
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cl2− chemical ionization mass spectrometry (Cl2-CIMS) for the measurement of acyl peroxy radicals†
Tyson C.
Berg
 ,
Michael F.
Link‡
and
Delphine K.
Farmer
*
,
Michael F.
Link‡
and
Delphine K.
Farmer
*
Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523, USA. E-mail: delphine.farmer@colostate.edu
First published on 28th April 2025
Abstract
Organic peroxy radicals (RO2) are produced in the atmosphere by oxidation of volatile organic compounds (VOCs) and, in some cases, VOC photolysis. However, photolytic sources of RO2 are often poorly understood, in part due to challenges in directly detecting RO2 in both ambient and laboratory settings. We investigated Cl2− as a chemical ionization mass spectrometry reagent ion (Cl2-CIMS) for measuring and speciating RO2 in a laboratory setting. Cl2-CIMS was more sensitive to the acetyl peroxy radical (CH3C(O)O2; 2.30 ± 0.04 ncps/ppt) than iodide CIMS (I-CIMS; 1.54 ± 0.03 ncps/ppt), but high backgrounds in our setup resulted in a slightly higher detection limit of 5 ppt (1 second integration) for Cl2-CIMS than I-CIMS (2 ppt). We demonstrate the application of Cl2-CIMS by quantifying the quantum yields of two radical products, CH3C(O) and C2H5C(O), from methyl ethyl ketone photolysis at 254 nm. We identified O2− and Cl− as possible secondary reagent ions that created unintended product ions in our experiments and thus could complicate the interpretation of Cl2-CIMS mass spectra for complex atmospheric samples. While several strategies may minimize these effects, Cl2-CIMS is suitable for measuring RO2 in controlled laboratory experiments.
Environmental significanceOrganic peroxy radicals are intermediates in the atmospheric oxidation cycles of organic molecules which impact production of atmospheric free radicals and ozone. Speciated measurements of organic peroxy radicals in the atmosphere are difficult but possible through recent advancements in chemical ionization mass spectrometry. We developed a chemical ionization mass spectrometry method using Cl2− as the primary reagent ion for detection of select organic peroxy radicals. We demonstrate this technique through laboratory measurements of the acetyl and propionyl peroxy radical quantum yields from methyl ethyl ketone photolysis. Cl2− reagent ion CIMS may be applicable to ambient measurements of organic peroxy radicals, if methods are developed for better isolation of Cl2− chemistry from reactions of secondary reagent ions. |
Introduction
Organic peroxy radicals (RO2) are key components of atmospheric oxidation chemistry but are challenging to measure due to their high reactivity and low ambient concentrations (∑RO2 ≤ 40 ppt).1,2 RO2 reactions control hydroxyl (OH) radical production and can contribute to radical termination reactions, thus influencing oxidative capacity of the atmosphere and impacting air quality.3 RO2 are primarily formed from the oxidation of volatile organic compounds, though certain moieties, such as aldehydes and ketones, can also undergo photolysis to produce RO2.4Approaches to measuring RO2 are primarily spectroscopic, including ultraviolet3 and infrared absorbance5 measurements and cavity ring-down spectroscopy.6 Laser-induced fluorescence measurements are possible following the conversion of RO2 to OH through reaction with CO and NO.7 Chemical amplification of RO2 is also possible through reaction with CO and NO, or C2H6 and NO, to produce and measure NO2.8,9 However, these spectroscopic techniques typically cannot quantify speciated radicals and are often limited to measuring HO2 in the lab10 (HO2 can be considered the smallest RO2 species, given its analogous sources and sinks) or summed concentrations of many RO2 in the field.11
There is a clear need for new analytical techniques to directly measure and speciate RO2. Ambient measurements would be improved through the development of a method that could detect individual RO2 species in the field. Ambient concentrations of RO2 other than CH3O2 and HO2 are typically a few ppt or less.12
Better RO2 measurements would also improve laboratory data on RO2 production from organic gas photolysis. Photolytic sources of RO2 become more important relative to oxidation reactions in the upper troposphere where oxidants (OH, O3, NO3, Cl) are scarce and the actinic flux is higher compared to the rest of the troposphere. For example, acetone photolysis is thought to produce HOx (OH and HO2) at a similar rate to the O(1D) + H2O reaction in the upper troposphere.13,14 However, the role of acetone in upper tropospheric HOx production is poorly understood, as laboratory studies of acetone photolysis quantum yields are limited to indirect measurements of the dominant products: acetyl peroxy (CH3C(O)O2) and methyl peroxy (CH3O2) radicals. This results in substantial uncertainties in acetone photolysis quantum yields.15 Prior studies typically measure either minor photolysis product channels16 or stable products (e.g., CO, CO2, and peroxyacetyl nitrate) of secondary reactions.17–19 Photolysis quantum yields for larger and more complex ketones are even more limited, as few techniques can speciate CH3C(O)O2 and CH3O2 radicals from RO2 of different structures with fast time resolution.10
One technique that can provide fast, direct, and speciated detection of RO2 is chemical ionization mass spectrometry (CIMS). CIMS has been used to measure RO2 in both laboratory and ambient settings. Direct CIMS detection methods include measurements of HO2 with I− (I-CIMS)20 and Br− reagent ions,21,22 acyl peroxy radicals (R(O)O2) and other RO2 with I-CIMS,23–26 and highly oxidized molecule RO2 with NH4+, CH3C(O)O−,27 or C3H7NH3+ reagent ions.28 Additional derivatization-aided detection methods exist for H3O+, NH4+,29 and NO3−, but are typically labor-intensive.12,30 We recently described the use of I-CIMS to measure ketone photolysis product quantum yields via direct detection of RC(O)O2 produced from photolysis of methyl ethyl ketone (MEK) and 2,3-butadione.23
However, available CIMS reagent ions, including iodide, are often subject to interferences that impact RO2 detection such as decreases in sensitivity due to small amounts of water vapor,22 unintended production of radicals inside the instrument,21 and secondary ion chemistry that complicates quantification.31 The sensitivities of CIMS reagent ions, such I− and Br−,22,25 can be temperature dependent as well. Current detection limits for RO2 at short integration times (∼1 s) are often too high (>5 ppt) for ambient measurements. Laboratory measurements of ketone photodissociation quantum yields are also challenged by high detection limits. Investigation into alternate CIMS reagent ions to measure speciated RO2 is warranted.
We describe a reagent ion system for CIMS with Cl2− as the primary reagent ion (Cl2-CIMS). We investigate the ion-molecule chemistry and demonstrate the application of Cl2-CIMS to measurements of RC(O)O2 from photolysis of acetone and MEK at 254 nm. Through organic acid and woodsmoke sampling, we evaluate the potential and limitations of Cl2-CIMS in ambient measurements.
Methods
The chemical ionization mass spectrometer
We used a time-of-flight chemical ionization mass spectrometer with atmospheric pressure interface (CIMS; Aerodyne Research, Inc.), which consists of a standard Aerodyne Research Inc. designed ion-molecule reaction region (IMR), ion optics, and a time-of-flight mass analyzer with a multi-channel plate detector.32,33 In the IMR, sample air interacts with a flow of reagent ions, which ionize analyte molecules through clustering, proton transfer, charge transfer, or other reactions. We operated the CIMS with a typical IMR pressure of 65 mbar. The ion optics consist of one small quadrupole and one large quadrupole, plus multiple skimmers and a segment of ion lenses, operated at increasing voltage (in this case, increasing negative voltages) as sample air moves from the high-to low-pressure regions. These sections maximize ion transport through the instrument as the total pressure decreases to 1 × 10−6 mbar in the time-of-flight region. The time-of-flight mass analyzer had a mass resolution of 3500 m/Δm (Δm = 0.04 at m/z 144.95, the m/z for Cl2(CH3C(O)O2)–), adequate for determining the elemental composition of detected ions in the size range described here.We introduced reagent ion flow through the ionizer port on the IMR (Fig. 1), with a sealed Po-210 α-particle source (Model P-2021; NRD, LLC) ion source. Cl2-CIMS reagent ions were formed by flowing trace concentrations of Cl2 through the ion source. We used 12 L glass bulbs filled to 1250 mbar with 100 to 400 ppm Cl2 in ultra-high purity N2 (99.999%; Airgas) and calibration gas cylinders containing 10 or 40 ppm Cl2 in N2 (Gasco) as the reagent Cl2 sources. A critical orifice limited Cl2 flows at 10 mL min−1 from the 12 L glass; flow from the Cl2 cylinders was adjusted by a precision needle valve. We used N2, mixtures of N2 and O2 (99.994%; Airgas), or ultra-zero air (UZA; 99.999%; Airgas) as carrier gases for the Cl2 reagent mixture. Needle valves controlled total carrier gas flow at >1250 mL min−1, which diluted Cl2 mixing ratios to ≤ 1 ppm in the ionizer. Unless otherwise stated, we used ∼100 ppb Cl2 in UZA carrier gas for Cl2-CIMS reagent ion generation.
 | ||
| Fig. 1 Schematic of flows to introduce Cl2− reagent ions into the IMR of the CIMS. Describes the major CIMS components and the ionizer; flows of Cl2, N2, humidified N2, zero air, and oxygen; and sample flow. The dashed lines and arrows denote the flow path used to produce I− reagent ions from CH3I in N2. This path was not permanently attached to the Cl2 flow system but replaced the Cl2 flow through the ionizer during I-CIMS operation. | ||
For I-CIMS, we generated I− by passing a constant flow of N2 over a CH3I permeation tube heated to 40 °C through the ionizer. The constant flow and temperature on the permeation tube were maintained when I-CIMS was not in use. We estimate CH3I concentration at the ionizer to be on the order of 500 ppb. We controlled water vapor partial pressure in the IMR through an additional port by changing the ratio of dry and water-saturated N2 while keeping total flow constant. The humidified line was typically maintained at 80% RH, providing ∼0.5 mbar H2O to the IMR.
Calibrations
We calibrated the Cl2-CIMS for ozone (O3), formic, acetic, propanoic, and isobutyric acids, and CH3C(O)O2. A calibrated O3 source (Model 306, 2B Technologies) generated O3 between 25-200 (±2.0%) ppb in a 1600 mL min−1 flow of air, which we diluted with UZA to 2 L min−1 before sampling by the Cl2-CIMS. Custom permeation tubes containing liquid organic acids were held at 40 °C and served as the acid calibration source. The permeation tube emission rates were determined gravimetrically. We used a custom peroxyacetyl nitrate (PAN) source34 for CIMS calibration to CH3C(O)O2. The PAN calibration source photolyzes acetone in UZA to form CH3C(O)O2 and CH3O2. These radicals then react with NO to form NO2, CH3C(O)O and CH3O. The NO2 reacts with the remaining CH3C(O)O2. In the source we mixed a 60 mL min−1 flow of 18 ppm acetone in UZA (Scott-Marin) with 2 mL min−1 of 2 ppm NO in N2. NO mixtures were prepared in 12 L glass bulbs. The light source was a phosphorus-coated Hg lamp (Jelight), with primary emission at 285 nm. Photolysis of the acetone-NO mixture occurred in a 200 cm3 quartz cell at 1070 mbar and approximately room temperature, producing a 60 mL min−1 flow containing 40 ppb of PAN. The PAN concentration output of the device was calibrated by GC-ECD prior to our use. Further details on the custom PAN source are available in Flocke et al.34 We diluted this PAN flow into 2 to 5 L min−1 N2 and thermally decomposed PAN at 150 °C to produce CH3C(O)O2 mixing ratios from 0.48 to 0.96 ppb. A heating rope around a 0.25 in O.D. glass sampling tube 1 cm from the CIMS inlet provided the necessary heating for PAN thermal decomposition.23 We calibrated the Cl2-CIMS to analytes at a Cl2 reagent mixture concentration of 100 ppb. Sensitivities are reported for IMR water partial pressure of 0.5 mbar and a total pressure of 65 mbar. All calibration standards were measured by the Cl2-CIMS as a function of (i) Cl2 ionizer concentrations (0.1 to 1.5 ppm), (ii) ion-molecule reactor (IMR) pressure between 50 and 100 mbar, (iii) IMR water partial pressure between 0 and 0.8 mbar, and (iv) de-clustering potential35 between the front of the second quadrupole and the adjacent (up-flow) skimmer plate (Fig. S1†).Sensitivities were calculated as the slope of the line of detected ion signal versus input concentration and reported as normalized ion counts per second per part per trillion (ncps/ppt; per 1 × 106 Hz of summed reagent ions). Unless indicated otherwise, Cl2-CIMS signals are normalized to the sum of Cl−, Cl2−, Cl3−, and O2−, the identified potential reagent ions. I-CIMS signals are normalized to the sum of I− and I(H2O)–, the two primary reagent ions for this chemistry. Typical total reagent ion counts for both chemistries were between one and two million Hz. However, the reagent ion counts for Cl2-CIMS varied, dependent on reagent gas O2 content, and other instrument conditions as described in calibration tests (i)–(iv) above. All signals were normalized to total reagent ion counts, as described here, for comparisons across changing instrument conditions. Limits of detection (1 s integration) are calculated as three times the background signal-to-noise ratio and assuming a poisson distribution for background count rates.23,32
Ketone photolysis product quantum yields
To evaluate whether Cl2− was a better reagent ion for laboratory radical measurements, we compared measurements of CH3C(O)O2 and propionyl peroxy radicals (C2H5C(O)O2), generated from photolysis of acetone and MEK, acquired with Cl2-CIMS versus I-CIMS. In these experiments, ketone photolysis generated acyl peroxy radicals ((R1a) and (R1b)):| RC(O)CH3 + hv → RC(O) + CH3 | (R1a) |
| RC(O) + O2 + M → RC(O)O2 | (R1b) |
We produced the MEK and acetone standards in 12 L glass bulbs containing percent-level ketone in N2 taken from the headspace of purified liquid ketones. Ketone samples in vacuum storage flasks were purified by freezing the liquid and pumping out the headspace. Ketone bulb concentration was determined through absorbance of 254 nm light (Hg Pen-Ray lamp; Analytik Jena) in a 50 cm glass cell with quartz end windows. We measured absorbance by bulb contents relative to background intensity in dry N2 in triplicate, using a lab-built photodiode detector.
The photolysis reactor was a 1 in O.D. glass tube with three 0.25 in O.D. ports for the addition of photolyte mixture and sampling by the CIMS and exhaust. A Hg Pen-Ray 254 nm lamp was inserted 3 in into the reactor in a 0.5 in O.D. quartz test tube. The CIMS sampled from the photolysis reactor 1 cm from the end of the Pen-Ray lamp through an inserted 0.25 in glass tube.
During photolysis experiments, 10 mL min−1 of ketone in N2 was mixed with 7 L min−1 of 21% O2 in N2 before entering the reactor adjacent to the Pen-Ray lamp housing. We measured the background flow of O2 and N2 before turning on the Pen-Ray Lamp for 100 s and then adding the ketone. After sampling during ketone photolysis for 5 min, we turned off the lamp, turned off the ketone flow, and sampled for at least one minute before turning the lamp on again. Experiments were run with both acetone and MEK. The Cl2-CIMS detected CH3C(O)O2 primarily as the cluster Cl2(CH3C(O)O2)–(R2):
| CH3C(O)O2 + Cl2− → Cl2(CH3C(O)O2)− | (R2) |
 | (E1) |
Woodsmoke experiments
The Cl2-CIMS sampled woodsmoke from a 1 m3 Teflon chamber that had ports for air sampling, injection of smoke, and a constant flow of 3 L min−1 zero air (produced by zero air generator; Model 7000, Environics). Following the method described in Li et al.,36 we burned ∼1 g of Douglas fir woodchips in a Breville (BSM600SILUSC) cocktail smoker and injected the smoke directly into the chamber. This injection produced approximately 300 μg m−3 of smoke aerosol along with concentrated gases that could be analyzed by the CIMS. We connected the CIMS to a sampling port through ∼1 m of PFA tubing (0.25 in O.D.) with a PTFE filter to remove particles. No lights were used to induce chemical aging of the smoke. A manual three-way valve allowed zero air to bypass the chamber directly to the CIMS. During measurements, we first flowed only zero air into the chamber for a background measurement before injecting smoke. The Cl2-CIMS sampled smoke from the chamber for 2 hours. We then switched to I-CIMS mode and performed the same woodsmoke experiment with a new smoke addition. For analysis of woodsmoke samples, we identified ions that increased during smoke sampling with m/z < 300 for Cl2-CIMS and < 350 for I-CIMS. We used a larger mass range for I-CIMS due to the difference in m/z of cluster ions formed with Cl2− and I−. Thus, we account for approximately the same range of analyte masses with both reagent ion chemistries.Results and discussion
Analyte detection
The Cl2-CIMS produced ions through a variety of mechanisms including proton abstraction; adduct formation with Cl−, Cl2−, and O2−; charge transfer; and secondary chemistry (Table 1 and Fig. S2†). Little fragmentation was observed. The dominant product ion varied by analyte. Cl2− cluster ions provided the largest sensitivity for formic acid and CH3C(O)O2, while product ions from secondary chemistry and O2− clustering provided the largest sensitivities for O3 and C2–C4 organic acids, respectively. CO3− is a known product ion of O3 for O2-CIMS and is formed from the reaction of O3− with CO2.37 Though C2–C4 organic acids were detected as O2− clusters with sensitivities on the order of 1 to 2 ncps/ppt, Cl2− clusters produced much lower sensitivities at <0.02 ncps/ppt. C2–C4 organic acid clusters with Cl− showed larger sensitivity than Cl2− at 1.2 ± 0.2 ncps/ppt for acetic acid, 0.215 ± 0.002 ncps/ppt for propanoic acid, and 0.142 ± 0.005 ncps/ppt for isobutyric acid.| Analyte | Molecular formula | Product ions | Primary product ion | Primary ion sens. (ncps/ppt)a | LOD (ppt) | Calibration range (ppb) |
|---|---|---|---|---|---|---|
| a Sensitivities normalized (per million ions) to sum of reagent ion signal (O2− + Cl− + Cl2− + Cl3−). b Sensitivity normalized (per million ions) to Cl2− signal. c General formulas of observed product ions for all organic acids. | ||||||
| Acetyl peroxy radical | CH3C(O)O2 | Cl2CH3C(O)O2− | Cl2CH3C(O)O2− | 2.30 ± 0.04 | 5 | 0.48–0.96 |
| 3.19 ± 0.05b | ||||||
| Ozone | O3 | O3−, CO3− | CO3− | 0.78 ± 0.02 | 291 | 18.4–147 |
| Formic acid | HC(O)OH | ClRC(O)OH−, Cl2RC(O)OH−, O2RC(O)OH−, RC(O)O−c | Cl2HC(O)OH− | 1.01 ± 0.02 | 73 | 33–65 |
| Acetic acid | CH3C(O)OH | O2CH3C(O)OH− | 1.6 ± 0.2 | 50 | 23–46 | |
| Propanoic acid | C2H5C(O)OH | O2C2H5C(O)OH− | 2.00 ± 0.01 | 9 | 8.7–17.2 | |
| Isobutyric acid | C3H7C(O)OH | O2C3H7C(O)OH− | 1.11 ± 0.01 | 22 | 0.97–1.9 | |
Sensitivities to all analytes were independent of IMR pressure within the range of 50-100 mbar (Fig. S3†) but did vary with IMR water partial pressure (0-0.6 mbar; Fig. S4 and S5†), and de-clustering voltage (Fig. S6†). The largest water partial pressure impacts were observed for Cl− and Cl2− clusters. Thus, we operated the Cl2-CIMS optimized for Cl2(RC(O)O2)– (∼0.5 mbar H2O) and overall cluster ions production (dV = 1.4).
Sensitivity to CH3C(O)O2, detected as Cl2(CH3C(O)O2)–, was 2.30 ± 0.04 ncps/ppt when normalized to the total reagent ion signal, and 3.19 ± 0.05 ncps/ppt when normalized to the Cl2− reagent ion signal. In contrast to other analytes, alternate ion formation pathways were negligible for the acetyl peroxy radical. The sensitivity of Cl2-CIMS was higher than that for I-CIMS (1.54 ± 0.03 ncps/ppt for ICH3C(O)O2−) under the same measurement conditions. However, the LOD for I-CIMS was lower (2 ppt) than we obtained for Cl2-CIMS (5 ppt) due to larger backgrounds for Cl2-CIMS.
We note that CIMS users employ various normalization techniques; either normalizing data to total ion counts to account for slight variations in instrument performance, or normalizing data to the relevant reagent ion signals acknowledging that variations in just those signals can modulate the sensitivity to an analyte.25,38,39 Different approaches to normalization impact the calculated sensitivity and detection limit. For example, we add a second Cl2-CIMS sensitivity to CH3C(O)O2 (normalized to the Cl2− counts only) in Table 1 and Fig. S7,† due to a consistent relationship between Cl2− and Cl2(CH3C(O)O2)– observed during CH3C(O)O2 calibration and MEK photolysis (Fig. S8†). This normalization method increased the normalized sensitivity, but the total sensitivity observed during the test remains the same. The Cl2-normalized sensitivity serves as both the maximum possible sensitivity in a reagent ion spectrum of entirely Cl2− and the most accurate method of calculating sensitivity to CH3C(O)O2 between tests. The CH3C(O)O2 sensitivity normalized to the sum of Cl−, Cl2−, Cl3−, and O2− did not reflect the difference of signal observed between Cl2-and I-CIMS during MEK photolysis.
MEK photolysis product quantum yield measurements at 254 nm
The Cl2-CIMS detected both CH3C(O)O2 and C2H5C(O)O2 from MEK photolysis as clusters with Cl2−. We assume that the Cl2-CIMS sensitivity to CH3C(O)O2 and C2H5C(O)O2 are equivalent based on voltage scanning data. Briefly, by changing the voltage difference between two components in the ion-focusing regions of the CIMS, we provide adequate energy to de-cluster reagent ion-analyte adducts.35,41 The voltage difference that corresponds to a 50% loss in adduct ion signal (dV50) correlates to the sensitivity of the instrument to the clustering mechanism.42 The voltage scan provides a dV50 of 4.58 ± 0.05 V for Cl2(CH3C(O)O2)– and 4.21 ± 0.05 V for Cl2(C2H5C(O)O2)– (Fig. S9†). This is quite close to the 0.3 V difference in dV50 found with I-CIMS,23 indicating that the sensitivities of Cl2-CIMS to CH3C(O)O2 and C2H5C(O)O2 are similar. This assumption is further supported by the agreement of quantum yields between Cl2-CIMS and I-CIMS and with prior literature.19,40Cl2-CIMS derived MEK photolysis quantum yields at 254 nm (ΦMEK, RC(O)254) were consistent with previous measurements19,23,40 and with I-CIMS ΦMEK, RC(O)254 obtained as part of this work. 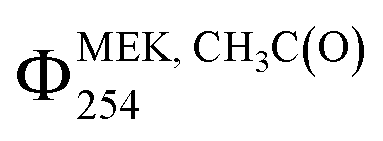 and ΣΦMEK, RC(O)254 from Cl2-CIMS and I-CIMS (Fig. 2) agreed within the measurement precision of 3%, while
and ΣΦMEK, RC(O)254 from Cl2-CIMS and I-CIMS (Fig. 2) agreed within the measurement precision of 3%, while 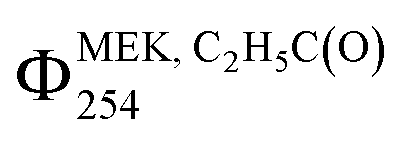 agreed within the experimental uncertainties of 26% and 24% calculated for Cl2-CIMS and I-CIMS, respectively. The experimental uncertainties were 20% for
agreed within the experimental uncertainties of 26% and 24% calculated for Cl2-CIMS and I-CIMS, respectively. The experimental uncertainties were 20% for 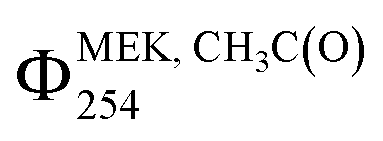 and 21% for ΣΦMEK, RC(O)254 for both Cl2-CIMS and I-CIMS. Signals for Cl2(CH3C(O)O2)– and Cl2(C2H5C(O)O2)– obtained by Cl2-CIMS during MEK photolysis were larger than the corresponding I− cluster signals from I-CIMS (Fig. S8†). Cl2-CIMS measured radical signals normalized to only Cl2− were a factor of 2.1 ± 0.1 larger than I-CIMS (normalized to the sum of I− and I(H2O)–), in agreement with the sensitivity ratio (2.07 ± 0.05) obtained during calibrations. This indicates that the Cl2-CIMS sensitivity to RC(O)O2 was dependent on Cl2− and not related to the total reagent ion count.
and 21% for ΣΦMEK, RC(O)254 for both Cl2-CIMS and I-CIMS. Signals for Cl2(CH3C(O)O2)– and Cl2(C2H5C(O)O2)– obtained by Cl2-CIMS during MEK photolysis were larger than the corresponding I− cluster signals from I-CIMS (Fig. S8†). Cl2-CIMS measured radical signals normalized to only Cl2− were a factor of 2.1 ± 0.1 larger than I-CIMS (normalized to the sum of I− and I(H2O)–), in agreement with the sensitivity ratio (2.07 ± 0.05) obtained during calibrations. This indicates that the Cl2-CIMS sensitivity to RC(O)O2 was dependent on Cl2− and not related to the total reagent ion count.
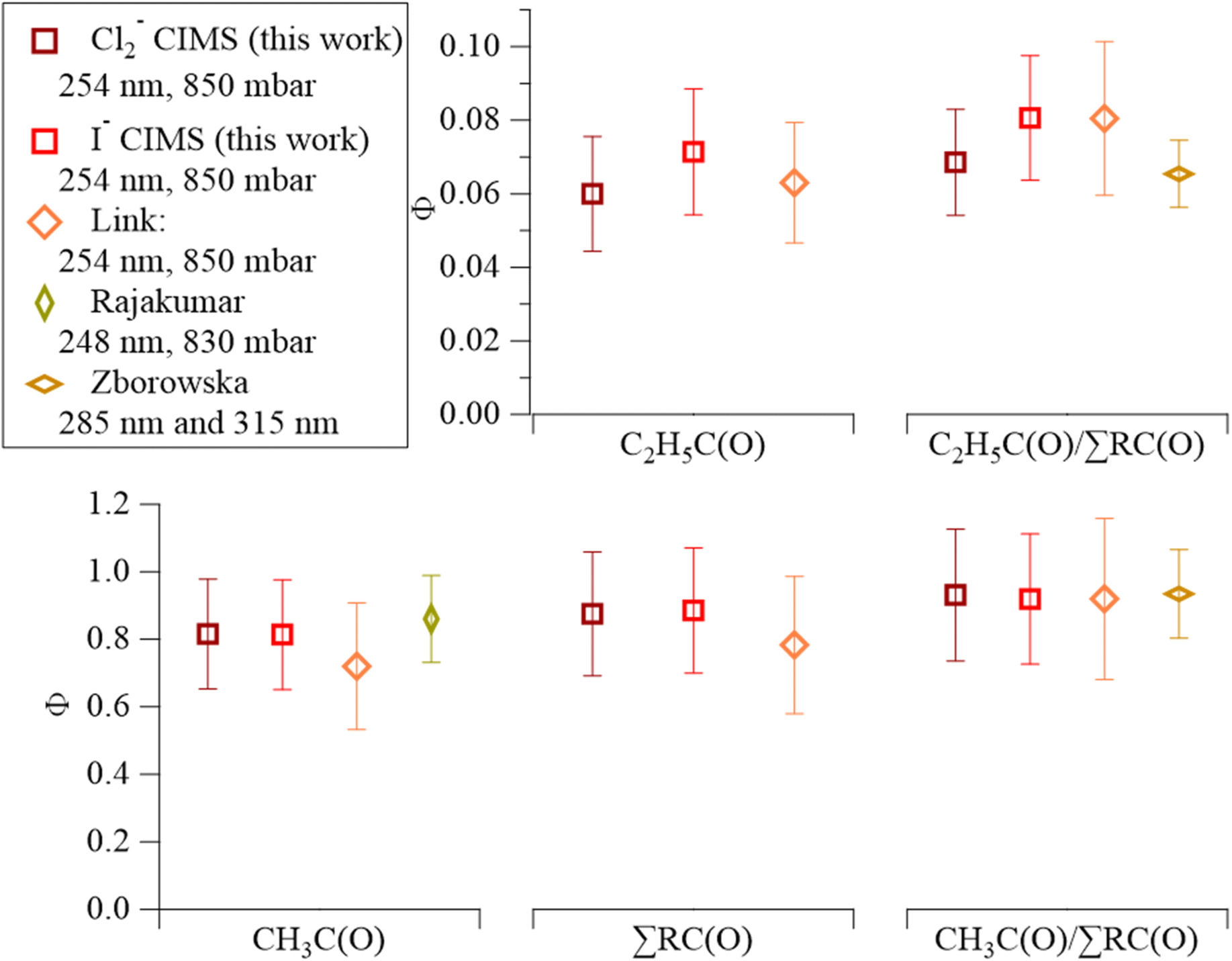 | ||
Fig. 2 Comparison of MEK photolysis product quantum yield values from the Cl2-CIMS (dark red) and I-CIMS (light red) from this study to recent literature values. The x-axis consists of segments for each MEK photolysis product: CH3C(O) on the bottom-left, C2H5C(O) on top-center, ΣRC(O) on the bottom-center, and branching ratios on the right (C2H5C(O)/∑RC(O) top; (CH3C(O)/∑RC(O) bottom) orange diamonds represent values from Link et al.;23 gold diamond represents the Rajakumar et al.40 value for ΦCH3C(O); light-brown diamonds represent branching ratios derived from C2H5C(O)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3C(O) signal ratios in Zborowska et al.19 Error bars represent experimental uncertainties. :CH3C(O) signal ratios in Zborowska et al.19 Error bars represent experimental uncertainties. | ||
Cl2-CIMS reagent ion chemistry
Cl2-CIMS reagent ion chemistry is a complex system of ionizing reactions involving Cl−, Cl2−, Cl3−, and O2−, which is best described by first examining the ion chemistry of Cl2 in N2. In a reagent gas mixture with dry N2, collisions of α-particles from the Po-210 source with N2 produce free electrons43(R3a). Cl2 is the primary molecule that attaches free electrons within the ionizer flow (R3b).| α + N2 → N2+ + e− + α | (R3a) |
| e− + Cl2 → Cl2−∗ | (R3b) |
Excited Cl2−* formed by electron attachment can decompose to Cl− and Cl (R4a),44 though the example reagent ion spectrum in N2 (Fig. 3(a)) shows a significant portion of Cl2−* in our system is stabilized by collisions (R4b). Cl2−* is the source of both Cl− and Cl2− in the reagent ion spectra through either decomposition or collisional stabilization.
| Cl2−∗ → Cl + Cl− | (R4a) |
| Cl2−∗ + M → Cl2− + M∗ | (R4b) |
| Cl + Cl− + M → Cl2− | (R5) |
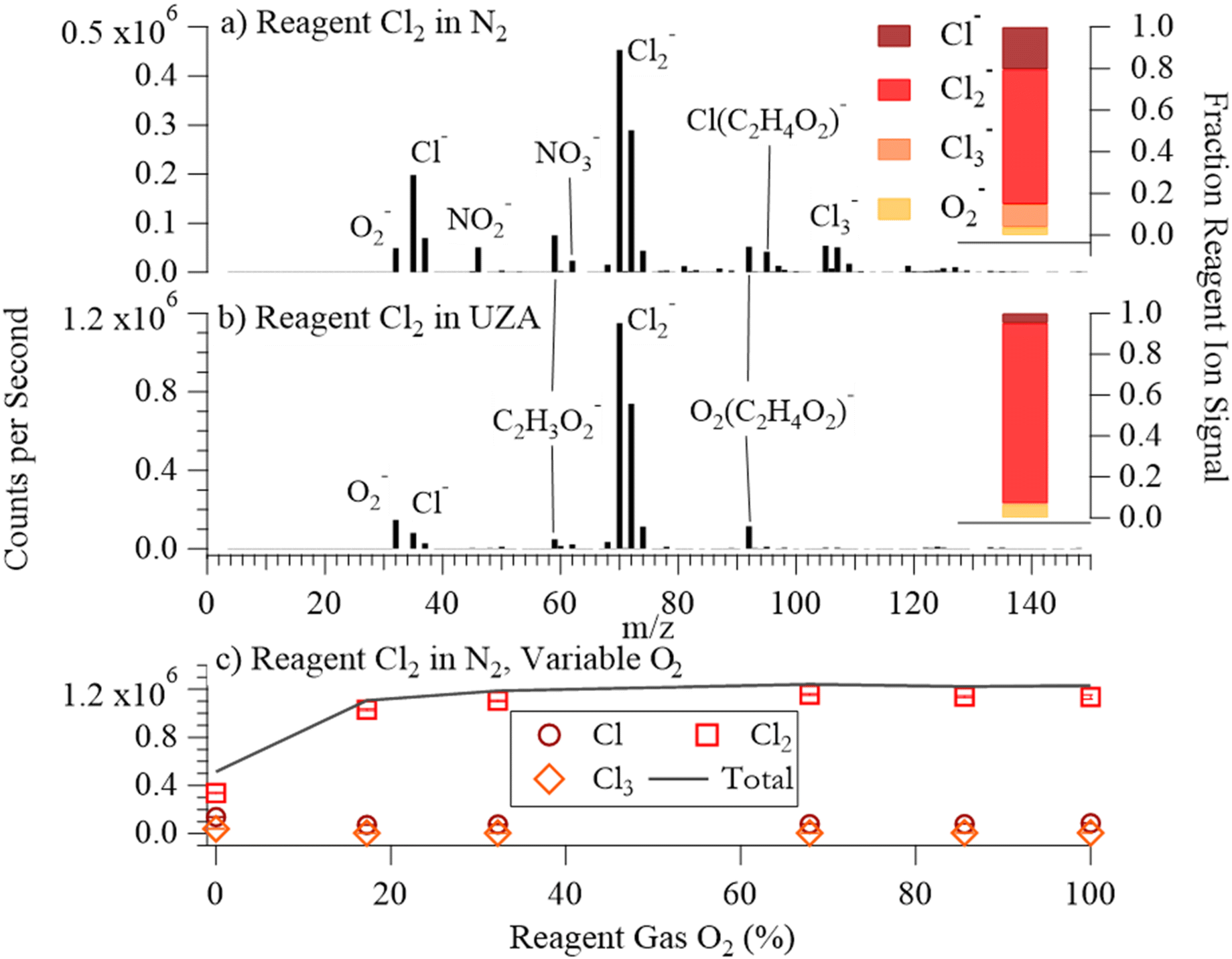 | ||
| Fig. 3 (a) Mass spectrum for dry N2 with a reagent ion flow of Cl2 in N2; fractional contributions of reagent ions (dark red: Cl−, light red: Cl2−, orange: Cl3−, yellow: O2−) to total reagent ion counts in the right-hand inset. (b) Mass spectrum for dry N2 with reagent ion flow of Cl2 in UZA and the respective fractional reagent ion contribution inset. (c) Ion counts for Cl−, Cl2− and Cl3− as a function of O2 mixing ratio (%) in the reagent gas mixture (dry N2 sampling). The black line represents the sum of these three ions. | ||
The recombination reaction (R5) is a significant source of Cl2− for liquid phase studies but is not expected to be important here.45 Instead, Cl− can go on to react with Cl2, forming the observed Cl3− signal (R6a), with Cl2− + Cl2(R6b) and Cl2− + Cl (R6c) reactions also being possible sources of Cl3−.46
| Cl− + Cl2 → Cl3− | (R6a) |
| Cl2− + Cl → Cl3− | (R6b) |
| Cl2− + Cl2 → Cl3− + Cl | (R6c) |
We found an upper limit of 400 ppb Cl2 in the reagent ion flow (Fig. 4(c)), above which point the Cl2− signal stopped increasing and Cl3− became the largest reagent ion signal. We were unable to isolate production of Cl2− through modulation of Cl2 concentration in N2 alone.
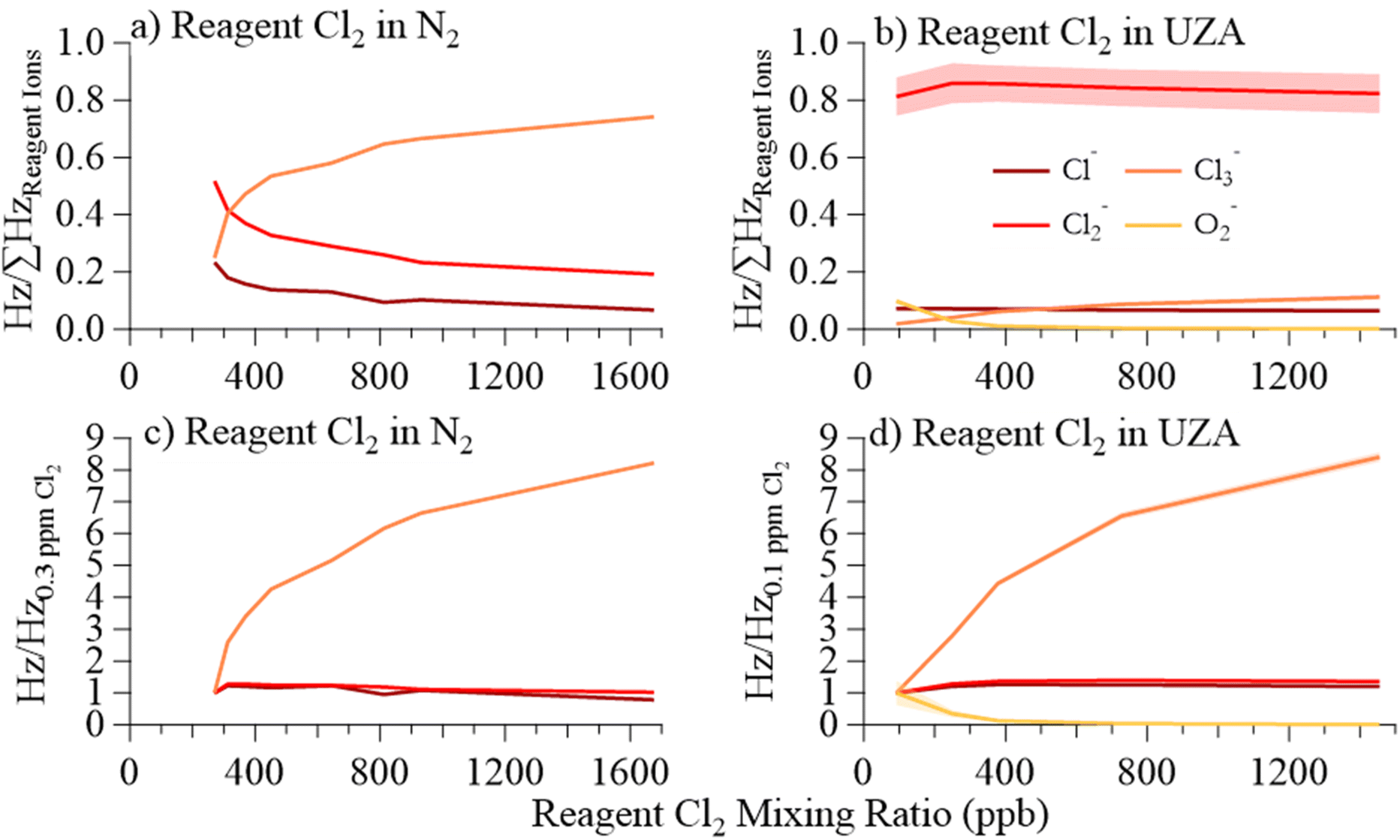 | ||
| Fig. 4 (a) Fraction of total reagent ion signal for Cl− (dark red), Cl2− (light red), and Cl3− (orange), as a function of reagent gas Cl2 mixing ratio (ppb) in N2. (b) Fraction of total reagent ion counts for Cl−, Cl2−, and Cl3−, and O2− (yellow) as a function of reagent gas Cl2 mixing ratio in UZA. (c) Reagent ion signal, relative to 300 ppb Cl2, as a function of reagent gas Cl2 mixing ratio in N2. (d) Reagent ion signal, relative to 100 ppb Cl2, as a function of reagent gas Cl2 mixing ratio in UZA. All reagent ion data was collected while sampling dry N2 to the CIMS inlet. | ||
Impacts of O2 and Cl2 concentrations
The reagent ion distribution changed with the addition of O2 (Fig. 3b and c). Adding ∼21% O2 to the ionizer flow increased total ion counts by 58%, increased the fraction of Cl2− ions in the total reagent ion signal from 60 to 90%, reduced Cl− and Cl3− signals, and increased O2−. Due to the direct dependence of Cl2(RC(O)O2)– on Cl2−, the net result of these changes was a factor of 2.4 increase in total sensitivity to RC(O)O2. Addition of O2 also caused a 32% decrease of non-reagent ions in background mass spectra (Fig. 3a and b), leading to overall improvements in LODs.The differences we observed in the reagent ion spectrum upon addition of O2 result from a change in the primary Cl2 ionization mechanism. At percent-level concentrations in the reagent gas mixture, O2 becomes the primary electron capturing molecule (R7).
| e− + O2 → O2− | (R7) |
Cl2− is a known product of Cl2 ionization by O2-CIMS,47 and we hypothesize Cl2− is primarily formed here in a multiple step process. Electrons formed by ionization in the Po-210 source may predominately attach to O2 molecules present in the reagent gas mixture at atmospheric mixing ratios. The electron affinities of Cl2 and O2 are 2.38 eV48 and 0.451 eV,49 respectively, indicating the following favorable electron transfer reaction as the primary source of Cl2− with O2 in the reagent gas mixture (R8).
| O2− + Cl2 → O2 + Cl2− | (R8) |
The reagent ion distribution impacts the chemical ionization mechanisms and thus sensitivity. We found that modulation of Cl2 reagent gas concentration in UZA produced a negative correlation between Cl2− and O2− (Fig. 4(d)). Analyte ions for the organic acids, O3, and CH3C(O)O2 responded to either Cl2− or O2−, depending on which ion chemistry produced each ion. The sensitivity of these analyte ions to changes in the Cl2 mixing ratio in the reagent gas mixture (in UZA) are shown in Fig. 5, with the slopes and r2 of the linear least squares fits in Table S10.† Cl− and Cl2− signals correlated well to both the Cl− and Cl2− cluster analyte ions (r2 > 0.73 for Cl− and > 0.59 for Cl2−). The exceptions were Cl2(C2H5C(O)OH)–, which had low sensitivity, and Cl2(CH3C(O)O2)– at Cl2 concentrations above 400 ppb (Fig. S11†). For Cl2 reagent gas concentrations below 400 ppb, Cl2− correlated to Cl2(CH3C(O)O2)– with slope = 0.53 and r2 = 0.99. Fitting of O2− cluster and H+ abstraction ion signals to Cl− and Cl2− resulted in negative slopes for all sampled acids. We also observed negative Cl− and Cl2− correlations for O3− and CO3− from O3 and for CH3C(O)O− and CH3C(O)O2− detected during CH3C(O)O2 sampling. The O2− clusters and H+ abstraction products, instead, correlated strongly to O2− signal, as did O3−, CO3−, and CH3C(O)O2− (r2 > 0.96; slopes = 1.0). Changes in Cl3− signal did not correlate to any analyte ion, returning slopes between 0.09 and −0.15.
 | ||
| Fig. 5 (a) Correlation plot of Cl− (diamonds) and Cl2− (hexagons) cluster ion signals for formic (dark red), acetic (light red), and propionic (orange) acids against background signals of Cl2− over the Cl2 concentration changes. Linear correlation fits shown with dashed (Cl− clusters) and solid (Cl2− clusters) lines. (b) Correlation plot of proton abstraction (circle) and O2− cluster (square) ion signals against background signals of O2− over the Cl2 concentration changes. Acid signals are indicated by the same colors as (a), with gray markers for O3 product ion correlations (squares for CO3− and circles for O3−). Linear fits are shown as dashed lines for proton abstraction and solid lines for O2− cluster. The y-axis represents ratios of the observed signal to the signal at the lowest Cl2 concentration; all error bars represent propagated uncertainty of one standard deviation in signal at each Cl2 concentration step. Isobutyric acid is left off to reduce graph clutter, as these signals had large uncertainty for Cl2− reactions. Fit information for isobutyric acid is provided in Table S10 and Fig. S12.† | ||
The formation of Cl− clusters from Cl− ionization is straightforward. However, Cl− cluster formation from Cl2− is also possible, due to the instability Cl2−. Here, the reaction between Cl2− and an analyte would act more like a ligand exchange, producing Cl-analyte ion and a Cl atom (R9).
| Cl2− + RC(O)OH → Cl + (RC(O)OH)Cl− | (R9) |
A similar ionization mechanism occurs with SF6− for some analytes.50 Cl3− formation acts as a sink of sensitivity in the Cl2-CIMS, as Cl3− signal did not show a positive correlation with any analyte ion. Cl3− is a cluster ion of Cl2 and Cl−, which has a high binding energy.51 This binding energy was larger than that of the acid-Cl- clusters (Fig. S6†). In addition, Cl3− has an electron affinity ≥ 3.8 eV,46 making electron transfer ionization unfavorable for most analytes.
The results in Fig. 5 demonstrate that analyte ion production is directly dependent on the availability of reagent ions that produce each analyte ion, and not necessarily on total reagent ion count. This observation suggests that the proper normalization for analyte sensitivities is to the corresponding reagent ion, as we provide for CH3C(O)O2 in Table 1. Sensitivities for all organic acid and O3 product ions are in Table S13.† Under the optimized conditions for Cl2− production in our system, the sensitivities normalized to Cl− and Cl2− shift only slightly in Table S13† from those in Table 1, as Cl2− accounted for ≥ 80% of total reagent ion counts across these experiments and the sum of Cl− and Cl2− accounted for ≥ 90%. Normalization to O2− increases the Cl2-CIMS sensitivity to proton abstraction and O2− cluster products by at least an order of magnitude compared to Table 1. The O2− normalized sensitivities are near the collision limit, but sensitivities to O2(CH3C(O)OH)– and CO3− are much larger than that limit and thus imply that additional ionization mechanisms or interferences are occurring.
O2− normalized sensitivities of 98 ± 12 ncps/ppt for O2(CH3C(O)OH)– and 56 ± 4 ncps/ppt for CO3− suggest that the measured O2− signals may not be representative of the O2− concentrations in the IMR. These inflated sensitivities may be due to changes in transmission efficiency with mass, as CIMS of similar design have lower transmission efficiencies at low m/z.52 If the reaction of O2− with Cl2 does not run to completion before sample introduction, analyte ions will appear at signals consistent with a larger O2− concentration than was measured, also explaining the surprisingly high O2− normalized sensitivities. Despite this problem, sensitivities in Table S13† may provide more consistent comparison of Cl2-CIMS response across changing instrument and sample conditions, as was the case for our CH3C(O)O2 calibration and MEK photolysis measurements.
Woodsmoke
While Cl2-CIMS is an effective detection technique for RO2 in controlled laboratory systems, woodsmoke demonstrates the problems of using Cl2-CIMS for complex mixtures. We use ion mass defects (defined as the difference between the exact m/z of an ion and the nearest unit mass) to visualize the differences in ions that appear in the Cl2-CIMS and I-CIMS mass spectra during woodsmoke experiments. Multiple reagent ions and potential reaction pathways produced more analyte ions with Cl2-CIMS (>300 ions; Fig. 6(a)). The I-CIMS spectra contained about half as many ions, which were mostly produced from iodide adduct formation (177 ions; Fig. 6(b)). Though more peaks appeared for Cl2-CIMS, these were not indicative of compounds unique to Cl2-CIMS detection. Instead, the larger peak count in Cl2-CIMS is a byproduct of multiple reagent ion chemistries, where a single analyte formula would appear in up to four product ions. We fit 103 Cl2− cluster analyte ions, which represented 39.8% of analyte ion signals measured with Cl2-CIMS. 82 Cl− clusters accounted for 21.6% of analyte ion signals, while 38.6% of analyte signal was from 128 ions which did not contain Cl. In contrast, the 111 identified I− containing ions accounted for 99.8% of analyte signals measured with I-CIMS. | ||
| Fig. 6 (a) Mass defect plot for identified ions from woodsmoke sampling with Cl2-CIMS. Blue circles indicate O2− cluster and non-clustered product ions, orange indicates Cl-containing, and red indicates Cl2-containing formulas. (b) Mass defect plot for detected ions during I-CIMS sampling of woodsmoke. Purple circles indicate peaks that did not contain iodide while green indicates I-containing formulas. Dot size represents the relative signal increase between background and smoke sampling steps. | ||
The largest signals for both reagent ions were small organic acids and peracids. Specifically, C2H4O2, CH2O2 and C3H6O3 were all measured at signals that were about two times greater than any other product by I-CIMS. These species were also among the most prominent Cl2-CIMS signals. However, the highest analyte signal for Cl2-CIMS was O2(CH4O2)– (∼200000 cps), corresponding to the oxygen cluster for acetic acid. This observation again indicates that the conditions in the IMR do not reflect the final spectra, and that the Cl2-CIMS acts largely as an O2-CIMS under polluted sampling conditions.
The appearance of Cl2−, Cl−, and O2− clusters, and molecular ions (or H+ abstraction products) led to an array of possible mass defects. Overlap of ions at the same mass-to-charge occurred more frequently for Cl2-CIMS. I-CIMS provided I− clusters with large mass defects and a lack of competing ion-molecule reactions. Thus, ions detected by I-CIMS were more often separated from interfering peaks than those detected by Cl2-CIMS. For example, nitrogen-containing products of biomass burning, including C2H5NO, CH3NCO, C3H7NO,53 could be identified with I-CIMS, but not clearly with Cl2-CIMS. This limitation was largely due to the tendency for overlap of Cl2− (even mass) clusters, which might contain N, with Cl− (odd mass) clusters, which did not (and vice versa). We saw little evidence for the identification of RO2 in our data due to spectral complexity. It is also possible in a complex sample that side reactions of Cl2-CIMS would produce ion formulas that are not representative of sampled molecular formulae, particularly if Cl radicals are formed from the decomposition of Cl2−. We could derive no clear evidence of this from the woodsmoke experiments but provide evidence of possible Cl radical impacts in Fig. S11.† Due to the detrimental complexities of the current Cl2-CIMS ion chemistry, we could draw little else from the analysis of these smoke samples without significant alterations to the CIMS IMR design.
Conclusion
Direct measurements of RO2 in both ambient and laboratory settings require low limits of detection (1 ppt). Cl2-CIMS is an effective reagent ion chemistry for laboratory measurement of CH3C(O)O2 and C2H5C(O)O2 with better sensitivity than existing I-CIMS measurements – capable of producing quantum yields for MEK photolysis consistent with results obtained from other measurement techniques in the literature. However, the reported LOD of 5 ppt, higher than that found for I-CIMS, is too high for ambient measurement of RO2. The sensitivity of Cl2-CIMS to CH3C(O)O2 and C2H5C(O)O2 would be increased through isolation of Cl2− ionization chemistry, without the non-selective ionization of O2− or the potential for secondary chemistry of Cl atoms in the IMR. Such alterations to Cl2-CIMS would lower LODs and expand its applications.Reactions of O2− may be avoided by using a different electron donor. For the electron transfer with Cl2 to occur, the electron donor must be more reactive than Cl2−. High purity air requires few considerations for use in CIMS reagent gas mixtures, compared to, for example, SF6 (electron affinity = 1.05 eV).50 In addition, O2− reacts with Cl2 in our system to form Cl2− with few notable secondary products other than Cl−. This is not a guarantee for other electron donors. A more effective means of isolating Cl2− chemistry may be to alter the IMR design to add an initial reaction chamber where the O2− + Cl2 reaction would run to completion prior to sample introduction, thus minimizing O2− chemistry. However, such modifications were outside the scope of this study. With the current instrumental design, the multiple reagent ion chemistries present in the Cl2-CIMS system prevent the reagent ion chemistry from being useful for ambient studies, as evidenced by the woodsmoke measurements. Alternatively, this work highlights the utility of dual reagent ions (Cl2− and O2−) to provide a selective measurement of acyl peroxy radicals.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest regarding the content of this manuscript.Acknowledgements
We thank Dr Kathryn Mayer for assisting with the woodsmoke sampling tests. We thank Dr Frank Flocke for providing the PAN source used for calibration tests. We thank the National Science Foundation (Grant 1922619) for funding.References
- C. A. Cantrell, R. E. Shetter and J. G. Calvert, Comparison of Perxoy Radical Concentrations at Several Contrasting Sites, J. Atmos. Sci., 1995, 52, 3408–3412 CrossRef.
- Z. Tan, A. Hofzumahaus, K. Lu, S. S. Brown, F. Holland, L. G. Huey, A. Kiendler-Scharr, X. Li, X. Liu, N. Ma, K. E. Min, F. Rohrer, M. Shao, A. Wahner, Y. Wang, A. Wiedensohler, Y. Wu, Z. Wu, L. Zeng, Y. Zhang and H. Fuchs, No Evidence for a Significant Impact of Heterogeneous Chemistry on Radical Concentrations in the North China Plain in Summer 2014, Environ. Sci. Technol., 2020, 54, 5973–5979 CrossRef CAS PubMed.
- G. S. Tyndall, R. A. Cox, C. Granier, R. Lesclaux, G. K. Moortgat, M. J. Pilling, A. R. Ravishankara and T. J. Wallington, Atmospheric chemistry of small organic peroxy radicals, J. Geophys. Res.:Atmos., 2001, 106, 12157–12182 CrossRef CAS.
- A. Mellouki, T. J. Wallington and J. Chen, Atmospheric Chemistry of Oxygenated Volatile Organic Compounds: Impacts on Air Quality and Climate, Chem. Rev., 2015, 115, 3984–4014 CrossRef CAS PubMed.
- S. Y. Chen and Y. P. Lee, Transient infrared absorption of t-CH3C(O)OO, c-CH3C(O)OO, and α-lactone recorded in gaseous reactions of CH3CO and O2, J. Chem. Phys., 2010, 132, 114303 CrossRef PubMed.
- M. George, M. Dolores Andrés Hernández, V. Nenakhov, Y. Liu and J. Philip Burrows, Airborne measurement of peroxy radicals using chemical amplification coupled with cavity ring-down spectroscopy: The PeRCEAS instrument, Atmos. Meas. Tech., 2020, 13, 2577–2600 CrossRef CAS.
- H. Fuchs, F. Holland and A. Hofzumahaus, Measurement of tropospheric RO2 and HO2 radicals by a laser-induced fluorescence instrument, Rev. Sci. Instrum., 2008, 79, 084104 CrossRef PubMed.
- E. C. Wood, B. L. Deming and S. Kundu, Ethane-based chemical amplification measurement technique for atmospheric peroxy radicals, Environ. Sci. Technol. Lett., 2017, 4, 15–19 CrossRef CAS.
- S. Kundu, B. L. Deming, M. M. Lew, B. P. Bottorff, P. Rickly, P. S. Stevens, S. Dusanter, S. Sklaveniti, T. Leonardis, N. Locoge and E. C. Wood, Peroxy radical measurements by ethane-nitric oxide chemical amplification and laser-induced fluorescence during the IRRONIC field campaign in a forest in Indiana, Atmos. Chem. Phys., 2019, 19, 9563–9579 CrossRef CAS.
- J. J. Orlando and G. S. Tyndall, Laboratory studies of organic peroxy radical chemistry: An overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- Z. Tan, H. Fuchs, K. Lu, A. Hofzumahaus, B. Bohn, S. Broch, H. Dong, S. Gomm, R. Häseler, L. He, F. Holland, X. Li, Y. Liu, S. Lu, F. Rohrer, M. Shao, B. Wang, M. Wang, Y. Wu, L. Zeng, Y. Zhang, A. Wahner and Y. Zhang, Radical chemistry at a rural site (Wangdu) in the North China Plain: Observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 2017, 17, 663–690 CrossRef CAS.
- R. S. Hornbrook, J. H. Crawford, G. D. Edwards, O. Goyea, R. L. Mauldin, J. S. Olson and C. A. Cantrell, Measurements of tropospheric HO2 and RO2 by oxygen dilution modulation and chemical ionization mass spectrometry, Atmos. Meas. Tech., 2011, 4, 735–756 CrossRef CAS.
- M. Neumaier, R. Ruhnke, O. Kirner, H. Ziereis, G. Stratmann, C. A. M. Brenninkmeijer and A. Zahn, Impact of acetone (photo)oxidation on HOx production in the UT/LMS based on CARIBIC passenger aircraft observations and EMAC simulations, Geophys. Res. Lett., 2014, 41, 3289–3297 CrossRef CAS.
- S. A. McKeen, T. Gierczak, J. B. Burkholder, P. O. Wennberg, T. F. Hanisco, E. R. Keim, R.-S. Gao, S. C. Liu, A. R. Ravishankara and D. W. Fahey, The photochemistry of acetone in the upper troposphere: a source of odd-hydrogen radicals, Geophys. Res. Lett., 1997, 24, 3177–3180 CrossRef CAS.
- P. O. Wennberg, T. F. Hanisco, L. Jaeglé, D. J. Jacob, E. J. Hintsa, E. J. Lanzendorf, J. G. Anderson, R. S. Gao, E. R. Keim, S. G. Donnelly, L. A. Del Negro, D. W. Fahey, S. A. McKeen, R. J. Salawitch, C. R. Webster, R. D. May, R. L. Herman, M. H. Proffitt, J. J. Margitan, E. L. Atlas, S. M. Schauffler, F. Flocke, C. T. McElroy and T. P. Bui, Hydrogen radicals, nitrogen radicals, and the production of O3 in the upper troposphere, Science, 1998, 279, 49–53 CrossRef CAS PubMed.
- M. A. Blitz, D. E. Heard, M. J. Pilling, S. R. Arnold and M. P. Chipperfield, Pressure and temperature-dependent quantum yields for the photodissociation of acetone between 279 and 327.5 nm, Geophys. Res. Lett., 2004, 31, 1–5 Search PubMed.
- T. Gierczak, J. B. Burkholder, S. Bauerle and A. R. Ravishankara, Photochemistry of acetone under tropospheric conditions, Chem. Phys., 1998, 231, 229–244 CrossRef CAS.
- M. Emrich and P. Warneck, Erratum: Photodissociation of acetone in air: dependence on pressure and wavelength, behavior of the excited singlet state, J. Phys. Chem. A, 2000, 104, 9436–9442 CrossRef CAS.
- A. G. Zborowska, C. Y. MacInnis, C. Z. Ye and H. D. Osthoff, On the photolysis branching ratio of methyl ethyl ketone, Atmos. Environ., 2021, 254, 118383 CrossRef CAS.
- P. R. Veres, J. M. Roberts, R. J. Wild, P. M. Edwards, S. S. Brown, T. S. Bates, P. K. Quinn, J. E. Johnson, R. J. Zamora and J. De Gouw, Peroxynitric acid (HO2NO2) measurements during the UBWOS 2013 and 2014 studies using iodide ion chemical ionization mass spectrometry, Atmos. Chem. Phys., 2015, 15, 8101–8114 CrossRef CAS.
- S. R. Albrecht, A. Novelli, A. Hofzumahaus, S. Kang, Y. Baker, T. Mentel, A. Wahner and H. Fuchs, Measurements of hydroperoxy radicals (HO2) at atmospheric concentrations using bromide chemical ionisation mass spectrometry, Atmos. Meas. Tech., 2019, 12, 891–902 CrossRef CAS.
- J. Sanchez, D. J. Tanner, D. Chen, L. G. Huey and N. L. Ng, A new technique for the direct detection of HO2 radicals using bromide chemical ionization mass spectrometry (Br-CIMS): initial characterization, Atmos. Meas. Tech., 2016, 9, 3851–3861 CrossRef CAS.
- M. F. Link, D. K. Farmer, T. Berg, F. Flocke and A. R. Ravishankara, Measuring Photodissociation Product Quantum Yields Using Chemical Ionization Mass Spectrometry: A Case Study with Ketones, J. Phys. Chem. A, 2021, 125, 6836–6844 CrossRef CAS PubMed.
- S. Iyer, X. He, N. Hyttinen, T. Kurtén and M. P. Rissanen, Computational and Experimental Investigation of the Detection of HO2 Radical and the Products of Its Reaction with Cyclohexene Ozonolysis Derived RO2 Radicals by an Iodide-Based Chemical Ionization Mass Spectrometer, J. Phys. Chem. A, 2017, 121, 6778–6789 CrossRef CAS PubMed.
- M. A. Robinson, J. Andrew Neuman, L. G. Huey, J. M. Roberts, S. S. Brown and P. R. Veres, Temperature-dependent sensitivity of iodide chemical ionization mass spectrometers, Atmos. Meas. Tech., 2022, 15, 4295–4305 CrossRef CAS.
- D. L. Slusher, L. G. Huey, D. J. Tanner, F. M. Flocke and J. M. Roberts, A thermal dissociation - chemical ionization mass spectrometry (TD-CIMS) technique for the simultaneous measurement of peroxyacyl nitrates and dinitrogen pentoxide, J. Geophys. Res.: Atmos., 2004, 109, 1–13 CrossRef.
- A. Hansel, W. Scholz, B. Mentler, L. Fischer and T. Berndt, Detection of RO2 radicals and other products from cyclohexene ozonolysis with NH4+ and acetate chemical ionization mass spectrometry, Atmos. Environ., 2018, 186, 248–255 CrossRef CAS.
- T. Berndt, B. Mentler, W. Scholz, L. Fischer, H. Herrmann, M. Kulmala and A. Hansel, Accretion Product Formation from Ozonolysis and OH Radical Reaction of α-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 2018, 52, 11069–11077 CrossRef CAS PubMed.
- A. Zaytsev, M. Breitenlechner, A. Novelli, H. Fuchs, D. A. Knopf, J. H. Kroll and F. N. Keutsch, Application of chemical derivatization techniques combined with chemical ionization mass spectrometry to detect stabilized Criegee intermediates and peroxy radicals in the gas phase, Atmos. Meas. Tech., 2021, 14, 2501–2513 CrossRef CAS.
- G. D. Edwards, C. A. Cantrell, S. Stephens, B. Hill, O. Goyea, R. E. Shetter, R. L. Mauldin, E. Kosciuch, D. J. Tanner and F. L. Eisele, Chemical Ionization Mass Spectrometer Instrument for the Measurement of Tropospheric HO2 and RO2, Anal. Chem., 2003, 75, 5317–5327 CrossRef CAS PubMed.
- W. Zhang and H. Zhang, Secondary Ion Chemistry Mediated by Ozone and Acidic Organic Molecules in Iodide-Adduct Chemical Ionization Mass Spectrometry, Anal. Chem., 2021, 8595–8602 CrossRef CAS PubMed.
- T. H. Bertram, J. R. Kimmel, T. A. Crisp, O. S. Ryder, R. L. N. Yatavelli, J. A. Thornton, M. J. Cubison, M. Gonin, D. R. Worsnop and A. field-deployable, chemical ionization time-of-flight mass spectrometer, Atmos. Meas. Tech., 2011, 4, 1471–1479 CrossRef CAS.
- P. Brophy and D. K. Farmer, A switchable reagent ion high resolution time-of-flight chemical ionization mass spectrometer for real-time measurement of gas phase oxidized species: Characterization from the 2013 southern oxidant and aerosol study, Atmos. Meas. Tech., 2015, 8, 2945–2959 CrossRef CAS.
- F. M. Flocke, A. J. Weinheimer, A. L. Swanson, J. M. Roberts, R. Schmitt and S. Shertz, On the measurement of PANs by gas chromatography and electron capture detection, J. Atmos. Chem., 2005, 52, 19–43 CrossRef CAS.
- P. Brophy and D. K. Farmer, Clustering, methodology, and mechanistic insights into acetate chemical ionization using high-resolution time-of-flight mass spectrometry, Atmos. Meas. Tech., 2016, 9, 3969–3986 CrossRef CAS.
- J. Li, M. F. Link, S. Pandit, M. H. Webb, K. J. Mayer, L. A. Garofalo, K. L. Rediger, D. G. Poppendieck, S. M. Zimmerman, M. E. Vance, V. H. Grassian, G. C. Morrison, B. J. Turpin and D. K. Farmer, The persistence of smoke VOCs indoors: Partitioning, surface cleaning, and air cleaning in a smoke-contaminated house, Sci. Adv., 2023, 9, eadh8263 CrossRef CAS PubMed.
- I. Dotan, J. A. Davidson, G. E. Streit, D. L. Albritton and F. C. Fehsenfeld, A study of the reaction O3−+CO2 ⇌ CO3− + O2 and its implication on the thermochemistry of CO3 and O3 and their negative ions, J. Chem. Phys., 1977, 67, 2874–2879 CrossRef CAS.
- M. Wang, X.-C. He, H. Finkenzeller, S. Iyer, D. Chen, J. Shen, M. Simon, V. Hofbauer, J. Kirkby, J. Curtius, N. Maier, T. Kurtén, D. Worsnop, M. Kulmala, M. Rissanen, R. Volkamer, Y. J. Tham, N. Donahue and M. Sipilä, Measurement of iodine species and sulfuric acid using bromide chemical ionization mass spectrometers, Atmos. Meas. Tech. Discuss, 2020, 1–23 Search PubMed.
- L. Xu, M. M. Coggon, C. E. Stockwell, J. B. Gilman, M. A. Robinson, M. Breitenlechner, A. Lamplugh, J. D. Crounse, P. O. Wennberg, J. A. Neuman, G. A. Novak, P. R. Veres, S. S. Brown and C. Warneke, Chemical ionization mass spectrometry utilizing ammonium ions (NH4+ CIMS) for measurements of organic compounds in the atmosphere, Atmos. Meas. Tech., 2022, 15, 7353–7373 CrossRef CAS.
- B. Rajakumar, T. Gierczak, J. E. Flad, A. R. Ravishankara and J. B. Burkholder, The CH3CO quantum yield in the 248 nm photolysis of acetone, methyl ethyl ketone, and biacetyl, J. Photochem. Photobiol., A, 2008, 199, 336–344 CrossRef CAS.
- J. M. Mattila, P. S. J. Lakey, M. Shiraiwa, C. Wang, J. P. D. Abbatt, C. Arata, A. H. Goldstein, L. Ampollini, E. F. Katz, P. F. Decarlo, S. Zhou, T. F. Kahan, F. J. Cardoso-Saldaña, L. H. Ruiz, A. Abeleira, E. K. Boedicker, M. E. Vance and D. K. Farmer, Multiphase Chemistry Controls Inorganic Chlorinated and Nitrogenated Compounds in Indoor Air during Bleach Cleaning, Environ. Sci. Technol., 2020, 54, 1730–1739 CrossRef CAS PubMed.
- F. D. Lopez-Hilfiker, S. Iyer, C. Mohr, B. H. Lee, E. L. D’ambro, T. Kurtén and J. A. Thornton, Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts, Atmos. Meas. Tech., 2016, 9, 1505–1512 CrossRef CAS.
- J. D. Crounse, K. A. McKinney, A. J. Kwan and P. O. Wennberg, Measurement of gas-phase hydroperoxides by chemical ionization mass spectrometry, Anal. Chem., 2006, 78, 6726–6732 CrossRef CAS PubMed.
- L. G. Christophorou and J. K. Olthoff, Electron Interactions With Cl2, J. Phys. Chem. Ref. Data, 1999, 28, 131–169 CrossRef CAS.
- J. C. Han, M. Suto, J. C. Lee and Z. L. Petrović, Transient signals induced by laser irradiation of negative ions in hollow electrode discharges of Cl2 and HCl in N2, J. Appl. Phys., 1990, 68, 2649–2656 CrossRef CAS.
- L. M. Babcock and G. E. Streit, Ion–molecule reactions of Cl2 with Cl− and F−, J. Chem. Phys., 1982, 76, 2407–2411 CrossRef CAS.
- B. D. Finley and E. S. Saltzman, Observations of Cl2, Br2, and I2 in coastal marine air, J. Geophys. Res.:Atmos., 2008, 113, D21301 CrossRef.
- W. A. Chupka, J. Berkowitz and D. Gutman, Electron affinities of halogen diatomic molecules as determined by endoergic charge transfer, J. Chem. Phys., 1971, 55, 2724–2733 CrossRef CAS.
- M. J. Travers, D. C. Cowles and G. B. Ellison, Reinvestigation of the Electron Affinities of O2 and NO, Chem. Phys. Lett., 1989, 164, 450–455 CrossRef.
- L. G. Huey, D. R. Hanson and C. J. Howard, Reactions of SF6− and I− with atmospheric trace gases, J. Phys. Chem., 1995, 99, 5001–5008 CrossRef CAS.
- C. Amelynck, E. Arijs, N. Schoon and A.-M. Van Bavel, Gas phase reactions of HNO3 with Cl−, Cl−H2O, and Cl−HCl, of Cl2 with Cl−H2O and Cl−HCl, and of HCl with Cl−H2O, Int. J. Mass Spectrom., 1998, 181, 113–121 CrossRef CAS.
- M. Heinritzi, M. Simon, G. Steiner, A. C. Wagner, A. Kürten, A. Hansel and J. Curtius, Characterization of the mass-dependent transmission efficiency of a CIMS, Atmos. Meas. Tech., 2016, 9, 1449–1460 CrossRef CAS.
- M. Priestley, M. Le Breton, T. J. Bannan, K. E. Leather, A. Bacak, E. Reyes-Villegas, F. De Vocht, B. M. A. Shallcross, T. Brazier, M. Anwar Khan, J. Allan, D. E. Shallcross, H. Coe and C. J. Percival, Observations of Isocyanate, Amide, Nitrate, and Nitro Compounds From an Anthropogenic Biomass Burning Event Using a ToF-CIMS, J. Geophys. Res.:Atmos., 2018, 123, 7687–7704 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ea00043b |
| ‡ Now at Engineering Laboratory, National Institute of Standards and Technology, Gaithersburg, Maryland 20899, USA. |
| This journal is © The Royal Society of Chemistry 2025 |