
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tunable broadband visible emission achieved by phase transformation-driven self-reduction of Eu3+ to Eu2+ in a calcium phosphate matrix†
Sapargali
Pazylbek‡
a,
Jonas
Stadulis‡
a,
Guna
Doke
 ab,
Andris
Antuzevics
b,
Vladimir
Pankratov
b,
Greta
Merkininkaite
a,
Arturas
Katelnikovas
a and
Aleksej
Zarkov
*a
ab,
Andris
Antuzevics
b,
Vladimir
Pankratov
b,
Greta
Merkininkaite
a,
Arturas
Katelnikovas
a and
Aleksej
Zarkov
*a
aInstitute of Chemistry, Vilnius University, Naugarduko 24, LT-03225 Vilnius, Lithuania. E-mail: aleksej.zarkov@chf.vu.lt
bInstitute of Solid State Physics, University of Latvia, Kengaraga 8, LV-1063 Riga, Latvia
First published on 21st April 2025
Abstract
In this work, we report the synthesis of Eu2+-doped alpha-tricalcium phosphate (α-TCP, α-Ca3(PO4)2) via a phase transformation of Eu3+-doped CaHPO4·2H2O. The phase conversion accompanied by a reduction of Eu3+ to Eu2+ occurred during the annealing of the starting material in a vacuum. The optical properties of the obtained α-TCP:Eu2+ were investigated by photoluminescence (PL), thermally stimulated luminescence, and persistent luminescence decay measurements. The obtained material exhibited tunable broadband PL with FWHM values ranging from 87 to 142 nm at room temperature. The PL can be tuned in terms of emission maximum and FWHM by varying the excitation wavelength. The broadband emission was achieved due to the multi-site occurrence of Eu2+ ions in the complex α-TCP matrix. Three types of traps were determined with activation energy values of 0.80, 0.75, and 0.68 eV. After irradiation with X-rays, an afterglow characterizable by an Eu2+ broadband spectrum with a maximum at around 480 nm can be detected for at least 10 h.
1. Introduction
The design of visible broadband emission phosphors is crucial for developing next-generation phosphor-converted white-light-emitting diodes (pc-WLEDs), which are considered efficient, green, and energy-saving light sources. Recent efforts have been focused on designing single-activator phosphors to overcome the disadvantages of combining phosphors with distinct emission colors.1 As one of the most common and highly efficient activators, the Eu2+ ion possesses broadband absorption in the UV to blue regions and multicolor emission. The optical properties of Eu2+-doped phosphors strongly depend on the local environment of Eu2+ ions.2 Consequently, the emission of Eu2+-doped materials can be tuned by site occupancy engineering, resulting in the emission maximum from blue to infrared,2 while the multiple-site occupancy of Eu2+ ions in a single host favors broadband emission.3 For this reason, a structural engineering strategy is often employed to develop single broadband white-emitting phosphors. Recently, broadband Eu2+ emission was achieved in various inorganic matrices such as Ba3ScB3O9,4 Ca3Sc2Si3O12,5 Ba3GeO4Br2,6 K2BaSr(PO4)2−x(BO3)x,7etc.Different phosphate-based matrices, including those of the calcium phosphate (CP) family, are widely employed to prepare lanthanide-activated phosphors.8–10 Recent works on β-Ca3(PO4)2-type (β-TCP) materials demonstrated the feasibility of the design of Eu2+-based broadband phosphors. For instance, Fan et al. achieved a broadband visible emission in Eu2+-activated β-TCP by employing chemical pressure.11 The occupation of Eu2+ ions in the crystal lattice and resulting emission were tuned through the co-substitution of Ca2+ by Y3+ and Mg2+. Full-spectrum white-light emission from Eu2+ ions occupying 3 inequivalent crystallographic sites was demonstrated for the Ca9−yMgyNaGd0.667(PO4)7:Eu2+ phosphor.12 Ultra-broadband emission reaching an FWHM of 330 nm was realized by Liu et al.1 in Ca9LiMg1−xAl2x/3(PO4)7:Eu2+; in this case, the Eu2+ ions occupied 4 different sites. β-TCP is a low-temperature polymorph with 5 inequivalent Ca sites, allowing multi-site doping. The crystal structure of the high-temperature α-TCP polymorph is more complicated, containing 18 Ca sites.13 This structural feature makes the α-TCP matrix highly attractive for developing broadband phosphors.
Most commonly, Eu2+-doped materials are synthesized in a reducing atmosphere;14 nevertheless, some specific inorganic matrices allow the stabilization of the Eu2+ oxidation state in air. The self-reduction phenomenon is typically observed in materials such as borates,15–17 silicates,18 and phosphates.19–21 In this light, CPs can also be considered suitable hosts for observing the self-reduction phenomenon.
CPs are a family of materials that can be classified by various parameters, including crystal structure, Ca/P ratio, and the presence of different structural species. Under particular conditions, the transformation from one phase to another phase can be achieved, including, but not limited to, polymorphism.22,23 Thermally induced phase transformations of different CP phases suggest the thermal treatment of the starting material at elevated temperatures. The Ca/P ratio in starting materials and final products is usually expected to be identical; however, recently, it was shown that the phase conversion between different CPs can occur with a drastic change in the Ca/P ratio from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Ca2P2O7) to 1.5:1 (Ca3(PO4)2) under certain conditions due to the evaporation of the phosphate species.24
:1 (Ca2P2O7) to 1.5:1 (Ca3(PO4)2) under certain conditions due to the evaporation of the phosphate species.24
Some reports on the preparation and investigation of Eu2+-doped α-TCP are already known; however, some contradictions can be found. For instance, the preparation of Ca3(PO4)2:Eu2+ reported by Ji et al.25 and Zhou et al.26 required a reducing atmosphere, while the optical properties of the products were tuned by varying the material cooling conditions during the synthesis or the Eu content. In the works by Li et al.,27,28 the partial self-reduction of Eu3+ to Eu2+ was observed in α-TCP synthesized by solid-state reaction under an air atmosphere; however, the emission spectra (λex = 393 nm) of the obtained materials were clearly dominated by the emission from Eu3+ ions indicating a low degree of reduction.
In the present work, we employed a phase transformation approach for preparing Eu2+-doped α-TCP from Eu3+-doped CaHPO4·2H2O (DCPD). The highly efficient self-reduction of Eu3+ to Eu2+ occurred during the phase conversion, resulting in multi-site doping and broadband PL that covered the entire visible spectrum. The tunable luminescence in terms of emission maximum and FWHM was achieved by varying the excitation wavelength.
2. Experimental
2.1. Synthesis
The starting Eu3+-doped DCPD powder was synthesized by the wet co-precipitation method modifying a previously reported procedure.24,29 For the synthesis, calcium nitrate tetrahydrate (Ca(NO3)2·4H2O, Carl Roth, >99%), europium(III) nitrate hexahydrate (Eu(NO3)3·6H2O, 99.9%, Acros Organics), and diammonium hydrogen phosphate ((NH4)2HPO4, Carl Roth, >98%) were used as starting materials. First, the metal nitrates were dissolved in deionized water, resulting in a 0.8 M solution, where the Eu-substitution level was adjusted to 0.1 mol% with respect to Ca2+ ions. The (NH4)2HPO4 solution of the same concentration was prepared in parallel. The solution containing phosphate ions was rapidly poured into the solution of metal ions under constant stirring, forming white precipitates. The as-prepared precipitates were aged in the reaction mixture for 5 min, and afterward, were vacuum filtered, washed with deionized water and dried in an oven at 50 °C overnight. For the synthesis of Eu-doped α-TCP, the as-prepared precipitates were annealed in a tube furnace in a vacuum (7.8 × 10−2 mbar) at 1300 °C for 24 h. After the annealing procedure, the furnace was allowed to cool down naturally.2.2. Characterization
Powder X-ray diffraction (XRD) analysis was performed using a Rigaku MiniFlex II diffractometer (Cu-Kα, λ = 1.5419 Å) working in Bragg–Brentano (θ/2θ) geometry. The data were collected within the 10–60° 2θ range with a step of 0.02° and a scanning speed of 5° min−1.The morphological features of the synthesized powder were analyzed by scanning electron microscopy (SEM) using a Hitachi SU-70 microscope.
A Bruker ELEXSYS-II E500 CW-EPR spectrometer was used for electron paramagnetic resonance (EPR) investigations of the sample. The spectral acquisition settings were: room temperature, 9.838 GHz (X-band) or 34.02 GHz (Q-band) microwave frequency, 10 mW microwave power, and 0.4 mT magnetic field modulation amplitude.
The photoluminescence excitation (PLE) and photoluminescence emission (PL) spectra were measured using an Edinburgh Instruments FLS980 spectrometer equipped with double excitation and emission monochromators, a 450 W Xe arc lamp, a cooled (−20 °C) single-photon counting photomultiplier (Hamamatsu R928P), and mirror optics for the powder samples. Emission spectra were recorded using a 0.5 nm emission slit width, a 3.5 nm excitation slit width, a step size of 0.5 nm, and an integration time of 0.2 s, and were averaged over three repetitions. Excitation spectra were acquired using a 1.5 nm emission slit width, a 0.5 nm excitation slit width, the same step size and integration time, and also averaged over three repetitions. The PL spectra were corrected by a correction file obtained from a tungsten incandescent lamp certified by the NPL (National Physics Laboratory, UK). The PLE spectra were corrected by a reference detector. PL decay curves were measured using the same Edinburgh Instruments FLS980 spectrometer. A 405 nm pulsed laser diode (Edinburgh Instruments, EPL-405) was used as an excitation source.
Thermally stimulated luminescence (TSL) and persistent luminescence decay measurements were performed using a Lexsyg research fully automated TSL/OSL reader from Freiberg Instruments GmbH equipped with a Hamamatsu R13456 photomultiplier tube. An Andor SR-303i-B spectrometer coupled with a DV420A-BU2 CCD camera was used to obtain a persistent luminescence spectrum. A VF-50 J/S X-ray tube (40 kV, 0.5 mA, W-anode) or Q-switched short-pulsed UV laser DTL-389QT (263 nm) from the Laser-compact Group was used as the irradiation source. The system was operated at a linear heating rate of 1 °C s−1. The afterglow decay was measured at a temperature of 25 °C. To eliminate any effects of previously stored energy, the samples were preheated to 300 °C before each measurement.
3. Results and discussion
The XRD pattern of the as-precipitated powder is shown in Fig. S1.† The major phase was identified as DCPD, in accordance with a previous study.30 A secondary monetite (DCPA, CaHPO4) phase was also detected; however, its presence is not considered critical since the Ca/P ratio in both DCPD and DCPA is the same (Ca/P = 1:1). Moreover, upon annealing in air, DCPD first decomposes exactly to DCPA with further transformation to Ca2P2O7 (CPP) at higher temperatures.31 The PL spectrum of the as-prepared Eu-doped DCPD exhibits several peaks ranging from approximately 520 to 720 nm (Fig. S2†). These peaks are ascribed to the Eu3+ radiation emitted by 4f–4f transitions, specifically 5D0 → 7F0–4,32,33 and there was no emission attributed to Eu2+.
The XRD pattern of the material annealed in a vacuum at 1300 °C is given in Fig. 1. Evidently, the phase transformation to α-TCP occurred. The diffraction peaks matched very well with the standard XRD data of monoclinic Ca3(PO4)2 (#00-070-0364). Such a transformation indicates the evaporation of phosphate species and formation of the CP phase with a significantly higher Ca/P ratio (Ca/P = 1.5:1). The SEM image (inset of Fig. 1) demonstrates the formation of monoliths with undefined shape and size varying in a microscopic range, which is typical for α-TCP prepared at high temperature.34
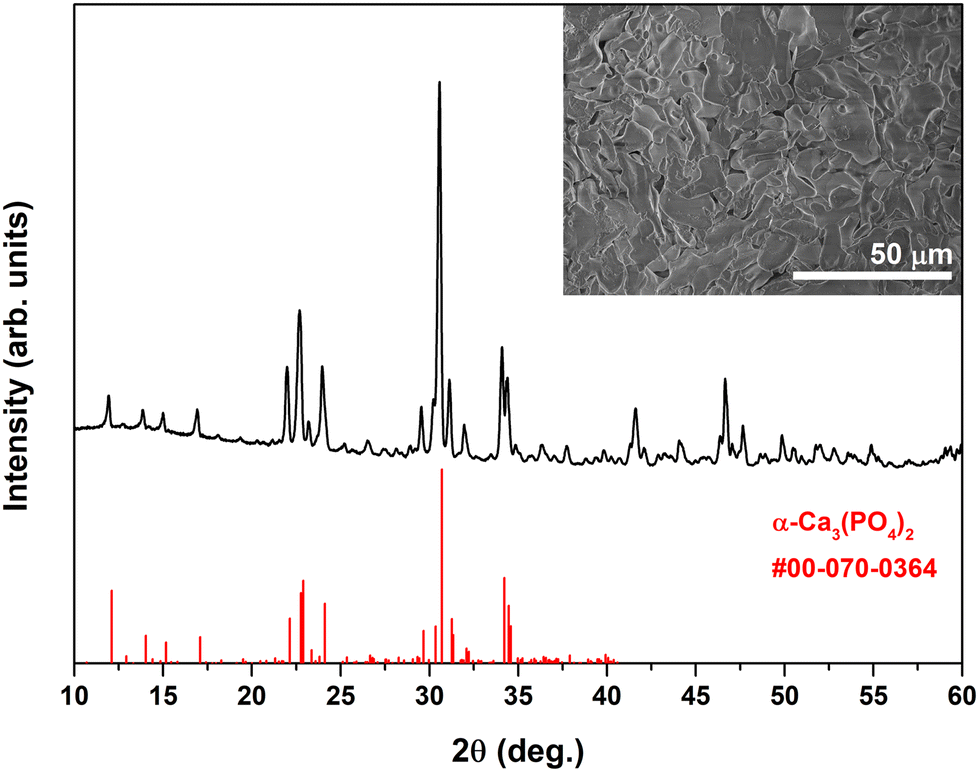 | ||
| Fig. 1 XRD pattern of Eu-doped α-TCP obtained after annealing of Eu-doped DCPD in a vacuum at 1300 °C. Inset: SEM micrograph of Eu-doped α-TCP. | ||
The normalized PLE spectra of Eu-doped α-TCP at emission wavelengths of 475 and 610 nm are shown in Fig. 2a. When monitored at λem = 475 nm, a broad band is seen, which is assigned to the 4f7 → 4f65d1 transition of Eu2+ ions; however, for λem = 610 nm, the shape of the PLE spectrum changed significantly. The difference in the shape of the PLE spectra suggests that Eu2+ ions occupy at least 2 different sites. The sharp lines are associated with the presence of a minor Eu3+ amount and indicate incomplete reduction. These lines originate from 7F0 → 5D4 (ca. 361.5 nm), 7F0 → 5G2 (ca. 368.5 nm), 7F0 → 5L6 (ca. 393 nm), 7F0 → 5D2 (ca. 464 nm), 7F0 → 5D1 (ca. 525 nm), and 7F1 → 5D1 (ca. 532 nm) intraconfigurational [Xe]4f6 → [Xe]4f6 transitions of Eu3+ ions.35
 | ||
| Fig. 2 Normalized PLE (λem = 475 and 610 nm) spectra (a); normalized PL (λex = 285 and 393 nm) spectra (b); normalized PL spectra obtained under different excitation wavelengths (c); dependence of the emission maximum and FWHM on the excitation wavelength (d); dependence of the CIE 1931 chromaticity coordinates on the excitation wavelength (e); and the decay curve (λex = 405 nm and λem = 475 nm) (f) for Eu-doped α-TCP. | ||
The PL spectrum obtained under 285 nm excitation exhibited a single broadband emission centered at 470 nm with an FWHM of 94 nm (4068 cm−1), which is attributed to the 4f65d1 → 4f7 transition typical of Eu2+ (Fig. 2b). The emission spectrum covers the entire range of visible light, while the asymmetric shape of the emission also suggests the presence of Eu2+ in multiple sites. In mixed-valent Eu-doped materials, the emission can be tuned by varying the excitation wavelength, which allows the observation of the emission of exclusively Eu2+ or Eu3+ ions or control of the intensity ratio of simultaneous emission.19,36,37 Such an effect allowing for direct control of the luminescence color is achieved due to the overlap in the excitation spectra of Eu2+ and Eu3+ within the range of 250–450 nm.18 Therefore, we also recorded the PL spectrum under an excitation wavelength of 393 nm. The emission lines associated with Eu3+ can be seen at ca. 590 nm (5D0 → 7F1), 610 nm (5D0 → 7F2), and 700 nm (5D0 → 7F4); however, the emission spectrum is still dominated by the broadband emission of Eu2+. The Eu2+ emission maximum was red-shifted to 503 nm, and a significant increase in the FWHM value to 142 nm (5268 cm−1) was observed. Considering these spectral changes, we investigated the dependence of emission spectra on the excitation wavelength, systematically changing λex from 250 to 420 nm with a step of 10 nm (Fig. 2c). This variation allowed tuning of the emission spectra in terms of emission maximum and FWHM (Fig. 2d). Two opposite trends were observed depending on the excitation wavelength: the increase of the excitation wavelength from 250 to 310 nm led to a blue shift accompanied by the narrowing of the emission band, while in the range from 310 to 420 nm, the effect was opposite. Summarizing, under specific excitation, the emission maximum can be shifted from 467 to 517 nm, and FWHM values can be varied from 87 to 142 nm. Yu et al.37 previously achieved similar spectral tuning in Eu2+/Eu3+-doped Ba2InTaO6. The authors associated the observed shift with the occupation of different crystallographic sites by Eu2+ ions. The observed dependence of the CIE 1931 chromaticity coordinates on the excitation wavelength was nearly linear (Fig. 2e), shifting from a blue to greenish-yellow region as the excitation wavelength increased from 310 to 420 nm. The exact coordinate values are summarized in Table S1.† Under specific excitation, a near-white emission can be obtained, demonstrating a color-adjustable emission. Fig. 2f shows the PL emission decay curve of Eu-doped α-TCP when the specimen was excited at 405 nm, and the emission was monitored at 475 nm. A two-exponential PL decay was observed with fast (τ = 0.16 μs) and slow (τ = 1.35 μs) components. The calculated effective PL lifetime value (τeff) was 1.27 μs. It has been previously reported that the appearance of the fast component could be linked to non-radiative energy migration between Eu2+ ions.38
The mechanism of Eu3+ self-reduction in the phosphates of divalent metals is usually explained by the formation of cationic vacancies due to substitution with aliovalent Eu3+ ions.28,39 Negatively charged vacancies act as electron donors and reduce Eu3+ ions to the divalent state, being further protected from the oxidizing air atmosphere by the phosphate framework. In our case, such a mechanism is reasonably accepted; however, during the transformation of CPP to α-TCP in a vacuum, the charged phosphate species are evaporated, which also could influence the oxidation state of Eu ions in the matrix. Some reports on the Eu partial self-reduction in α-TCP synthesized by solid-state reaction are known.27,28,40 The comparison of the emission spectra under 393 nm revealed that we achieved a significantly higher degree of reduction, which could be associated with the peculiarities of the synthetic approach.
The Eu-doped DCPD precursor was also annealed in air at an identical temperature for comparison. The XRD analysis (Fig. S3†) revealed that the major crystalline phase was α-CPP, which indicates that the Ca/P ratio in the as-prepared and annealed materials is nearly the same. A negligible amount of β-TCP was also identified as an impurity. The formation of α-CPP was expected since α-CPP is assumed to be a high-temperature CPP polymorph, and an annealing procedure was performed above the β- to α-CPP phase transition temperature.29 The PL spectrum of this sample is dominated by sharp emission lines originating from Eu3+, indicating that Eu ions mainly exist in the +3 oxidation state. Nevertheless, the emission band centered at ca. 412 nm was also observed, suggesting that the minor amount of Eu ions was reduced during the annealing. Previously, Doat et al.41 reported the partial stabilization of Eu2+ ions in α-CPP synthesized under an air atmosphere and thermally quenched in liquid nitrogen. The emission band arising from Eu2+ was observed at 415 nm, which agrees well with our results.
Temperature-dependent PL spectra of Eu-doped α-TCP obtained under excitation wavelengths of 285 and 393 nm are depicted in Fig. 3. Under 285 nm excitation, the PL spectra changed in terms of intensity, spectral shape, and emission maximum (Fig. 3a). At 77 K, two emission peaks centered at 457 and 505 nm are clearly seen; however, with an increase in temperature, the spectral shape transforms into an asymmetric single-peak band. The position of the emission maximum was nearly stable at 470 nm in the temperature range from 150 to 350 K, followed by a red shift at higher temperatures. The FWHM value exhibited a multidirectional trend depending on the temperature: in the range from 77 to 300 K, it decreased, while in the range from 300 to 450 K, it increased (Fig. 3b). The maximum value of 114.5 nm (4761 cm−1) was determined at 77 K. A nearly linear thermal quenching of integrated intensity was observed (Fig. 3b). At 400 K the integrated emission intensity constituted 51% of that at 300 K. The relatively strong thermal quenching of emission might be caused by thermal ionization (photoionization), thermally activated crossover to the ground state, multiphonon-relaxation, and energy transfer to quenching centers.42–44 The thermal quenching that occurs might limit the phosphor from high-temperature applications. The CIE chromaticity coordinates were located in the cyan-white region (Fig. 3c).
 | ||
| Fig. 3 Temperature-dependent PL spectra (a and d); dependence of integrated emission intensity and FWHM on temperature (b and e); dependence of the CIE 1931 chromaticity coordinates on temperature (c and f) for Eu-doped α-TCP excited at 285 nm (a–c) and 393 nm (d–f). | ||
In the case of 393 nm excitation, the emission spectra obtained were significantly broader (Fig. 3d). The FWHM value demonstrated a decreasing trend in the temperature range from 77 to 350 K and nearly stabilized at higher temperatures (Fig. 3e). The highest value of 282 nm (8942 cm−1) was also observed at 77 K. For the calculations of FWHM, only the emission of Eu2+ ions was involved; the emission of Eu3+ was ignored. The diversified dependence of FWHM values for both excitation wavelengths can be associated with the different behavior of Eu2+ ions occupying particular sites in the crystal lattice. The emission intensity decreased with the increasing temperature owing to the non-radiative decay at higher temperatures. The observed decrease was also nearly linear; in this case, at 400 K, it constituted 49% of that at 300 K. The thermal quenching in the red region was more pronounced, which is reflected in the gradual shift of CIE chromaticity coordinates from orange to greenish-white (Fig. 3f).
The deconvolution of broadband emission spectra with multiple peaks can ascribe the spectral components to the Eu2+ ions occupying specific crystallographic sites. For instance, such an approach was demonstrated by Liao et al.,9 who investigated the PL spectra of Eu2+-doped Rb0.5K1.5CaPO4(F, Cl). The authors concluded that there are 3 suitable sites for Eu2+ substitution in the analyzed crystal structure; consequently, 3 components of the PL spectrum were specifically ascribed. In our case, the determination of Ca sites occupied by Eu2+ ions is extremely challenging due to the peculiarities of the α-TCP crystal structure. This compound crystallizes in a monoclinic crystal structure with a P21/a space group having 18 inequivalent crystallographic Ca sites.45 Considering the doping level investigated in this work, the commonly employed Rietveld refinement of the XRD data does not seem to be a reliable tool for determining the Eu2+ position in the crystal lattice.
The theoretical work by Matsunaga et al. investigated the thermodynamic equilibrium between TCP polymorphs substituted with divalent cations having different sizes.46 One of the investigated ions was Sr2+. Taking into account the nearly identical ionic radii of Eu2+ and Sr2+,47 a simplified assumption can be made that the behavior of Eu2+ in the considered crystal structure could be identical to that of Sr2+. It was shown that 4 specific Ca sites in the α-TCP structure are energetically more favorable for Sr2+. The lowest defect formation energies of Sr2+ are in the Ca-5 (CN = 7), Ca-8 (CN = 7), Ca-14 (CN = 7), and Ca-17 (CN = 7) positions, having polyhedron volumes of 19.9, 19.9, 17.2, and 20.0 Å3, respectively.45
The fitting of emission spectra obtained at 77 and 300 K under 285 and 393 nm excitation is shown in Fig. 4. Regardless of the excitation wavelength and temperature, all spectra can be fitted with 3 sub-spectra corresponding to Eu2+ ions in at least 3 different environments. Another approach for ascribing the sub-spectra proposed by van Uitert considers the calculations involving the coordination number, radius of the cation replaced by Eu2+, and electron affinity of anion atoms.48 In the case of α-TCP:Eu2+, there is only one type of cation and anion in the matrix. If we assume the abovementioned calculations by Matsunaga et al.46 for Eu2+, the coordination number for all four positions is also the same. Thus, further investigation is needed to attribute the emission of Eu2+ centers to specific crystallographic sites.
 | ||
| Fig. 4 PL spectra and Gaussian fitting curves of Eu-doped α-TCP for λex = 285 nm (a and b) and λex = 393 nm (c and d) at 77 K (a and c) and 300 K (b and d). | ||
EPR spectroscopy was employed to confirm the multi-site occupancy of Eu2+ ions in α-TCP. The EPR spectra of the investigated α-TCP sample (Fig. 5) indicated the presence of Eu in the divalent state. The detected spectra consisted of many lines spread over a broad field range with maximum intensity at g ≈ 2.0 (356 mT for the X-band; 1221 mT for the Q-band), typical for Eu2+ in various crystalline hosts.49–52 The EPR spectrum of Eu2+ originates from zero-field splitting (ZFS) of the 8S7/2 ground state, which results in seven allowed orientation-dependent ZFS transitions.51,52 In powdered samples, the averaging across all possible single-crystal orientations and the overlap of signals from multiple possible sites generally lead to complex spectra, which are problematic to interpret unambiguously. The “EPR-silent” Eu3+ ions do not contribute to the spectrum.51
 | ||
| Fig. 5 X-band (a) and Q-band (b) EPR spectra of the Eu-doped α-TCP powder. | ||
The EPR spectra suggest the formation of several isolated Eu2+ centers in α-TCP. One component is quite symmetrically centered at g ≈ 2.0 in the spectra acquired at both frequency bands, indicating an intermediate ZFS magnitude for this site. Additional signals at lower fields are visible in the X-band spectrum (40, 120, and 255 mT), implying at least one more Eu2+ site with larger ZFS values. The absence of broad, structureless signals typically associated with Eu2+ clustering effects50 suggests that europium is sufficiently diluted to form isolated centers in α-TCP.
It was observed that when the Eu-doped α-TCP sample is exposed to ionizing radiation (X-rays and UV), the delocalized charge carriers are generated and subsequently trapped by several charge traps corresponding to different material defects. This effect is demonstrated by the TSL measurements shown in Fig. 6. The basic principle of the TSL method involves irradiation of the material with appropriate high-energy radiation at room or lower temperature; thus, some charge carriers will delocalize and become trapped. Then, the material is heated typically at a constant heating rate, and charges can be released if sufficient energy is accumulated. The luminescence signal may increase and decrease throughout all measuring processes, with increases corresponding to the traps with specific activation energy. In the end, a glow curve consisting of one or more glow peaks as I = f(T) will be obtained.53
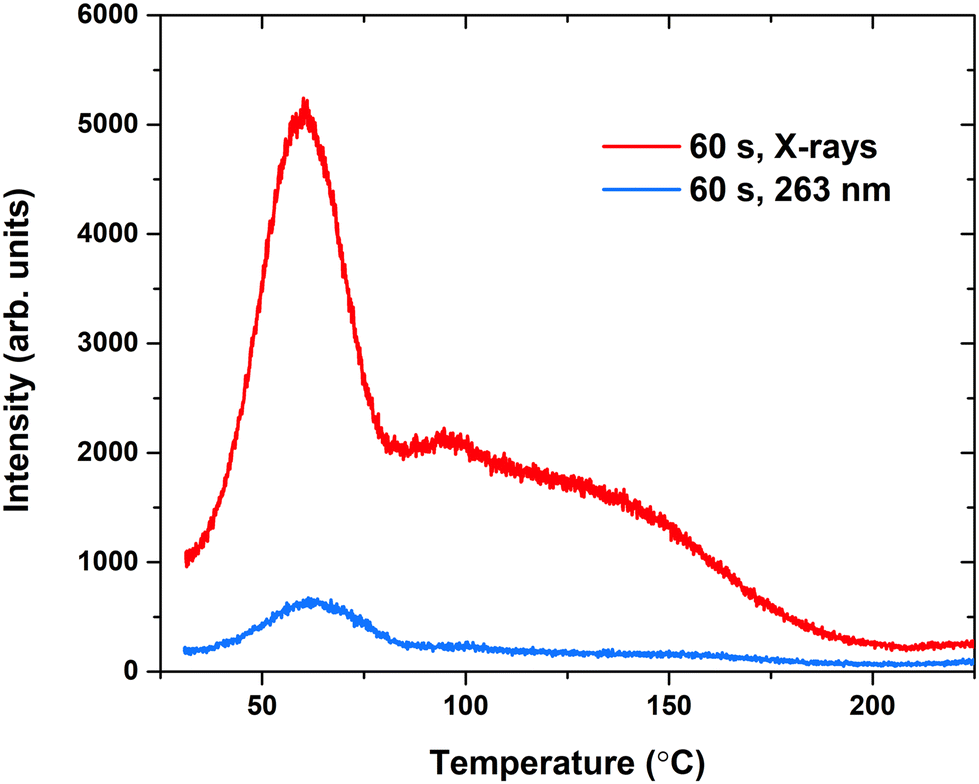 | ||
| Fig. 6 TSL glow curves of the Eu-doped α-TCP sample after irradiation with X-rays or UV for 60 s, heating rate 1 °C s−1. | ||
Fig. 6 depicts the TSL glow curves of α-TCP:Eu2+ after irradiation with X-rays and UV at 263 nm. In both cases, the same structure of glow peaks remains; thus, the same traps are filled regardless of the irradiation source. On the other hand, after exposure to the X-rays, a much stronger TSL signal is detected; thus, there is a higher filled trap concentration. For further analysis, X-ray irradiation was selected. The glow curve consists of multiple glow peaks, with the most prominent one having a maximum at around 60 °C; at higher temperatures, there are indistinguishable numbers of overlapping glow peaks.
For further analysis, the well-established and extensively used Tmax–Tstop and initial rise analysis (IRA) methods were applied.54–56 For Tmax–Tstop, a step of 5 °C from 35 to 150 °C was used. Measured TSL glow curves are shown in Fig. 7a, while Fig. 7b depicts IRA plots of the same glow curves. The data for Fig. 7c were obtained by analyzing these two plots. The Tmax–Tstop plot reveals that charge traps can be divided into three groups. Similarly, trap depth or activation energy (Ea) values can be divided into groups with approximate values of 0.80, 0.75, and 0.68 eV. However, counterintuitively, lower Ea values correspond to higher Tstop values. This goes against basic TSL theory, which predicts that glow peaks with maxima at higher temperatures correspond to deeper traps. Similar results have been observed previously but not explained indisputably.57,58 False shallow trap depth value appearance in IRA plots could be linked to de-trapping via thermally assisted tunneling, where the obtained Ea value would stand not for the energy necessary to breach the gap between the trap and the band as in the classical TSL process but for the energy difference between the ground level and the tunneling level of a trap.59,60 However, further investigation outside the scope of this research would be necessary for profound conclusions.
 | ||
| Fig. 7 TSL glow curves of the Eu-doped α-TCP sample after preheating up to different Tstop values between 35 and 150 °C with a 5 °C step. The sample was irradiated with X-rays for 60 s before each preheating (a); IRA plots of the glow curves obtained from the Tmax–Tstop experiment (b); the Tmax–Tstop plot and the Ea–Tstop plot (c). | ||
Previous studies have suggested that materials with trap depths of around 0.7 eV could lead to effective persistent luminescence at room temperature.61 This is also true for the Eu-doped α-TCP sample. After irradiation with X-rays, an afterglow characterizable by an Eu2+ typical broadband spectrum28,62,63 with maxima around 480 nm can be detected for at least 10 h (Fig. 8). The persistent luminescence spectrum is primarily associated with a single Eu2+ site. This further suggests tunneling as a key mechanism of persistent luminescence. The tunneling recombination process is highly sensitive to the spatial separation between traps and luminescent centers. Therefore, it is expected that persistent luminescence will be predominantly generated from Eu2 ions in close proximity to traps.
 | ||
| Fig. 8 The afterglow decay curve of the Eu-doped α-TCP sample after irradiation for 5 min with X-rays; inset: the corresponding Eu2+ spectrum. | ||
The α-TCP crystal structure is known to be highly susceptible to the presence of impurities and the conditions of thermal treatment.46,64 Moreover, thermal quenching is often used to prepare the phase-pure α-TCP; however, the annealing conditions are also closely related to the chemical purity. Unfortunately, we were not able to prepare α-TCP doped with a higher amount of Eu2+ due to the melting of the product. At the same time, the synthesis at lower temperatures resulted in the formation of β-TCP impurities. In exploring the self-reduction phenomenon, further attempts should be made to prepare Yb2+ and Sm2+-doped α-TCP in a similar manner. Both these ions are redox active; however, their reduction potentials are significantly more negative.65 Moreover, the distribution of Eu2+ ions within the α-TCP structure and, consequently, the optical properties could be tuned by partially substituting Ca2+ with Sr2+ or Ba2+. While smaller cations are hardly adopted by the α-TCP structure, Sr2+ or Ba2+ ions can be easily dissolved by occupying particular Ca sites.
4. Conclusions
In this work, Eu2+-doped α-TCP was successfully synthesized via a phase transformation of Eu3+-doped DCPD. The phase conversion, accompanied by a reduction of Eu3+ to Eu2+, was achieved by annealing the starting material at 1300 °C in a vacuum. The initial Ca/P ratio of 1:1 characteristic of DCPD changed to ca. 1.5:1 due to the selective evaporation of phosphate species. The obtained material exhibited a broadband emission covering the entire visible spectrum. The emission tunability was achieved by varying the excitation wavelength, resulting in the shift of the emission maximum and FWHM values ranging from 87 to 142 nm at room temperature. The broadband emission was achieved due to the multi-site occupancy of Eu2+ ions in a complex α-TCP matrix. The deconvolution of emission spectra indicated that Eu2+ ions occupy at least 3 inequivalent crystallographic sites. TSL measurements revealed the presence of three types of traps with activation energy values of 0.80, 0.75, and 0.68 eV. After irradiation with X-rays, an afterglow of the Eu2+ broadband spectrum could be detected for at least 10 h. The high degree of reduction and multi-site occupancy of Eu2+ ions in α-TCP achieved by the selected synthetic approach opens new possibilities for the development of color-tunable broadband phosphors.
Data availability
The data supporting this study have been included within the article and the corresponding ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the Research Council of Lithuania (LMTLT), agreement no. S-MIP-23-85. A. A. and G. D. acknowledge the donation of SIA “Mikrotīkls” (MikroTik) administered by the University of Latvia Foundation, project no. 2326. V. P. acknowledges the LZP grant lzp-2023/1-0063.References
- S. Liu, L. Li, X. Qin, R. Du, Y. Sun, S. Xie, J. Wang, M. S. Molokeev, S. Xi, J. G. Bunzli, L. Zhou and M. Wu, Adv. Mater., 2024, e2406164 CrossRef PubMed.
- Z. Yang, Y. Zhao, Y. Zhou, J. Qiao, Y.-C. Chuang, M. S. Molokeev and Z. Xia, Adv. Funct. Mater., 2022, 32, 2103927 CrossRef CAS.
- Y. Zhu, X. Wang, J. Qiao, M. S. Molokeev, H. C. Swart, L. Ning and Z. Xia, Chem. Mater., 2023, 35, 1432–1439 CrossRef CAS.
- Z. Tang, Q. Zhang, Y. Cao, Y. Li and Y. Wang, Chem. Eng. J., 2020, 388, 124231 CrossRef CAS.
- C. Jiang, Q. Liu, K. Li, Y. Feng, Y. Fu, Y. Li, Y. Zhang, X. Qian, B. Wei and P. Du, Chem. Eng. J., 2024, 496, 154360 CrossRef CAS.
- Z. Tang, F. Du, L. Zhao, H. Liu, Z. Leng, H. Xie, G. Zhang and Y. Wang, Laser Photonics Rev., 2023, 17, 2200911 CrossRef CAS.
- G.-D. Xie, J.-Y. Si, G.-H. Li, G.-M. Cai, Z.-P. Wang and X.-J. Wang, J. Mater. Chem. C, 2024, 12, 18066–18076 RSC.
- H. Chen, Z. Zhang, R. Mi, M. S. Molokeev, Y. Liu, B. Wang, L. Mei, M. Fang, X. Wu, X. Min and Y.-g. Liu, J. Lumin., 2024, 275, 120795 CrossRef CAS.
- M. Liao, F. Wu, D. Zhu, X. Zhang, H. Dong, Z. Lin, M. Wen and Z. Mu, Chem. Eng. J., 2022, 449, 137801 CrossRef CAS.
- D. Wu, J. Si, G. Xie, J. Huang and G. Cai, Ceram. Int., 2023, 49, 34805–34813 CrossRef CAS.
- Z. Fan, Y. Wang, Z. Leng, G. Gao, L. Li, L. Huang and G. Li, J. Am. Chem. Soc., 2024, 146, 11500–11506 CAS.
- Z. Luo, H. Zou, Z. Zheng, Y. Zhao, C. Wang, Q. Sun and Y. Song, Inorg. Chem., 2024, 63, 17449–17459 CrossRef CAS PubMed.
- R. G. Carrodeguas and S. De Aza, Acta Biomater., 2011, 7, 3536–3546 CrossRef CAS PubMed.
- M. Cao, J. Tian, W. Zhuang, R. Liu, Y. Liu, G. Chen, G. Zhou, L. Wang and J. Wang, Inorg. Chem., 2022, 61, 1805–1815 CrossRef CAS PubMed.
- Z. Meng, Y. Gao, J. a. Song, Z. Jiang, W. Lv, Q. Zeng, D. Wen and T. Hu, Dalton Trans., 2023, 52, 5443–5452 RSC.
- P. Liang, W.-L. Lian and Z.-H. Liu, Chem. Commun., 2021, 57, 3371–3374 RSC.
- M. Sójka, M. Runowski, T. Zheng, A. Shyichuk, D. Kulesza, E. Zych and S. Lis, J. Mater. Chem. C, 2022, 10, 1220–1227 RSC.
- W. Teng, H. Dong, C. Hu, X. Yang, J. Wang, X. Liang, B. Yang, J. Ru, B. Teng and D. Zhong, J. Lumin., 2024, 269, 120524 CrossRef CAS.
- H. Wang, K. Su, L. Mei, Q. Guo and L. Liao, Inorg. Chem., 2023, 62, 12468–12479 CrossRef CAS PubMed.
- H. Wang, K. Su, B. Ma, Y. Wang, L. Mei, Q. Guo and L. Liao, Chem. Eng. J., 2023, 478, 147361 CrossRef CAS.
- S. Li, Z. Qiu, Y. Mo, H. Zhang, H. Lian, J. Zhang and S. Lian, Dalton Trans., 2022, 51, 6622–6630 RSC.
- L. Sinusaite, I. Grigoraviciute-Puroniene, A. Popov, K. Ishikawa, A. Kareiva and A. Zarkov, Ceram. Int., 2019, 45, 12423–12428 CrossRef CAS.
- L. Sinusaite, A. Popov, E. Raudonyte-Svirbutaviciene, J.-C. Yang, A. Kareiva and A. Zarkov, Ceram. Int., 2021, 47, 12078–12083 CrossRef CAS.
- D. Griesiute, E. Raudonyte-Svirbutaviciene, A. Kareiva and A. Zarkov, CrystEngComm, 2022, 24, 1166–1170 RSC.
- H. Ji, Z. Huang, Z. Xia, M. S. Molokeev, M. Chen, V. V. Atuchin, M. Fang, Y. g. Liu and X. Wu, J. Am. Ceram. Soc., 2015, 98, 3280–3284 CrossRef CAS.
- W. Zhou, J. Han, X. Zhang, Z. Qiu, Q. Xie, H. Liang, S. Lian and J. Wang, Opt. Mater., 2015, 39, 173–177 CrossRef CAS.
- D. Luo, C. Tong, Y. Zhu, C. Xu and Y. Li, J. Lumin., 2020, 219, 116863 CrossRef CAS.
- C. Tong, Y. Zhu, C. Xu and Y. Li, Phys. B, 2016, 500, 20–23 CrossRef CAS.
- D. Griesiute, E. Garskaite, A. Antuzevics, V. Klimavicius, V. Balevicius, A. Zarkov, A. Katelnikovas, D. Sandberg and A. Kareiva, Sci. Rep., 2022, 12, 7116 CrossRef CAS PubMed.
- D. Griesiute, J. Gaidukevic, A. Zarkov and A. Kareiva, Sustainability, 2021, 13, 7859 CrossRef CAS.
- A. Antuzevics, G. Krieke, G. Doke, A. Zarins, L. Avotina, E. Sprugis, J. Stadulis, A. Beganskiene, A. Kareiva and A. Zarkov, J. Alloys Compd., 2024, 996, 174779 CrossRef CAS.
- X. Han, C. Xin, S. Wang, J. Wu, Z. Ye, H. Yu and H. Zhang, Inorg. Chem., 2023, 62, 9679–9686 CrossRef CAS PubMed.
- S. N. Jayaramu, D. Janardhana, L. J. B. Erasmus, E. Coetsee, D. E. Motaung and H. C. Swart, Dalton Trans., 2024, 53, 16557–16576 RSC.
- L. Sinusaite, A. Antuzevics, A. I. Popov, U. Rogulis, M. Misevicius, A. Katelnikovas, A. Kareiva and A. Zarkov, Ceram. Int., 2021, 47, 5335–5340 CrossRef CAS.
- Z. Hao, J. Zhang, X. Zhang and X. Wang, Opt. Mater., 2011, 33, 355–358 CrossRef CAS.
- X.-Y. Sun, X.-C. Le, Z. Xiao, X. Shi, W.-F. Wang, Z. Hu, Q.-M. Yang, R. Wei and H. Guo, J. Am. Ceram. Soc., 2020, 103, 3119–3125 CrossRef CAS.
- B. Yu, Y. Li, Y. Wang, N. Li, P. Xiao, D. Liu and L. Geng, Inorg. Chem., 2022, 61, 2463–2475 CrossRef CAS PubMed.
- W. Zhou, J. Han, F. Pan, J. Zhang, Q. Xie, S. Lian, L. Yu and J. Wang, J. Am. Ceram. Soc., 2014, 97, 3631–3635 CrossRef CAS.
- M. Peng, Z. Pei, G. Hong and Q. Su, J. Mater. Chem., 2003, 13, 1202–1205 RSC.
- C. Tong, Y. Zhu, C. Xu, L. Yang and Y. Li, Phys. B, 2017, 521, 153–157 CrossRef CAS.
- A. Doat, F. Pellé and A. Lebugle, J. Solid State Chem., 2005, 178, 2354–2362 CrossRef CAS.
- P. Dorenbos, J. Phys.:Condens. Matter, 2005, 17, 8103 CrossRef CAS.
- G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer-Verlag, Berlin, Heidelberg, 1994 Search PubMed.
- W. M. Yen and M. J. Weber, Inorganic phosphors: compositions, preparation and optical properties, CRC press, 2004 Search PubMed.
- M. Yashima and Y. Kawaike, Chem. Mater., 2007, 19, 3973–3979 CrossRef CAS.
- K. Matsunaga, T. Kubota, K. Toyoura and A. Nakamura, Acta Biomater., 2015, 23, 329–337 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- L. G. van Uitert, J. Lumin., 1984, 29, 1–9 CrossRef CAS.
- M. Buryi, T. Salamakha, V. Babin, J. Paterek, F. Hájek, Z. Remeš, L. Landová, E. Trusova and Y. Tratsiak, Ceram. Int., 2021, 47, 29232–29252 CrossRef CAS.
- L. Havlák, J. Bárta, M. Buryi, V. Jarý, E. Mihóková, V. Laguta, P. Boháček and M. Nikl, J. Phys. Chem. C, 2016, 120, 21751–21761 CrossRef.
- D. K. Patel, B. Rajeswari, V. Sudarsan, R. K. Vatsa, R. M. Kadam and S. K. Kulshreshtha, Dalton Trans., 2012, 41, 12023–12030 RSC.
- A. Antuzevics, M. Kemere, G. Krieke and R. Ignatans, Opt. Mater., 2017, 72, 749–755 CrossRef CAS.
- S. W. McKeever, Thermoluminescence of solids, Cambridge university press, 1985 Search PubMed.
- S. W. S. McKeever, Phys. Status Solidi A, 1980, 62, 331–340 CrossRef CAS.
- V. Pagonis, G. Kitis and C. Furetta, Numerical and practical exercises in thermoluminescence, Springer Science & Business Media, 2006 Search PubMed.
- G. Doke, G. Krieke, P. Rodionovs, D. Nilova and A. Antuzevics, J. Rare Earths, 2025, 43, 676–683 CrossRef CAS.
- S. Akca, M. Oglakci, Z. G. Portakal, N. Kucuk, M. Bakr, M. Topaksu and N. Can, Radiat. Phys. Chem., 2019, 160, 105–111 CrossRef CAS.
- K. H. Gavhane, M. S. Bhadane, A. S. Bhoir, P. P. Kulkarni, B. J. Patil, V. N. Bhoraskar, S. D. Dhole and S. S. Dahiwale, J. Alloys Compd., 2020, 817, 152805 CrossRef CAS.
- A. Vedda, M. Nikl, M. Fasoli, E. Mihokova, J. Pejchal, M. Dusek, G. Ren, C. R. Stanek, K. J. McClellan and D. D. Byler, Phys. Rev. B:Condens. Matter Mater. Phys., 2008, 78, 195123 CrossRef.
- M. Kitaura, A. Sato, K. Kamada, A. Ohnishi and M. Sasaki, J. Appl. Phys., 2014, 115, 083517 CrossRef.
- K. Van den Eeckhout, A. J. J. Bos, D. Poelman and P. F. Smet, Phys. Rev. B:Condens. Matter Mater. Phys., 2013, 87, 045126 CrossRef.
- J. Qiao, J. Zhao and Z. Xia, Opt. Mater.:X, 2019, 1, 100019 CAS.
- M. Leimane, K. Krizmane, I. Bite, J. Grube and V. Vitola, Materials, 2023, 16, 4416 CrossRef CAS PubMed.
- L. Sinusaite, A. Kareiva and A. Zarkov, Cryst. Growth Des., 2021, 21, 1242–1248 CrossRef CAS.
- M. R. MacDonald, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 9857–9868 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5dt00681c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |