
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterizing highly oxygenated organic molecules in limonene secondary organic aerosols: roles of temperature and relative humidity†
Yitong
Zhai
 *a,
Vasilios G.
Samaras
*b and
S. Mani
Sarathy
a
*a,
Vasilios G.
Samaras
*b and
S. Mani
Sarathy
a
aClean Energy Research Platform, Physical Sciences and Engineering (PSE) Division, King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Saudi Arabia. E-mail: yitong.zhai@kaust.edu.sa
bAnalytical Chemistry Core Lab, King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Saudi Arabia. E-mail: vasileios.samaras@kaust.edu.sa
First published on 28th March 2025
Abstract
Highly oxygenated organic molecules (HOMs) are significant contributors to the formation of secondary organic aerosols (SOAs) and new particles in the atmosphere. The process of HOM formation via autoxidation is highly dependent on several factors, such as temperature, relative humidity (RH), and initial ozone concentration, among others. The current work investigates how temperature and RH affect the formation of HOMs in SOAs from limonene ozonolysis. Experiments were conducted in a laminar flow tube reactor under different experimental conditions (T = 5 °C and 25 °C; RH = 15% and 75%). A scanning mobility particle sizer was used to measure the concentration and size distribution of generated SOA particles. Fourier transform ion cyclotron resonance mass spectrometry was used to detect and characterize HOMs and SOAs. Experimental results show that lower temperatures (i.e., T = 5 °C) and higher RH levels (e.g., RH = 75%) promote the generation of HOMs and SOAs. Limonene-oxidation-derived HOMs exhibit a preference for stabilization under low-temperature and high-RH conditions. Within this context, semi-volatile, low-volatile, and extremely low-volatile organic compounds play a significant role. Our experimental findings indicate that the formation of C10 compounds during limonene ozonolysis is strongly influenced by peroxy radical chemistry. Given that peroxy radicals are key intermediates in this process, their reactions—including autoxidation and bimolecular termination pathways—likely play a significant role in the formation and stabilization of HOMs in SOAs. The observed product distributions also suggest that these radicals contribute to the incorporation of multiple oxygen atoms, facilitating the formation of ELVOCs and LVOCs that ultimately drive particle-phase growth. The present work can improve our understanding of the generation of biogenic HOMs and SOAs at different temperatures and RH, which can be used in future exposure risk or climate models to provide more accurate air quality prediction and management.
Environmental significanceHighly oxygenated organic molecules (HOMs) are recognized as significant contributors to the formation of secondary organic aerosols (SOAs) and new particles in the atmosphere. Their production, especially through the autoxidation process, is influenced by various factors, including temperature and relative humidity (RH). However, the specific impacts of these factors on the chemical composition of HOMs and SOAs are poorly understood. In this study, we explore how the formation of HOMs and the molecular composition of SOAs generated from limonene ozonolysis—a key biogenic oxidation process—vary with temperature and RH. Our findings underscore the importance of characterizing molecular-level composition, particularly regarding how temperature and RH influence HOM and SOA formation. This knowledge can be applied in future exposure risk and climate models to improve the accuracy of air quality predictions and management strategies. |
1 Introduction
Organic aerosols (OAs) are small particles, either liquid or solid, that are suspended in the atmospheric environment. These particles constitute a predominant fraction of ambient fine particulate matter (PM2.5).1,2 OAs can interact directly with light via adsorption and scattering or indirectly by serving as cloud condensation nuclei. Therefore, OAs play an important role in many environmental processes at local and global scales.3,4 Atmospheric OAs are one of the significant factors that affect air quality, visibility, climate, and, therefore, public health.5 Several epidemiological studies have found a link between elevated atmospheric PM concentrations and increased morbidity and mortality.6–8 OAs have a major impact on atmospheric physicochemical and biochemical characteristics, such as toxicity, hygroscopicity, and radiative forcing.9,10 They originate from a wide range of sources and atmospheric processes. Consequently, OAs can be commonly categorized as either primary OAs (POAs) or secondary OAs (SOAs). In contrast, POAs are directly emitted from natural sources, such as vegetative detritus, and anthropogenic sources, such as wood burning, kitchen cooking, vehicular emissions, and industrial processes. Meanwhile the formation of SOAs is derived from multiple autoxidation steps of thousands of gas-phase precursors by ozone (O3), hydroxyl radicals (OH), and nitrate radicals (NO3).11,12 In the atmosphere, SOA mass loadings usually exceed those of POAs, dominating the OA burden. The organic compounds comprising SOAs are complex, complicating our understanding of their behavior. SOAs consist of a variety of organic oxygenates with different chemical functionalities that govern their volatilities.13,14 As the functionalization and the degree of oxygenation increase, the vapor pressure gradually decreases, allowing the gaseous compounds to further condense on existing particles or nucleate and form new compound particles.15,16Monoterpenes (C10H16), as the typical biogenic volatile organic compounds (BVOCs), play a crucial role as precursors for SOA formation.17,18 The ozonolysis of monoterpenes is a highly effective process for the formation of semi-volatile, low-volatile, and extremely low-volatile organic compounds (SVOCs, LVOCs, and ELVOCs) in the ambient environment, which are major contributors to SOA formation.19 It has been widely acknowledged that the presence of sulfuric acid is critical for initiating the majority of new particle formation (NPF) in the atmosphere. However, recent experiments have indicated that BVOCs can generate significant quantities of aerosol particles through the formation of ultra-low-volatility organic compounds (ULVOCs), even without the presence of sulfuric acid.20,21 The (extremely) low volatilities of these organic compounds allow them to irreversibly distribute onto the surface of aerosol particles, which could be enhanced.22 The role of ELVOCs is significant in the formation of atmospheric cloud condensation nuclei, which could promote the nucleation of new particles in most continental regions.23,24 Additionally, LVOCs are significant for the growth of particles larger than a few nanometers.19
As an important monoterpene, limonene is among the most abundantly emitted biogenic compounds in the atmosphere, with a global average emission rate of 11.4 Tg per year.25 Due to its widespread use in household products such as detergents and deodorizers, limonene is also one of the most commonly found VOCs in indoor air.26 The presence of two electron-rich C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds (i.e., exocyclic and endocyclic double bonds) renders limonene highly reactive, adding complexity to its behavior and fate in the atmosphere. Its rapid reaction with O3 can lead to a significant production of HOMs and even SOAs.27 The initial stage of limonene ozonolysis involves the reaction of O3 with the exocyclic/endocyclic CC double bond, forming the highly energetic primary ozonides (POZs). The POZs subsequently decompose into two groups of carbonyls and carbonyl oxides, collectively referred to as excited Criegee intermediates (CIs).28 After the formation, these energy-rich compounds can either stabilize to form stabilized CIs or decompose into an OH radical and an alkyl radical, with the alkyl radical quickly converting to RO2 radicals. The radical chain reaction of RO2 continues until it is terminated by either the ejection of an OH or HO2 radical or by bimolecular reactions with NO/HO2/RO2, resulting in the formation of closed-shell products that include peroxides, carbonyls, alcohols, and other potential functionalities.29
C double bonds (i.e., exocyclic and endocyclic double bonds) renders limonene highly reactive, adding complexity to its behavior and fate in the atmosphere. Its rapid reaction with O3 can lead to a significant production of HOMs and even SOAs.27 The initial stage of limonene ozonolysis involves the reaction of O3 with the exocyclic/endocyclic CC double bond, forming the highly energetic primary ozonides (POZs). The POZs subsequently decompose into two groups of carbonyls and carbonyl oxides, collectively referred to as excited Criegee intermediates (CIs).28 After the formation, these energy-rich compounds can either stabilize to form stabilized CIs or decompose into an OH radical and an alkyl radical, with the alkyl radical quickly converting to RO2 radicals. The radical chain reaction of RO2 continues until it is terminated by either the ejection of an OH or HO2 radical or by bimolecular reactions with NO/HO2/RO2, resulting in the formation of closed-shell products that include peroxides, carbonyls, alcohols, and other potential functionalities.29
Highly oxygenated organic molecules (HOMs), produced from the rapid autoxidation (i.e., successive intramolecular H-shift and O2 addition reactions) of peroxy radicals (RO2), are a diverse class of organic compounds that contain multiple functionalities and a high number of oxygen atoms. The presence of HOMs in the atmosphere exhibited a diverse range of redox functionalities,20,22 which are of significant importance in the initial stages of atmospheric organic aerosol growth.30,31 Also, the formation of aqueous radicals is closely associated with these HOMs.32 Because of the significance of SOAs in Earth's energy budget and air quality, the study of SOAs and their precursors (i.e., HOMs) has been a predominant research focus in atmospheric chemistry for several decades. During the autoxidation process, there are various opportunities for gas-phase chemistry to modify the HOM distribution and consequently influence SOA production, for example, the temperature and relative humidity (RH). Previous quantum computations indicated that the activation barrier for the intramolecular H-shift reaction of RO2 radicals is high, at 84 kJ mol−1 or greater.29,33,34 This leads to a strong temperature dependence on the autoxidation process. Also, a recent model study revealed that the rate-limiting step of total HOM formation is the first H-shift reaction in RO2 isomerization.33 Consequently, possible changes in the distribution of HOMs as functions of temperature are expected to result mainly from the alterations in the autoxidation process. Besides temperature, RH is also regarded as a crucial factor controlling the fate of HOMs and SOAs. For example, Kundu et al. found that the relative intensities of the most oxidized products and the molecular formulae with average carbon numbers were higher under humid conditions compared to those under dry conditions.35 Moreover, the changes in RH can influence the NPF by either regulating the formation of gas-phase precursors or by participating in cluster formation.36 A few studies have proposed that high water content may inhibit the formation of organics related to NPF resulting from the oxidation of biogenic precursors.37,38 Given the considerable variability of chemical constituents, sources, and possible chemical evolutions,39 comprehensive and detailed characterization of HOM and SOA components is crucial to elucidate the complicated formation processes and address the adverse effects of aerosols. Furthermore, a comprehensive understanding of the various autoxidation steps, as well as their temperature dependences and RH effects, is still lacking for the atmospheric monoterpene-oxidant systems.
To understand the mechanisms underlying SOA formation that depend on temperature and RH, it is crucial to gain a better understanding of the molecular composition of monoterpene-derived SOAs, especially the constituents that govern volatility and oxidation state. To achieve this, experiments were performed in a temperature-controlled flow tube reactor at different temperatures and RH. A Fourier transform ion cyclotron resonance mass spectrometer equipped with a 9.4 tesla superconducting magnet (9.4 T FT-ICR MS) was used to provide precise and accurate measurement of molecular composition and reaction mechanisms of polar SOA organic compounds. This technique enabled the clear assignment of molecular formulae of organic compounds, including carbon, hydrogen, and oxygen up to 950 Da, which helped to explore the differences in particulate HOMs derived from limonene oxidation under different experimental conditions.
2 Materials and methods
2.1 SOA generation and collection
A schematic diagram of the experimental setup used in the current work is shown in Fig. 1. Laboratory SOA generation was performed by the gas-phase ozonolysis of limonene in a laminar flow tube reactor. The reactor was made of borosilicate glass (length: 135 cm, inner diameter: 4.5 cm) and covered with a water jacket for temperature control, in which the temperature is governed by using water circulation from the chiller (−20–40 °C, Minichiller-NR, Huber). Approximately 2.0 mL of liquid R-(+)-limonene (Sigma-Aldrich, ≥99%) was stored in a homemade bubbler. The outer layer was also covered with a water jacket, and the bubbler temperature was controlled by another chiller (PolyScience, Inc.). Precursor vapor was produced by passing a certain amount of ultrahigh pure (UHP) N2 stream (purity >99.999%) over the liquid reactants at the predetermined temperature. The resulting vapor was then diluted with additional UHP N2 steam. O3 was generated using a commercial O3 generator (Ozone Solutions, Ins.) by passing the mixture of UHP N2 and O2. After the generation, the O2/O3/N2 mixture was introduced into the reactor with another carrier gas of UHP N2. The O3 concentration inside the reactor was measured by an O3 monitor (Model 205, 2B Technologies) after passing through a particle filter in the absence of limonene. To investigate the impact of humidity, there is another temperature-controlled bubbler filled with deionized water and covered with heating tape used to control the RH inside the reactor. The desired level of RH was achieved by regulating the temperature of the bubbler and the amount of N2. The final concentration of O3 was around 750 ± 25 ppb. The temperature and RH inside the reactor were monitored by a hygro-thermometer (Shenzhen Everbest Machinery Industry CO., Ltd, DT-8896). Based on the flow rate of N2 passing the VOC bubbler, the vapor pressure of the precursor at the selected temperature, and the total flow rate, the initial concentration of limonene is estimated to be ∼1.5 ppm. The gases containing the VOC precursor, O3, and synthetic air (80%:20% v/v) were continuously delivered to the reactor at a total flow rate of 4.0 standard L min−1. Therefore, the residence time in the reactor was estimated to be ∼33 s. Note that precursor concentrations used in this study are much higher than concentrations encountered under ambient atmospheric conditions. However, these concentrations can generate much more SOA materials, typically in the milligram range.40 Similar experimental conditions have been used in previously published investigations of SOA generation.41–47 Experiments to study the effect of temperature were conducted at 5 and 25 °C under dry conditions (RH < 1%). For the experiments to study the effect of RH, the temperature was fixed at 25 °C, and two different RH levels (i.e., 15% and 75%) were used. Detailed experimental conditions are listed in Table 1. Each experiment was repeated at least three times to ensure reliable and reproducible results. | ||
| Fig. 1 Schematic of the experimental setup for the laboratory SOA generation and collection. | ||
| Experimentsb | Initial conditions of the experimenta | |||
|---|---|---|---|---|
| Limonenec (ppm) | O3 (ppb) | RH (%) | Temperature (°C) | |
| a The residence time used in this work is 32–33 s. b Each experimental condition is repeated three times. c The initial concentration of limonene was estimated by the passing amount of the carrier gas and the total flow rate. d TSOA means that temperature is the control factor on the SOAs formed from limonene ozonolysis. e HSOA means that relative humidity is the control factor on the formed SOAs from limonene ozonolysis. | ||||
| Temperature | ||||
| TSOA-1d | 1.5 | 750 ± 25 | <2 | 25 ± 0.5 |
| TSOA-2 | 1.5 | 750 ± 25 | <2 | 5 ± 0.5 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Relative humidity | ||||
| HSOA-1e | 1.5 | 750 ± 25 | 15 | 25 ± 0.5 |
| HSOA-2 | 1.5 | 750 ± 25 | 75 | 25 ± 0.5 |
When the O3 concentration reached a stable level, the limonene precursor was introduced into the flow reactor. After 1 hour from the start of the ozonolysis experiments (i.e., when the size distribution of SOAs was stable), SOAs were collected onto a PTFE 47 mm filter (Sigma-Aldrich, 0.2 μm pore size). To increase the amount of SOA samples, a common mechanical pump was connected after the aerosol samplers with a flow rate of approximately 2 L min−1. In this study, a short-length sampling tube was directly connected to the reactor outlet and the SOA collector to minimize particle losses and ensure efficient aerosol transfer. Given the relatively high sampling flow rate and short residence time within the sampling tube, potential evaporation losses are expected to be minimal. The sampling process lasted for a duration time of 6 hours, and the collector was maintained at room temperature throughout the experiments. To assess the potential impact of evaporation during the SOA sampling process, we evaluated the volatility of major SOA constituents in relation to temperature-dependent partitioning effects. While semi-volatile components may exhibit some degree of loss, the dominant species identified in our experiments, including HOMs and oligomeric compounds, are known to have significantly lower volatility, reducing the likelihood of substantial evaporation during collection. However, recognizing the potential for sampling biases, we compared our SOA composition with data from studies employing real-time sampling techniques that mitigate evaporation effects.48 This comparison revealed strong consistency in the observed product distributions, indicating that sampling-induced volatilization does not significantly alter the final SOA composition under our experimental conditions. While we acknowledge that some degree of uncertainty may arise from temperature differences between the reactor and the sampling setup, the presence of low-volatility species suggests that our findings remain consistent.
Prior to collecting SOAs, filters were heated at 50 °C in a furnace for 2–3 hours to remove the adsorbed volatiles and kept in low-humidity environments. After the collection process, filters were immediately transferred to an amber glass vial, sonicated, and extracted using a mixture of Milli-Q water and methanol (LC-MS grade, Fisher) in a 4:1 volume ratio in an ice bath for 15 minutes. The glass vials containing the filter extracts were promptly transferred to a small storage box that was packed with dry ice. The samples were then delivered to the core lab for analysis using ultrahigh-resolution FT-ICR MS. To minimize potential secondary reactions and analyte losses, the filter extracts were not evaporated to dryness and were instead directly analyzed without further processing. Similarly, a blank filter was analyzed using the same procedure as the SOA samples. After each experiment, the reactor was carefully rinsed with acetone and then dried with UHP N2 gas for at least 4 hours to ensure that no residual VOC precursors/aerosol products were left behind. In addition, the size distribution and number concentration of the generated SOA were measured using a scanning mobility particle sizer (SMPS, TSI 3936), which consisted of a nano-differential mobility analyzer (DMA, TSI 3086) and an ultrafine condensation particle counter (CPC, TSI 3776).
During ozonolysis experiments of alkenes, a large number of highly reactive OH radicals can be produced.49 To prevent precursor autoxidation with OH radicals, high concentrations of OH scavengers such as cyclohexane, alcohols, and aldehydes are typically introduced into the O3/alkene system.50 However, the use of OH radical scavengers can affect the alkene autoxidation mechanism, and consequently, the distribution of oxidized products,51,52 which in turn makes the comparison of results difficult. Therefore, OH radical scavengers were not used in this study, avoiding any interference with the distribution of oxidized products. Note that although OH radicals are formed during limonene ozonolysis, their contribution to limonene oxidation is secondary to direct ozonolysis under our experimental conditions. While literature values for OH radical yield and reaction rate constants could provide an estimate of OH radical's contribution, such calculations involve uncertainties due to variations in experimental conditions and secondary oxidation effects. Instead, we assessed the influence of OH radicals by comparing our product distribution with previous studies on limonene ozonolysis.53 The strong similarity in observed SOA composition suggests that OH-mediated oxidation does not significantly affect the final molecular profile. These findings confirm that under our experimental conditions, direct ozonolysis remains the predominant oxidation pathway.
2.2 FT-ICR MS measurement and molecular formula assignment
The extracted samples from both SOA filters and blank filters were finally analyzed using an ultrahigh-resolution SolariX XR FT-ICR MS (Bruker Daltonik GmbH, Bremen, Germany) that was equipped with a 9.4 T refrigerated superconducting cryo-magnet and ParaCell™ analyzer. The chemical composition of SOA samples was analyzed using atmospheric pressure electrospray ionization (ESI) in positive ion mode. Spectra were acquired over a mass range of m/z 100–1200, with an ion accumulation time of 0.002 s and a data size of 8 M. Mass spectra were obtained by accumulating 300 individual mass scans. The drying gas temperature was set to 200 °C. The spectra were externally calibrated with sodium trifluoroacetate (NaTFA) clusters using a solution of 0.1 mg mL−1 NaTFA in extracts. After data acquisition, the raw data were further internally recalibrated with the exact masses of known compounds. The samples were infused directly into the ESI source at a flow rate of 4 μL min−1, with the vaporizer temperature set at 250 °C and a dry N2 gas flow of 4.0 L min−1.To ensure that ion-induced clustering did not affect the molecular characterization results, we conducted a series of control experiments. First, the optimization of electrospray ionization conditions was performed by analyzing blank samples and fully processed samples prepared in different solvent systems, including water, acetonitrile, and methanol, to fine-tune ionization parameters. Additionally, sodium trifluoroacetate (NaTFA) was introduced as an internal standard to monitor ionization efficiency and detect potential clustering effects. To further assess the impact of ion-induced clustering, a systematic dilution study was carried out, allowing us to differentiate real molecular ions from potential cluster artifacts, as clustering effects typically diminish at lower concentrations. Finally, spectral analysis was conducted to identify clustering signatures, such as systematic mass shifts, unusual peak distributions, or peak broadening. Internal mass calibration and generation of mass lists were performed using DataAnalysis 5.2 (Bruker Daltonics GmbH & Co. KG, Bremen, Germany). Data analysis, including the calculation of molecular formulae and relative abundances of compound classes, was performed using Composer software (Sierra Analytics, Pasadena, CA, USA). The formula calculator was set to calculate formulae in the mass range of 100–1200 Da, with elemental compositions up to 50 carbon (C), 150 hydrogen (H), and 30 oxygen (O) atoms with a tolerance of ± 1 ppm when only C, H, and O are investigated. To ensure the unambiguous assignment of chemical formulae to SOA constituents, softer ionization with higher mass resolution was employed.
2.3 Data processing
Significant quantities of molecules can be captured and identified using FT-ICR MS, which makes it difficult to perform a comprehensive analysis by sequential approaches. Therefore, graphical representation and statistical tools become more necessary. In this study, different data processing methods were considered.The assigned molecular formulae were examined by using the Kendrick mass defect (KMD) and double bond equivalent (DBE) series. To assess the saturation and oxidation degree of limonene SOAs, the value of DBE is calculated using eqn (1):
| DBE = nC − nH/2 + 1 | (1) |
The potential building units of the molecular formulae and their chemical characteristics were evaluated using Kendrick mass analysis.54 The observed m/z is converted to Kendrick masses with respect to any specific Kendrick base, as listed in eqn (2). Thereafter, the KMD is determined using eqn (3):
| Kendrick mass (base) = measured mass × [(nominal mass of base)/(exact mass of base)] | (2) |
| Kendrick mass defect (base) = |nominal mass − Kendrick mass (base)| | (3) |
The carbon oxidation state (OSc), which describes the composition of organic compounds undergoing autoxidation processes,55 is defined using eqn (4):
| OSc = 2nO/nC − nH/nC | (4) |
The weighted average of molecular weight (MW), O/C ratio, OSc, O atom, and DBE was computed using eqn (5):
| X = ∑(lnti × Xi)/∑lnti | (5) |
ti and Xi are the relative intensity and parameter value of each individual compound i, respectively. To estimate the contribution of carbonyl equivalent groups in the molecule with oxygen number greater than or equal to DBE, the maximum carbonyl ratio (MCR) was used, following the method described by Zhang et al.56 The MCR value was calculated as the ratio of the sum of double bond equivalents (DBEs) to the sum of oxygen atoms in the molecule, as shown in eqn (6). | (6) |
Therefore, HOMs were then categorized into four groups based on their MCR values as follows:
(I) Very highly oxidized organic compounds (VHOOCs; 0 ≤ MCR ≤ 0.2).
(II) Highly oxidized organic compounds (HOOCs; 0.2 < MCR ≤ 0.5).
(III) Intermediately oxidized organic compounds (IOOCs; 0.5 < MCR ≤ 0.9).
(IV) Oxidized unsaturated organic compounds (OUOCs; 0.9 < MCR ≤ 1).
These categories help to distinguish between different levels of oxidation in the HOMs and provide insights into the atmospheric processing of organic aerosols.
All the gas- and particle-phase compounds can be classified based on volatility, which determines their partitioning between the gas and particle phase. Directly measuring the volatilities of individual HOMs is extremely challenging due to their difficult synthesis and the fact that their vapor pressures are too low for current volatility measurement techniques. To describe the partitioning (e.g., chemical composition, molecular characteristics of molar mass, and structure) of complex OAs, saturation vapor pressure (C0, μg m−3) is used. Based on the calculated values of C0, organic compounds can be grouped into ranges of intermediate-volatility organic compounds (IVOCs), semi-volatility organic compounds (SVOCs), low-volatility organic compounds (LVOCs), ELVOCs, and ULVOCs.57 Compounds in each range are approximately as follows: 300 < C0 < 3 × 106 μg m−3 for IVOCs, 0.3 < C0 < 3 00 μg m−3 for SVOCs, 3 × 10−5 < C0 < 0.3 μg m−3 for LVOCs, 3 × 10−9 < C0 < 3 × 10−5 μg m−3 for ELVOCs, and C0 < 3 × 10−9 μg m−3 for ULVOCs. C0 is calculated using the 2D volatility basis set (2D-VBS) in eqn (7):
 | (7) |
C0, respectively. bCO represents the carbon–oxygen nonideality.15,58 All parameters used for the C0 calculation were adopted from previous work by Li and her co-authors.59 To estimate the temperature dependence on volatility, C0(T) can be described according to the Clausis–Clapeyron eqn (8): | (8) |
According to previous references,58,60 the evaporation enthalpy ΔHvapi can be approximated as follows:
| ΔHvapi [kJ mol−1] = −5.7 × log10C0i(300 K) + 129 | (9) |
Therefore, a change in the temperature range of 15 to 20 K can lead to a shift of the volatility bin by 1 order of magnitude. This work mainly focuses on the oxidation products that fall under the categories of ELVOCs and ULVOCs, which are responsible for initiating cluster growth and creating new particles. It is worth noting that under the high precursor concentration conditions used in this work, ELVOCs could enhance their participation in the nucleation process, contributing to the initial formation of new particles. Furthermore, ULVOCs are also highly efficient in nucleation. Due to the limited information available about the exact chemical structures and functional groups of the oxidation products, we assume an overall uncertainty of ± 1 bin in the volatility distribution (i.e., 1 order of magnitude in C0 at 300 K).
2.4 Definition of HOMs
In previous research, Ehn et al.17 identified that in the atmosphere of Hyytiälä, as well as in laboratory-generated α- and β-pinene SOAs, HOMs consistently exhibit an O/C ratio of at least 0.7. Tröstl et al.19 extended this understanding and proposed that α-pinene SOA-based HOMs can be characterized by chemical formulae of CxHyOz with x = 8–10, y = 12–16, and z ≥ 6 for monomers. For dimers, the suggested range is CxHyOz with x = 17–20, y = 26–32, and z ≥ 8. Building on this, Tu et al.61 further defined HOMs in biogenic SOAs from α-pinene, β-pinene, and limonene, classifying them based on assigned formulae with O/C ≥ 0.6 or higher, as well as OSC ≥ 0. The categories include highly oxygenated and highly oxidized HOMs (O/C ≥ 0.6 and OSC ≥ 0), highly oxygenated but less oxidized HOMs (O/C ≥ 0.6 but OSC < 0), and highly oxidized HOMs with a moderate level of oxygenation (OSC ≥ 0 but H/C < 1.2). These categorizations were introduced to explore the relative significance of oxygen content versus oxidation state in HOMs. Subsequent investigations62 have revealed that HOMs present in SOAs originating from monoterpenes are predominantly composed of ELVOCs, LVOCs, and a minor fraction of SVOCs.Specifically, HOMs with molecular formulae of C8–10H12–16O6–9 and C17–20H26–32O8–15 were considered in the present study. Furthermore, non-HOM monomers were filtered out based on an O/C ratio < 0.7.53 The formation pathways of HOMs were estimated to mainly involve the hydroperoxide channel and Criegee channel27,63 based on previous research. In this paper, monomers refer to C8–10 molecules, while dimers refer to C17–20 molecules.
3 Results and discussion
3.1 Effect of temperature and RH on the size distribution of limonene SOAs
The averaged particle size distributions and the number concentration of SOAs produced from the ozonolysis of limonene under different experimental conditions are shown in Fig. 2. In general, SOAs formed at 5 °C showed a broader size distribution. Accordingly, the dominant size range measured at T = 5 °C expanded from ∼20–100 to ∼10–100 nm, and the diameter of peak number concentration was ∼45 nm. In contrast, the peak size and number concentration appeared at 40 nm in experiments for both RH conditions, presenting no significant difference in the particle size distributions between high and low RH. Furthermore, the peak number concentration of limonene SOAs was higher under low-temperature and high-RH conditions, indicating a higher SOA yield. Notably, the peak of the generated particle number concentration decreased from 1.0 × 107 to 0.7× 107 cm−3 with the increased temperature from 5 °C to 25 °C and from 0.8 × 107 to 0.6× 107 cm−3 with the decreased RH from 75% to 15%. Likewise, the number concentration exhibited a trend to decrease with the increasing temperature and decreasing RH. | ||
| Fig. 2 Particle size and number concentration distributions of the SOA from the ozonolysis of limonene at different (a) temperatures and (b) RH levels. | ||
Some studies have investigated the effects of RH and temperature on the generated oxidized products from the ozonolysis of exocyclic and endocyclic organic compounds, with varying results.48,64–67 They have reported that the RH/temperature either suppresses or promotes effects on the particle formation process by measuring the number size distributions of SOA particles generated during the ozonolysis of monoterpenes using a SMPS. Based on the conclusion from Zhang et al.,68 the inhibiting/promoting effect on SOA mass concentration mainly relies on the maximum SOA growth point, which may be explained by the evaporation of previously condensed organic compounds on particles back into the gas phase or experimental setups such as the initial concentrations of monoterpene and O3, variation of temperature/RH, presence of OH scavengers, residence time inside reactors, and other factors. It is important to note that SOA mass concentration begins to decrease after this point.
The observed trend in the present work is possible due to the formation of high molecular weight and low-volatility compounds through the gas- or particle-phase reactions.69 The high SOA yield observed under low-temperature and high-RH conditions indicates that the lower temperature and higher RH level could promote the formation and growth of molecular clusters, eventually reaching cloud condensation nuclei-active sizes. Additionally, the particle size distribution of limonene SOAs exhibits a broader range and lower size at T = 5 °C than those in other experiments, indicating more complex partitioning and agglomeration kinetics. Therefore, to comprehensively understand the mechanisms governing the enhancement of NPF and SOA growth under different experimental conditions, chemical characterization of both gas-phase and particle-phase reaction products was conducted in this study.
3.2 Effect of temperature and RH on product distribution and composition
 | ||
| Fig. 3 Mass spectral fingerprint and signal intensity of limonene ozonolysis products at different temperatures and RH levels: T = 25 °C (a) in blue, T = 5 °C (b) in green, RH = 15% (c) in red, and RH = 75% (d) in purple. Specific masses, selected for representing HOMs, are highlighted in darker colors. | ||
The total formula number and intensity of limonene SOAs decreased from T = 25 °C to 5 °C and increased from RH = 15% to 75% (Fig. S1†). At room temperature, molecular weight in the MW range of 250–550 Da (i.e., HOM region) exhibited a high ratio, whereas a low portion was observed for those with molecular weight in the range of 650–950 Da (i.e., trimer and tetramer regions). However, at a high-RH of 75%, molecular weight in the MW range of 250–550 and 650–950 Da both presented a higher percentage. The twenty most abundant HOM peaks for each experiment observed in the present work are provided in Table S1.† The similarity in the major peaks observed between the low- and high-temperature, as well as the RH, indicates that the predominant HOMs are generated by a similar mechanism. However, the changes in HOM intensity also reveal the formation of HOMs that are sensitive to temperature and RH more or less.
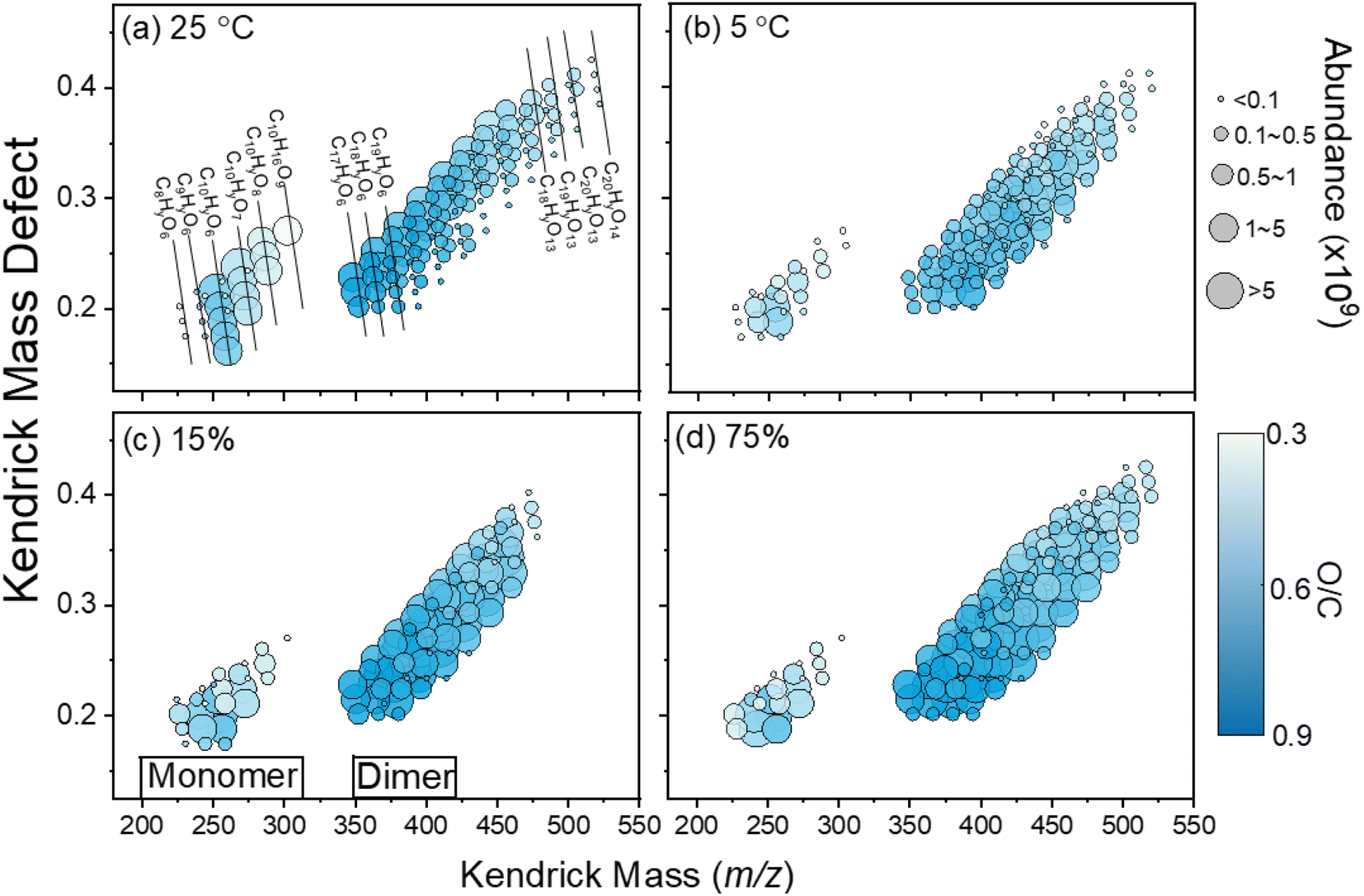 | ||
| Fig. 4 Mass defect plot of HOM signals from limonene ozonolysis measured from (a) T = 25 °C, (b) T = 5 °C, (c) RH = 15%, and (d) RH = 75%. | ||
Moreover, the product distribution of limonene SOAs was greatly affected by the temperature, with no obvious signal for trimers and tetramers at room temperature (T = 25 °C). The decrease in temperature leads to a visible reduction in the oligomer region, affecting the SOA composition. Specifically, the average SOA formula changed from C18.54H29.51O7.97 at room temperature to C19.96H33.01O8.46 under the low-temperature condition (T = 5 °C), as detailed in Table 2. At a low temperature, the average carbon, hydrogen, and oxygen numbers in the limonene SOA are higher than those at room temperature, suggesting the larger SOA components. At room temperature, the limonene SOA has an average O/C ratio of 0.42, an average H/C ratio of 1.60, and an average OSc ratio of −0.75. Under the low-temperature condition, however, the average O/C ratio, the average H/C ratio, and the average OSc ratio were 0.45, 1.66, and −0.77, respectively. Therefore, oxidized products generated at a low temperature tend to have higher H/C, O/C, and OSc values compared to the products formed at room temperature. The phenomenon above might be due to the presence of oligomers, as large amounts of high-molecular-weight compounds were observed at T = 5 °C. We speculated that the presence of oligomers in the mass spectrum may be because the low-temperature condition promotes the gas-particle partitioning and alters the particle-phase reactions. At room temperature, the less volatile products require more gas-phase oxidation steps to condense into the particle phase due to the high saturation vapor pressure caused by the high temperature. Therefore, oxidized products formed at room temperature have lower H/C, O/C, and OSc values. Conversely, less oxidized and more volatile products can readily condense into the particle phase under low-temperature conditions, allowing them to undergo particle-phase reactions such as oligomerization. These reactions can result in the formation of products with higher H/C, O/C, and OSc values. Furthermore, many HOMs contain thermally unstable functional groups, such as peroxy and hydroperoxide, which decompose more readily at higher temperatures. This could lead to the loss of oxygen and the formation of less oxygenated species, reducing the overall O/C ratio. At lower temperatures, HOMs remain more stable, allowing them to persist and undergo further oxidation, maintaining a higher O/C ratio. This temperature-dependent decomposition explains why oxidized products formed at room temperature have a lower average O/C ratio compared to those formed at a lower temperature. Note that literature reports consistently showed average O:C ratio values between 0.43 and 0.50, as determined from mass spectra analysis under oxidative conditions of 1–10 ppm for both O3 and limonene.28,71
| Experiments | C | H | O | H/C | O/C | OSc |
|---|---|---|---|---|---|---|
| Temperature | ||||||
| TSOA-1 | 18.54 | 29.51 | 7.67 | 1.60 | 0.42 | −0.75 |
| TSOA-2 | 19.96 | 33.01 | 8.90 | 1.66 | 0.45 | −0.77 |
|
||||||
| RH | ||||||
| HOSA-1 | 20.18 | 32.49 | 9.28 | 1.61 | 0.46 | −0.69 |
| HSOA-2 | 20.60 | 33.42 | 9.47 | 1.63 | 0.46 | −0.71 |
The mass spectra of the limonene SOA obtained at a RH of 15% and 75% are depicted, respectively (Fig. 3c and d). Although similar oxidized product distribution was observed in both spectra, their signal intensities varied, with higher RH resulting in stronger SOA signals. To better understand the composition of SOAs generated from limonene ozonolysis at different RH, we also assessed the chemical characteristics of the formed SOAs, which are summarized in Table 2. At RH = 15%, the limonene SOA had an average molecular formula of C20.18H32.49O9.28, compared to the formula of C20.60H33.22O9.47 at 75% RH. The average H/C ratio was around 1.61 at 15% RH, which is lower than the value of 1.63 at 75% RH. However, both had a similar average O/C ratio of approximately 0.46. The OSc also presented the same tendency as H/C with the increase of RH level, changing from −0.69 at 15% RH to −0.71 at 75% RH. Based on the above-mentioned results, it seems that the condensation reaction channel of limonene ozonolysis is more important under low temperature and high RH conditions, possibly due to the higher oxygen content in the organic matter produced. This may contribute to the formation of larger size particles of molecular clusters.
The MCR-van Krevelen (VK) diagram of the limonene SOA-containing HOMs collected on filters is presented in Fig. 5. Despite varying experimental conditions, the MCR-VK plots appear quite similar for the ozonolysis of limonene. The majority of oxidized products – around two-thirds of the total –are located in Region II, with Region III following, while only a few compounds are present in Regions I and IV. This distribution pattern highlights that most aerosolized oxidation products resulting from the limonene ozonolysis are categorized as HOOCs and IOOCs, with MCR falling within the range of 0.2–0.9. Notably, monoterpene oxidation products, oxidative aging products, and higher-generation oxidation products are typically found in this category.56 Interestingly, the detected compounds with high signal abundance are mainly HOOCs and IOOCs. In contrast, there is a notably limited presence of organic compounds exhibiting MCR values below 0.2 or above 0.9, indicating the relatively low prevalence of VHOOCs or OUOCs. Remarkably, the highest signal intensity of HOMs in limonene-derived SOAs exhibited an upward trend as RH increased and temperature decreased. This observation suggests that the lower temperature and higher RH might have a promotional impact on the formation of oxygenated organic compounds associated with limonene SOAs. This is because lower temperature is known to slow down the decomposition and fragmentation reactions, allowing intermediate products to remain intact longer and react to form larger and more complex molecules.72 Additionally, lower temperature could reduce the volatility of organic compounds, making them more likely to condense onto existing SOA particles.73 Higher RH can introduce more water vapor into the reaction system, facilitating hydrogen bonding reactions, which lead to the formation of more oxygenated species and promote the growth of oligomers.74 Furthermore, water can be absorbed by aerosol particles, potentially increasing their size, which may contribute to the formation of larger molecular clusters.75
 | ||
| Fig. 5 MCR-VK diagrams for the HOMs generated from the ozonolysis of limonene at different temperatures (5 °C and 25 °C) and RH levels (15% and 75%). The colors of the dots indicate the signal intensity of detected HOMs. The Roman numbers refer to the MCR classification, where I: VHOOCs, II: HOOCs, III: IOOCs, and IV: OUOCs. | ||
3.3 Volatility of HOMs in limonene SOAs
To enhance our understanding of the behavior and fate of HOMs derived from limonene ozonolysis in the atmosphere, we have categorized the identified limonene HOMs into distinct volatility classes. This categorization encompasses SVOCs, LVOCs, ELVOCs, and ULVOCs in the present work. By classifying the limonene HOMs into different volatility classes, we can gain insights into their varying vapor pressures, partitioning behavior between gas and particle phases, and their potential contributions to aerosol formation and composition. It is evident that the LVOC subgroup dominates the peak intensity among the limonene SOA-containing HOMs in experiments at different temperatures (Fig. 6). This pattern is consistent with the observations in the RH experiments (Fig. 6b). Furthermore, the peak intensities of ELVOCs were the second highest among the volatility classes, surpassing those of ULVOCs and SVOCs. Notably, only a limited number of ULVOCs were observed in the present work, suggesting that they are not the primary compounds in the distribution of HOMs resulting from limonene ozonolysis. Moreover, the formula count of LVOC-type HOMs constitutes a significant portion of the total number of identified HOMs, followed by ELVOC-type HOMs under all experimental conditions. This indicates that HOMs generated from limonene ozonolysis predominantly exist in the particle phase, with LVOCs and ELVOCs contributing significantly to the HOM composition in limonene-derived SOAs. Furthermore, the calculated results revealed that the majority of HOM monomers fell into the SVOCs or even LVOCs at a temperature of 25 °C, which was consistent with the experimental findings in α-pinene ozonolysis.48 Therefore, this indicated that the loss rate of these autoxidation compounds from the gas-phase stage is lower in comparison to the ELVOCs. It is noteworthy that, at these temperatures, HOM monomers exhibited the higher saturation vapor pressures (i.e., SVOCs), and none of them can be classified as ULVOCs. All of the observed ELVOCs and ULVOCs in this study are HOM dimers. As the temperature decreases, the saturation vapor pressures of the HOM monomers tend to shift toward the SVOC category. At these two temperatures, only the most oxygenated monomers have an extremely low saturation vapor pressure, while none of them fit the ULVOC class. All ELVOCs and ULVOCs observed in this work are HOM dimers. As the temperature decreases, the saturation vapor pressures of the HOM monomers shift towards the SVOC class.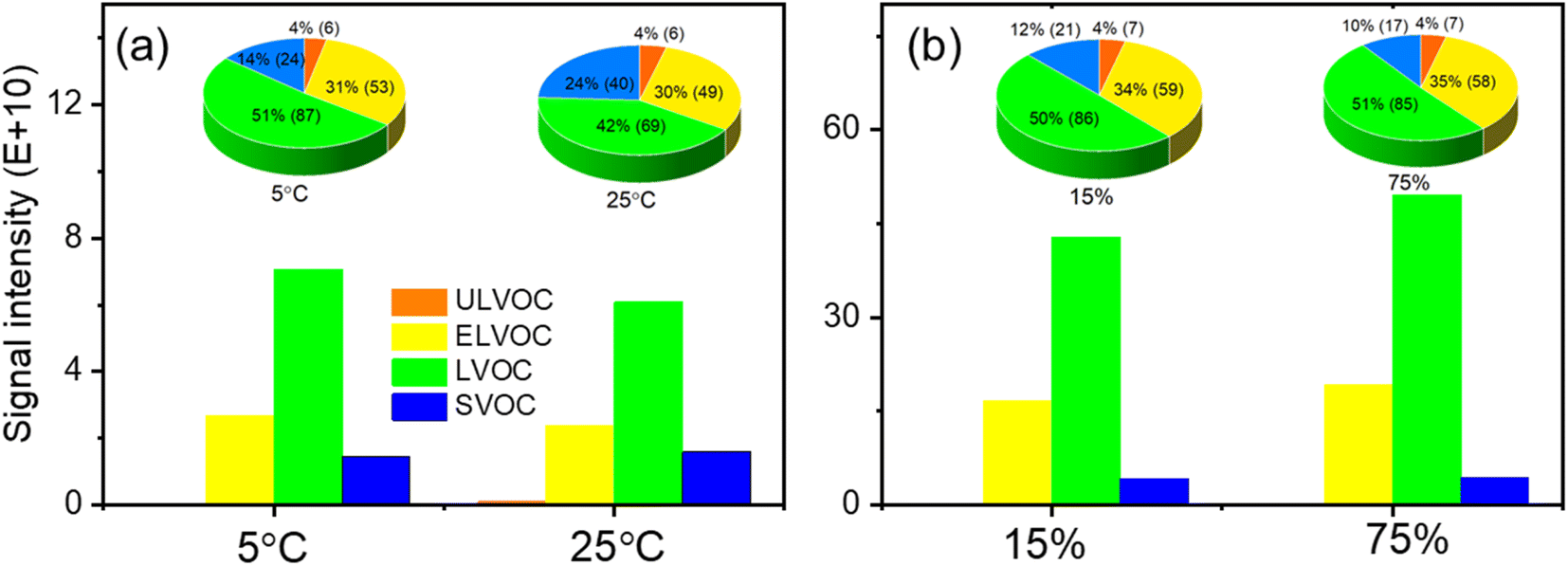 | ||
| Fig. 6 The peak intensity of HOMs with different volatilities in SOA particles from limonene ozonolysis at different (a) temperatures and (b) RH levels. The pie charts show the percentage and formula number of HOMs with different volatilities among all the identified HOMs. | ||
The mass variation of limonene SOAs during ozone chemistry was largely driven by LVOCs and ELVOCs (Fig. 6). These findings align with previous studies on limonene ozonolysis, which have demonstrated the predominance of LVOCs and ELVOCs in the ozonolysis of monoterpenes.53,76 The peak intensities and the formula counts of LVOC- and ELVOC-type HOMs were increased with the decreasing of temperature and the increasing of RH. Previous reports on monoterpene ozonolysis have indicated that HOM monomers are mainly of low volatility, with a small fraction being semi-volatile, while HOM dimers are all at least of low volatility and likely extremely low volatility.77 The low temperature and high RH promoted the formation of dimers from the combination of two monomers (as shown in Fig. 2), leading to the decreased intensity and formula count of SVOCs and the increased intensities and formula counts of LVOCs and ELVOCs in the current work.
The O/C ratio of limonene SOAs versus their estimated volatility is summarized in Fig. 7. It is apparent that ULVOCs constituted a large part of all SOA constituents. Also, the O/C ratio distribution of ULVOCs in limonene SOAs is more broadened at a lower temperature (T = 5 °C) and high RH (RH = 75%). Furthermore, there is a significant increase in the signal intensities of IVOCs, SVOCs, LVOCs, and ELVOCs with the decrease of RH level and the increase of temperature. This indicates that these compounds with higher O/C ratios and their corresponding signal strengths exhibited notable enhancements. Conversely, the abundance of compounds with lower O/C ratios decreased in comparison. This finding suggests that the ozonolysis of limonene resulted in the production of a greater proportion of SOAs, characterized by higher O/C ratios. These compounds, including IVOCs, SVOCs, LVOCs, and ELVOCs, demonstrated increased signal intensities, indicating their heightened presence in the generated aerosol particles at low temperature and high RH. Furthermore, the enrichment of high signal intensity ELVOCs in limonene SOAs confirmed that limonene has a greater propensity to form particulate ELVOCs and possibly even ULVOCs. This may be attributed to its higher reactivity, which is influenced by its intrinsic structural features such as the presence of two double bonds and an endocyclic structure.
 | ||
| Fig. 7 The oxygen-to-carbon ratios (O/C) versus the saturation mass concentration (C0) of limonene SOAs at different temperatures and RH levels: (a) T = 25 °C, (b) T = 5 °C, (c) RH = 15%, and (d) RH = 75%. The dot size is scaled to the logarithm of signal intensities. | ||
3.4 Potential formation mechanisms of HOMs and SOAs in limonene ozonolysis
Previous studies have indicated that the formation of HOMs in limonene ozonolysis involves various gas-phase reactions, including RO2 isomerization, RO2 + HO2, RO2 + RO2, and the reaction between stabilized Criegee intermediates and carboxylic acids. These reactions resulted in the production of gas-phase monomers and dimers, which contributed to the SOA formation process. However, ketones and aldehydes are typically converted to carboxyl and ester groups through Baeyer–Villiger reactions with peroxyacids and hydroperoxides in the particle phase. In contrast, peroxyacids and hydroperoxides are transformed into acids, alcohols, or ketones. These particle-phase reactions, driven by the high reactivity of the involved species, play a significant role in the transformation of organic compounds.The formation of HOM monomers in the limonene ozonolysis process can occur through two main channels: the hydroperoxide channel and the oxygen-increasing reactions (H-shift → O2 → RO2, OIR) of the Criegee channel.27,76,77 These mechanisms are illustrated in Fig. 8, depicting the possible pathways for the formation of C10 HOM monomers (e.g., C10H16O6 and C10H14O7). Note that these two monomers were observed in all experiments. In the former channel, the reaction involved the initial ozonolysis of limonene, leading to the formation of a primary ozonide. Subsequently, the primary ozonide underwent rearrangement and decomposition reactions, generating alkoxy radicals and hydroperoxides. The hydroperoxides can then react with RO2 radicals or undergo further decomposition, eventually resulting in the formation of C10 HOM monomers. The latter channel involved the reaction between SCIs and carboxylic acids. The SCIs can react with carboxylic acids, leading to the production of HOM monomers with an increase in oxygen content. It is expected that dimers with very low volatility, which are formed in the gas phase, will be directly distributed onto particles due to their high reactivity. These dimers were strongly influenced by particle-phase chemistry, which may involve accretion reactions (e.g., C8–10 + C8–10 → C17–20) and the decomposition of high molecular weight compounds (e.g., C20 → C17–19). The formation of HOM dimers was primarily driven by the accretion reaction between various HOM monomer RO2, as well as the termination reaction of dimer RO2 formed through further reactions of closed-shell dimers with O3. Additionally, these HOM dimers may also be formed through C10 reactions of RO2 with monoterpenes. While the formation of trimers through the accretion reaction of dimer RO2 and monomer RO2 is possible, it will not be extensively discussed in the subsequent analysis.
 | ||
| Fig. 8 Proposed gas-phase formation mechanism of HOMs from the ozonolysis of limonene via the hydroperoxide channel (purple), oxygen-increasing reactions of the Criegee channel (blue), and the accretion reaction process (orange).27,76 The molecules shown represent the hypothesized structures. | ||
Accretion (RO2 + RO2 → ROOR + O2) and esterification reactions (alcohol + acid → ester + H2O) played a crucial role in the transformation of mass from monomers to oligomers during the ozonolysis of unsaturated volatile organic compounds,71,78 resulting in the formation of larger compounds with an increased number of carbon atoms and converting semi-volatile molecules into higher-molecular weight compounds. As the molecular weight and carbon content increase, the saturation vapor concentrations of the resulting products decrease. This means that the newly formed oligomers have lower vapor pressures and are more likely to exist in the condensed phase, such as aerosol particles. These reactions not only contributed to the increase in molecular size and complexity but also exerted importance on altering the volatility characteristics of the oxidized products, which was suggested to have implications in the ambient environment.79
Apart from the accretion and esterification reactions suggested above, other potential paths cannot be excluded among the processes leading to oligomers. Several studies in the literature suggested that the ozonolysis of VOCs, such as monoterpenes, can be accompanied by dimerization and trimerization reactions facilitated by non-covalent hydrogen bonding. These reactions involved the participation of functional groups, including carboxylic groups, dicarboxylic groups, and functional groups resembling ketones or alcohols.80 These functional groups can form hydrogen bonds with each other, leading to the generation of larger and more complex molecules (Fig. S2†). This non-covalent bonding pathway is considered significant for the formation of SOAs in the atmosphere, as it contributed to the growth and composition of SOA particles. A recent study81 has already shown that a large number of products that possess similar functional groups can be generated in limonene ozonolysis, which enabled the formation of non-covalent bonds, leading to the formation of oligomers. Moreover, aldolisation and SCI reactions were also responsible for the oligomer formation. Considering limonene structures, the presence of two double bonds played a crucial role in the formation of trimers. One of the CC double bonds underwent an autoxidation process, yielding dimer compounds, while the remaining CC double bond provided a reactive site for the further autoxidation of the dimers, facilitating the generation of dimer RO2 radicals.21 Based on the mass spectral data in the present work, we concluded that low temperature and high RH promoted the progress of the oligomerization reaction in the particle phase through the above-mentioned reactions. Moreover, products with lower oxidation states and high volatilities are inclined to condense into the particle phase at a low temperature and high RH level and subsequently engage in particle-phase reactions, which would further accelerate the oligomerization reactions.
4 Conclusions
This work reported the experimental study of the HOM and SOA composition in limonene ozonolysis with respect to the roles of different temperatures (T = 5 and 25 °C) and RH levels (RH = 15 and 75%). The number concentration and size distribution of generated limonene SOA particles were monitored using a scanning mobility particle sizer. Moreover, the characterization and composition of HOMs and SOAs were analysed by Fourier transform ion cyclotron resonance mass spectrometry. The results showed that the variations in temperature and RH changed the product distribution of HOMs and SOAs more or less. Different volatilities and abundances of HOMs in limonene SOAs indicated the diverse molecular responses of particulate reaction products to the oxidation states of biogenic precursors. This variation in response contributes to the differences observed in the size and number concentration distribution profiles of the SOA formed from these precursors. Low-temperature and high-RH conditions were found to promote the formation of HOMs and ELVOCs. Under these conditions, the number and the intensity of unique organic molecules generated during the ozonolysis process were observed to be higher.In addition to temperature and RH, other oxidants present in the atmosphere can have varying effects on HOMs. Different oxidants may react with HOMs through different pathways, leading to diverse chemical transformations and alterations in their abundance and properties. For example, the presence of NO2 suppressed the formation of HOM dimers.82 Initially, NO promoted the generation of HOMs, but these molecules were further inhibited by the increase in NO concentration.83 Moreover, higher ozone concentration was found to be beneficial for HOM formation.53 However, the current capabilities of mass spectrometry techniques were limited to providing information solely on the molecular formula of HOMs. This limitation posed a challenge in studying the intricate formation mechanisms of numerous atmospheric precursors. To overcome this hurdle, it was essential to explore different experimental conditions and develop novel analytical techniques that can enable rapid and comprehensive characterization of HOMs.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
YTZ and SMS designed the experiments. YTZ performed the ozonolysis experiments. YTZ and VGS carried out the data analysis. YTZ, VGS, and SMS wrote the manuscript. SMS acquired the funding and supervised YTZ.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research under the award number OSR-2019-CRG7-4077. The authors also acknowledge the experimental facilities at CCRC and Core Labs at KAUST.References
- S. F. Maria, L. M. Russell, M. K. Gilles and S. C. Myneni, Organic aerosol growth mechanisms and their climate-forcing implications, Science, 2004, 306(5703), 1921–1924 CAS.
- M. Kanakidou, J. Seinfeld, S. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. Van Dingenen, B. Ervens, A. Nenes and C. Nielsen, Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 2005, 5(4), 1053–1123 CAS.
- M. Andreae and D. Rosenfeld, Aerosol–cloud–precipitation interactions. Part 1. The nature and sources of cloud-active aerosols, Earth-Sci. Rev., 2008, 89(1–2), 13–41 Search PubMed.
- P. S. Monks, C. Granier, S. Fuzzi, A. Stohl, M. L. Williams, H. Akimoto, M. Amann, A. Baklanov, U. Baltensperger and I. Bey, Atmospheric composition change–global and regional air quality, Atmos. Environ., 2009, 43(33), 5268–5350 CAS.
- U. Pöschl, Atmospheric aerosols: composition, transformation, climate and health effects, Angew. Chem., Int. Ed., 2005, 44(46), 7520–7540 Search PubMed.
- F. Laden, L. M. Neas, D. W. Dockery and J. Schwartz, Association of fine particulate matter from different sources with daily mortality in six US cities, Environ. Health Perspect., 2000, 108(10), 941–947 CAS.
- K. Ito, W. F. Christensen, D. J. Eatough, R. C. Henry, E. Kim, F. Laden, R. Lall, T. V. Larson, L. Neas and P. K. Hopke, PM source apportionment and health effects: 2. An investigation of intermethod variability in associations between source-apportioned fine particle mass and daily mortality in Washington, DC, J. Exposure Sci. Environ. Epidemiol., 2006, 16(4), 300–310 CAS.
- C. Billionnet, D. Sherrill and I. Annesi-Maesano, Estimating the health effects of exposure to multi-pollutant mixture, Ann. Epidemiol., 2012, 22(2), 126–141 Search PubMed.
- K. Y. Kondratyev, L. S. Ivlev, V. F. Krapivin and C. A. Varostos, Atmospheric Aerosol Properties: Formation, Processes and Impacts, Springer Science & Business Media, 2006 Search PubMed.
- I. Riipinen, T. Yli-Juuti, J. R. Pierce, T. Petäjä, D. R. Worsnop, M. Kulmala and N. M. Donahue, The contribution of organics to atmospheric nanoparticle growth, Nat. Geosci., 2012, 5(7), 453–458 CrossRef CAS.
- A. L. Robinson, N. M. Donahue, M. K. Shrivastava, E. A. Weitkamp, A. M. Sage, A. P. Grieshop, T. E. Lane, J. R. Pierce and S. N. Pandis, Rethinking organic aerosols: Semivolatile emissions and photochemical aging, Science, 2007, 315(5816), 1259–1262 CrossRef CAS PubMed.
- A. Calvo, C. Alves, A. Castro, V. Pont, A. Vicente and R. Fraile, Research on aerosol sources and chemical composition: Past, current and emerging issues, Atmos. Res., 2013, 120, 1–28 CrossRef.
- A. H. Goldstein and I. E. Galbally, Known and unexplored organic constituents in the earth's atmosphere, Environ. Sci. Technol., 2007, 41(5), 1514–1521 CrossRef CAS PubMed.
- B. H. Lee, C. Mohr, F. D. Lopez-Hilfiker, A. Lutz, M. Hallquist, L. Lee, P. Romer, R. C. Cohen, S. Iyer and T. Kurtén, Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(6), 1516–1521 CrossRef CAS PubMed.
- N. M. Donahue, J. Kroll, S. N. Pandis and A. L. Robinson, A two-dimensional volatility basis set–Part 2: Diagnostics of organic-aerosol evolution, Atmos. Chem. Phys., 2012, 12(2), 615–634 CrossRef CAS.
- E. Saukko, A. Lambe, P. Massoli, T. Koop, J. Wright, D. Croasdale, D. Pedernera, T. Onasch, A. Laaksonen and P. Davidovits, Humidity-dependent phase state of SOA particles from biogenic and anthropogenic precursors, Atmos. Chem. Phys., 2012, 12(16), 7517–7529 CrossRef CAS.
- M. Ehn, E. Kleist, H. Junninen, T. Petäjä, G. Lönn, S. Schobesberger, M. Dal Maso, A. Trimborn, M. Kulmala and D. Worsnop, Gas phase formation of extremely oxidized pinene reaction products in chamber and ambient air, Atmos. Chem. Phys., 2012, 12(11), 5113–5127 CrossRef CAS.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. Donahue, C. George and A. Goldstein, The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 2009, 9(14), 5155–5236 CrossRef CAS.
- J. Tröstl, W. K. Chuang, H. Gordon, M. Heinritzi, C. Yan, U. Molteni, L. Ahlm, C. Frege, F. Bianchi and R. Wagner, The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 2016, 533(7604), 527–531 CrossRef PubMed.
- J. Kirkby, J. Duplissy, K. Sengupta, C. Frege, H. Gordon, C. Williamson, M. Heinritzi, M. Simon, C. Yan and J. Almeida, Ion-induced nucleation of pure biogenic particles, Nature, 2016, 533(7604), 521–526 CrossRef CAS PubMed.
- Y. Guo, H. Shen, I. Pullinen, H. Luo, S. Kang, L. Vereecken, H. Fuchs, M. Hallquist, I.-H. Acir and R. Tillmann, Identification of highly oxygenated organic molecules and their role in aerosol formation in the reaction of limonene with nitrate radical, Atmos. Chem. Phys., 2022, 22(17), 11323–11346 CrossRef CAS.
- X. Zhang, A. T. Lambe, M. A. Upshur, W. A. Brooks, A. Gray Bé, R. J. Thomson, F. M. Geiger, J. D. Surratt, Z. Zhang and A. Gold, Highly oxygenated multifunctional compounds in α-pinene secondary organic aerosol, Environ. Sci. Technol., 2017, 51(11), 5932–5940 CrossRef CAS PubMed.
- V.-M. Kerminen, M. Paramonov, T. Anttila, I. Riipinen, C. Fountoukis, H. Korhonen, E. Asmi, L. Laakso, H. Lihavainen and E. Swietlicki, Cloud condensation nuclei production associated with atmospheric nucleation: a synthesis based on existing literature and new results, Atmos. Chem. Phys., 2012, 12(24), 12037–12059 CrossRef CAS.
- T. Jokinen, T. Berndt, R. Makkonen, V.-M. Kerminen, H. Junninen, P. Paasonen, F. Stratmann, H. Herrmann, A. B. Guenther and D. R. Worsnop, Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(23), 7123–7128 CrossRef CAS.
- A. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. Emmons and X. Wang, The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2. 1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 2012, 5(6), 1471–1492 Search PubMed.
- L. Morawska, C. He, G. Johnson, H. Guo, E. Uhde and G. Ayoko, Ultrafine particles in indoor air of a school: possible role of secondary organic aerosols, Environ. Sci. Technol., 2009, 43(24), 9103–9109 Search PubMed.
- S. Kundu, R. Fisseha, A. L. Putman, T. A. Rahn and L. R. Mazzoleni, High molecular weight SOA formation during limonene ozonolysis: insights from ultrahigh-resolution FT-ICR mass spectrometry characterization, Atmos. Chem. Phys., 2012, 12(12), 5523–5536 CrossRef CAS.
- M. L. Walser, Y. Desyaterik, J. Laskin, A. Laskin and S. A. Nizkorodov, High-resolution mass spectrometric analysis of secondary organic aerosol produced by ozonation of limonene, Phys. Chem. Chem. Phys., 2008, 10(7), 1009–1022 RSC.
- M. P. Rissanen, T. Kurten, M. Sipila, J. A. Thornton, J. Kangasluoma, N. Sarnela, H. Junninen, S. Jørgensen, S. Schallhart and M. K. Kajos, The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene, J. Am. Chem. Soc., 2014, 136(44), 15596–15606 CrossRef CAS PubMed.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipilä, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann and B. Lee, A large source of low-volatility secondary organic aerosol, Nature, 2014, 506(7489), 476–479 CrossRef CAS.
- M. Wang, D. Chen, M. Xiao, Q. Ye, D. Stolzenburg, V. Hofbauer, P. Ye, A. L. Vogel, R. L. Mauldin III and A. Amorim, Photo-oxidation of aromatic hydrocarbons produces low-volatility organic compounds, Environ. Sci. Technol., 2020, 54(13), 7911–7921 CrossRef CAS PubMed.
- H. Tong, Y. Zhang, A. Filippi, T. Wang, C. Li, F. Liu, D. Leppla, I. Kourtchev, K. Wang and H.-M. Keskinen, Radical formation by fine particulate matter associated with highly oxygenated molecules, Environ. Sci. Technol., 2019, 53(21), 12506–12518 CrossRef CAS PubMed.
- M. Schervish and N. M. Donahue, Peroxy radical chemistry and the volatility basis set, Atmos. Chem. Phys., 2020, 20(2), 1183–1199 CrossRef CAS.
- R. V. Otkjær, H. H. Jakobsen, C. M. Tram and H. G. Kjaergaard, Calculated hydrogen shift rate constants in substituted alkyl peroxy radicals, J. Phys. Chem. A, 2018, 122(43), 8665–8673 CrossRef PubMed.
- S. Kundu, R. Fisseha, A. L. Putman, T. A. Rahn and L. R. Mazzoleni, Molecular formula composition of β-caryophyllene ozonolysis SOA formed in humid and dry conditions, Atmos. Environ., 2017, 154, 70–81 CrossRef CAS.
- J. Merikanto, D. Spracklen, G. Mann, S. Pickering and K. Carslaw, Impact of nucleation on global CCN, Atmos. Chem. Phys., 2009, 9(21), 8601–8616 CrossRef CAS.
- S. Hyvönen, H. Junninen, L. Laakso, M. Dal Maso, T. Grönholm, B. Bonn, P. Keronen, P. Aalto, V. Hiltunen and T. Pohja, A look at aerosol formation using data mining techniques, Atmos. Chem. Phys., 2005, 5(12), 3345–3356 CrossRef.
- M. Boy and M. Kulmala, Nucleation events in the continental boundary layer: Influence of physical and meteorological parameters, Atmos. Chem. Phys., 2002, 2(1), 1–16 CrossRef CAS.
- J. L. Jimenez, M. Canagaratna, N. Donahue, A. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe and N. Ng, Evolution of organic aerosols in the atmosphere, Science, 2009, 326(5959), 1525–1529 CrossRef CAS PubMed.
- T. B. Nguyen, P. B. Lee, K. M. Updyke, D. L. Bones, J. Laskin, A. Laskin and S. A. Nizkorodov, Formation of nitrogen-and sulfur-containing light-absorbing compounds accelerated by evaporation of water from secondary organic aerosols, J. Geophys. Res.:Atmos., 2012, 117, D01207 Search PubMed.
- C. Wong, D. Vite and S. A. Nizkorodov, Stability of α-Pinene and d-Limonene Ozonolysis Secondary Organic Aerosol Compounds Toward Hydrolysis and Hydration, ACS Earth Space Chem., 2021, 5(10), 2555–2564 CAS.
- P. Venkatachari and P. K. Hopke, Characterization of products formed in the reaction of ozone with α-pinene: case for organic peroxides, J. Environ. Monit., 2008, 10(8), 966–974 CAS.
- K. J. Heaton, R. L. Sleighter, P. G. Hatcher, W. A. Hall IV and M. V. Johnston, Composition domains in monoterpene secondary organic aerosol, Environ. Sci. Technol., 2009, 43(20), 7797–7802 CAS.
- M. Duncianu, R. I. Olariu, V. r. Riffault, N. Visez, A. Tomas and P. Coddeville, Development of a new flow reactor for kinetic studies. Application to the ozonolysis of a series of alkenes, J. Phys. Chem. A, 2012, 116(24), 6169–6179 CAS.
- B. Witkowski and T. Gierczak, Early stage composition of SOA produced by α-pinene/ozone reaction: α-Acyloxyhydroperoxy aldehydes and acidic dimers, Atmos. Environ., 2014, 95, 59–70 CAS.
- N. M. Czoschke, M. Jang and R. M. Kamens, Effect of acidic seed on biogenic secondary organic aerosol growth, Atmos. Environ., 2003, 37(30), 4287–4299 CAS.
- M. P. Tolocka, K. J. Heaton, M. A. Dreyfus, S. Wang, C. A. Zordan, T. D. Saul and M. V. Johnston, Chemistry of particle inception and growth during α-pinene ozonolysis, Environ. Sci. Technol., 2006, 40(6), 1843–1848 CAS.
- K. Kristensen, L. Jensen, M. Glasius and M. Bilde, The effect of sub-zero temperature on the formation and composition of secondary organic aerosol from ozonolysis of alpha-pinene, Environ. Sci.:Processes Impacts, 2017, 19(10), 1220–1234 CAS.
- R. Atkinson, A. Winer and J. Pitts Jr, Rate constants for the gas phase reactions of O3 with the natural hydrocarbons isoprene and α-and β-pinene, Atmos. Environ., 1982, 16(5), 1017–1020 CAS.
- M. Keywood, J. Kroll, V. Varutbangkul, R. Bahreini, R. Flagan and J. Seinfeld, Secondary organic aerosol formation from cyclohexene ozonolysis: Effect of OH scavenger and the role of radical chemistry, Environ. Sci. Technol., 2004, 38(12), 3343–3350 CAS.
- K. S. Docherty and P. J. Ziemann, Effects of stabilized criegee intermediate and OH radical scavengers on aerosol formation from reactions of β-pinene with O3, Aerosol Sci. Technol., 2003, 37(11), 877–891 CAS.
- K. Kristensen, Å. K. Watne, J. Hammes, A. Lutz, T. Petäjä, M. Hallquist, M. Bilde and M. Glasius, High-molecular weight dimer esters are major products in aerosols from α-pinene ozonolysis and the boreal forest, Environ. Sci. Technol. Lett., 2016, 3(8), 280–285 CAS.
- D. Liu, Y. Zhang, S. Zhong, S. Chen, Q. Xie, D. Zhang, Q. Zhang, W. Hu, J. Deng and L. Wu, Large differences of highly oxygenated organic molecules (HOMs) and low volatile species in SOA formed from ozonolysis of β-pinene and limonene, Atmos. Chem. Phys., 2023, 23(14), 8383–8402 CAS.
- E. Kendrick, A mass scale based on CH2=14.0000 for high resolution mass spectrometry of organic compounds, Anal. Chem., 1963, 35(13), 2146–2154 CAS.
- J. H. Kroll, N. M. Donahue, J. L. Jimenez, S. H. Kessler, M. R. Canagaratna, K. R. Wilson, K. E. Altieri, L. R. Mazzoleni, A. S. Wozniak and H. Bluhm, Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 2011, 3(2), 133–139 CrossRef CAS PubMed.
- Y. Zhang, K. Wang, H. Tong, R. J. Huang and T. Hoffmann, The maximum carbonyl ratio (MCR) as a new index for the structural classification of secondary organic aerosol components, Rapid Commun. Mass Spectrom., 2021, 35(14), e9113 CrossRef CAS PubMed.
- N. M. Donahue, A. L. Robinson and S. N. Pandis, Atmospheric organic particulate matter: From smoke to secondary organic aerosol, Atmos. Environ., 2009, 43(1), 94–106 CrossRef CAS.
- N. M. Donahue, S. Epstein, S. N. Pandis and A. L. Robinson, A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 2011, 11(7), 3303–3318 CrossRef CAS.
- Y. Li, U. Pöschl and M. Shiraiwa, Molecular corridors and parameterizations of volatility in the chemical evolution of organic aerosols, Atmos. Chem. Phys., 2016, 16(5), 3327–3344 CrossRef CAS.
- S. A. Epstein, I. Riipinen and N. M. Donahue, A semiempirical correlation between enthalpy of vaporization and saturation concentration for organic aerosol, Environ. Sci. Technol., 2010, 44(2), 743–748 CrossRef CAS PubMed.
- P. Tu, W. A. t. Hall and M. V. Johnston, Characterization of Highly Oxidized Molecules in Fresh and Aged Biogenic Secondary Organic Aerosol, Anal. Chem., 2016, 88(8), 4495–4501 CrossRef CAS PubMed.
- A. L. Vogel, J. Schneider, C. Müller-Tautges, T. Klimach and T. Hoffmann, Aerosol chemistry resolved by mass spectrometry: Insights into particle growth after ambient new particle formation, Environ. Sci. Technol., 2016, 50(20), 10814–10822 CrossRef CAS PubMed.
- O. Horie and G. Moortgat, Decomposition pathways of the excited Criegee intermediates in the ozonolysis of simple alkenes, Atmos. Environ., Part A, 1991, 25(9), 1881–1896 CrossRef.
- X. Li, S. Chee, J. Hao, J. P. Abbatt, J. Jiang and J. N. Smith, Relative humidity effect on the formation of highly oxidized molecules and new particles during monoterpene oxidation, Atmos. Chem. Phys., 2019, 19(3), 1555–1570 CAS.
- Å. Jonsson, M. Hallquist and E. Ljungström, The effect of temperature and water on secondary organic aerosol formation from ozonolysis of limonene, Δ 3-carene and α-pinene, Atmos. Chem. Phys., 2008, 8(21), 6541–6549 CrossRef.
- H. Saathoff, K.-H. Naumann, O. Möhler, Å. M. Jonsson, M. Hallquist, A. Kiendler-Scharr, T. F. Mentel, R. Tillmann and U. Schurath, Temperature dependence of yields of secondary organic aerosols from the ozonolysis of α-pinene and limonene, Atmos. Chem. Phys., 2009, 9(5), 1551–1577 CrossRef CAS.
- C. Denjean, P. Formenti, B. Picquet-Varrault, M. Camredon, E. Pangui, P. Zapf, Y. Katrib, C. Giorio, A. Tapparo and B. Temime-Roussel, Aging of secondary organic aerosol generated from the ozonolysis of α-pinene: effects of ozone, light and temperature, Atmos. Chem. Phys., 2015, 15(2), 883–897 CrossRef.
- H. Zhang, J. Surratt, Y. Lin, J. Bapat and R. Kamens, Effect of relative humidity on SOA formation from isoprene/NO photooxidation: enhancement of 2-methylglyceric acid and its corresponding oligoesters under dry conditions, Atmos. Chem. Phys., 2011, 11(13), 6411–6424 CrossRef CAS.
- M. Shrivastava, C. D. Cappa, J. Fan, A. H. Goldstein, A. B. Guenther, J. L. Jimenez, C. Kuang, A. Laskin, S. T. Martin and N. L. Ng, Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 2017, 55(2), 509–559 CrossRef.
- X. Zhang, R. C. McVay, D. D. Huang, N. F. Dalleska, B. Aumont, R. C. Flagan and J. H. Seinfeld, Formation and evolution of molecular products in α-pinene secondary organic aerosol, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(46), 14168–14173 CrossRef CAS PubMed.
- A. P. Bateman, S. A. Nizkorodov, J. Laskin and A. Laskin, Time-resolved molecular characterization of limonene/ozone aerosol using high-resolution electrospray ionization mass spectrometry, Phys. Chem. Chem. Phys., 2009, 11(36), 7931–7942 RSC.
- P. E. Sheehan and F. M. Bowman, Estimated effects of temperature on secondary organic aerosol concentrations, Environ. Sci. Technol., 2001, 35(11), 2129–2135 CrossRef CAS PubMed.
- D. J. Price, M. Kacarab, D. R. Cocker, K. L. Purvis-Roberts and P. J. Silva, Effects of temperature on the formation of secondary organic aerosol from amine precursors, Aerosol Sci. Technol., 2016, 50(11), 1216–1226 CrossRef CAS.
- T. B. Nguyen, P. J. Roach, J. Laskin, A. Laskin and S. A. Nizkorodov, Effect of humidity on the composition of isoprene photooxidation secondary organic aerosol, Atmos. Chem. Phys., 2011, 11(14), 6931–6944 CrossRef CAS.
- P. J. Ziemann and R. Atkinson, Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 2012, 41(19), 6582–6605 RSC.
- S. Tomaz, D. Wang, N. Zabalegui, D. Li, H. Lamkaddam, F. Bachmeier, A. Vogel, M. E. Monge, S. Perrier and U. Baltensperger, Structures and reactivity of peroxy radicals and dimeric products revealed by online tandem mass spectrometry, Nat. Commun., 2021, 12(1), 300 CrossRef CAS PubMed.
- O. Peräkylä, M. Riva, L. Heikkinen, L. Quéléver, P. Roldin and M. Ehn, Experimental investigation into the volatilities of highly oxygenated organic molecules (HOMs), Atmos. Chem. Phys., 2020, 20(2), 649–669 CrossRef.
- T. Jokinen, M. Sipilä, S. Richters, V. M. Kerminen, P. Paasonen, F. Stratmann, D. Worsnop, M. Kulmala, M. Ehn and H. Herrmann, Rapid autoxidation forms highly oxidized RO2 radicals in the atmosphere, Angew. Chem., Int. Ed., 2014, 53(52), 14596–14600 CAS.
- T. Berndt, W. Scholz, B. Mentler, L. Fischer, H. Herrmann, M. Kulmala and A. Hansel, Accretion product formation from self-and cross-reactions of RO2 radicals in the atmosphere, Angew. Chem., Int. Ed., 2018, 57(14), 3820–3824 CrossRef CAS PubMed.
- M. S. Claflin, J. E. Krechmer, W. Hu, J. L. Jimenez and P. J. Ziemann, Functional group composition of secondary organic aerosol formed from ozonolysis of α-pinene under high VOC and autoxidation conditions, ACS Earth Space Chem., 2018, 2(11), 1196–1210 CrossRef CAS.
- F. Jacob, N. Houzel, P. Genevray, C. Clety, C. Coeur, E. Perdrix, L. Y. Alleman, S. Anthérieu, G. Garcon, G. Dhont and A. Cuisset, New insights into the chemical composition and formation mechanisms of secondary organic aerosols produced in the ozonolysis of limonene, J. Aerosol Sci., 2023, 173, 106214 CrossRef CAS.
- M. P. Rissanen, NO2 suppression of autoxidation–inhibition of gas-phase highly oxidized dimer product formation, ACS Earth Space Chem., 2018, 2(11), 1211–1219 CrossRef CAS PubMed.
- Z. Wang, M. Ehn, M. P. Rissanen, O. Garmash, L. Quéléver, L. Xing, M. Monge-Palacios, P. Rantala, N. M. Donahue and T. Berndt, Efficient alkane oxidation under combustion engine and atmospheric conditions, Commun. Chem., 2021, 4(1), 18 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ea00153b |
| This journal is © The Royal Society of Chemistry 2025 |