
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Macrocyclic transition-metal parashift complexes for MRI at clinical and pre-clinical magnetic fields†
Nicola J.
Rogers
 *a,
Chowan
Ashok Kumar
a,
Carlson
Alexander
a,
Daniel
Bowdery
b,
Galina
Pavlovskaya
c and
Peter
Harvey
cd
*a,
Chowan
Ashok Kumar
a,
Carlson
Alexander
a,
Daniel
Bowdery
b,
Galina
Pavlovskaya
c and
Peter
Harvey
cd
aDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong. E-mail: nicolarogers@hkbu.edu.hk
bDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK
cSir Peter Mansfield Imaging Centre, School of Medicine, University of Nottingham, University Park, Nottingham NG7 2RD, UK
dSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK
First published on 25th June 2025
Abstract
A series of macrocyclic transition-metal complexes, including Fe(II), Co(II), Ni(II), and Cu(II) complexes, have been evaluated for parashift MRI imaging applications, by assessing their paramagnetic NMR properties, including proton chemical shifts, nuclear relaxation rates, and any exchange dynamics in solution, at magnetic fields strengths relevant to clinical and pre-clinical imaging. Among the complexes studied, Fe(II) and Co(II) systems demonstrated significant paramagnetic shifts with desirable relaxation properties, making them potential candidates for lanthanide-free parashift molecular probes for MRI. Field-dependent nuclear relaxation rate analyses provided insights into electronic relaxation times, confirming the suitability of certain complexes for parashift imaging at lower magnetic fields. Phantom imaging experiments at 9.4 T further validated the feasibility of molecular imaging using a cyclen-based macrocyclic Fe(II) complex, making a significant advance toward developing transition metal-based MRI probes using biogenic metal ions, and offer promise for future responsive imaging due to the direct signal detection.
1. Introduction
Contrast enhanced MRI has revolutionised radiology and diagnostics over the last four decades, particularly for imaging tumour tissue and vascular diseases.1–3 More recently there has been a drive towards molecular imaging agents to complement current progress towards personalised medicine. Whilst conventional MRI contrast agents enable visualisation of pathologies, usually due to residing in blood pools and highlighting vascular structures, parashift probes (i.e. metal complexes that can be detected directly due to a paramagnetically-shifted proton resonance that is shifted outside of the diamagnetic window of the endogenous signal) have great potential as molecular probes.Using magnetic resonance spectroscopy (MRS) imaging, parashift agents offer scope to monitor molecular processes in the body and encode molecular-level information i.e. a calibrated response to specific biomolecules or physiological conditions, and to map and quantify biochemical changes associated with disease. The speciation of the molecular probe is reported by a signature chemical shift that is independent of probe concentration.
Lanthanide-containing 1H NMR parashift agents have been developed previously,4–9 with limits of detection possible in the 10 micromolar range, and acquisition times of just 1 minute per image.7 Despite reducing the lanthanide dose for the direct-detection method down to 0.04 mmol kg−1 (compared with 0.1 mmol kg−1 dosing for standard indirect gadolinium MRI contrast agents), it is desirable to replace the xenobiotic lanthanide ions altogether. Transition metal complexes with magnetic anisotropy offer attractive biogenic alternatives for parashift agents10 because essential ‘trace’ metals including cobalt can be metabolised, and hence better long-term toxicity profiles are envisaged.
First row transition metal ions have a long history of being used as shift agents (e.g. in metalloprotein work11), and whilst they have comparatively smaller magnetic moments than Ln(III) ions, shifts of up to 400 ppm have been observed, as well as significant relaxation enhancement effects.12 Transition metal complexes have been developed for paraCEST (paramagnetic chemical exchange saturation transfer) imaging in order to shift the exchangeable proton signal far from the exchanging pool of water protons to access CEST signals from protons with more rapid exchange, and enable the design of responsive systems.10,13–16 Paramagnetic transition metal complexes with low-lying excited states (ΔE ∼ kBT) are required for parashift probes, where magnetic susceptibility anisotropies can be quite large (giving large pseudocontact shifts) and Orbach electronic relaxation mechanisms can operate efficiently (otherwise long electronic relaxation times will shorten T1 and T2 too much for imaging).12
To optimise the detection sensitivity of a parashift agent, a large, sharp NMR signal is desired (i.e. several isochronous protons) that is shifted outside of the physiological diamagnetic NMR window for direct detection. The overall rigidity and symmetry of a parashift agent is therefore an important design criterion, as a single NMR environment is desired for the reporting protons, not a range of conformational isomers, as exchange broadening can arise from dynamic interconversions between conformers on the NMR timescale.
Fast longitudinal relaxation (short T1) of the NMR reporter group is also a sensitivity requirement (i.e. that the proton signal returns to thermal equilibrium quickly) so that fast imaging pulses can be used to enhance sensitivity;17 similar strategies are also used to enhance the sensitivity of 19F contrast agents.18–20 Fast imaging sequences require wide spectral bandwidths (>10 kHz),7 however, and therefore highly shifted signals (±40 ppm at 3 T) are also a sensitivity requirement to avoid signal overlap with endogenous protons.7,21 The sensitivity of a parashift probe is also determined by the transverse signal decay (T2), which reduces the overall signal-to-noise (SNR) due to line broadening, and hence a T2/T1 ratio close to unity is desired.7,21
Metal ions with a relatively short electronic relaxation time (i.e. <10 ps) are required to avoid significant T2-broadening, such as high-spin pseudo-octahedral Fe(II) and Co(II) complexes.12,22 The contact contributions (which are quite reasonably ignored for Ln(III) systems) can be significant in d-block complexes, even for nuclei several bonds away from the metal centre.23 In addition, some Co(II) complexes exhibit slightly longer electronic relaxation times (T1E = 1–10 ps) than most of the Ln(III) shift ions, which influences the dipolar relaxation mechanism that is important at the lower (clinically relevant) magnetic fields.12
In pursuit of lanthanide-free parashift agents for imaging at clinical (i.e. 1.5–3 T) and pre-clinical (i.e. 7–9.4 T) imaging fields, we have studied a set of transition metal complexes (Scheme 1). We evaluate the paramagnetic shifts and nuclear relaxation rates of protons within each complex at variable magnetic field strengths to aid future probe design.
 | ||
| Scheme 1 Schematic of model transition metal parashift complexes. | ||
2. Results and discussion
A series of water-soluble complexes [using Fe(II), Co(II), Ni(II), and Cu(II) chloride salts] has been prepared to evaluate their paramagnetic NMR properties at a range of magnetic fields (Fig. 1). Macrocyclic ligands bearing picolyl coordinating pendants were synthesised by alkylation of 1,4,7-triazacyclononane (TACN) or 1,4,7,10-tetraazacyclododecane (cyclen) with either 2-bromomethyl-6-methylpyridine,24 or (5-tert-butylpyridin-2-yl)methyl methanesulfonate.5 Divalent transition metal complexes were formed using the respective dichloride salt, and for comparison the diamagnetic Zn(II) analogues ([ZnL1a](ClO4)2 and [ZnL2a](ClO4)2) were synthesised using the perchlorate salt, as confirmed by high-resolution mass spectrometry (ESI). Macrocyclic ligands were chosen to impart thermodynamic stability and kinetic inertness with these labile first-row transition metal ions, and nitrogen donors were incorporated to favour the divalent high-spin states. Morrow and co-workers have previously reported the Fe(II) and Co(II) complexes of L1a and L2a, and found that the 6-methyl group on the picolyl unit is important for imparting rigidity into these structures; sharp signals were observed in the NMR spectra at 11.7 T.24 The 6-methyl group also facilitates the stabilisation of the 2+ oxidation states with L1a and the high spin states required for the desired paramagnetic shift and relaxation properties, due to the steric demand of the three intertwined methyl groups inducing M–Npy elongation, in the M–N bond lengths determined by X-ray crystallography vs. unsubstituted tripicolyl-TACN.24,25 | ||
| Fig. 1 1H NMR spectra of [ML1a]2+ (a) and [ML2a]2+ (b) where (M = Fe(II), Co(II), Ni(II), Cu(II)) at 60 MHz (1.4 T, 299 K) in D2O. Pyridyl proton resonances indicated by *. | ||
In this study, we have expanded the series to include Ni(II) and Cu(II), and assessed paramagnetic NMR properties and the conformational dynamics of the complexes in solution, at clinically relevant magnetic fields. We have also synthesised ligand frameworks with tert-butyl groups in the 5-position on the pyridyl unit (L1b and L2b) to assess whether the steric bulk at the 5-position could impart rigidity, and whether the protons of the tert-butyl could experience an enhanced paramagnetic shift at this distance (ca. 6.5 Å)5 from the metal centre.
2.1 Solution-state NMR and solid-state structures
The 1H NMR spectra of complexes of L1a and L2a were recorded at 1.4 T in D2O for M = Fe(II), Co(II), Ni(II), and Cu(II) (Fig. 1) and 9.4 T (see ESI, Fig. S83†), i.e. typical clinical and pre-clinical imaging fields respectively. The spectra were recorded using a large sweep width and short acquisition time as the paramagnetic complexes contain resonances that extend well beyond the conventional diamagnetic range and relax back to thermal equilibrium quickly. The 1H NMR spectra contain ten discernible resonances for compounds [FeL1a]2+and [CoL1a]2+, indicative of a single diastereomer in solution in each case, with high-spin dicationic metal centres, rigid structures, and three-fold symmetry. The 1H NMR spectrum of [NiL1a]2+ contains broad resonances (observable in the +220 to −20 ppm range), and that of [CuL1a]2+ contains further broadened resonances evident in the range +40 to −20 ppm. For [CoL1a]2+ and [FeL1a]2+ the pyridyl protons in the 1H NMR spectra were assigned in accordance with Morrow et al.24 (see * Fig. 1); if we assume that the relaxation rates follow pseudocontact Bloch–Redfield–Wangsness theory, such that R1 rates decrease steeply with the distance, r, from the paramagnetic centre (i.e. R1 is proportional to r−6) the three pyridyl protons are expected to be the sharpest signals in all complexes, with the slowest longitudinal relaxation rates, as they are furthest from the paramagnetic metal centre. The methyl peak was identified by its relative signal intensity.
The methyl resonance appears most shifted from the diamagnetic region for FeL1a (at +22 ppm, 299 K), with R1 = 290 ± 5 Hz and R2 = 440 ± 40 Hz at 1.4 T, and R1 = 356 ± 1 Hz and R2 = 600 ± 30 Hz at 9.4 T.
The manganese complex, [MnL1a]Cl2 was also synthesised but its NMR spectra were broad and featureless (see ESI, Fig. S15†) owing to the long electronic relaxation times associated with Mn(II).22 The sample also appeared to oxidise in solution (D2O) within the NMR tube (formation of brown precipitate – presumably MnO2 – was observed at 24 h), suggesting that the complex is not kinetically stable in water to oxidation, owing to the lack of crystal field stabilisation of a d5 metal ion.
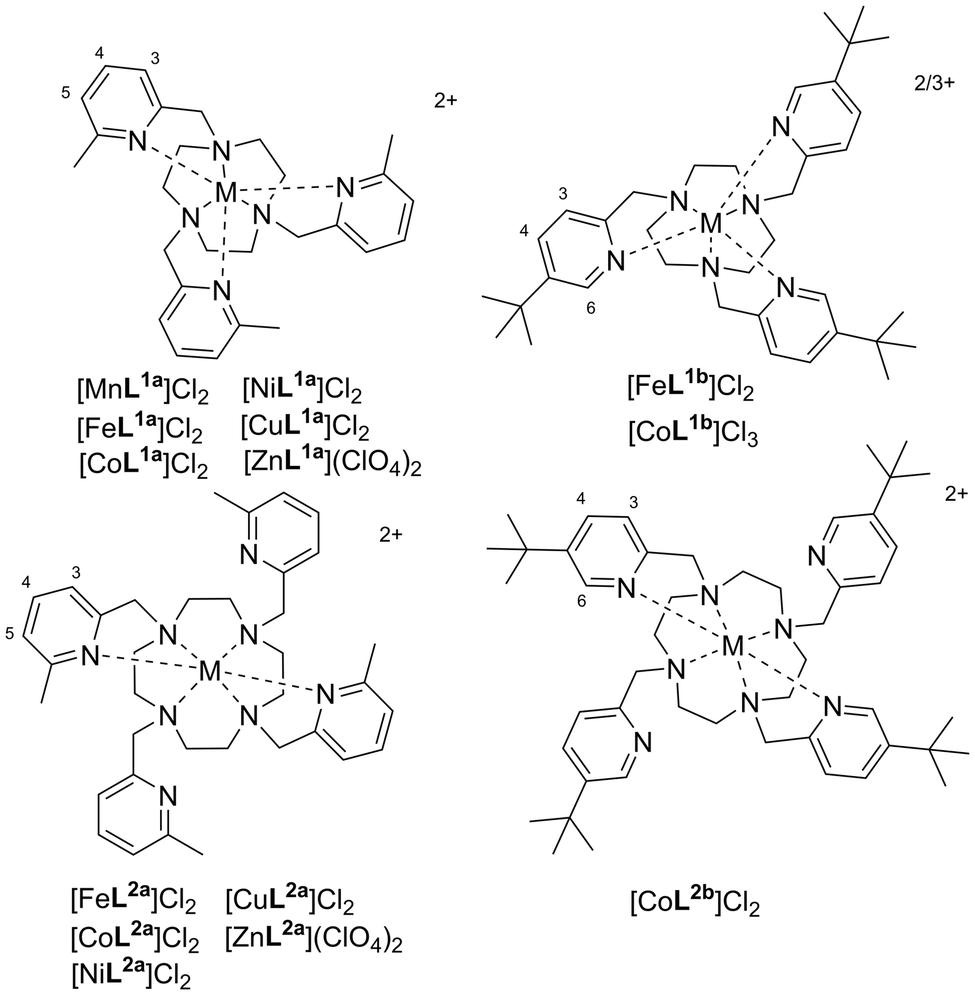
Crystallography data of the L1a complexes, including the previously reported structures of [FeL1a](CF3SO3)2 and [CoL1a](NO3)2,24 revealed six-coordinate metal centres with approximate C3-symmetry, containing an ‘N3TACN’ plane of coordinated nitrogen atoms of the macrocycle and an ‘N3Py’ plane of the three coordinated picolyl nitrogen atoms (Fig. 2). The pendant arms twist around the metal centre in a helical array (Δ or Λ configuration), whilst the macrocyclic chelate rings adopt cooperative helicity (either λλλ or δδδ).‡
 | ||
| Fig. 2 Structure of the Λ(δδδ) cationic complexes [ML1a]2+ both side-on and top-down (with ‘N3TACN’ plane highlighted in light blue spheres, ‘N3py’ plane highlighted with dark blue spheres) for (a) M = Zn2+, (b) M = Ni2+, and (c) M = Cu2+ (elongated axial Cu–N bonds are highlighted with spheres in the side-on view). H atoms and counterions removed for clarity. | ||
The diamagnetic [ZnL1a](ClO4)2 complex was also synthesised, and crystallised in the P21/c space group, with Δ(λλλ) and Λ(δδδ) enantiomers within the unit cell, and all Zn–N bond distances were 2.16–2.25 Å [Fig. 2(a)]. The complex geometry is best described as distorted octahedral, with average apical twist angles§ of 44.2(6)°. The [FeL1a]2+ and [CoL1a]2+ unit cells also contain analogous diastereomers with average twist angles of 43.6(8)° and 45.4(3)° respectively,24 although there are also two other conformations present in the [FeL1a]2+ structure, i.e. Δ(λλλ) with twist angle = 30.1(9)° and Λ(λλλ) with twist angle = 43.6(5)°, perhaps due to the crystal packing effects of neighbouring [Na(CF3SO3)4]3− anions.
Compounds [NiL1a](ClO4)2 and [CuL1a](ClO4)2 also crystallised in the P21/c space group, again with Δ(λλλ)and Λ(δδδ) enantiomers in the unit cell [Fig. 2(b) and (c)]. In the former case, the complex has distorted octahedral geometry with an average apical twist angle of 46.5(9)° and the Ni–N bonds were in the 2.10–2.21 Å range, whilst for the latter we observed distinct elongation in one axis, ascribed to Jahn–Teller distortion; four of the Cu–N bonds are in the 2.06–2.14 Å range, with two axially-related bonds at 2.24(1) Å and 2.47(1) Å and three apical twist angles of 41.5°, 44.3°, and 49.1°. The twist angles increase from Fe(II) to Ni(II) as the metal ion radius decreases across the period and varies to accommodate the tetragonal distortion for Cu(II).
The 1H NMR spectra of [FeL2a]2+ and [CoL2a]2+ contain 20 resonances, consistent with a single diastereomer of the previously reported C2-symmetry of the complexes in the solid state, in which the metal centres are six-coordinate with two trans-coordinated pyridine groups on the same face of the macrocycle and two uncoordinated pendant arms pointing away from the metal centre.24 The six-coordinate system is best described as a slightly distorted trigonal prism (unlike the distorted octahedra with L1a) and Λ(λλλλ)/Δ(δδδδ) helicity.24 The 1H NMR spectrum of [NiL2a]2+ is also consistent with a single diastereomer with average two-fold symmetry, whilst the 1H NMR spectra of [CuL2a]2+ is broadened significantly (like that of [CuL1a]2+). For the Fe(II) and Co(II) complexes, the CH3 resonances (from the coordinated picolyl unit) have greater linewidths for L2a complexes than L1a, whilst in general the resonances appear sharper in the spectra of [NiL2a]2+ than [NiL1a]2+. We were unsuccessful in growing crystals of [NiL2a]2+ suitable for crystallography, whilst a crystal structure of the protonated species [CuL2aH2](ClO4)4 (see ESI Fig. S6†) was obtained from the addition of Zn(ClO4)2 to [CuL2a]Cl2 in water, revealing a five-coordinate Cu(II) centre, in contrast to [FeL2a]2+ and [CoL2a]2+, although like the Fe(II) and Co(II) complexes two pyridines coordinate and two do not.
In the solid state we observe Δ(λλλ) and Λ(δδδ) enantiomers of [FeL1b]2+ in the crystal structure (Fig. S70, ESI†), with a larger twist angle of 47.9(5)° [cf. 43.6(8)°] and shorter Fe–N bonds of 1.98(2) Å [cf. 2.21–2.32 Å] than the [FeL1a]2+ complexes. The tert-butyl protons are ca. 6.5 Å from the metal centre but do not create enough steric bulk at the 5-position to favour high-spin Fe(II) or Co(II), even with the fac-oriented crowding of the three tert-butyl groups within the octahedral-type geometry of L1a.
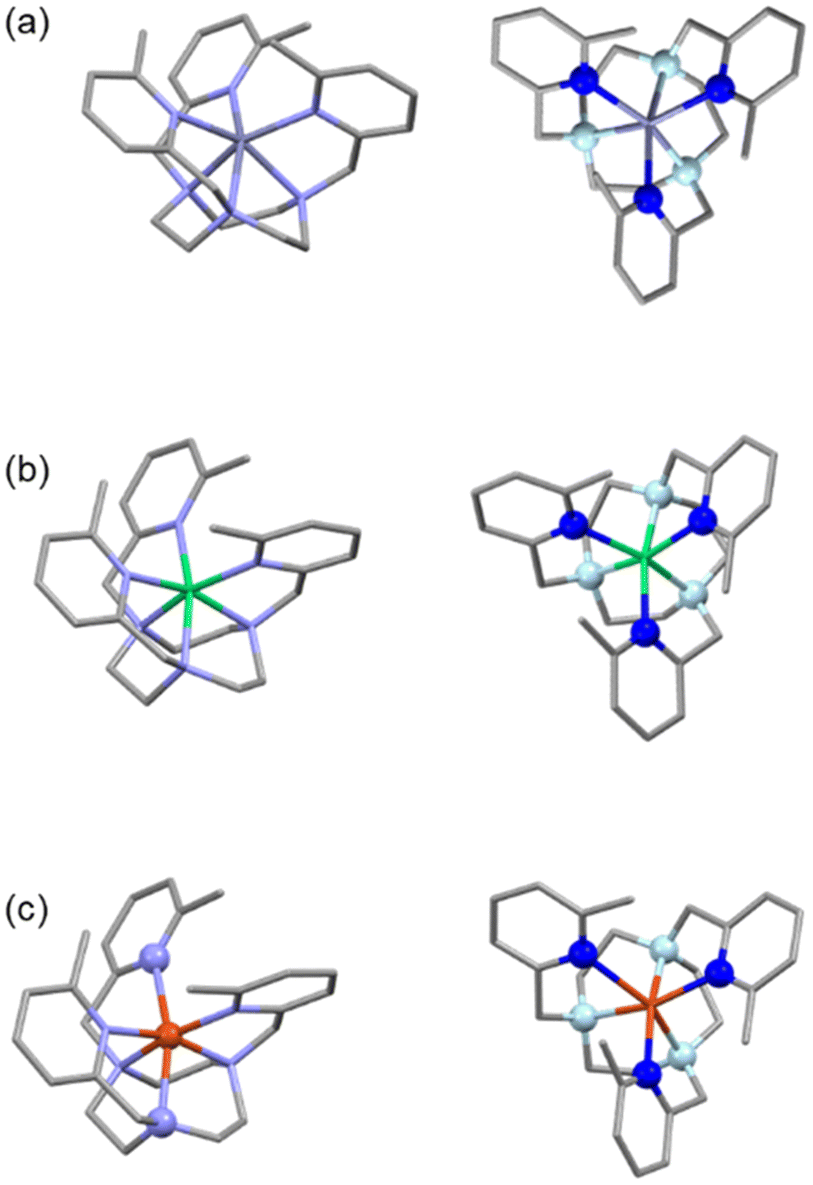
For complex [CoL2b]2+, unlike with the L1b ligand, the 1H NMR spectrum spans a +200 to −20 ppm range (Fig. 3a) and is indicative of a paramagnetic Co(II) metal centre, hence the larger cyclen ring favours Co2+ over the smaller Co3+ ion, as it cannot easily form the small-cavity octahedral donor environment that stabilised Co(III). However, the NMR spectral resonances are relatively broad with only half the number of signals as the L2a complex, consistent with a fluxional structure in solution with averaged four-fold symmetry on the NMR time scale, even at high field (500 MHz, 296 K), and a single tert-butyl resonance at −1 ppm. Variable temperature 1H NMR revealed signal sharpening with increased temperature (see ESI, Fig. S78†), as expected for a further increase in the exchange rates.
 | ||
| Fig. 3 Solution and solid-state structures of [CoL2b]2+. (a) 1H NMR (500 MHz, D2O, 296 K) of [CoL2b]Cl2 (inset showing zoomed in resonances in the +190 to +0 ppm region). (b) X-ray crystal structure of (b) [CoL2b]2+ and (c) [CoL2a]2+ for comparison (taken from ref. 24), H atoms and counterions removed for clarity. | ||
Vapour deposition of ethyl acetate into a methanolic solution of [CoL2b]Cl2 led to complex crystallisation in the P21/n space group with [CoCl4]2− counterions, as shown in Fig. 3(b). Unlike the crystal structure of CoL2a,24 [Fig. 3(c)] the four picolyl groups all point towards the Co(II) metal centre, with two sets of Co–Npy bond lengths of 2.18(1) and 3.2(1) Å. The unit cell contains Δ(δδδδ) and Λ(λλλλ) isomers, similar to [CoL2a]2+,24 but with a smaller twist angle around the C2 axis of 22.0(1)°, cf. 33.0(1)° for [CoL2a]2+. The six-coordinate system can be described as a distorted trigonal prism, similar to [CoL2a]2+, whereby the ‘pseudo-C3’-axis is perpendicular to the C2-symmetry axis of the molecule; apical twist angles of 28.2, 8.3, and 8.6° around the ‘pseudo-C3’-axis are similar to [CoL2a]2+ (cf. 18.6, 4.2, and 4.2°) but more distorted particularly at the two coordinated Npy atoms (i.e. the 28.2° twist angle).
2.2 Complex rigidity in solution
The diamagnetic analogues [ZnL1a](ClO4)2 and [ZnL2a](ClO4)2 were prepared in order to compare the conformational dynamics in solution by 1H NMR spectroscopy. The 1H NMR spectrum of [ZnL1a](ClO4)2 revealed narrow resonances at 9.4 T with clear J-coupling evident for the macrocycle ring protons (Fig. S35, ESI†), consistent with a rigid complex structure in (acetonitrile) solution, and formation of a single diastereomer; variable temperature 1H NMR (9.4 T) revealed no further species at 253 K, whilst slight broadening of the TACN ring and methylene protons was observed at 313 K and above, indicative of the ring and/or pendant arms wobbling at these elevated temperatures (see ESI, Fig. S37†).The 1H NMR spectrum of [ZnL2a](ClO4)2 shows exchange-broadening at room temperature at 9.4 T (see ESI, Fig. S61†), indicating less rigidity in the cyclen-based structures.
Although it is very common to observe exchange between the Δ(λλλλ)/Λ(δδδδ) and Λ(λλλλ)/Δ(δδδδ) diastereomeric configurations in lanthanide complexes based on cyclen macrocyclic frameworks,27 (due to ring inversion between δδδδ and λλλλ configurations as well as arm rotation that converts between Δ and Λ helicities) we do not observe major/minor conformational isomers in the 1H NMR spectra for the paramagnetic Fe(II)/Co(II) complexes of L1a; this exchange process would have to be incredibly fast to give rise to an average species in solution, rather than two sets of resonances at e.g. 9.4 T, with such large Δω differences expected between diastereomer proton pairs with the paramagnetic complexes.
Variable temperature 1H NMR spectra of [ZnL2a](ClO4)2 (at 9.4 T, see ESI, Fig. S63†) revealed one set of cyclen and pendent arm methylene protons at 253 K, and two types of pyridyl protons (one sharp and one broadened), consistent with a single structure with two-fold symmetry. Upon heating, the resonances broaden and coalesce to give a spectrum consistent with a four-fold-symmetric complex at 333 K, whilst at 1.4 T the NMR spectrum of [ZnL2a](ClO4)2 contains a single set of sharpened pyridyl signals at room temperature. Thus, we conclude that the L2a complexes exist with ‘on’–‘off’ dancing pendant arms, such that they are on average six-coordinate at all times at room temperature, whilst the complexes remain in their Δ(λλλλ) or Λ(δδδδ) preferred helicities, as a racemic mixture.
In the case of [CoL2b]2+ the tert-butyl groups are at the 5-position of the coordinating picolyl units (trans- to the methylene group that attaches to the macrocycle) and do not introduce steric hindrance to the pendent arm rotation as effectively as the methyl groups in the 6-position, and hence fast exchange dynamics are observed to give average four-fold symmetry in the paramagnetic 1H NMR; it is thus crucial that the picolyl pendants have substituents in the 6-position for these cyclen-based complexes, to render them sufficiently rigid in solution to give distinct NMR spectral resonances for use in parashift applications.
2.3 Evaluating the nuclear relaxation rates for MRI
The ideal transition metal parashift probe will contain reporting protons that appear as a single NMR-equivalent peak that is shifted significantly (ca. 50 ppm) so that the probe can be detected free of endogenous background proton signals. The relaxation rates of the reporter protons are key to optimising the SNR for detection of a parashift agent by MRI, by exploiting paramagnetic relaxation enhancement effects;7,21 fast longitudinal relaxation rates, R1 (i.e. T1−1) allow rapid pulsing when at the order of 100 Hz, such that the SNR can be increased by a factor of √R1 in a given scan duration. The sensitivity of a parashift probe is also influenced by the transverse relaxation rate, R2 (i.e. T2−1), which reduces the overall SNR due to line broadening (T2-losses during the echo time), and hence an R1/R2 ratio close to unity is desired. The rate at which the imaging can be performed is limited by the minimum possible repetition time (predominantly determined by the spectral width) and is also limited in vivo by the specific absorption rate of tissue, to prevent heating.28 Previously reported parashift imaging in vivo (preclinical murine imaging at 7 T) used image sequences with repetition times of approximately 8 ms range (with a 90° excitation pulse and a spectral width of 20 kHz), and echo times of ca. 1 ms.7 The limit at which increasing R1 enhances the SNR per unit time is ca. 400 Hz under similar experimental conditions (assuming the repetition time needs to be three times R1 for signal recovery); signals with R1 > 400 Hz will fully recover within the 3 ms repetition time but cannot be imaged any faster at the same spectral width and matrix size, whilst a greater proportion of signal loss in the 1 ms echo time scales with increasing R2. Faster repetition times are possible with larger spectral widths, but this is determined by the water/fat-parashift frequency difference (approximately 20 kHz for the signal 60 ppm away from the endogenous signals at 7 T), and risks tissue damage from heating. At a clinical field of 3 T, 60 ppm is only 8 kHz, therefore the minimum repetition time will approximately double with a similar setup and R1 > 200 Hz cannot be imaged any faster to improve SNR.To assess the L1a and L2a complexes for parashift imaging, relaxation measurements (see Table 1) were performed at 1.4 T (60 MHz) and 9.4 T (400 MHz) using an inversion recovery sequence to measure R1 and spectral linewidths to estimate R2, as any contributions to the linewidths from magnetic field inhomogeneity is small compared to the fast intrinsic R2 rates measured here (however, R2 estimates assume there are no chemical exchange processes leading to line broadening). The table displays values highlighted in bold where the relaxation properties or shifts are favourable for parashift imaging, i.e. R1 > 100 Hz, R1/R1 > 0.5, and |δ| > 50 ppm.
| Complex | Proton | r /Å | δ /ppm | 1.4 T | 1.4 T | R 1/R2 (1.4 T) | 9.4 T | 9.4 T | R 1/R2 (9.4 T) |
|---|---|---|---|---|---|---|---|---|---|
Est. R2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c/Hz c/Hz |
R
1c/Hz |
Est. R2d/Hz |
R
1d/Hz |
||||||
| a δ at 299 K. b Taken from crystal structure data. c T = 299 K. d T = 296 ± 1 K. e R 1 measurement not possible due to broad resonance and very fast R1 relaxation. f Difficult to measure due to overlapping resonances. R1 > 100 Hz, R1/R2 > 0.5, and |δ| > 50 ppm highlighted in bold. | |||||||||
| FeL1a | CH3 | 3.75 | +22 | 440 ± 40 | 290 ± 5 | 0.66 | 600 ± 30 | 356 ± 1 | 0.59 |
| py-H5 | 5.36 | +53 | 110 ± 10 | 24 ± 2 | 0.22 | 230 ± 20 | 34 ± 2 | 0.15 | |
| py-H3 | 5.00 | +34 | 70 ± 5 | 34 ± 4 | 0.49 | 220 ± 20 | 48 ± 1 | 0.22 | |
| py-H4 | 5.90 | −4 | 75 ± 5 | 16 ± 4 | 0.21 | 40 ± 5 | 20 ± 1 | 0.50 | |
| CoL1a | CH3 | 3.78 | +8.5 | 180 ± 10 | 160 ± 5 | 0.89 | 200 ± 10 | 140 ± 4 | 0.70 |
| py-H5 | 5.30 | +50 | 65 ± 5 | 19 ± 2 | 0.29 | 65 ± 5 | 18 ± 1 | 0.28 | |
| py-H3 | 4.98 | +34 | 60 ± 5 | 23 ± 2 | 0.38 | 65 ± 5 | 22 ± 1 | 0.34 | |
| py-H4 | 5.91 | +6 | 45 ± 5 | 11 ± 1 | 0.24 | 50 ± 10 | 7 ± 1 | 0.14 | |
| NiL1a | CH3 | 3.83 | −10 | ca. 2000 | —e | — | ca. 4000 | —e | — |
| py-H5/3 | 4.94/5.27 | +42 | 630 ± 60 | 420 ± 40 | 0.67 | 1400 ± 100 | 477 ± 4 | 0.34 | |
| py-H5/3 | 4.94/5.27 | +34 | 530 ± 50 | 380 ± 40 | 0.72 | 1400 ± 100 | 641 ± 9 | 0.49 | |
| py-H4 | 5.88 | +13 | 180 ± 20 | 130 ± 10 | 0.72 | 290 ± 30 | 219 ± 4 | 0.76 | |
| CuL1a | CH3 | 3.83 | −3 | ca. 1000 | —e | — | ca. 3000 | —e | — |
| py-H4 | 5.90 | +11 | 225 ± 15 | 90 ± 20 | 0.40 | 250 ± 20 | 133 ± 6 | 0.53 | |
| FeL2a | CH3 | 3.66 | −57 | 750 ± 150 | 760 ± 180 | ca. 1 | 1700 ± 200 | 875 ± 3 | 0.51 |
| — | — | +62 | 340 ± 30 | 305 ± 15 | 0.90 | 390 ± 10 | 229 ± 3 | 0.59 | |
| — | — | +57 | 120 ± 20 | 105 ± 5 | 0.88 | 170 ± 10 | 100 ± 2 | 0.59 | |
| CoL2a | CH3 | 3.69 | −122 | ca. 3000 | —e | — | ca. 3000 | —e | — |
| — | — | +56 | 570 ± 80 | 530 ± 10 | 0.93 | 505 ± 25 | 440 ± 10 | 0.87 | |
| — | — | +25 | 410 ± 60 | 330 ± 30 | 0.80 | 370 ± 10 | 275 ± 10 | 0.74 | |
| — | — | +134 | 720 ± 70 | —e | — | 1290 ± 100 | 396 ± 2 | 0.45 | |
| NiL2a | — | — | +63 | ca. 330 ± 30 | —f | — | ca. 1800 ± 200 | —f | — |
The methyl resonance of [CoL1a]2+ has a promising ratio of R1/R2 rates (at r = 3.75 Å), of 0.70 at 9.4 T (and 0.89 at 1.4 T), whilst the R1 of 160 Hz at low field would allow repetition times of 20 ms for imaging. Unfortunately, the resonance is not shifted very far from the diamagnetic window, however similar complexes with r = 3.75 Å have potential for imaging if the contact and/or pseudocontact shifts can be modulated, perhaps by functionalisation of the pyridine ring. In comparison, the R1/R2 ratio for [FeL1a]2+ is slightly lower at both fields, but the R1 rates are faster, allowing repetition times of 10 ms at low field (8 ms at 9.4 T). Again, the moderately shifted resonance at +22 ppm will only allow 5 kHz bandwidth at 7 T (2 kHz at 3 T), reducing the possible repetition times to ca. 30 ms for a similar imaging experiment to that previously described above,7 and 70 ms at 3 T. The pyridyl protons of [CoL1a]2+and [FeL1a]2+ have inferior R1/R2 ratios than the methyl resonances, and the R1 relaxation rates are much slower (ca. 20 Hz), limiting the minimum repetition times used to increase SNR in imaging experiments to ca. 150 ms; they are too slow for fast imaging sequences and are thus too far away from the paramagnetic centre.
The methyl proton resonances are much broader (FWHM ca. 1000 Hz) for [NiL1a]2+ and [CuL1a]2+, consistent with expected longer electronic relaxation rates that enhance dipolar relaxation mechanisms; long electronic relaxation times are expected as these metal complexes do not have low-lying electronic levels to facilitate efficient Orbach-type electronic relaxation mechanisms, due to an A1g electronic ground state in the case of octahedral Ni(II) and the lifted degeneracy of the Eg ground state [CuL1a]2+ due to Jahn–Teller distortion (see crystal structure, Fig. 2(c)). [NiL1a]2+ and [CuL1a]2+ contain resonances (at +13 and +11 ppm respectively) with appropriate relaxation properties for imaging, and we assume that these are the furthest pyridyl-H4 protons from the metal centres (ca. 5.9 Å away) as they are the sharpest peaks in each spectrum. There are only three equivalent protons per complex, however, and the chemical shifts are still not far enough from endogenous water for distinct imaging. In general, it will be difficult to enhance the hyperfine shift for Ni/Cu(II) complexes based on L1a, as the reporter group needs to be at these longer distances to reduce signal broadening, where the contact shift term diminishes.
In the case of both [FeL2a]2+ and [CoL2a]2+, the R1 and R2 rates of the methyl group on the coordinated picolyl unit are faster than their L1a analogues, significantly so in the case of Co(II) where resonances are so broad and the R1 is so fast that it could not be determined. The resonances observed at +62 ppm and +57 ppm for [FeL2a]2+ have ideal relaxation rates for imaging at both fields, as well as hyperfine shifts far enough from water for large spectral widths but only represent two equivalent protons per molecule in each case. In contrast, the resonances generally appear sharper for the [NiL2a]2+ than [NiL1a]2+, i.e. the resonances out at +50 to +300 ppm are readily observable for the former.
We have previously observed significant decreases in the relaxation rates at low field with many lanthanide parashift complexes (particularly Tm(III), Dy(III), and Tb(III) complexes)29 due to the substantial Curie terms and fast electronic relaxation rates, but with these transition metal complexes, particularly the Co(II) complexes, the Curie term does not dominate as much due to their comparatively moderate magnetic susceptibilities, as demonstrated in Fig. 4 (see also Table S1, ESI†).
 | ||
| Fig. 4 Variation of R1 with magnetic field in the range 1.4–16.4 T for (a) [FeL1a]2+ (red filled circles), [CoL1a]2+ (blue filled circles), [NiL1a]2+ (green filled circles), [FeL2a]2+ (red open circles), [CoL2a]2+ (blue open circles). (b) Zoomed in section of [FeL1a]2+ (red filled circles) and [CoL1a]2+ (blue filled circles). Multiparameter fitting of 1H R1 data (298 ± 3 K, D2O) to Bloch–Redfield–Wangsness theory, using the measured fixed values of μeff (lines). | ||
In the case of [FeL2a]2+, the R1 relaxation rate of the coordinated picolyl methyl protons only vary by 25% over the 1.4–16.4 T magnetic field range, and for [CoL2a]2+R1 in fact decreases at high field, with the dipolar term dominating the relaxation mechanism. This is particularly important when considering parashift probe development for imaging at clinical fields.
Fitting the field-dependent R1 relaxation rates to Bloch–Redfield–Wangsness theory (Fig. 4) using multiparameter fitting for r, τr and T1e and the experimental values of μeff, gives T1e values of 0.3 and 0.6 ps for [FeL1a]2+ and [FeL2a]2+ respectively, and 0.3 and 1.2 ps for [CoL1a]2+ and [CoL2a]2+ (Table 2). We also fitted the data for [NiL1a]2+, which estimates a much larger T1e of 40 ps, as expected for an A1g electronic ground state,22 hence the much broader NMR resonances cf. the Fe(II) and Co(II) analogues (Fig. 1). We acknowledge that Bloch–Redfield–Wangsness theory assumes a point dipole approximation, and neglects to account for contact effects and the relaxation effects of the magnetic anisotropy. The model assumes that the Zeeman interactions are much larger than the zero field splitting (ZFS) effects,22 which is not the case for transition metal complexes. Even with lanthanide complexes, previous studies have demonstrated an angular-dependence of the paramagnetic relaxation enhancement effects.30 It has also been shown recently that ZFS rhombicity can significantly slow down nuclear relaxation at low field.31 That being said, fitting the longitudinal relaxation data (Fig. 4 and Table 2) using the Bloch–Redfield–Wangsness theory returns r values similar to those determined experimentally (except for [CoL2a]2+where r is not known). The resulting rotational correlation times also appear reasonable when compared to literature values for macrocyclic lanthanide complexes32 as well as with approximations derived from the hydrodynamic diameters of the L1a and L2a complexes estimated from the crystal structures.¶
| Compound | Experimental data | Calculated values (using exp. μeff) | |||
|---|---|---|---|---|---|
| r /Å | μ eff/μB | r/Å | τ r/ps | T 1e/ps | |
| a From crystal structure. b Estimated using Evans method. c μ eff assumed to be similar to that measured by Evans method of [CoL2a](NO3)2. d Assignment unknown. e μ eff measured by Evans method of [CoL2a](NO3)2. | |||||
| [FeL1a]2+ (pyH5) | 5.36 | 5.4 ± 0.3b | 5.7 ± 0.1 | 200 ± 20 | 0.34 ± 0.02 |
| [CoL1a]2+ (pyH5) | 5.30 | 5.1c | 5.6 ± 0.2 | 80 ± 20 | 0.27 ± 0.08 |
| [NiL1a]2+ (pyH3/5) | 5–5.3 | 3.4 ± 0.1b | 5.3 ± 0.1 | 280 ± 15 | 39 ± 3 |
| [FeL2a]2+ (pyCH3) | 3.66 | 5.8 ± 0.7b | 3.6 ± 0.1 | 380 ± 60 | 0.59 ± 0.16 |
| [CoL2a]2+ | —d | 5.1 ± 0.7e | 4.2 ± 0.1 | 450 ± 25 | 1.16 ± 0.24 |
The simple point-dipole approximation correlates the faster low-field proton relaxation rates of the L2a complexes cf. their L1a analogues with slower T1e rates. When there are electronic energy levels within ca. 1000 cm−1 from the ground state vibrational-orbital couplings (Orbach mechanism) via phonons in the 50–1000 cm−1 energy range (at 300 K) can occur,12 which induces spin-changes and fast electronic relaxation times due to the spin–orbit coupling. It is plausible that the pseudo-octahedral L1a complexes have faster electronic relaxation rates with respect to the more trigonal-prismatic L2a analogues complexes due to greater orbital degeneracy in the ground state and therefore a higher density of low-lying energy levels to facilitate Orbach relaxation.
The six-fold methyl resonance at −57 ppm of [FeL2a]2+ presents the best option for imaging in the table, certainly at preclinical fields; at 7 T a 20 kHz spectral width is possible, thus for imaging with 8 ms repetition times R1 up to 400 Hz can enhance signal, although R2 is faster than desired.
2.4 Phantom MR imaging
Phantom imaging studies of [FeL2a]2+ were undertaken at 400 MHz (9.4 T) to assess the feasibility of imaging the resonance at −57 ppm at preclinical fields (Fig. 5, for unprocessed data see ESI, Fig. S84†). A 10 mM solution of [FeL2a]2+ (right hand side, Fig. 4) was prepared in D2O, in a 5 mm NMR tube, alongside a tube containing H2O (left hand side, Fig. 4), and the two phantoms were placed inside a 30 mm 1H imaging coil (Bruker, Germany). Imaging was performed by simultaneous signal acquisition of the diamagnetic water signal and the parashift reporter resonance of [FeL2a]2+ (i.e. the methyl group) achieved by exciting the signal at −57 ppm using a Gaussian pulse, followed by 2D gradient echo using localising gradients with amplitudes to achieve the field of view (FOV) shown in Fig. 5. Simultaneous acquisition of the water signal (excited by the sufficient excitation within the bandwidth of the −57 ppm pulse) was collected by sampling across a wide MRI FOV to generate a ‘double image’ with two independent imaging channels for water (bottom) and the [FeL2a]2+ complex (top) in Fig. 5. The water phantom (ca. 50 M) appears as a very intense image in the water (lower) channel, whilst the [FeL2a]2+ phantom is visible in both channels due to the residual protonated solvent in the D2O, as well as the resonance at −57 ppm. | ||
| Fig. 5 Non-slice selective 2D gradient echo MRI of two 5 mm tubes (cross-section) containing water (left) and 10 mM [FeL2a]2+ dissolved in D2O (right) at 9.4 T. TR = 40 ms, TE = 1.48 ms, NS = 512, SW = 249 ppm (100 kHz) matrix size = 256 × 32. Raw data processed using ROI mapping to remove noise. Frequency-resolved image in the vertical direction, showing the shifted image for [FeL2a]2+ (arrow) due to the resonance at −57 ppm. Image acquired in 12 min. | ||
3. Conclusions
This study presents the development of lanthanide-free macrocyclic transition-metal parashift complexes, evaluating their potential as MRI molecular probes at clinical and pre-clinical magnetic field strengths. The findings underscore the significance of molecular rigidity and strategic ligand design in optimizing nuclear relaxation rates and minimizing resonance broadening.The 1H NMR spectra of Fe(II) and Co(II) complexes of L1a and L2a displayed sharp resonances with significant hyperfine shifts, whilst those of the analogous Ni(II) and Cu(II) complexes contained much broader resonances, almost indiscernible in the case of Cu(II). In the solid state, the [ML1a]2+ complexes were pseudo-octahedral for M = Zn(II), Fe(II), Co(II), and Ni(II), whilst Jahn–Teller distortion was observed for Cu(II). The [ML2a]2+ complexes crystallise as six-coordinate trigonal prismatic systems for M = Fe(II), Co(II), whilst a five-coordinate complex was observed in the case of Cu(II).
In both the TACN and cyclen complexes studied the methyl group is required at the 6-position on the coordinating pyridines (complexes of L1a or L2a) to produce potential parashift complexes, but for different reasons; for the TACN complexes it is needed to extend the M–Npy bond length and prevent the formation of LS Fe(II) or LS Co(III) complexes (e.g. in the case of the L1b complexes), whereas in the trigonal prismatic cyclen complexes (where HS Co(II) is stabilised regardless, e.g. in the case of [CoL2b]2+), the bulky group serves to limit exchange broadening. We synthesised analogues of L1a and L2a with a tert-butyl group in the 5-position of the coordinating pyridines (rather than the 6-methyl group) but found that the steric bulk from the 5-pyridyl position cannot impart rigidity in the cyclen system (i.e. [CoL2b]2+) or influence the M–Npy distance in the L1b systems. It is not possible to put the tert-butyl at the 6-position, as this prevents metal coordination.
The nuclear relaxation rates (R1 and R2) of proton resonances within the L1a and L2a complexes were measured at a range of magnetic fields, to assess their suitability for imaging. Among the complexes studied, [FeL1a]2+ and [CoL1a]2+ have potential for imaging (based on the relaxation properties of the picolyl methyl group) if the contact shift can be modulated via functionalisation of the pyridine ring, such that the hyperfine shift is in the <−50 ppm or >+50 ppm range.
The electronic relaxation time (T1e) of [FeL1a]2+, [CoL1a]2+, [NiL1a]2+, [FeL2a]2+, and [CoL2a]2+ were estimated by fitting field-dependent nuclear relaxation rates of a given proton in each complex; T1e is a function of both the transition metal ion and the local ligand field and is an important parameter for the design of parashift agents for low field imaging, where the dipolar term tends to dominate the relaxation rates according to Bloch–Redfield–Wangsness theory. For the Fe(II) and Co(II) complexes investigated, the T1e values are slower in the trigonal prismatic L2a complexes than their pseudo-octahedral L1a analogues, likely due to the reduction in symmetry lifting orbital degeneracy of the ground states, reducing the density of low-lying states that facilitate Orbach relaxation mechanisms.
[FeL2a]2+ demonstrates promising imaging capability due to its highly shifted methyl resonance at −57 ppm, fast longitudinal relaxation rates, and sufficient complex stability over 24 hours. To our knowledge this is the first example of imaging a parashift complex that incorporates a biogenic metal centre, which could exploit the body's natural metal metabolism for clearance if trace metal leaches into the system. We have successfully imaged the parashift probe selectively, in the presence of phantom tubes containing both strong (control tube) and weak (parashift tune) water signals, even though the resonance is rather broad (FWHM is ca. 500 Hz at 9.4 T). The signal is distinct from even a high background of water within the same image. Future systems with slower relaxation rates (R1 and R2ca. 400 Hz) would reduce T2-losses whilst allowing fast imaging sequences with greater sensitivity.
Author contributions
Project conceptualisation and management was performed by N. J. R. Data collection was carried out by N. J. R., C. A. K., C. A., D. B., G. C., G. P., and P. H. Data curation, analyses, project supervision, and article writing was performed by N. R.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for [[CoL1b]3+, [CoL2b]2+, [CuL1a]2+, [CuL2a]2+, [FeL1b]2+, [NiL1a]2+, [ZnL1a]2+] have been deposited at the CCDC under deposition numbers 2446484–2446490.†Acknowledgements
NJR thanks HKBU, the Hong Kong Jockey Club Charities Trust, and the University of Warwick for support. CAK and CA thank the Hong Kong Jockey Club for support under the Global JC STEM Professorship scheme. PH was supported by a URKI Future Leaders Fellowship [MR/X034046/1]. We acknowledge Dr Andrew Coy and Dr Bertram Manz at Magritek and Dr Alan Kenwright for assistance with NMR, Dr Guy Clarkson for support with X-ray crystallography, and Professor David Parker and Professor Thomas Meersmann for useful discussions.References
- J. H. Duyn and A. P. Koretsky, Novel frontiers in ultra-structural and molecular MRI of the brain, Curr. Opin. Neurol., 2011, 24, 386–393 CrossRef PubMed
.
- J. C. Ferré, M. S. Shiroishi and M. Law, Advanced techniques using contrast media in neuroimaging, Magn. Reson. Imaging Clin. N. Am., 2012, 20, 699–713 Search PubMed
- C. S. Restrepo, S. Tavakoli and A. Marmol-Velez, Contrast-enhanced cardiac magnetic resonance imaging, Magn. Reson. Imaging Clin. N. Am., 2012, 20, 739–760 Search PubMed
- S. Aime, M. Botta, M. Fasano, E. Terreno, P. Kinchesh, L. Calabi and L. Paleari, A new ytterbium chelate as contrast agent in chemical shift imaging and temperature sensitive probe for MR spectroscopy, Magn. Reson. Med., 1996, 35, 648–651 CrossRef CAS PubMed
- P. Harvey, A. M. Blamire, J. I. Wilson, K.-L. N. A. Finney, A. M. Funk, P. K. Senanayake and D. Parker, Moving the goal posts: enhancing the sensitivity of PARASHIFT proton magnetic resonance imaging and spectroscopy, Chem. Sci., 2013, 4, 4251–4258 Search PubMed
- D. Coman, R. A. de Graaf, D. L. Rothman and F. Hyder, In vivo three-dimensional molecular imaging with Biosensor Imaging of Redundant Deviation in Shifts (BIRDS) at high spatiotemporal resolution, NMR Biomed., 2013, 26, 1589–1595 CrossRef CAS PubMed
- P. K. Senanayake, N. J. Rogers, K.-L. N. A. Finney, P. Harvey, A. M. Funk, J. I. Wilson, D. O'Hogain, R. Maxwell, D. Parker and A. M. Blamire, A new paramagnetically shifted imaging probe for MRI, Magn. Reson. Med., 2017, 77, 1307–1317 Search PubMed
- R. Schmidt, N. Nippe, K. Strobel, M. Masthoff, O. Reifschneider, D. D. Castelli, C. Höltke, S. Aime, U. Karst, C. Sunderkötter, C. Bremer and C. Faber, Highly Shifted Proton MR Imaging: Cell Tracking by Using Direct Detection of Paramagnetic Compounds, Radiology, 2014, 272, 785–795 CrossRef PubMed
- C. Harriswangler, F. Lucio-Martínez, L. Godec, L. K. Soro, S. Fernández-Fariña, L. Valencia, A. Rodríguez-Rodríguez, D. Esteban-Gómez, L. J. Charbonnière and C. Platas-Iglesias, Effect of Magnetic Anisotropy on the 1H NMR Paramagnetic Shifts and Relaxation Rates of Small Dysprosium(III) Complexes, Inorg. Chem., 2023, 62, 14326–14338 Search PubMed
- F. Slade, J. F. Collingwood and N. J. Rogers, Transition metal Parashift and ParaCEST MRI agents: Current progress and challenges, Coord. Chem. Rev., 2024, 516, 215940 Search PubMed
- I. Bertini, P. Turano and A. J. Vila, Nuclear magnetic resonance of paramagnetic metalloproteins, Chem. Rev., 1993, 93, 2833–2932 CrossRef CAS
-
I. Bertini, C. Luchinat, G. Parigi and E. Ravera, in NMR of Paramagnetic Molecules, ed. I. Bertini, C. Luchinat, G. Parigi and E. Ravera, Elsevier, Boston, 2nd edn, 2017, pp. 175–253, DOI:10.1016/B978-0-444-63436-8.00008-9
- S. K. Panda, A. Rai and A. K. Singh, Study of paraCEST response on six-coordinated Co(ii) and Ni(ii) complexes of a pyridine-tetraamide-based ligand, Dalton Trans., 2023, 52, 18407–18415 RSC
- P. Pérez-Lourido, E. Madarasi, F. Antal, D. Esteban-Gómez, G. Wang, G. Angelovski, C. Platas-Iglesias, G. Tircsó and L. Valencia, Stable and inert macrocyclic cobalt(II) and nickel(II) complexes with paraCEST response, Dalton Trans., 2022, 51, 1580–1593 RSC
- S. J. Dorazio, A. O. Olatunde, P. B. Tsitovich and J. R. Morrow, Comparison of divalent transition metal ion paraCEST MRI contrast agents, JBIC, J. Biol. Inorg. Chem., 2014, 19, 191–205 CrossRef CAS PubMed
- R. Uzal-Varela, A. Rodríguez-Rodríguez, D. Lalli, L. Valencia, M. Maneiro, M. Botta, E. Iglesias, D. Esteban-Gómez, G. Angelovski and C. Platas-Iglesias, Endeavor toward Redox-Responsive Transition Metal Contrast Agents Based on the Cross-Bridge Cyclam Platform, Inorg. Chem., 2024, 63, 1575–1588 CrossRef CAS PubMed
- F. Schmid, C. Höltke, D. Parker and C. Faber, Boosting 19F MRI—SNR efficient detection of paramagnetic contrast agents using ultrafast sequences, Magn. Reson. Med., 2013, 69, 1056–1062 Search PubMed
- J. Blahut, L. Benda, J. Kotek, G. Pintacuda and P. Hermann, Paramagnetic Cobalt(II) Complexes with Cyclam Derivatives: Toward 19F MRI Contrast Agents, Inorg. Chem., 2020, 59, 10071–10082 CrossRef CAS PubMed
- V. Herynek, M. Martinisková, Y. Bobrova, A. Gálisová, J. Kotek, P. Hermann, F. Koucký, D. Jirák and M. Hájek, Low-molecular-weight paramagnetic 19F contrast agents for fluorine magnetic resonance imaging, Magn. Reson. Mater. Phys., Biol. Med., 2019, 32, 115–122 Search PubMed
- Z. Garda, F. Szeremeta, O. Quin, E. Molnár, B. Váradi, R. Clémençon, S. Même, C. Pichon, G. Tircsó and É. Tóth, Small, Fluorinated Mn2+ Chelate as an Efficient 1H and 19F MRI Probe, Angew. Chem., Int. Ed., 2024, 63, e202410998 CAS
- K. H. Chalmers, A. M. Kenwright, D. Parker and A. M. Blamire, 19F-lanthanide complexes with increased sensitivity for 19F-MRI: Optimization of the MR acquisition, Magn. Reson. Med., 2011, 66, 931–936 CrossRef CAS PubMed
- E. Ravera, L. Gigli, L. Fiorucci, C. Luchinat and G. Parigi, The evolution of paramagnetic NMR as a tool in structural biology, Phys. Chem. Chem. Phys., 2022, 24, 17397–17416 Search PubMed
- B. Martin and J. Autschbach, Kohn–Sham calculations of NMR shifts for paramagnetic 3d metal complexes: protocols, delocalization error, and the curious amide proton shifts of a high-spin iron(II) macrocycle complex, Phys. Chem. Chem. Phys., 2016, 18, 21051–21068 RSC
- P. B. Tsitovich, J. M. Cox, J. B. Benedict and J. R. Morrow, Six-coordinate Iron(II) and Cobalt(II) paraSHIFT Agents for Measuring Temperature by Magnetic Resonance Spectroscopy, Inorg. Chem., 2016, 55, 700–716 Search PubMed
- L. Christiansen, D. N. Hendrickson, H. Toftlund, S. R. Wilson and C. L. Xie, Synthesis and structure of metal complexes of triaza macrocycles with three pendant pyridylmethyl arms, Inorg. Chem., 1986, 25, 2813–2818 CrossRef CAS
- Antimicrobial Peptide Database. Journal.
- J. Blahut, P. Hermann, Z. Tošner and C. Platas-Iglesias, A combined NMR and DFT study of conformational dynamics in lanthanide complexes of macrocyclic DOTA-like ligands, Phys. Chem. Chem. Phys., 2017, 19, 26662–26671 RSC
- S. Oh, A. G. Webb, T. Neuberger, B. Park and C. M. Collins, Experimental and numerical assessment of MRI-induced temperature change and SAR distributions in phantoms and in vivo, Magn. Reson. Med, 2010, 63, 218–223 CrossRef PubMed
- A. M. Funk, P. Harvey, K.-L. N. A. Finney, M. A. Fox, A. M. Kenwright, N. J. Rogers, P. K. Senanayake and D. Parker, Challenging lanthanide relaxation theory: erbium and thulium complexes that show NMR relaxation rates faster than dysprosium and terbium analogues, Phys. Chem. Chem. Phys., 2015, 17, 16507–16511 RSC
- E. A. Suturina, K. Mason, C. F. G. C. Geraldes, N. F. Chilton, D. Parker and I. Kuprov, Lanthanide-induced relaxation anisotropy, Phys. Chem. Chem. Phys., 2018, 20, 17676–17686 RSC
- J. C. Ott, E. A. Suturina, I. Kuprov, J. Nehrkorn, A. Schnegg, M. Enders and L. H. Gade, Observability of Paramagnetic NMR Signals at over 10 000 ppm Chemical Shifts, Angew. Chem., Int. Ed., 2021, 60, 22856–22864 CrossRef CAS PubMed
- A. M. Funk, P. H. Fries, P. Harvey, A. M. Kenwright and D. Parker, Experimental Measurement and Theoretical Assessment of Fast Lanthanide Electronic Relaxation in Solution with Four Series of Isostructural Complexes, J. Phys. Chem. A, 2013, 117, 905–917 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2446484–2446490. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt01149c |
| ‡ Λ/Δ denote pendant arm helicity with respect to the pseudo-C3 axis, λλλ/δδδ denote the configuration in each NCCN chelate ring. |
| § Apical twist angles measured from the crystal structure data, using the torsion angle between the nitrogen atom of a pyridine and its connected TACN nitrogen, around the axis comprising the centroids of the “N3-py” and “N3-TACN”. |
| ¶ Estimated hydrodynamic diameters of L1a and L2a complexes (10 and 15 Å respectively) from the crystal structures correspond to rotational correlation times of 100 and 400 ps at 298 K, using Stokes–Einstein law. |
| This journal is © The Royal Society of Chemistry 2025 |