
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ti-induced surface stabilization for enhanced capacity of aqueous-processed Ni-rich cathodes†
Heyin
Chen
 a,
Soham
Mukherjee
*b,
Tuan Thien
Tran
c,
Mikaela
Görlin
a,
Mahesh
Ramakrishnan
d,
Daniel
Primetzhofer
c,
William R.
Brant
a,
Haidong
Liu
*ae and
Maria
Hahlin
*ab
a,
Soham
Mukherjee
*b,
Tuan Thien
Tran
c,
Mikaela
Görlin
a,
Mahesh
Ramakrishnan
d,
Daniel
Primetzhofer
c,
William R.
Brant
a,
Haidong
Liu
*ae and
Maria
Hahlin
*ab
aDepartment of Chemistry –Ångström Laboratory, Uppsala University, Box 538, 751 20 Uppsala, Sweden. E-mail: maria.hahlin@kemi.uu.se; haidong.liu@kemi.uu.se
bDivision of X-ray Photon Science, Department of Physics and Astronomy, Uppsala University, Box 516, Uppsala SE-75120, Sweden. E-mail: soham.chem@gmail.com
cDivision of Materials Physics, Department of Physics and Astronomy, Uppsala University, Box 516, SE-571 20, Uppsala, Sweden
dMAX IV Laboratory, Lund University, Box 118, 221 00 Lund, Sweden
eDepartment of Technical Physics, University of Eastern Finland, 70210 Kuopio, Finland
First published on 22nd April 2025
Abstract
Aqueous processing offers an environmentally friendly alternative to conventional organic solvent-based methods for producing Ni-rich cathodes for lithium-ion batteries (LIBs). However, interaction with water can drive phase reconstruction at the cathode surface region, increasing activation polarization and charge transfer resistance, which can partially lower specific capacity. This study investigates the beneficial effect of Ti(IV) incorporation on single-crystalline Ni-rich LiNi0.8Mn0.1Co0.1O2 (SNMC) cathodes and demonstrates that the resulting Ti-rich, Ni-depleted surface layer enhances overall structural stability. Specifically, high-resolution scanning transmission electron microscopy images reveal that titanium suppresses undesirable rock-salt phase formation during aqueous processing, electrochemical impedance spectroscopy shows that Ti-incorporated SNMC cathodes exhibit lower charge transfer resistance compared to the conventional SNMC electrodes, and soft X-ray photoelectron spectroscopy exhibits a thinner cathode electrolyte interphase (CEI) layer with Ti incorporation. Improved cycling capacity was evidenced down to the atomic scale by utilizing operando X-ray absorption spectroscopy measurements, revealing stronger changes in the formal valence of all transition metals (or higher redox activity/charge compensation) during battery cycling for Ti-incorporated SNMC electrodes compared to the conventional SNMC electrodes. These results highlight that controlled surface modification using titanium can effectively stabilize the surface structure and reduce charge transfer resistance, offering a viable strategy to improve the structural and electrochemical performance of aqueous-processed Ni-rich cathode materials.
Broader contextWe are pleased to submit our original research titled “Ti-induced surface stabilization for enhanced capacity of aqueous-processed Ni-rich cathodes” for consideration for publication in the journal of EES batteries as a regular article.Aqueous processing offers a more sustainable alternative to conventional organic solvent-based methods for producing Ni-rich cathodes for lithium-ion batteries (LIBs). A major challenge to this method stems from undesirable phase reconstruction at the cathode surfaces upon interaction with water, increasing activation polarization and charge transfer resistance, which can partially lower specific capacity. Our study reveals how strategic Ti-incorporation in Ni-rich cathodes used in lithium-ion batteries effectively stabilizes the particle surface by suppressing the rock-salt phase formation and preserving the layered structure. Such surface-stabilization reduces charge transfer resistance, enhances cycling capacity, and results in the formation of a thinner cathode–electrolyte interphase layer. The EES batteries journal focuses on significant advancements in battery research, with a vision of transitioning toward a more sustainable energy future. In this context, our work addresses a major challenge related to the development for the aqueous processing method in manufacturing of the Ni-rich cathodes for lithium-ion batteries (LIBs), potentially enabling broader applications in sustainable battery production technologies. Given the highly topical nature of this work, we believe it will appeal to a broad audience in the sustainable battery technology communities, aligning well with the scope of EES batteries. |
Introduction
Ni-rich layered-structure LiNixMnyCo1−x−yO2 (NMC, x > 0.5) cathode materials in Li-ion batteries (LIBs) have been widely reported due to their high specific capacity and low cost.1–7 Traditionally, Ni-rich cathodes are produced using N-methyl-2-pyrrolidone (NMP) as a solvent in combination with polyvinylidene fluoride (PVDF) as a binder. To develop a more sustainable and environmentally friendly electrode manufacturing, aqueous processing is considered an alternative to the hazardous, volatile, and flammable NMP solvent.8–11 However, aqueous processing during electrode preparation introduces several challenges in the manufacturing of electrodes, which can lead to lower performance compared to conventional solvent-based processes. One significant issue is that the reaction between water and the NMC particle surface produces LiOH and/or induces the formation of carbonate species on the particle surface, which can negatively affect electrode performance.12 Also, during slurry coating on the Al current collector, the high pH of the slurry can lead to corrosion of the Al foil. Furthermore, Li-ions can leach out from the material and the induced Li/Ni disordering in the near-surface regions leads to surface reconstruction.9,12,13 These structural changes in the near-surface region increase activation polarization and charge transfer resistance, ultimately diminishing the specific capacity and cycling stability of the cathode material.To address these issues, a few mitigation approaches, like slurry additive, materials surface coating, and material doping have been reported. For instance, involving additives such as H3PO4 and Li2SO4 during slurry mixing lowers Al current collector corrosion. It has also been shown that H3PO4 can enhance the cycling performance to some degree by inhibiting the electrochemically inactive phase formation.14–16 However, H3PO4 does not form a uniform protective layer to prevent the reaction between NMC particles and H2O. Protective surface coatings, such as Al2O3, Li2O–B2O3, ZrO2, and spinel NiFe2O4, have been investigated to mitigate the interfacial degradation during aqueous processing.17–21 While this strategy offers a potential route for material protection during aqueous processing, the thickness of the coating material and the associated costs bring extra consideration to this approach. Additionally, bulk doping with Al, Ti and Zr have been utilized to stabilize the particle structure.22–25 Specifically, Ti doping is known to enhance the structural integrity of Ni-rich cathodes through stronger Ti–O bonds,26 thereby improving cycling stability. A targeted Ti-rich surface on the Ni-rich cathode particles was achieved to study its beneficial effect on electrochemical interface stability and cycling performance at high voltages.27 Another study identified the Ti surface-doped NMC material exhibits enhanced discharge capacity under low temperatures by changing the lattice parameters.28 The reported effects from Ti have the potential to be beneficial also in mitigating the challenges with water-based processing. To establish this, the role of Ti (surface) doping in suppressing Li/Ni cation disorder during water-based processing in the slurry during electrode preparation and its impact on the charge transfer kinetics needs to be clarified. For example; (1) how does Ti incorporation influence surface reconstruction post-aqueous processing? (2) how do the Li-ion transfer kinetics and electrochemical performance get enhanced with Ti incorporation? Moreover, the specific mechanisms of Ti doping in modifying the cathode–electrolyte interphase (CEI) and its effects on activation overpotential require further investigation. Addressing these knowledge gaps is critical for optimizing aqueous-processed Ni-rich cathodes, potentially enabling broader applications in sustainable battery production technologies.
In this study, we investigate the role of Ti(IV) in single-crystalline LiNi0.8Mn0.1Co0.1O2 (SNMC) materials in enhancing the electrochemical performance of the aqueous processed cathodes. We find that, as targeted, Ti is effectively localized in the near-surface regions of the Ti-incorporated SNMC (Ti-SNMC) particles, forming a thin, spatially homogeneous Ti-rich, Ni-depleted layer that suppresses the formation of the rock-salt phase during aqueous processing. The surface stabilization leads to reduced activation polarization and charge transfer resistance, resulting in enhanced initial capacities and electrochemical performance, as well as the formation of a thinner CEI layer. The improved surface stability with less charge transfer resistance for the aqueous-processed Ti-SNMC particle affects larger changes in the valence states of Ni, Co and Mn compared to the SNMC electrode cycled within the same voltage window. This confirms that the undesired rock-salt phase formed during aqueous processing is primarily responsible for hindering Li-ion transfer in the SNMC particles, and thereby reduces the specific capacity. In a broader perspective, our findings highlight how surface doping/incorporation in NMC cathodes can be utilized as a key design parameter to develop more sustainable and high-performance lithium-ion batteries in terms of electrode preparation.
Experimental section
Materials synthesis
The conventional SNMC particles were synthesized with procedures explained as follows: a stoichiometric ratio of lithium, nickel(II), manganese(II), and cobalt(II) acetate salts were mixed and dissolved in deionized water. During heating and stirring, lithium acetate (3 mol% excess lithium for the compensation of lithium loss) and citric acid (chelating agent/transition metal ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were added to the solution. The solution was continuously stirred and heated on a heating plate at ∼60 °C until complete dehydration was achieved. The obtained mixture was dried overnight at 100 °C in a ventilated oven. The resulting precursor powder was calcined at 500 °C for 5 hours and 850 °C for 12 hours in a tube furnace under O2 atmosphere with a heating rate of 5 °C min−1 to obtain the pristine SNMC particles. The Ti-incorporated material LiNi0.8Mn0.095Co0.1Ti0.005O2 was prepared by dissolving a stoichiometric amount of titanium source into the water with the required amounts of other transition metal salts as described above. Titanium(IV) isopropoxide (99.9%, Sigma-Aldrich), was used as the Ti source. The following steps were the same as the synthesis of the pristine SNMC811 as described above. Results from inductively coupled plasma atomic emission spectrometry (ICP-OES) measurements are displayed in Table S1 (ESI†), confirming the elemental composition and successful doping. The synthesized particles are confirmed to be single crystalline by the HR-TEM images and the corresponding Fourier transform patterns (see Fig. S1, ESI†).
:1) were added to the solution. The solution was continuously stirred and heated on a heating plate at ∼60 °C until complete dehydration was achieved. The obtained mixture was dried overnight at 100 °C in a ventilated oven. The resulting precursor powder was calcined at 500 °C for 5 hours and 850 °C for 12 hours in a tube furnace under O2 atmosphere with a heating rate of 5 °C min−1 to obtain the pristine SNMC particles. The Ti-incorporated material LiNi0.8Mn0.095Co0.1Ti0.005O2 was prepared by dissolving a stoichiometric amount of titanium source into the water with the required amounts of other transition metal salts as described above. Titanium(IV) isopropoxide (99.9%, Sigma-Aldrich), was used as the Ti source. The following steps were the same as the synthesis of the pristine SNMC811 as described above. Results from inductively coupled plasma atomic emission spectrometry (ICP-OES) measurements are displayed in Table S1 (ESI†), confirming the elemental composition and successful doping. The synthesized particles are confirmed to be single crystalline by the HR-TEM images and the corresponding Fourier transform patterns (see Fig. S1, ESI†).
Electrode preparation
For the aqueous casting, 90% SNMC or Ti-SNMC was mixed with 5% conductive carbon black (Super C65T, Imerys) and 5% carboxymethyl cellulose (Na-CMC, Dow Wolff Cellulosics) binder. The aqueous-based mixture was coated on a carbon-coated Al foil (SSNTM Co., Ltd), as the current collector. The coated electrodes were pre-dried at 70 °C for 5 hours, and then dried at 120 °C for 12 hours in a vacuum oven. The produced electrodes were stored in an Ar-filled glove box to prevent moisture and air exposure before cell assembly.Transmission electron microscopy (TEM) measurements
Prior to TEM analysis, electron transparent lamellae from the two samples (SNMC and Ti-SNMC) were prepared using a focused ion beam system (Zeiss FIB-SEM Crossbeam 550). The lamellae were sculpted from the samples and subsequently transferred to copper (Cu) TEM grids using an in situ micromanipulator. Once on the Cu grids, the lamellae were finally thinned down and polished to a thickness of ∼50 nm. TEM analysis was conducted with a Thermo Fisher Titan Themis 200 TEM system at Uppsala University. The system is equipped with a spherical aberration corrector for achieving a spatial resolution below 1 Å at an acceleration voltage of 200 kV. For HR-S/TEM, a double-tilt TEM sample holder was used for orienting one of the zone axes of the crystals parallel to the electron beam. For Energy-Dispersive X-ray spectroscopy (EDS) acquisition, a SuperX EDS detecting system of the TEM was used. Finally, the EDS data were analyzed using the Velox program (Thermo Fisher).Time-of-flight elastic recoil detection analysis (ToF-ERDA)
Depth profiles of the overall elemental composition of the SNMC and Ti-SNMC electrodes were nondestructively obtained using ToF-ERDA using the 5 MV pelletron accelerator at Uppsala University.29 A primary beam of 36 MeV iodine ions was employed to create recoils being detected in a detector telescope positioned under 45 degrees with respect to the incident beam axis.30 Data evaluation was performed using the CONTES software.31Ex situ photoelectron spectroscopy (PES) analysis
The pristine (uncycled) SNMC and Ti-SNMC electrodes were measured using the HAXPES instrument (Scienta Scientific) with the Ga Kα (9252.79 eV) sources. The electrodes were mounted on Omicron sample plates using conductive adhesive tabs. The pass energy and slit width were selected as 300 eV and 0.8 mm for the spectrometer, respectively. Ni 2p and Ti 2p spectra were collected with step size 0.1 eV. The cycled SNMC and Ti-SNMC were disassembled from the pouch cells after the initial cycle and were washed using dimethyl carbonate subsequently before the PES measurements. The cycled electrodes were measured using the AXIS Supra + (Kratos) instrument with the Al Kα (1486.7 eV) source. The samples were transferred to the instrument with an air-tight sample holder to avoid exposure to air. C 1s, F 1s, O 1s, and P 2p spectra were collected with step size 0.1 eV.All obtained spectra were curve-fitted using Igor Pro 9 software with combined Lorentzian and Gaussian functions, in which the full width at half maximum (FWHM) is fixed to 0.2 eV (core levels of light elements) and 0.4 eV (Ni 2p and Ti 2p, larger lifetime broadening) for the Lorentzian function contribution and variable in the Gaussian functions to converge the fitting. The spin-orbit splitting value in Ti 2p was set to 5.7 eV.32 A Shirley background was utilized in the Ni 2p spectrum, while a linear background was subtracted in the rest spectra. The binding energy was referenced to Ti 2p3/2 peak at 457.4 eV for the Ni 2p spectrum measured with the Ga-source, while for the spectra measured by Al Kα, the energy calibration was referenced to the hydrocarbon peak in C 1s at 285 eV.
Ex situ soft X-ray absorption spectroscopy (XAS) analysis
The soft X-ray absorption measurements were conducted at the FlexPES beamline at the MAX IV Laboratory, Lund.33 SNMC and Ti-SNMC were measured as powder, pristine electrodes, and after the first charge electrodes with partial electrode yield (PEY), total electron yield (TEY), and fluorescence yield (FY) modes, providing depth information ∼2 nm, ∼5 nm, and ∼100 nm respectively. NiO, LiNiO2, Li2CO3, and TiO2 powder were measured as well as references. All spectra were collected with an MCP (microchannel plate) detector with retardation voltage – 400 V, −300 V, and −600 V for O K-, Ti L2,3-, and Ni L2,3-edge absorption spectra in the PEY mode. The obtained spectra were normalized by the I0 signal from a clean gold sensor in the beamline, and the background was further corrected by subtracting a linear background to ensure constant pre-edge intensity. Further, the spectra were spectra were normalized to a point after the absorption edge.Cell preparation and electrochemical characterization
Cathode electrodes having 13 mm diameter were combined with 15 mm diameter Li disk (China Energy Lithium Co., Ltd) as the anode in pouch cells, separated by Celgard 2325 separators. 100 μL electrolyte containing 1 mol L−1 LiPF6 in a mixture of ethylene carbonate/ethyl methyl carbonate (EC/DEC 3:7 by volume, Solvionic) was added. Galvanostatic cycling performance was recorded using the Arbin battery testing system at 0.1C (20 mA g−1) between 3.0 and 4.3 V. Electrochemical impedance spectroscopy (EIS) was carried out using a VMP instrument (Bio-Logic) over a frequency range of 100 kHz to 1 mHz with a 5 mV amplitude perturbation. Three-electrode cells were prepared with SNMC or Ti-SNMC electrode as the cathode, Li disk as the anode, and Li foil as the reference electrode. The EIS spectra were recorded at 3.9 V (vs. Li/Li+) during charge at 0.1C (20 mA g−1) after 4 formation cycles (0.1C). Before collecting each EIS spectrum, the cells rested until the current was less than 1 uA.
Operando XAS analysis
The operando hard XAS measurements were carried out at the Balder beamline at the MAX IV Laboratory, Lund.34 During the measurements, two pouch cells were cycled at 0.1C with VMP Potentiostat (Bio-Logic) equipment, and Ni, Co, and Mn K-edge XAS spectra were collected in transmission mode over a longer energy range (−150 eV to +750 eV) for Ni to extract the EXAFS region, while Co and Mn data was limited to XANES (−80 eV to +180 eV). The incoming beam size was monochromatized by a Si (111) double crystal monochromator and focused (spot size ∼100 × 80 μm) on the pouch cells. Two cells were mounted in the customized pouch cell holder and measured sequentially. Multiple scans were collected at each transition metal (TM) edge in several loops to improve the statistics, with the beam being moved to a fresh spot every 20 min in the pouch cell to mitigate any radiation damage, as well as probing a larger electrode volume. The Athena software package35 was used to process the XAS data, including energy calibration, followed by two background subtractions: a linear background for the energy region below the absorption edge, and a third-order polynomial at energies above the absorption edge. The edge energy positions (referred to as E0 here onwards) were extracted from the 1st derivatives of the XANES spectra36 for all TM ions, as the higher noise level of the operando process restricted the linear combination approach37 to estimate E0 values. Energy calibration was performed using metallic Ni metal foils. The measured E0 was 1.8 eV higher than the reference energy of 8332.8 eV. All spectra were subsequently shifted by 1.8 eV to lower energies. EXAFS simulations were performed using Artemis, part of the IFEFFIT suite, and SimXLite (developed by Prof. Holger Dau and Petko Chernev at the Free University of Berlin).38 Similar energy calibrations were performed for Co (1.3 eV) and Mn (1.7 eV) by applying energy offsets calculated from the respective metal foils.Phase functions were generated using FEFF939 with a self-consistent field option activated and atomic coordinates from the LiNiO2 structure. The considered data range was 1.98–12.55 Å−1 (15–600 eV). The local atomic coordination numbers (CN) were constrained to 6, while the coordination distances (R) and Debye–Waller parameters (σ2) were fitted. An amplitude reduction factor (S02) of 0.85 was used. All scatterers were selected as Ni atoms for simplicity since Ni constitutes the major element in the material (∼80%) and the phase functions do not vary significantly between Mn, Co, and Ni. Optimization was performed using a least-squares fit and the Levenberg–Marquardt algorithm with numerical derivatives. The error associated with the fit parameters was estimated from the covariance matrix, corresponding to a 68% confidence level. The R-factor filtered (Rf) was used as a measure of the quality of the fit and is calculated as the percentage difference between the experimental and simulated EXAFS spectra. The bond valence sum (BVS) was used to estimate the oxidation states of the absorbing based on the M–O shell.
Results and discussion
Ti enrichment at the particle surface
TEM-EDS measurements were conducted to investigate the distribution of all transition metals (TM) within the SNMC and Ti-SNMC particles. The EDS mapping in Fig. 1 reveals the distribution of the TM from the particle surface towards the bulk, and the estimated composition values for the TM. The schematics to the left of Fig. 1 illustrate the experimental setup, including sample lamella thickness, the direction of the electron beam, and an arrow indicating the path from the particle's surface toward the bulk. In the SNMC particle (Fig. 1a), Ni, Co, and Mn were homogeneously distributed from the surface to the bulk, with compositions of Ni, Mn, and Co being 79.6 ± 2.9 at%, 9.1 ± 1.7 at%, and 11.3 ± 1.9 at% respectively, which are in close agreement with the ideal elemental stoichiometry (Ni:Co:Mn = 8:1:1 in NMC811). The addition of Ti (Fig. 1b) however, altered the TM composition in the near-surface region. Specifically, this near-surface region exhibits a decrease in Ni concentration, and a simultaneous increase in Co and Mn levels relative to the ideal composition which is still maintained in the bulk of the particle (close to Ni:Co:Mn = 8:1:1). Interestingly, the surface gets substantially enriched with Ti, while the bulk of the Ti-SNMC particle contains minimal Ti. This Ti-incorporated surface is reported to be more stable compared to the bare Ni-rich surface in the NMC particles.
 | ||
| Fig. 1 Measurement spot sketches, TEM-EDS element mapping, and transition metals composition of pristine/uncycled (a) SNMC and (b) Ti-SNMC electrodes. | ||
To obtain a more precise estimate of the concentration ratio of Ni to Ti at the surface regions up to depths of around 30 nm, PES measurements were carried out using the Ga Kα source. The collected Ti 2p and Ni 2p core level spectra are displayed in Fig. 2. The concentration ratios between Ti and Ni were calculated from the integrated peak area (Iarea) using the equation below,40
| Iarea = B × σ(hv,α) × T(Ek) × na × λ(Ek) |
:17, which is about an order of magnitude higher than the average Ti to Ni ratio of 1:170 (ICP results, Table S1 in ESI†) in the particles. In addition, the obtained composition depth profiles of relevant chemical species from the first approximately 300 nm from the sample surface are shown in Fig. S2 (ESI†) using ToF-ERDA. Apart from near-surface degradation caused by impurity contamination, the element composition remains uniform beneath the near-surface region, featuring an overall ratio of Ti to Ni of approximately 1:75. Combining all the four independent analytical approaches (EDS, ICP, PES and ToF-ERDA) provides a comprehensive description of the nature of Ti incorporation in NMC, confirming that significant Ti enrichment occurs in a rather thin layer close to the surface of the Ti-SNMC particles.
 | ||
| Fig. 2 The Ti 2p and Ni 2p spectra measured with a Ga Kα source from pristine Ti-SNMC electrodes. | ||
| Photon source | Cross-sections | Area intensity | Ratio (Ti:Ni) |
||
|---|---|---|---|---|---|
| Ti 2p | Ni 2p | Ti 2p | Ni 2p | ||
| Ga Kα | 4.7 × 10−4 | 1.7 × 10−3 | 1710 | 107600 |
∼1:17 |
Suppression of rock-salt phase formation
To investigate the effects of the aqueous processing on the NMC particle surface structure, HR-STEM images from SNMC and Ti-SNMC particles were obtained. Fig. 3a shows that the near-surface structure of SNMC particles has transformed into a different phase compared to the bulk. This altered phase is identified as the rock-salt phase, which can be attributed to the Li/Ni disorder induced during the aqueous processing.16,42,43 It has been reported that this rock-salt phase forms irreversibly and is primarily responsible for the capacity fade of the electrode and cracking in the particles.3,44 In contrast, no rock-salt phase is observed at the Ti-SNMC particle surface as shown in Fig. 3b. The fast Fourier transform (FFT) analysis reveals that the layered structure is preserved at the surface regions in Ti-NMC particles, similar to the crystal structure in the bulk, confirming that Ti is incorporated into the layered structure of NMC. The absence of a phase boundary suggests that the Ti-rich, Ni-depleted surface in Ti-NMC particles has higher structural stability than the Ni-rich surface of SNMC particles, thereby effectively alleviating rock-salt phase formation. | ||
| Fig. 3 (a) High-resolution STEM images of the uncycled SNMC sample. Fast Fourier transform (FFT) shows the particle has a layered crystal structure in the bulk and a rock-salt structure in the near-surface regions, in agreement with the literature.42 (b) High-resolution STEM images of the uncycled Ti-SNMC electrode. FFT of the image at the near-surface and the bulk region shows similar patterns, indicating identical crystal structures in both regions, with no evidence of rock-salt phase formation. XAS-PEY spectra of Ti L2,3-edge measured from Ti-SNMC powder, pristine and after 1st charge Ti-SNMC electrodes, and powder material TiO2 (c), Ni L2,3-edge (d) and Co L2,3-edge (e) spectra collected from SNMC and Ti-SNMC powder and (uncycled) electrodes. | ||
Ti L2,3-edge XAS (Fig. 3c) reveals four characteristic narrow peaks for Ti-SNMC particles, unlike TiO2 powder which exhibits a peak splitting at ∼460 eV. This implies that Ti is effectively incorporated into the near-surface region of the NMC structure,45,46 rather than existing as TiO2 deposited on the surface. Both XAS-FY spectra and more surface-sensitive XAS-TEY spectra (Fig. S3 in ESI†) reveal that Ti in the Ti-SNMC particles predominantly exists in the Ti4+ state (d0 configuration) under various charging conditions, indicating that Ti ions do not participate in the electrochemical reactions.46 With Ti4+ incorporated into the layered structure, charge neutrality can be maintained either by reducing the Li content or by lowering the valence state of the transition metals (Ni, Mn, and Co). While determining the concentration of Li in the near-surface region is non-trivial, soft XAS allows us to obtain the valence state information of the transition metals. The Ni and Co L2,3-edge spectra of Ti-SNMC powder reveal slightly stronger Ni2+ and Co2+ signals, compared to SNMC powder (Fig. 3d and e). This is evidenced by an increased peak intensity at ∼853 eV (L3 low) relative to the peak at ∼855 eV (L3 high) in the Ni L2,3-edge spectrum (Fig. 3d) and by a small peak intensity at ∼777 eV in the Co L2,3-edge spectrum (inset in Fig. 3e). However, no significant differences are seen in XAS-FY spectra, compared to the XAS-TEY and XAS-PEY spectra, between SNMC and Ti-SNMC (Fig. S4, ESI†), suggesting that Ti incorporation does not sufficiently influence the valence states of Ni and Co in the bulk of the Ti-SNMC particles. Similar conclusions can be drawn for the Mn L2,3-edge spectra which also show no noticeable changes from surface to bulk (Fig. S5, ESI†). For the aqueous-processed SNMC and Ti-SNMC electrodes, Ni and Co L2,3-edge spectra from the electrodes are presented at the bottom of Fig. 3(d and e). The observed differences in the Ni and Co L2,3-edge spectra of SNMC and Ti-SNMC are negligible, thus inferring that the aqueous processing induced a relatively larger increase of the Ni2+ and Co2+ in the case of SNMC electrodes. It is however noted that for both systems, the amount of Ni2+ state increases during the aqueous processing. This is possibly due to some degree of Li+ leaching and oxygen loss in both systems, however mitigated for Ti-SNMC. The above results thus show that although Ti incorporation changes the stoichiometry in the near-surface region of the Ti-SNMC particle, the Ti4+ ions remain redox inactive. Instead, Ti mainly enhances structural stability by retaining a continuous phase which likely benefits charge transfer.
Enhanced initial capacity and thinner CEI layer formation
The cycling performance of SNMC and Ti-SNMC electrodes is shown in Fig. 4. After 5 formation cycles, the delivered discharge capacities of the SNMC and Ti-SNMC electrodes are ∼160 and ∼180 mA h g−1, respectively, as shown in Fig. 4a. Capacity retentions after 55 cycles were recorded as 97.8% and 93.6% (compared to the 5th cycle) for SNMC and Ti-SNMC electrodes, respectively. The slightly higher capacity retention in the SNMC electrode is attributed to increased overpotential, which results in a narrower actual cycling window and reduces the extent of capacity fade. In the charge/discharge capacity profile of the 1st and the 55th cycles in Fig. 4b and c, the SNMC electrode displays a higher overpotential compared to the Ti-SNMC electrode, especially in the initial cycle. The overpotential in the initial cycle is most likely due to impurity species formed from reactions between the reactive SNMC electrode surface and atmospheric gases during water processing, slurry coating, and electrode storage.47,48 The overpotential is reduced after the initial cycle (see Fig. S6a†). However, it is still unclear what species were decomposed from the C 1s spectrum (Fig. S6b†), and the CO32− species seems to be stable during the initial charge. Phase transitions occurring in SNMC and Ti-SNMC electrodes during cycling are shown in the dQ/dV plot in Fig. 4d and e.25,44,49 Typically, Li-ion extraction from NMC cathodes is characterized by series of phase transitions, i.e., from the initial Hexagonal 1 (H1) phase to the Monoclinic phase (M) at ∼3.7 V, followed by M to Hexagonal 2 (H2) phase, and from H2 to Hexagonal 3 (H3) phase appearing at ∼4.0 V and ∼4.2 V, respectively. In the dQ/dV curves in Fig. 4d, the SNMC electrode exhibits a high overpotential (activation polarization) during the first charge, resulting in an incomplete phase transition from M to H2 and H2 to H3 phases, thereby limiting its capacity. The Ti-SNMC on the other hand shows only a minimal overpotential in the first cycle, allowing complete phase changes up to H3 at the high cut-off voltage (e). The (003) Bragg reflection from the XRD patterns (Fig. S7†) reveals that at the end of the charge, many Ti-SNMC particles assume close to the H3 phase. In contrast, the H2 to the H3 phase transformation is less complete in SNMC compared to Ti-SNMC.3,50,51 The reduced Li-ion extraction from the SNMC electrode during the charging process leads to fewer Li-ions available for reinsertion during the discharging process, explaining a lower initial discharge capacity compared to the Ti-SNMC electrode.52 Increased charge transfer resistance for the SNMC electrode, compared to the Ti-SNMC electrode, is confirmed by EIS measurements, as shown in Fig. 4f. Based on these electrochemical data, we therefore suggest that the Ti-rich surface in the Ti-SNMC particle reduces charge transfer resistance by mitigating rock-salt phase formation during water processing, which in turn facilitates a higher charge capacity. | ||
| Fig. 4 Electrochemical characterization of SNMC and Ti-SNMC electrodes with galvanostatic cycling in half cells at 0.1C with a cut-off voltage window between 3 and 4.3V vs. Li+/Li. (a) The specific discharge capacity of both electrodes for 55 cycles. Charge–discharge capacity profile of 1st (b) and 55th (c) cycle. The dQ/dV plot for pouch cells with cathode SNMC (d), and Ti-SNMC (e) of 1st, 18th, and 55th cycles. (f) EIS curves measured at 3.9 V during charge in 5th cycle at 0.1C. | ||
PES measurements were conducted on the SNMC and Ti-SNMC electrodes after the initial cycle to analyze the thickness and chemical composition of the initial CEI and link this to the surface stability of the uncycled electrodes. As displayed in Fig. 5, C1s, O 1s, F 1s, and P 2p core level spectra have been collected using Al Kα radiation for both electrodes at the discharge state after the initial cycle at 0.1C, with the different chemical species labelled at the peaks.53–55 Detailed peak assignments and peak positions in binding energies are shown in Table S2 (ESI†). In the C 1s spectrum, negligible differences are observed between SNMC and Ti-SNMC electrodes, except for a relatively higher intensity of C–H species in the SNMC electrode compared to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C peak, suggesting a greater accumulation of hydrocarbon species on the SNMC electrode surface. In the O1s spectrum, the Ti-SNMC electrode shows stronger M–O (related to the hexagonal crystal structure) signals from bulk. These observations indicate that either a thinner CEI layer formed on the Ti-SNMC electrode, or that the number of oxygen species in the surface region of the Ti-SNMC is higher. A thinner CEI is typically associated with lower Li-ion transfer resistance, which is consistent with the EIS measurements in the first semi-circle in Fig. 4f. Furthermore, the formation of a rock-salt structure in the near-surface region of the SNMC electrode coincides with a theoretical 50% decrease in oxygen atoms (LiTMO2vs. NiO), compared to Ti-SNMC. The relative ratio between the M–O and CC intensities differs between the samples, suggesting that both the above explanations are plausible. In the F 1s and P 2p spectra, increased peak intensity associated with LiPF6 species is observed in the Ti-SNMC electrode, which is most likely due to electrolyte residue on the electrode surface. Overall, a comparison of the SNMC (top) and Ti-SNMC (bottom) electrodes after the first cycle shows that Ti stabilizes the surface of the aqueous processed electrode, modifying the surface chemistry and consequently forming a thinner CEI layer.
C peak, suggesting a greater accumulation of hydrocarbon species on the SNMC electrode surface. In the O1s spectrum, the Ti-SNMC electrode shows stronger M–O (related to the hexagonal crystal structure) signals from bulk. These observations indicate that either a thinner CEI layer formed on the Ti-SNMC electrode, or that the number of oxygen species in the surface region of the Ti-SNMC is higher. A thinner CEI is typically associated with lower Li-ion transfer resistance, which is consistent with the EIS measurements in the first semi-circle in Fig. 4f. Furthermore, the formation of a rock-salt structure in the near-surface region of the SNMC electrode coincides with a theoretical 50% decrease in oxygen atoms (LiTMO2vs. NiO), compared to Ti-SNMC. The relative ratio between the M–O and CC intensities differs between the samples, suggesting that both the above explanations are plausible. In the F 1s and P 2p spectra, increased peak intensity associated with LiPF6 species is observed in the Ti-SNMC electrode, which is most likely due to electrolyte residue on the electrode surface. Overall, a comparison of the SNMC (top) and Ti-SNMC (bottom) electrodes after the first cycle shows that Ti stabilizes the surface of the aqueous processed electrode, modifying the surface chemistry and consequently forming a thinner CEI layer.
 | ||
| Fig. 5 The C 1s, O 1s, F 1s, and P 2p spectra collected from SNMC (top) and Ti-SNMC (bottom) electrodes at the discharge state after the initial cycle at 0.1C. All spectra were normalized by dividing each highest peak intensity. | ||
Enhanced Li-ions transfer kinetics with Ti incorporation
Operando XAS measurements were performed at K-edges of Ni, Co, and Mn to examine the effect of Ti doping in the TMs’ local environment. Delivered charge/discharge capacity in LIBs originates from the redox reactions of the transition metal environments (TM).56 Since absorption edge positions for K-XAS represent the binding energy of the 1s electrons of a given element, changes to the E0 values for TM K-XAS serve as an effective indicator to monitor redox processes for a given TM element, representing the state of charge/discharge.57Fig. 6(a and b) depicts how Ni K-XANES profiles for SNMC and Ti-SNMC electrodes (normalized) evolve as a function of applied potential during electrochemical cycling. The E0 positions extracted from Ni K-XANES (axis to the left) and the corresponding oxidation states of Ni (axis to the right) are displayed in Fig. 6c, where open and closed symbols denote the charging and discharging profiles, respectively. Fig. 6d shows the corresponding specific charge/discharge capacities within a cycling window of 3–4.3 V under the constant current rate of 0.1C (20 mA g−1) in this operando measurement. Fig. 6c clearly reveals the E0 values to increase/decrease monotonically (ΔE ∼ 3 eV) during charging–discharging, indicative of the reversible Ni3+ → Ni4+ redox process for both SNMC and Ti-SNMC, as also reported for NMC622 material.57 In Fig. 6d, at voltages above 3.8 V the change in E0 values for the Ti-SNMC electrode is greater than in the SNMC electrode during charging. This implies that at a given voltage during charging (∼3.8 V to ∼4.24 V), more Li-ions were extracted from the Ti-SNMC sample, most likely due to the reduced charge transfer resistance, as confirmed by our EIS measurements (see Fig. 4f). At the maximum applied voltage of 4.24 V, a larger shift in E0 is observed for the Ti-SNMC electrode, compared to SNMC, which explains the higher delivered specific capacity of the Ti-SNMC electrode. Although the Ni K-XANES for the Ti-SNMC electrode was recorded during discharge, it can be compared to the Ni K-XAS of the SNMC electrode since minimal electrochemical activity occurs during the discharge from 4.3 V to 4.24 V, as shown by the plateau in Fig. 6d.
 | ||
| Fig. 6 Normalized Ni K-edge XANES spectra collected from SNMC (a) and Ti-SNMC (b) electrodes at different voltages during charge/discharge from operando XAS measurements. (c) Comparative E0 for Ni ions and (d) specific capacities for SNMC (green) and Ti-SNMC (purple) cathodes as a function of voltage changes, cycled at 0.1C between 3.0–4.3 V. (e) Comparison of Ni K-edge EXAFS for SNMC (green) and Ti-SNMC (purple); solid and dotted lines representing the beginning of charge (BoC) at 3.7 V and at 4.24 V measured at close to the end of charge (∼EoC) and the beginning of discharge (∼BoD). (f) Comparison of Ni–O and Ni–Ni interatomic distances, and (g) corresponding pseudo-Debye–Waller factors (σ2) for all four electrodes denoted by the same color scheme. | ||
The variation of normalized K-XANES profiles with applied voltage for Co and Mn in SNMC (in green) and Ti-SNMC (in purple) electrodes are shown in Fig. S8(a and b) and S8(d and e) of the ESI,† respectively. The estimated E0 values plotted in Fig. S8c (for Co) and S8f (for Mn)† reveal smaller energy shifts for both Co and Mn as compared to Ni (Fig. 6c), indicating lower extents of charge compensation than Ni. However, the energy shifts were systematically higher for Co and Mn in case of Ti-SMNC (ΔE ∼ 2 eV) in comparison to SNMC (ΔE ∼ 1.5 eV), consistent with the observed differences in Ni redox profiles between the two electrodes. The larger uncertainties associated with the extracted E0 values for Co and Mn result from their lower concentrations compared to Ni in the NMC811 electrodes. Nonetheless, this finding highlights that Mn is electrochemically active during battery cycling, as opposed to some previous reports.58,59 We also note that Co becomes more electrochemically active above ∼3.9 V, similar to NMC622 materials.57 Such charge redistribution among the TM ions is not unexpected considering that Ni, Co, and Mn share a common crystallographic site and participate in the charge compensation process to varying extents. Thus, the rock-salt phase-induced increase in charge transfer resistance narrows down the range of oxidation state variation for all transition metals during battery cycling.
Comparing the operando Ni EXAFS data of SNMC and Ti-SNMC electrodes helps to understand how Ni charge compensation mechanisms affect the local Ni geometry during battery cycling and whether Ti affects such structural changes. Fig. 6e shows the modulus of the R-space (mod[χ(R)]) functions for SNMC and Ti-SNMC measured at 3.7 V (the beginning of charge, BoC) and at 4.24 V (close to the end of charge, ∼EoC and beginning of discharge, ∼BoD, respectively). It can be found that the overall χ(R) amplitudes increase upon charging, indicating a higher local order (lower σ2) around Ni sites in the charged state. This is particularly visible for the Ni–O6 octahedra (the first peak. i.e., the first coordination shell), and the change is relatively smaller for the second peak which corresponds to the TM ions (the second coordination shell).
Local atomic parameters were determined using feff simulations.39 The coordination numbers (CN) were fixed, while the bond lengths (R) and Debye–Waller factors (σ2) were optimized. Fits to the first (O) and second (TM) coordination shells allow extraction of the Ni–O and Ni–Ni interatomic distances (Fig. 6f) and the corresponding disorder values (Fig. 6g), respectively. Fits to mod[χ(R)] of Ni K-EXAFS for all four samples (SNMC and Ti-SNMC at 3.7 V and 4.24 V) are shown in Fig. S9 in the ESI.†Fig. 6f reveals that Ni–O bond lengths in both SNMC and Ti-SNMC contract by ∼0.07 Å from 1.94 to 1.87 Å upon charging, consistent with an increase by roughly 1 unit in oxidation state from Ni3+ to Ni4+ (Table S3, ESI†).57,60–62A contraction of both the Ni–O and the Ni–TM interatomic distances upon charging is consistent with lattice volume contractions during delithiation.57 As Li-ions are extracted from the structure, Ni oxidizes to compensate for the charge; the resultant higher charge density shrinks the local volume and improves the local order around Ni sites. During discharge, intercalation of Li-ions induces Ni reduction, expanding the local volume and decreasing local order back to its original state. These changes in R and σ2 are highly reversible and occur continuously over the entire voltage window preserving the structural integrity of the hexagonal structure. However, Fig. 6f and g reveals that the R and σ2 values for Ti-SNMC and SNMC are rather comparable. This is due to the low Ti content, which is mostly incorporated near the surfaces of these micron-sized particles and thus has minimal impact on the bulk structure of the active material. Instead, as illustrated in Fig. 7, the Ti-incorporated surface maintains the layered structure in the Ti-SNMC particle after aqueous processing, consequently enhancing Li-ion transfer kinetics. Such capacity enhancement by strategic Ti incorporation was evidenced down to the atomic-scale, affecting faster redox processes at the TM sites (Fig. 6c).
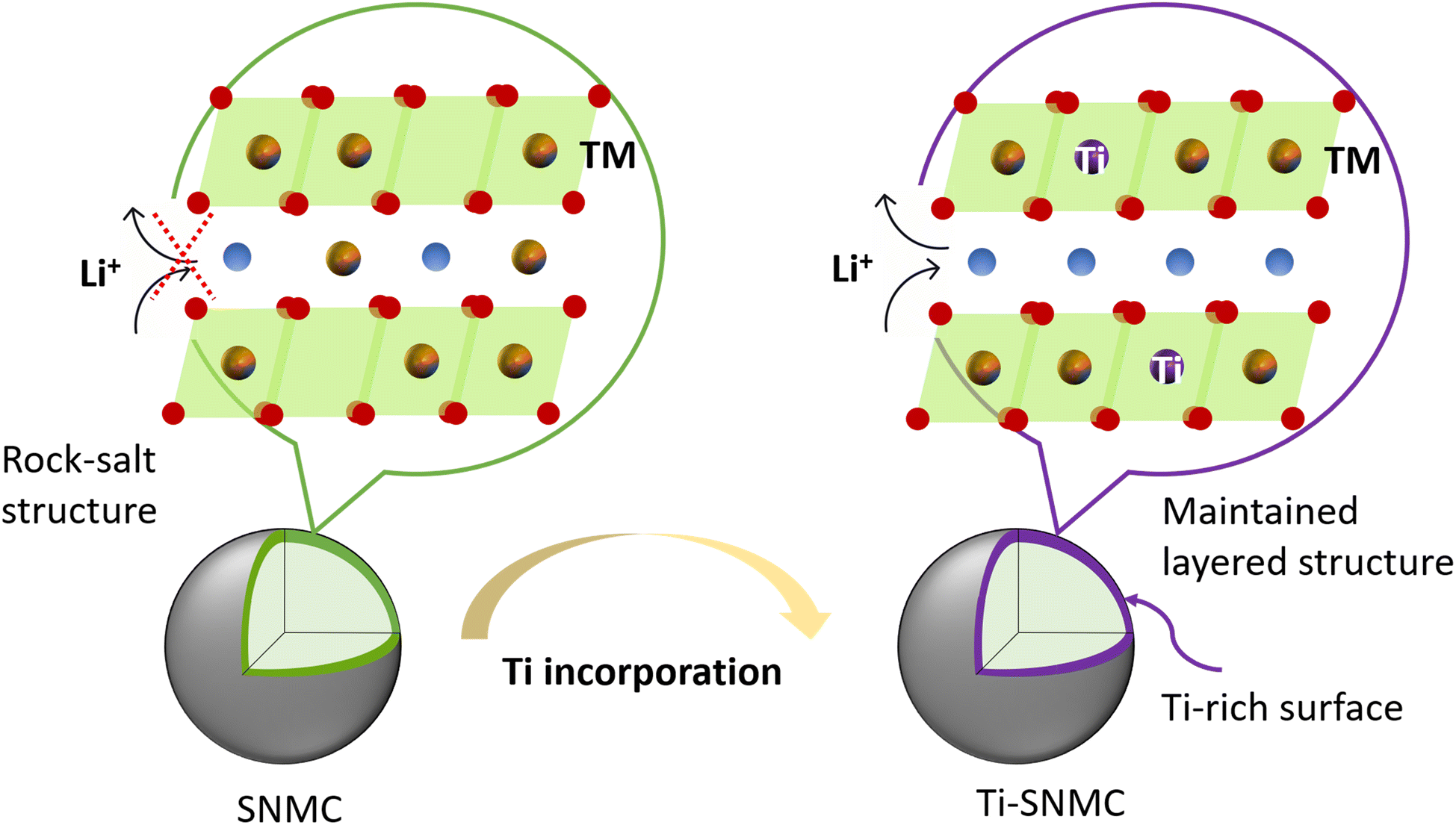 | ||
| Fig. 7 The illustration of Li-ion transfer kinetics in the aqueous-processed SNMC and Ti-SNMC particles. The rock-salt phase formation at the surface of the SNMC particle increased the charge transfer resistance, while the surface structure of the Ti-SNMC particle was well maintained due to Ti incorporation. | ||
Conclusions
We investigated the role of titanium in enhancing the capacity and cycling performance of aqueous-processed NMC electrodes. The Ti4+ (d0) ions are mostly localized in the near-surface regions, modifying the Ti-SNMC particle surfaces by suppressing the irreversible formation of the undesired rock-salt phase during electrode preparation using an aqueous solvent. This preserves the inherent single-phase layered structure, thereby aiding in reducing activation polarization during cycling. The surface stabilized Ti-SNMC electrode subsequently forms a thinner CEI layer after the initial cycle, compared to the SNMC electrode. During electrochemical cycling, the Ti-SNMC electrode shows a larger change in oxidation state for Ni compared to the SNMC electrode. Co and Mn contributions to the redox reactions, albeit lower than Ni, increase systematically upon Ti incorporation. Thus, although Ti4+ does not participate in the redox reactions and remains more confined to the particle surfaces, it improves TM redox activities. This stems from reduced charge transfer resistance in the Ti-SNMC electrode benefiting from the stability of the layer structure in the surface regions. Overall, our work demonstrates that localized Ti incorporation leads to improved electrochemical performance of aqueous-processed electrodes through a combination of factors: stabilizing the surface structure, reducing charge transfer resistance, and enhancing the reversibility of phase transitions. The study highlights that precise control of Ti incorporation can effectively address capacity degradation associated with the aqueous processing of Ni-rich cathodes. The ability to tailor such surface modification strategies is a key tool for advancing the design of doped/incorporated Ni-rich cathodes towards more sustainable battery technologies.Data availability
The data supporting this article have been included as part of the ESI.† This includes additional TEM, soft XAS, and operando XAS measurements on SNMC and Ti-SNMC powder and electrodes, ToF-ERDA measurements on uncycled SNMC and Ti-SNMC electrodes, ICP measurements on SNMC and Ti-SNMC powder, simulated k3-weighted Ni K-edge EXAFS spectra and parameter for SNMC and Ti-SNMC electrodes at BoC and ∼EoC, peak fitting parameter of the core-level spectra from SNMC and Ti-SNMC electrodes after the initial cycle.Conflicts of interest
There are no conflicts of interest to declareAcknowledgements
We gratefully acknowledge the Swedish Energy Agency (48678-1, P2020-90112, P2022-00055, and P2023-00603), and STandUP for energy for financial support. The financial support by VR (2018-06465, 2022-06076) is acknowledged by the authors, S.M. also acknowledges support by the Swedish government assignment COMPEL. We acknowledge MAX IV Laboratory for time on FlexPES Beamline (proposal #20230127 and #20230636), and Balder beamline (proposal #20230596). Research conducted at MAX IV Laboratory, a Swedish national user facility, is supported by the Swedish Research Council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. We acknowledge Myfab Uppsala for providing facilities and experimental support. Myfab is funded by the Swedish Research Council (2020-00207) as a national research infrastructure. Support by VR-RFI for accelerator operation at the Ion Technology Center national research infrastructure (contract #2019_00191) is gratefully acknowledged. We gratefully acknowledge Rebecka Lindblad (Uppsala University) for the training on the HAXPES instrument. We acknowledge Agnes-Matilda Mattsson (Uppsala University) for her help during the Balder beamtime.References
- X. Wang, Y. Ding, Y. Deng and Z. Chen, Ni–Rich/Co–Poor Layered Cathode for Automotive Li–Ion Batteries: Promises and Challenges, Adv. Energy Mater., 2020, 10(12), 1903864, DOI:10.1002/aenm.201903864.
- D. Hou, Z. Xu and Z. Yang, et al. Effect of the grain arrangements on the thermal stability of polycrystalline nickel-rich lithium-based battery cathodes, Nat. Commun., 2022, 13, 3437, DOI:10.1038/s41467-022-30935-y.
- C. Xu, K. Märker and J. Lee, et al., Bulk fatigue induced by surface reconstruction in layered Ni-rich cathodes for Li-ion batteries, Nat. Mater., 2021, 20, 84–92, DOI:10.1038/s41563-020-0767-8.
- C. M. Julien and A. Mauger, NCA, NCM811, and the Route to Ni-Richer Lithium-Ion Batteries, Energies, 2020, 13(23), 6363, DOI:10.3390/en13236363.
- Y. Bi, J. Tao and Y. Wu, et al., Reversible planar gliding and microcracking in a single-crystalline Ni-rich cathode, Science, 2020, 370(6522), 1313–1318, DOI:10.1126/science.abc3167.
- X. H. Meng, T. Lin and H. Mao, et al., Kinetic Origin of Planar Gliding in Single-Crystalline Ni-Rich Cathodes, J. Am. Chem. Soc., 2022, 144(25), 11338–11347, DOI:10.1021/jacs.2c03549.
- T. M. M. Heenan, A. Wade and C. Tan, et al., Identifying the Origins of Microstructural Defects Such as Cracking within Ni-Rich NMC811 Cathode Particles for Lithium-Ion Batteries, Adv. Energy Mater., 2020, 10, 2002655, DOI:10.1002/aenm.202002655.
- J. Li, Y. Lu, T. Yang, D. Ge, D. L. Wood and Z. Li, Water-Based Electrode Manufacturing and Direct Recycling of Lithium-Ion Battery Electrodes—A Green and Sustainable Manufacturing System, iScience, 2020, 23(5), 101081, DOI:10.1016/j.isci.2020.101081.
- M. Bichon, D. Sotta and N. Dupré, et al., Study of Immersion of LiNi0.5Mn0.3Co0.2O2 Material in Water for Aqueous Processing of Positive Electrode for Li-Ion Batteries, ACS Appl. Mater. Interfaces, 2019, 11(20), 18331–18341, DOI:10.1021/acsami.9b00999.
- S. Radloff, R. G. Scurtu, M. Hölzle and M. Wohlfahrt-Mehrens, Applying Established Water-Based Binders to Aqueous Processing of LiNi0.83Co0.12Mn0.05O2 Positive Electrodes, J. Electrochem. Soc., 2021, 168(10), 100506, DOI:10.1149/1945-7111/ac2861.
- H. Chen, A. M. Mattsson and L. King, et al., Study of degradation mechanisms in aqueous-processed Ni-rich cathodes for enhanced sustainability of batteries, J. Mater. Chem. A, 2024, 12(37), 25393–25406, 10.1039/D4TA03592E.
- S. Radloff, G. Carbonari, R. G. Scurtu, M. Hölzle and M. Wohlfahrt-Mehrens, Fluorine-free water-based Ni-rich positive electrodes and their performance in pouch- and 21700-type cells, J. Power Sources, 2023, 553, 232253, DOI:10.1016/j.jpowsour.2022.232253.
- L. Hartmann, D. Pritzl, H. Beyer and H. A. Gasteiger, Evidence for Li+/H+ Exchange during Ambient Storage of Ni-Rich Cathode Active Materials, J. Electrochem. Soc., 2021, 168(7), 70507, DOI:10.1149/1945-7111/ac0d3a.
- M. Bichon, D. Sotta, E. De Vito, W. Porcher and B. Lestriez, Performance and ageing behavior of water-processed LiNi0.5Mn0.3Co0.2O2/Graphite lithium-ion cells, J. Power Sources, 2021, 483, 229097, DOI:10.1016/j.jpowsour.2020.229097.
- M. Heidbüchel, T. Schultz and T. Placke, et al., Enabling Aqueous Processing of Ni–Rich Layered Oxide Cathode Materials by Addition of Lithium Sulphate, ChemSusChem, 2023, 16, e202202161, DOI:10.1002/cssc.202202161.
- H. Chen, A. M. Mattsson and L. King, et al., Study of degradation mechanisms in aqueous-processed Ni-rich cathodes for enhanced sustainability of batteries, J. Mater. Chem. A, 2024, 12(37), 25393–25406, 10.1039/D4TA03592E.
- W. Zhu, X. Huang and T. Liu, et al., Ultrathin Al2O3 Coating on LiNi0.8Co0.1Mn0.1O2 Cathode Material for Enhanced Cycleability at Extended Voltage Ranges, Coatings, 2019, 9(2), 92, DOI:10.3390/coatings9020092.
- S. N. Lim, W. Ahn, S. H. Yeon and P. S. Bin, Enhanced elevated-temperature performance of Li(Ni0.8Co0.15Al0.05)O2 electrodes coated with Li2O-2B2O3 glass, Electrochim. Acta, 2014, 136, 1–9, DOI:10.1016/j.electacta.2014.05.056.
- H. Zhang, J. Xu and J. Zhang, Surface-Coated LiNi0.8Co0.1Mn0.1O2 (NCM811) Cathode Materials by Al2O3, ZrO2, and Li2O-2B2O3 Thin-Layers for Improving the Performance of Lithium Ion Batteries, Front. Mater., 2019, 6, 309, DOI:10.3389/fmats.2019.00309.
- L. Qin, H. Yu, H. Zhang, Q. Cheng, L. Chen and H. Jiang, All-Dry Solid-Phase Synthesis of Single-Crystalline Ni-Rich Co-Poor Ternary Cathodes for Li-Ion Batteries, Ind. Eng. Chem. Res., 2024, 63(26), 11710–11716, DOI:10.1021/acs.iecr.4c01827.
- Q. Sun, X. Liu and L. Chang, et al., Promoting Durability of Ni-Rich Layered Oxide via Inverse Spinel NiFe2O4 Interface Layer for High-Voltage Lithium-Ion Batteries, ACS Appl. Energy Mater., 2024, 7(15), 6236–6247, DOI:10.1021/acsaem.4c00847.
- H. Zhang, K. Wu and N. Li, et al., Enhancing Thermal and High-Voltage Cycling Stability of Ni-Rich Layered Cathodes through a Ti-Doping-Induced Surface-Disordered Structure, ACS Appl. Energy Mater., 2022, 5(10), 12673–12681, DOI:10.1021/acsaem.2c02305.
- H. Bai, K. Yuan and C. Zhang, et al., Advantageous surface engineering to boost single-crystal quaternary cathodes for high-energy-density lithium-ion batteries, Energy Storage Mater., 2023, 61, 102879, DOI:10.1016/j.ensm.2023.102879.
- F. Bizzotto, W. Dachraoui and R. Grissa, et al., Modification of NMC811 with titanium for enhanced cycling and high-voltage stability, Electrochim. Acta, 2023, 462, DOI:10.1016/j.electacta.2023.142758.
- X. Ou, T. Liu and W. Zhong, et al., Enabling high energy lithium metal batteries via single-crystal Ni-rich cathode material co-doping strategy, Nat. Commun., 2022, 13, 2319, DOI:10.1038/s41467-022-30020-4.
- J. W. Heo, G. Lee, B. J. Lee, J. Kim and T. Yim, Preserving the structural stability of LiNi0.92Co0.04Mn0.04O2 cathode by Ti4+-ion doping for lithium-ion batteries, J. Electroanal. Chem., 2024, 960, 118206, DOI:10.1016/j.jelechem.2024.118206.
- J. D. Steiner, H. Cheng and J. Walsh, et al., Targeted Surface Doping with Reversible Local Environment Improves Oxygen Stability at the Electrochemical Interfaces of Nickel-Rich Cathode Materials, ACS Appl. Mater. Interfaces, 2019, 11(41), 37885–37891, DOI:10.1021/acsami.9b14729.
- G. Li, Z. Huang, Z. Zuo, Z. Zhang and H. Zhou, Understanding the trace Ti surface doping on promoting the low temperature performance of LiNi1/3Co1/3Mn1/3O2 cathode, J. Power Sources, 2015, 281, 69–76, DOI:10.1016/j.jpowsour.2015.01.173.
- P. Ström and D. Primetzhofer, Ion beam tools for nondestructive in situ and in-operando composition analysis and modification of materials at the Tandem Laboratory in Uppsala, J. Instrum., 2022, 17(4), P04011, DOI:10.1088/1748-0221/17/04/P04011.
- P. Ström, P. Petersson, M. Rubel and G. Possnert, A combined segmented anode gas ionization chamber and time-of-flight detector for heavy ion elastic recoil detection analysis, Rev. Sci. Instrum., 2016, 87(10), 103303, DOI:10.1063/1.4963709.
- M. S. Janson, in CONTES instruction manual, Uppsala Univ Uppsala, Sweden, 2004 Search PubMed.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Sc, Ti, V, Cu and Zn, Appl. Surf. Sci., 2010, 257(3), 887–898, DOI:10.1016/j.apsusc.2010.07.086.
- A. Preobrajenski, A. Generalov and G. Öhrwall, et al., FlexPES: a versatile soft X-ray beamline at MAX IV Laboratory, J. Synchrotron Radiat., 2023, 30(4), 831–840, DOI:10.1107/s1600577523003429.
- K. Klementiev, K. Norén, S. Carlson, K. G. V. S. Clauss and I. Persson, The BALDER Beamline at the MAX IV Laboratory, J. Phys.:Conf. Ser., 2016, 712, 012023, DOI:10.1088/1742-6596/712/1/012023.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12(4), 537–541, DOI:10.1107/S0909049505012719.
- S. Mukherjee, S. Riva and C. Comparotto, et al., Interplay between Growth Mechanism, Materials Chemistry, and Band Gap Characteristics in Sputtered Thin Films of Chalcogenide Perovskite BaZrS3, ACS Appl. Energy Mater., 2023, 6(22), 11642–11653, DOI:10.1021/acsaem.3c02075.
- S. Mukherjee, H. Ganegoda, A. Kumar, S. Pal, C. U. Segre and D. D. Sarma, Evolution of the Local Structure within Chromophoric Mn-O5 Trigonal Bipyramids in YMn1-xInxO3 with Composition, Inorg. Chem., 2018, 57(15), 9012–9019, DOI:10.1021/acs.inorgchem.8b00997.
- M. Risch, K. Klingan and J. Heidkamp, et al., Nickel-oxido structure of a water-oxidizing catalyst film, Chem. Commun., 2011, 47(43), 11912, 10.1039/c1cc15072c.
- J. J. Rehr, J. J. Kas, F. D. Vila, M. P. Prange and K. Jorissen, Parameter-free calculations of X-ray spectra with FEFF9, Phys. Chem. Chem. Phys., 2010, 12(21), 5503–5513, 10.1039/B926434E.
- R. K. Vempati, T. R. Hess and D. L. Cocke, in X-Ray Photoelectron Spectroscopy, 2018. DOI:10.2136/sssabookser5.3.c12.
- J. J. Yeh and I. Lindau, Atomic subshell photoionization cross sections and asymmetry parameters: 1 ≤ Z ≤ 103, At. Data Nucl. Data Tables, 1985, 32(1), 1–155, DOI:10.1016/0092-640X(85)90016-6.
- N. Li, M. Sun and W. H. Kan, et al., Layered-rocksalt intergrown cathode for high-capacity zero-strain battery operation, Nat. Commun., 2021, 12(1), 4–13, DOI:10.1038/s41467-021-22527-z.
- W. Lee, S. Lee and E. Lee, et al., Destabilization of the surface structure of Ni-rich layered materials by water-washing process, Energy Storage Mater., 2022, 44, 441–451, DOI:10.1016/j.ensm.2021.11.006.
- T. Liu, L. Yu and J. Lu, et al., Rational design of mechanically robust Ni-rich cathode materials via concentration gradient strategy, Nat. Commun., 2021, 12, 6024, DOI:10.1038/s41467-021-26290-z.
- R. Singh, M. Gupta, D. M. Phase and S. K. Mukherjee, Phase growth analysis of sputtered TiO2 thin films at low oxygen partial pressures using XANES and XRR, Mater. Res. Express, 2019, 6(11), 116449, DOI:10.1088/2053-1591/ab4e3f.
- Y. Cao, X. Liu and M. Kareev, et al., Engineered Mott ground state in a LaTiO3+δ/LaNiO3 heterostructure, Nat. Commun., 2016, 7, 10418, DOI:10.1038/ncomms10418.
- J. Sicklinger, M. Metzger, H. Beyer, D. Pritzl and H. A. Gasteiger, Ambient Storage Derived Surface Contamination of NCM811 and NCM111: Performance Implications and Mitigation Strategies, J. Electrochem. Soc., 2019, 166(12), A2322–A2335, DOI:10.1149/2.0011912jes.
- R. Jung, R. Morasch and P. Karayaylali, et al., Effect of Ambient Storage on the Degradation of Ni-Rich Positive Electrode Materials (NMC811) for Li-Ion Batteries, J. Electrochem. Soc., 2018, 165(2), A132–A141, DOI:10.1149/2.0401802jes.
- H. H. Ryu, K. J. Park, S. C. Yoon and Y. K. Sun, Capacity Fading of Ni-Rich Li[NixCoyMn1−x–y]O2 (0.6 ≤ x ≤ 0.95) Cathodes for High-Energy-Density Lithium-Ion Batteries: Bulk or Surface Degradation?, Chem. Mater., 2018, 30(3), 1155–1163, DOI:10.1021/acs.chemmater.7b05269.
- E. Bautista Quisbert, F. Fauth, A. M. Abakumov, M. Blangero, M. Guignard and C. Delmas, Understanding the High Voltage Behavior of LiNiO2 Through the Electrochemical Properties of the Surface Layer, Small, 2023, 19, 2300616, DOI:10.1002/smll.202300616.
- J. B. Adamo and A. Manthiram, Understanding the Effects of Al and Mn Doping on the H2−H3 Phase Transition in High-Nickel Layered Oxide Cathodes, Chem. Mater., 2024, 36(12), 6226–6236, DOI:10.1021/acs.chemmater.4c01033.
- P. Xiao, Capacity Decay in LiNiO2: An Atomistic Kinetic Picture, ACS Energy Lett., 2025, 10(2), 639–646, DOI:10.1021/acsenergylett.4c03271.
- M. Hekmatfar, A. Kazzazi, G. G. Eshetu, I. Hasa and S. Passerini, Understanding the Electrode/Electrolyte Interface Layer on the Li-Rich Nickel Manganese Cobalt Layered Oxide Cathode by XPS, ACS Appl. Mater. Interfaces, 2019, 11(46), 43166–43179, DOI:10.1021/acsami.9b14389.
- C. Yang, X. Liao and X. Zhou, et al., Phosphate–Rich Interface for a Highly Stable and Safe 4.6 V LiCoO2 Cathode, Adv. Mater., 2023, 35(14), 2210966, DOI:10.1002/adma.202210966.
- Z. S. Wu, A. Zhang and Z. Bi, et al., Regulating Electrode/Electrolyte Interfacial Chemistry Enables 4.6 V Ultra-Stable Fast Charging of Commercial LiCoO2, Energy Environ. Sci., 2024, 17, 3021–3031, 10.1039/D4EE00676C.
- A. Mikheenkova, S. Mukherjee and M. Hirsbrunner, et al., The role of oxygen in automotive grade lithium-ion battery cathodes: an atomistic survey of ageing, J. Mater. Chem. A, 2024, 12(4), 2465–2478, 10.1039/D3TA05516G.
- K. R. Tallman, G. P. Wheeler and C. J. Kern, et al., Nickel-rich Nickel Manganese Cobalt (NMC622) Cathode Lithiation Mechanism and Extended Cycling Effects Using Operando X-ray Absorption Spectroscopy, J. Phys. Chem. C, 2021, 125(1), 58–73, DOI:10.1021/acs.jpcc.0c08095.
- Y. Yu, P. Karayaylali and L. Giordano, et al., Probing Depth-Dependent Transition-Metal Redox of Lithium Nickel, Manganese, and Cobalt Oxides in Li-Ion Batteries, ACS Appl. Mater. Interfaces, 2020, 12(50), 55865–55875, DOI:10.1021/acsami.0c16285.
- C. D. Quilty, P. J. West and G. P. Wheeler, et al., Elucidating Cathode Degradation Mechanisms in LiNi0.8Mn0.1Co0.1O2 (NMC811)/Graphite Cells Under Fast Charge Rates Using Operando Synchrotron Characterization, J. Electrochem. Soc., 2022, 169(2), 020545, DOI:10.1149/1945-7111/ac51f5.
- D. Friebel, M. W. Louie and M. Bajdich, et al., Identification of highly active Fe sites in (Ni,Fe)OOH for electrocatalytic water splitting, J. Am. Chem. Soc., 2015, 137(3), 1305–1313, DOI:10.1021/ja511559d.
- M. Görlin, P. Chernev and J. F. De Araújo, et al., Oxygen evolution reaction dynamics, faradaic charge efficiency, and the active metal redox states of Ni-Fe oxide water splitting electrocatalysts, J. Am. Chem. Soc., 2016, 138(17), 5603–5614, DOI:10.1021/jacs.6b00332.
- Z. Bai, Z. Ying and F. Zhang, et al., Enabling High Stability of Co-Free LiNiO2 Cathode via a Sulfide-Enriched Cathode Electrolyte Interface, ACS Energy Lett., 2024, 9(6), 2717–2726, DOI:10.1021/acsenergylett.4c00652.
Footnote |
| † Electronic supplementary information (ESI) available: Additional TEM, soft XAS, and operando XAS measurements on SNMC and Ti-SNMC powder and electrodes, ToF-ERDA measurements on uncycled SNMC and Ti-SNMC electrodes, ICP measurements on SNMC and Ti-SNMC powder, simulated k3-weighted Ni K-edge EXAFS spectra and fit parameter for SNMC and Ti-SNMC electrodes at BoC and ∼EoC, peak fitting parameter of the core-level spectra from SNMC and Ti-SNMC electrodes after the initial cycle. See DOI: https://doi.org/10.1039/d4eb00043a |
| This journal is © The Royal Society of Chemistry 2025 |