
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Review of interface issues in Li–argyrodite-based solid-state Li–metal batteries†
Berhanu Degagsa
Dandena
ab,
Dah-Shyang
Tsai
 b,
She-Huang
Wu
cd,
Wei-Nien
Su
*cd and
Bing Joe
Hwang
*abde
b,
She-Huang
Wu
cd,
Wei-Nien
Su
*cd and
Bing Joe
Hwang
*abde
aNano-electrochemistry Laboratory, Department of Chemical Engineering, National Taiwan University of Science and Technology, Taipei 106, Taiwan. E-mail: bjh@mail.ntust.edu.tw
bDepartment of Chemical Engineering, National Taiwan University of Science and Technology, Taipei 106, Taiwan
cNano-electrochemistry Laboratory, Graduate Institute of Applied Science and Technology, National Taiwan University of Science and Technology, Taipei 106, Taiwan. E-mail: wsu@mail.ntust.edu.tw
dSustainable Electrochemical Energy Development (SEED) Center, National Taiwan University of Science and Technology, Taipei 106, Taiwan
eNational Synchrotron Radiation Research Center (NSRRC), Hsin-chu 30076, Taiwan
First published on 3rd June 2025
Abstract
Sulfide solid electrolyte-based all-solid-state Li-metal batteries (ASSLBs) offer increased safety, extended cycle life, reduced costs, and increased energy and power density. However, sulfide-based electrolytes exhibit poor interface stability when they are in contact with active materials (anodes or cathodes). Issues of low capacity, inefficient coulombic efficiency, huge polarization, and capacity decay in ASSLBs are primarily attributed to sluggish charge transfer kinetics at the interface caused by chemical interactions, electrochemical degradation, weakening interfacial mechanical integrity, etc. Identifying the factors that affect the interfaces, studying the properties of interfaces, and proposing a mechanism to solve the problem are very important, as these properties directly or indirectly impact battery performance. Although the precise mechanism underlying these interface issues remains incompletely understood, combining chemomechanical processes and physical and (electro)chemical properties regulates the charge transfer phenomena at the interfaces. In this review, we identify the factors that affect the interfacial phenomena and properties of Li–argyrodite-based ASSLBs. Meanwhile, we also summarize the strategies and propose future perspectives to advance Li–argyrodite-based ASSLBS.
Broader contextAll-solid-state batteries have garnered increasing interest due to their significant potential for enhanced safety and energy density. However, the only batteries that can currently power electrochemical devices are those that use organic liquid electrolytes. To make solid-state batteries competitive with established technologies, there is a strong need for (electro)chemically stable and superionic solid electrolytes. Sulfide solid electrolytes are promising candidates for all-solid-state lithium metal batteries (ASSLBs) due to their high ductility, improved ionic conductivity, and versatile synthesis methods. Recently, Li–argyrodites Li6PS5X (X = Cl, Br, I) were recognized as one of the most promising sulfide electrolytes, thanks to their intrinsically outstanding structures and electrochemical stability, addressing existing limitations. However, their advancement is hindered by significant interfacial incompatibility and rapid dendritic growth. Identifying the factors that influence the interfaces, examining their properties, and proposing mechanisms to address any issues are crucial, as these aspects directly or indirectly affect battery performance. In this review, we explore the factors influencing interfacial phenomena and properties in Li–argyrodite-based ASSLBs while summarizing strategies and proposing future directions for developing these batteries. |
1. Introduction
Because of their extended cycle life and greater energy density, lithium-ion batteries are currently highly desirable power sources for electronic equipment and car batteries. However, using an organic liquid electrolyte (LE) for some applications restricts working temperatures and poses safety concerns.1,2 To alleviate these problems, ASSLBs with an inorganic solid electrolyte are a safer substitute that can store more energy than micro-batteries while also having good thermal stability.3–6 The primary concerns with all-solid-state lithium batteries (ASSLBs) are increasing the ionic conductivity (σ) of solid electrolytes (SEs) and ensuring stability between the SE and electrodes.6–8 Due to their relatively fast ion transport at ambient temperature and a moderate electrochemical stability window (ESW), sulfides have attracted much interest among various types of SEs.8,9 Furthermore, sulfides offer an advantage over oxides in cell manufacturing because of their ability to be processed in solution and capacity to deform during cold pressing. The grain boundary resistance of sulfide SEs can be significantly lowered via cold pressing at ambient temperature, eliminating the need for elevated temperature sintering. In ASSLBs, the soft property of sulfide SEs helps to provide light contact at the electrode/SE interfaces, inducing a substantial enhancement in cycle performance. Li–argyrodites have favorable electrochemical characteristics for ASSLBs, including high σ.9 The electrolyte/electrode interface must be considered despite these various benefits. It was previously reported that Li–argyrodite Li6PS5X (X = Cl, Br, and I) SEs displayed interfacial reactivity toward layered oxide cathode materials during cycling. Li6PS5X can undergo oxidation to produce Li2Sn, elemental sulfur, P2S5, phosphates, and LiX.10–14 Moreover, when the sulfide SEs and layered oxide cathodes interact, space-charge layers (SCL) and elemental mutual diffusion occur. An unstable interface causes cell failure and capacity degradation within ASSLBs.15,16 Parallel to this, Li6PS5X's incompatibility with Li metal causes an unstable anodic interface, resulting in high interfacial resistance. This leads to unmanageable lithium deposition and results in short circuits. By taking advantage of superior σ, outstanding electrochemical properties, and excellent machinability, it is crucial to boost the compatibility of Li6PS5X and electrodes to inhibit interfacial resistance growth and cell short circuits. Consequently, it has been claimed that developing novel SEs with a tendency to make self-limiting interfaces will improve the interfacial stability of the SE/electrode.17Using Li–argyrodites in a solid-state system offers several interesting benefits over LEs in a liquid system.18 Firstly, Li–argyrodites can be twisted, punched, and perforated without worrying about safety issues. Secondly, Li–argyrodites have ultra-high σ (approximately 10−3 to 10−2 S cm−1),19,20 which is comparable to that of organic LEs (typically around 10−2 S cm−1, 25 °C).20,21 This suggests that SE materials have significant potential for next-generation batteries. Third, Li–argyrodites exhibit greater stability over a wider temperature range than organic LEs, allowing the ASSLBs to run effectively within a temperature interval of −30 to +160 °C.22 All these properties show that Li–argyrodites hold great promise for next-generation batteries. However, SE and electrode interfaces pose a significant scientific problem that will prevent the actual deployment of ASSLBs.
In this review, we assess interface issues of Li–argyrodite Li6PS5X (X = Cl, Br, and I) electrolytes for a range of essential aspects: the interface between Li–argyrodites and the anode, the interface between Li–argyrodites and the cathode, Li–argyrodite SE and electrode assembly issues, and Li–argyrodite and electrode contact issues. This review also summarizes several advanced interface measurement techniques. Finally, we introduce strategies to improve interface stability and provide our future perspectives.
2. Interface challenges
Argyrodite-type solid electrolytes, particularly those based on the Li6PS5X (X = Cl, Br, and I) family, are considered promising candidates for ASSLBs due to their high lithium-ion conductivity, ease of processability, and relatively low synthesis temperatures.23 However, despite these advantages, their practical application is hindered by significant interface-related issues.One of the primary challenges is chemical and electrochemical instability at the interfaces between the argyrodite electrolyte and electrode materials.23,24 When in contact with high-voltage cathodes (e.g., NMC, LCO), argyrodites tend to undergo interfacial degradation, forming resistive interphases due to the decomposition of thiophosphate species.25–27 This leads to increased interfacial impedance, reduced ion transport, and poor long-term cycling performance. In addition, at low potential, argyrodite SEs endure a reduction of phosphorus (around 1.08 V vs. Li/Li+) upon electrochemical lithiation and produce Li2S, LiX and P as a by-product, making them incompatible with Li metal.28–30 Argyrodite SEs also produce Li2S, LiX and Li3P as a by-product when in contact with Li metal (chemical decomposition).31
Furthermore, argyrodites often suffer from poor mechanical properties, which can result in interfacial contact loss during battery operation due to volume changes in active materials or insufficient densification during cell assembly. This mechanical mismatch contributes further to increased resistance and capacity fading.32
Therefore, addressing chemical incompatibility and mechanical instability at the electrolyte–electrode interfaces is critical for unlocking the full potential of argyrodite-based ASSLBs.
In ASSLBs, undesirable physical and chemical interactions at corresponding electrolyte/electrode interfaces generate large gaps between expected and actual performances. The major issues of electrolyte/electrode interfaces in ASSLBs are depicted in Fig. 1a.
 | ||
| Fig. 1 (a) Challenges of SE and electrode interfaces in ASSLBs.52 (b) Scheme illustrating the oxidation and reduction of Li6PS5X (X = Cl, Br, I) argyrodite SEs when in contact with electrodes during cycling. Interpretation of EIS spectra. (c) Equivalent circuit used for fitting the EIS data along with a schematic describing the impedance contributions from the bulk (RB), grain boundaries (RGB), SEI (RSEI) and charge transfer (RCT) processes.46,47 (d) Variation in interfacial resistance, Rint (RSEI + RCT), from fitted EIS data as a function of the amount of charge passed during Li plating at current densities, J2.5 = 2.5 mA cm−2, J0.5 = 0.5 mA cm−2, J0.05 = 0.05 mA cm−2 and J0.01 = 0.01 mA cm−2.46,47 (e) Schematic representation of the likely mechanism of SEI formation and Li plating as a function of applied current density, at J2.5 (JHigh) and J0.01 (JLow).45 | ||
Inadequate contact between the SE and the electrode can limit the effective interfacial area. For instance, many SEs are inherently lithiophobic due to their high interfacial energy with Li metal, often resulting in contact angles greater than 90° when in contact with Li. This poor wettability hinders intimate contact and reduces the area available for efficient Li-ion transport.33 Additionally, designing hierarchically interconnected microstructures in non-Li-based electrodes is challenging, as it demands meticulous control over the shape, size, and spatial arrangement of the electrolyte, active materials, and electronic conductors. If these microstructures are not properly optimized, the contact area for charge transfer at the electrolyte–electrode interface can be substantially diminished.34 Furthermore, the rigid nature of solid–solid interfaces prevents them from adapting to the volume changes of active materials during cycling, accumulating mechanical stress. Over time, this cycling stress can initiate and propagate cracks at the interface, progressively weakening the interfacial contact.35
Sluggish charge transfer kinetics at the SE/electrode interface can raise the area-specific resistance. Side reactions often exacerbate this, which may occur when the electrode's operating potential lies outside the electrolyte's electrochemical stability window.36 Such reactions irreversibly consume the SE and/or electrode materials, producing interphase layers that are electronically conductive but poorly conductive for Li-ions. These interphases may promote further parasitic reactions or serve as barriers that impede Li-ion transport across the interface.37 Additionally, mismatches in Li-ion chemical potential between the electrolyte and the electrode material can lead to the formation of space charge layers at the interface. These regions, often characterized by local lithium depletion and reduced ion diffusion coefficients, further hinder effective Li-ion transport.38
Li dendrite growth at the Li/SE interface or within the SE can lead to serious short circuits. Dendrite formation is driven by uneven electric fields, which are amplified near protrusions on the lithium surface due to the tip effect and non-uniform Li-ion flux.39 The resistance to dendrite growth arises from the strain energy needed to deform the SE and the interfacial energy associated with expanding the contact area. Dendrite growth can occur spontaneously when the driving forces surpass these resistive barriers. Furthermore, although ideal SEs would conduct only Li-ions, most current materials exhibit some degree of electronic conductivity. In highly electron-conductive electrolytes, electrons can migrate from the lithium metal into the electrolyte, lowering its local electrochemical potential. During charging, this may reduce the electrolyte's potential below 0 V vs. Li/Li+, facilitating dendrite nucleation and propagation through microstructural defects.40
In addition to the issues, the interfacial reactions induced by the thermodynamic instability between argyrodite SEs and the electrodes are also worth attention. Janek et al.31 used X-ray photoelectron spectroscopy (XPS) to confirm that Li6PS5X would decompose at the interface due to the strong reduction of lithium. Some of the Li6PS5X decomposed on the surface of lithium metal to form the SEI with the ingredients of decomposition products such as Li3P, Li2S, and LiX (Fig. 1b). The SEI leads to increased interfacial resistance and hindered the conduction of lithium ions. An additional study by Wagemaker et al.30 revealed that the thermodynamic decomposition reaction of argyrodite SEs with the anode side was not instantaneous. Taking argyrodite Li6PS5Cl as an example, it was first reduced to unstable Li11PS5Cl, and then Li11PS5Cl was further reduced to Li2S, LiCl, and Li3P. In this process, the intermediate phases generated, such as S, Li2S, and LiCl, not only produced a large volume expansion but also led to the destabilization of the kinetics. The continued decomposition of argyrodite SEs at the interface as the cycle proceeds is the main reason for the increased interfacial resistance. In the argyrodite SE and NCM interface, due to the affinity of oxygen to react with phosphorus and sulfur, phosphate (POx) and sulfite/sulfate (SOx) and polysulfide (Sx) fragments are regarded as indicators for oxygen-involved degradation.41–44
Narayanan et al.45 studied how interfacial impedance changed depending on the applied current density. To analyze this behavior, they used Nyquist plots and fitted the data using an equivalent circuit model illustrated in Fig. 1c.46 This circuit consists of a single resistor in series with three parallel circuits of a resistor and a constant-phase element (CPE) along with an additional Warburg diffusion (WS) component. Schlenker et al.47 attributed the inclusion of the latter to an impedance at low frequencies arising from a lithium vacancy diffusion gradient generated most likely at the interface between LiIn and LPSCl. In this equivalent circuit, one of the parallel circuits (R2||CPE2) in combination with the individual resistor R1 can be assigned to the bulk and grain boundary resistance, which typically exhibits low effective capacitances (correlating to the term QCPE ∼ 10−6 F sa−1, with a as the constant phase, as estimated from the CPE component of electrochemical impedance spectroscopy (EIS) spectral data fitting). The other circuit element pairs (R3||CPE3 and R4||CPE4) exhibiting relatively higher effective capacitances (QCPE ∼ 10−4–10−2 F sa−1) can be understood to represent the LPSCl–Li interface and charge transfer (CT) processes, respectively, as plating begins with the formation of an SEI.46–48 The variations in impedance can thus be attributed largely to the interface, which comprises the SEI and CT components represented by the spectra's low-frequency segment.46–48 The interfacial impedance (Rint) can then be approximated as Rint = RSEI + RCT,46 where RCT is related to the intrinsic kinetics of the system.49–51 Indeed, a plot of combined resistances from SEI and CT contributions (Fig. 1d) suggests that at low current densities (in this study, J0.5 and J0.05), the interfacial resistance asymptotically reaches a minimum. In stark contrast, for Li plating conducted at a significantly higher current density (J2.5), the interface attains the same minimum resistance almost as soon as Li begins plating. Notably, the differences in impedance evolution are most prominent over ∼50 μA h cm−2 of charge passed, beyond which the effect diminishes as expected, while plating proceeds to form a metallic Li layer in both cases. Thus, a rapid drop in interfacial resistance, combined with the appearance of fully reduced reaction products (Li3P in particular) and the presence of a greater fraction of metallic Li in XPS analyses, all observed within ∼10 μA h cm−2 of charge passed during initial stages of plating, strongly suggest the formation of a more uniform and homogeneous SEI layer as well, for Li plated at high current densities (Fig. 1e).
As mentioned above, the increase in impedance at the interface between argyrodite SEs and electrodes is mainly due to interfacial reactions and the accumulation of electrically insulating byproducts.31,53,54 These reactions, particularly at the anode, result in forming an SEI and possibly lithium dendrites, both disrupting lithium-ion movement and elevating resistance. The key mechanisms that lead to a continuous rise in impedance include:
i. Loss of effective contact area: The SEI layer, if not well-adhered and conductive, can reduce the effective contact area between the electrode and the SE, limiting ion transport.55
ii. Insulating reaction products: The formation of insulating byproducts from interfacial reactions can directly block ion transport pathways, increasing the overall resistance.53,54
iii. Dendrite growth and short circuits: As dendrites grow and penetrate the electrolyte, they create pathways for current to flow through a non-Faradaic process, significantly increasing impedance and potentially short circuits.53
iv. Grain boundary contributions: In SEs, grain boundaries may impede ion flow, particularly if they are poorly connected or contain structural defects.56,57
Furthermore, as particles within the electrode change volume during lithiation/delithiation, they can move and shift relative to each other, leading to loss of contact and increased resistance between particles.58,59 This disrupts the flow of lithium ions within the electrode. Phase changes in the electrolyte, such as the formation of SEIs, can create barriers to ion transport.60 These barriers increase the resistance within the electrolyte and impede lithium ion transfer between the electrode and the electrolyte. Phase transitions within the electrode material, such as the shift between different lithium intercalation stages, can introduce additional resistance.58,59 These phase transitions often involve structural changes that hinder the smooth diffusion of lithium ions. Therefore, these dynamic changes within the battery system create additional interfaces and hinder the efficient movement of lithium ions, resulting in increased internal resistance, SEI/CEI growth and instability, microstructural changes, capacity fading, and reduced power density.
Generally, each interface in an ASSLB can be divided into one of three primary classes61,62 based on stability (Fig. 2): Type I, a thermodynamically stable interface lacking a reaction driver. The interface between the solid electrolyte and the electrode is chemically stable under operating conditions, and there is no driving force (i.e., “reaction driver”) that would cause undesirable chemical reactions at the interface. This means the materials at the interface are stable and cannot undergo undesirable reactions that could degrade the battery's performance or safety. Type II, reacting to generate a mixed ionic–electronic conducting interphase (MCI), a non-passivating interphase exhibiting both ionic and electronic conductivity. An MCI can form through a reaction between the two materials in the interface between an SE and an electrode. This interphase is characterized by its ability to conduct both ions and electrons, and it is considered a non-passivating interphase. This means it does not block the flow of electrons or ions, potentially leading to undesirable side reactions and performance issues. Type III, reacting to produce stable SEI and negligible electronic conductivity, thereby preventing additional reactions. When the SE and electrode are not fully chemically or electrochemically compatible, leading to interfacial reactions that produce a new, passivating layer—the SEI. This SEI layer is a stable protective barrier that blocks electronic conductivity (acting as an insulator) and enables Li-ion transport. By preventing electronic conductivity, the SEI stops further reactions between the electrode and SE, contributing to battery stability and longevity. Only type I (stable) and type III (passivating) interfaces can be anticipated to provide long-range stable battery performances. In type III interfaces, σ of the SEI is essential for overall battery efficiency.63 Although Li–argyrodite SEs have demonstrated impressive scientific and industrial advances, difficulties are still presented when sulfide SEs are incorporated into ASSLBs, particularly when used with high-voltage cathodes and lithium anodes.63–65 The generalized concept of ASSLB cells is displayed in Fig. 3.
 | ||
| Fig. 2 Scheme illustrating the three possible types of interface between Li6PS5X (X = Cl, Br, I) and lithium. | ||
 | ||
| Fig. 3 The generalized concept of the Li–argyrodite-based ASSLB cell. | ||
Moreover, although SSBs perform well at room and moderate temperatures (5–20 °C),66 their performance significantly declines at low temperatures (below 0 °C) due to reduced ionic conductivity in SEs and slower solid–solid interfacial reaction rates.67–69 These limitations hinder overall battery efficiency, and their exact causes remain unclear.70,71 To address this, Lu et al.72 introduced a systematic test and analysis framework, which identified the primary limiting factors at low temperatures as sluggish ion movement through the interfacial reaction layer and impaired charge transport at the degraded surface of the LCO cathode.
To explore the behavior of SSBs under low-temperature conditions, simulation methods provide crucial insights into their working mechanisms, aiding the advancement of the field. Shen et al.73 built a two-dimensional model of SSBs using multi-physics simulation software and discovered that reducing the SE thickness could enhance the low-temperature rate performance. Specifically, with an electrolyte thickness of 0.6 μm, the battery retained 99.5% of its capacity even as the discharge rate increased from 3C to 8C. In interface analysis, Guan et al.74 introduced an interfacial contact model and an electrochemical–mechanical coupling model accounting for curvature effects. They found that increasing curvature and the contact factor could delay the time required to reach the cutoff voltage, thereby improving battery capacity. Additionally, Yan et al.75 developed a 2D model incorporating electrochemical–mechanical–thermal coupling to evaluate the multiplicative charging performance of SSBs at low temperatures, considering the impact of interfacial contact degradation. Their findings indicate that both poor interfacial contact and higher charging rates contribute significantly to capacity at low temperatures.
Similarly, exposing SSEs to elevated temperatures can also influence their performance. Shin et al.76 conducted electrochemical tests on ASSLBs at around 60 °C, as the biphasic solid electrolyte exhibited insufficient ionic conductivity at room temperature, making normal battery operation difficult. Jiang et al.77 observed that increasing the temperature from 25 °C to 65 °C significantly reduced the area-specific resistance, improving battery capacity. However, at even higher temperatures, three heat-related processes become closely interlinked. Typically, heat generation initiates the sequence that can lead to thermal failure. Over time, as the battery ages, the cumulative thermal side effects begin to negatively affect performance and compromise stability. This phenomenon is referred to as the aging effect. If heat generation surpasses the system's thermal tolerance and causes sufficient material degradation, it can eventually trigger thermal runaway.
2.1. Electrolyte–anode interface
Although much research is carried out to raise σ of lithium SEs, electronic transport is also crucial for ASSLB cycling stability, calendar life, and energy density. Only Li ions should be able to move between electrodes in an ideal SE, which would have minimal electronic conductivity and elevated σ. The outcome of electronic conduction in SEs would be the self-discharge of ASSLBs driven by electronic leakage.78 Li kinetics are ultimately hindered by the interfacial resistance and electronic conductivity of SEs generated, which also lead to performance degradation and uncontrollably growing Li dendrites.79,80 Interfacial resistance hinders Li-ion kinetics in batteries by impeding the transfer of Li+ ions across the interface between the electrode and the SE. This resistance arises from poor contact, an insulating SEI layer, and sluggish charge transfer reactions at the interface.81 In addition, if the SE conducts electrons, electrons can leak into regions where only ions should move. These electrons react with Li+ in the SE, causing electrolyte decomposition.52 The result is the formation of resistive interphases (e.g., Li2S, Li3P), which block or narrow Li+ pathways. If the SEs have high electronic conductivity, they can provide an easy pathway for electrons to reach the Li metal anode. This allows for rapid and potentially non-uniform Li ion reduction and deposition.40,82 The rapid and non-uniform deposition of Li can lead to the formation of dendritic structures on the Li metal surface.40 These dendrites can grow and penetrate the SE, potentially leading to short circuits within the battery.40,83 Dendrites can propagate along grain boundaries due to the inherent structure of the SE.84–86 As dendrites grow, they can cause mechanical stress and damage to the surrounding electrolyte, leading to crack propagation and further dendrite growth. SEs can form new phases with different ionic and electronic conductivities when decomposing. While some decomposition products might be more conductive than the original material, they are still likely to have a lower electronic conductivity compared to the ionic conductivity.86–88 The results from our group and Wang's group indicated that the SE with the lowest electronic conductivity demonstrated enhanced capability to suppress lithium dendrite formation.40,89For many years, graphite has been commonly utilized as an anode material. With a specific capacity greater than 3500 mA h g−1, silicon (Si) has become a viable substitute for the graphite anode with a specific capacity of about 370 mA h g−1. This transition aims to boost energy densities for numerous energy-storage applications, including portable electronics and electric cars.90,91 Despite its potential, the commercialization of the Si anode is hindered by issues related to cycling stability and shelf life. These limitations are aggravated by the significant volumetric expansion of silicon (over 300%) during lithiation. This expansion often leads to a decline in specific capacity upon cycling, which is linked to stress formation,92 mechanical fractures, and irreversible degradation reactions caused by the volume changes.93 Lithium metal is widely considered the best alternative anode material with its exceptionally high theoretical specific capacity (3860 mA h g−1) and a very low redox potential (−3.040 V versus SHE).94,95 Due to this, there is considerable interest in utilizing lithium metal as an anode in ASSLB configurations.96 In the following parts, the Li anode is generally referred to, although it is important to note that this analysis is also relevant to silicon and carbon anodes.
Metallic Li has been the main focus of most ASSLB studies to maximize cell energy densities.97,98 A great obstacle to improving the efficiency of ASSLMBs is the interface between SE and electrodes. This interface is crucial for understanding battery electrochemistry, as it is the site where lithium ions and electrons interact and are deposited in the electrode through processes such as intercalation, alloying, or plating.99–104 The interface is often complicated by the existence of passivation layers on the electrode. Characterization of this layer on negative electrodes started with Dey's findings for Li metal immersed in non-aqueous electrolytes.99 In 1979, Peled announced the idea of the SEI as a passivation layer that was electronically insulating and ionically conductive, emerged between the electrode and electrolyte, and functioned as an SE.100 Thus, it is called the SEI. This model was further enhanced with compositional information observed over two decades and summarized by Peled et al.101 in 1997 and Aurbach et al.102 in 1999. On one hand, a dense and undamaged SEI can hinder electron passage, thereby preventing further degradation of the SE, which is essential for the chemical and electrochemical stability of ASSLB. Conversely, the emergence and development of the SEI consume active Li and SE, leading to capacity loss, rising battery resistance, and reduced power density.105 Meanwhile, the uneven SEI may cause Li dendrite growth and safety concerns upon charging.
The composition and growth rate of the SEI layer between lithium metal and argyrodite-type solid electrolytes (e.g., Li6PS5X, where X = Cl, Br, I) can vary significantly, impacting interfacial resistance and overall battery performance.31,106 When lithium metal contacts argyrodite electrolytes, a chemical reaction occurs, leading to the formation of an SEI composed of Li3P, Li2S, and LiX. The rate at which interfacial resistance increases can differ among various argyrodite compositions. For example, Li6PS5Cl forms a protective interphase consisting of Li3P, Li2S, and LiCl when in contact with lithium metal. In contrast, Li6PS5I exhibits more reactive behavior, resulting in a different SEI composition and potentially higher interfacial resistance.12,31,106,107
In general, the composition of the SEI layer and the rate at which interfacial resistance increases between lithium metal and argyrodite electrolytes are influenced by several factors, including the specific halide component of the argyrodite, the growth mechanism of the SEI, and the condition of the lithium anode surface. Understanding these variables is crucial for optimizing the performance and longevity of solid-state batteries utilizing lithium metal anodes.
Thus, there are still plenty of challenges to overcome before Li metal anodes can be used in ASSLMBs: (1) unmanageable lithium dendrite development due to electrochemical and mechanical instability at the SE/Li interface causes serious safety issues;108 (2) the thermodynamic instability of lithium metal because of its high Fermi energy level can result in irreversible and continuous reaction between lithium and SE that produces a thick solid SEI layer on the lithium metal surface, consume lithium and SE, and elevate the internal resistance, thus shortening the cycling span; and (3) repeated plating and stripping operations can cause significant volumetric and morphological alterations to the lithium metal anode; however, the previously mentioned SEI coatings are very thin to inhibit such substantial alterations to the lithium metal anode completely.109–113 These adverse effects may result in significant safety risks and a loss of efficiency. These harmful effects might be mitigated by SEs that are chemically stable towards the lithium anode. Nevertheless, several unanswered questions remain about the kinetics and nature of ion transport at Li/SE interfaces. To realize energy-dense ASSLBs, the Li metal morphology must be controlled during stripping/plating processes at the Li/SE interface.46,114,115
At low potential, Li6PS5Cl undergoes a reduction of P (approximately 1.08 V vs. Li/Li+) during lithiation, resulting in the byproducts Li2S, LiCl, and P (Fig. 4a), which renders it unstable towards the lithium anode.28–30 Li6PS5Cl also produces Li2S, LiCl, and Li3P as byproducts when in contact with the lithium anode (chemical decomposition).31
 | ||
| Fig. 4 Decomposition pathway and XPS analysis. (a) Electrochemical/chemical degradation pathway of Li6PS5Cl.118 (b) The diagram illustrates the emergence of voids at the Li/Li–argyrodite SE interfaces.46 Progression of core-level XPS spectra during the virtual electrode plating process at the Li6PS5Cl surface for (c) Li 1s, (e) S 2p and (g) P 2p. Quantification of XPS spectra as a function of different quantities of charge passed at various CDs, showing compositional fractions of (d) metallic Li (Li0) in Li 1s, (f) Li2S in S 2p, and (h) Li3P in P 2p. A higher fragment of Li0 (shown in panel d) and Li3P (indicated by green area in panel h) at low charge levels and elevated CDs suggests that the reaction kinetics at the interface is more rapid, leading to a more immediate emergence and development of a metallic Li layer during plating.45 (i) XPS spectra for S 2p, P 2p, O 1s, and Cl 2p of a Li6PS5Cl SE as the quantity of accumulated Li metal rises (progressing from bottom to top).31 | ||
Narayanan et al. studied the development of the SEI during virtual electrode plating at the Li6PS5Cl surface via XPS measurements.45 In the left panel of Fig. 4c, the Li 1s spectra at an electron beam current (EBC) of 30 μA with jeq ≈ 0.15 mA cm−2 initially show the peak widening and then a gradual shift to lower binding energies. The observed peak broadening can be directly linked to the creation of Li2S, LixP, LiCl, and other byproducts from contaminants such as Li2O and Li2CO3.31,116 In addition, the authors determined that Li deposited more rapidly at greater current densities compared to lower ones (Fig. 4d). As shown in Fig. 4e, for the S 2p signal, a doublet feature associated with Li2S (with a 2p3/2 binding energy of around 160 eV, highlighted in brown in Fig. 4e) quickly emerges as more lithium is plated. This component of the Li–Li6PS5Cl SEI is well-documented and forms even under conditions where Li is deficient.31,116,117 A detailed analysis of the constitution from spectra collected at various EBCs (Fig. 4f) reveals that the reduction of Li6PS5Cl to Li2S occurs significantly faster at 0.15 mA cm−2 (greater CD). In contrast, at lower current densities (jeq ≤ 0.05 mA cm−2), only about 70% of the S 2p spectrum consists of the reduced sulfide families. This suggests that the reaction kinetics is slower at low CDs due to multiple reduction reactions competing for available reactants, such as plated lithium. Furthermore, analyzing the P 2p spectra shows that at the highest current density (jeq), for an equivalent charges passed value of less than 5 μA h cm−2, there is a rapid reduction to a low binding energy doublet feature (2p3/2 ≈ 126 eV), indicative of fully reduced Li3P (Fig. 4g, left panel, green area). In comparison, at lower current densities (jeq ≤ 0.05 mA cm−2) and the same extent of charge, more notably, the initial SEI consists of a broad spectral feature (126 eV < B.E. 2p3/2 < 131 eV), which is indicative of partly reduced LixP. Over time, continued plating at these lower current densities also leads to the formation of Li3P. However, this process is slower and followed by a notable reduction in the total P 2p spectral intensity, indicating the deposition of plated lithium metal. These findings indicate that a Li3P-rich SEI forms more quickly in the early stages of plating via large CDs, even with a low value of equivalent charges passed. The fragmentation of Li3P as a function of equivalent charges passed, especially for qA < 10 μA h cm−2 (Fig. 4h), supports this observation. Additionally, as seen in Fig. 4e and g, the XPS signal for pristine Li6PS5Cl components diminishes more rapidly at high current densities for the same value of equivalent charges passed (e.g., qA = 12.8 μA h cm−2).
In our group's studies, we noted that the Li 1s spectra at the Li/Li6PS5Cl interface after cycling revealed the emergence of LiCl. Furthermore, the S 2p spectra of Li6PS5Cl after cycling showed an increase in the signal intensity for polysulfide, P2Sn, accompanied by decomposition products, including lithium polysulfide (Li2Sn) and Li2S, resulting from sulfide decomposition.89 According to Zeier's group, Li6PS5X disintegrates into an interphase made of Li3P, Li2S, and LiX when it comes into contact with Li metal.31 This interphase acts as an SEI and raises the interfacial resistance. For instance, according to this group, after contact with Li metal, Li6PS5Cl decomposes into Li2S, Li3P, and Li2O (Fig. 4i). The degradation of SEs is prompted by increased interfacial resistance.
Another issue at the Li/Li argyrodite interface is the growth of Li dendrites.119–122 Lithium dendrite is broadly seen in various types of SEs.123 Dendrites typically expand in the direction of grain boundaries. Sulfide SEs exhibit apparent dendrites because of weaker adhesion between sulfide particles, which results in a lower resistance to dendrite formation than in oxide SEs.50 Irregular lithium deposition at the anodic interface enables lithium dendrites to pass through the grain boundaries or voids within the bulk of Li argyrodite SEs, inducing battery degradation. Kasemchainan et al. recently showed that the critical current density (CCD) was essential for the lithium plating/stripping characteristics utilizing Li6PS5Cl SEs.46 As seen in Fig. 4b, voids develop with lithium bulk around the interface with the SE when lithium is withdrawn from the interface at a CD that exceeds the rate of replenishment. These voids accumulate over the course of subsequent cycles. The margins of these voids concentrate greater CD than other areas, eventually causing the formation of Li dendrites.
The quantity of the CCD is crucial as it determines the power density of a cell. Small CCDs are typically attributed to uneven potential drops caused by SE/electrode interfacial impedance. This impedance is predominantly determined by chemical, electrochemical, and mechanical stability issues at the interface. Understanding these uneven loss mechanisms and interpreting CCD tests can be difficult since SEs may experience “soft shorts” that remain unnoticed in symmetric cell tests using thick Li metal (greater than 30 μm).115 Moreover, as the CCD depends on plating capacity and cell stack pressure, precise data interpretation requires the reporting of specific experimental conditions (such as Li–metal thickness and stack pressure).114
Critical stack pressure is a concept that was recently developed by Sakamoto and colleagues which emphasizes the dependence of CCD measurements on pressure.114 This measure is a crucial addition to the other metrics used to assess the characteristics of Li electrodes at SE interfaces. Dendrite nucleation may originate from pore formation in lithium metal during stripping and unequal lithium ion transport at the Li/SE interface.124–126 A minimal stack pressure can help prevent pore evolution during Li stripping. Nonetheless, Li plating causes Li filament development and propagation. In Li–Li cells, when these happen at the same time, the pressure will have a big impact on the CCD. Nevertheless, it remains uncertain how these effects will manifest in full cells, where plating and stripping operate independently. More investigation is required to reconcile discrepancies between full-cell and symmetric measurements.
Currently, ASSLMB's performance is still far from that of its target. In general, CCD is a crucial criterion for assessing ASSLMBs. It offers opportunities to fully comprehend failure processes, interfacial behavior, and other electrochemical performance features.50 The performance of Li/Li symmetric cell configurations utilizing Li–argyrodite SEs is summarized in Table 1. Apart from SE characteristics, the mechanical characteristics of Li metal also play a significant role in the existence of lithium filaments within SEs. Recent investigations on the bulk mechanical behavior of lithium metal have shown that power-law creep is the predominant deformation mechanism over a broad range of strain rates and temperatures.127,128 These findings provide a viscous-flow perspective that helps to explain how mechanical stresses change as lithium plating takes place on the SE surface.
| Cell configuration | Critical current density (mA cm−2) | Cut-off capacity (mA h cm−2) | Test temperature (°C) | Ref. |
|---|---|---|---|---|
| RT: room temperature. | ||||
| Li6.3P0.9Cu0.1S4.9Cl1.1 | 3.0 | 3.0 | 50 | 89 |
| Li6.25PS4.75ClN0.25 | 1.52 | 1.52 | RT | 146 |
| Li6PS5Cl0.3F0.7 | 6.37 | 5.0 | RT | 79 |
| Li6PS5Cl | 0.55 | 0.55 | RT | 147 |
| Li6PS4.7O0.3Br | 0.90 | 0.90 | RT | 148 |
| Li6PS5Br | 0.45 | — | RT | 148 |
| Li6.05P0.95Mo0.05S4.9O0.1Cl | 0.55 | — | RT | 149 |
| Li6.04P0.98Bi0.02S4.97O0.03Cl | 1.1 | 0.1 | RT | 150 |
The linked interactions between CD and strain rate directly affect stress build-up at the solid–solid interface. This has considerable indications for the model of Porz et al.,129 where the ejection of lithium metal from the heterogeneous surface of the SEs contributes to the CD-dependent mechanical failure of the SEs.
For the lithium metal anode to work in a reliable, reversible, and safe manner, we must resolve any problems driven by inherent morphological instability while plating and stripping Li metal at the SE interface.49,130 Recent work has significantly advanced our understanding of both dendrite emergence and development and pore formation during stripping, as shown in Fig. 5a and b, respectively.131,132 However, many of these studies operate under excessively high stack pressures, whereas the ideal pressure should be below 0.1 MPa to align with the stack pressures that are typical for lithium-ion batteries.
 | ||
| Fig. 5 Crucial concerns with the lithium metal anode. (a) Filament and dendrite development. (b) Pore and void development.140 (c) Scheme illustrating the main factors affecting CCD.50 | ||
The CCD in solid batteries is influenced by numerous internal and external factors, many of which are interrelated. Due to poor interfacial compatibility in solid-state systems, CCD is more sensitive to external influences than systems using LEs. The major issues are summarized in Fig. 5c. Establishing good physical contact is one of the most essential requirements for SSBs.133 The interfacial contacts are directly related to the interfacial physical contacts and chemical stability. In SSBs, where liquid components are absent, Li-ion pathways rely solely on solid–solid contact.134 Additionally, reactions between Li metal and SEs can cause volume changes and generate interfacial byproducts, further altering interfacial contact.135
The intrinsic characteristics of SEs, such as relative and compacted densities, chemical composition, and mechanical strength, also significantly influence CCD.136 Most SEs are polycrystalline, comprising multiple single crystals, grain boundaries, isolated particles, impurities, and inherent flaws. These structural features impact internal ionic flux and provide sites for Li filament nucleation and propagation. In ASSLB systems, increasing the areal capacity during Li stripping leads to significant contact loss. As reported, 1 mA h cm−2 corresponds to a Li thickness of 4.9 μm.137 For commercial viability, areal capacities above 4 mA h cm−2 are needed, which could create void layers nearly 20 μm thick upon full discharge, severely impacting the interfacial contact and performance, especially at high current densities.138 Additionally, large-scale Li cycling causes substantial volume expansion and worsening contact degradation. According to the terrace–ledge–kink model, it is difficult to recover lost active plating/stripping sites due to sluggish and uncontrollable surficial Li adatom diffusion according to the terrace–ledge–kink model. Li growth exhibits a complexity highly related to the initial morphology and current density, leading to poor CCD.
SSBs must also operate reliably across wide temperature ranges in real-world applications. Temperature affects CCD by altering both lithium's physical properties and electrochemical behavior, such as ionic conductivity and diffusion at the interface. Elevated temperatures improve CCD by enhancing Li+ mobility. In liquid systems, high current dendrite formation is often explained using diffusion-limited models; similar models are now applied to solid-state systems. According to Sakamoto et al., higher CCD reflects a higher Li+ flux across interface regions.139
Pressure is another key external factor influencing CCD. Lithium metal's mechanical properties, such as elasticity, plasticity, and creep behavior, enable it to deform under applied stress. Li shows pronounced creep at higher temperatures, especially under operational stress levels. Accurately predicting lithium's deformation behavior is essential for enabling its use in SSBs. Masias et al. systematically measured the Young's modulus, shear modulus, time-dependent deformation (creep), and stress-dependent deformation.128 The elastic and plastic properties are directly related to solid–solid contacts. Deformed Li gradually flows to the side boundary of batteries without a hydrostatic pressure, increasing the risk of short circuits. The pre-stressed system constrains the undesirable flow of Li metal. The deformation of Li metal also induces microstructural evolution, significantly reducing interfacial troubles and enhancing the CCD values. Pre-pressing Li also minimizes creep-induced shorting.128 Li metal is softer than most inorganic solid-state electrolytes in Li metal-based SSBs. The deformation of Li metal dominates the deformation-dependent impedance reduction. Thus, increasing pressure helps to expand the true contact area at the interface and stabilizes the battery performance.
Kazyak et al. investigated that at elevated current densities and areal capacities, there was an uneven depletion of lithium and accumulation of voids.138 It was demonstrated that the interfacial capacitances primarily stemmed from these voids at the interface. Microelectrodes were utilized to visualize void accumulation (Fig. 6a). Clear areas of lithium depletion were detected following lithium stripping at 1.07 and 1.47 mA h cm−2 (Fig. 6b and c). These depleted regions signify areas of contact loss, resulting in increased polarization (Fig. 6d) and interfacial impedance (Fig. 6e). The loss of electrode-electrolyte contact primarily stems from the accumulation of vacancies at the interface and the volume changes occurring during the continuous stripping of lithium metal.141
 | ||
| Fig. 6 Contact loss at the anode in ASSLMBs. (a) Microelectrode arrangement for experimental use. Depletion regions of (b) 1.07 mA h cm−2 stripping and (c) 1.47 mA h cm−2. (d) Increase in polarization following contact losses. (e) Rise of impedance with continuous stripping.138 Schematic illustrations demonstrate physical contact losses in ASSLBs (f) and dendrite evolution within ASSLMBs (g).141 | ||
Moreover, inadequate contacts present significant difficulties for ASSLBs and can stem from both physical and chemical factors. Contact loss typically manifests during battery fabrication and exacerbates over-cycling. Therefore, the electrochemically active contact area between the active materials and SE is a critical parameter in ASSLBs.87 In the case of active materials in LIB electrodes, most of the surface area of active materials is fully in contact with the LE except for the area covered by the electric conductor and polymeric binder; hence, it can be considered as an electrochemically active contact area. However, the surface of active materials is not sufficiently covered with SEs due to their original shape and mechanical stiffness. Therefore, the real specific contact area (as = Ac/Va),143,144 which is defined as the contact area (Ac) between the active material and SE divided by the bulk volume (Va) of the active material, is considered an important parameter to improve the design of ASSLBs. An adequate as is typically on the order of as ≥ 105 cm2 cm−3. For example, a high-performance ASSLB using a sulfide SE with nano-engineered composite cathodes might achieve as ≈ 106–107 cm2 cm−3. This corresponds to well-distributed and conformal contact between electrolyte particles and the active material.87 Physical contact issues predominantly affect bulk-type ASSLBs (Fig. 6f). Achieving atomic-scale and conformal contact is paramount for optimal performance. However, attaining these perfect contacts in practice is challenging due to particle–particle contact and the presence of pores.141
During the process of lithium stripping, the release of lithium atoms creates vacancy sites, contributing to vacancy accumulation. This accumulation results in the formation of pores and flaws at the operational interfaces. Over time, these flaws diminish the efficient interfacial areas, leading to a partially concentrated flux of lithium ions (Fig. 6g). Moreover, this concentrated lithium ion flux exacerbates the contact loss further. Additionally, during lithium plating, significant volume expansion leads to the formation of more cracks, thereby accelerating lithium pulverization.142 Recent research by Kasemchainan et al.46 revealed that the buildup of voids at the Li/Li6PS5Cl (LPSCl) interface during continual plating/stripping processes triggered the evolution of Li-dendrites. After multiple cycles of stripping and plating, voids were observed at the LPSCl–Li interface compared to the pristine interface. The formation of interfacial voids may result when Li filaments are removed more quickly than they can be replenished. Li-dendrite initiation begins at the triple junction where lithium metal, voids, and SE coexist, eventually leading to short circuits and failure in ASSLBs. Additionally, pre-existing flaws and cracks can serve as pathways for the development and spreading of Li-dendrites through the SEs.145 Understanding how micro-cracks influence Li-dendrite propagation through SEs is crucial, necessitating in situ operando prediction of microstructural alteration, particularly in micro-crack formation, during battery cycling.145
2.2. Electrolyte–cathode interface
In contrast to metallic anodes, cathode components consist of active material (AM) particles like LiCoO2 (LCO), LiMn2O4, LiNiMnCoO2, LiNiCoAlO2, and LiFePO4. These particles combine with a suitable SE (ionic conductor), carbon (electron conductor), and/or binder to improve ion and electron transportation. In an LE battery, the LE fills the voids between cathode components and provides intimate contact with the electrodes. In contrast, solid–solid interfaces in ASSLBs rely on physical contact. To analyze the impacts of interfaces, Luntz et al.151 utilized a symmetrical cell to assess the internal resistance (IR) drop at both cathode/SE and anode/SE interfaces. Fig. 7a and b presents the evaluation method and findings. In their experimental work, an Au/SE/Au configuration was used as the reference for IR drop measurements. A negligible IR drop was observed when using a Li/SE/Li configuration, indicating interface resistance between Li and SE. However, combining SE with carbon electrodes (Fig. 7c and d) led to a significantly higher IR drop compared to Li, showing greater interfacial resistances. The IR drop at the Li/SE interface was ascribed to SEI generation and inadequate contact. In contrast, the electrochemical processes at the carbon–electrolyte interface are more complex. Although their findings highlight the distinct contributions of interface resistance at both electrodes, they focused solely on carbon electrodes when concluding that the greatest interfacial resistance happened at the carbon/SE interfaces. | ||
| Fig. 7 Common cathode/SE interfaces in solid-state batteries. (a) Au/SE/Au. (b) Li/SE/Li. (c) Li/SE/C. (d) C|SE|C.151 Potential variation (U), carbon (C). (e) A schematic energy level diagram of a Li cell using LiCoO2 as the cathode and Li metal as the anode shows how the electromotive force (EMF) originates. In this diagram, the Fermi level of the lithium anode lies above the electrolyte's LUMO, which explains the typical reduction of the electrolyte upon contact with Li. Additionally, during Li deintercalation from the cathode, the Fermi level of LiCoO2 shifts downward, as indicated by a small arrow in the diagram.157 | ||
When two materials with differing Fermi levels come into contact, electrons will flow from the material with higher Fermi energy to the one with lower Fermi energy until equilibrium is reached. This electron transfer causes band bending at the interface, forming space charge regions and potential barriers that can impede charge carrier movement.
When gold (Au) and an SE come into contact, their Fermi levels (EF), representing the chemical potential for electrons, are typically not aligned initially. The electrons flow from one to the other and lead to electronic leakage, causing decomposition. This impacts charge transfer and interfacial stability.
In the case of carbon and SEs, the Fermi level of carbon may align differently with the conduction band of the SE. This misalignment can influence the charge transfer kinetics and the stability of the interface, potentially leading to issues such as interfacial resistance or degradation over time.
From a physical point of view, one may relate the cell voltage to the difference of the (electro-)chemical potential of the electrons between the cathode and anode as given by the position of their Fermi levels (see Fig. 7e). Note, however, that the chemical potential difference for lithium between the anode and cathode may be expressed as the sum of the chemical potential difference for both electrons (Δμe−) and lithium ions (ΔμLi+), eqn (1):152,153
| −eVOC = Δμe− + ΔμLi+ | (1) |
Therefore, considerations restricted to the Fermi level (electron chemical potential) of (single) electrode materials in discussing electrode potentials are approximate, albeit often resulting in reasonable accuracy.
Besides the active (intercalation) material, commercial Li-ion electrodes consist of a polymer binder and conductive additives, forming complex, often nano-sized, 2D or 3D composites. The performance of these composite electrodes is highly dependent on the distribution, ratio, and types of different constituents.154,155 From an idealized point of view, the voltage and capacity of Li-ion batteries are only determined by the thermodynamic bulk properties of the active phases. However, contact potentials and kinetic effects in real batteries strongly influence the practically achieved voltages, capacities, and current densities. As a consequence, all practical performance data (e.g., energy and power density, lifetime) are influenced by kinetic factors, which are often dominated by interface effects and continually evolve over time due to side reactions and other degradation phenomena.156
Nowadays, layered LiMO2 materials (M = Ni, Co, Mn) are widely recognized as standard cathode active materials (CAMs) for high-energy-density batteries. Particularly, LiNiaCobMncO2 (a + b + c = 1; NCM-abc) and LiNixCoyAl1−x−yO2 (NCA) are capable of achieving higher voltages and higher volumetric specific energies. As a result, they have established themselves as primary CAMs in electric vehicle applications.
For instance, Ni-rich NCM cathodes, such as LiNi0.8Mn0.1Co0.1O2 (NCM811), are highly attractive due to their competitive capacities and energy densities compared to advanced LIBs.158–161 However, integrating sulfide-based ASSLMBs with Ni-rich oxide cathodes faces significant difficulties: (1) sulfide SE degradation occurs at high voltages due to their restricted ESW; (2) undesirable interfacial reactions between sulfide SEs and NCM811 result in the existence of ionic-insulating degradation products; (3) an SCL forms between sulfide SEs and oxide cathode materials because of mismatched chemical potentials, leading to highly resistant lithium depletion layers at the sulfide SE side; (4) structural deterioration at the surface and grain boundaries of Ni-rich oxide cathode particles leads to capacity and voltage decay issues.52,162–165 Although raising the upper cutoff voltage can enhance the capacity of Ni-rich NCM811 cathode materials, it also risks material degradation due to issues such as cracking and oxygen loss. Grasping these degradation mechanisms is essential for facilitating high-voltage operation and enhancing the capacity of advanced materials.166,167 Recently, studies have focused on single-crystal (SC) NCM-based cathodes for ASSLBs. These materials typically have a lower nickel content to reduce volume changes during cycling, although this approach does come with the trade-off of decreased discharge capacity.168–170
Chemical decomposition of an SE can lead to contact loss between the cathode and SE due to the volume decrease associated with the chemical reaction, which can cause physical separation or cracking.25,171,172 The decomposition process can form new interphase layers or degrade existing ones, hindering the ionic conductivity and leading to resistance increases. In addition, chemical and mechanical degradation at the solid electrolyte/cathode interface can interact synergistically, accelerating performance decline in solid-state batteries.173–175 Chemical degradation, such as the formation of resistive reaction layers or the dissolution of cathode materials, can induce mechanical stresses at the interface, leading to cracking or delamination. Conversely, mechanical stress from cathode expansion/contraction during cycling can accelerate chemical degradation by exposing fresh reaction sites. This can lead to capacity loss, reduced rate capabilities, and a general decline in battery performance.
From a microstructural perspective, lattice mismatches present physical contact challenges. Such interfaces often arise around the contact area of solid materials with differing lattice parameters. This mismatch can happen around electrode/SE interfaces, inducing strain and the existence of super-lattices, consequently elevating interfacial resistance.141 In contrast, interfaces between materials with minimal lattice differences exhibit substantially lower interfacial resistances and higher σ compared to those with significant lattice deviations. It is important to note that lithium-ion transport throughout all ASSLBs is also affected by interface impedance and bulk σ. Indeed, weak Li+ transport may also manifest at the lattice-matched interface with inadequate bulk σ.
Theoretical calculations help to elucidate the electronic/atomic distributions around lattice mismatched interfaces. Utilizing DFT calculations, Jand and Kaghazchi investigated LiCoO2/SE interface structures.176 This investigation can be done by varying the biaxial compressive and tensile strain magnitudes. Their simulations identified three energetically favorable interfaces. Analysis of all three models revealed atomic rearrangements and a big strain at the interface due to lattice mismatch. While this model does not include ab initio molecular dynamics, it showcases the successful application of DFT methods in simulating and predicting interfacial characteristics in ASSLBs. Nevertheless, predicting interfacial behavior in ASSLBs remains complex. Therefore, a sophisticated computational technique employing an intelligent algorithm is necessary to find energetically favorable interface structures. This provides a comprehensive understanding of lattice-mismatch mechanisms.
Recently, Gao et al. introduced an estimation method to understand lattice-mismatched structures around interfaces using the particle swarm optimization (CALYPSO) method.177 This approach considered both lateral and vertical displacements. By combining the CALYPSO approach and DFT calculations, they identified 6 kinds of LiCoO2/Li3PS4 interface structures, as illustrated in Fig. 8a. Simulation results highlighted the existence of highly energetically favorable interfaces, such as cobalt sulfide and phosphorus oxide. Here, cation (cobalt/phosphorus) and anion (sulfur/oxygen) inter-diffusion occurs around interfaces. These theoretical findings align well with experimental observations.178Fig. 8b shows two different interfaces.141 On the left, two materials share the same crystal structure and similar lattice dimensions, creating a straightforward pathway for Li-ion migration. This leads to minimal interfacial resistance and high ionic conductivity. In contrast, the right-hand side illustrates a pairing with markedly mismatched lattice structures, which forms a convoluted Li-ion trajectory and significantly increases interfacial resistance. This issue is widespread in current ASSLBs, where interfaces typically exhibit considerable lattice mismatch. However, effective Li-ion transport within ASSLBs hinges on more than just interfacing crystal lattices. It also relies heavily on bulk ionic conductivity, grain boundary diffusion, and overall interface impedance. Even a well-aligned—i.e., lattice-matched—interface can suffer from sluggish Li-ion movement if the materials themselves possess inherently low ionic conductivity. A viable strategy to alleviate lattice variations is by employing materials with greater structural resemblance for both electrodes and SEs.
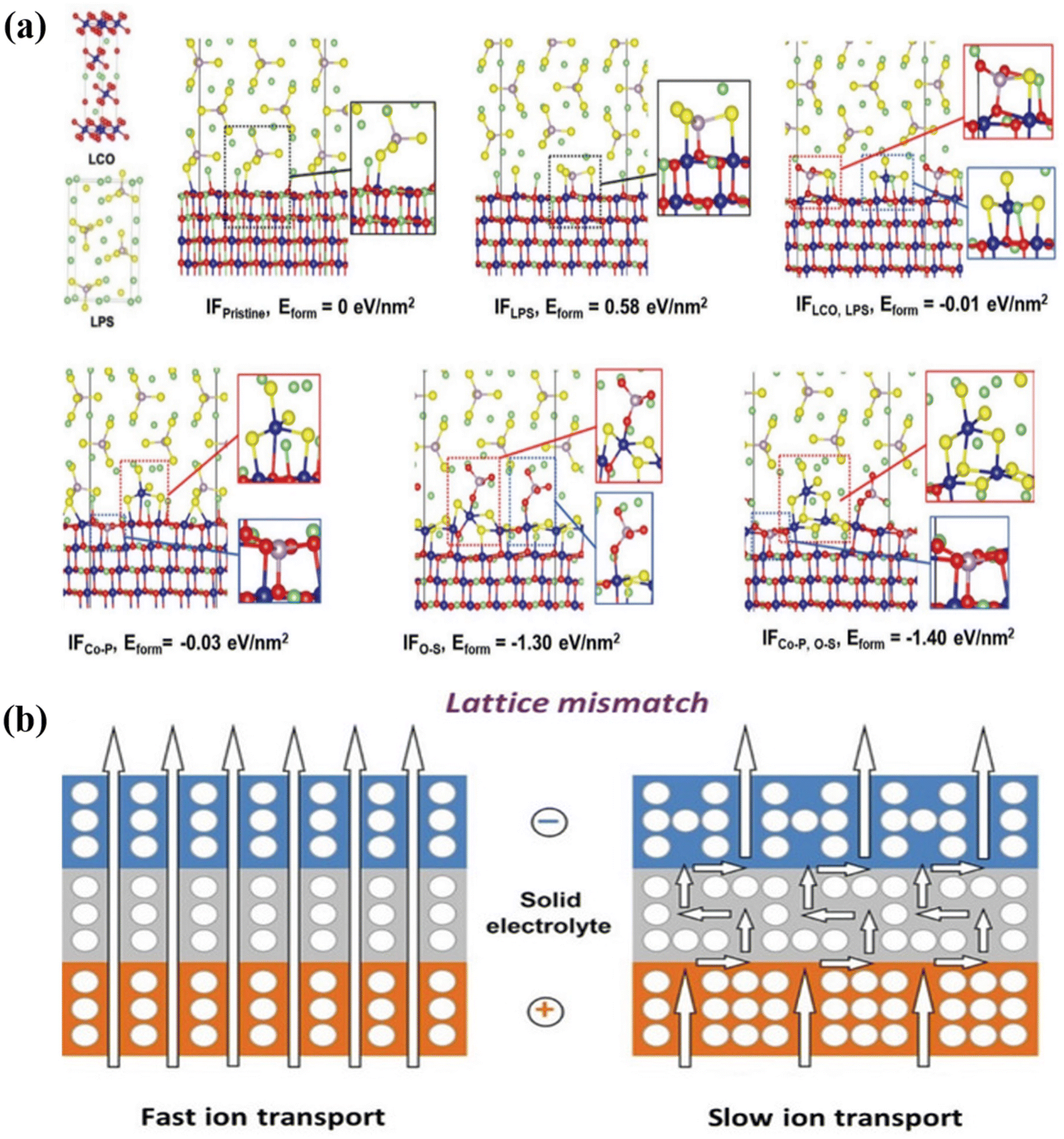 | ||
| Fig. 8 (a) Predicted low-energy interface structures between LiCoO2 and Li3PS4 for six different energy states.177 (b) Lattice (mis)match.141 | ||
Generally, when a sulfide SE comes into contact with layered oxide cathode materials with a low lithium ion chemical potential (μLi) vs. S2−/S, the sulfide SEs undergo oxidation even under open circuit voltage (OCV) conditions. This oxidation process contributes to the degradation of both the SEs and the CAMs, leading to structural damage.
Even though ASSLBs have attracted significant attention owing to their potential increased energy density and high safety, the thermal runaway of batteries cannot be mitigated just by utilizing nonflammable SEs. The interface between the cathode material and the electrolyte is a critical area for safety concerns.179–182 Safety issues in solid-state batteries, particularly with argyrodite SE and layered cathode materials, are often linked to interfacial phenomena like SO2 gas formation during exothermic reactions.183 These reactions can lead to heat buildup, decomposition of the SEI, and ultimately, thermal runaway. Safety hazards with exothermic behavior and combustion phenomena were demonstrated between sulfide SEs (glassy-ceramic and crystalline types) and the fully delithiated Ni-rich (LiNi0.8Co0.1Mn0.1O2, NCM) layer cathode. The heat generation of sulfide SEs + NCM was 900% larger than that of LE + NCM.183 The interaction between the cathode and electrolyte can also reduce the decomposition temperature and exacerbate safety concerns. Compared with comprehensive investigations on the safety of LIBs,184,185 it is imperative to reveal the comprehensive safety characteristics of ASSLBs with respect to multiple aspects, guiding the safety design of sulfide-based ASSLBs.
2.3. Mechanical properties
The mechanical characteristics of SEs represent a crucial aspect of the development of ASSLBs.32 For instance, SEs need to possess high processability to establish close solid–solid contact between electrode active materials (EAMs) and SEs.186–188 Achieving intimate contacts between solid particles is vital for ensuring high-performance metrics such as increased capacity, extended cycle life, and enhanced rate capability. In ASSLBs, where both EAMs and electrolytes are solid, establishing close contact between the electrode and SE is challenging compared to traditional batteries employing LEs. Moreover, maintaining this contact throughout charging and discharging is essential despite the volume alterations experienced by the EAMs during these processes. The mechanical properties of both the electrode and SEs play a vital role in determining the integrity of the electrode/electrolyte contacts upon cycling. The elastic modulus serves as a crucial mechanical property for evaluating the electrode/SE contact.189In ASSLBs, EAMs undergo expansion and contraction during the charge/discharge process. For example, silicon electrodes boast high theoretical capacities (3579 mA h g−1 for the Li15Si4 phase) but experience significant volume changes exceeding 400%.190–192 SEs play a crucial role in maintaining electrode/electrolyte contacts despite these volume changes in the EAMs. However, when SEs with very high elastic moduli are employed, the EAMs endure substantial stresses due to the volume alterations. Consequently, they may fracture and lose contact with the SEs, resulting in capacity degradation. The elastic modulus of SEs influences this fragmentation of electrode materials; ideally, SEs should possess an average elastic modulus that is neither excessively high nor extremely low.193 SEs with moderate elastic moduli can alleviate such large stresses, thereby enhancing the longevity of ASSLBs.
The physicochemical behavior of SEs differs significantly from that of LEs, especially in how they influence Li deposition. SEs are intrinsically inhomogeneous, containing grain boundaries and grains with distinct mechanical properties and carrier transport characteristics. Notably, the bulk electronic conductivity of SEs is higher than that of LEs, allowing Li ions to be reduced and deposited directly within the SE matrix. At the interface between SEs and electrodes, electrochemical reactions such as Li metal deposition occur at solid–solid contact points, enabling the transfer of charge carriers (either electrons or ions).194–196 These reactions induce mechanical changes in the SE, often resulting in localized lattice deformation.197 Typically, such deformations are confined to surface regions, creating localized stress fields. In most SEs, which typically exhibit linear elastic fracture behavior, the level of local stress directly influences the crack formation ability, as well as the dimensions, such as the length and width, of any cracks that develop.198–200 The severity of the resulting mechanical damage depends on the intensity of the stress and the dimensions of the developing cracks. Various theoretical models have been proposed to explain how Li protrusions grow within SEs, and all of them emphasize the strong relationship between Li metal electrochemical deposition and the generation of local mechanical stress.125
The different mechanical processes in SSBs, localized in different areas of the cell, are illustrated in Fig. 9a. Key processes include: losing contact at the electrode/electrolyte interfaces, detachment from current collectors (Cu or Al foils), void generation at interfaces, crack propagation inside SEs, and the application of external pressure. These processes are induced and influenced by many electrochemical, chemical, and external factors. First, the volume changes (expansion or shrinkage) in cathodes and anodes due to lithiation or de-lithiation have a profound effect on the solid/solid interface between the electrode and electrolyte. The second major factor is the external pressure applied to the packaged cell and/or module, which is inevitable in single-cell and large battery packs. Moreover, dendrites and byproducts generated at the interfaces can also add local pressure. These mechanical effects have a significant influence on further electrochemical processes, such as increasing impedance, uneven distribution of the lithium-ion flux and electrical field, fast decay in capacities, and short circuits. As a whole, the mechanical processes are intimately coupled with the electrochemical ones. The mechanical parameters for evaluating SSBs and corresponding characterization methods are summarized in Fig. 9b.
 | ||
| Fig. 9 (a) Schematic of mechanical processes and their causes in SSBs. (b) The mechanical parameters and corresponding characterization methods used in SSBs.211 (c) Schematic setups used to ensure ion-blocking and electron-blocking measurement conditions together with transmission line models (TLM) used to evaluate impedance data. (d) EIS data (circles) measured under ion- and electron-blocking conditions are exemplary for an NCM83–LPSCl composite with an NCM volume fraction of 40%. (e) Direct current (DC) polarization data measured under ion-blocking (circles) and electron-blocking (diamonds) conditions, as well as the corresponding fits (line) are exemplarily shown for an NCM83–LPSCl composite with a NCM volume fraction of 40%. (f) Resulting effective ionic and electronic conductivities as a function of NCM volume fraction. Effective electronic conductivities measured via DC polarization (circles) and EIS (squares) are shown in purple, whereas effective ionic conductivities are depicted in green. Each data point corresponds to a single measurement. Effective conductivities from simulations with the resistor network model are shown as dots connected with straight dashed lines as a guide to the eye.205 | ||
In addition, SSBs use composite electrodes made up of electrochemically active materials, SEs, and, when needed, additional components such as binders or conductive additives like vapor-grown carbon fibers (VGCFs). In these composite structures, achieving sufficiently high ionic conductivity (σion) and electronic conductivity (σe) is essential, as both charge carriers must effectively reach the active material during charging and discharging. A significant mismatch between ionic and electronic conductivity can result in uneven reaction rates across the electrode's thickness. It is crucial to balance these transport properties to ensure efficient electrode utilization and prevent localized reaction fronts during operation.201,202
The transport of charge carriers is strongly affected by the microstructure of the composite. For example, Froboese et al.203 showed how varying the size of inclusions impacts the composite's effective ionic conductivity. Additional research on NCM622-LPSCl201 and Si-LPSCl-C202 electrodes demonstrates that changes in active material particle size significantly influence battery performance, reinforcing the importance of microstructural control. Similarly, adjusting the SE particle size in LiNi0.83Co0.11Mn0.06 (NCM83)–LPSCl204 composites leads to more uniform ion flow and better electrode utilization. These findings highlight that the particle sizes of both the active material and the solid electrolyte are critical factors for optimizing electrode design.
Ketter et al.205 measured the effective ionic and electronic conductivities in NCM83–LPSCl composite cathodes by varying the volume fractions of NCM83 (φNCM) and LPSCl (φLPSCl), using EIS. To analyze electronic and ionic conductivity separately, they applied ion-blocking contacts for electronic measurements and electron-blocking contacts for ionic measurements (as shown in Fig. 9c). The impedance data were interpreted using a transmission line model (TLM), previously developed for similar NCM622–LPSCl composite systems.206 This TLM acts as an equivalent circuit representing ion and electron transport through interconnected pathways in the composite cathode. Although the model simplifies the complex nature of charge transport in such composites, it captures key behaviors and enables the accurate determination of total effective conductivities from EIS data.206
Fig. 9d illustrates typical impedance results and their TLM fits for a composite cathode with 40% NCM83. To further validate the TLM, researchers used an alternative equivalent circuit commonly applied to mixed ionic–electronic conductors207 to assess the same data, demonstrating consistent results. In addition, DC polarization experiments were performed to evaluate ionic and electronic currents by applying different voltages and blocking contacts to isolate each charge carrier type. These measurements, interpreted using Ohm's law (eqn (2)), yielded effective conductivity values. An exemplary dataset for φNCM = 40% is shown in Fig. 9e.
An equivalent relationship for the movement of charge carriers (ions and electrons) as a response to an electric field is given by Ohm's law (eqn (2)):208
| J = −σ∇ϕ | (2) |
The measured and simulated effective electronic and ionic conductivities are in good agreement, as shown in Fig. 9f. Consistent with previous findings,206 the effective conductivities of both ions and electrons vary dramatically depending on the ratio of NCM83 to LPSCl. Specifically, electronic conductivity spans from about 101 mS cm−1 to 10−2 mS cm−1, while ionic conductivity ranges from 101 mS cm−1 to 10−5 mS cm−1 across the compositions tested. The best ionic and electronic transport balance is observed at φNCM = 40%. However, as the content of either LPSCl or NCM83 becomes too low, both conductivities drop sharply due to a loss of percolation pathways. Since the percolation threshold is highly influenced by the material's microstructure, significant discrepancies between experimental and simulated conductivity values are more likely near the threshold as only simplified, virtual microstructures are assumed in the resistor network simulations.209,210 Differences in the measured and simulated effective ionic conductivities for compositions with φNCM > 40% may be attributed to this effect.
Recently, researchers utilized first-principles calculations to predict the elastic moduli of different SEs.212 These efforts underscore the significance of identifying the optimal mechanical properties for ASSLBs and advancing the development of SEs with enhanced mechanical characteristics.193 Key mechanical parameters to be taken into account for SEs are outlined in Table 2.
| Electrolyte | B (GPa) | G (GPa) | E (GPa) | v | G/B |
|---|---|---|---|---|---|
| Li6PS5Cl | 28.7 | 8.1 | 22.1 | 0.37 | 0.28 |
| Li6PS5Br | 29.0 | 9.3 | 25.3 | 0.35 | 0.32 |
| Li6PS5I | 29.9 | 11.3 | 30.0 | 0.33 | 0.38 |
Addressing interfacial resistance entails optimizing the contact between materials, a pursuit that has been investigated on a macro-scale through different synthesis and assembly methods.213 Enhanced adhesion contributes to prolonged mechanical lifespan in ASSLBs by ensuring consistent interface contact during cycling. Experimental evidence supports this notion, as demonstrated by the application of pressure to cells, facilitating improved wetting or adhesion of interfaces.214 In ASSLBs, expanding the contact area between materials is crucial for leveraging the bulk σ offered by Li–P–S systems.
3. Strategies to improve the interface stability
Because of Li metal's strong reactivity, limited materials are stable towards Li.63,215 The critical factor for effectiveness in LIBs is the self-passivating SEI layer formed at the LE/electrode interfaces. This concept recently started to be explored in the ASSLB domain. Advancing the discovery of non-reactive SEs at interfaces is a critical step for the development of next-generation batteries. However, this focus does not address the operational voltage or interfacial resistance criteria.216The strategies proposed to improve the interfacial stabilities include coating, electrolyte modification by doping, wetting, and multilayered approaches.
3.1. Coating
Applying coating materials is the best approach for tackling the interfacial problems, and enabling a wider range of combinations of SE and electrodes to be used for the success of the battery. Coatings must possess electrochemical stability windows that sufficiently cover the entire operational voltage range of the electrodes. Nevertheless, even if two materials are chemically inert and show stability in similar voltage ranges, they can still encounter significant interfacial resistance.217Kimura et al.218 systematically investigated how the thickness of a coating layer (CL) affected the protection of SEs, under fixed values for electronic conductivity (σele) and other related parameters. They assumed σele values of 1.0 × 10−11 S cm−1 for the SE and 1.0 × 10−14 S cm−1 for the CL, with the lithium-ion conductivity (σLi+) of the CL set at 1.0 × 10−6 S cm−1. Fig. 10a shows the lithium chemical potential (μLi) distribution within the SE and CL when the thickness of the CL is 10, 50, and 100 nm and the thickness of the SE is 100 μm. As the CL thickness increases, the μLi variation within the coating becomes larger, which raises μLi at the CL/SE interface. Notably, coatings thicker than 50 nm shift μLi above the SE's oxidation limit, making the SE thermodynamically stable. This supports the intuitive understanding that thicker coatings offer stronger protection, especially when ultra-low σele materials are not available.
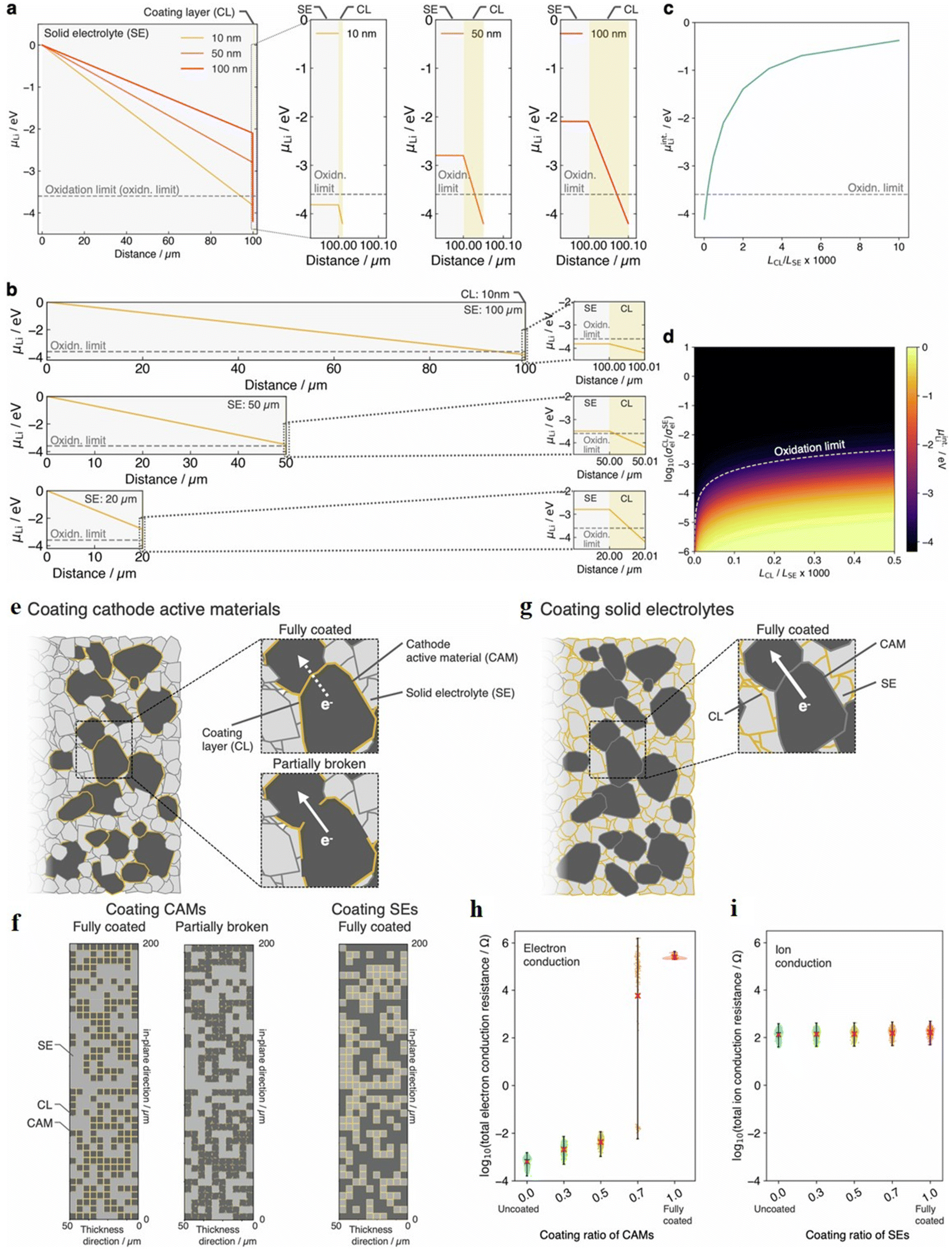 | ||
| Fig. 10 Influence of the thickness of the CL and SE on the protective capability of the CL. (a) μLi distribution within the SE and CL for CL thicknesses of 10, 50, and 100 nm, with the thickness of the SE constant at 100 μm. (b) μLi distribution with SE thicknesses of 100, 50, and 20 μm, maintaining the thickness of the CL at 10 nm. (c) μLi at the interface as a function of the thickness ratio of CL to SE. (d) Heat map illustrating μLi at the interface between the SE and CL across varying thickness ratios and σele ratios of CL to SE. Influence of the arrangement of the CL in composite electrodes on the ohmic resistance. (e) Schematic of a composite SSB electrode with CLs applied to CAMs. (f) 2D lattice model of the composite SSB electrode with CAMs either fully or partially covered by CLs (left) and SE-coated configuration (right). (g) Schematic of a composite SSB electrode with CLs applied to SEs. Violin plots of (h) the electron conduction resistance of the entire electrode under various CAM coating ratios and (i) the ion conduction resistance of the entire electrode under various SE coating ratios. The red cross in each plot represents the average resistance for each condition.218 | ||
However, the thickness of a CL required to effectively protect an SE varies considerably based on the thickness of the SE. As shown in Fig. 10b, the protective effect changes when the CL remains 10 nm thick but the SE thickness is varied (100, 50, and 20 μm). For the 100 μm SE, μLi at the interface remains below the oxidation limit, indicating instability, while SEs 50 μm or thinner show interfacial μLi values within the stable electrochemical window. Hence, the thickness of the SE directly impacts on how well a given CL performs.
Fig. 10c plots the relationship between μLi at the interface and the thickness ratio (LCL/LSE). A sharp increase in interfacial μLi is observed until the CL reaches about 0.2% of the SE thickness, stabilizing around 0.02%. This means that, for a 100 μm-thick SE, a coating thickness below 200 nm is often sufficient for protection. Crucially, this shows that the relative thickness, not the absolute thickness, of the CL determines its effectiveness. Consequently, a CL that performs well in thin-film SSBs may not offer the same protection in bulk-type batteries, and vice versa. Any comparison of CL performance across systems must consider the SE geometry.
Moreover, the ideal CL thickness also depends on the relative σele values of the CL and SE. Fig. 10d shows a heat map of μLi at the CL/SE interface under varying σele and thickness ratios. A white dashed line marks the oxidation stability limit. For a CL to protect the SE thermodynamically, both its thickness and σele must fall below this line. The map reveals that at very thin coatings (e.g., <10 nm for a 100 μm SE), small changes in thickness dramatically shift the required σele. Therefore, in practical battery design, achieving effective SE protection requires carefully balancing both the thickness and electronic conductivity of the coating layer.
As previously outlined, the protective effect of a CL on an SE in SSBs is influenced by both σele of CL and its thickness. While a lower σele and greater thickness are generally favorable for SE protection, practical considerations in composite electrodes, especially those without conductive additives, complicate this relationship. In composite electrodes, especially those lacking conductive additives, CAMs serve as the primary electron conduction pathways. Applying a thick CL with low σele, as illustrated in Fig. 10e, over the CAMs can significantly increase the electron conduction resistance across the entire electrode, potentially degrading SSB performance.219,220 However, in real composite electrodes with the CL, such a substantial increase in resistance is often mitigated by partial fracturing of the CL during electrode fabrication processes, such as pressing (Fig. 10e). These fractures can restore electron conduction pathways, reducing the overall resistance. The impact of the proportion of CLs is shown in Fig. 10f. When 70% of the CAM surface is coated, the resistance shows significant variation depending on the arrangement of the AM and SE lattices, likely because of differences in the availability of current percolation pathways that enable current to flow solely through the CAM regions. When the CL coverage is reduced to 50% or less, the increase in electron conduction resistance remains within acceptable limits, suggesting that partial coverage can balance SE protection and electron conduction. However, this means that over half of the interface remains unshielded, underscoring the challenge of achieving solid electrolyte protection and low resistance in practical composite solid-state battery electrodes.
An alternative strategy involves coating the SE instead of the CAMs. This approach maintains direct contact between CAMs, preserving electron conduction pathways, and typically results in a less pronounced difference in σLi+ between the SE and CL compared to the σele difference between CAMs and CL (Fig. 10g). Consequently, the overall ion conduction resistance of the electrode does not increase significantly, even with a fully coated SE. Fig. 10h presents violin plots illustrating the electron conduction resistance of the whole electrode under various CAM coating conditions. The total resistance in the case of fully coated CAMs is about 8 to 9 orders of magnitude higher than uncoated CAMs. This outcome clearly shows that even though the CL is much thinner than the CAM, a coating layer with low σele can significantly increase the overall electron conduction resistance of the electrode, indicating the strong influence of the CL on electrode performance. Fig. 10i presents the total ionic resistance of the composite electrode when the SEs are coated with a 10 nm-thick CL. In this model, σLi+ of the SE and CL are set to 0.5 × 10−3 and 1.0 × 10−6 S cm−1, respectively, with ion transport attributed solely to the SE and CL. Under these conditions, the increase in ionic resistance remains relatively minor, even with full coating of the SEs. This indicates that applying the coating to the SE rather than the CAM may be a promising approach for simultaneously protecting the SE and maintaining low internal resistance. However, some technical hurdles may need to be addressed.
These findings demonstrate the importance of considering not only the intrinsic properties of the CL but also its spatial arrangement within the composite electrode. The geometry and coverage of the CL relative to the SE and CAMs are critical design parameters that influence the performance and thermodynamic stability of SSBs.
Hood et al. examined the role of Al2O3 coating for Li6PS5Cl powders.226 To gain a comprehensive understanding of how atomic layer deposition (ALD) alumina coatings and their thickness influence the electrochemical performance of argyrodite, the authors assessed key properties such as ionic conductivity, Arrhenius activation energy, and electronic conductivity using pellets formed from coated powders. Prior research has shown that fine-tuning both bulk and surface chemistry is crucial for optimizing the electrochemical behavior of fast lithium-ion conducting electrolytes.118,227,228 Using EIS, the authors measured the total ionic conductivity of both coated and uncoated argyrodite pellets (see Fig. 11a and b). The uncoated (pristine) argyrodite exhibited a Li+ conductivity of 0.9 ± 0.05 × 10−3 S cm−1 at room temperature (25 °C), with an activation energy of 0.31 ± 0.03 eV. Interestingly, the materials coated with 1 and 10 cycles of ALD Al2O3 showed significantly improved ionic conductivities of 1.2 ± 0.05 × 10−3 S cm−1 and 1.7 ± 0.05 × 10−3 S cm−1, respectively. This enhancement was accompanied by a slight reduction in activation energy, to 0.30 ± 0.02 eV for 1 cycle and 0.28 ± 0.02 eV for 10 cycles, indicating that thin ALD coatings could enhance Li+ transport. However, when the coating thickness was increased to 100 ALD cycles, the ionic conductivity dropped sharply to 0.27 ± 0.05 × 10−3 S cm−1 at 25 °C, while the activation energy remained relatively low at 0.29 ± 0.02 eV. This decline in conductivity is likely due to the thicker Al2O3 layer acting as a barrier to Li+ movement across grain boundaries, given the poor lithium-ion conductivity of Al2O3. These results suggest that the optimal ALD coating thickness lies around 1–2 nm, beyond which Li+ transport becomes impeded.
 | ||
| Fig. 11 (a) Arrhenius plots, (b) ionic conductivity at 25 °C, (c) current–time curves (DC polarization at 200 mV, 25 °C) and (d) electronic conductivity at 25 °C for Li6PS5Cl pellets pressed from powders coated with 1, 10, and 100 ALD alumina cycles in comparison with pellets pressed from uncoated powders.226 | ||
The authors hypothesize that the enhanced electrochemical performance, specifically the increase in room-temperature ionic conductivity and the reduction in activation energy, is likely due to a combination of matrix and grain boundary effects. These effects arise from the redistribution of vacancies and interstitial sites, such as space-charge regions, at the interface between the ALD alumina coating and the argyrodite electrolyte.229–232 This behavior is similar to previously reported electrochemical results observed in β-Li3PS4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al2O3 composites.232 While ball milling-based composite formation can introduce layers of electronic and ionic point defects, the close interfacial contact formed via ALD can similarly influence ion transport. However, when the ALD coating is too thick and non-conductive, it acts as a barrier to Li+ movement, significantly lowering ionic conductivity, as seen with 100 ALD alumina cycle coatings. Thus, precise control over both the coating thickness and its chemical properties is essential to properly manipulate space-charge effects and enhance ionic transport. Applying highly conductive ALD coatings to Li argyrodite materials could be a promising strategy for improving total ionic conductivity.
:Al2O3 composites.232 While ball milling-based composite formation can introduce layers of electronic and ionic point defects, the close interfacial contact formed via ALD can similarly influence ion transport. However, when the ALD coating is too thick and non-conductive, it acts as a barrier to Li+ movement, significantly lowering ionic conductivity, as seen with 100 ALD alumina cycle coatings. Thus, precise control over both the coating thickness and its chemical properties is essential to properly manipulate space-charge effects and enhance ionic transport. Applying highly conductive ALD coatings to Li argyrodite materials could be a promising strategy for improving total ionic conductivity.
Besides aiding Li+ transport, ALD alumina coatings also reduce electronic conductivity in argyrodite powder-based pellets. Electronic conductivity was measured using symmetric cells under a 200 mV bias (Fig. 11c and d). For uncoated Li6PS5Cl, the electronic conductivity was 6.3 ± 0.05 × 10−9 S cm−1 at 25 °C. After applying 1, 10, and 100 ALD Al2O3 cycles, the electronic conductivity dropped significantly, reaching as low as 1.7 ± 0.05 × 10−9 S cm−1. These results show that carefully adjusting the ALD coating's thickness and composition enables control over both lithium-ion transport and undesirable electronic leakage in solid electrolyte membranes.
The authors concluded that coating improved the stability of argyrodites under humid and oxidizing conditions, effectively protecting them from reacting with Li metal. This improvement led to significantly enhanced overall electrochemical performance, enabling higher current densities, Li metal plating/stripping capacities, and extended cycle lifetimes.
Through computational work, Ransom et al. identified LiAl5O8 and LiAlSiO4 as leading coating candidates for various electrolyte systems.233 Their flexible morphologies enable simpler synthesis approaches and offer versatile avenues for optimization, including adjusting the lithium content. These coatings exhibited good adhesion characteristics, along with Erxn < 0.1 eV. The versatility in synthesizing LiAl5O8 material into nanowire composite sintered thin films and sol–gel coatings, each offering different electrochemical enhancements, provides ample opportunities for optimizing this compound.234–236 Despite its individual Li-ion conductivity being around ∼10−6 S cm−1, its capability to form films thinner than 10 nm can mitigate the effects of its low σ.237 LiAl5O8 also helps reduce electrochemical reaction and chemical decomposition of the cathode materials. Wang et al. further discovered that LiAl5O8 could inhibit lithium metal dendrite formation in ASSLBs.236 Its ability to optimize various electrochemical metrics makes LiAl5O8 highly promising for further development. LiAlSiO4 is similarly feasible for further research, having demonstrated improved capacity retention and been experimentally confirmed for synthesis and assembly into a battery.238 The synthesis approaches leverage the glassy nature of this material, with its amorphous phase enhancing σ.239 LiAlSiO4 may be produced and coated utilizing simpler solution and dry methods, making it an attractive option for further development. There is potential for optimizing lithium content, as studies have explored adjusting the weight ratios of the coatings and enhancing σ through thin film morphologies.240,241 Li4SiO4 and Li5GaSi2O8 share structural similarities with LiAlSiO4, suggesting that exploring the glassy phases of these coatings could enhance σ.242 Furthermore, Li2B6O9F2 has emerged as the most efficient coating specifically for sulfide SE systems and has been verified via computational investigations.243
Failures in lithium metal batteries often originate from the lithium anode. The initially shiny metallic lithium foil becomes black after several cycles due to the formation of a microporous, mossy structure (Fig. 12a).245 This mossy lithium is covered by a passivation layer (SEI), and the emergence of “dead lithium” reduces anode capacity.246 Additionally, the porous nature of lithium metal with a high surface area encourages a degradation reaction and rapidly depletes the SE, which raises cell impedance. Therefore, various approaches are proposed for enhancing the cycling span of lithium anodes, mainly focusing on reducing the degradation reaction between Li and SE while ensuring electrical contact among the deposited Li particles (Fig. 12c). One of the best approaches for addressing this issue is applying protective coatings.
 | ||
| Fig. 12 (a) Porous surface of the electroplated lithium anode after extended cycling. (b) The dense surface of the lithium anode is electroplated underneath the protective coating. (c) Approaches for enhancing the cycling span of lithium metal anodes. | ||
Protective coatings serve as ion-conductive layers that allow for the electrodeposition of lithium metal (Fig. 12b). As lithium ions move throughout coatings, the ion flux will be highly uniform around the electrode's surfaces, even promoting lithium deposition.246,247 Protective layers also minimize the electrolyte/lithium contact areas, reducing side reactions. Unlike the SEI layer that forms from side reactions within batteries, protective coatings are considered artificial SEI layers. The constitution of these coatings can be adjusted to enhance σ and mechanical strength.248,249
To stabilize the SE/Li interface, various kinds of materials, such as oxides and nitrides, have been employed. Compounds within the Li–Al–O chemical family have effectively protected different solid electrolytes from lithium metal.33,250,251 The computed stability window of Li5AlO4 ranges from 0.06 to 3.07 V, indicating strong stability of Al3+ in contact with Li, which aligns with XPS measurements at the SE/Li interfaces.252 Additionally, in situ-formed polyanionic compounds like Li3PO4 and LiH2PO4 have been utilized to stabilize SE/Li interfaces.253,254 DFT calculations predict that Li3PO4 will generate Li3P and Li2O when in contact with lithium, while LiH2PO4 will yield LiH. These reaction products help to create a passivating layer that facilitates stable cycling in Li symmetric cells.253 Furthermore, while investigating alternative anion chemistries for stabilizing the solid electrolyte against reduction by lithium, nitrides have shown the lowest calculated reduction limits among various anion types, making them promising for protecting the SEs on the anode side.255 Notably, boron nitride (BN) was recently reported to effectively safeguard the SE/Li interface,256 and an SE showed excellent stability with lithium, as evidenced by a stable cycle life in a Li symmetrical cell.257
The coatings with the highest stability for the sulfide system demonstrate good adhesion towards the lithium anode (Erxn of 0 eV). However, they exhibit less favorable adhesion towards the SEs. Recent studies have explored the combined use of LiCl and LiBr to enhance σ, as LiCl alone is found to have inadequate conductivity (approximately 10−6 S cm−1).258–260 Previously, Lutz et al. examined LiCl species as a coating material, although the potential of CsLiCl2 was not thoroughly investigated.260 Additionally, Calpa et al. prepared an argyrodite type Li6PS5Cl-based SE-coated graphite (graphite:Li6PS5Cl, 64:36 weight ratio) using a dissolution–reprecipitation method and investigated its application as a negative electrode in ASSLBs. The half-cell assembled with argyrodite-coated graphite and a carbon additive in a 100:1 weight ratio showed discharge capacities of 335 and 372 mA h g−1 at 8C at 25 °C and 100 °C, respectively.261
Besides the above observations, Zheng et al. developed a dual-purpose functional material that both protected Li metal from air/water corrosion and acted as an SE.262 As shown in Fig. 13a and b, bare lithium transforms from a smooth surface to a porous network after 30 min in a 60% humidity environment. In contrast, lithium protected by PVDC (Li@PVDC) retains its metallic shine and remains smooth and crack-free under the same conditions. Fig. 13c indicates that the Li/Li symmetric cell performance of Li@PVDC initially matches that of bare lithium, highlighting its effectiveness at facilitating lithium ion transport. However, after 30 min of air exposure, bare lithium significantly increases the plating/stripping overpotential, indicating diminished performance. In contrast, Li@PVDC maintains stable overpotential levels (Fig. 13d). The bare lithium cell experiences rapid polarization increases and short circuits around 155 h, while the Li@PVDC cell demonstrates consistent performance for over 250 h, underscoring the protective coating's role in enhancing stability and reducing dendrite formation during cycling.
 | ||
| Fig. 13 The role of interlayers in suppressing air corrosion in the lithium metal anode. SEM images of (a) Li@PVDC and (b) bare lithium before and after exposure to ambient air for 30 min. Voltage profiles of bare lithium and Li@PVDC symmetric cells (c) without and (d) with 30 min air exposure at 1 mA cm−2 and 1 mAh cm−2.262 | ||
The main approach to address the strict stability requirements is to apply an electronically insulating but ionically conductive coating. This coating introduces coating/electrode material and additional coating/solid electrolyte interfaces. The coating acts like a secondary electrolyte, necessitating stability at the electrode voltages and resistance to chemical interactions towards electrode and solid electrolyte. But, if imperfections in the coatings leave portions of the electrode exposed to the SE, undesirable interfacial reactions may still take place within the coated electrode system. Conversely, these imperfections might be essential for facilitating electron transportation at coated electrodes/CC interfaces, creating a dilemma in currently used coating strategies.219 LiAl5O8, used as a coating on nickel–manganese–cobalt cathodes, has been shown to enhance coulombic efficiency and capacity retention.234 In addition, ASSLBs using gradient Li3P1+xO4S4x coated NCM811 were reported to show an elevated reversible capacity of ∼160 mA h g−1 at 0.089 mA cm−2 at 25 ± 5 °C with excellent retention of 80% after 250 cycles when combined with the commercial sulfide SE.264Fig. 14 provides a schematic representation of the diverse range of interfaces found in cathode composites.
 | ||
| Fig. 14 Interfaces within cathode composites. A schematic representation of the different cathode composite interfaces within ASSLBs, both with and without cathode coatings.63 | ||
Furthermore, Jung et al. explored the distinct impact of cathode and sulfide SE chemical reactions in solid-state batteries.25 Their findings revealed that the impedance of the composite electrodes, including bare NCM and LiNbO3-coated NCM, exhibited two semicircles representing ionic and electronic pathways (Fig. 15a and b). Bare NCM showed an initial total impedance of approximately 100 Ω, lower than that of 300 Ω observed for the LiNbO3-coated NCM, but it increased more rapidly over time, suggesting that the coating helped to prevent chemical degradation. Upon initial charging, the specific capacities for bare NCM and LiNbO3-coated NCM were 247 mA h g−1 and 222 mA h g−1, respectively (Fig. 15c). However, after aging, bare NCM maintained 83% of its initial capacity, while the LiNbO3-coated version only saw a 5% decrease (205 mA h g−1), highlighting the coating's effectiveness at maintaining performance. Analysis indicated that the aged bare NCM experienced significant degradation, reducing active sites, even at a state of charge (SOC) of 0. In contrast, the coated NCM retained its capacity due to slower chemical reaction kinetics. Secondary ion mass spectrometry (SIMS) analysis confirmed the formation of a thick reaction layer in the bare NCM, primarily consisting of chlorine-based byproducts (Cl, LiCl, and LiCl2), while the thickness of the interface for the LiNbO3-coated NCM remained relatively unchanged (Fig. 15d and e).25
 | ||
| Fig. 15 Chemical and electrochemical performance of bare and coated NCM. Chemical decomposition between the cathode and SE at steady state. (a and b) EIS spectra of uncoated bare NCM or LiNbO3-coated NCM with the argyrodite SE. Impact of chemical instability at the cathode–SE interface on electrochemical performance. (c) Electrochemical performance of the first cycle for both a fresh cell (without rest) and an aged cell (with rest), with charge and discharge CDs of 16 and 7 mA g−1, respectively. ToF-SIMS profiles as a function of sputtering time of fresh (left) and aged cells (right) of (d) bare NCM and (e) LiNbO3-coated NCM composite pellet.25 | ||
In general, coatings help mitigate surface-related degradation issues in layered transition metal oxides (LTMOs), while dopants are primarily used to enhance structural integrity by reducing irreversible phase changes during cycling. Additionally, they can improve rate capability by enhancing electronic conductivity and σ, which results from expanded lithium diffusion channels and defect formation that lowers polarization resistances.265 In certain instances, dopants within LTMOs containing greater amounts of Ni can further increase σ by decreasing the number of unwanted defects (like  ). This suppresses oxygen release due to stronger M–O bonding energy.266 High-valence ions are commonly utilized to enhance the stability of nickel-rich LTMO cathodes.267,268 These ions typically occupy transition metal sites and raise the repulsive forces between interlayers.269 Compared to smaller ions like Ni3+, Mn4+, and Co3+ (0.53–0.56 Å), high-valence dopants are generally larger, which can enhance Li layer spacing and lattice parameters, thereby improving Li diffusion. Moreover, they form stronger M–O bonds that contribute to the stability of the layered structures upon cycling.
). This suppresses oxygen release due to stronger M–O bonding energy.266 High-valence ions are commonly utilized to enhance the stability of nickel-rich LTMO cathodes.267,268 These ions typically occupy transition metal sites and raise the repulsive forces between interlayers.269 Compared to smaller ions like Ni3+, Mn4+, and Co3+ (0.53–0.56 Å), high-valence dopants are generally larger, which can enhance Li layer spacing and lattice parameters, thereby improving Li diffusion. Moreover, they form stronger M–O bonds that contribute to the stability of the layered structures upon cycling.
For comparison, Xu et al. examined how coating and doping of NCM622 affected battery performance.270 The authors claimed that the coating significantly lowered electrochemical impedance and enhanced battery cycling performances. The doping ions minimize the mixing of Ni2+ and Li+ in the NCM622 cathode material, facilitating Li+ diffusion and thereby increasing the stability of the cathode while preventing structural damage to NCM622 during charging and discharging cycles.
3.2. Electrolyte modified by doping
 | ||
| Fig. 16 Scheme illustrating the role of cation-substituted Li6PS5X (X = Cl, Br, I) SEs. Anode side interface: (a) bare and (b) cation-doped SEs towards Li metal after cycling. Cathode side interface: (c) bare and (d) cation-doped electrolyte with layered oxide cathode materials after cycling. | ||
Doping is a useful approach for improving the compatibility of electrolyte/electrode interfaces.272–275 Recently, yttrium ions (Y3+) were introduced as dopants to replace phosphorus (P5+) in Li6PS5Cl, enhancing interfacial stability against lithium metal and inhibiting dendrite growth.276 Indium-doped Li6PS5I (Li6.5In0.25P0.75S5I) showed improved interfacial stability compared to its pristine form.277
The Sun group demonstrated that a Li//Li6.2P0.8Sn0.2S5I//Li symmetric cell could achieve ultra-stable lithium plating and stripping for over 700 h (350 cycles) at 0.1 mA cm−2 and 0.1 mA h cm−2 at ambient temperature. However, significant fluctuations and increasing overpotential suggested subpar kinetics at the Li/Li6PS5I interface.26 Meanwhile, Nazar's group reported that Li6.7Si0.7Sb0.3S5I exhibited excellent stability with lithium metal, maintaining a steady voltage profile for 600 h at 0.3 mA cm−2 and for 1000 h at a higher CD of 0.6 mA cm−2. Their findings indicated that a stable interphase formed between lithium metal and Li6.7Si0.7Sb0.3S5I, which was promising for developing long-lasting ASSLMBs.278
Using cation-doped Li–argyrodites rather than undoped versions may be beneficial, as these substitutions can facilitate the formation of a lithium–metal alloy (Li–M) at the interface (see Fig. 16b).23 The Li–M alloy could promote uniform lithium deposition and enhance cycling longevity. Additionally, cation substitution may influence these materials’ thermodynamic and kinetic stability, ultimately affecting the battery's long-term performance.
Furthermore, due to the rigid nature of SEs, voids and cracks are observed on the surface of solid electrolytes during pelletizing or battery assembly. This void and crack formation promotes the rapid development of dendrites, which reduces the lifetime of the battery (Fig. 17a). However, doping the SE with liquid metal wets the gaps between the electrolyte particles as well as the gaps between the electrode and electrolyte as physical contact is expected for solid–solid interfaces in ASSLBs. This avoids the formation of voids and cracks on the surface of the electrolyte and prevents dendrite formation (Fig. 17b). In addition, incorporating liquid metal as a dopant reduces the Young's modulus, helping to prevent early short-circuit formation in the battery. The presence of liquid dopant enables the formation of M–Li alloy, which is critical for facilitating uniform Li deposition at the interface and leads to outstanding long-term stable cycling (Fig. 17c). Furthermore, doping with liquid metal can boost the mechanical properties of SEs by enhancing wettability and sealing any voids and cracks (Fig. 17d).
 | ||
| Fig. 17 (a) Mechanism for the formation of a short circuit at the Li/Li–argyrodite interfaces. (b) A suggested pathway to prevent dendrite growth at the Li/Li–argyrodite interfaces via liquid metal dopant. The role of liquid metal dopant for cycling with (c) high capacity and (d) high current density. | ||
Apart from this, due to its unique rheological and metallic properties, low-melting point liquid metal has seen growing applications in thermal control processes.279–283
3.2.2.1. Interfacial stability of cation-doped Li6PS5X (X = Cl, Br, I) towards the sulfur cathode. Jiang et al. conducted a study using a sulfur cathode to evaluate the effectiveness of the Li5.6Cu0.2PS4.8Br1.2 SE.299 They reported that this sulfur cathode achieved an impressive initial discharge capacity of 931 mA h g−1 at a rate of 0.05C, along with strong cycling stability, retaining 83% of its capacity after 100 cycles at 0.1C. In contrast, a full-cell configuration using S/Li6PS5Br/Li exhibited a notable voltage polarization and a lower discharge capacity of 851 mA h g−1 at the same rate. The researchers attributed the reduced discharge capacity of Li6PS5Br to its lower σ compared to Li5.6Cu0.2PS4.8Br1.2. Furthermore, the S/Li6PS5Br/Li setup experienced a soft short circuit during the 20th cycle, primarily due to poor interfacial compatibility with the sulfur cathode, which negatively affected its cycle performance. Li5.6Cu0.2PS4.8Br1.2 demonstrated an excellent rate capability, recovering to 810 mA h g−1 when the CD was adjusted back to 0.1C. However, during rate capability testing, a short circuit was also observed in the S/Li6PS5Br/Li configuration. These results indicate that Li5.6Cu0.2PS4.8Br1.2 is highly stable when paired with the sulfur cathode, suggesting its potential for future applications in Li–S batteries.
3.2.2.2. Interfacial stability of cation-doped Li6PS5X (X = Cl, Br, I) towards layered oxide cathodes. Layered oxide cathodes are strong contenders for high-capacity, high-energy-density applications in advanced LIBs. However, integrating these cathodes with sulfide-based ASSLMBs presents challenges. Increasing the upper cutoff voltage for layered oxide cathodes can enhance their capacity but may lead to material degradation due to processes such as oxygen loss and cracking. This degradation can cause parasitic interactions at the interface with Li6PS5X (where X = Cl, Br, I), resulting in the formation of ion insulator byproducts like Li2S, SO42−, and PO43− (see Fig. 16c). Understanding these mechanisms is essential for enabling high-voltage operation and enhancing the performance of current materials. A promising solution is cation-substituted Li–argyrodite, which can bind strongly with sulfur and prevent oxygen from displacing it (Fig. 16d).
Liu et al. examined the cell performance of LNO@NCM/Li6P0.925Sb0.075S5Cl/Li, reporting an initial discharge capacity of 129.9 mA h g−1, which stabilized at 107.4 mA h g−1 after 60 cycles, achieving a magnificent capacity retention of 82.6%.27 In contrast, the ASSLB configuration using Li6PS5Cl (LNO@NCM/Li6PS5Cl/Li) showed a significant capacity drop (116.2 → 31.3 mA h g−1) after 60 cycles, resulting in only 26.3% retention. The authors attributed the better performance of Li6P0.925Sb0.075S5Cl to its superior σ and interfacial stability. EIS revealed that the impedance of LNO@NCM/Li6PS5Cl/Li increased sharply after cycling, while LNO@NCM/Li6P0.925Sb0.075S5Cl/Li showed a minimal change, indicating enhanced stability at the interface. Additionally, the rate capability of the Li6P0.925Sb0.075S5Cl configuration was evaluated, with a capacity recovery of 118.6 mA h g−1 when the CD returned to 0.2C, demonstrating its excellent performance.
In addition, Zhao et al. reported on Sn-doped Li6PS5I solid electrolytes, achieving a remarkable first-cycle coulombic efficiency of 91% with a capacity of 123.7 mA h g−1 in their Li/LPSI-20Sn//LGPS//LCO@LNO//LGPS setup. They attributed this high efficiency to the enhanced interfacial stability of LPSI-20Sn.26
For practical solid-state battery applications, thick electrode designs are essential for maximizing energy density.300–302 Zeier's group developed thick cathodes (160 μm) and anodes (160 μm) using Li6.6P0.4Ge0.6S5I solid electrolyte (450 μm).303 Their findings revealed that this configuration delivered an initial charging capacity of 120.8 mA h g−1 and an initial discharge capacity of 88.8 mA h g−1 at 0.25C at 60 °C.35,288,304 Notably, the cell operated stably over 50 cycles without carbon additives,289 exhibiting minimal capacity fading and high coulombic efficiency even at 1C. The total resistance of the cell remained below 13 Ω cm−2 at 60 °C; this was attributed to the outstanding interfacial compatibility between Li6.6P0.4Ge0.6S5I and NCM-622. The researchers concluded that employing excellent ionic conductor sulfide SEs combined with effective interphase formation resulted in superior battery performance.
 | ||
| Fig. 18 The role of cation dopant in thermal stability.305 | ||
3.3. Multi-layer electrolyte
Ye et al. developed a multilayer electrolyte system consisting of a less stable electrolyte positioned between two more stable SEs.306 This design effectively inhibits Li dendrite evolution through well-localized decomposition within the less stable layer. They conducted experiments using symmetric cells to evaluate the stability of Li10Ge1P2S12 (LGPS) and Li5.5PS4.5Cl1.5 (LPSCl) electrolytes in contact with Li metal. The symmetric cell featuring pure Li metal electrodes and LGPS electrolyte demonstrated rapid failure, as indicated by a voltage spark (Fig. 19a). While most sulfide SEs experience some level of decomposition upon contact with lithium, Li–argyrodite Li6−yPS5−yCl1+y exhibit greater stability in this context compared to LGPS.10,31,46,97,307 In tests, the LPSCl symmetric battery was able to operate for over 150 h (Fig. 19b) before ultimately experiencing a short circuit. Fig. 19a and b illustrates two common failure modes: electrolyte decomposition causing increased overpotential and a sudden voltage drop due to short-circuiting.46,308 XPS analysis revealed significant decomposition of LGPS to reduced sulfur and germanium, while LPSCl showed minimal decomposition, indicating that short-circuiting in the LPSCl battery was likely to be due to lithium dendrite penetration. The multilayer electrolyte design provided impressive cycling performance, lasting 1800 h at a CD of 0.25 mA cm−2 (Fig. 19c), significantly outperforming batteries using a single electrolyte type. Moreover, the multilayer symmetric battery demonstrated the ability to cycle at an exceptionally high CD of 20 mA cm−2 with a low overpotential of approximately 0.5 V without noticeable short-circuiting at 55 °C (Fig. 19d). | ||
| Fig. 19 Designing multi-layer SE. (a and b) Li–Li symmetric cells comprising LGPS and LPSCl SEs, respectively. (c and d) Long-term cycling using multi-layer SEs.306 (e) Charge–discharge profile of LiCoO2:Li3YCl6|Li6PS5Cl|Li–In at 10C. (f) Capacity retention and (g) charge–discharge profile of LiCoO2:Li3YCl6|Li3InCl6|Li6PS5Cl|Li–In (red) and LiCoO2:Li3YCl6|Li3YCl6|Li6PS5Cl|Li–In (blue) at 10C. (h) Scheme illustrating the suggested properties of the electrolyte required to fulfill the multi-layer design system.309 | ||
Samanta et al. proposed a bilayer SE comprising Li argyrodite LPSCl and halide SEs, in which halide SEs face the cathode, and LPSCl faces the anode.309 When LPSCl is placed at the cathode side, it aggressively reacts with LCO (Fig. 19e). They noted that the oxidative stability of the halide electrolytes enabled their use alongside high-voltage cathodes, addressing the instability of LPSCl under such conditions (Fig. 19f and g). Fig. 19h outlines the desired properties of electrolytes used in a multilayer design.
4. Techniques for exploring interfaces
The type and characteristics of interfaces in ASSLBs are crucial because of the significant impact of the extent of contact and a large charge transfer impedance at these interfaces.310–312 Therefore, to advance the development of next-generation ASSLBs that satisfy diverse industrial and consumer needs, it is vital to understand the phenomena of interfacial impedance, which can be studied through advanced characterization techniques that reveal morphological changes and electrochemical processes. While there are established methods for examining LE battery systems, investigating interfacial behavior in ASSLBs presents a significant challenge. The interfaces in ASSLBs are harder to isolate than those in LE systems. For instance, in LE systems, electrode materials can typically be extracted for post-experimental analysis without causing damage. In contrast, the fabrication of ASSLBs often involves techniques like hot-pressing and chemical or physical evaporation to achieve optimal interfacial contacts.313 As a result, these tightly integrated interfaces are not easily accessible without harming their surface conditions, making it challenging to gather critical information regarding interfacial reactions and kinetics, particularly for Li/SE interfaces.62 Thus, a new approach, ideally involving in situ measurements, is necessary.314 Traditional studies often rely on ex situ measurements conducted in coin cells. However, interfacial research can also be performed using hybrid cells,313 symmetric cells,151 or a combination of ex situ and in situ methods.The primary challenges in characterizing interfaces in ASSLBs include the limited exposed surface area and the sensitivity of these interfaces when extracted for analysis.62 As a result, many conventional techniques used for LE interfaces, such as infrared spectroscopy (IR), are not suitable for ASSLB interface characterization.315 Therefore, characterization methods that offer high measurement accuracy in a narrow area and protect the properties of samples during analysis are available for characterizing the interfaces in ASSLBs.
In situ characterization is vital for examining interfacial phenomena in SSBs, as it enables the observation of real-time changes in materials and interfaces during actual operating conditions like charging, discharging, or temperature variations. Unlike ex situ methods, which assess the system after these changes have occurred, potentially missing or altering transient interfacial states, in situ techniques are ideal for dynamic processes. However, ex situ approaches still play a key role by offering detailed structural insights (e.g., via TEM, ToF-SIMS), analyzing surface chemistry under ultra-high vacuum conditions (e.g., with XPS), and evaluating bulk properties after extended cycling periods.
Here, we discuss various characterization techniques such as chemical/electrochemical, diffraction and images.
4.1. Coulometric titration time analysis (CTTA)
The CTTA method is effective at quantitatively analyzing decomposition reactions between Li and argyrodite electrolytes, offering valuable insights into electrolyte stability.316 This versatile approach can also be applied to various electrodes and allows for the investigation of how key operational factors, such as temperature and CD, affect the characteristics of the SEI and cell degradation.Aktekin et al.316 developed the CCTA technique to quantify side reactions that occurred between redox-active electrode materials and SEs. Fig. 20 displays the results from a CTTA measurement. In an ideal scenario, where the electrolyte is stable, and no decomposition reactions occur, the cell potential would rapidly stabilize at E = 0 V after a period, as Li dictates the potential of both the working and counter electrodes, achieving a symmetric cell stage. This would result in an infinite potential at E = 0 V. However, if the SE is unstable and a decomposition reaction takes place, these unwanted reactions gradually consume the lithium metal that has been ‘titrated’. As long as some Li remains at the working electrode, the potential stays at E = 0 V. Eventually, once all the lithium metal is depleted, the potential at the working electrode becomes variable, leading to an increase in cell voltage. In LPSCl solid electrolyte, the rise in potential happens swiftly after the initial titration step (Fig. 20b). When the potential hits 0.05 V, an identical titration step is performed (marked in gray), resulting in the cell voltage dropping back below E = 0 V as fresh lithium is accumulated on the current collector. During the subsequent OCV period, it is observed that the time taken to consume all Li is greater than that during the preceding OCV period, indicating a reduced rate of side reactions following the second titration step. As illustrated in Fig. 20c, the consumption of a similar amount of Li metal takes progressively longer as the measurement continues, suggesting that side reaction products form a passivating SEI layer. Fig. 20d presents the total charge used for side reactions  , where the total is the number of titrations multiplied by the step charge) in relation to the duration of the experiment, enabling precise quantification of SEI growth over time. Fig. 20e indicates that this growth exhibits a linear relationship with the square root of time. For LPSCl, a charge of 1 μA h cm−2 (approximately 3.7 × 10−8 mol cm−2 Li) leads to an SEI thickness of about 9 nm, supposing a compact mixture of Li2S, LiCl, and Li3P, without any gaseous byproducts.
, where the total is the number of titrations multiplied by the step charge) in relation to the duration of the experiment, enabling precise quantification of SEI growth over time. Fig. 20e indicates that this growth exhibits a linear relationship with the square root of time. For LPSCl, a charge of 1 μA h cm−2 (approximately 3.7 × 10−8 mol cm−2 Li) leads to an SEI thickness of about 9 nm, supposing a compact mixture of Li2S, LiCl, and Li3P, without any gaseous byproducts.
 | ||
| Fig. 20 CTTA measurements. (a) Output for the LPSCl SE in a stainless steel/LPSCl/Li assembly at a temperature of 25 °C and pressure of approximately 13 MPa. The potential profiles are depicted for two distinct time intervals: an early stage (b) and a later stage (c) of the measurement. The total stored capacity over time is illustrated in (d) as a function of time and (e) as a function of the square root of time.316 | ||
4.2. X-ray photoelectron spectroscopy
XPS is a well-established method for chemical analysis,317 particularly effective at examining the composition of interfaces in ASSLBs. It offers high sensitivity, accuracy, resolution, and the ability to test various elements across the periodic table. This makes XPS especially valuable for obtaining valence band spectra, which are crucial for analyzing the electronic structure of surfaces and interfaces in these systems. The SE/electrode interfaces are vital in ASSLBs, yet ex situ XPS fails to accurately capture these interfaces’ chemical and electronic properties. In situ measurements are possible by connecting an ultrahigh vacuum transfer system to the XPS chamber,318 allowing for real-time analysis of interfacial properties during electrode deposition. Wood et al.117 utilized in situ XPS to investigate the interphase between lithium and sulfide solid electrolyte during electrochemical cycling, finding that a Li3PO4 phase formed initially through phase segregation of Li3POxS4−x and then evolved further (Li3−xP and Li2O2) during discharge.Aktekin et al.319 conducted an operando hard X-ray photoelectron spectroscopy (HAXPES) study on a sulfide-based SE, Li6PS5Cl, to examine its reduction reactions during lithiation and the resulting development of the SEI. As illustrated in Fig. 21a–d, the surface of the solid electrolyte pellet is coated with a 6 nm layer of nickel, which serves as both a CC and a working electrode (WE). Li metal is pressed in place on the pellet's opposite side, functioning as a counter electrode (CE) and a reference electrode (RE). This setup is termed an “anode-free” configuration due to the absence of an initial lithium reservoir in a specialized holder connected to a potentiostat. This arrangement allows for the gradual polarization of the WE to specific potentials, eventually leading to the electrochemical plating of lithium metal below 0 V vs. Li+/Li. Utilizing the high photon energies from a synchrotron source enables the detection of higher kinetic energy electrons, essential for analyzing the Ni|LPSCl interface beneath the 6 nm nickel layer within a proper timeframe. This methodology permits the investigation of the SEI formed beneath a thin metal film, like 6 nm nickel, in an electrochemical cell incorporating the Li6PS5Cl SE. The electrolyte begins to undergo reduction reactions at 1.75 V (vs. Li+/Li), leading to the significant formation of Li2S, particularly within the voltage range of 1.5–1.0 V. A heterogeneous and layered microstructure of the SEI is noted, with Li2O and Li2S deposits primarily found near the CC. Additionally, the study reveals the reversibility of decomposition products as Li2O and Li2S degrade within the 2–4 V potential range, producing oxidized sulfur species, sulfites, and sulfates.
 | ||
| Fig. 21 (a) Scheme illustrating the operando HAXPES experiment setup. (b) An enlarged view of the operando cell. (c) A confocal microscope image showing the Ni-coated stainless steel perforated CC support. (d) A top-down SEM image of the operando cell. Normalized HAXPES spectra, including peak fittings at various working electrode potentials, are presented for (e) S 1s, (f) Li 1s, and (g) O 1s core levels. Detailed normalized HAXPES spectral changes during polarization to elevated potentials, with peak fittings for (h) S 1s, (i) Li 1s, and (j) O 1s core levels.319 | ||
Additionally, Zhang et al. employed complementary in situ approaches, including atomic force microscopy (AFM) and XPS, to directly observe the morphological and chemical changes, Li plating and stripping processes, and SEI dynamics at the Li and sulfide SE interface.320 The morphology at the open circuit potential (OCP) was first analyzed using AFM (Fig. 22a). At the same time, XPS was employed to identify the functional groups present in LPS (Fig. 22g). After applying a discharge overpotential (0.1 V) for approximately 240 s, small particles began to show on the Li metal surface, as designated by the white arrows in Fig. 22b. After 600 s of Li plating, a few isolated particles were observed on the Li metal surface (Fig. 22c). In addition to morphological alterations, chemical evolution at the Li–SE interface during the Li plating process was investigated using in situ XPS (Fig. 22g). The emergence and increased intensity of signals corresponding to Li2S, LixP, and Li3P reveal the formation of the SEI containing these components, which occurs concurrently with the Li plating process. An overpotential of 0.1 V was applied to investigate the Li stripping processes. As indicated in Fig. 22d and e, the Li spheres began to dissolve upon application of the stripping potential. After 600 s of stripping, most of the deposited Li spheres had dissolved, leaving wrinkles at the sites where dissolution occurred (Fig. 22f). Analysis of the primary XPS signals for Li2S, LixP, and Li3P (Fig. 22g) indicated that these residual wrinkles were primarily due to the SEI present at the interface. The detailed Li plating and stripping processes were elucidated through in situ AFM and in situ XPS analyses. This approach allowed for a detailed examination of the morphological/chemical changes of the SEI. The findings indicate that the presence of the SEI, characterized by limited electron-conducting properties, plays a vital role in sustaining the interfacial stability of LPS. The dynamics of lithium plating/stripping within the LPS system were analyzed by examining the Li volumes derived from in situ AFM images. Fig. 22h shows the changes in Li volume throughout the plating and stripping processes, with the Li plating rate in the LPS system determined to be 0.15 μm3 s−1. Simultaneously, the evolution of species containing sulfur and phosphorus was investigated through quantitative analysis of in situ XPS spectra, as depicted in Fig. 22i and j, respectively. The authors concluded that the rate of lithium plating was not directly linked to the rate of SE decomposition or SEI formation.
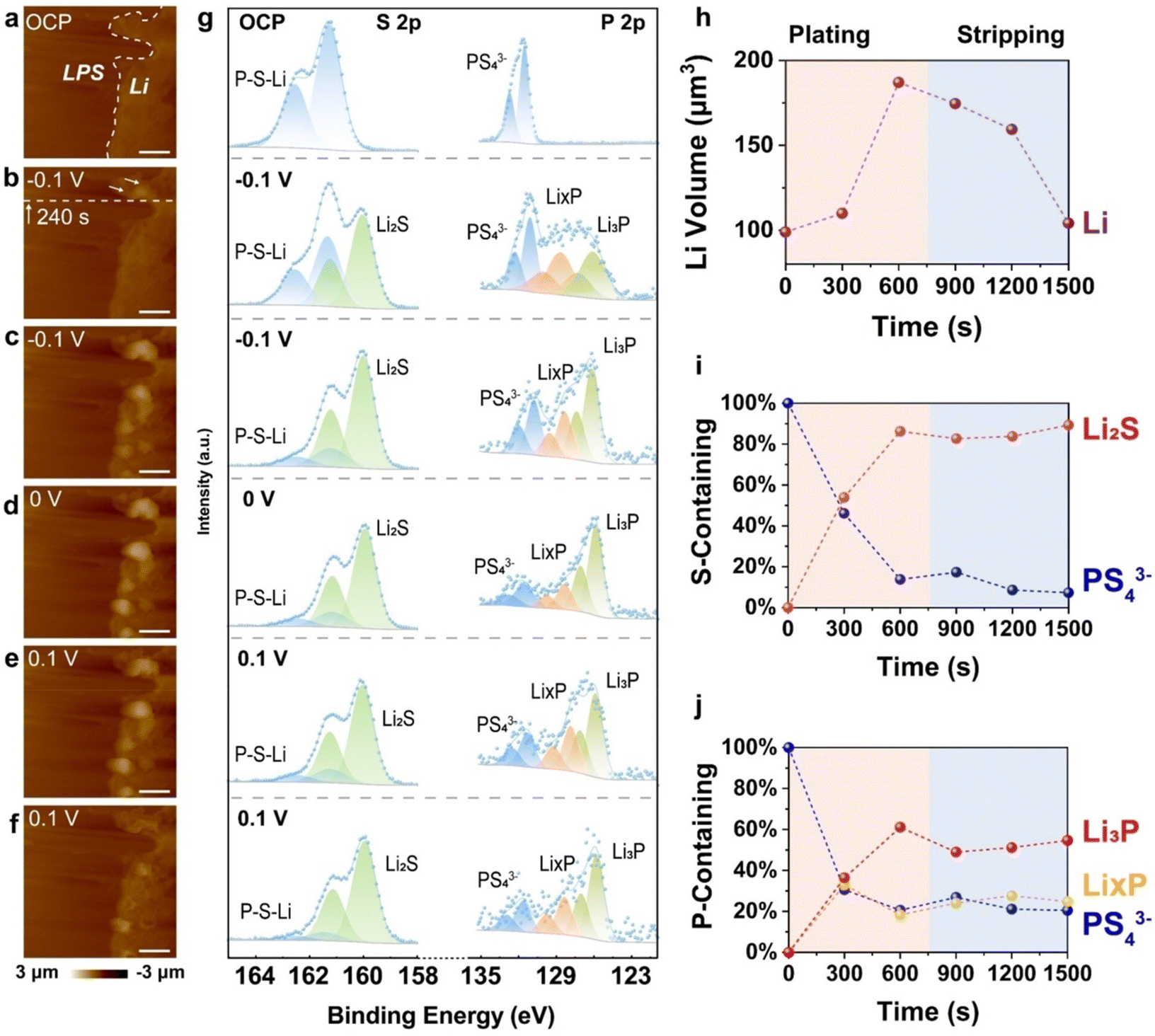 | ||
| Fig. 22 In situ studies of the morphological/chemical evolution and dynamics at the lithium and sulfide SE interface. In situ AFM images of the lithium and LPS interface captured at various potentials: (a) OCP, (b and c) cathodic −0.1 V, (d) 0 V, (e and f) anodic 0.1 V. (g) In situ XPS spectra of the lithium and LPS interface at different potentials. Each AFM image capture time and XPS spectrum collection time is 300 s. (h) The Li volume alterations during lithium plating/stripping. (i) Changes to the S-containing species during lithium plating/stripping. (j) Alterations to the P-containing species during Li plating and stripping.320 | ||
Despite the advantages of in situ XPS for monitoring electrochemical reactions at ASSLB interfaces, relevant studies remain limited. Integrating the charge/discharge testing apparatus with XPS equipment is a key challenge. Additionally, the electrode thickness required for in situ tests must be nanoscale, complicating the application of common coating methods.321 Therefore, effectively designing ASSLBs compatible with XPS technology is essential for advancing in situ XPS applications.
4.3. Raman spectroscopy
The Raman technique is a valuable tool for investigating various organic and inorganic compounds at the interfaces of ASSLBs, thanks to its suitability, greater accuracy, and nondestructive nature.322,323 Li et al.324 verified that decomposition reactions occurred at the interface between Ni-rich cathodes and sulfide SEs. At the same time, a Ni-poor coating layer can mitigate these reactions. Raman spectroscopy also illustrated structural alterations to the P4S16/C cathode during lithiation and delithiation, indicating the formation of LixPS4 and PS43− species during lithiation, while the P–S characteristic peak returned without the observation of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bonds after delithiation.325
S bonds after delithiation.325
Raman mapping, a derivative of Raman spectroscopy, offers an intuitive view of the phase composition at ASSLB interfaces. Luo et al. analyzed the surface of In foil after being in contact with Li6PS5Cl and compared it with fresh In foil without contact via Raman spectroscopy. The result obtained showed the formation of In2S3 after In foil was in contact with Li6PS5Cl.292 Rui et al. examined the thermal stability of a mixture of Li6PS5Cl and LiNi0.8Co0.1Mn0.1O2via Raman spectroscopy.183 The result indicates the formation of PO43−, SO42−, and Li2Sx decomposition products at 400–500 °C. Zhou et al. utilized in situ electrochemical Raman microscopy to analyze the interfacial degradation of Li6PS5Cl with lithium metal and LiCoO2, detecting Li2S during Li deposition and identifying polysulfides and P2Sx species at the Li6PS5Cl/LiCoO2 interface during charging (Fig. 23).326 Zeng et al. compared the stability of Li6.4PS5.4Cl0.6 and Li5.7PS4.7Cl1.3 towards Li metal via in situ Raman measurements.327 Their results showed that for the Li6.4PS5.4Cl0.6 cell, new peaks associated with P2S5 and Li2Sn emerged after charging to 3.6 V, with peak intensities increasing with voltage. In contrast, the Li5.7PS4.7Cl1.3 cell did not show decomposition product peaks, and the PS43− peak intensity only slightly decreased, indicating self-limiting interface characteristics for Li/Li5.7PS4.7Cl1.3.
 | ||
| Fig. 23 (a) The diagram illustrates the in situ Raman cell setup. In situ Raman spectra of the Li6PS5Cl/Li interface in the Li/Li6PS5Cl/Cu configuration (b) under a constant potential of −0.1 V vs. Li+/Li; (c) under varying potential. In situ Raman spectra of the LiCoO2/Li6PS5Cl interface during (d) charging to 4.2 V and (e) discharging to 2.5 V.326 | ||
4.4. Electrochemical impedance spectroscopy
EIS is a virtual approach for examining the interfacial behavior between electrodes and SEs, providing valuable insights into lithium-ion diffusion in ASSLMBs.328 It is observed that the interfacial stability and resistance during cycling can be assessed through changes to the semicircles observed in both the low- and high-frequency ranges.329 Thus, EIS is a valuable technique in electrochemical studies that is useful for analyzing materials, complete cell devices, and battery packs.330 Wei et al.331 examined how σ affected the impedance changes in ASSLBs during cycling through in situ EIS measurements. As illustrated in Fig. 24a, the bulk impedance of the 3Li2S–LiI/Li5.5PS4.5Cl1.5/Li–In cell was elevated during the initial charging process, with the increment in interfacial resistance being attributed to the decomposition of the sulfide solid electrolyte throughout cycling.332 In Fig. 24b, the 5Li2S–LiI/Li5.5PS4.5Cl1.5/Li–In cell exhibited similar changes in resistance at the interfaces of the AM and the SE. However, these resistances were noticeably greater than those observed in the 3Li2S–LiI/Li5.5PS4.5Cl1.5/Li–In cell during the first cycle, which could be ascribed to enhanced σ in the cathode mixture following the addition of LiI. Furthermore, Zhang et al. compared the interfacial stabilities of a single sulfide SE with the sandwiched sulfide SE in a Li–Li symmetric configuration via in situ EIS and associated DRT measurements (Fig. 24c and d). The results demonstrated the advantages of sandwich SEs, revealing reduced resistance and stable interfacial characteristics.320 | ||
| Fig. 24 In situ EIS measurements. In situ EIS performed on Li5.5PS4.5Cl1.5-based ASSLBs, utilizing cathode mixtures of 3Li2S–LiI (a) and 5Li2S–LiI (b) during the first cycle at a CD of 0.13 mA cm−2, within a voltage range of 0.4 to 3 V vs. Li–In at ambient temperature.331 (c) In situ EIS of Li|LPS-LGPS-LPS|Li cells operated at 0.1 mA cm−2 and 0.1 mA h cm−2. (d) DRT results from the in situ EIS of Li|LPS-LGPS-LPS|Li cells operated at 0.1 mA cm−2 and 0.1 mA h cm−2.320 | ||
4.5. Diffuse reflectance infrared Fourier-transform spectroscopy
In situ diffuse reflectance infrared Fourier-transform spectroscopy (in situ DRIFTS) is a method used to study the intricate changes in surface species of SEs.333 Our research team examined the behavior of functional groups and C–S bonding during CO2 adsorption on Li6PS5Cl using in situ DRIFTS (Fig. 25).334 The adsorption of CO2 was essential for enhancing both interfacial and electrochemical stability between Li and Li6PS5Cl. The emergence of the new S–CO2 bond is critical for altering interfacial behavior, as it improves interfacial stability between Li and Li6PS5Cl while decreasing cell resistance. Furthermore, the Li|CO2@ Li6PS5Cl|LTO configuration demonstrates an impressive performance, achieving 62% capacity retention and an extremely high coulombic efficiency of 99.91% after 1000 cycles. | ||
| Fig. 25 In situ DRIFTS measurements. The in situ DRIFTS findings of CO2 adsorption on Li6PS5Cl at 150 °C show: (a) the initial conditions under Ar purging and (b) the spectral alterations for Li6PS5Cl when CO2 is purged at a rate of 30 c.c. min−1. (c) A depiction of monodentate CO2 chemisorption on Li6PS5Cl is also provided.334 | ||
4.6. Pressure measurements
Wu et al.335 conducted operando pressure measurements to monitor pressure changes in Li1.2Ni0.13Mn0.54Co0.13O2-based ASSLBs during cycling, with the aim of systematically exploring the connection between internal stress and electrochemical performance. A zero-strain LTO anode was utilized as the CE, allowing the CAM to dominate the overall pressure changes. Both the polycrystal (PC)- and single crystal (SC)-Li1.2Ni0.13Mn0.54Co0.13O2 cathodes displayed similar pressure profiles (Fig. 26). The pressure decreased during charging and increased during discharging, indicating high reversibility. This pressure variation primarily results from volume contraction and expansion upon lithium extraction and intercalation from/into Li1.2Ni0.13Mn0.54Co0.13O2, respectively.336 However, it is noteworthy that the pressure fluctuations in PC-Li1.2Ni0.13Mn0.54Co0.13O2 were less pronounced than those in SC-Li1.2Ni0.13Mn0.54Co0.13O2. This difference can be attributed to two main factors: first, SC-Li1.2Ni0.13Mn0.54Co0.13O2 provides higher capacities, leading to more significant changes in lithium content and, consequently, more noticeable pressure variations. Second, uneven stress distribution in the PC-Li1.2Ni0.13Mn0.54Co0.13O2 cathode can cause structural collapse, forming gaps or voids that can partially accommodate the volume changes associated with electrode breathing. | ||
| Fig. 26 Operando pressure curves of (a) PC- and (b) SC-Li1.2Ni0.13Mn0.54Co0.13O2 in Li4Ti5O12|Li6PS5Cl|Li1.2Ni0.13Mn0.54Co0.13O2 cells.335 | ||
4.7. X-ray diffraction
X-ray diffraction (XRD) is a method used to analyze the structure of crystals. In this technique, an X-ray beam is directed at a sample, which diffracts the rays in various directions due to its crystalline structure. By measuring the angles and intensities of these diffracted beams, a crystallographer can create a three-dimensional representation of the electron density within the crystal. This allows for the extraction of information about the crystalline structure, including chemical bonds, crystallographic defects, and atomic distances based on the positions of atoms. XRD is favored as an analytical tool due to its straightforward operation, minimal contamination during detection, and ease of data processing, making it the most widely used method for determining crystalline structures.337Hau et al.338 explored the microstructure of manganese-based cathodes using in situ XRD measurements. They found that the nanoscale domain structure and short coherence length of partly disordered spinel-like domains are vital for the electrochemical performance of the δ-phase. These characteristics adequately mitigate the two-phase reaction typically seen in nanomaterials.339 Reducing the coherence length in a material can turn first-order phase transitions into second-order transitions or eliminate them.340 In the δ-phase, a short coherence length significantly impacts on the electrochemical performance by eliminating the 3 V plateau and converting the related phase change into a solid solution region (Fig. 27a). This attribute of the δ-phase removes phase change strain as a potential decomposition pathway, allowing manganese-based spinels to cycle across their entire theoretical capacity range. Furthermore, forming the δ-phase enhances rate performance, aligning with previous theoretical findings that spinel-like arrangements have a greater extensive low-barrier transition metal percolation network, facilitating lithium transport. In the δ-phase, a larger portion of the capacity is delivered at greater voltages, which is advantageous for power delivery in practical battery applications. The δ-phase exhibits spinel-like voltage profiles and rates while maintaining a sufficiently short coherence length to exist as a solid solution, thus inhibiting the two-phase reaction observed in ordered spinels (Fig. 27).
 | ||
| Fig. 27 In situ XRD measurements. In situ XRD measurements of Li0.7Mn0.65Ti0.1O1.9F0.1 (L07M65-DH) and LiMn2O4 during their initial cycles. Voltage profile (left) and in situ XRD patterns (right) for (a) L07M65-DH and (b) LiMn2O4. The current rate was 20 mA g−1, with X-ray scans performed every 30 min. Peaks corresponding to cubic spinel are highlighted in red, while those for tetragonal spinel are indicated in white.338 | ||
4.8. Electron backscatter diffraction
Electron backscatter diffraction (EBSD) is the most potent method for analyzing the microstructure of metals and tracking their evolution. This technique provides quantitative data on grain size, orientation, boundaries, and potential dislocations and strain within large single grains.341–343 However, it necessitates a well-defined sample surface regarding flatness, crystallinity, and chemical composition. Thin decomposition layers, often less than 50 nm, that form on lithium344,345 can obscure the electron diffraction pattern because EBSD has a limited probe depth of about 20 nm. Consequently, there are minimal studies utilizing EBSD on lithium foils.341,346–348Fuchs et al.349 developed a method to study metal growth and dissolution in anode-free solid-state batteries using in situ electron backscatter diffraction (EBSD). Their examination throughout cross-sectional deposition and dissolution provided insights into microstructural evolution (Fig. 28), revealing that small lithium grains undergo annealing akin to Ostwald ripening. They found that pore emergence during the discharge of the metal anode primarily happened at the interface between the grain bulk and the solid electrolyte. Notably, the areas where metal grain boundaries encounter the interface remain undamaged; this is because of the more rapid diffusion of metal and vacancies along these boundaries. The density of nucleation is impacted by factors like applied CD, temperature, and the surface characteristics of the SE.131,350,351 Furthermore, their in situ EBSD findings indicate that both grain nucleation and the growth process are crucial for controlling the resulting microstructure. The applied CD may also affect grain development during electrodeposition, and applying stack pressure could be an effective method for managing the lateral extension of developing grains.352 Overall, this analysis enhances the understanding of metal electrodes and their microstructural evolution.
 | ||
| Fig. 28
In situ EBSD approach for examining the microstructural changes occurring during the electrodeposition and electrodissolution of an alkali metal electrode in contact with a solid electrolyte. (a) A schematic representation of the in situ EBSD setup. (b) Voltage profiles for depositing 50 μm of Li at the Cu|Li|SE interface and for stripping Na until pore formation occurs at the Cu|Q-Na|SE interface. (c) Microstructural changes during Li plating. (1) shows an SEM image of the pristine cross-section of Cu|Li|SE. The lithium reservoir is not freshly deposited, and is also too thin to obtain EBSPs of sufficient quality to generate an IPF map. However, upon depositing around 10–15μm of additional lithium, the IPF map depicted in (2) was obtained. Herein, several grains around 10–30μm wide are visible. Surprisingly, after another ~5μm of lithium deposition, fewer but wider grains are visible in the next IPF map in (3). Two small blue grains close to an <111> orientation from the previous map apparently fused into larger neighbouring grains close to an <101> orientation (green). After another deposition step, IPF map (4) shows even wider grains, with another blue grain from the previous map being fused to a neighbouring green grain. After another long deposition step resulting in around 20μm of additional lithium, multiple grains have fused together forming a large grain with >100μm width, again close to a <101> orientation as visible in (5). (d) Microstructural changes during Na stripping. Starting from an optimal interfacial contact between sodium and SE (A), a dark region close to the interface emerges (B) after the first stripping interval, attributed to pores. Interestingly, the pores formed mainly within the green grain, while the vertical grain boundaries on both sides remain intact (C). As stripping progresses, another pore nucleates within the large blue grain starting from a grain boundary, supporting the previous description. Moreover, the pores within the green grain further grow into its bulk (D).349 | ||
4.9. Cryo-electron microscopy
Cryo-electron microscopy (Cryo-EM), originally developed for structural biology, is increasingly attracting interest from materials scientists who seek to exploit its capabilities for studying beam-sensitive materials.353,354 Battery materials, particularly after cycling, are often sensitive to air exposure, making cryogenic temperatures crucial for maintaining their native state and preserving inherent information. Cryo-EM enables the observation of battery materials with enhanced electron beam tolerance, minimizing radiation damage. For instance, Li et al. utilized cryo-EM to image the atomic structure of lithium metal dendrites formed through electrochemical deposition, as well as their interfaces with the SEI354 (Fig. 29a). They discovered variations depending on the electrolyte systems used, which aided in understanding the causes of failure within high-energy batteries. This approach can also be applied to other chemically reactive and beam-sensitive materials. Similarly, Wang et al.355 investigated the microstructure of electrochemically deposited lithium (EDLi) and its associated SEI at the atomic level (Fig. 29b), noting how different electrolyte additives impacted the surface properties of EDLi, thereby influencing coulombic efficiency. This demonstrates cryo-EM's effectiveness at examining micro-mechanisms related to lithium materials that affect battery efficiency. Zachman et al.355 also employed a novel cryo-FIB method to characterize and extract intact interfaces from coin cell batteries for cryo-STEM analysis. Cryo-STEM's structural/chemical mapping revealed insights into lithium dendrite formation mechanisms (Fig. 29c). These cryo-EM studies significantly enhance the perception of reaction intermediates and mechanisms in lithium-metal batteries. By merging cryo-STEM with cryo-FIB,355 new insights into interactions and causes of failure at the lithium/solid electrolyte interfaces may emerge, potentially leading to effective strategies for advancing lithium metal anode technology. A comprehensive review of cryo-EM applications in battery material research is available for further reference.356 | ||
| Fig. 29 (a) Atomic-resolution transmission electron microscopy images showcasing Li metal and its interface with the SEI.357 (b) Cryogenic transmission electron microscopy images depicting EDLi.355 (c) High-angle annular dark field cryo-scanning transmission electron microscopy images along with corresponding electron energy loss spectroscopy elemental mapping for two distinct types of dendrite. HAADF cryo-STEM imaging reveals an extended SEI layer on the type I dendrite (marked-c), but not on the type II dendrite (marked-d). EELS elemental mapping shows that both SEIs are oxygen-rich, but that the type II SEI contains no carbon. The type I dendrite has an appreciable oxygen content (marked-e), whereas the type II dendrite does not (marked-f).358 | ||
4.10. Scanning electron microscopy (SEM)
In contrast to ex situ characterization methods, in situ and operando techniques, such as SEM, enable real-time observation of interfacial structural changes and phase transitions in non-equilibrium states during charging and discharging. These methods provide immediate insights into lithiation and delithiation processes, aiding in understanding structure–performance relationships and electrochemical mechanisms in ASSLMBs. Recent studies have employed in situ/operando techniques to examine interfacial challenges within solid-state systems. For instance, in situ SEM characterization has shown that achieving uniform Li deposition on the SE and preventing dendrite formation is essential for facilitating highly reversible Li deposition/dissolution reactions.359Nagao et al.200 investigated the Li deposition/dissolution processes at varying CDs in sulfide-based ASSLMB using in situ SEM observations (Fig. 30). Fig. 30a presents schematics of the Li/SE/stainless steel cells before cycling, along with a magnified view of the SE and stainless steel interface during charging/discharging. As Li is deposited onto stainless steel during the charging process, the space between the SE and stainless steel enlarges due to the increasing thickness of accumulated Li. Conversely, during discharging, as Li ions dissolve in the SE, this space narrows. Fig. 30b illustrates the voltage curves for Li deposition/dissolution in Li/SE/stainless steel cells at 0.01 mA cm2 during the first and second cycles and at 0.05 mA cm2 during the third and fourth cycles. The cell maintains a relatively stable voltage of approximately +40 mV for only two cycles at 0.01 mA cm2. Fig. 30c shows the morphological changes at the SE/CC interface (first → second cycle). Unlike the morphology observed during high CD electrochemical tests, no new crack or pillared deposit appears across the SE surface during charge/discharge processes at a lower CD of 0.01 mA cm2.
 | ||
| Fig. 30 In situ SEM measurements at different CDs. (a) Schematic diagrams illustrating the Li/SE/SS cells before the cycle and a detailed view of the interface between the solid electrolyte and stainless steel during charging and discharging. (b) Li deposition and dissolution curves for Li/SE/SS are presented at a CD of 0.01 mA cm−2 during the first and second cycles and at 0.05 mA cm−2 during the third and fourth cycles. (c) SEM images of the interface show conditions (A) after the first charge, (B) after the first discharge, (C) after the second charge, and (D) after the second discharge. SEM images of the SE/stainless steel interface at a similar location within a similar configuration are shown (d) instantly following a short circuit, (e) during Li deposition via 5 mA cm−2 for 10 min after the short circuit, and (f) during Li dissolution via 20 mA cm−2 for 20 min shortly after (e).200 | ||
Fig. 30d–f displays SEM images of the interface morphology between the SE layer and stainless steel at the same location within the cell under different conditions: (d) immediately after a short circuit, (e) during Li deposition at 5 mA cm2 for 10 min following the short circuit, and (f) during Li dissolution at 20 mA cm2 for 20 min after (e). These images illustrate the morphological changes that occur when electrochemical measurements are conducted at high current densities after short circuits, indicating limited Li deposition and dissolution. In Fig. 30d, a pillared deposit is visible, emerging from big cracks in the SE layer's surface. This deposit differs from the SE layer's morphology, suggesting it is lithium. In Fig. 30e, lithium appears to grow slightly during the deposition process compared to Fig. 30d, with the dotted line measuring 6.6 mm in (d) and 12.8 mm in (e). A new big crack is observed on the right side of the pillared deposit, likely resulting from the elongation of Li deposits, demonstrating that lithium is extending from the inner part of the solid electrolyte layer. Fig. 30f reveals that the pillared deposit decreases following the Li dissolution reactions, with the dotted line in this image measuring 10.9 mm, which is smaller than the measurement in Fig. 30e, confirming lithium contraction.
In general, their findings visually demonstrated that the morphologies of Li deposition changed with CDs. When the CD exceeds 1 mA cm−2, localized Li deposition causes significant cracking, which reduces the reversibility of deposition/dissolution processes. Conversely, at a low CD of 0.01 mA cm−2, uniform deposition allows for reversible lithium processes and minimizes the formation of harmful cracks. These outcomes indicate that obtaining uniform Li deposition on the solid electrolyte and preventing Li metal development along grain boundaries within the solid electrolyte are crucial for facilitating repeated Li deposition/dissolution without compromising the integrity of the solid electrolyte.
4.11. Optical microscopy
Huang et al.360 examined lithium plating/stripping phenomena in argyrodite-based ASSLMBs using optical microscopy (OM). To explore the impact of current densities on the CE, they performed an in situ OM study to analyze lithium plating and stripping behaviors during the initial two cycles (Fig. 31a). As plating commences, small white spots appear on the surface of SE, indicating nucleation of metallic lithium. This nucleation is observed to be random and heterogeneous (Fig. 31b). As plating progresses, these white spots serve as seeds that enlarge. The central region becomes black during this growth while the surrounding area remains white. Ultimately, large white islands with a central black spot are randomly distributed across the SE surface (Fig. 31c). During the stripping phase, the white islands are anticipated to shrink. However, they unexpectedly expand even while lithium is being removed from them (Fig. 31d and e), likely due to internal pressure within the in situ cell. As plating continues, additional white spots appear beneath the existing islands (marked by blue circles in Fig. 31f and g). In the second stripping cycle, similar behavior was observed, as characterized by expansion of the outer white areas and shrinkage of the central dark regions (blue circles in Fig. 31h and i). Overall, lithium eventually forms isolated islands on the surface of the SE.361 Additionally, a cell's plating and stripping processes upon cycling at 200 μA cm−2 exhibited behavior similar to that observed at 10 μA cm−2. The initial lithium plating remains non-uniform and localized. With higher CD, more lithium islands form, featuring larger central black spots, and these islands grow at an accelerated rate.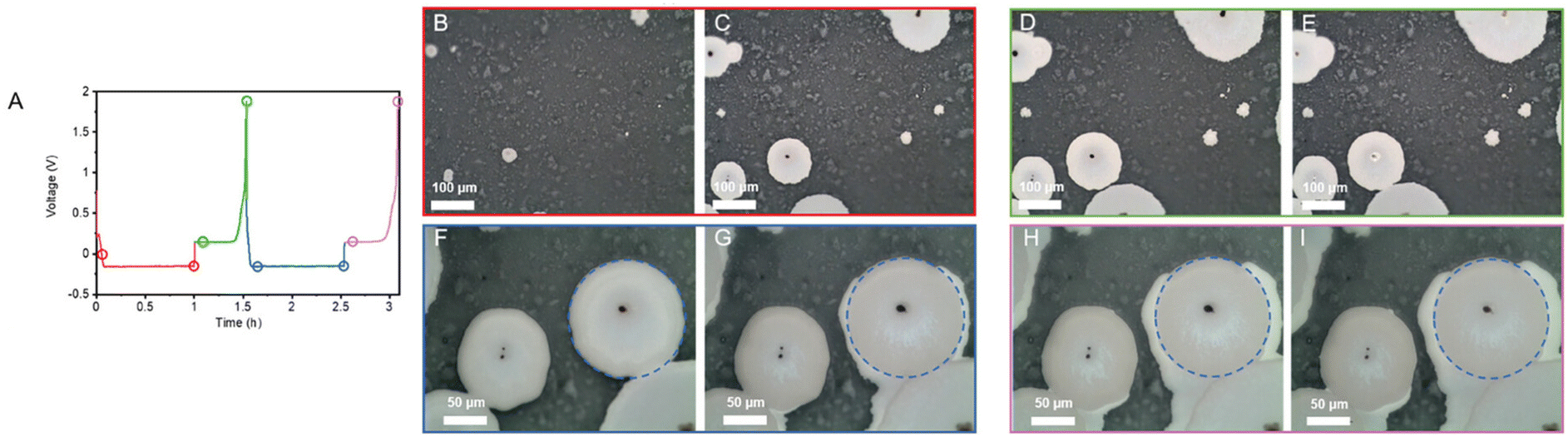 | ||
| Fig. 31 In situ OM measurements. (A) Voltage profiles at 10 μA cm−2 along with optical images taken at various time points, (B and C) during the first plating, (D and E) during the first stripping, (F and G) during the second plating, (H and I) during the second stripping.360 | ||
4.12. Time-of-flight secondary ion mass spectrometry (ToF-SIMS)
Huo et al. investigated the phenomena of particles in the Li6PS5Cl matrix before cycling and after 100 cycles via ToF-SIMS mass images (Fig. 32a and b).362 Specifically, after cycling, a layer exhibiting increased intensity of sulfur fragments was observed around the silicon particles (Fig. 32b). This is interpreted as strong confirmation of the Li2S-rich SEI at the Si|Li6PS5Cl interface. | ||
| Fig. 32 ToF-SIMS images of the Cl− fracture and the product of the LiS− and S− fractures in Si/Li6PS5Cl composites before cycling (a) and after 100 cycles (b).362 | ||
4.13. Kelvin probe force microscopy (KPFM)
Recently, Wan et al. utilized in situ Kelvin probe force microscopy (KPFM), a robust technique for monitoring potential variations, to visualize the lithium ion behavior in LPS|LGPS|LPS sandwich electrolyte interfaces (Fig. 33a). The LPS|LGPS|LPS sandwich electrolyte will be a model system to examine the interfacial interactions between distinct SEs. Fig. 33b shows the KPFM image of the LGPS|LPS interface at OCP. Upon applying an overpotential of −0.5 V, the potential at the interface increased, as depicted in Fig. 33c. Both LGPS and LPS, as inorganic SEs, have transference numbers close to unity, indicating that Li ions are nearly the only mobile ions in these SEs.64 Therefore, the increased interfacial potential was ascribed to the accumulation of Li ions at the interface. Amplification of the overpotential led to a further increase in interfacial potential (Fig. 33d), indicating a huge concentration of lithium ions at the interfaces. KPFM imaging successfully visualized the distribution of the lithium-ion field. Complementary COMSOL simulations (Fig. 33e–g) provided further insights into this phenomenon, confirming the results obtained from the in situ KPFM analysis. | ||
| Fig. 33 Li-ion behavior at the interface between different SEs. (a) A schematic illustrating the in situ KPFM setup and Li-ion interfacial enrichment at the sandwiched SE. In situ KPFM images of the LGPS/LPS interface at various overpotentials of (b) OCP, (c) cathodic −0.5 V, (d) cathodic −1V. The scale bars are 5 μm, and the sweep direction is from bottom to top for (b–d). The COMSOL simulation images of the LGPS/LPS interface at different overpotentials of (e) OCP, (f) cathodic −0.5V, (g) cathodic −1 V.320 | ||
Advanced in situ and ex situ characterization techniques are crucial for understanding solid interfacial reactions, including structural evolution, phase transitions, and fundamental ion-transport mechanisms at interfaces. These methods can help to address interfacial challenges, such as compatibility, stability, and resistance in solid-state systems, thus revealing the kinetics of reactions and mechanisms of decay in ASSLBs. A combination of various in situ and ex situ methods is essential for investigating interface evolution further.
5. Perspectives
Improving the efficiency of ASSLBs hinges on integrating SEs that exhibit significant σ at ambient temperature and enhanced stability at interfaces. While Li–argyrodites stand out as the most promising electrolytes for ASSLBs, substantial challenges persist concerning the material itself, electrode materials, and their interfaces, despite notable advancements achieved thus far. To realize Li–argyrodite-based ASSLBs, these problems should be solved first as follows.➢ Optimization of SEs based on interfacial stability: Despite the current focus in ASSLB research on developing high ionic conductivity SEs to match the conductivity of LEs, there is a notable lack of consideration for the electrode–SE charge transfer resistance in the design of these new SEs. While adding halides like I, Br, or Cl can enhance the ionic conductivity of Li argyrodites, they are incompatible with Li metal and high-voltage oxide cathode materials due to numerous anionic redox phenomena while charging. This leads to a thicker and unstable SEI and CEI layer with high interfacial resistance, making these electrolytes impractical. Hence, there is a need to prioritize and conduct the examination of SEs according to their interfacial characteristics with specific electrode materials. For instance, using a cation-doped argyrodite SE and optimizing the interface stability between lithium metal and the electrolyte through Li–M alloy formation leads to minimized interfacial resistance.
➢ Mechanical properties of SEs and electrodes: The mechanical characteristics of each component in ASSLBs are critical factors for ensuring successful cycle performance. This is particularly crucial for the soft lithium metal anode because the development of lithium metal dendrites and lithium plating/stripping kinetics are heavily influenced by mechanical integrity around the interface. Applying lithiophilic coatings on the lithium anode stabilizes the lithium and solid electrolyte interface, promoting non-dendritic lithium deposition but typically at small current densities. Nevertheless, questions remain regarding the long-term mechanical stabilities of these lithiophilic coating layers, especially with higher capacities of lithium depositions. For instance, a coating thickness of 1–2 nm is optimal for electronically insulating but chemically protective layers like Al2O3, while a thickness of 5–10 nm is more appropriate for lithium-ion conductive coatings. In addition, heat treatment (∼400–600 °C) generally increases the crystallinity of argyrodite SEs, leading to improved ionic conductivity. Pore size and distribution play crucial roles in shaping lithium-ion transport paths. They may act as stress concentration points, increasing the risk of fractures or lithium dendrite intrusion. Densification methods such as hot-pressing or sintering will be employed to reduce porosity and enhance mechanical integrity. Mechanical strain develops throughout battery cycling due to volumetric changes, requiring solid electrolytes to endure compressive and tensile stresses without fracturing. Incorporating dopants can enhance the ductility of solid electrolytes, helping to suppress crack initiation. At the interfaces between solid electrolytes and electrodes, lattice mismatch can cause interfacial strain and defects, compromising interfacial stability and elevating resistance. This challenge can be addressed through the use of interlayers or surface coatings. Additionally, interface defects such as cracks, voids, or delamination can obstruct ion transport and contribute to mechanical degradation. Optimization of microstructural design and the application of surface coatings can enhance interfacial contact and help prevent such degradation. Furthermore, the mechanical characteristics and structures of new interfaces toward battery performance require thorough investigation. Detailed mechanical and chemical modeling of interfaces under static and dynamic changes in electric fields is necessary. Additionally, mechanical stress in ASSLBs can extend over considerable distances. For instance, extensive lithium depositions (negative electrode side) can induce stress that propagates through the solid electrolyte toward cathodes. This impacts chemical and mechanical compatibility during cycling. Therefore, comprehensive stress characterization at the full cell level is essential, as it is more relevant than focusing solely on characterizing individual components in ASSLBs.
➢ Appropriate and optimized binders: Binders are commonly employed to address issues such as electrode or electrolyte disintegration and the disruption of conducting paths. These are essential for achieving thin film electrolytes with robust mechanical properties, which significantly impact on the battery performance. Traditional binders like PVDF, PTFE, or PEO are typically electrically insulating and ionically inactive, which can hinder lithium-ion transport, especially if they are unevenly distributed, potentially obstructing conductive pathways. To enhance overall transport performance, dual-conductive binders can support both ionic and electronic conduction. Additionally, binders may cause interfacial resistance or create voids at the electrode–electrolyte interface, impairing effective solid–solid contact that is essential for lithium-ion movement. Under cycling or thermal stress, some binders may swell or deform, disrupting interfacial pressure and stability. As a solution, hybrid inorganic/polymer binders will offer a good compromise by providing mechanical robustness and improved conductivity, thereby mitigating adverse effects on electrochemical transport. However, it is crucial to consider the interaction between Li argyrodites and binders as a design factor for ASSLB electrodes. Therefore, it is strongly advised that the adhesive properties of binders be thoroughly understood and the binder quantity optimized before cell assembly.
➢ Pressure: In ASSLB systems, the procedures related to external pressure during assembly are closely linked to the stabilities of materials at the atomistic/interface level. Applying external pressure on the cathode side enhances interfacial ion diffusion, decreases interfacial impedance, and boosts ion transport efficiency. Suitable pressure helps to eliminate interfacial voids on the anode side, preserving a continuous ion transport network. However, excessive pressure may lead to short circuits, as many anode materials exhibit creep behavior under high stress. Applying stack pressures in the range of 3–7 MPa can effectively minimize problems such as lithium metal deformation and improve the physical contact between the solid electrolyte and electrodes, lowering interfacial resistance and enhancing cycling performance. The concept of local mechanical constriction connects the macroscopic parameters of the device with the microscopic properties of materials. In such environments, degradation reactions with positive reaction strains, leading to volume extension during decomposition, may encounter a unique effect called constriction-induced meta-stability. This effect can cause a sudden change in the energy landscape related to these decomposition reactions. Therefore, understanding the local mechanical constriction effect and interfacial compatibility during cycling is crucial for exploring the potential to further design this effect for Li argyrodites. This understanding could lead to unlocking battery performance improvements beyond what is currently achieved in commercial batteries. Developing ductile electrodes, electrolytes, and interphases of a Li–argyrodite-based ASSLB is crucial for its practical application under reasonable pressure. For example, using a solid electrolyte with intrinsically ductile and low porosity can help lower both the fabrication pressure required during processing and the stack pressure needed during operation of the ASSLB.
➢ Artificial intelligence (AI) and machine learning (ML): Machine learning models are capable of forecasting key properties of solid electrolytes, such as ionic conductivity, chemical stability, and compatibility with electrode materials, based on their structural and compositional features. Moreover, ML can be utilized to simulate interfacial degradation phenomena, including the development of resistive interphase layers, by utilizing extensive molecular simulations supported by machine-learning-derived interatomic potentials. This approach enables the rapid evaluation of new material candidates before committing to time-intensive experimental testing. Additionally, AI techniques assist in interpreting intricate interfacial transport behavior, including lithium-ion diffusion and the dynamics of charged species.
AI and ML are poised to significantly enhance the development of battery technologies. To realize this potential, several challenges must be addressed.
i. Establishing standardized protocols: Developing widely accepted standards for data collection, processing, and reporting in battery research is essential. This includes defining data formats, measurement conditions, and performance metrics to ensure consistency and comparability across studies. Implementing systematic data disclosure practices will facilitate data sharing and collaboration within the research community.
ii. Identifying appropriate descriptors: Selecting the most relevant descriptors (features) for ML models is crucial. Descriptors should capture the underlying physics and chemistry of battery materials and interfaces to enable accurate predictions. Integrating domain-specific knowledge can aid in identifying these descriptors and improving model interpretability.
iii. Quantifying model uncertainty: Determining the associated error or uncertainty of ML models is vital for assessing their reliability and robustness. Implementing uncertainty quantification methods can help identify areas where models may be less confident and guide further data collection or model refinement.
Addressing these challenges will pave the way for AI and ML to play a transformative role in the advancement of battery technologies, leading to more efficient, durable, and sustainable energy storage solutions.
➢ Characterization techniques: While advanced characterization techniques have revealed crucial mechanisms related to interfaces, most of these methods are ex situ, providing limited real-time information. On the other hand, in situ/operando approaches are essential for understanding interface composition and structure, which are vital for ASSLB design. In situ characterization is essential for capturing the dynamic and coupled chemomechanical processes occurring at interfaces in SSBs. For example, in situ X-ray computed tomography (CT) facilitates the real-time observation of morphological changes within SSBs during cycling, such as the development of cracks and voids in the SE and at interfaces. These structural alterations can lead to capacity loss and failure. Identifying these changes is crucial for understanding failure modes and guiding design improvements. In addition, optical coherence tomography (OCT) is a non-invasive, high-resolution imaging technique that enables real-time, cross-sectional visualization of internal structures in SSBs. It has been effectively employed to monitor the morphology, growth, and evolution of lithium dendrites at the interfaces between electrodes and SEs during cycling under various conditions. This capability allows for the dynamic observation of dendrite formation and progression, providing valuable insights into the mechanisms affecting SSB performance and safety. At the same time, ex-situ methods serve as valuable complements by delivering high-resolution structural or compositional information after changes have occurred. However, ex-situ techniques offer only fixed, post-mortem views, which can miss short-lived or intermediate phenomena. Additionally, transferring samples to measurement setups can introduce artifacts, such as reactions with air or moisture, and these methods often struggle to accurately reflect the true interfacial chemistry, particularly in reactive materials like sulfide-based electrolytes. However, extracting solid–solid interfaces embedded in ASSLBs presents challenges. Moreover, due to the fragility of interfaces, solid electrolytes, and lithium metal, cryogenic protection is vital to reduce beam destruction and stabilize samples. Hence, developing and customizing new techniques with integrated functionalities is crucial for obtaining information on structure, composition, and kinetics. In general, computational modeling and experimental methods necessitate high-throughput screening, diagnosis, and interfacial engineering for ASSLBs.
Data availability
The data supporting this article have been included as part of the ESI.† Other data could be available upon kind request by contacting the corresponding authors.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
Financial support from the National Science and Technology Council of Taiwan (NSTC 113-2639-E-011-001-ASP, 113-2923-E-011-002, 113-2222-E-011-004; 112-2923-E-011-001, 112-2923-E-011-004-MY3; 112-2218-E-011-011) and the Ministry of Education of Taiwan (the Sustainable Electrochemical Energy Development Center (SEED Center) from the Featured Areas Research Center Program), and the supporting facilities from the National Taiwan University of Science and Technology (NTUST), the National Center for High Performance Computing (NCHC), and the National Synchrotron Radiation Research Centre (NSRRC), are all gratefully acknowledged.References
- S. Boulineau, M. Courty, J.-M. Tarascon and V. Viallet, Solid State Ionics, 2012, 221, 1–5 CrossRef CAS.
- B. Vinay, Y. Nikodimos, T. Agnihotri, S. A. Ahmed, T. M. Hagos, R. Hasan, E. B. Tamilarasan, W.-N. Su and B. J. Hwang, Energy Environ. Sci., 2025 10.1039/D4EE04820B.
- P. Birke, F. Salam, S. Döring and W. Weppner, Solid State Ionics, 1999, 118, 149–157 CrossRef CAS.
- A. Aboulaich, R. Bouchet, G. Delaizir, V. Seznec, L. Tortet, M. Morcrette, P. Rozier, J. M. Tarascon, V. Viallet and M. Dollé, Adv. Energy Mater., 2011, 1, 179 CrossRef CAS.
- C. Masquelier, Nat. Mater., 2011, 10, 649–650 CrossRef CAS PubMed.
- T. Chen, L. Zhang, Z. Zhang, P. Li, H. Wang, C. Yu, X. Yan, L. Wang and B. Xu, ACS Appl. Mater. Interfaces, 2019, 11, 40808–40816 CrossRef CAS PubMed.
- Y. Cheng, X. Ke, Y. Chen, X. Huang, Z. Shi and Z. Guo, Nano Energy, 2019, 63, 103854 CrossRef CAS.
- X.-B. Cheng, C.-Z. Zhao, Y.-X. Yao, H. Liu and Q. Zhang, Chem, 2019, 5, 74–96 CAS.
- R. Xu, X. Xia, S. Zhang, D. Xie, X. Wang and J. Tu, Electrochim. Acta, 2018, 284, 177–187 CrossRef CAS.
- J. Auvergniot, A. Cassel, J.-B. Ledeuil, V. Viallet, V. Seznec and R. Dedryvère, Chem. Mater., 2017, 29, 3883–3890 CrossRef CAS.
- J. Auvergniot, A. Cassel, D. Foix, V. Viallet, V. Seznec and R. Dedryvère, Solid State Ionics, 2017, 300, 78–85 CrossRef CAS.
- G. G. Serbessa, B. W. Taklu, Y. Nikodimos, N. T. Temesgen, Z. B. Muche, S. K. Merso, T.-I. Yeh, Y.-J. Liu, W.-S. Liao, C.-H. Wang, S.-H. Wu, W.-N. Su, C.-C. Yang and B. J. Hwang, ACS Appl. Mater. Interfaces, 2024, 16, 10832–10844 CrossRef CAS PubMed.
- Y. Nikodimos, C.-J. Huang, B. W. Taklu, W.-N. Su and B. J. Hwang, Energy Environ. Sci., 2022, 15, 991–1033 RSC.
- N. T. Temesgen, H. K. Bezabh, M. A. Weret, K. N. Shitaw, Y. Nikodimos, B. W. Taklu, K. Lakshmanan, S.-C. Yang, S.-K. Jiang, C.-J. Huang, S.-H. Wu, W.-N. Su and B. J. Hwang, J. Power Sources, 2023, 556, 232462 CrossRef CAS.
- H. Lee, P. Oh, J. Kim, H. Cha, S. Chae, S. Lee and J. Cho, Adv. Mater., 2019, 31, 1900376 CrossRef PubMed.
- J. Ma, B. Chen, L. Wang and G. Cui, J. Power Sources, 2018, 392, 94–115 CrossRef CAS.
- S. P. Culver, R. Koerver, T. Krauskopf and W. G. Zeier, Chem. Mater., 2018, 30, 4179–4192 CrossRef CAS.
- Y. Wang, W. D. Richards, S. P. Ong, L. J. Miara, J. C. Kim, Y. Mo and G. Ceder, Nat. Mater., 2015, 14, 1026–1031 CrossRef CAS PubMed.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- Y. Seino, T. Ota, K. Takada, A. Hayashi and M. Tatsumisago, Energy Environ. Sci., 2014, 7, 627–631 RSC.
- M. Nagao, H. Kitaura, A. Hayashi and M. Tatsumisago, J. Electrochem. Soc., 2013, 160, A819–A823 CrossRef CAS.
- B. D. Dandena, D.-S. Tsai, S.-H. Wu, W.-N. Su and B. J. Hwang, Energy Storage Mater., 2024, 103305 CrossRef.
- B.-Y. Tsai, S.-K. Jiang, Y.-T. Wu, J.-S. Yang, S.-H. Wu, P.-C. Tsai, W.-N. Su, C.-Y. Chiang and B. J. Hwang, J. Phys. Chem. C, 2023, 127, 14336–14343 CrossRef CAS.
- S.-K. Jung, H. Gwon, S.-S. Lee, H. Kim, J. C. Lee, J. G. Chung, S. Y. Park, Y. Aihara and D. Im, J. Mater. Chem. A, 2019, 7, 22967–22976 RSC.
- F. Zhao, J. Liang, C. Yu, Q. Sun, X. Li, K. Adair, C. Wang, Y. Zhao, S. Zhang, W. Li, L. Zhang, S. Zhao, S. Lu and X. Sun, Adv. Energy Mater., 2020, 10, 1903422 CrossRef CAS.
- H. Liu, Q. Zhu, Y. Liang, C. Wang, D. Li, X. Zhao, L. Gao and L.-Z. Fan, Chem. Eng. J., 2023, 462, 142183 CrossRef CAS.
- D. H. Tan, E. A. Wu, H. Nguyen, Z. Chen, M. A. Marple, J.-M. Doux, X. Wang, H. Yang, A. Banerjee and Y. S. Meng, ACS Energy Lett., 2019, 4, 2418–2427 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS PubMed.
- T. K. Schwietert, V. A. Arszelewska, C. Wang, C. Yu, A. Vasileiadis, N. J. de Klerk, J. Hageman, T. Hupfer, I. Kerkamm, Y. Xu, E. V. D. Maas, E. M. Kelder, S. Ganapathy and M. Wagemaker, Nat. Mater., 2020, 19, 428–435 CrossRef CAS PubMed.
- S. Wenzel, S. J. Sedlmaier, C. Dietrich, W. G. Zeier and J. Janek, Solid State Ionics, 2018, 318, 102–112 CrossRef CAS.
- B. D. Dandena, L.-T. Wu, T.-I. Yeh, C.-Y. Chang, J.-C. Jiang, D.-S. Tsai, M. Ihrig, G. G. Serbessa, S.-C. Yang, K. Lakshmanan, H.-S. Sheu, C.-H. Wang, S.-H. Wu, W.-N. Su and B. J. Hwang, Chem. Eng. J., 2025, 162450 CrossRef CAS.
- X. Han, Y. Gong, K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang, G. Rubloff, Y. Mo, V. Thangadurai, E. D. Wachsman and L. Hu, Nat. Mater., 2017, 16, 572–579 CrossRef CAS PubMed.
- S. Choi, M. Jeon, J. Ahn, W. D. Jung, S. M. Choi, J.-S. Kim, J. Lim, Y.-J. Jang, H.-G. Jung, J.-H. Lee, B.-I. Sang and H. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 23740–23747 CrossRef CAS PubMed.
- R. Koerver, I. Aygün, T. Leichtweiß, C. Dietrich, W. Zhang, J. O. Binder, P. Hartmann, W. G. Zeier and J. Janek, Chem. Mater., 2017, 29, 5574–5582 CrossRef CAS.
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Adv. Energy Mater., 2016, 6, 1501590 CrossRef.
- X. Fan, X. Ji, F. Han, J. Yue, J. Chen, L. Chen, T. Deng, J. Jiang and C. Wang, Sci. Adv., 2018, 4, eaau9245 CrossRef CAS PubMed.
- J. Haruyama, K. Sodeyama, L. Han, K. Takada and Y. Tateyama, Chem. Mater., 2014, 26, 4248–4255 CrossRef CAS.
- J. L. Barton and J. O. M. Bockris, Proc. R. Soc. London, Ser. A, 1962, 268, 485–505 CAS.
- F. Han, A. S. Westover, J. Yue, X. Fan, F. Wang, M. Chi, D. N. Leonard, N. J. Dudney, H. Wang and C. Wang, Nat. Energy, 2019, 4, 187–196 CrossRef CAS.
- F. Walther, R. Koerver, T. Fuchs, S. Ohno, J. Sann, M. Rohnke, W. G. Zeier and J. Janek, Chem. Mater., 2019, 31, 3745–3755 CrossRef CAS.
- F. Walther, S. Randau, Y. Schneider, J. Sann, M. Rohnke, F. H. Richter, W. G. Zeier and J. Janek, Chem. Mater., 2020, 32, 6123–6136 CrossRef CAS.
- F. Walther, F. Strauss, X. Wu, B. Mogwitz, J. Hertle, J. Sann, M. Rohnke, T. Brezesinski and J. Janek, Chem. Mater., 2021, 33, 2110–2125 CrossRef CAS.
- Y. Yamagishi, H. Morita, Y. Nomura and E. Igaki, ACS Appl. Mater. Interfaces, 2020, 13, 580–586 CrossRef PubMed.
- S. Narayanan, U. Ulissi, J. S. Gibson, Y. A. Chart, R. S. Weatherup and M. Pasta, Nat. Commun., 2022, 13, 7237 CrossRef CAS PubMed.
- J. Kasemchainan, S. Zekoll, D. S. Jolly, Z. Ning, G. O. Hartley, J. Marrow and P. G. Bruce, Nat. Mater., 2019, 18, 1105–1111 CrossRef CAS PubMed.
- R. Schlenker, D. Stepien, P. Koch, T. Hupfer, S. Indris, B. Roling, V. Miß, A. Fuchs, M. Wilhelmi and H. Ehrenberg, ACS Appl. Mater. Interfaces, 2020, 12, 20012–20025 CrossRef CAS PubMed.
- W. Jiang, X. Zhu, Y. Liu, S. Zhao, R. Huang, M. Ling, L. Wang and C. Liang, eTransportation, 2023, 17, 100246 CrossRef.
- T. Krauskopf, B. Mogwitz, H. Hartmann, D. K. Singh, W. G. Zeier and J. Janek, Adv. Energy Mater., 2020, 10, 2000945 CrossRef CAS.
- Y. Lu, C. Z. Zhao, H. Yuan, X. B. Cheng, J. Q. Huang and Q. Zhang, Adv. Funct. Mater., 2021, 31, 2009925 CrossRef CAS.
- Y. Liu, X. Xu, M. Sadd, O. O. Kapitanova, V. A. Krivchenko, J. Ban, J. Wang, X. Jiao, Z. Song, J. Song, S. Xiong and A. Matic, Adv. Sci., 2021, 8, 2003301 CrossRef CAS PubMed.
- Y. Pang, J. Pan, J. Yang, S. Zheng and C. Wang, Electrochem. Energy Rev., 2021, 4, 169–193 CrossRef CAS.
- L. Mazzapioda, A. Tsurumaki, G. Di Donato, H. Adenusi, M. A. Navarra and S. Passerini, Energy Mater., 2023, 3, 1–3025 Search PubMed.
- T. Tao, Z. Zheng, Y. Gao, B. Yu, Y. Fan, Y. Chen, S. Huang and S. Lu, Energy Mater., 2022, 2, 200036 CAS.
- T. Rahman and T. Alharbi, Batteries, 2024, 10, 220 CrossRef CAS.
- H. Sano, Y. Morino, A. Yabuki, S. Sato, N. Itayama, Y. Matsumura, M. Iwasaki, M. Takehara, T. Abe, Y. Ishiguro, T. Takahashi, N. Miyashita, A. Sakuda and A. Hayashi, Electrochemistry, 2022, 90, 037012–037012 CrossRef CAS.
- C. Yu, S. Ganapathy, E. R. van Eck, L. van Eijck, S. Basak, Y. Liu, L. Zhang, H. W. Zandbergen and M. Wagemaker, J. Mater. Chem. A, 2017, 5, 21178–21188 RSC.
- S. Y. Han, C. Lee, J. A. Lewis, D. Yeh, Y. Liu, H.-W. Lee and M. T. McDowell, Joule, 2021, 5, 2450–2465 CrossRef CAS.
- S. Lou, Q. Liu, F. Zhang, Q. Liu, Z. Yu, T. Mu, Y. Zhao, J. Borovilas, Y. Chen, M. Ge, X. Xiao, W.-K. Lee, G. Yin, Y. Yang, X. Sun and J. Wang, Nat. Commun., 2020, 11, 5700 CrossRef CAS PubMed.
- S. Wang, A. La Monaca and G. P. Demopoulos, Energy Adv., 2025, 11–36 RSC.
- S. Wenzel, S. Randau, T. Leichtweiß, D. A. Weber, J. Sann, W. G. Zeier and J. r. Janek, Chem. Mater., 2016, 28, 2400–2407 CrossRef CAS.
- S. Wenzel, T. Leichtweiss, D. Krüger, J. Sann and J. Janek, Solid State Ionics, 2015, 278, 98–105 CrossRef CAS.
- Y. Xiao, Y. Wang, S.-H. Bo, J. C. Kim, L. J. Miara and G. Ceder, Nat. Rev. Mater., 2020, 5, 105–126 CrossRef CAS.
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5, 229–252 CrossRef CAS.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- S. Wang, H. Song, X. Song, T. Zhu, Y. Ye, J. Chen, L. Yu, J. Xu and K. Chen, Energy Storage Mater., 2021, 39, 139–145 CrossRef.
- Y. Xuan, Y. Wang, B. He, S. Bian, J. Liu, B. Xu and G. Zhang, Chem. Mater., 2022, 34, 9104–9110 CrossRef CAS.
- J. Tan, X. Ao, H. Zhuo, L. Zhuang, X. Huang, C. Su, W. Tang, X. Peng and B. Tian, Chem. Eng. J., 2021, 420, 127623 CrossRef CAS.
- J. Xu, K. Liu, M. A. Khan, H. Wang, T. He, H. Zhao, D. Ye, Y. Tang and J. Zhang, Chem. Eng. J., 2022, 443, 136637 CrossRef CAS.
- P. Lu, S. Gong, C. Wang, Z. Yu, Y. Huang, T. Ma, J. Lian, Z. Jiang, L. Chen, H. Li and F. Wu, ACS Nano, 2024, 18, 7334–7345 CrossRef CAS PubMed.
- S. Deng, M. Jiang, N. Chen, W. Li, M. Zheng, W. Chen, R. Li, H. Huang, J. Wang, C. V. Singh and X. Sun, Adv. Funct. Mater., 2022, 32, 2205594 CrossRef CAS.
- P. Lu, Y. Wu, D. Wu, F. Song, T. Ma, W. Yan, X. Zhu, F. Guo, J. Lu, J. Peng, L. Chen, H. Li and F. Wu, Energy Storage Mater., 2024, 67, 103316 CrossRef.
- X. Shen, Y. Yang, X. Zhang and M. Chen, J. Energy Storage, 2023, 74, 109296 CrossRef.
- L. Guan, Y. Shi, C. Gao, T. Wang, J. Zhou and R. Cai, Electrochim. Acta, 2023, 440, 141669 CrossRef CAS.
- J. Yan, J. Liu, Z. Liu, Q. Zhang, Y. Feng, W. Xia and J. Zhou, J. Power Sources, 2025, 631, 236186 CrossRef CAS.
- H. S. Shin, W. G. Ryu, M. S. Park, K. N. Jung, H. Kim and J. W. Lee, ChemSusChem, 2018, 11, 3184–3190 CrossRef CAS PubMed.
- Z. Jiang, H. Xie, S. Wang, X. Song, X. Yao and H. Wang, Adv. Energy Mater., 2018, 8, 1801433 CrossRef.
- B. Shao, Y. Huang and F. Han, Adv. Energy Mater., 2023, 13, 2204098 CrossRef CAS.
- F. Zhao, Q. Sun, C. Yu, S. Zhang, K. Adair, S. Wang, Y. Liu, Y. Zhao, J. Liang, C. Wang, X. Li, X. Li, W. Xia, R. Li, H. Huang, L. Zhang, S. Zhao, S. Lu and X. Sun, ACS Energy Lett., 2020, 5, 1035–1043 CrossRef CAS.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Chem. Mater., 2016, 28, 266–273 CrossRef CAS.
- C. Wang, J. Liang, S. Hwang, X. Li, Y. Zhao, K. Adair, C. Zhao, X. Li, S. Deng, X. Lin, X. Yang, R. Li, H. Huang, L. Zhang, S. Lu, D. Su and X. Sun, Nano Energy, 2020, 72, 104686 CrossRef CAS.
- Q. Lv, Y. Jiang, B. Wang, Y. Chen, F. Jin, B. Wu, H. Ren, N. Zhang, R. Xu, Y. Li, T. Zhang, Y. Zhou, D. Wang, H. Liu and S. Dou, Cell Rep. Phys. Sci., 2022, 3, 100706 CrossRef CAS.
- D. Avadanii, S. Ganschow, M. Stypa, S. Müller, S. Lang, D. Kramer, C. Kirchlechner and R. Mönig, ACS Energy Lett., 2025, 10, 2061–2067 CrossRef CAS.
- B. Zhang, B. Yuan, X. Yan, X. Han, J. Zhang, H. Tan, C. Wang, P. Yan, H. Gao and Y. Liu, Nat. Commun., 2025, 16, 1906 CrossRef CAS PubMed.
- C. Zhu, T. Fuchs, S. A. Weber, F. H. Richter, G. Glasser, F. Weber, H.-J. Butt, J. Janek and R. Berger, Nat. Commun., 2023, 14, 1300 CrossRef CAS PubMed.
- H. Liu, Y. Chen, P.-H. Chien, G. Amouzandeh, D. Hou, E. Truong, I. P. Oyekunle, J. Bhagu, S. W. Holder, H. Xiong, P. L. Gor’kov, J. T. Rosenberg, S. C. Grant and Y.-Y. Hu, Nat. Mater., 2025, 1–8 Search PubMed.
- N. Sarfraz, N. Kanwal, M. Ali, K. Ali, A. Hasnain, M. Ashraf, M. Ayaz, J. Ifthikar, S. Ali, A. Hendi, N. Baig, M. F. Ehsan, S. S. Shah, R. Khan and I. Khan, Energy Storage Mater., 2024, 103619 CrossRef.
- M. K. Tufail, P. Zhai, W. Khokar, M. Jia, N. Zhao and X. Guo, Interdiscip. Mater., 2023, 2, 529–568 Search PubMed.
- B. W. Taklu, W.-N. Su, Y. Nikodimos, K. Lakshmanan, N. T. Temesgen, P.-X. Lin, S.-K. Jiang, C.-J. Huang, D.-Y. Wang, H.-S. Sheu, S.-H. Wu and B. J. Hwang, Nano Energy, 2021, 90, 106542 CrossRef CAS.
- Y. Jin, B. Zhu, Z. Lu, N. Liu and J. Zhu, Adv. Energy Mater., 2017, 7, 1700715 CrossRef.
- A. F. Gonzalez, N.-H. Yang and R.-S. Liu, J. Phys. Chem. C, 2017, 121, 27775–27787 CrossRef.
- V. A. Sethuraman, M. J. Chon, M. Shimshak, V. Srinivasan and P. R. Guduru, J. Power Sources, 2010, 195, 5062–5066 CrossRef CAS.
- X. Yuan, H. Liu and J. Zhang, Lithium-ion batteries: advanced materials and technologies, CRC Press, 2011 Search PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS.
- K. N. Wood, M. Noked and N. P. Dasgupta, ACS Energy Lett., 2017, 2, 664–672 CrossRef CAS.
- Y.-G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D.-S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, Nat. Energy, 2020, 5, 299–308 CrossRef CAS.
- R. Xu, J. Yue, S. Liu, J. Tu, F. Han, P. Liu and C. Wang, ACS Energy Lett., 2019, 4, 1073–1079 CrossRef CAS.
- A. N. Dey and B. P. Sulliva, J. Electrochem. Soc., 1970, 117, 222 CrossRef CAS.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047 CrossRef CAS.
- E. Peled, D. Golodnitsky and G. Ardel, J. Electrochem. Soc., 1997, 144, 8 CrossRef.
- D. Aurbach, B. Markovsky, M. Levi, E. Levi, A. Schechter, M. Moshkovich and Y. Cohen, J. Power Sources, 1999, 81, 95–111 CrossRef.
- M. Winter, Z. Phys. Chem., 2009, 223, 1395–1406 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- L. M. Riegger, S. Mittelsdorf, T. Fuchs, R. Rueß, F. H. Richter and J. Janek, Chem. Mater., 2023, 35, 5091–5099 CrossRef CAS.
- G. Chaney, A. Golov, A. van Roekeghem, J. Carrasco and N. Mingo, ACS Appl. Mater. Interfaces, 2024, 16, 24624–24630 CrossRef CAS PubMed.
- P. Albertus, V. Anandan, C. Ban, N. Balsara, I. Belharouak, J. Buettner-Garrett, Z. Chen, C. Daniel, M. Doeff, N. J. Dudney, D. Bruce, S. J. Harris, S. Herle, E. Herbert, S. Kalnaus, J. A. Libera, D. Lu, S. Martin, B. D. McCloskey, Y. S. Meng, J. Nanda, J. Sakamoto, E. C. Self, S. Tepavcevic, E. Wachsman, C. Wang, A. S. Westover, J. Xiao and T. Yersak, ACS Energy Lett., 2021, 6, 1399–1404 Search PubMed.
- Y. Lu, Z. Tu and L. A. Archer, Nat. Mater., 2014, 13, 961–969 CrossRef CAS PubMed.
- J.-G. Zhang, W. Xu and W. A. Henderson, Lithium metal anodes and rechargeable lithium metal batteries, Springer International Publishing Cham, 2017 Search PubMed.
- R. Cao, W. Xu, D. Lv, J. Xiao and J. Zhang, Adv. Energy Mater., 2015, 5, 1402273 CrossRef.
- N. W. Li, Y. Shi, Y. X. Yin, X. X. Zeng, J. Y. Li, C. J. Li, L. J. Wan, R. Wen and Y. G. Guo, Angew. Chem., 2018, 130, 1521–1525 CrossRef.
- S. H. Wang, Y. X. Yin, T. T. Zuo, W. Dong, J. Y. Li, J. L. Shi, C. H. Zhang, N. W. Li, C. J. Li and Y. G. Guo, Adv. Mater., 2017, 29, 1703729 CrossRef PubMed.
- M. J. Wang, R. Choudhury and J. Sakamoto, Joule, 2019, 3, 2165–2178 CrossRef CAS.
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Nat. Energy, 2018, 3, 16–21 CrossRef CAS.
- K. N. Wood and G. Teeter, ACS Appl. Energy Mater., 2018, 1, 4493–4504 CrossRef CAS.
- K. N. Wood, K. X. Steirer, S. E. Hafner, C. Ban, S. Santhanagopalan, S.-H. Lee and G. Teeter, Nat. Commun., 2018, 9, 2490 CrossRef PubMed.
- L. Zhou, N. Minafra, W. G. Zeier and L. F. Nazar, Acc. Chem. Res., 2021, 54, 2717–2728 CrossRef CAS PubMed.
- Q. Zhang, D. Cao, Y. Ma, A. Natan, P. Aurora and H. Zhu, Adv. Mater., 2019, 31, 1901131 CrossRef CAS PubMed.
- R. Chen, Q. Li, X. Yu, L. Chen and H. Li, Chem. Rev., 2019, 120, 6820–6877 CrossRef PubMed.
- L. Xu, S. Tang, Y. Cheng, K. Wang, J. Liang, C. Liu, Y.-C. Cao, F. Wei and L. Mai, Joule, 2018, 2, 1991–2015 CrossRef CAS.
- S. Xin, Y. You, S. Wang, H.-C. Gao, Y.-X. Yin and Y.-G. Guo, ACS Energy Lett., 2017, 2, 1385–1394 CrossRef CAS.
- C.-Z. Zhao, H. Duan, J.-Q. Huang, J. Zhang, Q. Zhang, Y.-G. Guo and L.-J. Wan, Sci. China: Chem., 2019, 62, 1286–1299 CrossRef CAS.
- C. Monroe and J. Newman, J. Electrochem. Soc., 2004, 151, A880–A886 CrossRef CAS.
- C. Monroe and J. Newman, J. Electrochem. Soc., 2005, 152, A396–A404 CrossRef CAS.
- P. Barai, K. Higa and V. Srinivasan, Phys. Chem. Chem. Phys., 2017, 19, 20493–20505 RSC.
- W. S. LePage, Y. Chen, E. Kazyak, K.-H. Chen, A. J. Sanchez, A. Poli, E. M. Arruda, M. Thouless and N. P. Dasgupta, J. Electrochem. Soc., 2019, 166, A89–A97 CrossRef CAS.
- A. Masias, N. Felten, R. Garcia-Mendez, J. Wolfenstine and J. Sakamoto, J. Mater. Sci., 2019, 54, 2585–2600 CrossRef CAS.
- L. Porz, T. Swamy, B. W. Sheldon, D. Rettenwander, T. Frömling, H. L. Thaman, S. Berendts, R. Uecker, W. C. Carter and Y. M. Chiang, Adv. Energy Mater., 2017, 7, 1701003 CrossRef.
- X. Liu, R. Garcia-Mendez, A. R. Lupini, Y. Cheng, Z. D. Hood, F. Han, A. Sharafi, J. C. Idrobo, N. J. Dudney, C. Wang, C. Ma, J. Sakamoto and M. Chi, Nat. Mater., 2021, 20, 1485–1490 CrossRef CAS PubMed.
- T. Krauskopf, R. Dippel, H. Hartmann, K. Peppler, B. Mogwitz, F. H. Richter, W. G. Zeier and J. Janek, Joule, 2019, 3, 2030–2049 CrossRef CAS.
- Z. Ning, D. S. Jolly, G. Li, R. De Meyere, S. D. Pu, Y. Chen, J. Kasemchainan, J. Ihli, C. Gong, B. Liu, D. L. R. Melvin, A. Bonnin, O. Magdysyuk, P. Adamson, G. O. Hartley, C. W. Monroe, T. J. Marrow and P. G. Bruce, Nat. Mater., 2021, 20, 1121–1129 CrossRef CAS PubMed.
- A. Banerjee, X. Wang, C. Fang, E. A. Wu and Y. S. Meng, Chem. Rev., 2020, 120, 6878–6933 CrossRef CAS PubMed.
- R. Usiskin and J. Maier, Adv. Energy Mater., 2021, 11, 2001455 CrossRef CAS.
- H. Tang, Z. Deng, Z. Lin, Z. Wang, I.-H. Chu, C. Chen, Z. Zhu, C. Zheng and S. P. Ong, Chem. Mater., 2018, 30, 163–173 CrossRef CAS.
- C. Wang, W. Ping, Q. Bai, H. Cui, R. Hensleigh, R. Wang, A. H. Brozena, Z. Xu, J. Dai and Y. Pei, Science, 2020, 368, 521–526 CrossRef CAS PubMed.
- C. Yang, H. Xie, W. Ping, K. Fu, B. Liu, J. Rao, J. Dai, C. Wang, G. Pastel and L. Hu, Adv. Mater., 2019, 31, 1804815 CrossRef PubMed.
- E. Kazyak, R. Garcia-Mendez, W. S. LePage, A. Sharafi, A. L. Davis, A. J. Sanchez, K.-H. Chen, C. Haslam, J. Sakamoto and N. P. Dasgupta, Matter, 2020, 2, 1025–1048 CrossRef.
- M. Wang, J. B. Wolfenstine and J. Sakamoto, Electrochim. Acta, 2019, 296, 842–847 CrossRef CAS.
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef.
- C. Chen, M. Jiang, T. Zhou, L. Raijmakers, E. Vezhlev, B. Wu, T. U. Schülli, D. L. Danilov, Y. Wei, R. A. Eichel and P. H. L. Notten, Adv. Energy Mater., 2021, 11, 2003939 CrossRef CAS.
- S. Guo, Y. Sun and A. Cao, Chem. Res. Chin. Univ., 2020, 36, 329–342 CrossRef CAS.
- M. Hansen and E. Wallström, J. Adhes., 1983, 275–286 CrossRef.
- R. M. Tiggelaar, V. Verdoold, H. Eghbali, G. Desmet and J. G. Gardeniers, Lab Chip, 2009, 9, 456–463 RSC.
- G. Bucci, T. Swamy, Y.-M. Chiang and W. C. Carter, J. Mater. Chem. A, 2017, 5, 19422–19430 RSC.
- Y. Liu, H. Su, M. Li, J. Xiang, X. Wu, Y. Zhong, X. Wang, X. Xia, C. Gu and J. Tu, J. Mater. Chem. A, 2021, 9, 13531–13539 RSC.
- G. Liu, W. Weng, Z. Zhang, L. Wu, J. Yang and X. Yao, Nano Lett., 2020, 20, 6660–6665 CrossRef CAS PubMed.
- Z. Zhang, L. Zhang, X. Yan, H. Wang, Y. Liu, C. Yu, X. Cao, L. van Eijck and B. Wen, J. Power Sources, 2019, 410, 162–170 CrossRef.
- Y. Subramanian, R. Rajagopal, S. Kang and K.-S. Ryu, J. Alloys Compd., 2022, 925, 166596 CrossRef CAS.
- H. Liu, Q. Zhu, C. Wang, G. Wang, Y. Liang, D. Li, L. Gao and L. Z. Fan, Adv. Funct. Mater., 2022, 32, 2203858 CrossRef CAS.
- A. C. Luntz, J. Voss and K. Reuter, J. Phys. Chem. Lett., 2015, 6, 4599–4604 CrossRef CAS PubMed.
- H. Gerischer, F. Decker and B. Scrosati, J. Electrochem. Soc., 1994, 141, 2297 CrossRef CAS.
- P. G. Bruce, Solid state electrochemistry, Cambridge University Press, 1997 Search PubMed.
- G. Liu, H. Zheng, A. Simens, A. Minor, X. Song and V. Battaglia, J. Electrochem. Soc., 2007, 154, A1129–A1134 CrossRef CAS.
- M. Manickam and M. Takata, Electrochim. Acta, 2003, 48, 957–963 CrossRef CAS.
- B. Stiaszny, J. C. Ziegler, E. E. Krauß, J. P. Schmidt and E. Ivers-Tiffée, J. Power Sources, 2014, 251, 439–450 CrossRef CAS.
- R. Hausbrand, G. Cherkashinin, H. Ehrenberg, M. Gröting, K. Albe, C. Hess and W. Jaegermann, J. Mater. Sci. Eng. B, 2015, 192, 3–25 CrossRef CAS.
- T. Liu, L. Yu, J. Liu, J. Lu, X. Bi, A. Dai, M. Li, M. Li, Z. Hu, L. Ma, D. Luo, J. Zheng, T. Wu, Y. Ren, J. Wen, F. Pan and K. Amine, Nat. Energy, 2021, 6, 277–286 CrossRef CAS.
- B. Xiao, H. Liu, N. Chen, M. N. Banis, H. Yu, J. Liang, Q. Sun, T. K. Sham, R. Li, M. Cai, G. A. Botton and X. Sun, Angew. Chem., Int. Ed., 2020, 59, 14313–14320 CrossRef CAS PubMed.
- Y. Zhang, X. Sun, D. Cao, G. Gao, Z. Yang, H. Zhu and Y. Wang, Energy Storage Mater., 2021, 41, 505–514 CrossRef.
- T. Li, X.-Z. Yuan, L. Zhang, D. Song, K. Shi and C. Bock, Electrochem. Energy Rev., 2020, 3, 43–80 CrossRef.
- S. P. Culver, R. Koerver, W. G. Zeier and J. Janek, Adv. Energy Mater., 2019, 9, 1900626 CrossRef.
- A. M. Nolan, Y. Zhu, X. He, Q. Bai and Y. Mo, Joule, 2018, 2, 2016–2046 CrossRef CAS.
- D. H. Tan, A. Banerjee, Z. Chen and Y. S. Meng, Nat. Nanotechnol., 2020, 15, 170–180 CrossRef CAS PubMed.
- Q. Xie, Z. Cui and A. Manthiram, Adv. Mater., 2021, 33, 2100804 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 RSC.
- Y. Mao, X. Wang, S. Xia, K. Zhang, C. Wei, S. Bak, Z. Shadike, X. Liu, Y. Yang, R. Xu, P. Pianetta, S. Ermon, E. Stavitski, K. Zhao, Z. Xu, F. Lin, X.-Q. Yang, E. Hu and Y. Liu, Adv. Funct. Mater., 2019, 29, 1900247 CrossRef.
- C. Wang, R. Yu, S. Hwang, J. Liang, X. Li, C. Zhao, Y. Sun, J. Wang, N. Holmes, R. Li, H. Huang, S. Zhao, L. Zhang, S. Lu, D. Su and X. Sun, Energy Storage Mater., 2020, 30, 98–103 CrossRef CAS.
- X. Li, W. Peng, R. Tian, D. Song, Z. Wang, H. Zhang, L. Zhu and L. Zhang, Electrochim. Acta, 2020, 363, 137185 CrossRef CAS.
- R. Koerver, W. Zhang, L. de Biasi, S. Schweidler, A. O. Kondrakov, S. Kolling, T. Brezesinski, P. Hartmann, W. G. Zeier and J. Janek, Energy Environ. Sci., 2018, 11, 2142–2158 RSC.
- C. Liu, F. Roters and D. Raabe, Nat. Commun., 2024, 15, 7970 CrossRef CAS PubMed.
- C. Park, J. Lee, S. Lee, Y. J. Han, J. Kim and S. K. Jung, Adv. Energy Mater., 2023, 13, 2203861 CrossRef CAS.
- T. Shi, Y.-Q. Zhang, Q. Tu, Y. Wang, M. Scott and G. Ceder, J. Mater. Chem. A, 2020, 8, 17399–17404 RSC.
- M. Clausnitzer, M. Ihrig, L. Cressa, S. Hein, M. Finsterbusch, S. Eswara, L.-Y. Kuo, T. Danner, P. Kaghazchi, D. Fattakhova-Rohlfing, O. Guillon and A. Latz, Energy Storage Mater., 2024, 67, 103262 CrossRef.
- H. Huo, Y. Bai, S. L. Benz, T. Weintraut, S. Wang, A. Henss, D. Raabe and J. Janek, Adv. Mater., 2025, 37, 2415006 CrossRef CAS PubMed.
- S. P. Jand and P. Kaghazchi, MRS Commun., 2018, 8, 591–596 CrossRef.
- B. Gao, R. Jalem, Y. Ma and Y. Tateyama, Chem. Mater., 2019, 32, 85–96 CrossRef.
- A. Sakuda, A. Hayashi and M. Tatsumisago, Chem. Mater., 2010, 22, 949–956 CrossRef CAS.
- S. Lee, Y. Kim, C. Park, J. Kim, J.-S. Kim, H. Jo, C. J. Lee, S. Choi, D.-H. Seo and S.-K. Jung, ACS Energy Lett., 2024, 9, 1369–1380 CrossRef CAS.
- P. Nzereogu, A. Oyesanya, S. Ogba, S. Ayanwunmi, M. Sobajo, V. Chimsunum, V. Ayanwunmi, M. Amoo, O. Adefemi and C. Chukwudi, Hybrid Adv., 2024, 100339 Search PubMed.
- S. J. Yang, J. K. Hu, F. N. Jiang, H. Yuan, H. S. Park and J. Q. Huang, InfoMat, 2024, 6, e12512 CrossRef CAS.
- T. Kim, K. Kim, S. Lee, G. Song, M. S. Jung and K. T. Lee, Chem. Mater., 2022, 34, 9159–9171 CrossRef CAS.
- X. Rui, D. Ren, X. Liu, X. Wang, K. Wang, Y. Lu, L. Li, P. Wang, G. Zhu, Y. Mao, X. Feng, L. Lu, H. Wang and M. Ouyang, Energy Environ. Sci., 2023, 16, 3552–3563 RSC.
- Y. Wang, X. Feng, W. Huang, X. He, L. Wang and M. Ouyang, Adv. Energy Mater., 2023, 13, 2203841 CrossRef CAS.
- Y. Wang, X. Feng, Y. Peng, F. Zhang, D. Ren, X. Liu, L. Lu, Y. Nitta, L. Wang and M. Ouyang, Joule, 2022, 6, 2810–2820 CrossRef CAS.
- A. Sakuda, A. Hayashi, T. Ohtomo, S. Hama and M. Tatsumisago, Electrochem. Solid-State Lett., 2010, 13, A73–A75 CrossRef CAS.
- A. Sakuda, A. Hayashi, T. Ohtomo, S. Hama and M. Tatsumisago, J. Power Sources, 2011, 196, 6735–6741 CrossRef CAS.
- A. Hayashi, K. Noi, A. Sakuda and M. Tatsumisago, Nat. Commun., 2012, 3, 856 CrossRef PubMed.
- A. Sakuda, A. Hayashi, Y. Takigawa, K. Higashi and M. Tatsumisago, J. Ceram. Soc. Jpn., 2013, 121, 946–949 CrossRef CAS.
- Y. Kubota, M. C. S. Escaño, H. Nakanishi and H. Kasai, J. Appl. Phys., 2007, 102, 053704 CrossRef.
- M. Ashuri, Q. He and L. L. Shaw, Nanoscale, 2016, 8, 74–103 RSC.
- M. Gu, Z. Wang, J. G. Connell, D. E. Perea, L. J. Lauhon, F. Gao and C. Wang, ACS Nano, 2013, 7, 6303–6309 CrossRef CAS PubMed.
- A. Sakuda, A. Hayashi and M. Tatsumisago, Sci. Rep., 2013, 3, 2261 CrossRef PubMed.
- J. Wu, S. Liu, F. Han, X. Yao and C. Wang, Adv. Mater., 2021, 33, 2000751 CrossRef CAS PubMed.
- M. K. Tufail, P. Zhai, M. Jia, N. Zhao and X. Guo, Energy Mater. Adv., 2023, 4, 0015 CrossRef CAS.
- M. Jia, M. K. Tufail and X. Guo, ChemSusChem, 2023, 16, e202201801 CrossRef CAS PubMed.
- W. Qin, T. Nagase, Y. Umakoshi and J. Szpunar, Philos. Mag. Lett., 2008, 88, 169–179 CrossRef CAS.
- R. Sudo, Y. Nakata, K. Ishiguro, M. Matsui, A. Hirano, Y. Takeda, O. Yamamoto and N. Imanishi, Solid State Ionics, 2014, 262, 151–154 CrossRef CAS.
- Y. Ren, Y. Shen, Y. Lin and C.-W. Nan, Electrochem. Commun., 2015, 57, 27–30 CrossRef CAS.
- M. Nagao, A. Hayashi, M. Tatsumisago, T. Kanetsuku, T. Tsuda and S. Kuwabata, Phys. Chem. Chem. Phys., 2013, 15, 18600–18606 RSC.
- R. Bradbury, G. F. Dewald, M. A. Kraft, T. Arlt, N. Kardjilov, J. Janek, I. Manke, W. G. Zeier and S. Ohno, Adv. Energy Mater., 2023, 13, 2203426 CrossRef CAS.
- Y. Kimura, M. Fakkao, T. Nakamura, T. Okumura, N. Ishiguro, O. Sekizawa, K. Nitta, T. Uruga, M. Tada, Y. Uchimoto and K. Amezawa, ACS Appl. Energy Mater., 2020, 3, 7782–7793 CrossRef CAS.
- L. Froboese, J. F. Van Der Sichel, T. Loellhoeffel, L. Helmers and A. Kwade, J. Electrochem. Soc., 2019, 166, A318–A328 CrossRef.
- A. Bielefeld, D. A. Weber and J. Janek, J. Phys. Chem. C, 2018, 123, 1626–1634 CrossRef.
- L. Ketter, N. Greb, T. Bernges and W. G. Zeier, Nat. Commun., 2025, 16, 1–9 Search PubMed.
- P. Minnmann, L. Quillman, S. Burkhardt, F. H. Richter and J. Janek, J. Electrochem. Soc., 2021, 168, 040537 CrossRef CAS.
- J. Zahnow, T. Bernges, A. Wagner, N. Bohn, J. R. Binder, W. G. Zeier, M. T. Elm and J. Janek, ACS Appl. Energy Mater., 2021, 4, 1335–1345 CrossRef CAS.
- D. K. Ferry, Science, 2012, 335, 45–46 CrossRef CAS PubMed.
- S. Kirkpatrick, Rev. Mod. Phys., 1973, 45, 574 CrossRef.
- M. E. Frary and C. A. Schuh, Phys. Rev. E:Stat., Nonlinear, Soft Matter Phys., 2007, 76, 041108 CrossRef PubMed.
- S. Hao, Q. Zhang, X. Kong, Z. Wang, X. P. Gao and P. R. Shearing, Adv. Energy Mater., 2025, 15, 2404384 CrossRef CAS.
- Z. Deng, Z. Wang, I.-H. Chu, J. Luo and S. P. Ong, J. Electrochem. Soc., 2015, 163, A67–A74 CrossRef.
- Z. Jiang, Q. Han, S. Wang and H. Wang, ChemElectroChem, 2019, 6, 2970–2983 CrossRef CAS.
- R. Schlenker, D. Stępień, P. Koch, T. Hupfer, S. Indris, B. Roling, V. Miß, A. Fuchs, M. Wilhelmi and H. Ehrenberg, ACS Appl. Mater. Interfaces, 2020, 12, 20012–20025 CrossRef CAS PubMed.
- G. G. Serbessa, Y. Nikodimos, B. W. Taklu, S. K. Merso, Z. B. Muche, B. D. Dandena, S. A. Vallal, T.-I. Yeh, F. Valencia, Y.-F. Hung, J.-H. Hsu, C.-M. Lee, S.-H. Wu, W.-N. Su, C.-C. Yang and B. J. Hwang, Energy Storage Mater., 2025, 104103 CrossRef.
- L. E. Camacho-Forero and P. B. Balbuena, J. Power Sources, 2018, 396, 782–790 CrossRef CAS.
- K. J. Kim and J. L. Rupp, Energy Environ. Sci., 2020, 13, 4930–4945 RSC.
- Y. Kimura, T. Fujisaki, T. Shimizu, T. Nakamura, Y. Iriyama and K. Amezawa, Commun. Mater., 2024, 5, 125 CrossRef CAS.
- Y. Xiao, L. J. Miara, Y. Wang and G. Ceder, Joule, 2019, 3, 1252–1275 CrossRef CAS.
- H.-S. Kim, Y. Kim, S.-I. Kim and S. W. Martin, J. Power Sources, 2006, 161, 623–627 CrossRef CAS.
- N. Ohta, K. Takada, L. Zhang, R. Ma, M. Osada and T. Sasaki, Adv. Mater., 2006, 18, 2226–2229 CrossRef CAS.
- T. Kato, T. Hamanaka, K. Yamamoto, T. Hirayama, F. Sagane, M. Motoyama and Y. Iriyama, J. Power Sources, 2014, 260, 292–298 CrossRef CAS.
- K. H. Kim, Y. Iriyama, K. Yamamoto, S. Kumazaki, T. Asaka, K. Tanabe, C. A. Fisher, T. Hirayama, R. Murugan and Z. Ogumi, J. Power Sources, 2011, 196, 764–767 CrossRef CAS.
- D. Rettenwander, R. Wagner, A. Reyer, M. Bonta, L. Cheng, M. M. Doeff, A. Limbeck, M. Wilkening and G. Amthauer, J. Phys. Chem. C, 2018, 122, 3780–3785 CrossRef CAS PubMed.
- J. A. Lewis, F. J. Q. Cortes, M. G. Boebinger, J. Tippens, T. S. Marchese, N. Kondekar, X. Liu, M. Chi and M. T. McDowell, ACS Energy Lett., 2019, 4, 591–599 CrossRef CAS.
- Z. D. Hood, A. U. Mane, A. Sundar, S. Tepavcevic, P. Zapol, U. D. Eze, S. P. Adhikari, E. Lee, G. E. Sterbinsky, J. W. Elam and J. G. Connell, Adv. Mater., 2023, 35, 2300673 CrossRef CAS PubMed.
- M. Balaish, J. C. Gonzalez-Rosillo, K. J. Kim, Y. Zhu, Z. D. Hood and J. L. Rupp, Nat. Energy, 2021, 6, 227–239 CrossRef CAS.
- J. C. Bachman, S. Muy, A. Grimaud, H.-H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- J. Maier, J. Electrochem. Soc., 1987, 134, 1524 CrossRef CAS.
- J. Maier, Prog. Solid State Chem., 1995, 23, 171–263 CrossRef CAS.
- E. Rangasamy, G. Sahu, J. K. Keum, A. J. Rondinone, N. J. Dudney and C. Liang, J. Mater. Chem. A, 2014, 2, 4111–4116 RSC.
- Z. D. Hood, H. Wang, Y. Li, A. S. Pandian, M. P. Paranthaman and C. Liang, Solid State Ionics, 2015, 283, 75–80 CrossRef CAS.
- B. Ransom, A. Ramdas, E. Lomeli, J. Fidawi, A. Sendek, T. Devereaux, E. J. Reed and P. Schindler, ACS Appl. Mater. Interfaces, 2023, 15, 44394–44403 CrossRef CAS PubMed.
- F. Yang, S. Lin, Z. Guo, Y. Shao, B. Zhang, X. Zhang, S. Yan and A. A. Volinsky, J. Alloys Compd., 2019, 805, 1034–1043 CrossRef CAS.
- E. Temeche, S. Indris and R. M. Laine, ACS Appl. Mater. Interfaces, 2020, 12, 46119–46131 CrossRef CAS PubMed.
- Y. Wu, D. Lei and C. Wang, Mater. Today Phys., 2021, 18, 100395 CrossRef CAS.
- S. Ito, S. Fujiki, T. Yamada, Y. Aihara, Y. Park, T. Y. Kim, S.-W. Baek, J.-M. Lee, S. Doo and N. Machida, J. Power Sources, 2014, 248, 943–950 CrossRef CAS.
- S. Miyakawa, S. Matsuda, N. Tanibata, H. Takeda, M. Nakayama, T. Saito and S. Fukuchi, Sci. Rep., 2022, 12, 16672 CrossRef PubMed.
- R. Johnson Jr, B. Morosin, M. Knotek and R. Biefeld, Phys. Lett. A, 1975, 54, 403–404 CrossRef.
- F. Shin-ichi, S. Satoshi, S. Kaduhiro and T. Hitoshi, Solid State Ionics, 2004, 167, 325–329 CrossRef CAS.
- Y. Sun, F. Li, Q. Qiao, J. Cao, Y. Wang and S. Ye, Electrochim. Acta, 2015, 176, 1464–1475 CrossRef CAS.
- P. Quintana, F. Velasco and A. West, Solid State Ionics, 1989, 34, 149–155 CrossRef CAS.
- C. Wang, K. Aoyagi, P. Wisesa and T. Mueller, Chem. Mater., 2020, 32, 3741–3752 CrossRef CAS.
- H. Zhou, S. Yu, H. Liu and P. Liu, J. Power Sources, 2020, 450, 227632 CrossRef CAS.
- C. Niu, H. Lee, S. Chen, Q. Li, J. Du, W. Xu, J.-G. Zhang, M. S. Whittingham, J. Xiao and J. Liu, Nat. Energy, 2019, 4, 551–559 CrossRef CAS.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M.-H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
- Q. Cheng, L. Wei, Z. Liu, N. Ni, Z. Sang, B. Zhu, W. Xu, M. Chen, Y. Miao, L.-Q. Chen, W. Min and Y. Yang, Nat. Commun., 2018, 9, 2942 CrossRef PubMed.
- Z. Tu, S. Choudhury, M. J. Zachman, S. Wei, K. Zhang, L. F. Kourkoutis and L. A. Archer, Joule, 2017, 1, 394–406 CrossRef CAS.
- Y. Gao, Z. Yan, J. L. Gray, X. He, D. Wang, T. Chen, Q. Huang, Y. C. Li, H. Wang, S. H. Kim, T. E. Mallouk and D. Wang, Nat. Mater., 2019, 18, 384–389 CrossRef CAS PubMed.
- Y. Liu, Q. Sun, Y. Zhao, B. Wang, P. Kaghazchi, K. R. Adair, R. Li, C. Zhang, J. Liu, L.-Y. Kuo, Y. Hu, T.-K. Sham, L. Zhang, R. Yang, S. Lu, X. Song and X. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 31240–31248 CrossRef CAS PubMed.
- L. Sang, K. L. Bassett, F. C. Castro, M. J. Young, L. Chen, R. T. Haasch, J. W. Elam, V. P. Dravid, R. G. Nuzzo and A. A. Gewirth, Chem. Mater., 2018, 30, 8747–8756 CrossRef CAS.
- P. Hartmann, T. Leichtweiss, M. R. Busche, M. Schneider, M. Reich, J. Sann, P. Adelhelm and J. Janek, J. Phys. Chem. C, 2013, 117, 21064–21074 CrossRef CAS.
- Y. Ruan, Y. Lu, X. Huang, J. Su, C. Sun, J. Jin and Z. Wen, J. Mater. Chem. A, 2019, 7, 14565–14574 RSC.
- Z. Zhang, S. Chen, J. Yang, J. Wang, L. Yao, X. Yao, P. Cui and X. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 2556–2565 CrossRef CAS PubMed.
- Y. Zhu, X. He and Y. Mo, Adv. Sci., 2017, 4, 1600517 CrossRef PubMed.
- Q. Cheng, A. Li, N. Li, S. Li, A. Zangiabadi, T.-D. Li, W Huang, A. C. Li, T. Jin, Q. Song, W. Xu, N. Ni, H. Zhai, M. Dontigny, K. Zaghib, X. Chuan, D. Su, K. Yan and Y. Yang, Joule, 2019, 3, 1510–1522 CrossRef CAS.
- M. Shigeno, K. Nagao, M. Deguchi, C. Hotehama, H. Kowada, A. Sakuda, A. Hayashi and M. Tatsumisago, Solid State Ionics, 2019, 339, 114985 CrossRef CAS.
- R. Court-Castagnet, C. Kaps, C. Cros and P. Hagenmuller, Solid State Ionics, 1993, 61, 327–334 CrossRef CAS.
- T. Tsujimura, S. Ito, K. Yoshida, Y. Higashiyama, Y. Aihara, N. Machida, Y. Park and D. Im, Solid State Ionics, 2022, 383, 115970 CrossRef CAS.
- H. Lutz, W. Schmidt and H. Haeuseler, J. Phys. Chem. Solids, 1981, 42, 287–289 CrossRef CAS.
- M. Calpa, N. C. Rosero-Navarro, A. Miura and K. Tadanaga, J. Sol-Gel Sci. Technol., 2022, 1–8 Search PubMed.
- Y. Zheng, W. Zhang, J. Gui, Q. Cao, Y. Wang, Y. Xu, Y. Liu and Y. Liang, Energy Storage Mater., 2023, 61, 102884 CrossRef.
- Y. Zhu, X. He and Y. Mo, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC.
- J. Liang, Y. Zhu, X. Li, J. Luo, S. Deng, Y. Zhao, Y. Sun, D. Wu, Y. Hu, W. Li, T.-K. Sham, R. Li, M. Gu and X. Sun, Nat. Commun., 2023, 14, 146 CrossRef CAS PubMed.
- J. Li, M. Zhang, D. Zhang, Y. Yan and Z. Li, Chem. Eng. J., 2020, 402, 126195 CrossRef CAS.
- Z. Ahaliabadeh, X. Kong, E. Fedorovskaya and T. Kallio, J. Power Sources, 2022, 540, 231633 CrossRef CAS.
- H. H. Sun, U.-H. Kim, J.-H. Park, S.-W. Park, D.-H. Seo, A. Heller, C. B. Mullins, C. S. Yoon and Y.-K. Sun, Nat. Commun., 2021, 12, 6552 CrossRef CAS PubMed.
- G.-T. Park, B. Namkoong, S.-B. Kim, J. Liu, C. S. Yoon and Y.-K. Sun, Nat. Energy, 2022, 7, 946–954 CrossRef CAS.
- D. Shumei, T. Dan, L. Ping, L. Huiqin, W. Fenyan and H. Zhang, J. Solid State Electrochem., 2023, 27, 1–23 CrossRef.
- L. Xu, F. Zhou, B. Liu, H. Zhou, Q. Zhang, J. Kong and Q. Wang, Int. J. Electrochem., 2018, 2018, 1–12 CrossRef.
- Q. Liu, Y. Zhang, J. Gao, Z. Zhou, H. Wang, K. Wang, X. Zhang, L. Li and J.-F. Li, Energy Environ. Sci., 2018, 11, 3531–3539 RSC.
- F. Han, J. Yue, X. Zhu and C. Wang, Adv. Energy Mater., 2018, 8, 1703644 CrossRef.
- G. Liu, D. Xie, X. Wang, X. Yao, S. Chen, R. Xiao, H. Li and X. Xu, Energy Storage Mater., 2019, 17, 266–274 CrossRef.
- N. Wang, K. Yang, L. Zhang, X. Yan, L. Wang and B. Xu, J. Mater. Sci., 2018, 53, 1987–1994 CrossRef CAS.
- A. Kato, M. Nagao, A. Sakuda, A. Hayashi and M. Tatsumisago, J. Ceram. Soc. Jpn., 2014, 122, 552–555 CrossRef CAS.
- Y. Xia, J. Li, J. Zhang, X. Zhou, H. Huang, X. He, Y. Gan, Z. Xiao and W. Zhang, J. Power Sources, 2022, 543, 231846 CrossRef CAS.
- Z. Jiang, H. Peng, Y. Liu, Z. Li, Y. Zhong, X. Wang, X. Xia, C. Gu and J. Tu, Adv. Energy Mater., 2021, 11, 2101521 CrossRef CAS.
- L. Zhou, A. Assoud, Q. Zhang, X. Wu and L. F. Nazar, J. Am. Chem. Soc., 2019, 141, 19002–19013 CrossRef CAS PubMed.
- V. Y. Prokhorenko, V. V. Roshchupkin, M. A. Pokrasin, S. Prokhorenko and V. Kotov, High Temp., 2000, 38, 954–968 CrossRef CAS.
- H. Ge, H. Li, S. Mei and J. Liu, Renewable Sustainable Energy Rev., 2013, 21, 331–346 CrossRef CAS.
- J. Liu, Y.-X. Zhou, Y.-G. Lv and T. Li, IMECE, 2005, 501–510 CAS.
- H. Ge and J. Liu, Front. Energy, 2012, 6, 207–209 CrossRef.
- H. Ge and J. Liu, J. Heat Transfer, 2013, 135, 054503 CrossRef.
- S. Chen, D. Xie, G. Liu, J. P. Mwizerwa, Q. Zhang, Y. Zhao, X. Xu and X. Yao, Energy Storage Mater., 2018, 14, 58–74 CrossRef.
- J. Lau, R. H. DeBlock, D. M. Butts, D. S. Ashby, C. S. Choi and B. S. Dunn, Adv. Energy Mater., 2018, 8, 1800933 CrossRef.
- Z. Gao, H. Sun, L. Fu, F. Ye, Y. Zhang, W. Luo and Y. Huang, Adv. Mater., 2018, 30, 1705702 CrossRef PubMed.
- W. Zhang, F. H. Richter, S. P. Culver, T. Leichtweiss, J. G. Lozano, C. Dietrich, P. G. Bruce, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2018, 10, 22226–22236 CrossRef CAS PubMed.
- R. Koerver, F. Walther, I. Aygün, J. Sann, C. Dietrich, W. G. Zeier and J. Janek, J. Mater. Chem. A, 2017, 5, 22750–22760 RSC.
- W. Zhang, T. Leichtweiß, S. P. Culver, R. Koerver, D. Das, D. A. Weber, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2017, 9, 35888–35896 CrossRef CAS PubMed.
- A. Banerjee, H. Tang, X. Wang, J.-H. Cheng, H. Nguyen, M. Zhang, D. H. Tan, T. A. Wynn, E. A. Wu, J.-M. Doux, T. Wu, L. Ma, G. E. Sterbinsky, M. S. D’Souza, S. P. Ong and Y. S. Meng, ACS Appl. Mater. Interfaces, 2019, 11, 43138–43145 CrossRef CAS PubMed.
- D. H. Tan, A. Banerjee, Z. Deng, E. A. Wu, H. Nguyen, J.-M. Doux, X. Wang, J.-h. Cheng, S. P. Ong, Y. S. Meng and Z. Chen, ACS Appl. Energy Mater., 2019, 2, 6542–6550 CrossRef CAS.
- S. Luo, Z. Wang, X. Li, X. Liu, H. Wang, W. Ma, L. Zhang, L. Zhu and X. Zhang, Nat. Commun., 2021, 12, 6968 CrossRef CAS PubMed.
- W. Fitzhugh, L. Ye and X. Li, J. Mater. Chem. A, 2019, 7, 23604–23627 RSC.
- W. Hao, M. Lee and G. S. Hwang, J. Power Sources, 2023, 560, 232689 CrossRef CAS.
- Z. Jiang, T. Liang, Y. Liu, S. Zhang, Z. Li, D. Wang, X. Wang, X. Xia, C. Gu and J. Tu, ACS Appl. Mater. Interfaces, 2020, 12, 54662–54670 CrossRef CAS PubMed.
- T. Chen, D. Zeng, L. Zhang, M. Yang, D. Song, X. Yan and C. Yu, J. Energy Chem., 2021, 59, 530–537 CrossRef CAS.
- H. Xu, G. Cao, Y. Shen, Y. Yu, J. Hu, Z. Wang and G. Shao, Energy Environ. Mater., 2022, 5, 852–864 CrossRef CAS.
- Z. Wang, Y. Jiang, J. Wu, Y. Jiang, W. Ma, Y. Shi, X. Liu, B. Zhao, Y. Xu and J. Zhang, Nano Energy, 2021, 84, 105906 CrossRef CAS.
- Z. Jiang, Y. Liu, H. Peng, J. Li, X. Xu, H. Su, Y. Zhong, X. Wang, C. Gu and J. Tu, Energy Storage Mater., 2023, 56, 300–309 CrossRef.
- Y. Kato, S. Shiotani, K. Morita, K. Suzuki, M. Hirayama and R. Kanno, J. Phys. Chem. Lett., 2018, 9, 607–613 CrossRef CAS PubMed.
- A. M. Stavola, X. Sun, D. P. Guida, A. M. Bruck, D. Cao, J. S. Okasinski, A. C. Chuang, H. Zhu and J. W. Gallaway, ACS Energy Lett., 2023, 8, 1273–1280 CrossRef CAS PubMed.
- D. Cao, Y. Zhao, X. Sun, A. Natan, Y. Wang, P. Xiang, W. Wang and H. Zhu, ACS Energy Lett., 2020, 5, 3468–3489 CrossRef CAS.
- M. A. Kraft, S. Ohno, T. Zinkevich, R. Koerver, S. P. Culver, T. Fuchs, A. Senyshyn, S. Indris, B. J. Morgan and W. G. Zeier, J. Am. Chem. Soc., 2018, 140, 16330–16339 CrossRef CAS PubMed.
- W. Zhang, D. A. Weber, H. Weigand, T. Arlt, I. Manke, D. Schröder, R. Koerver, T. Leichtweiss, P. Hartmann and W. G. Zeier, ACS Appl. Mater. Interfaces, 2017, 9, 17835–17845 CrossRef CAS PubMed.
- M. Otoyama, A. Sakuda, M. Tatsumisago and A. Hayashi, ACS Appl. Mater. Interfaces, 2020, 12, 29228–29234 CAS.
- L. Ye and X. Li, Nature, 2021, 593, 218–222 CrossRef CAS PubMed.
- P. Adeli, J. D. Bazak, K. H. Park, I. Kochetkov, A. Huq, G. R. Goward and L. F. Nazar, Angew. Chem., Int. Ed., 2019, 58, 8681–8686 CrossRef CAS PubMed.
- T. Krauskopf, F. H. Richter, W. G. Zeier and J. Janek, Chem. Rev., 2020, 120, 7745–7794 CrossRef CAS PubMed.
- S. Samanta, S. Bera, R. K. Biswas, S. Mondal, L. Mandal and A. Banerjee, ACS Energy Lett., 2024, 9, 3683–3693 CrossRef CAS.
- M. Sakuma, K. Suzuki, M. Hirayama and R. Kanno, Solid State Ionics, 2016, 285, 101–105 CrossRef CAS.
- L. Yang, Z. Wang, Y. Feng, R. Tan, Y. Zuo, R. Gao, Y. Zhao, L. Han, Z. Wang and F. Pan, Adv. Energy Mater., 2017, 7, 1701437 CrossRef.
- C. Yu, S. Ganapathy, E. R. v. Eck, H. Wang, S. Basak, Z. Li and M. Wagemaker, Nat. Commun., 2017, 8, 1086 CrossRef PubMed.
- T. Kato, R. Yoshida, K. Yamamoto, T. Hirayama, M. Motoyama, W. C. West and Y. Iriyama, J. Power Sources, 2016, 325, 584–590 CrossRef CAS.
- L. Mai, M. Yan and Y. Zhao, Nature, 2017, 546, 469–470 CrossRef CAS PubMed.
- S. A. Pervez, M. A. Cambaz, V. Thangadurai and M. Fichtner, ACS Appl. Mater. Interfaces, 2019, 11, 22029–22050 CrossRef CAS PubMed.
- B. Aktekin, L. M. Riegger, S.-K. Otto, T. Fuchs, A. Henss and J. Janek, Nat. Commun., 2023, 14, 6946 CrossRef CAS PubMed.
- Y. Huang, X. Li, J. Wang, L. Miao, C. Li, J. Han and Y. Huang, Energy Storage Mater., 2018, 15, 108–115 CrossRef.
- J. Yang, Z. Gao, T. Ferber, H. Zhang, C. Guhl, L. Yang, Y. Li, Z. Deng, P. Liu, C. Cheng, R. Che, W. Jaegermann, RenéHausbrand and Y. Huang, J. Mater. Chem. A, 2020, 8, 7828–7835 RSC.
- B. Aktekin, E. Kataev, L. M. Riegger, R. Garcia-Diez, Z. Chalkley, J. Becker, R. G. Wilks, A. Henss, M. Bär and J. Janek, ACS Energy Lett., 2024, 9, 3492–3500 CrossRef CAS.
- X.-S. Zhang, J. Wan, Z.-Z. Shen, S.-Y. Lang, S. Xin, R. Wen, Y.-G. Guo and L.-J. Wan, Angew. Chem., Int. Ed., 2024, e202409435 CAS.
- Y. Li, Z. Gao, F. Hu, X. Lin, Y. Wei, J. Peng, J. Yang, Z. Li, Y. Huang and H. Ding, Small Methods, 2020, 4, 2000111 CrossRef CAS.
- R.-J. Chen, Y.-B. Zhang, T. Liu, B.-Q. Xu, Y.-H. Lin, C.-W. Nan and Y. Shen, ACS Appl. Mater. Interfaces, 2017, 9, 9654–9661 CrossRef CAS PubMed.
- B. Liu, K. Fu, Y. Gong, C. Yang, Y. Yao, Y. Wang, C. Wang, Y. Kuang, G. Pastel, H. Xie, E. D. Wachsman and L. Hu, Nano Lett., 2017, 17, 4917–4923 CrossRef CAS PubMed.
- X. Li, Q. Sun, Z. Wang, D. Song, H. Zhang, X. Shi, C. Li, L. Zhang and L. Zhu, J. Power Sources, 2020, 456, 227997 CrossRef CAS.
- X. Li, J. Liang, M. N. Banis, J. Luo, C. Wang, W. Li, X. Li, Q. Sun, Y. Hu, Q. Xiao, T.-K. Sham, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li and X. Sun, Energy Storage Mater., 2020, 28, 325–333 CrossRef.
- Y. Zhou, C. Doerrer, J. Kasemchainan, P. G. Bruce, M. Pasta and L. J. Hardwick, Batteries Supercaps, 2020, 3, 647–652 CrossRef CAS.
- D. Zeng, J. Yao, L. Zhang, R. Xu, S. Wang, X. Yan, C. Yu and L. Wang, Nat. Commun., 2022, 13, 1909 CrossRef CAS PubMed.
- J. Dai, C. Yang, C. Wang, G. Pastel and L. Hu, Adv. Mater., 2018, 30, 1802068 CrossRef PubMed.
- P. Nellist, B. McCallum and J. M. Rodenburg, Nature, 1995, 374, 630–632 CrossRef CAS.
- P. Vadhva, J. Hu, M. J. Johnson, R. Stocker, M. Braglia, D. J. Brett and A. J. Rettie, ChemElectroChem, 2021, 8, 1930–1947 CrossRef CAS.
- C. Wei, C. Yu, L. Peng, Z. Zhang, R. Xu, Z. Wu, C. Liao, W. Zhang, L. Zhang, S. Cheng and J. Xie, Mater. Adv., 2022, 3, 1047–1054 RSC.
- C. Yu, J. Hageman, S. Ganapathy, L. van Eijck, L. Zhang, K. R. Adair, X. Sun and M. Wagemaker, J. Mater. Chem. A, 2019, 7, 10412–10421 RSC.
- L. H. Adem, B. N. Olana, B. W. Taklu, B. D. Dandena, G. G. Serbessa, B.-J. Hwang and S. D. Lin, Electrochim. Acta, 2024, 498, 144622 CrossRef CAS.
- S.-K. Jiang, S.-C. Yang, W.-H. Huang, H.-Y. Sung, R.-Y. Lin, J.-N. Li, B.-Y. Tsai, T. Agnihotri, Y. Nikodimos, C.-H. Wang, S. D. Lin, C.-C. Wang, S.-H. Wu, W.-N. Su and B. J. Hwang, J. Mater. Chem. A, 2023, 11, 2910–2919 RSC.
- Y. Wu, C. Li, X. Zheng, W. Zhao, H. Wang, J. Gu, Y. Cheng, Y. Lin, Y. Su, F. Ren, D. Feng, J. Liu, J. Peng, Z. Lv, Z. Wang, T. Brezesinski, Z. Gong and Y. Yang, ACS Energy Lett., 2024, 9, 5156–5165 CrossRef CAS.
- Z. Xu, X. Guo, W. Song, J. Wang, T. Qin, Y. Yuan and J. Lu, Adv. Mater., 2024, 36, 2303612 CrossRef CAS PubMed.
- Q. Zhang, Y. Huang, Y. Liu, S. Sun, K. Wang, Y. Li, X. Li, J. Han and Y. Huang, Sci. China Mater., 2017, 60, 629–636 CrossRef CAS.
- H.-M. Hau, T. Mishra, C. Ophus, T.-Y. Huang, K. Bustilo, Y. Sun, X. Yang, T. Holstun, X. Zhao, S. Wang, Y. Ha, G.-H. Lee, C. Song, J. Turner, J. Bai, L. Ma, K. Chen, F. Wang, W. Yang, B. D. McCloskey, Z. Cai and G. Ceder, Nat. Nanotechnol., 2024, 1–9 Search PubMed.
- M. Okubo, Y. Mizuno, H. Yamada, J. Kim, E. Hosono, H. Zhou, T. Kudo and I. Honma, ACS Nano, 2010, 4, 741–752 CrossRef CAS PubMed.
- H. Ji, J. Wu, Z. Cai, J. Liu, D.-H. Kwon, H. Kim, A. Urban, J. K. Papp, E. Foley, Y. Tian, M. Balasubramanian, H. Kim, R. J. Clément, B. D. McCloskey, W. Yang and G. Ceder, Nat. Energy, 2020, 5, 213–221 CrossRef CAS.
- J. Aspinall, D. E. Armstrong and M. Pasta, Mater. Today Energy, 2022, 30, 101183 CrossRef CAS.
- N. Brodusch, K. Zaghib and R. Gauvin, Microsc. Res. Tech., 2015, 78, 30–39 CrossRef CAS PubMed.
- A. Bastos, S. Zaefferer and D. Raabe, J. Microsc., 2008, 230, 487–498 CrossRef CAS PubMed.
- S.-K. Otto, Y. Moryson, T. Krauskopf, K. Peppler, J. Sann, J. Janek and A. Henss, Chem. Mater., 2021, 33, 859–867 CrossRef CAS.
- S.-K. Otto, T. Fuchs, Y. Moryson, C. Lerch, B. Mogwitz, J. Sann, J. Janek and A. Henss, ACS Appl. Energy Mater., 2021, 4, 12798–12807 CrossRef CAS.
- S. Kaboli, P. Noel, D. Clément, H. Demers, A. Paolella, P. Bouchard, M. L. Trudeau, J. B. Goodenough and K. Zaghib, Sci. Adv., 2020, 6, eabd5708 CrossRef CAS PubMed.
- H. Somekawa, K. Nishikawa, T. Moronaga and T. Ohmura, J. Power Sources, 2023, 569, 233019 CrossRef CAS.
- E. Darnbrough, J. Aspinall, M. Pasta and D. E. Armstrong, Mater. Des., 2023, 233, 112200 CrossRef CAS.
- T. Fuchs, T. Ortmann, J. Becker, C. G. Haslam, M. Ziegler, V. K. Singh, M. Rohnke, B. Mogwitz, K. Peppler, L. F. Nazar, J. Sakamoto and J. Janek, Nat. Mater., 2024, 1–8 Search PubMed.
- M. Motoyama, M. Hirota, T. Yamamoto and Y. Iriyama, ACS Appl. Mater. Interfaces, 2020, 12, 38045–38053 CrossRef CAS PubMed.
- T. Fuchs, J. Becker, C. G. Haslam, C. Lerch, J. Sakamoto, F. H. Richter and J. Janek, Adv. Energy Mater., 2023, 13, 2203174 CrossRef CAS.
- M. J. Wang, E. Carmona, A. Gupta, P. Albertus and J. Sakamoto, Nat. Commun., 2020, 11, 5201 CrossRef CAS PubMed.
- Y. Li, Y. Li and Y. Cui, Chem, 2018, 4, 2250–2252 CAS.
- Y. Li, W. Huang, Y. Li, A. Pei, D. T. Boyle and Y. Cui, Joule, 2018, 2, 2167–2177 CrossRef CAS.
- M. J. Zachman, Z. Tu, L. A. Archer and L. F. Kourkoutis, ACS Energy Lett., 2020, 5, 1224–1232 CrossRef CAS.
- X. Wang, Y. Li and Y. S. Meng, Joule, 2018, 2, 2225–2234 CrossRef CAS.
- Y. Li, Y. Li, A. Pei, K. Yan, Y. Sun, C.-L. Wu, L.-M. Joubert, R. Chin, A. L. Koh, Y. Yu, J. Perrino, B. Butz, S. Chu and Y. Cui, Science, 2017, 358, 506–510 CrossRef CAS PubMed.
- M. J. Zachman, Z. Tu, S. Choudhury, L. A. Archer and L. F. Kourkoutis, Nature, 2018, 560, 345–349 CrossRef CAS PubMed.
- A. Schwöbel, R. Hausbrand and W. Jaegermann, Solid State Ionics, 2015, 273, 51–54 CrossRef.
- D. Huang, G. Liu and W. Tong, Small, 2024, 2406862 CrossRef CAS PubMed.
- M. Motoyama, M. Ejiri and Y. Iriyama, J. Electrochem. Soc., 2015, 162, A7067–A7071 CrossRef CAS.
- H. Huo, M. Jiang, Y. Bai, S. Ahmed, K. Volz, H. Hartmann, A. Henss, C. V. Singh, D. Raabe and J. Janek, Nat. Mater., 2024, 1–9 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Interfacial challenges: Assembly, additives, and space charge layer. Strategies to improve the interface stability: Li metal coating techniques, protection layers, appropriate additives, appropriate assembling techniques, and utilizing the appropriate ratio of materials. Figures: XPS spectra of the S 2p and P 2p signals for cycled and uncycled composite cathodes with and without VGCF after surface cleaning, the influence of carbon on SE degradation, Li coating techniques, schematic representation of how protective layers help prevent lithium dendrite formation in SEs, scheme illustrating the criteria that binders are expected to fulfill, plots of ionic conductivity versus pressure for samples with various binder materials, and the impact of pressure on the battery assembly. Tables: Comparison of the electrochemical performance of coated/doped cathodes with bare cathodes, solvents and binders used for wet-slurry preparation of ASSLB electrodes using Li–argyrodite SEs, properties of Li6PS5Cl film with different binders, the Hansen solubility parameters for the chosen polymer binders and p-xylene solvent, and the conductivity of the Li–argyrodites as documented in the literature along with the preparation pressure, stack pressure, and electrode material. See DOI: https://doi.org/10.1039/d5eb00101c |
| This journal is © The Royal Society of Chemistry 2025 |