged in a triangular pattern. The (100) facets are marked by orange lines and have an interplanar spacing of 2.55 nm, corresponding to the reflection spot indicated by a blue circle in the FFT (inset). The magnified HRTEM image of SPCOF and the overlaid atomic structure model in Fig. 3b provide a clearer depiction of the pore structure formed by interlaced carbon atom chains. Simulated HRTEM image using the SPCOF model at an under focus value of 750 nm matches well with the experimental one (Fig. 3c and Fig. S11, ESI†). As a comparison, the experimental HRTEM images of SHCOF, shown in Fig. 3d–f and Fig. S12 (ESI†), show structural pore periodicity similar to SPCOF. However, SHCOF exhibits a slightly larger interplanar spacing of 2.6 nm and a pore diameter of approximately 1.7 nm, while 1.4 nm in SPCOF. This is strong structural evidence that the perfluorohexyl chains have modify the morphology of the pores, consistent with our previous explanations. In addition, low-temperature electron energy loss spectroscopy (cryo-EELS) confirmed the presence of perfluorohexyl chains in SPCOF, as evidenced by the characteristic edges of C, N, O, and F (the fingerprint of perfluorohexyl chains), as shown in Fig. 3g with uniform distribution Fig. S9c (ESI†). In contrast, the EELS spectrum and elemental maps of SHCOF demonstrate the absence of F signal (Fig. 3h and Fig. S10c, ESI†). The HRTEM-derived pore diameter of SPCOF agrees well with that determined by BET tests. The smaller pore size in SPCOF is due to the perfluorohexyloxy chains integrated within the COF matrix, resulting in a smaller BET surface area relative to SHCOF. Both COFs manifested pore sizes below the Debye length of Zn ions in the electrolyte.
 | ||
| Fig. 3 Microscopic structure and elemental characterization of SPCOF and SHCOF. (a and d), High-resolution lattice fringe images of SPCOF (a) and SHCOF (d) taken along the [001] zone axis at approximately 80 K, with a cumulative electron dose of less than 100 e− Å−2. The insets display their respective fast Fourier transform patterns. (b and e) Magnified high-resolution lattice fringe images of SPCOF (b) and SHCOF (e), with their respective atomic structure models overlaid. (c and f), Simulated high-resolution TEM images showcasing structures and contrasts consistent with the experimental results. (g) The EELS spectrum of SPCOF shows distinct onset features of C–K, N–K, O–K and F–K edges. The inset provides an enlarged view of the F–K edge. (h) The EELS spectrum of SHCOF indicates the presence of C, N and O elements, while no signal for F is detected. | ||
Fast dehydration of SPCOF layer
The COF layer was applied to the Zn anode using blade coating and rolling to make a dense SPCOF layer approximately 10 μm thick, as shown in the SEM images in Fig. S13 (ESI†). Thermogravimetric Analysis (TGA) curves of SHCOF and SPCOF demonstrate the thermal stability of samples during electrode modification and battery measurement (Fig. S14, ESI†). The modified electrodes, designated as SPCOF@Zn and SHCOF@Zn, were evaluated for their anti-corrosion properties. The low surface energy of the SPCOF layer effectively shields the Zn surface from direct water contact. This is evidenced by the high contact angles observed in digital images and measured in contact angle tests, as detailed in Fig.s S15 and S16 (ESI†). Durability tests in 2 M ZnSO4 solutions for 7 days revealed the largest amount of granular corrosion products on the bare Zn surface, followed by SHCOF@Zn anode, but notably absent on the SPCOF@Zn electrode, as confirmed by SEM in Fig. S17 (ESI†). Linear sweep voltammetry (LSV) in 1 M Na2SO4 electrolyte showed that SPCOF@Zn had the lowest current density, indicative of superior corrosion resistance (Fig. S18, ESI†).26 This is corroborated by LSV tests in 2 M ZnSO4 using a symmetrical battery setup, in which SPCOF@Zn exhibited a high corrosion potential of 22 mV and a low corrosion current of 10.07 mA cm−2, surpassing both SHCOF@Zn and bare Zn anodes (Fig. S19, ESI†). The dewetting capabilities of the nanochannels were also tested by using in situ gas chromatography (GC) to measure hydrogen production during zinc stripping/plating in symmetric cells (Fig. 4a). As a direct evidence of low HER activity (i.e. low water activity), SPCOF@Zn maintained hydrogen production below 2 ppm at a current density of 1 mA cm−2 and capacity of 1 mA h cm−2, in contrast to SHCOF@Zn and bare Zn electrodes that showed higher hydrogen concentration of 30–40 ppm and over 50 ppm, respectively. | ||
| Fig. 4 Dehydration behavior of SPCOF based interface. (a) In situ electrochemical GC profiles for different electrode cells at 5 mA cm−2 and 5 mA h cm−2. (b and c), XANES spectra and d Fourier-transformed EXAFS spectra comparing bulk electrolytes with electrolytes within COF layers. (d), RDF of zinc ion and oxygen in SPCOF confined electrolyte. (e) Comparison between coordination number of zinc ion and oxygen in different electrolyte. (f) Statistical binding energy in the simulated system between Zn2+ and H2O in different electrolyte. | ||
X-ray absorption near-edge structure (XANES) and Raman spectroscopy were utilized to examine the functionality of perfluorohexyl chains in SPCOFs and the Zn2+ solvation configuration in cycled electrodes (Fig. 4b and Fig. S20, ESI†). Pre-cycling electrodes were encased within battery cases sealed with Kapton tape to prevent water vaporization (Fig. S21, ESI†). XANES analysis revealed a shift towards lower energy levels in the electrolytes within COFs compared to pristine ZnSO4, indicating inhibited electron transfer from Zn to oxygen in water, which was supported by k3-weighted extended X-ray absorption fine structure (EXAFS) analysis (Fig. 4c). The primary Zn–O peak at 1.66 Å showed a slight redshift, confirming reduced oxygen coordination around Zn2+.27,28 The weak Zn–O interactions in the SPCOF layers reflects the desolvation of hydrated Zn ions, as corroborated by redshifted Raman peaks around 990 cm−1, indicative of lower water content in Zn solvation structures.29,30 MD simulations provided atomic-level insights into the structure of the solvation shell around the Zn ions, with radial distribution function (RDF) profiles and coordination numbers detailed in Fig. 4d and e and Fig. S22 and S23, ESI.† Confined electrolytes in SPCOF showed a pronounced peak at approximately 2.1 Å, indicating desolvation of Zn2+ from water, with significant reductions in coordination number and binding energy, enhancing the dehydration propensity of electrolytes within SPCOF channels.31,32 Electrochemical stability windows (ESW) evaluated in Fig. S24 (ESI†) showed that the SPCOF layer improved the ESW of ZnSO4 electrolyte to 3.1 V, significantly inhibiting side reactions like HER and OER, thus enhancing zinc battery cycle stability.
Low resistance Zn ions migration mechanism in SPCOF nanochannel
Ion migration dynamics within COF nanochannels were analyzed using density mappings of Zn2+, SO42−, and H2O (Fig. 5a–d and Fig. S25–S27, ESI†). In SHCOF, the adsorption of Zn2+ on zincophilic sites on the wall and the increased H2O density near the walls indicated a site-hopping migration mechanism. Conversely, in SPCOF channels, the presence of fluorine chains caused a more uniform Zn2+ distribution in the center of the channel, effectively reducing wall wetting and enhancing ion mobility. As illustrated in Fig. 5e and f, the migration behavior of partially hydrated Zn ions within the COF channel is governed by several forces: Fdrive propels the ion along the surface, while Fresistance opposes this motion. Fdrive is the electric force during charging and discharging processes. Considering the electrolyte in the nanochannel as consisting of several small electrolyte droplets, the main resistances include the viscous force between the electrolyte and the nanochannel Fviscous and the interfacial friction Fresistance occurring during this process.33,34 Thus, Fresistance can be expressed as:| Fresistance = Fviscous + Ffriction | (2) |
 | (3) |
 | ||
| Fig. 5 Low resistance migration mechanism of Zn ions in SPCOF channel. (a and b), 2D density contour displaying the distribution of Zn2+ in different COF channels: (a) Zn2+ in SHCOF; (b) Zn2+ in SPCOF. (c and d), 2D density contour displaying the distribution of H2O in different COF channels: (c) H2O in SHCOF; (d) H2O in SPCOF. (e and f) Schematic illustration of electrolyte immersion state in different COFs and force analysis: (e) SHCOF; (f) SPCOF. (g) Statistical binding energy in the simulated system between Zn2+/SO42−/H2O and different COFs. (h) Potential mean force as a function of the distance for Zn ions moving from the bulk solution to cross the nanochannel. (i) Simulated snapshots showing the migration of Zn ion across SPCOF. (j) Mean square displacement of Zn2+ within the SPCOF channel and COF channel. | ||
The reduction in viscous forces (Fviscous) of the electrolyte in SPCOF is reflected by a decrease in the statistical binding energy between the COF skeleton and the electrolyte species (Zn2+, SO42+, and H2O), detailed in Fig. 5g. The binding energies within the SPCOF-confined electrolyte are 1.24, 0.21, and 1.10 kcal mol−1, respectively, which are significantly lower than those observed in SHCOF, facilitating lower resistane ion migration. The potential mean force (PMF) analysis for Zn ions traversing few-layered COFs shows that they face a lower free energy barrier in SPCOF than in SHCOF, aided by fluorine chains that guide Zn ion migration along lower resistance pathways (Fig. 5h and i). This effect is further supported by tracking the mean square displacements (MSD) of Zn2+, SO42−, and H2O within various COF channels (Fig. 5j and Fig. S28, ESI†), revealing notably higher diffusion rates in SPCOF layers, thereby demonstrating the structural and chemical properties that enhance mobility.
Stabilized Zn metal anode with SPCOF
To verify that the low-resistance nanochannels in the SPCOF layer enhance fast ion migration, the activation energy (Ea) for the mass transfer of Zn ions was determined using the Arrhenius equation (Fig. 6a). The charge transfer resistance of Zn||Zn symmetric batteries was measured using electrochemical impedance spectroscopy (EIS) tests conducted across a temperature range of 25–75 °C, as illustrated in Fig. S29 (ESI†). SPCOF@Zn electrode exhibits a lower activation energy (0.125 eV) compared to SHCOF@Zn (0.270 eV) and bare Zn electrode (0.315 eV). This result implies faster Zn ion transport within the frictionless nanochannels provided by the SPCOF layer. Besides, SPCOF@Zn electrode also demonstrates Zn ion transference number of 0.425 (Fig. 6b), which is much higher than that of SHCOF@Zn electrode (0.308) and bare Zn electrode (0.199) (Fig. S30, ESI†). Cyclic voltammetry (CV) curves were collected to investigate the Zn plating/stripping process on both bare Ti foil and Ti foil coated with various COFs, as depicted in Fig. 6c. All electrodes exhibited reversible oxidation and reduction peaks, corresponding to the zinc plating and stripping processes, respectively. Notably, the SPCOF@Ti electrode demonstrated a distinct nucleation current below 0 V, indicating the lowest barrier for Zn migration and deposition. Furthermore, as is shown in Fig. S31 (ESI†), the highest ionic conductivity of SPCOF reaches 0.0589 S cm−1, which is higher than that of SHCOF (0.0225 S cm−1) and GF/D separator (0.0255 S cm−1). This observation provides additional evidence of the decreased resistance in SPCOFs, which facilitate efficient and rapid ion transport, ultimately enhancing the overall performance of the battery.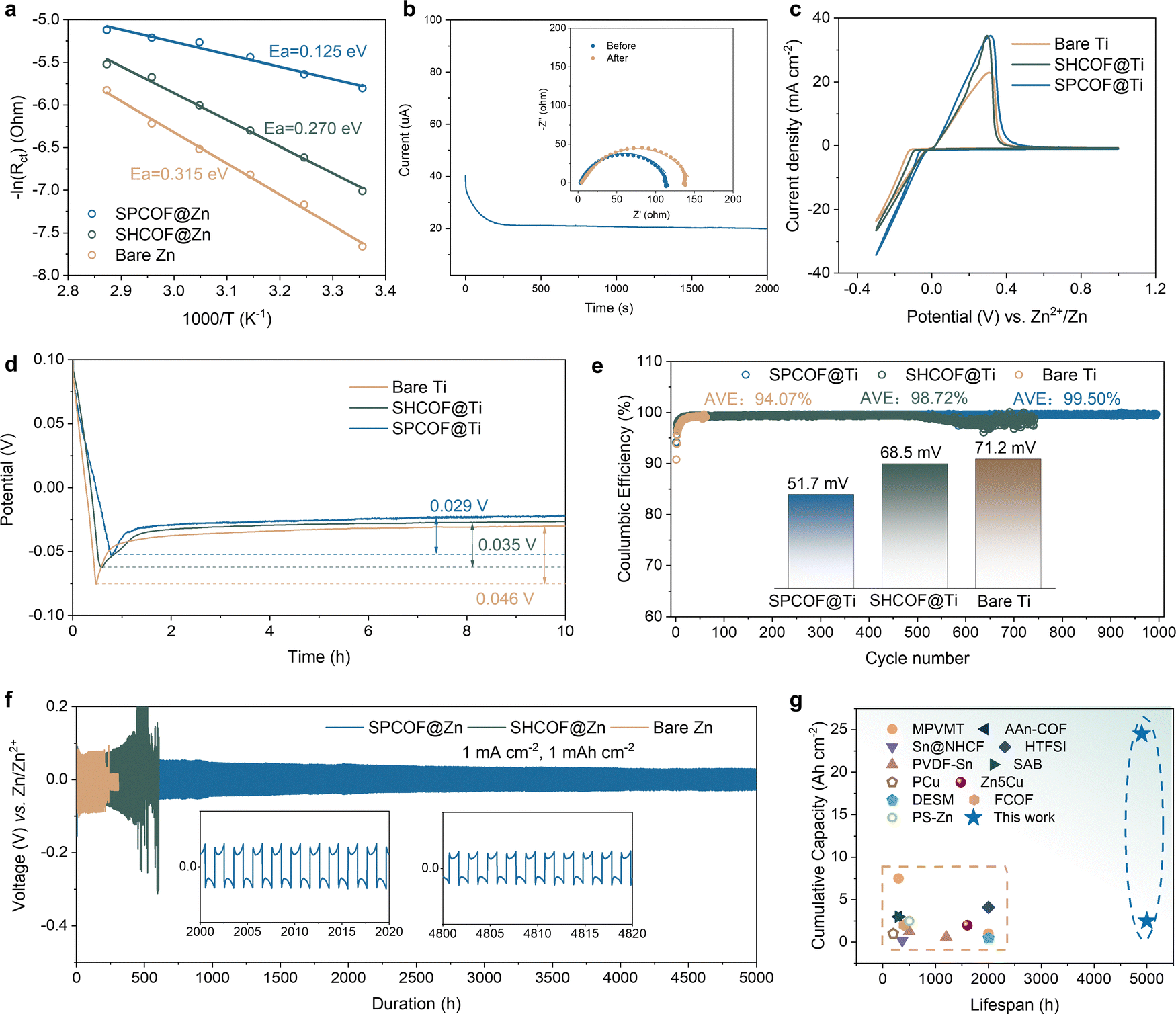 | ||
| Fig. 6 Improved kinetics and stability of Zn metal half cell. (a) Arrhenius plots showing inverse RCT values at varying temperatures (ranging from 25 to 75 °C) for the working electrode. (b) Current–time curves of Zn symmetric cells following a constant polarization of 10 mV for 2000 s with SPCOF@Zn. Inset: EIS result of the Zn symmetric cell before and after the transference number test. (c) CV curves of Ti-Zn asymmetric cells with different interfaces. (d) Nucleation overpotential of bare Ti, COF@Ti, and SPCOF@Ti. (e) CE of the Zn||Ti cells with a current density of 1mAcm−2 and a cut-off charging voltage of 0.4V. (f) Galvanostatic Zn stripping/plating behavior in Zn||Zn symmetric cells with different modification layers at 1 mA cm−2 with a 1 mA h cm−2 capacity. (g) Comparative analysis of cumulative plating capacity versus lifespan between this study and previously reported strategies. | ||
To evaluate the utilization and sustainability of the SPCOF@Zn anode, nucleation overpotentials on Ti foil with various modifications were measured. The SPCOF@Ti electrode showed a nucleation overpotential of 29 mV, lower than bare Ti (46 mV) and SHCOF@Ti (35 mV), as shown in Fig. 6d. In contrast, the bare Zn-Ti cell demonstrated a low average Coulombic efficiency (ACE) of 94.07% and failed before reaching 100 cycles at a current density of 1 mA cm−2 and capacity of 1 mA h cm−2 (Fig. 6e). The introduction of SHCOF interfaces extended the lifespan to over 500 cycles with 98.72% ACE, while SPCOF interphases improved longevity to 1000 cycles with an ACE of 99.5%. Additionally, the SPCOF-enhanced battery exhibits a reduced overpotential of 51.7 mV and stable cycling for over 2000 cycles with an ACE of 99.8% at a higher current density of 10 mA cm−2 (Fig. S32, ESI†). The homogeneous deposition of Zn facilitated by the SPCOF interphase was observed in situ by optical microscopy, SEM, chronoamperometry, XRD, and XPS analyses. The SPCOF@Zn anode displayed ultra-dense and uniform zinc deposition, favoring the crystallization of Zn films with faceted Zn (002) plane (Fig. S33–S42, ESI†).
Long-term galvanostatic cycling experiments were conducted on Zn||Zn symmetric cells to assess the stability of Zn anodes. The bare Zn cell, operating at a current density of 1 mA cm−2 and a capacity of 1 mA h cm−2, failed after just 330 hours due to a short-circuit (Fig. 6f). In contrast, a Zn battery equipped with SHCOF exceeded 700 hours of operation. Remarkably, introducing the SPCOF layer further enhanced performance, achieving over 5000 hours of stable cycling, demonstrating the SPCOF layer's capability to stabilize the Zn-electrolyte interface and effectively guide Zn deposition. The decrease in overpotential with cycles can be attributed to the increased surface area of the Zn electrode, which occurs during repeated cycling.40 Notably, the cumulative capacity enabled by the SPCOF@Zn symmetric cell are much higher than most of the previously reported values from Zn electrodes based on different modification strategies (Fig. 6g, see Table S1 for details, ESI†). Even under increased current densities of 10 mA cm−2, the SPCOF@Zn anode maintained this exceptional longevity and stability, whereas the bare Zn anode lasted less than 80 hours, showing significant voltage hysteresis (Fig. S43, ESI†). Additional tests at a high depth of discharge (50%) using thin Zn foils (20 μm) revealed that SPCOF@Zn anodes greatly improved cycling stability, enduring 350 hours, confirming the SPCOF@Zn electrode's suitability for prolonged lifespan applications under low N/P ratio conditions (Fig. S44, ESI†).
SPCOF layer for practical full cells
The full-cell capabilities of the SPCOF@Zn anode, paired with ZnxV2O5·nH2O (ZVO) cathodes in a coin cell configuration, were evaluated using standard elecrtrode preparation methods (Fig. S45, ESI†).41 These batteries, tested at a current density of 2 A g−1, exhibited superior capacity retention, maintaining over 153 mA h g−1 after 2000 cycles, significantly outperforming control cells with SHCOF@Zn and bare Zn anodes (Fig. S46, ESI†). Enhanced rate performance was also noted, and pairing with an I2 cathode allowed the SPCOF@Zn||I2 full cell to maintain stable capacities for up to 7000 cycles, far surpassing those with COF@Zn and bare Zn anodes (Fig. S47, ESI†).Using a high-loading ZVO cathode, the SPCOF@Zn||ZVO battery demonstrated exceptional cycling stability under rigorous conditions, sustaining over 800 and 500 cycles at N/P ratios of approximately 4 and 2, respectively (Fig. 7a and Fig. S48, ESI†), which outperforms most of the previous reported AZBs (Fig. 7b, see Table S2 (ESI†) for details). Note that variations in the initial capacity of high-loading Zn||ZVO full cells are due to slight differences in loading between cells. Additionally, the gradually increasing capacity during the initial hundreds of cycles can be attributed to improved wetting, which enhances ionic transport and electrode utilization. The scalability of the SPCOF@Zn anode was further proven in larger configurations. A 3 × 4 cm2 single-layer pouch cell maintained stable cycling for over 200 cycles at a current density of 4 mA cm−2 (0.2 A g−1), while the 7 × 8 cm2 SPCOF@Zn||ZVO pouch cells achieved capacities of 0.3 A h and 0.55 A h by employing one-layer and two-layer at a current density of 4 mA cm−2, respectively. These results highlight the enhanced performance and versatility of the SPCOF@Zn anode.
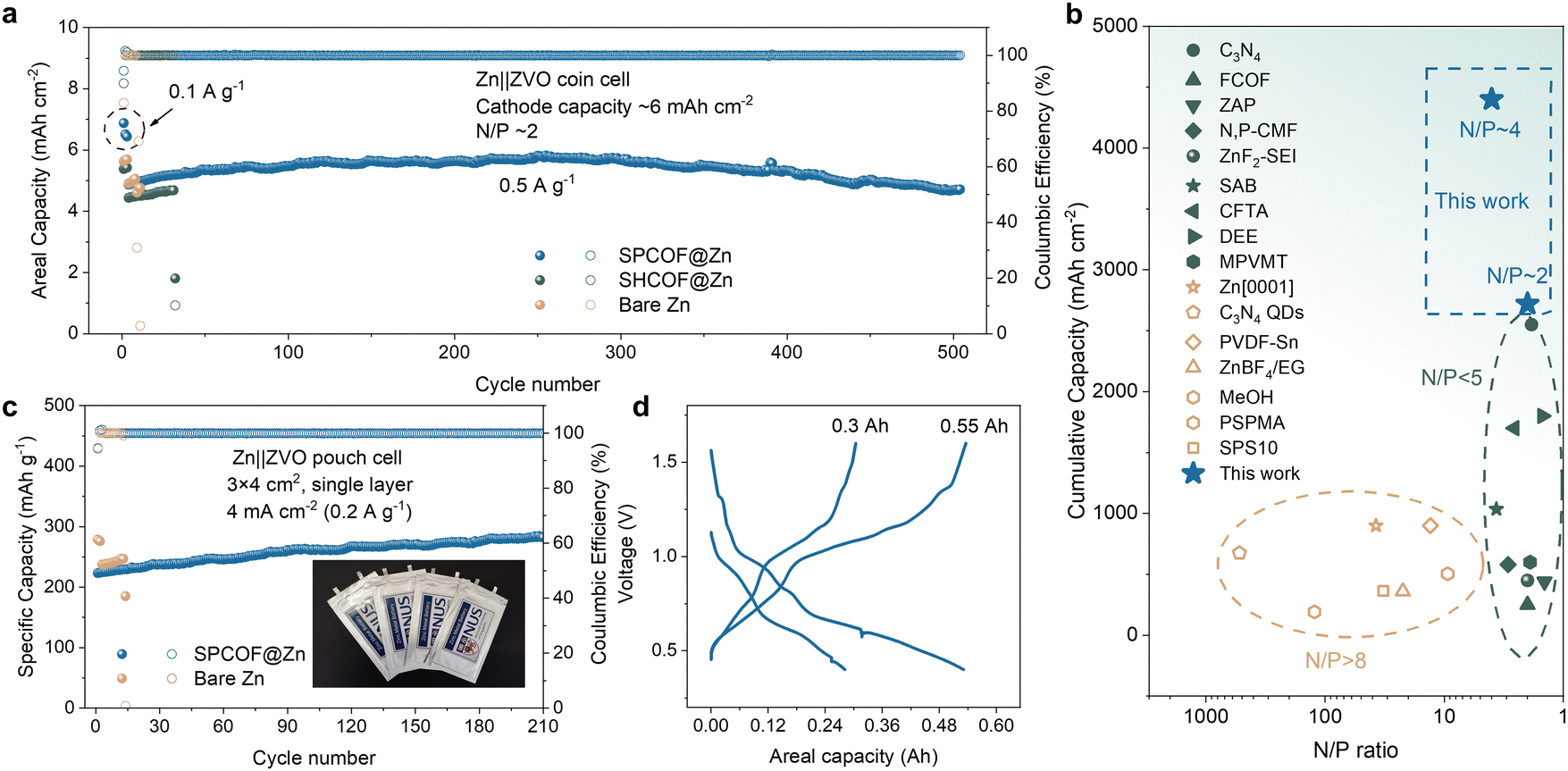 | ||
| Fig. 7 The performance of Zn metal full cell enabled by SPCOF layer. (a) Cycling performance of Zn||ZVO batteries with different anodes in a 2 M ZnSO4 electrolyte at a low N/P ratio of ∼2. The Zn thickness is 20 μm, corresponding to a capacity of ∼11.7 mA h cm−2, with a cathode capacity of ∼6 mA h cm−2. (b) Comparison of N/P ratio and cumulative capacity in this work with prior studies. (c) Cycling performance of 3 × 4 cm2 Zn||ZVO pouch cells. Inset is optical image of as prepared Zn||ZVO pouch cell. (d) Galvanostatic charge–discharge profiles of 0.3 A h and 0.55 A h Zn||ZVO pouch cells. | ||
Besides aqueous Zn battery, the SPCOF modification layer also can be used as an anode modifier in non-aqueous Li batteries. SPCOF significantly enhances Li deposition, resulting in a denser and smoother morphology (Fig. S51a–c, ESI†). Additionally, the SPCOF@Li electrode shows superior performance compared to the bare Li anode, featuring a reduced nucleation overpotential of 28 mV, a higher average coulombic efficiency of 99.97%, and an extended lifespan of over 2000 hours for the Li metal anode (Fig. S51d–f, ESI†). To further demonstrate its practical application in lithium-based battery systems, we also evaluated the performance of Li–S and Li–CO2 batteries incorporating the SPCOF modification layer (Fig. S52, ESI†). For the Li–S battery, the SPCOF layer facilitated improved cycling stability over 500 cycles, with a capacity decay of just 0.066% per cycle. In the Li–CO2 battery, the modification layer enhanced energy efficiency, reducing overpotentials to 0.72 V, 1.10 V, 1.38 V, and 1.61 V at current densities of 100 mA g−1, 200 mA g−1, 500 mA g−1, and 1000 mA g−1, respectively. Additionally, Na anode with the SPCOF modification layer demonstrated improved cycling stability, as shown in Fig. S53 (ESI†). These results highlight the versatility of the SPCOF layer as an anode modifier for wide-ranging energy storage system operating in both aqueous and non-aqueous electrolytes.
Conclusion
We demonstrate that integrating superhydrophobic covalent organic frameworks (SPCOFs) into AZBs promotes rapid Zn2+ migration and suppresses dendrite formation. Molecular dynamics simulations reveal that SPCOFs regulate the electrolyte environment near electrodes through their confined nanostructures and functionalized walls, which modulate interactions between channel walls, cations, and electrolyte species. By incorporating superhydrophobic fluorine chains into the COF framework, we mimic the functionality of Aquaporins, facilitating ion migration via a dewetting mechanism. The SPCOF-modified Zn anodes exhibited exceptional performance, achieving an operational longevity exceeding 5000 hours and sustaining up to 800 cycles across varied N/P ratios without significant degradation. These findings underscore that functionalized COFs, when applied as an anode modifier layer, enable precise control over solvation structures and electrolyte behavior through their confined channels, offering a novel approach to advanced battery engineering.Author contributions
K. P. L. supervised the project. K. P. L. and K. Z. led and conceive the project. K. Z. designed and conducted the experiment. K. Z. and Y. Y. synthesized the materials and analyzed the results. G. W. measured the high-resolution TEM of the COF under the supervision of J. L. L. M. helped to perform some battery data under the supervision of K. X. F. C. provided improtant insight in synthesis and ion migration mechanism. S. X. and C. W. conducted XAS measurement. J. L. and B. Z. helped to conduct in situ GC test. C. L., H. L. and C. L. helped to carry out some materials characterization, battery test and discuss the data. K. Z. and Y. Y. conducted theoretical simulations and structural simulations. K. Z. discussed the project with X. L., K. Z. Y. Y., G. W. and K. P. L. co-wrote the manuscript with comments and revisions from all the authors.Data availability
All data supporting the findings of this study are available within the paper and its ESI.† Data are also available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no competing interests.Acknowledgements
K. P. Loh thanks Singapore's Ministry of Education Tier 1 Grant number A80026710000 “A new class of metal free dielectrics.” G. W and J. L. acknowledge support from the National Natural Science Foundation of China (grant NO. 52473302, 12404017), the Guangdong Innovative and Entrepreneurial Research Team Program (grant NO. 2019ZT08C044), Guangdong Basic Science Foundation (2023B1515120039), the Shenzhen Science and Technology Program (NO. 20200925161102001), the Science, Technology and Innovation Commission of Shenzhen Municipality (NO. ZDSYS20190902092905285), the Postdoctoral Fellowship Program of CPSF (Grant NO. GZB20240295), the China Postdoctoral Science Foundation (Grant NO. 2024M751288), andQuantum Science Strategic Special Project from the Quantum Science Center of Guangdong-Hong Kong-Macao Greater Bay Area (GDZX2301006, SZZX2301004). K. X. acknowledge support from Open project of Shaanxi Laboratory of Aerospace Power (2022ZY2-JCYJ-01-09), full-depth-sea battery project (No. 2020-XXXX-XX-246-00) and the Innovation Team of Shaanxi Province. The authors also thank the kind help of Dr C.Y. Fang and Prof. S. Zhang from National University of Singapore for contact angles test.References
- C. Zhong, B. Liu, J. Ding, X. Liu, Y. Zhong, Y. Li, C. Sun, X. Han, Y. Deng, N. Zhao and W. Hu, Nat. Energy, 2020, 5, 440 CrossRef CAS.
- D. Dong, T. Wang, Y. Sun, J. Fan and Y.-C. Lu, Nat. Sustainability, 2023, 6, 1474 CrossRef.
- S. W. D. Gourley, R. Brown, B. D. Adams and D. Higgins, Joule, 2023, 7, 1415 CrossRef CAS.
- C. Yang, J. Xia, C. Cui, T. P. Pollard, J. Vatamanu, A. Faraone, J. A. Dura, M. Tyagi, A. Kattan, E. Thimsen, J. Xu, W. Song, E. Hu, X. Ji, S. Hou, X. Zhang, M. S. Ding, S. Hwang, D. Su, Y. Ren, X.-Q. Yang, H. Wang, O. Borodin and C. Wang, Nat. Sustainability, 2023, 6, 325 CrossRef.
- Z. Xu, Y. Li, G. Li, H. Zhang and X. Wang, Matter, 2023, 6, 3075 CrossRef CAS.
- H. Jiang, L. Tang, Y. Fu, S. Wang, S. K. Sandstrom, A. M. Scida, G. Li, D. Hoang, J. J. Hong, N.-C. Chiu, K. C. Stylianou, W. F. Stickle, D. Wang, J. Li, P. A. Greaney, C. Fang and X. Ji, Nat. Sustainability, 2023, 6, 806 CrossRef.
- H. Qiu, X. Du, J. Zhao, Y. Wang, J. Ju, Z. Chen, Z. Hu, D. Yan, X. Zhou and G. Cui, Nat. Commun., 2019, 10, 5374 Search PubMed.
- Z. Zhao, R. Wang, C. Peng, W. Chen, T. Wu, B. Hu, W. Weng, Y. Yao, J. Zeng, Z. Chen, P. Liu, Y. Liu, G. Li, J. Guo, H. Lu and Z. Guo, Nat. Commun., 2021, 12, 6606 CrossRef CAS PubMed.
- J. Huang, L. Yu, S. Wang, L. Qi, Z. Lu, L. Chen, D. Xu, H. Deng and C. J. I. M. Chen, Innov. Mater., 2023, 1, 100029 CrossRef.
- L. Han, Y. Guo, F. Ning, X. Liu, J. Yi, Q. Luo, B. Qu, J. Yue, Y. Lu and Q. Li, Adv. Mater., 2024, 36, e2308086 CrossRef.
- L. Yu, J. Huang, S. Wang, L. Qi, S. Wang and C. J. A. M. Chen, Adv. Mater., 2023, 35, 2210789 CrossRef CAS.
- Y. Zhao, K. Feng and Y. Yu, Adv. Sci., 2024, 11, e2308087 CrossRef.
- C. Guo, J. Zhou, Y. Chen, H. Zhuang, Q. Li, J. Li, X. Tian, Y. Zhang, X. Yao, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202210871 CrossRef CAS PubMed.
- L. Yang, Q. Ma, Y. Yin, D. Luo, Y. Shen, H. Dou, N. Zhu, R. Feng, Y. Kong, A. Yu, B. Cheng, X. Wang and Z. Chen, Nano Energy, 2023, 117, 108799 CrossRef CAS.
- B. Li, P. Ruan, X. Xu, Z. He, X. Zhu, L. Pan, Z. Peng, Y. Liu, P. Zhou, B. Lu, L. Dai and J. Zhou, Nanomicro. Lett., 2024, 16, 76 CAS.
- P. Zuo, C. Ye, Z. Jiao, J. Luo, J. Fang, U. S. Schubert, N. B. McKeown, T. L. Liu, Z. Yang and T. Xu, Nature, 2023, 617, 299 CrossRef CAS PubMed.
- H. Chen, C. Hu, X. Zhang and L. Chen, Acc. Mater. Res., 2024, 5, 1303 CrossRef CAS.
- D.-G. Wang, T. Qiu, W. Guo, Z. Liang, H. Tabassum, D. Xia and R. Zou, Energy Environ. Sci., 2021, 14, 688 RSC.
- H. Tan, M. Duan, H. Xie, Y. Zhao, H. Liu, M. Yang, M. Liu and J. J. S. A. Yang, Sci. Adv., 2024, 10, eade9520 CrossRef CAS PubMed.
- C. Sun, Q. Lyu, Y. Si, T. Tong, L. C. Lin, F. Yang, C. Y. Tang and Y. Dong, Environ. Sci. Technol., 2022, 56, 5775 CrossRef CAS PubMed.
- Y. Itoh, S. Chen, R. Hirahara, T. Konda, T. Aoki, T. Ueda, I. Shimada, J. J. Cannon, C. Shao and J. J. S. Shiomi, Science, 2022, 376, 738 CrossRef CAS PubMed.
- C. Yan, C. Lv, Y. Zhu, G. Chen, J. Sun and G. Yu, Adv. Mater., 2017, 29, 1703909 CrossRef PubMed.
- K. Zhang, W. Liu, Y. Gao, X. Wang, Z. Chen, R. Ning, W. Yu, R. Li, L. Li, X. Li, K. Yuan, L. Ma, N. Li, C. Shen, W. Huang, K. Xie and K. P. Loh, Adv. Mater., 2021, 33, e2006323 CrossRef PubMed.
- P. Liu, X. Y. Kong, L. Jiang and L. Wen, Chem. Soc. Rev., 2024, 53, 2972 RSC.
- K. Zhang, X. Li, L. Ma, F. Chen, Z. Chen, Y. Yuan, Y. Zhao, J. Yang, J. Liu, K. Xie and K. P. Loh, ACS Nano, 2023, 17, 2901 CrossRef CAS PubMed.
- L. Ma, Q. Li, Y. Ying, F. Ma, S. Chen, Y. Li, H. Huang and C. Zhi, Adv. Mater., 2021, 33, e2007406 CrossRef PubMed.
- W. Zhang, M. Dong, K. Jiang, D. Yang, X. Tan, S. Zhai, R. Feng, N. Chen, G. King, H. Zhang, H. Zeng, H. Li, M. Antonietti and Z. Li, Nat. Commun., 2022, 13, 5348 CAS.
- C. Li, R. Kingsbury, A. S. Thind, A. Shyamsunder, T. T. Fister, R. F. Klie, K. A. Persson and L. F. Nazar, Nat. Commun., 2023, 14, 3067 CrossRef CAS PubMed.
- H. Yang, Y. Qiao, Z. Chang, H. Deng, P. He and H. Zhou, Adv. Mater., 2020, 32, e2004240 CrossRef PubMed.
- D. Xu, X. Ren, H. Li, Y. Zhou, S. Chai, Y. Chen, H. Li, L. Bai, Z. Chang, A. Pan and H. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202402833 CrossRef CAS PubMed.
- F. Ming, Y. Zhu, G. Huang, A. H. Emwas, H. Liang, Y. Cui and H. N. Alshareef, J. Am. Chem. Soc., 2022, 144, 7160 CrossRef CAS PubMed.
- W. Xu, J. Li, X. Liao, L. Zhang, X. Zhang, C. Liu, K. Amine, K. Zhao and J. Lu, J. Am. Chem. Soc., 2023, 145, 22456 CAS.
- W. Liu, Y. Lu, Y. Shen, H. Chen, Y. Ni and Y. Xu, ACS Omega, 2023, 8, 16450 CrossRef CAS PubMed.
- P. Lenz and R. Lipowsky, Phy. Rev. Lett., 1998, 80, 1920 CAS.
- R. Sakamoto, Z. Izri, Y. Shimamoto, M. Miyazaki and Y. T. Maeda, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2121147119 CrossRef CAS PubMed.
- X. Li, F. Bodziony, M. Yin, H. Marschall, R. Berger and H. J. Butt, Nat. Commun., 2023, 14, 4571 CrossRef CAS PubMed.
- C. Luo, M. Xiang, X. Liu and H. Wang, Microfluid. Nanofluid., 2010, 10, 831 CrossRef.
- B. X. Zhang, S. L. Wang and X. D. Wang, Langmuir, 2019, 35, 662 CrossRef CAS.
- M. T. Rauter, S. K. Schnell and S. Kjelstrup, J. Phys. Chem. B, 2021, 125, 12730 CrossRef CAS PubMed.
- G. Bieker, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2015, 17, 8670 RSC.
- F. Wang, J. Zhang, H. Lu, H. Zhu, Z. Chen, L. Wang, J. Yu, C. You, W. Li, J. Song, Z. Weng, C. Yang and Q. H. Yang, Nat. Commun., 2023, 14, 4211 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00132c |
| ‡ These authors contributed equally: Zhang Kun, Yuan Yijia and Wang Gang. |
| This journal is © The Royal Society of Chemistry 2025 |