Investigation of organic hydrotrioxide (ROOOH) formation from RO2 + OH reactions and their atmospheric impact using a chemical transport model, STOCHEM-CRI†
Received
27th January 2025
, Accepted 5th March 2025
First published on 6th March 2025
Abstract
Incorporating the reactions of fifty peroxy radicals (RO2) with the hydroxyl radical (OH) into the global chemistry transport model, STOCHEM-CRI, affected the composition of the troposphere by changing the global burdens of NOx (−2.7 Gg, −0.5%), O3 (−2.3 Tg, −0.7%), CO (−3.2 Tg, −0.8%), HOx (+2.1 Gg, +7.7%), H2O2 (+0.5 Tg, +18.3%), RO2 (−8.0 Gg, −18.2%), RONO2 (−19.4 Gg, −4.7%), PAN (−0.1 Tg, −3.4%) HNO3 (−7.4 Gg, −1.3%) and ROOH (−96.9 Gg, −3.8%). The RO2 + OH addition reactions have a significant impact on HO2 mixing ratios in tropical regions with up to a 25% increase, resulting in increasing H2O2 mixing ratios by up to 50% over oceans. Globally, a significant amount of organic hydrotrioxides (ROOOH) (86.1 Tg per year) are produced from these reactions with CH3OOOH (67.5 Tg per year, 78%), isoprene-derived ROOOH (5.5 Tg per year, 6%) and monoterpene-derived ROOOH (4.2 Tg per year, 5%) being the most significant contributors. The tropospheric global burden of CH3OOOH is found to be 0.48 Gg. The highest mixing ratios of ROOOH, of up to 0.35 ppt, are found primarily in the oceans near the tropical land areas. The RO2 + OH reactions have a small, but noticeable, contribution to OH reactivity (∼5%) over tropical oceans. Additionally, these reactions have a significant impact on RO2 reactivity over tropical oceans where losses of the CH3O2 radical, isoprene derived peroxy radical (ISOPO2) and monoterpene derived peroxy radical (MONOTERPO2) by OH can contribute up to 25%, 15% and 50% to the total RO2 loss, respectively. The changes in RO2 reactivity influence the global abundances of organic alcohols (ROH) which are important species due to their crucial impact on air quality. The ROOOH generate secondary organic aerosol (SOA) of up to 0.05 μg m−3 which affects the Earth's radiation budget because of enhancing modelled organic aerosol by up to 5% and 2000% on land surfaces and the remote tropical oceans, respectively.
Environmental significance
Organic hydrotrioxides (ROOOH) formed from the oxidation of peroxy radicals (RO2) by OH in the troposphere play a significant role in the oxidising capacity of the atmosphere. Investigation of RO2 + OH reactions and their product distributions is important for understanding the impact of volatile organic compound (VOC) emission and their chemistry that degrades air quality in the troposphere. In this study, we quantify the production of ROOOH from both anthropogenic and biogenic VOCs and their role in secondary organic aerosol formation. The inclusion of RO2 + OH reactions in global Chemistry Transport Models (CTMs) and Earth System Models (ESMs) is highly recommended for the assessment of policies addressing global air quality and climate change.
|
1. Introduction
Organic peroxy radicals (RO2) are highly reactive intermediates and are ubiquitous in the troposphere.1–6 They are mainly formed from the oxidation of volatile organic compounds (VOCs) by hydroxyl radicals (OH) via reactions (R1) and (R2) and additionally by ozone (O3), nitrate radicals (NO3), stabilized Criegee intermediates (SCIs) and chlorine atoms (Cl). RO2 can be lost by several gas phase reactions depending on the nature of the tropospheric chemistry regime, whether it is low NOx (NOx = NO + NO2) (NO mixing ratios typically smaller than 10 pptv) or high NOx (NO mixing ratios typically higher than 1 ppbv). In high NOx environments, the dominant loss of tropospheric RO2 is through reaction with nitric oxide (NO) which produces an alkoxy radical (RO) and nitrogen dioxide (NO2), leading to HOx (= OH + HO2) propagation and NOx catalysed ozone generation (O3) (R3a–R7).3,4,7,8 A small branching ratio exists for the reaction between RO2 and NO (R3b) resulting in the formation of organic nitrates (RONO2). The yields of ozone and RONO2 strongly depend on the composition of VOCs and the amount of oxidants (OH, O3, Cl, SCIs or NO3) in the atmosphere.9–11 However, in low NOx environments, RO2 radicals preferentially react with other RO2, including hydroperoxy radicals (HO2),5 or isomerise or auto-oxidise to form different multi-functionalised oxygenated peroxy radicals.12 The highly oxygenated organic molecules (HOM) formed from the isomerisation or auto-oxidation of RO2 can condense on pre-existing particles and contribute to the formation of secondary organic aerosol (SOA).13 The peroxy–peroxy radical self- and cross-reactions lead to either formation of RO (R8a) or a chain terminating mechanism that ultimately results in carbonyl and alcohol production (R8b) or formation of dimers.4,14 The extent of the branching of this reaction is determined by the nature of the alkyl group incorporated into the RO2 radical.3,5 The RO formed predominantly from <C5 peroxy radicals proceed to react with O2 yielding HO2 which can go on to participate in a further reaction with RO2 ((R4) and (R9)) to form organic hydroperoxides (ROOH). ROOH act as temporary HOx reservoirs until they are subjected to photolysis or undergo reaction with OH. ROOH can also be lost from the troposphere via deposition as well as participating in the formation of SOA.15–20| | | R + O2 + M → RO2 + M (M = N2 or O2) | (R2) |
| | | RO2 + NO + M → RONO2 + M | (R3b) |
| | | RO + O2 → carbonyl product(s) + HO2 | (R4) |
| | | NO2 + hν → NO + O(3P) | (R6) |
| | | O(3P) + O2 + M → O3 + M | (R7) |
| | | RO2 + RO2 → 2RO + O2 | (R8a) |
| | | RO2 + RO2 → carbonyl product(s) + ROH + O2 | (R8b) |
| | | RO2 + RO2 → ROOR + O2 | (R8c) |
| | | RO2 + HO2 → ROOH + O2 | (R9) |
The rates at which RO2 react are directly related to their structure and the nature of their organic moiety. RO2 self-reactions act to slow the production of O3 from RO2, and, in some circumstances, O3 formation is eliminated completely if the rate of the self-reaction is fast.21 At night-time, the reaction between RO2 and NO3 is important which increases the concentrations of RO and HOxvia propagation reactions ((R10) and (R4)).2,22
| | | RO2 + NO3 → RO + NO2 + O2 | (R10) |
Experimental studies have shown that CH3O2 reacts rapidly with OH with a range of rate coefficients e.g., 2.8 × 10−10 cm3 molecule−1 s−1,23 8.4 × 10−11 cm3 molecule−1 s−1,24 1.6 × 10−10 cm3 molecule−1 s−1,25 and 1.0 × 10−10 cm3 molecule−1 s−1.26 The reactions of larger peroxy radicals, RO2 (C2H5O2 to C4H9O2) with OH showed the same behaviour but with larger rate constants e.g. 1.2 × 10−10 cm3 molecule−1 s−1 to 1.4 × 10−10 cm3 molecule−1 s−1.27,28 Large rate coefficients are also reported by Berndt et al.29 for the reactions of isoprene and monoterpene derived peroxy radicals with OH, e.g. 5.1 × 10−11 cm3 molecule−1 s−1 for HO-C5H8O2, 1.1 × 10−10 cm3 molecule−1 s−1 for HO-C5H8(O2)O2 and 1.8 × 10−10 cm3 molecule−1 s−1 for HO-C10H16(O2)2O2. So, the RO2 + OH reactions are competitive with reactions (R3), (R8) and (R10) and act as major sinks for RO2 in remote atmospheres.
The possible products of the RO2 + OH reactions are as follows:
| | | RO2 + OH → RO + HO2 | (R11a) |
| | | RO2 + OH → ROH + O2 | (R11b) |
| | | RO2 + OH → R(–H)O2 + H2O | (R11c) |
The estimation through theoretical approaches by Müller et al.30 and through experiments by Yan et al.24 and Assaf et al.27 showed a negligible product, SCI (R11c) with a fractional upper limit of 0.05 and different branching ratios for reaction (R11a) and (R11b) if R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH3. An experimental kinetic study of the reaction of CH3O2 + OH by Caravan et al.31 suggested that channel (R11b) is an insignificant product channel with yields of 0.09 and 0.06 at low and high pressure, respectively. A high yield of 0.9 for reaction (R11a) was determined experimentally at low pressure (67 hPa),32 which is consistent with the best theoretical estimate (0.92) determined by Müller et al.30 The yield for reaction (R11a) is highly dependent on the size of RO2 and is reduced to 0.75 for C2H5O2, 0.41 for C3H7O2 and 0.15 for C4H9O2.32 The decrease of the yield of reaction (R11a) with increasing size of the R group led to the increase of the stabilization of ROOOH (R11d).29 Müller et al.30 predicted a yield for stabilized trioxide (CH3OOOH) (R11d) of 0.07 at 760 torr. However, with increasing the size of the R, the formation of thermalized ROOOH is the dominant product from (R11).29 The ROOOH formed in (R11d) have several possible fates, among which are reaction with OH and uptake by aqueous aerosols followed by decomposition into ROH + O2 which could be the dominant channel.30
CH3. An experimental kinetic study of the reaction of CH3O2 + OH by Caravan et al.31 suggested that channel (R11b) is an insignificant product channel with yields of 0.09 and 0.06 at low and high pressure, respectively. A high yield of 0.9 for reaction (R11a) was determined experimentally at low pressure (67 hPa),32 which is consistent with the best theoretical estimate (0.92) determined by Müller et al.30 The yield for reaction (R11a) is highly dependent on the size of RO2 and is reduced to 0.75 for C2H5O2, 0.41 for C3H7O2 and 0.15 for C4H9O2.32 The decrease of the yield of reaction (R11a) with increasing size of the R group led to the increase of the stabilization of ROOOH (R11d).29 Müller et al.30 predicted a yield for stabilized trioxide (CH3OOOH) (R11d) of 0.07 at 760 torr. However, with increasing the size of the R, the formation of thermalized ROOOH is the dominant product from (R11).29 The ROOOH formed in (R11d) have several possible fates, among which are reaction with OH and uptake by aqueous aerosols followed by decomposition into ROH + O2 which could be the dominant channel.30
The reaction of RO2 with NO and HO2 and self- or cross-reactions with other RO2 radicals are included in most of the atmospheric chemistry transport models (CTMs), but RO2 + OH reactions are not explicitly considered in CTMs yet because of unexplored rate coefficients of the reaction of large RO2 radicals with OH, the yields of the products and the fate of the ROOOH formed from these reactions. We considered all RO2 + OH reactions taking into account the literature-based rate coefficients and product yields for channels (R11a–R11d) which led to an update of the burdens of the global tropospheric composition and their life cycles. Taking into account the lack of global modelling studies of RO2 with OH radicals and their potential impact in low NOx environments, we have incorporated these reactions in the STOCHEM-CRI model and investigated the impact of these reactions on tropospheric composition. The formation of individual ROOOH, their distribution throughout the troposphere and their environmental impacts in terms of SOA formation have been investigated in the study. We also showed the impact of RO2 + OH reactions on the loss rates of peroxy radicals and hydroxyl radicals focusing on the tropics and discussed their implications for the oxidising capacity of the atmosphere.
2. Model description
STOCHEM is a global three-dimensional transport model that utilises a 3-hour time step for the advection of its 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 constant mass air parcels that represent the Earth's troposphere.33 It is within these air parcels that the chemical reactions and photochemical dissociations leading to the loss and production of trace gases are taking place. The data concerning pressure, temperatures, winds, clouds, humidities, tropopause heights, precipitation, boundary layer depth and surface parameters are taken for the year 1998 from archived meteorological data from the UKMO Unified Model (UM).34 Meteorological data are based on a grid resolution of 1.25° longitude, 0.833° latitude and 12 unevenly spaced (with respect to altitude) vertical levels between the surface and an upper boundary of ∼100 mb.33 A Lagrangian approach for advection allows uncoupling of the chemistry and transport processes. The chemical mechanism used in STOCHEM is the Common Representative Intermediates (CRI) version 2 reduction 5 (CRI v2-R5) which was built using a series of five-day box model simulations on each compound, on a compound-by-compound basis. CRI v2-R5 was developed initially by Jenkin et al.35 from the Master Chemical Mechanism (https://mcm.york.ac.uk/MCM/) with subsequent improvement by Watson et al.,36 and the CRI scheme involves the most reduced mechanism, making it suitable for global modelling due to its traceability to the MCM. More details of the CRI v2-R5 mechanism can be found in Utembe et al.37,38 and recent updates of the CRI v2.2 mechanism can be found in Jenkin et al.39 STOCHEM-CRI calculates one-hourly photolysis rates for each reaction within each air parcel, before linearly interpolating the data with respect to time. This provides photolysis rates at a resolution of 5 minutes to be used in the chemical integration. The photolysis rates were calculated by integrating (over all wavelengths) the product of flux, absorption cross section and quantum yield33 which were included in the model as described in Khan et al.40 In addition to the gas phase chemical reaction and photolysis, the air parcels are also influenced by emissions and physical removal processes (dry and wet deposition). The prevalent physical removal processes for chemical species within air parcels within the boundary layer are dry deposition and wet deposition. Dry deposition rates depend on whether a Lagrangian air parcel is over land or ocean with appropriate species dependent deposition velocities. The dry deposition velocities used in STOCHEM were adapted from the annual mean values calculated using the MATCH global model.41 The removal of soluble species through dissolution in precipitation is known as wet deposition. These dissolved components can originate in the environment during cloud activation or incorporate into precipitation as it falls. The scavenging coefficients for convective and dynamic precipitation taken from Penner et al.42 were combined with precipitation rates and scavenging profiles to calculate the loss rates of species (wet deposition) from an air parcel.
000 constant mass air parcels that represent the Earth's troposphere.33 It is within these air parcels that the chemical reactions and photochemical dissociations leading to the loss and production of trace gases are taking place. The data concerning pressure, temperatures, winds, clouds, humidities, tropopause heights, precipitation, boundary layer depth and surface parameters are taken for the year 1998 from archived meteorological data from the UKMO Unified Model (UM).34 Meteorological data are based on a grid resolution of 1.25° longitude, 0.833° latitude and 12 unevenly spaced (with respect to altitude) vertical levels between the surface and an upper boundary of ∼100 mb.33 A Lagrangian approach for advection allows uncoupling of the chemistry and transport processes. The chemical mechanism used in STOCHEM is the Common Representative Intermediates (CRI) version 2 reduction 5 (CRI v2-R5) which was built using a series of five-day box model simulations on each compound, on a compound-by-compound basis. CRI v2-R5 was developed initially by Jenkin et al.35 from the Master Chemical Mechanism (https://mcm.york.ac.uk/MCM/) with subsequent improvement by Watson et al.,36 and the CRI scheme involves the most reduced mechanism, making it suitable for global modelling due to its traceability to the MCM. More details of the CRI v2-R5 mechanism can be found in Utembe et al.37,38 and recent updates of the CRI v2.2 mechanism can be found in Jenkin et al.39 STOCHEM-CRI calculates one-hourly photolysis rates for each reaction within each air parcel, before linearly interpolating the data with respect to time. This provides photolysis rates at a resolution of 5 minutes to be used in the chemical integration. The photolysis rates were calculated by integrating (over all wavelengths) the product of flux, absorption cross section and quantum yield33 which were included in the model as described in Khan et al.40 In addition to the gas phase chemical reaction and photolysis, the air parcels are also influenced by emissions and physical removal processes (dry and wet deposition). The prevalent physical removal processes for chemical species within air parcels within the boundary layer are dry deposition and wet deposition. Dry deposition rates depend on whether a Lagrangian air parcel is over land or ocean with appropriate species dependent deposition velocities. The dry deposition velocities used in STOCHEM were adapted from the annual mean values calculated using the MATCH global model.41 The removal of soluble species through dissolution in precipitation is known as wet deposition. These dissolved components can originate in the environment during cloud activation or incorporate into precipitation as it falls. The scavenging coefficients for convective and dynamic precipitation taken from Penner et al.42 were combined with precipitation rates and scavenging profiles to calculate the loss rates of species (wet deposition) from an air parcel.
Emissions are treated as an additional term to the source fluxes of each species during each integration time step.33,43 The emission profile for STOCHEM-CRI consists of three different categories: surface emissions, stratospheric sources and 3-dimensional emissions. Emissions that fall under surface emissions are anthropogenic, biomass burning, oceans, soils and vegetation which are mapped monthly from a two-dimensional source map at a resolution of 5° longitude × 5° latitude. The emission data employed in the base case STOCHEM-CRI model were adapted from the Precursor of Ozone and their Effects in the Troposphere (POET) inventory for the year 1998.44 More details about the emission data can be found in Khan et al.45 The emissions flux is implemented during the chemical timestep unless there are no Lagrangian cells present in a particular Eulerian grid cell, in which case the emissions are stored for implementation after the next advection step. Finally, all emissions are converted into units of molecules per second per grid square.
In this study, the base case experiment involves the STOCHEM being integrated with the CRI v2-R5 mechanism with the updated isoprene HOx recycling mechanism46 subsequently referred to as ‘STOCH-base’. A further simulation, ‘STOCH-RO2-OH was performed which involved the inclusion of all organic peroxy radicals (RO2) + OH reactions, with product yields of reaction weighted averages across different RO2 of 0.82, 0.07, 0.0, 0.11 for channels (R11a–R11d), respectively for each individual reaction to the STOCH-base scenario. A total of 200 reactions for 50 RO2 species are thus added in STOCHEM-RO2-OH. The rate coefficients of RO2 + OH reactions show no significant dependence on the size of the R group on the RO2, thus all kOH+RO2, were assumed to be 1.6 × 10−10 cm3 molecule−1 s−1 at 295 K which was selected on the basis of the experimental determination for the CH3O2 + OH reaction from the work of Assaf et al.25 The products of (R11a) are based on similar alkoxy radical formation as in the reactions of RO2 + NO already in the CRI mechanism. The unique products from the reactions (R11b–R11d) are considered as ROH, R(–H)OO and ROOOH. The loss of ROOOH to ROH + O2 with a rate coefficient of 5.0 × 10−3 s−1 was used which was estimated based on the studies of Müller et al.30 and Caravan et al.31 The losses of ROH and R(–H)OO were considered in the model based on the loss processes of C3H8OH and CH3CHOO already in the CRI mechanism, respectively. As the product yields of (R11) are highly variable with the size of the alkyl moiety of the peroxy radicals, we performed four further model simulations assuming 100% yield for each individual channel (R11a–R11d) referred to as STOCH-RO2-OH-A, STOCH-RO2-OH-B, STOCH-RO2-OH-C, and STOCH-RO2-OH-D, respectively. Another simulation, STOCH-CH3O2-OH, was performed including only the reaction of CH3O2 + OH to assess the impact of the reaction on the global budget of CH3OH and CH3OOOH. All simulations were conducted with meteorology from 1998 for a period of 24 months with the first 12 allowing the model to spin up. Analyses were performed on the subsequent 12 months of data. Using the loss of OH by RO2 and loss of RO2 by OH derived from the STOCH-RO2-OH model run for each model box in the tropical region, we calculated the fraction of this loss process compared with the total OH and RO2 losses. Formation of SOA from ROOOH is accounted for in the model by equilibrium partitioning between gas and aerosol phases, based on the Pankow absorption model.47 The vapor pressures of all ROOOH were calculated using the method of Nannoolal et al.48 in conjunction with species boiling points estimated by the method of Nannoolal et al.49 extracted directly from the University of Manchester multiphase system online property prediction (UManSysProp) facility.50 The method employed in the model to account for the partitioning of the gas-phase insertion products onto pre-existing primary organic aerosol has been documented previously.17,18 Nine ROOOH (RTN28OOOH, RTN26OOOH, RTN25OOOH, RTN24OOOH, RTN23OOOH, RTN14OOOH, RTX28OOOH, RU12OOOH, and NRU12OOOH) showed sufficiently low volatilities and a similar technique17 is utilised in this study to account for their role as nucleating agents for new particle formation.
3. Results and discussion
3.1. Impact of (R11) on the global burden of the species
The STOCHEM-RO2-OH model simulation results show that a significant amount of ROOOH (86.1 Tg per year) and ROH (30.2 Tg per year from direct reaction of (R11b) and 47.5 Tg per year through ROOOH decomposition) was formed from RO2 + OH reactions (Table 1 and ESI Table S1†). Among all ROOOH and ROH, the formation of CH3OOOH (67.5 Tg per year, 78%) and CH3OH (55.2 Tg per year, 71%) is found to be the highest. In a previous study utilising the same model, we reported a global budget of CH3OH by photochemical production of 48 Tg per year through the reaction of CH3O2 + RO2.45 In the STOCHEM-RO2-OH model, we found ∼50% decreased CH3OH production from CH3O2 + RO2 (23.5 Tg per year) similar to the study of Bates et al.51 (24.0 Tg per year). However, we also found a total secondary methanol production of 78.7 Tg per year, which is ∼60% and ∼30% higher than that reported by Khan et al.45 and Bates et al.,51 respectively. The reduction in CH3O2 due to its reaction with OH retarded the in situ production of CH3OH through the self-reaction of CH3O2 and its cross-reactions with other RO2 resulting in decreasing methanol production from CH3O2 + RO2.31 In the STOCHEM-RO2-OH model, the CH3O2 + OH reaction directly produced 21.5 Tg per year CH3OH and 67.5 Tg per year CH3OOOH formation followed by its decomposition to produce further 33.7 Tg per year CH3OH. In contrast, the study of Bates and co-workers51 considered only the primary formation of CH3OH (33 Tg per year) with a yield of 13% and did not consider the secondary formation via CH3OOOH. From the STOCH-CH3O2-OH model simulation results, the global burdens were found to be 5.56 Tg for CH3OH and 0.48 Gg for CH3OOOH. The atmospheric lifetime of CH3OH was found to be 6.8 days for CH3OH, which was within the range of the estimated lifetime by Jacob et al.52 After CH3OOOH and CH3OH, isoprene-derived ROOOH (5.5 Tg per year, 6%) and ROH (7.0 Tg per year, 9%), monoterpene-derived ROOOH (4.2 Tg per year, 5%) and ROH (5.8 Tg per year, 7%), peroxy acetyl-OOOH (5.4 Tg per year, 6%) and peroxy acetyl-OH (5.9 Tg per year, 8%) made up the most significant contributions to the total formation of ROOOH and ROH, respectively (Table 1). The isoprene and monoterpene derived ROH and ROOOH in the model were highly oxidized molecules and have a significant impact on SOA, which will be discussed in more detail in Section 3.5.
Table 1 Contribution of different ROOOH and ROH from STOCHEM-RO2-OH model simulation
| ROOOH/ROHa |
Total number of species |
Formation of ROOOH (Tg per year) |
Formation of ROH (Tg per year) |
|
Structures can be obtained using the species name and search facility on the MCM website (https://mcm.york.ac.uk/MCM/).
Small ROOOH/ROH species are specific C1–C3 structures that are derived from degradation of many VOCs.
ROOOH/ROH species are derived solely from isoprene degradation.
ROOOH/ROH species are derived solely from monoterpene (α-pinene and β-pinene) degradation.
Small RC(O)OOOH/RC(O)OH species are derived from degradation of many VOCs.
Other ROOOH/ROH species are derived from degradation of all other VOCs. More detailed information about individual contributions of ROOOH and ROH can be found in ESI Table S1.
|
| CH3OOOH/CH3OH |
1 |
67.5 |
55.2 |
| Small specific ROOOH/ROHb |
6 |
1.9 |
2.0 |
| Isoprene derived ROOOH/ROHc |
7 |
5.5 |
7.0 |
| α-,β-Pinene derived ROOOH/ROHd |
12 |
4.2 |
5.8 |
| RC(O)OOOH/RC(O)OHe |
4 |
5.4 |
5.9 |
| Other ROOOH/ROHf |
20 |
1.6 |
1.8 |
| Total ROOOH/ROH |
50 |
86.1 |
77.7 |
The detailed summary of the atmospheric composition simulated under the STOCH-base case and the changes resulting under the STOCH-RO2-OH scenario are shown in Table 2. The inclusion of the fifty RO2 + OH reactions in the STOCH-RO2-OH resulted in little change to the global burdens of NOx (−2.65 Gg, −0.5%), O3 (−2.3 Tg, −0.7%), CO (−3.2 Tg, −0.8%), CH3CHO (+2.1 Gg, +1.4%) and HNO3 (−7.4 Gg, −1.3%). However, compared with the STOCH-base case, the RO2 + OH reactions have significant impacts on the global trace gas burdens with increments of 2.1 Gg HOx (7.7%) and 0.53 Tg H2O2 (18.3%) and decrements of 8.0 Gg RO2 (18.2%), 19.4 Gg RONO2 (4.7%), 0.11 Tg PAN (3.4%), 34.9 Gg HCHO (3.3%) and 96.6 Gg ROOH (3.8%).
Table 2 Global annual mean tropospheric burden of selected species for the STOCH-base case and the change of annual global burden for the STOCH-RO2-OH casea
| Species |
Base global burden (Gg) |
Change in global burden (Gg) |
|
Global burdens are in Gg, percent changes are in brackets.
|
| HOx |
26.7 |
+2.1 (+7.7) |
| O3 |
310.8 × 103 |
−2.3 × 103 (−0.7) |
| CO |
401.2 × 103 |
−3.2 × 103 (−0.8) |
| NOx |
492.2 |
−2.7 (−0.5) |
| H2O2 |
2.9 × 103 |
+534.2 (+18.3) |
| HCHO |
1.1 × 103 |
−34.9 (−3.3) |
| CH3CHO |
146.6 |
+2.1 (+1.4) |
| HNO3 |
503.0 |
−7.4 (−1.5) |
| RONO2 |
411.754 |
−19.4 (−4.7) |
| PAN |
3.3 × 103 |
−112.9 (−3.4) |
| RO2 |
44.2 |
−8.0 (−18.2) |
| ROOH |
2.5 × 103 |
−96.6 (−3.8) |
Our previous study of the atmospheric impact of the CH3O2 + OH reaction31 showed that the reaction of methyl peroxy and hydroxyl radicals increased the global burden of HOx by 6.2%. The inclusion of all fifty RO2 + OH reactions in this study increased the global burden of HOx by 7.7%. These reactions proceed through oxygen atom transfer from RO2 to OH producing HO2, hence the large production of HO2 (569.6 Tg per year) and the significant loss of OH (186.4 Tg per year) from these reactions found in the STOCH-RO2-OH simulation. The burden change of HOx (=OH + HO2) was a balance between two opposite effects; however, the production of HO2 was outweighed by the loss of OH resulting in an increase in the global burden of HOx. H2O2 acts a reservoir of HOx; thus the annual mean global burden of H2O2 is increased by 534.2 Gg (18.3%) due to the increased H2O2 production (132.2 Tg per year, 18.2% from the STOCH-base case) from the HO2 + HO2 reaction. ROOH was another reservoir of HOx, but the global burden of ROOH is decreased by 96.6 Gg (3.8%) due to its reduced production (444.3 Tg per year, 13.9%) from the RO2 + HO2 reaction following on from the decreased RO2 level. Due to the increase of HO2 burden in the STOCH-RO2-OH, the production of ozone through HO2 + NO was increased by 319.5 Tg per year (7.8%). However, the decreased levels of RO2 have the effect of decreasing the production of ozone through the RO2 + NO channel by 325.8 Tg per year (10.0%). In addition, the increased HO2 enhanced the loss of ozone through the HO2 + O3 reaction by 68.7 Tg per year (5.7%). Overall, the ozone burden decreased by 2.3 Tg (0.7%). The decreased production of NO2 through the RO2 + NO reaction had the effect of decreasing the tropospheric burden of NOx (2.7 Gg, 0.5%). The combined effect of decreased RO2 and decreased NOx also resulted in lower productions of related NOx oxidation products, as the global burden of RONO2 and PAN decreased by 19.4 and 112.9 Gg, respectively. Additionally, the HNO3 global burden was decreased by 7.4 Gg (1.5%) due to its decreased production (4.5 Tg per year, 2.3%) through the OH + NO2 reaction. Acetaldehyde (CH3CHO) and formaldehyde (HCHO) are oxidation products of VOCs, including RO2 and ROOH. Hence, the decreased levels of RO2 and ROOH in STOCH-RO2-OH produced smaller amounts of HCHO (337.2 Tg per year) and CH3CHO (6.8 Tg per year). However, the oxidation of RO2 with OH produced additional amounts of HCHO (244.5 Tg per year) and CH3CHO (6.9 Tg per year). Overall, the global burden of HCHO decreased by 30.5 Gg (2.9%) and the global burden of CH3CHO increased by 0.87 Gg (0.2%), respectively.
3.2. Impact of (R11) on the surface distribution of the species
The percentage change in annual surface levels of OH, HO2, O3, H2O2, HCHO, CH3CHO, RO2, ROOH, NOx, HNO3, PAN and RONO2 in STOCH-RO2-OH compared with the STOCH-base case is shown in Fig. 1. The addition of fifty RO2 + OH reactions had an impact on OH and HO2 mixing ratios near the equatorial region with up to 12% decrease and 25% increase over the oceans, respectively. HO2 and RO2 are important during the daytime and are produced during the oxidation of trace gases. In the equatorial regions, the abundances of these RO2 species are high due to the increased levels of VOCs along with increased photochemistry. When these RO2 radicals reacted with OH, the mixing ratios of RO2 radicals were reduced (by up to 50%). However, these RO2 produced a significant amount of HO2, with a similar distribution to the decrease in RO2 in these regions (see Fig. 1). The inclusion of these reactions in the model led to an increase of H2O2 mixing ratios by up to 50%. The significant increase of H2O2 can enhance particle growth due to the increased oxidation of SO2 in cloud droplets formed on sulfuric acid seed aerosol.53 The change of surface distribution of H2O2 is driven by the increase in HO2 abundances (see Fig. 1). The inclusion of RO2 + OH reactions led to a decrease in NOx over tropical oceans by up to 6%. The decreased levels of NOx in the tropical ocean regions combined with decreased RO2 resulted in the decreased formation of organic nitrates resulting in decreased PAN mixing ratios by up to 10% and decreased RONO2 mixing ratios by up to 3.5% in the tropical ocean regions. A decrease in HNO3 by up to 12% was also observed over the tropical oceans due to decreased levels of NOx. Over the oceans where NOx was decreased, the O3 concentrations had decreased by up to 3.5% due to increased HO2 levels.
 |
| | Fig. 1 Annual surface percentage changes in (a) OH, (b) HO2, (c) O3, (d) H2O2, (e) HCHO, (f) CH3CHO, (g) RO2, (h) ROOH, (i) NOx, (j) HNO3, (k) PAN and (l) RONO2 on inclusion of RO2 + OH reactions. | |
The inclusion of the (R11) reaction set in the model produced up to 0.35 ppt ROOOH and an additional 150 ppt ROH in tropical regions. The spatial distributions of ROOOH and ROH mixing ratios were found to be similar to that of their precursors (OH and RO2). The highest mixing ratios of ROOOH were found mostly in the oceans adjacent to the tropical land areas, whereas the highest mixing ratios of ROH were found in the tropical forest regions (Fig. 2). The abundances of ROOOH in our study were found to be low compared with Fittschen et al’s study54 who used the UM-UKCA global chemistry-climate model with considering 100% yield of ROOOH from all RO2 + OH reactions, using a low removal rate of ROOOH (1 × 10−4 s−1 compared with our study, 5 × 10−3 s−1) and showing diurnal and seasonal peak concentrations of ROOOH in their calculation.
 |
| | Fig. 2 Annual averaged surface mixing ratios of (a) ROOOH and (b) ROH simulated by STOCH-RO2-OH. | |
Caravan et al.31 showed that CH3O2 + OH is a significant source of CH3OH over the tropical ocean if the yield of ROH (ϕROH) is higher than 0.15. In this study, we also found a significant increase of ROH over tropical regions using ϕROH = 0.18 (direct (0.07) + through decomposition of ROOOH (0.11)). Excluding CH3O2, the biogenic RO2 (especially isoprene derived peroxy radical (ISOPO2) and monoterpene derived peroxy radical (MONOTERPO2)) dominated over the tropical forest region; thus, the abundances of ROH were found to be highest in the tropical forest region.
3.3. Contribution of (R11) to the reactivity of OH and RO2
The RO2 + OH addition reactions increase the OH reactivity by up to 5% over remote tropical oceans (Fig. 3a), consistent with the finding of Ferracci et al.55 Considering the average loss fluxes of OH over the tropical region, it was found that RO2 + OH reactions represented as much as 0.05 s−1 (∼2% of the sinks) in the tropical regions (Fig. 3b and ESI Table S2†). This small OH reactivity contribution could not reduce the discrepancy of the model-measured OH reactivity with a mean bias of −11.9 s−1 and -20.4 s−1 for all locations and tropical regions, respectively (see ESI Table S3†). The significantly underestimated OH reactivity in the tropical forest area could be due to insufficient representation of reactive VOCs in the model, especially monoterpenes and sesquiterpenes.56 No measurements have been performed so far in the tropical oceanic area on OH reactivity, so it was not possible to validate the improvement of the model-measured OH reactivity biases in the tropical oceans.
 |
| | Fig. 3 (a) Percent increase in OH reactivity on inclusion of RO2 + OH reactions and (b) fraction of OH reactivity over the tropical region. | |
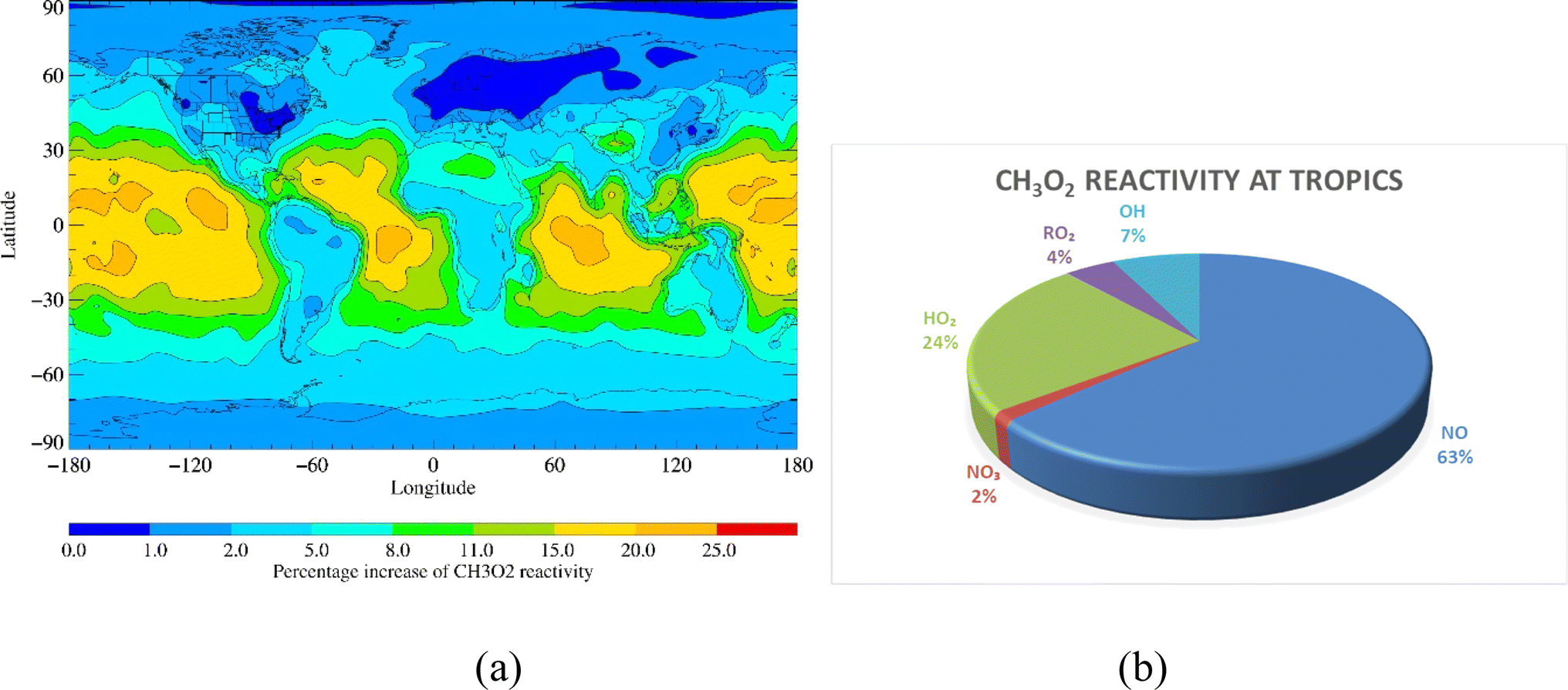
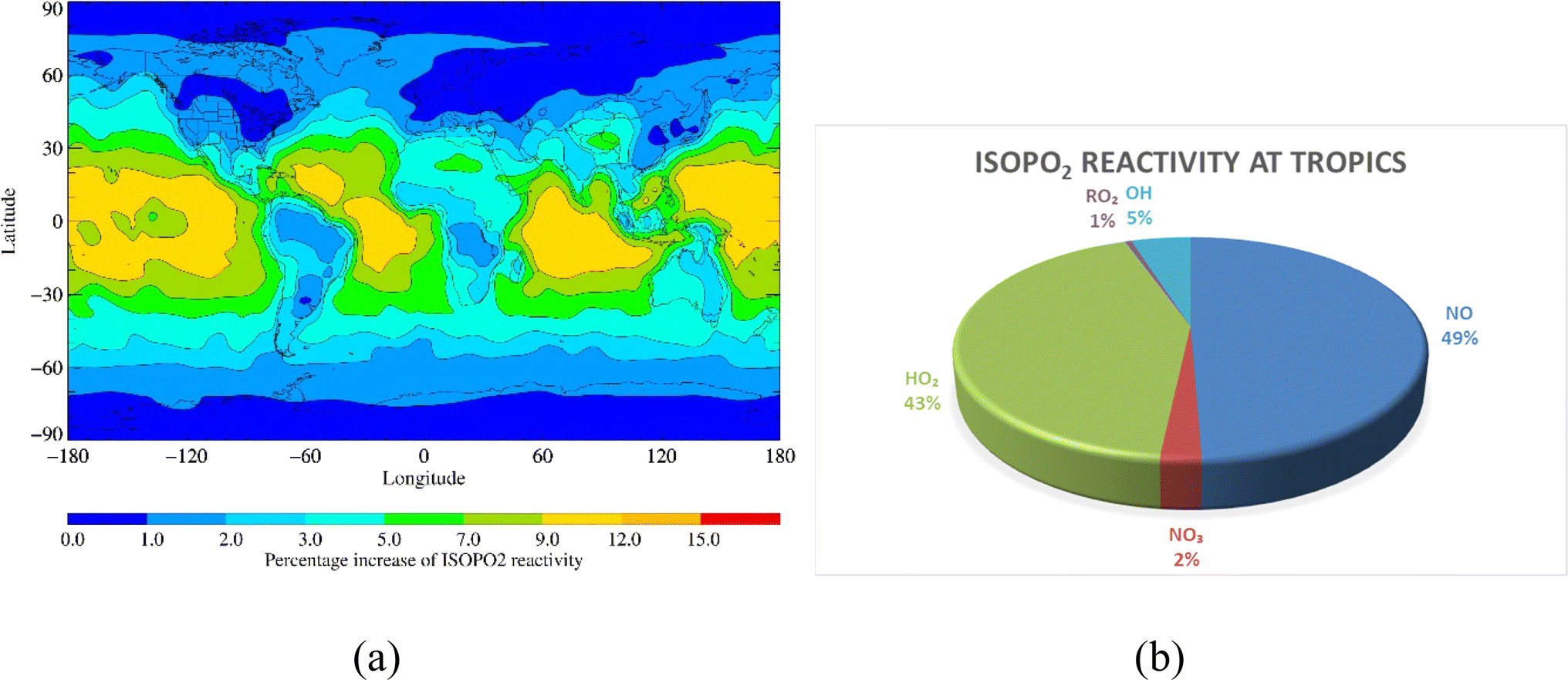
RO2 + OH reactions enhanced CH3O2, ISOPO2 and MONOTERPO2 reactivities by up to 25%, 15% and 50%, respectively, over remote tropical oceans (Fig. 4a, 5a and 6a). The annual loss fluxes of the reactions of RO2 with NO, NO3, RO2, HO2 and OH throughout the troposphere are shown in ESI (Table S4)† and rate coefficients used are summarised in references for the development of the CRI mechanism.35–39 Considering the average loss fluxes of CH3O2, ISOPO2 and MONOTERPO2 over the tropical region, it was found that CH3O2 + OH, ISOPO2 + OH and MONOTERPO2 + OH reactions represent as much as 3.7 × 10−4 s−1 (∼7%), 2.6 × 10−3 s−1 (∼5%) and 4.4 × 10−3 s−1 (∼7%) of the total losses in the tropical regions, respectively (Fig. 4b, 5b, 6b and ESI Table S5†). The RO2 + OH reactions dominated over RO2 + RO2 reactions in tropical regions, resulting in redistribution of product formation (e.g. ROH) based on the reaction channels (R11). ISOPOOOH and MONOTERPOOOH generated from ISOPO2 + OH and MONOTERPO2 + OH could be important for atmospheric aerosol formation and thus for the Earth's radiation budget.29
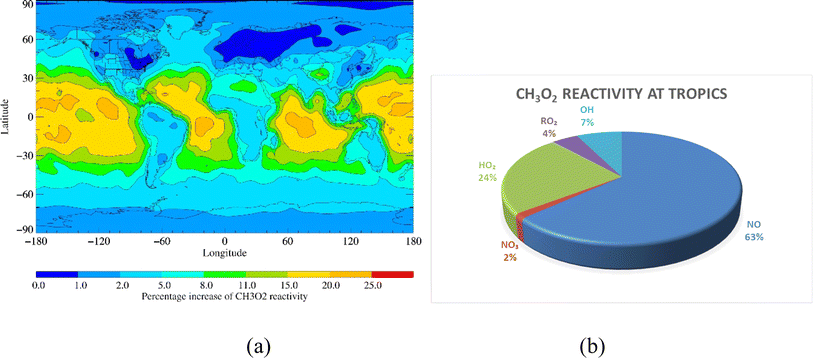 |
| | Fig. 4 (a) Percent increase in CH3O2 reactivity on inclusion of RO2 + OH reactions and (b) fraction of CH3O2 reactivity over the tropical region. | |
 |
| | Fig. 5 (a) Percent increase in ISOPO2 reactivity on inclusion of RO2 + OH reactions and (b) fraction of ISOPO2 reactivity over the tropical region. | |
 |
| | Fig. 6 (a) Percent increase in MONOTERPO2 reactivity on inclusion of RO2 + OH reactions and (b) fraction of MONOTERPO2 reactivity over the tropical region. | |
3.4. The product branching simulations of (R11) with an yield of 1 for each channel
The STOCH-RO2-OH-A simulation results showed that the mixing ratios of HO2 were further increased by up to 3.5% over remote oceans when the yield of (R11a) is increased from 0.82 to 1 (Fig. 7a). The yield increment of (R11a) did not have significant impacts (less than 1% change) on the mixing ratios of O3, OH, RO2, ROOH, RONO2, HNO3, NOx and other species. The reasons for the small impact of these species were similar to the STOCH-RO2-OH simulation results explained in the previous Section (3.2). The STOCH-RO2-OH-D simulation results showed significant amounts of ROOOH (up to 3 ppt; Fig. 7b) and ROH (up to 800 ppt; Fig. 7c) over tropical regions. The mixing ratios of ROH were found to be highest over tropical forest regions (e.g. Amazon and Congo) and the mixing ratios of ROOOH were found to be highest over the tropical oceans. The STOCH-RO2-OH-B simulation results also showed similar amounts (up to 800 ppt) and distribution of ROH (ESI Fig. S1a†) with a decrease of only 1% (∼5 ppt, ESI Fig. S1b†) compared with the STOCH-RO2-OH-D simulation. Overall, the variation of the product yields from (R11b) and (R11d) does not have significant impact on ROH mixing ratios. The STOCH-RO2-OH-C simulation results showed that up to ∼3000 molecules per cm3 SCIs were formed in the tropical oceanic regions (Fig. 7d). Experimental studies24,27 suggest that this channel is negligible for RCH3, however, for larger R, the scenario could be different. The STOCH-RO2-OH-C simulation produced ∼1500 molecules per cm−3 in the tropical forest region (Amazon) which added 8% to the total SCI concentrations.57 However, the SCIs in the tropical oceans influenced the atmospheric oxidation capacity in the marine boundary layer significantly.
 |
| | Fig. 7 (a) Annual HO mixing ratio change from STOCH-RO2-OH to STOCH-RO2-OH-A simulation, (b) annual ROOOH mixing ratios simulated by STOCH-RO2-OH-D, (c) annual ROH mixing ratios simulated by STOCH-RO2-OH-D, and (d) annual Criegee intermediate mixing ratios simulated by STOCH-RO2-OH-C. | |
3.5. SOA formation from ROOOH
The STOCH-RO2-OH scenario (which includes the partitioning parameters of nine ROOOH) produced a non-negligible amount of SOA (up to 0.05 μg m−3, Fig. 8a) which can enhance model organic aerosol by up to 5% on land surfaces (Fig. 8b). However, a significant enhancement (by up to 2000%) of organic aerosol was seen over the remote tropical oceans (Fig. 8b). When the partitioning parameters were included in the STOCH-RO2-OH-D model, the SOA formation was increased by up to 0.40 μg m−3 (∼10-fold higher than STOCH-RO2-OH; ESI Fig. S2†). Currently, the STOCHEM-CRI model underpredicts organic aerosol mass concentration in remote, biomass burning, anthropogenically polluted and both biomass burning plume and anthropogenically polluted regions with mean biases of −0.26, −1.69, −2.03 and −2.80 μg m−3, respectively.18 The SOA contribution from STOCH-RO2-OH and STOCH-RO2-OH-D simulations brought model into better agreement with observations, especially in remote regions with a bias reduction of −0.24 μg m−3 (7% improvement) and −0.10 μg m−3 (62% improvement), respectively (see ESI Fig. S3†).
 |
| | Fig. 8 (a) Annual SOA concentration simulated by STOCH-RO2-OH and (b) percentage annual increase of organic aerosol formation from STOCH-base. | |
4. Conclusion
Organic hydrotrioxides (ROOOH) are formed in the atmosphere through the gas-phase reaction of organic peroxy radicals (RO2) with hydroxyl radicals (OH). We simulated the reactions of RO2 + OH with four different possible product channels and investigated the formation of ROOOH, their abundances and the environmental impacts in terms of secondary organic aerosol (SOA) formation. CH3OOOH (67.5 Tg per year, 78%) is the most significant contributor of ROOOH followed by isoprene-derived ROOOH (5.5 Tg per year, 6%), monoterpene-derived ROOOH (4.2 Tg per year, 5%) and peroxy acetyl-OOOH (5.4 Tg per year, 6%). The model simulations showed that the reactions of RO2 by OH compete with their reactions by HO2, RO2, and NOx and change the global burdens of NOx (−2.65 Gg, −0.5%), O3 (−2.3 Tg, −0.7%), CO (−3.2 Tg, −0.8%), CH3CHO (+2.1 Gg, +1.4%), HNO3 (−7.4 Gg, −1.3%), HOx (+2.1 Gg, +7.7%) and 0.5 H2O2 (+0.53 Tg, +18.3%), RO2 (−8 Gg, −18.2%), RONO2 (−19.4 Gg, −4.7%), PAN (−0.11 Tg, −3.4%), HCHO (−34.9 Gg, −3.3%) and ROOH (−96.9 Gg, −3.8%). The surface RO2 and HO2 mixing ratios are found to be decreased by up to 50% and increased by up to 25%, respectively near the tropical oceanic region which are the important factors for changing the surface distribution of O3, H2O2, HCHO, CH3CHO, RO2, ROOH, NOx, HNO3, PAN and RONO2. RO2 + OH reactions represent as much as 0.05 s−1 (∼2% of the total OH sinks) in the tropical regions whereas CH3O2 + OH, ISOPO2 + OH and MONOTERPO2 + OH reactions represent as much as 3.7 × 10−4 s−1 (∼7% of the total CH3O2 sinks), 2.6 × 10−3 s−1 (∼5% of the total ISOPO2 sinks) and 4.4 × 10−3 s−1 (∼7% of the total MONOTERPO2 sinks) in the tropical regions, respectively. The sensitivity analysis with 100% yield of individual product channels showed a further increment of HO2 mixing ratios by up to 3.5% for (R11a), formation of ROOOH by up to 3 ppt for (R11d), formation of ROH by up to 800 ppt for (R11b) and formation of stabilized Criegee intermediates by up to 12 × 10−5 ppt for (R11c). The ROOOH generate secondary organic aerosol (SOA) of up to 0.05 μg m−3 in the tropical region. This value is likely a lower limit as the study used yield of ROOOH, øROOOH = 0.11 for all ROOOH, but with increasing size of the R, the formation of ROOOH could be the dominant product from (R11) and all these large sized ROOOH partition to form organic aerosol. Considering the yield = 1 for all large ROOOH, up to 0.4 μg per m3 SOA is formed in tropical regions.
In previous work, we have made an intercomparison of the major chemical mechanisms employed in chemistry-transport (CTM) and earth system (ESM) models to describe the pre-industrial, present day and future air quality and greenhouse gas composition.58 The composition data are essential to formulating global policies addressing global health impacts and global climate change. To our knowledge, few chemical mechanisms include any of the RO2 + OH chemical reactions. A prerequisite for understanding the importance of the RO2 + OH reactions is the inclusion of the RO2 + OH reaction rate coefficients, together with their temperature dependences and reaction products, in the evaluated chemical kinetic data compilations of JPL and IUPAC. We recommend the inclusion of the RO2 + OH reactions in global CTMs and ESMs used for the assessment of policies addressing global air quality and climate change.
Data availability
The data presented in this study are available on request from the corresponding authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank various NERC grants (NE/K004905/1; NE/J009008/1; NE/I014381/1, NE/G01972X/1, NER/A/S/2001/00438, and NER/A/S/2001/00374), Bristol ChemLabS and the Primary Science Teaching Trust under whose auspices various aspects of this work were carried out. C. J. P.'s work was carried out at Jet Propulsion Laboratory, California Institute of Technology, under contract with the National Aeronautics and Space Administration (NASA, 80NM0018D0004) and was supported by the Upper Atmosphere Research and Tropospheric Chemistry Programs.
References
- M. A. H. Khan, M. C. Cooke, S. R. Utembe, A. T. Archibald, R. G. Derwent, M. E. Jenkin, W. C. Morris, N. South, J. C. Hansen, J. S. Francisco, C. J. Percival and D. E. Shallcross, Global analysis of peroxy radicals and peroxy radical-water complexation using the STOCHEM-CRI global chemistry and transport model, Atmos. Environ., 2015, 106, 278–287 CrossRef CAS.
- U. Platt, G. LeBras, G. Poulet, J. P. Burrows and G. Moortgat, Peroxy radicals from night-time reaction of NO3 with organic compounds, Nature, 1990, 348, 147–149 CrossRef CAS.
- P. D. Lightfoot, R. A. Cox, J. N. Crowley, M. Destriau, G. D. Hayman, M. E. Jenkin, G. K. Moortgat and F. Zabel, Organic peroxy radicals: kinetics, spectroscopy and tropospheric chemistry, Atmos. Environ., 1992, 26, 1805–1961 CrossRef.
- J. J. Orlando and G. S. Tyndall, Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- G. S. Tyndall, R. A. Cox, C. Granier, R. Lesclaux, G. K. Moortgaat, M. J. Pilling, A. R. Ravishankara and T. J. Wallington, Atmospheric chemistry of small organic peroxy radicals, J. Geophys. Res., 2001, 106, 12157–12182 CrossRef CAS.
- R. Chhantyal-Pun, M. A. H. Khan, N. Zachhuber, C. J. Percival, D. E. Shallcross and A. J. Orr-Ewing, Impact of Criegee intermediate reactions with peroxy radicals on tropospheric organic aerosol, ACS Earth Space Chem., 2010, 4, 1743–1755 CrossRef.
- M. E. Jenkin and K. C. Clemitshaw, Ozone and other secondary photochemical pollutants: chemical processes governing their formation in the planetary boundary layer, Atmos. Environ., 2000, 34, 2499–2527 CrossRef CAS.
- W. Lei, R. Zhang, X. Tie and P. Hess, Chemical characterization of ozone formation in the Houston-Galveston area: A chemical transport model study, J. Geophys. Res.:Atmos., 2004, 109, D12301 CrossRef.
- J. L. Fry, D. C. Draper, K. C. Barsanti, J. N. Smith, J. Ortega, P. M. Winkle, M. J. Lawler, S. S. Brown, P. M. Edwards, R. C. Cohen and L. Lee, Secondary Organic Aerosol Formation and Organic Nitrate Yield from NO3 Oxidation of Biogenic Hydrocarbons, Environ. Sci. Technol., 2014, 48, 11944–11953 CrossRef CAS PubMed.
- A. E. Perring, S. E. Pusede and R. C. Cohen, An observational perspective on the atmospheric impacts of alkyl and multifunctional nitrates on ozone and secondary organic aerosol, Chem. Rev., 2013, 113, 5848–5870 CrossRef CAS PubMed.
- P. O. Wennberg, K. H. Bates, J. D. Crounse, L. G. Dodson, R. C. McVay, L. A. Mertens, T. B. Nguyen, E. Praske, R. H. Schwantes, M. D. Smarte, J. M. St Clair, A. P. Teng, X. Zhang and J. H. Seinfeld, Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 2018, 118, 3337–3390 CrossRef CAS PubMed.
- J. D. Crounse, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard and P. Wennberg, Autooxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Highly Oxygenated Organic Molecules (HOM) from
Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS PubMed.
- S. E. Murphy, J. D. Crounse, K. H. Møller, S. P. Rezgui, N. J. Hafeman, J. Park, H. G. Kjaergaard, B. M. Stoltz and P. O. Wennberg, Accretion product formation in the self-reaction of ethene-derived hydroxy peroxy radicals, Environ. Sci.: Atmos., 2023, 3, 882 CAS.
- B. Bonn, R. von Kuhlmann and M. G. Lawrence, High contribution of biogenic hydroperoxides to secondary organic aerosol formation, Geophys. Res. Lett., 2004, 31, L10108 CrossRef.
- F. Paulot, J. D. Crounse, H. G. Kjaergaard, J. H. Kroll, J. H. Seinfeld and P. O. Wennberg, Isoprene photooxidation: new insights into the production of acids and organic nitrates, Atmos. Chem. Phys., 2009, 9, 1479–1501 CrossRef CAS.
- S. R. Utembe, M. C. Cooke, A. T. Archibald, D. E. Shallcross, R. G. Derwent and M. E. Jenkin, Simulating secondary organic aerosol in a 3-D Lagrangian chemistry transport model using the reduced Common Representative Intermediates mechanism (CRI v2-R5), Atmos. Environ., 2011, 45, 1604–1614 CrossRef CAS.
- M. A. H. Khan, M. E. Jenkin, A. Foulds, R. G. Derwent, C. J. Percival and D. E. Shallcross, A modeling study of secondary organic aerosol formation from sesquiterpenes using the STOCHEM global chemistry and transport model, J. Geophys. Res., 2017, 122, 4426–4439 CrossRef CAS.
- M. Krapf, I. E. Haddad, E. A. Bruns, U. Molteni, K. R. Daellenbach, A. S. H. Prévôt, U. Baltensperger and J. Dommen, Labile peroxides in secondary organic aerosol, Chem, 2016, 1, 603–616 CAS.
- H. Li, Z. Chen, L. Huang and D. Huang, Organic peroxides' gas-particle partitioning and rapid heterogeneous decomposition on secondary organic aerosol, Atmos. Chem. Phys., 2016, 16, 1837–1848 CrossRef CAS.
- P. S. Monks, Gas-phase radical chemistry in the troposphere, Chem. Soc. Rev., 2005, 34, 295–376 RSC.
- S. Vaughan, C. E. Canosa-Mas, C. Pfrang, D. E. Shallcross, L. Watson and R. P. Wayne, Kinetic studies of reactions of the nitrate radical (NO3) with peroxy radicals (RO2): an indirect source of OH at night?, Phys. Chem. Chem. Phys., 2006, 8, 3749–3760 RSC.
- A. Bossolasco, E. P. Farago, C. Schoemaecker and C. Fittschen, Rate constant of the reaction between CH3O2 and OH radicals, Chem. Phys. Lett., 2014, 593, 7–13 CrossRef CAS.
- C. Yan, S. Kocevska and L. N. Krasnoperov, Kinetics of the Reaction of CH3O2 Radicals with OH Studied over the 292–526 K Temperature Range, J. Phys. Chem. A, 2016, 120, 6111–6121 CrossRef CAS PubMed.
- E. Assaf, B. Song, A. Tomas, C. Schoemaecker and C. Fittschen, Rate constant of the reaction between CH3O2 radicals and OH radicals revisited, J. Phys. Chem. A, 2016, 120, 8923–8932 CrossRef CAS PubMed.
- C. Fittschen, The reaction of peroxy radicals with OH radicals, Chem. Phys. Lett., 2019, 725, 102–108 CrossRef CAS.
- E. Assaf, L. Sheps, L. Whalley, D. Heard, A. Tomas, C. Schoemaecker and C. Fittschen, The reaction between CH3O2 and OH radicals: product yields and atmospheric implications, Environ. Sci. Technol., 2017, 51, 2170–2177 CrossRef CAS PubMed.
- E. P. Faragó, C. Schoemaecker, B. Viskolcz and C. Fittschen, Experimental determination of the rate constant of the reaction between C2H5O2 and OH radicals, Chem. Phys. Lett., 2015, 619, 196–200 CrossRef.
- T. Berndt, J. Chen, E. R. Kjærgaard, K. Møller, A. Tilgner, E. H. Hoffmann, H. Herrmann, J. D. Crounse, P. O. Wennberg and H. G. Kjaergaard, Hydrotrioxide (ROOOH) formation in the atmosphere, Science, 2022, 376, 6596 CrossRef PubMed.
- J.-F. Müller, Z. Liu, V. S. Nguyen, T. Stavrakou, J. N. Harvey and J. Peeters, The reaction of methyl peroxy and hydroxyl radicals as a major source of atmospheric methanol, Nat. Commun., 2016, 7, 13213 Search PubMed.
- R. L. Caravan, M. A. H. Khan, J. Zádor, L. Sheps, I. O. Antonov, B. Rotavera, K. Ramasesha, K. Au, M.-W. Chen, D. Rösch, D. L. Osborn, C. Fittschen, C. Schoemaecker, M. Duncianu, A. Grira, S. Dusanter, A. Tomas, C. J. Percival, D. E. Shallcross and C. A. Taatjes, The reaction of hydroxyl and methylperoxy radicals is not a major source of atmospheric methanol, Nat. Commun., 2018, 9, 4343 CrossRef PubMed.
- E. Assaf, C. Schoemacker, L. Vereecken and C. Fittschen, Experimental and theoretical investigation of the reaction of RO2 radicals with OH radicals: Dependence of the HO2 yield on the size of the alkyl group, Int. J. Chem. Kinet., 2018, 50, 670–680 CrossRef CAS.
- W. J. Collins, D. S. Stevenson, C. E. Johnson and R. G. Derwent, Tropospheric ozone in a Global-Scale Three-Dimensional Lagrangian Model and its response to NOx emission controls, J. Atmos. Chem., 1997, 26, 223–274 CrossRef CAS.
- M. J. Cullen, The unified forecast/climate model, Meteorol. Mag., 1993, 122, 81–94 Search PubMed.
- M. E. Jenkin, L. A. Watson, S. R. Utembe and D. E. Shallcross, A Common Representative Intermediate (CRI) mechanism for VOC degradation. Part-1: gas phase mechanism development, Atmos. Environ., 2008, 42, 7185–7195 CrossRef CAS.
- L. A. Watson, D. E. Shallcross, S. R. Utembe and M. E. Jenkin, A Common Representative Intermediate (CRI) mechanism for VOC degradation. Part 2: gas phase mechanism reduction, Atmos. Environ., 2008, 42, 7196–7204 CrossRef CAS.
- S. R. Utembe, L. A. Watson, D. E. Shallcross and M. E. Jenkin, A Common Representative Intermediates (CRI) mechanism for VOC degradation. Part 3: Development of a secondary organic aerosol module, Atmos. Environ., 2009, 43, 1982–1990 CrossRef CAS.
- S. R. Utembe, M. C. Cooke, A. T. Archibald, M. E. Jenkin, R. G. Derwent and D. E. Shallcross, Using a reduced Common Representative Intermediates (CRI v2-R5) mechanism to simulate tropospheric ozone in a 3-D Lagrangian chemistry transport model, Atmos. Environ., 2010, 13, 1609–1622 CrossRef.
- M. E. Jenkin, M. A. H. Khan, D. E. Shallcross, R. Bergström, D. Simpson, K. L. C. Murphy and A. R. Rickard, The CRI v2.2 reduced degradation scheme for isoprene, Atmos. Environ., 2019, 212, 172–182 CrossRef CAS.
- M. A. H. Khan, M. C. Cooke, S. R. Utembe, A. T. Archibald, P. Maxwell, W. C. Morris, P. Xiao, R. G. Derwent, M. E. Jenkin, C. J. Percival, R. C. Walsh, T. D. S. Young, P. G. Simmonds, G. Nickless, S. O'Doherty and D. E. Shallcross, A study of global atmospheric budget and distribution of acetone using global atmospheric model STOCHEM-CRI, Atmos. Environ., 2015, 112, 269–277 CrossRef CAS.
- R. von Kuhlmann, M. G. Lawrence and P. J. Crutzen, A model for studies of tropospheric ozone and nonmethane hydrocarbons: Model evaluation of ozone-related species, J. Geophys. Res., 2003, 108, 4729 Search PubMed.
-
J. E. Penner, C. S. Atherton and T. Graede, Global emissions and models of photochemically active compounds, in Global Atmospheric Biospheric Chemistry: OHOLO Conf. Ser. Books, ed. R. G. Prinn, Plenum, New York, 1994, pp. 223–247 Search PubMed.
- W. J. Collins, D. S. Stevenson, C. E. Johnson and R. G. Derwent, The European regional ozone distribution and its links with the global scale for the years 1992 and
2015, Atmos. Environ., 2000, 34, 255–267 CrossRef CAS.
-
C. Granier, J. F. Lamarque, A. Mieville, J. F. Muller, J. Olivier, J. Orlando, J. Peters, G. Petron, S. Tyndall, and S. Wallens, POET, a Database of Surface Emissions of Ozone Precursors, 2005, http://accent.aero.jussieu.fr/database_table_inventories.php Search PubMed.
- M. A. H. Khan, M. C. Cooke, S. R. Utembe, P. Xiao, R. G. Derwent, M. E. Jenkin, A. T. Archibald, P. Maxwell, W. C. Morris, N. South, C. J. Percival and D. E. Shallcross, Reassessing the photochemical production of methanol from peroxy radical self and cross reactions using the STOCHEM-CRI global chemistry and transport model, Atmos. Environ., 2014, 99, 77–84 CrossRef CAS.
- M. A. H. Khan, B.-L. Schlich, M. E. Jenkin, M. C. Cooke, R. G. Derwent, J. L. Neu, C. J. Percival and D. E. Shallcross, Changes to simulated global atmospheric composition resulting from recent revisions to isoprene oxidation chemistry, Atmos. Environ., 2021, 244, 117914 CrossRef CAS.
- J. F. Pankow, An absorption model of gas-particle partitioning involved in the formation of secondary organic aerosol, Atmos. Environ., 1994, 28, 189–193 CrossRef CAS.
- Y. Nannoolal, J. Rarey and D. Ramjugernath, Estimation of pure component properties. Part 3. Estimation of the vapor pressure of non-electrolyte organic compounds via group contributions and group interactions, Fluid Phase Equilib., 2008, 269, 117–133 Search PubMed.
- Y. Nannoolal, J. Rarey, D. Ramjugernath and W. Cordes, Estimation of pure component properties. Part 1. Estimation of the normal boiling point of non-electrolyte organic compounds via group contributions and group interactions, Fluid Phase Equilib., 2004, 226, 45–63 CrossRef CAS.
- D. Topping, M. Barley, M. K. Bane, N. Higham, B. Aumont, N. Dingle and G. McFiggans, UManSysProp v1.0: an online and open-source facility for molecular property prediction and atmospheric aerosol calculations, Geosci. Model Dev., 2016, 9, 899–914 CrossRef.
- K. H. Bates, D. J. Jacob, S. Wang, R. S. Hornbrook, E. C. Apel, M. J. Kim, D. B. Millet, K. C. Wells, X. Chen, J. F. Brewer, E. A. Ray, R. Commane, G. S. Diskin and S. C. Wofsy, The global budget of atmospheric methanol: New constraints on secondary, oceanic and terrestrial sources, J. Geophys. Res., 2021, 126, e2020JD033439 CrossRef CAS.
- D. J. Jacob, B. D. Field, Q. Li, D. R. Blake, J. de Gouw, C. Warneke, A. Hansel, A. Wisthaler, H. B. Singh and A. Guenther, Global budget of methanol: constraints from atmospheric observations, J. Geophys. Res., 2005, 110, D08303 Search PubMed.
- P. Caffrey, W. Hoppel, G. Frick, L. Pasternack, J. Fitzgerald, D. Hegg, S. Gao, R. Leaitch, N. Shantz, T. Albrechcinski and J. Ambrusko, In-cloud oxidation of SO2 by O3 and H2O2: cloud chamber measurements and modelling of particle growth, J. Geophys. Res., 2001, 106, 27587–27601 CrossRef CAS.
- C. Fittschen, M. A. Ajami, S. Batut, V. Ferracci, S. Archer-Nicholls, A. T. Archibald and C. Schoemaecker, ROOOH: a missing piece of the puzzle for OH measurements in low-NO environments, Atmos. Chem. Phys., 2019, 19, 349–362 CrossRef CAS.
- V. Ferracci, I. Heimann, N. L. Abraham, J. A. Pyle and A. T. Archibald, Global modelling of the total OH reactivity: investigations on the “missing” OH sink and its atmospheric implications, Atmos. Chem. Phys., 2018, 18, 7109–7129 CrossRef CAS.
- R. L. Caravan, T. J. Bannan, F. A. F. Winiberg, M. A. H. Khan, A. C. Rousso, A. W. Jasper, S. D. Worrall, A. Bacak, P. Artaxo, J. Brito, M. Priestley, J. D. Allan, H. Coe, Y. Ju, D. L. Osborn, N. Hansen, S. J. Klippenstein, D. E. Shallcross, C. A. Taatjes and C. J. Percival, Observational evidence for Criegee intermediate oligomerization reactions relevant to aerosol formation in the troposphere, Nat. Geosci., 2024, 17, 219–226 CrossRef CAS.
- R. Chhantyal-Pun, M. A. H. Khan, R. Martin, N. Zachhuber, Z. J. Buras, C. J. Percival, D. E. Shallcross and A. J. Orr-Ewing, Direct kinetic and atmospheric modelling studies of Criegee intermediate reactions with acetone, ACS Earth Space Chem., 2019, 3, 2363–2371 CrossRef CAS.
- R. G. Derwent, D. D. Parrish, A. T. Archibald, M. Deushi, S. E. Bauer, K. Tsigaridis, D. Shindell, L. W. Horowitz, M. A. H. Khan and D. E. Shallcross, Intercomparison of the representations of the atmospheric chemistry of pre-industrial methane and ozone in earth system and other global chemistry-transport models, Atmos. Environ., 2021, 248, 118248 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Rayne
Holland
a,
Asan
Bacak
bc,
Thomas J.
Bannan
*a,
Rayne
Holland
a,
Asan
Bacak
bc,
Thomas J.
Bannan