
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic H/D exchange of (hetero)arenes with early–late polyhydride heterobimetallic complexes: impact of transition metal pairs†
Abdelhak
Lachguar
a,
Till
Neumann
 a,
Andrey V.
Pichugov
a,
Erwann
Jeanneau
b,
Laurent
Veyre
a,
Chloé
Thieuleux
a and
Clément
Camp
*a
a,
Andrey V.
Pichugov
a,
Erwann
Jeanneau
b,
Laurent
Veyre
a,
Chloé
Thieuleux
a and
Clément
Camp
*a
aLaboratory of Catalysis, Polymerization, Processes and Materials (CP2 M UMR 5128), CNRS, Universite Claude Bernard Lyon 1, CPE-Lyon, Institut de Chimie de Lyon, 43 Bd du 11 Novembre 1918, F-69616 Villeurbanne, France. E-mail: clement.camp@univ-lyon1.fr
bCentre de Diffractométrie Henri Longchambon, Universite Claude Bernard Lyon 1, 5 Rue de la Doua, 69100 Villeurbanne, France
First published on 21st January 2025
Abstract
Metal-catalyzed hydrogen isotope exchange (HIE) has become a valuable method for incorporating deuterium and tritium into organic molecules, with applications in a wide range of scientific fields. This study explores the role of transition metal cooperativity in enhancing catalytic hydrogen/deuterium (H/D) exchange using early–late heterobimetallic polyhydride (ELHB) complexes. A series of four ELHB complexes, of general formula [M(CH2tBu)3(H)xM′Cp*], combining early transition metals (M = Hf, Ta) with late metals (M′ = Ir, Os), were synthesized and evaluated for their catalytic activity in HIE of (hetero)arenes. Hafnium–iridium and hafnium–osmium complexes showed a clear improvement in catalytic efficiency and reaction rate over monometallic analogues, suggestive of metal–metal synergy. Conversely, the tantalum-based heterobimetallic complexes showed lower catalytic performance, revealing that not all metal combinations are equally effective. These results underline the importance of careful metal selection to optimize transition metal cooperativity, and open up new possibilities for the design of more efficient H/D exchange catalysts.
Introduction
Catalytic hydrogen isotope exchange (HIE) has become a highly effective tool for labelling organic compounds with deuterium or tritium,1–9 isotopes that are invaluable in a wide range of applications.10–21 For instance, in drug design, deuterium substitution can be employed to enhance the metabolic stability and therapeutic efficacy of some pharmaceuticals, as demonstrated by drugs like VX-984, deucravacitinib or deutetrabenazine.11,16,22 Likewise, tritiation plays a critical role in radiolabeling, enabling detailed studies of drug metabolism, pharmacokinetics, and receptor binding interactions.5,6,23,24 This has driven significant research efforts toward developing more efficient and selective C–H activation strategies, in particular molecular and nano-catalytic systems based on transition metals.25–32 Heterobimetallic systems have recently emerged as an elegant solution for heterolytic activation of C–H bonds, where both metals cooperate synergistically.33–43 Early–late heterobimetallic (ELHB) complexes, featuring polarized metal–metal interactions that enable asymmetric substrate interactions,44–49 are particularly promising in this regard. However, the use of ELHB complexes in metal-catalyzed hydrogen isotope exchange remains very limited compared to monometallic systems, leaving much of their potential largely unexplored.To the best of our knowledge, the only ELHB complexes applied in catalytic HIE are those separately reported by the group of Suzuki50,51 and our group.33,36,52 Suzuki and colleagues reported a zirconium–iridium polyhydride heterobimetallic complex (Fig. 1, left) which was found to be active, at 120 °C, for the H/D exchange between methoxyarenes and C6D6 as solvent and deuterium source.50 Subsequently, the same group reported an analogue featuring and ansa-(cyclopentadienyl)(amide) to stabilize the zirconium fragment, which was found active catalyst for H/D exchange reaction between several arenes and C6D6 at 120 °C.51 The contribution of our group to this field has been to evidence that the well-defined silica-supported Ta/Ir polyhydride surface organometallic species, [![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiOTa(CH2tBu)H{IrH2(Cp*)}] (Fig. 1, center), abbreviated TaIr/SiO2, exhibits drastically enhanced catalytic performances in H/D exchange reactions with respect to (i) monometallic analogues as well as (ii) homogeneous systems.33,36 This TaIr/SiO2 catalyst is, for instance, active at room temperature for the perdeuteration of fluorobenzene using C6D6 or D2 as deuterium sources. In a subsequent study, using a hafnium–iridium complex immobilized on silica (Fig. 1, right), we discovered that the combination of hafnium with iridium significantly improved chemoselectivity in the perdeuteration of C(sp3)–H bonds in alkanes with D2.52 In contrast, the monometallic analogues lead to deuterogenolysis by-products under the same experimental conditions, through competitive C–C bond cleavage.52
SiOTa(CH2tBu)H{IrH2(Cp*)}] (Fig. 1, center), abbreviated TaIr/SiO2, exhibits drastically enhanced catalytic performances in H/D exchange reactions with respect to (i) monometallic analogues as well as (ii) homogeneous systems.33,36 This TaIr/SiO2 catalyst is, for instance, active at room temperature for the perdeuteration of fluorobenzene using C6D6 or D2 as deuterium sources. In a subsequent study, using a hafnium–iridium complex immobilized on silica (Fig. 1, right), we discovered that the combination of hafnium with iridium significantly improved chemoselectivity in the perdeuteration of C(sp3)–H bonds in alkanes with D2.52 In contrast, the monometallic analogues lead to deuterogenolysis by-products under the same experimental conditions, through competitive C–C bond cleavage.52
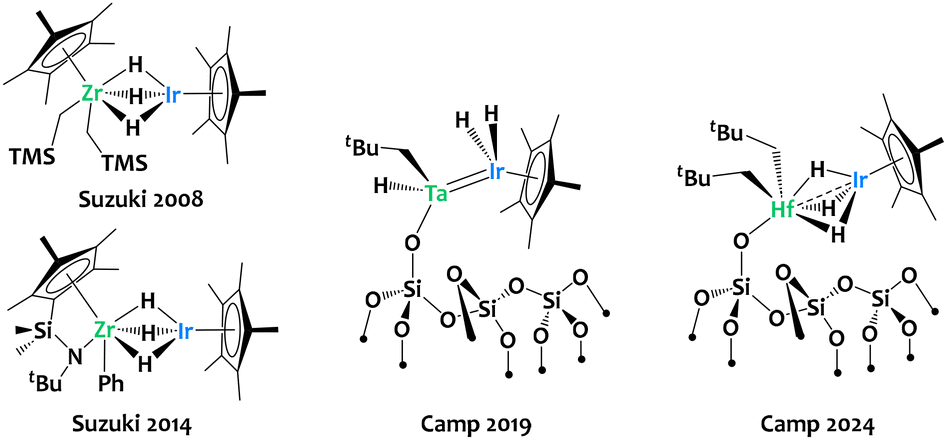 | ||
| Fig. 1 Relevant examples of polyhydride early–late heterobimetallic species applied in metal-catalyzed H/D exchange reactions.33,50–52 | ||
These initial studies have demonstrated the effectiveness of iridium-based ELHB compounds in promoting HIE. However, these catalytic investigations were conducted under varying experimental conditions, making direct comparisons difficult. Moreover, the impact of different metal pairings within similar ligand frameworks on C–H activation efficiency remains largely unexplored, and moving beyond iridium has proven challenging. Key questions remain regarding whether iridium performs better when paired with group 4 or group 5 elements, and if other late metal centers, such as osmium, could rival or surpass iridium's catalytic activity, potentially serving as viable alternatives. Accordingly, the objective of this study is to examine a broader range of transition metals combinations and to systematically compare the catalytic HIE performances of a series of related ELHB complexes and their monometallic counterparts, in order to identify the most promising metal–metal combinations to achieve cooperative effects in C–H activation catalysis.
Results and discussion
Synthesis and characterization
The protonolysis reaction between acidic metal hydride complexes and nucleophilic metal alkyl derivatives has been established as an effective approach for synthesizing heterobimetallic complexes via alkane elimination.35,53–58 In particular, our group previously reported the successful preparation of a series of early–late polyhydride heterobimetallic complexes through the reaction of early transition metal perhydrocarbyl complexes—Ta(CHtBu)(CH2tBu)3 (1)59 or Hf(CH2tBu)4 (2)60—with the late transition metal polyhydrides Cp*IrH4 (3)61,62 or Cp*OsH5 (4).63 This reaction yielded complexes [Ta(CH2tBu)3IrH2Cp*] (5),33 [Ta(CH2tBu)3(μ-H)3OsCp*] (6)64 and [Hf(CH2tBu)3(μ-H)3IrCp*] (7)65 in excellent yields (93% to 99%, see Scheme 1). Expanding on this strategy, we now report the preparation of the hafnium osmium complex in this series. Complex [Hf(CH2tBu)3(μ-H)4OsCp*], 8, was isolated in 99% yield from the equimolar reaction between Hf(CH2tBu)4 (2) and Cp*OsH5 (4), and fully characterized by NMR spectroscopy, Diffuse Reflectance Infrared Fourier Transform (DRIFT) spectroscopy, X-ray diffraction (XRD), and elemental analysis, enabling a comparison of the spectroscopic and structural properties throughout the series. | ||
Scheme 1 Synthesis of heterobimetallic complexes. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Yield from reported procedure.33,64,65 Yield from reported procedure.33,64,65 | ||
The 1H spectrum of 8 in C6D6 solution is in agreement with the proposed structure (Scheme 1), with the Cp* signal appearing at 1.97 ppm and a characteristic hydride resonance appearing at δ = −11.72 ppm. The Cp* protons and hydrides integrate in a 15:4 ratio in 8, indicating that 4 hydrides are preserved upon protonolysis. For comparison, the 1H NMR hydride signals are found at δ = −11.10 ppm (3H) for complex 7 (Hf–Ir)65 and δ = −11.97 ppm (2H) for complex 5 (Ta–Ir),33 while that for complex 6 (Ta–Os)64 appears at δ = −6.80 ppm (3H) (see Table 1). The Hf–CH213C resonance is found at δ = +112.0 in 8, which is a usual chemical shift for classical neopentyl ligands.52,65–67
| Compound | M1–M2 (Å) | FSR | M1–M2–Cp*centroid (°) | v (M–H) (cm−1) | 1H NMR (C6D6) (M–H) (ppm) |
|---|---|---|---|---|---|
| 3 (Ir) | — | — | — | 2150 | −15.43 (4H) |
| 4 (Os) | — | — | — | 2214, 2083 | −11.00 (5H) |
| 5 (Ta–Ir) | 2.3559(6) | 0.90 | 151.3 | 2056 | −11.97 (2H) |
| 6 (Ta–Os) | 2.4817(2) | 0.95 | 178.1 | 1961 | −6.80 (3H) |
| 7 (Hf–Ir) | 2.6773(4) | 0.99 | 179.2 | 1982 | −11.10 (3H) |
| 8 (Hf–Os) | 2.6780(2) | 0.99 | 177.7 | 2011 | −11.75 (4H) |
Single crystals of complex 8 (Hf–Os), suitable for X-ray diffraction, were obtained from a saturated pentane solution at −40 °C overnight. The solid-state structure of complex 8 (Hf–Os), shown in Fig. 2, reveals that the Hf–Os–Cp*centroid angle is nearly linear (177.7(1)°), as in complexes 6 and 7, in agreement with the presence of four bridging hydrides between the metals. The hafnium–osmium separation (2.6780(2) Å) is 0.019 Å shorter than the sum of the metallic radii of hafnium (1.442 Å) and osmium (1.255 Å).68 This results in a formal shortness ratio (FSR) slightly below unity (FSR = 0.99), which is similar to what was observed in the hafnium–iridium analogue (complex 7).65 We attribute this proximity to the presence of the bridging hydrides, although some interaction between the metals is possible. Note that the tantalum–osmium (6) and tantalum–iridium (5) derivatives exhibit much shorter metal–metal distances (see Table 1) and thus stronger metal–metal interactions.33,64,69 The Hf–C distances vary from 2.210(3) to 2.227(3) Å, falling in the expected range for Hf–neopentyl ligands.52,65,70 The latter are arranged in pseudo-tetrahedral manner around the hafnium center (average C–Hf–C angle of 102°).
 | ||
| Fig. 2 Solid-state molecular structure of 8 (50% probability ellipsoids). Hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Os1–Hf1 2.6780(2), Hf1–C16 2.210(3), Hf1–C11 2.220(3), Hf1–C21 2.227(3), Hf1–Os1–Cp*centroid 177.7(1). | ||
The DRIFT spectrum of compound 8 displays an intense metal-hydride stretching signal centered at 2011 cm−1. This represents a notable decrease in the hydride stretch wavenumber upon complexation with the hafnium center, compared to the monometallic polyhydride precursor, Cp*OsH5, which shows stretches at 2214 and 2083 cm−1 (see Table 1).63 This is in line with what found in other M–Os species featuring bridging hydrides, such as (CH2tBu)3Ta(μ-H)3OsCp* (compound 6), Th{(μ-H)4OsCp*}4, U{(μ-H)4OsCp*}4 and Cp2Zr(H)(μ-H)3Os(PMe2Ph)3 where hydride stretches appear at 1961, 1993, 1990 and 1970 cm−1, respectively.64,71,72 However, the shift observed in compound 8 is less pronounced.
Catalytic H/D exchange activity
As explained in the introduction, previous research has identified iridium-based polyhydride heterobimetallic complexes,34 both in homogeneous form48,50,51 and supported on silica,33,52,73 as efficient catalysts for hydrogen–deuterium (H/D) isotope exchange (HIE). However, the impact of varying metal pairings within similar ligands environments on the HIE catalytic activity remains underexplored. Building on these considerations, we evaluated the performances of the four heterobimetallic complexes 5–8 and their corresponding monometallic precursors 1–4 in homogeneous conditions. The benchmark reaction chosen for comparing catalytic activities was the perdeuteration of fluorobenzene, with toluene-d8 serving as the deuterium source. This reaction is ideal because the fluorine substituent enhances the activation of the arene C–H bonds, allowing even minimal catalytic activity to be detected. Additionally, reaction monitoring is conveniently achieved using both 19F NMR and 1H NMR spectroscopies.Kinetic reaction monitoring was carried out under argon at 110 °C in a toluene-d8 solution, using a fluorobenzene concentration of 0.13 M and 6 mol% of each complex as catalyst. As depicted in Fig. 3, the monometallic Hf and Ta complexes (1) and (2) showed almost no activity under these experimental conditions. In contrast, the late-metal complex Cp*OsH5, (4), demonstrated moderate activity, achieving 57% of deuterium incorporation after 163 hours, while the iridium complex Cp*IrH4, (3), reached a 95% deuterium incorporation in 70 hours. Note that the latter showed an induction period, which is ascribed to the decomposition of the initial complex into catalytically active Ir(0) nanoparticles (see below).
 | ||
| Fig. 3 Kinetic monitoring of fluorobenzene deuteration in toluene-d8 at 110 °C using complexes 1–8 as catalysts (6 mol%). | ||
Interestingly, the hafnium-containing heterobimetallic complexes significantly outperformed their monometallic counterparts. The Hf–Ir complex (7) reached 93% deuterium incorporation within just 27 hours, whereas the monometallic iridium complex (3) reached only a 25% deuterium incorporation in the same timeframe, requiring 70 hours to achieve a similar 95% yield. A comparable trend was observed with osmium analogue: the Hf–Os heterobimetallic complex (8) reached a 90% deuteration yield in 46 hours, compared to just 24% yield for the monometallic Os complex (4) (Fig. 3). This suggests that combining hafnium with late-metal hydrides is beneficial for H/D exchange catalysis, while the reactivity order Ir > Os is maintained.
In contrast, tantalum-based heterobimetallic complexes demonstrated markedly lower catalytic efficiency. The Ta–Ir complex (5) performed worse than the Hf–Ir complex, with the latter (7) reaching a 93% deuterium incorporation in less than 20 hours, whereas the Ta–Ir complex took nearly 140 hours to reach a comparable 93% deuteration yield. A similar trend was observed with osmium: after 46 h hours the hafnium–osmium catalyst (7) achieved 90% deuterium incorporation, versus only 12% for the Ta–Os complex (6) under the same conditions (Fig. 3).
To further investigate the comparative robustness of the two most active catalysts, we performed STEM analyses of the spent catalyst reaction mixtures. For the monometallic iridium catalyst, 3, large micrometer-scale iridium metal particles were observed after the reaction (Fig. S32†), correlating with the black coloration of the reaction mixture after 24 hours at 110 °C. This suggests that the progressive agglomeration of Ir metal particles is probably responsible for the prolonged inductive period observed in the H/D exchange rate, and that these nanoparticles are necessary for catalysis. In contrast, the catalytic reaction medium remains pale in colour after 24 hours of reaction at 110 °C and no nanoparticles were detected by electron microscopy in the spent catalysts 7 (Hf–Ir) and 8 (Hf–Os) (Fig. S33 and S34†). This suggests that the heterobimetallic catalysts 7 and 8 exhibit greater stability under the reaction conditions, and that the catalytically active species are most likely molecular in nature rather than aggregated metal particles in these cases. The robustness of catalyst 7 is further confirmed by 1H NMR spectroscopy, which shows that the signals corresponding to 7 persist after prolonged heating (up to 80 h) in a toluene-d8 solution at 110 °C (see Fig. S31†), suggesting that the bimetallic core structure is maintained under the catalytic conditions. Note that the monometallic osmium pentahydride complex 4 is also robust and does not decompose upon prolonged heating in aromatic solvents.63,74 Thus, the enhanced catalytic performance of the bimetallic Hf–Os system cannot solely be attributed to a reluctance to agglomerate. Instead, it is most likely due to a bimetallic C–H activation mechanism, as proposed for analogous systems in the literature.34,36,51
Complex 7 (Hf–Ir), which exhibited the highest efficiency and robustness in the H/D exchange reaction among the eight complexes tested, was subsequently evaluated for its versatility by performing perdeuteration on a range of arenes and heteroarenes (Scheme 2). The reactions were carried out at 100 °C using deuterated benzene as the deuterium source, with 6 mol% of the hafnium–iridium complex 7 (Hf–Ir), for a total of 135 hours. As shown in Scheme 2 (left), fluorobenzene underwent complete deuteration, but no regioselective labelling pattern was observed. In contrast, in naphthalene, position 2 exhibited much higher deuteration (96% deuteration incorporation) than position 1 (32%). Anisole showed more selective deuterium incorporation at the meta positions (84%) compared to 46% at the ortho and para positions. Deuterium incorporation at the methyl position was limited to only 3%, highlighting the good selectivity of catalyst 7 for sp2-hybridized C–H bonds. Toluene was preferentially deuterated at the meta/para position (99%), with no deuteration detected on the ortho position or on the methyl group. Catalyst 7 also showed high efficiency in deuterating the ethylenic C–H bonds in styrene, with 87–90% deuteration at the alkene positions and 29–32% at the aromatic ring. Importantly, no hydrogenation of the alkene moiety was observed, highlighting the catalyst's selectivity for H/D exchange. In heteroarenes, furan showed higher deuteration at position 2, with 51% of deuterium incorporation, compared to 32% at position 3. In pyridine, position 4 had the lowest labeling, with just 15% of deuterium, while positions 2 and 3 were more reactive, showing 32% and 37% of incorporation, respectively. Finally, high degree of deuteration of N-methyl indole was achieved (Scheme 2): up to 98% for sp2-CH bonds. This substrate scope study highlights the potential of catalyst 7, which shows tolerance to a variety of functional groups, including coordinating moieties, demonstrating its versatility in H/D exchange reactions. It should be noted, however, that 1H-indole led to complete deactivation of the catalyst, as no deuterium incorporation was detected in this substrate. This deactivation is likely due to the acidic N–H group present in 1H-indole, which is assumed to promote protonolysis of the Hf–neopentyl bond in complex 7, resulting in loss of catalytic activity.
 | ||
| Scheme 2 Substrate scope with the percentage of deuterium incorporated per position achieved through H/D exchange reaction catalyzed by the bimetallic Hf–Ir complex, 7 (left) and the monometallic iridium analogue, 3 (right). | ||
For comparison, the monometallic iridium complex 3 was evaluated under identical experimental conditions to assess its substrate scope (Scheme 2). In most cases, the overall deuterium incorporation achieved with complex 3 was significantly lower than that obtained with the heterobimetallic catalyst 7. For example, sp2 C–H bonds in fluorobenzene exhibited 98% deuteration with catalyst 7, compared to only 41% with catalyst 3. Similarly, anisole achieved 61% deuteration with 7versus 33% with 3; toluene, 59% versus 35%; and styrene, 47% versus 4%. Notably, for styrene, catalyst 3 produced some hydrogenation by-products, whereas 7 demonstrated high selectivity for H/D exchange. A notable exception, yet, is for furane and pyridine, where catalyst 3 showed better H/D exchange performances (99% and 96% overall deuteration, respectively, versus 42% and 32% for 7), offering a complementary alternative. Note that neither 3 nor 7 were able to deuterate mesitylene.
While it could be hypothesized that the bimetallic core might serve as a strategy to orient regioselectivity, for instance through the coordination of the Lewis-acidic Hf site to O- or N-coordinating moieties, the catalytic results indicate that catalyst 7 exhibits no specific site selectivity. When regioselectivity is observed, it aligns with that of the monometallic complex 3 in most cases and is most likely driven by classical stereoelectronic directing effects in arene reactivity.
Experimental
General considerations
Unless otherwise noted, all reactions were performed either using standard Schlenk line techniques or in an MBRAUN glovebox under an atmosphere of purified argon (<1 ppm of O2/H2O). Glassware and cannulas were stored in an oven at ∼100 °C for at least 16 h prior to use. THF and n-pentane were purified by passage through a column of activated alumina, dried over Na/benzophenone, vacuum-transferred to a storage flask, and freeze–pump–thaw degassed prior to use. Deuterated solvents (toluene-d8 and C6D6) were dried over Na/benzophenone, vacuum-transferred to a storage flask, and freeze–pump–thaw degassed prior to use. Compounds Ta(CH2tBu)3(CHtBu),59 Hf(CH2tBu)4,60,70 Cp*IrH4,61,62 Cp*OsH5,63,75 [Ta(CH2tBu)3IrH2(Cp*)],33,69 [Hf(CH2tBu)3(μ-H)3IrCp*],65 and [Ta(CH2tBu)3(μ-H)3OsCp*]64 were prepared according to the literature procedures. All other reagents were acquired from commercial sources and used as received. CAUTION: upon exposure to air the osmium derivatives may lead to OsO4 which is acutely toxic upon inhalation. As a precaution measure, the osmium-containing solutions were thus quenched using corn oil under a well-ventilated fumehood before appropriate disposal.IR spectroscopy
The sample was prepared in a glovebox (diluted in dry KBr powder), sealed under argon in a DRIFT cell fitted with KBr windows, then analyzed using a Nicolet 6700 FT-IR spectrometer equipped with a MCT detector.Elemental analyses
Elemental analyses were performed under an inert atmosphere at Mikroanalytisches Labor Pascher, Germany.Electron microscopy (TEM and STEM-HAADF)
Electron microscopy experiments were performed using a MET JEOL 2100F (FEG) microscope at the “Centre Technologique des Micro-structures”, CTμ Villeurbanne, France; equipped with an Oxford Instruments Energy-Dispersive Spectroscopy (EDS) SDD detector. Under an inert argon atmosphere within a glovebox, a drop (ca. 0.05 mL) of the crude reaction mixture (post-catalysis) was carefully deposited onto a TEM copper grid coated with ultrathin carbon film on lacey carbon. Once the solvent had evaporated, the prepared sample grids were transferred to the microscope for analysis, ensuring the inert argon atmosphere was maintained throughout the transfer process with specific holder.X-ray structural determinations
Experimental details regarding single-crystal XRD measurements are provided in the ESI.† CCDC 2395565 contain supplementary crystallographic data for this article.†NMR spectroscopy
Solution NMR spectra were recorded on Bruker AV-300 and AV-500 spectrometers using NMR tubes equipped with J-Young valves. 1H and 13C chemical shifts were measured relative to residual solvent peaks, which were assigned relative to an external TMS standard set at 0.00 ppm. 1H and 13C NMR assignments were confirmed by 1H–1H COSY, 1H–13C HSQC, and HMBC experiments.Synthesis of [Hf(CH2tBu)3(μ-H)4Os(Cp*)] 8
A colorless pentane solution (1 mL) of Cp*OsH5 (20 mg, 0.061 mmol, 1 equiv.) was added to a pentane solution (1 mL) of Hf(CH2tBu)4 (28 mg, 0.061 mmol, 1 equiv.). The reaction mixture was stirred at room temperature for 2 h. Volatiles were removed under vacuum affording pure complex 8 as a white powder (43 mg, 0.060 mmol, 99%). Single crystals of complex 8 suitable for X-ray diffraction were obtained through recrystallization in a minimal amount of pentane at −35 °C. 1H-NMR (500 MHz, C6D6, 298 K) δ ppm 1.97 (s, 15H, Cp*), 1.37 (s, 27H, 3 C(C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 3)3), 0.77 (s, 6H, 3 C2C(CH3)3), −11.72 (s, 4H, Os4). 13C{1H}-NMR (126 MHz, C6D6, 298 K) δ ppm 112.02 (s, 3C, 3 Hf
3)3), 0.77 (s, 6H, 3 C2C(CH3)3), −11.72 (s, 4H, Os4). 13C{1H}-NMR (126 MHz, C6D6, 298 K) δ ppm 112.02 (s, 3C, 3 Hf![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H2), 94.17 (s, 5C, 5(CH3)5), 36.06 (s, 3C, 3 HfCH2(CH3)3), 35.95 (s, 9C, 3 HfCH2C(H3)3), 11.61 (s, 5C, C5(H3)5). FTIR (293 K, cm−1)ν = 2959 (br s, νC–H), 2861 (s, νC–H), 2764 (s, νC–H), 2699 (s, νC–H), 2011 (s, νM–H), 1460 (s), 1383 (s), 1359 (s), 1229 (s), 1034(s), 751 (s). Anal. calcd for C25H52OsHf: C, 41.62; H, 7.27 found: C, 41.63; H, 7.29.
H2), 94.17 (s, 5C, 5(CH3)5), 36.06 (s, 3C, 3 HfCH2(CH3)3), 35.95 (s, 9C, 3 HfCH2C(H3)3), 11.61 (s, 5C, C5(H3)5). FTIR (293 K, cm−1)ν = 2959 (br s, νC–H), 2861 (s, νC–H), 2764 (s, νC–H), 2699 (s, νC–H), 2011 (s, νM–H), 1460 (s), 1383 (s), 1359 (s), 1229 (s), 1034(s), 751 (s). Anal. calcd for C25H52OsHf: C, 41.62; H, 7.27 found: C, 41.63; H, 7.29.
General procedure for catalytic H–D exchange reactions
In a J. Young NMR tube, using micropipettes, a 0.3 mL solution of complexes 1–8 (0.00462 mmol, 6 mol%) in toluene-d8 (or C6D6) was added in one portion to a 0.3 mL solution of toluene-d8 (or C6D6) containing the substrate (0.077 mmol) and hexamethyldisiloxane (0.02 mmol) as an internal standard. The reaction mixture was then heated at 110 °C (or 100 °C when using C6D6 as solvent) using an oil bath and monitored regularly by both 1H NMR and 19F NMR spectroscopy (for fluorobenzene).Conclusions
This study demonstrates the role of transition metal cooperativity in catalytic H/D exchange using early–late poly-hydride heterobimetallic complexes.34 The results revealed that the combination of hafnium to iridium or osmium significantly enhanced the catalytic activity, outperforming the monometallic analogues. In contrast, tantalum-based complexes, when combined with iridium or osmium polyhydrides, exhibited reduced catalytic performance compared to their monometallic counterparts. These results highlight that while some early–late metal combinations, such as hafnium–iridium, can significantly improve the efficiency of H/D exchange, not all metal pairs are beneficial. Another key insight from this study is the role of hafnium in enhancing catalyst stability by mitigating aggregation. The most active complex, [Hf(CH2tBu)3(μ-H)3IrCp*] (7), exhibited very good efficiency (up to 99%) in the H/D exchange reaction of a range of arenes as well as heteroarenes, and tolerance to a variety of functional groups including strongly coordinating moieties such as pyridine. Furthermore, the newly reported hafnium–osmium species, [Hf(CH2tBu)3(μ-H)4OsCp*] (8), while less active than its Hf–Ir analogue (7), significantly outperformed the Os monometallic complex (4). We hope this knowledge will be beneficial and serve as a guiding framework for the future design of novel early–late heterobimetallic catalysts.Author contributions
A. Lachguar performed the experimental synthesis and catalysis work and wrote the original draft of the manuscript. T. Neumann and A. V. Pichugov completed the substrate scope study and catalyst robustness screening. E. Jeanneau recorded and interpreted the XRD data. L. Veyre recorded and interpreted the electron microscopy data. C. Camp conceptualized the research, found the funds, administrated the project, revised and edited the second version of the manuscript. C. Camp and C. Thieuleux supervised the work. All authors have read, edited and agreed to the published version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† CCDC 2395565 contain supplementary crystallographic data for this article; they can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Anne Baudouin and Emmanuel Chefdeville are acknowledged for their help in acquiring the NMR data. Julien Petit is acknowledged for preliminary investigations on this topic. Funded by the European Union (ERC, DUO, 101041762). Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them.References
- S. R. Klei, J. T. Golden, T. D. Tilley and R. G. Bergman, J. Am. Chem. Soc., 2002, 124, 2092–2093 CrossRef CAS.
- J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS.
- S. Kopf, F. Bourriquen, W. Li, H. Neumann, K. Junge and M. Beller, Chem. Rev., 2022, 122, 6634–6718 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS.
- R. Pony Yu, D. Hesk, N. Rivera, I. I. Pelczer and P. J. Chirik, Nature, 2016, 529, 195–199 CrossRef CAS PubMed.
- Y. Y. Loh, K. Nagao, A. J. Hoover, D. Hesk, N. R. Rivera, S. L. Colletti, I. W. Davies and D. W. C. MacMillan, Science, 2017, 358, 1182–1187 CrossRef CAS PubMed.
- T. He, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2022, 144, 4734–4738 CrossRef CAS PubMed.
- M. Daniel-Bertrand, S. Garcia-Argote, A. Palazzolo, I. Mustieles Marin, P. F. Fazzini, S. Tricard, B. Chaudret, V. Derdau, S. Feuillastre and G. Pieters, Angew. Chem., Int. Ed., 2020, 59, 21114–21120 CrossRef CAS PubMed.
- A. Palazzolo, T. Naret, M. Daniel-Bertrand, D. A. Buisson, S. Tricard, P. Lesot, Y. Coppel, B. Chaudret, S. Feuillastre and G. Pieters, Angew. Chem., Int. Ed., 2020, 59, 20879–20884 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758–1784 CrossRef CAS.
- R. M. C. Di Martino, B. D. Maxwell and T. Pirali, Nat. Rev. Drug Discovery, 2023, 22, 562–584 CrossRef CAS PubMed.
- H. Tsuji, C. Mitsui and E. Nakamura, Chem. Commun., 2014, 50, 14870–14872 RSC.
- H. J. Bae, J. S. Kim, A. Yakubovich, J. Jeong, S. Park, J. Chwae, S. Ishibe, Y. Jung, V. K. Rai, W. J. Son, S. Kim, H. Choi and M. H. Baik, Adv. Opt. Mater., 2021, 9, 2100630 CrossRef CAS.
- T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276–5297 CrossRef CAS PubMed.
- G. Prakash, N. Paul, G. A. Oliver, D. B. Werz and D. Maiti, Chem. Soc. Rev., 2022, 3123–3163 RSC.
- S. H. DeWitt and B. E. Maryanoff, Biochemistry, 2018, 57, 472–473 CrossRef CAS.
- Z. Ma, L. Zhang, Z. Cui and X. Ai, Molecules, 2023, 28, 1–9 Search PubMed.
- X. Peng, C. H. Yeh, S. F. Wang, J. Yan, S. Gan, S. J. Su, X. Zhou, Y. X. Zhang and Y. Chi, Adv. Opt. Mater., 2022, 10, 1–10 Search PubMed.
- B. Reif, Chem. Rev., 2022, 122, 10019–10035 CrossRef CAS PubMed.
- W. J. S. Lockley, A. McEwen and R. Cooke, J. Labelled Compd. Radiopharm., 2012, 55, 235–257 CrossRef CAS.
- Y. Teng, H. Yang and Y. Tian, Molecules, 2024, 29, 4109 CrossRef CAS PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2017, 16, 305 Search PubMed.
- W. J. S. Lockley, A. McEwen and R. Cooke, J. Labelled Compd. Radiopharm., 2012, 55, 235–257 CrossRef CAS.
- D. B. Werz, C. Teja, S. Kolb, P. Colonna, J. Grover, G. K. Lahiri, G. Pieters, D. Maiti and S. Garcia-Argote, Angew. Chem., Int. Ed., 2024, 63, e202410162 CrossRef.
- W. J. S. Lockley and J. R. Heys, J. Labelled Compd. Radiopharm., 2010, 53, 635–644 CrossRef CAS.
- H. Yang and D. Hesk, J. Labelled Compd. Radiopharm., 2020, 63, 296–307 CrossRef CAS PubMed.
- E. Levernier, K. Tatoueix, S. Garcia-Argote, V. Pfeifer, R. Kiesling, E. Gravel, S. Feuillastre and G. Pieters, JACS Au, 2022, 2, 801–808 CrossRef CAS.
- M. Lepron, M. Daniel-Bertrand, G. Mencia, B. Chaudret, S. Feuillastre and G. Pieters, Acc. Chem. Res., 2021, 54, 1465–1480 CrossRef CAS PubMed.
- C. M. Stork, R. Weck, M. Valero, H. Kramp, S. Güssregen, S. R. Waldvogel, A. Sib and V. Derdau, Angew. Chem., Int. Ed., 2023, 62, e202301512 CrossRef CAS.
- J. D. Smith, G. Durrant, D. H. Ess, B. S. Gelfand and W. E. Piers, Chem. Sci., 2020, 11, 10705–10717 RSC.
- G. De Ruiter, S. Garhwal, A. Kaushansky, N. Fridman and L. J. W. Shimon, J. Am. Chem. Soc., 2020, 142, 17131–17139 CrossRef.
- C. Yao and C. Copéret, ChemRxiv, 2024, DOI:10.26434/chemrxiv-2024-k6jnq.
- S. Lassalle, R. Jabbour, P. Schiltz, P. Berruyer, T. K. Todorova, L. Veyre, D. Gajan, A. Lesage, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2019, 141, 19321–19335 CrossRef CAS.
- A. Lachguar, A. V. Pichugov, T. Neumann, Z. Dubrawski and C. Camp, Dalton Trans., 2023, 53, 1393–1409 RSC.
- R. Sun, Y. Jiang, H. R. Chen, X. Jiang, Y. C. Cao, S. Ye, R. Z. Liao, C. H. Tung and W. Wang, Inorg. Chem., 2024, 63, 6082–6091 CrossRef CAS PubMed.
- I. Del Rosal, S. Lassalle, C. Dinoi, C. Thieuleux, L. Maron and C. Camp, Dalton Trans., 2021, 50, 504–510 RSC.
- S. Deolka, O. Rivada-Wheelaghan, S. L. Aristizábal, R. R. Fayzullin, S. Pal, K. Nozaki, E. Khaskin and J. R. Khusnutdinova, Chem. Sci., 2020, 11, 5494–5502 RSC.
- J. Campos, J. Am. Chem. Soc., 2017, 139, 2944–2947 CrossRef CAS.
- N. Hidalgo, C. Maya and J. Campos, Chem. Commun., 2019, 55, 8812–8815 RSC.
- M. G. Alférez, J. J. Moreno, N. Hidalgo and J. Campos, Angew. Chem., Int. Ed., 2020, 59, 20863–20867 CrossRef.
- N. Gorgas, A. J. P. White and M. R. Crimmin, J. Am. Chem. Soc., 2022, 144, 8770–8777 CrossRef CAS PubMed.
- N. Gorgas, A. J. P. White and M. R. Crimmin, Chem. Commun., 2022, 58, 10849–10852 RSC.
- T. X. Gentner, M. J. Evans, A. R. Kennedy, S. E. Neale, C. L. McMullin, M. P. Coles and R. E. Mulvey, Chem. Commun., 2022, 58, 1390–1393 RSC.
- B. G. Cooper, J. W. Napoline and C. M. Thomas, Catal. Rev., 2012, 54, 1–40 CrossRef CAS.
- N. H. Hunter, E. M. Lane, K. M. Gramigna, C. E. Moore and C. M. Thomas, Organometallics, 2021, 40, 3689–3696 CrossRef CAS.
- J. Campos, Nat. Rev. Chem., 2020, 4, 696–702 CrossRef CAS PubMed.
- E. Bodio, M. Picquet and P. Le Gendre, in Topics in Organometallic Chemistry, Springer, Cham, 2015, vol. 59, pp. 139–186 Search PubMed.
- M. Oishi, M. Kino, M. Saso, M. Oshima and H. Suzuki, Organometallics, 2012, 31, 4658–4661 CrossRef CAS.
- S. Sinhababu, Y. Lakliang and N. P. Mankad, Dalton Trans., 2022, 51, 6129–6147 RSC.
- M. Oishi, T. Kato, M. Nakagawa and H. Suzuki, Organometallics, 2008, 27, 6046–6049 CrossRef CAS.
- M. Oishi, M. Oshima and H. Suzuki, Inorg. Chem., 2014, 53, 6634–6654 CrossRef CAS.
- A. V. Pichugov, L. Escomel, S. Lassalle, J. Petit, R. Jabbour, D. Gajan, L. Veyre, E. Fonda, A. Lesage, C. Thieuleux and C. Camp, Angew. Chem., 2024, 136, e202400992 CrossRef.
- M. V. Butovskii, C. Döring, V. Bezugly, F. R. Wagner, Y. Grin and R. Kempe, Nat. Chem., 2010, 2, 741–744 CrossRef CAS PubMed.
- A. P. Sobaczynski, J. Obenauf and R. Kempe, Eur. J. Inorg. Chem., 2014, 2014, 1211–1217 CrossRef CAS.
- L. Escomel, E. Jeanneau, C. Thieuleux and C. Camp, Inorganics, 2024, 12, 72 CrossRef CAS.
- L. Escomel, N. Soulé, E. Robin, I. Del Rosal, L. Maron, E. Jeanneau, C. Thieuleux and C. Camp, Inorg. Chem., 2022, 61, 5715–5730 CrossRef CAS PubMed.
- L. Escomel, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2021, 143, 4844–4856 CrossRef CAS PubMed.
- C. Z. Ye, I. Del Rosal, M. A. Boreen, E. T. Ouellette, D. R. Russo, L. Maron, J. Arnold and C. Camp, Chem. Sci., 2022, 14, 861–868 RSC.
- R. R. Schrock and J. D. Fellmann, J. Am. Chem. Soc., 1978, 100, 3359–3370 CrossRef CAS.
- P. J. Davidson, M. F. Lappert and R. Pearce, J. Organomet. Chem., 1973, 57, 269–277 CrossRef CAS.
- T. M. Gilbert, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1985, 107, 3508–3516 CrossRef CAS.
- T. M. Gilbert and R. G. Bergman, Organometallics, 1983, 2, 1458–1460 CrossRef CAS.
- C. L. Gross and G. S. Girolami, Organometallics, 2007, 26, 160–166 CrossRef CAS.
- A. Lachguar, C. Z. Ye, S. N. Kelly, E. Jeanneau, I. Del Rosal, L. Maron, L. Veyre, C. Thieuleux, J. Arnold and C. Camp, Chem. Commun., 2024, 60, 7878–7881 RSC.
- S. Lassalle, J. Petit, R. L. Falconer, V. Hérault, E. Jeanneau, C. Thieuleux and C. Camp, Organometallics, 2022, 41, 1675–1687 CrossRef CAS.
- R. Srivastava, E. A. Quadrelli and C. Camp, Dalton Trans., 2020, 49, 3120–3128 RSC.
- A. Lachguar, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2024, 146, 18306–18319 CrossRef CAS.
- L. Pauling, J. Am. Chem. Soc., 1947, 69, 542–553 CrossRef CAS.
- S. Lassalle, R. Jabbour, I. Del Rosal, L. Maron, E. Fonda, L. Veyre, D. Gajan, A. Lesage, C. Thieuleux and C. Camp, J. Catal., 2020, 392, 287–301 CrossRef CAS.
- G. Tosin, C. C. Santini, M. Taoufik, A. De Mallmann and J. M. Basset, Organometallics, 2006, 25, 3324–3335 CrossRef CAS.
- C. Z. Ye, I. Del Rosal, S. N. Kelly, I. J. Brackbill, L. Maron, C. Camp and J. Arnold, Chem. Sci., 2024, 15, 9784–9792 RSC.
- J. W. Bruno, J. C. Huffman, M. A. Green and K. G. Caulton, J. Am. Chem. Soc., 1984, 106, 8310–8312 CrossRef CAS.
- L. Escomel, D. Abbott, V. Mougel, L. Veyre, C. Thieuleux and C. Camp, Chem. Commun., 2022, 58, 8214–8217 RSC.
- C. L. Gross, S. R. Wilson and G. S. Girolami, J. Am. Chem. Soc., 1994, 116, 10294–10295 CrossRef CAS.
- T. Shima and H. Suzuki, Organometallics, 2005, 24, 3939–3945 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and DRIFT spectra, X-ray diffraction and electron microscopy data. CCDC 2395565. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03171g |
| This journal is © The Royal Society of Chemistry 2025 |