
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of ancillary ligands on the formation and functionality of oxovanadium(V) metallosupramolecular assemblies: advanced computational and catalytic analyses†
Edi
Topić
 a,
Josipa
Sarjanović
a,
Danijela
Musija
b,
Mirna
Mandarić
a,
Andrea
Cocut
a,
Tomica
Hrenar
a,
Dominique
Agustin
cd,
Jana
Pisk
*a and
Višnja
Vrdoljak
*a
a,
Josipa
Sarjanović
a,
Danijela
Musija
b,
Mirna
Mandarić
a,
Andrea
Cocut
a,
Tomica
Hrenar
a,
Dominique
Agustin
cd,
Jana
Pisk
*a and
Višnja
Vrdoljak
*a
aUniversity of Zagreb, Faculty of Science, Department of Chemistry, Horvatovac 102a, 10000 Zagreb, Croatia. E-mail: jana.pisk@chem.pmf.hr; visnja.vrdoljak@chem.pmf.hr
bUniversity of Zagreb, School of Medicine, Department of Chemistry and Biochemistry, Šalata 3, 10000 Zagreb, Croatia
cLCC-CNRS (Laboratoire de Chimie de Coordination), 205 Route de Narbonne, BP44099, CEDEX 4, 31077 Toulouse, France
dDepartment of Chemistry, IUT Paul Sabatier, Université Paul Sabatier, University of Toulouse, Av. G. Pompidou, CS20258, 81104 Castres, France
First published on 26th February 2025
Abstract
A coordination-driven self-assembly approach offers an opportunity for designing metallosupramolecular architectures with tailored properties. Applying this strategy, we present the synthesis and detailed characterization of tetranuclear and polynuclear vanadium(V) compounds with an aroylhydrazone ligand. These assemblies were obtained using the 3-methoxy-2-hydroxybenzaldehyde isonicotinoyl hydrazone ligand (H2VIH) and NH4VO3 in the presence of primary aliphatic alcohols with increasing carbon chain length (from one to five carbon atoms). Reactions of lower alcohols selectively yielded metallocyclic compounds [VO(VIH)(OR)]4 (1, 2, and 3, where R = CH3, C2H5, and C3H7, respectively), while reactions of higher alcohols afforded infinite zig-zag chain polymers [VO(VIH)(OR)]n (4 and 5, where R = C4H9 and C5H11, respectively). Their formation was studied experimentally and computationally. Quantum chemical calculations using density functional theory provided valuable insights into the stability of both cyclic and chain assemblies. The solid-state structures of 1–5 and pseudopolymorphs of [VO2(HVIH)] (6·0.5H2O·0.5CH3OH and 6·2H2O) were confirmed using single-crystal and powder X-ray diffraction methods. The oxovanadium(V) species were tested as catalysts for cyclooctene and benzyl alcohol oxidation, with results correlated with those for analogous molybdenum complexes [MoO2(VIH)]4 (7) and [MoO2(VIH)(C2H5OH)] (8). The influence of the metal centre, nuclearity, and ancillary ligand identity on catalytic performance was also investigated, revealing that the vanadium metal center influences the catalytic activity due to its capacity to form robust structures.
Introduction
Transition metal oxo complexes in high oxidation states, particularly those of Groups 5 and 6, play significant roles in various chemical and biological processes. Complexes containing VIV,V,1–7 MoVI,8–11 and WVI,12–14 are commonly used as (pre)catalysts in oxidation reactions with oxidants such as O2, H2O2, or tert-butyl hydroperoxide (TBHP). Notably, related discrete metal complexes can feature multiple coordination pockets which further enhance their catalytic versatility.15–17 Alternatively, {MoVIO2}2+ metallosupramolecular complexes are gaining attention for their effectiveness as epoxidation catalysts.18–22 Metal–organic frameworks STA-12(Co), STA-12(Ni), and STA12(Ni + 20% Co) effectively catalyse the oxidation of benzylic alcohols.23 A Zn-based MOF (ZIF-8) and its Co(II)- and Cu(II)-modified assemblies also showed good catalytic activity.24 Moreover, an Fe(phthalocyanine) complex demonstrated high effectiveness for alcohol oxidation using TBHP and H2O2 without organic solvents.25However, reports on oxovanadium(V) metallosupramolecular architectures involving polydentate organic ligands and {VO}3+ units remain relatively rare. The complex [{VO2(2,2′-bpy)}2(tp)]n is the first 1D vanadium(V) coordination polymer containing bipyridine and terephthalate ligands.26 Rare examples of VIVO assemblies include the cyclic complex [VO(3HPA)]4 (3HPA = 3-hydroxypicolinic acid) and coordination polymers [VO(dod)2]X2 (dod = 1,4-diazoniabicyclo[2,2,2] octane-1-4-diacetate).27,28 A compelling example is interconvertible vanadium-seamed hexameric pyrogallol[4]arene nanocapsules.29 To the best of our knowledge, oxovanadium(V) complexes with an isonicotinoyl or nicotinoyl moiety represent rare examples where the hydrazone ligand coordinates two metal centres to form a polymer.30–32
A coordination-driven self-assembly method is a powerful tool for developing metallosupramolecular architectures.33–37 The assembly outcomes are influenced by a variety of factors, including the metal ion's coordination geometry, ligands’ denticity, spacer unit flexibility, and donor atom positions.38,39 The process involves reversible metal–ligand bond formation and cleavage,40,41 resulting in diverse supramolecular isomers, from discrete entities to complex networks. Structures that maximize metal–ligand bonding interactions typically have increased enthalpy, while smaller assemblies may benefit from entropic effects,42–45 allowing for the design of functional materials with tailored properties.
Building on our prior research,21,22,46–49 we aimed to develop vanadium-based assemblies utilizing a coordination-driven self-assembly method. We selected 3-methoxy-2-hydroxy-benzaldehyde isonicotinoyl hydrazone (H2VIH, Scheme 1) to link vanadium mononuclear building units into higher dimensional structures. We aimed to determine whether primary alcohols ROH (CnH2n+1OH, n = 1–5) could facilitate the assembly of specific supramolecular structures. We conducted quantum chemical calculations to evaluate the stability of cyclic tetramer complexes and compared them to chain configurations. By examining their unique structural properties and interactions, we aimed to uncover their potential applications in various fields, including catalysis.
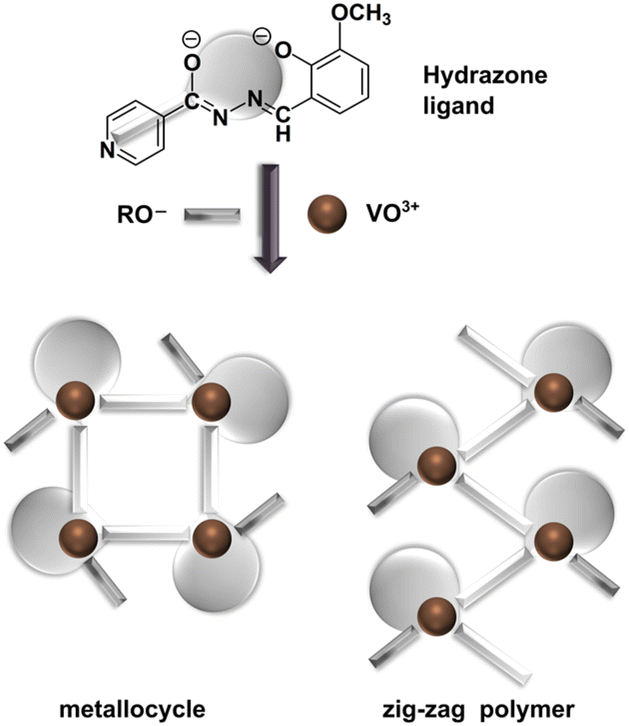 | ||
| Scheme 1 A schematic presentation of the doubly deprotonated ligand VIH2−, the ancillary ligand OR−, and metallosupramolecular assemblies [VO(VIH)(OR)]4 (1, 2, and 3, where R = CH3, C2H5, and C3H7, respectively) and [VO(VIH)(OR)]n (4 and 5, where R = C4H9 and C5H11, respectively). | ||
The preliminary evaluations of their catalytic potential were conducted through two oxidation reactions: cyclooctene epoxidation and benzyl alcohol oxidation. The production of epoxides is crucial in industry, especially in the production of plastics, adhesives and coatings, highlighting their significance in a variety of applications.50,51 On the other hand, benzaldehyde, utilized in fragrances and pharmaceuticals,52,53 is often produced through conventional synthesis methods that tend to generate harmful by-products.54,55 Therefore, exploring more sustainable synthesis pathways is essential for minimizing environmental impacts while meeting industrial needs.
Results and discussion
Metallosupramolecular assemblies
We have previously reported the synthesis of 3-methoxy-2-hydroxybenzaldehyde isonicotinoyl hydrazone (H2VIH) as part of our work on a series of aroylhydrazones.56 Refluxing a slight excess of H2VIH with one equivalent of NH4VO3 in the corresponding alcohol (CnH2n+1OH, n = 1–5) led to the isolation of dark red, almost black crystalline solids. These coordination assemblies were characterized by X-ray crystallography along with spectral and analytical methods. They were identified as metallosupramolecular tetranuclear [VO(VIH)(OR)]4 (1, 2, and 3, where R = CH3, C2H5, and C3H7, respectively) and polymeric products [VO(VIH)(OR)]n (4 and 5, where R = C4H9 and C5H11, respectively), Scheme 1.The three tetranuclear complexes 1, 2, and 3 crystallize in the tetragonal space group I41/a and exhibit remarkably similar crystal structures, as evidenced by powder XRD patterns (see the ESI, Fig. S1†) as well as the obtained structural model (Fig. 1a1, Table S1 and Fig. S2a–c, see the ESI†). The crystal structure of 2 has been reported previously (ref. code: CAZVIM).57 Each tetranuclear unit in these compounds consists of four [VO(VIH)] monomers linked through oxovanadium-pyridyl V–Npy coordination bonds, which are oriented approximately perpendicular to the chelation plane. This arrangement yields a robust metallocycle, representing a unique structure among the oxovanadium(V) compounds.
 | ||
| Fig. 1 (a1) Structural overlay of tetranuclear [VO(VIH)(OR)]4 species 1 (light green), 2 (light red) and 3 (light blue). (a2) Packing of tetramers in 1, showing significant crystal voids. (b1) Infinite polymeric chains found in 4. (b2) Packing of infinite polymeric chains of 4. (c) Coordination environment of the vanadium(V) core with mean and standard deviations of relevant bond lengths across compounds 1–5. (d) Monomer volume and logarithm of the spherical probe pore volume per monomer in relation to the ancillary chain length for compounds 1–5. Packing of molecules/polymeric chains in (a) 1 and (b) 4, shown in polyhedral style. Voids in 1 are represented by the yellow surface. | ||
In contrast, compounds 4 and 5, which feature disordered n-butoxy and n-pentoxy ancillary ligands, assemble differently. These compounds crystallize in the orthorhombic Pca21 space group and form isostructural 1D coordination polymers (Fig. 1b1, Table S2, Fig. S2d and e, see the ESI†), albeit with a similar overall geometry and connectivity to observed metallocycles – differing only by the relative orientation of the neighbouring monomers. The chains can be described with a ubiquitous 2C1 topology, i.e. a 2-node one-dimensional periodic net (Fig. 1b2).58
Among tetranuclear complexes, complex 1 is particularly noteworthy due to its significant porosity (Fig. 1a2 and Fig. S3, see the ESI†). The spherical probe pore volume is approximately 30 Å3 per monomer unit, corresponding to about 9.2% of the unit cell volume.59 This substantial pore space within the methoxy complex suggests a possible potential for guest inclusion or adsorption, although the nature of the included chemical species in the measured crystal could not be conclusively determined from the diffraction data. This pore volume contrasts with the more compact structures observed in compounds 2–5, which lack notable porosity, indicating that the smallest alkoxy ligand uniquely supports this open framework structure. Alongside, an interesting trend is noticed when observing monomer volumes and pore volumes for the whole series – while monomer volumes increased approximately linearly with the number of carbon atoms in the ancillary ligand, the pore volume decreased exponentially (Fig. 1d).
The relevant metal–ligand distances in the oxovanadium core (Fig. S4, see the ESI†) remain fairly consistent regardless of the selected ancillary ligand (Fig. 1c), with bond lengths varying by only a few percent from the mean values, reflecting the well-known robustness of the ONO coordination environment of the doubly deprotonated ligand in enolato-imino tautomeric form (Fig. S5, see the ESI†). However, in the tetranuclear complex 1, two specific deviations stand out: the vanadium–methoxy oxygen bond distance is notably shorter than those in the other compounds, while the vanadium–hydrazonato nitrogen bond distance is elongated. One factor that appears to be correlated with the formation of discrete polymeric compounds is the torsion angle between the pyridyl and aryl rings of the VIH2− ligand. For complexes 1, 2, and 3, the interring torsion angles are 9°, 24°, and 14°, respectively, suggesting a relatively small degree of distortion that enables the formation of a compact, closed assembly. In contrast, polymers 4 and 5 exhibit significantly larger interring torsion angles of approximately 44° and 40°. This increased torsional angle, probably mediated by the bulk of the ancillary ligand, might favour an open chain-like polymeric structure.
Regarding intermolecular interactions, no significant hydrogen bonding is observed within these structures – all potential hydrogen bond donors are lost through ligand deprotonation and coordination. However, numerous C–H⋯O contacts are observed throughout the crystal structures, particularly in the 1D polymers, where these contacts contribute to stabilizing the extended framework.

According to the calculated standard Gibbs energies of binding (in Table 1), tetramers for all structures should exist. However, besides electronic effects, the steric effects of packing should also be considered. The overlay of crystal structures for 1, 2, and 3 is presented in Fig. 2. It is clear that the smallest distance between hydrogen atoms in the crystal structure of 3 is 2.407 Å. The addition of another methylene –CH2– group would cause steric repulsion, confirming that steric effects are the most important for crystal packing.
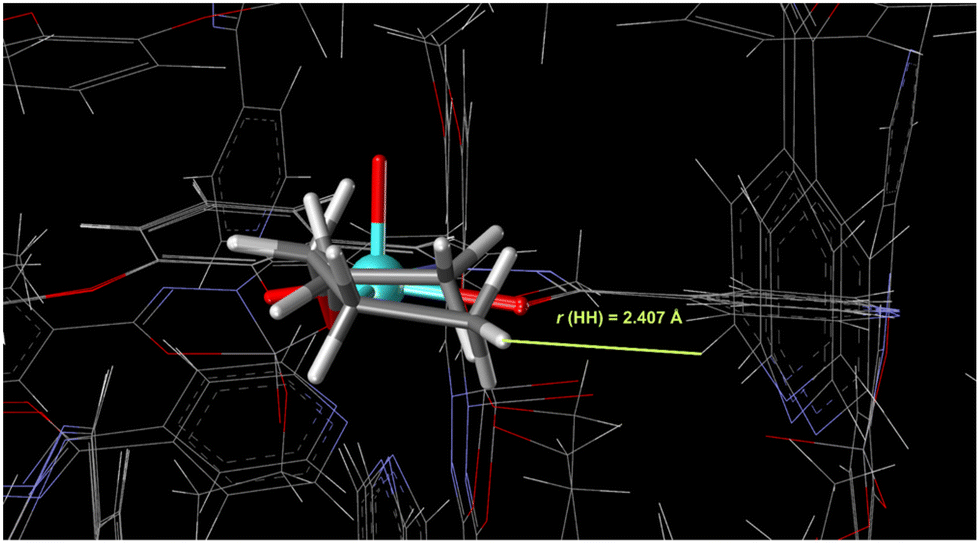 | ||
| Fig. 2 Overlaid crystal structures of 1, 2, and 3 where the distance from the hydrogen atom of the propyl group to the nearest atom is highlighted. Extension in the alkoxy group with an additional methylene –CH2– group in the same configuration is impossible due to the steric repulsions. | ||
| Ligand | ΔbG° (tetramer)/kJ mol−1 |
|---|---|
| OCH3 | –20.36 |
| OC2H5 | –23.24 |
| OC3H7 | –24.78 |
| OC4H9 | –32.97 |
| OC5H11 | –31.29 |
Stability was also cross-checked according to the chain polymer models. For each ligand, a chain tetramer model was created where the last binding place was filled up with a pyridine molecule. The difference in standard Gibbs energies of tetramer formation ΔΔfG° (cyclic–chain) was calculated as

Corresponding differences in standard Gibbs energies of tetramer formation are presented in Table 2. In each case, the cyclic tetramer structures were lower in energy, reflecting the fact that cyclic tetrameric structures with all ligands should exist. Therefore, the only possible explanation for different crystal packing is steric effects.
| Ligand | ΔΔfG° (cyclic–chain)/kJ mol−1 |
|---|---|
| OCH3 | –35.23 |
| OC2H5 | –29.55 |
| OC3H7 | –26.35 |
| OC4H9 | –46.62 |
| OC5H11 | –35.78 |
Mononuclear complexes
The reaction carried out in methanol at room temperature led to a dark-coloured solution after two weeks. Evaporation of the solution in an airstream yielded orange crystals of [VO2(HVIH)]·0.5H2O·0.5CH3OH (6·0.5H2O·0.5CH3OH). Upon prolonged exposure of the filtrate to air, a few crystals of [VO2(HVIH)]·2H2O (6·2H2O) also formed.The two pseudopolymorphs were characterized by SCXRD (ESI, Table S3, Fig. S1f and g†). The dihydrate 6·2H2O crystallizes in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group forming an intriguing supramolecular chain mediated by V
space group forming an intriguing supramolecular chain mediated by V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯water⋯water⋯Npy hydrogen bonds (ESI, Fig. S6†). On the other hand, hemihydrate hemimethanolate 6·0.5H2O·0.5CH3OH crystallizes in the monoclinic space group Cc with four symmetrically inequivalent molecules of [VO2(HVIH)] in the asymmetric unit. The relative orientation and difference density map between the neighbouring pyridyl fragments suggest partial protonation of the pyridyl moieties, forming a hydrogen bond between the pairs of neighbouring [VO2(HVIH)] molecules. Although the structure of solvent molecules could not be determined satisfactorily, the electron density of the refined solvent mask suggests the aforementioned composition. More precise conclusions about the nature of hydrogen bonding in 6·0.5H2O·0.5CH3OH would require a structural probe more sensitive to the hydrogen atom position. However, the molecular structures of the complexes are in line with the expectations – the {VO2}+ core coordinated by the ONO chelating pocket of a deprotonated ligand, in an enolato-imino form (Fig. S5, see the ESI†).
O⋯water⋯water⋯Npy hydrogen bonds (ESI, Fig. S6†). On the other hand, hemihydrate hemimethanolate 6·0.5H2O·0.5CH3OH crystallizes in the monoclinic space group Cc with four symmetrically inequivalent molecules of [VO2(HVIH)] in the asymmetric unit. The relative orientation and difference density map between the neighbouring pyridyl fragments suggest partial protonation of the pyridyl moieties, forming a hydrogen bond between the pairs of neighbouring [VO2(HVIH)] molecules. Although the structure of solvent molecules could not be determined satisfactorily, the electron density of the refined solvent mask suggests the aforementioned composition. More precise conclusions about the nature of hydrogen bonding in 6·0.5H2O·0.5CH3OH would require a structural probe more sensitive to the hydrogen atom position. However, the molecular structures of the complexes are in line with the expectations – the {VO2}+ core coordinated by the ONO chelating pocket of a deprotonated ligand, in an enolato-imino form (Fig. S5, see the ESI†).
Molybdenum(VI) assemblies
For comparison purposes in the catalytic part of this research, we prepared molybdenum complexes [MoO2(VIH)]4 (7) and [MoO2(VIH)(C2H5OH)] (8), known from our previous study.46 The crystal structure of [MoO2(VIH)(C2H5OH)] (8), not reported previously, has been described in short in the ESI (Fig. S6†).Principal component analysis
Next, we focused on gaining a deeper understanding of this reaction system. The reaction between NH4VO3 and H2VIH (with the initial concentrations of the reactants being 4.0 × 10−5 mol dm−3) was studied in methanol at room temperature by time-dependent UV-Vis spectrophotometry.Time-dependent UV-Vis spectra (Fig. 3) were decomposed using the 2nd-order tensor decomposition tool principal component analysis. The obtained results showed that the original dataset (389 spectra) could be correctly represented using only 2 principal components. These two components described 98% of the variance in the original data. The first principal component was responsible for 74.04% of the total variance and describes a major reaction involving the coordination of the hydrazonato ligand to vanadium. The second component describes 23.88% of the total variance and represents a secondary reaction in the system (Fig. 4). The analysis of the UV-Vis spectra of the V-hydrazonato complex (Fig. S7, see the ESI†) revealed a final transformation of the initially formed hydrazonato complex into the corresponding dioxovanadium(V) species in very diluted CH3OH solutions.
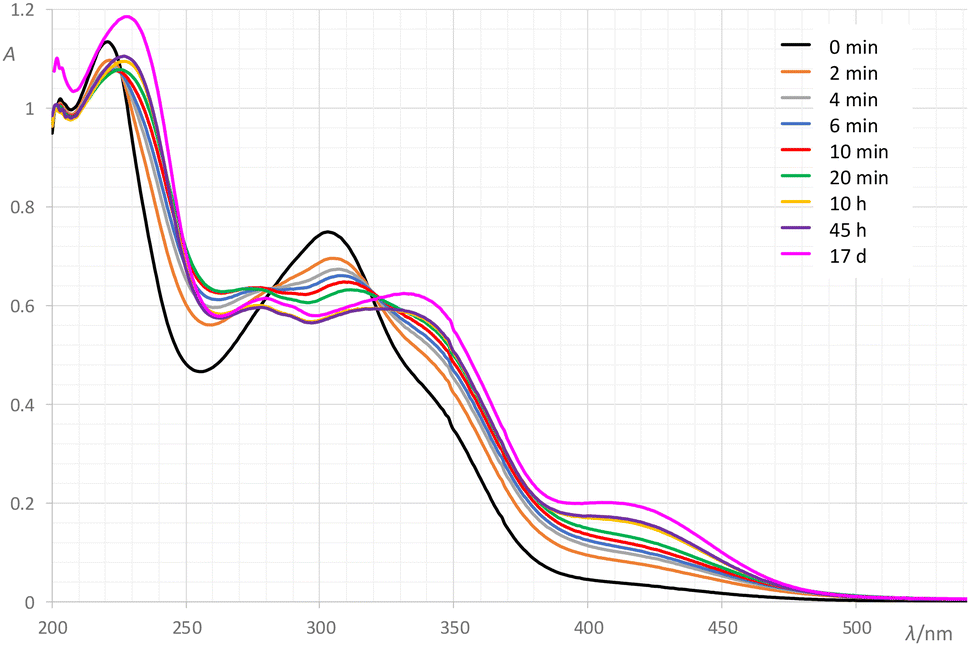 | ||
| Fig. 3 Time-dependent UV-Vis spectra of a reaction mixture of NH4VO3 and H2VIH in methanol at room temperature. | ||
 | ||
| Fig. 4 Principal component scores for the reaction of NH4VO3 and H2VIH in methanol at room temperature monitored in time by UV-Vis spectrophotometry. | ||
Spectroscopic characterization
In the UV-Vis spectrum of a methanolic solution of 7, the intraligand π → π* transitions are observed at 225 nm and 317 nm and n → π* at 365 nm, while the bands appearing at 280 nm and 420 nm can be assigned to LMCT transitions.6,63
N and OCH3 groups, respectively. The signals at 8.07 ppm and 8.68 ppm are due to isonicotinoyl protons (H6, H10 and H7, H9, respectively), whereas those in the range 7.26–6.91 ppm are assigned to aromatic ring protons. The most significant coordination-induced difference between signals is noticed for imine CHN, up to 0.18 ppm. A similar observation was also noted in the spectrum of the molybdenum complexes, showing greater differences in chemical shifts (up to 0.27 ppm for imine CHN).
In the 13C NMR spectra, the chemical shifts for C1, C4, and C12 carbons experience deshielding effects of up to 4.28 ppm, 6.78 ppm, and 6.84 ppm, respectively. The signals for the isonicotinoyl carbons C5–C10 do not exhibit an appreciable change in chemical shifts when compared to those of the free ligand (max Δδ = 0.64 ppm), confirming that the metallosupramolecular nature of complexes found in the crystal structures is not retained in very diluted CD3OD solutions.
Unfortunately, the low solubility of all investigated compounds prevented detailed NMR analysis in non-coordinated solvents. The 1H NMR spectra of the partially dissolved complexes in CDCl3 show characteristic resonances for the V–OCH3 or V–OCH2 protons on the alkoxide ligand. They are significantly shifted compared to the signals of the corresponding free alcohols. Specifically, the resonances are at 5.38 ppm for 1, 5.63 ppm for 2, 5.58 ppm for 3, 5.63 ppm for 4, and 5.62 ppm for 5.30 A representative 1H NMR spectrum of 3, partially dissolved in CDCl3 after 24 hours, is shown in Fig. S15, ESI.†
O stretching, respectively.56 These bands are absent in the spectra of the complexes (Fig. S16, see the ESI†), suggesting the occurrence of hydrazone tautomerization N–NH–CO ⇄ N–NC–OH and deprotonation. Moreover, the bands belonging to C–Ophenolato (at 1260 cm−1), CNimine (at 1615 cm−1), and C–Ohydrazonato (at 1350 cm−1) indicate ligand coordination to vanadium through ONO donor atoms.18–20 Weaker intensity bands for V–Nimine at 780 cm−1, V–Ohydrazonato at 580 cm−1, and V–Ophenolato at 570 cm−1 corroborate the ONO ligand coordination on the vanadium. The formation of metallosupramolecular assemblies is suggested by the emergence of a new absorption band at 710 cm−1 for the V–Npy bond, which results from the isonicotinoyl nitrogen atom binding at the vanadium-free site of the neighbouring complex. Additionally, the coordination of alkoxide is confirmed by a newly observed band in the range of 1034–1064 cm−1 for C–OOR,64 as well as a band for V–OOR at 590 cm−1. All assignments are supported by high level quantum chemical calculations.
The spectra also show a very intense peak at 962 cm−1 (for 1, 2, and 3) and 957 cm−1 (for 4 and 5), indicating the presence of the {VO}3+ core.65 It is noteworthy that these bands of vanadium(V) complexes are found at higher frequencies compared to those of the corresponding molybdenum(VI) compounds.18–20 Additionally, the presence of the dioxovanadium(V) VO2+ group is inferred from the band at 930 cm−1 and 890 cm−1 for 6,66 which is attributed to VO stretching.6,67,68
Thermogravimetric analyses
The compounds were thermally analysed in an oxygen stream. The gases that evolved during the analysis were monitored in real time using FT-IR. Fig. S17–S21 (see the ESI†) present the thermograms of compounds 1 and 5 along with a 3D graphical representation of the TG-FT-IR spectra. The thermograms of crystalline samples revealed the two-step decomposition of compounds 1–5. The temperature at which the alkoxide oxidation occurs (198 °C for 1, 183 °C for 2, 166 °C for 3, 185 °C for 4, and 162 °C for 5) suggests the stability of alkoxide OR ligands which decreases with the alkyl chain length in the metallocyclic and polymeric series. The thermograms clearly demonstrate the correlation between the release of gaseous products and the observed mass loss. Gaseous compound identification was facilitated through a library search based on the obtained FT-IR spectrum. The presence of the corresponding aldehyde was indicated by the main band found at approximately 1760 cm−1. These results are consistent with theoretical calculations published for the thermal decomposition of the vanadium alkoxo complex.69Furthermore, the partial decomposition of formaldehyde in the case of compound 1 was detected, likely due to the gas carrier tube being at 200 °C. In the second step, residuals 1*–5* exhibited significant mass loss with decomposition temperatures ranging from 229 °C to 244 °C. In all the cases, the final residual corresponds well with the formation of V2O5.
For 6·0.5CH3OH·0.5H2O, the thermal gravity curve displays three distinct steps. The initial two steps indicate the loss of methanol and water (the onset temperatures are 25 °C and 63 °C, respectively). The most significant mass loss, occurring between 158 °C and 409 °C, is attributed to the decomposition of hydrazone.
Catalytic studies
The initial screening of catalytic activity involved vanadium catalysts 1–6 in cyclooctene epoxidation, using TBHP in decane as an oxidant. This reaction was selected based on our prior work on epoxidation reactions with tetranuclear molybdenum-based catalysts derived from aroylhydrazones.18,21 The oxidant choice was based on a previous investigation with similar vanadium catalysts, prioritising high selectivity and yields towards the desired epoxide and high catalytic turnover.49 In this study, the performance of the vanadium catalysts was compared with those of the molybdenum analogue catalysts 7 and 8.As expected, cyclooctene conversion and selectivity towards the corresponding epoxide were higher with TBHP in decane, approximately 10% greater than with TBHP in water. According to the nature of the mixture, water might lower the reactivity since it can compete with TBHP by coordinating the vanadium center, thus slowing the oxygen transfer process. Regardless of the oxidant used, the measured parameters for all vanadium catalysts were relatively consistent (Fig. S22, see the ESI†). Cyclooctene conversion with TBHP in decane ranged from 80% to 85%, being the highest for catalyst 2 and lowest for catalysts 3 and 5, while in water, it ranged from 76% (catalyst 1) to 81% (catalyst 4). Epoxide selectivity with TBHP in decane was between 63% (catalysts 2 and 3) and 76% (catalyst 6), compared to 64% (catalyst 2) to 72% (catalysts 4 and 5) with TBHP in water. To compare with Mo catalyst 7, it is obvious that the conversion for 7 is higher either with TBHP in decane (93%) or in water (71%), while the selectivity towards epoxide is around 85% no matter the oxidant used. Catalytic profiles for V catalysts with TBHP in decane are presented in Fig. S23, ESI.† Catalyst 2 exhibits the slowest conversion to catalytically active species, whereas catalyst 6 converts the fastest, presumably linked to the nature of the coordination between molecular species, as discussed above.
The presented results with catalyst 6 can be correlated with the investigation performed previously, with VV mononuclear compounds obtained by pyridoxal-based ligands.49 Given that the catalyst loading was ten times less in the previous investigation, a corresponding decrease in catalytic performance was anticipated. This is reflected in the observed 31% conversion of cyclooctene and a notably lower epoxide selectivity of 10%. Furthermore, the presented investigation included different mechanistic pathways for the catalyzed oxygen atom transfer to olefins that were systematically investigated using density functional theory (DFT) calculations. Among the examined pathways, two were identified as energetically feasible. Notably, one of these pathways exhibited a lower energy span, corresponding to an H-bond-assisted oxygen transfer mechanism to an external olefin. A similar mechanism has been previously explored in the context of MoVI systems, but its applicability to VV systems has not been investigated prior to the published study (Fig. S25, see the ESI†).
The catalytic potential of vanadium catalysts 1–6 was further investigated towards benzyl alcohol oxidation. The study examined the effect of different oxidizing agents, including TBHP (commercially available in decane or water) and H2O2 (with acetonitrile addition). The use of H2O2 as an oxidant, without the addition of acetonitrile, was also explored. However, the results were negligible, with minimal catalytic activity and selectivity towards the desired product observed under these conditions. Due to the lack of significant outcomes, these results are not included in the present study. Reactions using TBHP as an oxidant were monitored for 5 hours. In contrast, when H2O2 with MeCN was employed as the oxidant, the reactions were followed for only 50 minutes due to the rapid conversion of the aldehyde into benzoic acid, a by-product of the studied reaction. The obtained results are compiled in Fig. 5 and Table 3. The comparison of benzyl alcohol conversion and selectivity towards benzaldehyde, as presented in Fig. 5 is discussed at two key time points: 20 minutes from the reaction start and at the end of the reaction. A reaction time of 20 min was chosen because alcohol conversion reaches its peak at this point and then decreases over time, suggesting subsequent conversion to other by-products of the reaction. The benzyl alcohol conversions using TBHP, whether dissolved in decane or water, exhibit very similar trends. Across all vanadium catalysts (1–6), the conversion values exceed 80% at 300 min and approach 60% or more at 20 min of the reaction.
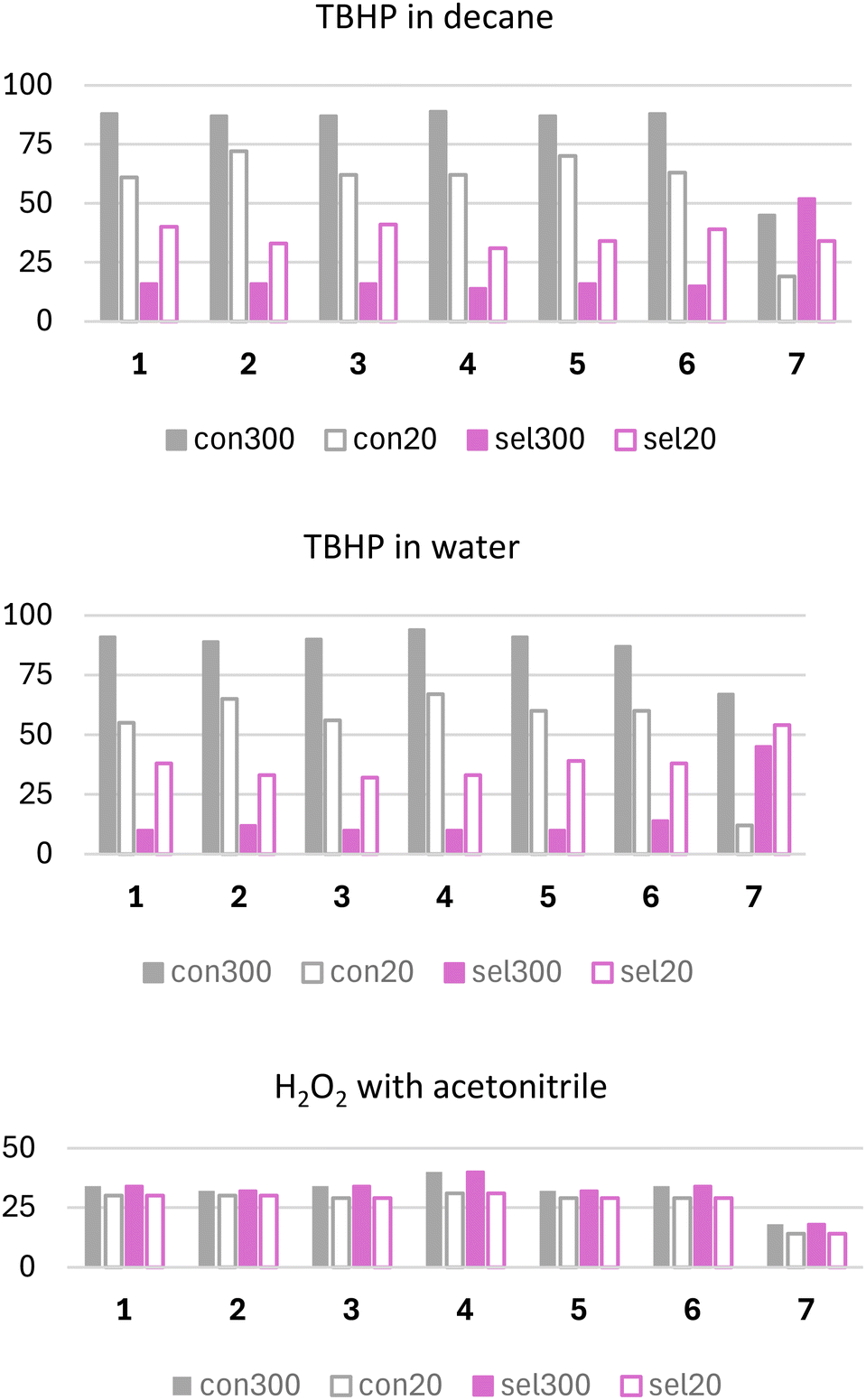 | ||
| Fig. 5 Comparison of alcohol conversion (grey-edged bar) and aldehyde selectivity (pink-edged bar) after 300 min (pink full-coloured bar) and 20 min (grey full-coloured bar) with vanadium and molybdenum-analogue catalysts, with different oxidants applied. | ||
| Catalyst | Oxidant | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TBHP in decane | TBHP in water | H2O2 with CH3CN | ||||||||||
| Cona/% | Selb/% | Cona/% | Selb/% | Cona/% | Selb/% | |||||||
| Reaction time/min | ||||||||||||
| 300 | 20 | 300 | 20 | 300 | 20 | 300 | 20 | 50 | 20 | 50 | 20 | |
| a Alcohol consumed at 20 min and at the end of the reaction. b n(alcohol) transformed/n(catalyst)/time(h) at 20 min and at the end of the reaction. | ||||||||||||
| [VO(VIH)(OCH3)]4 (1) | 88 | 61 | 16 | 40 | 91 | 55 | 10 | 38 | 34 | 30 | 34 | 30 |
| [VO(VIH)(OC2H5)]4 (2) | 87 | 72 | 16 | 33 | 89 | 65 | 12 | 33 | 32 | 30 | 32 | 30 |
| [VO(VIH)(OC3H7)]4 (3) | 87 | 62 | 16 | 41 | 90 | 56 | 10 | 32 | 34 | 29 | 34 | 29 |
| [VO(VIH)(OC4H9)]n (4) | 89 | 62 | 14 | 31 | 94 | 67 | 10 | 33 | 40 | 31 | 40 | 31 |
| [VO(VIH)(OC5H11)]n (5) | 87 | 70 | 16 | 34 | 91 | 60 | 10 | 39 | 32 | 29 | 32 | 29 |
| [VO2(HVIH)] (6) | 88 | 63 | 15 | 39 | 87 | 60 | 14 | 38 | 34 | 29 | 34 | 29 |
| [MoO2(VIH)]4 (7) | 45 | 19 | 52 | 34 | 67 | 12 | 45 | 54 | 18 | 14 | 18 | 14 |
It can be concluded that water, obtained by the decomposition of H2O2, influences the reaction environment. An increase in the water content seems to hinder benzyl alcohol oxidation. On the other side, during the reaction, TBHP decomposes into tert-butanol, an organic solvent, meaning that it is less likely to interfere with the oxidation reaction and might even act as a co-solvent.21,49
Benzaldehyde selectivity demonstrates a different trend. At 300 minutes, the selectivity values are approximately 16% with TBHP in decane and 10% with TBHP in water. These values are lower compared to those at 20 minutes, where selectivity ranges from 30 to 40% with TBHP in decane, 32–39% with TBHP in water, and around 30% with H2O2 across all vanadium catalysts (1–6), implying that after 20 min of the reaction, benzaldehyde is slowly converted to the corresponding carboxylic acid, confirmed by a characteristic peak in the chromatogram.
It seems that the presence of an organic solvent in the system facilitates the formation of the desired product by enhancing the solubility of the reactants and stabilizing catalytic intermediates. In contrast, the water content adversely impacts the reaction by potentially disrupting the catalyst activity and reducing the efficiency of substrate–catalyst interactions. The exact amount of benzoic acid was not quantified due to the instrumentation limitations. This phenomenon is more pronounced when employing hydrogen peroxide as an oxidizing agent. The conversion parameter remains largely consistent between 20 and 50 minutes, indicating that a plateau in the kinetic profile is established. Meanwhile, in contrast to catalysts 1–6, catalysts 7 and 8 exhibit a different pattern. While the benzyl alcohol conversion with TBHP as an oxidant is much lower compared to vanadium catalysts 1–6, the selectivity towards benzaldehyde is significantly higher, reaching approximately 50% for catalyst 7, but very low for 8. Conversely, Mo catalysts perform poorly with H2O2 as the oxidant, which is in contrast to the reported data with a similar class of ligands.70,71 As observed in Fig. 6, all vanadium catalysts (1–6) exhibit very similar kinetic profiles, achieving over 50% conversion at the onset of the reaction and improving further as the reaction progresses. Initial conversion values range from 55% for catalyst 1 to 67% for catalyst 4, indicating the highest TOF20 min for catalyst 4 (404) and the lowest TOF20 min for catalyst 1 (327). This further implies that all catalysts follow a similar reaction mechanism, transitioning to catalytically active species at comparable rates, depending on the alcohol coordinated to the vanadium metal centre. By the end of the reaction, all catalysts achieve conversion rates within a narrow range of 89–93%, resulting in similar TON values across all catalysts (176–190), Fig. 7.
 | ||
| Fig. 6 Kinetic profiles of benzyl alcohol conversion with V catalysts. The reaction temperature was 80 °C and TBHP in decane was used as the oxidizing agent. Reaction conditions: time, 5 h; temperature, 80 °C, n(catalyst)/n(cyclooctene)/n(oxidant) = 0.1 mmol/20 mmol/40 mmol. | ||
 | ||
| Fig. 7 Comparison of TOF20 min and TON values for V catalysts 1–6 and Mo catalysts 7 and 8, with TBHP in decane and water as the oxidizing agent, for benzyl alcohol oxidation. | ||
Fig. 6 and S24 (see the ESI†) illustrate that the kinetic profiles of catalysts 1–6 are remarkably similar, irrespective of whether TBHP is used in decane or water, respectively. The TOF20 min values are slightly elevated when TBHP is employed in water, with catalyst 2 exhibiting the highest TOF20 min value of 432, and catalyst 6 exhibiting the lowest value of 366. By the end of the reaction, the TON values range from 160 to 183, Fig. 7.
In comparison with Mo catalysts 7 and 8, all vanadium catalysts 1–6 have higher TOF20 min values, no matter the media in which TBHP is available. However, for catalyst 7, a TON of 122 in decane is lower in comparison with a TON of 181 in water, while the values for catalyst 8 are extremely low.
We extended our investigation to evaluate the catalytic potential of [VO(acac)2] and V2O5 as reference systems specifically for benzyl alcohol oxidation. The results from these reference systems are detailed in the ESI section (Table S7†). The results obtained using [VO(acac)2] as a catalyst with TBHP in water or H2O2 in MeCN as an oxidant closely mirror those achieved with vanadium catalysts 1–6. However, when employing TBHP in decane, the catalytic performance differed significantly. In this case, the benzyl alcohol conversion reached only 72% after 300 minutes of reaction, with a conversion of just 22% observed at the 20-minute mark. These findings suggest that the catalytic system requires a longer activation time than catalysts 1–6. Interestingly, the aldehyde selectivity was initially high at 71% after 20 minutes but decreased substantially to 37% by the end of the reaction. This trend further supports the hypothesis that TBHP is a more effective oxidant for this reaction than hydrogen peroxide, likely due to its superior ability to activate the catalyst and sustain high selectivity during the reaction. Furthermore, the results obtained with V2O5 as a catalyst, regardless of the oxidant employed, yielded outcomes that are consistent with those observed for catalysts 1–6. This indicates that V2O5 exhibits comparable catalytic efficiency and selectivity under similar reaction conditions.
The mechanistic pathway for benzyl alcohol oxidation has not been addressed in this manuscript and remains a focal point for future investigations.
Experimental
General details
3-Methoxy-2-hydroxybenzaldehyde isonicotinoyl hydrazone (H2VIH),56 [VO(acac)2],72 [MoO2(acac)2] (acac = acetylacetonate),73 [MoO2(VIH)]4,46 and [MoO2(VIH)(C2H5OH)]46 were synthesised following previously published procedures. 3-Methoxy-2-hydroxybenzaldehyde and isonicotinyl hydrazine were commercially available. All chemicals and solvents were used without further purification. They were purchased from Alfa Aesar or Aldrich. Additional characterization details (elemental and thermal analyses, ATR-IR, UV-Vis, NMR, PXRD, and SCXRD methods) and figures as noted in the text can be found in the ESI.†Synthesis of [VO(VIH)(OR)]4 (1, 2, and 3) and [VO(VIH)(OR)]n (4 and 5)
NH4VO3 (0.023 g, 0.2 mmol) was added to a solution of H2VIH (0.057 g, 0.21 mmol) in alcohol (30 mL). The reaction mixture was refluxed for 3 h and left overnight. The solution was concentrated under reduced pressure to a third of its volume and left to stand at room temperature. Dark red-black crystals formed over one week were isolated by filtration and dried in vacuo.N)imine, 1596 (CN), 1350 (C–Ohydrazone), 1262 (C–Ophenolato), 1043 (C–Oalkoxo), 970 (N–N), 962 (VO), 750 (V–Nimine), 708 (V–Npy), 602 (V–Oalkoxo), 580 (V–Ohydrazonato), 571 (V–Ophenolato).
N)imine, 1595 (CN), 1349 (C–Ohydrazone), 1262 (C–Ophenolato), 1034 (C–Oalkoxo), 971 (N–N), 961 (VO), 751 (V–Nimine), 710 (V–Npy), 600 (V–Oalkoxo), 578 (V–Ohydrazonato), 569 (V–Ophenolato).
N)imine, 1594 (CN), 1350 (C–Ohydrazone), 1261 (C–Ophenolato), 1060 (C–Oalkoxo), 971 (N–N), 962 (VO), 751 (V–Nimine), 707 (V–Npy), 598 (V–Oalkoxo), 574 (V–Ohydrazonato), 570 (V–Ophenolato).
N)imine, 1598 (CN), 1351 (C–Ohydrazone), 1258 (C–Ophenolato), 1064 (C–Oalkoxo), 974 (N–N), 957 (VO), 756 (V–Nimine), 708 (V–Npy), 599 (V–Oalkoxo), 574 (V–Ohydrazonato), 565 (V–Ophenolato).
N)imine, 1598 (CN), 1351 (C–Ohydrazone), 1256 (C–Ophenolato), 1067 (C–Oalkoxo), 972 (N–N), 958 (VO), 756 (V–Nimine), 709 (V–Npy), 599 (V–Oalkoxo), 574 (V–Ohydrazonato), 565 (V–Ophenolato).
Synthesis of [VO2(HVIH)]·0.5CH3OH·0.5H2O (6·0.5·0.5H2O)
NH4VO3 (0.023 g, 0.2 mmol) was added to a solution of H2VIH (0.057 g, 0.21 mmol) in methanol (30 mL). The reaction mixture was left to stand at room temperature for two weeks. The solution was concentrated in an airstream to a third of its volume and left to stand in a refrigerator. An orange powdered product formed over one week. Complex 6·0.5CH3OH·0.5H2O was isolated by filtration and dried. Upon further evaporation, a few crystals of [VO2(HVIH)]·2H2O (6·2H2O) also formed.N)imine, 1599 (CN), 1348 (C–Ohydrazone), 1249 (C–Ophenolate), 973 (N–N), 930, 890 (VO), 756 (V–Nimine), 579 (V–Ohydrazonato), 572 (V–Ophenolato).
Quantum chemical calculations
Optimizations of geometries of all computed complexes were performed using the hybrid functional B3LYP74 with the D3 version of Grimme's dispersion75 and Becke–Johnson dumping in combination with the Def2TZVP76 basis set. Initial geometries were taken from crystallographically determined structures. Harmonic frequency calculations were performed to confirm that the obtained geometries were local minima.77,78 The standard Gibbs energies of formation were calculated at T = 298.15 K and p = 101![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 325 Pa. Strengths of intramolecular interactions were estimated by the calculation of relative differences in the standard Gibbs energies of binding ΔbG°. All quantum chemical calculations were carried out using the Gaussian 16 program package.79
325 Pa. Strengths of intramolecular interactions were estimated by the calculation of relative differences in the standard Gibbs energies of binding ΔbG°. All quantum chemical calculations were carried out using the Gaussian 16 program package.79
Principal component analysis
Data obtained by time-dependent UV-Vis spectrophotometry of the monitored chemical reaction were exported to the ASCII format and arranged in the matrix (numbers written in a free format). The thus obtained 2nd-order data tensor with dimensions, number-of-spectra × number-of-wavelengths, was decomposed using the 2nd-order tensor reduction tool principal component analysis (PCA). PCA enables one to find the best linear projections for a high-dimensional set of data in the least-squares sense. Scores represent projections of the original sample points in the principal component (PC) direction and can be used for the representation of reactions in a reduced space. Therefore, each point in the score plots represents one sample UV-Vis spectrum. Data were mean-centered and PCA of the covariance matrix was performed using a NIPALS algorithm implemented in our own program Moonee.77,80Conclusions
This study explores the synthesis, characterization, and catalytic activity of a series of vanadium(V) metallosupramolecular complexes with an aroylhydrazone ligand, demonstrating the potential of these complexes in oxidation reactions. Employing 3-methoxy-2-hydroxybenzaldehyde isonicotinoyl hydrazone (H2VIH) and NH4VO3 with primary alcohols, we successfully synthesized and characterized both tetranuclear metallocycles and one-dimensional polymeric chains—resulting from varying the alkoxy ligand.Through quantum chemical calculations, we assessed the stability of these assemblies, providing insights into the energetic favorability of cyclic versus polymeric forms and identifying key structural influences, such as the torsion angle between ligand rings and metal–ligand bond lengths. These findings indicate that ligand steric effects and torsional flexibility can significantly impact the formation and stability of these supramolecular structures, suggesting avenues for tuning structural properties through ligand modification.
Catalytic testing of the vanadium complexes in the epoxidation of cyclooctene and the oxidation of benzyl alcohol revealed their efficacy and consistency, with minor variations in conversion and selectivity depending on the specific vanadium complex and oxidant used. When compared with molybdenum analogues, the vanadium catalysts demonstrated competitive activity, though molybdenum retained a slight advantage in conversion and selectivity. Additionally, mechanistic studies using time-dependent UV-Vis spectrophotometry and principal component analysis allowed us to monitor reaction intermediates and transformations, revealing that the primary reaction pathway involved hydrazonato ligand coordination to vanadium, followed by the formation of stable dioxo species under dilute conditions.
The findings in the described series of oxovanadium(V) coordination compounds illustrate how subtle changes in ancillary ligand size and flexibility can dictate not only the dimensionality of the structures but also influence critical geometric and packing features, as well as catalytic performance. Expanding the studies to even longer or branched alkoxy ancillary ligands might provide a way to control the assembly of monomers into tailor-made discrete or polymeric species as even more performant functional materials.
Author contributions
Edi Topić: investigation, writing – original draft preparation, review and editing. Josipa Sarjanović: investigation and formal analysis. Danijela Musija: investigation, formal analysis, and writing – original draft preparation. Mirna Mandarić: investigation and formal analysis. Andrea Cocut: quantum chemical calculation. Tomica Hrenar: quantum chemical calculation, chemometric analysis, data curation, writing – original draft preparation, and visualization. Dominique Agustin: supervision, review and editing. Jana Pisk: investigation, supervision, formal analysis, and writing – original draft preparation. Višnja Vrdoljak: conceptualization, supervision, funding acquisition, investigation, writing – original draft preparation, visualization, review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data sets for structures 1–5, 6·0.5H2O·0.5CH3OH, 6·2H2O, and 8 are available through the Cambridge Structural Database with deposition numbers CCDC 2367618–2367624 and 2400624.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Croatian Science Foundation under the project number HRZZ-IP-2022-10-7368. We acknowledge the support from project CIuK co-financed by the Croatian Government and the European Union through the European Regional Development Fund-Competitiveness and Cohesion Operational Programme (Grant KK.01.1.1.02.0016). LCC CNRS and the Chemistry Dept of IUT Paul Sabatier are acknowledged for providing the equipment and chemicals for catalytic experiments considering cyclooctene epoxidation reactions.References
- I. Gryca, K. Czerwińska, B. Machura, A. Chrobok, L. S. Shul'pina, M. L. Kuznetsov, D. S. Nesterov, Y. N. Kozlov, A. L. Pombeiro, I. A. Varyan and G. B. Shul'pin, High Catalytic Activity of Vanadium Complexes in Alkane Oxidations with Hydrogen Peroxide: An Effect of 8-Hydroxyquinoline Derivatives as Noninnocent Ligands, Inorg. Chem., 2018, 57(4), 1824–1839 CrossRef CAS PubMed.
- D. Dragancea, N. Talmaci, S. Shova, G. Novitchi, D. Darvasiová, P. Rapta, M. Breza, M. S. Galanski, J. Kožíšek, N. M. R. Martins, L. M. D. R. S. Martins, A. J. L. Pombeiro and V. B. Arion, Vanadium(V) Complexes with Substituted 1,5-bis(2-hydroxybenzaldehyde)carbohydrazones and Their Use As Catalyst Precursors in Oxidation of Cyclohexane, Inorg. Chem., 2016, 55, 9187–9203 CrossRef CAS PubMed.
- J.-Q. Wu and Y.-S. Li, Well-defined vanadium complexes as the catalysts for olefin polymerization, Coord. Chem. Rev., 2011, 255, 2303–2314 CrossRef CAS.
- G. B. Shul'pin and Y. N. Kozlov, Kinetics and mechanism of alkane hydroperoxidation with tert-butyl hydroperoxide catalysed by a vanadate anion, Org. Biomol. Chem., 2003, 1, 2303–2306 RSC.
- M. Sutradhar, L. M. D. R. S. Martins, M. F. C. G. da Silva and A. J. L. Pombeiro, Vanadium complexes: Recent progress in oxidation catalysis, Coord. Chem. Rev., 2015, 301–302, 200–239 CrossRef CAS.
- W. Trakarnpruk and P. Hoonsart, Oxidation of Alcohols over Vanadium Catalysts, Chin. J. Catal., 2007, 28, 290–292 CrossRef CAS.
- M. R. Maurya, N. Jangra, F. Avecilla, N. Ribeiro and I. Correia, Vanadium(V) and Molybdenum(VI) Complexes Containing ONO Tridentate Schiff Bases and Their Application as Catalysts for Oxidative Bromination of Phenols, ChemistrySelect, 2019, 4, 12743–12756 CrossRef CAS.
- J. Pisk, B. Prugovečki, D. Matković-Čalogović, R. Poli, D. Agustin and V. Vrdoljak, Charged dioxomolybdenum(VI) complexes with pyridoxal thiosemicarbazone ligands as molybdenum(V) precursors in oxygen atom transfer process and epoxidation (pre)catalysts, Polyhedron, 2012, 33, 441–449 CrossRef CAS.
- J. Pisk, D. Agustin, V. Vrdoljak and R. Poli, Epoxidation Processes by Pyridoxal Dioxomolybdenum(VI) (Pre)Catalysts Without Organic Solvent, Adv. Synth. Catal., 2011, 353, 2910–2914 CrossRef CAS.
- R. Bikas, V. Lippolis, N. Noshiranzadeh, H. Farzaneh-Bonab, A. J. Blake, M. Siczek, H. Hosseini-Monfared and T. Lis, Electronic Effects of Aromatic Rings on the Catalytic Activity of Dioxidomolybdenum(VI)–Hydrazone Complexes, Eur. J. Inorg. Chem., 2017, 999–1006 CrossRef CAS.
- M. R. Maurya, L. Rana and F. Avecilla, Catalytic oxidation of internal and terminal alkenes by oxidoperoxidomolybdenum(VI) and dioxidomolybdenum(VI) complexes, Inorg. Chim. Acta, 2015, 429, 138–147 CrossRef CAS.
- L. J. McCormick, B. F. Abrahams, B. A. Boughton, M. J. Grannas, T. A. Hudson and R. Robson, Synthesis, Structure and Cation-Binding Properties of Some [4 + 4] Metallocyclic MO22+ (M = Mo or W) Derivatives of 9-Phenyl-2,3,7-trihydroxyfluor-6-one, Inorg. Chem., 2014, 53, 1721–1728 CrossRef CAS PubMed.
- V. Vrdoljak, J. Pisk, D. Agustin, P. Novak, J. P. Vuković and D. Matković-Čalogović, Dioxomolybdenum(VI) and dioxotungsten(VI) complexes chelated with the ONO tridentate hydrazone ligand: synthesis, structure and catalytic epoxidation activity, New J. Chem., 2014, 38(12), 6176–6185 RSC.
- M. Z. Ćorović, F. Wiedemaier, F. Belaj and N. C. Mösch-Zanetti, Replacement of Molybdenum by Tungsten in a Biomimetic Complex Leads to an Increase in Oxygen Atom Transfer Catalytic Activity, Inorg. Chem., 2022, 61, 12415–12424 CrossRef PubMed.
- E. Topić, V. Damjanović, K. Pičuljan and M. Rubčić, Dinuclear Molybdenum(VI) Complexes Based on Flexible Succinyl and Adipoyl Dihydrazones, Crystals, 2024, 14, 135 CrossRef.
- M. Sutradhar, N. V. Shvydkiy, M. F. C. G. da Silva, M. V. Kirillova, Y. N. Kozlov, A. J. L. Pombeiro and G. B. Shul'pin, A new binuclear oxovanadium(V) complex as a catalyst in combination with pyrazinecarboxylic acid (PCA) for efficient alkane oxygenation by H2O2, Dalton Trans., 2013, 42, 11791–11803 RSC.
- M. R. Maurya, N. Kumar and F. Avecilla, Controlled Modification of Triaminoguanidine-Based μ3 Ligands in Multinuclear [VIVO]/[VVO2] Complexes and Their Catalytic Potential in the Synthesis of 2-Amino-3-cyano-4H-pyrans/4H-chromenes, Inorg. Chem., 2024, 63, 2505–2524 CrossRef CAS PubMed.
- V. Vrdoljak, M. Mandarić, T. Hrenar, I. Đilović, J. Pisk, G. Pavlović, M. Cindrić and D. Agustin, Geometrically Constrained Molybdenum(VI) Metallosupramolecular Architectures: Conventional Synthesis versus Vapor and Thermally Induced Solid-State Structural Transformations, Cryst. Growth Des., 2019, 19, 3000–3011 CrossRef CAS.
- D. Cvijanović, J. Pisk, G. Pavlović, D. Šišak-Jung, D. Matković-Čalogović, M. Cindrić, D. Agustin and V. Vrdoljak, Discrete mononuclear and dinuclear compounds containing a MoO22+ core and 4-aminobenzhydrazone ligands: synthesis, structure and organic-solvent-free epoxidation activity, New J. Chem., 2019, 43, 1791–1802 RSC.
- J. Pisk, M. Rubčić, D. Kuzman, M. Cindrić, D. Agustin and V. Vrdoljak, Molybdenum(VI) complexes of hemilabile aroylhydrazone ligands as efficient catalysts for greener cyclooctene epoxidation: an experimental and theoretical approach, New J. Chem., 2019, 43, 5531–5542 RSC.
- J. Pisk, D. Agustin and V. Vrdoljak, Tetranuclear molybdenum(VI) hydrazonato epoxidation (pre)catalysts: Is water always the best choice?, Catal. Commun., 2020, 142, 106027 CrossRef CAS.
- M. Mandarić, E. Topić, D. Agustin, J. Pisk and V. Vrdoljak, Preparative and Catalytic Properties of MoVI Mononuclear and Metallosupramolecular Coordination Assemblies Bearing Hydrazonato Ligands, Int. J. Mol. Sci., 2024, 25, 1503 CrossRef PubMed.
- A. Farrokhi, M. Jafarpour and R. Najafzade, Phosphonate-based Metal Organic Frameworks as Robust Heterogeneous Catalysts for TBHP Oxidation of Benzylic Alcohols, Catal. Lett., 2017, 147, 1714–1721 CrossRef CAS.
- U. S. F. Arrozi, R. Pratama, F. Soraya, Y. Permana, S. Hartina, E. Salduna, W. W. Lestari, W. Ciptonugroho and Y. P. Budiman, Solvent-Free Oxidation of Benzyl Alcohol Using Modified Zeolitic Imidazolate Frameworks-8 (ZIF-8) Catalysts, ChemistrySelect, 2024, 9, e202304882 CrossRef CAS.
- B. M. Peterson, M. E. Herried, R. L. Neve and R. W. McGaff, Oxidation of primary and secondary benzylic alcohols with hydrogen peroxide and tert-butyl hydroperoxide catalyzed by a “helmet” phthalocyaninato iron complex in the absence of added organic solvent, Dalton Trans., 2014, 43, 17899–17903 RSC.
- M. Y. Enbo, W. Ying, L. Shutao, W. Yangguang, L. Wang and C. Hu, A novel chain-like binuclear vanadium(V) coordination polymer containing mixed ligands: hydrothermal synthesis and crystal structure of [{VO2(2,2′-bipy)}2(tp)]∞ (tp=terephthalate), Inorg. Chim. Acta, 2003, 344, 257–261 CrossRef.
- E. Kiss, A. Benyei and T. Kiss, VO(IV) complexes of 3-hydroxypicolinic acid: a solution study and the structure of a supramolecular assembly in the solid state, Polyhedron, 2003, 22, 27–33 CrossRef CAS.
- X.-M. Zhang, M.-L. Tong, H. K. Lee and X.-M. Chen, The First Noncluster Vanadium(IV) Coordination Polymers: Solvothermal Syntheses, Crystal Structure, and Ion Exchange, J. Solid State Chem., 2001, 160, 118–122 CrossRef CAS.
- K. Su, M. Wu, D. Yuan and M. Hong, Interconvertible vanadium-seamed hexameric pyrogallol[4]arene nanocapsules, Nat. Commun., 2018, 9, 4941 CrossRef PubMed.
- S. P. Dash, A. K. Panda, S. Pasayat, R. Dinda, A. Biswas, E. R. T. Tiekink, S. Mukhopadhyay, S. K. Bhutia, W. Kaminsky and E. Sinn, Oxidovanadium(V) complexes of aroylhydrazones incorporating heterocycles: synthesis, characterization and study of DNA binding, photo-induced DNA cleavage and cytotoxic activities, RSC Adv., 2015, 5, 51852–51867 RSC.
- L. Zhang, Z. You and H.-Y. Qian, Synthesis, characterization and crystal structure of a novel tetranuclear oxidovanadium(V) complex derived from N′,-(2-hydroxy-3-methoxybenzylidene)isonicotinohydrazide with catalytic property, Inorg. Nano-Met. Chem., 2022, 1–5 Search PubMed.
- H. Kargar, A. Kaka-Naeini, M. Fallah-Mehrjardi, R. Behjatmanesh-Ardakani, H. Amiri Rudbari and K. S. Munawar, Oxovanadium and dioxomolybdenum complexes: synthesis, crystal structure, spectroscopic characterization and applications as homogeneous catalysts in sulfoxidation, J. Coord. Chem., 2021, 74, 1563–1583 CrossRef CAS.
- C. Lescop, Coordination-Driven Syntheses of Compact Supramolecular Metallacycles toward Extended Metallo-organic Stacked Supramolecular Assemblies, Acc. Chem. Res., 2017, 50, 885–894 CrossRef CAS PubMed.
- B. H. Northrop, H.-B. Yang and P. J. Stang, Coordination-driven self-assembly of functionalized supramolecular metallacycles, Chem. Commun., 2008, 5896–5908 RSC.
- T. R. Cook and P. J. Stang, Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- S. Leininger, B. Olenyuk and P. J. Stang, Self-assembly of discrete cyclic nanostructures mediated by transition metals, Chem. Rev., 2000, 100, 853–908 CrossRef CAS PubMed.
- B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Self-Organization in Coordination-Driven Self-Assembly, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef CAS PubMed.
- M. Ahmadi and S. Seiffert, Coordination Geometry Preference Regulates the Structure and Dynamics of Metallo-Supramolecular Polymer Networks, Macromolecules, 2021, 54, 1388–1400 CrossRef CAS.
- R. L. Borkar, M. Bhol, B. Shankar, J. Karges and M. Sathiyendiran, Solvent-Directed Rhenium(I) Metallocyclic Tubes, Dimeric Capsule, and Intercalated Dimers, Cryst. Growth Des., 2024, 24, 1987–1999 CrossRef CAS.
- M. S. McCreadya and R. J. Puddephatt, Self-assembly of isomeric clamshell dimers of platinum(II), Dalton Trans., 2012, 41, 12378–12385 RSC.
- C. H. Li and J. L. Zuo, Self-Healing Polymers Based on Coordination Bonds, Adv. Mater., 2019, 1903762 Search PubMed.
- N. Giuseppone, J. L. Schmitt and J. M. Lehn, Driven Evolution of a Constitutional Dynamic Library of Molecular Helices Toward the Selective Generation of [2 × 2] Gridlike Arrays under the Pressure of Metal Ion Coordination, J. Am. Chem. Soc., 2006, 128, 16748–16763 CrossRef CAS PubMed.
- C. Dietrich-Buchecker, B. Colasson, M. Fujita, A. Hori, N. Geum, S. Sakamoto, K. Yamaguchi and J. P. Sauvage, Quantitative Formation of [2]Catenanes Using Copper(I) and Palladium(II) as Templating and Assembling Centers: The Entwining Route and the Threading Approach, J. Am. Chem. Soc., 2003, 125, 5717–5725 CrossRef CAS PubMed.
- V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J. M. Lehn, Interconversion of single and double helices formed from synthetic molecular strands, Nature, 2000, 407, 720–723 CrossRef CAS PubMed.
- A. A. Levchenko, C. K. Yee, A. N. Parikh and A. Navrotsky, Energetics of Self-Assembly and Chain Confinement in Silver Alkanethiolates: Enthalpy−Entropy Interplay, Chem. Mater., 2005, 17, 5428–5438 CrossRef CAS.
- V. Vrdoljak, B. Prugovečki, D. Matković-Čalogović, R. Dreos, P. Siega and C. Tavagnacco, Zigzag Chain, Square Tetranuclear, and Polyoxometalate-Based Inorganic−Organic Hybrid Compounds - Molybdenum vs Tungsten, Cryst. Growth Des., 2010, 10, 1373–1382 CrossRef CAS.
- V. Vrdoljak, B. Prugovečki, D. Matković-Čalogović, J. Pisk, R. Dreos and P. Siega, Supramolecular Hexagon and Chain Coordination Polymer Containing the MoO22+ Core: Structural Transformation in the Solid State, Cryst. Growth Des., 2011, 11, 1244–1252 CrossRef CAS.
- V. Vrdoljak, B. Prugovečki, D. Matković-Čalogović, T. Hrenar, R. Dreos and P. Siega, Three Polymorphic Forms of a Monomeric Mo(VI) Complex: Building Blocks for Two Metal–Organic Supramolecular Isomers. Intermolecular Interactions and Ligand Substituent Effects, Cryst. Growth Des., 2013, 13, 3773–3784 CrossRef CAS.
- J. Pisk, J.-C. Daran, R. Poli and D. Agustin, Pyridoxal based ONS and ONO vanadium(V) complexes: Structural analysis and catalytic application in organic solvent free epoxidation, J. Mol. Catal. A:Chem., 2015, 403, 52–63 CrossRef CAS.
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981, vol. 9 Search PubMed.
- L. C. Hill, in Advances in Oxygenated Processes, ed. A. L. Baumstark, JAI Press Inc., London, 1988, vol. 1 Search PubMed.
- R. J. J. Jachuck, D. K. Selvaraj and R. S. Varma, Process intensification: oxidation of benzyl alcohol using a continuous isothermal reactor under microwave irradiation, Green Chem., 2006, 8, 29–33 RSC.
- C. Ragupathi, J. J. Vijaya, S. Narayanan, S. K. Jesudoss and L. J. Kennedy, Highly selective oxidation of benzyl alcohol to benzaldehyde with hydrogen peroxide by cobalt aluminate catalysis: a comparison of conventional and microwave methods, Ceram. Int., 2015, 41, 2069–2080 CrossRef CAS.
- C. Lu, J. Hu, M. Y. N. Meng, A. D. Zhou, F. Zhang and Z. B. Zhang, The synergistic effect of benzyl benzoate on the selective oxidation of toluene to benzaldehyde, Chem. Eng. Res. Des., 2019, 141, 181–186 CrossRef CAS.
- C. Miao, H. Zhao, Q. Zhao, C. Xia and W. Sun, NHPI and ferric nitrate: a mild and selective system for aerobic oxidation of benzylic methylenes, Catal. Sci. Technol., 2016, 6, 1378–1383 RSC.
- J. Pisk, T. Hrenar, M. Rubčić, G. Pavlović, V. Damjanović, J. Lovrić, M. Cindrić and V. Vrdoljak, Comparative studies on conventional and solvent-free synthesis toward hydrazones: application of PXRD and chemometric data analysis in mechanochemical reaction monitoring, CrystEngComm, 2018, 20, 1804–1817 RSC.
- L. Zhang, Z. You and H.-Y. Qian, Synthesis, characterization and crystal structure of a novel tetranuclear oxidovanadium(V) complex derived from N′-(2-hydroxy-3-methoxybenzylidene)isonicotinohydrazide with catalytic property, Inorg. Nano-Met. Chem., 2022, 54, 282–286 CrossRef.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Applied topological analysis of crystal structures with the program package ToposPro, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- As implemented in CCDC Mercury software, C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- S. Y. Ebrahimipour, M. Mohamadi, I. Sheikhshoaie, S. Suárez, R. Baggio and M. Khaleghi, A novel oxido-vanadium(V) Schiff base complex: synthesis, spectral characterization, crystal structure, electrochemical evaluation, and biological activity, Res. Chem. Intermed., 2016, 42, 611–623 CrossRef.
- Y.-H. Lu, Y.-W. Lu, C.-L. Wu, Q. Shao, X.-L. Chen and R. N. B. Bimbong, UV–visible spectroscopic study of the salicyladehyde benzoylhydrazone and its cobalt complexes, Spectrochim. Acta, Part A, 2006, 65, 695–701 CrossRef PubMed.
- H. Hosseini-Monfared, S. Alavi, R. Bikas, M. Vahedpour and P. Mayer, Vanadiumoxo–aroylhydrazone complexes: Synthesis, structure and DFT calculations, Polyhedron, 2010, 29, 3355–3362 CrossRef.
- R. Dinda, P. Sengupta, S. Ghosh, H. Mayer-Figge and W. S. Sheldrick, A family of mononuclear molybdenum-(VI), and -(IV) oxo complexes with a tridentate (ONO) ligand, J. Chem. Soc., Dalton Trans., 2002, 4434–4439 RSC.
- L. M. Martínez-Prieto, P. Palma, E. Álvarez and J. Cámpora, Nickel Pincer Complexes with Frequent Aliphatic Alkoxo Ligands [(iPrPCP)Ni-OR] (R = Et, nBu, iPr, 2-hydroxyethyl). An Assessment of the Hydrolytic Stability of Nickel and Palladium Alkoxides, Inorg. Chem., 2017, 56(21), 13086–13099 CrossRef PubMed.
- S. R. Patra, S. Mondal, D. Sinha and K. K. Rajak, Mono Versus Dinuclear Vanadium(V) Complexes: Solvent Dependent Structural Versatility and Electro Syntheses of Mixed-Valence Oxovanadium(IV/V) Entities in Solution, ACS Omega, 2022, 7, 11710–11721 CrossRef PubMed.
- J. A. Bonadies and C. J. Carrano, Vanadium phenolates as models for vanadium in biological systems. 1. Synthesis, spectroscopy, and electrochemistry of vanadium complexes of ethylenebis[(o-hydroxyphenyl)glycine] and its derivatives, J. Am. Chem. Soc., 1986, 108, 4088–4095 CrossRef CAS.
- M. R. Maurya, S. Khurana, C. Schulzke and D. Rehder, Dioxo- and Oxovanadium(V) Complexes of Biomimetic Hydrazone ONO, Donor Ligands: Synthesis, Characterisation, and Reactivity, Eur. J. Inorg. Chem., 2001, 779–788 CrossRef CAS.
- M. R. Maurya, B. Sarkar, F. Avecilla and I. Correia, Vanadium Complexes Derived from Acetyl Pyrazolone and Hydrazides: Structure, Reactivity, Peroxidase Mimicry and Efficient Catalytic Activity for the Oxidation of 1-Phenylethanol, Eur. J. Inorg. Chem., 2016, 4028–4044 CrossRef CAS.
- J. Döbler, M. Pritzsche and J. Sauer, Oxidation of Methanol to Formaldehyde on Supported Vanadium Oxide Catalysts Compared to Gas Phase Molecules, J. Am. Chem. Soc., 2005, 127, 10861–10868 CrossRef PubMed.
- L. Balapoor, R. Bikas and M. Dargahi, Catalytic oxidation of benzyl-alcohol with H2O2 in the presence of a dioxidomolybdenum(VI) complex, Inorg. Chim. Acta, 2020, 510, 119734 CrossRef CAS.
- S. L. Pandhare, R. R. Jadhao, V. G. Puranik, P. V. Joshi, F. Capet, M. K. Dongare, S. B. Umbarkar, C. Michon and F. Agbossou-Niedercorn, Molybdenum(VI) dioxo complexes for the epoxidation of allylic alcohols and olefins, J. Organomet. Chem., 2014, 772–773, 271–279 CrossRef CAS.
- B. E. Bryant, W. C. Fernelius, D. H. Busch, R. C. Stoufer and W. Stratton, Vanadium(IV) Oxy(acetylacetonate), in Inorganic Syntheses, ed. T. Moeller, John Wiley & Sons, Inc, 1957, vol. 5, pp. 113–116 Search PubMed.
- J.-J. Chen, J. W. McDonald and W. E. Newton, Synthesis of molybdenum(IV) and molybdenum(V) complexes using oxo abstraction by phosphines, Mechanistic implications, Inorg. Chem., 1976, 15, 2612 CrossRef.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 785 CrossRef PubMed; (b) R. Peverati and D. G. Truhlar, Phys. Chem. Chem. Phys., 2021, 14, 16187 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corsrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- (a) F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (b) F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- T. Hrenar, I. Primožič, D. Fijan and M. Majerić Elenkov, Conformational analysis of spiro-epoxides by principal component analysis of molecular dynamics trajectories, Phys. Chem. Chem. Phys., 2017, 19, 31706–31713 RSC.
- I. Primožič, T. Hrenar, K. Baumann, L. Krišto, I. Križić and S. Tomić, Mechanochemical and Conformational Study of N-heterocyclic Carbonyl-Oxime Transformations, Croat. Chem. Acta, 2014, 87, 153–160 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- O. Jović, T. Smolić, I. Primožič and T. Hrenar, Spectroscopic and Chemometric Analysis of Binary and Ternary Edible Oil Mixtures: Qualitative and Quantitative Study, Anal. Chem., 2016, 88, 4516–4524 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Powder diffraction patterns, additional figures for compounds, tables of selected bond distances and angles and of hydrogen bond parameters, UV-Vis spectra, NMR spectra, ATR-IR spectra, TGA curves, and crystallographic data sets for structures 1–5, 6·0.5H2O·0.5CH3OH, 6·2H2O, and 8. CCDC 2367618–2367624 and 2400624. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00105f |
| This journal is © The Royal Society of Chemistry 2025 |