
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical language models can generate biomolecules atom-by-atom†
Kevin
Zhu‡
 ab,
Daniel
Flam-Shepherd‡
ab and
Alán
Aspuru-Guzik
*abcde
ab,
Daniel
Flam-Shepherd‡
ab and
Alán
Aspuru-Guzik
*abcde
aDepartment of Computer Science, University of Toronto, Toronto, Ontario M5S 2E4, Canada. E-mail: aspuru.assistant@utoronto.ca
bVector Institute for Artificial Intelligence, Toronto, Ontario M5S 1M1, Canada
cDepartment of Chemistry, University of Toronto, Toronto, Ontario M5G 1Z8, Canada
dCanadian Institute for Advanced Research, Toronto, Ontario M5G 1Z8, Canada
eAcceleration Consortium, Toronto, Ontario M5S 3H6, Canada
First published on 10th July 2025
Abstract
Chemical language models are powerful generative models, capable of learning complex molecular distributions such as the largest molecules in Pubchem. In this work, we further show that chemical language models can learn atom-level representations of substantially larger molecules – scaling even to biomolecules like proteins. We show that chemical language models can generate entire biomolecules atom by atom – effectively learning the multiple hierarchical layers of molecular information from primary sequence to tertiary structure. Even further, we demonstrate that chemical language models can explore chemical space and protein space simultaneously by generating novel examples of protein-drug conjugates. The results demonstrate the potential for atom level biomolecular design with chemical language models.
Chemical language models are deep neural networks trained using masking or next-token prediction1 using atom-level linear sequences parsed from molecular graphs.1,2 These sequences completely represent the molecule including all atoms, bonds, rings, aromaticity, branching, and stereochemistry. The two most prominent sequence representations are SMILES strings3 or SELFIES strings4 which are completely robust and always valid.
Recently, chemical language models1 were found to have the ability to generate larger, complex molecules, relative to small drug-like molecules such as the largest molecules in PubChem. These molecules are still much smaller than proteins, but this indicates that atom-level protein generation with language models is feasible. In this work, we demonstrate that chemical language models are capable of generating entire proteins atom by atom, including biomolecules beyond protein space. Specifically, we train models on various biomolecules including proteins from the Protein DataBank. We also create a synthetic biomolecular dataset by attaching small molecules from the ZINC database5 to single-domain antibodies (sdAbs) obtained from the antibody structural database.6
We discover that chemical language models can learn the language of proteins entirely from scratch – by learning to generate atom-level sequences that define proteins with valid primary sequences that can correspond to meaningful secondary, and tertiary structure, which we check using AlphaFold7 structure predictions. Importantly, the language model learns valid protein backbones and natural amino acid structures as well as the primary sequence patterns in the training proteins. We further demonstrate that chemical language models can generate novel proteins and small molecules together at the same time as protein-drug conjugates. In particular, we find that the model learns both the protein space of the single domain antibodies and the chemical space defined by the ZINC molecules – generating antibody–drug conjugates with valid and novel protein sequences and structures attached to novel drug-like molecules warheads similar to the structures in ZINC.
In this study, the datasets are constructed by using small proteins from the Protein Data Bank (PDB) – specifically between 50 and 150 residues – as well as sdAbs obtained from the antibody structural database.6 We use atom-level graph representations of each protein so that sidechain modifications can be made directly. For training, each protein can be parsed to a linear string representation, and random data augmentation can be used to increase the training data size. We describe the main details and results for each dataset in the following sections.
1. Proteins
For the first dataset, which consists of proteins with standard amino acids and no sidechain modifications, we test the ability of the language model to explore protein space while maintaining protein structure and constraints. After training, as shown in Fig. 1(A) – we generate a thousand (1k) samples from the language model and evaluate their atom, residue, and protein-level properties. At the protein level, we determine if the generated samples are proteins by attempting to determine their primary sequence. If we can ascertain their primary sequence, we can use AlphaFold2 (ref. 7) to further evaluate if the model has learned the amino acid sequences that correspond to good structure predictions. Additionally, we study model samples for their distribution of amino acids and other atom-level properties that can be computed using RDKit.8 | ||
| Fig. 1 Proteins (A) dataset preparation. The training workflow for the model: training, generation, amino acid sequence determination, and AlphaFold visualization. (B) Comparison of amino acid distributions. (C) AlphaFold visualizations of model-generated proteins coloured by pLDDT, including the PDB ID of the closest protein and its % sequence overlap. | ||
We check if samples generated by the model are proteins by analyzing if they preserve the basic structure of the protein backbone and natural amino acids form. First, we perform a backbone structure search and then attempt to arrange the backbone from the N terminus to the C terminus while simultaneously classifying each sidechain using another substructure search for the standard set of amino acids. If this is successful and there are no discontinuities in the backbone or other side chain errors, then we classify the sample as a protein and parse the amino acid sequence. By this process, we determine roughly ∼68.2% of samples are proteins, furthermore, all the parsed amino acid sequences are unique (there are no duplicates and the model isn't repeating specific proteins) and novel (they are different from the training sequences).
We compare the distribution of amino acids in the training sequences to the distribution learned by the model based on the generated samples. We plot histograms, in Fig. 1(B), displaying the frequency of occurrence of every amino acid in samples from both the model and the training data – both distributions are very similar and mostly overlap although for some amino acids, the language model slightly underestimates the training frequencies.
Using AlphaFold,7 in Fig. 1(B), we visualize selected examples of proteins generated by the language model. In each sample, residues are color-coded according to pLDDT, which is a per-residue estimate of the model's confidence on a scale from 0 to 100. Regions with pLDDT > 90 are dark blue and have high accuracy. Regions with pLDDT from 90 down to 70 are still expected to be good predictions and are colored light blue that transitions to green (with decreasing confidence). Regions with pLDDT between 50 and 70 are lower confidence and are colored yellow to green. The regions with pLDDT < 50 are not confident and likely disordered – these are colored red.
On this scale, in Fig. 1(C), we see that the selected proteins generated by the model result in good structure predictions – ranging between 70 and 90 pLDDT. This indicates that the model can generate proteins with well-defined structures that are not disordered. For a simple baseline comparison, we considered sequences of random amino acids, the structure predictions for these consistently result in disordered proteins with low pLDDT < 50.
Additionally, in Fig. 1(C), the proteins generated by the language model contain a variety of secondary structures including alpha helices, beta sheets, and omega loops. Globally, the generated proteins combine many of these secondary structures into various and unique domains. We can conclude, based on these samples, and further examples in ESI Fig. S1,† that language models can generate proteins, atom by atom, not just with valid primary sequences but proteins with meaningful secondary and tertiary structure.
Furthermore, in Fig. 1(C) and ESI Fig. S1,† under each generated protein we label the primary sequence percentage overlap between the generated proteins and their most similar PDB training example – which ranges from 86% to 40% (excluding one other generated protein that has no nearest PDB training example). This is evidence that the model draws heavily from the amino acid sequence patterns in its training data but does not memorize them.
Also, in ESI Fig. S2,† we plot histograms comparing atom-level properties of the samples generated from the model with the training data. The model roughly approximates the training distribution of atoms but slightly underestimates some properties.
2. Antibody drug conjugates
Next, we test the ability of the language model to generate proteins attached to small molecules and simultaneously explore protein space and chemical space. One of the most promising examples of this kind of biomolecule with immense therapeutic potential are antibody–drug conjugates, which are a form of cancer therapy intended to target and kill cancer cells but spare healthy cells.9 Structurally, they are composed of an antibody attached to single or multiple anticancer drugs typically using some linker molecule. To construct a synthetic dataset of antibody–drug conjugates, as shown in Fig. 2(A), we attach a single drug-like molecule from the ZINC dataset5 to single-domain antibodies (sdAbs) from the structural antibody dataset6,10 to test the ability of language models to generate antibody–drug conjugates. We use two possible linkers for cysteine attachments and two other linkers for lysine attachments. Both linkers are selected from real antibody–drug conjugates described in ref. 9. The linker is randomly attached to the small molecule from ZINC and the specific lysine or cysteine residue for attachment is also randomly chosen. Since there are only 1k sdAbs in the structural antibody dataset we use data augmentation to expand the dataset size to 250k proteins that can be attached to every molecule in ZINC. | ||
| Fig. 2 Antibody drug conjugates – sdAbs (A) single domain antibody–drug conjugates dataset creation overview. (B) Comparison of amino acid distributions for training and model sdAbs. (C) Histogram comparing the size of training and model sdAbs (by number of residues). (D) Example sdAbs (with warheads excluded) generated by the language model visualized by AlphaFold and coloured by pLDDT. Under each, we include the PDB ID of the closest protein and its % sequence overlap. | ||
After training, we again generate 1k samples from the language model for evaluation, and first test the model's ability to explore protein space and learn the distribution of single-domain antibodies – results are shown in Fig. 2(B–D). Similar to the standard protein data, we compare the distribution of amino acids in the training sequences to the distribution learned by the model. We plot histograms, in Fig. 3(B), displaying the frequency of occurrence of every amino acid in samples from both the model and the training data – from these, we can see the language model accurately learns the training distribution of amino acids. Similarly, in Fig. 3(C), the model accurately learns the size of the training sdAbs.
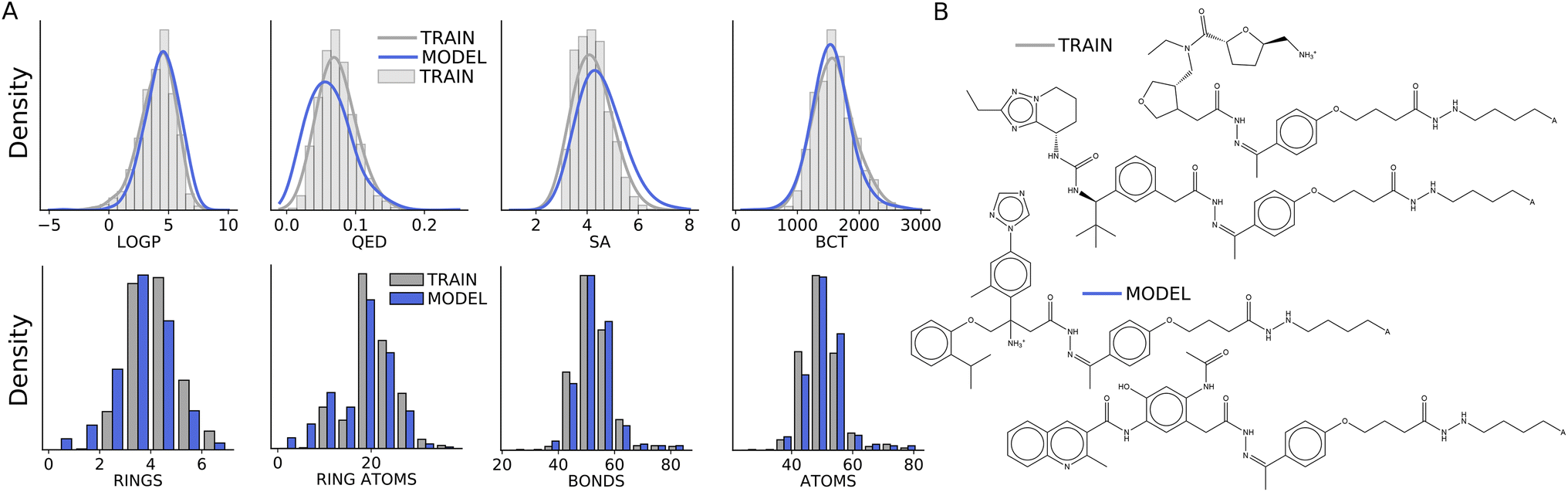 | ||
Fig. 3 Antibody drug conjugates – warheads (A) histograms and density plots of atom-level measures or properties of warheads. Density plots use a Gaussian kernel density estimator fit to log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, MW, and PSA values for the training molecules by tuning the bandwidth parameter. (B) Examples of model and train “warheads”. P, MW, and PSA values for the training molecules by tuning the bandwidth parameter. (B) Examples of model and train “warheads”. | ||
Similar to the standard proteins, we can attempt to determine the amino acid sequences of the single-domain antibodies (ignoring the warheads). We determine roughly ∼90.8% of samples are proteins and their primary sequences are unique and novel (there are no duplicates and all are different from training sequences).
Even further, examples of AlphaFold structure predictions,7 visualized in Fig. 2(D) and ESI S6,† confidently show that the language model can produce sequences that fold into the expected structure for single domain antibodies. Additionally, based on the primary sequence overlap of model samples with their nearest PDB training example in Fig. 1(C) and ESI Fig. S1,† the model learns to produce amino acid sequence structures that are similar to the training sdAbs. The primary sequence overlap with training examples ranges from 63% to 93% in the ESI.† Investigating further, we see that the model draws heavily from the sdAb sequences making new examples of sequences by memorizing small snippets of amino acids and using larger training snippets but with many single mutations randomly distributed throughout the snippet.
From the training examples and model samples, we detach and collect “warheads” which we expand the definition of to include the linker and sidechain in addition to the small molecule (warhead typically refers to just the small molecule). In Fig. 3(B), two examples of train and model warheads are shown as graphs to clarify this. Additional model and training warheads are shown as graphs in ESI Fig. S8 and S7† – as expected the same four linkers repeat across samples but the small molecules attached to them differ and are structurally similar to the ZINC molecules in the training warheads.
We also evaluate the language model's warheads in terms of their atom-level properties. In Fig. 3(A), the model captures the atom-level properties of the training warheads, specifically, it learns the continuous atom-level properties of the training warheads including logP,11 drug-likeness (QED),12 Synthetic Accessibility Score (SA) and molecular graph complexity (BCT) as well as the number of atoms, bonds, rings and atoms in rings. However, the model slightly underestimates the main modes for QED and SA as well as the number of rings per warhead.
Additionally, we assess the model warheads and compare them with the training warheads, we find that model warheads are unique (there are no duplicates and the model is not repeating a few examples) as well as novel (the model does not make exact copies of warheads from the training data). Given that the linkers are memorized, this indicates that the model is learning to generate new small molecules similar to ZINC molecules and effectively exploring chemical space at the same time it learns to explore the protein space defined by the sdAbs.
Also, in ESI Fig. S9,† we see that the model does learn the atom-level properties of the training antibody drug-conjugates. Additionally, in ESI Fig. S3–S5,† we show a single train antibody drug-conjugate and four model samples.
3. Discussion
In this work, we show that chemical language models can generate large biomolecules atom by atom including proteins and protein drug conjugates. By analyzing generated samples we find that language models learn multiple hierarchical layers of molecular information that define the training biomolecules. This includes atom-level molecular properties or residue-level constraints for backbone and amino acid structure as well as primary sequence patterns and motifs that define meaningful secondary and tertiary structure. Indeed, chemical language models learn to generate protein structures as sequence representations of atom-level graphs that are similar to the training proteins in the PDB.Effectively we demonstrate that chemical language models can also serve as biological language models – capable of learning the language of proteins atom by atom. Instead of only learning representations of amino acid sequences, chemical language models generate entire molecular graphs, enabling us to show that chemical language models can be used to explore not just chemical space but also both chemical and protein space at the same time. This work is an initial demonstration of the potential of chemical language models beyond the scale and space that they were designed for – more work is necessary to design real biomolecules that can be experimentally validated.
Further work should be done to enable the model to more consistently generate valid backbone and amino acid form. This will also assist the model in learning distributions consisting of larger biomolecules including structures with more than 150 residues and multiple domains. Using memorizing transformers13 may help the model generate valid protein sequences. Also, other architectures built for longer sequence lengths14 can increase the size and range of structures that the model can learn. Another limitation is that chemical sequence representations do not include the three-dimensional structure of the biomolecule. A potential solution could involve using a point cloud representation of every atom of the biomolecule, combined with reinforcement learning15 or bayesian optimization16 to guide the model to make sidechain modifications in 3D using energy. Additionally, incorporating hierarchical representations – such as group SELFIE – could provide a more robust encoding of the data's inherent structure, allowing the model to represent both amino acid building blocks and small molecules simultaneously.17
The goal of this work is to demonstrate the power of chemical language models and their ability to learn atom-level representations of biomolecules. We envision future language models will be able to explore any combinatorial space in chemistry or biology using any representation type the user wishes.18
4. Methods
4.1. Datasets
From the PDB we successfully parse around ∼10K proteins between 50 and 150 residues. In all datasets, we only parse proteins that conform to atom-level graphs with no more than 2 macrocycles (created by residue–residue connections) – this makes primary sequence determination more successful. Given this constraint, we parse around ∼10k and ∼5k proteins from the PDB for the first two training datasets. In order to increase the size of the training data, we randomize the atom orderings of each protein in RDKit to obtain multiple different random copies of each biomolecule as SMILES (and then SELFIES strings). Using this data augmentation we expand all training datasets to around ∼250k sequences. We use RDKit8 to represent each protein as atom-level graphs and make side-chain modifications. We also made use of Colabfold19 for quick visualization and NGLview20 for figure construction.4.2. Tokenization
We use SELFIES4 version one for the sequence representation of atom-level protein graphs. Other than special tokens like [BOS], [EOS], [PAD], [UNK], the vocab consists of standard selfies tokens, encoding all information in a molecular graph including: atom tokens {[C], [N],…}, bond tokens {[
consists of standard selfies tokens, encoding all information in a molecular graph including: atom tokens {[C], [N],…}, bond tokens {[![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C], [#N],…}, ring tokens: {[Ring1],[Ring2],…} branching tokens: {[Branch1_1], [Branch1_2],…}. In total for all datasets, the vocabulary is around ∼30 tokens.
C], [#N],…}, ring tokens: {[Ring1],[Ring2],…} branching tokens: {[Branch1_1], [Branch1_2],…}. In total for all datasets, the vocabulary is around ∼30 tokens.
4.3. Language modeling for molecular design
In language modeling for molecular design, we want to estimate the unsupervised distribution of the training molecules (mol1, mol2,…, moln) each composed of variable length sequences of tokens from a chemical language [CT]i where such that MOL=([CT]1, [CT]2,…,[CT]n). The joint probabilities over a single molecule can be written as
such that MOL=([CT]1, [CT]2,…,[CT]n). The joint probabilities over a single molecule can be written as | (1) |
4.4. Training
During training, we one-hot encode SELFIES sequences using a basic vocabulary that consists of 30 possible alphabet tokens. All language models are trained using next-token prediction conditioned on the entire sequence for context. The training data only uses sequences that have a maximum length of 1664 tokens. We trained language models with decoder only, GPT-like architecture22 with 4–8 attention heads. Language models are implemented in Python 3 with PyTorch.23 Molecules properties are computed using RDKit.8 Models were trained for 100–200 epochs on single A100 GPUS or roughly 24–48 hours. We used 8–12 transformer layers. Models are trained with batches of 4 SELFIES sequences using the ADAM optimizer with a small learning rate 5 × 10−4.Code availability
Code for training and evaluating the models is available at the following URL: https://github.com/kevqyzhu/chemical_lm_biomolecules. The corresponding archived version is available at: https://doi.org/10.5281/zenodo.15742662.Data availability
All PDB IDs, their corresponding SMILES representations, SMILES fragments for sidechain modification, ZINC molecules as SMILES, and code to generate the final training data as SELFIES are available at: https://github.com/kevqyzhu/chemical_lm_biomolecules. A snapshot of the data used in this study has been archived on Zenodo with the DOI: https://doi.org/10.5281/zenodo.15742662.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. A.-G. acknowledge funding from Dr Anders G. Frøseth. A. A.-G. also acknowledges support from the Canada 150 Research Chairs Program, the Canada Industrial Research Chair Program, and from Google, Inc. Models were trained using the Canada Computing Systems.24 This research was undertaken thanks in part to funding provided to the University of Toronto's Acceleration Consortium from the Canada First Research Excellence Fund.References
- D. Flam-Shepherd, K. Zhu and A. Aspuru-Guzik, Language models can learn complex molecular distributions, Nat. Commun., 2022, 13(1), 3293 CrossRef CAS.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling and D. Sheberla, et al., Automatic chemical design using a data-driven continuous representation of molecules, ACS Cent. Sci., 2018, 4(2), 268–276 CrossRef PubMed.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28(1), 31–36 CrossRef CAS.
- M. Krenn, F. Häse, A. Nigam, P. Friederich and A. Aspuru-Guzik, SELFIES: a robust representation of semantically constrained graphs with an example application in chemistry, arXiv, 2019, preprint, arXiv: 190513741 Search PubMed.
- J. J. Irwin and B. K. Shoichet, ZINC- a free database of commercially available compounds for virtual screening, J. Chem. Inf. Model., 2005, 45(1), 177–182 CrossRef CAS.
- C. Schneider, M. I. J. Raybould and C. M. Deane, SAbDab in the age of biotherapeutics: updates including SAbDab-nano, the nanobody structure tracker, Nucleic Acids Res., 2021, 50(D1), D1368–D1372, DOI:10.1093/nar/gkab1050.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov and O. Ronneberger, et al., Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596(7873), 583–589 CrossRef CAS PubMed.
- G. Landrum, RDKit: A software suite for Cheminformatics, Computational Chemistry, and Predictive Modeling, Academic Press, 2013 Search PubMed.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Strategies and challenges for the next generation of antibody–drug conjugates, Nat. Rev. Drug Discovery, 2017, 16(5), 315–337 CrossRef CAS.
- C. Schneider, M. I. Raybould and C. M. Deane, SAbDab in the age of biotherapeutics: updates including SAbDab-nano, the nanobody structure tracker, Nucleic Acids Res., 2022, 50(D1), D1368–D1372 CrossRef CAS.
- S. A. Wildman and G. M. Crippen, Prediction of physicochemical parameters by atomic contributions, J. Chem. Inf. Comput. Sci., 1999, 39(5), 868–873 CrossRef CAS.
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Quantifying the chemical beauty of drugs, Nat. Chem., 2012, 4(2), 90–98 CrossRef CAS PubMed.
- Y. Wu, M. N. Rabe, D. Hutchins and C. Szegedy, Memorizing Transformers, in: International Conference on Learning Representations, 2021 Search PubMed.
- R. Child, S. Gray, A. Radford and I. Sutskever, Generating long sequences with sparse transformers, arXiv, 2019, preprint, arXiv: 190410509 Search PubMed.
- D. Flam-Shepherd, A. Zhigalin and A. Aspuru-Guzik, Scalable Fragment-Based 3D Molecular Design with Reinforcement Learning, arXiv, 2022, preprint, arXiv: 220200658 Search PubMed.
- T. C. Wu, D. Flam-Shepherd and A. Aspuru-Guzik, Bayesian Variational Optimization for Combinatorial Spaces, arXiv, 2020, preprint, arXiv: 201102004 Search PubMed.
- A. H. Cheng, A. Cai, S. Miret, G. Malkomes, M. Phielipp and A. Aspuru-Guzik, Group SELFIES: a robust fragment-based molecular string representation, Digital Discovery, 2023, 2, 748–758, 10.1039/D3DD00012E.
- D. Flam-Shepherd and A. Aspuru-Guzik, Language models can generate molecules, materials, and protein binding sites directly in three dimensions as XYZ, CIF, and PDB files, arXiv, 2023, preprint, arXiv: 230505708 Search PubMed.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, ColabFold: making protein folding accessible to all, Nat. Methods, 2022, 19(6), 679–682 CrossRef CAS PubMed.
- H. Nguyen, D. A. Case and A. S. Rose, NGLview–interactive molecular graphics for Jupyter notebooks, Bioinformatics, 2018, 34(7), 1241–1242 CrossRef CAS PubMed.
- A. Radford, K. Narasimhan, T. Salimans and I. Sutskever, Improving language understanding by generative pre-training, 2018 Search PubMed.
- A. Radford, J. Wu, R. Child, D. Luan, D. Amodei and I. Sutskever, et al., Language models are unsupervised multitask learners, 2019.
- A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury and G. Chanan, et al., Pytorch: An imperative style, high-performance deep learning library, Adv. Neural Inf. Process. Syst., 2019, 32, 8026–8037 Search PubMed.
- S. Baldwin, Compute Canada: advancing computational research, in: Journal of Physics: Conference Series, vol. 341, IOP Publishing, 2012, p. 012001 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1: Examples of proteins generated by the model visualized by AlphaFold and colored by pLDDT. Fig. S2: Histograms of atom level properties for the basic proteins training data. These include exact molecular weight (MW), octanol–water partition coefficient (logP),11 molecular complexity (BCT), topological polar surface area (PSA), number of rings (RINGS), number of atoms (ATOMS), number of bonds (BONDS), number of fragments found by breaking up the molecule at rotatable bonds (FRAGS), number of carbons (C), number of nitrogens (N), number of oxygens (O), and number of any other atoms (OTHER). Fig. S3: Antibody–drug conjugates example antibody–drug conjugates from the training data (above) and one example produced by the language model (below) and plotted using ChemDraw as an atom-level graph. Fig. S4: Model antibody–drug conjugates example antibody–drug conjugates produced by the language model and plotted using ChemDraw as an atom-level graph. Fig. S5: Model antibody–drug conjugates example antibody–drug conjugates produced by the language model and plotted using ChemDraw as an atom-level graph. Fig. S6: Examples of single domain antibodies from samples of sdAb-drug conjugates (excluding warheads) generated by the model visualized by AlphaFold and colored by pLDDT. Fig. S7: examples of detached warheads from training single domain antibody–drug conjugates. Fig. S8: Examples of detached warheads from model generated single domain antibody–drug conjugates. Fig. S9: Histograms of atom level properties for the antibody–drug-conjugates training data. These include exact molecular weight (MW), octanol–water partition coefficient (logP),11 molecular complexity (BCT), topological polar surface area (PSA), number of rings (RINGS), number of atoms (ATOMS), number of bonds (BONDS), number of fragments found by breaking up the molecule at rotatable bonds (FRAGS), number of carbons (C), number of nitrogens (N), number of oxygens (O), and number of any other atoms (OTHER). See DOI: https://doi.org/10.1039/d5dd00107b |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |