
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Homogeneous catalytic hydrogenation of CO2 – amino acid-based capture and utilization
Yong
Peng
 a,
Elisabetta
Alberico
ab,
Henrik
Junge
*a and
Matthias
Beller
*a
a,
Elisabetta
Alberico
ab,
Henrik
Junge
*a and
Matthias
Beller
*a
aLeibniz-Institut für Katalyse e. V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: henrik.junge@catalysis.de; matthias.beller@catalysis.de
bIstituto di Chimica Biomolecolare – CNR, tr. La Crucca 3, 07100 Sassari, Italy
First published on 2nd May 2025
Abstract
In this review, we provide an overview of research efforts to integrate carbon dioxide capture specifically using amino acid-based sorbents with its thermocatalytic hydrogenation promoted by homogeneous metal complexes. Carbon capture and utilization (CCU) is a promising strategy for the production of fuels, chemicals and materials using CO2 scrubbed from point sources and the atmosphere as a C1 feedstock while mitigating CO2 emissions. Compared to established (alkanol)amines, amino acids offer some advantages as CO2 capture agents due to their lower volatility, higher oxygen stability and lower regeneration energies. We report how the structural diversity of amino acids and the possibility of combining them with cations in salts and ionic liquids have been exploited in the design of absorbers for improved absorption kinetics and capacity. Furthermore, we discuss selected examples from the literature illustrating the use of 1°/2° (poly)amines, since the 1°/2° amino groups are mainly responsible for CO2 chemisorption in amino acid-based capture media, the nature of the corresponding adducts, and the most promising catalysts capable of converting the latter to formate and methanol while regenerating the scrubber. General trends regarding the influence of catalyst structure and reaction parameters on the efficiency, productivity, and selectivity of such processes will be highlighted. We will detail how this knowledge has informed the design of novel processes in which CO2 is chemisorbed by amino acid-based solvents and hydrogenated in situ to formate and methanol, or alternatively used as a fuel to implement a “hydrogen battery” where, after metal-catalyzed H2 release from formate, CO2 is retained by the amino acid-based solvent in the “spent battery” which can then be recharged by hydrogenation of the retained CO2 promoted by the same catalyst. The topic is still in its infancy, and several issues have emerged that will be critically discussed in the final section of this review. These issues need to be addressed in order to improve performance and provide a playground for researchers whose interest we hope to have aroused with this review.
 Yong Peng | Yong Peng obtained his PhD degree from the Instituto de Tecnología Química (ITQ)-Universitat Politècnica de València (UPV) in 2021, under the supervision of Prof. Hermenegildo García. In 2022, he joined the “Catalysis for Energy” group led by Dr Henrik Junge in the Department of Applied Homogeneous Catalysis under Prof. Matthias Beller at LIKAT, Rostock, where he was supported first by an Alexander von Humboldt postdoctoral fellowship, and currently holds a Marie Skłodowska-Curie individual Fellowship. His research interests focus on the design of novel photocatalysts for applications including C–H activation and functionalization, CO2 valorization, N2 fixation and H2 production. |
 Elisabetta Alberico | Elisabetta Alberico obtained her doctorate at RWTH in Aachen, Germany, under the supervision of Prof. Albrecht Salzer. Since 2001 she has held a permanent position at the Institute of Biomolecular Chemistry of the National Research Council of Italy. Since 2011 she has been an associate researcher in the group “Catalysis for Energy” led by Dr Henrik Junge in the Department of Applied Homogeneous Catalysis led by Prof. Matthias Beller at LIKAT, Rostock. Her research interests are directed to the design of novel homogeneous catalysts for hydrogenation and dehydrogenation reactions, for hydrogen storage in liquid chemical compounds and hydrogen generation from renewable resources. |
 Henrik Junge | Henrik Junge studied chemistry at the University of Rostock, where he also completed his PhD. From 1995 he worked in the group of Prof. Günther Oehme, before he joined the group of Prof. Matthias Beller in 1999, both at the Institute for Organo-catalytic Research. Since 2009 Junge has been the Group Leader for “Catalysis for Energy” at LIKAT. From 2008 to 2014 he was the chairman of the board of the Hydrogen Technology Initiative Mecklenburg-Western Pomerania. In September 2015 Junge was a visiting professor at the Collage of Chemical Engineering at of the Zhejiang University of Technology in Hangzhou/China. Research interests of Junge include homogeneous as well as nano particle- and single atom-based heterogeneous catalysis and their application in energy related transformations, e.g. (de)hydrogenation and photocatalytic reactions. |
 Matthias Beller | Matthias Beller, born in Gudensberg (Germany) in 1962, obtained his PhD in 1989 working with Lutz F. Tietze at the University of Göttingen. After one year of postdoctoral research with Barry Sharpless at MIT (USA), from 1991 to 1995 he worked at Hoechst AG in Frankfurt. Then, he started his academic career at TU Munich. In 1998, he relocated to Rostock to head the Leibniz-Institute for Catalysis. Matthias Beller is also Vice-president of the Leibniz Association and a member of three German Academies of Sciences including the German National Academia “Leopoldina”. The research of his group has been published in more than 1200 original articles and reviews and focused on applying homogeneous and heterogeneous catalysis for the sustainable synthesis of fine/bulk chemicals as well as energy technologies. |
1. Introduction
The Paris Agreement, negotiated by 196 parties, finally entered into force in November 2016 with the goal of keeping global warming well below 2 °C above pre-industrial levels. There is growing public, scientific and political awareness of the impact of CO2 emissions from fossil fuel combustion on global warming and ecosystems. However, the development and deployment of renewable energy technologies has not yet reached a level where they can fully meet the high energy demands of modern society. Therefore, fossil fuels will remain the primary source of energy for the foreseeable future, and significant amounts of CO2 from their combustion will be emitted into the atmosphere. In this scenario, CO2 capture from point sources and from the atmosphere will be one of the key strategies to mitigate the adverse effects of CO2 emissions and to achieve the target set by the convention, along with the development of new technologies for improved energy efficiency and renewable energy production.Currently, CO2 capture technologies can be divided into four categories, including physical adsorption, chemical absorption, membrane and cryogenic separation. Among these technologies, chemical absorption is the most efficient and has been implemented in power plants using alkanolamines, such as monoethanolamine (MEA), diethanolamine (DEA), and N-methyl diethanolamine (MDEA), as absorbents, with MEA being widely used as the benchmark absorbent. The captured CO2 is then typically desorbed and compressed to either be used as a C1 feedstock producing chemicals and fuels, or sequestered underground. The alkanolamine based solvents have been shown to be quite efficient, capturing up to 90% of the CO2 present in power plant flue gas streams.1 However, the energy required for solvent regeneration (CO2 desorption) ranges between 2.4 and 4.2 GJ per ton of CO2, accounting for 60–70% of the total capital investment of the capture process.2
To circumvent the high energy penalty of the desorption and compression steps, processes are being investigated that can be collectively referred to as reactive CO2 capture (Scheme 1), where the captured CO2 is used directly as a C1 feedstock for the synthesis of value-added products. Currently, the fertilizer industry uses about 230 Mt CO2 per year for urea production (∼130 Mt) and enhanced oil recovery (∼80 Mt).3 Among the possible applications within the reactive CO2 capture concept, the direct hydrogenation of the captured CO2 to formate and methanol is the most promising from an economic point of view. One reason is that green H2 derived from water electrolysis using renewable energy or from photocatalysis is much more sustainable compared to the production of other precursors used in CO2 fixation, such as epoxide, alkenes and alkynes.4 In addition, formic acid and methanol can also be used as liquid organic hydrogen carriers (LOHC).5 Moreover, methanol is one of the most important organic feedstocks for the chemical industry6 which is produced annually on the order of 100 Mt. More specifically, methanol is used in the synthesis of chemicals including acetic acid, formaldehyde, ethylene, methyl methacrylate, and propylene via the methanol-to-olefin (MTO) route. It can also be used as a fuel, either by itself, in a blend with gasoline, to produce biodiesel, or in the form of methyl tert-butyl ether (MTBE) and dimethyl ether (DME). Although reactive CO2 capture is a relatively new topic, several research groups, for instance, Prakash,6b,7 Milstein,8 Leitner,9 Gademann,10 He,11 Nielsen,12 Sanford,13 and our group14 have advanced this field and demonstrated the applicability of the in situ conversion of captured CO2 for the synthesis of methanol, formates, oxazolidinones, and urea. In these studies, mostly organic amines were used as the base for CO2 capture and subsequent hydrogenation.
 | ||
| Scheme 1 Schematic illustration of the CO2 reactive capture concept. | ||
Although the alkanolamine-based capture technologies can be easily retrofitted to existing post-combustion power plants and have been shown to be highly efficient in capturing CO2 from flue gas, they suffer from some fatal drawbacks, such as volatile loss, thermal degradation by O2 in the flue gas and thus the formation of corrosive by-products. Therefore, many efforts have been made to develop alternative absorbents to alkanolamines. Among the developed absorbents, amino acid-based absorbents, whose amino groups can bind to CO2 in a similar manner to amines, show the advantages in terms of their low volatility due to their ionic properties, high oxygen resistance and low regeneration energies. There are more than 500 amino acids in nature, offering a wide range of possibilities in terms of basicity and steric hindrance, which have been shown to be the most important parameters determining absorption kinetics, type of CO2 adducts, and uptake capacity, etc.15 In addition, since the amino groups can only react with CO2 in their neutral form after the amino acid zwitterions have been neutralized/deprotonated, the choice of counter cations can also influence the absorption performance, and thus offer another possibility for absorbent design, as will be discussed in detail in Section 3.
Amino acids are the building blocks of carboxylases, the key enzymes in photosynthesis and the global carbon cycle, that capture CO2 from the atmosphere and convert it into organic biomass.16 Carboxylases promote the formation of a C–C bond between CO2, the electrophile, and a substrate, which act as the nucleophile. To overcome the thermodynamic and kinetic inertness of CO2, most carboxylases activate the substrate (for example, a thioester, α-ketoacid, or ketone) by transforming it into an enol(ate), which is a stronger nucleophile. On the other hand, the electrophilic CO2 molecule is closely positioned in the active site through interaction with amino acids to allow nucleophilic attack. This interaction relies on a variety of binding elements that exploit the difference in electronegativity between carbon and oxygen: CO2 binds through metals, hydrogen bonds and hydrophobic interactions.
Molecular dynamics calculations suggest that the diffusion of CO2 around and into carboxylases to access the active site is dominated by hydrophobic interactions with amino acids with small hydrophobic side chains, such as alanine (ALA), valine (VAL), leucine (LEU), and isoleucine (ILE), and the sulfur-containing cysteine (CYS) residue.16 Within the active site, catalysis requires tight control of the reaction between the enolate and CO2. This is realized by a network of hydrogen bonds, or more precisely acid/base motifs, provided by strategically located amino acid residues. Bioinformatic analysis shows that the highly basic amino acids, such as lysine (LYS) (pI 9.5), arginine (ARG) (pI 10.8), and histidine (pI 7.6), are the most abundant in the crystal structures of CO2 protein binding sites.17 In enoyl-CoA carboxylase/reductases, the carboxylases with the highest turnover frequencies, four amino acid residues anchor and position the CO2 molecule for attack by a reactive enolate formed during the catalytic cycle. The amide group of an asparagine residue (ASN) holds the CO2 in place from one side, while a water grid, starting from a water molecule secured between a residue of histidine (HIS) and glutamic acid (GLUA), serves as an additional anchoring point for CO2 from the opposite side.
The aromatic ring of a phenylalanine (PHE) residue actively prevents the diffusion of water into the active site which would trigger the competitive protonation/reduction of the enolate, the alternative reaction catalyzed by enoyl-CoA carboxylase/reductases in the absence of CO2. It is noteworthy that CO2 is bound to the protein environment through the two oxygen atoms leaving the carbon atom relatively free for any incoming substrate. This is an aspect that should be considered in the design and application of biomimetic catalysis for CO2 capture and utilization (vide infra). CO2 can also react with unprotonated N-primary amine or LYS-ε-amino groups in proteins affording the corresponding carbamates/carbamic acid.18 This post-translational modification plays a crucial role in regulating oxygen binding in hemoglobin and activating the CO2-fixing enzyme ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO).19 It is estimated that 400 Gt of CO2 are fixed annually by RuBisCO which is ultimately responsible for virtually all the organic carbon present in the biosphere.20 The active site of RuBisCO is centered on a magnesium ion which is held tightly by an ASN residue, a GLUA and the carbamate group formed by carbamylation of the ε-amino group of a LYS residue.21 A second CO2 molecule is then fixed through nucleophilic attack by the enolate of ribulose 1,5-bisphosphate, generated by H3 proton abstraction by the activated LYS carbamate (Fig. 1).22
 | ||
| Fig. 1 The active site of RuBisCO is centered on a magnesium ion. It is held tightly by three amino acids: an asparagine, glutamic acid, and the carbamate group formed by carbamylation of the ε-amino group of a lysine residue. The carbon dioxide molecule (left, dotted outline) attached to this lysine serves as an activator in the carbon fixing reaction. This activator carbon dioxide is different from the carbon dioxide molecule that is fixed to the ribulose 1,5-bisphosphate. Reproduced with permission from ref. 22: Image from the RCSB PDB (https://rcsb.org) November 2000 Molecule of the Month feature by David Goodsell. | ||
Inspired by the unique properties of amino acids in the natural process and some favorable features compared to alkanolamines, we believe that amino acid-based absorbents can be attractive alternatives for reactive CO2 capture. In recent years, our group has focused on using amino acid salts (AAS) and amino acid-based ionic liquids (AAILs) as the basis for CO2 hydrogenation, and productivities (TONs) for formate superior to amine and alkanolamine based systems have been achieved.23 Based on these promising results, we feel compelled to highlight this emerging field by summarizing the recent progress in CO2 capture and hydrogenation realized in amino acid-based systems.
In this review, we summarize recent advances in the application of amino acid-based absorbents for CO2 capture and subsequent in situ hydrogenation. As readers will note, amino acid-based reactive CO2 capture has only emerged in the last five years, whereas earlier developments were largely focused on conventional amine/hydroxide-based systems. To provide a comprehensive overview, we also briefly discuss advances in reactive CO2 capture using non-amino acid-based systems and the guiding principles behind these approaches. To facilitate the understanding of the relationship between CO2 properties and CO2 reactivity in capture and hydrogenation, we present the essential background information on the physical and chemical properties of CO2 in Section 2. Furthermore, we highlight the distinct advantages of amino acid-based absorbents by discussing recent advances in this field in Section 3. Following this, recent advances in reactive CO2 capture in non-amino acid-based systems are discussed in Section 4. Finally, the achievements of reactive CO2 capture with amino acid-based absorbents are highlighted in Section 5.
2. CO2 properties and activation
CO2 is a linear triatomic molecule in which the sp-hybridized central carbon is bonded to two peripheral oxygen atoms via σ and π bonds, making it highly thermodynamically stable (ΔGof = −396 kJ mol−1). The symmetric dipole moments and linear geometry determine the overall nonpolar property. However, due to the electronegativity differences between carbon and oxygen, the bonds are polar, resulting in a partial positive charge on the carbon atom and a partial negative charge at the oxygen atoms. The positive carbon center can thus act as electrophile, allowing nucleophilic attack by a base, for instance, hydroxide or amines, which is the basis for CO2 capture via chemical absorption.The Lewis basic and acidic properties of CO2 can also be explained by the orbital structure at its ground state, as shown in Fig. 2a. The HOMO (1πg) orbitals contain only the contribution from the 2p orbitals of the O atoms, corresponding to their out-of-phase orbitals including lone electron pairs. The LUMO  comprises contributions from the 2px and 2py orbitals of the C and O atoms, with significant carbon character and localization on the carbon atom, while the corresponding bonding orbitals (1πu) have an energy level slightly lower than the O 2p orbitals, due to the higher electronegativity of the O element.
comprises contributions from the 2px and 2py orbitals of the C and O atoms, with significant carbon character and localization on the carbon atom, while the corresponding bonding orbitals (1πu) have an energy level slightly lower than the O 2p orbitals, due to the higher electronegativity of the O element.
 | ||
| Fig. 2 (a) Molecular orbitals of CO2 at the ground state. (b) Common binding modes of the CO2 molecule with metal. Reproduced from ref. 24 and 25 with copyright permission. | ||
The disruption of the linear geometry and symmetric electron distribution of the molecule can be an indicator of CO2 activation. In general, chemisorption is the first step prior to the activation. As shown in Fig. 2b, CO2 can be coordinated to an isolated activation site by four different modes: (1) when the carbon center is bonded to a Lewis base or metal surface, the backward electron donation to the anti-bonding orbital of CO2 leads to the bending of the O–C–O angle to less than 180°, and to a lowering of the
leads to the bending of the O–C–O angle to less than 180°, and to a lowering of the  orbital energy;26 (2) when the O center is end-on adsorbed on a Lewis acid site, i.e., metal oxide or electron-deficient metal surface, the CO2 molecule can maintain its linear structure or be slightly distorted;27 (3) in the case of η2(C,O) side-on adsorption on the active site, both back-donation from the active site, and donation to the active site, occur at the same time, resulting in a bent O–C–O bonds; (4) both O atoms can simultaneously bind to the active center, forming a bidentate species. In a system with multiple active sites, for instance, metal clusters/nanoparticles, organometallic complexes, the mode III and mode IV in Fig. 2b can be further extended to a bridge-on absorption mode.
orbital energy;26 (2) when the O center is end-on adsorbed on a Lewis acid site, i.e., metal oxide or electron-deficient metal surface, the CO2 molecule can maintain its linear structure or be slightly distorted;27 (3) in the case of η2(C,O) side-on adsorption on the active site, both back-donation from the active site, and donation to the active site, occur at the same time, resulting in a bent O–C–O bonds; (4) both O atoms can simultaneously bind to the active center, forming a bidentate species. In a system with multiple active sites, for instance, metal clusters/nanoparticles, organometallic complexes, the mode III and mode IV in Fig. 2b can be further extended to a bridge-on absorption mode.
In the heterogenous hydrogenation of CO2, one pathway for further conversion of the adsorbed intermediates is the dissociation of CO2 into CO and O species, resulting from the weakening of the C–O bond. Further hydrogenation of the adsorbed CO can then result in formate, methanol, methane and C2+ hydrocarbons, depending on the reaction conditions and catalysts.28 Alternatively, the hydride adsorbed on the active surface can migrate and nucleophilic attack the carbon center of the bound CO2 to form adsorbed formate species. For example, a case study by Bowen and colleagues demonstrated the activation of CO2 by platinum hydride clusters using mass spectroscopy and photoelectron spectroscopy (Fig. 3a). Combining experimental data with DFT calculations, they confirmed that the insertion of CO2 into the Pt–H bond of [PtH3]− is energetically favorable, leading to the formation of formate anion/[PtH2] adducts (Fig. 3b). In contrast, [PtH]− was found to only activate CO2 (Fig. 3c).29
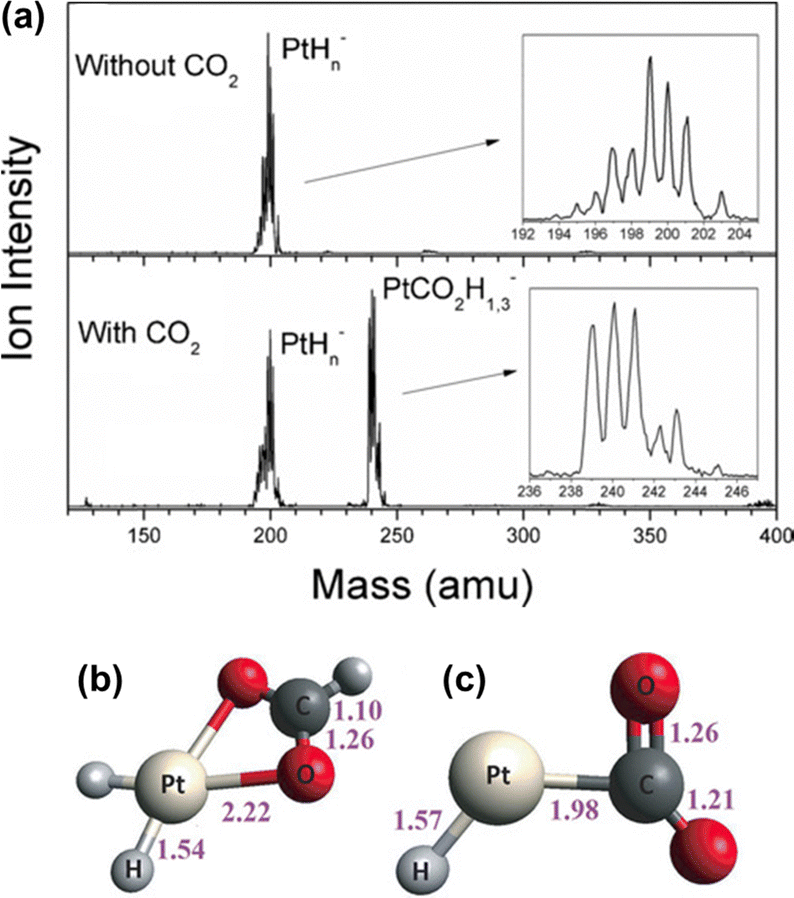 | ||
| Fig. 3 Illustration of the activation of CO2 by PtHn− cluster: (a) mass spectra with and without the presence of CO2, (b) PtCO2H3− and (c) PtCO2H−. Bond lengths (Å). Reproduced from ref. 29 with copyright permission. | ||
Compared to heterogeneous catalytic CO2 upgrading, the homogeneous pathway typically allows the conversion under milder conditions with easier control of selectivity.30 The general modes of CO2 coordination and subsequent activation on transition metal complexes are shown in Fig. 2b. The reduction of CO2 to formate requires the insertion of the CO2 molecule into a M–H bond. The respective mechanism is shown in Fig. 4, where a side-on coordination of CO2 to the metal center occurs (activation mode I in Fig. 4), followed by hydride transfer to the carbon center (nucleophilic attack).31 For a coordinatively saturated metal complex, a vacant site made available by the dissociation of an ancillary ligand may be a prerequisite for the CO2 coordination and subsequent insertion step into the M–H bond. Since the reaction for the formation of formic acid is endergonic  , a base is always added to shift the reaction equilibrium. It should be noted that the direct nucleophile attack by the hydride (activation mode II in Fig. 4) has also been proposed, which does not require coordination of CO2 to the metal, and is therefore possible also for coordinatively saturated metal catalysts such as the Nozaki pyridine-based PNP-ligated iridium(III) trihydride.32 More recently, functional ligands have been developed which incorporate a functional group besides the donor atoms. Hazari and co-workers demonstrated that the presence of an H-bond donor incorporated in the second coordination sphere of an Ir complex favors CO2 insertion and conversion.33 As shown in Fig. 5, the aliphatic PNP ligand in the iridium trihydride catalyst contains an –NH motif that can form a hydrogen bond with the oxygen of an incoming CO2 molecule, thereby increasing the electrophilicity of its central carbon to facilitate hydride transfer. DFT calculations were performed to explore the pathway for CO2 insertion. It was calculated that of the three available hydrides, axial Ha, syn to the N–H bond, is preferentially transferred (Fig. 6). The hydrogen bond, which is also present in the Ir–formate complex, makes the insertion thermodynamically and kinetically more favourable. Indeed, complex 1 (Fig. 5) was found to readily insert CO2 at room temperature, leading to the highly moisture and air stable formate–Ir complex 2. For the catalytic hydrogenation of CO2 in alkaline water promoted by 1, nucleophilic attack of CO2 by the hydride was calculated to be the rate-determining step, requiring no pre-coordination of CO2 to iridium. Similar transition states and intermediates have been calculated for the insertion of CO2 into the Ru–H bond of a ruthenium dihydride complex carrying an aliphatic bisphosphinoamino pincer ligand.34
, a base is always added to shift the reaction equilibrium. It should be noted that the direct nucleophile attack by the hydride (activation mode II in Fig. 4) has also been proposed, which does not require coordination of CO2 to the metal, and is therefore possible also for coordinatively saturated metal catalysts such as the Nozaki pyridine-based PNP-ligated iridium(III) trihydride.32 More recently, functional ligands have been developed which incorporate a functional group besides the donor atoms. Hazari and co-workers demonstrated that the presence of an H-bond donor incorporated in the second coordination sphere of an Ir complex favors CO2 insertion and conversion.33 As shown in Fig. 5, the aliphatic PNP ligand in the iridium trihydride catalyst contains an –NH motif that can form a hydrogen bond with the oxygen of an incoming CO2 molecule, thereby increasing the electrophilicity of its central carbon to facilitate hydride transfer. DFT calculations were performed to explore the pathway for CO2 insertion. It was calculated that of the three available hydrides, axial Ha, syn to the N–H bond, is preferentially transferred (Fig. 6). The hydrogen bond, which is also present in the Ir–formate complex, makes the insertion thermodynamically and kinetically more favourable. Indeed, complex 1 (Fig. 5) was found to readily insert CO2 at room temperature, leading to the highly moisture and air stable formate–Ir complex 2. For the catalytic hydrogenation of CO2 in alkaline water promoted by 1, nucleophilic attack of CO2 by the hydride was calculated to be the rate-determining step, requiring no pre-coordination of CO2 to iridium. Similar transition states and intermediates have been calculated for the insertion of CO2 into the Ru–H bond of a ruthenium dihydride complex carrying an aliphatic bisphosphinoamino pincer ligand.34
 | ||
| Fig. 4 General mechanism of CO2 hydrogenation by metal hydrides. Adapted from ref. 35 and 30 with copyright permission. | ||
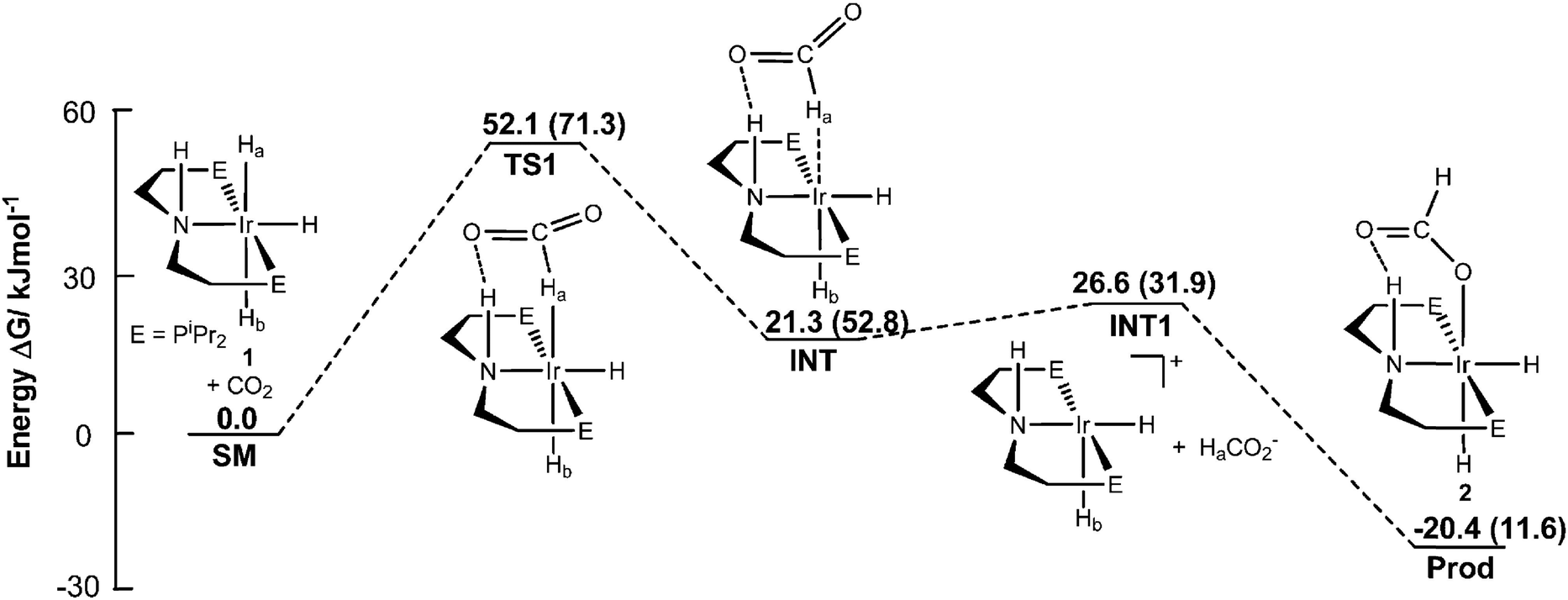 | ||
| Fig. 5 Reaction pathway for insertion of CO2 into 1. All energies are solvent corrected (THF) Gibbs free energies. Numbers in parentheses are for insertion into Hb. For which hydrogen bonding between the entering CO2 and the ligand NH is not available. E = PiPr2. Reproduced with permission from ref. 33. | ||
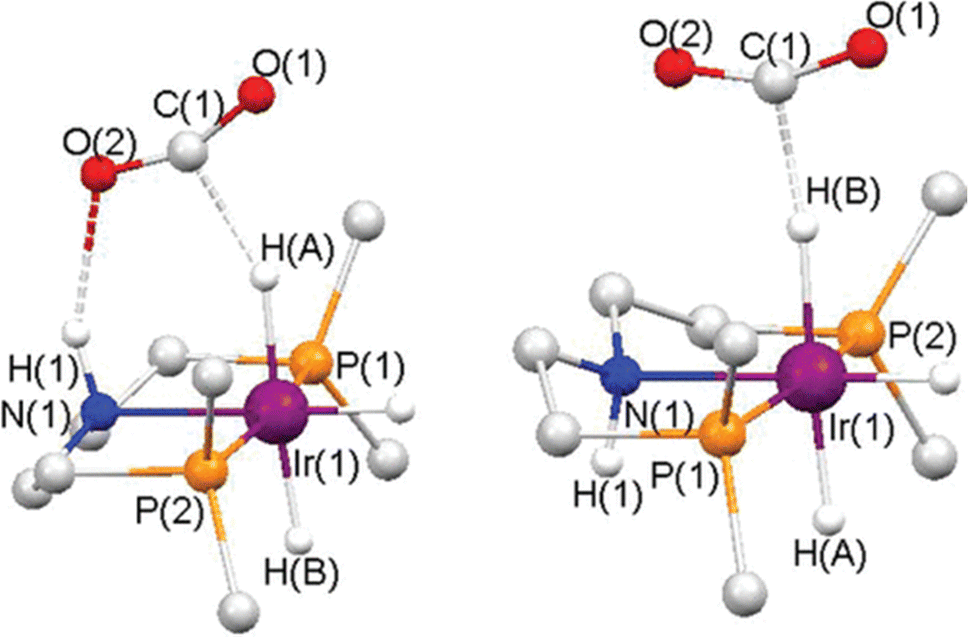 | ||
| Fig. 6 Calculated transition state for CO2 insertion into (a) Ha and (b) Hb respectively of complex 1 shown in Fig. 4. Selected hydrogen atoms and isopropyl groups have been removed for clarity. The smaller Ir–Ha–C bond angle for Ha attack on CO2 (152.2°) compared to the corresponding bond angle Ir–Hb–C for Hb attack on CO2 (172.4°) allows for a stabilizing H-bonding interaction between O(2) and H(1). As a result, the former process is thermodynamically and kinetically favoured. Reproduced with permission from ref. 33. | ||
Nucleophilic attack may not always be an activation when considering the target hydrogenation product. For example, the adsorption of CO2 onto an oxygen-rich surface, such as an oxide support, could form a stable and strongly adsorbed carbonate intermediate species, which may not be favorable for further hydrogenation. The same applies to CO2 absorbed in solution, either in water or an organic solvent, in the form of bicarbonate and/or carbamate, which are thermodynamically poorer hydride acceptors than CO2 itself. Yet, anionic carboxylates/carbamates can coordinate to cationic metal complexes and become charge neutral, allowing reduction. Alternatively, ion pairing can be used to tune the properties of the anions and should be considered in reaction design. Whether the hydrogenation within a given solvent proceeds on the CO2-adduct formed after CO2 scrubbing or on free CO2 in equilibrium with the latter under the experimental conditions of the hydrogenation reaction is questionable and specific to the system under investigation.36 In the case of bicarbonate hydrogenation, claims can be made in either case or a combination of both can be put forward based on either the successful hydrogenation of (bi)carbonate without CO2 overpressure and the detection of a metal-(bi)carbonate resting state,34,37 or the higher TON achieved in the presence of pressurized CO2.38 In natural photosynthesis catalyzed by RuBisCO, CO2 released from bicarbonate is the species which undergoes nucleophilic attack by the negatively charged enolates.22 It has recently been demonstrated that alkylcarbonates are more reactive toward ruthenium hydride complexes than CO2 under comparable conditions when the catalyst operates via an inner-sphere mechanism in which the substrate alkylcarbonate is coordinated to the metal center.39 It is likely that carbamates have a similar reactivity, although with coordinatively saturated metal hydrides which cannot bind the substrate, formate is more likely to result from preferential hydride transfer to free CO2 in equilibrium with carbamate, due to the higher electrophilicity of the former.13
3. CO2 capture with amino acid-based absorbents
As discussed in the introduction, current industrial-scale carbon capture, utilization, and storage (CCUS) technologies use alkanolamines as absorbents. However, these amine-based systems suffer from several disadvantages, such as high regeneration energy, low resistance to oxygen degradation, corrosiveness, and volatility. In comparison, amino acids could potentially overcome the above-mentioned disadvantages and have thus attracted considerable attention from both academia and industry. In addition to high CO2 loading capacity, low regeneration energy, resistance to O2 degradation, and high-boiling point, the amino acid-based absorbents are also biodegradable, and can be obtained from biomass, making them environmentally friendly. The evaluation criteria for CO2-capture and -storage solvents include the capture capacity/equilibrium constants, cyclic capacity, density and dynamic viscosity, heat capacity, dissociation constant, and CO2 mass transfer. However, since the focus of this review is on valorization of CO2, only the capture capacity, absorption kinetics and equilibrium constant will be considered and discussed.Amino acids can be considered as analog of amines when in the active form (Scheme 2a), as the amino group serves as the active center for CO2 capture, and thus, the capture mechanisms of amines can be applied to amino acid-based systems. Specifically, the dissolved CO2 undergoes nucleophilic attack by the amino group, forming a C–N bond (Scheme 2b, eqn (1)) in a zwitterionic form, after which, another base, either the amino acid itself, H2O, as Brønsted base, or a hydroxide, present in the solution, can abstract a proton from the protonated amino group to form a carbamate (Scheme 2b, eqn (2) and (3)). Thus, the maximum loading capacity is 0.5 moles CO2 per mole of amino acid in the carbamate form when another molecule of amino acid acts as the counter cation. In addition, if an additional base, usually hydroxide, or water, is present, it may also promote the formation of bicarbonate (Scheme 2b, eqn (4)) or carbamic acid (Scheme 2b, eqn (5)), and therefore, the theoretical maximum load can exceed 0.5 (eqn (3) and (5)) or even 1 (Scheme 2b, eqn (4)). However, the amino acids are inactive in CO2 capture over a wide pH range. For example, in a medium with a pH between pKa(–COOH) and pKa(–NH2), amino acids exist in a zwitterionic form with a net charge of zero, and the protons from the carboxyl groups protonate and deactivate the amino groups. Under more acidic conditions, the amino groups would be protonated by external protons with a net charge of +1, which would also result in a deactivated amino group. Therefore, to activate the amino groups, the amino acids must be neutralized, or ionized by a base, such as alkali metal hydroxides, or strongly basic amines. Scheme 2a illustrates the effect of pH on the transitions between different forms of amino acids. Based on the type of cations, the amino acid-based absorbents for CO2 capture can be classified into three categories, namely, amino acid salts, amino acid ionic liquids, and amino acids mixtures. In the following three subsections, the state-of-the-art reference work on amino acid-based absorbents for CO2 capture mainly published within the last 5 years will be discussed.
 | ||
| Scheme 2 (a) The dependence of aqueous amino acid form on the pH; (b) possible reactions for CO2 capture with active amino acid absorbents. | ||
3.1. Amino acid salts for CO2 capture
Amino acid salts (AAS) can be obtained simply by neutralizing the amino acids with an equimolar amount of alkali hydroxides in an aqueous solution. There are 20 α-amino acids that are the building blocks of proteins, with the amine and carboxylic group located on the same carbon. However, the properties and CO2-binding capacities vary depending on the side group. For example, potassium lysinate (LYSK), potassium argininate (ARGK), and potassium histidinate (HISK) are reported to have the highest capture capacity and by far exceed the performance of 30 wt% of MEA (benchmark), due to the high pKa of the additional primary or secondary amine groups on the side chain.15,40 Potassium prolinate (PROK) was also reported to possess high capacity due to the instability of the corresponding carbamate and its tendency to hydrolyze to bicarbonate, which, in turn, shifts the equilibrium toward amine-CO2 bond formation. To make it easier for the reader to compare the amino acid properties and their correlation with the CO2 capture performance of the corresponding salts, a list of the structure and pKa of the amino acids, as well as the reported CO2 loading capacities, and the pseudo-first-order reaction constant (Kov) are summarized in Table 1.| Name | Structure | pKa ata 298.15 K | Loading temperature and pressure | Concentration | Absorption kineticsc [Kov (S−1)] | Loading capacityb (molCO2 molAAS−1) | Ref. |
|---|---|---|---|---|---|---|---|
| a Relative to the most basic group in the amino acid. b The loading capacity means the maximum CO2 capture capacity of the amino acid under the given conditions in each entry. c All the absorption kinetic data are taken from ref. 50. | |||||||
| Alanine (ALA) |
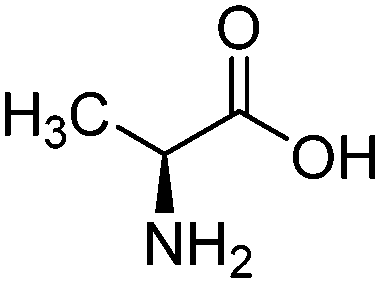
|
10.01 | 30 kPa | 2.5 M | — | 0.68 | 41 |
| 298–313 K | |||||||
| Arginine (ARG) |

|
13.8842 | 15 kPa | 1 M | 4203 (0.5 M) | 1.107 | 15 |
| 298–333 K | |||||||
| Lysine (LYS) |
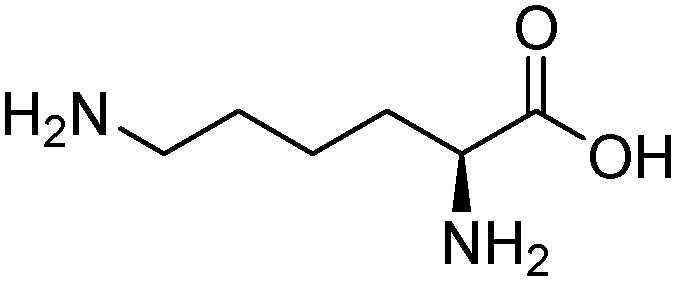
|
10.67 | 1 atm | 0.5–2.5 M | — | 0.17–1.063 | 43 |
| 298–313 K | |||||||
| Asparagine (ASN) |
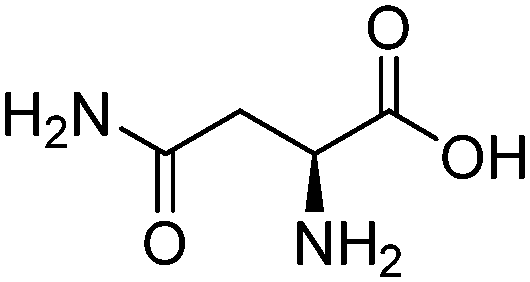
|
8.80 | 5–950 kPa | 8.5–34 wt% | 0.17–1.22 | 44 | |
| 313–353 K | |||||||
| Glutamic acid (GLU) |
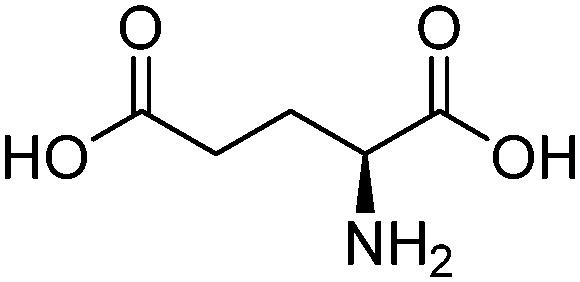
|
9.98 | 25–225 kPa | 2–10 wt% | 1442 (0.5 M) | 0.3–1.6 | 45 |
| 293–313 K | |||||||
| Glutamine (GLN) |

|
9.13 | 5–950 kPa | 9.2–36.8 wt% | — | 0.28–1.44 | 44 |
| 313–353 K | |||||||
| Glycine (GLY) |
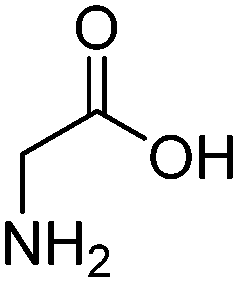
|
9.73 | 293–351 K | 0.1–3 M | — | 0.1–1.4 | 46 |
| 0.1–100 kPa | |||||||
| Histidine (HIS) |

|
9.17 | 313–353 K | 1–2 M | — | 0.6–2.4 | 47 |
| 154–4000 kPa | |||||||
| Phenyl-alanine (PHE) |
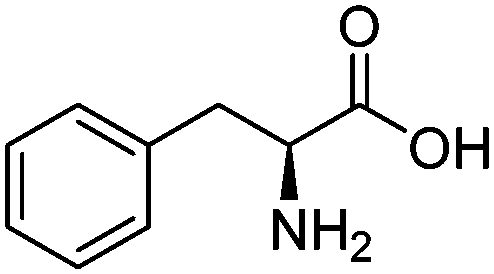
|
9.13 | 303–333 K | 10–25% | — | 0.2–1.9 | 48 |
| 200–2500 Pa | |||||||
| Proline (PRO) |

|
10.64 | 313–353 K | 7.5–27.4 wt% | 7218 (0.5 M) | 0.24–1.16 | 49 |
| 1–1000 kPa | |||||||
| Taurine (TAU) |

|
11.150 | 313 K | 1 M | — | 0.573 | 15 |
| 15 kPa | |||||||
| β-Alanine (BALA) |

|
10.24 | 313 K | 1 M | 1499 (0.5 M) | 0.721 | 15 |
| 15 kPa | |||||||
| Aminobutyric acid (AABA) |
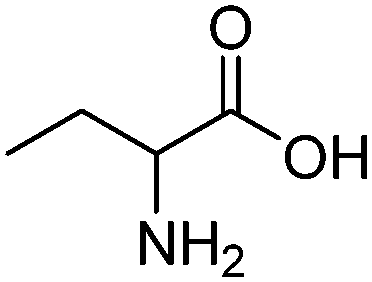
|
9.83 | 313 K | 1 M | — | 0.728 | 15 |
| 15 kPa | |||||||
| Serine (SER) |

|
9.15 | 313 K | 1 M | — | 0.619 | 15 |
| 15 kPa | |||||||
| Hydroxy proline (HYPRO) |

|
— | 313 K | 1 M | — | 0.655 | 15 |
| 15 kPa | |||||||
| Pyroglutamic acid (PGA) |

|
— | 313 K | 1 M | — | 0.224 | 15 |
| 15 kPa | |||||||
| Diglycine (DIGLY) |
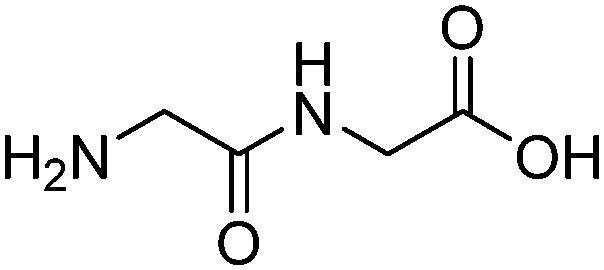
|
10.864 | 313 K | 1 M | — | 0.510 | 15 |
| 15 kPa | |||||||
| Cysteine (CYS) |

|
10.28 | 313 K | 1 M | — | 0.485 | 15 |
| 15 kPa | |||||||
| γ-Aminobutyric acid (GABA) |

|
10.556 | 313 K | 1 M | — | 0.749 | 15 |
| 15 kPa | |||||||
| 6-Aminohexanoic acid |

|
10.95 | 298 K | 0.5 M | 1266 | — | 51 |
| 0–10 kPa | |||||||
| Sarcosine (NMGLY) |

|
10.21 | 298 K | 0.5 M | 2973 | — | 51 |
| 0–10 kPa | |||||||
The CO2 absorption rate is one of the critical parameters for evaluating a good absorbent for CO2 capture, as fast kinetics results in a smaller absorption column size, or lower absorbent concentration, thus reducing the operating cost of the stripping process. The Hogendoorn's group studied the overall pseudo-first order reaction rate constant (Kov) of the potassium salt of various amino acids, including taurine (TAU), methionine (MET), glycine (GLY), GLUA, alanine (ALA), 6-aminohexanoic acid, sarcosine (N-methyl glycine, NMGLY), PRO, and ARG, as a function of the amino acid pKa.51 The results show that NMGLY, GLY and PRO exhibit comparatively higher Kov than MEA and DEA at a concentration of 0.5 M, demonstrating the great potential of amino acids salts as a replacement for conventional alkanolamines for industrial-scale application (Fig. 7). However, ARG exhibited a similar apparent rate constant despite having a pKa as high as 13.8.51 They further studied the influence of alkali cations on the apparent absorption rate and found that the rate is independent of the alkali counterions in the case of NMGLY, while the potassium salt exhibited a faster rate than the lithium salt with PRO as the counterion.
 | ||
| Fig. 7 (a) Kovvs. pKa for various potassium salts of amino acids; (b) and (c) results for (Kapp, pseudo-first-order rate constant, S−1) as a function of the potassium concentration of (b) sarcosine, (c) proline. (d) and (e) Results for (Kapp) as a function of the lithium concentration of (d) sarcosine, (e) proline. Figures reproduced from ref. 51 with copyright permission. | ||
Song et al. studied the influence of steric hindrance on the CO2 absorption and desorption rate, and the net cyclic capacity.15 As shown in Fig. 8a, the authors claimed that the stability of the zwitterionic products is influenced by the R groups attached to the amines, as they exert a steric repulsion on the two oxygen atoms of CO2. Therefore, sterically hindered amino acids allow for easier CO2 desorption. Based on this model, the authors tested 16 amino acids with different R groups, including, linear amino acids [GLY, TAU and β-alanine (BALA)], sterically hindered amino acids [ALA, aminobutyric acid (AABA), serine (SER), α-methyl alanine (AMALA) and CYS], cyclic amino acids [proline (PRO), hydroxyproline (HYPRO) and pyroglutamic acid (PGA)], and polyamino acids [(ASN, glutamine (GLN), diglycine (DIGLY) and ARG). The authors found that the ease of CO2 desorption increases when the distance between the CO2-bonded amino group and the carboxyl group decreases or in the presence of bulkier substituents, which is consistent with the predicted model (Fig. 8b and c). However, exceptional cases not following the rule were also observed: for example, the presence of a carbonyl group adjacent to the amino group (e.g., ASN and GLUA), leads to nitrogen lone pair delocalization and reduced nucleophilicity, which results in lower CO2 loading capacity. It should be noted that the net cyclic capacity of ARG is similar to that of NMGLY, despite its loading capacity being the highest among those of the tested amino acids. The authors speculated that the lower net cyclic capacity of arginate salts can be rationalized by the fact that they form stable carbamates which cannot be completely decomposed under the stripping conditions applied in this study.
 | ||
| Fig. 8 (a) Illustration of the influence of the amino acid side chain on the stability of corresponding carbamate species; (b) the relationship of the net cyclic capacity and the initial absorption rate of different amino acid salts; (c) the relationship of the net cyclic capacity and the initial desorption rate of different amino acid salts. Figures reproduced from ref. 15 with copyright permission. | ||
Clearly, the CO2 absorption rate and loading capacity varies among amino acid salts, influenced by factors such as steric hindrance, pKa value, and viscosity. Liao's group suggested that a blend of different amino acid salts in a solvent mixture could lead to a synergistic improvement, resulting in a higher CO2 loading capacity.52 To verify this hypothesis, four different types of amino acids, including GLY, with low steric hindrance on the amine group, ALA, with a methyl group in the vicinity of the amine group, PRO, with a cyclic secondary amine, and LYS, with two primary amine groups, were selected for their study. For aqueous solutions of a single amino acid salt, the percentage of CO2 captured as carbamate decreases with increasing steric bulk, with glycinate exhibiting the highest, and alaninate, with a methyl group on the alpha carbon, the lowest. Nevertheless, the absorption capacities were similar, about 0.58 molCO2 molAAS−1 for the three monoamine amino acids, and 0.74 molCO2 molAAS−1 for LYS. Interestingly, the blended mixture, containing combinations of different AAS, showed an absorption capacity of 0.91 molCO2 molAAS−1, which is over 19% higher than any of the single component systems at the same amino group concentration. The authors attributed the enhancement to a synergistic effect of the structure and basicity properties of each component, although a deeper understanding of the role of each component is needed. Based on these findings, Liao further studied the employment of microalgal biomass as the precursor for a blended mixture of AAS for CO2 capture.53 The compositional analysis revealed that ALA, GLY, GLUA, aspartic acid (ASP), LYS, LEU, and PRO are the main amino acids with a total amino acid concentration of 0.592 M obtained from microalgal hydrolysis. Most importantly, the cyclic capacity obtained from this system is as high as 1.27 molCO2 molamine−1, three times higher than the benchmark MEA solution.
A potential disadvantage of using an amino acid-based salt as an absorbent at high concentrations is the decreased solubility after CO2 loading, as this leads to increased viscosity and consequently to higher energy input, as well as reactor clogging and fouling. On the other hand, if the captured CO2 precipitates along with the amino acid salt, the equilibrium will be shifted towards the formation of carbamate, and thus higher CO2 loading capacity can be achieved. In addition, the CO2-rich phase can be separated for stripping, while the CO2-lean phase can be recirculated directly to the capture reactor. Such a strategy might contribute to a reduction of the capital cost for the regeneration process. The first example of using an amino acid salt-based biphasic system for CO2 capture was reported by Li's group.54 In this study, aqueous solutions of one of five different amino acid salts were tested as the absorbents, including ALA, ARG, LYS, SER, and ASP. After CO2 bubbling, only ALA underwent a phase change, forming a clear phase at the top and a milky phase at the bottom (Fig. 9a–c). Further studies on the composition of these two phases were performed by NMR spectroscopy, and by using tetramethylammonium chloride as an internal standard, it was confirmed that 90% of the adsorbed CO2 was concentrated in the milky phase in the form of NaHCO3 and carbamate. Benefiting from the self-concentration phenomenon, the cyclic capacity reached 0.62 molCO2 molALA−1. With such a system, only the CO2-rich phase needed the stripping process for solvent regeneration, while the CO2-lean phase could be directly reused (Fig. 9a), thus reducing the energy consumption for the regeneration step.
 | ||
| Fig. 9 (a) Illustration of the CO2 capture and regeneration process with a biphasic system, (b) the homogenous amino acid salt solution before, and (c) after CO2 loading, showing the formation of two phases. Reproduced from ref. 54 with copyright permission. | ||
Due to their high heat capacity, aqueous solutions always result in higher energy penalty during the regeneration process. The use of an organic solvent, such as ethanol, is advantageous because of the lower heat capacity and binding energy, thus, requiring less energy input during the CO2 stripping process. Based on this consideration, Shen et al. developed a prolinate potassium (PROK)/ethanol phase-change system for CO2 capture under post-combustion conditions.55 Compared to the aqueous PROK solvent, the ethanol/PROK exhibited a more than four-fold higher absorption rate, reaching 18.06 kmol m−2 s−1, which was confirmed to have benefited from the three-fold higher solubility of CO2 in the ethanol solution. During CO2 loading, the solution underwent a phase change, with the bottom phase enriched with 55–60% of the captured CO2, consisting of PRO carbamate, ethyl carbonate, and bicarbonate salts. To minimize solvent loss due to the low boiling point character of ethanol, Shen and co-workers reported in 2020 the alternative use of 2-methoxyethanol (EGME) and 2-ethoxyethanol (EGEE) as antisolvent to trigger the precipitation.56 The authors suggested that a lower dielectric constant favors the precipitation of hydrophilic CO2 captured products, and, consequently, the absorption capacity of both PROK and sarcosinate potassium (NMGLY-K)-based systems using EGME or EGEE solvent were enhanced by about 15–20% compared to their aqueous solutions. XRD and NMR analysis showed that 50–80% of the captured CO2 is precipitated in the form of bicarbonate and carbamate. Impressively, the PROK/EGME system exhibited a very low energy penalty for the regeneration of 1.87–2.61 GJ per t CO2, which is almost half the amount required for the benchmark MEA system, demonstrating the advantages of amino acid salt-based solvents for the CO2 capture applications.
A further improvement of the amino acid-based biphasic system was reported by Rahimpour and his colleagues in 2021.57 Using potassium glycinate in combination with either N,N-dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP), ethanol, or EGEE as the phase change solvent, all of the captured CO2 was concentrated in one phase while no bicarbonate or carbamate were detected in the CO2-lean phase. Additionally, they observed that the CO2 absorption capacity exhibited a non-linear response to variations in co-solvent percentages. For example, with NMP as the co-solvent, the loading capacity initially decreased with increasing amount of co-solvent compared to the neat aqueous potassium glycinate solution, scoring the lowest value of 0.60 molCO2 molAAS−1 at 10 wt% NMP. Further increasing the percentage of NMP resulted in an increased loading capacity, reaching the highest value of 0.74 molCO2 molAAS−1 at 50 wt% NMP. The authors suggested that the lower loading capacity of the solvent resulting from co-solvent addition in the range 0–30 wt% as compared to neat aqueous solution is due to a less favorable carbamate hydrolysis to bicarbonate in the presence of the co-solvent, while a further increase of the amount of added co-solvent would promote phase separation and precipitation, consequently shifting the reaction equilibrium and enhancing the loading capacity accordingly. It is clear from the above examples that by carefully choosing AAS and co-solvent, a biphasic system with higher CO2 capture capacity and lower re-generation energy penalty can be achieved.
3.2. Amino acid-based ionic liquid as the absorbent for CO2 capture
In addition to alkanolamines and amino acid salts, ionic liquids are another promising type of absorbent.58 Like amino acid salts, ionic liquids exhibit high thermal stability, low volatility, and excellent CO2 solubility.59 Unfunctionalized ILs capture CO2 by physical interaction and the absorption rate/capacity is very low, although low energy input is required for the regeneration.59b One common strategy to improve the absorption performance of ionic liquids is to functionalize the constituent cation or anion with amine groups, thus enabling the same chemical CO2 absorption that occurs in amine-based systems via the formation of carbamate and bicarbonate species.Among the functionalized ILs, AAILs have attracted considerable attention by the research community because they combine the advantages of both ILs and AAS. In 2015, Li and co-workers investigated the CO2 capture ability of an aqueous solution of the amino acid-based ionic liquid (AAIL) featuring hydroxyethyl methyl imidazole as the hydrophilic cation and GLY as the anion (denoted as [C2OHmim][GLY]).60 For comparison, solutions of MEA and sodium glycinate with the same concentration (0.4 M) were tested, and the results showed that the CO2 loading capacities of MEA, sodium glycinate, and [C2OHmim][GLY] are 0.457, 0.26, and 0.575 molCO2 molabsorbent−1 respectively, demonstrating the superior performance of the AAIL. In addition, the latter shows higher stability compared to the other two absorbents after a multiple regeneration process under high O2 concentration conditions. This demonstrates the high potential of this type of absorbents to overcome the disadvantages of amine-based solvents.
In 2014, Khanna and co-workers reported the application of a series of AAILs combining the butyl methyl imidazolium (BMIM)+ cation with nine different amino acids. It should be noted that these studies were performed under moisture-free conditions using the lean AAILs. The loading capacity increased in the order ARG > LYS > HIS > MET > LEU > GLY > VAL > ALA > PRO (Table 2). The authors speculated that the lowest CO2 loading capacity of PRO is due to the steric hindrance of its 5-membered ring, whereas the highest activity of AAILs based on ARG, LYS and HIS can be explained by the presence of an additional amine functional group in their side chains. According to the authors, the absorption as carbamate follows the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 model (see Scheme 2b), rather than the 1:1 one. Noteworthy, it has been demonstrated that the CO2 absorption capacity of ILs with amine-functionalized cations is lower than that of anion-functionalized ILs, which also include amino acid-based ILs, highlighting a further advantage of the use of AAILs for this application.
:1 model (see Scheme 2b), rather than the 1:1 one. Noteworthy, it has been demonstrated that the CO2 absorption capacity of ILs with amine-functionalized cations is lower than that of anion-functionalized ILs, which also include amino acid-based ILs, highlighting a further advantage of the use of AAILs for this application.
| Cation | Anion | Temperature, pressure | Solvent | CO2 loading capacity [mol CO2 per mol IL] | Ref. |
|---|---|---|---|---|---|
| [B4MPyr] | [L-ARG] | 298 K, 0.2–6 bar | Pure AAILs | 0.097–0.613 | 61 |
| [L-LYS] | 0.097–0.554 | ||||
| [L-HIS] | 0.062–0.524 | ||||
| [L-TYR] | 0.056–0.487 | ||||
| [GLY] | 0.070–0.475 | ||||
| [L-ALA] | 0.074–0.469 | ||||
| [L-VAL] | 0.066–0.414 | ||||
| [L-PRO] | 0.080–0.379 | ||||
| [VBIM] | [L-ARG] | 283–313 K, 0.2–4 bar | Pure AAILs | 0.061–0.990 | 62 |
| [L-LYS] | 0.053–0.761 | ||||
| [L-HIS] | 0.034–0.768 | ||||
| [GLY] | 0.048–0.602 | ||||
| [L-ALA] | 0.036–0.598 | ||||
| [L-VAL] | 0.048–0.507 | ||||
| [L-PRO] | 0.044–0.493 | ||||
| [CHO] | [L-PRO] | 298–313 K, atm pressure | 16 wt% in DMSO | 0.66–0.74 | 63 |
| [GLY] | 298 K, atm pressure | 16 wt% in DMSO | 0.86 | ||
| [BMIM] | [ARG] | 298 K, 2 bar | Pure AAILs | 0.62 | 64 |
| [LYS] | 0.48 | ||||
| [HIS] | 0.45 | ||||
| [MET] | 0.42 | ||||
| [LEU] | 0.38 | ||||
| [GLY] | 0.38 | ||||
| [VAL] | 0.39 | ||||
| [ALA] | 0.39 | ||||
| [PRO] | 0.32 | ||||
| [C2OHmim] | [GLY] | 303 K, 10 kPa | 0.4 M in water | 0.575 | 60 |
| [LYS] | 295 K, 1 bar | 50 wt% in water | 2.2 | 60 | |
| [PRO] | 308 K, 1 bar | 50 wt% PEG2000 | 0.61 | 65 | |
| [GLY] | 298 K, 1 bar | 50 wt% in DMSO | 0.78 | 66 | |
| [ALA] | 0.79 | ||||
| [SER] | 0.61 | ||||
| [PRO] | 0.84 | ||||
| [PHE] | 0.70 | ||||
| [SAR] | 0.71 | ||||
| [P4444] | [ALA] | 298 K, 1 bar | Pure AAILs | 0.77 | 67 |
| [ILE] | 0.85 | ||||
| [GLY] | 0.86 | ||||
| [MET] | 0.93 | ||||
| [PRO] | 0.88 | ||||
| [P66614] | [GLY] | 298 K, 1 bar | Pure AAILs | 0.97 | |
| [MET] | 0.99 | ||||
| [PRO] | 0.91 |
Comparing the behavior of different AAILs, it can be concluded that the CO2 absorption capacity is mainly determined by the number of amine groups and the structure of the amino acid side chain (Table 2): the more amino groups, the more potential binding sites for CO2 chemical sorption are present, which on the other hand, can be negatively affected by the bulkiness of the side chain, hindering the CO2 access to the amine groups. Thus, in most of the reports on AAILs, ARG shows the highest absorption capacity, followed by LYS and HIS, regardless of the cation which was either 1-butyl-4-methyl pyridinium,61 vinyl imidazolium, or butyl methyl imidazolium.62 PRO was reported to be the least active amino acid among those without a second amino functionality, due to the high steric hindrance of the five-membered ring. An exception was reported by Dai and co-workers: [P4444][PRO] showed a higher capacity compared to [P4444][GLY] and [P4444][ALA] due to the lowest stability of the deprotonated dianion of PRO-carbamic acid, and thus the absorption is dominated by a 1:1 reaction mechanism.67
The values of the CO2 loading capacities of different AAILs reported in the literature show that the cation also affects the performance, as well as the reaction mechanism. For example, Dai and co-workers systematically investigated the absorption mechanism of pure AAILs under moisture-free conditions.67 In this study, AAILs were prepared using tetra-alkyl phosphonium cation and amino acid-derived anions, including, ALA, isoleucine (ILE), GLY, MET and PRO. Contrary to the 2:1 absorption model observed in Khanna's work,64 they claimed that the 1:1 model dominates in all the cases, with carbamic acid as the main CO2 absorption product, which was confirmed by IR and NMR studies. [P4444][PRO] (P4444, tetrabutylphosphonium cation) shows the highest absorption capacity of (0.91 molCO2 molIL−1), followed by [P4444][GLY] (0.86 molCO2 molIL−1), and [P4444][ALA] (0.77 molCO2 molIL−1). Calculations indicate that the deprotonated form of the PRO-carbamate dianion has a strong repulsion between the two carboxyl groups, making it less stable compared to the ALA-carbamate and GLY-carbamate dianions, and consequently, less carbamate is formed following the 2:1 model (Table 2). This result is in contrast to that reported by Khanna and co-workers,64 who concluded that the [BMIM][PRO] exhibited the lowest capacity, demonstrating the important role of cations in determining the capture capacity. In another example, Riisager and co-workers analyzed the loading capacities of trihexyl(tetradecyl) ammonium lysinate [N66614][LYS] and trihexyl(tetradecyl) phosphonium lysinate [P66614][LYS], which reached 2.0 and 1.6 molCO2 molAAILs−1, respectively.68 The authors demonstrated that the basic carboxyl group of the amino acid acts as the catalyst for rapid CO2 absorption, and in the case of [P66614][LYS], a strong hydrogen bond interaction between the formed carbamic acid and the carboxyl group of the amino acid inhibits the catalytic effect, whereas in the case of [N66614][LYS], the interaction is much weaker, allowing the reaction to proceed in a stoichiometric manner (Fig. 10). Therefore, the above work demonstrates the importance of selecting the right cations when exploring new types of AAILs for CO2 capture applications.
 | ||
| Fig. 10 Structure differences in the CO2 captured adduct formed by [N66614][LYS] and [P66614][LYS] absorbents. Figure reprinted from ref. 68 with copyright permission. | ||
Choline (CHO) is naturally abundant and biodegradable, making it a potentially more environmentally friendly cation for AAILs compared to pyridine- and imidazole derivatives. In 2019, Bordiga and co-workers reported the synthesis of choline glycinate [CHO][GLY] and choline prolinate [CHO][PRO] AAILs from choline chloride, and their application in CO2 capture.63 In this study, the AAILs were diluted in dimethyl sulfoxide (DMSO) to overcome the problem of their high viscosity. Both [CHO][GLY] and [CHO][PRO] showed a CO2 loading capacity of about 0.70–0.86 molCO2 molAAILs−1, mostly in the form of carbamate (max. 0.5 molCO2 molAAILs−1), and carbamic acid species (max. 1 molCO2 molAAILs−1). The absorption mechanism was further confirmed by ATR-IR. For [CHO][GLY], the authors observed the formation of carbamate in the first step, and then, proton transfer occurred between the zwitterionic GLY and the carbamates, forming carbamic acid and choline glycinate, as shown in Fig. 11a. In the case of [CHO][PRO], carbamic acid was formed directly from [CHO][PRO] via a 1:1 mechanism, in parallel with the formation of carbamates. However, the formation of carbamic acid from carbamate was excluded (Fig. 11b). This work also showed that the steps involved in CO2 capture are different when using different anions, although the final CO2 loading is similar. In continuation of this work, Bordiga and Bocchini further evaluated the CO2 capacity of choline cation-based AAILs with different amino acids in cycling experiments, including PRO, GLY, ALA, SER, PHE, and NMGLY. They showed that [CHO][PRO] has the highest stability with only 15% of capacity loss after 10 cycles. Noteworthy, they have demonstrated that the [CHO][AA]-DMSO system requires a lower temperature (70–80 °C) for CO2 desorption compared to the classical aqueous solution (over 100 °C).69
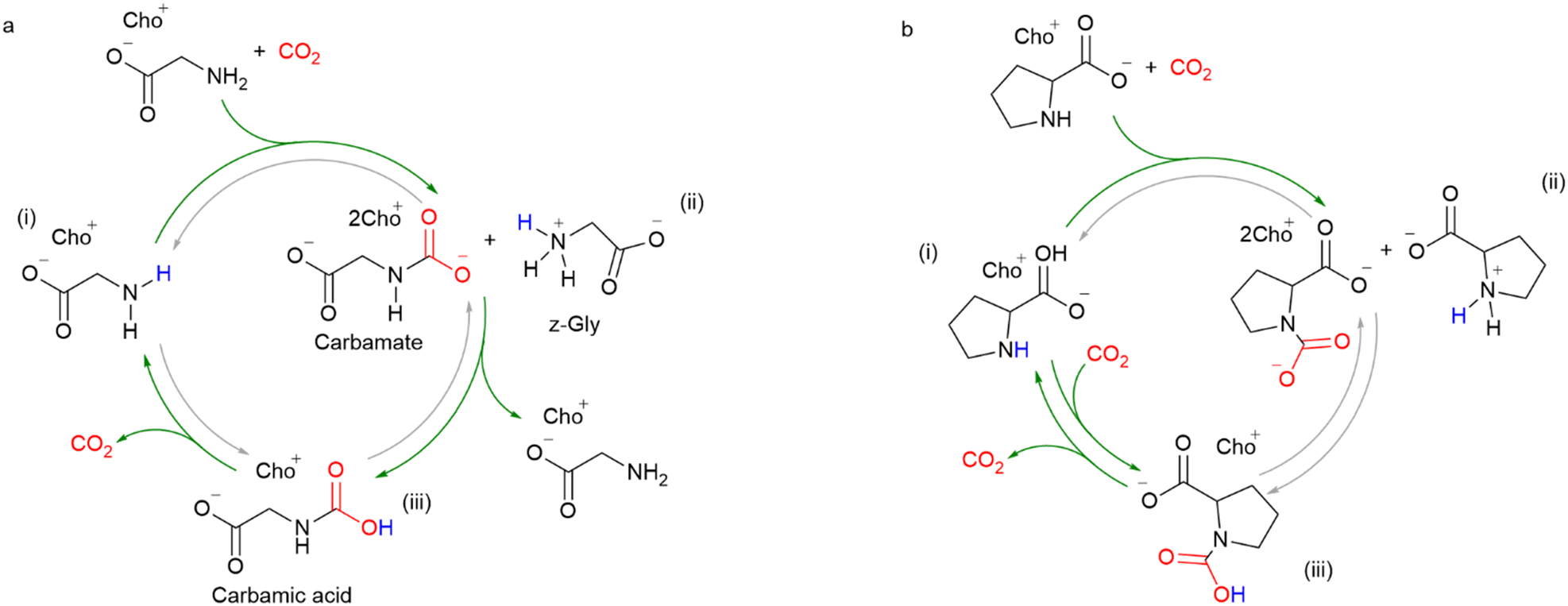 | ||
| Fig. 11 Confirmed reaction pathway by ATR-IR (green arrow) and reaction pathway not observed (grey arrow) of (a) [CHO][GLY] and (b) [Cho][Pro] for CO2 capture process. Reproduced from ref. 63 with copyright permission. | ||
3.3. Blended mixtures of amino acids and other absorbents
Amino acid-based sorbents often have high viscosity compared to the traditional amine solvent, which results in increased resistance to fluid flow, requiring more pumping power and potentially affecting the overall performance and efficiency. Although increasing the operating temperature can significantly reduce the viscosity in some cases, the energy required and the resulting reduction in CO2-absorption capacity cancels the benefits of using a pure amino acid-based solvent. Alternatively, blending the amino acids with other absorbents, such as amines, carbonates, and ILs, can provide a potential replacement for alkanolamine-based solvents. A good example is provided by the technology developed by Siemens,70 also known as the PostCap™ process, in which the mixture of MEA, ammonia, and amino acid salts in aqueous solution is used as absorbent. With the PostCap™ absorbent mixture, less than 20% of MEA was lost during one year of operation. Another study reported by Tan and co-workers employed piperazine (PZ) (15 wt%), diethylenetriamine (DETA) (15 wt%), and sodium aliphatic diamine sulphonate (NaADS) (10 wt%) as a blend for CO2 capture.71 The results show that, although the viscosity of this mixture is 1.48 times higher than that of the PZ (15 wt%)/DETA (15 wt%) mixture, similar CO2 absorption rate and capacity were achieved. In this case, an advantage of the amino acid addition is a 7.3% reduction in regeneration energy. Furthermore, the addition of an amino acid to the blend considerably slowed down the degradation rate of the absorbents, as the dissolved oxygen is half compared to the PZ (15 wt%)/DETA (15 wt%) mixture. Similarly, blending AAILs and conventional alkanolamines provides efficient CO2 capture reagents. For example, Zhang and co-workers demonstrated that the mixture of MDEA and [N1111][GLY] has a higher absorption rate compared to MDEA solvent.72 Zhou's group applied [N1111][GLY] as a promoter for the absorbent 2-amino-2-methyl-1-propanol (AMP), which showed a comparable acceleration effect to PZ, MEA, and DEA.73In a very recent work by Zhang ad co-workers, a novel absorbent formulation with significant industrial potential was developed.74 The optimal absorbent composition containing 5.02 wt% [N1111][Gly] + 17.06 wt% PZ + 20.00 wt% MDEA + 57.92 w% H2O, noted as IPMH, demonstrated superior capture capacity compared to a conventional 30 wt% MEA solution. More notably, it exhibited a 22.7% higher regeneration efficiency and a 30% reduction in regeneration energy consumption. The authors employed a potentiometric method to identify the absorbed species and found that in the IPMH system, 70% of the captured CO2 existed as bicarbonate, compared to only 10% in the MEA solution, which explains the lower regeneration energy requirement, as bicarbonate is more readily desorbed than carbamate.
Aqueous K2CO3 has been demonstrated as another alternative absorbent for CO2 capture due to its high thermal stability and low regeneration energy. However, the sluggish absorption rate hinders its practical large-scale application. In response, various promoters, such as piperazine, amine, and amino acids,75 have been added to improve the capture performance. Among these additives, amino acids are superior to the other two in terms of their thermal stability and low vapor pressure. In 2013, Stevens and co-workers showed that the addition of either GLY, NMGLY, or PRO to a 30 wt% K2CO3 aqueous solution with a final amino acid concentration of 1 M, increased the total CO2 uptake rates by 22, 45, and 14 times, respectively.76 In addition, they also demonstrated that the promoting effect of amino acids bearing either a primary or secondary amino group is comparable to that of common alkanolamines having a primary (in the case of MEA), or secondary amine (in the case of DEA), respectively. Hu and co-workers further demonstrated that the amino acid-based promoters are more effective than amines in boosting the CO2 desorption rate and cyclic capacity of K2CO3 solutions.77
Non-functionalized ILs are also promising absorbents for CO2 capture via a physical process, albeit with low capacity and high viscosity. On the other hand, the high solubility of CO2 in ILs can be advantageously exploited by combining it with the chemical sorption exhibited by amines and amino acids, Zhang and coworkers evaluated the use of sodium glycinate (GLY-Na) and butyl methyl imidazolium tetrafluoroborate ([BMIM][BF4]) as the absorbents and demonstrated that the blend has a better resistance to oxidative degradation compared to the benchmark MEA solution. In addition, the regeneration energy can be reduced compared to the pure sodium glycinate solution.2a
In summary, amino acids with a non-protonated amino group can serve as effective absorbents. The cation can be either a simple metal ion, such as potassium, or more complex ones, i.e., ammonium, phosphonium, imidazolium, to obtain an amino acid-based ionic liquid. Absorbents based on such ionic species exhibit low volatility and minimal solvent loss compared to those based on amines. In addition, their biocompatibility, biodegradability, comparatively high absorption capacity, and efficient capture kinetics make amino acid-based sorbents a promising alternative to traditional alkanolamine sorbents. On the other hand, the high viscosity of AAILs, which further increases after CO2 absorption, would increase the energy penalty, and thus hinder their practical application. The use of blends, combining AA/AAIL and other sorbents, and biphasic systems have been investigated to mitigate this drawback. However, a reactive CO2 capture system does not require the CO2 regeneration step. Instead, the captured CO2 is used directly for hydrogenation, forming formic acid/methanol while simultaneously releasing the absorbents, potentially circumventing the viscosity drawbacks. In the following section, the recent progress on the reactive capture of CO2 to methanol and formic acid will be discussed.
4. Conventional amine-based CO2 transformation to methanol and formates
4.1. Basis principles for CO2 hydrogenation to methanol
On an industrial scale, CH3OH is produced from fossil fuel-derived synthesis gas with added CO2 (CO/H2, and CO2) over a heterogeneous catalyst comprising copper and zinc oxides, supported on alumina (Cu/ZnO/Al2O3) at high pressure and elevated temperature (>200 °C).78 Alternatively, CH3OH can be obtained by hydrogenation of CO2 with hydrogen, preferably derived from water splitting using renewable energy.79 Both heterogeneous80 and homogeneous metal catalysts81 are being developed to catalyze this process. Homogeneous molecularly defined catalysts, while far from being ready for practical application, usually operate under milder conditions and are prone to easier structural modification in search of better efficiency and stability. A perusal of the literature shows that such catalysts can be divided into two broad categories, depending on whether they operate in a neutral or slightly acidic medium, or in a basic one. In the first case, HCOOH, formed by the initial reduction of CO2, is converted into a formate ester through reaction with an alcohol, used as the solvent, or in catalytic amount to facilitate the reaction in its initial stages. The alcohol can be CH3OH itself, thus self-breeding the reaction. Hydrogenation of the ester affords CH3OH and regenerates the assisting alcohol.82Base-compatible catalysts should be better suited for hydrogenation directly in the basic media used to scrub CO2, from flue gas or air. Thus, these catalysts have been more extensively explored for combined CO2 capture and hydrogenation. Here, the species present in solution following CO2 capture and the experimental conditions required for the conversion of CO2 to CH3OH depend on the nature of the absorbent, either an inorganic base, such as an alkali hydroxide, or an organic nitrogen base; and the solvent, either water or a water-lean system. In the presence of an organic solvent, a further distinction must be made between an alcohol or a non-protic solvent. Regardless of the reaction medium and the possible reaction intermediates, the hydrogenation of CO2 to CH3OH generates one equivalent of H2O and therefore the consequences of its presence must be considered, especially at higher conversions. Scheme 3 gives an overview of the species which are formed in solution after CO2 has been captured, depending on the type of base used. Only those cases are illustrated here that were combined with in situ homogeneously catalyzed hydrogenation of the captured CO2 to CH3OH. For a more general and in depth description of the various species formed in solution following CO2 capture with amines, their 1H and 13C NMR identification, the relative equilibria and how these are influenced by intrinsic properties (Lewis and Brønsted basicity) and concentration of the amine, by the nature of the solvent, the partial CO2 pressure and the reaction temperature, the reader is referred to the articles by Kortunov and co-workers.83
 | ||
| Scheme 3 CO2 capture with amines, either 1°/2° (path a) or 3° (path b), and alkali hydroxides MOH (path c) and integrated metal-catalyzed hydrogenation to formate and methanol. Depending on reaction conditions, formation of methanol can proceed through hydrogenolysis of formamide or ester. | ||
As mentioned in the introduction, large-scale post-combustion CO2 separation processes are mainly based on the deployment of aqueous solutions of amines and aminoalcohols such as MEA, DEA, and MDEA.84 Amines react with CO2 according to the equilibria depicted in Scheme 3, where a distinction is made between primary/secondary (path a) and tertiary amines (path b). When a primary/secondary amine is used, the nucleophilic amine attacks the electrophilic carbon atom of the CO2 molecule affording the zwitterionic of the resultant carbamic acid, which is then rapidly deprotonated by a second mole of free amine, acting as a Lewis base, to give a more stable ammonium carbamate (Scheme 3, path a).85 Under these conditions, the amine/CO2 ratio is 2:1, i.e., the maximum absorption capacity is 0.5 moles of CO2 per mole of amine. In the presence of water, the ammonium carbamate can be hydrolyzed, releasing the free amine, to afford the corresponding bicarbonate. In this case, the amine/CO2 ratio is 1:1, meaning that one mole of CO2 has been captured per mole of amine. The ratio between carbamate and (bi)carbonate products is influenced by various parameters, for instance the CO2 up-taking, the Brønsted and Lewis basicity of the amine, and its concentration (i.e., the amine/water stoichiometry). In the presence of a primary/secondary amine, a stepwise pathway to CH3OH can be envisioned. The first step is the metal-catalyzed reduction of the captured CO2 to ammonium formate. Ammonium formate can then be thermally dehydrated to formamide, whose further metal-catalyzed hydrogenolysis yields CH3OH and releases the amine for the next capture/hydrogenation cycle.86
Unlike primary and secondary amines, tertiary amines cannot form stable carbamates with CO2 in aqueous or non-aqueous solutions because they lack a transferable proton. They are weak Lewis bases but can be relatively strong Brønsted bases, thus promoting the formation of ammonium bicarbonate, where water acts as the nucleophile (Scheme 3, path b). When an alcohol is used as the solvent, an alkyl carbonate is the prevailing product. In this case, after the first hydrogenation step, further reduction to CH3OH can occur by hydrogenolysis of the formate ester, in equilibrium with ammonium formate due to the high alcohol concentration and despite the elevated temperature and the basic conditions.87 Because of the increasing steric bulk, tertiary amines have kinetically lower efficiencies for CO2 capture. However, reduction of formate esters should be energetically more favorable than the reduction of formamides, which explains the interest in this approach.82a
Due to their high CO2-binding affinities, alkali hydroxides are better suited to capture CO2 at the lower concentration present in air (direct air capture).88 If an alcohol is used in which both the metal hydroxide and the product of CO2 capture, the metal alkyl carbonate, are soluble, then this approach can be advantageously exploited to convert CO2 to CH3OH, again through the hydrogenation of an intermediate formate ester (Scheme 3, path c).89 Key contributions demonstrating the feasibility of the approach described in Scheme 3 are due to Sanford,13 Milstein,8 Ding,90 Prakash7a,91 and Wass.92
Chart 1 illustrates the catalysts and amines they used. The TONs in CH3OH are relative to single experiments under the reported conditions. There has been a proliferation of studies documenting efforts to improve solvent (physisorption) and base (chemisorption) CO2 capture capability, homogeneous catalyst efficiency, productivity and selectivity, and recyclability of active components. Excellent reviews have summarized these studies.6b,93 Our discussion is not intended to be comprehensive and will focus on selected examples which rely on the use of 1°/2° (poly)amines because primary amino groups are mainly responsible for CO2 chemisorption in amino acid-based capturing media. Besides, a perusal of the literature shows that Ru-MACHO-BH 1 has been used in several capturing media under a variety of conditions. The steric and electronic properties of this catalyst can be modulated by changing the substituents at the phosphorus and nitrogen of the pincer ligand (Chart 2). In few cases, similar PNP complexes, in which ruthenium was replaced by iron7a,94 and manganese94a,95 were tested as well under comparable conditions, allowing a direct comparison with Ru-MACHO-BH. Therefore, as an introduction for amino acid-aided CO2 capture and following catalytic hydrogenation, we would like to highlight some general trends which emerged from these studies. Here, Ru-MACHO-BH serves as an example how reaction conditions, absorbent medium, and catalyst structure influence the outcome of the integrated CO2 capture and hydrogenation and how all these parameters can be modified to ease product separation and absorbent/catalyst recyclability.
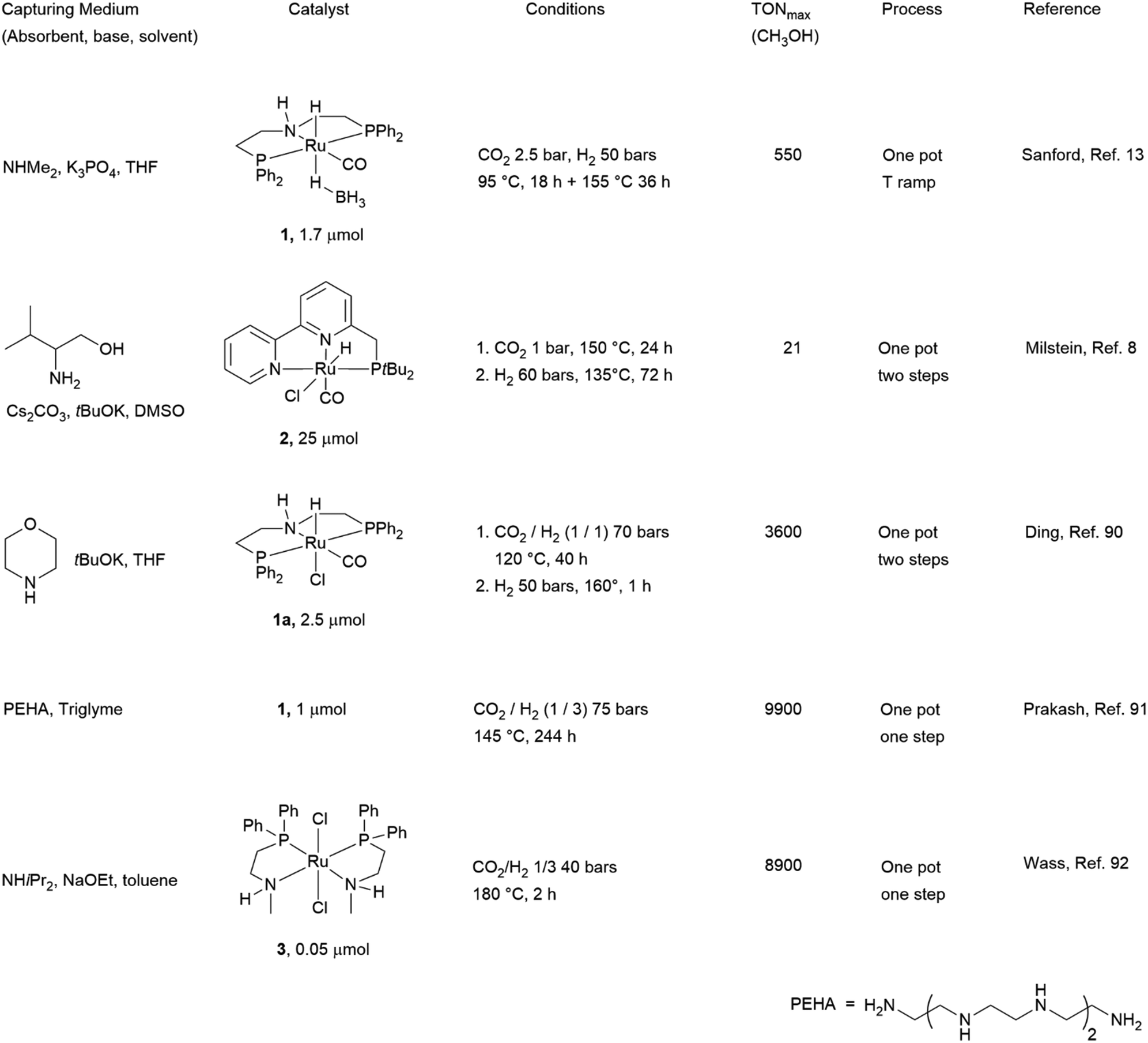 | ||
| Chart 1 Seminal studies demonstrating the feasibility of combined amine-based CO2 capture and homogeneously metal-catalysed hydrogenation to CH3OH. | ||
 | ||
| Chart 2 Range of catalysts belonging to the aliphatic PNP MACHO-type family which have been applied to the combined amine-based CO2 capture and homogeneously catalyzed hydrogenation to HCOO−/CH3OH. | ||
4.2. Influence of catalyst structure on the efficiency and selectivity in CO2 hydrogenation to methanol
Ru-MACHO-BH 1 belongs to the family of catalysts that realize their activity through metal–ligand cooperativity,96 due to the presence of a PNP ligand bearing an –NH group coordinated to ruthenium, which not only defines the steric and electronic properties of the metal center, but also contributes to substrate activation and facilitates catalysis. The –NH group can hydrogen-bond to the incoming substrate,97e.g. CO2, carbamate, and formamide, enhancing its electrophilicity and properly orienting it for the transfer of the metal hydride to the electrophilic carbon, and with the reduced product stabilizing it.98 This pathway may not require prior coordination of the substrate to the metal and proceeds via a so-called outer-sphere mechanism.99 Cooperativity may also contribute to the activation of H2. In a basic environment, the –HN group in the MACHO family catalyst precursor (Chart 2) can be deprotonated with the elimination of HX (X = BH4−, Cl−, Br−), producing a 16 electron metal-amido species (Scheme 4).100 Such species are able to heterolytically split H2, which adds to the metal–nitrogen bond, thereby regenerating the metal–hydride. Here, the ligand nitrogen acts as an internal base. A reduction in catalyst activity when the hydrogen on the nitrogen is replaced by an alkyl group suggests that ligand–metal cooperativity may indeed be operative, allowing an otherwise energetically more demanding transformation. Alternatively, if a vacant coordination site on the catalyst becomes available, H2 can coordinate to the metal forming a σ-adduct (Scheme 4). An exogenous base can then abstract a proton from the coordinated dihydrogen and regenerate the metal hydride. This reactivity is also open to catalyst 1e (Chart 2), which, because its ligand nitrogen is methylated, cannot engage in ligand–metal cooperativity but is still an efficient hydrogenation catalyst.101 Metal–ligand cooperativity is also possible for catalysts such as 2 (Chart 1), whose pincer ligands contain a pyridyl group that can undergo dearomatization upon deprotonation of a distal carbon (Scheme 4).102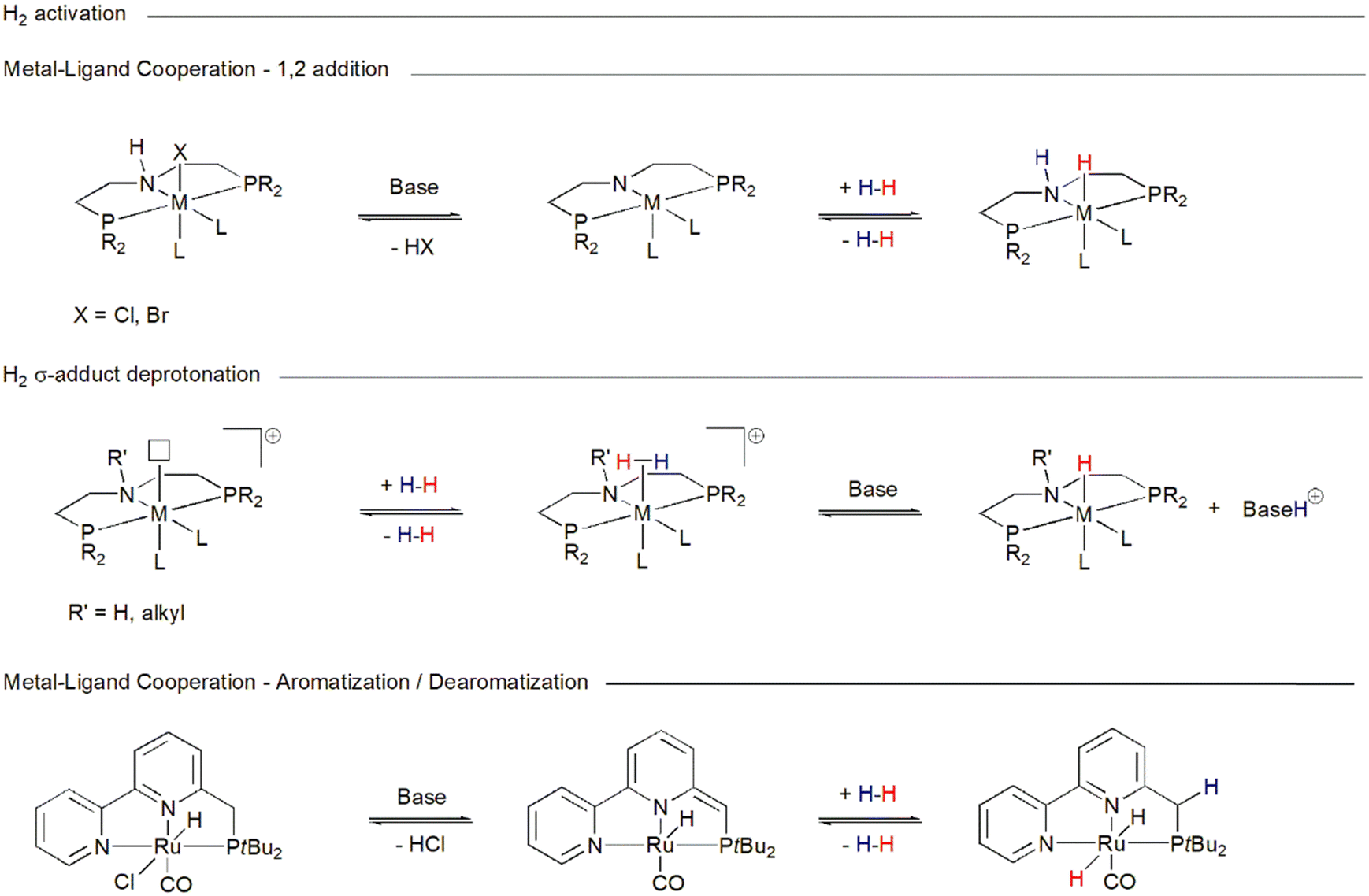 | ||
| Scheme 4 Pathways accessible to catalysts in Charts 1 and 2 for heterolytic H2 activation. | ||
In her seminal work reporting the first example of the combined use of a secondary amine Me2NH as a capturing agent for CO2 and Ru-MACHO-BH as a catalyst for the hydrogenation of the resulting carbamate to methanol via formamide, Sanford highlighted some key issues that need to be tackled when applying this approach.13Scheme 4 illustrates the main steps and the simplified underlying catalytic cycle, as proposed later on by Prakash and co-workers based on mechanistic studies.91 The reaction was carried out in THF, in the presence of an exogenous base, K3PO4, without which no catalyst turnover was observed. A temperature ramp from 95 (18 h) to 155 (36 h) °C was applied to optimize the methanol yield while minimizing catalyst decomposition at higher temperature: by applying 2.5 bar CO2 and 50 bar H2, a maximum TON of 550 in CH3OH and 1870 in formate/formamide were achieved. The first product, formate, likely arises from preferential reduction of free CO2 in equilibrium with dimethylammonium dimethylcarbamate, due to the higher electrophilicity of the former. The hydrogenation of CO2 to HCOOH is an endergonic process which becomes thermodynamically more favorable in the presence of dimethylamine which stabilizes HCOOH as dimethylammonium formate, thus shifting the equilibrium. This reaction is favored at low temperatures while hydrogenolysis of dimethyl formamide is the most energetically demanding step among those which comprise the reduction of captured CO2 to CH3OH and requires high hydrogen pressures. The temperature ramp proposed by Sanford represents the first solution to accommodate these contrasting requirements.13 155 °C defines the temperature at which Ru-MACHO-BH decomposition sets in: if the use of a smaller amount of catalyst warrants higher TONs, extension of reaction time to improve CH3OH yields should be performed at lower temperature.
Subsequently, Prakash and colleagues pioneered the direct capture of CO2 from the atmosphere and its subsequent hydrogenation to CH3OH using the same catalyst.7a This innovative method represented a significant advancement over the previous system, as it employed pentaethylenehexamine, PEHA (Chart 1), as the capturing agent. This was due to the high nitrogen content and high boiling point of PEHA, which rendered it a more suitable alternative to Me2NH. This choice is advantageous for substantial CO2 absorption, as evidenced by a high CO2/amine and subsequent CH3OH/mole amine ratio. PEHA is both affordable and readily available. Additionally, its combination with a high-boiling solvent is imperative for facilitating CH3OH separation and recovery while minimizing loss of the capturing phase. Through the optimization of reaction conditions, a TON of 1200 in CH3OH was achieved in a single experiment using Ru-MACHO-BH4 (20 μmol), PEHA (5.1 mmol), and triglyme (10 mL) at 145 °C for 200 hours with 75 bars of CO2/H2 (1/9). It was demonstrated that the TON could be elevated to 2150 through the execution of five recycling experiments, each with a duration of 40 hours, under conditions that were otherwise identical. Following each experiment, the produced CH3OH and H2O were distilled off, and the remaining solution, containing the catalyst and amine, was pressurized again. A number of aspects concerning the catalytic system were highlighted, too.91 In the absence of amine, traces of CH3OH were detected. However, when comparing different amines, higher CH3OH yields were obtained with 1,2-diamines, combining a primary and a secondary amine, or polyamines containing this structural motif as repeating unit. Furthermore, the CH3OH yield increased with higher amine content (up to a certain extent) and in the presence of a stronger base such as K3PO4. However, this was not a prerequisite for CH3OH formation, contrasting with Sanford's system.103 The gas phase analysis revealed the presence of 0.2–0.4% CO after the reaction. This amount was found to be reduced by decreasing the CO2/H2 ratio from 1/3 to 1/9, while maintaining a total pressure of 75 bars and a reaction temperature of 145 °C. Moreover, an increase in the H2 pressure, concomitant with a decrease in the CO2 pressure, has been observed to enhance the conversion of formamide to CH3OH, under conditions of constant total pressure.91 As illustrated in Table 3, the yields of formate, formamide, and methanol were affected by modifying the structure of Ru-MACHO-BH 1 by replacing the phenyl substituents at phosphorus with more electron-donating alkyl groups.91 Catalyst 1a, with a chloride in place of BH4−, is also included for a more consistent comparison with 1b (iPr), 1c (Cy) and 1d (tBu). While all catalysts promoted the formation of formate and formamide, which accumulates in solution, hydrogenation of the latter to CH3OH proceeded more efficiently with catalysts 1 and 1a with TON of 1050 and 1040, respectively (Table 3, entries 1 and 2), while was modest with 1b (TON 320, Table 3, entry 3) and poor with 1c (TON 50, Table 3, entry 4). No CH3OH was detected with 1d (Table 3, entry 5). Noteworthy, when comparing the performance of 1/1a and 1b, it appears that the latter is overall a better catalyst for the reduction of CO2 to formate and thus for the build-up of formamide. Table 3 also shows that the formation of higher amounts of CH3OH is accompanied by small amounts (0.22%) of CO. Scheme 5 illustrates the catalytic cycle underlying the amine-assisted CO2 hydrogenation to CH3OH, as proposed by Prakash and co-workers based on their mechanistic studies. According to the authors, CO likely arises from decarbonylation of formaldehyde, which is the product of formamide hydrogenolysis, and the reduction of which finally leads to CH3OH.91 CO can act as a poison to the catalysts because of the formation of cationic dicarbonyl species F (Scheme 5), which were identified as resting states with all the catalysts tested. F can be brought back into the catalytic cycle if dihydrogen is able to displace the extra carbonyl ligand. The latter is more labile in the resting state arising from 1/1a explaining the better performance of 1/1a in reducing CO2 to the CH3OH level. CO detachment becomes more and more difficult with catalysts 1b–d which, because of the more electron-donating substituents at phosphorus, possess a metal center that binds CO more tightly through enhanced back-donation. Therefore, even trace amounts of CO can turn F into a dead-end, so that less and less of the active catalyst is available for the reduction of formamide to CH3OH, as shown by the data in Table 3. Therefore, within this family of catalysts, Ru-MACHO-BH appears to be the catalyst of choice if the captured CO2 is to be hydrogenated to CH3OH. Its robustness was demonstrated by Prakash and co-workers in a long term experiment lasting more than 10 days, when with just 1 μmol of 1, 5.1 mmol PEHA in triglyme, 10 mL, at 145 °C, 75 bars CO2/H2 1/3, a total of 9.9 mmol MeOH was obtained, corresponding to a TON of 9900.91
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Formatea (mmol) | Formamidea (mmol) | CH3OHa (mmol) | COb (%) | TON formate + formamide | TON CH3OH |
| Reaction conditions: PEHA 5.1 mmol; catalyst 10 μmol; K3PO4 1 mmol; triglyme 10 mL; CO2/H2 1/3 75 bars; 145 °C, 40 h.a Yields were determined from 1H NMR spectra using 1,3,5-trymethoxybenzene (TMB) as internal standard.b CO detection limit 0.099%.c No K3PO4 was used. Adapted from ref. 91. | |||||||
| 1c | 1 | 1.2 | 8.0 | 10.5 | 0.21 | 920 | 1050 |
| 2 | 1a | 1.6 | 8.1 | 10.4 | 0.21 | 970 | 1040 |
| 3 | 1b | 1.1 | 22.6 | 3.2 | nd | 2370 | 320 |
| 4 | 1c | 1.0 | 14.7 | 0.5 | nd | 1570 | 50 |
| 5 | 1d | 1.6 | 17.5 | nd | nd | 1910 | 0 |

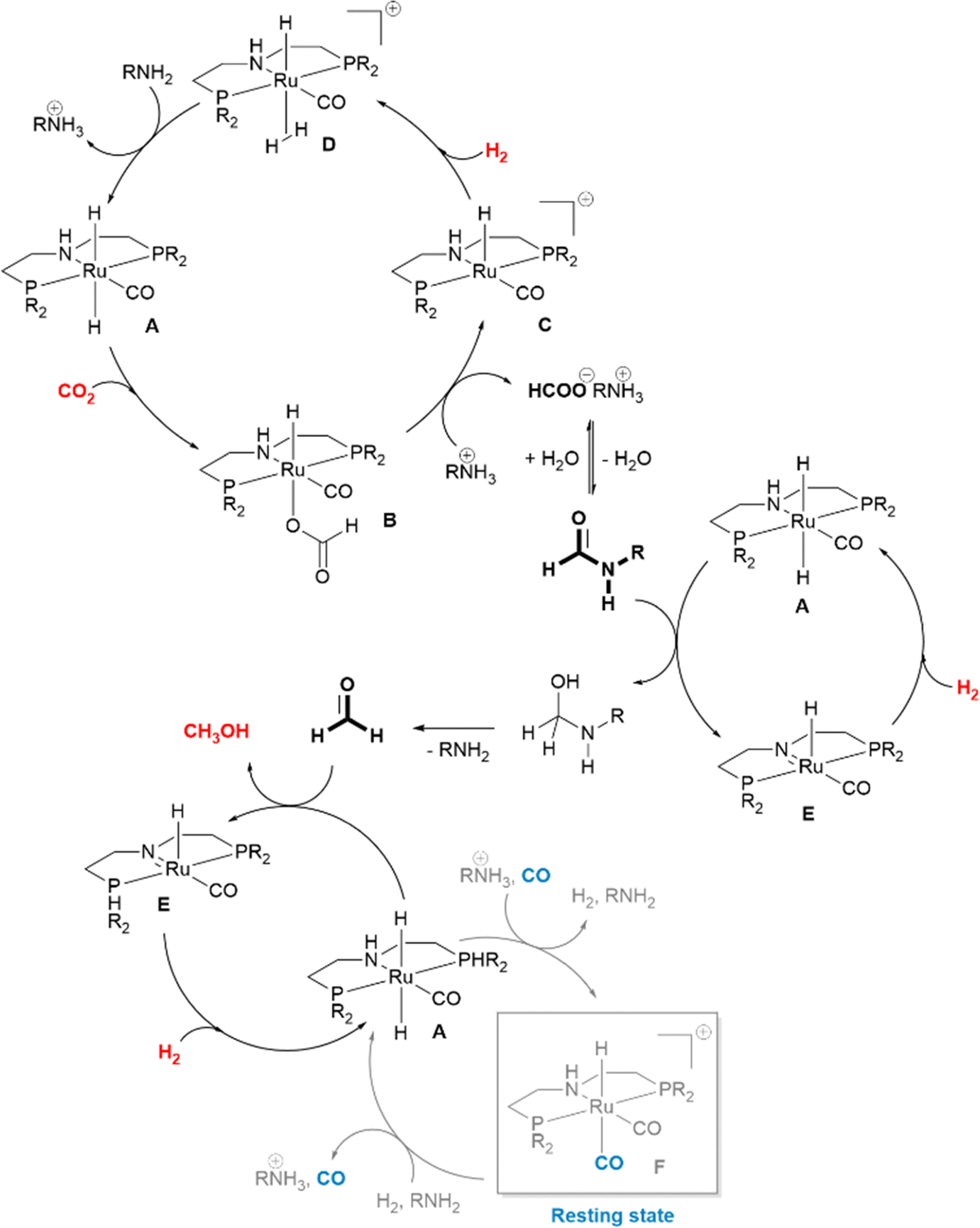 | ||
| Scheme 5 Proposed general mechanism for the amine-assisted hydrogenation of CO2 to methanol promoted by Ru-MACHO-type catalysts. Metal-mediated formaldehyde decarbonylation is a possible source of CO, explaining the formation of the observed resting state F. Adapted from ref. 91. | ||
Another interesting aspect emerges from a comparison of the performance of Ru-MACHO 1a with that of its congener 1e wherein the hydrogen at the ligand nitrogen has been replaced by a methyl group, thereby precluding the possibility of metal–ligand cooperativity. It has been shown that under comparable conditions, 1e exhibited a level of efficacy in promoting the hydrogenation of the PEHA-captured CO2 to formate, that was comparable to that of 1a. However, in contrast to 1a, 1e was unable to catalyze the hydrogenolysis of formamide to CH3OH.7a A similar behavior has been observed by Weiss and co-workers applying catalyst 3 (Chart 1). Under the conditions outlined in Chart 1, this catalyst has achieved one of the highest TONs, 8900, in the diisopropylamine-aided hydrogenation of CO2 to CH3OH.92 Analogous to Ru-MACHO, this catalyst can function through metal–ligand cooperativity. If the amino group at the ligand bears not one but two methyl groups, then the hydrogenation stops at the level of formate, resulting in the exclusive formation of formamide. These two examples support the notion that while for the base-promoted metal-catalyzed hydrogenation of CO2 to formate two catalytic pathways are open and equally productive, either metal-centred99 or ligand-assisted, for the metal-catalyzed hydrogenolysis to CH3OH the latter seems mandatory.
Pincer PNP complexes334 and {FeHBr(CO)[HN(CH2CH2PiPr2)2]} 5 (Chart 2) based on more abundant and inexpensive metals, were also tested for the integrative amine-assisted CO2 capture and hydrogenation to CH3OH.94a After capture with an aqueous solution of PEHA, CO2 was hydrogenated in a biphasic 2-MTHF/water system. Both catalysts were as efficient as the Ru-MACHO one for the hydrogenation of the captured CO2 to formate, but only small amounts or no methanol were obtained in the hydrogenation of the accumulated formamide with 4 and 5, respectively. The reasons behind the poor performance of 4 and 5 were highlighted in two dedicated studies by Prakash95a and Bernskoetter,94b respectively. It was demonstrated that, when using morpholine as the amine, the two catalysts can promote both the reduction of CO2 to formyl morpholine and the hydrogenolysis of the latter to methanol, if the two steps are operated separately. If the two steps are to be combined, then residual CO2 must be discharged after the first step, because CO2 negatively impacts the subsequent formamide hydrogenolysis. This obviously represents an obstacle for the implementation of practical CO2 capture and utilization processes. At the molecular level, for both systems, a metal–formato species, similar to the ruthenium-formato species B in Scheme 5, was identified as the main catalyst resting state. The strong coordination of the formato ligand, further enhanced by hydrogen bonding with the –NH group of the PNP ligand, renders these species stable. As shown in Scheme 5 for the analogous Ru-species B, formate must be displaced to allow H2 coordination and activation for further catalytic turnover and this process is assisted by the amine. Once the amine is “trapped” as formamide, collapse of the resting state proceeds through de-insertion of CO2 and reconstitution of the metal–hydride bond. This process is reversible and is influenced by CO2 in solution. The addition of LiOTf, in combination with DBU, 1,8-diazabiciclo[5.4.0]undec-7-ene, to assist and ease the expulsion of the formato ligand in the formamide hydrogenation step promoted by the iron catalyst 5 improved the yield of methanol, yet was not enough to mitigate the CO2 effect to realize a one-batch process.94b The use of a Lewis acid was not tested in the report by Prakash using the manganese catalyst 4.95a Although this might be beneficial, as recently highlighted by Leitner and co-workers, in a mechanistic investigation of the 4 Mn-promoted hydrogenation of CO2 to methanol assisted by alcohols, through the intermediate formation of formate esters.9b
The issues that limit the efficiency of catalysts 4 and 5 and prevent their successful application in the combined amine-aided CO2 capture and straightforward hydrogenation of the captured CO2 to methanol apparently do not affect catalyst [FeCl2{κ3-HC(pz)3] 6, reported by Martins and co-workers (Scheme 6).104 The catalyst was prepared by reaction of FeCl2 with hydrotris(1-pyrazolyl)methane, HC(pz)3, in water affording a pentacoordinated 16 electrons Fe(II) complex. The authors reported that in the presence of neat PEHA, without any added solvent, the catalyst (15 μmol) promoted the hydrogenation of CO2 (CO2/H2 1/3 75 atm) at 80 °C in a one-pot one-step process, achieving a remarkable TON in CH3OH of 2387 over 36 hours. After the reaction, a biphasic mixture was obtained consisting of an upper pale-yellow solution and a milky lower phase, probably containing unconverted formate and formamide, although no mention as to their possible detection and quantification is made. The two phases were separated and CH3OH recovered by simple distillation from the upper one. No recycling experiments for assessment of amine and catalyst reusability were described and the authors did not report mechanistic investigations to elucidate the structure of the active catalytic species in solution. Instead, they postulated that the activity of the catalyst could be ascribed to the presence of a free coordination site at the metal for any incoming substrate and a basic hemilabile scorpionate ligand. Thus, catalytically active hydride species would be generated through ligand-promoted heterolytic cleavage of coordinated dihydrogen and the protonated ligand could function as proton shuttle around the catalytic cycle. This hypothesis has been recently supported by DFT calculations.105
 | ||
| Scheme 6 Amine-aided hydrogenation of CO2 to CH3OH promoted by an iron(II)–C–scorpionate catalyst. | ||
The selected studies reported so far showed how the hydrogenation of the captured CO2 to CH3OH is affected by the catalyst structure and key reaction parameters such as pressure and temperature. For practical applications, the ease of product separation from the reaction medium and the recyclability of both the homogeneous catalyst and capturing amine are other important issues which need to be solved, too. In this respect, the use of high boiling solvents and ionic liquids, the application of biphasic systems to separate or immobilize the organometallic active species in a hydrophobic solvent as the catalyst phase, and the use of polymeric amines or the immobilization of amines on solid support have been described to address such issues and will be briefly summarized in the following section.
4.3. Influence of base and solvent on the CO2 hydrogenation to methanol
Ionic liquids, which are composed of ions with a melting point below 100 °C by definition, are thermally stable solvents with very low vapor pressure. For these reasons, they have been extensively investigated as CO2 capturing agents.106 Solubility of CO2 in ionic liquids can be modulated through proper choice and combination of the cation and the anion, although it is primarily affected by the anion. Tailored ionic liquids, modified by the introduction of functional groups, such as amino groups, on either the anion or the cation, can boost their CO2 sorption capacity through chemisorption.107 Relevant to the present discussion, their use as reaction medium, bare or in combination with another solvent, has proved to be beneficial to the catalytic hydrogenation of CO2 in some cases.108 In fact, in the metal-catalyzed hydrogenation of CO2, proper solvents, akin to formate salt formation, contribute to formic acid stabilization (thermodynamic drive) via hydrogen bonding and ionic interactions. Solvent stabilization of formic acid and formate salts increases in the order: ethers < alcohols < water < ionic liquid. Indeed, the application of imidazolium-based ionic liquids in the hydrogenation of CO2 using homogeneous ruthenium12,109 and iridium110 catalysts led to excellent productivities for formic acid formation.Recently, Prakash and co-workers have demonstrated the positive effect that the addition of the ionic liquid 1-butyl-3-methylimidazolium acetate (BMIM·OAc) has on the amount of of CH3OH accessible through the Ru-MACHO-BH 1 catalyzed hydrogenation of CO2, 4.96 mmol, TON 248, (1 20 μmol; PEHA 2.45 mmol, BMIM·OAc 0.5 mmol; THF 5 mL; CO2/H2 1/3 75 bars; 140 °C, 24 h) compared to the use of the sole polyamine PEHA, 3.20 mmol, TON 160, under otherwise identical conditions (Scheme 7, conditions b and a, respectively).111 After the first explorative experiments, the CH3OH yield, and catalyst productivity could be further improved to 13.2 mmol and TON 660, respectively through proper choice and amount of ionic liquid, solvent, amine and adjustment of reaction conditions (Scheme 7, conditions c). A few aspects of this work are worth mentioning: as to the anion in the BMIM-based ionic liquid composition, acetate outperformed bromide and chloride, while in the presence of hydroxide, comparable amounts of CH3OH as to the acetate were obtained. However, in the latter case large quantities of formate were obtained, suggesting that hydroxide negatively affects the further reduction to methanol. The reaction could be performed in the neat ionic liquid; however, the addition of a solvent to mitigate viscosity issues following CO2 capture proved beneficial. Among the tested solvents, triglyme performed best. Although no recycling experiments were described, the high boiling point of this solvent, in combination with the ionic liquid, should facilitate CH3OH separation and recovery of capturing medium and catalyst. Increasing the amount of PEHA increased the amount of CH3OH. The manganese catalyst 4 was also active, although to a lower extent than 1, in the combined CO2 capture and hydrogenation to CH3OH. According to the authors, this is likely due to the cation BMIM acting as Lewis acid, thus favoring the release of formate from a Mn–formate species for subsequent dihydrogen activation and catalyst turnover.
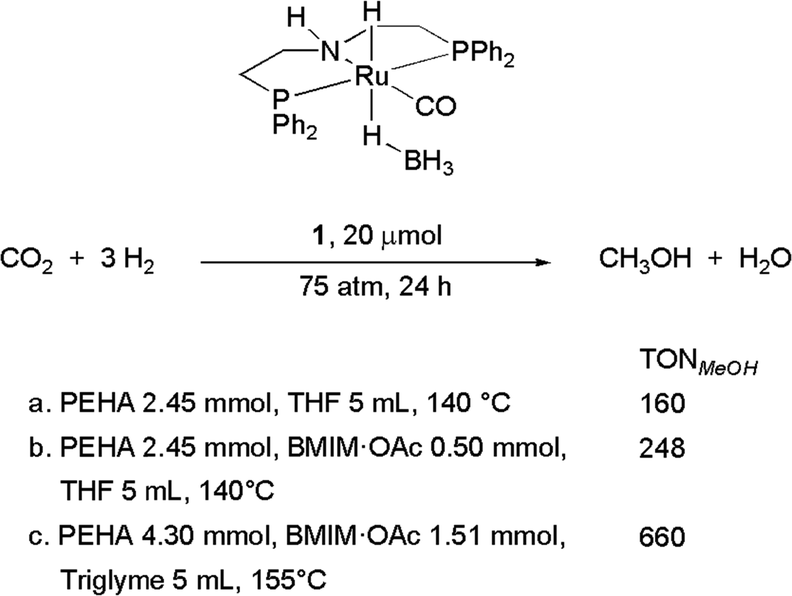 | ||
| Scheme 7 Polyamine-assisted Ru-MACHO-BH catalyzed hydrogenation of CO2 to methanol: influence of ionic liquid 1-butyl-3-methylimidazolium acetate BMIM·OAc. | ||
In the interest of enhancing recyclability, the development of a biphasic system that facilitates the segregation of reaction components, catalysts, absorbents, and products into distinct phases is of significant importance. Conventionally, aqueous solutions of amines or alkanolamines have been employed for the purpose of removing carbon dioxide from industrial gas streams. In contrast, homogeneous metal catalysts, unless specifically engineered to enhance hydrophilicity, exhibit preferential solubility in organic solvents. This disparity has been strategically capitalized upon by Prakash and colleagues, who have devised a recyclable system for the tandem capture of CO2 in water and its subsequent hydrogenation to MeOH, as depicted in Fig. 12.94a Here, CO2 was captured by exposing an aqueous solution of the amine at room temperature either to an atmosphere of pure CO2 at a constant pressure of 1 psi or to synthetic air containing a CO2 concentration of 408 ppm. CO2 was captured both as carbamate and carbonate/bicarbonate. Among the tested (poly)amines, PEHA offered the best trade-off between the amount of captured CO2 (0.43 mmol of CO2 per mol of N atom) and CH3OH yield. Using Ru-MACHO-BH 1, 10 μmol, in a Me-THF/H2O biphasic mixture containing 11 mmol of absorbed CO2, after 72 h at 145 °C under 70 bars H2, TON of 231 in HCOO−/formamide and 520 in CH3OH (yield 47%) were achieved, even though the presence of water as a co-solvent might negatively affect ammonium formate dehydration to formamide. CH3OH, which partitioned between the two phases, was recovered by distillation, after which both the catalyst and the amine could be recycled through phase separation. After hydrogenation, it was established that, within the detection limits of 31P NMR, no catalyst had leached into the aqueous phase. Therefore, the organic phase was separated, and, after removal of volatiles, the catalyst reused four times, with only minimal decrease of TON in subsequent applications, likely due to minute catalyst loss after each work up.
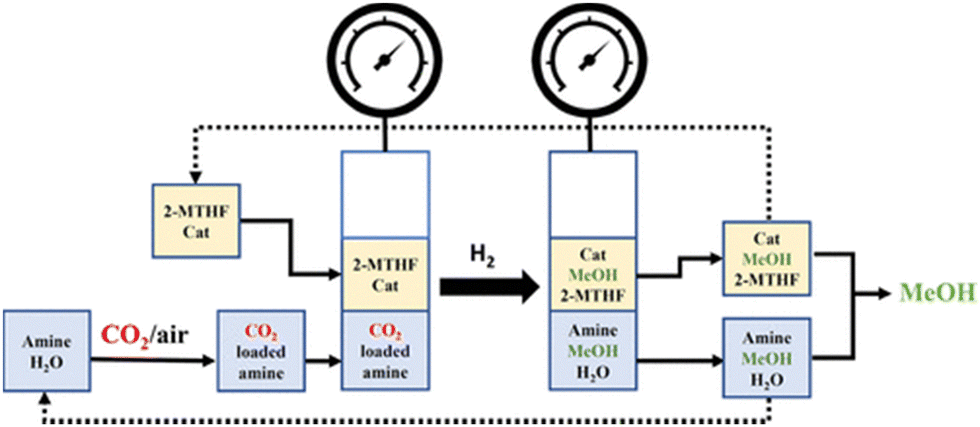 | ||
| Fig. 12 Schematic representation of a biphasic system for CO2 capture from air using an amine aqueous solution and hydrogenation of the captured CO2 to MeOH through combination of the absorbing aqueous solution with an organic phase containing the homogeneous hydrogenation catalyst. Reproduced with permission from ref. 94a. | ||
The use of polymeric amines or amines chemically grafted on fumed silica suspended in an organic solvent has also been explored as a mean to ease their recycling. PEI600 (CAS No. 25987-06-8, average Mw ∼ 800, average Mn ∼ 600) is a branched polyethylenimine containing primary, secondary, and tertiary amino groups. With PEI600 (1.5 mmol N) and Ru-MACHO-BH 1, 0.05% as to PEI nitrogen content, under optimized conditions, CO2/H2 1/3 80 bars, THF, 150 °C, 100 h, a maximum catalyst TON of 689 in CH3OH was achieved. After the reaction, the polymeric amine could be recovered by simple filtration.112
Alternatively, polyamines were immobilized on a solid support and the functionalized support was used neat for CO2 capture. More specifically, polyethylenimines were either physically impregnated on or covalently bound to fused silica. The CO2 loading capacity was directly correlated to the number of nitrogen atoms per gram of solid supported amine (SSA). The physically impregnated SSA afforded better yield in CH3OH than the chemically grafted ones, but this could be directly correlated to the concentration of amines leaching in the organic solvent for the former materials. Among the tested supported amines, the covalently attached SSA, obtained by reacting trimethoxysilylpropyl-modified polyethylenimine (Mw 1500–1800) with fused silica, was selected owing to its high leaching resistance and amine content. However, under optimized conditions, 500 mg of the SSA, Ru-MACHO-BH 1 10 μmol, THF 5 mL, CO2/H2 1/3 80 bars, 145 °C, 40 h, a maximum TON of just 90 in CH3OH was achieved, although the possibility of both SSA and catalyst recovery and reuse was demonstrated.113
4.4. Basic principles in CO2 hydrogenation to formate/formic acid
So far, we have described catalytic homogeneous systems which warrant access to CH3OH and, as such, are obviously competent for the reduction of CO2 to formate as well. Formic acid is an important bulk chemical in itself, the global market of which has reached around 750 thousand tons in 2022. Formic acid and its salts find applications in animal forage, grass silaging, leather tanning, and antifreezing,114 and in the synthesis of a range of products including dyes and finishes, drilling fluids, natural rubber and food additives. The production of formic acid on an industrial scale is mainly based on a two-stage process which comprises first the carbonylation of methanol to afford methyl formate, and then hydrolysis of the later. The by-product methanol is recycled back to the first step. Free formic acid can also be obtained from its salts, mainly sodium formate and calcium formate. The acidolysis is normally carried out with sulfuric acid or phosphoric acid. The atom economy of the process is obviously poor because of the formation of calcium salts as by-product. For this reason, the industrial scale of this process is quite limited.A greener route to formic acid is based on the hydrogenation of CO2, using green hydrogen, promoted by metal catalysts. Relevant to the current discussion, a plethora of homogenous catalysts, based mainly on noble metals such as iridium and ruthenium, have been described115 which are competent for such transformation, although examples which demonstrate the possibility to use non-precious metals have also been reported.116 These catalysts have been studied especially in the contest of a hydrogen economy where formic acid is intended as a sustainable storage medium and liquid vector for hydrogen, as it contains 4.4 wt% H2 and its volumetric storage density is 53 g H2 per L.
In the discussion so far, it has been shown that while catalysts which operate through metal–ligand cooperativity have so far proved better suited for converting CO2 to CH3OH, metal–ligand cooperativity is not mandatory if amine-aided CO2 hydrogenation is intended to stop at the formate level. Lower temperatures can and should then be applied as the hydrogenation of CO2 to formate in the presence of a base becomes an exergonic process, the equilibrium of which can be further shifted towards the products by applying high hydrogen pressures. The produced ammonium formate can be thermally cleaved to afford free HCOOH and recover the amine. A process based on this approach has been developed at BASF which relies on the use of [Ru(H)2(PnBu3)4] and a tertiary amine in alcohol.117 The formic acid–amine complex is thermally dissociated at 150 °C and recovered through distillation under reduced pressure.
Tertiary amines are commonly used for this purpose because formate salts of primary and secondary amines are thermally dehydrated to formamide at higher temperatures. Therefore, a profitable use of amino acids to capture CO2 and aid its subsequent metal-catalysed hydrogenation to formate should preferentially involve a direct application of the amino acid formate solution as, for example, the implementation of a “hydrogen battery” (vide infra), where following metal-catalysed H2 release from formate, CO2 is retained by the amine in the “spent battery” which can then be recharged through hydrogenation of the retained CO2 promoted by the same catalyst.118 This would have the added advantage that a separation step to remove CO2 before feeding H2 into a PEM fuel cell would not be necessary.
4.5. Recent examples of reactive CO2 capture to formates
Here we would like to highlight a few selected examples which, because of the use of 1°/2° amines to capture CO2 and aid its further metal-catalysed hydrogenation to formate and the use of aqueous solutions as reaction medium, can serve to highlight some key issues related to this transformation which are relevant to the use of amino acids as well.He and co-workers provided the first demonstration that CO2, captured by PEI 600 in methanol as ammonium carbamate and alkylcarbonate, could be directly hydrogenated in situ with a RhCl3·3H2O/PPh2Cy system in CH3OH, providing the corresponding ammonium formate with a TON as high as 852.119
Prakash and co-workers investigated the capturing ability of various amines in aqueous media and the hydrogenation of the captured CO2 to formate promoted by Ru-MACHO-BH. CO2 was bubbled through the amine aqueous solutions at room temperature and then an organic co-solvent such as THF or dioxane was added to improve catalyst solubility for the subsequent hydrogenation.120 Superbases such as 1,1,3,3-tetramethylguanidine TMG and 1,4-diazabicyclo[2.2.2]octane DABCO proved particularly efficient for both CO2 capture and hydrogenation of the in situ formed bicarbonate/carbonate. As with ammonium carbamate, evidence of whether hydrogenation proceeds through hydride transfer from the catalytic active species to a negatively charged bicarbonate species or free CO2 in equilibrium with the latter is lacking.36b
With TMG, 0.92 mmol of CO2 per mmol of base were captured as bicarbonate/carbonate. When using 17 mmol TMG, in an aqueous/dioxane solution, Ru-MACHO-BH 1 20 μmol, H2 50 bar, 50 °C, for 15 hours, a TON as high as 7375 was achieved, corresponding to a 95% yield in formate as to the captured CO2 (Scheme 8).120 In the same study, the authors reported the implementation of a biphasic water/Me-THF system for catalyst recovery and recycling, the optimization of which was however realised using the tertiary amine DABCO.87
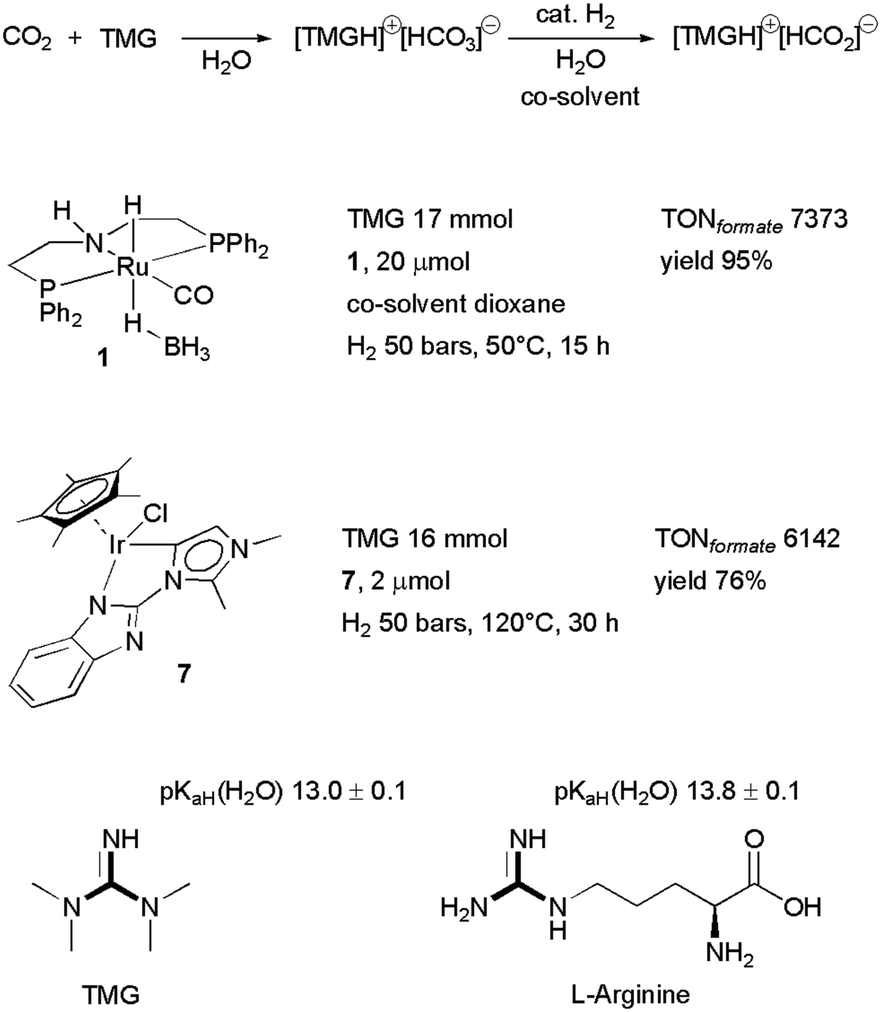 | ||
| Scheme 8 TMG-aided CO2 capture and hydrogenation to formate catalysed by Ru-MACHO-BH 1 and water-soluble Ir catalyst 7. A guanidino group is present in the basic side-chain of L-arginine which could operate in the same fashion. pKaH(H2O) from ref. 121 (TMG) and ref. 42 (L-arginine). | ||
TMG was likewise used as CO2 capturing agent in combination with a water-soluble iridium catalyst bearing a chelating N-heterocyclic carbene 7, Scheme 8, which therefore did not require the use of an organic co-solvent.122 Notably, the catalyst was found to be air-stable and could be handled without rigorous exclusion of air also in solution. When using 16 mmol TMG in water, 7 2 μmol, H2 50 bar, 120 °C, for 30 hours, a TON of 6142 was achieved, corresponding to a 76% yield in formate as to the captured CO2 (Scheme 8). Interestingly, a guanidino group is present in the basic side chain of the amino acid arginine (Scheme 8).
Finally, we would like to report about the study by Leitner and co-workers who implemented a biphasic system in combination with the ruthenium catalyst precursor cis-[Ru(dppm2Cl2)] (dppm = bis-diphenylphosphinomethane) to ease product and catalyst separation and recovery.9a Here, a few amine/organic solvent combinations were tested for CO2 hydrogenation to formate. High catalyst activity and stability were achieved using an aqueous solution of MEA and methyl isobutyl carbinol (MIBC). MEA is a prototypical example of a scrubbing amine applied on commercial scale and MIBC is a protic yet water-immiscible solvent with some favourable properties such as being a stable, medium boiling (bp 131.6 °C), affordable and industrially acceptable liquid. Hydrogenation was carried out at 70 °C on a 20% MEA aqueous solution, pre-saturated with CO2, under a total pressure of 90 bars (5/85 CO2/H2). After the reaction, the amine/formate adduct was almost completely retained in the aqueous phase and a TON of about 1550 in a single run could be achieved. The catalyst remained in the organic phase with less than 0.26% Ru and 1% P leaching in the aqueous phase as established by inductively coupled plasma mass spectrometry (ICP-MS). Based on these premises, a semicontinuous process was realized, to demonstrate the potential applicability and recyclability of the system. After each run, the product phase was removed through a valve at the bottom of the reactor, which was then reloaded with a fresh solution of MEA pre-saturated with CO2. Over 11 runs, a total turnover number (TTON) of approximately 150000 was achieved.
5. Recent developments of amino acid-based reactive CO2 capture to formate and methanol
As it has been discussed above, since one example reported by our group in 2011 on the hydrogenation of bicarbonate to formate, an analogue concept of reactive capture of CO2,14a and the first real example in 2015 reported by Sanford and co-workers,13 the last decade has witnessed significant progress in the optimization of processes for the combined CO2 capture and hydrogenation, as to (1) catalyst design, from the use of noble metal-based catalysts to the exploration of non-noble metal-based alternatives, (2) the application of different solvent media, especially biphasic systems, to ease catalyst separation and recyclability, (3) the development of various absorbents, among which a range of amines and hydroxides, for the efficient capture of CO2 and its selective hydrogenation to formates and methanol. However, most systems rely on amines as capturing agents which are volatile: to overcome this limitation strategies have been devised employing high molecular weight polyamines, tethering amines on solid supports, and replacing them with hydroxides. In 2021 our group reported the first example of the use of amino acids to combine CO2 capture with the direct hydrogenation of the resulting adducts to formate (Fig. 13).23a In this study, the CO2 capture efficiency of various amino acids in water was first evaluated. LYS, having a basic side chain for the presence of a second amino group, proved to be the best absorbent among the amino acids tested, even capable of direct CO2 capture from air, affording up to 98% of the initial molar amount of LYS in the form of bicarbonate and carbamate. Subsequently, the captured CO2 was hydrogenated in situ to formate using Ru-MACHO 1 as catalyst, reaching a TON of about 55000 (Lys 5 mmol, 1 0.02 μmol, H2O/THF 1/1 10 mL, H2 80 bar, 145 °C, 12 h), thus clearly demonstrating the potential of amino acids for the direct air capture and valorization. Here an organic co-solvent as THF, miscible with water, was added to help catalyst solubilization.
 | ||
| Fig. 13 Illustration of the direct air capture of CO2 using LYS as the absorbent/base and its hydrogenation to formate with the Ru-MACHO-BH catalyst. Reproduced from ref. 23a with copyright permission. | ||
Indeed such potential was further exploited by our group in 2022 with the development of a rechargeable “hydrogen battery” based on a non-noble Mn-pincer catalyst (Fig. 14a).23b To fulfil both the H2 storage and the H2 release steps within the same cell without switching the catalytic system, the applied catalyst must be active for both the charging (CO2 hydrogenation) and discharging (formate dehydrogenation) processes. A perusal of the relevant literature showed that only a few noble metal-based catalysts are able to promote both processes efficiently,123 while Mn-based ones show high activity either for the hydrogenation or the dehydrogenation process. Through informed screening, a Mn pincer complex bearing a triazine backbone pincer PNP ligand was selected which, in combination with LYS as base showed unprecedented high activity towards CO2 hydrogenation to formate with TON up to 230000 (Lys 5.0 mmol, Mn catalyst R = NH-C3H5 0.02 μmol; H2O/THF 1/1 10 mL, CO2:H2 20:60 bar, 115 °C, 12 h), and the reverse formic acid dehydrogenation with TON up to 29400 (Lys 5.0 mmol, Mn catalyst R = NH-C3H5 0.02 μmol, H2O/THF 1/1 10 mL, 90 °C, 12 h). In the dehydrogenation process, under the conditions reported above, 28% of the produced CO2 was released together with H2. The essential role of LYS in achieving high activity was also demonstrated, in fact with the widely used organic amines, like pentaethylenehexamine (PEHA), tetramethylguanidine (TMG), 1,4-diazabicyclo[2.2.2]octane (DABCO) and 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU), much lower activities were scored. In order to combine the two steps in the “hydrogen battery” and increase the CO2 absorbing capacity of the reaction medium, potassium lysinate, obtained by treating lysine with one equivalent of KOH, was used instead of LYS. In the developed system, CO2 can be directly captured from air with potassium lysinate as absorbent, and the subsequent hydrogenation results in over 90% of formate yield. In the dehydrogenation phase, 99.9% of CO2 is retained in solution which has a two-fold advantage: pure uncontaminated hydrogen is produced, and no CO2 recharge step is required. Mechanistic studies revealed that potassium lysinate facilitates catalyst activation via N–H deprotonation and dearomatization of the triazine moiety (Fig. 14b). Furthermore, it was demonstrated that the combined presence of the α-amino acid moiety and a suitable basic side chain in the amino acid molecule is essential for the Mn-catalysed CO2 hydrogenation reaction, as well as for the CO2 capture processes.
 | ||
| Fig. 14 (a) Potassium lysinate promoted H2 storage and release cycle in a carbon-neutral fashion. (b) Proposed mechanism for catalyst activation and formic acid/CO2 (de)hydrogenation promoted by a Mn pincer complex with a triazine backbone PNP ligand. Reproduced from ref. 23b with copyright permission. | ||
Inspired by the fact that amino acids are common supplements of poultry and pig feed, and formic acid/formats are used for the preservation of animal feeds, Leitner and co-workers proposed to integrate the CO2 capture with its hydrogenation to formate using amino acids.124 The resulting formate/amino acid mixture can be directly used as additive for animal feeds. For this specific purpose, metal contamination of the product must be avoided. The use of a biphasic reaction medium, whereby the catalyst can be retained in the organic phase while the product stays in the aqueous phase, was applied. Initially the catalyst precursor cis-[Ru(dppm)2Cl2] (dppm = bis-diphenylphosphinomethane) was used in the biphasic mixture of methylisobutylcarbinol (MIBC) and water. The reactions were carried out under 90 bar total pressure at room temperature (CO2:H2 1:2) and a reaction temperature of 70 °C. The proteinogenic amino acids ARG, HIS, and LYS were used in the aqueous phase because they carry basic side groups which contribute further to CO2 capture and HCOOH stabilization, rendering the reaction thermodynamically more favourable. In the screening tests, ARG secured both the highest activity and conversion with values comparable to those obtainable with MEA under similar conditions. The HIS based system exhibited relative lower reaction rate. LYS, despite its high pKa value, provided the lowest activity and formate yield (Fig. 15). The authors found that, after the reaction, 2.2% of the catalyst had cross-dissolved into the aqueous phase, which would increase the cost for downstream purification. To reduce the catalyst solubility in water and ensure its exclusive partitioning in the organic phase, catalyst [Ru(C12-dppm)2Cl2] containing the lipophilic-tagged ligand bis(bis(4-dodecylphenyl)phosphanyl)methane (C12-dppm) was used. After a screening of various organic solvents, they found that dodecanol meets the system design criteria of high catalytic activity, biocompatibility and no cross-solubility into the aqueous solution. Based on the dodecanol/H2O biphasic system, the authors further tested the reactive capture of CO2 to formate, using a CO2 saturated aqueous solution of 6 M Arg: the capture capacity of Arg was determined as 0.94 mol CO2 per mol Arg under 2 bar. This solution was fed into an autoclave as a part of a semi-batch process. Under 70 bars H2 at 70 °C, after 5 hours no further pressure drop was detected. The aqueous phase could be removed for analysis under pressure at 70 °C and a new batch of aqueous CO2-saturated ARG recharged. The process was repeated ten times affording a final formic acid-to-amino acid ratio of 1:1 (average of 0.91 ± 0.03:1, and 9.0–10.8 wt% of FA) demonstrating a quantitative conversion of the captured CO2 to FA with practical reusability. They also showed that the tenfold cycle production of FA corresponds to a total TON of 102282, and the production of FA (in kg) per g of ruthenium is as high as 46.6. It should also be noted that only 3.3% of the Ru catalyst had leached out to the product phase with a total loss of only 1% in runs 2–10, demonstrating excellent catalyst retainability.
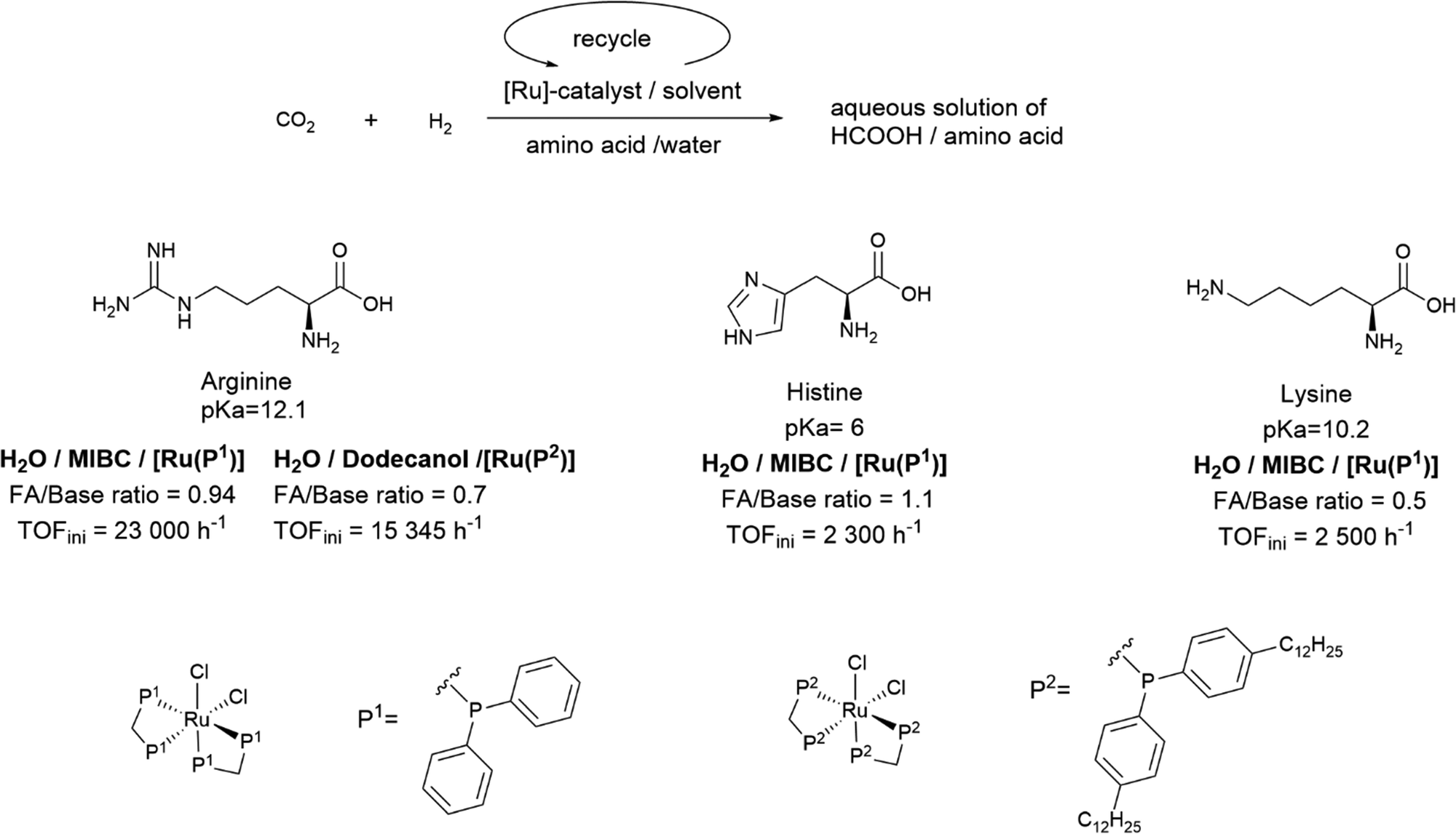 | ||
| Fig. 15 The reaction system for the CO2 hydrogenation to product HCOOH/amino acids mixture as the animal feeds additive using a biphasic system with cis-[Ru(dppm)2Cl2] (P1 ligand) and cis-[Ru(C12-dppm)2Cl2] (P2 ligand) catalysts. MIBC, methyl isobutyl carbinol. Reproduced from ref. 124 with copyright permission. | ||
AAILs have been shown to be a potential absorbent for CO2 capture, but it was not until 2022 that our group published the first report on the use of such a system for the reactive capture and conversion of CO2 to formates.23c In this study, it was shown that the CO2 capture capacity of the AAIL is influenced by both the cation and the anion. With the ionic liquid [TBA][ARG], prepared by reaction of ARG with an aqueous solution of tetrabutyl ammonium (TBA) hydroxide, a capture capacity of 1.94 molCO2 molabsorbent−1 was achieved under 2 bars of CO2. [TBA][LYS], prepared in the same way from LYS, showed a capturing capacity of 1.80 molCO2 molabsorbent−1. The loading capacity was evaluated after dissolving the AAIL in water to alleviate the problem of increased viscosity after CO2 loading, which would limit gas diffusion and mass transfer. It should be noted that the loading capacity of ARG in the ILs form is even higher than that of the corresponding potassium salt and [TBA][OH], demonstrating again the advantage of using amino acids as components of ionic liquids for CO2 capture. The subsequent hydrogenation step was carried out using Ru-MACHO-BH 1, achieving a conversion of over 60% from captured CO2 to formate (CO2 capture step: [TBA][ARG] 5 mmol, CO2 20 bars, abosrbed CO2 10.1 mmol; hydrogenation step: Ru-MACHO-BH 1 10 μmol), H2O/THF 1/1 10 mL, H2 60 bars, 80 °C, 6 h). If no CO2 overpressure is applied and CO2 is directly captured from air at room temperature, then a final formate to absorbent ratio of 0.5 can be achieved demonstrating the general capability of amino acid-based ionic liquids for the reactive capture of CO2.
The use of amino acid as the absorbent for capture and in situ hydrogenation has just been studied within the last 5 years, but it has showcased the high potential of such biocompatible and thermostable absorbents for achieving highly efficient CO2 hydrogenation transformation. The water-soluble iridium catalyst 7 described in Scheme 8, reported in 2022 by Choudhury and co-workers, while performing best in combination with TMG, was likewise tested with LYS as absorbent. After CO2 capture from air (7.5 mmol captured with 10 mmol LYS after 1 h at room temperature), hydrogenation of the loaded aqueous solution with 7 (7 4.3 μmol, H2O 10 mL, H2 50 bars, 120 °C, 8 h) afforded a 71% yield in formate as to the captured CO2.122
Very recently, Prakash has demonstrated that also ARG, beside PEHA, in combination with the ionic liquid 3-butyl-1-methyl imidazolium acetate (BMIMOAc) can aid the Ru-MACHO-BH 1-catalyzed hydrogenation of CO2 to methanol. However, only a meager TON of 42.5 was recorded compared to that achieved with PEA, 410, under otherwise identical conditions (1 20 μmol, BMIMOAc 1.5 mmol, L-Arg 2.6 mmol, triglyme 5 mL, 75 bar CO2:3H2, 155 °C, 24 hours).111 Very recently our group could greatly improve this result achieving a maximum TON in methanol of 700 (1 2 μmol, [TBA][ARG] 5.0 mmol, THF 10 mL, 80 bar CO2:3H2, 150 °C, 24 hours) by directly incorporating the ARG anion in the structure of the ionic liquid [TBA][ARG] used as the CO2 capturing medium.125 The study revealed the peculiar advantage provided by ARG: in fact, although the two basic amino acid ARG and LYS when combined with the TBA cation were equally effective in the capture of CO2 and its further 1-catalyzed hydrogenation to formate, more methanol could be produced with [TBA][ARG]. The main difference between the two amino acids is the presence of the highly basic guanidino group in ARG which, being protonated under the conditions of the AAIL synthesis and application, can likely establish hydrogen bonding interactions with the intermediate formamide and enhance its electrophilic character thus favoring its reduction. Yet the system suffers from some limitations such as the limited stability of the tetrabutylammonium cation under the harsher conditions required for CO2 hydrogenation beyond the formate stage.
While a proof of concept has been demonstrated, further investigation is obviously needed to disclose the potential of amino acids-based CO2-capturing media for the reactive conversion of the captured CO2.
6. Discussion
As demonstrated by the aforementioned examples, the process of amino acid-assisted CO2 capture126 and in situ hydrogenation to either formate or methanol, while still in its nascent stages, is both viable and feasible. The former method has exhibited a higher degree of success compared to the latter. Research in this domain has successfully substantiated the concept, yet further investigation is imperative to address the various challenges that have surfaced during the review of existing publications and to achieve practical applications with high levels of selectivity and efficiency.It has been demonstrated that CO2 captured directly from air at room temperature with aqueous solutions of basic amino acids such as LYS and ARG can be directly pressurized with hydrogen in the presence of selected catalysts to yield aqueous solutions of formate. Productivities (TON) are positively affected by higher CO2 pressures and/or concentrations (up to a certain level). Therefore, the same approach could, at least in theory, be applied to reactive CO2 capture from flue gases after combustion (0.03–0.14 bar CO2 partial pressure) and from industrial sources such as cement production (0.14–0.33 bar) and stainless steel plants (0.2–0.6 bar) and hydrogen production (3–5 bar), all of which have higher CO2 content and partial pressure than air.127 In addition, the high temperature of post-combustion CO2 flue gas streams could be advantageously used to support subsequent hydrogenation if the capture process can be performed at such a temperature.
One problem, however, is related to the kinetics of CO2 absorption: measurements of the capturing ability of aqueous solutions of LYS showed that in air at room temperature up to 4 days are required for reaching a 0.5/1.0 CO2/LYS molar ratio, with CO2 trapped in solution as the exclusive carbamate.23b However, the process of CO2 capture can be accelerated and gradually increased to nearly 1/1 CO2/LYS molar ratio with increasing CO2 pressure up to 20 bar by forming bicarbonate besides carbamate. Such an approach to improve kinetics and process productivity would require an additional energy input if to be applied to a post-combustion CO2 capture process. Alternatively, when the basic amino acids LYS and ARG are applied in the form of the corresponding ionic liquids23c in combination with the TBA cation, the CO2/AA ratio can be increased up to 1.80 and 1.94 for LYS and ARG, respectively when CO2 is applied at 2 bars. While the rate of CO2 uptake falls far short of what would be required for a potential practical application, the use of AAIL, which has an additional basic site in the amino acid side chain, expands the scavenging capacity of the IL, thereby increasing the local concentrations of the putative CO2 source and theoretically increasing the rate of conversion of the captured CO2 into products. In addition, the careful selection of the cations for the amino acid anion in the form of salt, ILs, or mixtures of different types of absorbents could serve as another strategy to modulate the CO2 uptake rate and capacity. However, it is imperative to consider compatibility with the subsequent hydrogenation process. Additional properties of capturing agents to be assessed for reactive CO2 capture include stability under the reducing conditions necessary for conversion chemistry and compatibility with the catalyst. Further investigation is necessary into issues such as sorbent integrity after hydrogenation and its interaction with the catalyst metal, which can reduce its efficiency.
Two of the catalysts applied in the AA-assisted CO2 capture and hydrogenation, Ru-MACHO-BH 1 and the N-heterocyclic carbene-ligated iridium catalyst 7, have been used directly for the hydrogenation of CO2 captured from air in water. These catalysts have demonstrated tolerance to both oxygen and water at the reaction temperature of 155 °C and 120 °C, respectively. It is imperative that catalysts engineered for reactive CO2 capture exhibit stability against oxygen and water, as these elements are also present in flue gases in varying amounts.128 Additionally, flue gases contain other contaminants, such as NOx and SOx, against which catalysts must be evaluated to ensure a more realistic assessment. Ru-MACHO-BH 1 is a very robust catalyst; however, it is almost insoluble in water. Its use in combination with amino acid-based absorbents requires the addition of a miscible organic solvent to improve its efficiency. While this may be a limitation, the use of a water-insoluble organic solvent to solubilize the catalyst in a biphasic system can be advantageously exploited to confine and recycle the catalyst while facilitating product separation.
The lipophilicity of the catalyst can be further enhanced by modifying its ligand structure through the introduction of long alkyl chains, as demonstrated by Leitner and coworkers.129 The same research group has synthesized a Ru-MACHO-BH complex tagged with linear C12 alkyl chains at the para position of the phosphorus-phenyl substituents and demonstrated its efficiency in catalyzing the aforementioned reaction. Disappointingly, in a biphasic n-decane/water system, while the overall TON for CO2 conversion in the presence of N,N′-dimethylethane-1,2-diamine was close to that exhibited by the self-separating system, however, a significantly lower selectivity of approximately 50% for complete hydrogenation to methanol was observed, resulting in the accumulation of significant amounts of formamide at the reaction. This suggests that excess water hinders the reduction of captured CO2 to methanol, as water is a by-product and an excessive amount could adversely impact the equilibrium position of such a conversion. The employment of water-lean solvents, such as ionic liquids, may offer a solution to this challenge.130 In the sole instance of CO2 hydrogenation to formate in AAIL,23c the ionic liquid was dissolved in water to alleviate the rise in viscosity triggered by CO2 absorption. Consequently, further research is necessary to reconcile these two conflicting requirements by exploring amino acid-based ionic liquids that are less viscous and by identifying suitable alternative solvents to water that would not affect the conversion of CO2 to methanol. In this respect, the knowledge established in amino acid-based CO2 capture systems could be revisited. In terms of catalysts, both noble and non-noble metals have been successfully applied to the reactive capture of CO2 by conversion to formate.
With one exception, catalysts based on non-noble metals,104 at least those recalled in this review and supported by pincer ligands, are less efficient for integrated CO2 capture and hydrogenation to methanol because of the formation of highly stable metal formate adducts. These adducts require the use of additives to return them to the catalytic cycle in an active form. Replacement of noble metals by non-noble metals is urged by economic considerations and the limited availability of the former, although even manganese has been identified as a critical element due to its increased use.131 It must be said that, at least for the MACHO-type catalysts, regardless of the metal, the ligand contributes significantly to the overall cost. Nevertheless, the cost of the metal and ligand may not be an issue if the catalyst can be retained and recycled.
Although this review deals with homogeneous catalysts, it is worth mentioning that heterogenization of homogeneous catalysts has been implemented for the sake of catalyst recovery and reuse, and although not directly applied to amino acid-based solvents, examples have been reported concerning the amine-based hydrogenation of CO2 to either formate132 or methanol,133 which has been realized by anchoring iridium metal to a bidentate PN moiety within a suitably modified polyimine PEI. Alternatively, the hyper-crosslinking polymerization (HCP) technique has been used to prepare a single-site heterogenized version of of Ir-132b and Ru133-based molecular catalysts whose ligand carries a phenyl substituent. In the presence of triphenylmethane as copolymerizing arene monomer and formaldehyde dimethylacetal as crosslinking agent, aryl moieties of ligand units could be linked together by the Friedel–Crafts alkylation reaction using FeCl3 as catalyst in 1,2-dichloroethane as solvent. Such HCP catalysts have shown activities superior to those of their homogeneous counterparts in CO2 hydrogenation to formate132b and methanol,133 combined with excellent recyclability. Interestingly, a similar heterogenization has also been applied to Ru-MACHO 1a, and the heterogenized version has been applied to aqueous reforming to methanol,134 with activity comparable to that of 1a under otherwise identical conditions.135 To the best of our knowledge, no application to amine-assisted hydrogenation of CO2 with such a catalyst has yet been reported.
7. Conclusions and outlook
The preceding discourse delineated advancements and unresolved challenges in the domain of reactive CO2 capture employing amino acid-based solvents. These challenges encompass sluggish absorption kinetics, constrained capacity, reliance on CO2 sources and partial pressure, substantial catalyst expense, product selectivity, and concerns pertaining to catalyst recyclability. The surmounting of these limitations is imperative to enhance the overall performance of amino acid-based reactive CO2 capture systems and facilitate their large-scale implementation. In light of the aforementioned discussion, the authors propose the following research directions to address these critical challenges and enhance the long-term prospects of reactive CO2 capture technologies:• It is imperative to investigate further scalable and inexpensive systems for reactive CO2 capture.
• Furthermore, there is a need to develop more efficient solvents for reactive CO2 capture, e.g. amino acid-based ones, able to operate under low CO2 pressure and to withstand hydrogenation conditions.
• Additionally, there is a need to develop more efficient, active, stable, and selective catalysts that are tolerant to O2, NOx and SOx.
• There is a need to develop strategies to facilitate product separation and efficiently recycle the sorbent and catalyst.
• Finally, techno-economic and life cycle assessment of CO2 capture and hydrogenation should be performed on a regular basis, preferentially, as Leitner and coworkers suggested, for all the stages, from “cradle-to-grave”.136 Despite the potential of reactive CO2 capture to circumvent the CO2 desorption and compression steps, the separation of the product may require an additional energy input. Therefore, the development of technologies to efficiently recover the final upgraded product from the absorbent medium with minimal energy expenditure is necessary. Additionally, a comparison of the total energy consumption and cost of integrated and non-integrated CO2 capture and hydrogenation is recommended, as Bardow and coworkers demonstrated, if the CO2 source is too diluted, the integration may indeed increase the capital investment compared to the non-integrated process.137
In conclusion, amine- and amino acid-based reactive CO2 capture represents a promising and central system for CO2 utilization and valorization. We believe that this review can serve as a valuable guide for researchers in this field in their efforts to respond to the ongoing search for effective strategies to reduce CO2 emissions and achieve sustainability.
List of acronyms
| MEA | Monoethanolamine |
| DEA | Diethanolamine |
| MDEA | N-Methyl diethanolamine |
| DETA | Diethylenetriamine |
| NaADS | Sodium aliphatic diamine sulphonate |
| LOHC | Liquid organic hydrogen carriers |
| RuBisCO | Ribulose-1,5-bisphophate-carboxylase/oxygenase |
| AAILs | Amino acid based ionic liquids |
| GLY | Glycine |
| TAU | Taurine |
| BALA | β-Alanine |
| VAL | Valine |
| LEU | Leucine |
| ALA | Alanine |
| AABA | Aminobutyric acid |
| NMGLY | Sarcosine |
| SER | Serine |
| AMALA | α-Methyl alanine |
| PHE | Phenylalanine |
| CYS | Cysteine |
| PRO | Proline |
| GABA | γ-Aminobutyric acid |
| HYPRO | Hydroxyproline |
| PGA | Pyroglutamic acid |
| ASN | Asparagine |
| GLN | Glutamine |
| GLU | Glutamic acid |
| DIGLY | Diglycine |
| ARG | Arginine |
| ILE | Isoleucine |
| MET | Methionine |
| PZ | Piperazine |
| EGME | 2-Methoxyethanol |
| EGEE | 2-Ethoxyethanol |
| DMF | N,N-Dimethylformamide |
| NMP | N-Methyl-2-pyrrolidone |
| BMIM | Butyl methyl imidazolium |
| N66614 | Trihexyl(tetradecyl) ammonium |
| P66614 | Trihexyl(tetradecyl) phosphonium |
| C2OHmim | Hydroxyethyl methyl imidazole |
| PEHA | Pentaethylenehexamine |
| TMG | Tetramethylguanidine |
| DABCO | 1,4-Diazabicyclo[2.2.2]octane |
| DBU | 1,8-Diazabicyclo(5.4.0)undec-7-ene |
| TBA | Tetrabutyl ammonium |
| MIBC | 4-Methyl-2-pentanol |
| CHO | Choline |
Author contributions
Y. P. and E. A.: writing – original draft. M. B. and H. J.: conceptualization, writing – review & editing.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support from the Federal Ministry of Education and Research (BMBF) and the State of Mecklenburg-Vorpommern. This project also received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 101058872.References
- F. Barzagli, F. Mani and M. Peruzzini, Environ. Sci. Technol., 2016, 50, 7239–7246 CrossRef CAS PubMed.
- (a) W. Li, X. L. Zhang, B. H. Lu, C. Sun, S. J. Li and S. H. Zhang, Int. J. Greenhouse Gas Control, 2015, 42, 400–404 CrossRef CAS; (b) Q. Zheng, G. J. O. Martin and S. E. Kentish, Energy Environ. Sci., 2016, 9, 1074–1082 RSC.
- CO 2 Capture and Utilisation, 2024, https://www.iea.org/energy-system/carbon-capture-utilisation-and-storage/co2-capture-and-utilisation#tracking.
- (a) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (b) H. O. LeClerc, H. C. Erythropel, A. Backhaus, D. S. Lee, D. R. Judd, M. M. Paulsen, M. Ishii, A. Long, L. Ratjen, G. Gonsalves Bertho, C. Deetman, Y. Du, M. K. M. Lane, P. V. Petrovic, A. T. Champlin, A. Bordet, N. Kaeffer, G. Kemper, J. B. Zimmerman, W. Leitner and P. T. Anastas, ACS Sustainable Chem. Eng., 2025, 13, 5–29 CrossRef CAS.
- (a) S. Sarp, S. Gonzalez Hernandez, C. Chen and S. W. Sheehan, Joule, 2021, 5, 59–76 CrossRef CAS; (b) I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS; (c) A. Kumar, P. Daw and D. Milstein, Chem. Rev., 2022, 122, 385–441 CrossRef CAS PubMed.
- (a) R. E. Siegel, S. Pattanayak and L. A. Berben, ACS Catal., 2023, 13, 766–784 CrossRef CAS; (b) S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed.
- (a) J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2016, 138, 778–781 CrossRef CAS PubMed; (b) Z. Suhail, C. J. Koch, A. Goeppert and G. K. S. Prakash, Langmuir, 2024, 40, 5401–5408 CrossRef CAS PubMed; (c) S. Kar, A. Goeppert, V. Galvan, R. Chowdhury, J. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2018, 140, 16873–16876 CrossRef CAS PubMed.
- J. R. Khusnutdinova, J. A. Garg and D. Milstein, ACS Catal., 2015, 5, 2416–2422 CrossRef CAS.
- (a) M. Scott, B. Blas Molinos, C. Westhues, G. Franciò and W. Leitner, ChemSusChem, 2017, 10, 1085–1093 CrossRef CAS PubMed; (b) D. A. Kuß, M. Hölscher and W. Leitner, ChemCatChem, 2021, 13, 3319–3323 CrossRef; (c) S. Wesselbaum, U. Hintermair and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 8585–8588 CrossRef CAS PubMed.
- J. M. Hanusch, I. P. Kerschgens, F. Huber, M. Neuburger and K. Gademann, Chem. Commun., 2019, 55, 949–952 RSC.
- (a) A.-H. Liu, R. Ma, C. Song, Z.-Z. Yang, A. Yu, Y. Cai, L.-N. He, Y.-N. Zhao, B. Yu and Q.-W. Song, Angew. Chem., Int. Ed., 2012, 51, 11306–11310 CrossRef CAS PubMed; (b) Z.-Z. Yang, L.-N. He, Y.-N. Zhao, B. Li and B. Yu, Energy Environ. Sci., 2011, 4, 3971–3975 RSC.
- L. Piccirilli, B. Rabell, R. Padilla, A. Riisager, S. Das and M. Nielsen, J. Am. Chem. Soc., 2023, 145, 5655–5663 CrossRef CAS PubMed.
- N. M. Rezayee, C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS PubMed.
- (a) A. Boddien, F. Gärtner, C. Federsel, P. Sponholz, D. Mellmann, R. Jackstell, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6411–6414 CrossRef CAS PubMed; (b) C. Ziebart, C. Federsel, P. Anbarasan, R. Jackstell, W. Baumann, A. Spannenberg and M. Beller, J. Am. Chem. Soc., 2012, 134, 20701–20704 CrossRef CAS PubMed.
- H.-J. Song, S. Park, H. Kim, A. Gaur, J.-W. Park and S.-J. Lee, Int. J. Greenhouse Gas Control, 2012, 11, 64–72 CrossRef CAS.
- I. Bernhardsgrütter, G. M. M. Stoffel, T. E. Miller and T. J. Erb, Curr. Opin. Biotechnol, 2021, 67, 80–87 CrossRef PubMed.
- T. R. Cundari, A. K. Wilson, M. L. Drummond, H. E. Gonzalez, K. R. Jorgensen, S. Payne, J. Braunfeld, M. De Jesus and V. M. Johnson, J. Chem. Inf. Model., 2009, 49, 2111–2115 CrossRef CAS PubMed.
- D. T. King, S. Zhu, D. B. Hardie, J. E. Serrano-Negrón, Z. Madden, S. Kolappan and D. J. Vocadlo, Nat. Chem. Biol., 2022, 18, 782–791 CrossRef CAS PubMed.
- L. I. Blake and M. J. Cann, Front. Mol. Biosci., 2022, 9, 825706 CrossRef CAS PubMed.
- R. Phillips and R. Milo, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21465–21471 CrossRef CAS PubMed.
- B. Stec, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18785–18790 CrossRef CAS PubMed.
- D. Goodsell DOI:10.2210/rcsb_pdb/mom_2000_11 .
- (a) D. Wei, H. Junge and M. Beller, Chem. Sci., 2021, 12, 6020–6024 RSC; (b) D. Wei, R. Sang, P. Sponholz, H. Junge and M. Beller, Nat. Energy, 2022, 7, 438–447 CrossRef CAS; (c) A. Moazezbarabadi, D. Wei, H. Junge and M. Beller, ChemSusChem, 2022, 15, e202201502 CrossRef CAS PubMed.
- (a) W. Taifan, J.-F. Boily and J. Baltrusaitis, Surf. Sci. Rep., 2016, 71, 595–671 CrossRef CAS; (b) A. Paparo and J. Okuda, Coord. Chem. Rev., 2017, 334, 136–149 CrossRef CAS.
- P. Sreejyothi and S. K. Mandal, Chem. Sci., 2020, 11, 10571–10593 RSC.
- (a) A. Álvarez, M. Borges, J. J. Corral-Pérez, J. G. Olcina, L. Hu, D. Cornu, R. Huang, D. Stoian and A. Urakawa, ChemPhysChem, 2017, 18, 3135–3141 CrossRef PubMed; (b) U. J. Etim, C. Zhang and Z. Zhong, Nanomaterials, 2021, 11, 3265 CrossRef CAS PubMed.
- (a) H. Li, J. Zhao, L. Luo, J. Du and J. Zeng, Acc. Chem. Res., 2021, 54, 1454–1464 CrossRef CAS PubMed; (b) G.-C. Wang, L. Jiang, Y. Morikawa, J. Nakamura, Z.-S. Cai, Y.-M. Pan and X.-Z. Zhao, Surf. Sci., 2004, 570, 205–217 CrossRef CAS.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- X. Zhang, G. Liu, K.-H. Meiwes-Broer, G. Ganteför and K. Bowen, Angew. Chem., Int. Ed., 2016, 55, 9644–9647 CrossRef CAS PubMed.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- (a) P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 344–355 CrossRef CAS; (b) Y.-y Ohnishi, T. Matsunaga, Y. Nakao, H. Sato and S. Sakaki, J. Am. Chem. Soc., 2005, 127, 4021–4032 CrossRef CAS PubMed.
- (a) C. Bo and A. Dedieu, Inorg. Chem., 1989, 28, 304–309 CrossRef CAS; (b) S. Sakaki and K. Ohkubo, Inorg. Chem., 1989, 28, 2583–2590 CrossRef CAS; (c) R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed; (d) R. Tanaka, M. Yamashita, L. W. Chung, K. Morokuma and K. Nozaki, Organometallics, 2011, 30, 6742–6750 CrossRef CAS; (e) M. S. G. Ahlquist, J. Mol. Catal. A: Chem., 2010, 324, 3–8 CrossRef CAS; (f) X. Yang, ACS Catal., 2011, 1, 849–854 CrossRef CAS.
- T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 CrossRef CAS PubMed.
- J. Kothandaraman, M. Czaun, A. Goeppert, R. Haiges, J.-P. Jones, R. B. May, G. K. S. Prakash and G. A. Olah, ChemSusChem, 2015, 8, 1442–1451 CrossRef CAS PubMed.
- H. Schwarz, Coord. Chem. Rev., 2017, 334, 112–123 CrossRef CAS.
- (a) D. J. Heldebrant, J. Kothandaraman, N. M. Dowell and L. Brickett, Chem. Sci., 2022, 13, 6445–6456 RSC; (b) E. S. Wiedner and J. C. Linehan, Chem. – Eur. J., 2018, 24, 16964–16971 CrossRef CAS PubMed.
- T. J. Geldbach, G. Laurenczy, R. Scopelliti and P. J. Dyson, Organometallics, 2006, 25, 733–742 CrossRef CAS.
- C. Federsel, R. Jackstell, A. Boddien, G. Laurenczy and M. Beller, ChemSusChem, 2010, 3, 1048–1050 CrossRef CAS PubMed.
- D. B. Lao, B. R. Galan, J. C. Linehan and D. J. Heldebrant, Green Chem., 2016, 18, 4871–4874 RSC.
- R. Ramezani, S. Mazinani and R. D. Felice, Rev. Chem. Eng., 2022, 38, 273–299 CrossRef CAS.
- J.-a Lim, D. H. Kim, Y. Yoon, S. K. Jeong, K. T. Park and S. C. Nam, Energy Fuels, 2012, 26, 3910–3918 CrossRef CAS.
- C. A. Fitch, G. Platzer, M. Okon, B. Garcia-Moreno E. and L. P. McIntosh, Protein Sci., 2015, 24, 752–761 CrossRef CAS PubMed.
- S. Mazinani, R. Ramazani, A. Samsami, A. Jahanmiri, B. Van der Bruggen and S. Darvishmanesh, Fluid Phase Equilib., 2015, 396, 28–34 CrossRef CAS.
- Z.-W. Chen, R. B. Leron and M.-H. Li, Fluid Phase Equilib., 2015, 400, 20–26 CrossRef CAS.
- A. Zarei, A. Hafizi, M. R. Rahimpour and S. Raeissi, J. Mol. Liq., 2020, 301, 111743 CrossRef CAS.
- A. F. Portugal, J. M. Sousa, F. D. Magalhães and A. Mendes, Chem. Eng. Sci., 2009, 64, 1993–2002 CrossRef CAS.
- A. Syalsabila, A. S. Maulud, H. Suleman and N. A. Hadi Md Nordin, Int. J. Chem. Eng., 2019, 9428638 CAS.
- S. Garg, A. M. Shariff, M. S. Shaikh, B. Lal, A. Aftab and N. Faiqa, J. Nat. Gas Sci. Eng., 2016, 34, 864–872 CrossRef CAS.
- Y.-T. Chang, R. B. Leron and M.-H. Li, J. Chem. Thermodyn., 2015, 83, 110–116 CrossRef CAS.
- J. D. Madura, J. B. Lombardini, J. M. Briggs, D. L. Minor and A. Wierzbicki, Amino Acids, 1997, 13, 131–139 CrossRef CAS.
- J. v Holst, G. F. Versteeg, D. W. F. Brilman and J. A. Hogendoorn, Chem. Eng. Sci., 2009, 64, 59–68 CrossRef.
- S. Uludag-Demirer, A. Smerigan, P. J. Hsiao, A. Marks, M. R. Smith and W. Liao, Ind. Eng. Chem. Res., 2023, 62, 4064–4072 CrossRef CAS.
- A. Smerigan, S. Uludag-Demirer, A. Cutshaw, A. Marks and W. Liao, J. CO2 Util., 2023, 69, 102394 CrossRef CAS.
- X. F. Wang, N. G. Akhmedov, D. Hopkinson, J. Hoffman, Y. H. Duan, A. Egbebi, K. Resnik and B. Y. Li, Appl. Energy, 2016, 161, 41–47 CrossRef CAS.
- S. Shen, Y. Bian and Y. Zhao, Int. J. Greenhouse Gas Control, 2017, 56, 1–11 CrossRef CAS.
- H. Li, H. Guo and S. F. Shen, ACS Sustainable Chem. Eng., 2020, 8, 12956–12967 CrossRef CAS.
- E. Olyaei, A. Hafizi and M. R. Rahimpour, J. Mol. Liq., 2021, 336, 116286 CrossRef CAS.
- (a) J. L. Anthony, E. J. Maginn and J. F. Brennecke, J. Phys. Chem. B, 2002, 106, 7315–7320 CrossRef CAS; (b) N. M. Yunus, M. I. A. Mutalib, Z. Man, M. A. Bustam and T. Murugesan, Chem. Eng. J., 2012, 189–190, 94–100 CrossRef CAS.
- (a) S. Sarmad, J.-P. Mikkola and X. Ji, ChemSusChem, 2017, 10, 324–352 CrossRef CAS PubMed; (b) M. Aghaie, N. Rezaei and S. Zendehboudi, Renewable Sustainable Energy Rev., 2018, 96, 502–525 CrossRef CAS; (c) S. Lian, C. Song, Q. Liu, E. Duan, H. Ren and Y. Kitamura, J. Environ. Sci., 2021, 99, 281–295 CrossRef CAS PubMed.
- B. Lv, Y. Xia, Y. Shi, N. Liu, W. Li and S. Li, Int. J. Greenhouse Gas Control, 2016, 46, 1–6 CrossRef CAS.
- N. Noorani, A. Mehrdad and I. Ahadzadeh, Fluid Phase Equilib., 2021, 547, 113185 CrossRef CAS.
- N. Noorani, A. Mehrdad and R. Z. Diznab, Fluid Phase Equilib., 2022, 557, 113433 CrossRef CAS.
- G. Latini, M. Signorile, V. Crocellà, S. Bocchini, C. F. Pirri and S. Bordiga, Catal. Today, 2019, 336, 148–160 CrossRef CAS.
- Y. S. Sistla and A. Khanna, Chem. Eng. J., 2015, 273, 268–276 CrossRef CAS.
- X. Li, M. Hou, Z. Zhang, B. Han, G. Yang, X. Wang and L. Zou, Green Chem., 2008, 10, 879–884 RSC.
- G. Latini, M. Signorile, F. Rosso, A. Fin, M. D'Amora, S. Giordani, F. Pirri, V. Crocellà, S. Bordiga and S. Bocchini, J. CO2 Util., 2022, 55, 102285 Search PubMed.
- Q. W. Yang, Z. P. Wang, Z. B. Bao, Z. G. Zhang, Y. W. Yang, Q. L. Ren, H. B. Xing and S. Dai, ChemSusChem, 2016, 9, 806–812 CrossRef CAS PubMed.
- S. Saravanamurugan, A. J. Kunov-Kruse, R. Fehrmann and A. Riisager, ChemSusChem, 2014, 7, 897–902 CrossRef CAS PubMed.
- G. Latini, M. Signorile, F. Rosso, A. Fin, M. d’Amora, S. Giordani, F. Pirri, V. Crocellà, S. Bordiga and S. Bocchini, J. CO2 Util., 2022, 55, 101815 CrossRef CAS.
- D. A. Kuettel, B. Fischer, S. Hohe, R. Joh, M. Kinzl and R. Schneider, Energy Procedia, 2013, 37, 1687–1695 CrossRef CAS.
- H. Chen, T.-C. Tsai and C.-S. Tan, Int. J. Greenhouse Gas Control, 2018, 79, 127–133 CrossRef CAS.
- Z. Feng, F. Cheng-Gang, W. You-Ting, W. Yuan-Tao, L. Ai-Min and Z. Zhi-Bing, Chem. Eng. J., 2010, 160, 691–697 CrossRef.
- G. Jing, L. Zhou and Z. Zhou, Chem. Eng. J., 2012, 181–182, 85–92 CrossRef CAS.
- H. Meng, C. Lu, J. Yang, Y. Fan, Y. Huang, X.-F. Kong and F. Zhang, ACS Sustainable Chem. Eng., 2025, 2097–2106 CrossRef CAS.
- (a) A. Izaddoust and P. Keshavarz, Energy Fuels, 2017, 31, 9790–9799 CrossRef CAS; (b) Q. Li, Z. Bao, N. G. Akhmedov, B. A. Li, Y. Duan, M. Xing, J. Wang, B. I. Morsi and B. Li, Ind. Eng. Chem. Res., 2022, 61, 12545–12554 CrossRef CAS; (c) G. Hu, K. H. Smith, Y. Wu, S. E. Kentish and G. W. Stevens, Energy Fuels, 2017, 31, 4280–4286 CrossRef CAS.
- H. Thee, N. J. Nicholas, K. H. Smith, G. da Silva, S. E. Kentish and G. W. Stevens, Int. J. Greenhouse Gas Control, 2014, 20, 212–222 CrossRef CAS.
- Y. Li, L. a Wang, Z. Tan, Z. Zhang and X. Hu, Sep. Purif. Technol., 2019, 219, 47–54 CrossRef CAS.
- (a) J. Ott, V. Gronemann, F. Pontzen, E. Fiedler, G. Grossmann, D. B. Kersebohm, G. Weiss and C. Witte, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2012 DOI:10.1002/14356007.a16_465.pub3; (b) S. S. Tabibian and M. Sharifzadeh, Renewable Sustainable Energy Rev., 2023, 179, 113281 CrossRef CAS; (c) https://www.methanol.org/applications/ (accessed Mai 14th, 2024).
- Non-renewable fossil fuels can also be considered as source of hydrogen. However, their combustion will produce additional CO2. To achieve a zero CO2 emission system, the fossil fuel must supply additional hydrogen for utilizing CO2 emissions after consuming its own carbon. Thus, not all fossil fuels are suitable. With methane, which has the highest H to C ratio, only formic acid, formaldehyde, and acetic acid can be produced under the zero emission principle. For an in depth discussion see: S. Vasudevan, S. Aggarwal, S. Farooq, I. A. Karimi and M. C. G. Quah, Technoenergetic and Economic Analysis of CO2 Conversion, in An Economy based on Carbon Dioxide and Water, ed. M. Aresta, I. Karimi and S. Kawi, Springer Nature, Switzerland AG, 2020, pp. 413–430.
- (a) Y. Xu, L. Wang, Q. Zhou, Y. Li, L. Liu, W. Nie, R. Xu, J. Zhang, Z. Cheng, H. Wang, Y. Huang, T. Wei, Z. Fan and L. Wang, Coord. Chem. Rev., 2024, 508, 215775 CrossRef CAS; (b) X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed; (c) A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- (a) R. Sen, A. Goeppert and G. K. Surya Prakash, Angew. Chem., Int. Ed., 2022, 61, e202207278 CrossRef CAS PubMed; (b) N. Onishi and Y. Himeda, Chem Catal., 2022, 2, 242–252 CrossRef CAS; (c) S.-T. Bai, G. De Smet, Y. Liao, R. Sun, C. Zhou, M. Beller, B. U. W. Maes and B. F. Sels, Chem. Soc. Rev., 2021, 50, 4259–4298 RSC.
- (a) P. A. Dub and T. Ikariya, ACS Catal., 2012, 2, 1718–1741 CrossRef CAS; (b) S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS.
- (a) P. V. Kortunov, M. Siskin, L. S. Baugh and D. C. Calabro, Energy Fuels, 2015, 29, 5919–5939 CrossRef CAS; (b) P. V. Kortunov, M. Siskin, L. S. Baugh and D. C. Calabro, Energy Fuels, 2015, 29, 5940–5966 CrossRef CAS; (c) P. V. Kortunov, L. S. Baugh, M. Siskin and D. C. Calabro, Energy Fuels, 2015, 29, 5967–5989 CrossRef CAS; (d) P. V. Kortunov, M. Siskin, M. Paccagnini and H. Thomann, Energy Fuels, 2016, 30, 1223–1236 CAS.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS PubMed.
- R. B. Said, J. M. Kolle, K. Essalah, B. Tangour and A. Sayari, ACS Omega, 2020, 5, 26125–26133 CrossRef CAS PubMed.
- J. R. Cabrero-Antonino, R. Adam, V. Papa and M. Beller, Nat. Commun., 2020, 11, 3893 CrossRef CAS PubMed.
- R. Sen, C. J. Koch, A. Goeppert and G. K. S. Prakash, ChemSusChem, 2020, 13, 6318–6322 CrossRef CAS PubMed.
- M. Zanatta, ACS Mater. Au, 2023, 3, 576–583 CrossRef CAS PubMed.
- R. Sen, A. Goeppert, S. Kar and G. K. S. Prakash, J. Am. Chem. Soc., 2020, 142, 4544–4549 CrossRef CAS PubMed.
- L. Zhang, Z. Han, X. Zhao, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2015, 54, 6186–6189 CrossRef CAS PubMed.
- S. Kar, R. Sen, J. Kothandaraman, A. Goeppert, R. Chowdhury, S. B. Munoz, R. Haiges and G. K. S. Prakash, J. Am. Chem. Soc., 2019, 141, 3160–3170 CrossRef CAS PubMed.
- M. Everett and D. F. Wass, Chem. Commun., 2017, 53, 9502–9504 RSC.
- (a) R. Sen, A. Goeppert and G. S. Prakash, Aldrichimica Acta, 2020, 53, 39–56 Search PubMed; (b) R. Bhardwaj and J. Choudhury, Chem. – Eur. J., 2025, 20, e202401327 CAS.
- (a) S. Kar, R. Sen, A. Goeppert and G. K. S. Prakash, J. Am. Chem. Soc., 2018, 140, 1580–1583 CrossRef CAS PubMed; (b) E. M. Lane, Y. Zhang, N. Hazari and W. H. Bernskoetter, Organometallics, 2019, 38, 3084–3091 CrossRef CAS.
- (a) S. Kar, A. Goeppert, J. Kothandaraman and G. K. S. Prakash, ACS Catal., 2017, 7, 6347–6351 CrossRef CAS; (b) C. J. Koch, A. Goeppert and G. K. Surya Prakash, ChemSusChem, 2024, 17, e202301789 CrossRef CAS PubMed.
- M. A. Stevens and A. L. Colebatch, Chem. Soc. Rev., 2022, 51, 1881–1898 RSC.
- B. Zhao, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744–4788 CrossRef CAS PubMed.
- (a) P. A. Dub and J. C. Gordon, Nat. Rev. Chem., 2018, 2, 396–408 CrossRef CAS; (b) P. A. Dub and J. C. Gordon, ACS Catal., 2017, 7, 6635–6655 CrossRef CAS.
- O. Eisenstein and R. H. Crabtree, New J. Chem., 2013, 37, 21–27 RSC.
- W. Kuriyama, T. Matsumoto, Y. Ino and O. Ogata, WO/2011/048727, 2011. (It should be noted that when X is a halide, an equivalent of base to the catalyst is required to generate the catalytically active species. With Ru-MACHO-BH, thermal activation suffices to access the corresponding ruthenium dihydride through loss of the BH3 moiety).
- P. A. Dub, B. L. Scott and J. C. Gordon, J. Am. Chem. Soc., 2017, 139, 1245–1260 CrossRef CAS PubMed.
- (a) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed; (b) T. P. Gonçalves, I. Dutta and K.-W. Huang, Chem. Commun., 2021, 57, 3070–3082 RSC.
- (a) J. Kothandaraman, D. J. Heldebrant, J. S. Lopez and R. A. Dagle, Front. Energy Res., 2023, 11, 1158499 CrossRef; (b) W. Yang, T. Y. Kalavalapalli, A. M. Krieger, T. A. Khvorost, I. Y. Chernyshov, M. Weber, E. A. Uslamin, E. A. Pidko and G. A. Filonenko, J. Am. Chem. Soc., 2022, 144, 8129–8137 CrossRef CAS PubMed; (c) C. L. Mathis, J. Geary, Y. Ardon, M. S. Reese, M. A. Philliber, R. T. VanderLinden and C. T. Saouma, J. Am. Chem. Soc., 2019, 141, 14317–14328 CrossRef CAS PubMed; (d) R. J. Hamilton and S. H. Bergens, J. Am. Chem. Soc., 2006, 128, 13700–13701 CrossRef CAS PubMed.
- A. P. C. Ribeiro, L. M. D. R. S. Martins and A. J. L. Pombeiro, Green Chem., 2017, 19, 4811–4815 RSC.
- C. Zhu, C. D'Agostino and S. P. de Visser, Chem. – Eur. J., 2023, 29, e202302832 CrossRef CAS PubMed.
- (a) S. Zeng, X. Zhang, L. Bai, X. Zhang, H. Wang, J. Wang, D. Bao, M. Li, X. Liu and S. Zhang, Chem. Rev., 2017, 117, 9625–9673 CrossRef CAS PubMed; (b) M. I. Qadir and J. Dupont, Angew. Chem., Int. Ed., 2023, 62, e202301497 CrossRef PubMed.
- G. Cui, J. Wang and S. Zhang, Chem. Soc. Rev., 2016, 45, 4307–4339 RSC.
- T. Sasaki, Curr. Opin. Green Sustainable Chem., 2022, 36, 100633 CrossRef CAS.
- (a) A. Weilhard, S. P. Argent and V. Sans, Nat. Commun., 2021, 12, 231 CrossRef CAS PubMed; (b) A. Weilhard, M. I. Qadir, V. Sans and J. Dupont, ACS Catal., 2018, 8, 1628–1634 CrossRef CAS; (c) A. Weilhard, K. Salzmann, M. Navarro, J. Dupont, M. Albrecht and V. Sans, J. Catal., 2020, 385, 1–9 CrossRef CAS.
- W. Ma, J. Hu, L. Zhou, Y. Wu, J. Geng and X. Hu, Green Chem., 2022, 24, 6727–6732 RSC.
- C. J. Koch, A. Goeppert and G. K. Surya Prakash, ChemSusChem, 2024, 17, e202301789 CrossRef CAS PubMed.
- A. Yoshimura, R. Watari, S. Kuwata and Y. Kayaki, Eur. J. Inorg. Chem., 2019, 2375–2380 CrossRef CAS.
- S. Kar, A. Goeppert and G. K. S. Prakash, ChemSusChem, 2019, 12, 3172–3177 CrossRef CAS PubMed.
- J. Hietala, A. Vuori, P. Johnsson, I. Pollari, W. Reutemann and H. Kieczka, Ullmann's Encyclopedia of Industrial Chemistry, 2016, pp. 1–22 Search PubMed.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- T. Singh, S. Jalwal and S. Chakraborty, Asian J. Org. Chem., 2022, 11, e202200330 CrossRef CAS.
- (a) T. Schaub and R. A. Paciello, Angew. Chem., Int. Ed., 2011, 50, 7278–7282 CrossRef CAS PubMed; (b) T. Schaub, R. Paciello, K. Mohl, D. Schneider, M. Schaefer and S. Rittinger, WO2010149507A2, 2010.
- D. Wei, R. Sang, A. Moazezbarabadi, H. Junge and M. Beller, JACS Au, 2022, 2, 1020–1031 CrossRef CAS PubMed.
- Y.-N. Li, L.-N. He, A.-H. Liu, X.-D. Lang, Z.-Z. Yang, B. Yu and C.-R. Luan, Green Chem., 2013, 15, 2825–2829 RSC.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. Surya Prakash, Green Chem., 2016, 18, 5831–5838 RSC.
- S. Tshepelevitsh, A. Kütt, M. Lõkov, I. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, Eur. J. Org. Chem., 2019, 6735–6748 CrossRef CAS.
- R. Bhardwaj, A. Kumar and J. Choudhury, Chem. Commun., 2022, 58, 11531–11534 RSC.
- S. Kushwaha, J. Parthiban and S. K. Singh, ACS Omega, 2023, 8, 38773–38793 CrossRef CAS PubMed.
- N. Guntermann, G. Franciò and W. Leitner, Green Chem., 2022, 24, 8069–8075 RSC.
- A. Moazezbarabadi, A. Kammer, E. Alberico, H. Junge and M. Beller, ChemSusChem, 2025, 18, e202401813 CrossRef CAS PubMed.
- V. Sang Sefidi and P. Luis, Ind. Eng. Chem. Res., 2019, 58, 20181–20194 CrossRef CAS.
- X. Wang and C. Song, Front. Energy Res., 2020, 8, 560849 CrossRef.
- J. M. Kolle, M. Fayaz and A. Sayari, Chem. Rev., 2021, 121, 7280–7345 CrossRef CAS PubMed.
- T. Diehl, P. Lanzerath, G. Franciò and W. Leitner, ChemSusChem, 2022, 15, e202201250 CrossRef CAS PubMed.
- D. J. Heldebrant, P. K. Koech, V.-A. Glezakou, R. Rousseau, D. Malhotra and D. C. Cantu, Chem. Rev., 2017, 117, 9594–9624 CrossRef CAS PubMed.
- (a) Asahi Glass Sustainability Report 2024 https://www.agc.com/en/sustainability/pdf/2004.pdf; (b) M. Grohol and C. Veeh, https://data.europa.eu/doi/10.2873/725585, 2023.
- (a) N. D. McNamara and J. C. Hicks, ChemSusChem, 2014, 7, 1114–1124 CrossRef CAS PubMed; (b) T. Mandal, A. Kumar, J. Panda, T. Kumar Dutta and J. Choudhury, Angew. Chem., Int. Ed., 2023, 62, e202314451 CrossRef CAS PubMed.
- R. Kumar, T. Mandal, A. Bhattacherya, M. K. Pandey, J. K. Bera and J. Choudhury, ACS Catal., 2024, 14, 13236–13245 CrossRef CAS.
- S. Padmanaban, G. H. Gunasekar and S. Yoon, Inorg. Chem., 2021, 60, 6881–6888 CrossRef CAS PubMed.
- H. Park and S. Yoon, ACS Sustainable Chem. Eng., 2023, 11, 12036–12044 CrossRef CAS.
- (a) J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed; (b) H. Lamberts-Van Assche and T. Compernolle, Clean Technol. Environ. Policy, 2022, 24, 467–491 CrossRef.
- C. M. Jens, L. Müller, K. Leonhard and A. Bardow, ACS Sustainable Chem. Eng., 2019, 7, 12270–12280 CAS.
| This journal is © The Royal Society of Chemistry 2025 |