
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrocyclic control of electron transfer to high valent uranium in heterobimetallic complexes†
Amit
Kumar‡§
a,
Riddhi R.
Golwankar‡
 a,
Mikaela M. F.
Pyrch¶
b,
Fynn L.
Cooper
a,
Grant A.
Arehart
a,
Korey P.
Carter
b,
Allen G.
Oliver
c,
Victor W.
Day
a,
Tori Z.
Forbes
*b and
James D.
Blakemore
*a
a,
Mikaela M. F.
Pyrch¶
b,
Fynn L.
Cooper
a,
Grant A.
Arehart
a,
Korey P.
Carter
b,
Allen G.
Oliver
c,
Victor W.
Day
a,
Tori Z.
Forbes
*b and
James D.
Blakemore
*a
aDepartment of Chemistry, University of Kansas, 1567 Irving Hill Road, Lawrence, Kansas 66045, USA. E-mail: blakemore@ku.edu
bDepartment of Chemistry, University of Iowa, Iowa City, Iowa 52242, USA. E-mail: tori-forbes@uiowa.edu
cDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA
First published on 12th March 2025
Abstract
The redox properties of the uranyl ion, UO22+, influence the chemistry required for nuclear fuel reprocessing, but little spectroscopic insight is available to support design strategies for influencing the redox properties of uranium complexes. Here, structural studies with X-ray diffraction analysis, electrochemical methods, and Raman spectroscopy have been used to examine one strategy for influencing uranyl redox chemistry, namely co-encapsulation of UO22+ and secondary metal cations (Cs+, Rb+, K+, Na+, Li+, and Ca2+) in macrocyclic ligands. Two ligands are compared in this work that differ in the denticities of their secondary cation binding sites (pentadentate vs. hexadentate), enabling direct quantification of influences on the redox and vibrational properties of the uranyl moiety. The UVI/UV thermodynamic reduction potential is correlated with the effective Lewis acidity of the secondary metal cations; solid-state and solution-phase Raman spectra show that this effect can be attributed to electrostatics that effectively drive diminished electron donation to uranium in adducts of more strongly Lewis acidic cations. The heterogeneous electron transfer (ET) rates for UVI/UV redox processes, however, depend on both the strength of cation binding in the macrocycles and the Lewis acidity of the cations, suggesting opportunities for molecular design in development of reagents for nuclear fuel reprocessing/separations.
Introduction
Nuclear power based on fission of uranium is an important source of low-carbon electricity, but unresolved issues related to fuel recycling and waste management have hampered further development of this remarkable global energy resource.1 Virtually all nuclear fuel recycling schemes involve redox-driven changes in actinide solubility and speciation that are used to separate useful uranium from other components of irradiated “used” fuel.2 This is true of both established fuel reprocessing chemistries, like the Plutonium Uranium Redox EXtraction (PUREX) process,3 and next-generation approaches involving totally non-aqueous conditions4 and/or molten salt chemistries5 to achieve isolation of useful uranium for further use in power generation.The chemistry of uranium under most conditions is dominated by the uranyl dication, UO22+.6 This species forms quite readily under oxidizing and/or aqueous conditions, including those found in the conventional fuel recycling chemistry of the PUREX process.3 UO22+ features high-valent uranium in the formal +VI oxidation state, but under most conditions, this species is rather difficult to reduce. This is consistent with the E°(UVI/UV) value of 0.16 V vs. the standard hydrogen electrode (SHE), in that under nearly all conditions,7 electro-reduction of UO22+ is challenging and associated with significant overpotentials. Indeed, electrolysis of UO22+-containing solutions can lead to significant parasitic reactions.8 Considering that reduction of UO22+ is required for fuel reprocessing and that UO22+ redox chemistry is also important in environmental speciation of uranium, understanding and ultimately controlling the redox chemistry of uranium could be useful for development of next-generation nuclear fuel recycling schemes.
Inspired by work in transition metal chemistry,9 a portion of our interest in recent years has been drawn to the strategy of using effectively Lewis acidic secondary metal cations for tuning the redox chemistry of uranium.10 In our work, we have used macrocyclic ligands to prepare complexes that can incorporate a diverse range of secondary metal cations, including lanthanide cations,10–14 and found for the case of UO22+ complexes that the UVI/UV potential could be tuned reliably by incorporation of cations. In that work, the Lewis acidity of the cations, quantified through the pKa values of the corresponding metal aqua complexes, served as a useful descriptor for the achievable reduction-potential modulation, revealing a relationship of −61 ± 9 mV/pKa for UVI/UV cycling. Hallmarks of this relationship have been observed in several other classes of compounds,15,16 beyond ours based on macrocyclic ligands. Prior work has tended to focus, however, on systems in which uranyl oxo ligands serve as bridging atoms to secondary metal cations. The groups of Arnold17 and Mazzanti,18 have mapped the properties of complexes of this type, in particular, finding that cations stabilize lower-valent forms of uranium (Fig. 1). Borane electrophiles can be used as well, as shown by Sarsfield,19 Hayton,20 and others, representing a particularly attractive set of reagents for stabilizing U(V). Blakemore and co-workers have recently been exploring boranes, including BPh3, electrode-driven uranyl functionalization as well.21,22
 | ||
| Fig. 1 Structures of complexes in which secondary cations influence the measured or apparent redox properties of the uranyl motif. From ref. 17a, 18 and 10. | ||
However, little spectroscopic data is available to help interpret the origins of redox tuning effects. Quantitative insights into the influence of secondary cations on actinide metal centers could be impactful for developing predictive models and design of new ligands. Along this line, the role of macrocyclic structure in achieving optimal tuning behavior has also not received the attention that it deserves, but we have anticipated that the macrocycle-promoted structural placement of secondary metal cations could influence cation-driven tuning.23–25 Such concepts are sensible in the general context of Lewis acid–base chemistry, but to the best of our knowledge, no studies in macrocyclic chemistry have directly interrogated how the structure of a supporting ligand framework modulates heterometallic redox tuning. Agapie and co-workers have shown, however, that non-macrocyclic ligand changes are quite impactful in iron oxo cluster chemistry.15 We thus hypothesized that the supramolecular fit of the secondary cations in ligands would influence the redox chemistry, building on the known propensity of macrocyclic ligands to override natural bonding preferences.26 Given the broad utility of Raman spectroscopy in actinide chemistry,27 we anticipated that Raman data for macrocyclic systems would afford insights relevant to development of controlled uranyl redox chemistry and next-generation nuclear fuel recycling strategies. Data examining cation-induced redox effects from the perspective of Raman spectroscopy would also usefully build on established correlations between first- and second-sphere interactions and observed vibrational frequencies.28–31
Here, we report a comparison of the structural, spectroscopic, and electrochemical properties of two related families of heterobimetallic complexes (L5UO2M and L6UO2M) that incorporate secondary redox-inactive metal ions M (where M is Cs+, Rb+, K+, Na+, Li+, or Ca2+, see Fig. 2). The consequences of binding the secondary metal ions have been mapped in order to delineate the influences of cation Lewis acidity, macrocyclic ligand structure, and relevant cation association constants on the vibrational and redox properties of the uranyl motif. X-ray diffraction analysis (XRD) revealed that the coordination properties of the secondary metal ion binding site (penta- vs. hexadentate) controls placement of the cations relative to the uranium center in the complexes, resulting in unique tuning sensitivities for both reduction potentials and symmetric U–O vibrational frequencies. However, the electron transfer rate is optimized only by balancing the influences of both Lewis acidity and macrocyclic structure, resulting in unique “volcano plots” for each ligand explored here that can be understood to arise from the molecular design features implemented in this work.
 | ||
| Fig. 2 Heterobimetallic complexes of the uranyl dication. | ||
Results
Characterization and comparison of the macrocyclic ligands
Two ligands were used in this work to support assembly of two unique families of heterobimetallic complexes. Ligands of the type used here were developed by Reinhoudt and co-workers in the late 1980s32 and subsequently elaborated upon by Vigato and co-workers.33 The ligands themselves, denoted as L5H2 and L6H2, feature a similar Schiff-base site in both cases that supports UO22+ binding (Fig. 2). Importantly, each ligand offers a second binding site, based upon a polyether motif, for incorporation of secondary metal cations. L5H2 provides a 15-crown-5-like pentadentate moiety for cation binding, while L6H2 offers a larger 18-crown-6-like hexadentate moiety. These polyether sites were designed to be proximal to the site for uranyl binding, encouraging interaction of the metal cations with the uranyl motif not by direct interaction with the terminal oxo ligands but rather via the phenoxide donor groups of the macrocycle. L5H2 has not previously been used to prepare heterobimetallic complexes, but we previously utilized L6H2 to prepare a small set of four crystallographically characterized heterobimetallic complexes.10Building on this experience, we recognized that monometallic uranyl complexes of the macrocyclic ligands, denoted L5UO2 and L6UO2, would need to be prepared in order to access divergent synthesis of a range of bimetallic complexes. To access these monometallic complexes, we prepared precursors in which the neutral ligands L5H2 and L6H2 were templated and stabilized by coordination to barium, affording species denoted BaPenta and BaHexa, respectively (see Experimental section in ESI† for details).10,331H and 19F nuclear magnetic resonance data confirmed the successful isolation of both these known precursors as desired (Fig. S1 and S2;† fluorine derived from triflate counteranions).
XRD analysis carried out on suitable crystals of BaPenta revealed, however, that this complex prefers to crystallize as a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 L5H2:Ba2+ adduct in which Ba2+ is sandwiched between the crown-ether-like sites of the L5H2 units (Fig. 3a and Fig. S164–166†) with the triflate counteranions in the outer sphere. The Ba2+ center adopts the high coordination number (C.N.) of 10 by interacting with all ten macrocyclic O-atoms present in the two equivalents of L5H2 present in the structure; this is sensible given the large ionic radius of 1.52 Å for Ba2+ with C.N. = 10.34 The formulation of the macrocycles as L5H2 units (rather than their deprotonated analogues) was confirmed by 1H NMR spectroscopy (Fig. S1†), as well as by successful location and free refinement of the phenol-associated H protons. These protons appear acidified by Ba2+ coordination, however, with the nearby imine N-atoms assisting in formation of strong [N–H⋯O] H-bonds.
:1 L5H2:Ba2+ adduct in which Ba2+ is sandwiched between the crown-ether-like sites of the L5H2 units (Fig. 3a and Fig. S164–166†) with the triflate counteranions in the outer sphere. The Ba2+ center adopts the high coordination number (C.N.) of 10 by interacting with all ten macrocyclic O-atoms present in the two equivalents of L5H2 present in the structure; this is sensible given the large ionic radius of 1.52 Å for Ba2+ with C.N. = 10.34 The formulation of the macrocycles as L5H2 units (rather than their deprotonated analogues) was confirmed by 1H NMR spectroscopy (Fig. S1†), as well as by successful location and free refinement of the phenol-associated H protons. These protons appear acidified by Ba2+ coordination, however, with the nearby imine N-atoms assisting in formation of strong [N–H⋯O] H-bonds.
 | ||
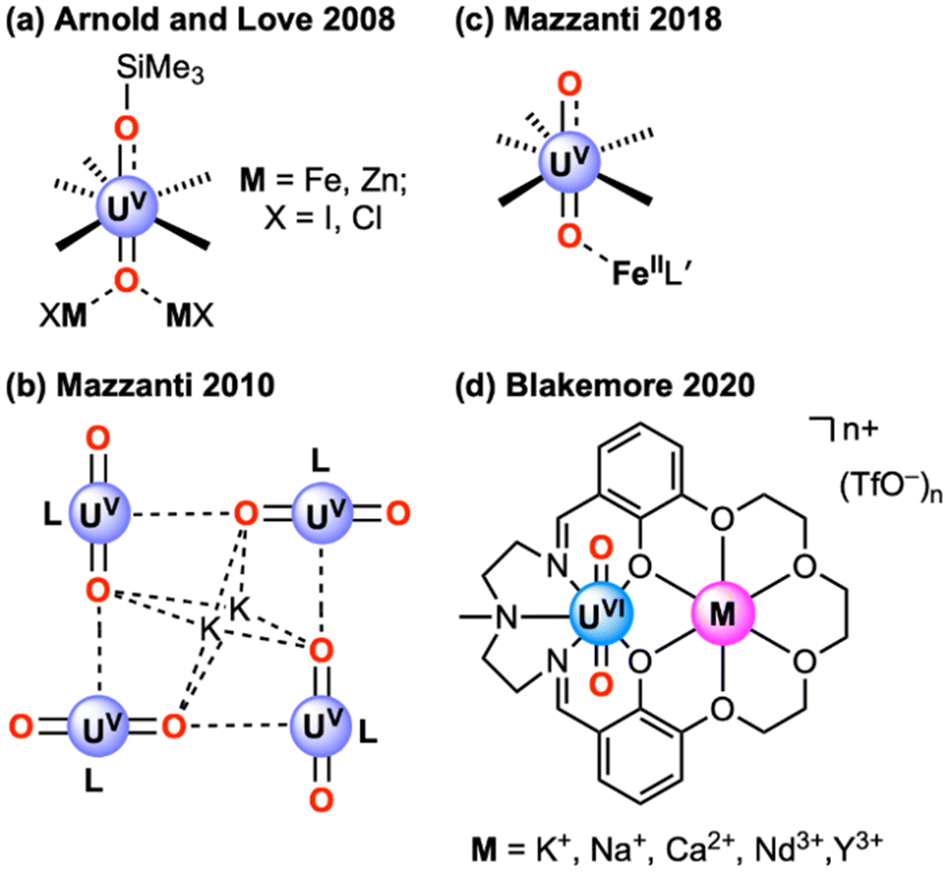
| Fig. 3 Solid-state structures from XRD of (a) BaPenta and (b) L5UO2. Outer sphere triflate counteranions and co-crystallized diethyl ether molecules associated with BaPenta, a co-crystallized CH3CN molecule associated with L5UO2, minor components of disorder, and all hydrogen atoms omitted for clarity. Displacement ellipsoids are shown at 20% probability level. The phenolic protons H1A, H2A, H1AA, and H2AA were included as independent atoms that were freely refined in the final structural model. | ||
The structure of BaPenta was compared with the available prior structure of BaHexa.10 The structure of BaHexa revealed a 1:1 L6H2:Ba adduct, with the Ba2+ ion coordinated to the six macrocyclic O-atoms of L6H2. Ba2+ adopts the C.N. of 10 by binding two triflate counteranions in the inner sphere, both in the κ2-mode, in line with the large size of Ba2+ and the apparent preference of this dication to adopt C.N. = 10 in both 18- and 15-crown systems. Perhaps the most important difference between the structures is that the Ba2+ ion does not bind near the centroid of either crown ether cavity to which it is bound in BaPenta. The phenomenon of Ba2+ being significantly displaced above the crown ether cavities was quantified by the ψM parameter, defined as the root mean square deviation of the Ba atom above the mean plane defined by the crown oxygen atoms (Table S12†). The ψM values for BaPenta and BaHexa are 1.570 and 0.048 Å, respectively. This finding is attributable to the larger size of the 18-crown-6-like cavity in L6H2, which enables Ba2+ to nestle comfortably into the site (Fig. S168†). The separation between two phenoxide O atoms (O1⋯O2) is smaller for BaPenta than BaHexa (3.328(4) Å vs. 3.641(3) Å), consistent with the presence of a more constricted and planar cavity that results in displacement of Ba2+ above the plane of the 15-crown-5-like site.
Using literature methods, BaPenta and BaHexa were converted into the monometallic uranyl-containing complexes L5UO2 and L6UO2.10,331H NMR and elemental analysis confirmed isolation of the desired complexes in each case (see Experimental section in ESI† for details). Single crystals of L5UO2 were grown for XRD analysis, and the resulting structure confirmed coordination of UO22+ in the Schiff base site of the macrocycle, similar to the case of L6UO2 as revealed in a prior structure.10 Both the new structure of L5UO2 and the older structure of L6UO2 confirm that the crown ether-like sites are poised to bind secondary metal cations (Fig. S173†). The crown ether moieties in L5UO2 and L6UO2 appear distinctive in each case, however, with significantly greater co-planarity of the oxygen atoms in L5UO2. As in our prior work,11 the ωcrown parameter was used to quantify the planarity of the crown-ether O-atoms (see Table 1). The value of this parameter is 0.055 for L5UO2 and 0.358 for L6UO2 (Table S14†), consistent with the notion that the O-atoms in L5UO2 form a more compact platform of donor atoms on top of which secondary metal cations could bind. This contrasts with the case of L6UO2, which features a crown site with sufficient flexibility and inter-O-atom spacing to encapsulate secondary cations.
| Compound | L5UO2Li | L6UO2Li | L5UO2Na |
L6UO2Nab |
L5UO2Ca |
L6UO2Cab |
|---|---|---|---|---|---|---|
| a From ref. 34. b Structural data taken from ref. 10, CCDC 1960629 (ref. 36), and CCDC 1960628 (ref. 37). c Defined as the average interatomic distance between the noted metal and the relevant oxygen/nitrogen atoms. Stated estimated standard deviations (e.s.d.'s) on distances were taken as the largest of the individual values in the refined data for the independent bond distance. d Defined as the root mean square deviation (RMSD) of the positions of crown atoms O1, O2, O3, O4, and O5 from the mean plane of their positions for the species ligated by L5. O6 was included in the calculation for the species ligated by L6. e Defined as the root mean square deviation (RMSD) of O1, O2, N1, N2, and N3 from the mean plane of their positions. f Absolute value of the distance between U and the mean plane of O1, O2, N1, N2, and N3. g Absolute value of the distance between M and the mean plane of O1, O2, O3, O4, and O5 for the species ligated by L5. O6 was included in the calculation of the mean plane for the species ligated by L6. h Values are reported as the mean of the relevant individual values corresponding to two disordered orientations of O3 (O3′) and O4 (O4′) occupying the same volume of the asymmetric unit. Atom labels are consistent with those given in the raw crystallographic data (see pp. S178–S179† for details). | ||||||
| pKa of [M(H2O)m]n+ | 13.8 | 14.8 | 12.6 | |||
| C.N. of M | 6 | 6 | 6 | 7 | 7 | 9 |
| Ionic radius of M (Å)a | 0.76 | 0.76 | 1.02 | 1.12 | 1.06 | 1.18 |
| U⋯M (Å) | 3.489(4) | 3.304(10) | 3.605(2) | 3.668(3) | 3.690(1) | 3.923(1) |
| O1⋯O2 (Å) | 2.840(2) | 2.821(5) | 2.945(6) | 2.973(7) | 2.818(7) | 2.948(8) |
| U–O7oxo (Å) | 1.778(2) | 1.782(4) | 1.761(5) | 1.782(5) | 1.772(5) | 1.795(4) |
| U–O8oxo (Å) | 1.784(2) | 1.786(4) | 1.770(4) | 1.780(5) | 1.776(5) | 1.783(4) |
| U–Ooxo (avg)c (Å) | 1.783(2) | 1.784(4) | 1.766(5) | 1.781(5) | 1.774(5) | 1.789(4) |
| U–O1phenoxide (Å) | 2.260(2) | 2.229(4) | 2.258(5) | 2.247(6) | 2.303(4) | 2.310(4) |
| U–O2phenoxide (Å) | 2.267(2) | 2.258(4) | 2.272(4) | 2.262(6) | 2.289(4) | 2.294(4) |
| U–Ophenoxide (avg)c (Å) | 2.264(2) | 2.244(4) | 2.265(5) | 2.255(6) | 2.297(4) | 2.302(4) |
| U–N1imine (Å) | 2.555(2) | 2.550(5) | 2.543(6) | 2.538(8) | 2.520(6) | 2.471(6) |
| U–N2imine (Å) | 2.533(2) | 2.552(5) | 2.544(6) | 2.555(8) | 2.514(6) | 2.527(6) |
| U–Nimine (avg)c (Å) | 2.544(2) | 2.551(5) | 2.544(6) | 2.547(8) | 2.517(6) | 2.499(6) |
| M–O1phenoxide (Å) | 2.216(4) | 2.119(10) | 2.403(5) | 2.584(7) | 2.398(5) | 2.525(4) |
| M–O2phenoxide (Å) | 2.288(4) | 2.086(10) | 2.444(4) | 2.435(8) | 2.419(4) | 2.715(4) |
| M–Ophenoxide (avg)c (Å) | 2.252(4) | 2.103(10) | 2.424(5) | 2.510(8) | 2.409(5) | 2.620(4) |
| ω crown (Å) | 0.367 | 0.701 | 0.366 | 0.717 | 0.332h | 0.609 |
| ω salben (Å) | 0.095 | 0.051 | 0.112 | 0.157 | 0.116 | 0.203 |
| ψ U (Å) | 0.013 | 0.011 | 0.032 | 0.045 | 0.017 | 0.049 |
| ψ M (Å) | 0.585 | 0.440 | 0.838 | 0.652 | 1.123h | 0.322 |
In situ synthesis and characterization of the heterobimetallic complexes
With the monometallic complexes L5UO2 and L6UO2 in hand, we turned to the preparation of the corresponding groups of heterobimetallic complexes. For the work reported here, we studied monovalent alkali metal cations (Cs+, Rb+, K+, Na+, Li+) and a single divalent ion (Ca2+), as these cations represent a wide acidity range of around four orders of magnitude, as estimated by consideration of the pKa values of corresponding metal aqua complexes. This series also features cations that could be reasonably anticipated to display satisfactory binding behavior with 15-crown-5 or 18-crown-6 moieties. We found that L5UO2 or L6UO2 could be treated with 1 equiv. of the desired metal triflate salt in acetonitrile, resulting in a virtually instantaneous change in the color of the solution that is concomitant with cation binding in the crown moieties. 1H NMR spectra for all the complexes formed in situ displayed uniform and sensible shifts in the positions of resonances that were consistent in all cases with binding of the metal cations to the oxygen atoms of the crown-ether-like cavities (see ESI, pp. S15–S33†). Isolation of powdered samples was generally not pursued; instead, samples of the complexes were generated in situ as needed to execute the experimental work. Titration studies were also performed to measure the equilibrium constants for cation binding (vide infra), and these studies indicate generally tight capture of the cations in the crown moieties as anticipated from the NMR data.Crystals of the adducts of L5UO2 with Li+, Na+, and Ca2+ suitable for X-ray diffraction analysis were grown in order to understand the structural properties of the complexes. Efforts were made to grow diffraction-quality crystals of L5UO2K, L5UO2Rb and L5UO2Cs, but none could be obtained; this could be due to the larger size of these cations.34 To facilitate the needed comparisons, we have tabulated the structural data for structures in the L5UO2M and L6UO2M families in Table 1; structures of the complexes L6UO2M were previously reported.10 Our previous work, however, did not include structural data for the L6UO2Li complex; a new structure of this compound is reported here. In the course of our work, we explored various solvent systems and note that we obtained four different crystalline polymorphs of the Na+ adduct of L5UO2 (two of which are were found to be nearly isomorphous, see p. S157 and S169†) and two different crystalline polymorphs of the Ca2+ adduct of L5UO2. The structural metrics among the related cases were quite similar (see Tables S15, S16, and Fig. S184, S194, S199†), and thus we discuss below in detail only one of the structures for L5UO2Na and L5UO2Ca in each case that lack co-crystallized outer-sphere solvent. (One structure of L5UO2Na and the structure of L5UO2Li were also found to be nearly isomorphous. See p. S170.†)
In all cases, XRD analysis confirmed assembly of the desired [UO2(μ2-Ophenoxide)2Mn+] cores. In the L5UO2M structures, the secondary metal cations ions are incorporated at the 15-crown-5-like site in all cases and ligated by the five macrocyclic O-atoms. Each of the structures with Li+ and Na+ feature one κ1-triflate while the structure with Ca2+ features two κ1-triflates (see Fig. 4 and Fig. S174–S199†). Li+ and Na+ adopt C.N. = 6 despite their disparate ionic radii; this observation is in line with an analogous structure of the L6UO2Na complex10 in which the secondary cation adopts C.N. = 7, suggesting that the size of the macrocyclic crown ether moieties, in both cases, influence the bonding preferences of the cations.
 | ||
| Fig. 4 Solid-state structures (from XRD) of (a) L5UO2Li, (b) L5UO2Na, and (c) L5UO2Ca. All hydrogen atoms, co-crystallized solvent molecules, and minor components of disorder are omitted for clarity. Displacement ellipsoids are shown at the 50% probability level. | ||
When Li+ binds to the 18-crown-6-like site in L6UO2Li, however, only five crown O-atoms interact with the cation, leaving one ethereal O-atom uncoordinated. The crown moiety is nonplanar in this structure, a feature attributable to the flexible nature of the crown moiety and a mismatch of the small Li+ cation's ionic radius with the larger 18-crown-6 moiety (Fig. S174–S177†). The result is the formation of a dimer of the desired L6UO2Li species in the solid state, wherein an Ooxo of the uranyl unit of one equivalent of L6UO2Li bridges to the Li+ of the adjacent equivalent of the heterobimetallic species (see Fig. 5). Interestingly, the O3⋯O6 separation of 3.947 Å results in a nearly square array of oxygens ideal for binding of Li+ at its center. This also allows O5 to coordinate above the plane but leaves a vacant trans-coordination site below the plane which can be accommodated by uranyl upon dimerization to give overall octahedral lithium centers. Despite the unique chemical environments of terminal oxo ligands O7 and O8, the U–O bond lengths are not different within the conventional 3σ criterion (see Table 1). This result is consistent with findings from Rissanen and co-workers in their characterization of a ditopic uranyl salophen complex.35 However, we note that direct interactions of actinyl oxos with secondary metals are still rather rare, especially for U(VI), and thus we anticipate that the properties of L6UO2Li could be worthy of further investigation.
 | ||
| Fig. 5 Solid-state dimeric structure from XRD of L6UO2Li. All hydrogen atoms, outer-sphere triflate counteranions, and minor components of disorder about the U–N3 bond are omitted for clarity. Displacement ellipsoids are shown at the 50% probability level. | ||
The U–Ooxo distances in all the structures reported here are consistent with the retention of the U(VI) formal oxidation state. Unsurprisingly, the U–Ophenoxide and U–Nimine distances span narrow ranges, consistent with the uranyl motif being firmly bound in structurally similar sites in all cases for both the L5UO2M and L6UO2M species. This is in accord with the high degree of similarity for the uranyl sites in the corresponding monometallic species (see Fig. S173†). Also unsurprisingly, comparing the structures of the L5UO2M and L6UO2M species reveals that the structures of the crown sites vary systematically across the series of complexes depending on the size and charge of the incorporated secondary cation. More specifically, the value of ωcrown increases in each case upon cation binding from the values measured for the monometallic complexes L5UO2 and L6UO2 of 0.055 and 0.358, respectively. Notably, the ωcrown parameter is ca. 2× larger in all cases for the 18-crown-6-containing derivatives, indicating that the larger crown behaves more flexibly in all cases. The ψM parameters for the 15-crown-5-based derivatives is systematically greater than those found for the 18-crown-6-based derivatives. Consequently, the smaller ligand can be concluded to be less able to encapsulate the secondary cations both because of the relative smaller size of the ring in relation to the ionic radii of the secondary cations. As a result, the cations tend to bind above the plane of the macrocyclic ligand, a feature that could be enhanced by coordination of oxygen from triflate. In the larger 18-crown-6-based ligand, however, the cations can be encapsulated within the crown moiety as it is able to flex around incorporated cations. This tendency for the cations in the L5UO2M series to be bound above the macrocyclic plane contrasts with the positions of the U centers that are much closer to the macrocyclic plane and have an ideal fit with the Schiff-base moiety (quantified by ψU).
Electrochemical studies
With the ability to prepare the heterobimetallic complexes in the L5UO2M or L6UO2M seriesm, we next explored the influence of the secondary metal cations on the thermodynamic E1/2(UVI/UV) reduction potentials of the complexes. This could be achieved by adding one equivalent of the corresponding metal triflate salt to a solution of the desired monometallic uranyl complex (either L5UO2 or L6UO2) in CH3CN and subsequently recording cyclic voltammetry (CV) data. To begin, however, the CV profiles of L5UO2 and L6UO2 are unremarkable, displaying chemically reversible reduction at E1/2 = −1.57 V and −1.53 V vs. ferrocenium/ferrocene (denoted hereafter as Fc+/0), respectively. The peak-to-peak separations (ΔEp) of 77 mV and 73 mV, respectively, are consistent with fast electron transfer (at 100 mV s−1) for our configuration (see Table 2). The similar E1/2 values in both cases are no doubt a consequence of the quite similar ligand environments of the uranyl moieties in the complexes, as confirmed by the XRD analysis (vide supra). On the basis of prior spectroscopic work with uranyl complexes of the Schiff-base type used here, the measured reductions can be assigned as UVI/UV couples in both cases. And, as the anodic and cathodic waves are linearly proportional to the square root of scan rate, both the UVI and UV forms of L5UO2 and L6UO2 are soluble and freely diffusing as anticipated (see ESI, Fig. S80–S93†).38| L5UO2M | L6UO2M | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| M | E 1/2/mV vs. Fc+/0 | ΔEp/mV | k 0/cm s−1 (X 10−3) | K a/M−1 | Δδmax1Ha/ppm |
E 1/2/mV vs. Fc+/0 | ΔEp/mV | k 0/cm s−1 (X 10−3) | K a/M−1 | Δδmax1Ha/ppm |
| a The uncertainty associated with measurements of 1H NMR chemical shifts, δ1H, in our experiments is ±0.003 ppm (reported as ±1σ) based on replicate linewidth measurements. See ESI, Fig. S163 and Table S11 on p. 139.† | ||||||||||
| None | –1.57 | 77 | 9.5 ± 4.0 | — | — | –1.53 | 74 | 14.6 ± 2.5 | — | — |
| Cs+ | –1.40 | 108 | 3.4 ± 1.0 | 550 ± 80 | 0.04 | –1.38 | 113 | 5.7 ± 1.9 | >105 | 0.03 |
| Rb+ | –1.40 | 100 | 4.9 ± 1.1 | 1370 ± 130 | 0.05 | –1.37 | 89 | 6.9 ± 1.4 | >105 | 0.04 |
| K+ | –1.37 | 91 | 5.2 ± 1.8 | 1840 ± 260 | 0.07 | –1.36 | 83 | 14.3 ± 1.5 | >105 | 0.04 |
| Na+ | –1.27 | 86 | 7.4 ± 1.3 | >105 | 0.11 | –1.35 | 79 | 13.5 ± 2.1 | >105 | 0.09 |
| Li+ | –1.25 | 130 | 2.0 ± 0.3 | >105 | 0.12 | –1.23 | 185 | 0.9 ± 0.1 | 1370 ± 180 | 0.21 |
| Ca2+ | –0.83 | 293 | 0.3 ± 0.1 | 9730 ± 5900 | 0.17 | –0.88 | 348 | 0.2 ± 0.1 | — | 0.15 |
Addition of one equivalent of the secondary cations (Cs+, Rb+, K+, Na+, Li+, and Ca2+) results in a shift of the E1/2 value to a more positive potential. Across the series of complexes, the more positive shifts are associated with the more Lewis acidic cations, such that Li+ engenders shifts in E1/2 of +310 mV for L5UO2Li and +300 mV for L6UO2Li, while Ca2+ engenders shifts of +740 for L5UO2Ca and +650 mV for L6UO2Li. In all cases, scan rate-dependent studies confirmed that the heterobimetallic species remain fully diffusional in both the U(V) and U(VI) oxidation states. Plotting all the measured E1/2 values for the heterobimetallic species obtained with the monometallic cations in each ligand family as a function of the pKa values of the corresponding metal aqua complexes reveals linear correlations in both cases of −63 ± 10 and −49 ± 14 mV/pKa, respectively (see Fig. 7). These values are indistinguishable within the uncertainty of the least-squares fitting used to determine the sensitivity of the relationship, and similar trend lines result from inclusion of the data for Ca2+ and/or using the cathodic peak potentials (Ep,c) in place of the formal E1/2 values (see ESI, Fig. S94–S96†). Consequently, we conclude that the cation-induced shift in solution redox chemistry is indistinguishable across the L5UO2M and L6UO2M series.
In order gain further insight into the cation-modulated electrochemical properties of the heterobimetallic species reported here, electrochemical titrations were carried out (see ESI, pp. S95–S110†). In this work, solutions containing L6UO2 were titrated with a standard series of triflate salts (KOTf, NaOTf, LiOTf, and Ca(OTf)2) in order to understand the interactions that may be occurring between the as-prepared U(VI) and electrogenerated formally U(V) forms of the complexes. Additionally, we were interested in understanding if flooding the system with excess metal cation in each case would lead to significant changes in the accessible redox chemistry. As direct cation-uranyl interactions could influence the heterogeneous electron transfer kinetics studied in this work, the nature of the binding of cations to the U(VI) and U(V) forms is particularly pertinent to this study and can be interrogated through these titrations as well.
The data corresponding to titration of L6UO2 with KOTf confirm key features of the chemical behavior of the resulting L6UO2K. Generally speaking, binding of K+ in the 18-crown-6-like site appears tight; there is a virtually isosbestic conversion of the starting monometallic complex to the expected bimetallic species up to addition of 1 equiv. of K+ (see Fig. S97†). Importantly, over this range, there is not a gradual shift in the value of E1/2 that would indicate rapid exchange on the electrochemical timescale. Instead, the redox wave for the parent monometallic complex diminishes cleanly and a new wave for L6UO2K grows in cleanly as the cation concentration increases. This suggests that K+ binding to both the formal U(VI) and U(V) forms of the complex is fast and irreversible on the timescale explored here. Additionally, the redox waves for both the monometallic and bimetallic complexes compare well with the data shown in Fig. 6, suggesting that the presence of U:M ratios less or greater than 1:1 do not result in chemical reactivity beyond 1:1 cation binding (see Table S5†). Importantly, the presence of excess K+ (up to 10 equiv.) does not contribute to changes in the appearance of the redox wave for the bimetallic species, suggesting little tendency of the uranyl oxo moieties in the electrogenerated, formally U(V) form to bind K+ under these conditions. Similar profiles were measured in this data regardless of the number of cycles of voltammetry carried out, further underscoring the conclusion that K+ does not interact strongly with either the U(VI) or U(V) forms of L6UO2 beyond binding in the 18-crown-6-like site.
 | ||
| Fig. 6 In situ cyclic voltammetry experiments for uranium complexes used in this study. L5UO2 and L5UO2M complexes are represented by solid colors; L6UO2 and L6UO2M complexes are represented by faded colors. Electrolyte: 0.1 M TBAPF6 in CH3CN, scan rate: 100 mV s−1, electrode: highly oriented pyrolytic graphite. | ||
 | ||
| Fig. 7 Plot of E1/2(UVI/V) vs. pKa of [M(H2O)m]n+ for L5UO2M complexes (a, upper panel) and L6UO2M complexes (b, lower panel). | ||
The results from titrations involving NaOTf (pp. S100–S102†), LiOTf (pp. S103–S106†), and CaOTf (pp. S107–S110†) are similar, suggesting tight and irreversible binding of 1 equiv. of M to the starting L6UO2 in each case. Additions of each cation beyond 1 equiv. were not associated with significant changes in the electrochemical profiles measured, apart from the anticipated dilution of the uranium-containing solution that led to diminished reductive and oxidative peak currents (ip,c and ip,a values) in the voltammograms associated with the solutions containing the greatest cation concentrations. Consequently, we conclude that all of the tested cations interact strongly with both U(VI) and U(V) by binding in the 18-crown-6-like site.
As formation of insoluble oxo-bridged multimetallic uranium species is a recognized reactivity mode of U(V) complexes, we particularly searched for evidence of formation of such species in the titration studies reported here. No significant electrode fouling was encountered in any case as confirmed by appropriate wipe tests (see Fig. S104 and S108†) and multicycling experiments (Fig. S98, S101, S105, and S109†). A very minor feature that appears to be associated with electrode modification was observable after significant numbers of cycling experiments, but as this feature was not found to vary significant with cation identity (Fig. S112 and S113†), we do not anticipate that it is associated with a soluble, molecular uranium-containing species. Consequently, based on the overall results from the titration studies, there is virtually no evidence of formation of oxo-bridged heterobimetallic species in solution either in the U(VI) or U(V) oxidation states. This conclusion is in accord with Raman studies carried out on the 1:1 U:M species in solution (vide infra) and with the general finding that the U(VI)/U(V) reduction potential does not depend on the added cation concentration beyond 1 equiv. (see Fig. S111†). And, all this suggests that the formation of oxo-bridged species is not likely to drive the measured changes in heterogeneous electron transfer rates as a function of cation (M) identity, since the availability of free cation in solution (not bound in the 18-crown-6-like site) could be anticipated to encourage formation of such species more readily than when the only cation available for formation of oxo-bridged species is already bound in the 18-crown-6-like site.
Raman spectroscopic interrogation of the uranyl moieties
We anticipated that a more detailed examination of the properties of the uranyl moiety in each heterobimetallic complex should be pursued. This is because the shift in UVI/UV reduction potential measured for both families of complexes is attributable to the electrostatic influence of the secondary cations on the uranyl moiety in both the U(VI) and U(V) oxidation states. Considering this, it would be virtually impossible to reliably attribute changes in the reduction potential to structural or electrostatic considerations known only for the oxidized form of the complexes, because the bound cations should influence the properties of both the U(VI) and U(V) forms. We anticipated, however, that it would be quite feasible to measure the dependence of vibrational features of the uranyl system in each ligand as a function of the incorporated secondary metal cation. With this approach, we anticipated that we could cross-correlate cation-dependent properties for the isolated complexes available to us in the U(VI) oxidation state. In this case, Raman spectroscopy appealed to us immediately as this technique has been firmly established as a useful probe of the uranium-oxo symmetric stretching mode (commonly denoted ν1 or νsym in the literature). From the Raman data, we envisioned that we might measure modulation of the donor power of the bridging phenoxide ligands to the uranyl motif induced by incorporation of the secondary metal cations to the complexes L5UO2M and L6UO2M.Raman spectra were thus collected on crystalline samples of the heterobimetallic complexes.|| Derivatives in the L5UO2M series where M = Rb+, K+, Na+, Li+, and Sr2+ were investigated, while work with the L6UO2M system was conducted with M = Rb+, K+, Na+, Sr2+, Ca2+, and La3+. Spectra were also collected on crystalline samples of L5UO2 and L6UO2. In the case of L5UO2 and L6UO2, νsym appeared at 812 and 813 cm−1, respectively (see ESI, Fig. S39, S46, and Tables S1, S2†). For the heterobimetallic L5UO2M and L6UO2M complexes, νsym frequencies for the U–Ooxo units were measured across the range of 812–819 cm−1 for the L5UO2M complexes and across a wider range from 807–828 cm−1 for the L6UO2M complexes (see ESI, Tables S1 and S2†). The similar νsym values for the monometallic complexes can be attributed to the similar uranium coordination environments in both cases, as confirmed by the XRD analysis. Incorporation of secondary metal cations into either L5UO2 or L6UO2 generally resulted in either no significant shift (including minor red-shifts) of the νsym or a more substantial blue shift; the shift measured appears to depend on the identity and characteristics of the particular incorporated cation (see ESI, pp. S34–S63†). Blue shifting, in this context, can be interpreted as being indicative of a relative strengthening of the U–Ooxo bonding upon formation of the heterobimetallic species. This phenomenon can be attributed to a charge density effect induced by interaction of the secondary metal cations with the phenoxide ligands that bridge to the uranium center. Of course, metal cations that are less charge dense would be anticipated to afford little to no blue shift in the measured value of νsym, owing to the meager power of those particular cations to modulate the donor power of the bridging phenoxide moieties to the uranium center.13
Plotting the symmetric stretching frequencies for each series of the heterobimetallic complexes as a function of the pKa values of the corresponding metal aqua complexes revealed a linear relationship for both the L5UO2M and L6UO2M series (Fig. 8). The trend line in the data for the L5UO2M series has a shallower dependence of −1.21 ± 0.51 cm−1/pKa than that measured for the L6UO2M series at −4.21 ± 0.70 cm−1/pKa, when comparing the data available for the mono- and divalent cations. (The result is similar when including data for L6UO2La as well; see ESI, Fig. S54.†) The substantial difference in the slopes of the trend lines (greater than their combined uncertainties) can be readily attributed to the distinguishing feature of the two series of complexes that was observed in the XRD analysis; namely, in the solid state, the secondary cations are bound well above the macrocyclic plane in the L5UO2M series, whereas in the L6UO2M series, the metal cations are effectively encapsulated by the 18-crown-6-like site. This conclusion, quantified based on the tabulated ψM parameters for the complexes, is notable when compared to findings in a recent analysis of the influence of secondary cations on the asymmetric stretching frequency of the vanadyl ion, [VO]2+, measured with infrared spectroscopy.13 In both cases, there is an apparent linear relationship between the tendency for the secondary metal cations to diminish the donor ability of the bridging phenoxides to the uranium center, driving the generally blue-shifted vibrational characteristics in both cases. These spectroscopic shifts depend only on the structures of the U(VI) forms of the complexes, in contrast to the electrochemical work in which U(V) forms also must be considered. This is noted as a key difference between the spectroscopic and electrochemical work reported here (Table 3).
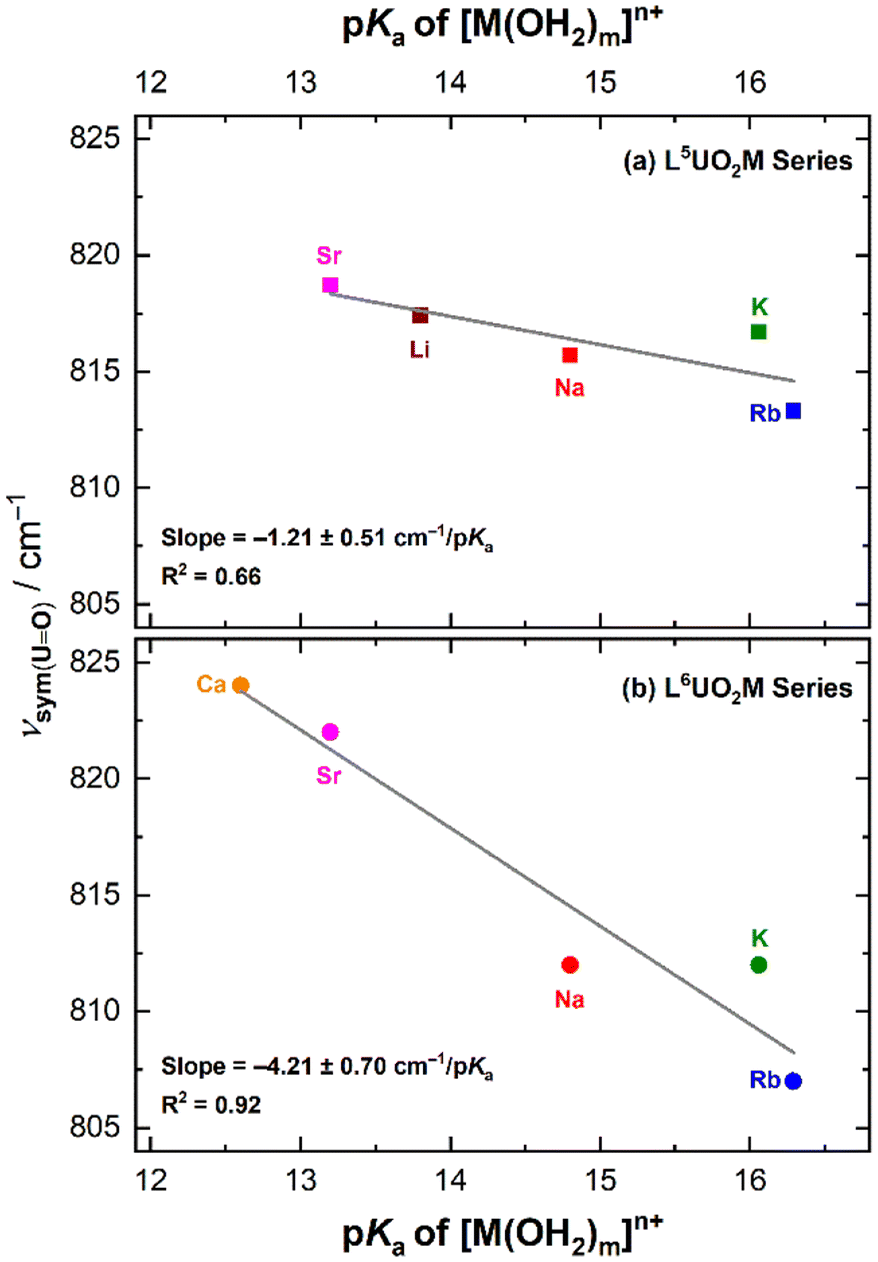 | ||
Fig. 8 Dependence of the U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O symmetric stretching frequency from Raman spectroscopy of the L5UO2M (a, upper panel) and L6UO2M (b, lower panel) complexes on the Lewis acidity (pKa) of the corresponding metal cation aqua complexes. O symmetric stretching frequency from Raman spectroscopy of the L5UO2M (a, upper panel) and L6UO2M (b, lower panel) complexes on the Lewis acidity (pKa) of the corresponding metal cation aqua complexes. | ||
Along this line, the solid-state Raman spectrum collected for crystalline L6UO2Li revealed νsym at 811 cm−1. This value does not align with the trend line measured for the other heterobimetallic complexes, a finding attributable to the dimeric nature of the complex in the solid state as observed by XRD. We anticipate that the direct U–Ooxo⋯Li interactions causes this deviation, a notion supported by observation of a distinctive signal for L6UO2Li at 897 cm−1 (see ESI, Fig. S50†) that is absent in the spectra of other complexes studied. The feature at 897 cm−1 can be assigned as the asymmetric stretching mode (commonly denoted ν3 or νas), observable under these conditions due to lowered site symmetry of the [UO2] motif upon direct interaction with Li+. The νas feature for L6UO2Li was also observed by infrared spectroscopy at a similar estimated energy of 892 cm−1 (see p. S67†). However, in line with the clean electrochemical behavior observed by cyclic voltammetry, a Raman spectrum collected on a solution of the L6UO2Li complex revealed a feature that corresponds to νsym at 821 cm−1 (see ESI, Fig. S59†) This frequency compares well with the νsym values of the other complexes in the L6UO2M family in solution (Table S3†), indicating that the cations remain bound in solution but that the dimeric form of L6UO2Li can be readily cleaved into individual heterobimetallic species in solution. To further interrogate the possibility of dimerization of L6UO2Li in solution, we collected Raman spectra in CH3CN solution across a range of concentrations from 1–25 mM (see ESI, pp. S62–S63†). There was no significant change in the established νsym value of 821 cm−1 for the compound across this range, suggesting that dimerization and/or other speciation of this system does not occur under these conditions. We anticipate that these observations are all consistent with the coordinating nature of CH3CN, which appears suitable for breaking apart the dimeric species observed in the solid-state XRD data.
In accord with all these observations, a tight linear correlation of −1.64 ± 0.31 cm−1/pKa between νsym and the pKa of [M(OH2)m]n+ was observed upon comparing all the solution-phase spectra (Fig. S63†), confirming that the cations’ influence over the properties of the uranyl system are persistent in solution. At this stage, we have not assigned an interpretation to the magnitudes of the correlations for the L6UO2M series in the solution vs. in the solid state, although we anticipate that the difference is attributable to solvation of the complexes upon dissolution.
Heterogeneous electron transfer kinetics
The trends measured among the reduction potentials and symmetric O–U–O stretching modes for the heterobimetallic complexes confirm that incorporation of the secondary metal cations results in more electron-poor uranium centers. This change in charge density is reflected in both the increased energies for the O–U–O stretching modes and the more positive reduction potentials. However, inspection of the electrochemical data revealed another set of cation-dependent behaviors. Namely, there is a distinct change in the peak-to-peak separations (ΔEp) of the redox waves across each series of complexes. As shown in Table 2, the ΔEp values for the L5UO2M complexes increase (at 100 mV s−1) going from the derivatives incorporating Cs+, Rb+, K+, and Na+, but then decrease again from Na+ to Li+ to Ca2+ (see Table 2). The results for the L6UO2M series are similarly non-monotonic, but in the case of that series of complexes, as the ΔEp values are similar for Na+ and K+ and for Li+ and Ca2+. Observing this, we initially imagined that the Lewis acidity (or another property) of the incorporated secondary metal cation, the cation's binding affinity, and/or geometric rearrangements on reduction could drive these changes in ΔEp.In order to examine this trend in a quantitative fashion, we determined the standard heterogeneous electron transfer rate constant, denoted k0, for each complex using the traditional method of Nicholson.41 In our method (see ESI, p. S140† for details), we measured the ΔEp values for the complexes in each series at six different scan rates, and then determined an average k0 value (given in Table 2). As expected, there was little variability in the measured k0 values across scan rates; this is reflected in the low uncertainty associated with the average k0 values and is indicative of k0 being invariant with scan rate as expected from electrochemical theory.42
Plotting the k0 values for each series of complexes as a function of the Lewis acidity of the incorporated secondary metal cation revealed “volcano plots” in each case (see Fig. 9). As expected on the basis of the ΔEp values, the complexes containing the most Lewis acidic cations in our series, Li+ and Ca2+, display the slowest electron transfer kinetics (reflected in small k0 values). In the L5UO2M series, L5UO2Na displays the fastest electron transfer kinetics, whereas in the L6UO2M series, both L6UO2Na and L6UO2K display fast kinetics that are indistinguishable within the uncertainty of our measurements. These findings suggest that the interplay of factors mentioned above could govern the electrochemical reversibility of the UVI/UV reduction manifold. Lewis acidity does appear to be an influence, a finding attributable to the tendency of a strongly Lewis acidic cations to engender chemical reactivity. However, on the basis of the titration experiments, chemical reorganization of the complexes upon generation of U(V) involving direct interaction of the secondary metal cations with the oxo ligands of the uranyl system upon reduction is unlikely.
 | ||
| Fig. 9 Plots of k0vs. pKa of [M(H2O)m]n+ for L5UO2M complexes (a, upper panel) and L6UO2M complexes (b, lower panel). | ||
Tight binding of the cations in the crown-ether-like sites of L5UO2 and L6UO2 would likely improve electrochemical reversibility by minimizing reorganization before/after reduction. Along this line, the diminished electron transfer kinetics associated with the derivatives of both L5UO2 and L6UO2 incorporating Rb+ and Cs+ are suggestive of a poor size match, and thus weaker binding, of these cations for/in the 15-crown-5- and 18-crown-6-like sites. As result, the cations are likely more easily lost from the crown sites, particularly upon uranium-centered reduction. We were unable to grow diffraction-quality crystals of the Rb+ and Cs+ complexes despite many attempts, a finding in accord the poor size match of these cations with the crown sites in the complexes.
Quantification of cation binding strength
To provide data for interpreting the cation influence over the electron transfer kinetics, we measured the binding constants (Ka values) of the cations in the monometallic complexes in CD3CN. The Ka values were measured with titrations relying on 1H nuclear magnetic resonance (NMR) spectroscopy; in our method, the chemical shift value (δ1H) associated with the three equivalent protons of the methyl group of each complex was found to be correlated in each case with increasing concentrations of the triflate salts of the individual cations. The change in the δ1H value (Δδ1H) was thus used to determine the value of Ka by fitting to the 1:1 binding isotherm.43 As discussed in prior work on use of titrations with phosphine-oxide probes to study cation behaviors, satisfactory fits to this isotherm provide support for 1:1 binding stoichiometry.39 The titration data also provide a readout of the maximum achievable change in chemical shift (Δδmax1H) as well, representing detailed information of the influence of the given secondary metal cation on the heterobimetallic species that is independent of its binding constant (Ka value). Stated another way, the Δδmax1H values describe the maximum possible de-shielding of the methyl protons, specifically at the condition where the metal ion of interest is maximally occupying its binding site.
In each set of titration data, the Δδ1H value initially undergoes significant changes at lower titrant cation equivalencies but eventually levels off at higher equivalencies (see ESI, pp. S114–S137†). The plateauing behavior is indicative of clean binding of 1 equiv. of cation in the crown-like-site in each case, even when large excesses (>5 equiv.) of the metal cations were added. For some cases, very tight binding was implied in the data, and for these cases, fitting to the 1:1 binding isotherm was not possible; the data in these cases suggest only a lower estimate of the Ka value.
The Ka values for binding of Cs+, Rb+, K+, and Na+ in the crown site of L5UO2 increase in the same order, a trend that is in agreement with the ionic radii of these cations. Similar to findings for related, purely organic 15-crown-5 derivatives, the Na+ ion binds tightly to L5UO2.44 Li+ also binds quite tightly to L5UO2, a finding in accord with the results of XRD analysis for L5UO2Li that confirmed the ability of the 15-crown-5-like site in this case to bind Li+. For Ca2+, titration results indicate more modest binding; the weaker binding in comparison to Na+ and Li+ could be attributable to the steric bulk associated with the two triflate counteranions accompanying Ca2+, as in the XRD structure, both triflates are restricted to binding to Ca2+ on the same hemisphere or face of the overall heterobimetallic species.
Tight associations of Cs+, Rb+, K+, and Na+ with L6UO2 were measured in all cases, in accord with the larger size the 18-crown-6-like site in comparison to the 15-crown-5-like site in L5UO2. However, the binding of these cations is sufficiently strong to preclude detailed comparisons among them (Table 2 and Fig. S162†). Such large binding coefficients (>105 M−1) are sensible, however, given the recognized ability of 18-crown-6 species to bind a variety of metal cations.
We also examined the Δδmax1H values from the titrations for any trends. Plotting the Δδmax1H values as a function of the pKa values resulted in reasonable linear correlations in both cases (Fig. 10). These data show that the more acidic cations result in more significant charge-density-driven perturbations in the heterobimetallic complexes. The observation of reasonable linear correlations in the NMR titration data and values of Δδmax1H for both the L5UO2M and L6UO2M series of complexes is commensurate with the other linear trends measured in this work.
 | ||
| Fig. 10 Plots of Δδmax1H vs. pKa of [M(H2O)m]n+ for L5UO2M complexes (a, upper panel) and L6UO2M complexes (b, lower panel). | ||
Discussion
The heterogeneous electron transfer rate constants for both the L5UO2M and L6UO2M series of complexes appear to be influenced by both the association constants for cation binding and the Lewis acidities of the cations. For L5UO2M, the smaller 15-crown-5 like site engenders behavior in which only Na+ and Li+ are bound very tightly. This result is in agreement with observation of the fastest ET kinetics for L5UO2Na. Li+ and Ca2+ are more Lewis acidic cations, but the origin of the diminished k0 values for these cations is not yet clear; reactivity with nascent uranium(V) species is not supported by related electrochemical titration data. However, the binding of Na+ is significantly stronger than that of Li+ in the polyether site; comparing to the organic crown literature, our system follows the natural preference of 15-crown-5-type species for binding of Na+.23,45 This could suggest that the lithium system undergoes greater reorganization after reduction, perhaps involving displacement of the bound triflate counteranion.For the heterobimetallic species in the L6UO2M family, similar results were obtained, but with a shift in the trends that is correlated with tightest binding of both Na+ and K+. Our data do not distinguish between these cations in terms of binding strength, but the fast electron transfer kinetics in both cases is consistent with the tight binding of these ions. As in the L5UO2M series, use of the more Lewis acidic cations Li+ or Ca2+ or the anticipated weaker binding Cs+ or Rb+ was found to be associated with slower electron transfer kinetics, but the precise mechanisms underpinning this behavior is not yet clear. Reorganization penalties associated with displacement of bound triflate, or other ligands, are possible in the L6UO2M series as in L5UO2M.
This study has generated a set of electrochemical and spectroscopic data suitable for quantitatively interpreting the rational influence that secondary metal cations exert over the uranyl moiety in heterobimetallic complexes. The pKa-dependence of the UVI/UV reduction potentials measured by cyclic voltammetry and the uranyl symmetric stretching vibrations measured by Raman spectroscopy suggest that the U(VI) centers in the heterobimetallic complexes become more electron deficient upon incorporation of secondary metal cations. A quantitative dependence on Lewis acidity of this type (involving vibrational spectroscopic and electrochemical evidence) has not been reported previously, to the best of our knowledge. However, this finding is reminiscent of recent work from Blakemore and co-workers13 that examined the use of a crown-containing ligand for the vanadyl dication, [VO]2+, as a spectroscopically and electrochemically addressable probe molecule in a set of heterobimetallic complexes; evidence was assembled in support of the hypothesis that the influence of secondary cations could be understood to arise from modulation of the donor strength of phenoxide ligands that bridged to the vanadium center. Here, Raman spectroscopy was used to probe the more symmetrical trans-dioxo uranyl system, and results in accord with cation-driven changes in bridging ligand donor power were obtained.
The secondary cations in the heterobimetallic complexes studied here appear to modulate the donor strength of the phenoxide moieties that interact with both the secondary cation and the uranium center in each species. Use of pKa values as the descriptor for Lewis acidity is sensible given the ability for secondary cations to polarize bound water molecules, resulting in the unique pKa value for each metal cation.46 The robust trends shown in Fig. 7 and 8 show that the pKa values can be used, similarly, to describe the polarization the phenoxide moieties in the heterobimetallic complexes studied here. Any other effects wrought by the incorporated cations appear subordinate to this first coordination-sphere effect.
In light of the consistent trends in the electrochemical and spectroscopic data presented in this manuscript, use of tailored macrocyclic ligand systems in concert with Lewis acidic metal cations can be concluded to be a useful strategy for controlling the redox properties of the uranyl dication. In the systems studied here, the ligand systems H2L5 and H2L6 enable formation of heterobimetallic species that would, most likely, not otherwise form in solution or persist for chemical investigations. Future work could focus on structural and spectroscopic investigations of the formally U(V) forms of the complexes, building on the useful features encountered during study of the U(VI) forms reported here. We anticipate that detailed study of the U(V) species would provide an even richer platform upon which to study cation-modulated actinide redox chemistry, considering the greater reactivity of U(V) in comparison to U(VI). Investigations along this line are currently underway in our laboratories.
Conclusions
Here, structural changes to supporting macrocyclic ligands have been shown to tune the ability of secondary metal cations to influence the electronic properties of the uranyl dication, UO22+. The association constants of the secondary cations with the parent uranyl-containing species were tuned by the ligand design, giving a structural handle for harnessing cations’ inherent Lewis acidity properties. Raman spectroscopy and electrochemistry have been used to demonstrate quantitative relationships between cation Lewis acidity and diminished bridging-ligand donation to uranium, affording a spectroscopic counterpart to the measured redox. Taken together, these studies demonstrate the broad usefulness of Lewis acidity as a descriptor for the properties of multimetallic actinide complexes, and suggest further opportunities for ligand design to impact studies of multimetallic actinide chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Justin Douglas and Sarah Neuenswander for assistance with NMR spectroscopy, and Michael Lemon and Alice Dale for assistance with radiation safety protocols. This work was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences through the Early Career Research Program (DE-SC0019169). The authors also acknowledge the US National Institutes of Health for support of the NMR instrumentation (S10OD016360 and S10RR024664) used in this study. T. Z. F. and M. K. P. acknowledge U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences through the Heavy Element Chemistry Program (DE-SC0021420) for the spectroscopic experiments that were performed at the University of Iowa. K. P. C. was supported by start-up funding from the College of Liberal Arts and Sciences and the Department of Chemistry at the University of Iowa. Solid-state mid- and far-IR spectra were collected on an instrument funded by a Nuclear Regulatory Commission Faculty Development Grant (NRC 31310018M0042).References
- B. M. T. Costa Peluzo and E. Kraka, Uranium: The nuclear fuel cycle and beyond, Int. J. Mol. Sci., 2022, 23, 4655 CAS.
- J. D. Navratil and W. W. Schulz, Actinide Separations, American Chemical Society, 1980, vol. 117 Search PubMed.
- (a) J. B. Starks, Purex process, DPSPU-77-11-1, TRN: 78-007289, United States, 1977 Search PubMed; (b) J. M. Mckibben, Chemistty of the Purex Process, Radiochim. Acta, 1984, 36, 3–16 CAS; (c) H. A. C. McKay, The PUREX process, CRC Press Inc., United States, 1990 Search PubMed.
- (a) M. F. Simpson and A. M. Casella, in Engineering Separations Unit Operations for Nuclear Processing, CRC Press, 2019, pp. 305–321 Search PubMed; (b) M. J. Martínez-Rodríguez, L. C. Olson, J. R. Gray and B. L. García-Díaz, Non-Aqueous Electrochemical Fluorination of Used Nuclear Fuel as an Advanced Separation Process, J. Electrochem. Soc., 2019, 166, E231 CrossRef.
- D. LeBlanc, Molten salt reactors: A new beginning for an old idea, Nucl. Eng. Des., 2010, 240, 1644–1656 CrossRef CAS.
- (a) R. G. Denning, Electronic structure and bonding in actinyl ions, Complexes, Clusters and Crystal Chemistry, 1992, pp. 215–276 Search PubMed; (b) R. G. Denning, Electronic Structure and Bonding in Actinyl Ions and their Analogs, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS PubMed.
- A. Bard, R. Parsons and J. Jordan, Standard Potentials in Aqueous Solution, M Dekker, New York, 1985 Search PubMed.
- J. W. Freiderich, E. Wanigasekara, X.-G. Sun, R. A. Meisner, H. M. Meyer, H. Luo, L. H. Delmau, S. Dai and B. A. Moyer, Direct Electrodeposition of UO2 from Uranyl Bis(trifluoromethanesulfonyl)imide Dissolved in 1-Ethyl-3-methylimidazolium Bis(trifluoromethanesulfonyl)imide Room Temperature Ionic Liquid System, Electrochim. Acta, 2014, 115, 630–638 CrossRef CAS.
- (a) S. Fukuzumi, Electron-transfer properties of high-valent metal-oxo complexes, Coord. Chem. Rev., 2013, 257, 1564–1575 CrossRef CAS; (b) W. Nam, Y.-M. Lee and S. Fukuzumi, Tuning reactivity and mechanism in oxidation reactions by mononuclear nonheme iron(IV)-oxo complexes, Acc. Chem. Res., 2014, 47, 1146–1154 CrossRef CAS PubMed; (c) S. Fukuzumi, K. Ohkubo, Y. M. Lee and W. Nam, Lewis acid coupled electron transfer of metal–oxygen intermediates, Chem. – Eur. J., 2015, 21, 17548–17559 CrossRef CAS PubMed; (d) J. Park, Y. Morimoto, Y.-M. Lee, W. Nam and S. Fukuzumi, Unified view of oxidative C–H Bond cleavage and sulfoxidation by a nonheme Iron(IV)–Oxo complex via lewis acid-promoted electron transfer, Inorg. Chem., 2014, 53, 3618–3628 CrossRef CAS PubMed.
- A. Kumar, D. Lionetti, V. W. Day and J. D. Blakemore, Redox-inactive metal cations modulate the reduction potential of the uranyl ion in macrocyclic complexes, J. Am. Chem. Soc., 2020, 142, 3032–3041 CrossRef CAS PubMed.
- A. Kumar, D. Lionetti, V. W. Day and J. D. Blakemore, Trivalent Lewis acidic cations govern the electronic properties and stability of heterobimetallic complexes of nickel, Chem. – Eur. J., 2018, 24, 141–149 CrossRef CAS PubMed.
- R. R. Golwankar, A. Kumar, V. W. Day and J. D. Blakemore, Revealing the Influence of Diverse Secondary Metal Cations on Redox–Active Palladium Complexes, Chem. – Eur. J., 2022, 28, e202200344 CrossRef CAS PubMed.
- C. M. Dopp, R. R. Golwankar, S. R. Kelsey, J. T. Douglas, A. N. Erickson, A. G. Oliver, C. S. Day, V. W. Day and J. D. Blakemore, Vanadyl as a Spectroscopic Probe of Tunable Ligand Donor Strength in Bimetallic Complexes, Inorg. Chem., 2023, 62, 9827–9843 CrossRef CAS PubMed.
- J. P. Karnes, A. Kumar, J. A. Hopkins Leseberg, V. W. Day and J. D. Blakemore, Trivalent Cations Slow Electron Transfer to Macrocyclic Heterobimetallic Complexes, Inorg. Chem., 2024, 63, 8710–8729 CrossRef CAS PubMed.
- (a) J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, A synthetic model of the Mn3Ca subsite of the oxygen-evolving complex in photosystem II, Science, 2011, 333, 733–736 CrossRef CAS PubMed; (b) E. Y. Tsui, J. S. Kanady and T. Agapie, Synthetic Cluster Models of Biological and Heterogeneous Manganese Catalysts for O2 Evolution, Inorg. Chem., 2013, 52, 13833–13848 CAS; (c) E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Redox-inactive metals modulate the reduction potential in heterometallic manganese–oxido clusters, Nat. Chem., 2013, 5, 293–299 CAS; (d) D. Lionetti, S. Suseno, E. Y. Tsui, L. Lu, T. A. Stich, K. M. Carsch, R. J. Nielsen, W. A. Goddard III, R. D. Britt and T. Agapie, Effects of Lewis acidic metal ions (M) on oxygen-atom transfer reactivity of heterometallic Mn3MO4 cubane and Fe3MO(OH) and Mn3MO(OH) clusters, Inorg. Chem., 2019, 58, 2336–2345 CAS.
- (a) A. H. Reath, J. W. Ziller, C. Tsay, A. J. Ryan and J. Y. Yang, Redox Potential and Electronic Structure Effects of Proximal Nonredox Active Cations in Cobalt Schiff Base Complexes, Inorg. Chem., 2017, 56, 3713–3718 CAS; (b) S. Maity, S. Ghosh and A. Ghosh, Elucidating the secondary effect in the Lewis acid mediated anodic shift of electrochemical oxidation of a Cu(II) complex with a N0O2 donor unsymmetrical ligand, Dalton Trans., 2019, 48, 14898–14913 CAS; (c) T. K. Ghosh, S. Maity, S. Ghosh, R. M. Gomila, A. Frontera and A. Ghosh, Role of Redox-Inactive Metal Ions in Modulating the Reduction Potential of Uranyl Schiff Base Complexes: Detailed Experimental and Theoretical Studies, Inorg. Chem., 2022, 61, 7130–7142 CAS.
- (a) P. L. Arnold, D. Patel, C. Wilson and J. B. Love, Reduction and selective oxo group silylation of the uranyl dication, Nature, 2008, 451, 315–317 CAS; (b) B. E. Cowie, J. M. Purkis, J. Austin, J. B. Love and P. L. Arnold, Thermal and photochemical reduction and functionalization chemistry of the uranyl dication, [UVIO2]2+, Chem. Rev., 2019, 119, 10595–10637 CAS.
- (a) R. Faizova, S. White, R. Scopelliti and M. Mazzanti, The effect of iron binding on uranyl(v) stability, Chem. Sci., 2018, 9, 7520–7527 RSC; (b) V. Mougel, P. Horeglad, G. Nocton, J. Pécaut and M. Mazzanti, Cation–cation complexes of pentavalent uranyl: from disproportionation intermediates to stable clusters, Chem. – Eur. J., 2010, 16, 14365–14377 CrossRef CAS PubMed.
- M. J. Sarsfield and M. Helliwell, Extending the chemistry of the uranyl ion: Lewis acid coordination to a UO oxygen, J. Am. Chem. Soc., 2004, 126, 1036–1037 CAS.
-
(a) D. D. Schnaars, G. Wu and T. W. Hayton, Reduction of pentavalent uranyl to U (IV) facilitated by oxo functionalization, J. Am. Chem. Soc., 2009, 131, 17532–17533 CAS;
(b) T. W. Hayton and G. Wu, Exploring the Effects of Reduction or Lewis Acid Coordination on the UO Bond of the Uranyl Moiety, Inorg. Chem., 2009, 48, 3065–3072 CAS.
- R. R. Golwankar, M. Z. Makoś, N. Cajiao, M. L. Neidig, A. G. Oliver, C. S. Day, V. W. Day, V.-A. Glezakou and J. D. Blakemore, Activation and Functionalization of the Uranyl Ion by Electrochemical Reduction, Inorg. Chem., 2024, 63, 24542–24553 CrossRef CAS PubMed.
- E. R. Mikeska, M. Z. Makoś, G. A. Arehart, V.-A. Glezakou and J. D. Blakemore, Distinguishing Desirable and Undesirable Reactions in Multicomponent Systems for Redox Activation of the Uranyl Ion, Inorg. Chem., 2025, 64, 5827–5845 CAS.
- C. J. Pedersen, Cyclic polyethers and their complexes with metal salts, J. Am. Chem. Soc., 1967, 89, 7017–7036 CAS.
- A. H. Reath, J. W. Ziller, C. Tsay, A. J. Ryan and J. Y. Yang, Redox Potential and Electronic Structure Effects of Proximal Nonredox Active Cations in Cobalt Schiff Base Complexes, Inorg. Chem., 2017, 56, 3713–3718 CrossRef CAS PubMed.
- (a) J. R. Robinson, P. J. Carroll, P. J. Walsh and E. J. Schelter, The impact of ligand reorganization on cerium(III) oxidation chemistry, Angew. Chem., Int. Ed., 2012, 40, 10159–10163 CrossRef PubMed; (b) J. R. Robinson, Z. Gordon, C. H. Booth, P. J. Carroll, P. J. Walsh and E. J. Schelter, Tuning reactivity and electronic properties through ligand reorganization within a cerium heterobimetallic framework, J. Am. Chem. Soc., 2013, 135, 19016–19024 CrossRef CAS PubMed; (c) N. T. Rice, J. Su, T. P. Gompa, D. R. Russo, J. Telser, L. Palatinus, J. Bacsa, P. Yang, E. R. Batista and H. S. La Pierre, Homoleptic imidophosphorane stabilization of tetravalent cerium, Inorg. Chem., 2019, 58, 5289–5304 CrossRef CAS PubMed.
- (a) G. Milburn, M. R. Truter and B. Vickery, The crystal structure of a sodium complex, sodium perchlorate–bis-[NN′-ethylenebis (salicylideneiminato) copper(II)], Chem. Commun., 1968, 1188–1188 RSC; (b) M. R. Truter, in Alkali Metal Complexes with Organic Ligands, Springer, 2005, pp. 71–111 Search PubMed; (c) C. Floriani, F. Calderazzo and L. Randaccio, Crystal and molecular structure of a cationic sodium complex: the sodium tetraphenylborate adduct of NN′-ethylenebis (salicylideneiminato) cobalt(II). Cobalt(II), nickel(II), and copper(II) complexes of quadridentate Schiff bases as complexing agents for lithium, sodium, and ammonium cations, J. Chem. Soc., Chem. Commun., 1973, 384–385 RSC.
- (a) B. Guillaume, G. Begun and R. Hahn, Raman spectrometric studies of” cation-cation” complexes of pentavalent actinides in aqueous perchlorate solutions, Inorg. Chem., 1982, 21, 1159–1166 Search PubMed; (b) G. Lu, T. Z. Forbes and A. J. Haes, Evaluating best practices in Raman spectral analysis for uranium speciation and relative abundance in aqueous solutions, Anal. Chem., 2016, 88, 773–780 CrossRef CAS PubMed; (c) M. Autillo, R. E. Wilson, M. Vasiliu, G. F. de Melo and D. A. Dixon, Periodic Trends within Actinyl (VI) Nitrates and Their Structures, Vibrational Spectra, and Electronic Properties, Inorg. Chem., 2022, 61, 15607–15618 CrossRef CAS PubMed; (d) J. Bullock, Raman and infrared spectroscopic studies of the uranyl ion: the symmetric stretching frequency, force constants, and bond lengths, J. Chem. Soc. A, 1969, 781–784 RSC.
- (a) L. H. Jones and R. A. Penneman, Infrared Spectra and Structure of Uranyl and Transuranium(V) and (VI) Ions in Aqueous Perchloric Acid Solution, J. Chem. Phys., 1953, 21, 542–544 CrossRef CAS; (b) L. H. Jones, Systematics in the vibrational spectra of uranyl complexes, Spectrochim. Acta, 1958, 10, 395–403 CrossRef CAS; (c) L. H. Jones, Determination of U-O bond distance in uranyl complexes from their infrared spectra, Spectrochim. Acta, 1959, 15, 409–411 CrossRef.
- (a) D. D. Schnaars and R. E. Wilson, Structural and Vibrational Properties of U(VI)O2Cl42− and Pu(VI)O2Cl42− Complexes, Inorg. Chem., 2013, 52, 14138–14147 CAS; (b) D. D. Schnaars and R. E. Wilson, Lattice Solvent and Crystal Phase Effects on the Vibrational Spectra of UO2Cl42−, Inorg. Chem., 2014, 53, 11036–11045 CAS; (c) D. D. Schnaars and R. E. Wilson, Synthesis, Structure, and Vibrational Properties of [Ph4P]2NpO2Cl4 and [Ph4P]2PuO2Cl4 Complexes, Inorg. Chem., 2018, 57, 3008–3016 CAS; (d) M. Autillo, R. E. Wilson, M. Vasiliu, G. F. de Melo and D. A. Dixon, Periodic Trends within Actinyl(VI) Nitrates and Their Structures, Vibrational Spectra, and Electronic Properties, Inorg. Chem., 2022, 61, 15607–15618 CrossRef CAS PubMed.
- (a) R. G. Surbella III, L. C. Ducati, K. L. Pellegrini, B. K. McNamara, J. Autschbach, J. M. Schwantes and C. L. Cahill, Transuranic Hybrid Materials: Crystallographic and Computational Metrics of Supramolecular Assembly, J. Am. Chem. Soc., 2017, 139, 10843–10855 CrossRef PubMed; (b) K. P. Carter, M. Kalaj, R. G. Surbella III, L. C. Ducati, J. Autschbach and C. L. Cahill, Engaging the Terminal: Promoting Halogen Bonding Interactions with Uranyl Oxo Atoms, Chem. – Eur. J., 2017, 23, 15355–15369 CAS; (c) K. P. Carter, M. Kalaj, A. Kerridge and C. L. Cahill, Probing hydrogen and halogen-oxo interactions in uranyl coordination polymers: a combined crystallographic and computational study, CrystEngComm, 2018, 20, 4916–4925 CAS.
- (a) M. Basile, E. Cole and T. Z. Forbes, Impacts of Oxo Interactions on Np(V) Crown Ether Complexes, Inorg. Chem., 2018, 57, 6016–6028 CAS; (b) J. L. Bjorklund, M. M. Pyrch, M. C. Basile, S. E. Mason and T. Z. Forbes, Actinyl-cation interactions: experimental and theoretical assessment of [Np(VI)O2Cl4]2− and [U(VI)O2Cl4]2− systems, Dalton Trans., 2019, 48, 8861–8871 CAS.
- (a) C. J. Van Staveren, D. E. Fenton, D. N. Reinhoudt, J. Van Eerden and S. Harkema, Co-complexation of urea and UO22+ in a Schiff base macrocycle: a mimic of an enzyme binding site, J. Am. Chem. Soc., 1987, 109, 3456–3458 CAS; (b) C. J. Van Staveren, J. Van Eerden, F. C. Van Veggel, S. Harkema and D. N. Reinhoudt, Cocomplexation of neutral guests and electrophilic metal cations in synthetic macrocyclic hosts, J. Am. Chem. Soc., 1988, 110, 4994–5008 CAS.
- (a) P. Zanello, A. Cinquantini, P. Guerriero, S. Tamburini and P. Vigato, Electrochemical behaviour of acyclic and macrocyclic complexes of nickel(II), copper(II) and uranyl (VI), Inorg. Chim. Acta, 1986, 117, 91–96 CrossRef CAS; (b) U. Casellato, S. Tamburini, P. Tomasin, P. A. Vigato, S. Aime and M. Botta, Synthesis, X-ray structure, and solution NMR studies of Ln(III) complexes with a macrocyclic asymmetric compartmental Schiff base. Preference of the Ln(III) ions for a crown-like coordination site, Inorg. Chem., 1999, 38, 2906–2916 CrossRef CAS PubMed; (c) N. Brianese, U. Casellato, S. Tamburini, P. Tomasin and P. Vigato, Asymmetric compartmental macrocyclic ligands and related mononuclear and hetero-dinuclear complexes with d-and/or f-metal ions, Inorg. Chim. Acta, 1999, 293, 178–194 CrossRef CAS; (d) U. Casellato, S. Tamburini, P. Tomasin and P. Vigato, Uranyl (VI) complexes with [1 + 1] asymmetric compartmental ligands containing a Schiff base and a crown ether-like chamber, Inorg. Chim. Acta, 2002, 341, 118–126 CrossRef CAS.
- (a) R. D. Shannon and C. Prewitt, Revised values of effective ionic radii, Acta Crystallogr., Sect. B, 1970, 26, 1046–1048 CrossRef CAS; (b) R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- T. Makela, M.-E. Minkkinen and K. Rissanen, Ion pair binding in the solid-state with ditopic crown ether uranyl salophen receptors, Inorg. Chem., 2016, 55, 1339–1346 CrossRef CAS PubMed.
- A. Kumar, D. Lionetti, V. W. Day and J. D. Blakemore, CCDC 1960629: Experimental Crystal Structure Determination, 2020, DOI:10.5517/ccdc.csd.cc23t638.
- A. Kumar, D. Lionetti, V. W. Day and J. D. Blakemore, CCDC 1960628: Experimental Crystal Structure Determination, 2020, DOI:10.5517/ccdc.csd.cc23t627.
- J.-M. Savéant, Elements of Molecular and Biomolecular Electrochemistry, Wiley-VCH, New Jersey, 2006 Search PubMed.
- A. Kumar and J. D. Blakemore, On the Use of Aqueous Metal-Aqua pKa Values as a Descriptor of Lewis Acidity, Inorg. Chem., 2021, 60, 1107–1115 CAS.
- D. D. Perrin, Ionisation Constants of Inorganic Acids and Bases in Aqueous Solution, Elsevier, 2016 Search PubMed.
- R. S. Nicholson, Theory and application of cyclic voltammetry for measurement of electrode reaction kinetics, Anal. Chem., 1965, 37, 1351–1355 CAS.
- A. J. Bard and L. R. Faulkner, Electrochemistry: Fundamentals and Applications, John Wiley and Sons Inc., Hoboken, 2nd edn, 2001 Search PubMed.
- P. Thordarson, Determining association constants from titration experiments in supramolecular chemistry, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- G. Salinas, A. Villarroel Marquez, M. Idir, S. Shinde, B. A. Frontana-Uribe, M. Raoux, J. Lang, E. Cloutet and A. Kuhn, Sodium-Ion Selectivity Study of a Crown-Ether-Functionalized PEDOT Analog, ChemElectroChem, 2020, 7, 2826–2830 CrossRef CAS.
- J. D. Blakemore, R. Chitta and F. D'Souza, Synthesis and study of crown ether-appended boron dipyrrin chemosensors for cation detection, Tetrahedron Lett., 2007, 48, 1977–1982 CrossRef CAS.
- G. Wulfsberg, Principles of Descriptive Inorganic Chemistry, University Science Books, Mill Valley, Calif., 1991 Search PubMed.
- J. E. Newbery, Vibrational spectra of the [UO2Cl4]2− and the [UO2Br4]2− ions, Spectrochim. Acta, Part A, 1969, 25, 1699–1702 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2344717–2344726. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03503h |
| ‡ These authors contributed equally to this work. |
| § Current address: Heraeus Precious Metals, 15524 Carmenita Road, Santa Fe Springs, California 90670, USA. |
| ¶ Current address: Department of Chemistry, University of California, Berkeley, Berkeley, California 94720, USA. |
| || IR spectra for the L6UO2M series were also collected, revealing features from 884 to 893 cm−1 (Fig. S66–S72†) that could be associated with asymmetric U–Ooxo stretching. However, there is no systematic trend in the data, suggesting that intra-ligand vibrations could cloud identification of U–Ooxo stretching. Similarly, IR spectra were collected in the far, low-energy region, revealing similar features in all cases from 252 to 256 cm−1 (Fig. S73–S79†) that could be associated with U–Ooxo bending modes,47 but no trend in the measured frequencies was identified. |
| This journal is © The Royal Society of Chemistry 2025 |