
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple and effective preparation of highly efficient heterogeneous base catalysts†
Oleg
Kikhtyanin
 *a,
Valeriia
Korolova
b,
Evgeniya
Grechman
b,
Miloslav
Lhotka
c,
Martin
Veselý
d,
Francisco
Ruiz Zepeda
e and
David
Kubička
ab
*a,
Valeriia
Korolova
b,
Evgeniya
Grechman
b,
Miloslav
Lhotka
c,
Martin
Veselý
d,
Francisco
Ruiz Zepeda
e and
David
Kubička
ab
aUniversity of Chemistry and Technology Prague, Technopark Kralupy, nám. G. Karse 7, 278 01 Kralupy nad Vltavou, Czechia. E-mail: kikhtyao@vscht.cz
bUniversity of Chemistry and Technology Prague, Department of Petroleum Technology and Alternative Fuels, Technická 5, 166 28 Prague, Czechia
cUniversity of Chemistry and Technology Prague, Department of Inorganic Technology, Technická 5, 166 28 Prague, Czechia
dUniversity of Chemistry and Technology Prague, Department of Organic Technology, Technická 5, 166 28 Prague, Czechia
eNational Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia
First published on 16th January 2025
Abstract
The simple hydration of physically mixed Mg and Al oxides results in the formation of an LDH (layered double hydroxide) phase via the dissolution–precipitation–crystallization mechanism. Nevertheless, the change in the properties and catalytic performances of these materials with respect to the hydration conditions has not been well explained to date. Therefore, in this study, we hydrated physically mixed Mg and Al oxides at T = 25 °C or 75 °C within a time range of 5 minutes to 7 days. Subsequently, we characterized the properties of the resulting materials using an array of appropriate methods and assessed their performances in aldol condensation of furfural and acetone. An increase in the hydration time and temperature promoted the formation of the desired LDH phase. Nevertheless, the performance of the catalysts did not improve accordingly. We assumed that the surprisingly inferior performance of the LDH-rich catalysts was due to the deposition of Al species on their external solid surface instead of being incorporated in the LDH phase, and these species blocked the access of reactants to the catalytically active sites in the HTC phase, resulting in a decreased catalytic activity. Thus, the maximum activity of the catalysts was observed at a relatively low concentration of water-soluble Al species and, accordingly, at a relative content of LDH phase of 20–25%. These results are beneficial for understanding the relationship between the hydration conditions of MgO + Al2O3 physical mixtures and the properties of the derived LDH-phase containing solids and have potential in the design of green, efficient and stable solid base catalysts.
1. Introduction
The sustainability of modern society depends largely on the successful introduction of technologies based on net-zero principles.1,2 Accordingly, the use of bio-derived sources in the production of valuable products has significantly contributed to this field. Among the renewable sources, lignocellulosic biomass, especially various residual materials, is the most abundant, which can contribute to the production of higher value-added compounds. The processing of biomass by hydrolysis, fractionation, extraction, etc. yields different aldehydes, ketones, acids, phenols, etc., which are suitable substrates for further chemical transformations.3,4 However, the direct use of these bio-derived molecules is limited owing to their unsuitable composition, for example, the limited number of C atoms or large number of O atoms in these molecules. Nonetheless, they can be used as building blocks in various reactions to form larger molecules. Aldol condensation, i.e. the reaction between an aldehyde and a ketone, is a well-known reaction that results in the formation of C–C bonds and, hence, helps in valorising small bio-derived molecules.5–8 Nevertheless, the innovative design and development of efficient catalysts are essential for the development of this valorisation pathway.Heterogeneous catalysts are more sustainable than homogeneous catalysts for aldol condensation as they dramatically decrease the amount of the processed wastewater. They also offer technological benefits, such as prevention of equipment corrosion and easy separation and reusability. Among the various solid catalysts, MgAl layered double hydroxides (LDHs) have been demonstrated to be the most promising catalysts for aldol condensation owing to their high activity even under ambient conditions.9 Generally, LDHs are referred to as solids with the general formula of [M2+nM3+m(OH)2(n+m)]m+[Ax−]m/x·yH2O, where M2+ and M3+ are divalent (Mg, Ca, and Zn) and trivalent (Al, Ga, and Fe) metal cations, respectively. Usually, the M3+/M2+ ratio is close to 3, although phase-pure hydrotalcites can be synthesized in a wider range of M3+/M2+ ratios.10 In their initial carbonate form, MgAl LDHs (also known as MgAl hydrotalcites, HTCs) do not exhibit high activity in catalytic applications, such as aldol condensation. However, they can be converted into mixed oxides by calcination, which is accompanied by decomposition of their interlayer carbonates and release of CO2 and water. Consequently, Al atoms are homogeneously distributed throughout the MgO matrix framework in the formed MgAl mixed oxides, and the material possesses strong Lewis basic sites according to the CO2-TPD method, and consequently good activity in aldol condensation.11,12
The interaction of the freshly calcined MgAl mixed oxides with decarbonated water results in reconstruction of the LDH layered structure (so-called “memory effect”). The reconstructed MgAl HTCs contain exchangeable OH− hydroxyls in their interlayer and have pronounced Brønsted base character. Consequently, they possess high activity in different base-catalyzed organic reactions, including aldol condensation,7,9,13–17 which is higher than that of the corresponding mixed oxides. Nevertheless, only the sites located on the edges of LDH platelets are operative in catalytic transformations,18,19 while the interlayer basic sites in the bulk are largely inaccessible to reactant molecules. This underscores the high intrinsic activity of the accessible sites and opens immense potential to further increase the catalytic activity of LDH-derived catalysts.
Consequently, alternatives to the conventional LDH materials prepared by co-precipitation that possess only a small fraction of accessible basic sites are desirable. The focus is particularly on creating surface layers of LDH phase on suitable supports. Creasey, et al.20 impregnated Al2O3 with a magnesium methoxide solution, followed by calcination and hydrothermal treatment (at T = 125 °C with stirring for 21 h) steps. The resulting LDH/Al2O3 composites outperformed conventional bulk hydrotalcites in the transesterification of C4–C18 triglycerides to fatty acid methyl ester (FAME). This was attributed to the improved accessibility of bulky triglycerides to the active surface base sites on the large-area alumina support. Alternatively, Korolova et al.21 prepared MgAl samples with a broad range of Mg/Al ratios of 2–30 via the impregnation of an MgO support with aluminum isopropoxide solution, followed by calcination and hydration (for 20 min at room temperature). The optimized LDH/MgO catalyst (Mg/Al = 20) was more active (1.5 times higher TOF) than the conventional MgAl HTC (Mg/Al = 3–5). This was explained by the maximized efficiency of aluminum atoms to generate accessible Brønsted basic sites.
Beside deposition strategies, materials with required MgAl LDH structure can be prepared via the direct hydration of physically mixed Mg and Al oxides.22–24 In accordance with the ideas proposed by Xu and Lu,22 and later developed by other researchers,23,24 MgAl LDH materials can be prepared without using Mg and Al salts and alkaline solutions, which significantly reduces the water consumption given that washing of the counter-anions/cations from the precursors can be avoided. Xu and Lu22 suggested that MgO and Al2O3 were initially hydrated into Mg(OH)2 and Al(OH)3, respectively. Then, under the basic conditions created by the dissociation of Mg(OH)2 to Mg2+ and OH−, Al(OH)3 was transformed to Al(OH)4− species, which were deposited on the surface of Mg(OH)2/MgO. As a result, a MgAl pre-LDH material was formed and further crystallized upon continuous heating via the diffusion of metal ions in the solid lattice. Later Lee, et al.23 confirmed that the MgAl LDHs were formed by the reaction of Mg2+ and Al(OH)4−, resulted from the hydration of both metal oxides. They prepared a phase-pure LDH structure after several days of the reaction between MgO and Al2O3 at T = 60 °C or 80 °C. They also suggested that the prepared LDH-based catalysts demonstrated considerable activity in the isomerization of glucose to fructose. In turn, Warmuz and Madej24 determined the relationship between the properties of the initial metal oxide powder (susceptibility to hydration, grain size and specific surface area) and hydration kinetics. Thus, these studies show that the desired LDH phase can be formed via the simple hydration of Mg and Al oxides, and this synthetic approach shows interesting potential for the further development of catalysts or their precursors for different base-catalyzed applications directly from the respective metal oxides.
Recently, we recently proposed13 that the extra-framework or partially framework Al-rich species formed during the preparation of reconstructed phase-pure LDH materials can prevent the reactant molecules from accessing the catalytically active sites. Similarly, the formation of the LDH structure during the hydration of physically mixed Mg and Al oxides starts with the deposition of Al(OH)4− species on the surface of Mg(OH)2/MgO.22–24 Accordingly, the influence of the deposited pre-LDH phase on the physicochemical properties and catalytic performance of the resulting LDH-base material can also be assumed. Obviously, more studies are needed to identify factors that contribute to understanding the prospects of using modified LDH compounds in different applications.
For instance, the physico-chemical properties of MgAl solids prepared via the hydration of physically mixed Mg and Al oxides and the chemical composition of the liquid phase remaining after the synthesis depending on the hydration conditions have not been monitored to date. This knowledge can allow the rational design of LDH-phase containing catalysts. Moreover, a correlation between the hydrating conditions (time and temperature) and the performance of the resulting catalysts in the aldol condensation of furfural and acetone, which can help in evaluating the number of catalytically active sites, also has not been reported. Therefore, in this work, we prepared a series of LDH-based materials by hydrating physically mixed Mg and Al oxides and varying the hydration time and temperature. The properties of the different prepared solids and remaining liquid phase were determined using several characterization methods, and the performance of resulting MgAl-based catalysts was evaluated in the aldol condensation of furfural and acetone.
2. Experimental
2.1. Preparation of catalysts
A conventional MgAl hydrotalcite with LDH structure (HTC, Mg/Al = 3) was prepared using the conventional coprecipitation method, as described in detail in our previous studies.13,17,21 The produced MgAl HTC was calcined at T = 450 °C for 3 h, and the formed MgAl mixed oxide was rehydrated using deionized water under static conditions at T = 25 °C for 20 min to obtain the reconstructed MgAl HTC. Both the as-prepared and rehydrated materials were used as reference samples in the study.An alternative synthesis strategy was employed using a physical mixture of MgO and Al2O3. The hydration of the physically mixed Mg and Al oxides was carried out using commercial MgO (≥99%, ∼325 mesh, Sigma-Aldrich) and Pural SB hydrated alumina (boehmite, 75% Al2O3, Sasol Germany GmbH), unless specified otherwise. In a typical procedure, 8 g of MgO and 4.45 g of Pural SB (Mg/Al = 3) were thoroughly mixed in a porcelain mortar to get a homogeneous powder. For the hydration step, 2 g of this physical mixture was first calcined at T = 450 °C for 3 h, and then added to a 20 mL glass vial filled with deionized water. For homogeneity, the content of the vial was shaken several times. After that, the vial was kept under specific hydration conditions (temperature of either 25 °C or 75 °C and time ranging from 5 min to 7 days) without additional shaking. In a comparative experiment, the content of the vial was stirred at 400 RPM and T = 25 °C using a magnetic stirrer Witeg MSH-30D (Germany). After completing the hydration step, the resulting solid was filtered using a Buchner funnel equipped with a vacuum pump. The prepared hydrated samples were further denoted as S“T”“nt”, where S represents the sample name, T stands for the hydration temperature, n stands for the hydration time and t means either min (m), h (h), or days (d).
Additional comparative experiments on the calcination/hydration of physically mixed Mg and Al reactants were performed using Mg nitrate (Mg(NO3)2·6H2O, 99.9%, Lach:Ner, Czech Republic), Mg ethoxide (98%, Sigma-Aldrich), Al nitrate (Al(NO)3·9H2O, 98.8%, Lach:Ner, Czech Republic), Al isopropoxide (>98%, Alfa Aesar), and Al acetylacetonate (99%, Sigma-Aldrich).
2.2. Physico-chemical characterization
All the prepared samples were characterized using a comprehensive set of physico-chemical methods. The research instruments used for XRD, chemical analysis, N2 physisorption, FTIR, TGA/DTG, SEM and TEM investigations were the same as that used in our previous studies, and their specification was described elsewhere.21The relative content of crystalline components in the hydrated samples was estimated from the area of the signals in their XRD patterns at the diffraction angles (2c) of about 43°, 18.5° and 11.2° for the MgO, Mg(OH)2 and MgAl LDH phases, respectively. In this case, the initial physical mixture of Mg and Al oxides was employed as the reference sample with 100% of MgO; Mg(OH)2 prepared by the hydration of MgO at T = 25 °C for 7 days and mixed with Pural SB to get Mg/Al ratio = 3 was employed as the reference sample with 100% of Mg(OH)2; and the conventionally prepared MgAl HTC (Mg/Al = 3) was employed as the reference sample with 100% LDH.
2.3. Catalytic tests
Acetone (99.9%, penta, Czech Republic) and furfural (99%, Sigma-Aldrich) were used as the reactants in all the catalytic experiments. As-received furfural was distilled using a rotary evaporator to exclude the impact of acidic admixtures,25 and then stabilized with 2,6-di-tert-butyl-4-methylphenol (DBMP, 99%, Sigma-Aldrich) and stored in a fridge, as described previously.13,21Aldol condensation of the stabilized furfural with acetone was carried out in a 100 mL glass reactor at temperature of 25 °C and under stirring at 400 rpm at ambient pressure using a mixture of 35.0 g of acetone and 11.5 g of furfural (acetone to furfural molar ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The loss of acetone during the catalytic experiments was prevented by using a cooler-condenser above the reactor. For each experiment, 1.0 g of a starting physical mixture of Mg and Al oxides was used (the mass of a catalyst after the calcination step was about 0.95 g). The hydrated sample was immediately transferred to the reactor after the filtration step to exclude (or at least limit) the contact of the hydrated catalyst with CO2 in the air. After the addition of the catalyst, the reaction was carried out for 180 min at T = 25 °C and 400 rpm. It was previously established that the reaction was not limited by external or internal mass transfer under the chosen reaction conditions (in tests with changing stirring rate and catalyst particle size21). Liquid reaction products were periodically withdrawn from the reactor during the experiment, filtered, diluted with methanol (1:25 by volume) and analyzed using an Agilent 7820 GC unit equipped with a flame ionization detector (FID) and an HP-5 capillary column (30 m/0.32 mm ID/0.25 μm). The catalytic results from the aldol condensation of furfural and acetone were described by conversion and selectivity parameters, which were calculated as follows:12,21
:1). The loss of acetone during the catalytic experiments was prevented by using a cooler-condenser above the reactor. For each experiment, 1.0 g of a starting physical mixture of Mg and Al oxides was used (the mass of a catalyst after the calcination step was about 0.95 g). The hydrated sample was immediately transferred to the reactor after the filtration step to exclude (or at least limit) the contact of the hydrated catalyst with CO2 in the air. After the addition of the catalyst, the reaction was carried out for 180 min at T = 25 °C and 400 rpm. It was previously established that the reaction was not limited by external or internal mass transfer under the chosen reaction conditions (in tests with changing stirring rate and catalyst particle size21). Liquid reaction products were periodically withdrawn from the reactor during the experiment, filtered, diluted with methanol (1:25 by volume) and analyzed using an Agilent 7820 GC unit equipped with a flame ionization detector (FID) and an HP-5 capillary column (30 m/0.32 mm ID/0.25 μm). The catalytic results from the aldol condensation of furfural and acetone were described by conversion and selectivity parameters, which were calculated as follows:12,21
| Reactant conversion (t) (mol%) = 100 × (reactantt=0 − reactantt=t)/reactantt=0, where t stands for reaction time |
| Selectivity to product I = 100 × (mole of reactant converted to product i)/(total moles of reactant converted) |
Acetone conversion under the same reaction conditions by its self-condensation was evaluated in separate experiments. It was below 5% in all cases, and thus we excluded it from further discussion.
The carbon balance was monitored in all experiments as the total number of carbon atoms detected in each organic compound with Cn atoms (where n = 3, 5, 8…) divided by the initial number of carbon atoms in the F + Ac feed, as follows:
| C balance (%) = (3 mol C3 + 5 mol C5… + nmol Cn)/(3 mol C3 (t = 0) + 5 mol C5 (t = 0)) |
The carbon balance was ≥96% in all the catalytic experiments. Each catalytic experiment was repeated several (at least 3) times to prove the reproducibility of the obtained results (with the experimental error evaluated as ±5% relative).
3. Results and discussion
3.1. Physico-chemical characterization
Fig. 1 depicts the XRD patterns of the materials obtained by the hydration of the calcined MgO + Al2O3 physical mixture under different hydration conditions (temperature was either 25 °C or 75 °C, time varied from 5 min to 7 days). Only a few comparative samples were selected for illustrative purposes. Keeping the suspension at T = 25 °C for only 5 min resulted in the partial transformation of MgO to Mg(OH)2, as evidenced by the appearance of reflections at 2θ ≈ 18.7°, 38°, and 51° in the XRD pattern of the S25-5m sample (Fig. 1A). At the same time, reflections at 2θ ≈ 11.5°, 23°, 34.8° also appeared in the XRD pattern of this sample, thus evidencing the formation of the LDH phase after 5 min of hydration treatment. After 1 day hydration, the XRD reflections from the MgO phase almost disappeared (S25-1d). Instead, the intensity of the reflections from the Mg(OH)2 phase increased significantly (Fig. 1A). The relative content of the LDH phase in S25-1d also increased, as could be judged by the change in the intensity of the corresponding reflections. A further increase in the duration of the hydration treatment to 3 and 7 days at T = 25 °C resulted in a minor increase in the intensity of the reflections from the LDH phase and corresponding decrease in the intensity of the reflections from Mg(OH)2 in the XRD patterns of S25-3d and S25-7d, respectively (Fig. 1A).
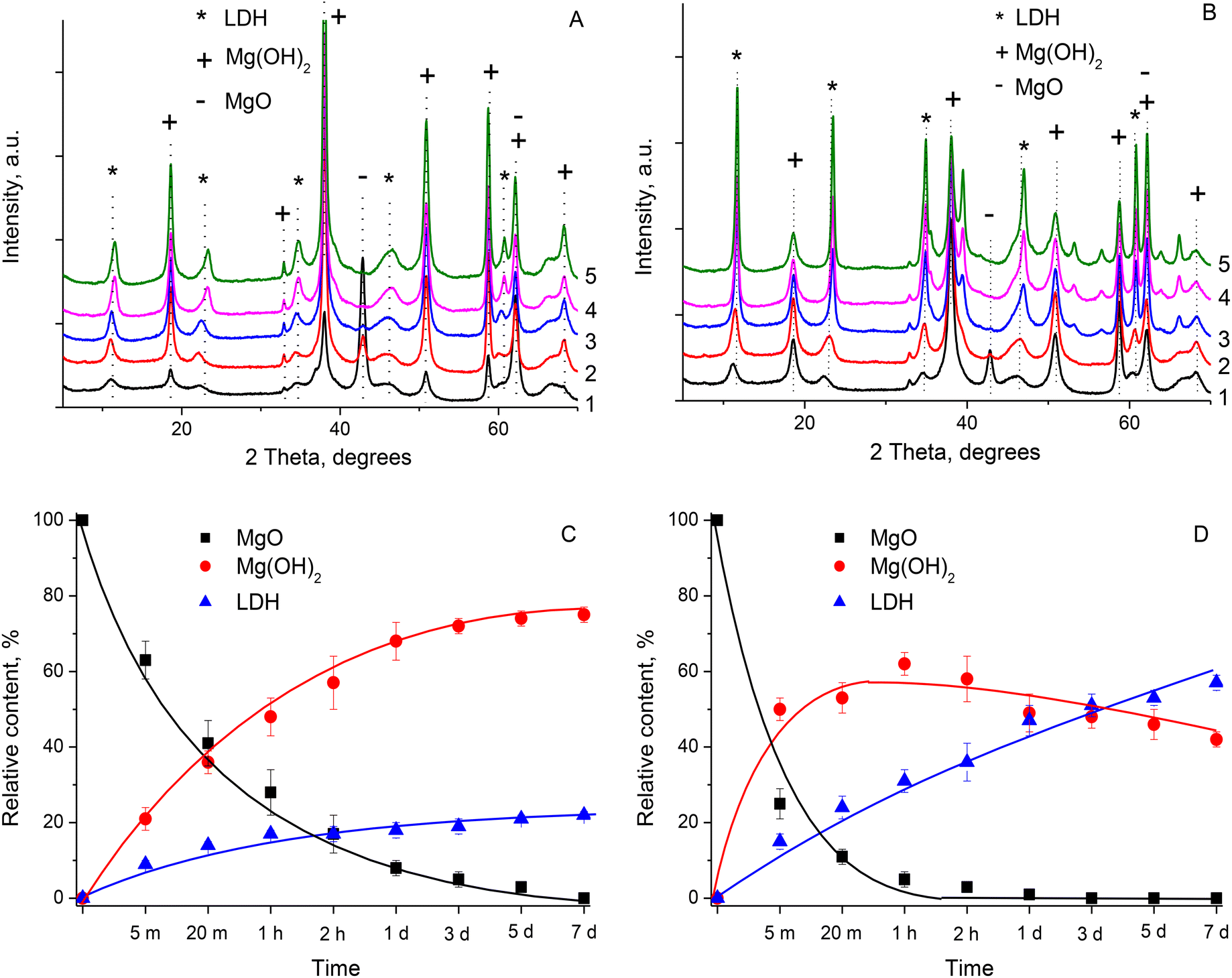 | ||
| Fig. 1 XRD patterns of materials prepared by the hydration treatment of pre-calcined MgO + Al2O3 physical mixture at T = 25 °C (A) and 75 °C (B). Hydration time: 1–5 minutes; 2–2 hours; 3–1 day; 4–3 days; and 5–7 days. Relative content of crystalline phases in the samples prepared at T = 25 °C (C) and 75 °C (D). | ||
An increase in the treatment temperature from T – 25 °C to 75 °C favoured the faster transformation of the starting MgO + Al2O3 mixture to Mg(OH)2 and LDH phases, as evidenced by the change in the intensity of the corresponding reflections in the XRD patterns of the samples prepared at T = 75 °C (Fig. 1B). After 2 h of hydration treatment at 75 °C, the reflections from the MgO phase almost disappeared (S75-2h). This was accompanied by an increase in the intensity of the reflections from the Mg(OH)2 and LDH phases. With a further increase in the hydration time, the intensity of the reflections from Mg(OH)2 reached the maximum, while the intensity of the reflections from the LDH phase gradually increased and reached the maximum after 7 days (Fig. 1B).
Fig. 1C and D demonstrate the change in the relative content of crystalline phases in the prepared samples as a function of the hydration time at T = 25 °C and T = 75 °C, respectively (here the results obtained upon repeating the experiments are shown with error bars, where the deviation in the values reflects the inhomogeneity of the mixture). At T = 25 °C, the relative content of the MgO phase successively decreased from the initial 100% to zero after 7 days-long hydration (Fig. 1C). At the same time, the relative content of the Mg(OH)2 phase gradually increased to about 75%. The relative content of the LDH phase increased to about 10% after 5 min, followed by a moderate increase to about 22% after 7 days. At T = 75 °C, the relative content of the MgO phase was negligible after 2 h of reaction, while the relative content of the Mg(OH)2 phase was about 60% after 1–2 days. The relative content of the LDH phase gradually increased to 56–60%, while the relative content of Mg(OH)2 decreased to 39–44% after 7 days of treatment (Fig. 1D). The total percentage of Mg-containing phases, i.e. MgO + Mg(OH)2 + LDH, was in the range of 87–103%, thus proving the correctness of the approach used. This also evidenced that Mg was present in the amorphous phase only marginally (if at all). In contrast, the presence of Mg(OH)2 in the XRD pattern of S75-7d indicated that Al was partially present either in the amorphous phase or HTC phase with an Mg/Al atomic ratio below 3 (Fig. 2).
 | ||
| Fig. 2 Correlation between the relative content of LDH phase in the prepared samples and the value of the basal spacing between LDH layers d(003). | ||
The mechanism of Mg–Al LDH formation by hydrating the mixture of Mg and Al oxides was proposed previously.22,23 According to the proposed scheme, Mg(OH)2 was first formed on the surface of MgO during the hydration process, and subsequently dissociated into Mg2+ and OH−. In turn, Al2O3 was hydrated to Al(OH)3, and then created Al(OH)4− species in a basic solution, which resulted from the partial dissociation of Mg(OH)2. Xu and Lu22 suggested that under basic conditions, the deposition of Al(OH)4− species on the Mg(OH)2 lattice occurred, thus forming the surface MgAl pre-LDH phase. The simultaneous bilateral diffusion of Mg and Al homogenized the cation distribution, producing a well-defined LDH phase. Thus, the XRD results obtained in our study agreed well with the proposed scheme of LDH formation.22,23 At first, MgO was hydrated to Mg(OH)2, and this initiated the Mg(OH)2 → Mg2+ + OH− dissociation, as reflected in the fast increase in the pH value of the reactive aqueous mixture to about 10. Consequently, Al(OH)4− species were also formed at the beginning of the reaction. Its deposition on the Mg(OH)2 surface resulted in the formation of the LDH phase with the relative content of 10% after 5 min of treatment at T = 25 °C (Fig. 1A and C). Nevertheless, the relative content of the Mg(OH)2 phase was above 30% and the relative content of the LDH phase did not exceed 65% even after 7 days of reaction at T = 75 °C. It can be assumed that the newly formed surface LDH phase prevented the diffusion of Al species into the bulk of the MgO/Mg(OH)2 material, similar to the suggestion by Xu and Lu.22 As evidenced in Fig. 1, the diffusion of Al species promoted the formation of the LDH phase, which was more limited at a low hydration temperature. In contrast, Lee, et al.23 successfully prepared phase-pure LDH samples by performing the hydration step for several days (usually 7–9 days) at T = 60–80 °C. The difference in the phase composition of the samples prepared in our study and in the study by Lee et al.23 can be explained by both the difference in reaction conditions and reactivity of the initial Mg and Al sources, as suggested by Warmuz and Madej.24
Based on results from earlier studies,12,26,27 the diffraction peak at 2θ ≈ 11.5° ascribed to the (003) reflection was used to calculate the basal spacing between the LDH layers d(003) in the prepared samples. Fig. 2 demonstrates that the calculated d(003) values almost linearly decreased with an increase in the relative content of the LDH phase. In case of the samples prepared at T = 25 °C, the moderate increase in the relative content of LDH phase from about 10% to about 20% resulted in a decline in the d(003) value from 7.90–7.95 Å to 7.65 Å, while in the case of the samples prepared at T = 75 °C (Fig. 2), the linear trend was more gentle. Specifically, the d(003) value decreased to below 7.60 Å only when the relative content of the LDH phase was above 40% (Fig. 2). Similar to the calculations for the conventionally prepared 3MgAl HTC (above), the d(003) value was used to evaluate the actual Mg/Al ratio in the platelets of the LDH phase. The d(003) value of about 7.90–7.95 Å observed for samples with the small relative content of LDH phase corresponded to the actual Mg/Al ratio in the LDH layers in the range of 4–5 (Fig. 2). This evidenced that at the beginning of the reaction, the LDH phase was deficient in Al atoms. With an increase in the hydration time, the d(003) value decreased to 7.65 Å and to 7.55–7.60 Å for the samples prepared at T = 25 °C and 75 °C, respectively (Fig. 2). This corresponds to the increase in the actual number of Al atoms in the LDH platelets, and thus the decline in the Mg/Al atomic ratio to 1.5–2.5 or even lower (Fig. 2). These calculations suggested that with an increase in the hydration time, Al atoms substituted Mg atoms in the already formed LDH phase, rather than participating in the formation of new LDH regions. This was most evident for the samples prepared at T = 25 °C with a small relative content of LDH phase. It also agreed with the suggestion that unreacted MgO/Mg(OH)2 was buried underneath the freshly formed MgAl LDH.22 Thus, the actual Mg/Al ratio below 3 (i.e. the Mg/Al ratio in initial reactive mixture) in the formed LDH platelets evidenced that the Mg(OH)2 phase partially remained intact even with a longer hydration time because of the non-homogeneous distribution of Al atoms. Consequently, the XRD results indicate that the newly formed LDH phase is present on the surface of the MgO/Mg(OH)2 particles where it should be well accessible to the aldol condensation reactants.
The pH value of the aqueous phase increased from about 7 to 9.5–9.8 just after adding the MgO + Al2O3 mixture to water. With an increase in the treatment time, the pH value of the aqueous phase increased only moderately to 10.0–10.2. Both Mg and Al were detected in the filtrate liquids, and their concentration depended on the hydration conditions (Fig. 3A and B). In the experiments at T = 25 °C, the concentration of Mg reached the maximum value of 1.5 mmol L−1 (63 mgMgO L−1) in the filtrate obtained after 5 min hydration (Fig. 3A). This value was very close to the solubility of MgO in water at room temperature (86 mg L−1) reported by Huang et al.28 When increasing the hydration time, the Mg concentration decreased to 0.6–0.9 mmol L−1 independent of the hydration temperature, reflecting the gradual transformation of MgO to Mg(OH)2, which has significantly lower solubility at room temperature of 12 mg L−1.28 At the same temperature, the concentration of Al did not exceed 0.2 mmol L−1, but with a definite increasing trend as the hydration time increased (Fig. 3A). These results evidenced the very small dissolution of Al oxide in aqueous medium at T = 25 °C and pH of about 10, even at prolonged hydration, and this was consistent with the low relative content of LDH phase in the resulting samples. In the experiments at T = 75 °C, the concentration of Mg also decreased from about 1.0 mmol L−1 after 5 min to below 0.4 mmol L−1 after hydration for 1 h (Fig. 3B), thus following the trend observed in the experiments at T = 25 °C (Fig. 3A). However, at longer hydration times, the concentration of Mg gradually increased to about 1.7 mmol L−1 after 7 days of reaction (Fig. 3B). This change was likely caused by the change in the phase composition of the samples prepared at T = 75 °C (Fig. 1). The concentration of Al was very low (below 0.1 mmol L−1) at short hydration times (5–20 min), but it substantially increased to the value of 2.2 mmol L−1 after 7 days. The observed high concentration of both elements in the aqueous phase was plausibly related to the growth in the relative LDH content in the samples prepared at T = 75 °C. Indeed, Bernard et al.29 found that the Al concentration in aqueous solution was 0.36 mmol L−1 in the case of carbonated MgAl HTC, but it increased to 11 mmol L−1 for the MgAl HTC with hydroxyls as interlayer anions. We also performed a series of repeated experiments for the determination of Mg and Al in the aqueous phase, which remained after the rehydration of the 3MgAl mixed oxide prepared by the calcination of the conventional 3MgAl HTC. The concentration of Mg and Al in the aqueous phase after 20–60 min of the rehydration step was 22.5–38.8 mmol L−1 and 7.5–13.3 mmol L−1, respectively. The Mg/Al ratio of the water-soluble species detected in the aqueous phase was about 3, i.e. close to the value in the 3MgAl mixed oxide used in the rehydration step. Nevertheless, the Mg and Al concentration determined in the aqueous phase after the rehydration of the conventional 3MgAl mixed oxide was substantially larger than the Mg and Al concentration in the filtrate liquids remaining after the hydration of the MgO + Al2O3 physical mixture because of the difference in the content of LDH phase in the hydrated products.
 | ||
| Fig. 3 Concentration of Mg and Al determined by AAS in the aqueous phase during the hydration of MgO + Al2O3 mixture at T = 25 °C (A) and 75 °C (B) as well as change in the Mg/Al ratio in the aqueous phase (C) as a function of hydration time. Dependence of Mg/Al ratio in the LDH phase (evaluated from d003 values) on the Mg/Al ratio in the aqueous phase (D). | ||
The Mg/Al ratio in the aqueous phase as a function of the hydration time was also calculated. Fig. 3C shows that the initial Mg/Al ratio was about 30 in the aqueous phase after 5 min at T = 25 °C. It gradually decreased to 4.7–7.7 as the reaction was continued for 1–7 days. The initial Mg/Al ratio in the experiments at T = 75 °C was about 12, but it decreased to about 1 for hydration times in the range of 1–7 days (Fig. 3C). The low Mg/Al ratio in the aqueous phase at longer hydration times contributed to the formation of the LDH phase enriched in Al, as can be inferred from the decreased d(003) values (Fig. 2). Indeed, the actual Mg/Al ratio in the LDH phase (evaluated from d(003) values) correlated well with the Mg/Al ratio determined in the aqueous phase (Fig. 3D).
As evidenced in Fig. 3A, the formation of the water-soluble Al(OH)4− species was limited at T = 25 °C, but even this low Al concentration was sufficient for the formation of the MgAl LDH phase (Fig. 1A and C). The concentration of soluble Al species was also low at T = 75 °C at the beginning of the reaction, as shown in Fig. 3B, but an increase in the hydration temperature favoured the deposition of the Al(OH)4− species on the surface of Mg(OH)2 and the diffusion of Al to the bulk of Mg(OH)2 with the formation of the MgAl LDH phase.22,23 At the same time, the increase in the relative content of MgAl LDH phase in the solids induced an increase in the concentration of Mg and Al in the aqueous phase (Fig. 3B), which further contributed to their interaction with the surface of Mg(OH)2 and the formation of the MgAl LDH phase enriched in Al (Fig. 2). In total, the Mg/Al ratio in the aqueous and solid phases were interrelated, where the larger the concentration of Al(OH)4− anions in the solution (Fig. 3A and B), the more effective the interaction of Mg and Al species with the formation of the MgAl LDH phase (Fig. 1), the lower the Mg/Al ratio in this phase (Fig. 3C), and the larger dissolution of this phase in the aqueous medium.
Fig. 4S (see the ESI†) depicts the FTIR spectra of MgO and Al2O3, as well as their physical mixture (Mg/Al = 3) before and after calcination at 450 °C. The FTIR spectrum of MgO is consistent with the data reported for similar materials.35–37 Both H2O and CO2 molecules are easily chemisorbed on the surface of MgO when exposed to open air.38 This is why we pre-calcined the oxides before performing a hydration step. The FTIR spectrum of the commercial Al source with a boehmite structure was also in accordance with the results obtained for similar materials.39,40 The FTIR spectrum of the MgO + Al2O3 mixture represents a superposition of the signals observed in the FTIR spectra of its individual oxides. As expected, the calcination of the mixture resulted in the decomposition of the surface OH groups and removal of the surface water molecules. This was reflected in the decrease in the intensity of the FTIR signals (Fig. 4S in the ESI†).
Fig. 4 depicts the FTIR spectra of the samples prepared by hydration of the physically mixed and calcined 3MgAl oxides at different temperatures and treatment times (only selected spectra are shown). The spectra of the samples prepared at T = 25 °C contained signals at 3700, 3470, 1630, 1485 and 1408 cm−1 (Fig. 4A). The sharp band at 3700 cm−1 is characteristic of Mg–OH groups, which agrees with the XRD evidencing the presence of the Mg(OH)2 phase in the prepared samples (Fig. 1A). The bands at 1408 cm−1 and 1485 cm−1 are attributed to the different forms of carbonates CO32− directly interacting with Mg2+ cations,13,21,26,34 which are also consistent with the XRD data, evidencing the presence of MgO and Mg(OH)2 (Fig. 1A). Other bands, such as the broad band at about 3400 cm−1 attributed to the stretching mode of hydrogen-bonded hydroxy groups from the brucite-like layer and interlayer water,13,21,26,34 confirmed the presence of the LDH phase in the samples hydrated at T = 25 °C. Besides, the band at about 1630 cm−1 evidenced presence of water molecules in the LDH interlayer space. Nevertheless, the characteristic band at about 1370 cm−1 typical for interlayer carbonates as charge-compensating anions between LDH platelets13,21,26,34 was present in the spectra only as a shoulder to the main band at 1408 cm−1 in the samples hydrated for 20 and 120 min, but it became more evident in the sample hydrated for 24 h (Fig. 4(A)). The low intensity of the characteristic band at 1370 cm−1 reflected the small content of LDH phase in the samples prepared at T = 25 °C independent of the hydration time, as indicated by the XRD data (Fig. 1A and C).
 | ||
| Fig. 4 FTIR spectra of samples prepared by hydration of 3MgAl mixed oxide at T = 25 °C (A) and T = 75 °C (B) as a function of hydration time. | ||
The FTIR profiles changed when the hydration temperature was increased to 75 °C (Fig. 4B). The band at 1370 cm−1 representing interlayer carbonates became more expressed, and also increased in intensity with an increase in the hydration time. The intensity of the broad band centered at near 3430 cm−1 also increased significantly, reflecting an increased content of hydrogen-bonded hydroxy groups connected with the brucite-like layers.13,21,26,34 Thus, the observed changes in the intensity of the bands were consistent with the XRD data, which showed that an increase in both the hydration temperature and hydration time increased the relative content of LDH phase in the prepared samples. The sharp band at about 785 cm−1 that appeared in the FTIR spectra of the samples prepared at T = 75 °C for 24 h can be assigned either to the lattice vibrations associated with hydroxide sheets,41 the translation vibration mode of the hydroxyl groups influenced by the Al3+ ions,34 or the stretching vibrations of Al–O–Al in the distorted AlO6.42 Additionally, the appearance of a band at about 550 cm−1 in the FTIR spectrum of the S75-1d sample reflects the presence of the boehmite AlO(OH) structure, as reported previously.42 The appearance of bands at about 785 cm−1 and 550 cm−1 in the FTIR spectra of the hydrated samples indirectly supports the existence of extra-framework Al-rich species formed by the deposition of the water-soluble Al(OH)4− species on the surface of Mg(OH)2, as discussed above (chapter 3.1.2.). Nevertheless, this suggestion may require additional FTIR experiments to be performed with the adsorption of specific probe molecules, which is outside the scope of the present study.
According to the XRD data, S25-2h consisted predominantly of Mg(OH)2 with a low content of LDH phase and residual MgO (Fig. 1A and C). The SEM image of this sample evidenced that the hydration of the starting MgO + Al2O3 mixture resulted in the transformation of MgO small particles to well-defined hexagonal platelets with the size of 0.3–1 μm (Fig. 5A). Considering the XRD data for this sample, these small particles were Mg(OH)2 platelets, which were formed by MgO hydration. A similar morphology of Mg(OH)2 particles was reported previously.43,44 Additionally, Fig. 5A shows that the surface of some hexagonal particles became defective with multiple cracks, which was likely a consequence of the incorporation of Al atoms into the Mg(OH)2 structure and the formation of the LDH phase.
 | ||
| Fig. 5 SEM images of S25-2h (A), S25-1d (B), S25-7d (C), S75-2h (D), S75-1d (E) and S75-7d (F). | ||
The morphology of S25-1d remained virtually the same as that of S25-2h (Fig. 5B), which consisted of numerous hexagonal platelets with only several of them having defective and cracked surface. This also agrees with the XRD data, where an increase in hydration time at this temperature did not result in a significant increase in the relative content of LDH phase.
After 7 days of hydration at T = 25 °C, the hexagonal arrangement of the platelets in S25-7d was maintained, while the number of the defective particles increased (Fig. 5C). Besides, the individual platelets, as clearly visible in the images of S25-2h and S25-1d (Fig. 5A and B, respectively), became more closely packed as if glued together (Fig. 5C).
According to XRD, the hydration of the initial mixture at T = 75 °C for 2 h resulted in the total consumption of MgO with the corresponding formation of Mg(OH)2 and LDH phases with their relative content of about 40% and 35% (Fig. 1B and D), respectively. It can be inferred that the agglomerated particles visible in the SEM image in S75-2h were composed of individual platelets having Mg(OH)2 and LDH compositions (Fig. 5D). In addition, these platelets became apparently thinner and more curved, thus reflecting the change in their phase and chemical composition in comparison with the samples prepared at T = 25 °C.
With an increase in the hydration time to 1 day at T = 75 °C, SEM image of S75-1d substantially changed (Fig. 5E). Individual platelets became less expressed due to their stacking, thus forming agglomerated particles with a size above 1 μm. Alternatively, with an increase in the hydration time, the phase composition of the samples did not change significantly (Fig. 1B and D). Thus, it can be assumed that the dense packing of hexagonal platelets visible in the SEM image of the S75-1d (Fig. 5E) may limit the access of Al atoms to the bulk of the material, thereby preventing the more efficient conversion of residual Mg(OH)2 to LDH phase. In addition, the chemical analysis of the aqueous phase after hydration experiments at T = 75 °C evidenced a high concentration of water-soluble Mg and Al species (Fig. 3B). Therefore, the visible adhesion of the individual platelets to each other can be attributed to the deposition of the water-soluble species on the surface of the solid phase, thus forming a surface non-crystalline layer. Additionally, the deposited non-structural Al atoms could not be incorporated into the crystalline LDH structure because the Mg/Al ratio in the already formed LDH phase was in the range of 1.5–2.5 (Fig. 2), i.e. already lower than necessary for the formation of the desired phase.
This hypothesis is further supported by the SEM image of the S75-7d sample (Fig. 5F). The individual platelets became even more shapeless and densely packed, with a lot of white extensive non-crystalline layer on the surface of the platelets. Considering the high concentration of water-soluble Mg and Al species after the experiment under these hydration conditions (Fig. 3B) and the limitation of the incorporation of Al species into the LDH crystalline framework, the deposition of the X-ray amorphous layer on the surface of the crystalline phase could not be excluded.
The hydration of this mixture at T = 25 °C for 2 h changed the TEM image of S25-2h (Fig. 6A). Instead of packed and dense particles visible in the image of the physically mixed 3MgAl oxides (Fig. 6S in the ESI†), individual particles with a length of 0.5–1 μm and thickness of several tens of nanometers were detected. Considering the XRD data (Fig. 1), the individual particles possessed an Mg(OH)2 structure, although the LDH platelets also present in S25-2h could not be distinguished because both possessed a layered structure. The increase in hydration time at this temperature did not result in any significant change in the morphology of S25-1d, although individual platelets were more visible (Fig. 6B). The similarity in the TEM images of S25-2h and S25-1d reflected the small difference in the phase composition of these samples, as evidenced by the XRD data (Fig. 1A and C).
 | ||
| Fig. 6 TEM images of S25-2h (A), S25-1d (B), S75-2h (C) and S75-1d (D). | ||
The increase in both the hydration temperature and hydration time promoted the growth in the content of LDH phase in the hydrated materials (Fig. 1B and D). Accordingly, the morphology of the samples also changed after hydration at T = 75 °C. Fig. 6C shows that the individual particles visible in the TEM image of S75-2h became more detectable in comparison with the samples prepared at 25 °C (Fig. 6A and B), respectively. The observed change in the image of the platelets can be explained by the increase in the contribution from a component with LDH structure to the total phase composition of the samples prepared at T = 75 °C (Fig. 1). The detection of individual platelets was further improved with an increase in the hydration time, as evident from the image of S75-1d (Fig. 6D). It can be inferred that the needle-like particles clearly visible in Fig. 6C and D were characteristic of the component with an LDH structure, although the presence of Mg(OH)2 particles could not be excluded, as evidenced by the different lattice spacings in the TEM images of S75-2h (Fig. 7S in the ESI†) and supported by XRD data (Fig. 1).
3.2. Catalysis
The general scheme for aldol condensation between furfural and acetone is described in Scheme 1.33 | ||
| Scheme 1 Reaction route of aldol condensation between furfural and acetone.33 | ||
Both conventionally prepared 3MgAl HTC catalysts (rehydrated at either 25 °C or 75 °C for 20 min) demonstrated an excellent performance in the aldol condensation of furfural and acetone with the furfural conversion of above 90% after 40 min of reaction (Fig. 8S(A), see the ESI†). The increase in the rehydration temperature from 25 °C to 75 °C resulted in an increase in furfural conversion. These results are in accordance with that obtained in our previous studies using similar materials.13,21,33
FAc–OH, FAc and F2Ac were the main reaction products, and the change in their selectivity depending on furfural conversion was similar to that observed in earlier studies.13,21,33 The selectivity for FAc–OH decreased from above 75% to below 20% with an increase in furfural conversion, while the selectivity for both FAc and F2Ac correspondingly increased. This was especially expressed when the furfural conversion approached 100% (Fig. 8S(B) in the ESI†).
The furfural conversion over individual MgO and Al2O3, their physical mixture and hydrated MgO was very low even after 180 min (Fig. 9S, in the ESI†). Among these oxides, MgO demonstrated the highest furfural conversion of about 6% because of its basic character,12 while Al2O3 was the least active.
The hydration of the physical mixture resulted in a significant increase in furfural conversion over the resulting catalysts. The performance of the catalysts hydrated at T = 25 °C and 75 °C for 5 min up to 7 days in the aldol condensation of furfural and acetone is summarized in Fig. 10S (in the ESI†). For comparison, Fig. 7 presents the furfural conversion over the hydrated physical mixture only after 60 min of reaction.
 | ||
| Fig. 7 Furfural conversion after 60 minutes of aldol condensation over MgAl catalysts (atomic ratio Mg/Al = 3) hydrated at T = 25 °C (A) and 75 °C (B) as a function of hydration time. The mass of the MgAl physical mixture used for the calcination/hydration steps was 1.0 g. Reaction conditions: Treac. = 25 °C, furfural mass = 11.5 g, acetone mass = 35 g, F:Ac = 1:5 (mol/mol), and RPM = 250. | ||
In the case of the samples hydrated at 25 °C, the furfural conversion gradually increased from 10% to 83% with an increase in the hydration time from 5 min to 1 day (Fig. 7A). Fig. 9S (in the ESI†) evidences that the furfural conversion over the calcined oxides and their physical mixture was very poor, and thus the significant improvement in the performance of the catalysts was attributed to the change in their phase composition due to their hydration, i.e. the formation of LDH phase (Fig. 1C). Considering the data on the physico-chemical characterization of the hydrated mixed oxides, one can assume that the performance of the active sites in the LDH phase in the hydrated samples was similar to that in the conventional rehydrated MgAl HTC catalysts. Nevertheless, a further increase in the hydration time from 1 day to 7 days at 25 °C resulted in a decline in furfural conversion from 83% to 55% (Fig. 7A), which did not correlate with the relative content of LDH phase (Fig. 8A).
 | ||
| Fig. 8 Change in furfural conversion observed over catalysts hydrated at T = 25 °C or 75 °C after 60 minutes of aldol condensation depending on the relative content of LDH phase in the samples (A) and concentration of water-soluble Al species (B). | ||
Furfural conversion of over 82% after 60 min of reaction was observed on the catalyst hydrated at T = 75 °C for only 5 min (Fig. 7B). Nevertheless, the positive effect due to the increase in hydration time, and consequently the increase in the relative content of the LDH phase in the hydrated samples were limited to the time range of 5 to 20–60 min at this temperature. Nonetheless, it resulted in only a small increase in furfural conversion from 82% to 88–89% (Fig. 7B). The further increase in hydration time dramatically decreased the furfural conversion to 34% over the catalysts hydrated for 5 and 7 days at T = 75 °C (Fig. 7B). Similar to the samples hydrated at T = 25 °C, no correlation was observed between the performance of the catalysts hydrated at T = 75 °C and their relative content of LDH phase (Fig. 8A).
Fig. 11S (see the ESI†) demonstrates the change in product selectivity with furfural conversion observed over the catalysts hydrated at 25 °C and 75 °C. Regardless of the hydration time, the change in the composition of the reaction products exhibited a similar trend, where the FAc–OH selectivity decreased, while both the FAc and F2Ac selectivity increased with an increase in furfural conversion. Thus, it can be assumed that the characteristics of the active sites in the hydrated catalysts were similar without any dependence on either time or temperature of hydration. For example, independent of the hydration conditions, the selectivity for FAcOH, FAc and F2Ac was in the range of 73–80%, 9–12% and 8–13%, respectively, at the furfural conversion of about 60% (Fig. 11S in the ESI†).
The performance of the catalysts prepared by the hydration of the MgO + Al2O3 mixture was compared with that observed for the rehydrated catalysts prepared using the conventional 3MgAl HTC by calculating the initial specific activity, i.e. mmol of furfural converted per mass of a calcined mixed oxide (before a (re)hydration step) per time, A = mmolF gcat.−1 min−1. For this purpose, the catalytic results obtained for the conventional rehydrated 3MgAl HTCs (Fig. 8S in the ESI†) and the most active hydrated MgO + Al2O3 catalysts, i.e. S25-24h and S75-20m, were used.
The initial activity of the conventional rehydrated MgAl HTC catalysts was 16.9–21.3 mmolF g−1 min−1 (Table 1). It decreased to 7.9–8.5 mmolF g−1 min−1 in the case of the hydrated MgAl samples (Table 1), i.e. they exhibited 2–2.7-times lower activity than the rehydrated conventional HTCs. However, it needs to be considered that the samples prepared from the physical mixture consisted largely of inactive oxides and Mg(OH)2. Then, we suggest that the performance of the prepared catalysts can explained by presence of active sites in the LDH phase and that the properties of the active sites in both the conventionally prepared MgAl HTCs and hydrated MgAl mixed oxides are similar. Of course, there is no direct evidence that the reaction rates scale with the number of accessible sites in the LDH phase. Nevertheless, it can be assumed that the activity of a catalyst is proportional to the number of accessible active sites in the LDH phase. Thus, it can be inferred that the number of accessible active sites is 2–2.7-times lower in the catalysts prepared by the hydration of the MgO + Al2O3 mixture. Accordingly, the relative content of the LDH phase in the catalysts should be decisive for the catalyst activity. Of course, this statement is valid if the accessibility of the active sites in the LDH phase is comparable. Consequently, it is related to the characteristics of the precursors employed in the calcination/(re)hydration steps, i.e. the distribution of Mg and Al atoms in the samples. The calcination of the conventionally prepared MgAl HTC resulted in the formation of an MgAl mixed oxide, which represents a solid solution with Al atoms nearly homogeneously distributed in the MgO structure. Hence, the rehydration of the mixed oxide resulted in the fast reorganization of its crystalline framework to the original LDH structure (so-called “memory effect”).13,34 Accordingly, the generation of interlayer anions in the newly formed LDH structure was also very fast. In the case on the freshly rehydrated HTC samples, these interlayer anions are hydroxyls, which represent catalytically active sites for base-catalyzed reactions, although only those accessible to the reactants, i.e. those located at the edges of the LDH platelets, are operative in the catalytic transformations.18,19 In contrast, the hydration of the MgO + Al2O3 mixture and the formation of the LDH phase occurred through the formation of water-soluble Mg and Al species, followed by the formation of an LDH structure. This process is slower than the reconstruction process in the conventional samples, and after the hydration of the oxidic mixture at T = 25 °C, the relative content of the LDH phase barely exceeded 20% even after treatment for 7 days (Fig. 1C). Thus, according to the catalytic data, the LDH component in the hydrated samples contained active sites similar to that found in the conventional reconstructed 3MgAl HTCs, as mentioned above and proven by the results in Fig. 11S in the ESI.† Consequently, these sites catalyze the aldol condensation of furfural and acetone at a high rate. Moreover, when expressing (normalizing) the catalytic activity per content of LDH phase (Fig. 1C and D), the intrinsic activity of the hydrated oxide catalysts exceeds that of the conventional rehydrated HTCs (Table 1). This indicates that the hydrated MgAl catalysts have more active sites in the LDH phase accessible to reactant molecules, i.e. an increase in the number of sites at the LDH edges with respect to the overall number of sites.
| Sample | (Re)hydration conditions | A, mmolF gcat.−1 min−1 | A norm.,a mmolF gcat.−1 min−1 |
|---|---|---|---|
| a A norm. – normalized specific initial activity recalculated based on the LDH phase content in the samples as estimated from the XRD data. | |||
| HTCconv. | 25 °C, 20 min | 16.9 | 16.9 |
| HTCconv. | 75 °C, 20 min | 21.3 | 21.3 |
| MgAl25-24h | 25 °C, 24 hours | 8.5 | 51.5 |
| MgAl75-20m | 75 °C, 20 min | 8.2 | 41.7 |
The increase in hydration time contributed to an increase in the relative content of LDH phase, which was especially evident at T = 75 °C (Fig. 1C and D). Nevertheless, this increase was not reflected in the corresponding increase in the activity of the catalysts (Fig. 8A), even assuming that the characteristics of the active sites remain the same, which is consistent with the comparable product selectivity at equal furfural conversion (Fig. 11S in the ESI†). The decline in furfural conversion clearly indicates a decrease in the number of accessible active sites. Considering the ratio between the maximum furfural conversion obtained and that observed after 7 days (Fig. 7), the number of accessible active sites decreased by 30% and 60% for the samples hydrated at T = 25 °C and T = 75 °C, respectively. The discrepancy between the phase composition and catalytic performance of the hydrated samples can be explained by considering the chemical analysis of the aqueous phase during the hydration of the Mg and Al oxides (Fig. 3A and B). It is postulated that the formation of the LDH phase by hydrating the mixture of Mg and Al oxides proceeds through the formation of water-soluble Mg and Al species.22–24 At T = 25 °C, the relative content of LDH phase was about 10% just after 5–20 min of treatment (Fig. 1C). This means that even the relatively low concentration of Al species in the aqueous phase, i.e. 0.05–0.07 mmol L−1 (Fig. 3C), is sufficient to initiate the fast formation of the desired LDH phase. The increase in the hydration time to 7 days increased the Al concentration in the aqueous phase to about 0.2 mmol L−1 (Fig. 3A), i.e. in 3–4 times; nevertheless, the relative content of the LDH phase only increased to 20–22%, i.e. only twice, with an increase in the Al content in the LDH platelets (Fig. 2). Accordingly, an increasing amount of water-soluble Al species remained unutilized for the formation of the LDH phase by their interaction with Mg(OH)2. At T = 75 °C, the difference in the Al concentration in the aqueous phase and the relative content of the LDH phase was even more significant. After hydration for 1 h, the Al concentration in the aqueous phase of 0.13 mmol L−1 (Fig. 3B) resulted in the formation of about 30% of LDH phase (Fig. 1D). After 7 days, the Al concentration increased to 2.2 mmol L−1, i.e. about 15 times, but the relative LDH content increased to about 60%, i.e. only twice. This means that the concentration of soluble species is larger than that required for the effective participation of Al atoms in the formation of the LDH phase. Thus, the formation of the LDH phase is determined not only by the concentration of the water-soluble Mg and Al species, but also by the efficiency of their participation in the formation of the desired LDH phase. Xu and Lu22 suggested that during the hydration process, the deposition of Al(OH)4− on the surface of Mg(OH)2 results in the formation of the MgAl pre-LDH phase. After that, the simultaneous bilateral diffusion homogenizes the cation distribution, thus forming a well-defined LDH phase. Nevertheless, this diffusion is a slow process, and thus some non-hydrated MgO is buried underneath the freshly formed MgAl LDH, as observed by XRD. In summary, at a low hydration temperature and time, the limited simultaneous diffusion of elements is equilibrated with their low concentration in the aqueous phase. Consequently, the furfural conversion after 1 h of reaction initially increases (Fig. 8B), i.e. as long as the Al atoms effectively participate in the formation of the desired LDH phase. As the hydration time increases, the excessive Al species are deposited on the surface of the newly formed LDH layer rather than diffusing into the bulk of the material. As a result, the surface Al species start blocking the access of the reactants to the active sites, i.e. the hydroxyls in the LDH interlayer. Accordingly, the furfural conversion decreases with the increase in the hydration time (Fig. 7B) and with the increase in Al concentration in the aqueous phase (Fig. 8B), even if the relative content of the LDH component increases (Fig. 8A).
A similar negative effect of the surface or extra-framework Al-rich species was also observed in the case of the rehydrated MgAl HTC catalysts prepared from the conventional MgAl HTC materials. Previously, we proposed that the difference in the catalytic performance of either repeatedly rehydrated HTCs33 or HTC catalysts with different crystal sizes13 can also be explained by the formation of Al-rich species, which blocked the access of the reactants to the catalytically active sites. Thus, the results obtained in the present study further underline the importance of the optimal hydration conditions to maximize the catalytic activity of LDH-based catalysts.
3.3. The performance of hydrated catalysts in repeated hydration–reaction–regeneration cycles
Previously, we showed that rehydrated catalysts prepared from conventional MgAl HTCs demonstrated good stability in repeated reaction cycles (aldol condensation–calcination–rehydration).13,17,21 However, in the present study, the aldol condensation catalysts were prepared via a non-conventional synthesis method, and thus it was necessary to assess their stability.In these repeated experiments, the starting calcined physical mixture of Mg and Al oxides (atomic ratio Mg/Al = 3) was hydrated under specific conditions, filtered and added to the furfural + acetone reaction mixture. After completing the reaction, the catalyst was separated from the reaction mixture by filtration, washed with acetone, dried, and then calcined at T = 450 °C, followed by re-hydration of the calcined sample under the same conditions (as during the hydration). Then, the rehydrated catalyst was used again in the reaction. The inevitable loss of catalyst during these reaction–regeneration cycles was compensated by adjusting the mass of reactants, i.e. the catalyst/reactant mass ratio was kept constant.
The physical mixture of oxides (atomic ratio Mg/Al = 3) was hydrated at either T = 25 °C or T = 75 °C for two different treatment times, 20 min and 24 h. Accordingly, the name of the catalysts in this series of experiments included not only hydration temperature and hydration time, but also the cycle number, i.e. S“T”“nt”“Rm”, where “m” represents the cycle number. Fig. 9 presents the furfural conversion after 20 min of reaction observed over the hydrated catalysts in repeated hydration–reaction–regeneration cycles (complete conversion curves showing the change in furfural conversion as a function of the reaction time are available in Fig. 12S in the ESI†).
 | ||
| Fig. 9 Furfural conversion after 20 minutes of aldol condensation over different hydrated MgAl catalysts (atomic ratio Mg/Al = 3) in successive reaction cycles at Treac. = 25 °C. Hydration conditions: A – 20 min at 25 °C (S25-20m-Rm series), B – 20 min at 75 °C (S75-20m-Rm series), C – 24 hours at 25 °C (S25-24h-Rm series), and D – 24 hours at 75 °C (S75-24h-Rm series). | ||
The furfural conversion after 20 min over S25-20m-R1 was about 20% (Fig. 9A), i.e. very similar to that observed in a previous experiment using a catalyst prepared under the same hydration conditions (Fig. 10S(A)†). With an increase the number of reaction cycles, the activity of the re-used catalysts gradually increased, and furfural conversion increased from 20% to 48–53% for S25-20m-R1 and S25-20m-R4/S25-20m-R5, respectively (Fig. 9A). The performance of the catalysts hydrated at T = 75 °C for 20 min also improved in the repeated runs, and the furfural conversion increased from 64% over S75-20m-R1 to 87–91% observed in the third-fifth cycles (Fig. 9B).
A similar trend in the change in furfural conversion with an increase in number of reaction cycles was observed for the samples hydrated for 24 h, both at T = 25 °C (Fig. 9C) and T = 75 °C (Fig. 9D). In both cases, the furfural conversion increased after the first cycle and remained apparently stable in the subsequent reaction cycles. It should be noted that at this hydration time, the catalysts hydrated at T = 25 °C slightly outperformed those prepared at T = 75 °C (Fig. 9C and D), respectively. Again, the excessive Al-rich species blocking the access of reactants to the active sites were hypothesized to be responsible for the observed catalytic performance, as discussed above.
Additionally, the Mg and Al concentrations in the aqueous phase after each hydration step were determined. Their concentration did not change in the repeated cycles, which was within the range obtained earlier in the experiments performed under similar hydration conditions (Fig. 3). Similar to previous studies,17,21 the plausible influence of metal leaching on the performance of the catalysts was evaluated by determining the Mg and Al content in the organic reaction mixtures after completing the reactions. The concentration of these elements was negligible, thus confirming that the leached metals had a negligible effect on the catalytic experiments, if at all.
Comparing the XRD patterns of the different hydrated samples after the first and the sixth hydration (i.e. calcined and hydrated after the fifth reaction cycle), it was found that the intensity of the reflections from the LDH structure increased, while that from Mg(OH)2 decreased (Fig. 13S in the ESI†). Hence, the results reveal that the successful transformation of the Mg + Al oxide mixture to the LDH phase was promoted by the successive treatment/reaction cycles. Accordingly, the relative content of the LDH phase increased in all cases, with the maximum value of 77% observed for the MgAl75-24h-5 sample after the sixth hydration step at T = 75 °C for 24 h (Table 2).
| Sample | Hydration conditions | Relative content of LDH phase, % | A, mmolF gcat−1 min−1 | A norm.,a mmolF gcat−1 min−1 |
|---|---|---|---|---|
| a A norm. – normalized specific initial activity recalculated based on the LDH phase content in the samples as estimated from the XRD data. | ||||
| MgAl25-20m-1 | 25 °C, 20 min, 1 cycle | 13 | 0.4 | 3.3 |
| MgAl25-20m-5 | 25 °C, 20 min, 6 cycles | 18 | 7.7 | 42.7 |
| MgAl25-24h-1 | 25 °C, 24 h, 1 cycle | 16 | 6.6 | 40.9 |
| MgAl25-24h-5 | 25 °C, 24 h, 6 cycles | 48 | 8.0 | 16.7 |
| MgAl75-20m-1 | 75 °C, 20 min, 1 cycle | 26 | 9.2 | 35.5 |
| MgAl75-20m-5 | 75 °C, 20 min, 6 cycles | 59 | 13.9 | 23.6 |
| MgAl75-24h-1 | 75 °C, 24 hours, 1 cycle | 43 | 4.2 | 9.8 |
| MgAl75-24h-5 | 75 °C, 24 hours, 6 cycles | 77 | 10.4 | 13.5 |
The increase in the content of LDH phase with an increase the number of treatment cycles can only partially explain the improvement in the performance of the catalysts in repeated reaction cycles. The specific activity of the differently hydrated catalysts (A, mmolF gcat−1 min−1) increased in the fifth reaction cycle (Table 2), i.e. together with an increase in the relative content of LDH phase. Nevertheless, the normalized specific activity of the catalysts (Anorm., recalculated based on the LDH phase content) decreased in the case of the samples from the MgAl25-24h and MgAl75-20m series. This evidenced that the newly formed LDH phase participated in the reaction only partially, i.e. not all the potentially active sites of the layered component were involved. This can be explained either by the inaccessibility of the active sites located in the bulk of the LDH phase, as was proposed for the conventionally prepared HTC catalysts,18,19,21,33 or by the remaining partial blocking of the active sites on the edges of the LDH platelets with extra-framework species, as discussed above. In any case, the performed experiments evidenced that the catalysts prepared by the hydration of the MgO + Al2O3 mixture were stable in the repeated reaction cycles, regardless of the hydration conditions. Hence, these experiments proved the application potential of these materials as catalysts for base-catalyzed reactions.
3.4. Factors influencing the performance of hydrated MgO + Al2O3 catalysts
Besides the hydration conditions, the influence of additional factors was also investigated, such as stirring, pre-calcination, mixing of oxides and raw material selection. All the experiments on the impact of the hydration conditions described above were carried out in static mode, i.e. without mechanical stirring of the aqueous mixture, because no significant difference was observed in the catalytic performance of the prepared catalysts by either static or stirring hydration procedure (Fig. 14S in the ESI†). The pre-calcination of the starting MgO + Al2O3 mixture at T = 450 °C (i.e. at temperature usually used for the transformation of MgAl HTC to a corresponding mixed oxide) prior to the hydration step was necessary to prepare an active catalyst for aldol condensation (Fig. 15S in the ESI†). Apparently, the pre-calcination of the mixture helped to remove carbonate species from the surface of MgO, which prevented the successful hydration of the oxide, followed by Mg2+ + 2OH− dissociation.22 The proper mixing of both components of the reactive mixture, MgO and Al2O3, in a mortar before the hydration step was also very beneficial for increasing the activity of the resulting catalyst. Adding both oxide components to water separately also resulted in a catalyst with a reasonable activity in the reaction, but furfural conversion was obviously lower than when close contact between the oxides was ensured by proper mixing when preparing the catalyst (Fig. 16S in the ESI†). Finally, further experiments demonstrated that a broad range of Mg and Al sources could be used to prepare hydrated catalysts (Table 3). The reagents for the experiments were selected based on their ability to decompose at T ≤ 450 °C and form the corresponding oxides. Both reactants where properly mixed, calcined at T = 450 °C and hydrated at T = 75 °C for 20 min. Table 3 evidences that both the nature of the reactants and the ratio between them influenced the performance of the resulting hydrated catalysts in aldol condensation. Moreover, the furfural conversion in the reaction was different, even when starting reactive mixtures with a similar Mg/Al ratio were employed for the hydration step (Table 3). The observed difference in the activity of the prepared catalysts can be related to the formation rate of water-soluble Mg and Al species during the hydration step and with the incorporation rate of the water-soluble Al atoms into the parent Mg(OH)2 structure, thus forming the desired LDH phase. In this case, the size of the primary oxide particles and their textural characteristics should significantly affect the dissolution–diffusion–substitution process, similar to the suggestions reported by Warmuz and Madej.24 Obviously, the ratio between the Mg and Al sources, as well as conditions to hydrate the calcined MgO + Al2O3 mixture should be optimized and strictly controlled in each case. Nevertheless, the performed experiments clearly evidence that a wide range of different Mg and Al sources can be used to prepare LDH-based solid catalysts via the simple hydration of a physical mixture of Mg + Al oxides, making these catalysts a promising object for further studies in various applications.:Ac = 1:5 (mol), and rpm = 250
| Initial reagents | Mg:Al ratio (mol) |
Furfural conversion, %a | |
|---|---|---|---|
| Mg | Al | ||
| a Furfural conversion after 60 minutes of the reaction. | |||
| MgO (S-A) | AlOOH (Pural) | 1 | 80.1 |
| MgO (S-A) | AlOOH (Pural) | 3 | 88.4 |
| MgO (S-A) | AlOOH (Pural) | 10 | 45.3 |
| MgO (S-A) | Hydrated Al oxide (S-A) | 3 | 63.9 |
| MgO (S-A) | Hydrated Al oxide (S-A) | 10 | 30 |
| MgO (S-A) | Al isopropoxide (Alfa Aesar) | 3 | 76.5 |
| MgO (S-A) | Al(NO3)3·9H2O (Lach:Ner) | 3 | 56.4 |
| MgO (S-A) | Al acetylacetonate (S-A) | 3 | 52.4 |
| Mg ethoxide (S-A) | AlOOH (Pural) | 3 | 9.6 |
| Mg(NO3)2·6H2O | AlOOH (Pural) | 3 | 43.6 |
4. Conclusion
In this study, we investigated in detail the properties of samples prepared via the hydration of a physical mixture of Mg and Al oxides at T = 25 °C and 75 °C lasting from 5 min up to 7 days. Besides, we also assessed the catalytic performance of the prepared hydrated materials in the aldol condensation of furfural and acetone. At both treatment temperatures, the formation of an active LDH phase was observed after only 5 min, which was associated with the formation of water-soluble Al species, as detected by AAS. The increase in both the hydration temperature and treatment time promoted the formation of the LDH phase, which was monitored by XRD, FTIR, SEM, and TEM. Nevertheless, even the hydration of the physically mixed oxides for 7 days at T = 75 °C resulted in the formation of a hydrated material with a relative content of the targeted LDH phase of about 60%. This was explained by the difficulty of the water-soluble Al species to react with the internal Mg(OH)2 moieties to be inserted in the crystalline framework. As a consequence, the excessive water-soluble Al species were incorporated into the already formed surface LDH phase, thus decreasing the actual Mg/Al ratio in the LDH platelets to below 2. This was concluded from the XRD data, which evidenced the change in the crystallographic parameters of the crystalline LDH framework. Besides, excessive Al species were also adsorbed on the external surface of the formed LDH phase, where they formed a non-crystalline layer, as confirmed by SEM and N2 physisorption experiments.Among the prepared hydrated materials, the catalysts with only about 20–25% of the relative content of the LDH phase demonstrated the best performance in the aldol condensation of furfural and acetone. The calculations showed that the initial activity of these solids (i.e. expressed per 1 g of calcined MgO + Al2O3 mixture employed in the hydration step) was 2–2.5 times lower than that of rehydrated catalysts prepared from the conventional MgAl HTC. Nevertheless, when the catalytic performance was associated only with the LDH phase (i.e. normalized per the content of LDH phase in the catalyst), the normalized activity of the catalyst prepared by the hydration of mixed Mg and Al oxides was 2 to 3 times higher. Assuming that the intrinsic activity of the active sites in the LDH phase is similar, it can be inferred that one half up to two thirds of the active sites in the conventional HTC catalysts are not accessible to reactants and there is significant room for the enhancement of the productivity when using LDH-base catalysts.
The further increase in the relative content of LDH phase in the catalysts prepared via the hydration of mixed Mg and Al oxides was not accompanied by a corresponding increase in furfural conversion, which was presumably because their catalytically active sites were blocked by the extra framework Al species deposited on the LDH surface. Thus, it was assumed that the formation of the LDH phase by the hydration of mixed Mg and Al oxides is necessary but not sufficient to prepare an effective catalyst for base-catalyzed reactions. In this case, strict control of the hydration conditions leading to the formation of the desired LDH phase with catalytically active sites, while not generating excessive water-soluble Al species that deposit on the surface and block the active base sites is of great importance.
The application potential of the catalytic materials prepared via the hydration of physically mixed Mg and Al oxides as catalysts for organic reactions was also demonstrated by their stable performance in five consecutive hydration–reaction–regeneration cycles, regardless of their hydration temperature and time. Finally, additional experiments using various Mg and Al precursors to obtain the corresponding oxides showed that the catalytic performance of the resulting hydrated mixture of Mg and Al oxides was largely determined by the origin of the precursors and the ratio between them. Therefore, each MgO + Al2O3 mixture prepared using different sources requires individual optimization of the hydration conditions to obtain the final catalysts with the optimal performance.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Oleg Kikhtyanin: conceptualization, writing-original draft, writing-review & editing. Valeriia Korolova: experiments, data processing & analysis. Evgeniya Grechman: experiments, data processing & analysis. Miloslav Lhotka: investigation, data processing & analysis. Martin Veselý: investigation, data processing & analysis. Francisco Ruiz Zepeda: investigation, data processing & analysis. David Kubička: supervision, funding acquisition, writing-review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The financial support by the Czech Science Foundation (grant number 19-22978S) is greatly acknowledged. This work was also supported from the grant of specific university research A1_FTOP_2024_004. The authors are also grateful to R. Pažout for XRD analyses and Dana Pokorná for AAS analyses (both from UCT Prague, Czech Republic).References
- A. Rashedi, T. Khanam and M. Jonkman, Energies, 2020, 13, 6048 CrossRef CAS.
- https://climate.ec.europa.eu/eu-action/climate-strategies-targets/2050-long-term-strategy_en .
- M. Mujtaba, L. F. Fraceto, M. Fazeli, S. Mukherjee, S. M. Savassa, G. A. de Medeiros, A. do Espírito Santo Pereira, S. D. Mancini, J. Lipponen and F. Vilaplana, J. Cleaner Prod., 2023, 402, 136815 Search PubMed.
- S. Takkellapati, T. Li and M. A. Gonzalez, Clean Technol. Environ. Policy, 2018, 20, 1615–1630 CrossRef CAS PubMed.
- M. J. Climent, A. Corma, H. Garcia, R. Guil-Lopez, S. Iborra and V. Fornes, J. Catal., 2001, 197, 385–393 CrossRef CAS.
- X. Li, J. Sun, S. Shao, X. Hu and Y. Cai, Fuel Process. Technol., 2021, 215, 106768 CrossRef CAS.
- O. Kikhtyanin, D. Kadlec, R. Velvarská and D. Kubička, ChemCatChem, 2018, 10, 1464–1475 CrossRef CAS.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS.
- V. Korolova, O. Kikhtyanin, E. Grechman, V. Russo, J. Warnå, D. Yu. Murzin and D. Kubička, Catal. Today, 2023, 423, 114272 CrossRef CAS.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, Catal. Rev.: Sci. Eng., 2001, 43, 443–488 CrossRef CAS.
- P. Kustrowski, D. Sulkowska, L. Chmielarz and R. Dziembaj, Appl. Catal., A, 2006, 302, 317–324 CrossRef CAS.
- O. Kikhtyanin, L. Čapek, L. Smolaková, Z. Tišler, D. Kadlec, M. Lhotka, P. Diblíková and D. Kubička, Ind. Eng. Chem. Res., 2017, 56, 13411–13422 CrossRef CAS.
- V. Korolova, L. Dubnová, M. Veselý, M. Lhotka, O. Kikhtyanin and D. Kubička, Appl. Clay Sci., 2024, 249, 107263 CrossRef CAS.
- D. P. Debecker, E. M. Gaigneaux and G. Busca, Chem. – Eur. J., 2009, 15, 3920–3935 CrossRef CAS PubMed.
- S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2013, 15, 2026–2042 RSC.
- A. Tampieri, C. Russo, R. Marotta, M. Constantí, S. Contreras and F. Medina, Appl. Catal., B, 2021, 282, 119599 CrossRef CAS.
- V. Korolova, O. Kikhtyanin, M. Veselý, D. Vrtiška, I. Paterová, V. Fíla, L. Čapek and D. Kubička, Catalysts, 2021, 11, 992 CrossRef CAS.
- J. C. A. A. Roelofs, D. J. Lensveld, A. J. van Dillen and K. P. de Jong, J. Catal., 2001, 203, 184–191 CrossRef CAS.
- L. Faba, E. Díaz and S. Ordónez, Appl. Catal., B, 2012, 113–114, 201–211 CrossRef CAS.
- J. J. Creasey, A. Chieregato, J. C. Manayil, C. M. A. Parlett, K. Wilson and A. F. Lee, Catal. Sci. Technol., 2014, 4, 861–870 RSC.
- V. Korolova, F. Ruiz Zepeda, M. Lhotka, M. Veselý, O. Kikhtyanin and D. Kubička, Appl. Catal., A, 2024, 669, 119506 CrossRef CAS.
- Z. P. Xu and G. Q. Lu, Chem. Mater., 2005, 17, 1055–1062 CrossRef CAS.
- G. Lee, J. Y. Kang, N. Yan, Y.-W. Suh and J. C. Jung, J. Mol. Catal. A: Chem., 2016, 423, 347–355 CrossRef CAS.
- K. Warmuz and D. Madej, Appl. Clay Sci., 2021, 211, 106196 CrossRef CAS.
- O. Kikhtyanin, V. Korolova, A. Spencer, L. Dubnová, B. Shumeiko and D. Kubička, Catal. Today, 2021, 367, 248–257 CrossRef CAS.
- J. I. Di Cosimo, V. K. Díez, M. Xu, E. Iglesia and C. R. Apesteguia, J. Catal., 1998, 178, 499–510 CrossRef CAS.
- S. K. Yun and T. J. Pinnavaia, Chem. Mater., 1995, 7, 348–354 CrossRef CAS.
- L. Huang, Z. Yang and S. Wang, Constr. Build. Mater., 2020, 262, 120776 CrossRef CAS.
- E. Bernard, W. J. Zucha, B. Lothenbach and U. Mäder, Cem. Concr. Res., 2022, 152, 106674 CrossRef CAS.
- R. Zăvoianu, R. Bîrjega, E. Angelescu and O. D. Pavel, C. R. Chim., 2018, 21, 318–326 CrossRef.
- O. D. Pavel, R. Bîrjega, M. Che, G. Costentin, E. Angelescu and S. Şerban, Catal. Commun., 2008, 9, 1974–1978 CrossRef CAS.
- A. J. Marchi and C. R. Apesteguıa, Appl. Clay Sci., 1998, 13, 35–48 CrossRef CAS.
- V. Korolova, O. Kikhtyanin, F. Ruiz Zepeda, M. Lhotka, M. Veselý and D. Kubička, Appl. Catal., A, 2022, 632, 118482 CrossRef CAS.
- S. Abelló, F. Medina, D. Tichit, J. Pérez-Ramírez, J. C. Groen, J. E. Sueiras, P. Salagre and Y. Cesteros, Chem. – Eur. J., 2005, 11, 728–739 CrossRef PubMed.
- Md. H. Zahir, M. M. Rahman, K. Irshad and M. M. Rahman, Nanomaterials, 2019, 9, 1773 CrossRef CAS PubMed.
- C. M. Janet, B. Viswanathan, R. P. Viswanath and T. K. Varadarajan, J. Phys. Chem. C, 2007, 111, 10267–10272 CrossRef CAS.
- N. C. S. Selvam, R. T. Kumar, L. J. Kennedy and J. Vijaya, J. Alloys Compd., 2011, 509, 9809–9815 CrossRef CAS.
- M. G. Gardeh, A. A. Kistanov, H. Nguyen, H. Manzano, W. Cao and P. Kinnunen, J. Phys. Chem. C, 2022, 126, 6196–6206 CrossRef CAS PubMed.
- B. Veriansyah, R. F. Susanti, A. Nugroho, B. K. Min and J. Kim, Mater. Lett., 2011, 65, 772–774 CrossRef CAS.
- G. K. Priya, P. Padmaja, K. G. K. Warrier, A. D. Damodaran and G. Aruldhas, J. Mater. Sci. Lett., 1997, 16, 1584–1587 CrossRef CAS.
- F. R. Costa, A. Leuteritz, U. Wagenknecht, D. Jehnichen, L. Häußler and G. Heinrich, Appl. Clay Sci., 2008, 38, 153–164 CrossRef CAS.
- T. Kolar, B. Mušič, R. Cerc Korošec and V. Kokol, Cellulose, 2021, 28, 9441–9460 CrossRef CAS.
- Y. Chen, T. Zhou, H. Fang, S. Li, Y. Yao, B. Fan and J. Wang, Particuology, 2016, 24, 177–182 CrossRef CAS.
- H. Pang, G. Ning, W. Gong, J. Yea and Y. Lin, Chem. Commun., 2011, 47, 6317–6319 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy01454e |
| This journal is © The Royal Society of Chemistry 2025 |