
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cross-disciplinary perspectives on the potential for artificial intelligence across chemistry
Austin M.
Mroz
 ab,
Annabel R.
Basford
a,
Friedrich
Hastedt
c,
Isuru Shavindra
Jayasekera
d,
Irea
Mosquera-Lois
e,
Ruby
Sedgwick
f,
Pedro J.
Ballester
g,
Joshua D.
Bocarsly
h,
Ehecatl
Antonio del Río Chanona
c,
Matthew L.
Evans
ijk,
Jarvist M.
Frost
a,
Alex M.
Ganose
a,
Rebecca L.
Greenaway
a,
King
Kuok (Mimi) Hii
a,
Yingzhen
Li
f,
Ruth
Misener
f,
Aron
Walsh
e,
Dandan
Zhang
bg and
Kim E.
Jelfs
*a
ab,
Annabel R.
Basford
a,
Friedrich
Hastedt
c,
Isuru Shavindra
Jayasekera
d,
Irea
Mosquera-Lois
e,
Ruby
Sedgwick
f,
Pedro J.
Ballester
g,
Joshua D.
Bocarsly
h,
Ehecatl
Antonio del Río Chanona
c,
Matthew L.
Evans
ijk,
Jarvist M.
Frost
a,
Alex M.
Ganose
a,
Rebecca L.
Greenaway
a,
King
Kuok (Mimi) Hii
a,
Yingzhen
Li
f,
Ruth
Misener
f,
Aron
Walsh
e,
Dandan
Zhang
bg and
Kim E.
Jelfs
*a
aDepartment of Chemistry, Imperial College London, London W12 0BZ, UK. E-mail: k.jelfs@imperial.ac.uk; Tel: +44 20759 43438
bI-X Centre for AI in Science, Imperial College London, London W12 0BZ, UK
cDepartment of Chemical Engineering, Imperial College London, London SW7 2AZ, UK
dDepartment of Mathematics, Imperial College London, London SW7 2AZ, UK
eDepartment of Materials, Imperial College London, London SW7 2AZ, UK
fDepartment of Computing, Imperial College London, London SW7 2AZ, UK
gDepartment of Bioengineering, Imperial College London, London SW7 2AZ, UK
hDepartment of Chemistry and Texas Center for Superconductivity, University of Houston, Houston, USA
iUCLouvain, Institute of Condensed Matter and Nanosciences (IMCN), Chemin des Étoiles 8, Louvain-la-Neuve 1348, Belgium
jMatgenix SRL, A6K Advanced Engineering Center, Charleroi, Belgium
kDatalab Industries Ltd, King's Lynn, Norfolk, UK
First published on 25th April 2025
Abstract
From accelerating simulations and exploring chemical space, to experimental planning and integrating automation within experimental labs, artificial intelligence (AI) is changing the landscape of chemistry. We are seeing a significant increase in the number of publications leveraging these powerful data-driven insights and models to accelerate all aspects of chemical research. For example, how we represent molecules and materials to computer algorithms for predictive and generative models, as well as the physical mechanisms by which we perform experiments in the lab for automation. Here, we present ten diverse perspectives on the impact of AI coming from those with a range of backgrounds from experimental chemistry, computational chemistry, computer science, engineering and across different areas of chemistry, including drug discovery, catalysis, chemical automation, chemical physics, materials chemistry. The ten perspectives presented here cover a range of themes, including AI for computation, facilitating discovery, supporting experiments, and enabling technologies for transformation. We highlight and discuss imminent challenges and ways in which we are redefining problems to accelerate the impact of chemical research via AI.
Introduction
Artificial intelligence (AI) is undeniably revolutionising scientific research, enabling researchers to explore chemical phenomena at scales and speeds that would otherwise be unattainable. Indeed, chemistry faces several challenges that are well-suited to data-driven approaches; these challenges largely stem from the massive search spaces that chemists have at their disposal. Consider, for example, the vastness of chemical space; there are estimated to be 1060 candidate small organic molecules that could feasibly be synthesised. This does not account for the variety of methods, protocols, and procedures that may be used to make them, nor does it account for the number of subsequent materials for which they could serve as building blocks. Indeed, this ‘needle-in-a-haystack’ problem possesses many challenging layers, ranging from high-dimensional search spaces to many non-linear, often stochastic, relationships between structure and function. Yet, the ability of AI to assist in chemistry is not limited to searching chemical space; there is opportunity for AI to accelerate discovery through improving computational models, data characterisation pipelines, as well as providing support for automation of experimental methods.The ability of AI to transform and accelerate research has been successfully demonstrated in numerous publications across chemistry. Indeed, these efforts have been highlighted in numerous reviews across sub-disciplines in chemistry.1–7 This recent surge in publications featuring AI to accelerate chemistry, in addition to the well-established chemical benchmarking datasets in the AI community, has led to collaborations across disciplines in ways not previously present in the literature. Ultimately, this has led to the application of state-of-the-art AI models to cutting-edge scientific research, featuring profound impact and implications for our ability to tackle complex scientific challenges.
With this collaboration between chemistry and AI, there is a beneficial increase in the diversity of perspectives within chemistry. Indeed, this discourse is prevalent across chemistry – from theoretical and computational chemistry, to experimental chemistry, as well as broader chemical initiatives that span research tools (e.g. molecular and materials discovery). Here, we present ten different perspectives on the impact of AI in chemical research coming from those with a range of backgrounds from experimental chemistry, computational chemistry, computer science, engineering and across different areas of chemistry, including drug discovery, catalysis, chemical automation, chemical physics, materials chemistry. The perspectives broadly covers the impact of AI on computation, discovery, experimentation, and its transformative role linking these through new technologies. We delve into the transformative potential of AI, highlighting many of the challenges we face, and offering potential strategies to address them.
1 AI for quantum chemistry
All of chemistry is an emergent property from the solution of the Schrödinger equation. As Dirac said, “the difficulty lies only in the fact that application of these laws leads to equations that are too complex to be solved.”For nearly 100 years, quantum chemistry has seen the development of ever more accurate approximate solutions to the Schrödinger equation. These methods have grown in lockstep with the consistently exponential increase in digital computer power during the last eight decades. Due to electron correlation (which physicists call quantum entanglement), the direct exact solution of the Schrödinger equation scales with the number of electrons as 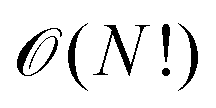 (in this asymptotic ‘big O’ notation, the computational effort scales as the expression in the brackets). Practical quantum chemistry methods have, therefore, mainly been concerned with developing approximate solutions at a lower computational cost. These are often expressed on a Jacob's ladder from a fully correlated top rung (the ‘heaven of chemical accuracy’), to a totally uncorrelated bottom. Here, we discuss the impact of AI in quantum Monte-Carlo, density functional theory (DFT), and semi-empirical quantum chemistry.
(in this asymptotic ‘big O’ notation, the computational effort scales as the expression in the brackets). Practical quantum chemistry methods have, therefore, mainly been concerned with developing approximate solutions at a lower computational cost. These are often expressed on a Jacob's ladder from a fully correlated top rung (the ‘heaven of chemical accuracy’), to a totally uncorrelated bottom. Here, we discuss the impact of AI in quantum Monte-Carlo, density functional theory (DFT), and semi-empirical quantum chemistry.
1.1 Neural network wavefunction ansatz and quantum Monte-Carlo (QMC)
In principle, quantum Monte-Carlo calculations can evaluate a quantum-mechanical observable (such as the total energy) exactly with the usual stochastic error reducing as the number of samples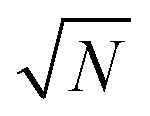 . In Bosonic systems, the errors are, therefore, under full control. In Fermionic systems (such as electrons in quantum chemistry), the fluctuating sign of the contributions to these integrals (due to the antisymmetry under exchange requirement of the Fermionic wavefunction, ψ(…, r1, …, r2, …) = −ψ(…, r2, …, r1, …)) exponentially slows down this
. In Bosonic systems, the errors are, therefore, under full control. In Fermionic systems (such as electrons in quantum chemistry), the fluctuating sign of the contributions to these integrals (due to the antisymmetry under exchange requirement of the Fermionic wavefunction, ψ(…, r1, …, r2, …) = −ψ(…, r2, …, r1, …)) exponentially slows down this 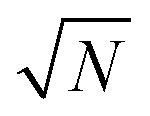 convergence, limiting study to small systems and effectively imposing
convergence, limiting study to small systems and effectively imposing  scaling.
scaling.
Adding knowledge about the wavefunction being integrated allows the use of importance sampling in the Monte-Carlo procedure. With perfect knowledge of the wavefunction, quantum Monte-Carlo becomes polynomial in time. Of course, if one had exact knowledge of the wavefunction, the Monte-Carlo calculation would be unnecessary!
One attractive aspect of quantum Monte-Carlo (and most post Hartree–Fock methods in quantum chemistry), is that they can generally be constructed in a variational manner. In a variational theory, any optimisation or adjustment which reduces the total energy takes the solution closer to the true value. This gives enormous freedom in the algorithmic approach to improve the solution, and a direct evaluation of the impact of any improvement. No external data is needed to evaluate the improvement, and so the approach can iterate between improving the guess to the wavefunction, and then using this wavefunction in Monte-Carlo evaluation of the energy. Such ‘self-play’ in the setting of symmetric games (such as chess) have enabled some of the most notable examples of superhuman AI.
The first application of neural networks (NNs) to represent many-body wavefunctions was by Carleo and Troyer8 in the context of a spin–lattice. To be used for quantum chemistry, this general approach needs to be extended to consider electrons in three-dimensional space.
There are two broad categories of quantum Monte-Carlo approaches:
1. Those constructed in first-quantisation directly consider the wavefunction over three-dimensional space ψ(x, y, z).
2. Those constructed in second-quantisation instead consider the wavefunction in terms of an occupation number over a finite basis, most often the Slater-determinants which result from a mean-field Hartree–Fock quantum-chemical calculation.
A notable benefit of first-quantisation is that no choice of basis set has to be made. Instead, the fundamental and general ψ(x, y, z) is being constructed. In systems where the chemical behaviour is not known a priori, this has the considerable advantage of not biasing the solution to what is expected. The complexity is that the methods used to predict the wavefunction have to correctly describe the antisymmetry present. A mean-field wavefunction in first-quantisation depends solely on the particle positions. Making the wavefunction dependent dynamically on the position of the other particles (configuration dependence) includes many-body correlation in the wavefunction. These are known as backflow wavefunctions, as the first application was the inclusion of an analytic hydrodynamic backflow contribution to the wavefunction in the study of liquid helium,9 which was later generalised and used to improve the solutions for electron gas calculations.10 The approach of using NNs to directly specify the backflow transformation11 with more flexibility then enabled with a fitted analytic model. PauliNet12 continues this approach further, producing a highly physically motivated NN with explicit Slater–Jastrow and Backflow components. FermiNet13 and Psiformer14 take a more maximally machine learning (ML) approach, giving more generalisability to the NN method. Both these approaches now enable state-of-the-art quantum Monte-Carlo calculations of small molecules (molecules featuring less than 100 electrons).
A major benefit of the second-quantisation is that the anti-symmetrisation of the wavefunction has been pushed into the use of Slater determinants (or mathematically similar functions); this means that the ML challenge can now use standard NNs and approaches. Here, one is trying to predict well-behaved occupation numbers of the second quantisation basis. A recent review focuses on NN ansatz in accelerating quantum Monte-Carlo calculations, with more technical detail.15
One issue with these techniques is that they tend to concentrate on the ground state energy of the system in question. From an experimental or molecular/material design point of view, this observable is not particularly interesting. Methods are much less developed, compared to quantum-chemistry approaches with a finite basis, to calculate response functions of the systems. There has been some interesting recent work in modelling excited states16 and unusual positronic chemistries17 with NN wavefunction approaches.
1.2 Machine learnt density functional theory (DFT)
The most successful method for quantum chemistry is density functional theory (DFT). These methods are based on the Hohenberg–Kohn mathematical proof that the same information that is present in the multi-dimensional electron wave function is equally present in the three-dimensional electron density. Practical Kohn–Sham implementations of this theory (where the kinetic energy is evaluated with a set of orbitals) rely on simple parameterisations for the correlation energy of the homogeneous electron gas, which in turn come from QMC calculations undertaken in the 1980s. The promise of ML approaches applied to DFT are that more powerful parameterisations could be developed which lead to more accurate solutions.There are two mechanisms by which the machine-learnt DFT can be trained. The first is to use a training dataset, typically derived from a higher-level quantum-chemistry approach (such as CCSD). The functional is then modified to reproduce the reference energies (and sometimes densities). The second approach is to use the number of exact constraints on the electron wave function which can be analytically specified and, therefore, introduced into the training set used to train these more expressive functionals. These can be challenging to include into an interpretable analytic functional, but one can hope to correctly reproduce them with a more expressive machine learnt functional. An example is the fractional electron condition as used in the training of the DM21 functional,18 improving considerably on the fictitious charge delocalisation usually present in density functionals. Ultimately, developing ML density functionals is highly attractive, as there is considerable community expertise in using the techniques, and community codes in which the methods can be implemented.
Alternatively, orbital-free DFT dispenses with the Kohn–Sham orbitals to calculate the electron kinetic energy, and instead directly constructs kinetic energy as a functional of the density. This approach is more computationally efficient, as you avoid the 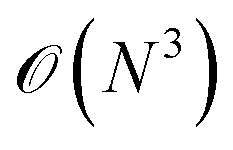 cost of orthogonalising orbitals, permitting enormously large calculations compared to standard Kohn–Sham DFT. There has been some recent success in constructing this functional with NN approaches.19
cost of orthogonalising orbitals, permitting enormously large calculations compared to standard Kohn–Sham DFT. There has been some recent success in constructing this functional with NN approaches.19
1.3 Machine learnt tight binding (TB) and semi-empirical quantum chemistry
Tight-binding (TB) and semi-empirical quantum chemistry are the most simple (and therefore computationally efficient) models that directly represent the electronic structure of molecules and materials. These methods use a minimalist basis set (often just atomic orbitals), and include electron correlation effects via effective parameters. Thus, the methods typically scale with the cost of orthogonalising these orbitals. However, the size of the basis is much smaller than an ab initio basis set, and there are further methods, such as bond order potentials, which can use the same parameters without an explicit orbital representation. In order to simulate structure and dynamics, tight-binding models also require a (mostly repulsive) pair–wise interaction potential to prevent the atoms unphysically overlapping.
cost of orthogonalising these orbitals. However, the size of the basis is much smaller than an ab initio basis set, and there are further methods, such as bond order potentials, which can use the same parameters without an explicit orbital representation. In order to simulate structure and dynamics, tight-binding models also require a (mostly repulsive) pair–wise interaction potential to prevent the atoms unphysically overlapping.
Two recent general tight-binding parameterisations that are seeing widespread adoption are the open-source DFTB (density functional tight-binding),20 and xTB (extended tight-binding)21 methods. These methods are semi-empirical, including atomic overlaps evaluated at the density functional theory level, with empirical parameters. Early work including machine learning in this area directly represented key steps in the Hamiltonian construction as a NN, enabling back-propagation of gradients, and, therefore, tuning.22 However, more recent work23,24 has generally used modern ML approaches (particularly, gradient optimisation and back propagation) to optimise standard parameters (based on the Slater–Koster analytic evaluation of atomic orbital overlaps), which enables a direct interpretation of the results and can more easily integrate with standard theoretical chemistry work processes. From this point of view, we can understand these approaches as building on the rich 80-year history of theoretical chemists building bespoke minimal parameter models, with the software-engineering and computational statistics of the big-data era.25
An alternative, hybrid, approach, is to use a delta-Machine-Learning technique to correct the results of a tight-binding model, which can be extended as a principal into a hierarchy of such corrections,26 or to use a tight-binding model as a computationally efficient way to provide a quantum-mechanically informed feature vector for a machine-learning model.27 However, research into these rather complex architectures has reduced recently due to the increasing power of force-field models.
A future development that is likely to have increasing importance is the use of equivariant and other more powerful basis sets (as developed for empirical ML potentials, see next section) to calculate the Hamiltonian. Zhang et al. applied the Atomic Cluster Expansion (ACE) to fit Hamiltonian matrix elements with greater accuracy and using less data.28 Generalising these methods to use a machine-learnt atomic feature vector would enable the construction of ‘foundational’ machine-learning tight-binding models of the entire periodic table.
1.4 Future outlook
There are unifying themes within the development of ML approaches for quantum chemistry. A motivating factor is that we can consider a quantum description of matter to be a strong inductive bias on the ML technique. Inductive biases lead to models that generalise better; by having a fundamentally quantum mechanical description, one would expect to have a model that has the correct long-range behaviour, and which extrapolates to larger systems.Perhaps the most surprising aspect of applying ML to quantum chemistry, is that these methods have not come to dominate. So far, though the methods do offer improvements on the state-of-the-art, the gains are relatively marginal, and come with significant costs in terms of additional expertise required to undertake the calculations, and the underlying uncontrolled approximations inherent to the methods. Fundamentally, we have not yet seen a method with such a large performance increase (in terms of accuracy versus computation) that it dominates. In many ways, this mirrors the human-led development of DFT functionals, where the majority of researchers use relatively simple few-parameter functionals developed 30–40 years ago. One possibility is that this is an example of Sutton's Bitter Lesson,29 which states that more simple ML methods that can leverage larger amounts of data and compute will inevitably dominate in the long-term.
2 Scaling atomistic simulations with ML force fields
Simulating chemical systems at the atomic scale requires a model of how atoms interact. The traditional trade-off between accuracy and computational cost is being disrupted by ML techniques that combine the quality of quantum mechanical methods discussed in the preceding section with the speed of traditional interatomic force fields. There are several extensive reviews on this topic,30–33 but here we focus on some important developments and ongoing directions in ML force fields (MLFFs) for materials.The potential energy surface of atomic configurations can be represented by empirical force fields – analytic models that approximate the forces between atoms as an expansion of two-body (distances) and higher-order (angles, dihedrals, etc.) terms. These models can be parameterised for specific systems (e.g. the TIP4P model for water34,35) or more general chemistries (e.g. the AMBER force fields for biomolecules36). Due to their fixed functional forms, such models are less accurate and transferable compared to quantum mechanical approaches. However, they allow simulations to be performed with length (nm–μm) and time scales (ps–μs) far beyond those accessible with methods such as DFT. The length and time scales afforded by these methods can describe rare processes (e.g. reactions in a catalytic cycle or crystal nucleation/degradation) and collective phenomena (e.g. self-assembly or spinodal decomposition), allowing for the study of emergent behaviours and material transformations.
2.1 Data-driven interatomic interactions
The training of force fields can be treated as a supervised learning task, where the input is the chemical structure. For crystalline materials, the outputs (labels) are usually the potential energy (E), atomic force (F), and cell stress (S). The quality and diversity of the EFS (potential energy, atomic force, and cell stress) training data determine the reliability of MLFFs in describing the thermodynamic and kinetic properties of compounds. For example, highly correlated systems (e.g. quantum spin liquids) would require labels from beyond-DFT methods, while polymorphic systems (e.g. perovskite crystals) require sampling of multiple structural configurations.The architecture of an MLFF is defined by the combination of representation and regression. A structural representation that is equivariant with respect to geometric operations like rotations or translations is favoured to produce robust models that require less training data.37–39 Most representations start from atom-centred functions that describe the distribution of neighbours around a given site. Radial and angular basis functions are used in several schemes such as the smooth overlap of atomic positions (SOAP),40 moment tensor potentials (MTP),41 and atomic cluster expansion (ACE).42,43 The role of the regression model is to map between the structural representation and the EFS outputs. While early models were built on feed-forward NNs44 or Gaussian process regression,45 graph neural networks (GNNs) are now widely used including the open-source Nequip,38 Allegro,39 and MACE46,47 architectures.
2.2 Facilitating chemical insights
The speed of MLFFs (typically 102–103 times faster than DFT) makes them attractive for use as surrogate models to tackle large compositional or configurational spaces, like crystal structure prediction48 or transition state searches.49 Beyond ideal systems, MLFFs have also been used to accelerate more realistic models of catalytic surface adsorbates50,51 or imperfect crystals. For example, a recent study of point defects trained a model on structural environments for 50 chalcogenide crystals (Fig. 1a) and showed a 70% reduction in the number of first-principles calculations required to identify the lowest-energy defect structure.52 More generally, surrogate models are being used to assess the structures, energies, and properties of novel compounds as part of materials discovery campaigns, which is the focus of the Matbench Discovery53 suite of benchmarks.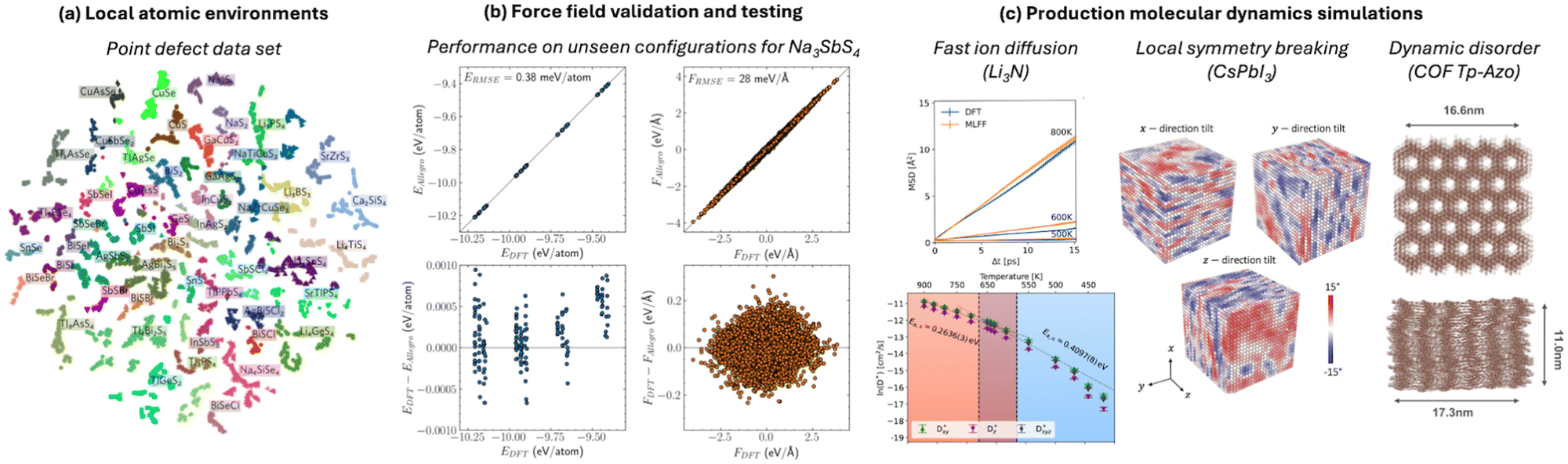 | ||
| Fig. 1 ML force fields involve (a) sampling of atomic environments (point defect dataset,52) (b) validation and testing on unseen configurations (parity plots and error distributions from an Allegro model,54) and (c) application to chemically interesting problems such as ion diffusion,55 symmetry breaking,56 and dynamic disorder.57 All figures are reproduced under a Creative Commons license. | ||
The scaling of these methods (often 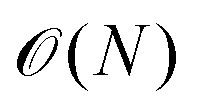 with system size rather than
with system size rather than  scaling for standard DFT) enables compositionally complex systems to be tackled. For instance, the ionic conductivity of Na3SbS4 is enhanced when doped by W to create Na vacancies. A realistic description of systems such as Na3−xWxSb1−xS4 would be prohibitively expensive using standard approaches; however, the Allegro architecture39 was used to train a predictive model (Fig. 1b) that was applied to system sizes up to 27
scaling for standard DFT) enables compositionally complex systems to be tackled. For instance, the ionic conductivity of Na3SbS4 is enhanced when doped by W to create Na vacancies. A realistic description of systems such as Na3−xWxSb1−xS4 would be prohibitively expensive using standard approaches; however, the Allegro architecture39 was used to train a predictive model (Fig. 1b) that was applied to system sizes up to 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 atoms for nanoscale-length simulations.54 Here, the test errors of <1 meV atom−1 approach the precision of the underlying DFT training data. Other examples from our work have included superionic phase transitions in Li3N,55 the formation of low symmetry phases in the halide perovskite CsPbI356 and dynamic layer displacements in covalent organic frameworks57 (Fig. 1c).
600 atoms for nanoscale-length simulations.54 Here, the test errors of <1 meV atom−1 approach the precision of the underlying DFT training data. Other examples from our work have included superionic phase transitions in Li3N,55 the formation of low symmetry phases in the halide perovskite CsPbI356 and dynamic layer displacements in covalent organic frameworks57 (Fig. 1c).
2.3 Next-generation force fields
The development of MLFFs can be divided into several classes:• Short-range potential. The first generation of MLFFs predict EFS by training on datasets derived from DFT calculations. The total energy of a system is expressed as a sum of the contributions from individual atoms  . The simple form has also allowed pre-trained foundation models for the entire periodic table47,58–63 with the number of parameters and performance listed on Matbench Discovery. Medium-range interactions (≈10–15 Å) can be captured through message-passing operations, which enable information exchange between neighbouring atoms.
. The simple form has also allowed pre-trained foundation models for the entire periodic table47,58–63 with the number of parameters and performance listed on Matbench Discovery. Medium-range interactions (≈10–15 Å) can be captured through message-passing operations, which enable information exchange between neighbouring atoms.
• Long-range electrostatics. Explicit long-range interactions are essential in some cases; for example, in describing the electric double layer at solid–liquid interfaces. Electrostatic MLFFs have been developed that combine a short-range potential with an electrostatic potential (e.g. calculated using an Ewald summation). Ongoing developments are assessing different long-range descriptors64 as well as how charges are assigned (e.g. Mulliken, Hirshfeld) and redistributed during reactions, ranging from fixed point charges to charge equilibration schemes.65–67 The torch-PME package68 has been designed to support such developments by providing a framework to compute long-range interactions built on the PyTorch library.69
• Property prediction. There are ongoing efforts to extend predictions beyond energy and include other important physical properties, such as dipole moments,70 polarizabilities,71 electron density,72,73 wavefunctions,74,75 and even spectroscopic features.76,77 One notable frontier is the direct prediction of electronic Hamiltonians, which enables electronic studies of large-scale systems with the accuracy of hybrid DFT that would otherwise be prohibitive.22,78,79
MLFFs are quickly becoming essential tools in computational chemistry and materials science, enabling large-scale simulations over long timescales. Developments in more powerful model architectures, more diverse datasets,80 and the integration of uncertainty in both model training and deployment,81 are ongoing.
3 Generative AI
A major challenge in computational chemistry is the identification of molecules and materials with specific properties that are stable and synthetically viable.82 The most common discovery paradigm screens large libraries of known compounds for novel functionalities.83 ‘Inverse design instead starts with a target property and then aims to determine the specific atomic arrangement and composition needed to achieve it ref. 84. Genetic algorithms, particle swarm optimisation, random structure searching, and fragment-based screening can assist in the exploration of chemical space and to propose candidate structures.85Generative AI offers a route to chemical discovery through probabilistic models that produce novel data.86 Driven by successes in inverse tasks across natural language, image, video, and audio generation,87 these methods are gaining increasing prominence in the chemical sciences. Crucially, they enable a natural coupling of structure generation with property constraints, directly allowing for the inverse design of molecules and materials.88 Despite the relatively early stage of development, a wide range of models have been trialled with varying success. While further innovations will be essential for practical use, these methods hold great promise for driving autonomous scientific discovery.
3.1 The evolution of generative models for chemistry
The key considerations of generative models for chemistry include (i) representation of chemical information in a latent space, for example, text or graph embeddings,89 (ii) inversion of the latent space to molecular and chemical structures, and (iii) generation of novel compounds and their properties through sampling a probability distribution.90Initial approaches employed generative adversarial networks (GANs). GANs support property-guided exploration by modifying the generator with a multi-objective loss function91 or through combination with reinforcement learning to produce hybrid generative models.92 GANs have employed SMILES93 and graph representations94 for molecules, and compositional embeddings95 and 3D voxel grids96 for materials. In practice, training property-guided GANs is hampered by the sensitivity toward hyperparameters and the training protocol.97,98 Improved methodologies such as Wasserstein GANs can alleviate some of these issues and stabilise model training.99
Variational autoencoders (VAEs) aim to learn a low-dimensional representation of data (the latent space), through chemical encoders and decoders.100 Concurrently training a property prediction model to organise the latent space enables property-guided generation.101 The continuous representation also allows interpolation across chemical space. The first application of VAEs employed SMILES strings102 and graphs103 for molecules, while voxel grids104 and invertible crystallographic descriptions were applied for materials.105,106 VAEs enable property-driven molecular design through controlled modifications in the latent space (e.g. interpolations), making it possible to generate and optimize compounds with desired properties, a key advantage in molecular discovery.101 Outstanding issues include the high data requirements and the susceptibility toward discarding data variations.107
Similar to VAEs, normalising flows and diffusion models produce new data by sampling latent space. By applying an invertible (in normalising flows108) or stochastic function (in diffusion models109) they gradually transform noise into chemical representations. Initial implementations include GraphAF110 for autoregressive end-to-end molecule generation and CDVAE111 which combines VAEs with a diffusive decoder for crystals. Recent work has focussed on incorporating symmetry equivariances,112,113 property-guided generation,114–118 support for molecular fragments,119 and complex multidimensional constraints such as electrostatics and pharmacophores.120 Diffusion models have further been applied to accelerate structural relaxations by learning of smoother pseudo potential energy surfaces.121
Autoregressive Large Language Models (LLMs) (discussed further in later sections) generate data sequentially, with the transformer architecture being the most prominently used. Such LLMs enable the direct output of atomic structures in common text formats such as XYZ,122 crystallographic information files (CIFs),123,124 and SLICES.125 Fine-tuning of open-source foundation models can improve generation performance,126 and property-guidance is enabled through prompt-engineering.127 Many approaches have been investigated, from generating symmetry inequivalent units,128 to retrieval-augmented generation of known chemical libraries,129 and application of structured state space sequence models for drug design.130 Despite the inherent lack of invariances in text representations (e.g. permutation invariance) some models based on LLMs still achieve competitive performance.131 Hybrid models combining LLMs with other deep learning approaches are also common, including integration Riemannian flow matching,132 diffusion models,131,133 and contrastive learning.134
3.2 Challenges and opportunities
Generative modelling in the chemical sciences is still in its infancy, with many hurdles to overcome before it is used regularly for scientific discoveries. Below, we outline some of the main challenges and opportunities for the field:• Beyond bulk materials and small molecules. To date, most attempts have been constrained to the generation of small molecules or crystals with less than 20 atoms in the unit cell.135 Technologically and pharmaceutically relevant compounds often are much larger and can contain disorder or defects.136 The field is already pushing in this direction, with generation of proteins,137 surfaces,138 porous materials,139 multi-component alloys,140 and metallic glasses.141 However, a recent benchmark showcased the limitations of existing models on interfaces and amorphous structures, highlighting the need for further developments.142
• Validation and benchmarking. Meaningfully evaluating the predictions from generative models is a major challenge.135 Metrics such as diversity and uniqueness are quick to evaluate but miss the main objectives of realistic and high-performance candidates. Similarly, structural validity assessment through charge neutrality and minimum interatomic distances is a poor proxy for kinetic and thermodynamic stability.143 Benchmarking platforms exist across the molecular144,145 and materials146 domains, however, an obvious gap is the absence of standardised multi-objective benchmarks for property-guided generation.147 While DFT has been employed for candidate evaluation116,134 it is computationally expensive and difficult to scale. Machine learned forcefield and property models offer an alternative route,47 but may be inaccurate for out-of-sample predictions.
• Synthesisability. The utility of generative models depends on their ability to suggest synthetically feasible candidates. One strategy involves human ranking of candidates to target in experiments.148 Another option is to bias generation towards accessible compounds by including a loss term for synthesisability.149 Challenges include the scarcity of widely applicable synthesisability metrics and the difficulty of balancing high-performance vs synthetic accessibility.150 A fully automated approach will be essential to enable closed-loop discovery. However, such platforms will likely have access to limited sets of reactants and processing conditions, thereby further constraining the range of accessible compounds with non-trivial impacts on synthesisability metrics.151
• Interpretability. Interpretable models are essential for expanding our understanding of the structure–property relationships across chemical space,152 and have implications for ethical and safe AI.153 State-of-the-art generative models are essentially black boxes and do not provide insights into why a particular compound was proposed.154 A key issue is the high dimensionality of embedded representations. While dimensionality reduction techniques can reveal the internal structure of latent spaces,143 they provide little information on the origin of proposed geometric arrangements. Emerging approaches for interpretable GNNs may be one strategy forward given their ubiquity in many generative models.155,156
3.3 Conclusions
Generative models have the potential to transform the Edisonian trial-and-error approach to chemical discovery. While the promise of efficient closed-loop workflows powered by generative models and self-driving experimentation is evident, generative approaches have a long way to come before this dream is realised. Few studies have reported the experimental verification of novel high-performing compounds proposed by a generative model. Success stories across biochemistry,157 antibiotics,158 and organic photovoltaics159 offer a tantalising glimpse of the impacts to come. However, as noted by Anstine and Isayev,135 even in failure, generative models can inspire human creativity and broaden our understanding of the chemical sciences.4 AI for drug discovery
AI is already having a substantial impact on drug discovery,160 leading to improvements in overall pharmaceutical R&D productivity.161,162 Such productivity is expected to nearly double upon the successful completion of ongoing clinical trials.160 Here, we review the factors that have been contributing to this ongoing transformation with a focus on preclinical efforts, which account for over 43% of overall R&D expenditure.163 Indeed, while the preclinical stages of a successful project are less costly than the clinical stages, the high failure rate during preclinical development strongly contributes to overall R&D expenditure. For example, GSK reported data showing that 93% of their projects did not achieve an antibiotic drug lead, with half of the remaining 7% projects being stopped for not overcoming the remaining preclinical requirements.164 The latter is guided by ADMET (absorption, distribution, metabolism, excretion, toxicity) modelling of the drug lead, which is also important for reducing potential adverse effects in the subsequent clinical stages.4.1 The impact of boosted funding
One key factor has been a substantial increase in private funding. Billions of pounds per year are invested in AI-driven drug discovery companies, sourced from partnership deals with pharmaceutical and biotechnology firms as well as private investors. This funding has enabled the development and prospective evaluation of AI models across drug discovery stages.Another important factor has been sustained public funding, which supports the generation, collection, curation, and redistribution of data,165,166 along with the development of reusable software tools. This has resulted in a wealth of well-documented AI algorithms that can be combined with relevant domain knowledge.167 Notably, self-supervised learning algorithms, which pre-train deep learning models on large amounts of unlabelled chemical structures and then fine-tune them using much smaller labelled datasets of molecules.
Self-supervised learning is being used to build small-data AI models for drug lead discovery and potency optimisation. For instance, an LLM model pre-trained on over 77 million SMILES strings was fine-tuned to predict molecular properties.168 These approaches are also being adapted to leverage high-dimensional structured data.169 There are already proof-of-concept prospective studies for structure-based prediction of protein–ligand binding affinities using pre-trained language models.170 Similarly, GNNs have shown promise in phenotypic virtual screening, including applications to human pathogens171 and cancer cell lines.172 Beyond deep learning, methods for uncertainty quantification, such as Gaussian Processes173 and conformal prediction,174 have also proven their potential in this area.
Small-data AI modelling is also being investigated for a range of ADMET properties.5 For instance, using advanced feature extraction175 or multi-task learning to leverage data from similar molecules and/or properties.176 These studies typically build upon existing datasets and generic learning algorithms and further advance the field by releasing processed datasets and AI models for use in future projects.
4.2 The need for better benchmarks
A concerning trend is the proliferation, and often excessive hype, of publications describing new AI algorithms. While the number of these methods applied to retrospective benchmarks for drug discovery is rapidly increasing, only a small fraction of them have demonstrated their value in prospective applications. This highlights the need for benchmarks that are more closely aligned with the real-world demands of drug discovery. For example, MoleculeNet177 is a popular suite of benchmarks aiming at evaluating molecular property prediction (a relatively new umbrella term for virtual screening, binding affinity prediction or ADMET end point prediction). However, it is unrealistic for a number of reasons, e.g. using ROC AUC to evaluate performance in early recognition problems178 or employing unrealistically easy training-test data splits179 (scaffold split or even random split).Progress in this area includes emphasising the use of more realistic data splits,179,180 employing performance metrics better suited to the specific tasks,180,181 leveraging true negatives in classification models to reduce false positives,182 and developing more comprehensive and representative benchmarks.183 These advancements will help address the gap between retrospective validation and prospective utility.
4.3 Recent technological breakthroughs expanding chemical and target spaces
Relatively recent technological breakthroughs have yet to reach their full potential. The first of them expands the chemical space via ultralarge libraries of molecules,184 synthesised on demand with success rates now exceeding 85%, which represents a major advancement.185 A key benefit of this technology is the unprecedented chemical diversity it provides, challenging the notion of an ‘undruggable’ target (was a target truly undruggable, or was the screened library simply too small to contain any drug lead?). The other key benefit is that screening larger libraries tends to yield a higher number of increasingly potent actives for a given target.186,187 However, a major roadblock is that screening the largest libraries remains accessible only to those with extensive computational resources, especially when docking is required to guide virtual screening. Encouragingly, new approaches are emerging to reduce the required resources,188,189 paving the way to democratise the screening of ultralarge libraries for any target.The other technological breakthrough was AlphaFold2,190 which is expanding the 3D target space. AlphaFold represents a multidisciplinary effort combining AI, computational chemistry, structural bioinformatics, and well-aligned benchmarks. This method can predict the ligand-free 3D structure of a target from its amino-acid residue sequence. Therefore, it is particularly useful for the many targets lacking any experimentally determined 3D structure or even reliable homology models. Rigorous retrospective studies predicted AlphaFold2s utility for structure-based drug design,191 a prediction now validated in prospective applications as well.192 A far more challenging drug discovery application of AlphaFold3 to generate ligand-bound structures of unseen targets.193 The ambition is to be able to do this for any user-supplied molecule and target sequence without having to specify the binding site residues, with an output including the correct location, orientation and binding strength of the molecule. This has so far only been achieved partly for the handful of ligands well represented in the Protein Data Bank (PDB), which therefore form part of training sets complexed with seen targets. Many AI models building upon AlphaFold's principles have also been presented.194
4.4 The enhanced prospective performance of AI models
A growing number of prospective studies are revealing the immense potential of AI in drug discovery. For example, GNN models have been used to identify novel molecules with whole-cell activity against E. coli,195A. baumannii196 or S. aureus.197 The discovery of antibiotics for these drug-resistant pathogens is both urgent and critical in addressing the antimicrobial resistance crisis.171 AI models for drug response prediction are also advancing in other disease models, while ML scoring functions for structure-based drug design have made significant strides since their inception.198 Among these, virtual screening remains their most challenging and impactful application.199–201 AtomNet, an ensemble of convolutional NNs, is currently the most successful ML scoring function for virtual screening.202 Atomwise, the company behind AtomNet, applied it prospectively on 318 targets as part of their Artificial Intelligence Molecular Screen (AIMS) programme. This ambitious effort involved partnerships with 482 academic labs and screening centres from 257 institutions across 30 countries. Despite focusing on hard targets and testing only molecules dissimilar from known actives, submicromolar actives were identified in approximately 60% of targets through dose–response experiments. Remarkably, this was achieved by synthesising and testing an average of just 85 molecules per target.2024.5 Next steps
To make AI-driven drug discovery more resource-efficient, future research must focus on improving our ability to create retrospective benchmarks that reliably predict prospective success. These benchmarks are critical, as they guide the selection of AI models for prospective applications. Another roadblock is data being repurposed from often heterogeneous datasets originally generated for different objectives, which introduces several issues, such as bias, inconsistency, or limited data size.203 A promising recent trend involves generating training data for the target using the same experimental protocol later employed to validate prospective predictions in vitro.195–197 This could circumvent some of these issues in AI for drug discovery.5 Synthesis route planning and selection via deep learning
With recent advances in generative ML, it has become possible to computationally design promising molecules for diverse applications in the (bio-) chemical and medicinal sciences. Whenever a novel molecule, for example a candidate drug molecule, is designed or generated (via ML), it must pass the synthesisability test. In other words, it does not matter whether one can computationally design the perfect molecule in terms of properties, if it cannot be synthesised in the laboratory, the molecule will always remain virtual.5.1 Retrosynthetic search
The first requirement to produce the real molecule through experimentation is a synthesis path connecting the molecule (product) to purchasable building blocks through a series of reactions. This path, also known as a synthesis route, is traditionally mapped out by experienced chemists in a backwards fashion – known as retrosynthesis.204 Retrosynthesis is a laborious and tedious process that highly depends on the chemists’ expertise for specific reaction types.To automate retrosynthesis, Corey et al.205 encoded reaction rules into a machine-readable format. His pioneering work in 1972 led to the development of expert systems that perform retrosynthesis in an autonomous fashion.206 With increased applications of ML in chemistry, the rule-based systems have slowly been outnumbered in favour of deep-learning approaches.207 Below, we provide a short overview, outlining current challenges and opportunities.
Data-driven retrosynthesis is constituted of two distinct parts: (i) single-step predictions208,209 and (ii) multi-step route generation.210,211 Single-step models predict a single-step reaction, that is, to find plausible reactants for a given product. Multi-step algorithms apply single-step models recursively to build synthesis routes of N single-step reactions ending in a set of purchasable molecules. Combining these two parts, one can generate several synthesis routes for a single product, referred to as a synthesis tree.212Fig. 2 provides an overview of this concept, showing how a target molecule can be broken down into multiple possible precursor combinations.
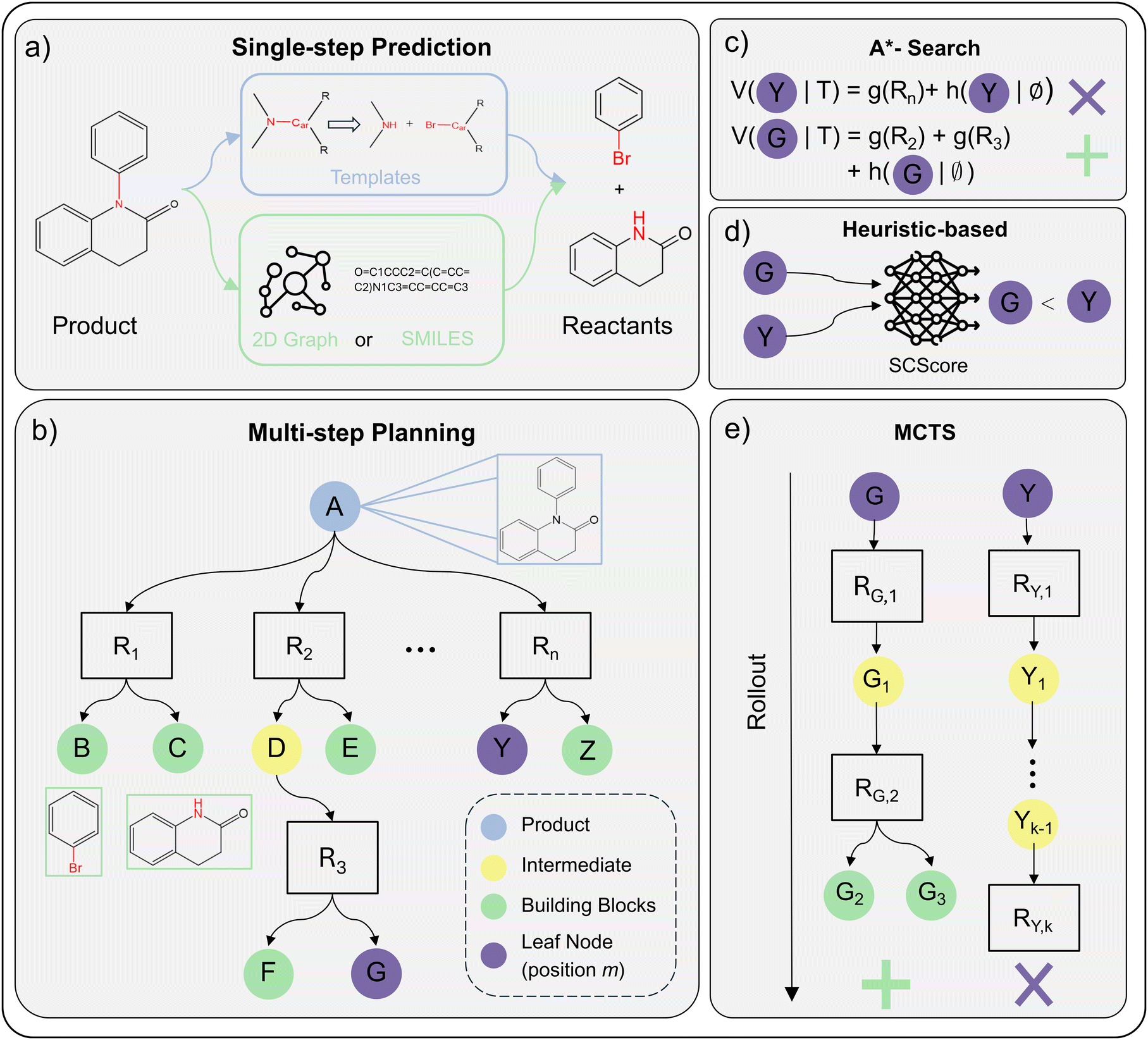 | ||
| Fig. 2 A holistic overview of ML-driven retrosynthesis (Section 5.1) (a) example of performing single-step reaction prediction on a product molecule. The reaction is either predicted via reaction templates (template-based) or in a data-driven fashion using SMILES or 2D Graphs as the molecular featurisation (semi-template/template-free). (b) AND-OR search tree for multi-step planning. Ri denotes a specific reaction that is applied to the parent node (molecule). Children nodes are precursors to Ri. Leaf nodes (purple) are open positions m in the tree that will be expanded by the single-step model. (c)–(e) Strategies for leaf node (position m) selection/prioritisation. Subplots assume same retrosynthesis tree as shown in b. Furthermore, we assume that node G is preferable to node Y. (c) A*-Search calculates the value of the open position m as a sum between reaction cost g(Ri) and future cost h(m|∅). As G is assumed to be preferable, h(G|∅) ≪ h(Y|∅) and/or g(R2) + g(R3) ≪ g(Rn) (d) heuristic-based search uses a pre-defined heuristic to assign a value. In this example, we assume that the SCScore213 heuristic prefers position G over Y. (e) Monte-Carlo Tree Search traditionally uses rollout. For node G, the rollout leads to building blocks g(G2) and g(G3). For Y, the rollout is unsuccessful and terminates at after k sampled reactions. Thus, the reward is given to position G and it is preferred for selection. | ||
First, template-based models utilise reaction rules.214–218 These rules define which bonds to break in the product and leaving groups to attach within the reaction centre (Fig. 2a). Reactions rules are usually obtained from literature precedence.219 Recently, researchers explored generative approaches to create reaction template, overcoming the limited reaction space covered by literature precedence.220–222
Second, semi-template models mainly split the predictions into two sub-tasks of reaction centre identification and synthon completion.223–227 The model identifies the reaction centre as the atom and bonds participating in electron rearrangement in a reaction.228 The output of this first step is a group of molecules (synthons)224 that are not chemically valid. To validate the synthons, the second step adds atoms224,229 or motifs/leaving groups230,231 to the synthons iteratively. Upon completion, a set of reactants is returned. Since semi-template models perform direct edits on the molecular graph, GNNs are the preferred ML architectures.
Third, template-free models perform sequence translation to generate the reactants token-by-token, mainly in their SMILES strings.232,233 The input to the model is either the product SMILES234–236 or 2D molecular graph.237–239 Since the nature of the problem is generative, the (graph) Transformer is the preferred.232,238 Furthermore, owing to its generative nature, template-free models can also predict reagents (solvents/catalysts) in addition to reactants given only the product.232,240,241
However, the combined problem of reactant and reagent prediction is difficult in nature. Most single-step retrosynthesis algorithms therefore focus on predicting reactants. Following the prediction, a separate (standalone) model recommends suitable conditions such as yield,242 catalysts, solvents, and temperature, framed as a multitask prediction problem.243,244 Unlike the combined problem, the reaction condition model takes the full reaction string (reactants → products) as input.
The search algorithms construct a synthesis tree/graph T with the product molecule as the root and purchasable molecules as terminal leaf nodes. Each branch of the tree is a distinct synthesis route consisting of several single-step reactions. The single-step reaction are represented in the tree by AND nodes. The parent of the reaction node is the product (outcome) of the single-step reaction. Precursors to the reaction are added as children (OR) nodes. This AND-OR assignment of nodes follows intuition: for a reaction to happen, all precursors must be available (boolean AND). On the other hand, a molecule can be synthesised as long as there exists one feasible reaction (boolean OR). For other types of retrosynthesis trees/graphs (e.g. OR tree), we refer the reader to previous publications.210,240
Fig. 2b shows an example of a partially explored synthesis tree. The algorithm constructs the tree/graph by selecting a (non-terminal) leaf node m and querying the single-step model to propose additional n reactions along with their precursors. The precursors are then added to the tree/graph, referred to as expansion. In Fig. 2b, node G or Y would be expanded next by the single-step model.
Most importantly, the search algorithms should be able to discern a good leaf node (position) m in T from a bad position. A good position is ideally expanded and exploited, while a bad one is abandoned. In other words, one wants to explore promising reaction pathways instead of wasting resources on potentially unfeasible pathways. For this purpose, researchers have proposed three different strategies: Monte-Carlo Tree Search (MCTS),210,245–247 A*-Search,211,248,249 and heuristic-based exploration.240,250 Below, we describe the node selection strategies for open positions m in T. From a synthetic chemistry perspective, this involves choosing which intermediate molecules m in the synthesis pathway should be prioritised for further retrosynthetic analysis. Some intermediates may resemble readily available commercial compounds, while others require additional synthetic steps to reach simpler precursors.
• MCTS evaluates position m by rollout (iteratively expanding a node until termination).210 Rollout iteratively samples random reactions from the single-step model (Fig. 2e). If the “random walk” terminates in purchasable building blocks, a reward is assigned to position m rendering m preferred to be expanded. As rollout is computationally expensive, and researchers proposed to use a model (usually a NN) trained from experience247,251 to replace rollout.
• A*-Search evaluates position m using a value function combining the cost of reactions in the existing tree (g(m|T)) with an estimated cost of future reactions (h(m|T)) as shown in Fig. 2c. As the cost of future reaction is not known for leaf nodes a priori, one must approximate it as h(m|∅) (the cost of synthesising m).211 Using cost estimates from existing synthesis trees, one can learn h(m|∅) in a supervised fashion.211,248,249,251 Otherwise, seeing the retrosynthesis planning problem as a single-player game,252 one can learn h(m|∅) online using self-play, also referred to as reinforcement learning.253,254
• Heuristics-based evaluates a position, as the name suggests, on user-defined search heuristics. Popular heuristics include accessibility metrics such as SCScore213 (Fig. 2d) or SAScore,255 the overall route length or molecule disconnection preferences.246
The search guidance provided by these algorithms definitely biases the search towards purchasable building blocks, but there is no theoretical guarantee that these will in fact be reached. Recently, Yu et al.256 proposed bi-directional search to alleviate this problem. By simultaneously building two synthesis trees, one going backwards from the product and one going forwards from the building blocks, they ensure constraint satisfiability.
The constraint of synthesis route feasibility is harder to achieve. Herein, feasibility refers to the likelihood of the synthesis plan to be validated through experimentation. This is because vital information such as yield, selectivity and reaction conditions are generally missing from the synthesis plan. Tripp et al.257 addressed this shortcoming by changing the search goal to (most probably) include at least one feasible synthesis route in the synthesis tree. Another idea is to propose synthetic routes that closely resemble existing routes in literature.258
5.2 Challenges and opportunities for synthesis planning
Rapid advances in the field of retrosynthesis planning has triggered the development of an overwhelming choice of different model architectures. Despite this, the community needs to overcome several challenges to yield a fully functional retrosynthesis tool.• Interpretability and reliability. Arguably the key cornerstone to a successful synthesis framework is the reliability of the single-step model. A reaction proposed by the model should ideally yield the product through experimentation. However, researchers often evaluate single-step models using model recall, known as top-k accuracy. By only focussing on recalling the reaction in the existing database using top-k accuracy, one can easily forget about the quality of all other reactions proposed by the model.207,262 This is indeed a problem, since the single-step model adds k reactions to the multi-step search tree during each expansion phase. To improve the reliability of the single-step model, one could use a post-hoc filter removing poor reactions.210,240 Otherwise, during model training, one can augment the dataset with negative (non-feasible) reactions.222 While both methodologies can improve the reliability, they are not rigorous. A more rigorous approach would build on thermodynamic insight, exemplified by the work of Ree et al.263 To further increase interpretability in the single-step models, one could augment the predicted global reaction with mechanistic insight.264 Lastly, researchers should prioritise evaluating their ML models on existing benchmarks207,262,265 and reconsider the overreliance on the top-k accuracy as a performance metric. Standardising evaluation practices not only facilitates the identification of model limitations, but also promotes transparency and clarity in reporting.
• Route selection strategies. Little research has addressed the (post) selection of synthesis route following the multi-step search. The multi-step algorithm returns N different synthesis routes to the end-user, where N depends on the time/iteration budget. The search algorithm ranks these routes by considering route length, number of reactions and/or overall route cost (single-step confidence).245 However, these are not clear indicators to confidentially claim that one route is better than another. Unfortunately, one does not have access to informative metrics such as overall route yield or the actual (physical) cost of carrying out the reactions. Badowski et al.266 assumed a fixed yield and fixed cost for each reaction, circumventing the problem. Fromer and Coley267 propose to select synthesis routes that maximise the utility of synthesising a batch of molecules (e.g. for virtual screening routines). Yujia et al.268 trained a model to select synthesis routes that are most likely feasible according to human expertise. All these approaches are good starting points and can be extended by considering other factors such as route ‘greenness’269 and scale-up potential.270
• Sustainability. Instead of selecting green routes a posteriori as suggested above, researchers have attempted to bake in sustainable aspects into the retrosynthesis framework. The first idea is to include biosynthetic/enzymatic reactions in the single-step model271,272 to bias search towards sustainable, energetically favourable reactions. Another idea is to preferably select routes utilising green solvents within reactions.273 As we strive towards greener chemistry, this field of research holds a lot of promise, and yet, there are still several challenges to address.274
• Implementation and adoption. Implementation and adoption is eased with existing open-source software and user-friendly interfaces.259,260 Nonetheless, the amount of papers reporting the synthesis of novel and/or complex molecules264,275 remains limited. This is partly attributed to a lack of interpretability and reliability, as discussed above. As models become more reliable in the future, we can expect increased adoption by scientists.
• Data sources. All points mentioned above are somewhat dependent on an improvement of current datasets and better data availability. Predominantly, the open-source USPTO database is used for model training and testing.276,277 The database is known to be scarce in terms of reaction conditions, often not reporting reagents, yields and selectivity.278 Commercial databases such as Reaxys279 are well-documented containing millions of substances and reactions, but are locked behind paywalls. This led to the development of collaborative initiatives to build a database through community engagement. Most well-known is the Open Reaction Database (ORD),278,280 which encourages chemists to contribute and upload their datasets. This initiative is still in its infancy and most of its entries are currently from the USPTO. Better awareness and integration is therefore needed to improve current databases.
6 Data-rich and data-led experimentation to support development of accurate and predictive models
The Data, Information, Knowledge, Wisdom (DIKW) hierarchy (Fig. 3a), also known as the knowledge pyramid, is widely evoked in AI as a model to represent the progression from data to wisdom.281 At the base of the pyramid is (raw) data, which may consist of unprocessed facts and figures. As we move up the pyramid, data is organised and classified; transforming into information that can subsequently be analysed to afford understanding and insights (‘knowledge’). Finally, at the pinnacle of the pyramid is wisdom, where the knowledge is applied to make informed decisions.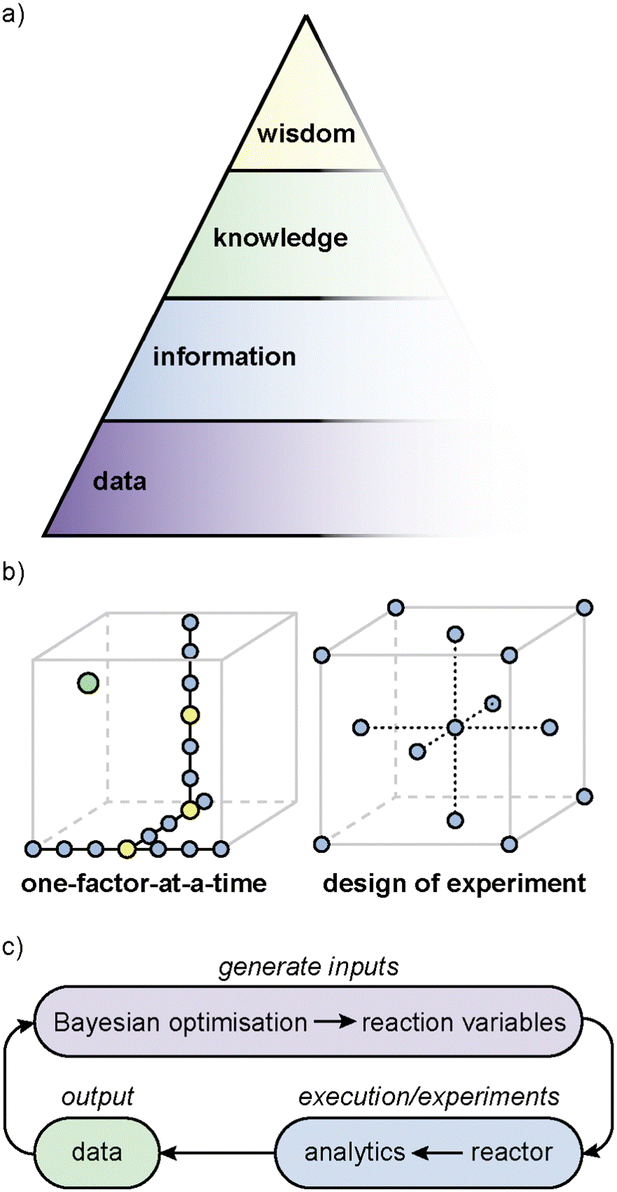 | ||
| Fig. 3 (a) Data-information-knowledge-wisdom pyramid. (b) One-factor-at-a-time (OFAT) and design of experiment (DoE) approaches to optimisation. The optimal point is shown in green, and the points that are the optimal along a certain parameter are highlighted in yellow. (c) An example closed-loop optimisation workflow. | ||
In ML, data is needed to train, validate and improve the AI model. The accuracy and reliability of AI depend on the availability of data collected from experiments. For the chemical sciences, there are generally two types of data:
1. Compound (characterisation) data. Typically collected after the reaction with the isolated compound or simulated computationally in silico. These data are needed to confirm the chemical composition and structure, as well as physical and chemical properties. There are many different types of characterisation data, ranging from discrete values (e.g. melting/boiling points, bandgap energy, emission wavelength), a dataset, or ‘fingerprint’ (e.g. NMR, IR spectroscopy), or images (e.g. morphology and particle sizes recorded using microscopy). Accordingly, these data are also highly heterogeneous, by nature. Characterisation data are closely associated with the molecular composition and structure, they are particularly useful for the prediction of chemical/physical properties, e.g. for the discovery of new materials; or biological properties, for applications in the discovery of new drugs, for example.
2. Reaction data. This is acquired during the reaction using either ex situ or in situ quantitative process analytical tools to monitor either the rate of formation or consumption of reaction components (typically reactants and products). The temporal progress of the reaction (kinetics) is particularly important to understand reactivity. Kinetic data is essential to elucidate reaction mechanism, enzyme kinetics (for designing more effective drugs), material degradation (improve safety and sustainability), and development of a commercially viable process (scaling up), for example. In contrast to characterisation data, kinetic data are much more uniform and ‘continuous’ in nature (either concentration vs. time or rate vs. temperature). A chemical reaction also involves several closely associated discrete and continuous variables that synergistically influence the reaction outcome; for example: reactants stoichiometry, catalyst, pH, additives, solvent, temperature, and pressure. The ‘robustness’ of a chemical reaction denotes its capacity to withstand variations in these variables without detrimental effects. Understanding the impact of these variables on the reaction outcome is essential not only for elucidating the reaction mechanism, but also for designing a process that ensures consistent product quality (‘Quality-by-Design’, QbD), which is particularly important for highly regulated industries such as pharmaceutical products.282
Traditional practices in chemistry have long relied on hands-on experimentation and observation, where experimental design and the interpretation of the results are still largely based on ‘chemical intuition’, acquired through empirical observations over many years (‘experience’). Under such conditions, workflows often follow one-factor-at-a-time (OFAT) experiments, where the effect of one factor (or variable) is studied while keeping the other factors at fixed values (Fig. 3b). Although this approach can be effective for optimising the yield of a simple reaction, it does not take into account interdependencies between the reaction variables, and could potentially miss the optimal point (Fig. 3b, green dot indicated). As the end point is arrived at empirically, it is impossible for such an approach to be able to predict the outcome of similar reactions; neither can it tackle multi-objective optimisations.
In the past decade or so, there has been increased interest in the use of statistical methods for optimising chemical processes. One of these is design of experiments (DoE), a popular statistical method that can be used to interrogate relationships between the reaction parameters (‘factors’) and outcomes (‘responses’), systematically (Fig. 3b).283 The approach starts with identifying the objective, which could include maximising yield, improving selectivity, or shortening reaction time, etc. The researcher then determines the factors that might influence these outcomes (‘responses’). Common factors include temperature, reactants concentration, catalyst loading, choice of solvent and reaction temperature. Using either a full- or partial-factorial design, different combinations of factors and levels are generated, and the experiments are randomised to minimise the effect of uncontrolled variables (for example, catalyst deactivation). The responses are analysed using Analysis of Variance (ANOVA) or regression analysis to produce F- and p-values against each factor, and also combination of factors (quadratic). If the F-value is significant and the p-value <0.05, the specific factor, or combination of factors, are considered to be statistically significant. The model is then used to produce a validation set of experiments, which is performed to test the accuracy, before the model is used to predict the final optimal outcome, which can be a balance between different objectives. In recent years, DoE has been applied successfully in optimising several chemical processes.284,285 Typically, DoE can require a large number of experiments, which can be costly and time-consuming. However, this has been largely addressed by advances in laboratory automation, to enable high-throughput experimentation286 and analytical techniques.287
In contrast, Bayesian optimisation utilises a surrogate model (typically a Gaussian process) to approximate the objective function and an acquisition function, to determine the next experiment to perform.288 This is particularly suitable for high-dimensional problems and also provides a measure of uncertainty. Bayesian optimisation will require a ‘close-loop’ experimentation, where the predictive algorithm is integrated with experiments on an autonomous robotic platform (or ‘self-driving lab’) (Fig. 3c).6,289 While this may minimise the number of experiments, it is also computationally more expensive. Furthermore, the integration of expert knowledge is also often needed to select the appropriate parameters to fine-tune the black-box functions.290
7 LLMs and multimodal models for chemistry
Large language models (LLMs)291–293 represent the latest ground-breaking developments in the field of natural language processing (NLP) with profound impact in general AI research. LLMs are pre-trained on web-scale corpus of text data (including natural language text and code, etc.) with the goal of learning rich internal representations of documented knowledge during human history.294,295 After pre-training, LLMs can then be used as a general-purpose AI for a diverse range of downstream tasks (such as sentiment analysis296 question-answering,297 and generative tasks,298,299) via fine-tuning on downstream task data, and/or in-context learning where the model learns to solve a task purely from the relevant context provided by the prompts.300 Furthermore, the ability to interact with an AI model via human language allows for descriptions of more complex tasks and reduces the barrier of AI expertise for using powerful AI models.301,3027.1 Chemical representations with LLMs
LLMs have attracted increasing attention in AI for chemistry research through the development of so-called “molecular” or “chemical” language models.168,303–305 These models largely inherit the transformer-based network architectures292,295 and pre-training strategies from popular LLMs in the NLP domain, except that they operate on text-based chemical data, such as SMILES strings.306,307 Similar to natural language-based LLMs, molecular LLMs learn to represent the underlying structural properties of molecules by understanding the unique grammar of chemical textual data. This again enables diverse applications with pre-trained molecular LLMs, including molecular property prediction (MPP),168,303,308 conditional molecule or material structure generation,304,309,310 and retrosynthesis.233,311,3127.2 Multimodal models for chemistry
Though increasingly used, current molecular LLMs fall short in fully representing the structural information and equivariance properties of molecular data. In general, molecules are challenging objects to describe: for instance molecular orbital theory313 and valence bond theory314 offer contrasting descriptions of a molecule. Therefore, limiting the representation of a molecule to a single form will not capture the full behaviour of the molecule. This motivates the creation for multimodal frontier models that can capture richer representations of molecules315,316 by incorporating molecular graphs, coordinate information, sequential data and other features to improve their performance on various downstream tasks such as property prediction.305,317–3207.3 LLMs for scientific workflows
LLM-assisted workflows are of particular interest for LLM developers and users since ChatGPT's release.321,322 In particular, LLMs are efficient at digesting, summarising323,324 and retrieving information from large documents;325,326 question-answering from prompt inputs;297 as well as performing domain-specific tasks such as translation,327 and computer programming.328 Also, very recent developments regarding reasoning and planning complex tasks with LLMs have shown promising results.329–331The diverse capabilities of LLMs offer many exciting opportunities in improving the workflows of scientific research in chemistry domain. To understand how LLMs can assist scientific discovery, an analysis of essential workflow steps with potential uses of LLMs is provided below.
• Idea formulation. LLMs trained on scientific publications and chemistry textbooks can assist scientists in formulating innovative research ideas. Via prompting332 and retrieval augmented generation techniques,333,334 LLMs can efficiently retrieve and summarise existing scientific knowledge in published/proprietary documents regarding a scientific question of interest.335 This qualitative information obtained via LLM-assisted search complements the quantitative information extracted from existing data-mining tools for chemistry data,336,337 contributing to a holistic overview of the scientific question to be addressed.
• Lab experiment troubleshooting. With access to electronic lab notebooks and chemistry literature, a domain-specific LLM can be used to troubleshoot specific issues during the lab experiment process.338 The natural language understanding capabilities of LLMs are especially useful in analysing text descriptions of experimental conditions that can vary in style across documents. Also, anomalous results may be described to an LLM via natural language, where the LLM can then retrieve relevant papers describing similar issues and provide a natural language explanation for the results. LLMs plays a growing role in teaching,322 and this may extend into the lab where students can use LLMs as a resource to aid in laboratory technique and troubleshoot issues.
• Experiment design. With access to external computational tools, LLM-hybrid models can perform complex planning tasks,339e.g., AlphaProof and AlphaGeometry340 for solving mathematics problems. Thus an LLM can be prompted to suggest plausible experimental procedures based upon existing lab data, for which scientists can then verify within a lab. LLM-assisted planning can complement existing experimental design algorithms (e.g., Bayesian optimisation341): the former can better utilise scientific knowledge and data presented in natural language form,342 while the latter can provide precise quantitative parameters for setting up the lab experiment. Fully autonomous chemical research may be possible with LLMs planning the high-level experimental steps (i.e. a sequence of action primitives)343 and autonomous lab robots executing the planned actions.344,345
7.4 Challenges for LLMs and frontier models in chemistry
Despite the aforementioned exciting opportunities, a number of profound challenges remain to be solved for training and utilising LLMs and frontier models for chemistry research and applications.• Mitigating hallucinations. LLMs are prone to hallucinations – they can generate responses that do not make sense for the given task.346,347 In molecular generation, hallucinations can lead to invalid molecular structures: for example, an atom having too many bonds.348 Hallucinations can also lead to inconsistent results when retrieving scientific information from research documents via LLMs. Efficient mitigation of hallucinations is key for the reliability of LLM usage in chemistry, e.g., experimental conditions should be retrieved precisely without removing important information or adding false data.349
• Data collection and data-efficient training. A critical challenge in modelling molecules is the high complexity of the data space containing all valid molecules. For instance, activity cliffs exist in such space, whereby a small change in molecular structure can result in large changes in molecular properties.350 Therefore, labelled data collection (with large quantity and high quality) remains a major bottleneck in molecular property prediction and generation tasks.351 Solutions to this challenge should focus on better data collection pipelines, as well as making frontier model training more data-efficient.
• Alternative molecular representations. In the domain of MPP, there are alternatives to the textual molecular representations used by LLMs. In particular, graph-based representations such as the molecular graph are used by graph neural networks (GNNs), which are a popular approach to MPP.352,353 Indeed, many state-of-the-art GNNs, can achieve comparable or improved prediction accuracy in-comparison to LLMs.354 Whilst LLMs have the advantage that they can be applied to domains beyond MPP to assist in areas such as scientific workflows, it remains a challenge for LLMs and frontier models to surpass the predictive performance obtained by models specialised to MPP.
• Advancing multimodal frontier models. Beyond multiple representations of molecules,305,318–320 there is a great potential for multimodal frontier models in chemistry to incorporate further related information broadly defined, including lab notebooks, scientific publications, experimental results, images of molecules, spectra information, etc.317 In fact frontier AI models such as Gemini355 and GPT-4356 have already incorporated visual, audio and text information to answer complex questions.
• Ethical use & development of frontier models. It is crucial that the development of chemical frontier models follows rigorous scientific process and adheres to research ethical policies. Open science and reproducibility should be promoted via suitable open-source practices.357 Meanwhile, there must be measures to prevent misuse of frontier models for creating dangerous molecules and materials.358,359
8 Experimental design for discrete and mixed input spaces
Despite recent successes of large foundation models, many chemistry applications remain challenging due to expensive and difficult data acquisition. In these scenarios, ML-aided experimental design can ensure effective data collection, thereby reducing the number of experiments required.288 Bayesian optimisation and active learning are popular approaches for designing experiments.360–362 The former aims to optimise the black-box function which is the experiment itself, while the latter seeks to learn the whole function. Both build a surrogate model of the black-box function and then use a decision policy to optimise or learn about the function, respectively. An effective decision policy balances search space exploration and exploitation of the areas expected to be most optimal.360For campaigns to accurately balance exploration and exploitation, the surrogate model needs to incorporate a measure of prediction certainty. This is relatively easy on continuous spaces, as the most popular surrogate models, for example, Gaussian processes, have uncertainty built in ref. 363. However, in many scenarios, input variables are discrete or a mixture of discrete and continuous.290 Even within these categories, there is much heterogeneity in the types of discrete variables. This means there is no one-size-fits-all solution to which surrogate model and decision policy should be used, and how uncertainty should be handled. Therefore, selecting an approach reflecting the specific characteristics of the type of variable(s) and the information that can be transferred between variables is essential.
8.1 Heterogeneity of problem classes
One challenge of discrete variables is the diversity of problem types requiring different treatments as shown in panel (a) of Fig. 4. We categorise discrete variables into four types: categorical, ordinal, combinatorial or mixed. Categorical variables take inputs that have no obvious ordering. For example, this might be enzyme cofactors,364 solvent type365,366 or additives.367 A special case of categorical variables are dichotomous variables that can only take two values, such as binary variables,368 or on/off. One hot encoding is a technique converting each discrete variable into a unique vector with a single high (1) for the value the variable takes and all other values low (0). This common approach for encoding discrete variables is often used for categorical variables, but can be used for other variable types.366,369,370 | ||
| Fig. 4 An overview of types of discrete variables (Section 8.2) and surrogate models (Section 8.1). (a) There are four main categories of discrete variables: categorical, ordinal, mixed and combinatorial. (b) Parallel surrogates method, fitting a different surrogate model for each discrete variable. (c) Continuous relaxation where the discrete variables are converted to a continuous one, in this case by using their molar mass. (d) A decision tree-based method where solvents are split into different leaves. (e) String kernel method where the molecules are first converted into SMILES strings, then a string kernel is used to determine their similarity. (f) latent variable methods, where an encoder is used to convert the discrete variable to a continuous latent space, a Gaussian process is fitted to the latent space and optimisation is conducted, then a decoder is used to retrieve the discrete variables again. (g) Graph approach for combinatorial variables, where each node represents a different combination. | ||
Ordinal variables are those that can be put in order, such as counts of atoms,371 aspect ratios of reactors,372 or the number of base-pairs in DNA molecules.373 Combinatorial variables take a set of discrete, often finite combinations. Some of the most common combinatorial variables are biological sequences,374–378 such as the CDRH3 region of an antibody,379,380 or various molecules.102,381,382 Combinatorial inputs may or may not have a set length.379 González-Duque et al.383 recently conducted a study comparing many high-dimensional Bayesian optimisation techniques for discrete sequences.
8.2 Modelling discrete variables with uncertainty
Another challenge of discrete inputs is determining how much information should be shared between different levels of a discrete variable. For example, if an extra data point is observed that falls in one leaf of a decision tree, how much should that influence the prediction of data points that fall in other leaves? This question has two aspects: how does the new information change the expected value of our prediction and how does it affect the certainty in this prediction? Uncertainty estimates are especially important for experimental design tasks as they guide the exploration of regions where the model is uncertain.It is also possible to learn the similarity between the separate models using transfer learning, such as multi-output Gaussian processes, which learn a covariance function over outputs.386,387 This has been used for learning the similarity between cell lines388 and DNA molecules. While the parallel surrogates approach is easily implementable, it is generally only feasible for a small number of discrete variables, as the computational cost and amount of data needed scales with the number of values the discrete variables can take.
8.3 Decision policies
Parallel surrogates, continuous relaxations and latent space approaches all map discrete variables into a continuous space, enabling the application of well-established continuous experimental design techniques.366,372,408,409,412 These methods typically employ a surrogate model, normally a Gaussian process, to estimate the mean response and the associated uncertainty. An acquisition function then combines the mean and uncertainty into a single metric guiding experimental design. For Bayesian optimisation, where the aim is optimising a target variable, common acquisition functions are expected improvement and upper confidence bound.360,361 Other experimental design strategies include active learning, which aims to learn the whole function,362 and Bayesian quadrature, which seeks to learn an integral.418Applying continuous optimisation methods to ordinal variables and selecting the closest integer value can lead to the same points being repeatedly sampled, wasting the experimental budget. This can be compensated for by altering the acquisition function when this occurs,389 or transforming the inputs before calculating the acquisition function.419 Continuous methods can be applied to parallel surrogate models, although this gets expensive when there are many continuous spaces or the continuous spaces are high dimensional. To reduce this computational cost, multi-armed bandits can be used to select which surrogates are most likely to offer improvements.420,421 Latent space approaches generally assume smoothness,381 allowing for Bayesian optimisation or active learning. In these cases, a Gaussian process is fitted to the latent space, where the output of the Gaussian process is the objective function.407,408 If the latent space is high dimensional, trust-region methods can be used to guide exploration of the space.408
For methods that do not convert discrete variables to continuous inputs, the biggest challenge is often exploring the space, as gradient-based optimisation methods can no longer be applied to the acquisition function. In combinatorial spaces, evolutionary or random walk algorithms can be used. For example, Khan et al.379 use random walk to explore a trust region, evaluating the acquisition function at each point.379,422 Bayesian optimisation for tree models can be done by optimising each leaf of the tree and picking the best one,423 using local search where a step is taken in one parameter at a time424 and global optimisation of the acquisition function using mixed integer programming.425
When specialist kernels, such as string kernels and Tanimoto kernels are used, genetic algorithms can explore the search space.382 Recent work has also demonstrated how transformer neural processes, a meta-learning model, where models use knowledge from previous datasets to learn a new task, can skip fitting a surrogate and directly meta-learn the acquisition model.380
8.4 Outlook
Experimental design over discrete and mixed inputs is challenging due to the heterogeneity of problem types, difficulty modelling uncertainty and lack of gradients for optimising acquisition functions. To mitigate these challenges, it is important to identify the types of discrete variable(s) present in a problem and select the right surrogate model. The methods outlined here have all been proven to work for several chemistry applications, however, uptake of such methods is slow. Several software packages have been developed to help experimentalists apply these approaches to their experimentalists: BOtorch,426 BOFire,427 and BayBe428 are all Bayesian optimisation packages; WebBO429 is a modular platform that can be integrated into electronic lab book frameworks; Atlas,430 Anubis,431 and Dragonfly432 are all packages for self-driving labs that integrate experimental design methodologies. To ensure its proper use it is important software incorporates educational aspects that help experimentalists, who may not be well versed in ML, to map their problem to the available methods and understand the assumptions being made.From a methodological perspective, future research directions include meta-learning of the acquisition function to amortise inference and to skip the need for a surrogate model altogether,380,433 dealing with systems where decisions that change with time434 and using experimental design to uncover causal relationships.435–437
9 AI for robotics in chemistry
Traditional chemistry laboratories rely heavily on human labour for repetitive, time-intensive, and sometimes hazardous tasks, such as chemical synthesis, sample preparation, and data analysis.438 This reliance on manual processes not only reduces operational efficiency but also exposes scientists to potentially harmful environments. The integration of robotics and automation into laboratory environments has emerged as a promising solution, which enables improved process optimisation, greater precision, and the potential for continuous operations without human intervention.Recent advancements in AI, combined with access to large-scale datasets and sophisticated laboratory automation tools, such as systems for synthesis, separation, purification, and characterisation,439 have enabled the development of ‘robot chemists’. These systems utilise AI as the cognitive engine, empowering robotic platforms to autonomously conduct experiments and transform traditional workflows in chemistry. AI-driven robotics are transforming laboratory practices by addressing inefficiencies and introducing advanced capabilities that streamline scientific research. These systems optimise workflows through continuous, autonomous operations, significantly reducing the time required for experimental iterations while enhancing productivity far beyond human limitations. By standardizing processes and minimising errors, they ensure consistent, reproducible results, fostering greater confidence in experimental outcomes. A key advantage of AI-driven robotics is their ability to handle hazardous chemicals and conduct high-risk reactions, thereby safeguarding human researchers and mitigating safety risks. Moreover, these systems excel in scalability, making them invaluable for large-scale research endeavours such as high-throughput screening and combinatorial studies. They could theoretically manage vast experimental conditions with remarkable speed and accuracy, enabling the exploration of expansive chemical and parameter spaces that would be infeasible manually.
These advancements, when adopted and employed, have effectively transformed traditional laboratories into automated discovery platforms, thereby significantly increasing the autonomy of scientific experimentation. The integration of ‘robot chemists’ (systems capable of automated learning, reasoning, and experimentation), has accelerated the discovery of new molecules, materials, and systems. By leveraging diverse data sources and modalities, these intelligent systems are able to operate continuously, make decisions under uncertainty, and generate reproducible data enhanced with comprehensive metadata and real-time sharing capabilities. This paradigm shift not only improves precision, efficiency, and scalability but also minimises manual errors and broadens the generalisability of research across a wide range of applications.438 Here, we discuss the prospective impact of integrating AI and robotics in chemistry. We begin by classifying ‘Robotic Chemists’ based on their levels of autonomy and highlighting their contributions to self-driving laboratories (SDLs). Finally, we outline a future roadmap for the development of AI and robotics in chemistry.
9.1 Classification of ‘robotic chemists’ based on autonomy levels
There are five levels of autonomy: (i) assistive automation, (ii) partial automation, (iii) conditional automation, (iv) high automation, and (v) full automation. Here, we discuss each level of autonomy as it relates to self-driving laboratories. Table 1 and Fig. 5 present an overview of the levels of autonomy and concrete examples in chemistry.| Autonomy level | Description | Example | Ref. |
|---|---|---|---|
| A1: Assistive automation | Automates single tasks; humans perform the majority of work. | Automated liquid handling systems that perform repetitive aspiration and dispensing tasks, reducing manual labour and minimising errors. | 440 |
| A2: Partial automation | Automates multiple sequential steps; requires human setup and supervision. | Setups where robotic arms handle the transfer of reactants between different stages of a reaction sequence, creating a distributed automation system. | 441 |
| A3: Conditional automation | Fully automates synthesis and characterisation processes; human intervention needed for unexpected conditions. | The ‘RoboChem’ platform developed by the University of Amsterdam autonomously performs chemical syntheses and optimises reaction conditions using AI-driven ML. | 442–444 |
| A4: High automation | Automates entire workflows, including setup and adaptation to unusual conditions; minimal human input. | The mobile robotic chemist developed by the University of Liverpool autonomously navigates laboratory environments and conducts experiments across various areas of chemical synthesis. | 343,445,446 |
| A5: Full automation | Completely autonomous systems capable of handling all tasks, including self-maintenance and safety hazard resolution. | This is an active area of research within chemistry, and will be powerful for chemical tasks where human input is not necessary. |
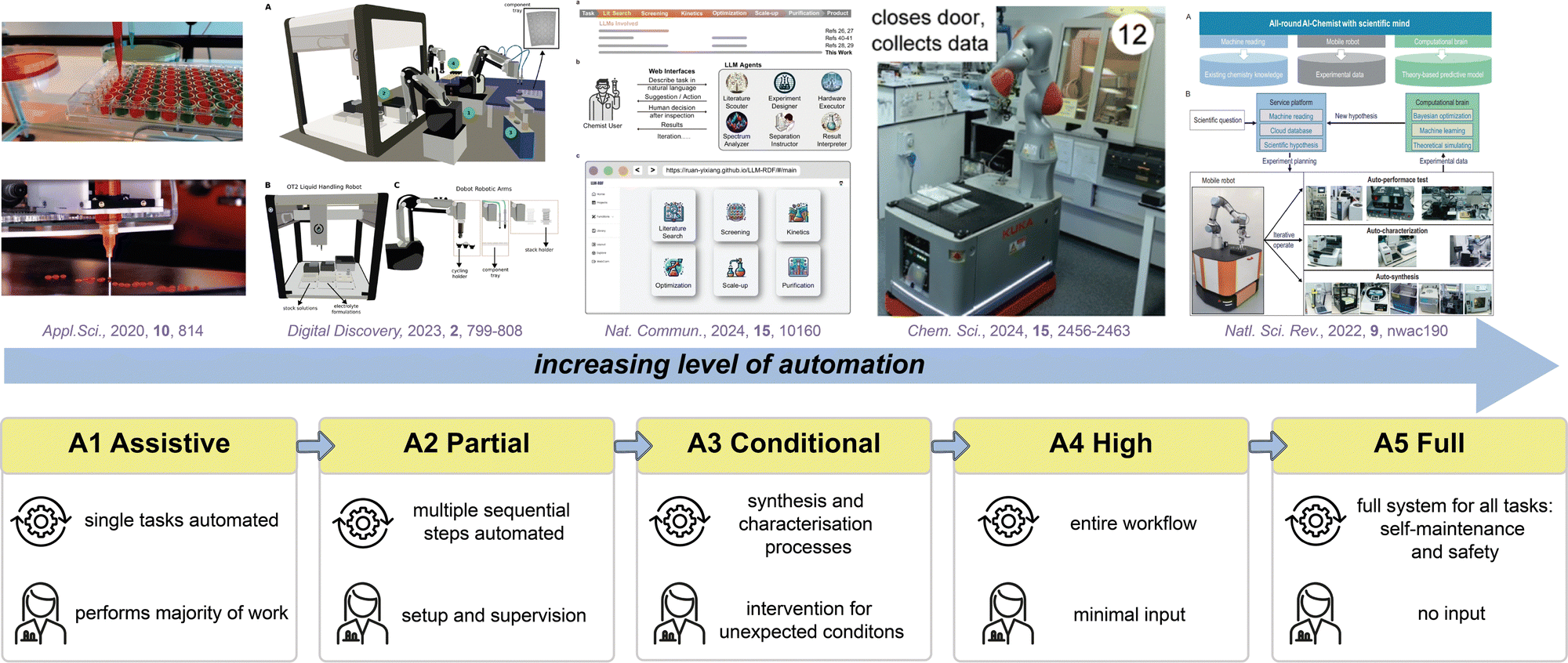 | ||
| Fig. 5 The five levels of autonomy differ in the automated steps and the level of human intervention. Select examples are depicted spanning the range of automation spanned by the levels of autonomy. Figures in the top panel are reproduced under a Creative Commons license.445,447–450 | ||
The RoboChem platform,442 developed by the University of Amsterdam, is an autonomous chemical synthesis robot that incorporates an AI-driven ML module. This platform can autonomously conduct chemical syntheses, optimise reaction conditions, and iteratively refine processes with minimal human involvement. The RoboChem platform has demonstrated superior performance compared to human chemists in terms of speed and accuracy, significantly accelerating the discovery of new molecules for pharmaceutical and industrial applications. The AlphaFlow system,443 designed to handle multistep synthesis and characterisation in flow chemistry, utilises reinforcement learning to optimise reaction pathways, monitors real-time data, and adjusts parameters to enhance efficiency and reproducibility. By integrating ML with advanced automation, AlphaFlow exemplifies the potential of conditional automation to streamline complex chemical processes. Another example is the ORGANA robotic assistant, a system designed to automate a wide range of chemistry experiments. ORGANA leverages LLMs to plan and execute experiments, interact with chemists using natural language, and adapt to diverse experimental requirements.444
9.2 Applications of self-driving laboratories
AI-driven robotics are making significant contributions to labs in chemistry, materials science, and biochemistry, such as complex reaction optimisation, high-throughput screening, and hazardous material handling. More specifically, advancements in AI and robotics are reshaping laboratories into self-driving labs (SDLs),452 which support advanced scientific discovery with minimal human intervention. Globally, SDLs are under active development in numerous laboratories, certainly too many to name here. SDLs are also described as materials acceleration platforms,453 Lab 4.0,454 the Internet of Laboratory Things,455 Robot Scientists,456 the autonomous research system (ARES),457 and autonomous experimentation systems.458 SDLs can conduct experimental design and execution, real-time data analysis, and parameter optimisation in an iterative process. This approach consists of three core components: a robotic system to conduct and analyse reactions, software to interpret analytical data, and an algorithm that correlates reaction outcomes with experimental parameters. The essence of SDLs lies in their ability to run closed-loop experiments, which utilises online analytics, real-time feedback from each experiment, and decision-making algorithms to inform subsequent actions.459–461Unlike conventional, human-dependent laboratories in chemistry and materials science, SDLs overcome three major limitations of traditional laboratories: (1) the slow and inefficient exploration of experimental space; (2) the lack of integration across different experimental stages; and (3) delays between the completion of one experiment and the initiation of the next.462 Here, some typical examples of applications are summarised, including high-throughput experimentation (HTE), self-optimising reaction systems, automated discovery platforms, and protein engineering. Importantly, there are many additional examples across chemistry.
HTE aims to rapidly screen and analyse chemical compounds through advanced automation and AI integration. One example is the HTE OS framework, developed specifically for robotic platforms, demonstrates the potential of AI-driven experimentation.463 By combining advanced scheduling algorithms, data processing techniques, and natural language processing (NLP) tools, these systems achieve parallel experimentation, significantly reducing the time required to evaluate large chemical libraries or complex reaction matrices.464 Autonomous robotic platforms can also be tailored for specific tasks, for example, for electrolyte formulation and coin cell assembly in high-throughput lithium-ion battery research.448 Additionally, high-throughput synthesis (HTS) enhances throughput by enabling researchers to synthesise multiple materials simultaneously through automated parallel processing.465
Self-optimising reaction systems leverage AI algorithms to dynamically adjust reaction parameters in real time, optimising critical outcomes such as yield or selectivity. Both autonomous and semi-autonomous robotic systems have contributed to the development of novel chemical synthesis methodologies.344,443,460,462 One example is from Schwalbe-Koda et al.,466 who describe a platform that autonomously optimises polymer synthesis using self-optimising flow reactors. These reactors iteratively adjust key variables, such as temperature and pH, based on real-time reaction monitoring. This approach has been further refined for applications such as photoinitiated RAFT polymerisation, where fully automated systems leverage real-time feedback to enhance process outcomes continuously.467
Data-driven ML algorithms are transforming materials and catalyst discovery, enabling the rapid analysis of experimental data to identify optimal candidates or refine reaction conditions. These systems autonomously explore novel chemical transformations, accelerating reaction discovery while minimising risks associated with reactive materials. Robotic systems, operating under optimised safety protocols, enable safe and efficient experimentation. For instance, the ‘Schlenkputer’ system executes reactive chemical transformations autonomously, employing AI algorithms to prioritise experimental pathways based on predicted reactivity.468
Protein engineering holds significant potential for applications in chemistry. However, the development of new proteins with enhanced or novel functions has traditionally been slow, labour-intensive, and inefficient. The Self-driving autonomous machines for protein landscape exploration (SAMPLE) platform represents a breakthrough in this domain. This fully autonomous system integrates an intelligent agent that analyses protein sequence-function relationships, generates new protein designs, and coordinates with an automated robotic system to experimentally test these designs. Feedback from the robotic system enables the intelligent agent to refine its understanding and optimise the protein engineering process iteratively.378
9.3 Future research directions
As discussed in the earlier sections, LLMs are further transforming the field of chemistry by extracting and interpreting complex chemical information from vast scientific literature. The integration of advanced multi-language large models (MLLMs) into robotics offers significant potential for laboratory automation. These models enhance the adaptability of robotic systems, allowing them to address diverse research challenges. To make these systems accessible to chemists, who often lack robotics expertise, there is a focus on creating user-friendly tools that simplify programming, data analysis, and experimental setup adaptation.474
Mixed-use laboratories, where humans and robots work side by side, will set new standards for safety and efficiency. These environments will integrate advanced monitoring systems and adaptive technologies to ensure secure and harmonious operations. Intuitive interfaces—such as voice commands and generative AI tools—will further enhance the accessibility and usability of automated systems, enabling smoother interactions and fostering a productive partnership between humans and machines.
10 AI-accelerated data management for digital chemistry
The proliferation of AI- and data-driven approaches in the physical sciences have the potential to not only accelerate scientific discovery, but also allow us to tackle qualitatively different problems. However, in order to really exploit this potential, we need to significantly improve the quality, quantity, and accessibility of the data captured in the modern research lab. In a recent survey by the UK's Physical Sciences Data Infrastructure (PSDI), less than 20% of respondents digitally managed all of their laboratory data and experiments.480 Among those, a variety of software packages were used, with varying levels of machine accessibility to the data. Ultimately, the large majority of laboratory data currently produced is not stored in such a way that it can be readily actioned upon by AI tools. There is, therefore, a timely need for data infrastructure that can help researchers to capture, organise, and share their data along with its provenance, metadata, and scientific context. Here, we discuss current and projected capabilities of laboratory data management for AI, and discuss our own efforts to integrate both AI assistants and agents into experimental materials chemistry research within the open source481datalab electronic laboratory notebook platform. We envision that the development and adoption of interoperable data management platforms that reproducibly store and make available diverse laboratory data will be necessary to reach the full potential of AI tools for scientific research.10.1 The role of AI-powered assistants and agents
As the quantity and diversity of our scientific data grows, researchers find themselves spending an increasing amount of time and effort managing data: organising connected experiments, converting between file formats, and performing analysis.480,482,483 The recent advent of capable ML models, in particular multi-modal large language models291,292,484–486 provides an extraordinary opportunity – for researchers and tool-builders alike – to build capable AI-driven agents and assistants that can meaningfully accelerate science by aiding experimental researchers in these data management and analysis tasks.487,488 Today's LLM-based tools generally fall into two categories: assistants and agents. Assistants, typified by the first iteration of ChatGPT released in November 2022, present the user with a chat-based interface to an (M)LLM that can answer user questions and perform basic tasks. The data for an assistant can either be pre-loaded directly into the prompt, or can be fetched as needed using search tools (i.e., Retrieval-augmented generation, RAG). On the other hand, autonomous LLM agents go a step further by allowing the LLM to iteratively take actions, observe the outcome, and then react further to accomplish a task. For example, an AI agent may have the ability to access web APIs, write arbitrary code, execute it, parse the output, and perform further actions. In principle, an AI agent could complete very complex research data management and analysis tasks that require multiple conversions, comparisons, visualisation, and information synthesis. However, while LLM-based assistants are currently well established, truly capable general-purpose agents are still early in their development.Both assistants and agents have the potential to greatly aid in scientific laboratory research. Assistants may read large quantities of (potentially multimodal) laboratory data from machine-accessible electronic laboratory notebooks in order to quickly summarise results, draw connections, extract data, or even propose new experiments. Assistants can also integrate various third-party tools to perform queries and simple analysis.489 Agents, on the other hand, can be tasked with more complex data-handling tasks that require multiple steps. For example, a researcher developing new battery electrodes may pose the following prompt: “Based on all the cathodes developed in this lab in the last 5 years, determine whether there is a correlation between particle size (by scanning electron microscopy (SEM) analysis) and battery performance”. While this question would be very time-consuming for a human researcher answer, a capable AI agent may attempt to tackle this problem autonomously by (1) writing code to search the groups electronic notebook for all relevant samples, (2) using vision capabilities, or specialist software provided in a machine-actionable way to the agent,490 to view SEM images from these samples, (3) writing code to analyse for any correlations and create a useful visualisation for the researchers. In this way, an AI agent could dramatically speed up human-driven research, by allowing researchers to quickly and easily ask important scientific questions that were inaccessible before.
10.2 The importance of user interfaces for data capture
The full utility of AI assistants and agents will only be realised if the scientific data, metadata, protocols, and observations that we collect daily in the lab are stored in a manner where they can easily be accessed by machine agents. For example, our open-source laboratory data management platform, datalab,481 stores scientific data along with relevant metadata and context in a database, and provides both GUI (human-friendly) and API (machine–friendly) interfaces. Within the GUI, a LLM-powered assistant, “whinchat”,487 can read, summarise, and answer questions about the recorded experiments. For more complex data management tasks, we have developed an external AI-powered agent, “yeLLowhaMmer”.488 YeLLowhaMmmer is pre-prompted with the datalab API documentation, so that it can iteratively write and execute Python code to access, filter, and process data as needed. A future area of development is capturing and storing the results of AI queries and automated analysis so that it can be reused and shared across a lab. It will be especially important to mark AI-generated content as such, so that it can be appropriately verified by humans.As we have discussed, the usefulness of AI-based tools hinges on the availability of scientific data. To make use of AI-based tools, laboratory data should be stored digitally, with all the metadata and context needed to make it experimentally useful. Researchers should strongly consider open, machine-accessible formats483 – ideally on a platform that allows programmatic access. No single platform or data management strategy will suit all research use cases, but developers should prioritise those with open APIs, schemas and code to enhance interoperability among tools.482,491–496 In our own work, we have found that a “semi-normalised” data model provides the best balance between rigidity and flexibility for laboratory work and for interaction with LLMs. Data sets are recorded with schemas that specify common fields and their data types, but free-text fields are also provided so that users can easily record information or observations that do not fit neatly into the predetermined schemas. From this base, LLMs will likely also find use in mapping to richer semantic data formats that can be used to readily exchange data in an interoperable way for use in knowledge graphs and other applications.496,497 Importantly, the use of machine-accessible data management platform not only enables the use of state-of-the art AI tools in experiments, but also makes it possible for researchers to contribute their data to train or fine-tune the next generation of ML models, if they should choose to share their data in this manner.
Future outlook
Retrospective benchmarks that reliably predict prospective success are necessary to improve the efficiency of AI-driven discovery, including drug discovery. Indeed, these advancements will also have implications for the application of generative models for chemical discovery. While few studies have experimentally validated high-performing compounds proposed by generative models, these methods have already demonstrated their unique ability to inspire human creativity. Similarly, retrosynthesis tools face challenges related to reliability, route selection, data quality, and adoption – motivating additional research in these areas.Beyond retrosynthetic planning, frontier models, including LLMs, are set to play an important role in experimental workflows. Yet, there exist several challenges to overcome, including mitigating hallucinations, advancing data-efficient raining, advancing multimodal models, and ensuring ethical use of frontier models in chemistry. Indeed, integrating LLMs in chemical workflows extends to their use in robotics and automation equipment.
The role of automation in experimental chemistry is continuing to improve. We note that, while there is a general move towards fully automated setups, that the value of human input and intervention should not be underestimated. Indeed, human-in-the-loop initiatives leverage the productivity of robotic automation, the efficiency of autonomous decision making, with the insight of human chemists. Future progress will depend on the development of: (i) open source tools; (ii) modular and scalable systems; (iii) cost-effective and accessible platforms; and (iv) advanced human-AI collaborative systems. Beyond these advancements, additional development of sensors and chemometrics that facilitate in situ analysis that does not require additional units of operation are paramount. Indeed, while most procedures can already be highly automated, it is the analytical tools that provide the data needed for ML. High-throughput analysis poses a challenge – while it can be fairly easy to automate sample preparation, a large proportion of characterisation techniques are still carried out offline, albeit equipped with autosamplers for handling larger numbers, and still carry out measurements in a sequential one-by-one manner which can be time-consuming. In addition to robotics and frontier models, AI-driven decision making algorithms have also started to redefine how experiments are planned and executed. Improvements in each of these areas are paramount to realising a fully autonomous chemical research workflow.
Underpinning all of these advancements is the critical need for accessible and robust data infrastructure, facilitating the creation of high-quality scientific data. The success of any AI model is dependent on the quality of the data on which it is trained. Thus, efforts to unify metadata standards, improve open-access data repositories and databases, as well as initiatives to ensure that laboratory data are stored digitally with associated metadata and made available in machine-readable formats through open platforms and APIs is critical to advancing AI-driven chemical research.
There is, undoubtedly, immense potential of AI to accelerate chemical research. However, realising the full impact requires addressing technical, methodological, and physical challenges. Sustained interdisciplinary collaboration and a commitment to open science and discourse are necessary to overcome these challenges, and advance both fundamental understanding of chemical phenomena and acceleration of fundamental research to real-world applications.
Author contributions
J. M. F. contributed AI for Quantum Chemistry. I. M. L. and A. W. contributed Scaling Atomistic Simulations with ML Force Fields. A. M. G. contributed Generative AI. P. J. B. contributed AI for Drug Discovery. F. H. and E. A. D. R. C. contributed Synthesis Route Planning and Selection via Deep Learning. K. K. H. contributed Data-rich and Data-led Experimentation to Support Development and Accurate Predictive Models. I. S. J. and Y. L. contributed LLMs and Multimodal Models for Chemistry. R. S. and R. M. contributed Experimental Design for Discrete and Mixed Input Spaces. D. Z. wrote the first draft of AI for Robotics in Chemistry. A. R. B. and R. L. G. finalized the AI for Robotics in Chemistry section. M. L. E. and J. D. B. contributed AI-Accelerated Data Management for Digital Chemistry. A. M. M. co-conceived the article focus and collated and organized the sections within the manuscript, and contributed the first draft of the introduction and conclusions. K. E. J. co-conceived the article and organized topics. All authors contributed to the editing of the final version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
M. L. E. is a shareholder and Director of datalab industries ltd.Acknowledgements
We acknowledge the AI for Chemistry: AIchemy hub for funding (EPSRC grant EP/Y028775/1 and EP/Y028759/1). A. M. M. is supported by the Eric and Wendy Schmidt AI in Science Postdoctoral Fellowship, a Schmidt Sciences program. P. J. B. thankfully acknowledges the support of the Wolfson Foundation and the Royal Society for a Royal Society Wolfson Fellowship (UK, RSWF R1 221005). R. L. G. thanks the Royal Society for a University Research Fellowship. M. L. E. thanks the BEWARE scheme of the Wallonia-Brussels Federation for funding under the European Commission's Marie Curie-Skłodowska Action (COFUND 847587). A. R. B and F. H. thank the React CDT for funding (EP/S023232/1). I. S. J. acknowledges the StatML CDT (EP/S023151/1), and ICONIC grant with BASF (EP/X025292/1). R. S. is supported by the Wellcome Trust (222836/Z/21/Z). J. D. B. acknowledges support from The Welch Foundation (E-2179-20240404).References
- Z. J. Baum, X. Yu, P. Y. Ayala, Y. Zhao, S. P. Watkins and Q. Zhou, J. Chem. Inf. Model., 2021, 61, 3197–3212 CrossRef CAS PubMed.
- R. L. Greenaway, K. E. Jelfs, A. C. Spivey and S. N. Yaliraki, Nat. Rev. Chem., 2023, 7, 527–528 CrossRef PubMed.
- J. A. Keith, V. Vassilev-Galindo, B. Cheng, S. Chmiela, M. Gastegger, K.-R. Müller and A. Tkatchenko, Chem. Rev., 2021, 121, 9816–9872 CrossRef CAS PubMed.
- L. Yang, Q. Guo and L. Zhang, Chem. Commun., 2024, 60, 6977–6987 RSC.
- B. Dou, Z. Zhu, E. Merkurjev, L. Ke, L. Chen, J. Jiang, Y. Zhu, J. Liu, B. Zhang and G.-W. Wei, Chem. Rev., 2023, 123, 8736–8780 CrossRef CAS PubMed.
- G. Tom, S. P. Schmid, S. G. Baird, Y. Cao, K. Darvish, H. Hao, S. Lo, S. Pablo-García, E. M. Rajaonson, M. Skreta, N. Yoshikawa, S. Corapi, G. D. Akkoc, F. Strieth-Kalthoff, M. Seifrid and A. Aspuru-Guzik, Chem. Rev., 2024, 124, 9633–9732 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, F. Sandfort, M. H. S. Segler and F. Glorius, Chem. Soc. Rev., 2020, 49, 6154–6168 RSC.
- G. Carleo and M. Troyer, Science, 2017, 355, 602–606 CrossRef CAS PubMed.
- R. P. Feynman and M. Cohen, Phys. Rev., 1956, 102, 1189–1204 CrossRef.
- Y. Kwon, D. M. Ceperley and R. M. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 12037–12046 CrossRef CAS PubMed.
- D. Luo and B. K. Clark, Phys. Rev. Lett., 2019, 122, 226401 CrossRef CAS PubMed.
- J. Hermann, Z. Schätzle and F. Noé, Nat. Chem., 2020, 12, 891–897 CrossRef CAS PubMed.
- D. Pfau, J. S. Spencer, A. G. D. G. Matthews and W. M. C. Foulkes, Phys. Rev. Res., 2020, 2, 033429 CrossRef CAS.
- I. von Glehn, J. S. Spencer and D. Pfau, A Self-Attention Ansatz for Ab-initio Quantum Chemistry, arXiv, 2023, preprint DOI:10.48550/arXiv.2211.13672.
- J. Hermann, J. Spencer, K. Choo, A. Mezzacapo, W. M. C. Foulkes, D. Pfau, G. Carleo and F. Noé, Nat. Rev. Chem., 2023, 1–18 CAS.
- D. Pfau, S. Axelrod, H. Sutterud, I. von Glehn and J. S. Spencer, Science, 2024, 385, eadn0137 CrossRef CAS PubMed.
- G. Cassella, W. M. C. Foulkes, D. Pfau and J. S. Spencer, Nat. Commun., 2024, 15, 1–7 Search PubMed.
- J. Kirkpatrick, B. McMorrow, D. H. P. Turban, A. L. Gaunt, J. S. Spencer, A. G. D. G. Matthews, A. Obika, L. Thiry, M. Fortunato, D. Pfau, L. R. Castellanos, S. Petersen, A. W. R. Nelson, P. Kohli, P. Mori-Sánchez, D. Hassabis and A. J. Cohen, Science, 2021, 374, 1385–1389 CrossRef CAS PubMed.
- K. Ryczko, S. J. Wetzel, R. G. Melko and I. Tamblyn, J. Chem. Theory Comput., 2022, 18, 1122–1128 CrossRef CAS PubMed.
- M. Gaus, Q. Cui and M. Elstner, J. Chem. Theory Comput., 2011, 7, 931–948 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- H. Li, C. Collins, M. Tanha, G. J. Gordon and D. J. Yaron, J. Chem. Theory Comput., 2018, 14, 5764–5776 CrossRef CAS PubMed.
- F. Hu, F. He and D. J. Yaron, J. Chem. Theory Comput., 2023, 19, 6185–6196 CrossRef CAS PubMed.
- A. McSloy, G. Fan, W. Sun, C. Hölzer, M. Friede, S. Ehlert, N.-E. Schütte, S. Grimme, T. Frauenheim and B. Aradi, J. Chem. Phys., 2023, 158, 034801 CrossRef CAS PubMed.
- G. R. Jenness, C. G. Bresnahan and M. K. Shukla, J. Chem. Theory Comput., 2020, 16, 6894–6903 CrossRef CAS PubMed.
- P. O. Dral, A. Owens, A. Dral and G. Csányi, J. Chem. Phys., 2020, 152, 1–12 CrossRef PubMed.
- Z. Qiao, M. Welborn, A. Anandkumar, F. R. Manby and T. F. Miller, J. Chem. Phys., 2020, 153, 1–11 CrossRef PubMed.
- L. Zhang, B. Onat, G. Dusson, A. McSloy, G. Anand, R. J. Maurer, C. Ortner and J. R. Kermode, npj Comput. Mater., 2022, 8, 1–14 CrossRef.
- R. Sutton, The Bitter Lesson, 2019, http://incompleteideas.net/IncIdeas/BitterLesson.html Search PubMed.
- O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K. T. Schütt, A. Tkatchenko and K.-R. Müller, Chem. Rev., 2021, 121, 10142–10186 CrossRef CAS PubMed.
- I. Poltavsky and A. Tkatchenko, J. Phys. Chem. Lett., 2021, 12, 6551–6564 CrossRef CAS PubMed.
- A. P. Bartók, M. C. Payne, R. Kondor and G. Csányi, Phys. Rev. Lett., 2010, 104, 136403 CrossRef PubMed.
- V. L. Deringer, A. P. Bartók, N. Bernstein, D. M. Wilkins, M. Ceriotti and G. Csányi, Chem. Rev., 2021, 121, 10073–10141 CrossRef CAS PubMed.
- J. L. F. Abascal, E. Sanz, R. García Fernández and C. Vega, J. Chem. Phys., 2005, 122, 234511 CrossRef CAS PubMed.
- J. L. F. Abascal and C. Vega, J. Chem. Phys., 2005, 123, 234505 CrossRef CAS PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- F. Musil, A. Grisafi, A. P. Bartók, C. Ortner, G. Csányi and M. Ceriotti, Chem. Rev., 2021, 121, 9759–9815 CrossRef CAS PubMed.
- S. Batzner, A. Musaelian, L. Sun, M. Geiger, J. P. Mailoa, M. Kornbluth, N. Molinari, T. E. Smidt and B. Kozinsky, Nat. Commun., 2022, 13, 2453 CrossRef CAS PubMed.
- A. Musaelian, S. Batzner, A. Johansson, L. Sun, C. J. Owen, M. Kornbluth and B. Kozinsky, Nat. Commun., 2023, 14, 579 CrossRef CAS PubMed.
- A. P. Bartók, R. Kondor and G. Csányi, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 184115 CrossRef.
- A. V. Shapeev, Multiscale Model. Simul., 2016, 14, 1153–1173 CrossRef.
- R. Drautz, Phys. Rev. B, 2019, 99, 014104 CrossRef CAS.
- G. Dusson, M. Bachmayr, G. Csányi, R. Drautz, S. Etter, C. van der Oord and C. Ortner, J. Comput. Phys., 2022, 454, 110946 CrossRef CAS.
- J. Behler and M. Parrinello, Phys. Rev. Lett., 2007, 98, 146401 CrossRef PubMed.
- J. Vandermause, S. B. Torrisi, S. Batzner, Y. Xie, L. Sun, A. M. Kolpak and B. Kozinsky, npj Comput. Mater., 2020, 6, 1–11 CrossRef.
- I. Batatia, D. P. Kovacs, G. Simm, C. Ortner and G. Csanyi, Advances in Neural Information Processing Systems, 2022, pp. 11423–11436 Search PubMed.
- I. Batatia, P. Benner, Y. Chiang, A. M. Elena, D. P. Kovács, J. Riebesell, X. R. Advincula, M. Asta, M. Avaylon, W. J. Baldwin, F. Berger, N. Bernstein, A. Bhowmik, S. M. Blau, V. Cărare, J. P. Darby, S. De, F. D. Pia, V. L. Deringer, R. Elijošius, Z. El-Machachi, F. Falcioni, E. Fako, A. C. Ferrari, A. Genreith-Schriever, J. George, R. E. A. Goodall, C. P. Grey, P. Grigorev, S. Han, W. Handley, H. H. Heenen, K. Hermansson, C. Holm, J. Jaafar, S. Hofmann, K. S. Jakob, H. Jung, V. Kapil, A. D. Kaplan, N. Karimitari, J. R. Kermode, N. Kroupa, J. Kullgren, M. C. Kuner, D. Kuryla, G. Liepuoniute, J. T. Margraf, I.-B. Magdău, A. Michaelides, J. H. Moore, A. A. Naik, S. P. Niblett, S. W. Norwood, N. O'Neill, C. Ortner, K. A. Persson, K. Reuter, A. S. Rosen, L. L. Schaaf, C. Schran, B. X. Shi, E. Sivonxay, T. K. Stenczel, V. Svahn, C. Sutton, T. D. Swinburne, J. Tilly, C. van der Oord, E. Varga-Umbrich, T. Vegge, M. Vondrák, Y. Wang, W. C. Witt, F. Zills and G. Csányi, A foundation model for atomistic materials chemistry, arXiv, 2024, preprint DOI:10.48550/arXiv.2401.00096.
- C. J. Pickard, Phys. Rev. B, 2022, 106, 014102 CrossRef CAS.
- S. Heinen, G. F. von Rudorff and O. A. von Lilienfeld, J. Chem. Phys., 2022, 157, 221102 CrossRef CAS PubMed.
- M. Schaarschmidt, M. Riviere, A. M. Ganose, J. S. Spencer, A. L. Gaunt, J. Kirkpatrick, S. Axelrod, P. W. Battaglia and J. Godwin, Learned Force Fields Are Ready For Ground State Catalyst Discovery, arXiv, 2022, preprint DOI:10.48550/arXiv.2209.12466.
- J. Lan, A. Palizhati, M. Shuaibi, B. M. Wood, B. Wander, A. Das, M. Uyttendaele, C. L. Zitnick and Z. W. Ulissi, npj Comput. Mater., 2023, 9, 172 CrossRef CAS.
- I. Mosquera-Lois, S. R. Kavanagh, A. M. Ganose and A. Walsh, npj Comput. Mater., 2024, 10, 1–9 CrossRef.
- J. Riebesell, R. E. A. Goodall, P. Benner, Y. Chiang, B. Deng, G. Ceder, M. Asta, A. A. Lee, A. Jain and K. A. Persson, Matbench Discovery – A framework to evaluate machine learning crystal stability predictions, arXiv, 2024, preprint DOI:10.48550/arXiv.2308.14920.
- J. Klarbring and A. Walsh, Chem. Mater., 2024, 36, 9406–9413 CrossRef CAS PubMed.
- G. Krenzer, J. Klarbring, K. Tolborg, H. Rossignol, A. R. McCluskey, B. J. Morgan and A. Walsh, Chem. Mater., 2023, 35, 6133–6140 CrossRef CAS.
- W. J. Baldwin, X. Liang, J. Klarbring, M. Dubajic, D. Dell'Angelo, C. Sutton, C. Caddeo, S. D. Stranks, A. Mattoni, A. Walsh and G. Csányi, Small, 2024, 20, 2303565 CrossRef CAS PubMed.
- J. Huang, S.-J. Shin, K. Tolborg, A. M. Ganose, G. Krenzer and A. Walsh, Mater. Horiz., 2023, 10, 2883–2891 RSC.
- C. Chen and S. P. Ong, Nat. Comput. Sci., 2022, 2, 718–728 CrossRef PubMed.
- B. Deng, P. Zhong, K. Jun, J. Riebesell, K. Han, C. J. Bartel and G. Ceder, CHGNet: Pretrained universal neural network potential for charge-informed atomistic modeling, arXiv, 2023, preprint DOI:10.48550/arXiv.2302.14231.
- Y. Park, J. Kim, S. Hwang and S. Han, J. Chem. Theory Comput., 2024, 20, 4857–4868 CrossRef CAS PubMed.
- L. Barroso-Luque, M. Shuaibi, X. Fu, B. M. Wood, M. Dzamba, M. Gao, A. Rizvi, C. L. Zitnick and Z. W. Ulissi, Open Materials, 2024, (OMat24) Inorganic Materials Dataset and Models, arXiv, 2024, preprint DOI:10.48550/arXiv.2410.12771.
- M. Neumann, J. Gin, B. Rhodes, S. Bennett, Z. Li, H. Choubisa, A. Hussey and J. Godwin, Orb: A Fast, Scalable Neural Network Potential, arXiv, 2024, preprint DOI:10.48550/arXiv.2410.22570.
- A. Bochkarev, Y. Lysogorskiy and R. Drautz, Phys. Rev. X, 2024, 14, 021036 CAS.
- A. Grisafi and M. Ceriotti, J. Chem. Phys., 2019, 151, 204105 CrossRef PubMed.
- S. A. Ghasemi, A. Hofstetter, S. Saha and S. Goedecker, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 045131 CrossRef.
- T. W. Ko, J. A. Finkler, S. Goedecker and J. Behler, Acc. Chem. Res., 2021, 54, 808–817 CrossRef CAS PubMed.
- J. Thomas, W. J. Baldwin, G. Csányi and C. Ortner, Self-consistent Coulomb interactions for machine learning interatomic potentials, arXiv, 2024, preprint DOI:10.48550/arXiv.2406.10915.
- P. Loche, K. K. Huguenin-Dumittan, M. Honarmand, Q. Xu, E. Rumiantsev, W. B. How, M. F. Langer and M. Ceriotti, Fast and flexible range-separated models for atomistic machine learning, arXiv, 2024, preprint DOI:10.48550/arXiv.2412.03281.
- A. Paszke, S. Gross, S. Chintala, G. Chanan, E. Yang, Z. DeVito, Z. Lin, A. Desmaison, L. Antiga and A. Lerer, NeurIPS 2017 Workshop on Autodiff, 2017.
- M. Rinaldi, M. Mrovec, A. Bochkarev, Y. Lysogorskiy and R. Drautz, npj Comput. Mater., 2024, 10, 1–12 CrossRef.
- S. Falletta, A. Cepellotti, A. Johansson, C. W. Tan, A. Musaelian, C. J. Owen and B. Kozinsky, Unified Differentiable Learning of Electric, Response, arXiv, 2024, preprint DOI:10.48550/arXiv.2403.17207.
- A. Fabrizio, A. Grisafi, B. Meyer, M. Ceriotti and C. Corminboeuf, Chem. Sci., 2019, 10, 9424–9432 RSC.
- A. M. Lewis, A. Grisafi, M. Ceriotti and M. Rossi, J. Chem. Theory Comput., 2021, 17, 7203–7214 CrossRef CAS PubMed.
- O. T. Unke, M. Bogojeski, M. Gastegger, M. Geiger, T. Smidt and K.-R. Müller, SE(3)-equivariant prediction of molecular wavefunctions and electronic densities, arXiv, 2021, preprint DOI:10.48550/arXiv.2106.02347.
- K. T. Schütt, M. Gastegger, A. Tkatchenko, K.-R. Müller and R. J. Maurer, Nat. Commun., 2019, 10, 5024 CrossRef PubMed.
- J. Schuetzke, N. J. Szymanski and M. Reischl, npj Comput. Mater., 2023, 9, 1–12 CrossRef.
- A. Angulo, L. Yang, E. S. Aydil and M. A. Modestino, Digital Discovery, 2022, 1, 35–44 RSC.
- Z. Tang, H. Li, P. Lin, X. Gong, G. Jin, L. He, H. Jiang, X. Ren, W. Duan and Y. Xu, Nat. Commun., 2024, 15, 8815 CrossRef CAS PubMed.
- Y. Zhong, H. Yu, M. Su, X. Gong and H. Xiang, npj Comput. Mater., 2023, 9, 1–13 CrossRef.
- C. Ben Mahmoud, J. L. A. Gardner and V. L. Deringer, Nat. Comput. Sci., 2024, 1–4 Search PubMed.
- A. Thomas-Mitchell, G. Hawe and P. L. Popelier, Mach. Learn.: Sci. Technol., 2023, 4, 045034 Search PubMed.
- H. Park, Z. Li and A. Walsh, Matter, 2024, 7, 2355–2367 CrossRef CAS.
- S. G. Louie, Y.-H. Chan, F. H. Da Jornada, Z. Li and D. Y. Qiu, Nat. Mater., 2021, 20, 728–735 CrossRef CAS PubMed.
- A. Zunger, Nat. Rev. Chem., 2018, 2, 0121 CrossRef CAS.
- A. R. Oganov, C. J. Pickard, Q. Zhu and R. J. Needs, Nat. Rev. Mater., 2019, 4, 331–348 CrossRef.
- H. Wang, T. Fu, Y. Du, W. Gao, K. Huang, Z. Liu, P. Chandak, S. Liu, P. Van Katwyk, A. Deac, A. Anandkumar, K. Bergen, C. P. Gomes, S. Ho, P. Kohli, J. Lasenby, J. Leskovec, T.-Y. Liu, A. Manrai, D. Marks, B. Ramsundar, L. Song, J. Sun, J. Tang, P. Velicković, M. Welling, L. Zhang, C. W. Coley, Y. Bengio and M. Zitnik, Nature, 2023, 620, 47–60 CrossRef CAS PubMed.
- Z. Epstein, A. Hertzmann, the Investigators of Human Creativity, M. Akten, H. Farid, J. Fjeld, M. R. Frank, M. Groh, L. Herman, N. Leach, R. Mahari, A. S. Pentland, O. Russakovsky, H. Schroeder and A. Smith, Science, 2023, 380, 1110–1111 CrossRef CAS PubMed.
- Y. Du, A. R. Jamasb, J. Guo, T. Fu, C. Harris, Y. Wang, C. Duan, P. Liò, P. Schwaller and T. L. Blundell, Nat. Mach. Intell., 2024, 6, 589–604 CrossRef.
- A. Onwuli, A. V. Hegde, K. V. T. Nguyen, K. T. Butler and A. Walsh, Digital Discovery, 2023, 2, 1558–1564 RSC.
- Q. Vanhaelen, Y.-C. Lin and A. Zhavoronkov, ACS Med. Chem. Lett., 2020, 11, 1496–1505 CrossRef CAS PubMed.
- G. L. Guimaraes, B. Sanchez-Lengeling, C. Outeiral, P. L. C. Farias and A. Aspuru-Guzik, Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models, arXiv, 2018, preprint DOI:10.48550/arXiv.1705.10843.
- E. Putin, A. Asadulaev, Y. Ivanenkov, V. Aladinskiy, B. Sanchez-Lengeling, A. Aspuru-Guzik and A. Zhavoronkov, J. Chem. Inf. Model., 2018, 58, 1194–1204 CrossRef CAS PubMed.
- B. Sanchez-Lengeling, C. Outeiral, G. L. Guimaraes and A. Aspuru-Guzik, Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC), 2017 DOI:10.26434/chemrxiv.5309668.v3.
- N. D. Cao and T. Kipf, MolGAN: An implicit generative model for small molecular graphs, arXiv, 2022, preprint DOI:10.48550/arXiv.1805.11973.
- S. Kim, J. Noh, G. H. Gu, A. Aspuru-Guzik and Y. Jung, ACS Cent. Sci., 2020, 6, 1412–1420 CrossRef CAS PubMed.
- Y. Zhao, M. Al-Fahdi, M. Hu, E. M. D. Siriwardane, Y. Song, A. Nasiri and J. Hu, Adv. Sci., 2021, 8, 2100566 CrossRef CAS PubMed.
- J. Gui, Z. Sun, Y. Wen, D. Tao and J. Ye, IEEE Trans. Knowledge Data Eng., 2023, 35, 3313–3332 Search PubMed.
- M. Arjovsky and L. Bottou, Towards Principled Methods for Training Generative Adversarial Networks, arXiv, 2017, preprint DOI:10.48550/arXiv.1701.04862.
- M. Arjovsky, S. Chintala and L. Bottou, Wasserstein GAN, arXiv, 2017, preprint DOI:10.48550/arXiv.1701.07875.
- D. P. Kingma and M. Welling, Auto-Encoding Variational Bayes, arXiv, 2022, preprint DOI:10.48550/arXiv.1312.6114.
- J. Zhou, A. Mroz and K. E. Jelfs, Digital Discovery, 2023, 2, 1925–1936 RSC.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed.
- Q. Liu, M. Allamanis, M. Brockschmidt and A. L. Gaunt, Constrained Graph Variational Autoencoders for Molecule Design, arXiv, 2019, preprint DOI:10.48550/arXiv.1805.09076.
- J. Noh, J. Kim, H. S. Stein, B. Sanchez-Lengeling, J. M. Gregoire, A. Aspuru-Guzik and Y. Jung, Matter, 2019, 1, 1370–1384 CrossRef.
- Z. Ren, S. I. P. Tian, J. Noh, F. Oviedo, G. Xing, J. Li, Q. Liang, R. Zhu, A. G. Aberle, S. Sun, X. Wang, Y. Liu, Q. Li, S. Jayavelu, K. Hippalgaonkar, Y. Jung and T. Buonassisi, Matter, 2022, 5, 314–335 CrossRef CAS.
- R. Zhu, W. Nong, S. Yamazaki and K. Hippalgaonkar, Matter, 2024, 7, 3469–3488 CrossRef CAS.
- B. Dai, Z. Wang and D. Wipf, The Usual Suspects? Reassessing Blame for VAE Posterior Collapse, arXiv, 2019, preprint DOI:10.48550/arXiv.1912.10702.
- G. Papamakarios, E. Nalisnick, D. J. Rezende, S. Mohamed and B. Lakshminarayanan, Normalizing Flows for Probabilistic Modeling and Inference, arXiv, 2021, preprint DOI:10.48550/arXiv.1912.02762.
- J. Ho, A. Jain and P. Abbeel, Denoising Diffusion Probabilistic Models, arXiv, 2020, preprint DOI:10.48550/arXiv.2006.11239.
- C. Shi, M. Xu, Z. Zhu, W. Zhang, M. Zhang and J. Tang, GraphAF: a Flow-based Autoregressive Model for Molecular Graph Generation, arXiv, 2020, preprint DOI:10.48550/arXiv.2001.09382.
- T. Xie, X. Fu, O.-E. Ganea, R. Barzilay and T. Jaakkola, Crystal Diffusion Variational Autoencoder for Periodic Material Generation, arXiv, 2022, preprint DOI:10.48550/arXiv.2110.06197.
- Y. Song, J. Gong, Y. Qu, H. Zhou, M. Zheng, J. Liu and W.-Y. Ma, Unified Generative Modeling of 3D Molecules via Bayesian Flow Networks, arXiv, 2024, preprint DOI:10.48550/arXiv.2403.15441.
- R. Jiao, W. Huang, Y. Liu, D. Zhao and Y. Liu, Space Group Constrained Crystal Generation, arXiv, 2024, preprint DOI:10.48550/arXiv.2402.03992.
- C.-Y. Ye, H.-M. Weng and Q.-S. Wu, Comput. Mater. Today, 2024, 1, 100003 CrossRef.
- X. Luo, Z. Wang, P. Gao, J. Lv, Y. Wang, C. Chen and Y. Ma, npj Comput. Mater., 2024, 10, 254 CrossRef.
- C. Zeni, R. Pinsler, D. Zügner, A. Fowler, M. Horton, X. Fu, S. Shysheya, J. Crabbé, L. Sun, J. Smith, B. Nguyen, H. Schulz, S. Lewis, C.-W. Huang, Z. Lu, Y. Zhou, H. Yang, H. Hao, J. Li, R. Tomioka and T. Xie, MatterGen: a generative model for inorganic materials design, arXiv, 2024, preprint DOI:10.48550/arXiv.2312.03687.
- S. Yang, K. Cho, A. Merchant, P. Abbeel, D. Schuurmans, I. Mordatch and E. D. Cubuk, Scalable Diffusion for Materials Generation, arXiv, 2024, preprint DOI:10.48550/arXiv.2311.09235.
- T. Weiss, E. Mayo Yanes, S. Chakraborty, L. Cosmo, A. M. Bronstein and R. Gershoni-Poranne, Nat. Comput. Sci., 2023, 3, 873–882 CrossRef PubMed.
- I. Igashov, H. Stärk, C. Vignac, A. Schneuing, V. G. Satorras, P. Frossard, M. Welling, M. Bronstein and B. Correia, Nat. Mach. Intell., 2024, 6, 417–427 CrossRef.
- K. Adams, K. Abeywardane, J. Fromer and C. W. Coley, ShEPhERD: Diffusing shape, electrostatics, and pharmacophores for bioisosteric drug design, arXiv, 2024, preprint DOI:10.48550/arXiv.2411.04130.
- K. Kahouli, S. S. P. Hessmann, K.-R. Müller, S. Nakajima, S. Gugler and N. W. A. Gebauer, Mach. Learn.: Sci. Technol., 2024, 5, 035038 Search PubMed.
- J. Wang, R. Qin, M. Wang, M. Fang, Y. Zhang, Y. Zhu, Q. Su, Q. Gou, C. Shen, O. Zhang, Z. Wu, D. Jiang, X. Zhang, H. Zhao, X. Wan, Z. Wu, L. Liu, Y. Kang, C.-Y. Hsieh and T. Hou, Token-Mol 1.0: Tokenized drug design with large language model, arXiv, 2024, preprint DOI:10.48550/arXiv.2407.07930.
- L. M. Antunes, K. T. Butler and R. Grau-Crespo, Crystal Structure Generation with Autoregressive Large Language Modeling, arXiv, 2024, preprint DOI:10.48550/arXiv.2307.04340.
- D. Flam-Shepherd and A. Aspuru-Guzik, Language models can generate molecules, materials, and protein binding sites directly in three dimensions as XYZ, CIF, and PDB files, arXiv, 2023, preprint DOI:10.48550/arXiv.2305.05708.
- Y. Chen, X. Wang, X. Deng, Y. Liu, X. Chen, Y. Zhang, L. Wang and H. Xiao, MatterGPT: A Generative Transformer for Multi-Property Inverse Design of Solid-State Materials, arXiv, 2024, preprint DOI:10.48550/arXiv.2408.07608.
- N. Gruver, A. Sriram, A. Madotto, A. G. Wilson, C. L. Zitnick and Z. Ulissi, Fine-Tuned Language Models Generate Stable Inorganic Materials as Text, arXiv, 2024, preprint DOI:10.48550/arXiv.2402.04379.
- K. Choudhary, J. Phys. Chem. Lett., 2024, 15, 6909–6917 CrossRef CAS PubMed.
- Z. Cao, X. Luo, J. Lv and L. Wang, Space Group Informed Transformer for Crystalline Materials Generation, arXiv, 2024, preprint DOI:10.48550/arXiv.2403.15734.
- Q. Ding, S. Miret and B. Liu, MatExpert: Decomposing Materials Discovery by Mimicking Human Experts, arXiv, 2024, preprint DOI:10.48550/arXiv.2410.21317.
- R. Özçelik, S. De Ruiter, E. Criscuolo and F. Grisoni, Nat. Commun., 2024, 15, 6176 CrossRef PubMed.
- S. Yang, S. Batzner, R. Gao, M. Aykol, A. L. Gaunt, B. McMorrow, D. J. Rezende, D. Schuurmans, I. Mordatch and E. D. Cubuk, Generative Hierarchical Materials, Search, arXiv, 2024, preprint DOI:10.48550/arXiv.2409.06762.
- A. Sriram, B. K. Miller, R. T. Q. Chen and B. M. Wood, FlowLLM: Flow Matching for Material Generation with Large Language Models as Base Distributions, arXiv, 2024, preprint DOI:10.48550/arXiv.2410.23405.
- N. Kazeev, R. Zhu, I. Romanov, A. E. Ustyuzhanin, S. Yamazaki, W. Nong and K. Hippalgaonkar, AI for Accelerated Materials Design – NeurIPS 2024, 2024.
- H. Park, A. Onwuli and A. Walsh, Exploration of crystal chemical space using text-guided generative artificial intelligence, 2024 DOI:10.26434/chemrxiv-2024-rw8p5.
- D. M. Anstine and O. Isayev, J. Am. Chem. Soc., 2023, 145, 8736–8750 CrossRef CAS PubMed.
- A. Subramanian, W. Gao, R. Barzilay, J. C. Grossman, T. Jaakkola, S. Jegelka, M. Li, J. Li, W. Matusik, E. Olivetti, C. W. Coley and R. Gomez-Bombarelli, An MIT Exploration of Generative AI, 2024 DOI:10.21428/e4baedd9.92e511e3.
- S. Yang and R. Gómez-Bombarelli, Chemically Transferable Generative Backmapping of Coarse-Grained Proteins, arXiv, 2023, preprint DOI:10.48550/arXiv.2303.01569.
- N. Rønne, A. Aspuru-Guzik and B. Hammer, Phys. Rev. B, 2024, 110, 235427 CrossRef.
- J. Park, A. P. S. Gill, S. M. Moosavi and J. Kim, J. Mater. Chem. A, 2024, 12, 6507–6514 RSC.
- Z. Li, W. Nash, S. O'Brien, Y. Qiu, R. Gupta and N. Birbilis, J. Mater. Sci. Technol., 2022, 125, 81–96 CrossRef CAS.
- Z. Zhou, Y. Shang, X. Liu and Y. Yang, npj Comput. Mater., 2023, 9, 15 CrossRef.
- A. X. B. Yong, T. Su and E. Ertekin, Digital Discovery, 2024, 3, 1889–1909 RSC.
- H. Park, A. Onwuli, K. T. Butler and A. Walsh, Faraday Discuss., 2025, 256, 601–613 RSC.
- D. Polykovskiy, A. Zhebrak, B. Sanchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V. Aladinskiy, M. Veselov, A. Kadurin, S. Johansson, H. Chen, S. Nikolenko, A. Aspuru-Guzik and A. Zhavoronkov, Front. Pharmacol., 2020, 11, 565644 CrossRef CAS PubMed.
- M. Thomas, N. M. O'Boyle, A. Bender and C. De Graaf, J. Cheminf., 2024, 16, 64 CAS.
- S. G. Baird, H. M. Sayeed, J. Montoya and T. D. Sparks, JOSS, 2024, 9, 5618 CrossRef.
- J. C. Fromer and C. W. Coley, Patterns, 2023, 4, 100678 CrossRef CAS PubMed.
- S. Bennett, F. T. Szczypiński, L. Turcani, M. E. Briggs, R. L. Greenaway and K. E. Jelfs, J. Chem. Inf. Model., 2021, 61, 4342–4356 CrossRef CAS PubMed.
- J. Guo and P. Schwaller, It Takes Two to Tango: Directly Optimizing for Constrained Synthesizability in Generative Molecular Design, arXiv, 2024, preprint DOI:10.48550/arXiv.2410.11527.
- A. Davariashtiyani, B. Wang, S. Hajinazar, E. Zurek and S. Kadkhodaei, Mach. Learn.: Sci. Technol., 2024, 5, 040501 Search PubMed.
- S. Seo, J. Lim and W. Y. Kim, Adv. Sci., 2023, 10, 2206674 CrossRef CAS PubMed.
- J. A. Esterhuizen, B. R. Goldsmith and S. Linic, Nat. Catal., 2022, 5, 175–184 CrossRef.
- J. Jiménez-Luna, F. Grisoni and G. Schneider, Nat. Mach. Intell., 2020, 2, 573–584 CrossRef.
- Z. Wu, J. Chen, Y. Li, Y. Deng, H. Zhao, C.-Y. Hsieh and T. Hou, J. Chem. Inf. Model., 2023, 63, 7617–7627 CrossRef CAS PubMed.
- Z. Wu, J. Wang, H. Du, D. Jiang, Y. Kang, D. Li, P. Pan, Y. Deng, D. Cao, C.-Y. Hsieh and T. Hou, Nat. Commun., 2023, 14, 2585 CrossRef CAS PubMed.
- Q. Yuan, F. T. Szczypiński and K. E. Jelfs, Digital Discovery, 2022, 1, 127–138 RSC.
- M. Korshunova, N. Huang, S. Capuzzi, D. S. Radchenko, O. Savych, Y. S. Moroz, C. I. Wells, T. M. Willson, A. Tropsha and O. Isayev, Commun. Chem., 2022, 5, 129 CrossRef PubMed.
- K. Swanson, G. Liu, D. B. Catacutan, A. Arnold, J. Zou and J. M. Stokes, Nat. Mach. Intell., 2024, 6, 338–353 CrossRef.
- J. D. Tan, B. Ramalingam, V. Chellappan, N. K. Gupta, L. Dillard, S. A. Khan, C. Galvin and K. Hippalgaonkar, ACS Energy Lett., 2024, 9, 5240–5250 CrossRef CAS.
- M. Kp Jayatunga, M. Ayers, L. Bruens, D. Jayanth and C. Meier, Drug Discovery Today, 2024, 29, 104009 CrossRef CAS PubMed.
- A. Schuhmacher, M. Hinder, A. Von Stegmann Und Stein, D. Hartl and O. Gassmann, Drug Discovery Today, 2023, 28, 103726 CrossRef PubMed.
- A. Schuhmacher, M. Hinder, A. Dodel, O. Gassmann and D. Hartl, Nat. Rev. Drug Discovery, 2023, 22, 781–782 CrossRef CAS PubMed.
- Congressional Budget Office, Research and Development in the Pharmaceutical Industry, 2021, https://www.cbo.gov/publication/57126.
- D. J. Payne, M. N. Gwynn, D. J. Holmes and D. L. Pompliano, Nat. Rev. Drug Discovery, 2006, 6, 29–40 CrossRef PubMed.
- B. Zdrazil, E. Felix, F. Hunter, E. J. Manners, J. Blackshaw, S. Corbett, M. de Veij, H. Ioannidis, D. M. Lopez, J. F. Mosquera, M. P. Magarinos, N. Bosc, R. Arcila, T. Kizilören, A. Gaulton, A. P. Bento, M. F. Adasme, P. Monecke, G. A. Landrum and A. R. Leach, Nucleic Acids Res., 2024, 52, D1180–D1192 CrossRef CAS PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2023, 51, D1373–D1380 CrossRef PubMed.
- P. J. Ballester, Nature, 2023, 624, 252 CrossRef CAS PubMed.
- J. Ross, B. Belgodere, V. Chenthamarakshan, I. Padhi, Y. Mroueh and P. Das, Nat. Mach. Intell., 2022, 4, 1256–1264 CrossRef.
- D. Chen, J. Liu and G.-W. Wei, Nat. Mach. Intell., 2024, 6, 799–810 CrossRef.
- R. Singh, S. Sledzieski, B. Bryson, L. Cowen and B. Berger, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2220778120 CrossRef CAS PubMed.
- F. Wong, C. De La Fuente-Nunez and J. J. Collins, Science, 2023, 381, 164–170 CrossRef CAS PubMed.
- S. Vishwakarma, S. Hernandez-Hernandez and P. J. Ballester, Biol. Methods Protoc., 2024, 9, bpae065 CrossRef PubMed.
- B. Hie, B. D. Bryson and B. Berger, Cell Systems, 2020, 11, 461–477.e9 CrossRef CAS PubMed.
- S. Hernandez-Hernandez, S. Vishwakarma and P. Ballester, Proceedings of the Eleventh Symposium on Conformal and Probabilistic Prediction with Applications, 2022, pp. 92–108.
- Y. Xu, X. Liu, W. Xia, J. Ge, C.-W. Ju, H. Zhang and J. Z. H. Zhang, J. Chem. Inf. Model., 2024, 64, 8440–8452 CrossRef CAS PubMed.
- J. Jiang, R. Wang, M. Wang, K. Gao, D. D. Nguyen and G.-W. Wei, J. Chem. Inf. Model., 2020, 60, 1235–1244 CrossRef CAS PubMed.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. Pande, Chem. Sci., 2018, 9, 513–530 RSC.
- J.-F. Truchon and C. I. Bayly, J. Chem. Inf. Model., 2007, 47, 488–508 CrossRef CAS PubMed.
- Q. Guo, S. Hernandez-Hernandez and P. J. Ballester, Artificial Neural Networks and Machine Learning – ICANN 2024, Springer Nature Switzerland, Cham, 2024, vol. 15025, pp. 58–72 Search PubMed.
- V.-K. Tran-Nguyen, M. Junaid, S. Simeon and P. J. Ballester, Nat. Protoc., 2023, 18, 3460–3511 CrossRef CAS PubMed.
- Q. Guo, S. Hernandez and P. Ballester, UMAP-clustering split for rigorous evaluation of AI models for virtual screening on cancer cell lines, 2024 DOI:10.26434/chemrxiv-2024-f1v2v.
- P. Gómez-Sacristán, S. Simeon, V.-K. Tran-Nguyen, S. Patil and P. J. Ballester, J. Adv. Res., 2025, 67, 185–196 CrossRef PubMed.
- K. Huang, T. Fu, W. Gao, Y. Zhao, Y. Roohani, J. Leskovec, C. W. Coley, C. Xiao, J. Sun and M. Zitnik, Nat. Chem. Biol., 2022, 18, 1033–1036 CrossRef CAS PubMed.
- O. O. Grygorenko, D. S. Radchenko, I. Dziuba, A. Chuprina, K. E. Gubina and Y. S. Moroz, iScience, 2020, 23, 101681 CrossRef CAS PubMed.
- C. Gorgulla, Annu. Rev. Biomed. Data Sci., 2023, 6, 229–258 CrossRef PubMed.
- F. Liu, O. Mailhot, I. S. Glenn, S. F. Vigneron, V. Bassim, X. Xu, K. Fonseca-Valencia, M. S. Smith, D. S. Radchenko, J. S. Fraser, Y. S. Moroz, J. J. Irwin and B. K. Shoichet, Nat. Chem. Biol., 2025 DOI:10.1038/s41589-024-01797-w.
- L. Fresnais and P. J. Ballester, Briefings Bioinf., 2021, 22, bbaa095 CrossRef PubMed.
- F. Gentile, J. C. Yaacoub, J. Gleave, M. Fernandez, A.-T. Ton, F. Ban, A. Stern and A. Cherkasov, Nat. Protoc., 2022, 17, 672–697 CrossRef CAS PubMed.
- G. Zhou, D.-V. Rusnac, H. Park, D. Canzani, H. M. Nguyen, L. Stewart, M. F. Bush, P. T. Nguyen, H. Wulff, V. Yarov-Yarovoy, N. Zheng and F. DiMaio, Nat. Commun., 2024, 15, 7761 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- F. Wong, A. Krishnan, E. J. Zheng, H. Stärk, A. L. Manson, A. M. Earl, T. Jaakkola and J. J. Collins, Mol. Syst. Biol., 2022, 18, e11081 CrossRef CAS PubMed.
- J. Lyu, N. Kapolka, R. Gumpper, A. Alon, L. Wang, M. K. Jain, X. Barros-Álvarez, K. Sakamoto, Y. Kim, J. DiBerto, K. Kim, I. S. Glenn, T. A. Tummino, S. Huang, J. J. Irwin, O. O. Tarkhanova, Y. Moroz, G. Skiniotis, A. C. Kruse, B. K. Shoichet and B. L. Roth, Science, 2024, 384, eadn6354 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C.-C. Hung, M. O'Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Ždek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Nature, 2024, 630, 493–500 CrossRef CAS PubMed.
- Nat. Methods, 2023, 20, 163 Search PubMed.
- J. M. Stokes, K. Yang, K. Swanson, W. Jin, A. Cubillos-Ruiz, N. M. Donghia, C. R. MacNair, S. French, L. A. Carfrae, Z. Bloom-Ackermann, V. M. Tran, A. Chiappino-Pepe, A. H. Badran, I. W. Andrews, E. J. Chory, G. M. Church, E. D. Brown, T. S. Jaakkola, R. Barzilay and J. J. Collins, Cell, 2020, 180, 688–702.e13 CrossRef CAS PubMed.
- G. Liu, D. B. Catacutan, K. Rathod, K. Swanson, W. Jin, J. C. Mohammed, A. Chiappino-Pepe, S. A. Syed, M. Fragis, K. Rachwalski, J. Magolan, M. G. Surette, B. K. Coombes, T. Jaakkola, R. Barzilay, J. J. Collins and J. M. Stokes, Nat. Chem. Biol., 2023, 19, 1342–1350 CrossRef CAS PubMed.
- F. Wong, E. J. Zheng, J. A. Valeri, N. M. Donghia, M. N. Anahtar, S. Omori, A. Li, A. Cubillos-Ruiz, A. Krishnan, W. Jin, A. L. Manson, J. Friedrichs, R. Helbig, B. Hajian, D. K. Fiejtek, F. F. Wagner, H. H. Soutter, A. M. Earl, J. M. Stokes, L. D. Renner and J. J. Collins, Nature, 2024, 626, 177–185 CrossRef CAS PubMed.
- Q. U. Ain, A. Aleksandrova, F. D. Roessler and P. J. Ballester, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 405–424 CAS.
- H. Li, K.-H. Sze, G. Lu and P. J. Ballester, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1478 Search PubMed.
- C. Shen, J. Ding, Z. Wang, D. Cao, X. Ding and T. Hou, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1429 CAS.
- T. Harren, T. Gutermuth, C. Grebner, G. Hessler and M. Rarey, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2024, 14, e1716 CAS.
- The Atomwise AIMS Program, I. Wallach, D. Bernard, K. Nguyen, G. Ho, A. Morrison, A. Stecula, A. Rosnik, A. M. O'Sullivan, A. Davtyan, B. Samudio, B. Thomas, B. Worley, B. Butler, C. Laggner, D. Thayer, E. Moharreri, G. Friedland, H. Truong, H. Van Den Bedem, H. L. Ng, K. Stafford, K. Sarangapani, K. Giesler, L. Ngo, M. Mysinger, M. Ahmed, N. J. Anthis, N. Henriksen, P. Gniewek, S. Eckert, S. De Oliveira, S. Suterwala, S. V. K. PrasadPrasad, S. Shek, S. Contreras, S. Hare, T. Palazzo, T. E. O'Brien, T. Van Grack, T. Williams, T.-R. Chern, V. Kenyon, A. H. Lee, A. B. Cann, B. Bergman, B. M. Anderson, B. D. Cox, J. M. Warrington, J. M. Sorenson, J. M. Goldenberg, M. A. Young, N. DeHaan, R. P. Pemberton, S. Schroedl, T. M. Abramyan, T. Gupta, V. Mysore, A. G. Presser, A. A. Ferrando, A. D. Andricopulo, A. Ghosh, A. G. Ayachi, A. Mushtaq, A. M. Shaqra, A. K. L. Toh, A. V. Smrcka, A. Ciccia, A. S. De Oliveira, A. Sverzhinsky, A. M. De Sousa, A. I. Agoulnik, A. Kushnir, A. N. Freiberg, A. V. Statsyuk, A. R. Gingras, A. Degterev, A. Tomilov, A. Vrielink, A. A. Garaeva, A. Bryant-Friedrich, A. Caflisch, A. K. Patel, A. V. Rangarajan, A. Matheeussen, A. Battistoni, A. Caporali, A. Chini, A. Ilari, A. Mattevi, A. T. Foote, A. Trabocchi, A. Stahl, A. B. Herr, A. Berti, A. Freywald, A. G. Reidenbach, A. Lam, A. R. Cuddihy, A. White, A. Taglialatela, A. K. Ojha, A. M. Cathcart, A. A. L. Motyl, A. Borowska, A. D'Antuono, A. K. H. Hirsch, A. M. Porcelli, A. Minakova, A. Montanaro, A. Müller, A. Fiorillo, A. Virtanen, A. J. O'Donoghue, A. Del Rio Flores, A. E. Garmendia, A. Pineda-Lucena, A. T. Panganiban, A. Samantha, A. K. Chatterjee, A. L. Haas, A. S. Paparella, A. L. S. John, A. Prince, A. ElSheikh, A. M. Apfel, A. Colomba, A. O'Dea, B. N. Diallo, B. M. R. M. Ribeiro, B. A. Bailey-Elkin, B. L. Edelman, B. Liou, B. Perry, B. S. K. Chua, B. Kováts, B. Englinger, B. Balakrishnan, B. Gong, B. Agianian, B. Pressly, B. P. M. Salas, B. M. Duggan, B. V. Geisbrecht, B. W. Dymock, B. C. Morten, B. D. Hammock, B. E. F. Mota, B. C. Dickinson, C. Fraser, C. Lempicki, C. D. Novina, C. Torner, C. Ballatore, C. Bon, C. J. Chapman, C. L. Partch, C. T. Chaton, C. Huang, C.-Y. Yang, C. M. Kahler, C. Karan, C. Keller, C. L. Dieck, C. Huimei, C. Liu, C. Peltier, C. K. Mantri, C. M. Kemet, C. E. Müller, C. Weber, C. M. Zeina, C. S. Muli, C. Morisseau, C. Alkan, C. Reglero, C. A. Loy, C. M. Wilson, C. Myhr, C. Arrigoni, C. Paulino, C. Santiago, D. Luo, D. J. Tumes, D. A. Keedy, D. A. Lawrence, D. Chen, D. Manor, D. J. Trader, D. A. Hildeman, D. H. Drewry, D. J. Dowling, D. J. Hosfield, D. M. Smith, D. Moreira, D. P. Siderovski, D. Shum, D. T. Krist, D. W. H. Riches, D. M. Ferraris, D. H. Anderson, D. R. Coombe, D. S. Welsbie, D. Hu, D. Ortiz, D. Alramadhani, D. Zhang, D. Chaudhuri, D. J. Slotboom, D. R. Ronning, D. Lee, D. Dirksen, D. A. Shoue, D. W. Zochodne, D. Krishnamurthy, D. Duncan, D. M. Glubb, E. L. M. Gelardi, E. C. Hsiao, E. G. Lynn, E. B. Silva, E. Aguilera, E. Lenci, E. T. Abraham, E. Lama, E. Mameli, E. Leung, E. Giles, E. M. Christensen, E. R. Mason, E. Petretto, E. F. Trakhtenberg, E. J. Rubin, E. Strauss, E. W. Thompson, E. Cione, E. M. Lisabeth, E. Fan, E. G. Kroon, E. Jo, E. M. García-Cuesta, E. Glukhov, E. Gavathiotis, F. Yu, F. Xiang, F. Leng, F. Wang, F. Ingoglia, F. Van Den Akker, F. Borriello, F. J. Vizeacoumar, F. Luh, F. S. Buckner, F. S. Vizeacoumar, F. B. Bdira, F. Svensson, G. M. Rodriguez, G. Bognár, G. Lembo, G. Zhang, G. Dempsey, G. Eitzen, G. Mayer, G. L. Greene, G. A. Garcia, G. L. Lukacs, G. Prikler, G. C. G. Parico, G. Colotti, G. De Keulenaer, G. Cortopassi, G. Roti, G. Girolimetti, G. Fiermonte, G. Gasparre, G. Leuzzi, G. Dahal, G. Michlewski, G. L. Conn, G. D. Stuchbury, G. R. Bowman, G. M. Popowicz, G. Veit, G. E. De Souza, G. Akk, G. Caljon, G. Alvarez, G. Rucinski, G. Lee, G. Cildir, H. Li, H. E. Breton, H. Jafar-Nejad, H. Zhou, H. P. Moore, H. Tilford, H. Yuan, H. Shim, H. Wulff, H. Hoppe, H. Chaytow, H.-K. Tam, H. Van Remmen, H. Xu, H. M. Debonsi, H. B. Lieberman, H. Jung, H.-Y. Fan, H. Feng, H. Zhou, H. J. Kim, I. R. Greig, I. Caliandro, I. Corvo, I. Arozarena, I. N. Mungrue, I. M. Verhamme, I. A. Qureshi, I. Lotsaris, I. Cakir, J. J. P. Perry, J. Kwiatkowski, J. Boorman, J. Ferreira, J. Fries, J. M. Kratz, J. Miner, J. L. Siqueira-Neto, J. G. Granneman, J. Ng, J. Shorter, J. H. Voss, J. M. Gebauer, J. Chuah, J. J. Mousa, J. T. Maynes, J. D. Evans, J. Dickhout, J. P. MacKeigan, J. N. Jossart, J. Zhou, J. Lin, J. Xu, J. Wang, J. Zhu, J. Liao, J. Xu, J. Zhao, J. Lin, J. Lee, J. Reis, J. Stetefeld, J. B. Bruning, J. B. Bruning, J. G. Coles, J. J. Tanner, J. M. Pascal, J. So, J. L. Pederick, J. A. Costoya, J. B. Rayman, J. J. Maciag, J. A. Nasburg, J. J. Gruber, J. M. Finkelstein, J. Watkins, J. M. Rodríguez-Frade, J. A. S. Arias, J. J. Lasarte, J. Oyarzabal, J. Milosavljevic, J. Cools, J. Lescar, J. Bogomolovas, J. Wang, J.-M. Kee, J.-M. Kee, J. Liao, J. C. Sistla, J. S. Abrahão, K. Sishtla, K. R. Francisco, K. B. Hansen, K. A. Molyneaux, K. A. Cunningham, K. R. Martin, K. Gadar, K. K. Ojo, K. S. Wong, K. L. Wentworth, K. Lai, K. A. Lobb, K. M. Hopkins, K. Parang, K. Machaca, K. Pham, K. Ghilarducci, K. S. Sugamori, K. J. McManus, K. Musta, K. M. E. Faller, K. Nagamori, K. J. Mostert, K. V. Korotkov, K. Liu, K. S. Smith, K. Sarosiek, K. H. Rohde, K. K. Kim, K. H. Lee, L. Pusztai, L. Lehtiö, L. M. Haupt, L. E. Cowen, L. J. Byrne, L. Su, L. Wert-Lamas, L. Puchades-Carrasco, L. Chen, L. H. Malkas, L. Zhuo, L. Hedstrom, L. Hedstrom, L. D. Walensky, L. Antonelli, L. Iommarini, L. Whitesell, L. M. Randall, M. D. Fathallah, M. H. Nagai, M. L. Kilkenny, M. Ben-Johny, M. P. Lussier, M. P. Windisch, M. Lolicato, M. L. Lolli, M. Vleminckx, M. C. Caroleo, M. J. Macias, M. Valli, M. M. Barghash, M. Mellado, M. A. Tye, M. A. Wilson, M. Hannink, M. R. Ashton, M. V. C. Cerna, M. Giorgis, M. K. Safo, M. S. Maurice, M. A. McDowell, M. Pasquali, M. Mehedi, M. S. M. Serafim, M. B. Soellner, M. G. Alteen, M. M. Champion, M. Skorodinsky, M. L. O'Mara, M. Bedi, M. Rizzi, M. Levin, M. Mowat, M. R. Jackson, M. Paige, M. Al-Yozbaki, M. A. Giardini, M. M. Maksimainen, M. De Luise, M. S. Hussain, M. Christodoulides, N. Stec, N. Zelinskaya, N. Van Pelt, N. M. Merrill, N. Singh, N. A. Kootstra, N. Singh, N. S. Gandhi, N.-L. Chan, N. M. Trinh, N. O. Schneider, N. Matovic, N. Horstmann, N. Longo, N. Bharambe, N. Rouzbeh, N. Mahmoodi, N. J. Gumede, N. C. Anastasio, N. B. Khalaf, O. Rabal, O. Kandror, O. Escaffre, O. Silvennoinen, O. T. Bishop, P. Iglesias, P. Sobrado, P. Chuong, P. O'Connell, P. Martin-Malpartida, P. Mellor, P. V. Fish, P. O. L. Moreira, P. Zhou, P. Liu, P. Liu, P. Wu, P. Agogo-Mawuli, P. L. Jones, P. Ngoi, P. Toogood, P. Ip, P. Von Hundelshausen, P. H. Lee, R. B. Rowswell-Turner, R. Balaña-Fouce, R. E. O. Rocha, R. V. C. Guido, R. S. Ferreira, R. K. Agrawal, R. K. Harijan, R. Ramachandran, R. Verma, R. K. Singh, R. K. Tiwari, R. Mazitschek, R. K. Koppisetti, R. T. Dame, R. N. Douville, R. C. Austin, R. E. Taylor, R. G. Moore, R. H. Ebright, R. M. Angell, R. Yan, R. Kejriwal, R. A. Batey, R. Blelloch, R. J. Vandenberg, R. J. Hickey, R. J. Kelm, R. J. Lake, R. K. Bradley, R. M. Blumenthal, R. Solano, R. M. Gierse, R. E. Viola, R. R. McCarthy, R. M. Reguera, R. V. Uribe, R. L. Do Monte-Neto, R. Gorgoglione, R. T. Cullinane, S. Katyal, S. Hossain, S. Phadke, S. A. Shelburne, S. E. Geden, S. Johannsen, S. Wazir, S. Legare, S. M. Landfear, S. K. Radhakrishnan, S. Ammendola, S. Dzhumaev, S.-Y. Seo, S. Li, S. Zhou, S. Chu, S. Chauhan, S. Maruta, S. R. Ashkar, S.-L. Shyng, S. G. Conticello, S. Buroni, S. Garavaglia, S. J. White, S. Zhu, S. Tsimbalyuk, S. H. Chadni, S. Y. Byun, S. Park, S. Q. Xu, S. Banerjee, S. Zahler, S. Espinoza, S. Gustincich, S. Sainas, S. L. Celano, S. J. Capuzzi, S. N. Waggoner, S. Poirier, S. H. Olson, S. O. Marx, S. R. Van Doren, S. Sarilla, S. M. Brady-Kalnay, S. Dallman, S. M. Azeem, T. Teramoto, T. Mehlman, T. Swart, T. Abaffy, T. Akopian, T. Haikarainen, T. L. Moreda, T. Ikegami, T. R. Teixeira, T. D. Jayasinghe, T. H. Gillingwater, T. Kampourakis, T. I. Richardson, T. J. Herdendorf, T. J. Kotzé, T. R. O'Meara, T. W. Corson, T. Hermle, T. H. Ogunwa, T. Lan, T. Su, T. Banjo, T. A. O'Mara, T. Chou, T.-F. Chou, U. Baumann, U. R. Desai, V. P. Pai, V. C. Thai, V. Tandon, V. Banerji, V. L. Robinson, V. Gunasekharan, V. Namasivayam, V. F. M. Segers, V. Maranda, V. Dolce, V. G. Maltarollo, V. C. Scoffone, V. A. Woods, V. P. Ronchi, V. Van Hung Le, W. B. Clayton, W. T. Lowther, W. A. Houry, W. Li, W. Tang, W. Zhang, W. C. Van Voorhis, W. A. Donaldson, W. C. Hahn, W. G. Kerr, W. H. Gerwick, W. J. Bradshaw, W. E. Foong, X. Blanchet, X. Wu, X. Lu, X. Qi, X. Xu, X. Yu, X. Qin, X. Wang, X. Yuan, X. Zhang, Y. J. Zhang, Y. Hu, Y. A. Aldhamen, Y. Chen, Y. Li, Y. Sun, Y. Zhu, Y. K. Gupta, Y. Pérez-Pertejo, Y. Li, Y. Tang, Y. He, Y.-C. Tse-Dinh, Y. A. Sidorova, Y. Yen, Y. Li, Z. J. Frangos, Z. Chung, Z. Su, Z. Wang, Z. Zhang, Z. Liu, Z. Inde, Z. Artía and A. Heifets, Sci. Rep., 2024, 14, 7526 CrossRef CAS PubMed.
- G. Ghislat, S. Hernandez-Hernandez, C. Piyawajanusorn and P. J. Ballester, Expert Opin. Drug Discovery, 2024, 19, 1297–1307 CrossRef CAS PubMed.
- E. J. Corey and W. T. Wipke, Science, 1969, 166, 178–192 CrossRef CAS PubMed.
- E. J. Corey, W. T. Wipke, R. D. Cramer III and W. J. Howe, J. Am. Chem. Soc., 1972, 94, 421–430 CrossRef CAS.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef PubMed.
- F. Hastedt, R. M. Bailey, K. Hellgardt, S. N. Yaliraki, E. A. D. R. Chanona and D. Zhang, Digital Discovery, 2024, 3, 1194–1212 RSC.
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, ACS Cent. Sci., 2017, 3, 1103–1113 CrossRef CAS PubMed.
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2017, 3, 1237–1245 CrossRef CAS PubMed.
- M. H. S. Segler and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed.
- B. Chen, C. Li, H. Dai and L. Song, The 37th International Conference on Machine Learning (ICML 2020), 2020.
- M. Pasquini and M. Stenta, J. Chem. Inf. Model., 2024, 64, 1765–1771 CrossRef CAS PubMed.
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, J. Chem. Inf. Model., 2018, 58, 252–261 CrossRef CAS PubMed.
- M. H. Segler and M. P. Waller, Chem. – Eur. J., 2017, 23, 5966–5971 CrossRef CAS PubMed.
- H. Dai, C. Li, C. Coley, B. Dai and L. Song, Advances in Neural Information Processing Systems, 2019, pp. 8870–8880 Search PubMed.
- P. Seidl, P. Renz, N. Dyubankova, P. Neves, J. Verhoeven, J. K. Wegner, M. Segler, S. Hochreiter and G. Klambauer, J. Chem. Inf. Model., 2022, 62, 2111–2120 CrossRef CAS PubMed.
- S. Xie, R. Yan, J. Guo, Y. Xia, L. Wu and T. Qin, Proceedings of the Thirty-Seventh AAAI Conference on Artificial Intelligence and Thirty-Fifth Conference on Innovative Applications of Artificial Intelligence and Thirteenth Symposium on Educational Advances in Artificial Intelligence, 2023.
- S. Chen and Y. Jung, JACS Au, 2021, 1, 1612–1620 CrossRef CAS PubMed.
- C. W. Coley, W. H. Green and K. F. Jensen, J. Chem. Inf. Model., 2019, 59, 2529–2537 CrossRef CAS PubMed.
- C. Yan, P. Zhao, C. Lu, Y. Yu and J. Huang, Biomolecules, 2022, 12, 1325 CrossRef CAS PubMed.
- Y. Shee, H. Li, P. Zhang, A. M. Nikolic, W. Lu, H. R. Kelly, V. Manee, S. Sreekumar, F. G. Buono, J. J. Song, T. R. Newhouse and V. S. Batista, Nat. Commun., 2024, 15, 7818 CrossRef CAS PubMed.
- P. Gaiński, M. Koziarski, K. Maziarz, M. Segler, J. Tabor and M. Śmieja, RetroGFN: Diverse and Feasible Retrosynthesis using GFlowNets, arXiv, 2024, preprint DOI:10.48550/arXiv.2406.18739.
- C. Yan, Q. Ding, P. Zhao, S. Zheng, J. Yang, Y. Yu and J. Huang, Proceedings of the 34th International Conference on Neural Information Processing Systems, Red Hook, NY, USA, 2020.
- C. Shi, M. Xu, H. Guo, M. Zhang and J. Tang, Proceedings of the 37th International Conference on Machine Learning, 2020-03-28, 2020.
- X. Wang, Y. Li, J. Qiu, G. Chen, H. Liu, B. Liao, C.-Y. Hsieh and X. Yao, Chem. Eng. J., 2021, 420, 129845 CrossRef CAS.
- Z. Chen, O. R. Ayinde, J. R. Fuchs, H. Sun and X. Ning, Commun. Chem., 2023, 6, 102 CrossRef PubMed.
- Y. Wang, C. Pang, Y. Wang, J. Jin, J. Zhang, X. Zeng, R. Su, Q. Zou and L. Wei, Nat. Commun., 2023, 14, 6155 CrossRef CAS PubMed.
- Z. Lan, Z. Zeng, B. Hong, Z. Liu and F. Ma, Pattern Recognit., 2024, 150, 110318 CrossRef.
- F. N. Baker, Z. Chen, D. Adu-Ampratwum and X. Ning, J. Chem. Inf. Model., 2024, 64, 6723–6735 CrossRef CAS PubMed.
- M. Sacha, M. Błaż, P. Byrski, P. D. Dąbrowski-Tumański, M. Chromiński, R. Loska, P. Włodarczyk-Pruszyński and S. Jastrzebski, J. Chem. Inf. Model., 2021, 61, 3273–3284 CrossRef CAS PubMed.
- L. Fang, J. Li, M. Zhao, L. Tan and J.-G. Lou, Nat. Commun., 2023, 14, 2446 CrossRef CAS PubMed.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, ACS Central Sci., 2019, 5, 1572–1583 CrossRef CAS PubMed.
- I. V. Tetko, P. Karpov, R. Van Deursen and G. Godin, Nat. Commun., 2020, 11, 5575 CrossRef CAS PubMed.
- R. Irwin, S. Dimitriadis, J. He and E. J. Bjerrum, Mach. Learn.: Sci. Technol., 2022, 3, 015022 Search PubMed.
- Z. Zhong, J. Song, Z. Feng, T. Liu, L. Jia, S. Yao, M. Wu, T. Hou and M. Song, Chem. Sci., 2022, 13, 9023–9034 RSC.
- A. Toniato, A. C. Vaucher, P. Schwaller and T. Laino, Digital Discovery, 2023, 2, 489–501 RSC.
- S.-W. Seo, Y. Y. Song, J. Y. Yang, S. Bae, H. Lee, J. Shin, S. J. Hwang and E. Yang, Proceedings of the AAAI Conference on Artificial Intelligence, 2021, pp. 531–539.
- Z. Tu and C. W. Coley, J. Chem. Inf. Model., 2022, 62, 3503–3513 CrossRef CAS PubMed.
- K. Zeng, B. Yang, X. Zhao, Y. Zhang, F. Nie, X. Yang, Y. Jin and Y. Xu, J. Cheminf., 2024, 16, 80 Search PubMed.
- P. Schwaller, R. Petraglia, V. Zullo, V. H. Nair, R. A. Haeuselmann, R. Pisoni, C. Bekas, A. Iuliano and T. Laino, Chem. Sci., 2020, 11, 3316–3325 RSC.
- F. Zipoli, Z. Ayadi, P. Schwaller, T. Laino and A. C. Vaucher, Mach. Learn.: Sci. Technol., 2024, 5, 025071 Search PubMed.
- P. Schwaller, A. C. Vaucher, T. Laino and J.-L. Reymond, Mach. Learn.: Sci. Technol., 2021, 2, 015016 Search PubMed.
- H. Gao, T. J. Struble, C. W. Coley, Y. Wang, W. H. Green and K. F. Jensen, ACS Central Sci., 2018, 4, 1465–1476 CrossRef CAS PubMed.
- J. Lu and Y. Zhang, J. Chem. Inf. Model., 2022, 62, 1376–1387 CrossRef CAS PubMed.
- S. Genheden, A. Thakkar, V. Chadimová, J.-L. Reymond, O. Engkvist and E. Bjerrum, J. Cheminf., 2020, 12, 70 Search PubMed.
- S. Ishida, K. Terayama, R. Kojima, K. Takasu and Y. Okuno, J. Chem. Inf. Model., 2022, 62, 1357–1367 CrossRef CAS PubMed.
- S. Hong, H. H. Zhuo, K. Jin, G. Shao and Z. Zhou, Commun. Chem., 2023, 6, 1–14 CrossRef PubMed.
- P. Han, P. Zhao, C. Lu, J. Huang, J. Wu, S. Shang, B. Yao and X. Zhang, Proceedings of the AAAI Conference on Artificial Intelligence, 2022, pp. 4014–4021.
- S. Xie, R. Yan, P. Han, Y. Xia, L. Wu, C. Guo, B. Yang and T. Qin, Proceedings of the 28th ACM SIGKDD Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 2022, pp. 2120–2129.
- D. Kreutter and J.-L. Reymond, Chem. Sci., 2023, 14, 9959–9969 RSC.
- D. Zhao, S. Tu and L. Xu, Commun. Chem., 2024, 7, 1–12 CrossRef PubMed.
- J. S. Schreck, C. W. Coley and K. J. M. Bishop, ACS Cent. Sci., 2019, 5, 970–981 CrossRef CAS PubMed.
- G. Liu, D. Xue, S. Xie, Y. Xia, A. Tripp, K. Maziarz, M. Segler, T. Qin, Z. Zhang and T.-Y. Liu, Proceedings of the 40th International Conference on Machine Learning, 2023, pp. 22266–22276.
- Y. Yu, Y. Wei, K. Kuang, Z. Huang, H. Yao and F. Wu, Advances in Neural Information Processing Systems, 2022, pp. 10257–10268 Search PubMed.
- P. Ertl and A. Schuffenhauer, J. Cheminf., 2009, 1, 8 Search PubMed.
- K. Yu, J. Roh, Z. Li, W. Gao, R. Wang and C. W. Coley, Double-Ended Synthesis Planning with Goal-Constrained Bidirectional Search, arXiv, 2024, preprint DOI:10.48550/arXiv.2407.06334.
- A. Tripp, K. Maziarz, S. Lewis, M. Segler and J. M. Hernández-Lobato, Retro-fallback: retrosynthetic planning in an uncertain world, arXiv, 2024, preprint DOI:10.48550/arXiv.2310.09270.
- F. Zipoli, C. Baldassari, M. Manica, J. Born and T. Laino, npj Comput. Mater., 2024, 10, 1–14 CrossRef.
- L. Saigiridharan, A. K. Hassen, H. Lai, P. Torren-Peraire, O. Engkvist and S. Genheden, J. Cheminf., 2024, 16, 57 Search PubMed.
- ASKCOS MIT, https://askcos.mit.edu.
- IBM RXN for Chemistry, https://rxn.app.accelerate.science.
- K. Maziarz, A. Tripp, G. Liu, M. Stanley, S. Xie, P. Gaiński, P. Seidl and M. H. S. Segler, Faraday Discuss., 2024, 256, 568–586 RSC.
- N. Ree, A. H. Göller and J. H. Jensen, Digital Discovery, 2024, 3, 347–354 RSC.
- J. F. Joung, M. H. Fong, J. Roh, Z. Tu, J. Bradshaw and C. W. Coley, Angew. Chem., Int. Ed., 2024, 63, e202411296 CAS.
- P. Torren-Peraire, A. K. Hassen, S. Genheden, J. Verhoeven, D.-A. Clevert, M. Preuss and I. V. Tetko, Digital Discovery, 2024, 558–572 RSC.
- T. Badowski, K. Molga and B. A. Grzybowski, Chem. Sci., 2019, 10, 4640–4651 RSC.
- J. C. Fromer and C. W. Coley, Nat. Comput. Sci., 2024, 1–11 Search PubMed.
- G. Yujia, M. Kabeshov, T. H. D. Le, S. Genheden, G. Bergonzini, O. Engkvist and S. Kaski, A Deep Learning with Expert Augmentation Approach for Route Scoring in Organic Synthesis, 2024 DOI:10.26434/chemrxiv-2024-tp7rh.
- J. M. Weber, Z. Guo, C. Zhang, A. M. Schweidtmann and A. A. Lapkin, Chem. Soc. Rev., 2021, 50, 12013–12036 RSC.
- A. A. Lapkin, P. K. Heer, P.-M. Jacob, M. Hutchby, W. Cunningham, S. D. Bull and M. G. Davidson, Faraday Discuss., 2017, 202, 483–496 RSC.
- S. Zheng, T. Zeng, C. Li, B. Chen, C. W. Coley, Y. Yang and R. Wu, Nat. Commun., 2022, 13, 3342 CrossRef CAS PubMed.
- I. Levin, M. Liu, C. A. Voigt and C. W. Coley, Nat. Commun., 2022, 13, 7747 CrossRef CAS PubMed.
- X. Wang, Y. Qian, H. Gao, C. W. Coley, Y. Mo, R. Barzilay and K. F. Jensen, Chem. Sci., 2020, 11, 10959–10972 RSC.
- G. Gricourt, P. Meyer, T. Duigou and J.-L. Faulon, ACS Synth. Biol., 2024, 13, 2276–2294 CrossRef CAS PubMed.
- B. Mikulak-Klucznik, P. Gołebiowska, A. A. Bayly, O. Popik, T. Klucznik, S. Szymkuć, E. P. Gajewska, P. Dittwald, O. Staszewska-Krajewska, W. Beker, T. Badowski, K. A. Scheidt, K. Molga, J. Mlynarski, M. Mrksich and B. A. Grzybowski, Nature, 2020, 588, 83–88 CrossRef CAS PubMed.
- D. M. Lowe, PhD thesis, University of Cambridge, 2012.
- S. Genheden and E. Bjerrum, Digital Discovery, 2022, 1, 527–539 RSC.
- D. S. Wigh, J. Arrowsmith, A. Pomberger, K. C. Felton and A. A. Lapkin, J. Chem. Inf. Model., 2024, 64, 3790–3798 CrossRef CAS PubMed.
- Reaxys|An Expert-Curated Chemical Database|Elsevier, https://www.reaxys.com/.
- S. M. Kearnes, M. R. Maser, M. Wleklinski, A. Kast, A. G. Doyle, S. D. Dreher, J. M. Hawkins, K. F. Jensen and C. W. Coley, J. Am. Chem. Soc., 2021, 143, 18820–18826 CrossRef CAS PubMed.
- J. Rowley, J. Inf. Sci., 2007, 33, 163–180 CrossRef.
- L. X. Yu, G. Amidon, M. A. Khan, S. W. Hoag, J. Polli, G. K. Raju and J. Woodcock, The AAPS J., 2014, 16, 771–783 CrossRef CAS PubMed.
- B. Jones and D. C. Montgomery, Design of Experiments: A Modern Approach, Wiley, Hoboken, NJ, 2020 Search PubMed.
- S. A. Weissman and N. G. Anderson, Org. Process Res. Dev., 2015, 19, 1605–1633 CrossRef CAS.
- V. Nori, A. Sinibaldi, F. Pesciaioli and A. Carlone, Synthesis, 2022, 4246–4256 CAS.
- J. A. Selekman, J. Qiu, K. Tran, J. Stevens, V. Rosso, E. Simmons, Y. Xiao and J. Janey, Ann. Rev. Chem. Biomol. Eng., 2017, 8, 525–547 CrossRef PubMed.
- N. Vervoort, K. Goossens, M. Baeten and Q. Chen, Anal. Sci. Adv., 2021, 2, 109–127 CrossRef PubMed.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Nature, 2021, 590, 89–96 CrossRef CAS PubMed.
- N. J. Szymanski, Y. Zeng, H. Huo, C. J. Bartel, H. Kim and G. Ceder, Mater. Horiz., 2021, 8, 2169–2198 RSC.
- Y. Wu, A. Walsh and A. M. Ganose, Digital Discovery, 2024, 3, 1086–1100 RSC.
- T. Brown, B. Mann, N. Ryder, M. Subbiah, J. D. Kaplan, P. Dhariwal, A. Neelakantan, P. Shyam, G. Sastry, A. Askell, S. Agarwal, A. Herbert-Voss, G. Krueger, T. Henighan, R. Child, A. Ramesh, D. Ziegler, J. Wu, C. Winter, C. Hesse, M. Chen, E. Sigler, M. Litwin, S. Gray, B. Chess, J. Clark, C. Berner, S. McCandlish, A. Radford, I. Sutskever and D. Amodei, Advances in Neural Information Processing Systems, 2020, pp. 1877–1901 Search PubMed.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, L. Kaiser and I. Polosukhin, Attention Is All You Need, arXiv, 2023, preprint DOI:10.48550/arXiv.1706.03762.
- P. Kumar, Artif. Intell. Rev., 2024, 57, 260 CrossRef.
- A. Radford, Technical Report, Open AI, 2018.
- J. D. M.-W. C. Kenton and L. K. Toutanova, NAACL, 2019, p. 2.
- K. Pipalia, R. Bhadja and M. Shukla, 2020 9th International Conference System Modeling and Advancement in Research Trends (SMART), 2020, pp. 411-415.
- K. Nassiri and M. Akhloufi, Appl. Intell., 2023, 53, 10602–10635 CrossRef.
- H. Y. Koh, J. Ju, M. Liu and S. Pan, ACM Comput. Surv., 2022, 55, 1–35 CrossRef.
- S. Aswani, K. Choudhary, S. Shetty and N. Nur, J. Autonomous Intell., 2024, 7, 1–14 Search PubMed.
- Q. Dong, L. Li, D. Dai, C. Zheng, J. Ma, R. Li, H. Xia, J. Xu, Z. Wu and B. Chang, et al., Proceedings of the 2024 Conference on Empirical Methods in Natural Language Processing, 2024, pp. 1107–1128.
- S. T. Arasteh, T. Han, M. Lotfinia, C. Kuhl, J. N. Kather, D. Truhn and S. Nebelung, Nat. Commun., 2024, 15, 1–12 Search PubMed.
- B. Lund, InfoScience Trends, 2024, 1, 4–7 CrossRef.
- B. Fabian, T. Edlich, H. Gaspar, M. Segler, J. Meyers, M. Fiscato and M. Ahmed, Machine Learning for Molecules Workshop at NeurIPS, 2020 Search PubMed.
- V. Bagal, R. Aggarwal, P. K. Vinod and U. D. Priyakumar, J. Chem. Inf. Model., 2021, 62, 2064–2076 CrossRef PubMed.
- Q. Zhang, K. Ding, T. Lyv, X. Wang, Q. Yin, Y. Zhang, J. Yu, Y. Wang, X. Li, Z. Xiang, K. Feng, X. Zhuang, Z. Wang, M. Qin, M. Zhang, J. Zhang, J. Cui, T. Huang, P. Yan, R. Xu, H. Chen, X. Li, X. Fan, H. Xing and H. Chen, arXiv, 2024, preprint DOI:10.48550/arXiv.2401.14656.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- M. Krenn, Q. Ai, S. Barthel, N. Carson, A. Frei, N. C. Frey, P. Friederich, T. Gaudin, A. A. Gayle, K. M. Jablonka, R. F. Lameiro, D. Lemm, A. Lo, S. M. Moosavi, J. M. Nápoles-Duarte, A. Nigam, R. Pollice, K. Rajan, U. Schatzschneider, P. Schwaller, M. Skreta, B. Smit, F. Strieth-Kalthoff, C. Sun, G. Tom, G. Falk Von Rudorff, A. Wang, A. D. White, A. Young, R. Yu and A. Aspuru-Guzik, Patterns, 2022, 3, 100588 CrossRef CAS PubMed.
- V. Korolev and P. Protsenko, Patterns, 2023, 4, 100803 CrossRef PubMed.
- Z. Xu, X. Lei, M. Ma and Y. Pan, Big Data Mining Anal., 2023, 7, 142–155 Search PubMed.
- N. Fu, L. Wei, Y. Song, Q. Li, R. Xin, S. S. Omee, R. Dong, E. M. D. Siriwardane and J. Hu, Mach. Learn.: Sci. Technol., 2023, 4, 015001 Search PubMed.
- P. Karpov, G. Godin and I. V. Tetko, International Conference on Artificial Neural Networks, 2019, pp. 817–830.
- Y. Wan, C.-Y. Hsieh, B. Liao and S. Zhang, International Conference on Machine Learning, 2022, pp. 22475–22490.
- J. A. Pople and D. L. Beveridge, Approximate Molecular Orbital Theory, McGraw-Hill, New York, 1970 Search PubMed.
- D. L. Cooper, J. Gerratt and M. Raimondi, Advances in chemical physics: ab initio methods in quantum chemistry part 2, 1987, 69, 319–397.
- N. Fei, Z. Lu, Y. Gao, G. Yang, Y. Huo, J. Wen, H. Lu, R. Song, X. Gao and T. Xiang, et al. , Nat. Commun., 2022, 13, 3094 CrossRef CAS PubMed.
- S. Yin, C. Fu, S. Zhao, K. Li, X. Sun, T. Xu and E. Chen, Nat. Sci. Rev., 2024, nwae403 CrossRef PubMed.
- Z. Zhao, B. Chen, J. Li, L. Chen, L. Wen, P. Wang, Z. Zhu, D. Zhang, Y. Li, Z. Dai, X. Chen and K. Yu, Sci. China Inf. Sci., 2024, 67, 67220109 Search PubMed.
- Z. A. Rollins, A. C. Cheng and E. Metwally, J. Cheminf., 2024, 16, 56 Search PubMed.
- S. Sakhinana and V. Runkana, NeurIPS 2023 Workshop on Instruction Tuning and Instruction Following, 2023.
- M. Livne, Z. Miftahutdinov, E. Tutubalina, M. Kuznetsov, D. Polykovskiy, A. Brundyn, A. Jhunjhunwala, A. Costa, A. Aliper and A. Aspuru-Guzik, et al. , Chem. Sci., 2024, 15, 8380–8389 RSC.
- J. G. Meyer, R. J. Urbanowicz, P. C. Martin, K. O'Connor, R. Li, P.-C. Peng, T. J. Bright, N. Tatonetti, K. J. Won and G. Gonzalez-Hernandez, et al. , BioData Mining, 2023, 16, 20 CrossRef PubMed.
- E. Kasneci, K. Seßler, S. Küchemann, M. Bannert, D. Dementieva, F. Fischer, U. Gasser, G. Groh, S. Günnemann and E. Hüllermeier, et al. , Learn. Individual Differences, 2023, 103, 102274 CrossRef.
- L. Tang, Z. Sun, B. Idnay, J. G. Nestor, A. Soroush, P. A. Elias, Z. Xu, Y. Ding, G. Durrett and J. F. Rousseau, et al. , npj Digital Med., 2023, 6, 158 CrossRef PubMed.
- T. Zhang, F. Ladhak, E. Durmus, P. Liang, K. McKeown and T. B. Hashimoto, Trans. Assoc. Comput. Ling., 2024, 12, 39–57 Search PubMed.
- F. Petroni, T. Rocktäschel, S. Riedel, P. Lewis, A. Bakhtin, Y. Wu and A. Miller, Proceedings of the 2019 Conference on Empirical Methods in Natural Language Processing and the 9th International Joint Conference on Natural Language Processing (EMNLP-IJCNLP), 2019, pp. 2463–2473.
- Y. Zhu, H. Yuan, S. Wang, J. Liu, W. Liu, C. Deng, H. Chen, Z. Liu, Z. Dou and J.-R. Wen, Large Language Models for Information Retrieval: A Survey, arXiv, 2024, preprint DOI:10.48550/arXiv.2308.07107.
- D. Vilar, M. Freitag, C. Cherry, J. Luo, V. Ratnakar and G. Foster, Proceedings of the 61st Annual Meeting of the Association for Computational Linguistics (Volume 1: Long Papers), 2023, pp. 15406–15427.
- X. Hou, Y. Zhao, Y. Liu, Z. Yang, K. Wang, L. Li, X. Luo, D. Lo, J. Grundy and H. Wang, ACM Trans. Software Eng. Methodol., 2023, 33, 1–79 Search PubMed.
- OpenAI, Learning to Reason with Llms, https://openai.com/index/learning-to-reason-with-llms/, 2024.
- K. Valmeekam, M. Marquez, S. Sreedharan and S. Kambhampati, Adv. Neural Inf. Process. Syst., 2023, 36, 75993–76005 Search PubMed.
- K. Valmeekam, M. Marquez, A. Olmo, S. Sreedharan and S. Kambhampati, Adv. Neural Inf. Process. Syst., 2024, 36, 1–13 Search PubMed.
- M. P. Polak and D. Morgan, Nat. Commun., 2024, 15, 1569 CrossRef CAS PubMed.
- J. Lála, O. O'Donoghue, A. Shtedritski, S. Cox, S. G. Rodriques and A. D. White, PaperQA: Retrieval-Augmented Generative Agent for Scientific Research, arXiv, 2023, preprint DOI:10.48550/arXiv.2312.07559.
- N. H. Park, T. J. Callahan, J. L. Hedrick, T. Erdmann and S. Capponi, Leveraging Chemistry Foundation Models to Facilitate Structure Focused Retrieval Augmented Generation in Multi-Agent Workflows for Catalyst and Materials Design, arXiv, 2024, preprint DOI:10.48550/arXiv.2408.11793.
- V. Fan, Y. Qian, A. Wang, A. Wang, C. W. Coley and R. Barzilay, J. Chem. Inf. Model., 2024, 64, 5521–5534 CrossRef CAS PubMed.
- M. C. Swain and J. M. Cole, J. Chem. Inf. Model., 2016, 56, 1894–1904 CrossRef CAS PubMed.
- J. Mavracic, C. J. Court, T. Isazawa, S. R. Elliott and J. M. Cole, J. Chem. Inf. Model., 2021, 61, 4280–4289 CrossRef CAS PubMed.
- M. Jalali, Y. Luo, L. Caulfield, E. Sauter, A. Nefedov and C. Wöll, Mater. Today Commun., 2024, 40, 109801 CrossRef CAS.
- S. Kambhampati, K. Valmeekam, L. Guan, M. Verma, K. Stechly, S. Bhambri, L. Saldyt and A. Murthy, LLMs Can't Plan, But Can Help Planning in LLM-Modulo Frameworks, arXiv, 2024, preprint DOI:10.48550/arXiv.2402.01817.
- T. H. Trinh, Y. Wu, Q. V. Le, H. He and T. Luong, Nature, 2024, 625, 476–482 CrossRef CAS PubMed.
- R. Garnett, Bayesian Optimization, Cambridge University Press, 2023 Search PubMed.
- T. Liu, N. Astorga, N. Seedat and M. van der Schaar, The Twelfth International Conference on Learning Representations, 2024.
- D. A. Boiko, R. MacKnight, B. Kline and G. Gomes, Nature, 2023, 624, 570–578 CrossRef CAS PubMed.
- D. Caramelli, J. M. Granda, S. H. M. Mehr, D. Cambié, A. B. Henson and L. Cronin, ACS Cent. Sci., 2021, 7, 1821–1830 CrossRef CAS PubMed.
- J. M. Granda, L. Donina, V. Dragone, D.-L. Long and L. Cronin, Nature, 2018, 559, 377–381 CrossRef CAS PubMed.
- T. Guo, B. Nan, Z. Liang, Z. Guo, N. Chawla, O. Wiest and X. Zhang, et al. , Adv. Neural Inf. Process. Syst., 2023, 36, 59662–59688 Search PubMed.
- L. Weidinger, J. Uesato, M. Rauh, C. Griffin, P.-S. Huang, J. Mellor, A. Glaese, M. Cheng, B. Balle and A. Kasirzadeh, et al., Proceedings of the 2022 ACM Conference on Fairness, Accountability, and Transparency, 2022, pp. 214–229.
- L. Zhao, C. Edwards and H. Ji, NeurIPS 2023 AI for Science Workshop, 2023.
- W. Zhang, Q. Wang, X. Kong, J. Xiong, S. Ni, D. Cao, B. Niu, M. Chen, Y. Li and R. Zhang, et al. , Chem. Sci., 2024, 15, 10600–10611 RSC.
- M. Cruz-Monteagudo, J. L. Medina-Franco, Y. Pérez-Castillo, O. Nicolotti, M. N. D. Cordeiro and F. Borges, Drug Discovery Today, 2014, 19, 1069–1080 CrossRef CAS PubMed.
- D. Van Tilborg, A. Alenicheva and F. Grisoni, J. Chem. Inf. Model., 2022, 62, 5938–5951 CrossRef CAS PubMed.
- X. Fang, L. Liu, J. Lei, D. He, S. Zhang, J. Zhou, F. Wang, H. Wu and H. Wang, Nat. Mach. Intell., 2022, 4, 127–134 CrossRef.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley and M. Mathea, et al. , J. Chem. Inf. Model., 2019, 59, 3370–3388 CrossRef CAS PubMed.
- E. Heid, K. P. Greenman, Y. Chung, S.-C. Li, D. E. Graff, F. H. Vermeire, H. Wu, W. H. Green and C. J. McGill, J. Chem. Inf. Model., 2023, 64, 9–17 CrossRef PubMed.
- G. Team, R. Anil, S. Borgeaud, J.-B. Alayrac, J. Yu, R. Soricut, J. Schalkwyk, A. M. Dai, A. Hauth and K. Millican, et al., Technical Report, Google Deepmind, 2023.
- OpenAI, Technical Report, OpenAI, 2023.
- S. Wankowicz, P. Beltrao, B. Cravatt, R. Dunbrack, A. Gitter, K. Lindorff-Larsen, S. Ovchinnikov, N. Polizzi, B. Shoichet and J. Fraser, AlphaFold3 Transparency and Reproducibility, 2024, https://zenodo.org/doi/10.5281/zenodo.11391920.
- S. Shankar and R. N. Zare, Nat. Mach. Intell., 2022, 4, 314–315 CrossRef.
- F. Urbina, F. Lentzos, C. Invernizzi and S. Ekins, J. Chem. Inf. Model., 2023, 63, 691–694 CrossRef CAS PubMed.
- J. Snoek, H. Larochelle and R. P. Adams, Advances in Neural Information Processing Systems, 2012 Search PubMed.
- B. Shahriari, K. Swersky, Z. Wang, R. P. Adams and N. de Freitas, Proc. IEEE, 2016, 104, 148–175 Search PubMed.
- B. Settles, Active Learning Literature Survey, University of wisconsin-Madison department of computer sciences technical Report, 2009.
- C. K. Williams and C. E. Rasmussen, Gaussian Processes for Machine Learning, MIT Press, Cambridge, MA, 2006, vol. 2 Search PubMed.
- S. S. Rosa, D. Nunes, L. Antunes, D. M. F. Prazeres, M. P. C. Marques and A. M. Azevedo, Biotechnol. Bioeng., 2022, 119, 3127–3139 CrossRef CAS PubMed.
- B. J. Reizman and K. F. Jensen, Chem. Commun., 2015, 51, 13290–13293 RSC.
- C. J. Taylor, K. C. Felton, D. Wigh, M. I. Jeraal, R. Grainger, G. Chessari, C. N. Johnson and A. A. Lapkin, ACS Central Sci., 2023, 9, 957–968 CrossRef CAS PubMed.
- B. Ranković, R.-R. Griffiths, H. B. Moss and P. Schwaller, Digital Discovery, 2023, 3, 654–666 RSC.
- S. Daulton, X. Wan, D. Eriksson, M. Balandat, M. A. Osborne and E. Bakshy, Adv. Neural Inf. Process. Syst., 2022, 35, 12760–12774 Search PubMed.
- F. Häse, M. Aldeghi, R. J. Hickman, L. M. Roch and A. Aspuru-Guzik, Appl. Phys. Rev., 2021, 8, 1–17 Search PubMed.
- J. Zhang, N. Sugisawa, K. C. Felton, S. Fuse and A. A. Lapkin, React. Chem. Eng., 2024, 9, 706–712 RSC.
- A. Tran, J. Tranchida, T. Wildey and A. P. Thompson, J. Chem. Phys., 2020, 153, 074705 CrossRef CAS PubMed.
- S. Park, J. Na, M. Kim and J. M. Lee, Comput. Chem. Eng., 2018, 119, 25–37 CrossRef CAS.
- R. Sedgwick, J. P. Goertz, M. M. Stevens, R. Misener and M. van der Wilk, Biotechnol. Bioeng., 2024, 122, 189–210 CrossRef PubMed.
- J. A. Valeri, K. M. Collins, P. Ramesh, M. A. Alcantar, B. A. Lepe, T. K. Lu and D. M. Camacho, Nat. Commun., 2020, 11, 5058 CrossRef CAS PubMed.
- G. Chen, Z. Shen, A. Iyer, U. F. Ghumman, S. Tang, J. Bi, W. Chen and Y. Li, Polymers, 2020, 12, 163 CrossRef CAS PubMed.
- P. A. Romero, A. Krause and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E193–E201 CrossRef CAS PubMed.
- C. R. Freschlin, S. A. Fahlberg and P. A. Romero, Curr. Opin. Biotechnol., 2022, 75, 102713 CrossRef CAS PubMed.
- J. T. Rapp, B. J. Bremer and P. A. Romero, Nat. Chem. Eng., 2024, 1, 97–107 CrossRef PubMed.
- A. Khan, A. I. Cowen-Rivers, A. Grosnit, D.-G.-X. Deik, P. A. Robert, V. Greiff, E. Smorodina, P. Rawat, R. Akbar, K. Dreczkowski, R. Tutunov, D. Bou-Ammar, J. Wang, A. Storkey and H. Bou-Ammar, Cell Rep. Methods, 2023, 3, 100374 CrossRef CAS PubMed.
- A. M. Maraval, M. Zimmer, A. Grosnit and H. B. Ammar, Thirty-seventh Conference on Neural Information Processing Systems, 2023.
- S. Stanton, W. Maddox, N. Gruver, P. Maffettone, E. Delaney, P. Greenside and A. G. Wilson, Proceedings of the 39th International Conference on Machine Learning, 2022, pp. 20459–20478.
- H. Moss, D. Leslie, D. Beck, J. González and P. Rayson, Advances in Neural Information Processing Systems, 2020, pp. 15476–15486 Search PubMed.
- M. González-Duque, R. Michael, S. Bartels, Y. Zainchkovskyy, S. Hauberg and W. Boomsma, A survey and benchmark of high-dimensional Bayesian optimization of discrete sequences, arXiv, 2024, preprint DOI:10.48550/arXiv.2406.04739.
- J. A. Manson, T. W. Chamberlain and R. A. Bourne, J. Global Optim., 2021, 80, 865–886 CrossRef.
- O. J. Kershaw, A. D. Clayton, J. A. Manson, A. Barthelme, J. Pavey, P. Peach, J. Mustakis, R. M. Howard, T. W. Chamberlain, N. J. Warren and R. A. Bourne, Chem. Eng. J., 2023, 451, 138443 CrossRef CAS.
- M. A. Álvarez, L. Rosasco and N. D. Lawrence, Found. Trends Mach. Learn., 2012, 4, 195–266 CrossRef.
- Z. Dai, M. Álvarez and N. Lawrence, Advances in Neural Information Processing Systems, 2017 Search PubMed.
- C. Hutter, M. von Stosch, M. N. Cruz Bournazou and A. Butté, Biotechnol. Bioeng., 2021, 118, 4389–4401 CrossRef CAS PubMed.
- P. Luong, S. Gupta, D. Nguyen, S. Rana and S. Venkatesh, AI 2019: Advances in Artificial Intelligence, Cham, 2019, pp. 473–484.
- J. Hill, A. Linero and J. Murray, Annu. Rev. Stat. Appl., 2020, 7, 251–278 CrossRef.
- H. A. Chipman, E. I. George and R. E. McCulloch, Ann. Appl. Stat., 2010, 4, 266–298 Search PubMed.
- C.-Y. Liu, S. Ye, M. Li and T. P. Senftle, J. Chem. Phys., 2022, 156, 164105 CrossRef CAS PubMed.
- M. Maia, K. Murphy and A. C. Parnell, Comput. Stat. Data Anal., 2024, 190, 107858 CrossRef.
- M. Balog, B. Lakshminarayanan, Z. Ghahramani, D. M. Roy and Y. W. Teh, The Mondrian Kernel, arXiv, 2016, preprint DOI:10.48550/arXiv.1606.05241.
- L. Wang, R. Fonseca and Y. Tian, Advances in Neural Information Processing Systems, 2020, pp. 19511–19522 Search PubMed.
- A. Davies and Z. Ghahramani, The Random Forest Kernel and other kernels for big data from random partitions, arXiv, 2014, preprint DOI:10.48550/arXiv.1402.4293.
- T. Boyne, J. P. Folch, R. M. Lee, B. Shafei and R. Misener, BARK: A Fully Bayesian Tree Kernel for Black-box Optimization, arXiv, 2025, preprint DOI:10.48550/arXiv.2503.05574.
- H. Lodhi, C. Saunders, J. Shawe-Taylor, N. Cristianini and C. Watkins, J. Mach. Learn. Res., 2002, 2, 419–444 Search PubMed.
- N. Cancedda, E. Gaussier, C. Goutte and J. M. Renderssss, J. Mach. Learn. Res., 2003, 3, 1059–1082 Search PubMed.
- C. Leslie and R. Kuang, J. Mach. Learn. Res., 2004, 5, 1435–1455 Search PubMed.
- Y. Xie, S. Zhang, J. Qing, R. Misener and C. Tsay, BoGrape: Bayesian optimization over graphs with shortest-path encoded, arXiv, 2025, preprint DOI:10.48550/arXiv.2503.05642.
- T. T. Tanimoto, An Elementary Mathematical Theory of Classification and Prediction, International Business Machines Corporation, 1958 Search PubMed.
- A. Tripp, S. Bacallado, S. Singh and J. M. Hernández-Lobato, Adv. Neural Inf. Process. Syst., 2024, 36, 1–31 Search PubMed.
- K. Korovina, S. Xu, K. Kandasamy, W. Neiswanger, B. Poczos, J. Schneider and E. Xing, Proceedings of the Twenty Third International Conference on Artificial Intelligence and Statistics, 2020, pp. 3393–3403.
- N. Brandes, D. Ofer, Y. Peleg, N. Rappoport and M. Linial, Bioinformatics, 2022, 38, 2102–2110 CrossRef CAS PubMed.
- R.-R. Griffiths, L. Klarner, H. Moss, A. Ravuri, S. Truong, Y. Du, S. Stanton, G. Tom, B. Rankovic and A. Jamasb, et al. , Adv. Neural Inf. Process. Syst., 2024, 36, 1–16 Search PubMed.
- N. Maus, Y. Zeng, D. A. Anderson, P. Maffettone, A. Solomon, P. Greenside, O. Bastani and J. R. Gardner, Inverse Protein Folding Using Deep Bayesian Optimization, arXiv, 2023, preprint DOI:10.48550/arXiv.2305.18089.
- N. Maus, H. Jones, J. Moore, M. J. Kusner, J. Bradshaw and J. Gardner, Adv. Neural Inf. Process. Syst., 2022, 35, 34505–34518 Search PubMed.
- A. Deshwal and J. Doppa, Advances in Neural Information Processing Systems, 2021, pp. 8185–8200 Search PubMed.
- Q. Zhang, P. Chien, Q. Liu, L. Xu and Y. Hong, J. Quality Technol., 2021, 53, 410–420 CrossRef.
- A. Iyer, Y. Zhang, A. Prasad, P. Gupta, S. Tao, Y. Wang, P. Prabhune, L. S. Schadler, L. C. Brinson and W. Chen, Mol. Syst. Des. Eng., 2020, 5, 1376–1390 RSC.
- C. Ma, A. Leroy and M. Alvarez, Latent Variable Multi-output Gaussian Processes for Hierarchical Datasets, arXiv, 2023, preprint DOI:10.48550/arXiv.2308.16822.
- S. Chithrananda, G. Grand and B. Ramsundar, ChemBERTa: Large-scale Self-Supervised Pretraining for Molecular Property Prediction, 2020 Search PubMed.
- E. C. Alley, G. Khimulya, S. Biswas, M. AlQuraishi and G. M. Church, Nat. Methods, 2019, 16, 1315–1322 CrossRef CAS PubMed.
- C. Oh, J. Tomczak, E. Gavves and M. Welling, Advances in Neural Information Processing Systems, 2019 Search PubMed.
- A. Deshwal, S. Belakaria and J. R. Doppa, International Conference on Machine Learning, 2021, pp. 2632–2643.
- M. Zhu, A. Mroz, L. Gui, K. E. Jelfs, A. Bemporad, E. A. del Río Chanona and Y. S. Lee, Digital Discovery, 2024, 3, 2589–2606 RSC.
- A. O'Hagan, J. Stat. Plann. Inference, 1991, 29, 245–260 CrossRef.
- E. C. Garrido-Merchán and D. Hernández-Lobato, Neurocomputing, 2020, 380, 20–35 CrossRef.
- D. Nguyen, S. Gupta, S. Rana, A. Shilton and S. Venkatesh, Proc. AAAI Conf. Artif., Intell., 2020, 34, 5256–5263 Search PubMed.
- B. Ru, A. Alvi, V. Nguyen, M. A. Osborne and S. Roberts, International Conference on Machine Learning, 2020, pp. 8276–8285 Search PubMed.
- X. Wan, V. Nguyen, H. Ha, B. Ru, C. Lu and M. A. Osborne, Think Global and Act Local: Bayesian Optimisation over High-Dimensional Categorical and Mixed Search Spaces, arXiv, 2021, preprint, https://arxiv.org/abs/2102.07188.
- X. Ma and M. Blaschko, Proceedings of the Twenty Third International Conference on Artificial Intelligence and Statistics, 2020, pp. 1015–1025.
- J.-C. Lévesque, A. Durand, C. Gagné and R. Sabourin, 2017 International Joint Conference on Neural Networks (IJCNN), 2017, pp. 286–293.
- A. Thebelt, C. Tsay, R. M. Lee, N. Sudermann-Merx, D. Walz, B. Shafei and R. Misener, 36th Conference on Neural Information Processing Systems, 2022.
- M. Balandat, B. Karrer, D. Jiang, S. Daulton, B. Letham, A. G. Wilson and E. Bakshy, Adv. Neural Inf. Process. Syst., 2020, 33, 21524–21538 Search PubMed.
- J. P. Dürholt, T. S. Asche, J. Kleinekorte, G. Mancino-Ball, B. Schiller, S. Sung, J. Keupp, A. Osburg, T. Boyne, R. Misener, R. Eldred, W. S. Costa, C. Kappatou, R. M. Lee, D. Linzner, D. Walz, N. Wulkow and B. Shafei, BoFire: Bayesian Optimization Framework Intended for Real Experiments, arXiv, 2024, preprint DOI:10.48550/arXiv.2408.05040.
- A. H. M. Fitzner, A. Šošić and A. Lee, BayBE.
- A. M. Mroz, P. N. Toka, E. A. Del Río Chanona and K. E. Jelfs, Faraday Discuss., 2024, 256, 221–234 RSC.
- R. Hickman, M. Sim, S. Pablo-García, I. Woolhouse, H. Hao, Z. Bao, P. Bannigan, C. Allen, M. Aldeghi and A. Aspuru-Guzik, Atlas: A Brain for Self-driving Laboratories, 2023 DOI:10.26434/chemrxiv-2023-8nrxx.
- R. Hickman, M. Aldeghi and A. Aspuru-Guzik, Anubis: Bayesian optimization with unknown feasibility constraints for scientific experimentation, 2023 DOI:10.26434/chemrxiv-2023-s5qnw.
- K. Kandasamy, K. R. Vysyaraju, W. Neiswanger, B. Paria, C. R. Collins, J. Schneider, B. Poczos and E. P. Xing, J. Mach. Learn. Res., 2020, 21, 1–27 Search PubMed.
- J. Qing, B. D. Langdon, R. M. Lee, B. Shafei, M. van der Wilk, C. Tsay and R. Misener, System-Aware Neural ODE Processes for Few-Shot Bayesian Optimization, arXiv, 2024, preprint DOI:10.48550/arXiv.2406.02352.
- T. Gao and X. Bai, J. Intell. Rob. Syst., 2022, 105, 91 CrossRef.
- A. Dhir, R. Sedgwick, A. Kori, B. Glocker and M. van der Wilk, Continuous Bayesian Model Selection for Multivariate Causal Discovery, arXiv, 2024, preprint DOI:10.48550/arXiv.2411.10154.
- V. Aglietti, X. Lu, A. Paleyes and J. González, International Conference on Artificial Intelligence and Statistics, 2020, pp. 3155–3164.
- A. Hägele, J. Rothfuss, L. Lorch, V. R. Somnath, B. Schölkopf and A. Krause, AISTATS, 2023.
- A. Angelopoulos, J. F. Cahoon and R. Alterovitz, Sci. Rob., 2024, 9, eadm6991 CrossRef PubMed.
- R. W. Epps, A. A. Volk, M. Y. Ibrahim and M. Abolhasani, Chem, 2021, 7, 2541–2545 CAS.
- F. Kong, L. Yuan, Y. F. Zheng and W. Chen, J. Lab. Autom., 2012, 17, 169–185 CrossRef CAS PubMed.
- K. Thurow, Anal. Bioanal. Chem., 2023, 415, 5057–5066 CrossRef CAS PubMed.
- A. Slattery, Z. Wen, P. Tenblad, J. Sanjosé-Orduna, D. Pintossi, T. den Hartog and T. Noël, Science, 2024, 383, eadj1817 CrossRef CAS PubMed.
- A. A. Volk, R. W. Epps, D. T. Yonemoto, B. S. Masters, F. N. Castellano, K. G. Reyes and M. Abolhasani, Nat. Commun., 2023, 14, 1403 CrossRef CAS PubMed.
- K. Darvish, M. Skreta, Y. Zhao, N. Yoshikawa, S. Som, M. Bogdanovic, Y. Cao, H. Hao, H. Xu, A. Aspuru-Guzik, A. Garg and F. Shkurti, arXiv, 2025 DOI:10.48550/arXiv.2401.06949.
- Q. Zhu, F. Zhang, Y. Huang, H. Xiao, L. Zhao, X. Zhang, T. Song, X. Tang, X. Li and G. He, et al. , Natl. Sci. Rev., 2022, 9, nwac190 CrossRef CAS PubMed.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li and R. Clowes, et al. , Nature, 2020, 583, 237–241 CrossRef CAS PubMed.
- A. Faiña, B. Nejati and K. Stoy, Appl. Sci., 2020, 10, 814 CrossRef.
- J. T. Yik, L. Zhang, J. Sjölund, X. Hou, P. H. Svensson, K. Edström and E. J. Berg, Digital Discovery, 2023, 2, 799–808 RSC.
- Y. Ruan, C. Lu, N. Xu, Y. He, Y. Chen, J. Zhang, J. Xuan, J. Pan, Q. Fang, H. Gao, X. Shen, N. Ye, Q. Zhang and Y. Mo, Nat. Commun., 2024, 15, 1–16 Search PubMed.
- A. M. Lunt, H. Fakhruldeen, G. Pizzuto, L. Longley, A. White, N. Rankin, R. Clowes, B. Alston, L. Gigli and G. M. Day, et al. , Chem. Sci., 2024, 15, 2456–2463 RSC.
- X. Chu, H. Fleischer, N. Stoll, M. Klos and K. Thurow, 2015 IEEE International Instrumentation and Measurement Technology Conference (I2MTC) Proceedings, 2015, pp. 500–504.
- M. Seifrid, R. Pollice, A. Aguilar-Granda, Z. Morgan Chan, K. Hotta, C. T. Ser, J. Vestfrid, T. C. Wu and A. Aspuru-Guzik, Acc. Chem. Res., 2022, 55, 2454–2466 CrossRef CAS PubMed.
- M. M. Flores-Leonar, L. M. Mejía-Mendoza, A. Aguilar-Granda, B. Sanchez-Lengeling, H. Tribukait, C. Amador-Bedolla and A. Aspuru-Guzik, Curr. Opin. Green Sustainable Chem., 2020, 25, 100370 CrossRef.
- S. Peschisolido, Lab 4.0: Making Digital Transformation Work for Your Laboratory|Technology Networks Search PubMed.
- T. Perraudin, Internet of Laboratory Things Makes Life Better at Work, 2020, https://www.paperlesslabacademy.com/2020/02/06/internet-of-laboratory-things/.
- K. Williams, E. Bilsland, A. Sparkes, W. Aubrey, M. Young, L. N. Soldatova, K. De Grave, J. Ramon, M. De Clare and W. Sirawaraporn, et al. , J. R. Soc., Interface, 2015, 12, 20141289 CrossRef PubMed.
- J. R. Deneault, J. Chang, J. Myung, D. Hooper, A. Armstrong, M. Pitt and B. Maruyama, MRS Bull., 2021, 46, 566–575 CrossRef.
- E. Stach, B. DeCost, A. G. Kusne, J. Hattrick-Simpers, K. A. Brown, K. G. Reyes, J. Schrier, S. Billinge, T. Buonassisi and I. Foster, et al. , Matter, 2021, 4, 2702–2726 CrossRef.
- S. Lo, S. G. Baird, J. Schrier, B. Blaiszik, N. Carson, I. Foster, A. Aguilar-Granda, S. V. Kalinin, B. Maruyama and M. Politi, et al. , Digital Discovery, 2024, 3, 842–868 RSC.
- A. M. Schweidtmann, A. D. Clayton, N. Holmes, E. Bradford, R. A. Bourne and A. A. Lapkin, Chem. Eng. J., 2018, 352, 277–282 CrossRef CAS.
- N. Cherkasov, Y. Bai, A. J. Expósito and E. V. Rebrov, React. Chem. Eng., 2018, 3, 769–780 RSC.
- M. Abolhasani and E. Kumacheva, Nat. Synth., 2023, 2, 483–492 CrossRef CAS.
- R. Rauschen, M. Guy, J. E. Hein and L. Cronin, Nat. Synth., 2024, 3, 488–496 CrossRef CAS.
- G. Wuitschik, V. Jost, T. Schindler and M. Jakubik, Org. Process Res. Dev., 2024, 28, 2875–2884 CrossRef CAS.
- H. Hysmith, E. Foadian, S. P. Padhy, S. V. Kalinin, R. G. Moore, O. S. Ovchinnikova and M. Ahmadi, Digital Discovery, 2024, 3, 621–636 RSC.
- M. Rubens, J. H. Vrijsen, J. Laun and T. Junkers, Angew. Chem., Int. Ed., 2019, 58, 3183–3187 CrossRef CAS PubMed.
- J. Lee, P. Mulay, M. J. Tamasi, J. Yeow, M. M. Stevens and A. J. Gormley, Digital Discovery, 2023, 2, 219–233 RSC.
- N. L. Bell, F. Boser, A. Bubliauskas, D. R. Willcox, V. S. Luna and L. Cronin, Nat. Chem. Eng., 2024, 1, 180–189 CrossRef.
- A. R. Basford, S. K. Bennett, M. Xiao, L. Turcani, J. Allen, K. E. Jelfs and R. L. Greenaway, Chem. Sci., 2024, 15, 6331–6348 RSC.
- A. Basford, A. H. Bernardino, P. Teeuwen, B. Egleston, J. Humphreys, K. Jelfs, J. Nitschke, I. Riddell and R. Greenaway, ChemRxiv, 2024 DOI:10.26434/chemrxiv-2024-hl427-v4.
- M. Seifrid, F. Strieth-Kalthoff, M. Haddadnia, T. C. Wu, E. Alca, L. Bodo, S. Arellano-Rubach, N. Yoshikawa, M. Skreta and R. Keunen, et al. , Digital Discovery, 2024, 3, 1319–1326 RSC.
- N. Yoshikawa, K. Darvish, M. G. Vakili, A. Garg and A. Aspuru-Guzik, Digital Discovery, 2023, 2, 1745–1751 RSC.
- C. Arnold, Nature, 2022, 606, 612–613 CrossRef CAS PubMed.
- H. Fakhruldeen, G. Pizzuto, J. Glowacki and A. I. Cooper, 2022 International Conference on Robotics and Automation (ICRA), 2022, pp. 6013-6019.
- D. Jones, C. Snider, A. Nassehi, J. Yon and B. Hicks, CIRP J. Manuf. Sci. Technol., 2020, 29, 36–52 CrossRef.
- S. Li, Y. Huang, C. Guo, T. Wu, J. Zhang, L. Zhang and W. Ding, Chemistry3D: Robotic Interaction Benchmark for Chemistry Experiments, arXiv, 2024, preprint DOI:10.48550/arXiv.2406.08160.
- N. Yoshikawa, M. Skreta, K. Darvish, S. Arellano-Rubach, Z. Ji, L. Bjørn Kristensen, A. Z. Li, Y. Zhao, H. Xu and A. Kuramshin, et al. , Autonomous Robots, 2023, 47, 1057–1086 CrossRef.
- J. Boyd, Science, 2002, 295, 517–518 CrossRef CAS PubMed.
- T. Savage and E. A. del Rio Chanona, Comput. Chem. Eng., 2024, 189, 108810 CrossRef CAS.
- S. Kanza, C. Willoughby, N. J. Knight, C. L. Bird, J. G. Frey and S. J. Coles, Digital Discovery, 2023, 2, 602–617 RSC.
- M. L. Evans and J. D. Bocarsly, Datalab, Zenodo, 2024 Search PubMed.
- L. C. Brinson, L. M. Bartolo, B. Blaiszik, D. Elbert, I. Foster, A. Strachan and P. W. Voorhees, MRS Bull., 2024, 49, 12–16 CrossRef PubMed.
- M. Scheffler, M. Aeschlimann, M. Albrecht, T. Bereau, H.-J. Bungartz, C. Felser, M. Greiner, A. Groß, C. T. Koch, K. Kremer, W. E. Nagel, M. Scheidgen, C. Wöll and C. Draxl, Nature, 2022, 604, 635–642 CrossRef CAS PubMed.
- A. D. White, Nat. Rev. Chem., 2023, 7, 457–458 CrossRef CAS PubMed.
- K. M. Jablonka, P. Schwaller, A. Ortega-Guerrero and B. Smit, Nat. Mach. Intell., 2024, 6, 161–169 CrossRef.
- N. Alampara, M. Schilling-Wilhelmi, M. Ríos-García, I. Mandal, P. Khetarpal, H. S. Grover, N. M. A. Krishnan and K. M. Jablonka, Probing the limitations of multimodal language models for chemistry and materials research, arXiv, 2024, preprint DOI:10.48550/arXiv.2411.16955.
- K. M. Jablonka, Q. Ai, A. Al-Feghali, S. Badhwar, J. D. Bocarsly, A. M. Bran, S. Bringuier, L. C. Brinson, K. Choudhary, D. Circi, S. Cox, W. A. De Jong, M. L. Evans, N. Gastellu, J. Genzling, M. V. Gil, A. K. Gupta, Z. Hong, A. Imran, S. Kruschwitz, A. Labarre, J. Lála, T. Liu, S. Ma, S. Majumdar, G. W. Merz, N. Moitessier, E. Moubarak, B. Mouriño, B. Pelkie, M. Pieler, M. C. Ramos, B. Ranković, S. G. Rodriques, J. N. Sanders, P. Schwaller, M. Schwarting, J. Shi, B. Smit, B. E. Smith, J. Van Herck, C. Völker, L. Ward, S. Warren, B. Weiser, S. Zhang, X. Zhang, G. A. Zia, A. Scourtas, K. J. Schmidt, I. Foster, A. D. White and B. Blaiszik, Digital Discovery, 2023, 2, 1233–1250 RSC.
- Y. Zimmermann, A. Bazgir, Z. Afzal, F. Agbere, Q. Ai, N. Alampara, A. Al-Feghali, M. Ansari, D. Antypov, A. Aswad, J. Bai, V. Baibakova, D. D. Biswajeet, E. Bitzek, J. D. Bocarsly, A. Borisova, A. M. Bran, L. C. Brinson, M. M. Calderon, A. Canalicchio, V. Chen, Y. Chiang, D. Circi, B. Charmes, V. Chaudhary, Z. Chen, M.-H. Chiu, J. Clymo, K. Dabhadkar, N. Daelman, A. Datar, W. A. de Jong, M. L. Evans, M. G. Fard, G. Fisicaro, A. S. Gangan, J. George, J. D. C. Gonzalez, M. Götte, A. K. Gupta, H. Harb, P. Hong, A. Ibrahim, A. Ilyas, A. Imran, K. Ishimwe, R. Issa, K. M. Jablonka, C. Jones, T. R. Josephson, G. Juhasz, S. Kapoor, R. Kang, G. Khalighinejad, S. Khan, S. Klawohn, S. Kuman, A. N. Ladines, S. Leang, M. Lederbauer, S.-L. Liao, H. Liu, X. Liu, S. Lo, S. Madireddy, P. R. Maharana, S. Maheshwari, S. Mahjoubi, J. A. Márquez, R. Mills, T. Mohanty, B. Mohr, S. M. Moosavi, A. Moæhammer, A. D. Naghdi, A. Naik, O. Narykov, H. Näsström, X. V. Nguyen, X. Ni, D. O'Connor, T. Olayiwola, F. Ottomano, A. B. Ozhan, S. Pagel, C. Parida, J. Park, V. Patel, E. Patyukova, M. H. Petersen, L. Pinto, J. M. Pizarro, D. Plessers, T. Pradhan, U. Pratiush, C. Puli, A. Qin, M. Rajabi, F. Ricci, E. Risch, M. Ríos-García, A. Roy, T. Rug, H. M. Sayeed, M. Scheidgen, M. Schilling-Wilhelmi, M. Schloz, F. Schöppach, J. Schumann, P. Schwaller, M. Schwarting, S. Sharlin, K. Shen, J. Shi, P. Si, J. D'Souza, T. Sparks, S. Sudhakar, L. Talirz, D. Tang, O. Taran, C. Terboven, M. Tropin, A. Tsymbal, K. Ueltzen, P. A. Unzueta, A. Vasan, T. Vinchurkar, T. Vo, G. Vogel, C. Völker, J. Weinreich, F. Yang, M. Zaki, C. Zhang, S. Zhang, W. Zhang, R. Zhu, S. Zhu, J. Janssen, C. Li, I. Foster and B. Blaiszik, Reflections from the 2024 Large Language Model (LLM) Hackathon for Applications in Materials Science and Chemistry, arXiv, 2025, preprint DOI:10.48550/arXiv.2411.15221.
- A. M. Bran, S. Cox, O. Schilter, C. Baldassari, A. D. White and P. Schwaller, ChemCrow: Augmenting large-language models with chemistry tools, arXiv, 2023, preprint DOI:10.48550/arXiv.2304.05376.
- M. L. Evans, G.-M. Rignanese, D. Elbert and P. Kraus, Metadata, automation, and registries for extractor interoperability in the chemical and materials sciences, arXiv, 2024, preprint DOI:10.48550/arXiv.2410.18839.
- D. Allan, T. Caswell, S. Campbell and M. Rakitin, Synchrotron Radiation News, 2019, 32, 19–22 CrossRef.
- C. W. Andersen, R. Armiento, E. Blokhin, G. J. Conduit, S. Dwaraknath, M. L. Evans, Á. Fekete, A. Gopakumar, S. Gražulis, A. Merkys, F. Mohamed, C. Oses, G. Pizzi, G.-M. Rignanese, M. Scheidgen, L. Talirz, C. Toher, D. Winston, R. Aversa, K. Choudhary, P. Colinet, S. Curtarolo, D. Di Stefano, C. Draxl, S. Er, M. Esters, M. Fornari, M. Giantomassi, M. Govoni, G. Hautier, V. Hegde, M. K. Horton, P. Huck, G. Huhs, J. Hummelshøj, A. Kariryaa, B. Kozinsky, S. Kumbhar, M. Liu, N. Marzari, A. J. Morris, A. A. Mostofi, K. A. Persson, G. Petretto, T. Purcell, F. Ricci, F. Rose, M. Scheffler, D. Speckhard, M. Uhrin, A. Vaitkus, P. Villars, D. Waroquiers, C. Wolverton, M. Wu and X. Yang, Sci. Data, 2021, 8, 217 CrossRef PubMed.
- M. L. Evans, J. Bergsma, A. Merkys, C. W. Andersen, O. B. Andersson, D. Beltrán, E. Blokhin, T. M. Boland, R. Castañeda Balderas, K. Choudhary, A. Díaz Díaz, R. Domínguez García, H. Eckert, K. Eimre, M. E. Fuentes Montero, A. M. Krajewski, J. J. Mortensen, J. M. Nápoles Duarte, J. Pietryga, J. Qi, F. D. J. Trejo Carrillo, A. Vaitkus, J. Yu, A. Zettel, P. B. De Castro, J. Carlsson, T. F. T. Cerqueira, S. Divilov, H. Hajiyani, F. Hanke, K. Jose, C. Oses, J. Riebesell, J. Schmidt, D. Winston, C. Xie, X. Yang, S. Bonella, S. Botti, S. Curtarolo, C. Draxl, L. E. Fuentes Cobas, A. Hospital, Z.-K. Liu, M. A. L. Marques, N. Marzari, A. J. Morris, S. P. Ong, M. Orozco, K. A. Persson, K. S. Thygesen, C. Wolverton, M. Scheidgen, C. Toher, G. J. Conduit, G. Pizzi, S. Gražulis, G.-M. Rignanese and R. Armiento, Digital Discovery, 2024, 3, 1509–1533 RSC.
- M. D. Hanwell, W. A. De Jong and C. J. Harris, J. Cheminf., 2017, 9, 55 Search PubMed.
- S. Lehtola, J. Chem. Phys., 2023, 159, 180901 CrossRef CAS PubMed.
- S. J. Coles, J. G. Frey, C. L. Bird, R. J. Whitby and A. E. Day, J. Cheminf., 2013, 5, 52 Search PubMed.
- S. Clark, F. L. Bleken, S. Stier, E. Flores, C. W. Andersen, M. Marcinek, A. Szczesna-Chrzan, M. Gaberscek, M. R. Palacin, M. Uhrin and J. Friis, Adv. Energy Mater., 2022, 12, 2102702 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |