
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved stereocontrol in reductive aminases through steric modification of residues around substrate and cofactor binding pockets†
Jake
Gooderham
a,
Beatrice-Maria
Zabava
a,
David D.
Aleku
a,
Julie
Vignot
a,
Zuoye
Xie
a,
Ruth T.
Bradshaw Allen
a,
Mario
Prejanò
 *b and
Godwin A.
Aleku
*a
*b and
Godwin A.
Aleku
*a
aInstitute of Pharmaceutical Science, Franklin-Wilkins Building, King's College London, 150 Stamford Street, London SE1 9NH, UK. E-mail: godwin.aleku@kcl.ac.uk
bDepartment of Chemistry and Chemical Technologies, University of Calabria, Via Pietro Bucci 14/C, 87036 Arcavacata di Rende, CS, Italy. E-mail: mario.prejano@unical.it
First published on 10th April 2025
Abstract
Asymmetric reductive amination catalysed by reductive aminases (RedAms) provides a green and direct route to 2° and 3° chiral amines. Identifying residues or motifs in these enzymes that facilitate stereocontrol is essential for designing highly desirable enantiodivergent RedAm systems. In this work, we have identified key residues within both the cofactor and substrate binding pockets in a fungal reductive aminase (MaRedAm) and a bacterial imine reductase (AoIRED) that enable stereocontrol through steric modification. In MaRedAm, removing steric bulk at the cofactor binding pocket via W33A or R35A mutation improved (R)-selectivity towards the synthesis of (R)-rasagiline, achieving up to 95% enantiomeric excess (e.e.). Conversely, the W211A mutation at the substrate binding pocket of MaRedAm inverted the stereoselectivity, yielding (S)-rasagiline (42% e.e.). Likewise, varying steric bulk at position N241 in AoIRED allowed for enantiodivergency. Notably, modifying the N241 position significantly improved AoIRED's solution stability and storability. The wild-type enzyme typically precipitates out of solution within 8 h after purification, even when stored at 4 °C, whereas its N241H and N241Y variants remain in solution for up to >1 week. Molecular dynamics (MD) simulations provided detailed insights into the effect of steric modification on stereoselectivity at the cofactor and substrate binding pockets. MaRedAm W33A and W35A mutations induced reorganisation and downsizing of the active site, enhancing (R)-selectivity. In contrast, the W211A mutation enlarged the substrate binding pocket, increasing flexibility for substrate rotation. These findings contribute to the ongoing effort to establish the functional roles of key residues to allow efficient rational engineering of stereoselectivity in IREDs/RedAms.
Introduction
Reductive amination of prochiral carbonyl compounds is a frequently employed transformation to construct chiral amine-based active pharmaceutical ingredients (APIs).1,2 Enzymatic reductive amination reactions such as those catalysed by imine reductases (IREDs) and reductive aminases (RedAms)3–7 are particularly attractive as they offer a highly enantioselective approach.8–11 IREDs/RedAms' amination route provides direct access to enantiopure 2° and 3° chiral amines without the need to use toxic alkylating reagents or stoichiometric amounts of reducing agents. Given that chiral amines are among the most prevalent scaffolds in APIs, developing enantiodivergent RedAm methodologies will have enormous synthetic relevance.12 However, IREDs/RedAms, similar to other redox enzymes, may on occasion catalyse the amination of prochiral ketones to yield the ‘wrong’ enantiomer of the target product or produce the desired enantio-enriched product in suboptimal optical purity. Achieving perfect enantioselectivity for a synthetic target remains challenging, often requiring extensive directed evolution campaigns,13 which are impractical within the time constraints of drug development projects.14Recent studies have sought to pinpoint the origin of stereoselectivity in IRED-catalysed reduction of cyclic imines based on the residue occupying the standard catalytic 187 position (SkIRED numbering, Fig. 1).15–17 From a wealth of empirical evidence, the so-called D-type IREDs which contain Asp at the 187 equivalent position are predominantly (R)-selective, and reduce simple cyclic imines such as 1-methyl-3,4-tetrahydroisoquinoline 1, 2-methylpyrroline, and 2-methylpiperideine to the corresponding (R)-amines whereas the so-called Y-type IREDs feature Tyr at the 187 position, are predominantly (S)-selective toward these substrates.18–23 However, some IREDs do not follow these selectivity patterns or contain residues other than Asp or Tyr at the standard Y187 position.15,17,24 In addition, other regulatory factors, such as electrostatic interactions between the anionic side chains of active site residues and the iminium cation, have been proposed to be critical to the stereochemical outcome of IRED-catalysed imine reduction.17
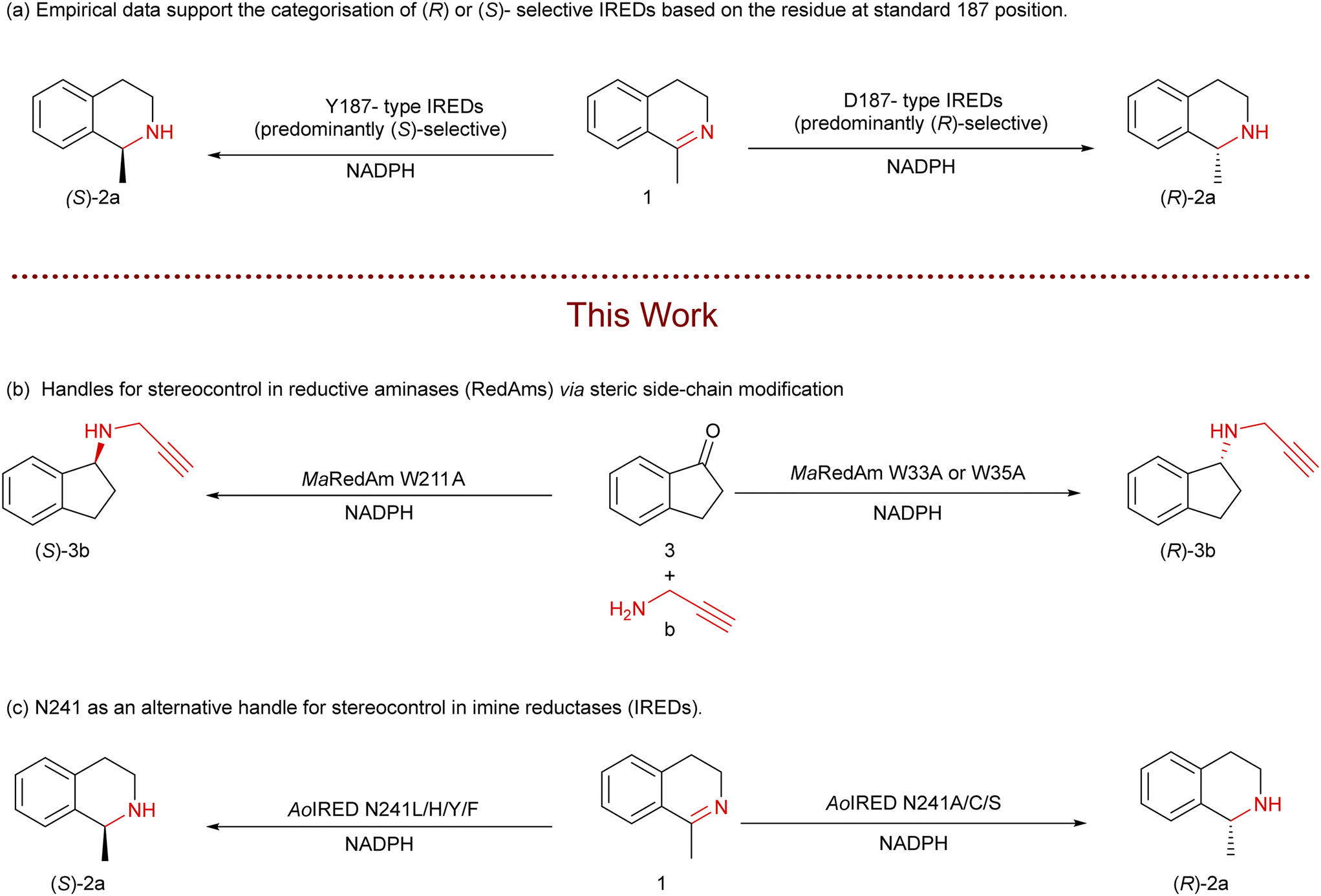 | ||
| Fig. 1 Stereoselectivity patterns of imine reductases (IREDs) towards the reduction of prochiral cyclic imines and an overview of work performed in this study. | ||
Despite the significant interest in engineering stereoselectivity in RedAms,4,13,25–28 the regulatory factors governing stereoselectivity in RedAm-catalysed reductive amination remain poorly understood. Unlike for IRED-catalysed reduction of cyclic imines where available empirical data can guide prediction of the stereoselectivity of an IRED of interest, there is no clear pattern for the categorisation of (S)-type or (R)-type enzymes in IRED/RedAm-catalysed reductive amination of ketones based on the residue occupying the catalytically important position 187. IREDs acting in the RedAm mode must bind NADPH, ketone and amine to form the corresponding iminium cation,4,29 and the size and nature of both the amine and ketone coupling partners can contribute to the stereochemical outcome.4,29–31
Steric modification of the side chains of key residues in many NAD(P)H-dependent redox enzymes, including IREDs, can directly or indirectly alter substrate and cofactor binding modes,24 with its attendant effects on enzymatic activity. While such modifications are less frequently reported or investigated for IREDs/RedAms,4,26 alteration of the substrate and cofactor binding modes in this manner can influence whether hydride delivery from NAD(P)H to the iminium ion occurs on the Si- or Re-face, which in turn can affect the stereochemical outcome. In this study, using a fungal reductive aminase (MaRedAm)31 and a bacterial imine reductase (AoRED),32 we systematically screened and identified multiple residues that enable stereocontrol in these enzymes through steric modification of their side chains.
Results and discussion
Handles in a reductive aminase for stereocontrol via modifying the side chain's steric bulk
To quickly assess the mutational effects of bulky conserved residues on RedAm's stereoselectivity, we employed alanine scanning site-directed mutagenesis, targeting conserved bulky residues (W, Y, F, and R) with a conservation score of >90%, regardless of whether these residues interact with the substrates or the cofactor. Six such positions were identified through sequence alignment of >1200 MaRedAm homologues (ESI,† Table S1). Alanine variants were successfully constructed, expressed in E. coli and the freshly prepared cell-free lysate (CFE) was used as the aminase catalyst solution to perform two amination transformations: 1-indanone 3 with propargylamine b and 4-phenyl-2-butanone 4 with b. Stereoselectivity of the product was assessed via chiral HPLC (Table 1).|
|
||||
|---|---|---|---|---|
|
|
|
|||
| MaRedAm and variants | Conv. (%) | e.e.% (abs. conf.) | Conv. (%) | e.e.% (abs. conf.) |
| a Absolute configuration was assigned based on a comparison with the elution patterns of the N-methyl analogue. n.d. = not determined. | ||||
| MaRedAm WT | 31 | 74 (R) | >99 | 63 (R)a |
| W33A | 43 | 95 (R) | >99 | 97 (R)a |
| R35A | 33 | 92 (R) | >99 | 91 (R)a |
| Y118A | 25 | 79 (R) | 61 | 92 (R)a |
| Y139A | 31 | 64 (R) | >99 | 25 (R)a |
| Y181A | <1 | n.d. | 7 | >99 (R)a |
| W211A | 30 | 42 (S) | 95 | 36 (S)a |
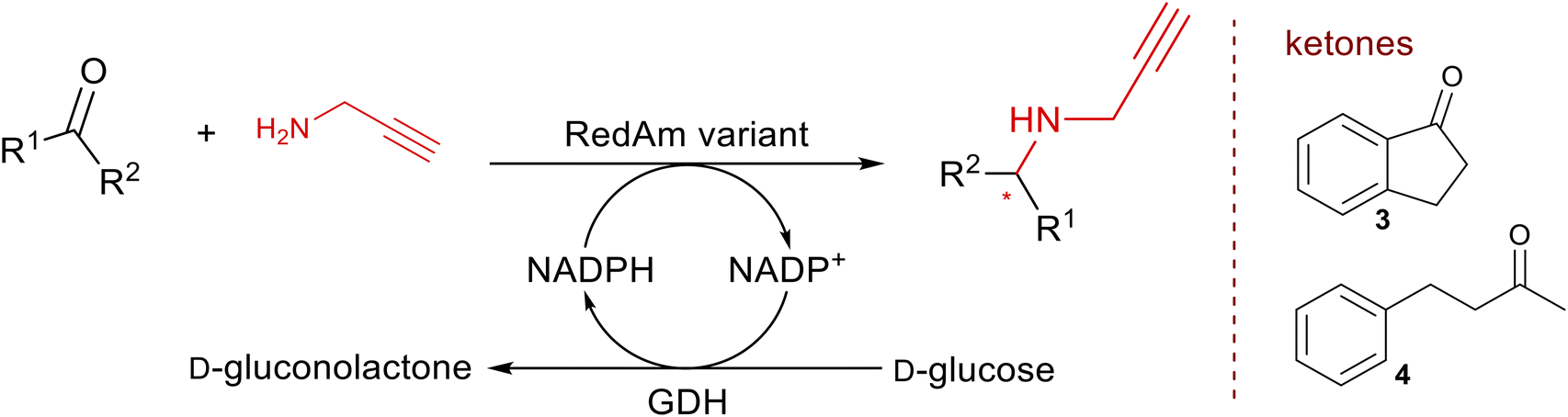

Mutations W33A, R35A, Y139A, and Y181A in MaRedAm (Scheme 1) led to observable changes in the stereoselectivity. The neighbouring variants W33A and R35A exhibited improvements in e.e. values, yielding, for example, (R)-rasagiline 3b with up to 95% e.e., compared to 74% e.e. achieved by the wild-type enzyme (Table 1). In contrast, the Y139A mutation led to a marked decrease in e.e. (64% e.e., (R)-4b). Similar trends were observed with these mutants for the synthesis of 4b, where W33A, R35A, Y118A, and Y181A all achieved significant improvement in e.e. of up >97% compared to 63% e.e. achieved by the wild-type enzyme. Again, Y139A mutation led to a significant decrease in e.e. value (25%). Replacing the bulky protic residues with alanine, such as in Y118A, Y139A, and Y181A, resulted in reduced activity and lower conversion values. Notably, the MaRedAm mutation, W211A, confers a profound switch in stereoselectivity, resulting in the formation of the opposite enantiomer for both transformations (3b, W211A yielding 42% e.e. (S) versus 74% e.e. (R) for wild-type; 4b, W211A gave 52% e.e. (S) versus 63% e.e. (R) for wild-type). Biotransformation reactions with purified enzyme preparation for selected variants (wild-type, W35A, and W211A) across three transformations confirmed this pattern of changes in stereoselectivity (ESI,† Table S2). In addition to the emerging evidence of the role of W211 in stereoselectivity as demonstrated in MaRedAm, AspRedAm4,33 and other RedAms,26 we identified two residues, W33 and W35, which are excellent handles for improving the stereoselectivity of MaRedAm. To our knowledge, the role of W33 and W35 in stereocontrol in this enzyme family has not been previously reported, and neither has any residue in this structural loop been linked to stereocontrol in RedAms. Our work, therefore, highlights these residues as key handles for improving stereoselectivity in MaRedAm and by extension, these residues may serve as handles for modifying stereoselectivity in other RedAms.
 | ||
| Scheme 1 AspRedAm structure in complex with NADP(H) and the amine product rasagiline (PDB: 5G6S) and an AlphaFold-generated structural model of MaRedAm, showing equivalent positions that affect enantioselectivity | ||
Molecular dynamics simulations reveal structural reorganisations of the active site
To investigate the rationale behind the observed changes in stereoselectivity, we performed molecular dynamics (MD) simulations on the AlphaFold model of MaRedAm-WT (ESI,† Fig. S1) and its variants, W33A, R35A, and W211A. Using MaRedAm-WT as the reference, the trajectories and structural changes resulting from these mutations were followed and analysed to detect any structural contributions in the binding step of the catalytic mechanism. In each simulation, NADP(H) and the substrates (indanone 3 + propargylamine b, for the formation of rasagiline 3b) were explicitly included in the models.Analysis of the MD trajectories of W33A and R35A mutations revealed a slight shift of NADP(H) towards W211, resulting in a downsizing of the substrate's binding pocket (Fig. 2). The space created by the loss of the bulky indole group of Trp is now occupied by the α-helix formed by amino acid residues V56-S63 in the W33A mutant. This movement, evidenced by the analysis of root mean square deviation (see 1.0 < RMSD (Å) < 2.0 vs. 0.5 < RMSD (Å) < 1.5 value distributions in Fig. 2a), indicates a shift of NADP(H) towards W211 (Fig. 2a and ESI,† S2 and S4). A similar effect was observed with the R35A mutant mainly due to the loss of the stabilising ionic interaction between the guanidinium group of R35 (found in WT) and the phosphate moiety of the cofactor, as highlighted by the radial distribution function (RDF) calculated for the R35-NADP(H)/R35A-NADP(H) pairs (see absence of peak at ca. 2 Å for MaRedAm-R35A, Fig. 2b). The cofactor shifts with respect to W211 by ∼1.0 Å, moving away from the R35A position, thus affecting the volume of the substrate binding site (see NADP(H)-W211 distance variations in Fig. S4†).
 | ||
| Fig. 2 RMSD of V56-S63 α-helix obtained from molecular dynamics (MD) simulations of MaRedAm-WT and MaRedAm-W33A systems (a). RDFs of R35-NADP(H) pair calculated from MD simulations of MaRedAm-WT and MaRedAm-R35A systems (b). For clarity, hydrogens were omitted, and only relevant species were reported. In the figures, atoms in transparent-background representation are for the wild-type enzyme. | ||
While W33A and W35A mutations led to the reduction in the volume of the substrate binding pocket, we observed an opposite behaviour in MaRedAm-W211A simulations, where the substrate's binding site was enlarged. The pocket generated by the W211A mutation was filled by water molecules, as shown by RDF calculated for W211-Ow/W211A-Ow pairs (Fig. 3a). In the case of the mutation-containing system, two peaks appeared at approximately 3.5 Å and 7 Å, corresponding to the first and second hydration shells surrounding the amino acid residue. The influx of water molecules in the site is also linked to a reorganisation of the surrounding protein domain, such as the L123-V136 loop, which, shifting from the equilibrium conformation in MaRedAm-WT, contributes to the further opening and enlargement of the active site pocket (see RMSD values in Fig. 3b). Regarding the NADP(H)-W211 distance, all these effects cumulated in an increase of up to 5 Å with respect to MaRedAm-WT (see Fig. S5†).
 | ||
| Fig. 3 RDFs of the W211-OW pair calculated from molecular dynamics (MD) simulations (a) and RMSD of the L123-V136 loop (b) obtained from MaRedAm-WT and MaRedAm-W211A systems. For clarity, hydrogens were omitted, and only relevant species were reported. Atoms in the transparent background are those of the wild-type enzymes, while water molecules are depicted as red spheres. | ||
These atomistic insights suggest that steric modulation of these bulky residues resulted in structural reorganisations of the cofactor and substrate binding pockets and their interactions, which in turn affected the stereoselectivity of MaRedAm. On the one hand, removing steric bulk around the cofactor binding pocket via mutation caused a shift of NADP(H) and the reorganisation of surrounding residues, leading to a downsize of the active site. This reconfiguration may limit the flexibility of the iminium intermediate within the reduced active site volume, ultimately enhancing the pro-(R) enantioselectivity of the wild-type, as observed with W33A and W35A mutations. On the other hand, removing steric bulk by mutations in the substrate's binding site creates new pockets due to the space created by the mutation itself or by shifts of the surrounding protein domains. Thus, the iminium ion may rotate more freely, positioning it for Si-face hydride delivery, as demonstrated by the switch in the stereoselectivity observed with W211A.
N241 in a bacterial imine reductase (AoIRED) enables stereocontrol via steric modification
Encouraged by these findings, we aimed to determine if steric modulation is equally significant in the enantioselective reduction of cyclic imines. In selecting a model IRED for this investigation, we considered the underexplored IRED block. Asp and Tyr are highly conserved at the standard 187 position in IREDs, resulting in most characterised IREDs broadly falling into the D-type and Y-type categories. However, a smaller proportion of IREDs contain alternative residues, including those featuring Asn at the standard 187 position, herein referred to as the N-type IREDs. As the stereoselectivity categorisation does not extend to the N-type IREDs, we first investigated the selectivity pattern of a small panel of N-type IREDs: AoIRED, AmIRED, ArIRED and SeIRED, retrieved through genome mining (Table 2). A multiple sequence alignment of over 500 homologues of these selected enzymes showed that Asn is conserved in >95% of the sequences, underscoring the N-type enzymes as an important IRED subgroup. Each of the four cloned enzymes was heterologously expressed in E. coli BL21 (DE3). Biotransformation with fresh lysate preparation of each enzyme converted imine 1 to the corresponding (S)-2a, affording >90% conversion and 40–87% e.e. (Table 2). This observation suggests that N-type IRED block are predominantly (S)-selective towards the enantioselective reduction of imine 1 and related structures, and a further study with a large panel of N-type IREDs is needed to establish the stereoselectivity pattern of the IRED block.| Investigating the stereoselectivity of IREDs containing Asn at the 187 position | |||
|---|---|---|---|

|
|
|
|||
|---|---|---|---|
| Enzymes | GenBank no. | Conversion [%] | e.e. [%] (R or S) |
| AoIRED | 5A9S_B | >99% | 85 (S) |
| AmIRED | WP_125690082.1 | 90 | 40 (S) |
| ArIRED | WP_257488618.1 | >99 | 69 (S) |
| SeIRED | WP_211898848.1 | >99 | 77 (S) |

Position 187 (N171 in AoIRED) has been shown to play an important role in stereocontrol in multiple IREDs, including the N-type IREDs. However, mutations at this position frequently lead to concurrent activity loss, as demonstrated with several IREDs.4,17,20,29,32,34 Previous studies have also shown that the Y179A mutation in AoIRED markedly altered the stereoselectivity towards 2,24,32 however, mutations at this position are likewise associated with a significant reduction in activity across various IREDs, including AoIRED.4,29,32 Hence, we sought to identify an alternative residue that can serve as a handle for stereocontrol with minimal impact on IRED activity. We previously demonstrated that AoIRED N241A mutant displays enhanced substrate tolerance towards bulky substrates such as dibenzazepines,35 implicating N241 in substrate recognition/specificity. The N241A mutant was also shown to exhibit opposite selectivity compared to the wild type,32 and this position has recently been shown to play a similarly critical role in engineering stereoselectivity inversion in D- and Y-types IREDs.36
Encouraged by these observations, we examined the effect of sterically varied residue substitutions at N241 on the stereoselectivity of AoIRED (Scheme 2). Eight defined point mutants at N241 comprising characteristically distinct amino acid residues were constructed, including N241A, N241S, N241C, N241Y, N241F, N241H, N241L, and N241W, and each variant was expressed in E. coli to generate recombinant whole-cell biocatalysts. Given the stereoselectivity changes that can occur with purified AoIRED upon storage,32 bioreduction of 2 was performed using resting E. coli whole cells expressing an AoIRED mutant. Analysis of these biotransformation reactions via chiral HPLC revealed that variants containing amino acid residues with small side chains, including N241A, N241S, and N241C, produced the (R)-2a product, albeit with low to moderate e.e. values ranging from 4% to 40% (Table 3). These results represent an inversion in stereopreference, as AoIRED WT yielded (S)-2a, 85% e.e. In contrast, variants bearing amino acids larger than Asn (N241Y, N241F, N241L, and N241H) retained the wild-type stereoselectivity, yielding (S)-2a, but with significantly improved enantiomeric excess (e.e.) values, ranging from 92% to >99% e.e. (Table 3). The N241W substitution was associated with activity loss, likely due to unproductive binding of the substrate caused by steric hindrance from the bulky Trp (Table 3).
 | ||
| Scheme 2 AoIRED in complex with amine 2a, and NADP(H) showing the targeted N241 residue (PDB: 5FWN). While N171 and Y179 have been shown to affect the stereoselectivity of IREDs, mutations at these positions are associated with marked loss of activity. This study examines the role of N241 in stereocontrol without compromising activity. | ||
|
|
||
|---|---|---|
| AoIRED variants | Conv. [%] | e.e. [%] (R or S) |
| WT | 98 | 85 (S) |
| N241L | >99 | 98 (S) |
| N241H | 87 | 92 (S) |
| N241Y | >99 | >99 (S) |
| N241F | 98 | 97 (S) |
| N241W | <5% | n.d. |
| N241A | 96 | 42 (R) |
| N241S | >99 | 13 (R) |
| N241C | 92 | 4 (R) |

The pattern of the stereoselectivity of AoIRED N241X variants indicates a critical role of steric bulk modification in determining the stereoselectivity of this enzyme. Residues with bulky hydrophobic and aromatic side chains at N241 would stabilise and ‘cage’ the imine substrate, resulting in less flexibility such that hydride attack predominantly occurs at one face, in this case, the Si-face. As a consequence, the (S)-configured amine product was obtained with excellent e.e. values, ranging from 92 to >99%. In contrast, a less intimate interaction may allow flexibility for hydride delivery to both Si-face and Re-face when a smaller amino acid occupies this position, as in N241A, N241S, and N251C. This is evidenced by the inversion of stereoselectivity and the poor to moderate e.e. values (4–42%) with these substitutions.
Using imine 1 as the prototype substrate, we determined the Michaelis–Menten constants of AoIRED WT, N241A, N241H, and N241S variants. The catalytic efficiency, kcat/Km (s−1 mM−1) of these variants were 1.034, 1.934 (∼1 fold improvement over parent (FIOP)), 2.054 (∼1 FIOP), 4.730 s−1 mM−1 (∼4 FIOP), respectively (ESI,† Table S3). These improvements were primarily driven by a decrease in the Km, except for N241S, which showed improvements in both Km and kcat (ESI,† Table S3). The N241 position, therefore, enables the fine-tuning of stereoselectivity and simultaneously enhances the catalytic efficiency of this enzyme.
N241Y/H in AoIRED exhibits significantly improved solution behaviour and storability
In our hands, AoIRED wild-type was shown to aggregate and precipitate out of solution between 6–8 h after purification when stored (4 °C) at concentrations above 4 mg mL−1. We purified N241X variants to homogeneity and investigated their solution behaviour (Table 4). Notably, we observed that N241X variants bearing polar residues/residues capable of engaging in hydrogen bonding interaction such as N241S, N241Y, and N241H, showed dramatically improved solution behaviour and storability.| Variants | Residues | Hydropathy index (Kyte-Doolittle) | Time to precipitate |
|---|---|---|---|
| AoIRED Wt | Asn | −3.5 | <8 h |
| N241F | Phe | 2.8 | <6 h |
| N241C | Cys | 2.5 | <8 h |
| N241A | Ala | 1.8 | 12 h |
| N241S | Ser | −0.8 | 24 h |
| N241Y | Tyr | −1.3 | 72 h |
| N241H | His | −3.2 | >1 week |
While the N241F variant precipitated within 6 h of purification, the N241S, N241Y and N241H remained homogeneous for up to 18 h, 72 h, and >168 h (>7 days), respectively, indicating a substantial improvement in solution behaviour. Analysis of the pattern of substitution in relation to the Kyte–Doolittle hydropathy index37 revealed that residues with negative hydropathy scores and capable of hydrogen bonding interactions, such as those in N241S, N241Y, and N241H, exhibited enhanced stability (Table 4). Protein aggregation can hinder the industrial exploitation of promising but aggregation-prone biocatalysts, such as AoIRED. It is therefore remarkable and novel for this enzyme family that single-point mutants at the N241 position, particularly N241H, dramatically enhanced solution behaviour. This position can thus be explored to improve the storability and stability of purified AoIRED and, by extension, other aggregation-prone IREDs/RedAms.
Conclusion
In summary, our findings demonstrate that steric modulation of residues at both the cofactor and substrate binding sites can induce stereocontrol in IREDs/RedAms, enabling the improvement as well as inversion of stereoselectivity through rational fine-tuning of the steric bulk of these residues. The residues identified in this study: W211 (in MaRedAm) and Q241 (N241 in AoIRED) at the substrate binding pocket and W33 and W35 at the cofactor binding site, can serve as key handles for fine-tuning stereoselectivity in these enzymes to achieve the desired stereochemical outcomes, especially through steric modulation. The comparative molecular dynamics outcomes obtained from the enzyme-substrate complex simulations have enabled the qualitative detection of structural details underlying the origin of MaRedAm's enantioselectivity and explain the effect of steric modulation resulting from these mutations. Remarkably, exploring substitutions at the N241 position also significantly enhanced the solution behaviour and storability of AoIRED, from less than 8 hours to over 1 week. This improvement not only enhances the practical application of AoIRED but also suggests that strategic mutations can have a significant impact on enzyme stability. This work lays an important foundation towards making engineering stereoselectivity in IREDs/RedAms predictable, faster, and cost-effective. Future efforts will focus on mapping additional residues that may act as handles for stereocontrol and pinpointing the origin of enantioselectivity in RedAms through quantitative calculations of accurate energy profiles for the catalytic reaction, as well as analysis of related intermediates and transition states.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualisation: G. A. A.; methodology: G. A. A. and M. P.; investigation and formal analysis: all authors; writing – initial draft: G. A. A., M. P.; supervision: G. A. A. All authors have read and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received support from the Leverhulme Trust through a Leverhulme Early Career Fellowship to G. A. A. (ECF-2020-694), and further support was provided by a Royal Society Grant to G. A. A. (RGS\R1\231514). G. A. A. is also grateful to the Institute of Pharmaceutical Science at King's College London for its financial support of this project. M. P. is grateful to the Department of Chemistry and Chemical Technologies, Università della Calabria, for financial support and to ISCRA-Leonardo for supercomputing resources (project ID: HP10CEZM7C).References
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed.
- G. A. Aleku, ACS Catal., 2024, 14, 14308–14329 CrossRef CAS.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961–969 CrossRef CAS PubMed.
- R. Kumar, M. J. Karmilowicz, D. Burke, M. P. Burns, L. A. Clark, C. G. Connor, E. Cordi, N. M. Do, K. M. Doyle, S. Hoagland, C. A. Lewis, D. Mangan, C. A. Martinez, E. L. McInturff, K. Meldrum, R. Pearson, J. Steflik, A. Rane and J. Weaver, Nat. Catal., 2021, 4, 775–782 CrossRef CAS.
- M. Schober, C. MacDermaid, A. A. Ollis, S. Chang, D. Khan, J. Hosford, J. Latham, L. A. F. Ihnken, M. J. B. Brown, D. Fuerst, M. J. Sanganee and G.-D. Roiban, Nat. Catal., 2019, 2, 909–915 CrossRef CAS.
- T. Liu, Z. Xu, J. Feng, S. Yu, M. Wang, P. Yao, Q. Wu and D. Zhu, Org. Lett., 2023, 25, 2438–2443 CrossRef CAS PubMed.
- L. Ducrot, M. Bennett, G. Grogan and C. Vergne-Vaxelaire, Adv. Synth. Catal., 2021, 363, 328–351 CrossRef CAS.
- A. K. Gilio, T. W. Thorpe, N. Turner and G. Grogan, Chem. Sci., 2022, 13, 4697–4713 RSC.
- T. Knaus, W. Böhmer and F. G. Mutti, Green Chem., 2017, 19, 453–463 RSC.
- B. Yuan, D. Yang, G. Qu, N. J. Turner and Z. Sun, Chem. Soc. Rev., 2024, 53, 227–262 RSC.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- E. J. Ma, E. Siirola, C. Moore, A. Kummer, M. Stoeckli, M. Faller, C. Bouquet, F. Eggimann, M. Ligibel, D. Huynh, G. Cutler, L. Siegrist, R. A. Lewis, A.-C. Acker, E. Freund, E. Koch, M. Vogel, H. Schlingensiepen, E. J. Oakeley and R. Snajdrova, ACS Catal., 2021, 12433–12445 CrossRef CAS.
- M. D. Truppo, ACS Med. Chem. Lett., 2017, 8, 476–480 CrossRef CAS PubMed.
- S. Fademrecht, P. N. Scheller, B. M. Nestl, B. Hauer and J. Pleiss, Proteins: Struct., Funct., Bioinf., 2016, 84, 600–610 CrossRef CAS PubMed.
- X.-X. Zhu, W.-Q. Zheng, Z.-W. Xia, X.-R. Chen, T. Jin, X.-W. Ding, F.-F. Chen, Q. Chen, J.-H. Xu, X.-D. Kong and G.-W. Zheng, Nat. Commun., 2024, 15, 10330 CrossRef CAS PubMed.
- K. Wu, J. Yan, Q. Liu, X. Wang, P. Wu, Y. Cao, X. Lu, Y. Xu, J. Huang and L. Shao, Chem. Sci., 2024, 15, 1431–1440 RSC.
- S. Hussain, F. Leipold, H. Man, E. Wells, S. P. France, K. R. Mulholland, G. Grogan and N. J. Turner, ChemCatChem, 2015, 7, 579–583 CrossRef CAS PubMed.
- F. Leipold, S. Hussain, D. Ghislieri and N. J. Turner, ChemCatChem, 2013, 5, 3505–3508 CrossRef CAS.
- H. Man, E. Wells, S. Hussain, F. Leipold, S. Hart, J. P. Turkenburg, N. J. Turner and G. Grogan, ChemBioChem, 2015, 16, 1052–1059 CrossRef CAS PubMed.
- H. Man, E. Wells, S. Hussain, F. Leipold, S. Hart, J. P. Turkenburg, N. J. Turner and G. Grogan, ChemBioChem, 2015, 16, 1052–1059 CrossRef CAS PubMed.
- P. N. Scheller, S. Fademrecht, S. Hofelzer, J. Pleiss, F. Leipold, N. J. Turner, B. M. Nestl and B. Hauer, ChemBioChem, 2014, 15, 2201–2204 CrossRef CAS PubMed.
- S. Velikogne, V. Resch, C. Dertnig, J. H. Schrittwieser and W. Kroutil, ChemCatChem, 2018, 10, 3236–3246 CrossRef CAS PubMed.
- M. Prejanò, X. Sheng and F. Himo, ChemistryOpen, 2022, 11, e202100250 CrossRef PubMed.
- H. Zhou, P. Chuang, L. Xu and Q. Wu, Org. Lett., 2023, 25, 6688–6692 CrossRef CAS PubMed.
- A. R. Casamajo, Y. Yu, C. Schnepel, C. Morrill, R. Barker, C. W. Levy, J. Finnigan, V. Spelling, K. Westerlund, M. Petchey, R. J. Sheppard, R. J. Lewis, F. Falcioni, M. A. Hayes and N. J. Turner, J. Am. Chem. Soc., 2023, 145, 22041–22046 CrossRef CAS PubMed.
- P. Stockinger, N. Borlinghaus, M. Sharma, B. Aberle, G. Grogan, J. Pleiss and B. M. Nestl, ChemCatChem, 2021, 13, 5210–5215 CrossRef CAS PubMed.
- J. Zhang, Y. Ma, F. Zhu, J. Bao, Q. Wu, S.-S. Gao and C. Cui, Chem. Sci., 2023, 14, 4265–4272 RSC.
- M. Sharma, J. Mangas-Sanchez, S. P. France, G. A. Aleku, S. L. Montgomery, J. I. Ramsden, N. J. Turner and G. Grogan, ACS Catal., 2018, 8, 11534–11541 CrossRef CAS.
- G.-D. Roiban, M. Kern, Z. Liu, J. Hyslop, P. L. Tey, M. S. Levine, L. S. Jordan, K. K. Brown, T. Hadi, L. A. F. Ihnken and M. J. B. Brown, ChemCatChem, 2017, 9, 4475–4479 CrossRef CAS.
- G. A. Aleku and F. Hollfelder, Chem Catal., 2024, 4, 101160 CrossRef CAS PubMed.
- G. A. Aleku, H. Man, S. P. France, F. Leipold, S. Hussain, L. Toca-Gonzalez, R. Marchington, S. Hart, J. P. Turkenburg, G. Grogan and N. J. Turner, ACS Catal., 2016, 6, 3880–3889 CrossRef CAS.
- G. A. Aleku, J. Mangas-Sanchez, J. Citoler, S. P. France, S. L. Montgomery, R. S. Heath, M. P. Thompson and N. J. Turner, ChemCatChem, 2018, 10, 515–519 CrossRef CAS.
- M. Rodríguez-Mata, A. Frank, E. Wells, F. Leipold, N. J. Turner, S. Hart, J. P. Turkenburg and G. Grogan, ChemBioChem, 2013, 14, 1372–1379 CrossRef PubMed.
- S. P. France, G. A. Aleku, M. Sharma, J. Mangas-Sanchez, R. M. Howard, J. Steflik, R. Kumar, R. W. Adams, I. Slabu, R. Crook, G. Grogan, T. W. Wallace and N. J. Turner, Angew. Chem., Int. Ed., 2017, 56, 15589–15593 CrossRef CAS PubMed.
- Y. Li, Y. Yang, M. Zhang, X. Yue, R.-T. Guo, Z. Huang and F. Chen, ACS Catal., 2025, 15, 2192–2199 CrossRef CAS.
- J. Kyte and R. F. Doolittle, J. Mol. Biol., 1982, 157, 105–132 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The supporting information, which can be found online, contains extended data, detailed experimental procedures, Chiral HPLC chromatograms for product analysis, sequence data, and MD simulations setup strategy. See DOI: https://doi.org/10.1039/d5cy00308c |
| This journal is © The Royal Society of Chemistry 2025 |