
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revolutionizing lithium-ion batteries: exploiting liquid crystal electrolytes†
Sijie
Liu‡
 *ab,
Le
Zhou‡
c,
Yun
Zheng
*d and
Kristiaan
Neyts
*c
*ab,
Le
Zhou‡
c,
Yun
Zheng
*d and
Kristiaan
Neyts
*c
aResearch Institute of Tsinghua University in Shenzhen, Shenzhen, Guangdong 518000, P. R. China. E-mail: liusijie@tsinghua.edu.cn
bInstitute of Nuclear and New Energy Technology, Tsinghua University, Beijing, 100084, P. R. China
cState Key Laboratory of Advanced Displays and Optoelectronics Technologies (SKLADT), State Key Laboratory of Displays and Opto-electronics, Clear Water Bay, Hong Kong, P. R. China. E-mail: eeneyts@ust.hk
dInstitute of New Energy Materials and Engineering, College of Materials Science and Engineering, Fuzhou University, Fuzhou, 350108, P. R. China. E-mail: yunzheng@fzu.edu.cn
First published on 2nd July 2025
Abstract
With the growing global demand for clean energy and sustainable development, the need for advanced battery technologies with high energy density, improved safety, and extended operational cycle life becomes increasingly critical. Lithium-ion batteries (LIBs) have become the dominant power source for a wide range of applications, from portable electronics to electric vehicles, thanks to their superior energy densities and efficiencies. However, the safety concerns and performance limitations of conventional liquid electrolytes in LIBs highlight the need to explore innovative alternatives. Liquid crystal electrolytes (LCEs) have emerged as a promising alternative to traditional electrolytes in LIBs, offering solutions to the inherent safety vulnerabilities and performance limitations of conventional liquid electrolytes. This review highlights recent advancements in the field of LCEs, with a focus on their classifications and structural characteristics to elucidate their ionic transport mechanisms. LCEs are primarily categorized based on their ion-transport architectures: one-dimensional (1D) linear channels, two-dimensional (2D) layered frameworks, and three-dimensional (3D) interconnected networks, all formed through molecular self-assembly processes; secondly, LCEs can be classified as ionic or non-ionic based on their molecular structures. Additionally, molecular design innovations that simultaneously increase ionic conductivity and optimize Li+ compatibility are crucial for advancing battery performances, particularly in energy density and cycling stability. This review concludes with an outlook on the future directions of LCE studies, emphasizing the potential of these materials to revolutionize energy storage solutions and enable the development of batteries with higher energy density, extended cycle life, and improved safety. By offering comprehensive analysis, valuable insights are provided, aiming to catalyze further researches and innovations in the applications of liquid crystal (LC) systems in energy storage technologies.
 Sijie Liu | Sijie Liu received his PhD degree in the Department of Materials Science and Engineering from Tsinghua University and his research focused on solid-state battery materials and devices. He is currently deputy director and associate research fellow of the Key Laboratory of Low-carbon Energy and Energy-saving Technologies at the Research and Development Center of Tsinghua University Research Institute in Shenzhen. He also does postdoctoral work in Tsinghua University and Tsinghua Research Institute in Shenzhen. His current research focuses on the development of all-solid-state lithium battery materials and industrial applications. |
 Le Zhou | Le Zhou is a research associate in the State Key Laboratory of Advanced Displays and Optoelectronics Technologies (SKL-ADT), Department of Electronic & Computer Engineering (ECE), School of Engineering, The Hong Kong University of Science and Technology (HKUST). She received her PhD degree from Peking University in 2019. She then continued her research as a postdoctoral scholar in Tsinghua University. Her research interest is on the liquid crystals. |
 Yun Zheng | Zheng Yun is a professor at Fuzhou University, a Ph.D. supervisor, and a “Minjiang Scholar” of Fujian Province. He has been engaged in research on solid-state electrochemical energy materials for many years, with specific focus on solid-state lithium metal batteries, proton exchange membrane fuel cells, and solid oxide fuel cells. He has published over 80 SCI papers, including more than 40 as the first author or corresponding author (including co-authorship) in prestigious journals such as Chem. Soc. Rev., Adv. Mater., PNAS, Joule, Adv. Energy Mater., Adv. Funct. Mater., and Nano Energy, with several high-citation papers, hot articles, and cover articles. He has also authored two academic books, applied for/been granted 16 invention patents, and led or participated in numerous national-level research projects. |
 Kristiaan Neyts | Kristiaan Neyts received the MSc degree in 1987 and the PhD degree in 1992, both from Ghent University, in physics engineering. He became assistant professor in 2000 and full senior professor in 2012 at Ghent University in the Electronics and Information Systems department, after post-docs at Ghent University and UC Berkeley. He is co-author of over 300 scientific papers and promotor or co-promotor of 30 completed PhDs. In 2023 he became Director of the State Key Laboratory of Advanced Displays and Optoelectronics Technologies (SKLADT) and professor at the Hong Kong University of Science and Technology (HKUST) in the ECE Department. |
Broader contextAs the demand for sustainable energy solutions continues to grow, lithium-ion batteries (LIBs) have become essential for powering a variety of devices, from portable electronics to electric vehicles. However, the widespread adoption faces several challenges, including safety concerns and limitations in performances, primarily due to the conventional liquid electrolytes used. The increasing need for higher energy densities, enhanced safety, and longer cycle lives has led to the exploration of alternative materials and technologies. Liquid crystal electrolytes (LCEs) present a promising solution by addressing these critical issues. LCEs offer improved ionic conductivity and enhanced safety features, making them a viable candidate for next-generation energy storage systems. However, the full potential of LCEs remains to be fully realized, as significant advancements are still needed in their designs and integrations within battery systems. This review delves into the structural and transport mechanisms of LCEs, aiming to advance their applications in LIBs. By highlighting current innovations and future directions, this paper seeks to provide insights that could drive further researches and accelerate the development of more efficient, safer, and durable battery technologies. |
1. Introduction
With the expanding global demand for clean energy and sustainable development-particularly in areas such as electric vehicles, renewable energy storage, and smart grid technologies, the need for batteries offering high energy density, enhanced safety, and extended operational lifetime has become increasingly urgent.1–36 Lithium-ion batteries (LIBs) have become the leading energy storage technology, famous for their exceptional energy densities, prolonged cycle life, and environmentally friendly characteristics, leading to their widespread adoption in portable electronic devices and large-scale energy storage systems.37,38Despite significant advancements in traditional liquid LIBs over the past several decades, their inherent safety risks and limitations in energy density have become increasingly apparent.39,40 These batteries typically rely on volatile and flammable liquid organic electrolytes, making them highly susceptible to thermal runaway under extreme conditions such as elevated temperatures, overcharging, and mechanical shock.41–43 Such conditions can lead to catastrophic outcomes, including fires and explosions.44–47 Furthermore, the stability and ionic conductivity of these electrolytes are crucial factors that directly impact the overall performance of LIBs.43
Consequently, the development of electrolytes that combine high ionic conductivity with non-volatility has become critically important.48–51 Such electrolytes have the potential to significantly enhance the electrochemical performance of LIBs while also mitigating the safety risks associated with traditional liquid electrolytes.52,53 Researchers are striving to develop materials that can maintain reliable performance across broad operational conditions through the meticulous optimization of chemical compositions and structural designs.54,55 This effect is crucial for tackling the dual challenges of safety and efficiency in modern battery technologies, paving the way for more reliable and high-performance energy storage solutions.56,57
To meet the increasing demands for improved performances in LIBs, liquid crystal electrolytes (LCEs) have emerged as a promising class of electrolytes.6,58,59 This innovative approach benefits from the unique molecular design of liquid crystal (LC) systems,60–64 providing exceptional thermodynamic and electrochemical stability across the operational temperature range of LIBs.65–71 These materials respond to external stimuli,72 including light,73,74 electric fields, heat,75 and magnetic fields, resulting in changes to molecular alignment and the emergence of unique optoelectronic effect.76–81 The molecular interactions within LCs are critical to their structural and functional properties.82–84 Through precious molecular engineering, LC materials can achieve a synergistic balance of high ionic conductivity, excellent thermal stability, and an expanded electrochemical window.85 To explore the fundamental principles and involution of LCEs, we also offer a brief overview of their historical evolution, as illustrated in Fig. 1. For a comparative perspective on the commercialization trajectory of LCEs relative to their research milestones, a detailed chronological summary is presented in Table S2 (ESI†). In 1996, Felix B. Dias et al. reported the first synthesis of LCE.19 In 2004, C. T. Imrie, et al. firstly demonstrated that ionic conduction can take place in essentially solid-like LCEs.29 In 2009, R. L. Kerr, et al. developed a new type of Li ion conductor with three-dimensional (3D) interconnected nanopores via polymerization of a liquid organic electrolyte-filled lyotropic LC.20 Shortly after that, in 2010, Yazaki and collaborators constructed LC nanostructures composed of ion-conductive and electronic charge transport layers through the association of π-conjugated molecules with ionic groups.86 These LCs display nanostructured smectic A phases, in which high ionic conductivities are observed. In 2012, Xu and colleagues investigated interdigitated bilayer smectic A structures in ionic LCEs.26 The study demonstrated an inverse correlation between fluorinated unit count (n) in the molecular architecture and the mesophase's thermal stability window. Furthermore, both elevated n values and temperature conditions were found to systematically diminish interlayer spacing. In the crystalline phase, two structural types with varying layer spacings are identified. Parallel (σ∥) and perpendicular (σ⊥) ionic conductivities of the smectic layers increase with the n value, while the anisotropy of these conductivities (σ∥/σ⊥) remains constant, as the thickness of the insulating layer from the dodecyl group is nearly stable. In 2015, Sakuda et al. have successfully developed thermotropic LCEs for LIBs for the first time.25 They utilized a rod-shaped LC molecule containing a cyclic carbonate group to create self-assembled two-dimensional (2D) pathways that facilitate ion conductivity with lithium salts. Three years later, Wang and colleagues developed a novel flexible discotic LC (DLC)-based cross-linked solid polymer electrolyte by employing a one-pot photopolymerization method.49 This process involved oriented reactive discogen, poly(ethylene glycol) diacrylate, and lithium salt to create adjustable ion-conducting channels. Experimental findings demonstrated that the macroscopic alignment of self-assembled columns within such LCE was successfully achieved through annealing and effectively stabilized via ultraviolet (UV) photopolymerization. One year later, Yuan et al. developed a novel thermotropic ionic LC lithium salt that features highly ordered and fast ion-conductive nano-pathways.46 Combining the advantages of LCs, excellent compatibility and high ionic conductivity from the complete dissociation of the electrolyte salt are provided. The approach that LCEs integrate the benefits of solid electrolytes, such as preventing leakage of volatile organic solvents, ensuring good mechanical stability and flexibility, and inhibiting lithium dendrite growth, is a promising strategy. Surprisingly, Wang et al. described molecular ionic composite electrolytes that consist of an aligned LC combined with ionic liquids and lithium salt in 2021.28 This solid electrolyte exhibits high strength (200 MPa) and non-flammability, demonstrating remarkable Li+ conductivity of 1 mS cm−1 at 25 °C and excellent electrochemical stability of 5.6 V against Li|Li+. It effectively suppresses dendrite growth and shows low interfacial resistance (32 Ω cm2) along with minimal overpotentials (≤120 mV at 1 mA cm−2) during cycling in lithium symmetric cells. In 2024, Zeng et al. demonstrated that rapid ion conduction is achieved through the synergistic effects of oriented LCs and electron-deficient boron atoms within a multiblock copolymer electrolyte for advanced solid-state lithium batteries (SSLBs).40 It was not until 2025 that Wang et al. first used LC elastomer materials as electrolytes of LIBs.44 The novel LCE exhibits the ability to intelligently adjust its rigidity and flexibility. Although the study of LCEs is still in its early stage, there have been numerous important advancements in recent years. These ongoing innovations provide optimism for more efficient and sustainable battery technologies, particularly in the context of LIBs. Consequently, it is crucial for researchers in this area to focus on fundamental studies.
 | ||
| Fig. 1 Some key points in the history of the development of LCEs. In 1996, the first synthesis of LCE.19 In 2004, the first demonstration of ionic conduction in solid-like LCE.29 In 2009, the cross-linked organic bilayer structure of LCE.20 In 2010, nanostructured LCEs combining ionic and electronic functions.86 In 2012, nematic LCEs with highly ordered ion channels.26 In 2015, the first application of thermotropic LCs as electrolytes.25 In 2018, self-Assembly of discotic LCEs.49 In 2019, the first application of thermotropic LCs as electrolytes.46 In 2021, flexible LCEs with ordered fast Li-ion-conductive nano-pathways for solid-state battery.46 In 2021, solid-state rigid-rod LCE with nanocrystalline.28 In 2024, LC molecule-modified copolymer with rapid ion conduction.40,62 In 2025, the first LC elastomer used in LIBs.61,68–71 | ||
For example, the strategic incorporation of functional groups, such as ethylene carbonate, into the structures of LC molecules markedly improves their solubility characteristics.87 Additionally, the method can significantly improve their compatibilities with Li+ and Na+.50,88 These enhancements in ionic solvation dynamics significantly boost the overall ionic conductivity of the LCE,89–92 positioning LCEs as a promising solution for advancing key battery performance parameters, including energy density, charge/discharge rates, and cycling stability.93–96 Further researches into the properties and behaviors of LCEs could lead to advancements in battery technology, broadening their applications in various energy storage sectors.68,97–103 One notable advantage of LCEs is their inherent fluidity, which significantly enhances ionic conductivity compared to solid-state electrolytes (SSEs).104 SSEs offer several advantages, including enhanced safety and excellent electrochemical stability.105 Their excellent wettability with electrodes further aids in mitigating dendrite formation.106 Thus, using LCEs in SSEs, the mesophase characteristics of LCEs effectively address common drawbacks of SSEs.107 By carefully tailoring their molecular structures, these electrolytes can achieve high ionic conductivity, thereby improving the performance of SSLBs.108 This unique combination of properties establishes LCEs as a highly promising option for advancing next-generation energy storage technologies.
It should be highlighted that LCE design fundamentally relies on creating molecules containing distinct ion-transporting portions (typically polar or ionic groups like cyclic carbonates or PEO segments to facilitate salt dissociation and ion conduction) and non-ionic portions (mesogenic units and alkyl chains to control the self-assembled nanostructure). Key desired characteristics arising from this design and the unique properties of LCs include achieving high ionic conductivity, particularly at room temperature (ideally ≥10−3 S cm−1), through the formation of well-ordered nanochannels (columnar, smectic, or cubic phases, see details in Table S1†); enhancing safety via non-flammability and inherent stability compared to liquid electrolytes; suppressing lithium dendrite growth by leveraging the anchoring energy of LC molecules on lithium electrode surfaces or the mechanical integrity provided by rigid LC structures; ensuring a wide electrochemical window for high-voltage operation; providing mechanical flexibility suitable for applications like wearable electronics; and enabling thermal stability. The overarching goal of LCE design is to integrate these functionalities to create electrolytes that overcome the limitations of conventional liquid and solid electrolytes, balancing high ionic conductivity with excellent safety and stability.
In recent years, the application and research of LCEs in the field of LIBs have attracted significant attention, inspiring a growing number of researchers to focus on the development and exploration of advanced LCEs.109–111 This growing interest is primarily attributed to the unique physicochemical properties of LC materials, which exhibit remarkable performance in electrochemical characteristics, structural stability, and ease of processing.112 Among these, double-continuous cubic LCs are considered one of the most promising candidates for quasi-SSEs, as they seamlessly integrate the advantages of both LCEs and SSEs.113 These materials uniquely merge the dynamic fluidity of LC phases with long-range order, enabling simultaneous achievement of high ionic conductivity and good electrode–electrolyte interface stability during repeated charge/discharge cycles.114 These attributes significantly enhance their potential for use as electrolytes in LIBs.115 By integrating these advanced materials into electrochemical systems, researchers aim to achieve groundbreaking progress in energy storage efficiency and sustainability.116 As research continues to evolve, the potential of LCEs to revolutionize LIB performance remains highly promising, paving the way for future innovations in energy storage solutions. However, despite the growing interest, there remains a lack of comprehensive reviews on the applications of LCEs within this context, which impedes the utilization of knowledge in this area.
Therefore, this review provides a comprehensive analysis of LCEs from several key perspectives. Firstly, the fundamental concepts and classifications of LCEs are explored, along with their structural characteristics and mechanisms underlying their ionic conductivities. Secondly, the specific applications of LCEs in LIBs are analyzed, focusing on their contributions to improving battery performance, extending cycle life, and improving safety. Finally, the review explores the future development trends of LCEs in LIB technologies, addressing both potential challenges and emerging opportunities. This comprehensive analysis offers valuable insights for further research on LCEs, thereby advancing the innovation and application of novel battery materials.
2. Classifications of LCs
When a substance changes from a crystalline state to a liquid state under changing external conditions, it first loses its structural order, leading to enhanced fluidity.117 However, certain anisotropic orientation characteristics of the crystalline state may still be retained,118 resulting in an intermediate phase that exhibits properties of both solids and liquids. This intermediate phase is known as a LC88,119 Derived from the Greek word “mesos” (meaning “intermediate”), this phase exhibits partial molecular order while retaining fluidity.79,120,121 The distinctive combination of anisotropic and fluidic properties is crucial for achieving precise control over molecular arrangements.122 The requisite control over molecular arrangements is achieved through several established techniques. Surface-induced alignment leverages specialized coatings that anchor liquid crystalline molecules near interfaces through physical surface patterns, enforcing directional order. Alternatively, photoalignment methods enable precise orientation control in photosensitive systems by translating optical patterns into molecular alignment. Further refinement is possible through localized modification of rubbing directions on alignment substrates, permitting spatially tuned reorientation of the liquid crystalline order. At the molecular level, LCs can form various mesophases, allowing tuning of their physical properties,123 as demonstrated in Fig. 2. In the LC phase, molecules retain orientational and/or positional long-range order in at least one direction, enabling mobility without fixed positions. This orientational order minimizes excluded volume and maximizes intermolecular interactions, distinguishing it from the disordered liquid state. LC materials exhibit anisotropic properties (e.g., refractive index, dielectric permittivity, conductivity) that are direction-dependent, manifesting distinct values along the director axis versus orthogonal orientations. Their molecular mobility allows responsiveness to external stimuli (e.g., electric field). Calamitic mesogens are rod-like molecules with a rigid central core and terminal groups, which can include flexible chains (enhancing molecular mobility) or polar groups. These molecules often exhibit a permanent dipole moment or anisotropic polarization, crucial for mesophase behavior. The nematic (N) phase which have no positional order but tend to point in the same direction, defined by the director n.124 This creates one-dimensional (1D) order. In bulk samples, the n fluctuates, forming microdomains with varying orientations.125 High aspect ratio mesogens favor N phase formation. Introducing chirality leads to a chiral nematic (cholesteric) phase, where the n rotates helically along a perpendicular axis, creating a helical superstructure. Blue phase LCs are characterized by 3D cubic defect structures, which arise from the interplay between molecular packing and chiral forces (as shown in Fig. 2).126–128 In blue phases, the mesogenic molecules adopt a “double-twist” configuration along both the x- and y-axes, forming what is known as a double-twisted cylinder.129,130 The molecular orientation in the space between these double-twisted cylinders cannot continuously align, leading to the formation of energetically unfavorable disclinations within the cubic lattice.7 As a result, blue phase LCs typically appear in a narrow temperature range of approximately 0.5–2.0 °C, situated between the isotropic and cholesteric phases in highly chiral LCs.131,132 Upon cooling process, three distinct blue phases are observed in sequence: blue phase I, blue phase II, and blue phase III.133,134 Blue phase I and blue phase II exhibit body-centered cubic (BCC) and simple cubic structures, respectively, while blue phase III is generally considered to be amorphous.135–138 Smectic phases (Sm) exhibit 1D positional order, with molecules or aggregates forming layered structures. The layer thickness, d, defines the periodicity, though layers may lack sharp boundaries.139 Smectic phases, however, arise largely from nano segregation effects, emphasizing the role of molecular organization beyond rigidity.140 The smectic A phase is the simplest smectic structure, characterized by molecules aligning their long axes parallel to the layer normal, though slight random tilts may occur. In the smectic C phase, molecules tilt within the layers along a preferred direction, adjacent layers share the same tilt direction (synclinic correlation).141 | ||
| Fig. 2 Schematic diagram of LC phases constructed from LC molecules. | ||
As shown in Fig. 2, discotic mesogens feature a disk-like structure, typically comprising a rigid, flat aromatic core surrounded by at least three flexible chains forming a “soft” periphery.142,143 When the columns are packed together to form a 2D crystalline array, columnar phases (Col) are formed. Similar to calamitic mesogens, discotic mesogens can form a nematic phase (ND) by aligning their short molecular axes along n, creating orientational order without positional order.144 Alternatively, they can stack into 1D columns, aligning parallel to each other but lacking lateral positional correlations, forming a columnar nematic phase (NC). These columns may arise from charge-transfer interactions between electron donors and acceptors or strong electrostatic interactions between cations and anions. The rectangular columnar phase (Colrec) exhibits a 2D rectangular lattice (p2mm symmetry) formed by disk-shaped molecules organized into columns with anisotropic intermolecular interactions, enabling direction-dependent charge transport.145–148 In contrast, the hexagonal columnar phase (Colhex), common in radially symmetric discotics like triphenylene derivatives, adopts a hexagonal close-packed arrangement (p6mm symmetry) with isotropic lateral packing and efficient 1D charge-carrier mobility.149–153 Lower-symmetry phases include the oblique columnar phase (Colobl), characterized by a parallelogrammic lattice (p2 symmetry) arising from asymmetric molecular packing, which induces tilted columns and birefringence,154–158 and the square columnar phase (Colsqu), stabilized by fourfold symmetric cores (e.g., metallophthalocyanines), featuring a tetragonal lattice (p4mm symmetry) that supports isotropic planar charge transport.21,78,159,160
3. Classifications of LCEs
3.1. Classifications by ionic transport mechanism
LC, with their unique anisotropic properties, impart LCEs with distinctive mechanisms for Li+ transport.161 Specifically, the self-assembly behavior of LCEs results in the formation of nanostructures such as columnar, smectic, and double-continuous cubic phases, which provide highly ordered channels for Li+ conduction.162 This order stems from the highly organized spatial arrangement of LC molecules, facilitating the formation of nanoporous channels that significantly enhance both the transport rate and stability of Li+, playing a crucial role in shaping the electro-chemical performance of LIBs.163,164Based on the mechanisms of ionic conduction within LCEs, these can be further categorized into three distinct types:165 firstly, the 1D transport channels formed by self-assembly, which provide linear pathways for the movement of Li+ through their 1D arrangements; secondly, the 2D transport channels, which allow for more flexible migration of Li+ within a 2D plane; and thirdly, the 3D transport channels, where a complex network is constructed in 3D space, effectively improving Li+ transport efficiency. These distinct mechanisms of ionic conduction not only shape the dynamics of ion migration but also directly influence the overall performance of LIBs.166 This understanding serves as both a theoretical foundation and practical guidance for optimizing the applications of LCEs in electrochemical energy storage devices.167 Consequently, systematically categorizing the ion conduction mechanisms of LCEs is crucial for understanding their conduction mechanisms and for designing LC molecules with superior performance characteristics.168,169
The 1D ionic transport mechanisms of LCEs in LIBs are illustrated in Fig. 3a–c.25 Yoshio and colleagues have introduced a novel class of LCEs that display fluidic self-organizing structures, as illustrated in Fig. 3a.23 The molecular formulas and structural schematics of the LC materials are shown in Fig. 3a, experimental results indicated that LCs maintain hexagonal columnar phases over a wide temperature range, including at room temperature. Remarkably, uniaxially oriented columnar LC materials, for the first time, demonstrated 1D ion conduction. Through the self-assembly of LC molecules, disk-like molecules can stack to form columnar LC phases, thereby creating 1D ion transport channels. This mechanism underlies the ion conduction in 1D LCEs.170–172 As shown in Fig. 3b, Yoshio et al. have developed 1D ion-conductive LCE films that contain ion nanochannels oriented perpendicular to the surface of the film.31 The macroscopic alignment of the columns occurs along uniaxial directions, with their orientation axes maintained orthogonally relative to the functionalized substrates composed of glass and indium tin oxide (ITO) undergoing surface treatment by using 3-(aminopropyl)triethoxysilane. The sequence of molecular self-ordering, macroscopic alignment, and photopolymerization in columnar imidazolium LCEs produces nanostructured films with 1D molecular organization. These LCEs demonstrate thickness-oriented ionic mobility, creating ordered charge transport channels perpendicular to the membrane plane.31 Such process establishes the fundamental formation mechanism for 1D ionic transport channels in LCE films. Shimura et al. have fabricated a fan-shaped molecule containing a propylene carbonate group, which facilitates the formation of nanosegregated columnar LC phases through complexation with lithium triflate (Fig. 3c).24 By applying alternating current (AC) electric field, these columnar structures can be macroscopically aligned.
 | ||
| Fig. 3 Some representative examples of 1D LCE in LIBs: (a) schematic illustration of the chemical structure and self-assembly of 1D LCE in LIBs, serving as a general example.23 (b) Schematic illustration of 1D ion-conductive polymeric films: self-assembly and subsequent photopolymerization of columnar LC.31 (c) The molecular structures and schematic illustration of the alignment of the columnar LC under electric field depict a 1D transport mechanism.24 (d) Molecular structures used in the cited 1D LCE works.19,23,24,31,46,49,50,54,56,173 | ||
There are still lots of LCs that can form 1D ion conductive channels, as illustrated in Fig. 3d. The complexation of a Col LC propylenecarbonate derivative and a lithium salt can form 1D Li+ conductor, showing the ionic conductivity of 2.2 × 10−8 S cm−1 at 22 °C and become 4.3 times when applying an AC electric field (2.5 V μm−1, 1 kHz) for 90 min.24 The efficient redox materials based on LC consist of π-conjugated mesogens and ionic moieties can be designed to induce Smectic A (SmA) phases, effective ionic transport occurs along the smectic layers formed by the nanosegregation.173 The ionic conductivities of these LCE can be ∼10−4 S cm−1, the combination of the ionic and electronic functions in the nanostructured LC phases can be useful in LIBs. Ionic LC based lithium salt combined with polymer (e.g. polyethylene oxide (PEO)) can form 1D ion-conductive channel with high ion conductivity of 0.45 × 10−3 S cm−1 at 30 °C.46 Smectic LC polymers with helical polyether backbones (e.g., C16O5/C16O6 bearing long alkyl side chains) complexed with Li+, Na+, or Mg2+ salts can form 1D ion-conductive channels via layered organization, where lithium salt complexes exhibit the highest ionic conductivity (approaching ∼10−3 S cm−1 near 100 °C), enhanced by mechanical shearing to align the conductive pathways.19 By designing an in situ formed LC interphase, the dual-electrode-free battery achieves enhanced deposition/stripping reversibility and extended cycle life (80% capacity retention over 950 cycles).50 The DLC triblock copolymer can be structured as a main-chain polymeric discotic core flanked by terminal PEO blocks, forming a hierarchical architecture for directional ion transport.54 Magnetic field alignment of the LC phase optimizes conducting pathway orientation, achieving a conductivity of ∼10−3 S cm−1 at 60 °C.54 The star-branched amphiphilic LC copolymers, synthesized via atom-transfer radical polymerization (ATRP), can self-assemble into ordered morphologies that enable efficient ion transport pathways.56 After LC-state annealing, the optimized structure achieves a maximum ionic conductivity of 5.39 × 10−5 S cm−1 at 25 °C. LCEs can suppress lithium dendrite formation. For example, a flexible DLC-based cross-linked solid polymer electrolyte can be fabricated via one-pot photopolymerization, achieving an 1D ion conductivity of 5.48 × 10−5 S cm−1 at 30 °C.49 These LC molecules that can form 1D channels play an optimizing role in enhancing ionic conductivity, electrochemical stability, and suppressing lithium dendrite formation.
These 1D self-assembled structures not only facilitate efficient ionic transport but also significantly improve charge carrier mobility during phase separation processes.160 For instance, the formation of polymeric phase-separated structures through hydrogen bonding between LC materials and fibers has resulted in an impressive threefold enhancement in the charge mobility of phenylene compounds.54 For example, when Li+ are combined with block copolymers such as PEO, LC hexagonal columnar phases can be achieved. Within this structure, the PEO chains form ionic conduction channels within the columnar phase, effectively enhancing the solubility and transport of Li+. Meanwhile, the carbon chains integrated into the molecular structure serve as ionic insulators, blocking undesired electronic migration and ensuring effective ionic transport.174 Furthermore, the integration of amphiphilic LC materials with lithium salts can yield stable 1D ionic transport channels, which enhance ion mobility and overall stability and safety of the electrolyte. The strategic design and optimization of such LCEs hold significant potential for the development of next-generation high-performance lithium batteries, particularly by enabling higher energy densities and prolonged cycling stabilities.56,175
As early as the 1990s, researchers have already investigated 1D transport channels based on the self-assembly of LCEs. F. B. Dias et al. synthesized a 1D LC molecular material, as depicted in Fig. 4a, to be used as an electrolyte.19 The study revealed that the –C16H33 side chains in the material melt at approximately 40 °C, transitioning into a LC phase. The LC-to-isotropic transition temperature varies slightly depending on the polymer and stoichiometry, with a value of 89 °C for the C16O5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :LiBF4 (1:1) complex. The ionic conductivity as a function of temperature exhibits distinct transitions corresponding to phase changes, while the complex impedance behavior also highlights the material's multiple states. The ionic conductivity of the 1:1 complex exceeds that of the 1:0.5 complex and is comparable to that of PEO:LiBF4 in both the crystalline and isotropic phases, though it is slightly lower within the LC phase temperature range. Further investigations have validated the mechanism underlying the 1D conductivity, as shown in the lower part of Fig. 4a.27 The slightly lower melting temperature of the side chains for C16O6 can be partially attributed to the reduced tendency for coalescence between adjacent side chains, which are separated by a longer sequence of the oligoether skeletal segment. In the crystalline state, the C6H33 side chains arrange into hexagonal lattices, while the polyether backbones adopt helical conformations.
:LiBF4 (1:1) complex. The ionic conductivity as a function of temperature exhibits distinct transitions corresponding to phase changes, while the complex impedance behavior also highlights the material's multiple states. The ionic conductivity of the 1:1 complex exceeds that of the 1:0.5 complex and is comparable to that of PEO:LiBF4 in both the crystalline and isotropic phases, though it is slightly lower within the LC phase temperature range. Further investigations have validated the mechanism underlying the 1D conductivity, as shown in the lower part of Fig. 4a.27 The slightly lower melting temperature of the side chains for C16O6 can be partially attributed to the reduced tendency for coalescence between adjacent side chains, which are separated by a longer sequence of the oligoether skeletal segment. In the crystalline state, the C6H33 side chains arrange into hexagonal lattices, while the polyether backbones adopt helical conformations.
 | ||
| Fig. 4 (a) A molecular formula for 1D LCE features R as a long chain alkyl substituent such as –C16H33.19,27 Energy-minimized molecular models of C16O5 at 20 °C, along with schematic diagrams of C16O5-lithium salt (1:1) complexes at 20 °C, (+) Li+, (−) (ClO4−). d denotes the spacing between layers.27 (b) Schematic depiction of the arrangement of the block copolymer under a vertically applied magnetic field. Free mesogens co-assemble with the covalently bound mesogens in the LC block, aligning their long axis along the field and parallel to the interface with the PEO domain. Lithium ions are confined within the PEO domain, which also contains PAA chains (not shown) that are closely associated with the PEO backbone.35 (c) Molecular structures of LCs 1–3.86 (d) Schematic illustration of a possible structure in the SmA phases for LCs 1–3. Green cylinders, red ellipsoids, and orange spheres represent π-conjugated moieties, triflate anions, and imidazolium cations, respectively. The ionic and π-conjugated moieties are arranged into segregated layers for enhanced structural organization.86 (e) A one-pot fabrication process for flexible DLC-based cross-linked solid polymer electrolyte with controlled ion-conductive pathways is achieved through photopolymerization under UV irradiation.49 (f) A schematic influence of the mechanism of PEO content on the crystallized structure of LCEs.46 | ||
In recent years, numerous researchers have reported on semi-crystalline materials based on PEO block copolymers with LC segments. Gopinadhan et al. investigated 1D copolymers, analyzing the influence of LCs on the crystallization of PEO.35 They utilized magnetic field to orient the polymers and discovered a significant enhancement in Li+ conductivity within the ordered flexible segments. Their findings demonstrated that magnetic field induced alignment can effectively organize the self-assembled lamellar and hexagonal domains of PEO in LC block copolymers. This alignment arises from the paramagnetic anisotropy driven by the layered order of the LC blocks, rather than from the crystallization of the ethylene oxide units. The crystallization of PEO blocks is notably suppressed due to the formation of polymer complexes with polyacrylic acid (PAA) and the addition of lithium perchlorate, the latter also interacting with the PEO chains, as illustrated in Fig. 4b. The incorporation of these dopants enhances the phase separation between the ethylene oxide (EO) and the acrylic acid/LCs segments, leading to an improvement in the long-range order of the microphase-separated structures, accompanied by an increase in the order–disorder transition temperature of the system. Importantly, the presence of these dopants does not adversely affect the capability of the magnetic field to align the system. The research team further demonstrated that rotational annealing could effectively disrupt the degeneracy of the alignment of the lamellar microdomains, theoretically allowing for the generation of lamellar arrangements corresponding to any direction of the substrate by simply controlling the orientations of the substrate relative to the magnetic field. In this process, custom-synthesized materials will be employed to independently access the lamellar and cylindrical formation regions in the phase diagram without the need for blending. The ability to achieve flexible alignment of self-assembled structures at a large scale is crucial for the applications of block copolymers in various emerging fields. Magnetic field-driven alignment is particularly well-suited for controlling ordering in situations where strong flow fields or complex topological constraints cannot be applied to the samples. The control over PEO alignment is especially noteworthy, as the ordered PEO microdomains hold potential applications as ion-conducting media in solid-state battery electrolyte films.
Yazaki et al. successfully synthesized three new molecular materials that combine ionic and electronic functions, utilizing LCs made of terthiophene-based mesogens and terminal imidazolium groups (Fig. 4c).86 These compounds exhibit thermotropic smectic A phases. Nano segregation of the π-conjugated mesogens and ionic imidazolium groups forms layered liquid-crystalline structures. These materials serve as efficient electrochromic redox systems, showing coupled electrochemical reduction and oxidation in their ordered bulk states. For instance, compound 1, containing a terthienylphenylcyanoethylene mesogen and an imidazolium triflate group, forms a smectic LC nanostructure and exhibits reversible electrochromic behavior between 0 and 2.5 V at 160 °C, without the need for additional electrolytes. In contrast, compounds 2 (with a tetrafluorophenylterthiophene group) and 3 (with a phenylterthiophene group) show irreversible cathodic reduction and reversible anodic oxidation in the smectic A phase. The use of poly(3,4-ethylenedioxythiophene)-poly(4-styrene sulfonate) (PEDOT-PSS) as an electron-accepting layer on the cathode enhances the electrochromic responses for compounds 2 and 3. These findings demonstrate the potential of self-organized molecular redox systems based on nano-segregated π-conjugated LCs containing imidazolium groups, which can be utilized in batteries, as shown in Fig. 4d. Wang et al. developed a DLC-based cross-linked solid polymer electrolyte (DLCCSPE) for LIBs, featuring controlled ion-conducting channels and enhanced safety (Fig. 4e).49 The LCE structure is formed by aligning DLCs and incorporating flexible poly(ethylene glycol) diacrylate (PEGDA), followed by photopolymerization to cross-link the DLCs with PEGDA. The macroscopic alignment of DLC nanostructures within the polymer films improves overall electrochemical performance, including increasing ionic conductivity and electrochemical stability. The resulting 1D solid-state LCE with nanochannels provides more efficient ion transport compared to unstructured films. LIBs assembled with these oriented electrolytes demonstrate excellent cycling performance, with stable discharge capacities of 143, 135, and 149 mAh g−1 at rates of 0.5, 1, and 0.2C, respectively. Yuan et al. developed a novel thermotropic ionic LC lithium salt (lithium bis(modified imidazole)borate (LiBMB)) with highly ordered, fast ion-conductive nano-pathways.46 The molecular structure and thermotropic LC behavior of LiBMB were characterized by various techniques. The material has a melting point of 31.2 °C and a clearing point of 42.7 °C, suggesting that it can offer more flexible Li-ions in the electrolyte. Flexible LiBMB/PEO SSEs, with LiBMB content ranging from 20–75 wt%, were prepared. The effect of LiBMB content on the crystalline structure of PEO is depicted in Fig. 4f. The resulting SSEs exhibited optimal ionic conductivity (0.45 × 10−3 S cm−1), ion transport number (0.54), DC conductivity (2.33 × 10−7 cm2 s−1), and electrochemical stability (7.2 V) at 30 °C. This performance is attributed to the maintained ordered ion-conductive nano-pathways, with a quasi-period value (Tp) under 100 ns. These LiBMB/PEO electrolytes were used to assemble LiFePO4|electrolyte|Li all-solid-state cells, with the optimal cell showing an initial discharge capacity of 145.5 mAh g−1 at 0.1C and stable performance across multiple cycles.
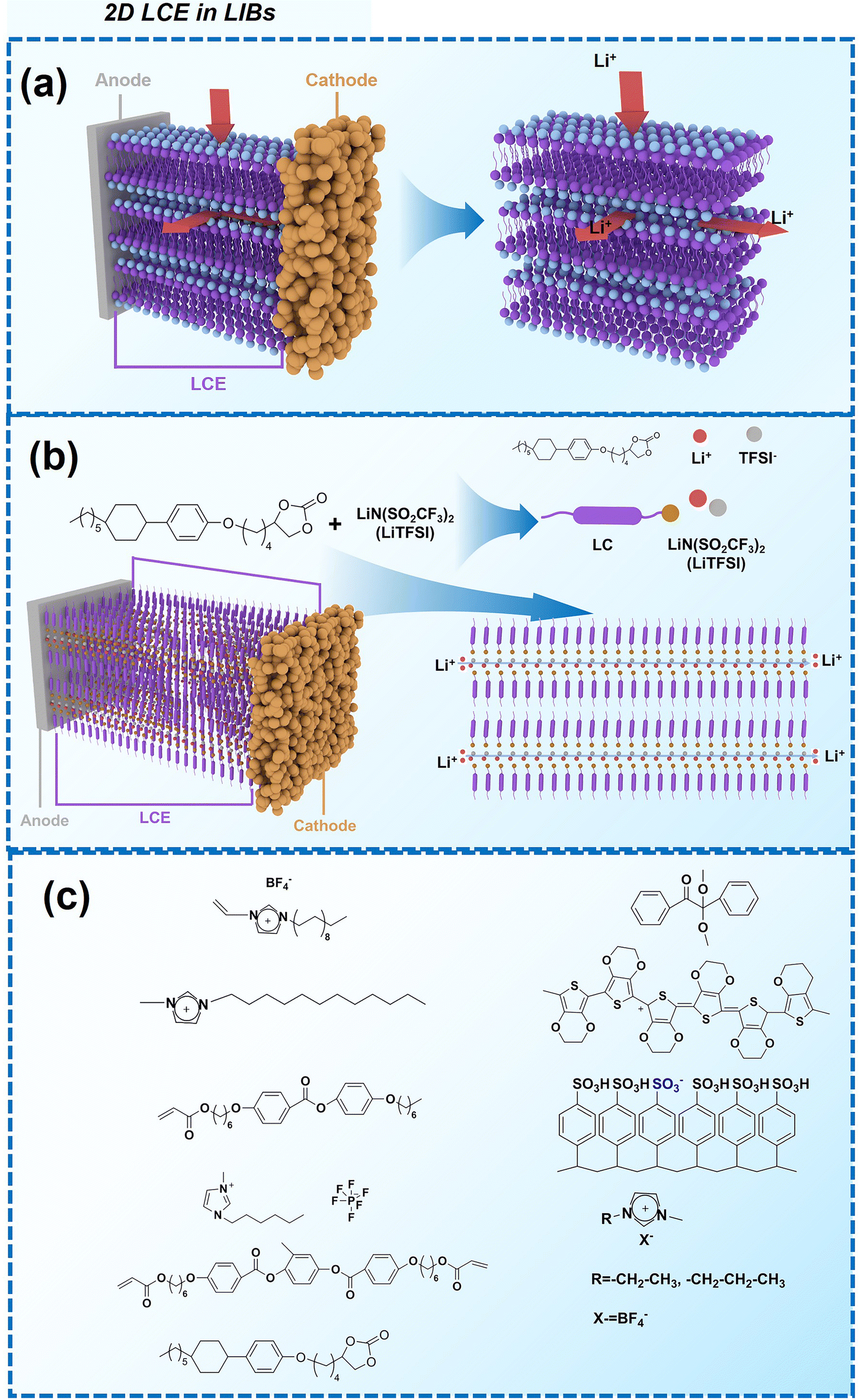 | ||
| Fig. 5 (a) Schematic illustration of the mechanism of 2D LCE in LIBs. (b) Molecular structures of LC compound and the lithium salt, and corresponding schematic illustration of 2D LCE in LIBs.25 (c) Molecular structures utilized in the referenced 2D LCE works.25,26,32,40,48,181,182 | ||
When designing and synthesizing LCE materials, it is crucial to incorporate specific ionic transport moieties, often derived from polyether or ethylene carbonate groups. These moieties promote the formation of ordered ionic transport channels through self-assembly processes and can substantially enhance the ionic conductivity of the material when combined with lithium salts such as LiTFSI. There are many LCEs can form 2D ion-conductive channels, as shown in Fig. 5c. The LC 1,4-bis(4-(60-acryloxy-hexyloxy)benzoyloxy)-2-toluene (C6M) combines with an ionic liquid and a lithium salt, showing the ionic conductivity of 2.14 × 10−2 S cm−1 at room temperature.32 The combination of an ionic LC 1-vinyl-3-octadecylimidazolium tetrafluoroborate ([C18VIm]BF4) with poly(ethylene glycol) diacrylate (PEGDA), poly(ethylene glycol) dimethyl ether (PEGDE), and LiBF4 produces a solid polymer electrolyte exhibiting high ionic conductivity (1.96 × 10−4 S cm−1 at 30 °C) and a Li+ transference number of 0.6.176 The combination of a star-shaped LC copolymer, 3-arm-poly(10-[(4-cyano-4′-biphenyl)oxy]decyl methacrylate)-block-poly[methoxy-poly(ethylene glycol) methacrylate] (3PMALC-PPEGMA), and a small-molecule mesogen, 4-cyano-4′-([(10-hydroxydecyl)oxy]biphenyl) (10-BPCN), with PEO and LiClO4 forms a composite solid electrolyte.180 This ternary blend (70% PEO, 25% 3PMALC-PPEGMA, 5% 10-BPCN) achieves an ionic conductivity of 1.3 × 10−5 S cm−1 at 25 °C after annealing, enhanced by two orders of magnitude compared to pristine PEO/LiClO4. The ionic LC 1-dodecyl-3-methylimidazolium fluorohydrogenate salts (C12MIm(FH)nF, n = 1.0–2.3) can form a SmA interdigitated bilayer structure, achieving anisotropic ionic conductivity with σ∥ ≈ 7.37 mS cm−1 at 25 °C (n = 1.0) parallel to the smectic layers, exceeding perpendicular conductivity over ten times.26 Star-shaped amphiphilic LC copolymers combining hydrophobic poly(10-[(4-cyano-4′-biphenyl)oxy]decyl methacrylate) (PMALC) with hydrophilic poly(methoxy-poly(ethylene glycol) methacrylate) (PPEGMA) achieve ordered ion-conducting pathways via mesogen alignment.180 The optimized 3PPEGMA-PMALC copolymer, with mesogens positioned exteriorly and annealed in the liquid-crystalline state, exhibits an ionic conductivity of 1.0 × 10−4 S cm−1 at 25 °C, surpassing non-mesogen counterparts. A six-arm star polymer based on a DLC (triphenylene core) combined with polystyrene (PS) and poly(ethylene glycol) methyl ether methacrylate (PPEGMA) segments forms ordered ion-conductive channels through self-assembly.183 The optimized electrolyte achieves an ionic conductivity of 1.46 × 10−4 S cm−1 at 30 °C, along with a wide electrochemical window (5.1 V) and enhanced lithium-ion transference number (0.37) after annealing, outperforming linear copolymer counterparts. The ionic LC 1-hexadecyl-3-methylimidazolium tetrafluoroborate ([C16min]BF4) combined with a poly(ionic liquid) (PMOBlm-BF4) and PEGDA cross-linker forms lamellar structures via co-assembly, creating ordered 2D ion-conductive pathways.184 The optimized PMPC0.5 electrolyte achieves an ionic conductivity of 7.14 × 10−5 S cm−1 at 25 °C and 2.17 × 10−3 S cm−1 at 95 °C, with a wide electrochemical window (4.2 V) and enhanced Li+ transference number (0.24). The construction of these 2D transport channels not only improves the mobility of ions within the material but also optimizes the performance of the electrolyte in LIBs. 2D LCEs demonstrate significant potential in terms of ionic transport efficiency and overall performance.183,184
Xu et al. investigated the thermal, structural, and ion-conductive properties of 1-dodecyl-3-methylimidazolium fluorohydrogenate 2D LCEs, C12MIm(FH)nF (n = 1.0–2.3), as shown in Fig. 6a.26 These LCEs exhibit a SmA interdigitated bilayer structure in their liquid crystalline phase, with the mesophase temperature range decreasing as the n value increases. The layer spacing of the bilayer structure reduces with higher temperature or increasing n value. A mixed crystal system was observed in C12MIm(FH)nF, with two distinct crystal structures: type I (1.0 ≤ n < 1.9) and type II (1.9 < n ≤ 2.3). The ionic conductivity increases with the n value in both directions-parallel and perpendicular to the smectic layers-while the anisotropy remains constant, as the thickness of the insulating dodecyl alkyl chain layer remains largely unchanged.
 | ||
| Fig. 6 (a) Phase behavior of C12MIm(FH)nF, n = 1.0–2.3 and their anisotropic ionic conductivities as LCEs.26 (b) Synthesis route of the LC monomer and preparation of the free-standing composite solid-state electrolyte film using LC and IL with ordered ion channels.32 (b) Reproduced with permission.32 Copyright 2019, The Royal Society of Chemistry. (c) The schematic illustration exhibiting the mechanism of rapid Li-ion conduction occurred in LCEs.40 (d) Lamellar nanostructured LCEs with liquid-like conducting behavior and solid-like instinct stability in LIBs.48 | ||
Wang et al. developed a novel solid electrolyte by combining a nematic LC with an ionic liquid (IL),32,185–188 as shown in Fig. 6b.32 The LC, with its ordered layered nanostructure, was polymerized and stabilized through UV irradiation, while the IL was incorporated into the structured ion channels to facilitate fast ion transport. Notably, this free-standing electrolyte film maintained stable, ordered 2D channels. The resulting SSE demonstrated high ionic conductivity (2.14 × 10−2 S cm−1) at room temperature, a broad electrochemical window (4.8 V), and excellent compatibility with lithium metal.
Recent studies have also focused on the development of 2D transport channels through the self-assembly of LCEs. Zeng et al. synthesized a novel type of boron-containing LC-based ABCBA multiblock copolymer electrolytes (BCPEs) for the first time.40 These BCPEs exhibit significantly improved Li+ conductivity. The highly oriented LCs with mesogenic –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N form ion-conductive channels that facilitate rapid Li+ transport, while the –CN groups enhance the oxidative resistance of the electrolytes. The introduction of electron-deficient boron atoms helps trap TFSI− derived from dissociated lithium salts, effectively adsorbing impurities and reducing concentration polarization, thus maintaining interface stability. The mechanism behind efficient Li+ transport in this system is shown in Fig. 6c. As a result, the multiblock copolymer electrolytes display excellent performance, including a high ionic conductivity of 1.13 × 10−4 S cm−1 at 30 °C, a broad electrochemical stability window of 4.85 V, an increasing Li-ion transference number of 0.311, and improved mechanical properties compared to similar materials. Ruan et al. explored the use of lamellar lyotropic LCs as SSEs for LIBs, demonstrating high voltage stability, efficient charge transport, and thermal safety.48 The lyotropic LCs were prepared through the self-assembly of 1-hexadecyl-3-methylimidazolium tetrafluoroborate ([C16Mim][BF4]) in LiBF4/PC liquid solutions. The nanosegregation of [C16Mim][BF4] led to the formation of layered structures, creating dynamic lamellar ion-conducting pathways (Fig. 6d). As the liquid content increased, these pathways expanded, enhancing the ionic conductivity of the lyotropic LCEs. The lyotropic lamellar nanostructures allowed for liquid-like ion conductivity at room temperature and provided superior stability, enabling the electrolytes to withstand high voltage and reduce flammability compared to LiBF4/PC liquid electrolytes.
N form ion-conductive channels that facilitate rapid Li+ transport, while the –CN groups enhance the oxidative resistance of the electrolytes. The introduction of electron-deficient boron atoms helps trap TFSI− derived from dissociated lithium salts, effectively adsorbing impurities and reducing concentration polarization, thus maintaining interface stability. The mechanism behind efficient Li+ transport in this system is shown in Fig. 6c. As a result, the multiblock copolymer electrolytes display excellent performance, including a high ionic conductivity of 1.13 × 10−4 S cm−1 at 30 °C, a broad electrochemical stability window of 4.85 V, an increasing Li-ion transference number of 0.311, and improved mechanical properties compared to similar materials. Ruan et al. explored the use of lamellar lyotropic LCs as SSEs for LIBs, demonstrating high voltage stability, efficient charge transport, and thermal safety.48 The lyotropic LCs were prepared through the self-assembly of 1-hexadecyl-3-methylimidazolium tetrafluoroborate ([C16Mim][BF4]) in LiBF4/PC liquid solutions. The nanosegregation of [C16Mim][BF4] led to the formation of layered structures, creating dynamic lamellar ion-conducting pathways (Fig. 6d). As the liquid content increased, these pathways expanded, enhancing the ionic conductivity of the lyotropic LCEs. The lyotropic lamellar nanostructures allowed for liquid-like ion conductivity at room temperature and provided superior stability, enabling the electrolytes to withstand high voltage and reduce flammability compared to LiBF4/PC liquid electrolytes.
The advantages of 3D transport channels extend beyond mere conductivity, they also contribute to enhanced mechanical stability and thermal resilience of the electrolytes. This is particularly relevant for applications in energy storage technologies, where the durability and performance of electrolytes under varying operational conditions are critical. The self-assembly of cubic phase LC structures into 3D ionic transport channels represents a transformative approach in the design of ion-conductive materials. Future research should focus on optimizing the composition and structural parameters of these LCEs to further enhance their ionic transport efficiency and broaden their applicability in next-generation electrochemical devices. The exploration of novel materials and self-assembly techniques will be instrumental in unlocking the full potential of 3D ionic transport channels in LCEs.
R. L. Kerr et al. have developed a novel nanostructured polymer material filled with LC that combines the advantageous properties of gelled polymers and LC-based electrolytes.20 This material exhibits stable ion conductivity (10−4 to 10−3 S cm−1 at room temperature), bicontinuous cubic phase, resembling liquid-like behavior over a broad temperature range, a feature not typically seen in either gelled polymers or LC-based electrolytes (as shown in Fig. 7a). Ongoing investigations are focusing on conductivity and Nuclear Magnetic Resonance (NMR) Diffusion-Ordered Spectroscopy (NMR DOSY) analyses over an extended temperature range. Additionally, efforts are being made to enhance the material's conductivity by eliminating the surface crust, increasing the concentration of PC solution and Li salt in the films, and experimenting with different types of liquid electrolytes. These findings further support the observation that bicontinuous cubic phases, which feature 3D interconnected nanochannels, offer superior ion conductivity compared to other LC phases. Yao et al. synthesized novel solid polymer electrolytes through in situ polymerization and the incorporation of a nematic LC into a poly(vinylidene fluoride-co-hexafluoropropene)-hexafluoropropylene (P(VDF-HFP)) matrix, achieved under UV irradiation in the presence of an ionic liquid (IL) (Fig. 7b).30 The enhanced migration of Li+ ions in the resulting SPEs was attributed to the formation of a poly(LC) (PLC) network, a mechanism supported by both FTIR analysis and DFT calculations. The resulting SPEs demonstrate exceptionally high ionic conductivity of 1.79 mS cm−1 at 20 °C, a Li+ transference number of 0.64, and an impressive electrochemical stability window extending to 5.0 V. As illustrated in Fig. 7c, Zeng et al. developed a series of semi-interpenetrating network (semi-IPN) polymer electrolytes by integrating a novel LC C6M with poly(ethylene glycol) diglycidyl ether (PEGDE) through UV irradiation for the first time.33 The incorporation of LCs not only significantly enhances the mechanical properties of the electrolyte membranes by forming a network structure with PEGDE, but also facilitates the creation of stable ion conduction pathways. Consequently, the resulting free-standing, flexible solid polymer electrolyte demonstrates excellent ionic conductivity of 5.93 × 10−5 S cm−1 at 30 °C, a wide electrochemical stability window of 5.5 V, remarkable thermal stability with decomposition temperatures exceeding 360 °C, and the ability to suppress the growth of lithium dendrites. Furthermore, LCEs can effectively address the challenges of weak polymerization kinetics and low polymerization degree in fluorinated solid-state polymer electrolytes (SPEs), which are used to form stable solid-electrolyte interphase (SEI) on Li metal. Qin, et al. developed a partially fluorinated SPE reinforced with meltblown cloth using LCs, which was synthesized through UV curing method (Fig. 7d).189 SEM revealed that the meltblown cloth was composed of randomly arranged polypropylene (PP) nanofibers with an average diameter of 5 μm and pores larger than 20 μm. This porous network facilitated precursor solution infiltration and provided robust mechanical support for the SPEs. Following infiltration and UV curing, the resulting partially fluorinated, meltblown cloth-reinforced SPE (PFMC-SPE) exhibited a smooth and uniform surface, which improved interfacial properties in battery assemblies. This PFMC-SPE exhibited significantly enhanced performance, owing to the improved mechanical properties from the meltblown cloth and the optimized electrochemical characteristics resulting from partial fluorination. The PFMC-SPE demonstrated excellent room temperature ionic conductivity (1.0 mS cm−1) and a broad electrochemical stability window (up to 5 V). Moreover, when used in a Li//LiFePO4 cell, it maintained stable cycling for over 750 cycles at a 1.0C rate, with a capacity retention of 84.7%, dropping from 141.2 mAh g−1 to 119.6 mAh g−1 at 30 °C. These further demonstrate that the use of LCEs can effectively promote the formation of a stable SEI on the lithium metal surface, thereby significantly improving the electrode–electrolyte interfacial contact in LIBs. Wang et al. used in situ polymerization to develop a new class of electrolytes based on LC elastomers, as shown in the synthesis diagram in Fig. 7e.44 These LC elastomers-based solid polymer electrolytes exhibit tunable rigidity and flexibility. The study revealed a distinct correlation between the structure, performance, and electrochemical properties of the 2D LCEs. These electrolytes demonstrated impressive ionic conductivity (≈3 mS cm−1) and a wide electrochemical stability window (≈5 V). Additionally, LiFePO4 batteries using the LCEs showed excellent cycling stability, retaining over 90% of their capacities after 450 cycles at 1C.
 | ||
| Fig. 7 (a) Li ion-conducting solid–liquid nanocomposite based on crosslinking of a bicontinuous cubic phase formed by molecule 1 with a PC-LiClO4 solution.20 (b) Synthetic route of the LC-IL solid polymer electrolytes.30 (c) The preparation process of semi-IPN electrolytes using C6M LC, PEGDE, and LiTFSI with directional network structure.33 (d) Fabrication process of PFMC-SPE.189 (e) Schematic diagram of LCEs manufacturing process.44 | ||
Wang et al. describe molecular ionic composite electrolytes (MICs) that combine aligned liquid crystalline polymers with ionic liquids and concentrated lithium salts. These solid-state rigid-rod polymer composite electrolytes feature nanocrystalline Li+ pathways and 3D transport channels, which are formed through the self-assembly of LCEs.28 The fabrication of Li-loaded MICs (LiMIC) involves a two-step process: (1) forming a polymer-ionic liquid network, and (2) ion exchange to load lithium ions. As shown in Fig. 8, the raw MIC (RMIC) is prepared by ion-exchanging sulfonated polyamide (Li-form PBDT) with the ionic liquid 1-ethyl-3-methyl imidazolium tetrafluoroborate (C2mimBF4). PBDT forms an ordered nematic LC phase at concentrations above 2 wt%, providing mechanical stability and nanoscale structure. RMICs are labeled RMIC-5 and RMIC-15, with PBDT weight percentages of 5% and 15%, respectively.28 In the second step, RMICs are immersed in an ionic liquid electrolyte (N-propyl-N-methylpyrrolidinium bis(fluorosulfonyl)imide, C3mpyrFSI with 50 mol% LiFSI), promoting ion exchange and Li+ incorporation, resulting in LiMICs. The SEM images of the RMICs, revealing locally aligned PBDT grains and interconnected grain boundaries. These boundaries form a conductive network that aids in Li+ transport. The LiMICs are macroscopically isotropic but exhibit local alignment due to the rigid PBDT chains, which can be observed using polarized optical microscopy. Fig. 8 further highlight the structure and ion distribution. It depicts the micrometer-scale organization of the LiMICs, where PBDT grains are interspersed with a nanocrystalline ionic phase that enhances ion conduction. Ion exchange at the grain boundaries increases Li+ density and transport speed, while the morphology of the PBDT grains and their nanocrystalline boundaries facilitates enhanced ion mobility across the electrolyte. This 3D LCE, which is both highly durable (200 MPa) and non-flammable, exhibits excellent performance as a SSE. It demonstrates a high Li+ conductivity of 1 mS cm−1 at 25 °C and remarkable electrochemical stability with a voltage window of 5.6 V versus Li|Li+. Additionally, it effectively suppresses the growth of lithium dendrites. When used in lithium symmetrical cells, the electrolyte shows a low interfacial resistance of 32 Ω cm2 and a reduced overpotential, with a value of less than 120 mV at a current density of 1 mA cm−2.
 | ||
| Fig. 8 Fabrication steps to form LiMICs.28 Step 1 involves creating the RMIC through electrostatic interactions, facilitated by an interfacial ion exchange between a water-soluble ionic liquid (e.g., C2mimBF4) and an aqueous solution of Li-form PBDT polyelectrolyte (Li-PBDT in water). The image shows a sliced transparent RMIC sample. In Step 2, the RMIC is immersed in an ILE consisting of C3mpyrFSI with 50 mol% LiFSI. During this process, C3mpyr+ cations tend to segregate into the PBDT-rich phase, while FSI− and BF4− anions preferentially associate with Li+, precipitating into a nanoscale heterogeneous structure at the grain boundaries. The resulting sliced LiMIC sample appears iridescent. After Step 2, the grain boundaries predominantly consist of a condensed salt phase, forming a nanocrystalline structure that supports fast Li+ transport. The aligned LC grains feature PBDT double-helical rods filled mainly with mobile IL cations, with a spacing of approximately 2 nm between the rods.28 | ||
3.2. Classification by types of molecular structures
The classification of LCEs based on the molecular structure of their constituent LC components is essential for elucidating their ionic transport mechanisms and optimizing their performance in electrochemical applications, particularly in LIBs. This classification delineates two primary categories: ionic LCEs and non-ionic LCEs, each exhibiting distinct properties and behaviors that influence their utilities as electrolytes. The classification of LCEs into ionic and non-ionic types provides valuable insights into their designs and functionalities as electrolytes. Each category offers unique advantages that can be leveraged to enhance ion transport mechanisms, making them critical materials in the pursuit of more efficient and high-performing energy storage systems. Understanding these distinctions not only informs the development of advanced LC electrolytes but also opens new avenues for research and innovation in the field of electrochemistry.Functional groups, such as imidazolium, ammonium, and phosphonium, play a pivotal role in the design of ionic LCEs. These groups are strategically integrated into the LC framework to create a favorable electrostatic environment for ion transport. The presence of these ionic moieties allows for the establishment of strong ion-dipole interactions, leading to the formation of structured ionic pathways within the liquid crystalline matrix. Such pathways facilitate both 1D and 2D ion transport, which is crucial for improving the ionic conductivity required for high-performance LIBs. The unique self-assembly behavior of ionic LCEs enables the formation of ordered mesophases, such as columnar, smectic, or cubic phases, which are conducive to ionic mobility. The nanoscale dimensions of the ionic channels created by self-assembly processes enhance the effective diffusion of lithium ions, thereby reducing the energy barriers associated with ion transport. Consequently, ionic LCEs exhibit superior electrochemical performance, making them attractive candidates for future electrolyte formulations in advanced energy storage systems. Fig. 9a highlights the strengths of ionic liquids (ILs) and ionic LCEs in energy storage and conversion systems, emphasizing their fundamental properties and interface behavior.193 A key feature of ILs is their inherent low volatility and nonflammability, which can be attributed to the unique zigzag (Z) bond present in their molecular structure. This Z-bond is formed through both coulombic interactions and hydrogen bonding between the cations and anions. The resulting bond is characterized by its strong energy, long lifespan, and distinct Z motif, which collectively contribute to the thermal stability, low volatility, and fire resistance of ILs. These characteristics address the increasing demand for safer, environmentally friendly materials in energy-related applications.
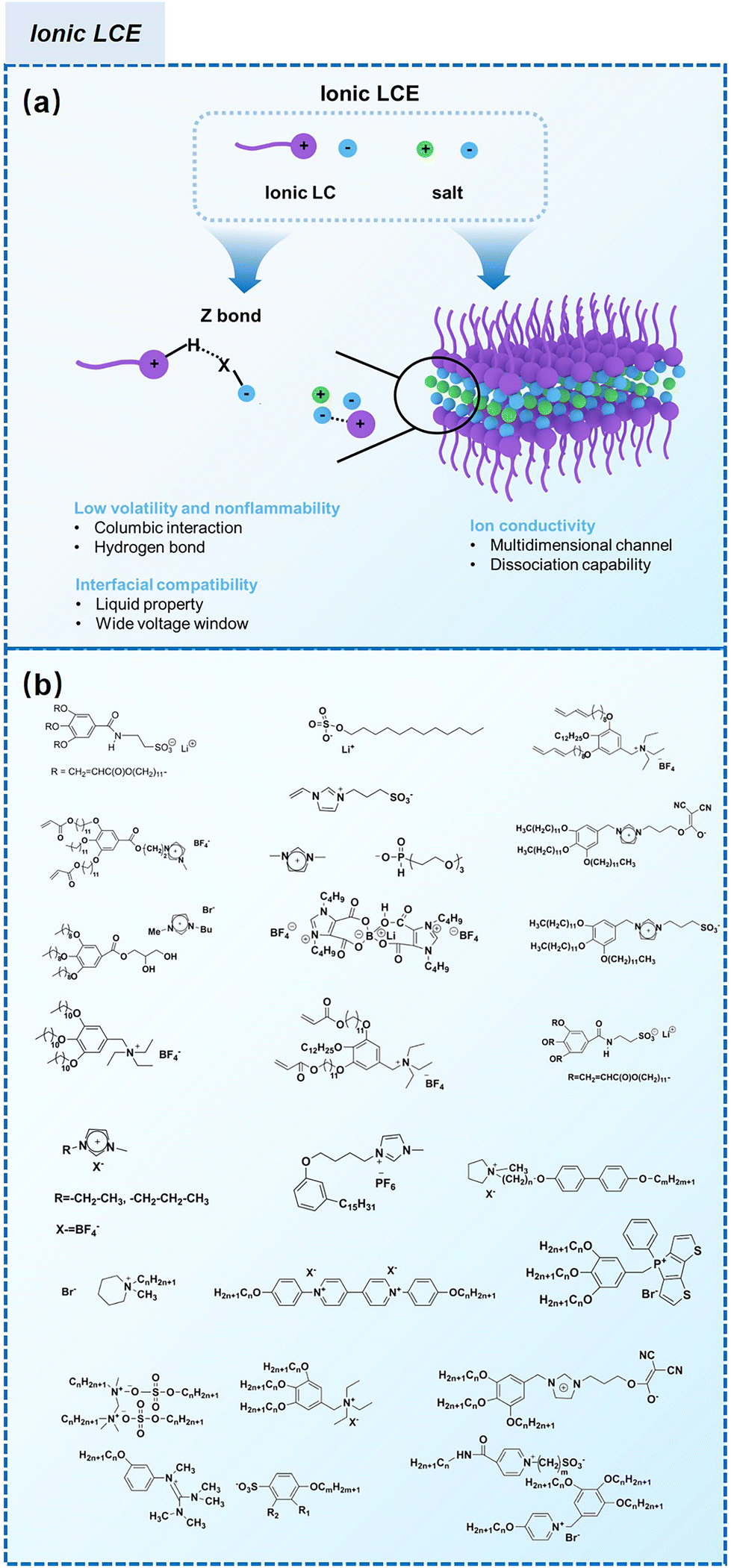 | ||
| Fig. 9 (a) Schematic illustration of ionic LCE structures and their advantages in electrochemical energy storage devices.21 (b) Summary of ionic LCEs.62,194–204 | ||
The foundational materials for ionic LCEs are the concept first explored in 1938 by G. A. Knight and B. D. Shaw,205 who discovered long-chain alkylpyridines and their derivatives as novel ionic LCEs. Since that time, ionic LCEs have garnered significant interest due to the growing recognition of ionic liquids (ILs). A key advantage of ionic LCEs lies in their availability and the flexibility they offer in terms of design. By varying the cation species, researchers can achieve a range of distinct physicochemical properties. Fig. 9b summarizes the molecular formulas of various ionic LCEs.
Lu et al. synthesized ionic LCEs composed of the conventional ionic liquid [C14Mim] [BF4] and LiBF4, designed specifically for solvent-free LIBs. The[C14Mim] [BF4] demonstrated excellent compatibility with LiBF4, facilitating the formation of a smectic phase.22 In this phase, LiBF4 was highly concentrated, creating dynamic 2D ion-conducting channels. These dynamic channels enabled the solvent-free electrolytes to achieve significant ionic conductivities in the range of ∼10−4 to ∼10−3 S cm−1 when in the smectic state. These ionic LCEs exhibited robust cycling stability, high charge/discharge efficiency, and intrinsic thermal safety in LIBs. The batteries showed a charge/discharge capacity of 154.7 mAh g−1 at a current density of 1C and maintained stable performance over 100 charge/discharge cycles across a wide temperature range. An interesting feature of the electrolytes was their abilities to thermally control the operation of the battery by transitioning between different phases. In the crystal phase, the electrolytes exhibited no charge/discharge behavior; however, once the electrolytes transitioned into the smectic phase, the batteries functioned efficiently. This demonstrated for the first time the concept of a thermal switch in ionic LCEs, offering the ability to “turn on” or “off” battery performance based on phase transitions, as shown in Fig. 10a. This work highlights the potential of ionic LCs to serve as advanced, high-performance electrolytes for solvent-free LIBs, with the added benefit of smart, phase-controlled operation.
 | ||
| Fig. 10 (a) SSEs based on lamellar nanostructured ionic LCs for LIBs with a thermal switch on/off performance.22 (a) Reproduced with permission.22 Copyright 2023, Elsevier. (b) Front and (c) side view SEM images of MIM-3, showing ordered microporous structure. (d) Magnified SEM image of MIM-3 displaying nanochannels along the pore walls. (e) Microporous structure under atomic force microscopy (AFM), (f), (g) magnified AFM images of MIM-3. (h) Phase image of MIM-3 showing well-oriented surface for effective charge transport characteristics. Ionic LECs were fabricated by varying the composition of PMIMP and P(VDF-HFP), and are designated as MIM-1, MIM-2, MIM-3, and MIM-4 individually.34 (b)–(g) Reproduced with permission.34 Copyright 2017, Wiley-VCH. (i) Schematic representation of the proposed mechanism of anisotropic Li mass transport effect towards dendrite suppression when compared to the isotropic transport. (j) Electrochemical Li plating/stripping of symmetric Li|ILC/LITFSI|Li and Li|PC/LiTFSI|Li cells with a current density of 0.1 mA cm−2; postmortem analysis (FE-SEM) of the cycled Li electrode of (k) PC/LiTFSI and (l) ILC/LiTFSI cells. Ionic liquid crystals denoted as ILCs.45 (i)–(l) Reproduced with permission.45 Copyright 2021, The Royal Society of Chemistry. (m)–(x) POM images and illustration of the structure of the isotropic, planar, homeotropic, and hybrid samples (first to fourth columns from left to right). Top row: Top view of iLCEs under cross-polarized light in transmission mode scale bar for all four textures is shown in (m). Middle row: Cross section of iLCEs in reflection mode (the thickness is bottom to the top of the image). Bottom row: Illustration of the side views of the director structure of the LCE (brown ellipsoids) and the ionic channels (blue lines) of the corresponding samples.5 (m)–(x) Reproduced with permission.5 Copyright 2022, AIP Publishing. | ||
In ionic LCEs, the incorporation of LCs allows for precise control over the structure of ILs. Additionally, the formation of well-ordered, porous channels within the membrane plays a crucial role in enhancing electrolyte uptake and improving wettability. This is reflected in a reduced contact angle and a significant improvement in the transport of lithium ions. These structural features contribute to the overall efficiency and performance of the electrolyte by facilitating better interaction between the ionic liquid and the electrode surfaces. Sasi, et al. developed a bio-based ionic LC-derived solid polymer electrolyte membrane composed of a polymer backbone, lithium salt, cardanol-derived ionic LCs, and a conventional plasticizer.34 The membrane exhibits high porosity and improved electrolyte absorption, which is attributed to its well-defined porous network structure. SEM analysis (Fig. 10b) reveals a highly ordered network with pore sizes averaging 6–7 μm. A side-view SEM image (Fig. 10c) shows the orientation of the porous network, which contributes to the excellent electrolyte uptake. High magnification SEM (Fig. 10d) further reveals that the micropores are surrounded by smaller nanopores of approximately 150 nm, contributing to the membrane's enhanced absorption capacity. AFM imaging (Fig. 10e) confirms the hierarchical structure, showing well-defined micropores like the SEM results. At higher magnifications (Fig. 10f and g), AFM images reveal that each pore is surrounded by nanostructured walls, formed due to the self-assembly of ionic liquid crystalline molecules. This self-assembly process, especially during the film drying phase, results in the formation of oriented nanoporous walls that improve electrolyte uptake. The phase profile (Fig. 10h) shows the extensive phase ordering within the membrane, which is essential for smooth ion transport and improved ionic conductivity. The uniform phase distribution observed in the phase image further indicates efficient charge transport across the membrane. The final ionic LCE membrane, produced with varying ratios of 3-(4-(3-pentadecylphenoxy)butyl)-1-methylimidazole-3-ium hexafluorophosphate (PMIMP) and PVdF-HFP (labeled MIM-1 to MIM-4), exhibits high conductivity, porosity, and wettability. Its anisotropic channels facilitate efficient Li+ transport, making it suitable for high-performance battery applications.
Anisotropic ionic LCEs have been explored for their ability to suppress lithium dendrite growth by effectively modulating the ion concentration gradient near the dendrite tips. Gopalakrishnan et al. proposed a novel approach where ionic LCEs serve as both the electrolyte and a pseudo-separator, utilizing their anisotropic properties to regulate Li-ion transport and mitigate dendrite formation, as illustrated in Fig. 10i.45 The role of these properties in dendrite suppression was investigated by examining the ionic transport dynamics at the electrode–electrolyte interface. To assess dendrite growth, galvanostatic cycling of Li|ILC–LiTFSI|Li symmetric cells was conducted, with lithium stripping performed at a constant current of 0.032 mA. These cells, which consisted of Li counter/reference and working electrodes separated by a Teflon spacer filled with the ionic LCEs, were cycled at a current density of 0.1 mA cm−2 and a temperature of 60 °C to evaluate their long-term electrochemical stability. The Li/ILC–LiTFSI/Li cell demonstrated stable plating and stripping processes, with a gradual increase in voltage hysteresis, a reflection of the polarization due to current density, interfacial resistance, and charge transfer characteristics. This hysteresis was notably more stable compared to symmetric cells using conventional LiTFSI/propylene carbonate (PC) electrolytes, which developed internal short circuits due to dendrite growth after 400 hours (Fig. 10j). In contrast, the cell with the LCE maintained stable cycling for over 850 hours, indicating that the anisotropic nature of the LCE facilitated more uniform Li+ transport, leading to compact and controlled dendrite growth. Further analysis using FE-SEM (Fig. 10k and l) revealed distinct differences between the two cells. The Li metal cycled in the PC/LiTFSI cell showed a rough, uneven surface with large lithium chunks, indicative of irregular deposition during plating/stripping. On the other hand, the Li metal cycled in the ILC/LiTFSI cell had a much smoother surface with only minor corrugations, suggesting more stable cycling and reduced lithium loss. The smoother surface corresponds to lower polarization and superior cell performance, attributable to the more uniform lithium deposition facilitated by the anisotropic electrolyte.
Rajapaksha et al. demonstrated the potential of ionic LC elastomers as solid electrolytes for flexible, substrate-free organic electrochemical transistors.5 A key aspect of their study was the manipulation of the LC elastomer's director alignment, which was used to fine-tune both the steady-state and transient responses of the devices. Fig. 10m–x illustrate the optical and structural characteristics of LC elastomers samples with different alignments. The first column of images (Fig. 10m–p) shows POM images of planar, homeotropic, hybrid, and isotropic samples, each with a 200 μm thickness. The top row (Fig. 10m–p) displays the top-view POM images in transmission. The isotropic sample (Fig. 10m) is entirely dark, confirming its optical isotropy. In contrast, the samples in Fig. 10n–p show bright droplets in a dark background, indicating phase separation due to spinodal decomposition between the ionic liquid-rich and nematic LCE-rich phases. This separation was triggered by the isotropic-nematic transition, resulting from the incompatibility between ionic liquids and nematic LCEs. The phase separation was found to depend on how long the sample remained in the nematic phase before cross-linking. After cross-linking, phase separation stopped, and the size of the birefringent and isotropic domains remained unchanged. To optimize phase separation, cross-linking was carried out at 50 °C, just below the I–N transition temperature. This approach contrasts with previous studies on colloidal LC gels by N. L. Abbott, although the microstructure differed due to variations in thickness and chemical composition between the two systems.206–210 While the top-view textures of the nematic samples were similar, significant differences were observed in the cross-sectional side views (Fig. 10q–t). The side view of the isotropic sample (Fig. 10q) displayed a uniform texture, while the planar (Fig. 10r), homeotropic (Fig. 10s), and hybrid (Fig. 10t) samples exhibited inhomogeneous textures. Notably, the planar and planar-side hybrid samples showed brighter reflections compared to the homeotropic or homeotropic-side hybrid samples, suggesting that the ionic channels, which influence light reflection, have larger cross-sections when oriented normal to the LCE director. This observation supports the idea that phase-separated ionic liquid channels align perpendicular to the director to minimize contact with the hydrophobic terminal chains. The director and ionic channel orientations, corresponding to this model, are shown in the bottom row (Fig. 10u–x). The brown ellipsoids represent the LCE director, while the blue lines indicate the orientation of the ionic channels, consistent with the assumed perpendicular alignment to the LCE director. This structural configuration plays a crucial role in the device's electrochemical performance. The observed phase separation, resulting from spinodal decomposition between ionic liquid-rich and nematic LCE-rich phases, is triggered by the isotropic-nematic transition due to inherent incompatibility between ionic liquids and nematic LCEs. To control this phase separation, the concentration ratio of ionic liquid to nematic LCEs serves as a critical governing parameter. Additionally, fabrication methodologies for composite films, such as casting, electro-spinning, hot-pressing, and microfluidics, provide practical strategies for modulating phase separation behavior.
Through the performance reported in these studies, it is evident that ionic conductivity plays a pivotal role in the effectiveness of electrolytes for energy storage applications. The ionic conductivity of ILs and ionic LCEs depends on ion density, charge, and mobility, with ionic LCEs exhibiting enhanced conductivity due to their ability to self-organize into structured, nanoscale domains. Under specific conditions of temperature and solvent concentration, these materials form 1D, 2D, or even 3D ion-conducting pathways. The self-assembly is driven by multiple intermolecular forces, such as the Z-bond, van der Waals interactions, and hydrophobic forces. This structural organization significantly increases ion mobility, improving conductivity. Moreover, the ionic pairs in ionic LCEs can also facilitate salt dissociation through coulombic interactions, which helps increase the concentration of free ions, further enhancing ionic conductivity. Besides, the interfacial properties of ionic LCEs are crucial for their performance in energy storage systems, particularly regarding charge transfer efficiency. The liquid nature of ionic LCEs contributes to their fluidity, reducing interfacial contact resistance and enabling more efficient charge movement. Additionally, the wide electrochemical stability window of these materials minimizes parasitic side reactions, allowing for the formation of stable and favorable interfaces that are critical for long-term device stability. Taken together, these properties make ionic LCEs promising candidates for use as electrolytes in energy conversion and storage technologies.
 | ||
| Fig. 11 Summary of non-ionic LCEs.1–4,64,90,183,211–214 | ||
The self-assembly of non-ionic LCEs leads to the formation of structured ionic channels that facilitate the transport of lithium ions. The interaction between the non-ionic LC matrix and the lithium salt results in the establishment of a dynamic ionic environment that promotes ion migration. The formation of 2D ionic channels within the liquid crystalline phase is particularly beneficial, as it allows for a more efficient pathway for Li+ diffusion, enhancing the overall ionic conductivity of the electrolyte. Moreover, the tunable properties of non-ionic LCEs enable researchers to optimize their electrochemical performance for specific applications. By varying the molecular structures and compositions of the LC components, it is possible to tailor the thermal and electrochemical stability of the electrolyte, as well as its ionic conductivity. This adaptability positions non-ionic LCEs as a versatile and promising option for a wide range of energy storage applications, including next-generation LIBs.
Shimura et al. synthesized a new type of fan-shaped LC molecule incorporating a high dielectric constant cyclic carbonate as an ion-conductive functional group.24 This LC was mixed with lithium trifluoromethanesulfonate (CF3SO3Li) to prepare a LCE. XRD analysis revealed that the electrolyte adopts a hexagonal columnar phase, which can form 1D ion conduction channels. As shown in Fig. 12a, at 22 °C without the application of alternating current (AC), the LC electrolyte exhibits a multi-domain structure with an ion conductivity of 2.2 × 10−8 S cm−1. After applying AC for 90 minutes, the columnar LC structure undergoes further ordering, aligning parallel to the electric field and transitioning into a single-domain structure. This alignment results in a 4.3-fold increase in ion conductivity compared to the unmodified LCE. These findings suggest that the single-domain columnar phase, aligned with the electric field, significantly enhances the transport of Li+ ions. Sakuda et al. developed the first thermotropic LCEs, which also serve as non-ionic LCEs for LIBs.25 These electrolytes were created using a rod-like LC molecule with a cyclic carbonate group, which self-assembles to form 2D ion-conductive pathways when mixed with lithium salts. Fig. 12b illustrates the ionic conductivities of mixtures of compound 1 and LiTFSI as the system is cooled from the isotropic phase. In the SmA phase, compound 1 exhibits homeotropic alignment, which results in the formation of 2D ion-conductive pathways parallel to the substrate surface. The ionic conductivities in the SmA phases range from 10−6 to 10−4 S cm−1, which are significantly higher than those observed in carbonate-based columnar LCEs, which show conductivities around 10−8 S cm−1. This improved conductivity is likely due to the lower viscosity and the larger area of conduction pathways in the SmA phase. In contrast, the lithium ions in the columnar structures are confined to 1D nanospaces, where they can only be transported along the column axes.
 | ||
| Fig. 12 (a) POM images of the complex of 1 and 2 (the molar ratio 10:1) in the columnar state at 22 °C under an electric field (2.5 V μm−1, 1 kHz). The time after the electric field was applied is indicated in the inset.24 (a) Reproduced with permission.24 Copyright 2009, Wiley-VCH. (b) Ionic conductivities of the mixtures of LC (molecule structure as 1) and LiTFSI in the 9:1 (black), 8:2 (blue), 7:3 (red), and 6:4 (green) molar ratios.25 (b) Reproduced with permission.25 Copyright 2014, Wiley-VCH. (c) Rate performances of LFP/LCE-SPEx/Li at various current densities at room temperature; (d) specific discharge capacity of LFP/LCE-SPEx/Li during 50 cycles at 0.2C at room temperature; (e) specific discharge capacity of LFP/LCE-SPEx/Li for 300 cycles at 1C at room temperature; (f) voltage profiles of symmetric Li/LCE-SPEx/Li cells at a current density of 0.1 mA cm−2 and a cycling capacity of 0.1 mAh cm−2; (g) specific discharge capacity and Coulomb efficiency of LFP/LCE-SPE2/Li for 450 cycles at 1C at room temperature; (h) selected charge/discharge curves of LFP/LCE-SPE2/Li at room temperature during cycling; (i) rate performance of the LFP/LCE-SPE2/Li cells at different C-rates at room temperature; (j) selected charge/discharge curves of LFP/LCE-SPE2/Li at room temperature during the multiplier test.44 (c)–(j) Reproduced with permission.44 Copyright 2025, Elsevier. | ||
Non-ionic LCEs have demonstrated excellent electrochemical performance when used in battery assembly, showcasing their potential as effective materials for energy storage applications. Wang et al. introduced LC elastomer materials as a novel electrolyte for LIBs.44 By modifying the ratio of LC monomers and chain extenders, they developed a series of LCE-SPE(x) membranes for battery assembly (Fig. 12c–j).44 The LFP/LCE-SPE2/Li batteries demonstrated excellent cycling stability, retaining over 90% of their capacity after more than 400 cycles at room temperature and a 1C charge/discharge rate (Fig. 12g). Additionally, LCE-SPE(x) (where x = 0, 1, 2, 3) exhibited long-term stability in lithium plating/stripping, with Li//Li symmetrical cells achieving over 1000 hours of reversible cycles at a current density of 0.1 mA cm−2 at 25 °C. LiFePO4 batteries using LCE-SPE-based electrolytes also showed outstanding performance, maintaining over 90% capacity retention after 450 cycles at 1C. The development of LCE-SPE is expected to lead to the integration of more advanced non-ionic LC/polymer composite properties in future electrolyte technologies. To conclude, Table 1 summarizes relevant information for various LCEs that have been reported.
| Number | Chemical formula | Ion transport mechanism | Ionic/non-ionic | Ionic conductivity (σ) | Remarks under specific conditions | Ref. |
|---|---|---|---|---|---|---|
| 1 |

|
1D | Non-ionic | 2.2 × 10−8 S cm−1 (22 °C) | 4.3 times when applying an AC electric field (2.5 V μm−1, 1 kHz) for 90 min | 242 |
| 2 |
|
1D | Ionic | 4.7 × 10−4 S cm−1 at 160 | 243 | |
| 3 |

|
1D | Ionic | 1.2 × 10−4 S cm−1 for 2 at 130 °C | 243 | |
| 4 |

|
1D | Ionic | 3.9 × 10−4 S cm−1 for 3 at 120 °C | 243 | |
| 5 |

|
1D | Ionic | 0.45 × 10−3 S cm−1 at 30 °C | This case is combined with PEO | 244 and 245 |
| 6 |
|
1D | Non-ionic | ∼10−3 S cm−1 near 100 °C | 244 and 245 | |
| 7 |

|
1D | Non-ionic | — | By designing an in situ formed LC interphase, this enhancement subsequently increases the performance of whole battery | 50 |
| 8 |

|
1D | Non-ionic | ∼10−3 S cm−1 at 60 °C | 54 | |
| 9 |
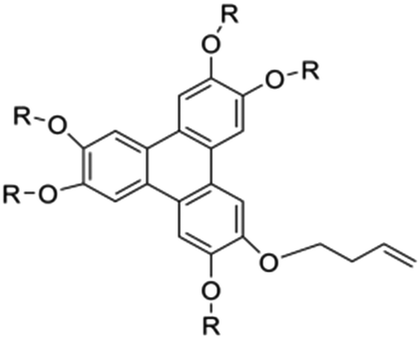
|
1D | Non-ionic | 5.48 × 10−5 S cm−1 at 30 °C | 49 | |
| 10 |

|
1D | Ionic | 1.2 × 10−2 S cm−1 at 150 °C | 31 | |
| 11 |
|
1D | Ionic | 4.8 × 10−4 S cm−1 at 130 °C | 244 and 245 | |
| 12 |

|
2D | Non-ionic | 10−6–10−4 S cm−1 at room temperature | 25 | |
| 13 |
|
2D | Non-ionic | 2.14 × 10−2 S cm−1 at room temperature | 32 | |
| 14 |

|
2D | Non-ionic | 3 ± 0.5 × 10−3 S cm−1 at room temperature | 246 | |
| 15 |

|
2D | Ionic | 1.96 × 10−4 S cm−1 at 30 °C | 176 | |
| 16 |

|
2D | Non-ionic | 7.37 mS cm−1 at 25 °C | 247 | |
| 17 |

|
2D | Non-ionic | 6.0 × 10−7 S cm−1 at 80 °C | This material lies in its high ionic conductivity achieved through the combination of polyethylene oxide a star-shaped liquid-crystalline copolymer (3PMALC-PPEGMA) | 248 |
| 18 |
|
2D | Ionic | 1.4 mS cm−1 at 30 °C | This material combines the amphiphilic feature of imidazolium 1-hexadecyl-3-methylimidazolium tetrafluoroborate with propylene carbonate and LiBF4, distinguishing it from other conventional electrolytes | 249 |
| 19 |

|
2D | Non-ionic | 1.0 × 10−7 S cm−1 at 25 °C | This material has been combined with LC or hydrophobic blocks to further improve its properties, setting it apart from other conventional polymer electrolytes | 180 |
| 20 |

|
2D | Non-ionic | 1.13 × 10−4 S cm−1 at 30 °C | These boron-containing LC polymer electrolytes are distinguished from other materials by their integration of LC | 250 |
| 21 |
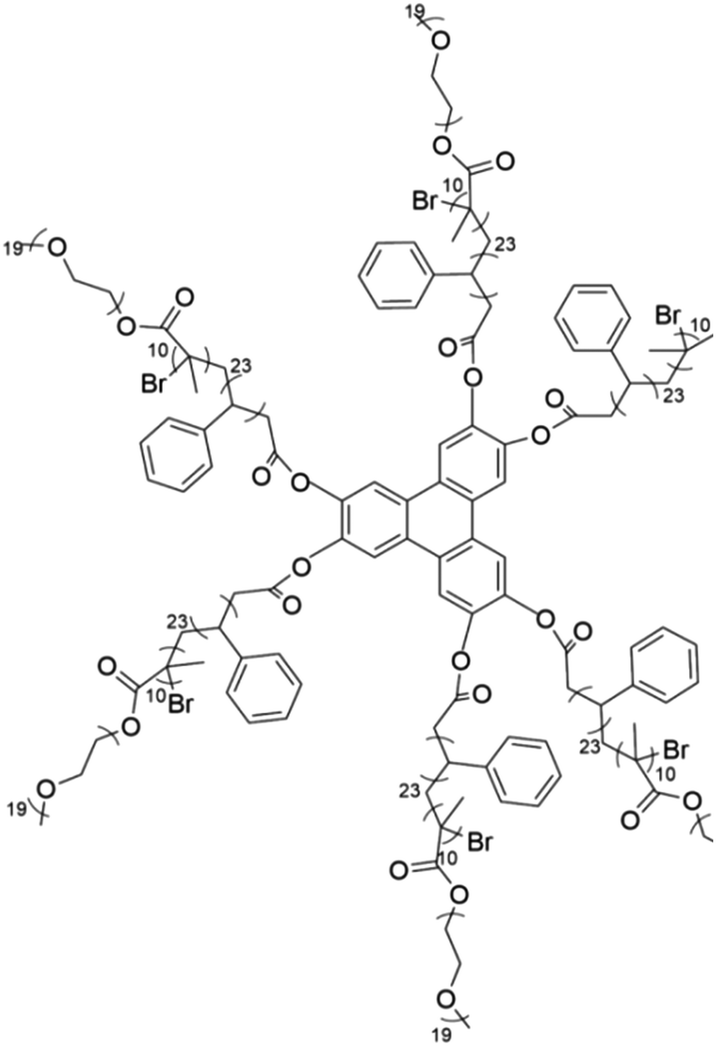
|
2D | Non-ionic | 1.46 × 10−4 S cm−1 at 30 °C | These boron-containing LC polymer electrolytes are distinguished from other materials by their integration of LC, which facilitate the construction of ordered ion channels for rapid Li-ion transport, and by their ability to anchor anions and adsorb impurities, thereby enhancing ion transport capability and electrochemical stability | 183 |
| 22 |

|
2D | Non-ionic | 7.14 × 10−5 and 2.17 × 10−3 S cm−1 at 25 and 95 °C | 184 | |
| 23 |
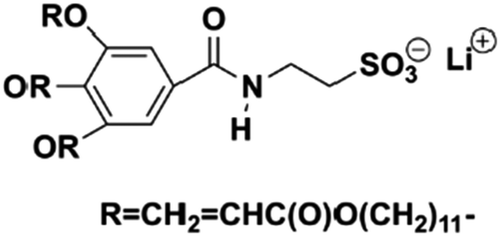
|
3D | Ionic | 10−4 to 10−3 S cm−1 at room temperature | This material integrates liquid electrolyte-filled polymer components, distinguishing it from traditional polymer electrolytes | 251 |
| 24 |

|
3D | Non-ionic | 1.79 mS cm−1 at 20 °C | This material is combined with ionic liquids (IL) to form poly(vinylidene fluoride-hexafluoropropylene)-poly(liquid crystal ion liquid), which exhibits enhanced electrochemical properties compared to other electrolytes | 30 |
| 25 |

|
3D | Non-ionic | 5.93 × 10−5 S cm−1 at 30 °C | 33 | |
| 26 |

|
3D | Ionic | 5 × 10−9 S cm−1 at 100 °C | 194 | |
| 27 |

|
3D | Ionic | 1 mS cm−1 at 25 °C | 252 | |
| 28 |
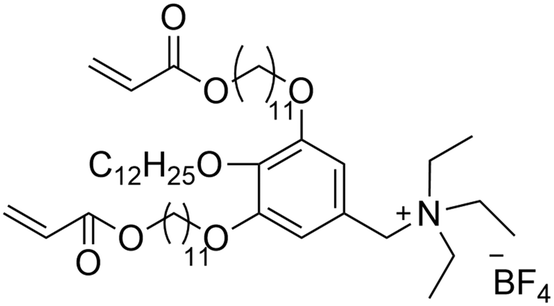
|
1D | Ionic | 8.8 × 10−6 S cm−1 (columnar phase), 3.1 × 10−6 S cm−1 (cubic phase) | 253 | |
| 29 |

|
1D | Ionic | 10−4 S cm−1 at 100 °C | 194 | |
| 30 |

|
1D | Non-ionic | 10−7 S cm−1 at 22 °C | 24 | |
| 31 |

|
2D | Non-ionic | 3.02 × 10−3 S cm−1 at 30 °C | 254 | |
| 32 |
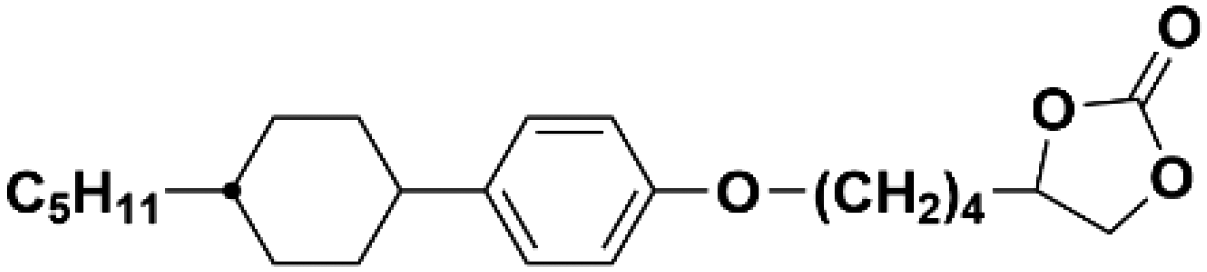
|
2D | Non-ionic | 10−4 S cm−1 at 60 °C | 25 | |
| 33 |

|
3D | Non-ionic | 5.93 × 10−5 S cm−1 at 30 °C | The semi-interpenetrating polymer network electrolyte, incorporating liquid crystal-induced ordered ion channels and a cross-linked framework, delivers thermal stability, mechanical strength, and dendrite suppression for advanced solid-state lithium batteries | 33 |
| 34 |

|
1D | Ionic | 10−4 S cm−1 at 100 °C | 194 | |
| 35 |

|
2D | Non-ionic | 10−4 S cm−1 at 93 °C | 33 | |
| 36 |
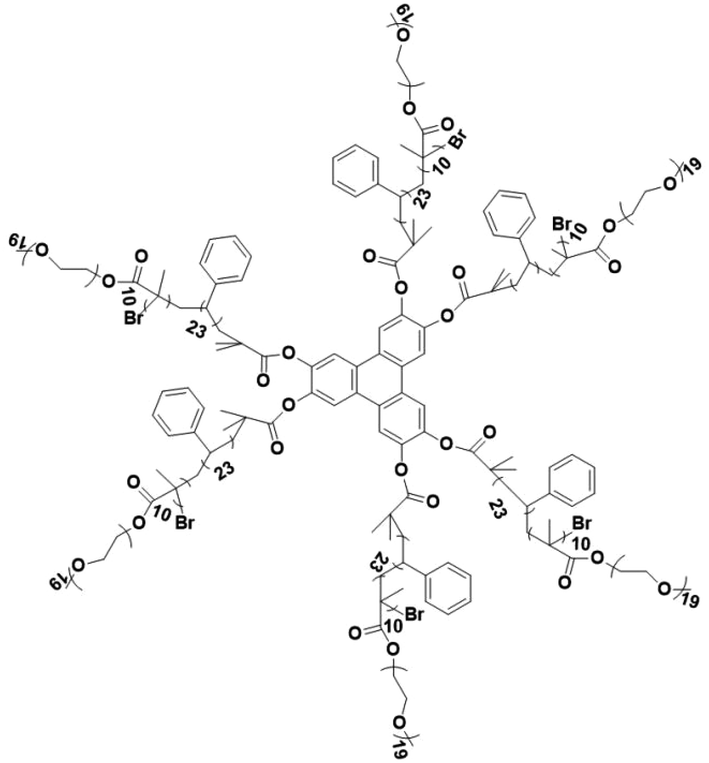
|
2D | Non-ionic | 1.94 × 10−4 S cm−1 at 30 °C | 255 | |
| 37 |

|
1D | Non-ionic | 2.2 × 10−8 S cm−1 at 22 °C | 24 | |
| 38 |

|
2D | Ionic | 3.02 × 10−3 S cm−1 at 30 °C | 62 | |
| 39 |

|
1D | Non-ionic | — | 211 | |
| 40 |

|
2D | Non-ionic | 5.5 × 10−4 S cm−1 at 60 °C | 64 | |
| 41 |
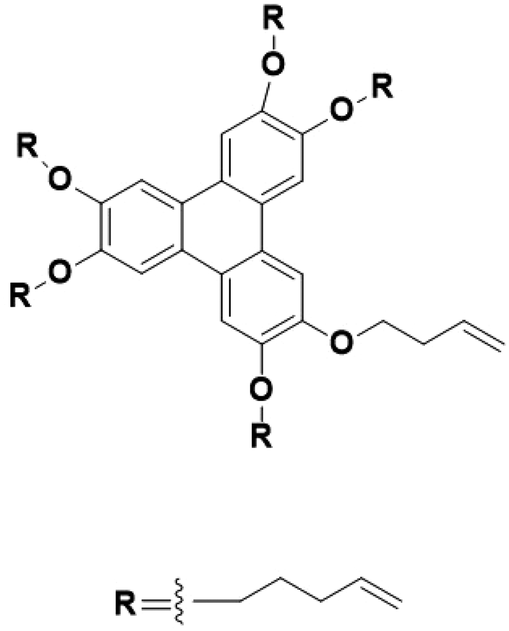
|
2D | Non-ionic | 5.48 × 10−5 S cm−1 at 30 °C | 256 | |
| 42 |

|
2D | Non-ionic | 7.14 × 10−5 S cm−1 at 25 °C, 2.17 × 10−8 S cm−1 at 95 °C | 257 | |
| 43 |

|
2D | Non-ionic | 10−5 S cm−1, −10−6 S cm−1 at 30 °C | 258 | |
| 44 |
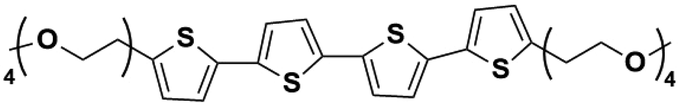
|
2D | Non-ionic | 5.2 × 10−4 S cm−1 at 70 °C | 259 | |
| 45 |

|
2D | Ionic | 0.45 × 10−3 S cm−1 at 30 °C | 257 | |
| 46 |

|
3D | Ionic | 2.1 × 10−3 S cm−1 at 25 °C | - | 28 |
| 47 |

|
2D | Non-ionic | 1.96 × 10−4 S cm−1 at 30 °C | 260 | |
| 48 |
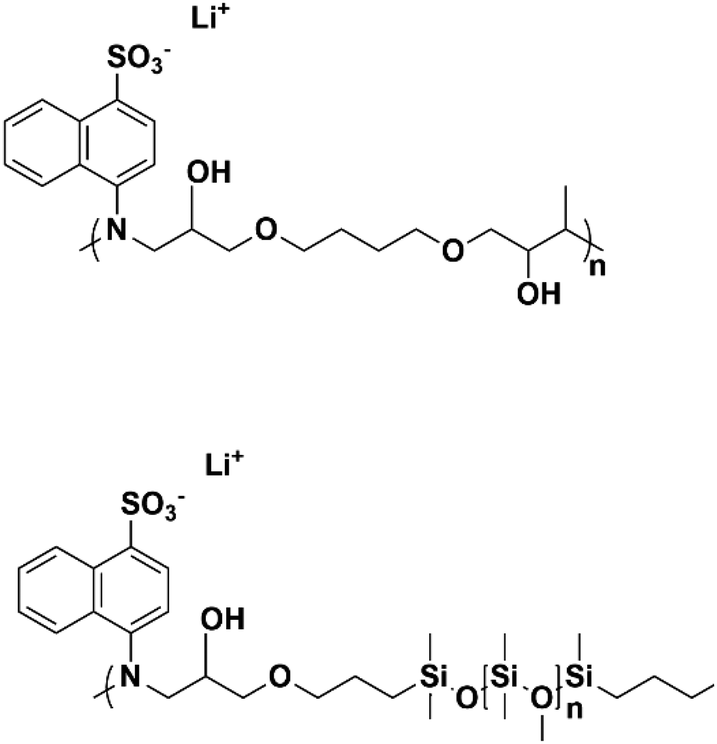
|
2D | Non-ionic | 5 × 10−9 S cm−1 at 100 °C | 261 | |
| 49 |

|
2D | Non-ionic | 1.3 × 10−4 S cm−1 at room temperature | 262 | |
| 50 |
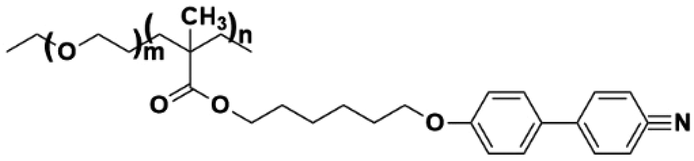
|
2D | Non-ionic | — | 212 | |
| 51 |
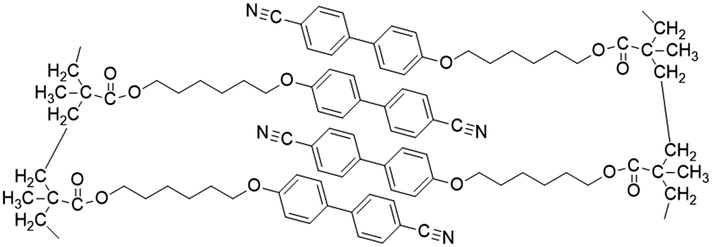
|
2D | Non-ionic | 6.2 × 10−5 S cm−1 at 25 °C, 5.6 × 10−4 S cm−1 at 95 °C | 213 | |
| 52 |

|
2D | Non-ionic | 10−6 S cm−1 at 72 °C | 239 | |
| 53 |

|
3D | Ionic | 10−3 S cm−1 at room temperature, 10−4 S cm−1 at −35 °C | 195 | |
| 54 |
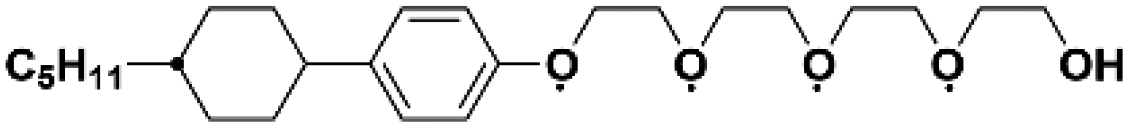
|
2D | Non-ionic | 10−4 S cm−1 at 60 °C | 25 | |
| 55 |
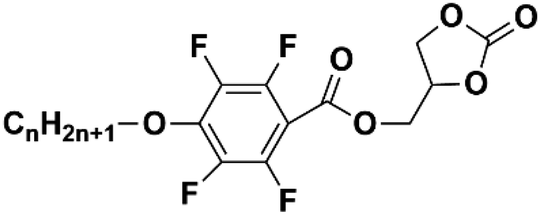
|
2D | Non-ionic | 1.5 × 10−5 S cm−1 at 45 °C, 2.7 × 10−4 S cm−1 at 100 °C | 90 | |
| 56 |

|
2D | Non-ionic | 10−4 S cm−1 at 40 °C | 263 | |
| 57 |

|
3D | Ionic | — | 264 | |
| 58 |
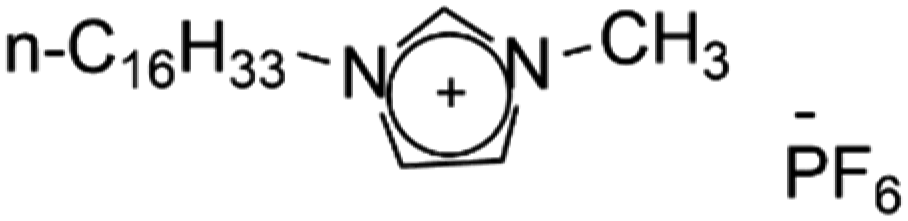
|
1D | Ionic | — | 265 | |
| 59 |
|
2D | Ionic | — | 266 | |
| 60 |

|
2D | Ionic | — | 267 | |
| 61 |

|
2D | Ionic | — | 268 | |
| 62 |
|
2D | Ionic | — | 268 | |
| 63 |

|
2D | Ionic | — | 269 | |
| 64 |

|
2D | Ionic | — | 269 | |
| 65 |
|
2D | Non-ionic | — | 270 | |
| 66 |

|
3D | Non-ionic | — | 270 | |
| 67 |
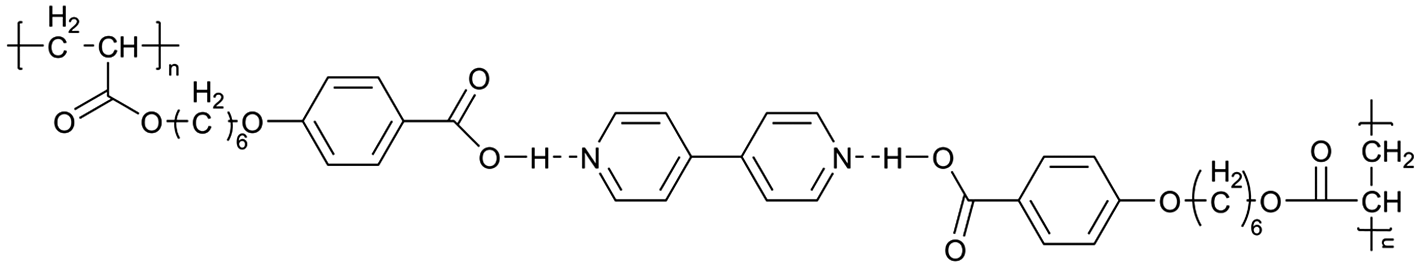
|
3D | Non-ionic | 270 | ||
| 68 |

|
1D | Non-ionic | 270 | ||
| 69 |
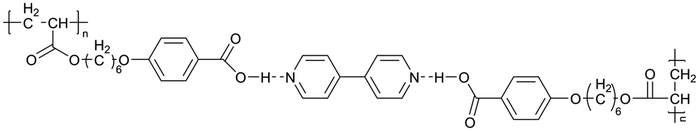
|
2D | Non-ionic | — | 270 | |
| 70 |

|
2D | Ionic | — | 270 | |
| 71 |

|
2D | Ionic | — | 270 | |
| 72 |
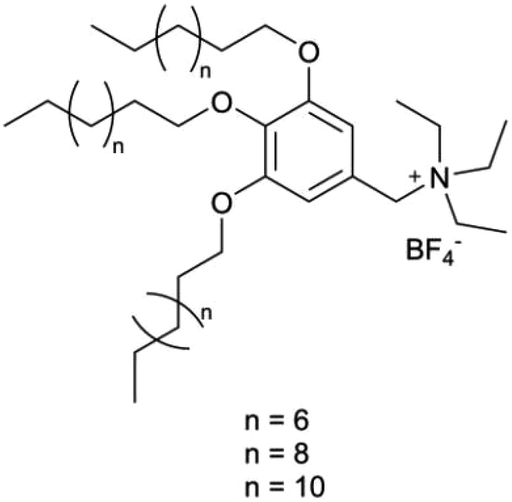
|
3D | Ionic | — | 270 | |
| 73 |
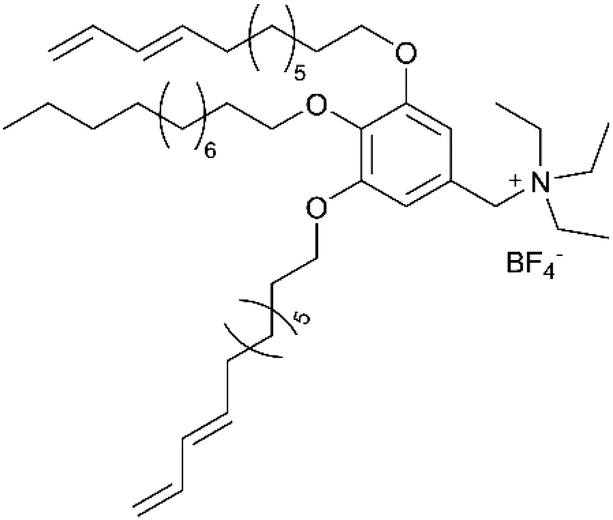
|
3D | Ionic | — | 270 | |
| 74 |

|
2D | Ionic | — | 270 | |
| 75 |
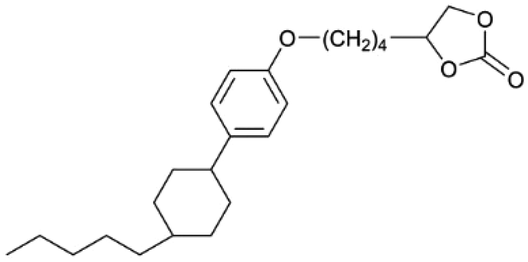
|
2D | Ionic | — | 270 | |
| 76 |

|
2D | Ionic | — | 271 | |
| 77 |

|
2D | Ionic | — | 271 | |
| 78 |

|
3D | Ionic | 3.1 × 10−4 S cm−1 at 90 °C, 7.75 × 10−5 S cm−1 at 90 °C, 8.8 × 10−6 S cm−1 at 90 °C | 272 | |
| 79 |

|
1D | Non-ionic | — | 182 | |
| 80 |

|
1D | Ionic | — | 273 | |
| 81 |

|
3D | Ionic | — | 273 | |
| 82 |
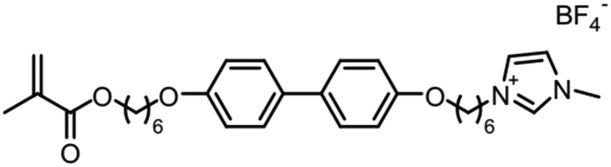
|
2D | Ionic | — | 273 | |
| 83 |

|
2D | Ionic | — | 274 | |
| 84 |

|
2D | Ionic | — | 274 | |
| 85 |

|
3D | Ionic | 10−4 S cm−1 at 130 °C | 275 | |
| 86 |

|
1D or 2D | Ionic | 10−4–10−3 S cm−1 from 25 °C to 100 °C | The material uses ionic LC dendrimers to create tunable 1D/2D proton-conductive channels stabilized by coumarin photocrosslinking | 276 |
| 87 |

|
1D | Ionic | — | 277 | |
| 88 |

|
2D | Ionic | — | 278 | |
| 89 |
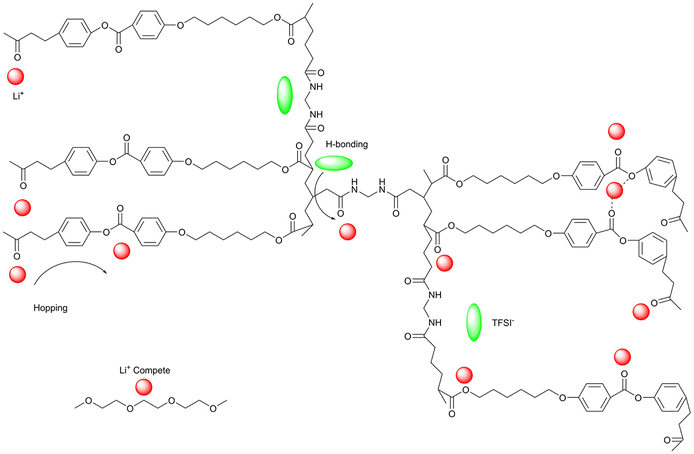
|
3D | Non-ionic | 4.86 × 10−4 S cm−1 at 30 °C | 279 | |
| 90 |

|
3D | Non-ionic | — | 280 | |
| 91 |

|
2D | Ionic | 4.5 × 10−6 S cm−1–4.54 × 10−3 S cm−1 at 30 °C | 281 | |
| 92 |
|
3D | Non-ionic | 0.014 mS cm−1 at 25 °C | 282 | |
| 93 |

|
1D | Ionic | — | 283 | |
| 94 |

|
3D | Ionic | — | 284 | |
| 95 |
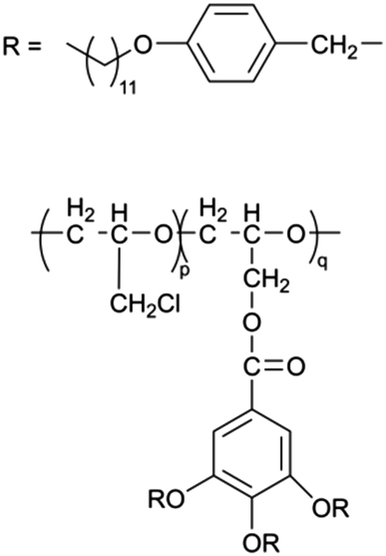
|
2D | Non-ionic | — | 8 |
4. Design rules for LCEs for LIBs
The distinctive molecular architecture and inherent properties of LC molecules enable them to self-organize into well-defined 3D nanostructures, as shown in Fig. 13a.5–8 These organized nanostructures play a crucial role in their application as electrolytes in LIBs. Based on their structural arrangement, LCEs can be categorized into three primary phases: columnar, smectic, and cubic, which correspond to the formation of 1D, 2D, and 3D nanoporous ion-transport channels, respectively. The presence of these nanopores significantly enhances the mobility of Li+, leading to improved overall battery performance and increased ionic conductivity. | ||
| Fig. 13 (a) Self-assembling nanostructures of LCEs for LIBs.5–8 (b) Schematic representation of a strategy to enhance Li+ transport through nanostructured LC materials. These materials are formed by the self-assembly of a zwitterionic LC, a lithium salt, and PC. Green arrows represent the direction of ion transport. Ionic channels are depicted in yellow, lilac, or purple, depending on their composition, while the aliphatic regions are shown in blue. The proposed molecular organization and the arrangement of salt within the ionic channels are illustrated in the zoomed-in side views.14–18 | ||
To develop LCEs with optimal self-assembly behavior suitable for LIBs, a well-designed molecular framework is essential.215 These molecules typically consist of two key components: an ion-transporting segment and a nonionic structural segment. The ion-conducting segment is generally composed of polar or ionic functional groups, such as cyclic carbonates and PEO chains, which facilitate salt dissociation and ion transport. In contrast, the nonionic portion, which commonly includes mesogenic cores and alkyl chains, governs the self-assembled nanostructure of the electrolyte, ultimately influencing ion migration efficiency. A higher dielectric constant in the polar groups typically results in more efficient salt dissociation, thereby ensuring the smooth conduction of lithium ions throughout the electrolyte matrix.216 Generally, hydrophilic functional groups are incorporated into the ion-transporting domain, while hydrophobic moieties are utilized in the nonionic segment. The interplay between these two segments is critical for promoting the formation of well-defined nanochannels that support efficient ion transport.
The development of LCEs can be further advanced by combining different-dimensional Li+ transport mechanisms, such as 1D and 3D channels, to enhance ionic conductivity. One effective approach is the design of imidazolium-based zwitterions, which can co-assemble with lithium bis(trifluoromethylsulfonyl)imide (LiTFSI) and propylene carbonate (PC) to form structured ionic pathways that support lithium transport (Fig. 13b).14–18,194 Zwitterionic LCs, with their charge-delocalized anionic structures, can self-organize into defined ionic channels, but in their unmodified state, they do not contain transportable ions (Fig. 13b, left). When lithium salts are introduced, interactions between the zwitterionic groups and the salt facilitate dissociation, enabling lithium ions to migrate through the established ionic pathways (Fig. 13b, middle). To further optimize Li+ conductivity, PC, a polar additive, can be incorporated to promote the movement of ions (Fig. 13b, right). The key to enhancing the ionic conductivity lies in the precise adjustment of the ratio between the zwitterions, lithium salt, and PC. This balance is believed to favor the formation of cubic phases, which contribute to the development of both 1D and 3D ionic channels. As a result, the combination of these components can significantly improve the ion transport properties of the LCE, thus advancing its performance for LIB applications.
Beyond molecular design, supramolecular interactions, including hydrogen bonding and non-covalent forces, have emerged as vital mechanisms for directing the formation of nanoscale electrolyte structures. In recent years, these interactions have gained significant attention as they contribute to the stabilization and enhancement of nanomaterial assembly, further optimizing ionic conductivity. Additionally, some LC molecules feature a third functional component-a polymerizable group-typically located at the molecular terminus. This polymerizable moiety enables in situ photopolymerization under UV irradiation, thereby solidifying the electrolyte structure while preserving ion-transport pathways. This strategy holds significant promise for achieving superior ionic conductivity in solid and semi-solid electrolytes.
LC molecules can be classified into ionic and nonionic types based on their molecular characteristics, which often adopt rod-like, fan-like, or discotic shapes. Beyond their primary function of enhancing ionic conductivity, rationally designed LCEs can also contribute to improved thermal stability, redox reversibility, and the expansion of the electrochemical stability window. As a result, the integration of these advanced LCEs into LIBs is expected to significantly elevate overall electrochemical performance, making them promising candidates for next-generation energy storage applications.
The unique anchoring properties of LC molecules make them highly suitable for applications in electrolytes (Fig. 14).9–13 LIBs are a promising energy storage technology, and lithium metal has emerged as an ideal anode material due to its exceptionally high specific capacity-more than ten times greater than that of commercial graphite anodes.217 However, the widespread adoption of lithium metal anodes is hindered by severe challenges, particularly the uncontrolled growth of lithium dendrites and the consequent loss of coulombic efficiency caused by non-uniform lithium electrodeposition.218–221 Various approaches have been explored to suppress dendritic growth, including the use of artificial solid electrolyte interphases, nanostructured surface modifications, electrolyte additives, and solid-state polymer or inorganic electrolytes.222–227 A particularly promising strategy involves leveraging mechanical stresses at the electrolyte/electrode interface, such as employing low-shear-modulus ceramic electrolytes or high-shear-modulus polymer electrolytes.228–231 However, achieving an optimal balance between softness and mechanical rigidity remains a critical challenge, as conventional polymer-based and ceramic-based electrolytes often fail to provide sufficient mechanical stability.232–236
 | ||
| Fig. 14 Schematic of an interface between Li anode and LCE. n(r) is the director field of the LC. The LC molecules (size exaggerated) orient along the surface of the electrode due to anchoring energy.9–13 | ||
A notable feature of LCEs is their exceptional ability to suppress dendrite growth, driven by their unique self-organization and anchoring effects, which make them particularly suited for LIB applications.237,238 Unlike traditional approaches, LCEs introduce an additional energy contribution derived from the anchoring and distortion properties of LC molecules.239,240 The reorientation and self-alignment tendencies of anisotropic LC molecules create an ordered electrolyte structure that resists dendrite formation. Compared to other dendrite inhibition strategies, LCE-based systems simplify material synthesis, facilitate integration into battery architectures, and function without the need for external mechanical constraints, such as applied pressure. This makes LCEs a highly attractive electrolyte candidate for LIBs. Additionally, the anchoring characteristics of LC molecules-already extensively studied in optoelectronic applications such as LC displays, molecular electronics, and lithography: are now being explored for their potential impact on electrochemical systems. The alignment of LC molecules on solid surfaces has been well-characterized, with research focusing on factors such as surface treatment, molecular orientation control, and the effects of interfacial interactions.
Recent advances have demonstrated that Li-containing LCEs offer a combination of high ionic conductivity, organized ion transport pathways (1D, 2D, or 3D), and enhanced electrochemical performance. These materials exhibit desirable properties such as a high lithium transference number, low flammability, cost-effective manufacturing, and stability over a broad temperature range. Compared with amorphous electrolytes, crystalline LCEs facilitate faster ionic transport due to their well-defined conduction channels. Computational studies, including phase-field simulations and density functional theory (DFT) calculations, have provided further insight into how LCEs influence electrodeposition. Ahmad et al. modeled metal electrodeposition in the presence of LCEs, incorporating bulk distortion and anchoring energy effects into their kinetic framework.241 Their findings indicate that LCEs with sufficient anchoring strength at the metal anode interface can promote uniform lithium deposition and significantly mitigate dendrite growth. By establishing three quantitative metrics related to interface shape, growth behavior, and location evolution over time, they proposed fundamental design principles for LCE architectures capable of suppressing dendrites. Further, their DFT analysis identified key molecular descriptors for anchoring energy and interface stability. They determined that the adsorption strength of LC molecules on the lithium metal surface serves as a reliable predictor of anchoring strength, while the highest-occupied molecular orbital (HOMO) level provides an estimate of oxidative stability. These insights contribute to the rational design of next-generation LCEs tailored for high-performance LIBs.
Thus, LCEs present a promising new avenue for mitigating lithium dendrite growth. The mechanism by which LCs inhibit dendrite formation is primarily driven by the inherent twisting and anchoring properties of the liquid crystal molecules. These anisotropic molecules have a natural tendency to rearrange and reorient, which results in the generation of additional energy within the electrolyte. This energy plays a key role in impeding the uncontrolled growth of lithium dendrites. Compared to traditional dendrite suppression techniques, this approach offers several advantages, including simplified material synthesis, easier fabrication, and more seamless integration of components within the battery system. One of the standout features of LC materials is their ability to spontaneously suppress dendrite formation without the need for external forces, such as applied pressure or mechanical constraints. This characteristic positions LCs as promising candidates for use as additives in electrolytes, further advancing the potential for stable and efficient LIBs. Overall, the design rules for LCEs in LIBs focus on molecular engineering and structural control to optimize ion transport and stability:
(i) Molecular architecture with dual components: LCE molecules must incorporate two distinct parts:
An ion-transporting portion composed of polar or ionic groups (e.g., cyclic carbonates like EC/PC, PEO segments) to facilitate lithium salt dissociation and ion mobility.
A non-ionic portion (e.g., mesogenic units, alkyl chains) responsible for directing the self-assembly into ordered nanostructures (columnar, smectic, cubic phases) that define the ion-conduction pathways. The interaction between these hydrophilic (ion-transport) and hydrophobic (non-ionic) components drives the formation of nanochannels.
(ii) Nanostructure dimensions and order control: the dimensionality (1D columnar, 2D smectic, 3D cubic) and degree of order within the self-assembled nanostructure critically determine ionic conductivity of Li+. Higher-dimensional structures (e.g., 3D cubic) generally offer superior conductivity due to greater channel density and interconnectivity compared to lower-dimensional ones (e.g., 1D columnar). Achieving a single-domain, ordered state (over a multidomain, disordered state) is essential for optimal performance. External stimuli like electric or magnetic fields can be applied to align the LC domains and enhance channel order, thereby boosting conductivity.
(iii) Incorporation of polymerizable groups: introducing polymerizable groups (e.g., acrylates, dienes) near the molecular termini allows for in situ photopolymerization (e.g., via UV irradiation). This immobilizes the beneficial self-assembled nanostructure within a solid or semi-solid matrix, preserving the ion-conduction pathways while improving mechanical stability and safety.
(iv) Leveraging anchoring energy: the intrinsic anchoring energy of LC molecules at the surface of lithium metal electrodes can be harnessed to promote uniform lithium electrodeposition and suppress detrimental lithium dendrite growth. This property is particularly valuable for enhancing interfacial stability with lithium metal anodes.
(v) Functional group modification: strategic modifications of molecular fragments can enhance specific properties. For example, fluorination of aromatic rings or alkyl chains is highlighted as a design strategy to potentially improve oxidative stability, thermal performance, and safety by promoting the formation of stable LiF-rich SEI layers, although its impact on ionic conductivity requires careful optimization.
5. Conclusions
5.1. Summary
In this review, we explore the research advancements and application potential of LCEs in the field of LIBs. LCEs exhibit outstanding ionic conductivity and high stability due to their self-assembled, 2D, and 3D ion transport channels, making them strong candidates for high-performance LIB electrolytes.285 The classification of LCEs is based on their ionic transport mechanisms and molecular structural types, specifically including Col, Sm, and Cub phases formed through self-assembly, as well as the distinct characteristics of ionic and non-ionic LCEs. In summary, the application of LCEs in LIBs demonstrates the following advantages:(1) High ionic conductivity: compared to traditional polymeric and inorganic electrolytes, LCEs provide effective pathways for lithium ion (Li+) transport through their self-assembled nanostructured channels.286,287 This structure not only enhances ion mobility but also improves the energy and power density of the batteries. Such characteristics are particularly beneficial for high-rate charge and discharge applications, catering to the demands for rapid charging and high energy density in modern electric vehicles and portable devices.
(2) Chemical stability: the high chemical stability of LCEs significantly mitigates the risk of short circuits caused by lithium dendrite formation. Dendrite growth is one of the primary safety concerns in LIBs, and the stability of LCEs can substantially reduce this risk, thus enhancing the safety and cycling life of the batteries. Additionally, LCEs exhibit good compatibility with electrode materials, effectively minimizing performance degradation due to electrolyte decomposition and interactions with electrode materials.
(3) Excellent cycling performance: LIBs utilizing LCEs demonstrate good charge and discharge cycling performance, indicating their reliability in practical applications. Research shows that these batteries can maintain a high capacity after hundreds of charge–discharge cycles, surpassing traditional electrolyte systems. This characteristic provides a significant advantage for LCEs in applications requiring long-term usage.
5.2. Challenges faced
Despite the promising advantages of LCEs, several significant challenges impede their practical implementation in lithium-ion batteries. Foremost among these is the inadequate ionic conductivity at room temperature; while ordered nanostructures facilitate ion transport, most reported LCE systems (e.g., polymer composites like PEO/E8) exhibit conductivities well below the commercially required threshold of 10−3 S cm−1, particularly under ambient conditions. Additionally, the high synthesis cost of LC monomers and intermediates (e.g., C6M) poses economic barriers to scalability, though large-scale production could mitigate this. Critically, current research predominantly targets liquid or quasi-solid-state systems, leaving the development of LCEs for all-solid-state batteries underexplored. Further limitations include the narrow thermal stability windows observed in thermotropic LCEs, which undergo phase transitions within restricted temperature ranges, and the inherent difficulty in achieving uniform, long-range alignment of nanochannels—a process often requiring external fields or complex processing. Moreover, comprehensive data on long-term electrochemical performance, interfacial compatibility with electrodes, and cycling stability in full-cell configurations remain scarce, highlighting unresolved practical constraints. To sum up, several challenges remain:(1) Enhancing ionic conductivity: currently, the ionic conductivity of LCEs is relatively low (approximately 10−4 S cm−1).288 A critical research direction is to optimize molecular structures, such as compounding polymers with LCs to facilitate ion transport and reduce crystallinity. This approach can improve phase compatibility and enhance ionic mobility. Enhancing ionic conductivity is vital not only for improving overall battery performance but also for expanding their applications in both low and high-temperature environments.
(2) Compatibility and performance evaluation: the compatibility of LCEs with electrode materials and the overall performance evaluation of the batteries require further in-depth researches. Different electrode materials may exert a significant influence on the performance of LCEs, necessitating more empirical studies to acquire reliable data. Furthermore, the performance of LCEs in various battery configurations still needs systematic exploration.
(3) Development of room temperature LCEs: the number of reported room temperature LCEs is currently limited. A key focus for future research will be to manipulate the molecular structure of LCEs to achieve effective LC states at room temperature. Room temperature LCEs will better meet practical application needs, enhancing battery usability and market acceptance.
(4) Challenges in commercial productions: despite the excellent performances of LCEs, their complex preparation processes and relatively high production costs pose barriers to commercialization. Developing efficient, cost-effective production processes and optimizing LCE formulations will be crucial factors in promoting the commercialization of LCEs.
5.3. Future prospects
Looking ahead, LCEs represent an emerging class of ionic transport materials, offering immense developmental potential due to their distinctive mesophase structures and the diversity of molecular designs. The prospects for LCEs in the research and applications of LIBs appears particularly promising, with several key areas deserving focused attention:(1) Future research efforts could focus on integrating LCEs with other functional materials to create multifunctional composites. For instance, combining LCEs with conductive polymers or inorganic nanomaterials could enhance their conductivities and thermal stabilities. Such multifunctional composite materials are expected to exhibit synergistic effects in terms of battery performance, energy density, and safety, thereby meeting higher application demands.
(2) With advancements in solid-state battery technology, LCEs have the potential to become crucial components in solid-state batteries.289 Solid-state batteries promise enhanced safety and energy density, and the flexibility and tunability of LCEs make them suitable for such applications.290,291 Future investigations could explore effective integration of LCEs with SSEs to further improve their overall performance.292 The integration of thermotropic LCEs with polymers represents a significant strategy for enhancing quasi-solid and solid-state lithium-ion batteries. Discotic LCs, characterized by their disc-like molecular structure and central π-conjugated electron clouds, offer distinct advantages in composite polymer electrolytes. Incorporating LCEs into polymeric matrices improves electrochemical stability and optical properties while substantially boosting ionic conductivity. This enhancement arises from the formation of efficient pathways for ionic conduction facilitated by the LCE phase. Furthermore, when ionic LCs act as inducers within poly(ionic liquid) composites, they promote nanoscale phase separation and establish a lamellar structure in the liquid crystalline state, optimizing ion transport dynamics. Supporting this approach, solution-cast polyethylene oxide (PEO)/E8 LC composite films incorporating NaIO4 have demonstrated a notable room-temperature ionic conductivity of 1.05 × 10−7 S cm−1, exemplifying the practical efficacy of such composites in solid electrolyte design.293
(3) In the context of increasing global emphasis on sustainable development, the synthesis of LCE materials derived from renewable resources emerges as an important research direction. Utilizing bio-based materials, such as natural polymers, to synthesize LCEs could not only lower production costs but also mitigate environmental impacts. Research into eco-friendly LCEs will provide novel solutions for the sustainable advancement of the battery industry.
(4) Leveraging modern computational techniques and machine learning methodologies can expedite the discovery and optimization processes of LCEs. Through high-throughput screening and data-driven design strategies, researchers can rapidly identify novel LCE materials with outstanding performance characteristics. This approach is expected to significantly enhance research efficiency and shorten material development timelines.
(5) Beyond their applications in LIBs, LCEs can be extended to other types of batteries and energy storage systems, such as sodium-ion batteries and lithium–sulfur batteries. By optimizing the designs and applications of LCEs according to the specific characteristics of different battery systems, new market opportunities can be opened within a broader range of energy storage technologies.
LCEs represent a novel class of electrolyte materials that exhibit exceptional ionic conductivities and remarkable chemical stabilities, showcasing significant application potential in LIBs. The unique mesophase structures and molecular designs of LCEs confer substantial advantages in ionic transport, effectively enhancing the energy densities and cycle lives of batteries. However, despite these promising attributes, the practical implementation of LCEs encounters several technical challenges, including compatibility with electrode materials, stability of ionic conductivity at elevated temperatures, and the complexity of manufacturing processes. Nevertheless, with ongoing advancements in materials science, chemical engineering, and computational science, the research and application of LCEs are poised to experience unprecedented opportunities. Utilizing state-of-the-art materials synthesis techniques and computational models, researchers can gain deeper insights into the behavior of LCEs and optimize their performance. Furthermore, interdisciplinary collaborations facilitate the development of more efficient synthesis routes and refined material design strategies, significantly propelling the application of LCEs in battery technologies. In the future, sustained research and development are expected to enable LCEs to play an increasingly vital role in energy storage technologies. This progress will not only contribute to the realization of more efficient battery technologies but also enhance battery safety, addressing the growing demands in sectors such as electric vehicles and renewable energy storage. The prospects for LCEs are promising, and there is an anticipation for their substantial contributions to the forthcoming energy technology revolution, thereby supporting the achievement of sustainable development goals.
Author contributions
S. J. Liu: writing – review & editing, writing – original draft, resources, conceptualization. L. Zhou: writing – review & editing, investigation, resources. Y. Zheng: writing – review & editing, investigation, funding acquisition. K. Neyts: writing – review & editing, investigation, resources.Conflicts of interest
There are no conflicts to declare.Data availability
No data was used for the research described in this article.Acknowledgements
This work was supported by the Shenzhen Lithium Battery Electrode Active Materials Engineering Laboratory Enhancement, funded by the Shenzhen Development and Reform Commission (Contract No. XMHT20240108002), the Natural Science Foundation of Fujian Province (2024J01261), the Educational Research Projects for Young and Middle-aged Teachers in Fujian Province (JZ230002), Fujian Province 100 Talents Program, Fujian Province Minjiang Scholar Program. We acknowledgement funding from the Research Grants Council (GRC) under No. 16204624 and the support of the State Key Lab (SKLADT) at HKUST.References
- X. Li, L. Zhang, H. Zhang, B. Zhang and G. Cui, Energy Storage Sci. Technol., 2020, 9, 1595–1604 Search PubMed.
- M. Wang, Y. Li, H. Yang, Z. Ding and R. Liu, J. Energy Storage, 2024, 100, 113781 CrossRef.
- X. Wang, Y. Huang, W. Ren, C. Luo, X. Xu, Y. Wang, Y. Wang, C. Zhang, Z. Zhao, L. Liu, X. Li, M. Wang and H. Cao, Chem. Eng. J., 2024, 502, 158155 CrossRef CAS.
- J. Espejo, C. O. Zellmann-Parrotta, D. Sarkar, A. Che, V. K. Michaelis, V. E. Williams and C. C. Ling, Chem. – Eur. J., 2024, 10(30), e202403232 CrossRef PubMed.
- C. P. Hemantha Rajapaksha, P. R. Paudel, P. M. S. G. Kodikara, D. Dahal, T. M. Dassanayake, V. Kaphle, B. Lüssem and A. Jákli, Appl. Phys. Rev., 2022, 9, 011415 CAS.
- T. Kato, Science, 2002, 295, 2414–2418 CrossRef CAS PubMed.
- H. Kikuchi, M. Yokota, Y. Hisakado, H. Yang and T. Kajiyama, Nat. Mater., 2002, 1, 64–68 CrossRef CAS PubMed.
- J. C. Ronda, J. A. Reina and M. Giamberini, J. Polym. Sci., Part A: Polym. Chem., 2003, 42, 326–340 CrossRef.
- S. Bao, S. Li, Z. Tang, Y. Wan, G. Zhou, Z. Yang, D. Sun, D. Ye and L. Zhu, J. Mater. Chem. C, 2025, 13, 9453–9464 RSC.
- I. Dierking, S. Yoshida, T. Kelly and W. Pitcher, Soft Matter, 2020, 16, 6021–6031 RSC.
- C. T. Imrie and P. A. Henderson, Chem. Soc. Rev., 2007, 36, 2096–2124 RSC.
- A. Ahmad, H. Dai, S. Feng, Z. Chen, Z. Mohmaed, A. A. Khan, X. Hao, Y. Wang, N. Iqbal and D. Mehvish, J. Mater. Chem. C, 2025, 13, 2606–2619 RSC.
- N. Khatun, A. C. Nkele and K. Bagchi, Phys. Chem. Chem. Phys., 2025, 27, 6408–6424 RSC.
- H. Wang, Z. Chen, Y. Yuan and H. Zhang, Soft Matter, 2022, 18, 5483–5491 RSC.
- S. Dutt, P. F. Siril and S. Remita, RSC Adv., 2017, 7, 5733–5750 RSC.
- H. K. Bisoyi and S. Kumar, Chem. Soc. Rev., 2011, 40, 306–319 RSC.
- M. Hird, Chem. Soc. Rev., 2007, 36, 2070–2095 RSC.
- H. K. Bisoyi and S. Kumar, Chem. Soc. Rev., 2010, 39, 264–285 RSC.
- F. B. Dias, S. V. Batty, J. P. Voss, G. Ungar and P. V. Wright, Solid State Ionics, 1996, 85, 43–49 CrossRef CAS.
- R. L. Kerr, S. A. Miller, R. K. Shoemaker, B. J. Elliott and D. L. Gin, J. Am. Chem. Soc., 2009, 131, 15972–15973 CrossRef CAS PubMed.
- Q. Ruan, M. Yao, D. Yuan, H. Dong, J. Liu, X. Yuan, W. Fang, G. Zhao and H. Zhang, Nano Energy, 2023, 106, 108087 CrossRef CAS.
- K. Lu, H. Ruan, S. Meng, Q. Zhao, J. Ji, Y. Wu, C. Wang and S. Tan, Chem. Eng. J., 2023, 475, 146418 CrossRef CAS.
- M. Yoshio, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2004, 126, 994–995 CrossRef CAS PubMed.
- H. Shimura, M. Yoshio, A. Hamasaki, T. Mukai, H. Ohno and T. Kato, Adv. Mater., 2009, 21, 1591–1594 CrossRef CAS.
- J. Sakuda, E. Hosono, M. Yoshio, T. Ichikawa, T. Matsumoto, H. Ohno, H. Zhou and T. Kato, Adv. Funct. Mater., 2015, 25, 1206–1212 CrossRef CAS.
- F. Xu, K. Matsumoto and R. Hagiwara, J. Phys. Chem. B, 2012, 116, 10106–10112 CrossRef CAS PubMed.
- F. B. Dias, S. V. Batty, A. Gupta, G. Ungar, J. P. Voss and P. V. Wright, Electrochim. Acta, 1998, 43, 1217–1224 CrossRef CAS.
- Y. Wang, C. J. Zanelotti, X. Wang, R. Kerr, L. Jin, W. H. Kan, T. J. Dingemans, M. Forsyth and L. A. Madsen, Nat. Mater., 2021, 20, 1255–1263 CrossRef CAS PubMed.
- C. T. Imrie, R. T. Inkster, Z. Lu and M. D. Ingram, Mol. Cryst. Liq. Cryst., 2004, 408, 33–43 CrossRef CAS.
- M. Yao, H. Zhang, K. Dong, B. Li, C. Xing, M. Dang and S. Zhang, J. Mater. Chem. A, 2021, 9, 6232–6241 RSC.
- M. Yoshio, T. Kagata, K. Hoshino, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2006, 128, 5570–5577 CrossRef CAS PubMed.
- S. Wang, Q. Zeng, A. Wang, X. Liu, J. Chen, Z. Wang and L. Zhang, J. Mater. Chem. A, 2019, 7, 1069–1075 RSC.
- Q. Zeng, Y. Lu, P. Chen, Z. Li, X. Wen, W. Wen, Y. Liu, S. Zhang, H. Zhao, H. Zhou, Z.-x. Wang and L. Zhang, J. Energy Chem., 2022, 67, 157–167 CrossRef CAS.
- R. Sasi, B. Chandrasekhar, N. Kalaiselvi and S. J. Devaki, Adv. Sustainable Syst., 2017, 1, 1600031 CrossRef.
- M. Gopinadhan, P. W. Majewski and C. O. Osuji, Macromolecules, 2010, 43, 3286–3293 CrossRef CAS.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 1800561, DOI:10.1002/adma.201800561.
- K. Takada, Solid State Ionics, 2004, 172, 25–30 CrossRef CAS.
- Q. Zeng, Y. Liu, B. Wujieti, Z. Li, A. Chen, J. Guan, H. Wang, Y. Jiang, H. Zhou, W. Cui, S. Wang and L. Zhang, Chem. Eng. J., 2024, 486, 150298 CrossRef CAS.
- K. S. Ngai, S. Ramesh, K. Ramesh and J. C. Juan, Ionics, 2016, 22, 1259–1279 CrossRef CAS.
- M. Park, X. Zhang, M. Chung, G. B. Less and A. M. Sastry, J. Power Sources, 2010, 195, 7904–7929 CrossRef CAS.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 RSC.
- X. Wang, Z. He, R. Yan, H. Niu, W. He and Z. Miao, Chem. Eng. J., 2025, 503, 158552 CrossRef CAS.
- D. Gopalakrishnan, S. Alkatie, A. Cannon, S. Rajendran, N. K. Thangavel, N. Bhagirath, E. M. Ryan and L. M. R. Arava, Sustainable Energy Fuels, 2021, 5, 1488–1497 RSC.
- F. Yuan, L. Yang, X. Zou, S. Dong, S. Chi, J. Xie, H. Xing, L. Bian, L. Bao and J. Wang, J. Power Sources, 2019, 444, 227305 CrossRef CAS.
- S. Rana, R. C. Thakur and H. S. Dosanjh, Solid State Ionics, 2023, 400, 116340 CrossRef CAS.
- H. Ruan, K. Lu, S. Meng, Q. Zhao, H. Ren, Y. Wu, C. Wang and S. Tan, Small, 2024, 20, e2310186 CrossRef PubMed.
- S. Wang, X. Liu, A. Wang, Z. Wang, J. Chen, Q. Zeng, X. Jiang, H. Zhou and L. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 25273–25284 CrossRef CAS PubMed.
- Y. Li, X. Zheng, E. Z. Carlson, X. Xiao, X. Chi, Y. Cui, L. C. Greenburg, G. Zhang, E. Zhang, C. Liu, Y. Yang, M. S. Kim, G. Feng, P. Zhang, H. Su, X. Guan, J. Zhou, Y. Wu, Z. Xue, W. Li, M. Bajdich and Y. Cui, Nat. Energy, 2024, 9, 1350–1359 CrossRef CAS.
- Q. Li, Q. Tang, N. Du, Y. Qin, J. Xiao, B. He, H. Chen and L. Chu, J. Power Sources, 2014, 248, 816–821 CrossRef CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287 RSC.
- Z. Stoeva, Z. Lu, M. D. Ingram and C. T. Imrie, Electrochim. Acta, 2013, 93, 279–286 CrossRef CAS.
- Y. Domi, H. Usui, Y. Shindo, S. Yodoya, H. Sato, K. Nishikawa and H. Sakaguchi, Electrochemistry, 2020, 88, 548–554 CrossRef CAS.
- Y. Tong, L. Chen, Y. Chen and X. He, Appl. Surf. Sci., 2012, 258, 10095–10103 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- C. Liu, S. Cao and M. Yoshio, Adv. Funct. Mater., 2023, 33, 2300538 CrossRef CAS.
- T. Ohtake, M. Ogasawara, K. Ito-Akita, N. Nishina, S. Ujiie, H. Ohno and T. Kato, Chem. Mater., 2000, 12, 782–789 CrossRef CAS.
- L. Zhou, S. Liu and T. Zhong, Nanoscale, 2023, 15, 1484–1492 RSC.
- N. Sundaresan, T. Thomas, T. J. Thomas and C. K. S. Pillai, Macromol. Biosci., 2005, 6, 27–32 CrossRef PubMed.
- F. Yuan, S. Chi, S. Dong, X. Zou, S. Lv, L. Bao and J. Wang, Electrochim. Acta, 2019, 294, 249–259 CrossRef CAS.
- S. K. Park, K. S. Han, J. H. Lee, V. Murugesan, S. H. Lee, C. M. Koo, J. S. Lee and K. T. Mueller, J. Phys. Chem. C, 2019, 123, 20547–20557 CrossRef CAS.
- T. Onuma, E. Hosono, M. Takenouchi, J. Sakuda, S. Kajiyama, M. Yoshio and T. Kato, ACS Omega, 2018, 3, 159–166 CrossRef CAS PubMed.
- V. Molahalli, P. Hirankittiwong, A. Sharma, H. Laeim, A. Shetty, N. Chattham and G. Hegde, Mater. Sci. Eng., B, 2024, 305, 117369 CrossRef CAS.
- Y. Luo, N. Marets and T. Kato, Chem. Sci., 2018, 9, 608–616 RSC.
- Y. Tong, L. Chen, X. He and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4341–4350 CrossRef CAS.
- Y. Xie, W. Zhang, J. Gao, Z. Yao, Y. Yan, X. Zou, G. Yang, Y.-M. Zhang and S. X.-A. Zhang, Chem. Eng. J., 2025, 504, 158818 CrossRef CAS.
- J. C. Y. Lin, C.-J. Huang, Y.-T. Lee, K.-M. Lee and I. J. B. Lin, J. Mater. Chem., 2011, 21, 8110 RSC.
- P. Prathumrat, M. Okhawilai, C. Likitaporn and H. Uyama, Mater. Res. Bull., 2025, 181, 113084 CrossRef CAS.
- K. T. Nam, D.-W. Kim, P. J. Yoo, C.-Y. Chiang, N. Meethong, P. T. Hammond, Y.-M. Chiang and A. M. Belcher, Science, 2006, 312, 885–888 CrossRef CAS PubMed.
- J. Zhao, L. Zhang and J. Hu, Adv. Intell. Syst., 2021, 4, 2100065 CrossRef.
- X. Zhang, Y. Xu, C. Valenzuela, X. Zhang, L. Wang, W. Feng and Q. Li, Light: Sci. Appl., 2022, 11, 223 CrossRef CAS PubMed.
- D. Wang, Y.-L. Li, F. Chu, N.-N. Li, Z.-S. Li, S.-D. Lee, Z.-Q. Nie, C. Liu and Q.-H. Wang, Light: Sci. Appl., 2024, 13, 16 CrossRef CAS PubMed.
- Y. Yang, C. Liu, Y. Ding, B. Ding, J. Xu, A. Liu, J. Yu, L. Grater, H. Zhu, S. S. Hadke, V. K. Sangwan, A. S. R. Bati, X. Hu, J. Li, S. M. Park, M. C. Hersam, B. Chen, M. K. Nazeeruddin, M. G. Kanatzidis and E. H. Sargent, Nat. Energy, 2024, 9, 316–323 CrossRef CAS.
- R. Zhang, Z. Zhang, J. Han, L. Yang, J. Li, Z. Song, T. Wang and J. Zhu, Light: Sci. Appl., 2023, 12, 2047–7538 Search PubMed.
- J. P. F. Lagerwall, C. Schütz, M. Salajkova, J. Noh, J. Hyun Park, G. Scalia and L. Bergström, NPG Asia Mater., 2014, 6, e80–e80 CrossRef CAS.
- W. Feng, Q. He and L. Zhang, Adv. Mater., 2024, 37, 2312313 CrossRef PubMed.
- A. Rešetič, Commun. Chem., 2024, 7, 56 CrossRef PubMed.
- S. J. Woltman, G. D. Jay and G. P. Crawford, Nat. Mater., 2007, 6, 929–938 CrossRef CAS PubMed.
- B. X. Dong, Z. Liu, M. Misra, J. Strzalka, J. Niklas, O. G. Poluektov, F. A. Escobedo, C. K. Ober, P. F. Nealey and S. N. Patel, ACS Nano, 2019, 13, 7665–7675 CrossRef CAS PubMed.
- Z. Zhang, Z. You and D. Chu, Light: Sci. Appl., 2014, 3, e213–e213 CrossRef.
- A. Yoshizawa, Polym. J., 2012, 44, 490–502 CrossRef CAS.
- R. Rondla, J. C. Y. Lin, C. T. Yang and I. J. B. Lin, Langmuir, 2013, 29, 11779–11785 CrossRef CAS PubMed.
- T. Ichikawa, M. Yoshio, A. Hamasaki, J. Kagimoto, H. Ohno and T. Kato, J. Am. Chem. Soc., 2011, 133, 2163–2169 CrossRef CAS PubMed.
- S. Yazaki, M. Funahashi, J. Kagimoto, H. Ohno and T. Kato, J. Am. Chem. Soc., 2010, 132, 7702–7708 CrossRef CAS PubMed.
- B. Soberats, M. Yoshio, T. Ichikawa, S. Taguchi, H. Ohno and T. Kato, J. Am. Chem. Soc., 2013, 135, 15286–15289 CrossRef CAS PubMed.
- K. Goossens, K. Lava, C. W. Bielawski and K. Binnemans, Chem. Rev., 2016, 116, 4643–4807 CrossRef CAS PubMed.
- Y. Cai, T. Xu, X. Meng, N. von Solms, H. Zhang and K. Thomsen, Electrochim. Acta, 2022, 424, 140679 CrossRef CAS.
- A. Eisele, K. Kyriakos, R. Bhandary, M. Schönhoff, C. M. Papadakis and B. Rieger, J. Mater. Chem. A, 2015, 3, 2942–2953 RSC.
- P. Judeinstein and F. Roussel, Adv. Mater., 2005, 17, 723–727 CrossRef CAS.
- T. Ohtake, Y. Takamitsu, K. Ito-Akita, K. Kanie, M. Yoshizawa, T. Mukai, H. Ohno and T. Kato, Macromolecules, 2000, 33, 8109–8111 CrossRef CAS.
- A. Yamashita, M. Yoshio, B. Soberats, H. Ohno and T. Kato, J. Mater. Chem. A, 2015, 3, 22656–22662 RSC.
- A. Kuwabara, M. Enomoto, E. Hosono, K. Hamaguchi, T. Onuma, S. Kajiyama and T. Kato, Chem. Sci., 2020, 11, 10631–10637 RSC.
- S. Qin, Y. Yu, J. Zhang, Y. Ren, C. Sun, S. Zhang, L. Zhang, W. Hu, H. Yang and D. Yang, Adv. Energy Mater., 2023, 13, 2301470 CrossRef CAS.
- M. Chen, W. Wang, X. Liang, S. Gong, J. Liu, Q. Wang, S. Guo and H. Yang, Adv. Energy Mater., 2018, 8, 1800171 CrossRef.
- M. Chen, Y. Cao, C. Ma and H. Yang, Nano Energy, 2021, 81, 105640 CrossRef CAS.
- M. Wang, H. Zhang, Y. Li, R. Liu and H. Yang, Chem. Eng. J., 2023, 476, 146658 CrossRef CAS.
- K. Wang, H. Ruan, Q. Yu, Q. Zhao, Y. Wu, C. Wang and S. Tan, J. Power Sources, 2025, 632, 236294 CrossRef CAS.
- P. Prathumrat, M. Okhawilai, C. Likitaporn and H. Uyama, Mater. Res. Bull., 2025, 181, 113084 CrossRef CAS.
- E. Jónsson, Energy Storage Mater., 2020, 25, 827–835 CrossRef.
- S.-M. Hao, S. Liang, C. D. Sewell, Z. Li, C. Zhu, J. Xu and Z. Lin, Nano Lett., 2021, 21, 7435–7447 CrossRef CAS PubMed.
- Z. Siddiquee, H. Lee, W. Xu, T. Kyu and A. Jákli, Batteries, 2025, 11, 106 CrossRef CAS.
- X. Bai, F. Xie, Z. Zhang, M. Cao, Q. Wang, S. Wang, C. Liu, X. Su, Z. Lin and G. Cui, Adv. Energy Mater., 2024, 14, 2401336 CrossRef CAS.
- W. Wu, Y. Chu, T. Zhang, T. He and J. Hao, Small, 2023, 20, 2305463 CrossRef PubMed.
- X. Miao, C. Song, W. Hu, Y. Ren, Y. Shen and C. w. Nan, Adv. Mater., 2024, 36, 2401473 CrossRef CAS PubMed.
- J. Dong, S. Nimori, K. Kawabata and H. Goto, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1894–1899 CrossRef CAS.
- Z. Liu, B. X. Dong, M. Misra, Y. Sun, J. Strzalka, S. N. Patel, F. A. Escobedo, P. F. Nealey and C. K. Ober, Adv. Funct. Mater., 2018, 29, 1805220 CrossRef.
- Y. Ye, L. Guo and T. Zhong, Polymers, 2023, 15, 2962 CrossRef CAS PubMed.
- I. S. Klein, Z. Zhao, S. K. Davidowski, J. L. Yarger and C. A. Angell, Adv. Energy Mater., 2018, 8, 1801324 CrossRef.
- X. Li, Z. Zhao, J. Zhou, C. Wang, Y. Wu and S. Tan, Chem. Eng. Sci., 2020, 221, 115710 CrossRef CAS.
- S. Zhu, Y. Su, Y. Liu, X. Niu, W. Zhang and W. Chen, Opt. Mater., 2024, 149, 115007 CrossRef CAS.
- M. S. Khan, A. Van Roekeghem, S. Mossa, F. Ivol, L. Bernard, L. Picard and N. Mingo, Phys. Chem. Chem. Phys., 2024, 26, 4338–4348 RSC.
- K. Shikinaka, N. Taki, K. Kaneda and Y. Tominaga, Chem. Commun., 2017, 53, 613–616 RSC.
- S. Meng, H. Ruan, Q. Yu, Q. Zhao, C. Wang, Y. Wu and S. Tan, J. Power Sources, 2025, 628, 235866 CrossRef CAS.
- S. Paladugu, C. Conklin, J. Viñals and O. D. Lavrentovich, Phys. Rev. Appl., 2017, 7, 034033 CrossRef.
- T. J. White and D. J. Broer, Nat. Mater., 2015, 14, 1087–1098 CrossRef CAS PubMed.
- P. Chen, B. Y. Wei, W. Hu and Y. Q. Lu, Adv. Mater., 2019, 32, 1903665 CrossRef PubMed.
- V. Nazarenko, M. V. Kurik, G. V. Klimusheva, Z. Y. Gotra, V. M. Sorokin and L. M. Lisetski, J. Mol. Liq., 2018, 267, 29–33 CrossRef CAS.
- L. Wang, A. M. Urbas and Q. Li, Adv. Mater., 2018, 32, 1801335 CrossRef PubMed.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2021, 122, 4887–4926 CrossRef PubMed.
- J. Xiong and S.-T. Wu, eLight, 2021, 1, 3 CrossRef.
- M. Pilz da Cunha, M. G. Debije and A. P. H. J. Schenning, Chem. Soc. Rev., 2020, 49, 6568–6578 RSC.
- J. Uchida, B. Soberats, M. Gupta and T. Kato, Adv. Mater., 2022, 34, 2109063 CrossRef CAS PubMed.
- K. Salikolimi, A. A. Sudhakar and Y. Ishida, Langmuir, 2020, 36, 11702–11731 CrossRef CAS PubMed.
- T. Paul and J. Saha, Sci. Rep., 2020, 10, 16016 CrossRef CAS PubMed.
- I. Bala, J. T. Plank, B. Balamut, D. Henry, A. R. Lippert and I. Aprahamian, Nat. Chem., 2024, 16, 2084–2090 CrossRef CAS PubMed.
- F. Castles, S. M. Morris, J. M. C. Hung, M. M. Qasim, A. D. Wright, S. Nosheen, S. S. Choi, B. I. Outram, S. J. Elston, C. Burgess, L. Hill, T. D. Wilkinson and H. J. Coles, Nat. Mater., 2014, 13, 817–821 CrossRef CAS PubMed.
- T.-H. Lin, D.-Y. Guo, C.-W. Chen, T.-M. Feng, W.-X. Zeng, P.-C. Chen, L.-Y. Wu, W.-M. Guo, L.-M. Chang, H.-C. Jau, C.-T. Wang, T. J. Bunning and I. C. Khoo, Nat. Commun., 2024, 15, 7038 CrossRef CAS PubMed.
- W. Cao, A. Muñoz, P. Palffy-Muhoray and B. Taheri, Nat. Mater., 2002, 1, 111–113 CrossRef CAS PubMed.
- W. Hu, L. Wang, M. Wang, T. Zhong, Q. Wang, L. Zhang, F. Chen, K. Li, Z. Miao, D. Yang and H. Yang, Nat. Commun., 2021, 12, 1440 CrossRef CAS PubMed.
- Y. Yang, L. Wang, H. Yang and Q. Li, Small Sci., 2021, 1, 459–464 Search PubMed.
- K. R. Schlafmann and T. J. White, Nat. Commun., 2021, 12, 4916 CrossRef CAS PubMed.
- M. Wang, H. Yang, X. Li, X. Zhang and H. Yang, Adv. Opt. Mater., 2025, 13, 2402581 CrossRef CAS.
- A. Yoshizawa, RSC Adv., 2013, 3, 25475 RSC.
- D. C. Wright and N. D. Mermin, Rev. Mod. Phys., 1989, 61, 385–432 CrossRef CAS.
- G. Chen, H. Pei, X. Zhang, W. Shi, M. Liu, C. F. J. Faul, B. Yang, Y. Zhao, K. Liu, Z. Lu, Z. Nie and Y. Yang, Nat. Commun., 2022, 13, 5549 CrossRef CAS PubMed.
- J. A. Martínez-González, Y. Zhou, M. Rahimi, E. Bukusoglu, N. L. Abbott and J. J. de Pablo, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13195–13200 CrossRef PubMed.
- R. Kádár, S. Spirk and T. Nypelö, ACS Nano, 2021, 15, 7931–7945 CrossRef PubMed.
- T. Kato, J. Uchida, T. Ichikawa and T. Sakamoto, Angew. Chem., Int. Ed., 2018, 57, 4355–4371 CrossRef CAS PubMed.
- M. Yoshio, C. H. Wu and C. Liu, Adv. Funct. Mater., 2024, 34, 2314087 CrossRef CAS.
- T. Kato, T. Yasuda, Y. Kamikawa and M. Yoshio, Chem. Commun., 2009, 729, 10.1039/b816624b.
- J. W. Goodby, R. J. Mandle, E. J. Davis, T. Zhong and S. J. Cowling, Liq. Cryst., 2015, 42, 593–622 CrossRef CAS.
- V. Percec and D. Sahoo, Giant, 2022, 12, 100127 CrossRef CAS.
- N. Maringa, J. Lenoble, B. Donnio, D. Guillon and R. Deschenaux, J. Mater. Chem., 2008, 18, 1524 RSC.
- D. T. Do and A. R. Schmitzer, ChemistrySelect, 2016, 1, 2448–2453 CrossRef CAS.
- M. Masuda, T. Kobayashi, M. Kohri and K. Kishikawa, Mater. Lett., 2022, 307, 131055 CrossRef CAS.
- A. B. G. M. Leferink op Reinink, E. van den Pol, D. V. Byelov, A. V. Petukhov and G. J. Vroege, J. Phys.: Condens. Matter, 2012, 24, 464127 CrossRef CAS PubMed.
- M. G. Reddy, N. P. Lobo, A. Roy, K. V. Ramanathan and T. Narasimhaswamy, Phys. Chem. Chem. Phys., 2020, 22, 23986–23997 RSC.
- X. J. Ma, Y. L. Yang, K. Deng, Q. D. Zeng, C. Wang, K. Q. Zhao, P. Hu and B. Q. Wang, ChemPhysChem, 2007, 8, 2615–2620 CrossRef CAS PubMed.
- S. R. McLaren, D. J. Tate, O. R. Lozman, G. H. Mehl and R. J. Bushby, J. Mater. Chem. C, 2015, 3, 5754–5763 RSC.
- J. Chen, Y. Sun, W. Zhao, J. Liu, J. Fang, T. Xu and D. Chen, J. Mater. Chem. C, 2021, 9, 3871–3881 RSC.
- C.-M. Young, C. L. Chang, Y.-H. Chen, C.-Y. Chen, Y.-F. Chang and H.-L. Chen, Soft Matter, 2021, 17, 397–409 RSC.
- S. i. Kawano, M. Kato, S. Soumiya, M. Nakaya, J. Onoe and K. Tanaka, Angew. Chem., Int. Ed., 2017, 57, 167–171 CrossRef PubMed.
- X. Li, B. Li, L. Chen, J. Hu, C. Wen, Q. Zheng, L. Wu, H. Zeng, B. Gong and L. Yuan, Angew. Chem., Int. Ed., 2015, 54, 11147–11152 CrossRef CAS PubMed.
- R. K. Gupta, S. K. Pathak, J. De, S. K. Pal and A. S. Achalkumar, J. Mater. Chem. C, 2018, 6, 1844–1852 RSC.
- P. Kumar, D. S. Shankar Rao, S. Krishna Prasad and N. Jayaraman, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3665–3678 CrossRef CAS.
- N. Canilho, E. Kasëmi, A. D. Schlüter, J. Ruokolainen and R. Mezzenga, Macromol. Symp., 2008, 270, 58–64 CrossRef CAS.
- C. Wang, X. Li, J. Zhou, W. Tian, J. Ji, S. Tan and Y. Wu, Nanoscale, 2020, 12, 22202–22209 RSC.
- D. Yuan, X. Li, H. Yao, Y. Li, X. Zhu, J. Zhao, H. Zhang, Y. Zhang, E. T. J. Jie, Y. Cai and M. Srinivasan, Adv. Sci., 2023, 10, e2206469 CrossRef PubMed.
- R. L. Kerr, J. P. Edwards, S. C. Jones, B. J. Elliott and D. L. Gin, Polym. J., 2016, 48, 635–643 CrossRef CAS.
- A. S. Shaplov, R. Marcilla and D. Mecerreyes, Electrochim. Acta, 2015, 175, 18–34 CrossRef CAS.
- X. Tang, S. Lv, K. Jiang, G. Zhou and X. Liu, J. Power Sources, 2022, 542, 231792 CrossRef CAS.
- H. J. Xie, B. Gélinas and D. Rochefort, Electrochem. Commun., 2016, 66, 42–45 CrossRef CAS.
- T. Kato, Angew. Chem., Int. Ed., 2010, 49, 7847–7848 CrossRef CAS PubMed.
- Q. X. Shi, Q. Xia, X. Xiang, Y. S. Ye, H. Y. Peng, Z. G. Xue, X. L. Xie and Y. W. Mai, Chem. – Eur. J., 2017, 23, 11881–11890 CrossRef CAS PubMed.
- K. Sato, T. Yamasaki, T. Mizuma, K. Oyaizu and H. Nishide, J. Mater. Chem. A, 2016, 4, 3249–3252 RSC.
- S. M. Said, A. Z. S. Zulkifli, M. A. Kamarudin, A. Mainal, B. Subramanian and N. S. Mohamed, Eur. Polym. J., 2015, 66, 266–272 CrossRef CAS.
- C.-Y. Liu, C.-F. Yen, Y.-H. Hung, C.-M. Tu, G.-Y. Wu and H.-Y. Chen, J. Mater. Chem. C, 2021, 9, 16672–16681 RSC.
- B. Soberats, M. Yoshio, T. Ichikawa, X. Zeng, H. Ohno, G. Ungar and T. Kato, J. Am. Chem. Soc., 2015, 137, 13212–13215 CrossRef CAS PubMed.
- B. H. Tan, M. Yoshio and T. Kato, Chem. – Asian J., 2008, 3, 534–541 CrossRef CAS PubMed.
- M. Yoshio, T. Ichikawa, H. Shimura, T. Kagata, A. Hamasaki, T. Mukai, H. Ohno and T. Kato, Bull. Chem. Soc. Jpn., 2007, 80, 1836–1841 CrossRef CAS.
- S. Yazaki, M. Funahashi, J. Kagimoto, H. Ohno and T. Kato, J. Am. Chem. Soc., 2010, 132, 7702–7708 CrossRef CAS PubMed.
- W. Ren, Z. Hou, L. Su, X. Gao, Y. Chu, L. Zheng and F. Lu, Chem. Eng. J., 2024, 501, 157794 CrossRef CAS.
- N. Zhao, Y. Liu, X. Zhao and H. Song, Nanoscale, 2016, 8, 1545–1554 RSC.
- S. Wang, X. Liu, A. Wang, Z. Wang, J. Chen, Q. Zeng, X. Wang and L. Zhang, Polym. Chem., 2018, 9, 4674–4682 RSC.
- C. Wang, Y. Sun, Z. Wang, W. Chen, D. Liu, J. Strzalka, S. N. Patel, P. F. Nealey, C. K. Ober and F. A. Escobedo, Adv. Funct. Mater., 2025, 35, 2423541 CrossRef CAS.
- Z. Wang, C. Wang, Y. Sun, K. Wang, J. W. Strzalka, S. N. Patel, P. F. Nealey, C. K. Ober and F. A. Escobedo, ACS Nano, 2022, 16, 20714–20729 CrossRef CAS PubMed.
- J. Sakuda, M. Yoshio, T. Ichikawa, H. Ohno and T. Kato, New J. Chem., 2015, 39, 4471–4477 RSC.
- Y. Tong, L. Chen, X. He and Y. Chen, J. Power Sources, 2014, 247, 786–793 CrossRef CAS.
- S. Wang, L. Zhang, J. Li, Q. Zeng, X. Liu, P. Chen, W.-Y. Lai, T. Zhao and L. Zhang, Electrochim. Acta, 2019, 320, 134560 CrossRef CAS.
- J. Lugger, D. Mulder, R. Sijbesma and A. Schenning, Materials, 2018, 11, 104 CrossRef PubMed.
- S. Wang, A. Wang, C. Yang, R. Gao, X. Liu, J. Chen, Z. Wang, Q. Zeng, X. Liu, H. Zhou and L. Zhang, J. Power Sources, 2018, 395, 137–147 CrossRef CAS.
- X. Chen, Y. Xie, Y. Ling, J. Zhao, Y. Xu, Y. Tong, S. Li and Y. Wang, Mater. Des., 2020, 192, 108760 CrossRef CAS.
- T. Stettner and A. Balducci, Energy Storage Mater., 2021, 40, 402–414 CrossRef.
- D. Suzuki, Y. Nonoguchi, Y. Kuwahara, T. Saito and N. Terasaki, Appl. Phys. Express, 2024, 17, 071002 CrossRef CAS.
- F. C. Tavares, C. M. Cholant, E. C. Kohlrausch, G. R. Bolzan, P. F. B. Gonçalves, E. S. Gil, S. Khan, J. Dupont, C. O. Avellaneda and M. J. Leite Santos, J. Electrochem. Soc., 2023, 170, 084501 CrossRef CAS.
- J. Zhang, Q. Wang and Z. Cao, Chin. Phys. B, 2020, 29, 087804 CrossRef CAS.
- S. Qin, Z. Wang, Y. Ren, Y. Yu, Y. Xiao, J. Chen, J. Zhang, S. Zhang, C. Sun, J. Xiao, L. Zhang, W. Hu and H. Yang, Nano Energy, 2024, 119, 109075 CrossRef CAS.
- D. Bresser, M. Leclere, L. Bernard, P. Rannou, H. Mendil-Jakani, G. T. Kim, T. Zinkevich, S. Indris, G. Gebel, S. Lyonnard and L. Picard, ChemSusChem, 2020, 14, 655–661 CrossRef PubMed.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- L. Zhou, S. Liu, X. Miao, P. Xie, N. Sun, Z. Xu, T. Zhong, L. Zhang and Y. Shen, ACS Mater. Lett., 2023, 5, 2760–2775 CrossRef CAS.
- S. Qin, Y. Cao, J. Zhang, Y. Ren, C. Sun, S. Zhang, L. Zhang, W. Hu, M. Yu and H. Yang, Carbon Energy, 2023, 5, e316 CrossRef CAS.
- B. Soberats, M. Yoshio, T. Ichikawa, H. Ohno and T. Kato, J. Mater. Chem. A, 2015, 3, 11232–11238 RSC.
- S. Jeong, W. Y. Kang, C. K. Song and J. S. Park, Dyes Pigm., 2012, 93, 1544–1548 CrossRef CAS.
- K. Goossens, K. Lava, P. Nockemann, K. Van Hecke, L. Van Meervelt, P. Pattison, K. Binnemans and T. Cardinaels, Langmuir, 2009, 25, 5881–5897 CrossRef CAS PubMed.
- T. Mihelj, J. Popović, Z. Skoko and V. Tomašić, Thermochim. Acta, 2014, 591, 119–129 CrossRef CAS.
- T. Ichikawa, M. Yoshio, A. Hamasaki, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2007, 129, 10662–10663 CrossRef CAS PubMed.
- R. T. Wang, G. H. Lee and C. K. Lai, J. Mater. Chem. C, 2018, 6, 9430–9444 RSC.
- Y. Ren, W. H. Kan, M. A. Henderson, P. G. Bomben and C. P. Berlinguette, J. Am. Chem. Soc., 2011, 133, 17014–17026 CrossRef CAS PubMed.
- L. M. Antill, M. M. Neidhardt, J. Kirres, S. Beardsworth, M. Mansueto, A. Baro and S. Laschat, Liq. Cryst., 2014, 41, 976–985 CrossRef CAS.
- Y. Zhao, X. Yue, X. Wang and X. Chen, J. Colloid Interface Sci., 2013, 389, 199–205 CrossRef CAS PubMed.
- T. Matsumoto, T. Ichikawa, J. Sakuda, T. Kato and H. Ohno, Bull. Chem. Soc. Jpn., 2014, 87, 792–796 CrossRef CAS.
- A. Pa
![[n with combining breve]](https://www.rsc.org/images/entities/char_006e_0306.gif) a, F. L. Badea, M. Ilis, T. Staicu, M. Micutz, I. Pasuk and V. Cîrcu, J. Mol. Struct., 2015, 1083, 245–251 CrossRef.
a, F. L. Badea, M. Ilis, T. Staicu, M. Micutz, I. Pasuk and V. Cîrcu, J. Mol. Struct., 2015, 1083, 245–251 CrossRef. - G. A. Knight and B. D. Shaw, J. Chem. Soc., 1938, 682–683 RSC.
- A. Agarwal, E. Huang, S. Palecek and N. L. Abbott, Adv. Mater., 2008, 20, 4804–4809 CrossRef CAS.
- H. Diestra-Cruz, E. Bukusoglu, N. L. Abbott and A. Acevedo, ACS Appl. Mater. Interfaces, 2015, 7, 7153–7162 CrossRef CAS PubMed.
- E. Bukusoglu, S. K. Pal, J. J. de Pablo and N. L. Abbott, Soft Matter, 2014, 10, 1602 RSC.
- S. K. Pal, A. Agarwal and N. L. Abbott, Small, 2009, 5, 2589–2596 CrossRef CAS PubMed.
- A. Agarwal, S. Sidiq, S. Setia, E. Bukusoglu, J. J. de Pablo, S. K. Pal and N. L. Abbott, Small, 2013, 9, 2785–2792 CrossRef CAS PubMed.
- K. A. Bogdanowicz, P. Gancarz, M. Filapek, D. Pociecha, M. Marzec, I. Chojnacka and A. Iwan, Dalton Trans., 2018, 47, 15714–15724 RSC.
- P. W. Majewski, M. Gopinadhan and C. O. Osuji, Polymers, 2019, 11, 887 CrossRef CAS PubMed.
- X. Cao, J. Cheng, X. Zhang, D. Zhou and Y. Tong, Int. J. Electrochem. Sci., 2020, 15, 677–695 CrossRef CAS.
- J. Sakuda, E. Hosono, M. Yoshio, T. Ichikawa, T. Matsumoto, H. Ohno, H. Zhou and T. Kato, Adv. Funct. Mater., 2015, 25, 1206–1212 CrossRef CAS.
- X. Wang, R. Yan, H. Niu, Z. He, W. He and Z. Miao, J. Energy Storage, 2024, 100, 113687 CrossRef.
- X. Li, P. Hu, J. Jiang, J. Pan, C. W. Nan and Y. Shen, Adv. Mater., 2025, 37, 2411507 CrossRef CAS PubMed.
- W. Z. Huang, P. Xu, X. Y. Huang, C. Z. Zhao, X. Bie, H. Zhang, A. Chen, E. Kuzmina, E. Karaseva, V. Kolosnitsyn, X. Zhai, T. Jiang, L. Z. Fan, D. Wang and Q. Zhang, MetalMat, 2023, 1, e6 CrossRef.
- Z. Hao, Q. Zhao, J. Tang, Q. Zhang, J. Liu, Y. Jin and H. Wang, Mater. Horiz., 2021, 8, 12–32 RSC.
- Y. Tang, L. Zhang, J. Chen, H. Sun, T. Yang, Q. Liu, Q. Huang, T. Zhu and J. Huang, Energy Environ. Sci., 2021, 14, 602–642 RSC.
- Y. Huang, H. Yang, Y. Gao, G. Chen, Y. Li, L. Shi and D. Zhang, Mater. Chem. Front., 2024, 8, 1282–1299 RSC.
- H. Zhang and Y. Qi, Sustainable Energy Fuels, 2022, 6, 954–970 RSC.
- H. Hong, N. A. R. Che Mohamad, K. Chae, F. Marques Mota and D. H. Kim, J. Mater. Chem. A, 2021, 9, 10012–10038 RSC.
- H. Kim, G. Jeong, Y.-U. Kim, J.-H. Kim, C.-M. Park and H.-J. Sohn, Chem. Soc. Rev., 2013, 42, 9011 RSC.
- D.-H. Liu, Z. Bai, M. Li, A. Yu, D. Luo, W. Liu, L. Yang, J. Lu, K. Amine and Z. Chen, Chem. Soc. Rev., 2020, 49, 5407–5445 RSC.
- M.-S. Tu, Z.-H. Wang, Q.-H. Chen, Z.-P. Guo, F.-F. Cao and H. Ye, Energy Environ. Sci., 2025, 18, 2873–2882 RSC.
- T. Wu, M. Jia, Y. Lu, J. Ye, D. Yang, Y. Zhang, S. Xie, D. Kang, L. Duan, H. Xie and J. Liu, J. Mater. Chem. A, 2025, 13, 7196–7206 RSC.
- F. Lan, H. Zhao, Y. Jiang, C. Jin, G. Zhao and L. Li, J. Mater. Chem. A, 2025, 13, 7357–7370 RSC.
- J. Sun, Y. Du, Y. Liu, D. Yan, X. Li, D. H. Kim, Z. Lin and X. Zhou, Chem. Soc. Rev., 2025, 54, 2543–2594 RSC.
- M. Wang, X. Guo, R. Luo, X. Jiang, Y. Tang and T. Wei, Chem. Commun., 2025, 61, 3777–3793 RSC.
- Y.-X. Yao, L. Xu, C. Yan and Q. Zhang, EES Batteries, 2025, 1, 9–22 RSC.
- H. H. Osman, P. Rodríguez-Hernandez, A. Munoz and F. J. Manjón, J. Mater. Chem. C, 2025, 13, 3774–3803 RSC.
- Z. Ning, D. S. Jolly, G. Li, R. De Meyere, S. D. Pu, Y. Chen, J. Kasemchainan, J. Ihli, C. Gong, B. Liu, D. L. R. Melvin, A. Bonnin, O. Magdysyuk, P. Adamson, G. O. Hartley, C. W. Monroe, T. J. Marrow and P. G. Bruce, Nat. Mater., 2021, 20, 1121–1129 CrossRef CAS PubMed.
- Y. Chen, F. Huo, S. Chen, W. Cai and S. Zhang, Adv. Funct. Mater., 2021, 31, e6 Search PubMed.
- L. Ye and X. Li, Nature, 2021, 593, 218–222 CrossRef CAS PubMed.
- P. Lu, L. Liu, S. Wang, J. Xu, J. Peng, W. Yan, Q. Wang, H. Li, L. Chen and F. Wu, Adv. Mater., 2021, 33, 2100921 CrossRef CAS PubMed.
- Y. Su, L. Ye, W. Fitzhugh, Y. Wang, E. Gil-González, I. Kim and X. Li, Energy Environ. Sci., 2020, 13, 908–916 RSC.
- X. Su, W. Jia, H. Ji and Y. Zhu, J. Energy Storage, 2021, 41, 102830 CrossRef.
- M. Monduzzi, G. Musu, M. Grosso, C. Carucci, B. Lindman, O. Söderman and A. Salis, Eur. Polym. J., 2022, 181, 111707 CrossRef CAS.
- H. K. Koduru, Y. G. Marinov, G. B. Hadjichristov, A. G. Petrov, N. Godbert and N. Scaramuzza, J. Non-Cryst. Solids, 2018, 499, 107–116 CrossRef CAS.
- Q. Wang, H. Fan, L.-Z. Fan and Q. Shi, Electrochim. Acta, 2013, 114, 720–725 CrossRef CAS.
- Z. Ahmad, Z. Hong and V. Viswanathan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 26672–26680 CrossRef CAS PubMed.
- S. Yazaki, M. Funahashi, J. Kagimoto, H. Ohno and T. Kato, J. Am. Chem. Soc., 2010, 132, 7702–7708 CrossRef CAS PubMed.
- S. Yazaki, M. Funahashi, J. Kagimoto, H. Ohno and T. Kato, J. Am. Chem. Soc., 2010, 132, 7702–7708 CrossRef CAS PubMed.
- F. B. Dias, S. V. Batty, J. P. Voss, G. Ungar and P. V. Wright, Solid State Ionics, 1996, 85, 43–49 CrossRef CAS.
- F. B. Dias, S. V. Batty, A. Gupta, G. Ungar, J. P. Voss and P. V. Wright, Electrochim. Acta, 1997, 43, 1217–1224 CrossRef.
- X. Wang, Z. He, R. Yan, H. Niu, W. He and Z. Miao, Chem. Eng. J., 2025, 158552 CrossRef CAS.
- F. Xu, K. Matsumoto and R. Hagiwara, J. Phys. Chem. B, 2012, 116, 10106–10112 CrossRef CAS PubMed.
- Y. Tong, L. Chen, X. He and Y. Chen, Electrochim. Acta, 2014, 118, 33–40 CrossRef CAS.
- H. Ruan, K. Lu, S. Meng, Q. Zhao, H. Ren, Y. Wu, C. Wang and S. Tan, Small, 2023, 20, 2310186 CrossRef PubMed.
- Q. Zeng, Y. Liu, B. Wujieti, Z. Li, A. Chen, J. Guan, H. Wang, Y. Jiang, H. Zhou, W. Cui, S. Wang and L. Zhang, Chem. Eng. J., 2024, 486, 2310186 Search PubMed.
- R. L. Kerr, S. A. Miller, R. K. Shoemaker, B. J. Elliott and D. L. Gin, J. Am. Chem. Soc., 2009, 131, 15972–15973 CrossRef CAS PubMed.
- S. Qin, Z. Wang, Y. Ren, Y. Yu, Y. Xiao, J. Chen, J. Zhang, S. Zhang, C. Sun, J. Xiao, L. Zhang, W. Hu and H. Yang, Nano Energy, 2024, 119, 109075 CrossRef CAS.
- N. Sun, F. Lu, A. Mariani, S. Passerini, X. Gao and L. Zheng, J. Membr. Sci., 2020, 605, 118113 CrossRef CAS.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS PubMed.
- S. Wang, X. Liu, A. L. Wang, Z. N. Wang, J. Chen, Q. H. Zeng, X. R. Jiang, H. H. Zhou and L. Y. Zhang, ACS. Appl. Mater. Interfaces, 2018, 10(30), 25273–25284 CrossRef CAS PubMed.
- S. Wang, A. Wang, C. Yang, R. Gao, X. Liu, J. Chen, Z. N. Wang, Q. H. Zeng, X. F. Liu, H. H. Zhou and L. Y. Zhang, J. Power Sources, 2018, 395, 137–147 CrossRef CAS.
- X. Chen, Y. Xie, Y. Ling, J. Zhao, Y. Xu, Y. Tong, S. Li and Y. Wang, Mater. Des., 2020, 192, 108760 CrossRef CAS.
- T. Onuma, M. Yoshio, M. Obi, K. Kashiwagi, S. Tahara and T. Kato, Polym. J., 2018, 50, 889–898 CrossRef CAS.
- Z. W. Liu, B. X. Dong, M. Misra, Y. Y. Sun, J. Strzalka, S. N. Patel, F. A. Escobedo, P. F. Nealey and C. K. Ober, Adv. Funct. Mater., 2019, 29(2), 1805220 CrossRef.
- S. Wang, X. Liu, A. Wang, Z. Wang, J. Chen, Q. Zeng, X. Wang and L. Zhang, Polym. Chem., 2018, 14, 655–661 Search PubMed.
- D. Bresser, L. Leclere, L. Bernard, P. Rannou, H. Mendil-Jakani, G. T. Kim, T. Zinkevich, S. Indris, G. Gebel, S. Lyonnard and L. Picard, ChemSusChem., 2020, 14, 655–661 CrossRef PubMed.
- J. Li, F. Huo, Y. Yang, T. Chen, Y. Cui, Y. Cai and H. Zhang, Chem. Eng. J., 2022, 433, 133562 CrossRef CAS.
- A. Martinez-Felipe, D. Zaton, M. Castillo-Vallés, A. Baldini, J. Pease, N. Leader, N. F. Kamalul Aripin, M. Giacinti-Baschetti and M. Blanca Ros, J. Mol. Liq., 2023, 390, 123100 CrossRef CAS.
- A. E. Frise, T. Ichikawa, M. Yoshio, H. Ohno, S. V. Dvinskikh, T. Kato and I. Furó, Chem. Commun., 2010, 46, 728–730 RSC.
- Y. Uchida, T. Matsumoto, T. Akitaa and N. Nishiyamaa, J. Mater. Chem. C, 2015, 3, 6144–6147 RSC.
- S. Padmajan Sasikala, J. Lim, I. H. Kim, H. J. Jung, T. Yun, T. H. Han and S. O. Kim, Chem. Soc. Rev., 2018, 47, 6013–6045 RSC.
- J. Dai, K. Qing Zhao, B.-Q. Wang, P. Hu, B. Heinrich and B. Donnio, J. Mater. Chem. C, 2020, 8, 4215–4225 RSC.
- T. Sakamoto, K. Asakura, N. Kang, R. Kato, M. Liu, T. Hayashi, H. Katayama and T. Kato, J. Mater. Chem. A, 2023, 11, 22178–22186 RSC.
- K. Hamaguchi, R. Ichikawa, S. Kajiyama, S. Torii, Y. Hayashi, J. Kumaki, H. Katayama and T. Kato, ACS Appl. Mater. Interfaces, 2021, 13, 20598–20605 CrossRef CAS PubMed.
- T. Kato, M. Gupta, D. Yamaguchi, K. P. Gan and M. Nakayama, Bull. Chem. Soc. Jpn., 2021, 94, 357–376 CrossRef CAS.
- D. Högberg, B. Soberats, R. Yatagai, S. Uchida, M. Yoshio, L. Kloo, H. Segawa and T. Kato, Chem. Mater., 2016, 28, 6493–6500 CrossRef.
- T. Ichikawa, M. Yoshio, A. Hamasaki, J. Kagimoto, H. Ohno and T. Kato, J. Am. Chem. Soc., 2011, 133, 2163–2169 CrossRef CAS PubMed.
- D. Kuo, T. Sakamoto, S. Torii, M. Liu, H. Katayama and T. Kato, Polym. J., 2022, 54, 821–825 CrossRef CAS PubMed.
- D. Kuo, M. Liu, K. R. S. Kumar, K. Hamaguchi, K. P. Gan, T. Sakamoto, T. Ogawa, R. Kato, N. Miyamoto, H. Nada, M. Kimura, M. Henmi, H. Katayama and T. Kato, Small, 2020, 16, 2001721 CrossRef CAS PubMed.
- B. Soberats, M. Yoshio, T. Ichikawa, S. Taguchi, H. Ohno and T. Kato, J. Am. Chem. Soc., 2013, 135, 15286–15289 CrossRef CAS PubMed.
- A. Concellón, T. Liang, A. P. H. J. Schenning, J. L. Serrano, P. Romero and M. Marcos, J. Mater. Chem. C, 2018, 6, 1000–1007 RSC.
- C.-H. Wu, W. Meng and M. Yoshio, ACS Mater. Lett., 2021, 4, 153–158 CrossRef.
- S. Yazaki, M. Funahashi and T. Kato, J. Am. Chem. Soc., 2008, 130, 13206–13207 CrossRef CAS PubMed.
- Y. Jiang, L. Liu, Y. Liu, J. Guan, H. Wang, M. Zhang, L. Chen, Y. Cao, R. Li, Y. Zhou, Q. Zeng, Z. Li, W. Liu, X. Li and L. Zhang, Adv. Funct. Mater., 2025, 2502613 CrossRef.
- F. Baskoro, P.-C. Chiang, Y.-C. Lu, J. N. Patricio, S. D. Arco, H.-C. Chen, W.-S. Kuo, L.-L. Lai and H.-J. Yen, Electrochim. Acta, 2022, 434, 141306 CrossRef CAS.
- X. Gao, F. Lu, L. Shi, H. Jia, H. Gao and L. Zheng, ACS Appl. Mater. Interfaces, 2013, 5, 13312–13317 CrossRef CAS PubMed.
- J.-Y. Seo, Y.-H. Lee, J.-H. Kim, Y.-K. Hong, W. Chen, Y.-G. Lee and S.-Y. Lee, Energy Storage Mater., 2022, 50, 783–791 CrossRef.
- M. Moriya, H. Kitaguchi, E. Nishibori, H. Sawa, W. Sakamoto and T. Yogo, Chem. – Eur. J., 2012, 18, 15305–15309 CrossRef CAS PubMed.
- F. Xie, F. Lu, C. Liu, Y. Tian, Y. Gao, L. Zheng and X. Gao, Colloids Surf., A, 2023, 658, 130731 CrossRef CAS.
- D.-Z. Zhang, Y.-y. Ren, Y. Hu, L. Li and F. Yan, Chin. J. Polym. Sci., 2020, 38, 506–513 CrossRef CAS.
- C. Wang, J. Zhou, J. Luo, K. Lu, H. Ruan, Q. Zhao, J. Ji, Y. Wu and S. Tan, Ind. Eng. Chem. Res., 2022, 61, 17930–17936 CrossRef CAS.
- C. Liu and M. Yoshio, ACS Appl. Mater. Interfaces, 2024, 16, 27750–27760 CrossRef CAS PubMed.
- C. Wang, D. Liu, T. Ma, Z. Wang, W. He and J. Liu, Phase Transitions, 2024, 97, 846–856 CrossRef CAS.
- S. Liu, L. Zhou, J. Han, K. Wen, S. Guan, C. Xue, Z. Zhang, B. Xu, Y. Lin, Y. Shen, L. Li and C. W. Nan, Adv. Energy Mater., 2022, 12, 2200660 CrossRef CAS.
- S. Liu, L. Zhou, T. Zhong, X. Wu and K. Neyts, Adv. Energy Mater., 2024, 14, 2403602 CrossRef CAS.
- H. Peng, X. Fang, W. Huang, W. Liu, Y. Yang, Q. Zhou and Y. Li, ACS Appl. Mater. Interfaces, 2024, 16, 44350–44360 CrossRef CAS PubMed.
- Y. An, X. Han, Y. Liu, A. Azhar, J. Na, A. K. Nanjundan, S. Wang, J. Yu and Y. Yamauchi, Small, 2021, 18, 2103617 CrossRef PubMed.
- I. Álvarez Moisés, M. Król, G. Keus, Z. He, A. Innocenti, S. Passerini, J. Ruokolainen and J.-F. Gohy, J. Am. Chem. Soc., 2025, 147, 20347–20358 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5eb00082c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |