
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntrinsic mechanisms for structural coherency and electrochemical excellence in layered-spinel Li–Mn–O cathodes†
Jihyeon
Gim‡
 a,
Jinhyup
Han‡
b,
Hacksung
Kim
*ac,
Qianqian
Li§
d,
Jinsong
Wu¶
d,
Vinayak P.
Dravid
d and
Eungje
Lee
*a
a,
Jinhyup
Han‡
b,
Hacksung
Kim
*ac,
Qianqian
Li§
d,
Jinsong
Wu¶
d,
Vinayak P.
Dravid
d and
Eungje
Lee
*a
aChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, IL 60439, USA. E-mail: eungje.lee@anl.gov; hskim@anl.gov
bDepartment of Chemical Engineering, School of Chemical Engineering and Applied Chemistry, Kyungpook National University, Daegu, South Korea
cCenter for Catalysis and Surface Science, Northwestern University, Evanston, IL 60208, USA
dNUANCE Center, Northwestern University, Evanston, IL 60208, USA
First published on 26th June 2025
Abstract
As the demand for lithium-ion batteries rises in consumer electronics, electric vehicles and stationary energy storage industries, there is both an opportunity and a need to innovate the cathode materials. Manganese-rich metal oxide cathodes have the potential to replace state-of-the-art cobalt- and nickel-rich layered electrode systems, which rely on metals that are scarce, high-cost, toxic, and unsafe. One approach is to use structurally compatible, manganese-based components such as layered Li2MnO3 and spinel Li1+yMn2−yO4 to reduce the Co and Ni content, thereby stabilizing lithium- and manganese-rich with nickel–manganese–cobalt (LMR-NMC) electrodes. A current study reports a detailed structural investigation of the baseline “layered-spinel” system xLi2MnO3·(1 − x)Li1+yMn2−yO4 (0 ≤ y ≤ 0.33) using synchrotron X-ray diffraction, high-resolution transmission electron microscopy, and high-resolution Raman spectroscopy. This provides insights into the complexity of this system and reveals clues that may assist in improving the electrochemical properties and stability of structurally integrated “layered-layered-spinel” LMR-NMC electrodes for high-energy lithium-ion battery systems.
Broader contextAs the demand for lithium-ion batteries (LIBs) continues to grow, there is an urgent need for sustainable and high-performance cathode materials, such as lithium- and manganese-rich (LMR) layered cathodes. This study focuses on the model system of Li2MnO3–LiMn2O4 to investigate how the integration of a spinel component can improve layered oxide cathodes. Our research delves into the atomic-scale coherency between layered and spinel crystal domains, which is essential for optimized performance. Through advanced characterization techniques, including synchrotron X-ray diffraction and high-resolution Raman spectroscopy, we uncover how these materials dynamically adjust their chemical composition and modulate crystal lattice parameters to reduce interfacial strain. This intrinsic mechanism enhances structural stability and electrochemical performance, offering valuable insights into the design of sustainable and efficient cathode materials. By elucidating the fundamental interactions between layered and spinel phases, our findings contribute to the broader understanding of how to effectively integrate different crystal structures to advance LIB technology. This work supports the ongoing transition to more environmentally friendly and economically viable energy storage solutions, aligning with global efforts to achieve a low-carbon economy. |
1. Introduction
In recent years, the adoption of layered nickel-rich oxides such as LiNi0.8Co0.15Al0.05O2 (NCA) and LiNi1−x−yMnxCoyO2 (NMC) as cathodes in advanced lithium-ion batteries (LIB) has been prevalent. These materials are favored for their high capacity, which approaches 200 mAh g−1, aligning with the energy density and driving range requirements of electric vehicles.1,2 The landscape of LIB technology is continuously evolving. Current research and industry trends focus on developing cathode materials that are not only high-performing but also sustainable and cost-effective. As such, there has been a marked shift towards exploring earth-abundant cathode compositions, particularly those rich in manganese (Mn), with reduced reliance on nickel (Ni) and cobalt (Co).3–5 This shift is primarily driven by the need for battery solutions that are more environmentally friendly and economically viable, as well as to alleviate the concerns associated with the limited availability and ethical sourcing of Ni and Co.Within this context, lithium- and manganese-rich (LMR) cathodes, often designated in layered-layered composite notation as xLi2MnO3·(1 − x)LiMO2 (M = Ni, Mn, Co), have regained significant attention. The formula represents the unique character of these two-component “layered-layered” materials, in which nanoscale domains of Li2MnO3 and LiMO2 are structurally integrated in highly complex arrangements within the crystal lattice.6,7 While the precise structural configuration of LMR cathodes remains under active investigation–including evaluations of alternative structural frameworks such as supercell and defect-based models8–these materials are particularly attractive due to their high capacity and energy density, which stem from their unique layered structure that facilitates high lithium-ion intercalation. These cathodes can deliver specific capacities of 250 mAh g−1 or more when the Li2MnO3 component is electrochemically activated above 4.5 V (versus Li0) during the initial charge. This process involves extracting lithium from the structure with concomitant loss of oxygen.6,9
However, these cathodes are not without limitations. Their structural instability is one concern; over prolonged use the structure gradually degrades, leading to voltage fade and reduced cycle life. This instability is caused when the transition metal ions (predominantly manganese ions) migrate from the transition-metal-rich layers to the lithium layers, which is reminiscent of the well-known layered-to-spinel phase transition of layered LiMnO2 electrodes.10 Therefore, it is critical to make the structure of LMR cathodes more robust if these materials are to be developed for practical LIB systems. Another critical issue in these materials is the nontrivial impedance, particularly at a low state-of-charge (SOC).11,12
A promising strategy to address these challenges is embedding a LiM2O4 spinel component into the layered-layered structure.3 This integration is hypothesized to fortify the cathode structure overall, as well as introducing efficient three-dimensional lithium-diffusion channels. The rationale behind this strategy is twofold: (1) the spinel structure, which is roust and thermally stable, can support the layered matrix, countering its inherent structural weaknesses; and (2) the three-dimensional pathways of the spinel phase can enhance lithium-ion diffusion, potentially improving the rate capability and overall electrochemical performance of the cathode.13,14 Thackeray et al.15,16 have demonstrated that the stability of layered–layered–spinel (LLS) composites can be improved by optimizing the concentration of the spinel component. Additionally, the incorporation of spinel into the layered–layered (LMR) framework enhances the rate performance of the cathode material.13,16–18
The benefits of integrated composite structure depend on achieving an intimate atomic-level integration between the layered and spinel phases.15,17,19,20 Therefore, the nature of layered-spinel integration is particularly intriguing, given that despite sharing a common oxygen sublattice (cubic-close-packed system), these phases represent fundamentally different structural types: a two-dimensional anisotropic structure and a cubic framework structure. This difference raises complex questions, particularly at the interface of these two domains. The extent of lattice mismatch, strain, and compositional redistribution at the interface are critical factors that remain largely unexplored, but they may hold the key to unlocking the full potential of these composite materials.
In light of these considerations, this study investigates a series of (1 − x)Li2MnO3·xLiMn2O4 materials, a simple model layered-spinel composite system, to delve into the intricacies of lattice integration, strain, and compositional dynamics within these composites. To study the detailed crystallographic relationships and compatibility between the layered and spinel components, we employed synchrotron X-ray diffraction (XRD), high-resolution transmission electron microscopy (HR-TEM), and high-resolution Raman spectroscopy analysis. By unraveling these structural complexities, this research seeks to contribute significantly to the future development of next-generation, sustainable cathode materials.
2. Experimental
A series of lithium-manganese-oxide materials with various Li/Mn ratios was prepared by solid-state synthesis. Appropriate amounts of lithium carbonate (Li2CO3, >99%, Sigma Aldrich) and manganese carbonate (MnCO3, >99%, Sigma Aldrich) precursor powders were thoroughly mixed in a mortar and fired at 850 °C for 24 h in air. The heating rate was 5 °C min−1 while the cooling rate was uncontrolled (taking about 6.5 h to reach 100 °C).Powder X-ray diffraction (XRD) patterns were collected on a Rigaku MiniFlex 600 laboratory diffractometer (Cu-Kα radiation) between 10 and 80° 2θ with a step size of 0.02° 2θ. High-resolution synchrotron XRD (HR-SXRD) data were collected using the 11-BM beamline at the Advanced Photon Source (APS) at Argonne National Laboratory. Scans of the cathode samples in spinning Kapton capillary tubes were collected in transmission mode. The calibrated X-ray wavelength was 0.413369 Å, and step size was 0.001° 2θ. Structural refinements of the XRD data were conducted using the Rietveld method and the TOPAS software package.
For high-resolution Raman spectroscopy, an excitation laser wavelength of 458 nm was provided by a Melles Griot Ar+ ion Laser System.21 A collimated laser light was focused onto the sample, and the scattered light from the sample refocused using a home-made 90° off-axis ellipsoidal reflector with the backscattering geometry onto the entrance slit of a triple-grating spectrometer (Princeton Instruments, Trivista 555) where Rayleigh light was filtered out and stray light significantly suppressed. The Raman light was collected by a liquid N2-cooled CCD detector (Princeton Instruments, SPEC-10). A home-made, in situ fluidized bed Raman cell in flowing helium (≈100 mL min−1) at room temperature combined with a very low laser power of 1 mW at the sample was used to avoid possible laser-induced sample degradation.21 Cyclohexane, chloroform, and trichloroethylene were used as Raman frequency standard materials. The accuracy of Raman shifts was estimated to be ±1 cm−1.
Samples for transmission electron microscopy (TEM) analyses were prepared by dispersing the powder samples directly onto lacey carbon-coated Cu 200 mesh TEM grids (Ted Pella) in an Ar-filled glovebox. Exposure of the samples to air while loading them into the microscope was minimized. Nanobeam electron diffraction (NED) and HR-TEM images were collected by a JEOL JEM-2100 FasTEM equipped with a Schottky field emission gun operating at 200 keV. Fast Fourier Transforms (FFTs) of the HR-TEM images were obtained using DigitalMicrograph software and indexed with the aid of SingleCrystal software.
The electrochemical tests were conducted using 2032-type coin cells. Electrode laminates were prepared by mixing a slurry of the active material, carbon black (Super P Li, Timcal), and polyvinylidene difluoride (PVDF, Solvay) in N-methyl pyrrolidone (NMP) solvent in a mass ratio of 84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:8. Electrode laminates were cast on aluminum foil with subsequent drying and calendaring process. 2032-type coin cells with metallic lithium anodes were assembled in an inert Ar glovebox (both water and oxygen levels ≤1 ppm). The electrolyte was a 1.2 M LiPF6 solution with a 3:7 (by weight) mixture of ethylene carbonate (EC) and ethylmethyl carbonate (EMC) solvents. Electrochemical tests were performed and recorded on a battery cycler system (MACCOR, Inc.) at room temperature; cells were cycled between 2 and 5 V vs. Li at a current rate of 10 mA g−1.
:8:8. Electrode laminates were cast on aluminum foil with subsequent drying and calendaring process. 2032-type coin cells with metallic lithium anodes were assembled in an inert Ar glovebox (both water and oxygen levels ≤1 ppm). The electrolyte was a 1.2 M LiPF6 solution with a 3:7 (by weight) mixture of ethylene carbonate (EC) and ethylmethyl carbonate (EMC) solvents. Electrochemical tests were performed and recorded on a battery cycler system (MACCOR, Inc.) at room temperature; cells were cycled between 2 and 5 V vs. Li at a current rate of 10 mA g−1.
3. Results and discussion
To gain insights into the structural and electrochemical properties of layered-spinel composite materials, a series of lithium-manganese-oxide LizMnOδ materials was synthesized with various Li/Mn ratios (0.5 ≤ z ≤ 2; 2 ≤ δ ≤ 3). For convenience, the formular can be conceptualized in composite notation as (1 − x)Li2MnO3·xLiMn2O4 assuming that the composition of the spinel component formed at 850 °C is LiMn2O4 for all Li/Mn ratios – an assumption that was not strictly accurate, as discussed later in this paper. With this assumption, materials with Li/Mn ratio (i.e., z in LizMnOδ) of 2.0, 1.75, 1.5, 1.25, 1.0, and 0.5 correspond to (1 − x)Li2MnO3·xLiMn2O4 compounds, in which x is 0, 0.1, 0.2, 0.33, 0.5 and 1.0, respectively, followed by the “layered-spinel tie-line” displayed in Fig. 1(b). | ||
| Fig. 1 (a) Compositional Li2MnO3-MnO-λ-MnO2 phase diagram and (b) stoichiometric tie-line for (1 − x)Li2MnO3·xLiMn2O4 with conceptional tie-line of (1 − x)Li2MnO3·xLi1+xMn2−yO4 (adapted and edited from Thackeray et al.3). | ||
3.1. X-ray diffraction analysis: evolution of composite structures
Fig. 2(a) shows the XRD patterns of the as-prepared (1 − x)Li2MnO3·xLiMn2O4 powder samples. The x = 0 sample shows typical pattern for the phase pure layered rock-salt Li2MnO3 (space group, S.G. 12, C2/m) structure. As x increases (i.e., decreasing Li/Mn ratio), the peaks related to spinel structure start to evolve at the expense of the peaks from Li2MnO3 until the LiMn2O4 spinel single phase (S.G. 227, Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, a = 8.244 Å) is obtained at x = 1. The formation of a spinel domain was not clearly observed for the x = 0.1 initially. However, careful observation reveals the development of a spinel structural motif at ∼36.4° 2θ, as shown in Fig. 2(b), indicating (311)s of the spinel structure. More complex diffraction patterns of the x = 0.2, 0.333, and 0.5 samples can be clearly indexed to both layered C2/m and spinel Fdm phases. Interestingly, these XRD data show no evidence of peak splitting into (001)L and (111)S reflections at ∼18.5° 2θ in the composite samples. To obtain better peak resolution, the x = 0.5 sample was examined by HR-SXRD (λ = 0.413369 Å, step size in 2θ = 0.001°). The HR-SXRD data in Fig. 2(c) further confirms the single peak shape at the 2θ positions for the (001)L//(111)S and (002)L//(222)S reflections (see Fig. 2(c) insets). These (001)L and (111)S reflections correspond to the oxygen close-packed planes in each structure. The overlap of the two main peaks thus indicates that interplanar distances are almost identical in the corresponding planes, and further suggests the crystallographic coherency between the layered and spinel phase domains to the direction of the oxygen closed packed planes.
m, a = 8.244 Å) is obtained at x = 1. The formation of a spinel domain was not clearly observed for the x = 0.1 initially. However, careful observation reveals the development of a spinel structural motif at ∼36.4° 2θ, as shown in Fig. 2(b), indicating (311)s of the spinel structure. More complex diffraction patterns of the x = 0.2, 0.333, and 0.5 samples can be clearly indexed to both layered C2/m and spinel Fdm phases. Interestingly, these XRD data show no evidence of peak splitting into (001)L and (111)S reflections at ∼18.5° 2θ in the composite samples. To obtain better peak resolution, the x = 0.5 sample was examined by HR-SXRD (λ = 0.413369 Å, step size in 2θ = 0.001°). The HR-SXRD data in Fig. 2(c) further confirms the single peak shape at the 2θ positions for the (001)L//(111)S and (002)L//(222)S reflections (see Fig. 2(c) insets). These (001)L and (111)S reflections correspond to the oxygen close-packed planes in each structure. The overlap of the two main peaks thus indicates that interplanar distances are almost identical in the corresponding planes, and further suggests the crystallographic coherency between the layered and spinel phase domains to the direction of the oxygen closed packed planes.
 | ||
| Fig. 2 (a and b) XRD patterns of (1 − x)Li2MnO3·xLiMn2O4 and (c) HR-SXRD pattern of the x = 0.5 sample. The diffraction peaks are indexed as (hkl)L for the layered structure (C2/m) and (hkl)S for the spinel structure (Fdm). | ||
A quantitative phase analysis of the XRD data was conducted by Rietveld refinement (see Table 1 and Tables S1–S5 in the ESI†). Note that the spinel phase in the x = 0.1 sample is excluded from the calculation because the corresponding peaks are too weak for a reliable analysis. The visual examination of the fitted profiles and the low R-factors (Rwp < 7) confirm that the quality of Rietveld refinement is satisfactory (Fig. S1–S5†). Fig. 3(a) plots the XRD measured spinel content as a function of the nominal spinel content, x in (1 − x)Li2MnO3·xLiMn2O4. The two corresponding spinel content values are nearly identical to each other, indicating that the overall layered-spinel phase segregation of composite materials occurs according to the Li2MnO3–LiMn2O4 (blue-dotted layered-spinel tie-line) in the displayed phase diagram (Fig. 1(b)). However, the refined structural data further suggests that the composition and structure of the spinel phase in composite materials deviate from the stoichiometric LiMn2O4. For example, the lattice parameter of the spinel phase decreases from 8.244 Å for the pure LiMn2O4 (x = 1.0) to 8.189 Å for the x = 0.5 composite sample. This decrease in the lattice parameter could be attributed to the stabilization of off-stoichiometric defect spinel phase with excess of Li on 16d sites, such as Li1+yMn2−yO4, because the lattice parameters are in between those of LiMn2O4 (y = 0; a = 8.244 Å) and Li4Mn5O12 (i.e., Li1.333Mn1.667O4; y = 0.333; a = 8.128 Å). The y ≈ 0.16 is estimated for x = 0.5 applying the linear relationship between y, off-stoichiometric defect and lattice parameter in Li1+yMn2−yO4.22
 | ||
| Fig. 3 (a) XRD spinel content and (b) interplanar distances of oxygen closed packed planes (d(001) for layered and d(111) for spinel) in (1 − x)Li2MnO3·xLiMn2O4 composites. | ||
| Li/Mn ratio | Nominal spinel content, x | XRD spinel content | Layered Li2MnO3 (C2/m) | Spinel LiMn2O4 (Fdm) |
|||||
|---|---|---|---|---|---|---|---|---|---|
| a [Å] | b [Å] | c [Å] | β [°] | d (001) [Å] | a [Å] | d (111) [Å] | |||
| a Spinel phase in the x = 0.1 sample is excluded from the Rietveld refinement because the corresponding peaks are too weak for a reliable analysis. | |||||||||
| 2 | 0 | 0 | 4.926 | 8.527 | 5.019 | 109.1 | 4.741 | — | — |
| 1.75 | 0.1 | 4.926 | 8.523 | 5.021 | 109.2 | 4.741 | |||
| 1.5 | 0.2 | 0.173 | 4.926 | 8.525 | 5.014 | 109.0 | 4.740 | 8.189 | 4.728 |
| 1.25 | 0.333 | 0.377 | 4.926 | 8.525 | 5.017 | 109.0 | 4.743 | 8.209 | 4.739 |
| 1 | 0.5 | 0.484 | 4.924 | 8.525 | 5.013 | 109.0 | 4.741 | 8.214 | 4.742 |
| 0.5 | 1 | 1 | — | — | — | — | — | 8.244 | 4.759 |
Notably, stabilizing the spinel component into the form of partially over-lithiated spinel Li1+yMn2−yO4 appears to be more effective in establishing a structurally coherent composite with layered Li2MnO3. Fig. 3(b) shows the interplanar distances of the oxygen closed packed planes in the layered Li2MnO3 (d(001)) and spinel Li1+yMn2−yO4 (d(111)) phases. Compared to pure LiMn2O4, which has a larger d(111), the Li1+yMn2−yO4 (0 < y < 0.333) phase in the composite structure has a smaller d(111) that is similar to the d(001) of Li2MnO3. Such compatible interplanar distances would help reduce the lattice strain and the interface energy between Li2MnO3 and Li1+yMn2−yO4 when the two structural domains form a coherent composite.
Previously, Johnson and Thackeray et al. reported a low-temperature form of layered-spinel composite material, 0.7Li2MnO3·0.3Li4Mn5O12.15 The interplanar distances of the Li2MnO3 (d(001)) and Li4Mn5O12 (d(111)) components in this low-temperature composite material were 4.707 and 4.696 Å, respectively. In this example, the interplanar distance of the low-temperature (i.e., 400 °C) Li2MnO3 component is smaller than that of the high-temperature Li2MnO3. This results in a low lattice misfit energy between the low-temperature Li2MnO3 and Li4Mn5O12. In contrast, the high-temperature synthesis in this study stabilizes Li2MnO3 with larger d(001) and the lattice coherency between the layered and spinel structures is maintained by the formation of Li1+yMn2−yO4 type spinel phase (y < 0.33) with an increased planar distance of d(111), which is still a smaller lattice parameter than that of a stoichiometric LiMn2O4 spinel.
3.2. Raman spectroscopy analysis: partially over-lithiated Li1+yMn2−yO4 spinel component
Vibrational Raman spectroscopy (RS) probes molecular vibrations by analyzing the inelastic (i.e., Raman) scattering of light and provides useful molecular structural information such as chemical bonds formation, breakage, elongation, etc. RS has notable advantage in probing the short-range environments of oxygen coordination around the cations in oxide lattices whose vibrations appear in the low frequency region, <≈1100 cm−1. The local cation configurations in the composite materials can also be sensitively probed by RS where various cationic environments are present.21,23Fig. 4 compares Raman spectra of a series of as-prepared powder samples. For monoclinic Li2MnO3 structure (x = 0) with a space group of C2/m (= C2h3), 15 Raman active modes (7Ag + 8Bg) are expected.24–26 In Fig. 4, at least 12 Raman bands of Li2MnO3 were found in the 200–700 cm−1 region. The most intense Raman band at 616 cm−1 is assigned to the symmetric Mn–O stretching vibrations essentially with oxygen atom shifts of MnO6 octahedra.25 On the basis of Li isotope effect in the Raman spectra of 7LiNbO3 and 6LiNbO3, only the frequencies in the 270–400 cm−1 range are influenced by Li cation displacements (i.e., Li–O stretching and O–Li–O bending modes) in LiO6 octahedra.27 Similarly, the weak Raman bands in the 300–400 cm−1 region in Fig. 4 can be assigned to the Li–O stretching and O–Li–O bending vibrations of LiO6 octahedra. In addition, the Raman bands in the 400–520 cm−1 range are assigned to the O–Mn–O bending vibrations that are likely coupled to Li–O stretching and O–Li–O bending vibrations similar to a strong coupling of O–Nb–O bending with the Li–O and O–Li–O vibrations for LiNbO3.27,28 The high-resolution Raman spectra with exceptionally distinct 12 Raman bands for Li2MnO3 were realized by a triple-grating spectrometer (see section 2, Experimental, for details).21 The triple spectrometer allows to significantly reject unwanted stray and Rayleigh light, and to obtain high resolution Raman spectra, that is coupled to a supersensitive liquid Nitrogen-cooled charge-coupled device (CCD) detector which provides an ultralow dark current and a high quantum efficiency in the whole spectral region. Such highly resolved Raman bands enabled the analysis of the local structural features in the layered-spinel composite system with greater detail and clarity. Specifically, it is found that as the spinel content, x increases from 0 to 0.5, additional Raman bands centered at ∼650 cm−1 (notable, broad) and ∼486 cm−1 (weak, broad) gradually develop on top of the characteristic Li2MnO3 bands. We attribute the two additional bands to spinel components. For LiMn2O4 and Li4Mn5O12 (i.e., Li1.333Mn1.667O4) spinel structures, the Raman bands become broader and the main peak corresponding to Mn–O stretching vibration indicates blue shifts to 627 cm−1 and ∼640 cm−1, respectively, from 616 cm−1 for Li2MnO3.28 | ||
| Fig. 4 Raman spectra of the (1 − x)Li2MnO3·xLiMn2O4 (0 ≤ x ≤ 1.0) and Li4Mn5O12 (i.e., Li1.333Mn1.667O4) samples; (a) x = 1.0, (b) x = 0.5, (c) x = 0.333, (d) x = 0.2 and (e) x = 0. | ||
To gain a deeper understanding of the spinel component embedded within the layered-spinel composite structure, the Raman spectra for ideal composites of Li2MnO3·LiMn2O4 and Li2MnO3·Li1.333Mn1.667O4 were derived through linear combination fitting of the experimentally obtained Raman spectra for the corresponding single-phase materials, i.e., Li2MnO3 + LiMn2O4 and Li2MnO3 + Li1.333Mn1.667O4. In Fig. 5(a) and (b), the two modeled spectra are generally well aligned with the observed spectrum for x = 0.5 across most wavelengths. The exception is the ∼650 cm−1 region, where a noticeable difference is apparent. In this region, the processed Raman spectrum of Li2MnO3 + LiMn2O4 underestimates the normalized intensity while that of Li2MnO3 + Li1.333Mn1.667O4 overestimates it. Fig. 5(c) shows the excellent fitting result with the Raman spectrum fitted with a combination of three phases as Li2MnO3 + LiMn2O4 + Li1.333Mn1.667O4, with the ratio of 50:26:24, corresponding to 0.5Li2MnO3·0.5Li1+yMn2−yO4 (y ∼ 0.16). This result is consistent with the HR-SXRD data, which suggests that the spinel component in the x = 0.5 sample is a partially over-lithiated Li1+yMn2−yO4, with y approximately 0.16.
 | ||
| Fig. 5 Comparison between the experimentally obtained Raman spectra for the x = 0.5 sample and the composite spectra derived from linear combination of the single-phase spectra; (a) Li2MnO3 + LiMn2O4 (1:1 ratio), (b) Li2MnO3 + Li1.333Mn1.667O4 (1:1), and (c) Li2MnO3 + LiMn2O4 + Li1.333Mn1.667O4 (50:26:24). | ||
3.3. Transmission electron microscopy: coherency and geometrical distribution
High-resolution transmission electron microscopy (HR-TEM) images of selected layered-spinel composite samples (x = 0.5 and 0.2) were obtained to investigate the crystallographic relationship between the layered and spinel local atomic structural domains. Fig. 6(a) shows an aggregate of primary particles (∼0.5 μm in size) for the x = 0.5 sample in the low magnification TEM image. Fig. 6(b) presents the corresponding electron diffraction pattern (EDP), which is indexed as Li2MnO3 with [110] or [100] zone axis, and LiMn2O4 with [−12−1] zone axis. The (001) plane of Li2MnO3 and the (111) plane of the spinel are indexed in the same spots. This suggests coherent arrangements of two crystallographic planes, which are oxygen closed-packed planes, and well matched with the HR-SXRD results (Fig. 2(c)). In Fig. 6(c), the HR-TEM image clearly shows distinct crystal domains for the layered and spinel structures where the lattices for both structure are coherently arranged into the direction of oxygen closed packed plane, (001)L and (111)S. A coherent interface region that connects those two phases is also clearly visible. Fast Fourier Transformed (FFT) images of the interface region correspond to the mixture of the adjacent layered and spinel single domains, indexed as [110]L and [1−2−1]S, respectively. | ||
| Fig. 6 TEM analysis of (a–c) 0.5Li2MnO3·0.5LiMn2O4 and (d and e) 0.8Li2MnO3·0.2LiMn2O4: (a) Low magnification image showing aggregated primary particles of ∼0.5 μm. Coherent lattice arrangement of the layered and spinel crystal domains are observed by (b) electron diffraction pattern (EDP) and (c and d) high resolution transmission electron microscopy (HR-TEM) images. (e) Dark field image constructed from a spinel spot (marked in the inset EDP image) exhibits the presence of spinel domains both in bulk and surface of a particle. | ||
The formation of such layered-spinel composite structure is also confirmed for the x = 0.2 sample, where the amount of spinel phase is reduced to 20% (Fig. 6(d)). Again, the (111)S planes of the spinel are structurally integrated with the (001)L planes of Li2MnO3 despite severe stacking faults to the c-axis direction in Li2MnO3 domains. In additiona, a dark field (DF) image is constructed by selecting only the diffraction spot that corresponds to the spinel phase (marked in the inset EDP image) in order to distinguish the positions of spinel components in a composite particle. In Fig. 6(e), the DF image for x = 0.2 shows bright particle edges indicating a spinel phase located at the particle edge regions. Meanwhile, there are also many bright bands and/or streaks shown in bulk regions as well (marked by yellow arrows). Therefore, our electron microscopy data confirms that the coherently integrated spinel structural domains uniformly exist in the bulk of a layered-spinel composite particle while the particle surface is preferentially saturated with spinel structure probably due to its lower surface energy.29 We presume that the reduced lattice mismatch and interface energy by partial over-lithiation in the spinel phase promotes the homogeneous bulk integration of the spinel nano-domains in Li2MnO3. These coherent crystallographic relationships will enable more effective lithium-ion diffusion within the materials as the interfaces between spinel and layered phase do not generate a diffusion barrier for lithium ions.
3.4. Electrochemical properties
Fig. 7 shows the initial (black solid line) and second (red dashed line) charge/discharge voltage profiles of the (1 − x)Li2MnO3·xLiMn2O4 composite electrodes. The x = 0 sample exhibits typical electrochemistry of Li2MnO3 that is characterized with the activation plateau at ∼5 V vs. Li0 during the initial charge and the sloping voltage curves during subsequent cycles. The initial charge and discharge capacities are 110 and 50 mAh g−1, respectively. The obtained initial capacity is substantially smaller than theoretical capacity (∼450 mAh g−1 for 2Li+ removal) due to the low electronic conductivity of Li2MnO3 and relatively long Li diffusion pathways for the sintered particles synthesized at a high temperature.30 The voltage profile for x = 0.1 is similar to that of Li2MnO3 and no characteristic voltage signature that can be attributed to a spinel phase was observed. Nevertheless, the x = 0.1 sample has a larger activation plateau and increased specific capacity (165 and 84 mAh g−1, respectively for the initial charge and discharge). The degree of Li2MnO3 activation (and following specific capacities) is largely limited by the poor electrical conductivity and Li transport of the Li2MnO3 material.30 Therefore, the increased activation capacity of the x = 0.1 sample corroborates the XRD data (Fig. 1(b)) that suggested the presence of embedded spinel nano-domains in the x = 0.1 sample, which facilitates the electrochemical Li reaction.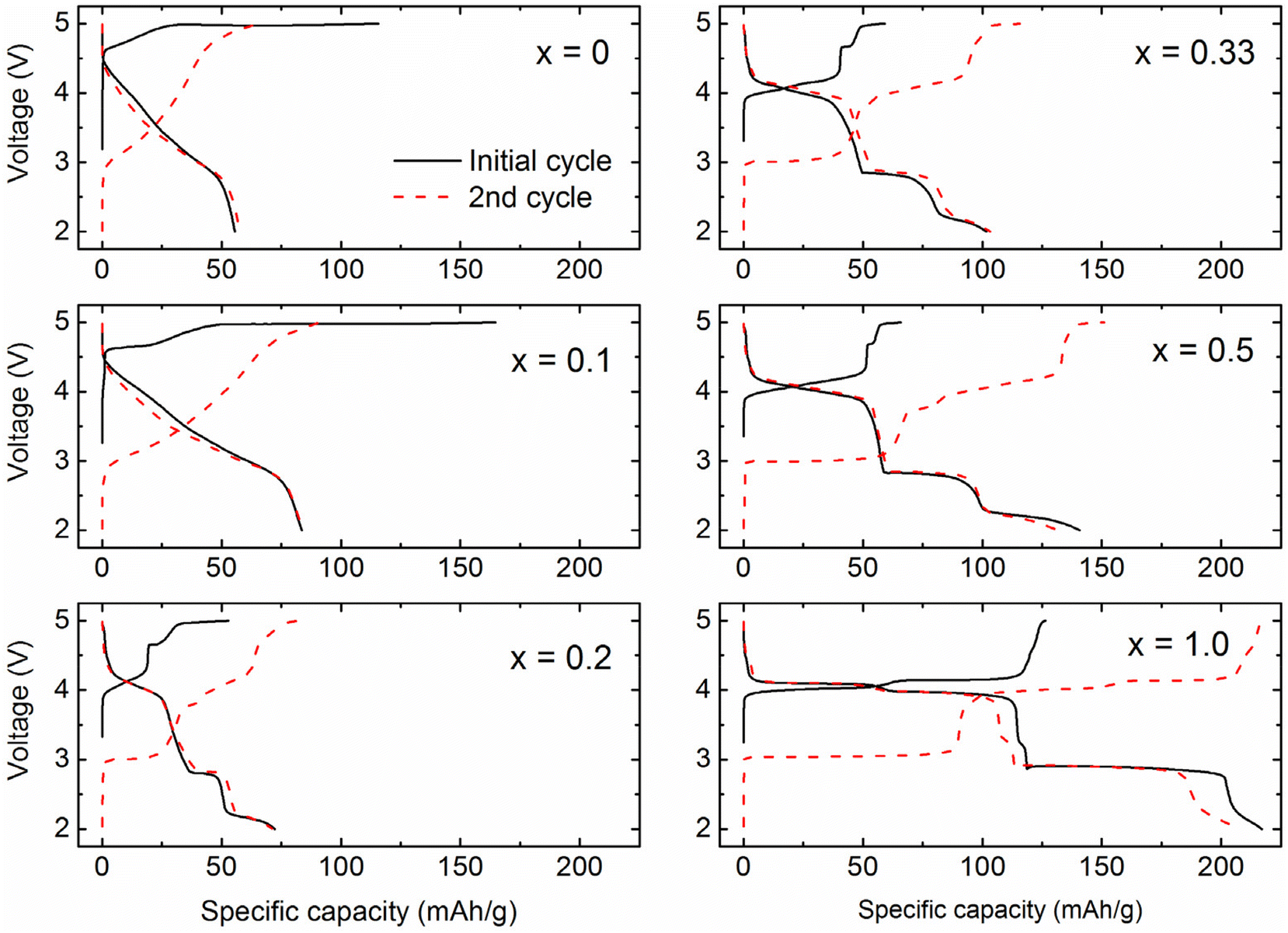 | ||
| Fig. 7 Voltage profiles of (1 − x)Li2MnO3·xLiMn2O4 cycled between 5.0 and 2.0 V vs. Li0 at the current rate of 10 mA g−1. | ||
Clear voltage profile features corresponding to a spinel phase start to appear from x ≥ 0.2; the voltage plateaus at ∼4 V and below 3 V correspond to lithium (de)intercalation to 8a tetrahedral sites and 16c octahedral sites of LiMn2O4 spinel, respectively.31 The significant decrease in the length of the ∼5 V activation plateau with increasing the spinel content (x ≥ 0.2) can be attributed to (1) the lower fractional amount of Li2MnO3 phase, and (2) preferential surface saturation of spinel phase and resultant isolation of Li2MnO3 domains in the bulk of the particles, which hampers the removal of oxygen to the surface. The isolation of Li2MnO3 domains in the bulk of the composite leads to stagnant activation. This aligns with the observation that it takes several more consecutive charge cycles to complete the activation process of the composite materials.
4. Conclusions
We conducted a systematic investigation into layered-spinel composite cathodes in the Li–Mn–O system by carefully analyzing high-quality characterization data. The study confirms that the composite structure evolves linearly as the Li/Mn ratio changes, which indicates that a stable layered-spinel composite system is formed. However, these composite phases do not adhere strictly to the linear tie-line between Li2MnO3 and stoichiometric LiMn2O4 in the phase diagram. Instead, they shift towards the Li4Mn5O12 (i.e., Li[Li1/3Mn5/3]O4) spinel phase, resulting in a partially over-lithiated Li1+yMn2−yO4 spinel component. The important compositional adjustment is crucial to reduce the lattice mismatch with the Li2MnO3 structure, particularly in the direction of the oxygen close-packed planes, ensuring excellent coherency between the two phases. Furthermore, HR-TEM data confirms that the spinel component is homogeneously distributed within the bulk of the composite particles while it preferentially forms on the particle surface. The bulk incorporation of the spinel phase improves the lithium reaction kinetics of the Li2MnO3 component, thereby enhancing the electrochemical properties of the composite cathode. However, the excessive presence of spinel on the surface can suppress oxygen removal from the particle surface, subsequently inhibiting Li2MnO3 activation.These findings underscore the importance of control over the spinel properties and its spatial arrangement to boost electrochemical performance by protecting the cathode against deleterious oxygen loss from the surface. Although the absence of a control Li2MnO3·LiMn2O4 sample with lattice mismatch and high interfacial strain limited direct measurement of kinetic data and performance comparison in the current study, a follow-up investigation using particle-level modeling of transport kinetics could provide valuable insights into the role of lattice coherency in lithium diffusion. This work outlines a promising pathway for optimizing the design and synthesis of sustainable, manganese-rich cathodes, paving the way for improved stability and enhanced electrochemical performance.
Conflicts of interest
The authors declare no conflict of interest.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
Support from the Office of Vehicle Technologies of the U.S. Department of Energy, particularly from the Earth-abundant Cathode Active Materials (EaCAM) consortium under Carine Steinway, Tina Chen, Tien Duong, and Brian Cunningham is gratefully acknowledged. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The Raman work at Argonne National Laboratory was supported by the U.S. Department of Energy Office of Basic Energy Sciences (under Grant Number DE-FG02-03ER15457), and Office of Vehicle Technologies, particularly from Haiyan Croft. Part of the work performed by J. H. was supported by the National Research Foundation (NRF) grant funded by the Ministry of Education (MOE) of Korea (no. 21202414153, BK21 Program for Low-Carbon Advanced Materials Innovation). This work made use of the EPIC facility of Northwestern University's NUANCE Center, which has received support from the ShyNE Resource (NSF ECCS-2025633), the IIN, and Northwestern's MRSEC program (NSF DMR-2308691).The submitted manuscript has been created by UChicago Argonne, LLC, Operator of Argonne National Laboratory (“Argonne”). Argonne, a U.S. Department of Energy Office of Science laboratory, is operated under Contract No. DE-AC02-06CH11357. The U.S. Government retains for itself, and others acting on its behalf, a paid-up, nonexclusive, irrevocable worldwide license in said article to reproduce, prepare derivative works, distribute copies to the public, and perform publicly and display publicly, by or on behalf of the Government.
References
- G. Zubi, R. Dufo-López, M. Carvalho and G. Pasaoglu, Renewable Sustainable Energy Rev., 2018, 89, 292–308 CrossRef.
- X. Zeng, M. Li, D. Abd El-Hady, W. Alshitari, A. S. Al-Bogami, J. Lu and K. Amine, Adv. Energy Mater., 2019, 9, 1900161 CrossRef.
- M. M. Thackeray, J. R. Croy, E. Lee, A. Gutierrez, M. He, J. S. Park, B. T. Yonemoto, B. R. Long, J. D. Blauwkamp, C. S. Johnson, Y. Shin and W. I. F. David, Sustainable Energy Fuels, 2018, 2, 1375–1397 RSC.
- R. Benedek, J. Electrochem. Soc., 2022, 169, 050505 CrossRef CAS.
- A. Masias, J. Marcicki and W. A. Paxton, ACS Energy Lett., 2021, 6, 621–630 CrossRef CAS.
- M. M. Thackeray, S. H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 RSC.
- P. K. Nayak, E. M. Erickson, F. Schipper, T. R. Penki, N. Munichandraiah, P. Adelhelm, H. Sclar, F. Amalraj, B. Markovsky and D. Aurbach, Adv. Energy Mater., 2018, 8, 1702397 CrossRef.
- A. Celeste, M. Tuccillo, A. S. Menon, W. Brant, D. Brandell, V. Pellegrini, R. Brescia, L. Silvestri and S. Brutti, Small Methods, 2024, 8, 2301466 CrossRef CAS.
- E. M. Erickson, F. Schipper, T. R. Penki, J.-Y. Shin, C. Erk, F.-F. Chesneau, B. Markovsky and D. Aurbach, J. Electrochem. Soc., 2017, 164, A6341–A6348 CrossRef CAS.
- J. R. Croy, M. Balasubramanian, K. G. Gallagher and A. K. Burrell, Acc. Chem. Res., 2015, 48, 2813–2821 CrossRef CAS.
- J. Chen, A. Gutierrez, M. T. Saray, R. S. Yassar, M. Balasubramanian, Y. Wang and J. R. Croy, J. Electrochem. Soc., 2021, 168, 080506 CrossRef CAS.
- A. Gutierrez, D. Tewari, J. Chen, V. Srinivasan, M. Balasubramanian and J. R. Croy, J. Electrochem. Soc., 2023, 170, 030509 CrossRef CAS.
- J. R. Croy, A. Gutierrez, M. He, B. T. Yonemoto, E. Lee and M. M. Thackeray, J. Power Sources, 2019, 434, 226706 CrossRef CAS.
- H. Xie, J. Cui, Z. Yao, X. Ding, Z. Zhang, D. Luo and Z. Lin, Chem. Eng. J., 2022, 427, 131978 CrossRef CAS.
- C. S. Johnson, N. Li, J. T. Vaughey, S. A. Hackney and M. M. Thackeray, Electrochem. Commun., 2005, 7, 528–536 CrossRef CAS.
- S. H. Park, S. H. Kang, C. S. Johnson, K. Amine and M. M. Thackeray, Electrochem. Commun., 2007, 9, 262–268 CrossRef CAS.
- D. Kim, G. Sandi, J. R. Croy, K. G. Gallagher, S. H. Kang, E. Lee, M. D. Slater, C. S. Johnson and M. M. Thackeray, J. Electrochem. Soc., 2013, 160, A31–A38 CrossRef CAS.
- J. R. Croy, J. S. Park, Y. Shin, B. T. Yonemoto, M. Balasubramanian, B. R. Long, Y. Ren and M. M. Thackeray, J. Power Sources, 2016, 334, 213–220 CrossRef CAS.
- I. Belharouak, G. M. Koenig Jr, J. Ma, D. P. Wang and K. Amine, Electrochem. Commun., 2011, 13, 232–236 CrossRef CAS.
- B. R. Long, J. R. Croy, J. S. Park, J. G. Wen, D. J. Miller and M. M. Thackeray, J. Electrochem. Soc., 2014, 161, A2160–A2167 CrossRef CAS.
- H. Kim, K. M. Kosuda, R. P. V. Duyne and P. C. Stair, Chem. Soc. Rev., 2010, 39, 4820–4844 RSC.
- W. Zhang, D. M. Cupid, P. Gotcu, K. Chang, D. Li, Y. Du and H. J. Seifert, Chem. Mater., 2018, 30, 2287–2298 CrossRef CAS.
- R. Baddour-Hadjean and J.-P. Pereira-Ramos, Chem. Rev., 2010, 110, 1278–1319 CrossRef CAS PubMed.
- Q. Wu, V. A. Maroni, D. J. Gosztola, D. J. Miller, D. W. Dees and W. Lu, J. Electrochem. Soc., 2015, 162, A1255–A1264 CrossRef CAS.
- R. E. Ruther, H. Dixit, A. M. Pezeshki, R. L. Sacci, V. R. Cooper, J. Nanda and G. M. Veith, J. Phys. Chem. C, 2015, 119, 18022–18029 CrossRef CAS.
- H. Koga, L. Croguennec, P. Mannessiez, M. Ménétrier, F. Weill, L. Bourgeois, M. Duttine, E. Suard and C. Delmas, J. Phys. Chem. C, 2012, 116, 13497–13506 CrossRef CAS.
- Y. Repelin, E. Husson, F. Bennani and C. Proust, J. Phys. Chem. Solids, 1999, 60, 819–825 CrossRef CAS.
- C. M. Julien and M. Massot, Mater. Sci. Eng., B, 2003, 100, 69–78 CrossRef.
- J. C. Garcia, J. Bareño, G. Chen, J. R. Croy and H. Iddir, Phys. Chem. Chem. Phys., 2020, 22, 24490–24497 RSC.
- J. R. Croy, J. S. Park, F. Dogan, C. S. Johnson, B. Key and M. Balasubramanian, Chem. Mater., 2014, 26, 7091–7098 CrossRef CAS.
- T. Ohzuku, M. Kitagawa and T. Hirai, J. Electrochem. Soc., 1990, 137, 769–775 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5eb00072f |
| ‡ These authors equally contributed. |
| § Current address: Materials Genome Institute, Shanghai University, Shanghai, China. |
| ¶ Current address: State Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan, China. |
| This journal is © The Royal Society of Chemistry 2025 |