
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidorhenium(V) complexes of apigenin: a rare example of catalysis with O,O-bidentate ligands obtained from a naturally renewable resource†
Milan R.
Milovanović
a,
Stefan R.
Nikolić
a,
Antoine
Dupé
b,
Jörg A.
Schachner
 *b and
Ljiljana E.
Mihajlović
*a
*b and
Ljiljana E.
Mihajlović
*a
aInnovation Center of the Faculty of Chemistry, Belgrade, Studentski trg 12–16, 11158 Belgrade, Serbia
bInstitute of Chemistry, University of Graz, Schuberstr. 1, 8010 Graz, Austria
First published on 2nd July 2025
Abstract
When Re(V) precursor [ReOCl3(PPh3)2] (P1) is reacted with apigenin (HL1, Hapg), the flavone coordinates as an O,O-bidentate ligand via the deprotonated 5-OH group and its neighboring ketone to the Re center. Novel oxidorhenium(V) complex [ReOCl2(PPh3)(L1)] (1) was successfully synthesized in boiling acetone in sufficient yield. To increase the solubility of apigenin and its respective complexes, the other two –OH groups in the 7 and 4′-position of the apigenin structure were protected. To do so, well-established acetyl- and TBDMS- (tBuMe2Si-) protecting groups were introduced to the apigenin frame, resulting in the novel ligands HL2 and HL3, respectively. Resulting oxidorhenium(V) complexes [ReOCl2(PPh3)(L2)] (2) and [ReOCl2(PPh3)(L3)] (3) showed much increased solubilities in standard organic solvents. All three complexes 1–3 were subsequently tested in catalytic olefin epoxidation, which is the first time for a Re complex with an O,O-bidentate ligand. All three complexes showed activity for substrate cyclooctene, with complex 2 giving a high activity for such complexes (TON = 300). The novel ligands HL2 and HL3 as well as all three novel complexes 1–3 were fully characterized by NMR and IR spectroscopy, MS spectrometry and elemental analysis. In addition, the solid-state structures of HL2, HL3 and 1–3 were determined by single-crystal X-ray diffraction analysis.
Introduction
The rich coordination chemistry of rhenium is based on a wide range of its oxidation states spanning from −1 to +7 due to its position in Group VII of the periodic table, where it combines properties of both early and late transition metals. The most investigated complexes in modern literature contain rhenium in the oxidation states of Re(I), Re(V) and Re(VII).1 These coordination complexes showed a broad field of applications in homogenous catalysis,2–4 bioinorganic and medicinal chemistry5,6 as well as small molecule activation.7 In parallel to the progress of Re synthetic chemistry grew its applicative potential. But among all applicative prospects, Re complexes have distinguished themselves either as radiopharmaceuticals for nuclear/bioimaging6,8,9 or as efficient catalysts in homogeneous catalytic epoxidation of olefins, most prominently with the discovery of methyltrioxorhenium(VII) (MTO).10The low oxidation state Re complexes have been primarily studied in the light of their favorable kinetic stability and good biodistribution, which enabled them to be used as photosensitizers and bioimaging agents.9,11 They are typically stabilized by π-acceptor ligands such as CO, PR3, and RCN. The most distinguished class is a Re(I) group of kinetically stable and inert compounds with the fac-[Re(CO)3]+ core, primarily reported by the group of Walter Hieber.12 The well-established field of Re(I) carbonyl-containing complexes then triggered the development of Re complexes in higher oxidation states, dominantly Re(V) and Re(VII). A milestone work of Herrmann in 1991 put the focus on MTO due to its remarkable catalytic activity in olefin epoxidation and oxidation.13 With turnover numbers (TONs) of over 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, MTO has been positioned as the most active Re-based epoxidation catalyst to date.3 Yet, the compound manifested a major drawback due to the often-observed hydrolysis of acid-sensitive epoxides. To address this challenge, oxidorhenium(V) complexes with tetra- and bidentate Schiff-base ligands were suggested in Herrmann's research group as potential epoxidation catalysts.14 Indeed, further efforts in the following two decades were directed toward developing oxidorhenium(V) compounds and their promotion as homogenous epoxidation catalysts of cyclooctene.2,15
000, MTO has been positioned as the most active Re-based epoxidation catalyst to date.3 Yet, the compound manifested a major drawback due to the often-observed hydrolysis of acid-sensitive epoxides. To address this challenge, oxidorhenium(V) complexes with tetra- and bidentate Schiff-base ligands were suggested in Herrmann's research group as potential epoxidation catalysts.14 Indeed, further efforts in the following two decades were directed toward developing oxidorhenium(V) compounds and their promotion as homogenous epoxidation catalysts of cyclooctene.2,15
Apigenin (4′,5,7-trihydroxyflavone, Hapg, HL1) is a naturally occurring, bioactive flavone-type of molecule, commonly present in a variety of fruits, vegetables, and medicinal plants. Its applicative potential is well-reported nowadays, especially having in mind its proven anti-allergic, antibacterial, anticancer, anti-inflammatory and antiviral effects.16 There is a growing body of published research concerning flavonoid ligands in transition metal complexes,17 but only a small number of them are apigenin complexes and none are of rhenium.18 In many cases, full characterization or isolation of the reported coordination compounds is lacking, especially X-ray confirmation of the solid state structure. From a strictly chemical point of view, Hapg shares coordination features with the acac moiety (Fig. 1). However, in contrast to the large number of Re complexes with various N,O,19O,P,20 or O,S and O,C21 ligands, the chemistry with O,O22 ligand donor sets is surprisingly scarce. So far, only a few Re-acetylacetonates (acac)23 and related compounds24 have been synthesized and structurally characterized. Also, to the best of our knowledge, such O,O-ligands have never been tested as epoxidation catalysts. These surprising gaps of knowledge further motivated us to investigate the naturally renewable molecule apigenin as a potential ligand for rhenium and the resulting chemistry of these complexes.
 | ||
| Fig. 1 Structure of apigenin HL1 showing the numbering of the 3 OH-groups; novel ligands HL2 and HL3. | ||
Within this publication, we report the synthesis and full characterization of the first three examples of oxidorhenium(V) complexes [ReOCl2PPh3(L1–3)] (1–3) containing the apigenin ligand HL1 and two newly synthesized apigenin derivatives HL2–3 (Fig. 1). To enhance the very low solubility of both apigenin HL1 and the resulting complexes 1, substituents on 4′-OH and 7-OH based on established protecting group chemistry (–OAc in HL2; –Si(tBu)Me2 or TBDMS in HL3) were introduced. As part of these chemical investigations, the three new oxidorhenium(V) complexes 1–3 were tested in a classical chemical catalysis, namely olefin epoxidation.
Results and discussion
Apigenin (HL1) itself is only soluble in the strong polar solvents DMSO and DMF, and poorly soluble in acetone (Table 1), a phenomenon often observed in flavonoid chemistry.25 This low solubility severely complicated synthesis of complexes, even more so as the use of DMSO was excluded because of reaction of these oxidorhenium(V) complexes with DMSO due to their Oxygen-Atom-Transfer (OAT) capabilities. Therefore, the synthesis of O-protected ligands HL2 and HL3 became necessary due to the low solubility of Apigenin HL1 (Fig. 1). For the synthesis of protected apigenin ligands HL2 and HL3, we turned to literature-established protocols. Acetylated ligand HL2 was synthesized via a modified Eichhorn acetylation26 using acetic anhydride and pyridine as solvent, thereby avoiding purification by column chromatography. We had observed that HL2 is partly deprotected again on silica gel. Silylated ligand HL3 was synthesized by adopting a literature procedure using TBDMSCl and Et3N in CH2Cl2.27 After the reaction, the crude product was extracted with pentane to remove Et3NCl. Single crystals of HL3 could also be grown from a concentrated pentane solution. Again, column chromatography was avoided, as partial hydrolysis of a silyl-protecting group did occur. In none of our experiments, the unwanted protection of the –OH group at the 5-position was observed, potentially due to a strong H-bond to the neighboring ketone and a resulting lower acidity at that position.| [mg ml−1] | 1 | 2 | 3 |
|---|---|---|---|
| DMF | 1.01 | 1.50 | 5.33 |
| Acetone | 0.31 | 1.38 | 9.86 |
| THF | 0.21 | 1.36 | 4.32 |
| Acetonitrile | <0.05 | 1.01 | 5.56 |
| MeOH | <0.05 | <0.05 | 0.78 |
| EtOH | <0.05 | <0.05 | <0.05 |
| DCM | <0.05 | 4.47 | 4.46 |
| Chloroform | <0.05 | 1.37 | 1.25 |
| Diethylether | <0.05 | <0.05 | <0.05 |
| Toluol | <0.05 | <0.05 | 0.77 |
The synthesis of novel Re complexes 1–3 was achieved by reaction of the precursor complex [ReOCl3(PPh3)2] (P1) and an equimolar amount of the respective ligand in boiling acetone. Reaction times of 1–2 hours were found to be enough to achieve a sufficient conversion (up to 80%). Here, the low solubility of parent apigenin HL1 in acetone required a pre-dissolution of HL1 in boiling acetone. To this solution, P1 was added and further heated to refluxing temperatures. For the more soluble ligands HL2 and HL3, the synthesis was started by mixing P1 and the respective ligand at 25 °C in acetone. The removal of the PPh3 by-product was most effective via a short flash chromatography column in cyclohexane/ethyl acetate (see Experimental section). Single crystals of 1 were also obtained from that solvent mixture (Scheme 1).
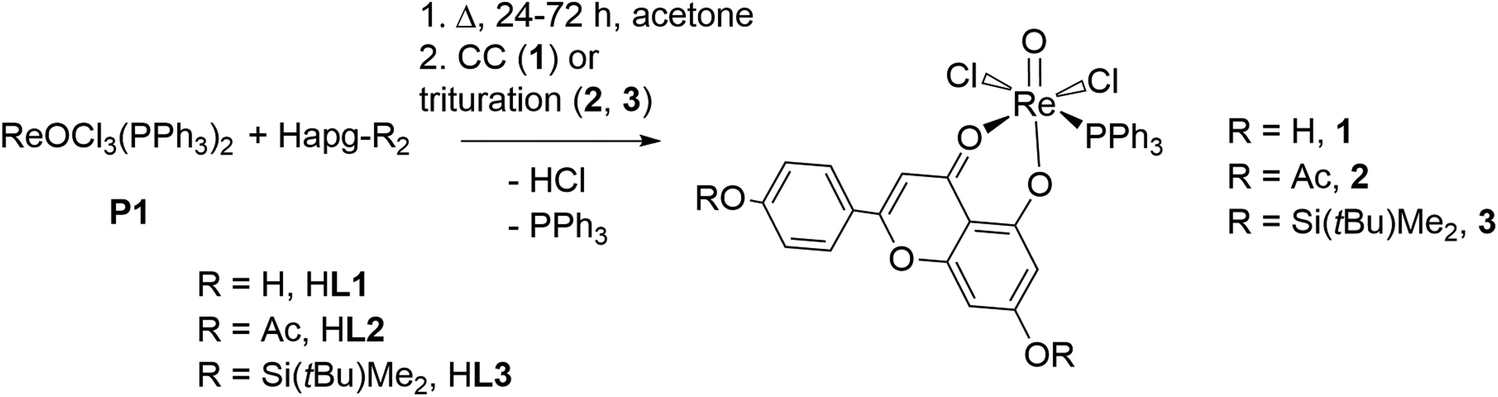 | ||
| Scheme 1 Synthesis of complexes 1–3 (CC = column chromatography, cyclohexane, EtOAc 1/1). Yields: 1 – 52%, 2 – 57% and 3 – 73%. | ||
Complex 1 showed the expected analytical data for an oxidorhenium(V) complex. Whereas the changes in the 1H NMR spectrum between free ligand HL1 and complex 1 were the least pronounced of all the NMR data, the color of 1 was an unusual dark brown. Mostly, oxidorhenium(V) complexes are green, as are 2 and 3. The resonance for the coordinated PPh3 ligand in 1 shifted significantly to higher field at −18.7 ppm (acetone-d6), similar to other published Re(V) complexes.28 The Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond was found at 943 cm−1 in the typical region of the IR spectrum for such rhenium-oxo bonds. X-ray diffraction analysis also confirmed an octahedral cis-Cl,Cl arrangement around the Re center, with the phenolate-O atom of the L1 moiety trans to the oxido ligand. Interestingly, the mass of 1 could not be measured by EI-MS, as only fragments even below the mass of HL1 were obtained. Only with a soft ionization technique like ESI-MS, the mass spectrum of 1 could be obtained. Complex 2 was synthesized from P1 and HL2 in acetone, albeit here simple mixing at rt and then boiling for only 2 h was sufficient. The green crude product of 2 was most efficiently purified by trituration of concentrated crude solution in diethyl ether. Single crystals of 2 were obtained by re-crystallization from acetone (see Experimental section). Similar structural parameters than for 1 were also observed for 2. The PPh3 ligand resonated at −17.7 ppm in the 31P NMR spectrum (acetone-d6), and a meaningful MS could again only be acquired by ESI-MS. In the IR spectrum however, the ReO bond was found at a rather unusual high shift of 980 cm−1. X-ray diffraction analysis also confirmed an octahedral cis-Cl,Cl arrangement around the Re center, with the phenolate-O atom of the L2 moiety trans to the oxido ligand. Complex 3 finally was synthesized under similar conditions as 2. As complex 3 suffered from partial loss of the silyl protecting group on silica, excess PPh3 could be removed by drop-wise addition of a concentrated crude solution of 3 in acetone into a large excess of n-pentane under fast stirring (trituration). A re-crystallization from acetone/diethylether finally yielded 3 in pure form. The coordinated PPh3 ligand in 3 resonated at −18.7 ppm in the 31P NMR spectrum (acetone-d6), the two inequivalent silicon atoms of 3 were detected at 25.13 and 23.43 ppm in the 29Si NMR spectrum. A meaningful mass spectrum could be acquired by ESI-MS. In the IR spectrum, the ReO bond was found at 969 cm−1. Complex 1 showed low to no solubility in the standard organic solvents, except for acetone, THF and DMF (Table 1). We assume this low solubility is due to an extended network of H-bridges that forms between the two –OH groups present in the backbone of the apigenin moiety of complex 1. In contrast to 1, complexes 2 and 3, where the –OH groups are protected, solubilities are largely increased in polar to medium polar solvents. An approximation of solubilities, done by visual checking of dissolution in the respective solvent, can be found in Table 1. All three complexes decompose in DMSO, most likely via OAT.
O bond was found at 943 cm−1 in the typical region of the IR spectrum for such rhenium-oxo bonds. X-ray diffraction analysis also confirmed an octahedral cis-Cl,Cl arrangement around the Re center, with the phenolate-O atom of the L1 moiety trans to the oxido ligand. Interestingly, the mass of 1 could not be measured by EI-MS, as only fragments even below the mass of HL1 were obtained. Only with a soft ionization technique like ESI-MS, the mass spectrum of 1 could be obtained. Complex 2 was synthesized from P1 and HL2 in acetone, albeit here simple mixing at rt and then boiling for only 2 h was sufficient. The green crude product of 2 was most efficiently purified by trituration of concentrated crude solution in diethyl ether. Single crystals of 2 were obtained by re-crystallization from acetone (see Experimental section). Similar structural parameters than for 1 were also observed for 2. The PPh3 ligand resonated at −17.7 ppm in the 31P NMR spectrum (acetone-d6), and a meaningful MS could again only be acquired by ESI-MS. In the IR spectrum however, the ReO bond was found at a rather unusual high shift of 980 cm−1. X-ray diffraction analysis also confirmed an octahedral cis-Cl,Cl arrangement around the Re center, with the phenolate-O atom of the L2 moiety trans to the oxido ligand. Complex 3 finally was synthesized under similar conditions as 2. As complex 3 suffered from partial loss of the silyl protecting group on silica, excess PPh3 could be removed by drop-wise addition of a concentrated crude solution of 3 in acetone into a large excess of n-pentane under fast stirring (trituration). A re-crystallization from acetone/diethylether finally yielded 3 in pure form. The coordinated PPh3 ligand in 3 resonated at −18.7 ppm in the 31P NMR spectrum (acetone-d6), the two inequivalent silicon atoms of 3 were detected at 25.13 and 23.43 ppm in the 29Si NMR spectrum. A meaningful mass spectrum could be acquired by ESI-MS. In the IR spectrum, the ReO bond was found at 969 cm−1. Complex 1 showed low to no solubility in the standard organic solvents, except for acetone, THF and DMF (Table 1). We assume this low solubility is due to an extended network of H-bridges that forms between the two –OH groups present in the backbone of the apigenin moiety of complex 1. In contrast to 1, complexes 2 and 3, where the –OH groups are protected, solubilities are largely increased in polar to medium polar solvents. An approximation of solubilities, done by visual checking of dissolution in the respective solvent, can be found in Table 1. All three complexes decompose in DMSO, most likely via OAT.
Solid state structures
As expected, there are no significant structural changes to the apigenin core in HL2 and HL3 (for structure discussion of ligands HL2 and HL3, please see the ESI†) compared to HL1 caused by the protecting groups (Table 2). The three bond lengths of the 5-OH group C1–O1 and its neighboring keto group C7–O2 are the same within experimental error.| [Å] | Re1O1 |
Re1–P1 | Re1–Cl1 | Re1–Cl2 | Re1–O2 | Re1–O3 |
| 1 | 1.709(2) | 2.4626(8) | 2.3361(7) | 2.4260(7) | 1.944(2) | 2.088(2) |
| 2 | 1.6788(15) | 2.4482(5) | 2.3441(5) | 2.3986(5) | 1.9912(14) | 2.0765(14) |
| 3 | 1.6814(17) | 2.4597(6) | 2.3488(6) | 2.3995(6) | 1.9741(15) | 2.1051(16) |
| [°] | O1Re1–O2 |
O1Re1–P1 |
Cl1–Re1–Cl2 | |||
| 1 | 168.94(9) | 89.42(8) | 90.98(3) | |||
| 2 | 166.61(7) | 91.66(5) | 89.605(18) | |||
| 3 | 166.14(8) | 88.19(6) | 87.89(2) |
Currently, complexes 1–3 are the first examples of Re-apigenin complexes characterized by single-crystal X-ray spectroscopy. All three complexes 1 (Fig. 2), 2 (Fig. 3) and 3 (Fig. 4) show a distorted octahedral coordination around the Re center in the solid state. The respective phenolate-oxygen of the apigenin moiety is located trans to the oxido ligand, and the two chlorido ligands are in cis orientation. Bond angles and distances for complexes 1–3 were all within expected range (Table 2, also see ESI†). Single crystals of 1 were obtained by re-crystallization from a cyclohexane/ethylacetate solvent mixture, of 2 from acetone and of 3 from acetone/diethylether. The presence of acetyl-groups in 2 or the TBDMS-groups in 3 does not affect the distorted octahedral geometry around the Re center. Also the bond lengths around Re are more less the same. The ReO bond in 1 is somewhat elongated (1.712(2) Å) compared to 2 (1.6788(15) Å) and 3 (1.6814(17) Å) (Table 2). One structural feature of all three complexes is interesting to note, that is the now more similar bond lengths of Re1–O2 and Re1–O3 bonds. This proves a significant bond delocalization, as is also observed in an acac-chelate ring, in contrast to the initially localized C–OH single- and CO double-bond of the respective apigenin ligands.
 | ||
| Fig. 2 ORTEP plot (50% probability) of complex 1 (H atoms except for the 7-OH and 4′-OH groups are omitted). | ||
 | ||
| Fig. 3 ORTEP plot (50% probability) of complex 2 (H atoms are omitted for clarity). | ||
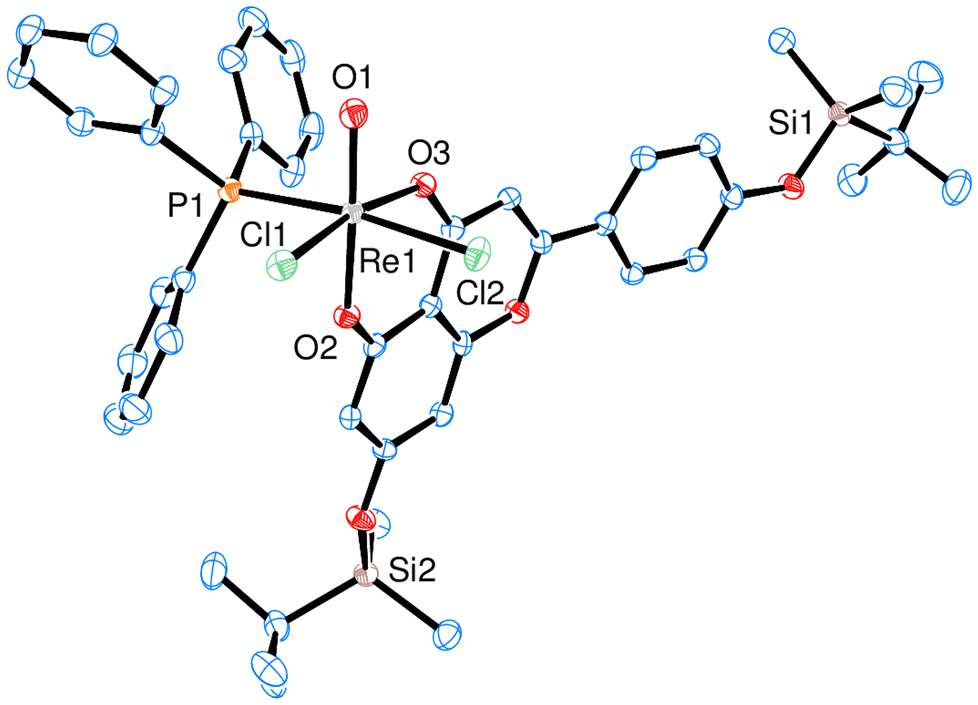 | ||
| Fig. 4 ORTEP plots (50% probability) of complex 3 (H atoms are omitted for clarity). | ||
By close visual inspection of crystal packing of HL2 one can recognize that most dominant non-covalent intermolecular interactions are stacking interactions between aromatic and resonance-assisted hydrogen-bridged (RAHB) rings, C–H⋯O (carbonyl) and C–H⋯π interactions. In case of HL3 most dominant non-covalent intermolecular interactions are π⋯π stacking interactions, C–H⋯O and C–H⋯π interactions. The similar interactions seem to be important in crystal packing of the corresponding Rhenium(V) complexes. Namely, in the case of 1 non-covalent interactions that play a role in crystal packing are π⋯π stacking interactions, C–H⋯π interactions and hydrogen bonds (O–H⋯O and O–H⋯Cl); while in the case of 2 seemingly there is no stacking interactions, but C–H⋯π, C–H⋯O and C–H⋯Cl interactions; interactions with a co-crystalized molecules of chloroform represent a significant portion of intermolecular interaction in the case of 2. In the case of 3 most dominant are π⋯π stacking and C–H⋯Cl interactions.
 | ||
| Scheme 2 Epoxidation catalysis with complexes 1–3 and olefinic substrates S1–3. | ||
An overview of epoxidation results with S1 is given in Table 3. In a first round of testing, all three complexes 1–3 showed activity for epoxidation of cyclooctene S1 with tertbutylhydroperoxide (TBHP) as oxidant in chloroform, but with pronounced different activities (Fig. 5). Complex 1 showed the lowest activity with a TON = 46 over 24 h reaction time. This rather low activity can most likely be attributed to the low solubility of 1 in CHCl3 (Table 1). After the 24 h experiment time, undissolved material of 1 was still visible in the reaction mixture. Under the same conditions, the fully dissolved complexes 2 and 3 showed a better reactivity. Complex 2 reached a TON of 73 after already four hours, 3 showed a similar activity to 2 reaching a TON of 60 after four hours. At a reduced catalyst loading of 0.1 mol%, complex 2 still gave 30% conversion to epoxide, resulting in a TON = 300.
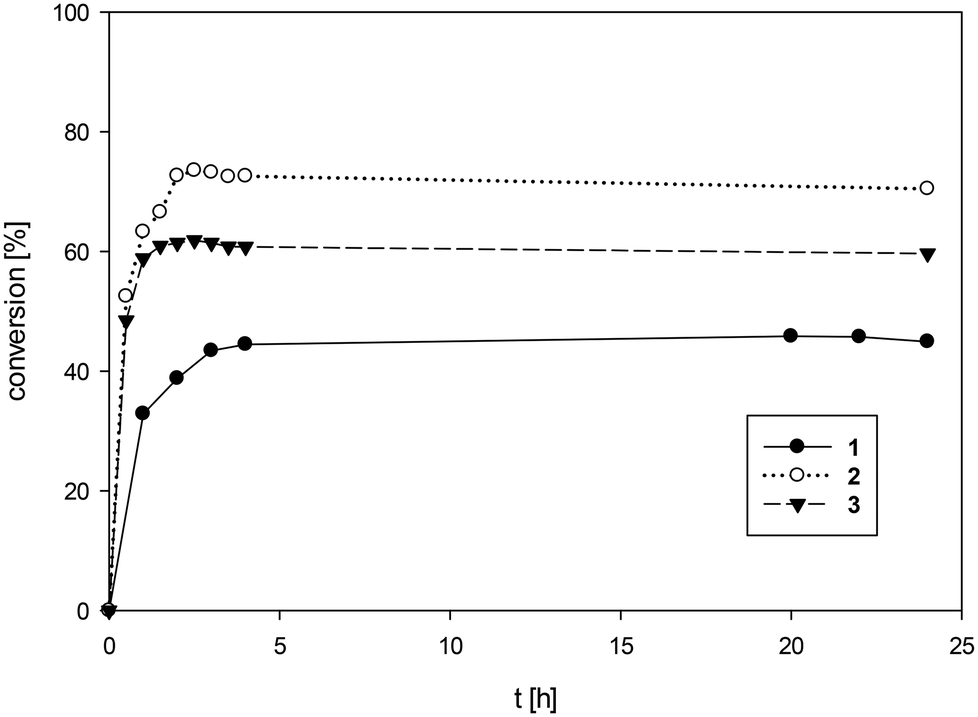 | ||
| Fig. 5 Comparison of epoxidation activity of complexes 1–3 for cyclooctene S1 (CHCl3, 50 °C, 1 mol%, 3 equiv. TBHP). | ||
| TBHP | H2O2 | ||
|---|---|---|---|
| 1 mol% | S1, CHCl3 | S1, acetone | S1, CHCl3 |
| 1 mol% catalyst loading, 50 °C; TOF in h−1 at time of maximum conversion. | |||
| 1 | 46 (1.9) | 34 (1.4) | <10 |
| 2 | 73 (18.25) | 28 (14) | <10 |
| 3 | 60 (15) | 30 (3.75) | 67 (22) |
In a second set of experiments, complexes 1–3 were tested with TBHP in acetone as a solvent. Caution has to be applied, as oxidizers and acetone can, in principle, form explosive acetone peroxides.29 However, reactions were carried out under TBHP dilution, in the absence of protons and the reaction mixture was shaken, not stirred by magnetic stirring. Whereas the solubilities of 1–3 are higher in acetone compared to CHCl3, the epoxidation activities were lower in all three cases (Table 3). Potentially, acetone is a too strongly coordinating solvent, which is able to compete with the TBHP for coordination on the Re center. In addition, complexes 2 and 3 were also tested in THF and MeOH, but both remained unreactive in these solvents. Probably, the donating nature of these solvents competes with TBHP for a coordination site on Re, thereby shutting down epoxidation activity.
In a second round of experiments, the aqueous oxidant H2O2 was tested for cyclooctene S1, as H2O2 is considered a greener oxidant compared to TBHP.30 We hoped that the presence of two –OH groups in the ligand backbone of complex 1 would be beneficial for the use of aqueous oxidant H2O2. Tests were performed only in CHCl3 as the mixture of acetone and H2O2 can lead to formation of explosive acetone peroxides.29 Somewhat surprisingly though, this time only complex 3, with the silyl-protected apigenin backbone, showed a remarkable activity of TON = 67 (Table 3). Even at a lowered catalyst loading of 0.1 mol%, complex 3 still reached a conversion of 17%, equivalent to a TON = 170 with H2O2 as the oxidant.
For the other two substrates S2–3, complexes 1–3 showed the same unsatisfactory, but often observed catalyst profile. For 1-octene S2, low activities were observed, leaving the substrate more or less unchanged over 24 hours. Parent apigenin Re-complex 1 also showed no activity for the epoxidation of styrene S3, whereas acetylated and silylated complexes, 2 and 3, respectively, completely over-oxidized S3 to various products, mostly benzaldehyde. The non-reactivity of complex 1 for styrene S3 is rather unusual for oxidorhenium(V) complexes.
As complexes 1–3 are the first examples of epoxidation catalysts containing an O,O-bidentate ligand, a comparison can only be made to other ligand systems of previously published oxidorhenium(V) catalysts [ReOCl2(PPh3)(LL)] (LL = mono-anionic, bidentate ligand). The largest class of those catalysts does contain O,N-bidentate ligands and were reviewed in 2020 by Wolff and Machura.2 Reported TONs for substrate S1 for 22 catalysts ranged between 47–97, with the four most active ones containing ligands LL1–LL4 (Fig. 6).31,32 Complexes [ReOCl2(PPh3)(LL1–3)] also showed activity with aqueous H2O2 as oxidant (TON = 60, 53 and 74, respectively), albeit in 1,2-dichloroethane at 80 °C. In comparison, the TON of 67 for 3 achieved already at 50 °C (CHCl3) can be considered to be a respectable result. The fantastic activities of MTO however (TON > 20000)3 also remain unchallenged by the three novel complexes reported herein.
 | ||
| Fig. 6 Three most active previously published ReOCl2(PPh3)(LL) complexes for S1 epoxidation (1mol% cat., 50 °C, CHCl3, 3 equiv. TBHP). | ||
The beneficial effect of ligands LL1–4 as well as the exact mechanism of Re(V) complexes vs. Re(VII) MTO-like complexes is still not fully understood. In general, the current mechanistic understanding follows the Sharpless mechanism of a concerted three component reaction between the complex, the oxidant and the olefin, where the olefin does not coordinate to the metal center.33 Furthermore, the nature of the OAT is considered to be an electrophilic attack of the oxygen atom onto the olefin. Hence, any measures to enhance the electrophilicity of that coordinated oxygen atom before OAT should result in more active epoxidation catalysts and must therefore also be a result of the other coordinated ligands on the Re center. From our own research, we cannot conclude that a simple −I effect of the ligand is required, as the methoxy substituent in ligand moiety LL1 should show a small +I effect. Potentially, it is the overall polarity of the ligands and the resulting complex that play a significant role for the concerted, three-membered Sharpless-type polar transition state. The three apigenin moieties L1–3 described herein probably also exert an overall −I effect on the rhenium center, due to the presence of four oxygen atoms in the backbone and three aromatic phenyl rings. And complexes 1–3 are definitely highly polar molecules, as evidenced by their low solubilities in organic solvents. Overall, these semi-empirical guidelines should make up for an acceptable oxidorhenium(V) epoxidation catalysts, which was evidenced by the catalytic experiments.
Conclusions. The results presented here are the first examples of the synthesis of oxidorhenium(V) complexes equipped with a flavonoid-type, O,O-bidentate ligand and their use in epoxidation catalysis. The majority of epoxidation catalysts contain O,N-bidentate ligands.15 Here, one general trend, that could be observed, was the beneficial influence of electron-withdrawing substituents and/or polarity of the complex on epoxidation activity.31,34 The presence of a two aromatic ring systems and OH/alkoxide groups in the ligand backbone of HL1 and HL2–3, respectively, overall effect a −I influence as a ligand. Complex 1 certainly suffers from its overall low solubility and hence results in lower catalytic activities. If the two remaining –OH groups of 1 are protected, both solubility as well as catalytic activity of resulting complexes 2 and 3 increase. Complex 3 in addition showed epoxidation activity with aqueous H2O2, also rarely observed for oxidorhenium(V) complexes.
Experimental section
General
Rhenium precursor [ReOCl3(PPh3)2] (P1) has been synthesized according to literature.35 All other chemicals were purchased from commercial sources and were used without further purification. NMR spectra were recorded with a Bruker (300 MHz) instrument. Chemical shifts are given in ppm and are referenced to residual protons in the solvent. Signals are described as s (singlet), bs (broad singlet), d (doublet), dd (doublet of doublet), t (triplet), qd (quaternary doublet), m (multiplet) and coupling constants (J) are given in Hertz (Hz). Mass spectra were recorded with an Agilent 5973 MSD – Direct Probe using the EI ionization technique or HR-MS (ESI) measurements were recorded on an Agilent Technologies 6230 TOF LC/MS with ESI or APCI mass spectrometer in positive and negative ion mode, the used solvent was acetonitrile. Samples for infrared spectroscopy were measured on a Bruker Optics ALPHA FT-IR Spectrometer equipped with an ATR diamond probe head. GC-MS measurements were performed on an Agilent 7890 A with an Agilent 19091J–433 column coupled to a mass spectrometer type Agilent 5975 C. Elemental analyses were carried out using a Heraeus Vario Elementar automatic analyzer at the University of Technology Graz and performed in duplicate.Crystal structures determination
Single-crystal X-ray data were obtained using a XtaLAB Synergy, Dualflex, HyPix-Arc 100 diffractometer (HL2, HL3, 3) or a Bruker APEX-II CCD diffractometer (1) or a Bruker D8 Venture Photon II diffractometer (2). Reflection data were collected at T = 100 K using monochromatized Mo Kα radiation (HL3, 1, 3), Cu Kα radiation (HL2) or Ga Kα radiation (2). Data reduction, scaling and absorption corrections were performed using the CrysAlisPro software35 (HL2, HL3, 3) or APEX software36 or APEX software37 (1, 2). For HL2, HL3 and 3, a numerical absorption correction based on Gaussian integration over a multifaceted crystal model and an empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm of CrysAlisPro, were performed. For 1 and 2, a semi-empirical absorption correction was performed using SADABS.38The structures were solved with the ShelXT 2018/239 structure solution program using the intrinsic phasing solution method and by using Olex240 as the graphical interface. The models were refined with version 2019/3 of ShelXL41 using full-matrix least-squares techniques against F2. All non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were calculated geometrically and refined using a riding model, except the H atoms of the OH groups which were found on a difference Fourier map and refined freely. For HL2, the O1–H1 distance was fixed at 0.88 Å with a DFIX constraint. For complex 3, DFIX, SADI, RIGU, SIMU and SUMP constraints and restraints were used to model the disorder of the solvent molecules. For complex 2, since the acetone solvent molecule lay on an inversion center and was disordered, the electron densities were removed from the electron density map by the SQUEEZE treatment using the OLEX2 solvent mask command. A solvent mask was calculated and 32 electrons were found in a volume of 222 Å3 in 1 void per unit cell. This is consistent with the presence of 1 molecule acetone per unit cell (Z = 2), which accounts for 32 electrons.
CCDC 2407661 (HL2), 2407662 (HL3), 2407663 (1), 2407664 (2) and 2407665 (3)† contain the supplementary crystallographic data for this paper.
O ester), 1649.80 (s, CO carbonyl), 1614.86 (s, CC), 1592.02 (m, CC aromatic), 1488.38 (m, CC aromatic), 1367.75 (m), 1338.08 (m), 1289.10 (m), 1216.15 (m), 1189.54 (s, O–H), 1169.38 (s), 1134.80 (s), 1109.57 (m), 1017.65 (s), 920.59 (m), 894.25 (m), 822.85 (m), 714.16 (m), 626.60 (m), 585.54 (w), 500.17 (w), 460.14 (w), 421.01 (w); EI-MS: 354.2 (M+); Elemental analysis calculated for C19H14O7 (354.3 g mol−1): calc. C 64.41%, H 3.98%; found: C 63.75%, H 3.79%.
O carbonyl), 1598.57 (m, CC aromatic), 1507.90 (m, CC aromatic), 1422.46 (m), 1340.58 (m), 1264.31 (s, Si–O), 1158.64 (s, O–H), 1113.05 (s), 1097.08 (w), 1029.39 (w), 983.93 (s), 908.62 (s), 825.21 (s), 781.42 (s), 711.25 (s), 674.09 (w), 546.30 (w), 479.09 (w); EI-MS: 498.5 (M+); Elemental Analysis calculated for C27H38O5Si2 (498.77 g mol−1): calc. C 65.02%, H 7.68%; found: C 64.72%, H 7.69%.
O carbonyl), 1581 (s, CC aromatic), 1528 (s), 1499 (s), 1435 (m), 1359 (m), 1242 (m), 1167 (s), 1129 (m), 943 (s, ReO), 846 (m), 824 (m), 690 (m, Re-P), 676 (m), 528 (s), 510 (s), 496 (s); (+)ESI-MS: 804.0 (M+); Elemental Analysis calculated for C33H24Cl2O6PRe (804.6 g mol−1): C 49.26%, H 3.18%; found: C 49.19%, H 2.96%.
O ester), 1622 (m, CO carbonyl), 1540 (m, CC aromatic), 1423 (m), 1341 (w), 1193 (s), 1169 (s), 1138 (s), 1094 (s), 1005 (s), 980 (s, ReO), 758 (m), 690 (m, Re–P), 529 (m), 509 (m); (+)ESI-MS: 852.9 (M+ − Cl− − H+); Elemental Analysis calculated for C37H28Cl2O8PRe (888.7 g mol−1): C 50.01%, H 3.18%; found: C 49.19%, H 2.96%.
O carbonyl), 1587 (s), 1483 (m), 1352 (m), 1244 (m, Si–O), 1173 (s), 1096 (m), 1038 (m), 969 (s, ReO), 904 (m), 827 (s), 782 (s), 691 (s, Re-P), 526 (s), 508 (m); (+)ESI-MS: 1034 (M+); Elemental Analysis calculated for: C 52.31%, H 5.07%; found: C 51.46%, H 4.89%.
Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
The authors declare no competing financial interests.Data availability
Data for this article, including files for NMR, IR and mass spectroscopic data, are available at Zenodo at https://zenodo.org/uploads/14883918; https://doi.org/10.5281/zenodo.14883918.Acknowledgements
The following granting agencies and institutions are acknowledged: This work was supported by the European Research Executive Agency through the MET-EFFECT project under grant no. 101086373. Furthermore, by the Ministry of Science, Technological Development and Innovation of Republic of Serbia, grant number 451-03-66/2024-03/200288 and the Innovative Centre Faculty of Chemistry Belgrade Ltd, Studentski trg 12-16, Belgrade, Serbia. Also, NAWI Graz is also acknowledged for financial support. Assoc. Prof. W. Gössler from Analytical Chemistry, Institute of Chemistry and Dr G. Rechberger from Department of Molecular Biosciences, University of Graz are thanked for acquiring ESI-MS spectra. Ass. Prof. M. Haas, Institute of Inorganic Chemistry, Graz University of Technology is thanked for acquiring 29Si NMR spectra.References
- J. R. Dilworth, Coord. Chem. Rev., 2021, 436, 213822 CrossRef CAS.
- M. Wolff and B. Machura, Rev. Inorg. Chem., 2020, 40, 47–73 Search PubMed.
- J. W. Kück, R. M. Reich and F. E. Kühn, Chem. Rec., 2016, 16, 349–364 CrossRef PubMed.
- (a) F. E. Kühn, J. Organomet. Chem., 2023, 122824 CrossRef; (b) S. C. A. Sousa and A. C. Fernandes, Coord. Chem. Rev., 2015, 284, 67–92 CrossRef CAS.
- (a) E. B. Bauer, A. A. Haase, R. M. Reich, D. C. Crans and F. E. Kühn, Coord. Chem. Rev., 2019, 393, 79–117 CrossRef CAS; (b) P. V. Simpson, N. M. Desai, I. Casari, M. Massi and M. Falasca, Future Med. Chem., 2019, 11, 119–135 CrossRef CAS PubMed; (c) C. C. Konkankit, S. C. Marker, K. M. Knopf and J. J. Wilson, Dalton Trans., 2018, 47, 9934–9974 RSC; (d) A. Leonidova and G. Gasser, ACS Chem. Biol., 2014, 9, 2180–2193 CrossRef CAS PubMed.
- M. A. Klenner, T. Darwish, B. H. Fraser, M. Massi and G. Pascali, Organometallics, 2020, 39, 2334–2351 CrossRef CAS.
- (a) W. Guan, F. B. Sayyed, G. Zeng and S. Sakaki, Inorg. Chem., 2014, 53, 6444–6457 CrossRef CAS PubMed; (b) L. Alig, K. A. Eisenlohr, Y. Zelenkova, S. Rosendahl, R. Herbst-Irmer, S. Demeshko, M. C. Holthausen and S. Schneider, Angew. Chem., Int. Ed., 2021, 61, e202113340 CrossRef PubMed; (c) I. Klopsch, M. Finger, C. Würtele, B. Milde, D. B. Werz and S. Schneider, J. Am. Chem. Soc., 2014, 136, 6881–6883 CrossRef CAS PubMed; (d) Q. J. Bruch, G. P. Connor, C.-H. Chen, P. L. Holland, J. M. Mayer, F. Hasanayn and A. J. M. Miller, J. Am. Chem. Soc., 2019, 141, 20198–20208 CrossRef CAS PubMed; (e) J. L. Smeltz, P. D. Boyle and E. A. Ison, J. Am. Chem. Soc., 2011, 133, 13288–13291 CrossRef CAS PubMed.
- S. Hostachy, C. Policar and N. Delsuc, Coord. Chem. Rev., 2017, 351, 172–188 CrossRef CAS.
- J. J. Wilson, in Biomedical applications of inorganic photochemistry, ed. P. C. Ford and R. van Eldik, Academic Press, Cambridge, MA, San Diego, CA, Oxford, London, 2022, p. 1 Search PubMed.
- (a) W. A. Herrmann, J. Organomet. Chem., 1995, 500, 149–173 CrossRef CAS; (b) F. E. Kühn, A. Scherbaum and W. A. Herrmann, J. Organomet. Chem., 2004, 689, 4149–4164 CrossRef.
- (a) E. Wolcan, Inorg. Chim. Acta, 2020, 509, 119650 CrossRef CAS; (b) J. Palion-Gazda, K. Choroba, A. M. Maroń, E. Malicka and B. Machura, Molecules, 2024, 29, 1631 CrossRef CAS PubMed.
- (a) W. Hieber, in Advances in organometallic chemistry, ed. F. A. Stone and R. West, Elsevier, Burlington, 1970, pp. 1 Search PubMed; (b) W. A. Herrmann, J. Organomet. Chem., 1990, 383, 21–44 CrossRef; (c) J. E. Ellis and W. Beck, Angew. Chem., Int. Ed. Engl., 1995, 34, 2489–2491 CrossRef CAS.
- W. A. Herrmann, R. W. Fischer and D. W. Marz, Angew. Chem., Int. Ed. Engl., 1991, 30, 1638–1641 CrossRef.
- (a) W. A. Herrmann, M. U. Rauch and G. R. J. Artus, Inorg. Chem., 1996, 35, 1988–1991 CrossRef CAS; (b) F. E. Kühn, M. U. Rauch, G. M. Lobmaier, G. R. J. Artus and W. A. Herrmann, Chem. Ber., 1997, 130, 1427–1431 CrossRef.
- B. Machura, M. Wolff and I. Gryca, Coord. Chem. Rev., 2014, 275, 154–164 CrossRef CAS.
- (a) D. Patel, S. Shukla and S. Gupta, Int. J. Oncol., 2007, 233–245 CAS; (b) R. Liu, H. Zhang, M. Yuan, J. Zhou, Q. Tu, J.-J. Liu and J. Wang, Molecules, 2013, 18, 11496–11511 CrossRef CAS PubMed.
- (a) E. Halevas, B. Mavroidi, G. Zahariou, M. Pelecanou and A. G. Hatzidimitriou, Inorg. Chim. Acta, 2023, 546, 121325 CrossRef CAS; (b) A. Yang, C. Liu, H. Zhang, J. Wu, R. Shen and X. Kou, Eur. J. Med. Chem., 2022, 233, 114216 CrossRef CAS PubMed; (c) E. Halevas, B. Mavroidi, M. Pelecanou and A. G. Hatzidimitriou, Inorg. Chim. Acta, 2021, 523, 120407 CrossRef CAS; (d) M. Kozsup, X. Zhou, E. Farkas, A. C. Bényei, S. Bonnet, T. Patonay, K. Kónya and P. Buglyó, J. Inorg. Biochem., 2021, 217, 111382 CrossRef CAS PubMed; (e) E. Halevas, A. Pekou, R. Papi, B. Mavroidi, A. G. Hatzidimitriou, G. Zahariou, G. Litsardakis, M. Sagnou, M. Pelecanou and A. A. Pantazaki, J. Inorg. Biochem., 2020, 208, 111083 CrossRef CAS PubMed; (f) K. Karami, Z. Mehri Lighvan, H. Farrokhpour, M. Dehdashti Jahromi and A. A. Momtazi-Borojeni, J. Biomol. Struct. Dyn., 2018, 36, 3324–3340 CrossRef CAS PubMed; (g) Y. Liu and M. Guo, Molecules, 2015, 20, 8583–8594 CrossRef CAS PubMed; (h) Z.-T. Zhang and J. Shi, J. Coord. Chem., 2007, 60, 1485–1491 CrossRef CAS; (i) Y. He, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, m469–m471 CrossRef PubMed; (j) A.-C. Munteanu, A. Notaro, M. Jakubaszek, J. Cowell, M. Tharaud, B. Goud, V. Uivarosi and G. Gasser, Inorg. Chem., 2020, 59, 4424–4434 CrossRef CAS PubMed; (k) X. Han, M. R. Kumar, A. Hoogerbrugge, K. K. Klausmeyer, M. M. Ghimire, L. M. Harris, M. A. Omary and P. J. Farmer, Inorg. Chem., 2018, 57, 2416–2424 CrossRef CAS PubMed; (l) S. Roy, R. Das, B. Ghosh and T. Chakraborty, Mol. Carcinog., 2018, 57, 700–721 CrossRef CAS PubMed; (m) D. Ravishankar, M. Salamah, A. Attina, R. Pothi, T. M. Vallance, M. Javed, H. F. Williams, E. M. S. Alzahrani, E. Kabova, R. Vaiyapuri, K. Shankland, J. Gibbins, K. Strohfeldt, F. Greco, H. M. I. Osborn and S. Vaiyapuri, Sci. Rep., 2017, 7, 5738 CrossRef PubMed.
- (a) M. Khater, D. Ravishankar, F. Greco and H. M. Osborn, Future Med. Chem., 2019, 11, 2845–2867 CrossRef CAS PubMed; (b) G. Erdogan, R. Karadag and A. Eler, Main Group Met. Chem., 2010, 33, 283–299 CrossRef CAS; (c) M. Tan, J. Zhu, Y. Pan, Z. Chen, H. Liang, H. Liu and H. Wang, Bioinorg. Chem. Appl., 2009, 2009, 347872 CrossRef PubMed; (d) J. J. Martínez Medina, L. G. Naso, A. L. Pérez, A. Rizzi, N. B. Okulik, E. G. Ferrer and P. A. Williams, J. Photochem. Photobiol., A, 2017, 344, 84–100 CrossRef.
- (a) V. S. Sergienko, Russ. J. Inorg. Chem., 2019, 64, 583–591 CrossRef CAS; (b) V. S. Sergienko and A. V. Churakov, Russ. J. Inorg. Chem., 2018, 63, 631–641 CrossRef CAS; (c) V. S. Sergienko and A. V. Churakov, Russ. J. Inorg. Chem., 2018, 63, 753–763 CrossRef CAS.
- V. S. Sergienko and A. V. Churakov, Russ. J. Inorg. Chem., 2017, 62, 1327–1342 CrossRef CAS.
- V. S. Sergienko, Russ. J. Inorg. Chem., 2017, 62, 751–759 CrossRef CAS.
- (a) V. S. Sergienko and A. V. Churakov, Russ. J. Inorg. Chem., 2016, 61, 1708–1726 CrossRef CAS; (b) C. Scholtysik, C. Njiki Noufele, A. Hagenbach and U. Abram, Inorg. Chem., 2019, 58, 5241–5252 CrossRef CAS PubMed.
- (a) C. J. L. Lock and C. Wan, Can. J. Chem., 1975, 53, 1548–1553 CrossRef CAS; (b) M. Irmler and G. Meyer, Z. Naturforsch., B: J. Chem. Sci., 1990, 45, 1103–1104 CrossRef CAS; (c) A. Brink, H. G. Visser and A. Roodt, Acta Crystallogr., Sect. E:Struct. Rep. Online, 2010, 67, m34–m35 CrossRef PubMed.
- C. Triantis, T. Tsotakos, C. Tsoukalas, M. Sagnou, C. Raptopoulou, A. Terzis, V. Psycharis, M. Pelecanou, I. Pirmettis and M. Papadopoulos, Inorg. Chem., 2013, 52, 12995–13003 CrossRef CAS PubMed.
- (a) L. Wei, J. Zhao, Y. Meng, Y. Guo and C. Luo, LWT, 2020, 118, 108874 CrossRef CAS; (b) F. J. Osonga, J. O. Onyango, S. K. Mwilu, N. M. Noah, J. Schulte, M. An and O. A. Sadik, Tetrahedron Lett., 2017, 58, 1474–1479 CrossRef CAS; (c) Q.-Q. Wang, J.-B. Shi, C. Chen, C. Huang, W.-J. Tang and J. Li, Bioorg. Med. Chem. Lett., 2016, 26, 1460–1465 CrossRef CAS PubMed.
- In Comprehensive organic name reactions and reagents, ed. Z. Wang, Wiley, Chichester, pp. 967 Search PubMed.
- C. Qi, Y. Xiong, V. Eschenbrenner-Lux, H. Cong and J. A. Porco, J. Am. Chem. Soc., 2016, 138, 798–801 CrossRef CAS PubMed.
- (a) B. Machura, M. Wolff, E. Benoist, J. A. Schachner, N. C. Mösch-Zanetti, K. Takao and Y. Ikeda, Polyhedron, 2014, 69, 205–218 CrossRef CAS; (b) J. A. Schachner, B. Terfassa, L. M. Peschel, N. Zwettler, F. Belaj, P. Cias, G. Gescheidt and N. C. Mösch-Zanetti, Inorg. Chem., 2014, 53, 12918–12928 CrossRef CAS PubMed; (c) S. C. A. Sousa, J. R. Bernardo, M. Wolff, B. Machura and A. C. Fernandes, Eur. J. Org. Chem., 2014, 1855–1859 CrossRef CAS; (d) B. Machura, M. Wolff, E. Benoist, J. A. Schachner and N. C. Mösch-Zanetti, Dalton Trans., 2013, 42, 8827–8837 RSC.
- N. A. Milas and A. Golubovic, J. Am. Chem. Soc., 1959, 81, 6461–6462 CrossRef CAS.
- R. A. Sheldon, Chem. Commun., 2008, 44, 3352 RSC.
- N. Zwettler, J. A. Schachner, F. Belaj and N. C. Mösch-Zanetti, Inorg. Chem., 2014, 53, 12832–12840 CrossRef CAS PubMed.
- J. A. Schachner, F. Belaj and N. C. Mösch-Zanetti, Dalton Trans., 2020, 49, 11142–11149 RSC.
- D. V. Deubel, G. Frenking, P. Gisdakis, W. A. Herrmann, N. Rösch and J. Sundermeyer, Acc. Chem. Res., 2004, 37, 645–652 CrossRef CAS PubMed.
- J. A. Schachner, B. Berner, F. Belaj and N. C. Mösch-Zanetti, Dalton Trans., 2019, 48, 8106–8115 RSC.
- G. Rouschias and G. Wilkinson, J. Chem. Soc. A, 1967, 993–1000 RSC.
- Rigaku Oxford Diffraction, CrysAlisPro:PX018, Rigaku Corporation, 2024 Search PubMed.
- Bruker AXS Inc., APEX2, Bruker, Madison, Wisconsin, USA, 2014 Search PubMed.
- Bruker AXS Inc., SADABS, Bruker, Madison, Wisconsin, USA, 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2407661–2407665. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt01095k |
| This journal is © The Royal Society of Chemistry 2025 |