
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and structure–function interplay in covalent organic frameworks for photocatalytic CO2 reduction
Shibani Mohata
 *ab,
Poulami Majumder
ab and
Rahul Banerjee
*abc
*ab,
Poulami Majumder
ab and
Rahul Banerjee
*abc
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research, Kolkata, Mohanpur 741246, India. E-mail: shibanimohata@gmail.com; r.banerjee@iiserkol.ac.in
bCentre for Advanced Functional Materials, Indian Institute of Science Education and Research, Kolkata, Mohanpur 741246, India
cCollege of Science, Korea University, 145 Anam-ro Seongbuk-gu, South Korea
First published on 21st May 2025
Abstract
The escalating global energy demands and the need to alleviate the rapid rise in greenhouse gases have led to colossal interest in designing efficient catalytic systems for photocatalytic CO2 reduction. While inorganic semiconductors have been the frontrunners for a long time, porous photocatalysts, particularly covalent organic frameworks (COFs), are gaining traction due to their atomically precise structures, enabling tuning their structural and chemical properties. Designed using the principles of reticular chemistry, the building units of COFs can be modulated to incorporate catalytically active sites periodically using robust covalent bonds to endow them with high efficiency, selectivity, and stability. Unlike the non-porous congeners, COFs, with their high porosity and precisely defined pore channels, allow for quicker diffusion of substrates and products, enabling the utilization of deeply buried photocatalytic sites. Our approach is to comprehend the significant roadblocks that must be overcome for designing state-of-the-art catalysts for photocatalytic CO2 reduction. Building upon that, we highlight the key strategies devised to design COF-based CO2RR photocatalysts. A fundamental understanding of the structure–property relationship is quintessential for utilizing the precision of COF chemistry for developing next-generation materials combining activity, selectivity, and efficiency in a single system. Throughout this review, we have taken a closer look at how the critical design aspects and molecular engineering reciprocate towards augmenting the bulk photocatalytic properties of efficiency and selectivity. Understanding molecular engineering and structure–property relationships will be conducive to developing sophisticated systems to solve global crises in this burgeoning area of research.
 Shibani Mohata | Dr Shibani Mohata received her PhD degree from the Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, India, in September 2024, under the supervision of Prof. Rahul Banerjee. She completed her BSc degree at SKBU University and received her MS degree from IISER Kolkata, India. Her research focuses on the design and synthesis of organic porous materials, 2D covalent organic framework membranes, and their applications in the development of heterogeneous photocatalytic CO2 reduction catalysts. |
 Poulami Majumder | Dr Poulami Majumder completed her PhD in the Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, India, under the supervision of Prof. Rahul Banerjee. She completed her BSc degree at Burdwan University and received her MSc degree from the Indian Institute of Science Education and Research (IISER) Kolkata, India. Her research interest is in exploring heterogeneous photocatalysis by using the covalent organic framework. |
 Rahul Banerjee | Prof. Rahul Banerjee has been a professor at IISER Kolkata since 2017. His research interests include crystallography, reticular chemistry and organic material synthesis. His group has developed unique methodologies of synthesizing covalent organic frameworks with various morphologies and exceptional chemical stability. His group further utilized the covalent organic frameworks (COFs) for carbon sequestration, water purification, energy storage and conversion, and separation applications. He has published more than 180 research articles, which have attracted 37 |
1. Introduction
The industrial revolution has led to a drastic increase in CO2 emissions from various anthropogenic sources, disrupting the carbon cycle.1 CO2 reduction to value-added products is an attractive route to mitigate this environmental crisis and achieve the coveted carbon-neutral economy.2 This has led to colossal interest in developing efficient catalytic processes and systems. Among the primary strategies for CO2 reduction, the most prominent include (a) electrocatalysis, (b) thermocatalysis, (c) biocatalysis, and (d) photocatalysis.3–5 While highly efficient and selective, biological catalysts are inherently fragile and constrained by stringent operational conditions, such as specific temperature and pH requirements.6 Thermocatalysis has demonstrated considerable progress; however, it typically necessitates elevated temperatures (≥500 K) and high pressures (≥10 atm).7 Electrocatalysis, a widely studied alternative, offers high efficiency but relies heavily on external electricity sources, rendering the process less sustainable and economically viable.8,9 In this regard, photocatalytic CO2 reduction has come to the forefront as a method to develop greener, cost-effective, and more sustainable approaches to advance the field of CO2 reduction. Compared to the conventional methods, photocatalytic transformation bestows several advantages: (1) it can be achieved at ambient temperatures and pressures, (2) using renewable (sun) light energy and comparatively simplistic reactors, and most importantly, (3) it can generate chemical fuels using only water and CO2 as feeds without introducing secondary pollution.10,11Designing an efficient photocatalyst with high activity and selectivity necessitates meeting specific criteria. The photocatalyst should possess a high density of exposed active sites to facilitate reactions, exhibit structural robustness to endure multiple catalytic cycles and maintain a well-defined structure to clearly understand structure–property relationships.12,13 The catalyst should be tunable to accommodate varying application requirements, ensuring versatility and optimization for specific processes.14 Covalent organic frameworks (COFs) serve as ideal platforms for achieving this within a single system.15–19 They are bestowed with periodic structures with abundant active sites, high surface areas, and well-defined pore channels conducive to facile diffusion of substrates to the active sites, resulting in high catalytic activity. Reticular chemistry enables the selection of tailored building units inspired by selective molecular catalysts, imparting them with high selectivity.20,21 Their extended structures render them insoluble in most common organic solvents, ensuring easy isolation through centrifugation. Moreover, these strong covalent linkages provide remarkable stability, allowing the materials to be reused across several reaction cycles. Significant efforts have been made to optimize the optoelectronic properties of COFs in terms of the light harnessing capability, charge separation and transfer, and band positions, imparting high photocatalytic efficiency.22–24 Although great strides have been made in developing COF-based catalysts for photocatalytic CO2 reduction, a gap persists in understanding the structure–property relationship and how the catalytic performance correlates with their molecular architecture.
This review delves into the intriguing research on engineering covalent organic frameworks for photocatalytic CO2 reduction. We highlight the diverse porous materials established as outstanding platforms for a broad spectrum of photocatalytic applications, with particular emphasis on the fundamental mechanisms of COFs in photocatalysis. A special focus has been placed on understanding the challenges associated with developing an ideal catalyst for photocatalytic CO2 reduction. We have emphasized the need to tackle key challenges, including improving product selectivity, mitigating parasitic hydrogen evolution in water, reducing dependence on metals, enhancing the oxidation half-reaction, addressing product overestimation, and preventing catalyst decomposition. We identify the properties of porous materials that set them apart from other semiconductor photocatalysts as promising materials for photocatalysis. Our discourse extends to the various strategies developed to design covalent organic frameworks for photocatalytic CO2 reduction reactions. COF-based photocatalysts are divided into two subclasses: (i) metalated: COFs bearing metal centres that act as active sites for CO2 reduction, enhancing catalytic efficiency and (ii) metal-free: systems relying on organic components to facilitate CO2 reduction. Ideally, such systems should also be free of metal-based photosensitizers or co-catalysts. We have extensively discussed the various important developments in these two classes. Our approach focuses on understanding the fundamental structure–function relationship to determine catalyst efficiency and selectivity. Developing a thorough understanding of the design principles affecting the photophysical and physiochemical properties of COFs will open new avenues for future research on developing tailor-made photocatalysts for CO2 reduction.
2. Porous photocatalysts and their applications
Photocatalysis has emerged as a greener and more cost-effective alternative, leveraging renewable solar energy under ambient temperature and pressure conditions while minimizing secondary pollution. This advancement has spurred the development of various homogeneous and heterogeneous semiconductor photocatalysts, including metals (e.g., Cu, Au, and Ag), titanium dioxide, molecular transition-metal complexes, metal oxides and chalcogenides, perovskites, and MXenes.25–30 Despite their promise, these materials face challenges such as limited band gap tunability, restricted light absorption, reliance on expensive noble metals, and surface deactivation. Additionally, their low porosity confines reactions to surface-exposed sites, thereby reducing efficiency.Homogeneous photocatalysts are widely studied for their well-defined chemical structures and the ability to fine-tune their activity by modifying ligands, metals, and coordination spheres, providing fundamental insights into reaction mechanisms.31,32 Conversely, heterogeneous photocatalysts, while more challenging to characterize in terms of active sites or intermediates, offer advantages such as higher activity, stability, and recyclability.33,34
In this context, porous materials engineered through the principles of reticular chemistry bridge the gap between these approaches.35 Their high surface area and tunable pore environments facilitate efficient mass transport, charge transfer, and accessibility to active sites.36 These features significantly enhance the efficiency and selectivity of photochemical transformations, providing an optimal blend of functionality and performance.
Porous materials span from zero-dimensional molecular cages and one-dimensional nanotubes to two and three-dimensional metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and hydrogen-bonded frameworks (HOFs) (Fig. 1a).37,38 They differ based on their crystallinity and porosity, based on which they can be categorized as amorphous with ill-defined structures like polymers of intrinsic microporosity (PIMs) and crystalline with well-ordered structure and composition. Nonetheless, both categories offer properties such as functional tunability, structural diversity, and robustness, which are essential for photochemical applications.39,40 Owing to these advantages, porous materials have been used for a wide range of photochemical applications, which span from the hydrogen evolution reaction (HER), oxygen evolution reaction (OER), oxygen reduction reaction (ORR), carbon dioxide reduction reaction (CO2RR), and nitrate reduction reaction (NO3RR) to various organic transformations (Fig. 1b).41–46 A detailed discussion of these categories of porous materials and various applications is beyond the scope of this review. Hence, we will be focusing on the photocatalytic CO2RR using covalent organic frameworks.
 | ||
| Fig. 1 (a) Schematic representation of the various categories of porous heterogeneous photocatalysts constructed using precise building units. (b) Illustration of the different photocatalytic applications of porous materials. | ||
3. Photocatalytic CO2 reduction mechanism
Photocatalysis involves the conversion of light energy into chemical energy via charge separation across the bandgap of the semiconductor to facilitate the redox transformation of chemical moieties on their surface. The photocatalytic transformation on a semiconductor photocatalyst consists of three fundamental steps:• Visible light absorption: the efficient harnessing of visible rays necessitates that the energy of the irradiated radiation should be greater than the band gap of the photocatalyst (Fig. 2).47,48 Therefore, for harnessing solar light, which mainly consists of visible and infrared radiation (95%, UV = 5%), the bandgap should be between 1.7 and 3.1 eV.
 | ||
| Fig. 2 Mechanism of photocatalytic CO2 reduction on a semiconductor photocatalyst. | ||
• Generation of charge carriers (electron–hole pairs): light absorption leads to the generation of photoexcited e−–h+ pairs. Notably, the lifetime time of the excited electrons should be sufficient for surface catalytic transformations that typically occur in 10−8 to 10−1 s compared to charge recombination, which is much faster and requires ∼10−9 s.49 Hence, it is ubiquitous to design materials to maximize light absorption and the lifetime of the excited states. The generated electrons and holes then migrate to the interface of the photocatalyst where the redox reaction takes place, and
• Redox reaction on the photocatalyst surface: alongside the bandgap, the band position of the photocatalyst is pivotal for CO2 reduction. The LUMO and HOMO of the photocatalyst must straddle the reduction potential of CO2 and the oxidation potential of the electron donor. In other words, the bottom of the conduction band must be more negative than the reduction potential of CO2, and the top of the valence band must be at a more positive potential relative to the electron donor including water, triethanolamine (TEOA), and triethylamine (TEA).
Photosensitive units and active sites (oxidation and reduction units) can be incorporated into the COF framework, wherein the COF can act simultaneously as an intrinsic photocatalyst to harness light and perform catalysis. The periodic structure of COFs can provide ample active sites, thereby maximizing performance with respect to the molecular congeners.50 The route followed in such a system is simplistic and the same as described above. However, most COFs have limited light-harnessing capability, necessitating an external photosensitizer typically consisting of Ir and Ru complexes. These complexes are excited upon irradiation, and if the bands are appropriately aligned with the COF, the photogenerated e− migrates to the reduction site on the COF, where the CO2RR takes place. The holes in the photosensitive complex participate in the oxidation reaction. Likewise, if the COF lacks an active centre, chelating ligands can be introduced on the COF backbone to bind with metal atoms (Co, Ni, Fe, Re, etc.), acting as the catalytic site. Herein, the COF acts as a photosensitizer, and upon illumination, the electrons from the CB of the COF migrate to the active centre where the CO2RR takes place while the sacrificial electron donor quenches the holes left. In other cases, the COF backbone can be used as a template to construct heterojunctions with semiconductors like nanoparticles and polyoxometalates that depict a better catalysis than the individual materials. The excited state electron transfer in such hybrid materials is complex and determined through advanced spectroscopic techniques.
4. Challenges of photocatalytic CO2 reduction
Despite the recent progress in developing CO2 photoreduction catalysts, several challenges need to be overcome. The challenges are tuning product selectivity, overcoming the parasitic hydrogen evolution reaction (HER), using metals and sacrificial agents, challenging oxidation half-reaction, overestimating product yield and catalyst decomposition.4.1. Product selectivity
CO2 is chemically inert and less reactive due to a high bond energy of 750 kJ mol−1.51 The reduction of CO2 is thermodynamically non-spontaneous (ΔG > 0) and is an uphill process. The one-electron reduction of CO2 has a high reduction potential (E(CO2/CO2˙−) = −1.85 V vs. SHE) due to the change in geometry from linear to bent, requiring high reorganizational energy, making it challenging to achieve under normal conditions. By contrast, proton-coupled electron transfer (PCET) has much lower reduction potentials that are positive with respect to the conduction band of numerous semiconductors.52 Hence, the photoreduction of CO2 proceeds through the PCET pathway. Here, it is worth noting that the reduction potentials of CO2 to various products such as CO, HCHO, HCOOH, CH4, CH3OH, and others are close and encompass a narrow range of −0.7 V to −0.2 V at pH = 7.53 This often leads to a mixture of products that are hard to separate owing to their similar properties (Fig. 3a). | ||
| Fig. 3 Challenges of photocatalytic CO2 reduction. (a) Selectivity of the product formed, (b) competitive hydrogen evolution reaction, (c) use of metals, (d) challenging oxidation half-reaction, (e) overestimation of the product yield, and (f) decomposition of the catalyst under the reaction conditions. | ||
The mechanism of formation of these products occurs via multi-step PCET and is still ambiguous. However, the first step is the adsorption of CO2 through the binding sites in the framework, leading to the subsequent formation of *OCHO or *COOH intermediates.54 Further protonation and desorption lead to the formation of HCOOH, while CO can be formed by reduction. Most COF-based photocatalysts have a weak *CO binding energy, usually leading to the formation of CO as the major product. Stronger binding of *CO may favor further PCET steps generating *CHO and *COH, ultimately forming CH3OH and CH4. Meanwhile, the two-step H+/e− transfer from the *OCHO or *COOH intermediates promotes formaldehyde generation (HCHO). Notably, photocatalytic conversion of CO2 to C2+ products like C2H5OH, CH3CHO, and CH3COOH is rarely achieved.
4.2. Hydrogen evolution reaction (HER)
Water is an ideal solvent to carry out the CO2RR as it surpasses the need for undesirable organic solvents, making the overall process much more sustainable. Also, it can simultaneously act as an electron donor to quench the holes and act as a hydrogen source during PCET. The reduction potential of water to hydrogen is E(H2O/H2) = −0.41 V vs. SHE at pH = 7, and it falls in the range of CO2 reduction to value-added products.55 Moreover, the reduction of CO2 to CH3OH, CH4, HCHO, etc. involves a multielectron transfer, whereas the HER is a two-electron transfer process, making it more feasible. Another concern is the limited solubility of CO2 in water, with only 1 molecule of water available for 1300 CO2 molecules.56 Thus, the HER is generally thermodynamically and kinetically feasible compared to the CO2RR. Hence, the HER is a parasitic reaction to the CO2RR, particularly for liquid phase reactions (Fig. 3b). This compromises the overall selectivity and efficiency of the process, and attempts need to be made to bypass the HER.4.3. Use of metals
Significant efforts to design photocatalysts have led to the development of highly efficient CO2 reduction catalysts. However, many of these photocatalysts rely on transition metals with multiple oxidation states and often require co-catalysts made from expensive metals to enhance their performance (Fig. 3c). Additionally, when COFs exhibit poor light-harvesting capabilities, external photosensitizers containing costly and scarce noble metals like Ru and Ir are frequently employed.57 This reliance on expensive materials poses a significant limitation, reducing scalability and making the process unsustainable. Furthermore, transition metal-based catalysts often favor hydrogen evolution reactions (HERs), compromising selectivity. They are also prone to self-oxidation and photo-corrosion and exhibit lower hydrolytic stability. Metal-free systems can be an excellent alternative for their abundance, relatively cheaper cost, environmental friendliness, and sustainability.58,594.4. Oxidation half-reaction
The holes generated during the illumination of the photocatalyst can be consumed in the oxidation of water, generating O2 or hydroxyl radicals (OH˙) as the oxidation half-reaction to the CO2RR. While the valence band of most semiconductor photocatalysts is more positive than water oxidation (EO2/H2O = 0.81 vs. NHE at pH = 7), making the oxygen evolution reaction (OER) thermodynamically feasible, being a 4-electron process that makes it challenging. Besides, the accumulation of the evolving oxidizing species OH˙ or O2 on the photocatalyst surface interrupts the CO2RR. To surpass this challenge, people often use sacrificial agents like alcohol and amines, making the process unsustainable (Fig. 3d).60 The catalyst design must be improved to promote water oxidation as a counter-reaction.4.5. Product overestimation
To improve the solubility of CO2, the dispersibility of the heterogeneous photocatalysts, and recyclability, alternative organic solvents like acetonitrile (ACN), ethyl acetate (EAA), and dimethylformamide (DMF) are often used instead of water. Solvents may exhibit photoactivity and undergo decomposition under certain conditions, such as specific light wavelengths, solvent combinations, temperature, and pressure (Fig. 3e). This can lead to generating one of the CO2 reduction products, causing inaccurate assessment of the catalyst's efficiency and making the overall process ambiguous.61As discussed, sacrificial agents like triethanol amine (TEOA) and triethyl amine (TEA) are used as sacrificial electron donors. It has also been reported that they can catalyze CO2 photoreduction to CO and CH4 through hydrogen atom transfer (HAT). In situ FTIR studies and density functional theory (DFT) calculations strongly support the idea that the terminal −OH group in TEOA can behave as the active centre.62,63 Unfortunately, possible solvent decomposition and sacrificial agent participation are often unnoticed when considering solvent and electron donor selection as a parameter for enhancing efficiency.
4.6. Catalyst decomposition
It has been confirmed that graphitic carbon nitride (g-C3N4) undergoes self-decomposition in light to generate CO, CO2, NO2, and NO2−/NO3 under the reaction conditions (Fig. 3f).64 The rate of CO production in the argon atmosphere was almost equal to that in the CO2 atmosphere. The mechanistic studies revealed that the instability was due to the electronic interaction between the adsorbed hydroxyl group and g-C3N4, which reduces the stability of the C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. Unfortunately, there are limited studies on the behavior of COF-based photocatalysts, and they need to be looked into in-depth, as the organic components in COFs are prone to photodegradation.
C bonds. Unfortunately, there are limited studies on the behavior of COF-based photocatalysts, and they need to be looked into in-depth, as the organic components in COFs are prone to photodegradation.
5. Favorable properties of porous materials as photocatalysts
Compared to the traditional non-porous congeners, where only surface sites are accessible for photocatalysis, the porous photocatalyst offers certain advantages. We will be discussing it in this section.5.1. Structural tailorability
COFs are designed through the principle of reticular chemistry, enabling atomically precise control of the chemical and topological structure that sets them apart from other classes of semiconductor photocatalysts like metallic oxides and porous polymers (Fig. 4a).65,66 The predetermined connections enable the design of a framework with a high surface area, desired pore environment, and hierarchical structures. Also, for similar topology, the pore sizes can be controlled simply by varying the length of the building blocks. The geometry of the linkers can be varied to fabricate three-dimensional structures, enhancing the accessibility of the active sites.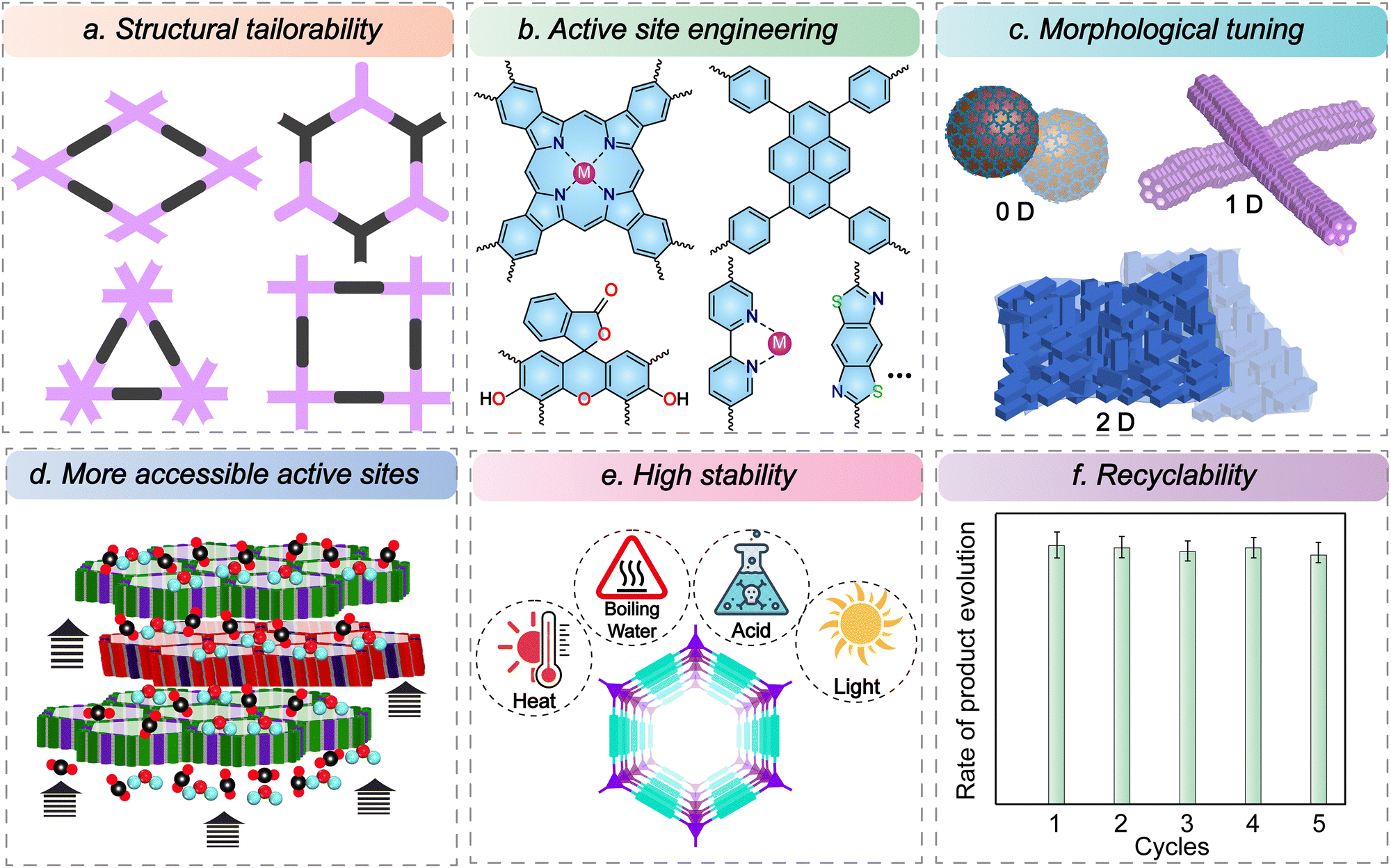 | ||
| Fig. 4 Favorable properties of porous materials as photocatalysts. | ||
5.2. Active site engineering
The precursors can be designed to integrate the catalytically active moieties in the framework, which improves photocatalytic efficiency by decreasing the overall inactive components.67,68 The building blocks are chosen to enhance the light-harnessing capability, tune the band gap, maximize CO2 absorption, and promote mass transfer (Fig. 4b). The functional components with high π-conjugation can be incorporated to manipulate the bandgap and promote charge separation and transfer. Alternatively, COFs with poor photosensitivity or lacking active sites can also be modified by post-synthetic modification to incorporate active components or by using the nanoconfinement effect of COFs.Broadly, these linkers can be classified into metalated and metal-free analogues. The building units can be engineered to anchor various photoactive metals as single atom sites, which helps greatly enhance performance by using a lesser quantity of metals than the bulk metallic catalyst. The strategy has allowed the incorporation of photoactive units to anchor the metal atoms, such as porphyrin, phthalocyanine, bipyridine, salen, and diol. Another pathway is incorporating building units that are highly conjugated, which acts as a photo absorber, thereby precluding the requirement of expensive photosensitizers. These include units like pyrene, eosin, perylene, and thiafulvalene. These moieties are excellent light absorbers and efficiently generate charge separation. Also, the principle of reticular chemistry can be used to engineer electron-rich donor and electron-poor acceptor sites that promote HOMO–LUMO separation and intramolecular charge transfer via the push–pull effects. Furthermore, incorporating heteroatoms helps in the adsorption and conversion of CO2.
5.3. Morphological tuning
The morphological effect is one critical yet often overlooked parameter in tuning the catalytic performance. Dimensionality can have a profound impact on the catalytic performance. The reticular synthesis enables complete structural control of the porous materials. Beyond that, supramolecular nanosynthesis allows for some degree of morphological tuning. Different morphologies can be obtained with the same set of linkers and common topology. The most common are 0D spheres, 1D fibres/tubes/rods, and 2D sheets (Fig. 4c).69–71 The morphology greatly influences the final processability and properties of the material.72 For example, the spheres can have colloidal stability and dispersibility for otherwise insoluble COFs. Likewise, fibres and tubes have high aspect ratios, enhancing the active surface areas. The synthetic methodology enables control of the size of the 0D spheres as well as particle size uniformity. Recent works have also led to the development of COFs with monolayers, a few layers to films of several micrometre thickness. Besides, they can also be grown on different substrates depending upon the application. The two-dimensional materials can also be exfoliated to synthesize nanosheets with much higher mass transport and ease of accessing active sites. Because of a higher degree of morphological control, these materials can be synthesized as powders, films, and foams, based on the application requirement. However, there is a lack of comprehensive studies exploring the impact of morphology and dimensionality on catalytic performance and light penetration, highlighting the need for further investigation.5.4. More accessible active sites
The unique design of COFs bestows them with periodic and well-defined pore channels. This promotes the mass transfer and diffusion of the substrates and products throughout the bulk, and the catalysis is not restricted to the surface-exposed active sites. The porous frameworks facilitate the confinement of suitable substrates, bringing them in proximity and promoting reaction rates compared to the non-porous congeners. The accessibility to the active sites can be further increased by fabricating ultrathin covalent organic nanosheets (CONs) with only a few layers of thickness (Fig. 4d).73 This can be achieved by either a top-down approach where a 2D COF is exfoliated or a bottom-up approach where structuring tuning and reaction methodology enable the growth of one or a few layers of CONs. In either case, the synthesized CONs have an even higher active surface area and shorter diffusion lengths than the bulk counterparts, leading to enhanced catalytic performance. Another vital strategy to augment active site accessibility is changing the material dimensionality from 2D to 3D. Although the synthesis of 3D COFs is comparatively more challenging, they can accomplish much higher surface areas and pore sizes, leading to an improvement in CO2 mass transfer and making the active sites more accessible.74,755.5. High stability
Despite achieving high catalytic performances using transition metal complexes and other metallic systems, one of the significant drawbacks is their poor hydrolytic and photostability. In this context, COFs linked through robust covalent bonds are much more stable (Fig. 4e). Traditionally, COFs were synthesized using reversible reactions such as boronate ester condensation and imine linkages. While these methods produced highly crystalline structures, they often exhibited poor stability, especially in water and under light irradiation.76 However, several methodologies, like post-synthetic modifications and interlayer hydrogen bonding, have been devised to improve their stability. Alternatively, β-ketoenamine COFs synthesized by the combination of reversible and irreversible pathways allowed the construction of COFs that were simultaneously crystalline and stable.77 However, long-range conjugation is impeded in such systems, compromising their charge transfer and, eventually, catalytic performances in some cases. In this context, the –CC– linked COFs that are exceptionally chemically stable and have extended π-conjugated systems come to the rescue.78,79 Nonetheless, the crystallization of COFs based on irreversible reactions remains challenging. Hence, decades of research on COFs enables the choice from a wide range of linkages like imidazole, olefin, keto-enamine, and benzoxazole, which are highly promising as robust platforms for CO2 photoreduction.80–83 It is worth mentioning that there are limited studies on the degradation mechanisms of these COFs under photoirradiation, and it largely remains unclear.
5.6. Recyclability
One crucial parameter that sets apart homogeneous catalysts from their heterogeneous analogues is recyclability. Porous materials like COFs are generally insoluble in most organic solvents. They can be easily recovered from the reaction medium by centrifugation or filtration.84 Hence, they can be used for several cycles, making the overall process cost-effective and sustainable (Fig. 4f). The post-synthetic structural and morphological studies have shown that COFs can maintain their catalytic performance and selectivity over several cycles while maintaining structural integrity.The ability to independently adjust these factors strengthens the potential of reticular chemistry in shaping the development of advanced next-generation CO2 reduction catalysts.
6. COFs as heterogeneous platforms for photocatalytic CO2 reduction
The covalently linked frameworks of covalent organic frameworks (COFs) impart exceptional structural robustness and extended π-conjugation, enhancing charge transport and stability under reaction conditions. Additionally, COFs facilitate the anchoring of single-atomic metallic sites via specific binding sites on their backbone, significantly reducing noble metal usage and promoting sustainability. The ability to engineer COFs in diverse morphologies supports their integration into advanced device architectures, paving the way for next-generation photocatalytic systems.Based on the properties of COFs, they can behave as photosensitizers and/or active catalysts. Generally, COFs have poor catalytic efficiency; hence, the COF structure is tailored to incorporate active metal sites for CO2 photoreduction. Based on this, COF-based photocatalysts can be divided into two sub-classes: (i) metalated and (ii) metal-free. Among the metalated COF catalysts, there are three essential classes: (1) COFs with predesigned inherited active sites: inspiration can be taken from well-studied homogeneous molecular catalysts wherein the building blocks of the COFs can be judiciously designed to incorporate the active sites (Fig. 5a). Due to cooperative effects, multiple active sites within a single framework in proximity can give rise to a better performance than the homogeneous counterparts. Moreover, they are easier to recover and reuse, making them appealing for large-scale applications. These features create a foundation for designing highly active photocatalysts and investigating atomic-level structure–property relationships. The tuning of the metal centres and their oxidation state can vary the selectivity and efficiency of the process. (2) Post-synthetic modification to introduce single-atomic metal sites: porous materials can be used as a porous support for anchoring single-atom active sites (Fig. 5b). Along with grafting metal centres within the building blocks used to synthesize photoactive COFs, another important strategy is the post-synthetic incorporation of precise active sites using various coordinating sites in the framework. The local environment of the metal atoms can be easily tuned by meticulous choice of linkers based on heteroatom donors like N, O, and S. Besides, the same linkers can be used to incorporate various metallic sites. Dual metal coordination is another critical approach wherein two atomically dispersed metal sites work in synergy. In this regard, Ni–Co, La–Ni, and Cu–Ni sites have been incorporated to tune the performance and improvise over single metal coordination. This strategy opens new avenues for single-atom catalysis using porous materials. (3) COFs as porous organic supports for encapsulation of homogeneous catalysts: COFs can act as excellent scaffolds for encapsulating active catalysts within their pores to form heterostructures for photocatalytic CO2 reduction (Fig. 5c). COFs can provide a hydrophobic microenvironment and promote catalysis in the aqueous environment, even for catalysts with poor hydrolytic stability, while the porous nature will simultaneously promote the rapid diffusion of substrates and products. Moreover, the COF pore enables the proximity of catalysts and substrates, enhancing the reaction rate. The catalyst can be immobilized by both covalent and non-covalent interactions. Significant progress has been made in this regard, and various catalysts, such as MOFs, polyoxometalates, metal/metal oxide nanoparticles, quantum dots, and carbon nanodots, have been encapsulated within the COF backbone.
 | ||
| Fig. 5 Various strategies for fabricating metalated covalent organic frameworks for photocatalytic CO2 reduction. | ||
Another important category of COFs for CO2 photoreduction is the metal-free analogues. These COFs usually act as both photoabsorbers and active catalysts. The optoelectronic properties can be tuned by selecting the building blocks from a large pool of π-conjugated and aromatic systems to enable efficient charge separation and migration. Moreover, heteroatom sites can be incorporated into the framework to enhance CO2 uptake through CO2 polarisation through the dipole–quadrupole interactions. We will be discussing these strategies in detail in the subsequent sections.
6.1. Metalated COFs as single-atom heterogeneous catalysts for photocatalytic CO2 reduction
The large surface areas, well-defined pore channels, and abundant periodic linkers make COFs excellent porous scaffolds for introducing metallic sites for developing single-atom heterogeneous catalysts for photocatalytic CO2 reduction. The excellent photo-absorbing capability of COFs can be coupled with the inherent catalytic efficiency of the metal centres. Reticular chemistry enables designing COFs with inherent metallic sites or the post-synthetic introduction using chelating ligands meticulously engineered on the COF backbone. The metal atoms control the selectivity and efficiency of the photocatalytic CO2 reduction by the variation in the binding affinities of CO2 and the subsequent intermediates generated during CO2 transformation. This can be tuned by varying the metal centres and their coordination environments. Over the years, several ligands have been designed to incorporate active metal sites in the COF framework, like bipyridine, catechol, porphyrin, salen, and acenaphthylene-1,2-diimine. They play a pivotal role in modulating the coordination environment of metal centers in COFs. The different combinations of chelating groups like N4, N3O, N2O2, N3O, and O4 can influence the strength and performance of the co-ordinated active metal sites. For instance, chelating ligands that create a more rigid coordination environment may stabilize the metal centre and improve its longevity during photocatalytic cycles, while the presence of specific functional groups on the ligand may tune the redox potential of the metal, affecting the photocatalytic performance. Ligands like porphyrin have inherent light harnessing capability and the robust N-macrocyclic core provides an ideal platform for co-ordinating a wide range of metal sites like Cu, Co, Ni, Fe, and Zn among others both pre- and post-synthetically.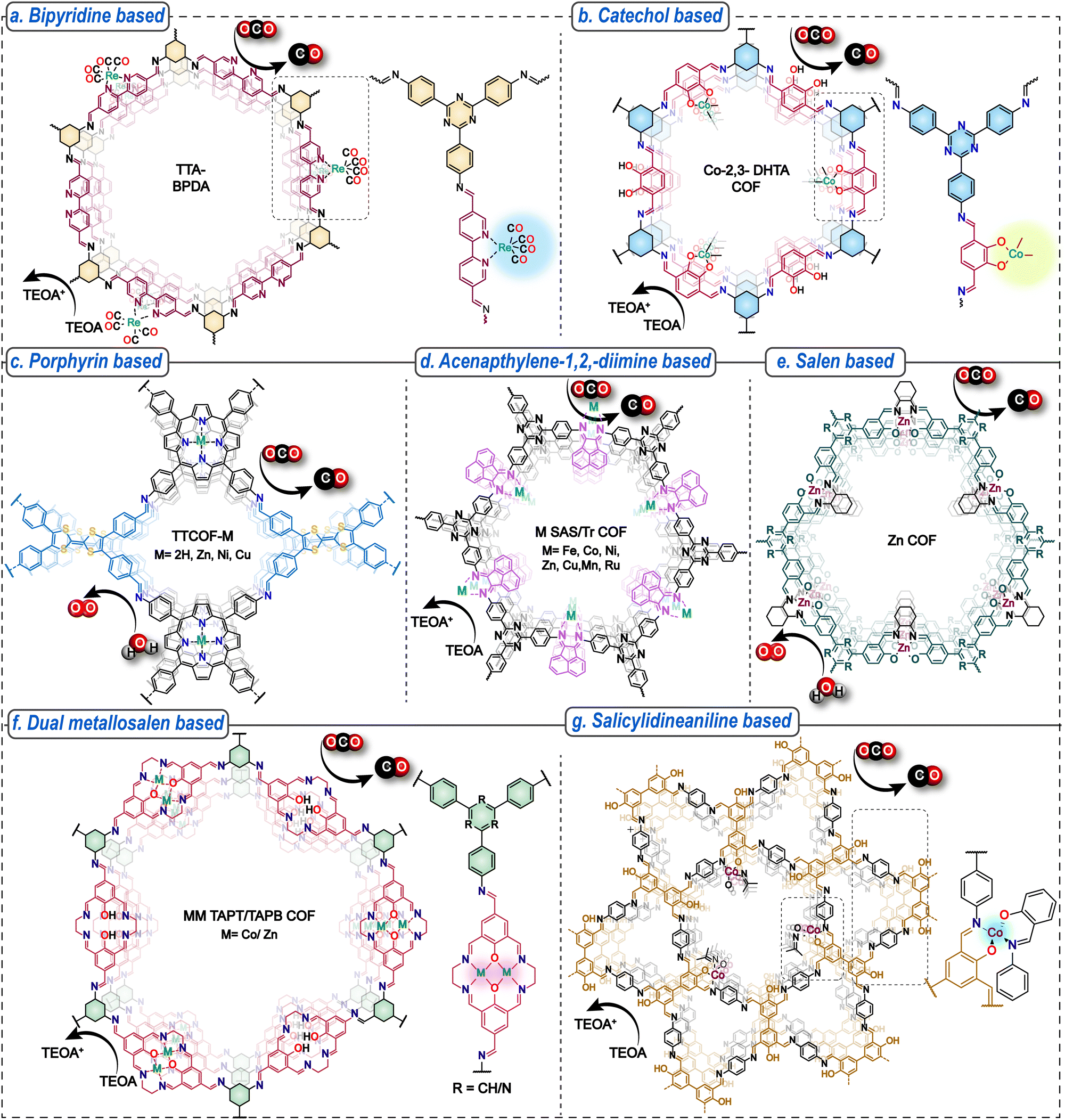 | ||
| Fig. 6 COF-based single-atom catalysts for photocatalytic CO2 reduction. The chemical structures of the COFs representing the ligand system and active metals involved. | ||
The concept was further extended by designing a –CC– bonded Re-Bpy-sp2c-COF synthesized by the condensation of 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetrabenzaldehyde (TFPPy) and 5,5′-bis(cyanomethyl)-2,2′-bipyridine and loading with [Re(CO)5Cl].87 Compared to imine COF congeners, the sp2c-COF has broader light absorption due to extended conjugation and higher structural robustness due to stronger bonds. Under the optimized conditions, the Re-Bpy-sp2c-COF converted CO2 to CO at 1040 μmol g−1 h−1 with 81% selectivity and a TON of 18.7. The material maintains its crystallinity and catalytic performance effectively for up to 17.5 hours of photocatalysis. However, after prolonged irradiation beyond 50 hours, a notable reduction in crystallinity and catalytic efficiency is observed. The homogeneous molecular catalyst [Re(bpy)(CO)3Cl] showed a much lower TON of 10.3 and was deactivated after three hours. Interestingly, the CO production rates can be further enhanced to 1400 μmol h−1 g−1 with 86% selectivity for over 5 h by using photoactive dyes in conjunction with the Re-Bpy-sp2c-COF. Notably, loading the Re-Bpy-sp2c-COF with varying amounts of Pt co-catalyst can generate tunable syngas with the CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 ratio ranging from 4:1 to 1:10. Another –CC– bonded COF viCOF-bpy-Re was formed through condensing trimethyltriazine and 2,2′-bipyridyl-5,5′-dialdehyde. The rational incorporation of Re as the reduction site and triazine core as the oxidation site promoted efficient charge separation, leading to simultaneous CO2 reduction to CO with a 190.6 μmol g−1h−1 rate and 100% selectivity and water oxidation to O2.88 The robust sp2 linkage led to excellent catalytic stability for over 50 h.
:H2 ratio ranging from 4:1 to 1:10. Another –CC– bonded COF viCOF-bpy-Re was formed through condensing trimethyltriazine and 2,2′-bipyridyl-5,5′-dialdehyde. The rational incorporation of Re as the reduction site and triazine core as the oxidation site promoted efficient charge separation, leading to simultaneous CO2 reduction to CO with a 190.6 μmol g−1h−1 rate and 100% selectivity and water oxidation to O2.88 The robust sp2 linkage led to excellent catalytic stability for over 50 h.
Furthermore, a β-keto-enamine-linked TpBpy COF was explored for photocatalytic CO2 reduction by decorating it with Ni active sites.89 The photocatalytic performance was evaluated in an aqueous medium using [Ru(bpy)3]Cl2 as the photosensitizer and TEOA as the sacrificial agent. Ni-TpBpy generated 4057 μmol g−1 CO in 5 h with 96% selectivity compared to pristine TpBpy, which produces only 4 μmol h−1 CO with 83% selectivity. Interestingly, an imine-bonded Ni bipyridine COF shows a much inferior performance at 0.7 μmol of CO and SCO/H2 = 58%, highlighting the importance of keto formation in stabilizing the key intermediates by H-bonding interactions during catalysis. The works provides important insights into tuning the structure to improve selectivity over the competing hydrogen evolution reaction.
Thus, the bipyridine core stands out for its facile synthesis and the ease of integration into different COF systems, connected via various linkages such as imine, vinylene, and β-keto-enamine. This variation in linkages can influence the conjugation length, which in turn affects the optoelectronic properties and long-term recyclability of the materials. The adjacent N atoms in 2,2′-bipyridine are susceptible to functionalization with most transition metals including Re, Ni, Co, Cu, Ru, Ir, etc., making them excellent candidates for a range of photocatalytic reactions.
000 μmol g−1 h−1, SCO/H2 = 95.7%, and a TOF of 111.8 h−1 in 4:1 MeCN/water solution using [Ru(bpy)3]Cl2 as a photosensitizer and TEOA as an ED under visible light irradiation.90 Notably, the CO production rate of Co-2,3-DHTA-COF was 23.6 times that of the pristine 2,3-DHTA-COF. Interestingly, another keto-enamine linked Co-TP-TAPT COF having a coordination mode of Co–O3N showed much lower RCO = 11600 μmol g−1 h−1 and SCO/H2 = 76.6%. The results highlight the role of the microenvironment in determining the CO2 reduction efficiency and selectivity, with the Co–O4 coordination mode being more active than the Co–O3N site.In another report, a series of Co-porphyrin donor–acceptor COFs were synthesized by varying the charge-carrier density and π-conjugation length of the donor group to gain essential insights into the role of electronic structure in CO2 reduction performance. Three COFs were synthesized by the Schiff base condensation of Co-5,10,15,20-tetrakis-(4-aminophenyl)-porphyrin acceptor and donor moieties based on benzodithiophene (BDT), thienothiophene (TT), or phenyl (TA).93 Subsequently, the photocatalytic CO2 reduction performance was monitored by dispersing the COF photocatalyst in MeCN and using Re-(bpy)(CO)3Cl as a photosensitizer and TEOA as a SED. Clearly, a BDT bearing COF showed the best CO evolution rate of 1424 μmol g−1 h−1. The other COFs bearing thienothiophene or phenyl groups demonstrated much inferior performance of RCO (Co-Por-TT) = 1182 μmol g−1 h−1 and RCO (Co-Por-TT) = 642 μmol g−1 h−1. The results highlight that that longer conjugation lengths and better donation ability are associated with higher charge carrier mobility and reduced exciton binding energy, leading to better performance.
Recently, a robust imidazole-linked photoactive M-Por-Py-COF (M = Co2+, Ni2+, Cu2+, and Zn2+) was demonstrated for photocatalytic CO2 conversion.94 Besides structural robustness, the imidazole group also shows a high affinity towards CO2 uptake. The photocatalytic CO2 reduction by dispersing the catalyst in a MeCN/water mixture, using Ru(bpy)3Cl2·6H2O as the photosensitizer and TEOA as the SED showed that the Co-Por-Py-COF gave the best CO production rate of 9645 μmol g−1 h−1 with 96.7% selectivity, more than 45 times higher than the pristine COF. Interestingly, the Ni-Por-Py-COF further tandem catalyzes the subsequent reduction of the generated CO to CH4 at the rate of 463.2 μmol g−1 h−1. The metal centres act as active sites for CO2 photoreduction by promoting CO2 adsorption and stabilizing the reaction intermediates and the final desorption of the produced CO.
The above study suggests that acenaphthylene-1,2-diimine presents a promising platform for CO2 photoreduction as it can bypass the use of predesigned metal co-ordinating ligands in the building blocks. Hence, more studies are required in this direction to unlock its full potential.
In another fascinating report, mononuclear (M-N2O2) and binuclear salen (M-N2O2–M-N2O2) metal sites, where M = Zn/Co, were incorporated in COFs for tunable syngas production by controlling the metal centres and their coordination environments. The crystalline M(salen)-COF-1 was obtained by the one-step Schiff base reaction of 1,4-hydroxyisophthalaldehyde (or 2-hydroxybenzene-1,3,5-tricarbaldehyde), ethylenediamine, metal acetate, and C3 amine (1,3,5-tris(4-aminophenyl)benzene (TAPB)/2,4,6-tris(4-aminophenyl)-1,3,5-triazine (TAPT)) (Fig. 6f).97 Likewise, M(salen)-COF-2 was synthesized by the two-step reaction involving the synthesis of MM-CHO/M-CHO followed by the Schiff base reaction with TAPT or TAPB amine. Subsequently, the photocatalytic CO2 reduction performance of Zn-TAPB-COFs, ZnZn-TAPB-COFs, Co-TAPB-COFs, and Co-TAPT-COFs was evaluated to determine the role of the synthetic protocol, co-ordination mode, metallic site, and ligand. COFs with Zn metal sites (Zn-TAPB and ZnZn-TAPB COFs) demonstrated lower CO and higher H2 production rates. In contrast, COFs bearing Co centres (Co-TAPB and Co-TAPT COFs) generated much higher amounts of both CO and H2. Notably, Co-TAPT-COF-1 showed a much higher photocatalytic activity than Co-TAPT-COF-2, bearing a similar chemical structure, highlighting the role of the synthetic protocol in the reaction efficiency. The superior performance of Co-TAPT-COF-1 can be attributed to its higher crystallinity and the nanotubular superstructure. Moreover, Co-TAPT-COF-1 demonstrated higher syngas production rates than Co-TAPB-COF-1, suggesting the importance of ligands in tuning the catalytic performance. Interestingly, tuning these parameters can control the H2/CO ratios in the resulting syngas from 1:1 to 30:1.
Although metallo-salen-based COFs are versatile in terms of metal binding, one significant drawback is the limited stability of the salen linkage. Thus, large scale applications of these materials require improvement in their structural stability.
In another report, the heterometallic hydrazone-linked COF (USTB-11(Cu,Ni)) was designed by incorporating the Cu3 cluster and the post-synthetic treatment to incorporate Ni. USTB-11 (Cu) was developed by the reaction of 4,4′,4′′-nitrilo-tribenzhydrazide (NTB-NH2) and tris(μ2-4-carboxaldehyde-pyrazolato-N,N′)-tricopper (Cu3Py3) (Cu3(PyCA)3) and the PSM with the Ni2+ ions generates USTB-11(Cu,Ni).100 As a proof of concept, a metal-free analogue USTB-12 was also synthesized by the condensation of 1,3,5-tris(4-formylphenyl)benzene (TBZ-CHO) and NTB-NH2 and further PSM with Ni2+ produces USTB-12(Ni). The heterometallic catalyst USTB-11(Cu,Ni) demonstrates a high CO production rate of 22130 μmol g−1 h−1 with 98% selectivity, 7 times higher than USTB-12(Ni), highlighting the role of the synergy of Cu3Py3 and Ni in determining the catalytic performance. In contrast, the other catalysts, USTB-11(Cu) and USTB-12, with only Cu clusters or no metal, show negligible photocatalytic activity. Mechanistic investigation with advanced spectroscopic techniques revealed the CuI/CuII mixed oxidation state in the Cu3Py3 cluster with bistable Cu34+(CuI2CuII): Cu35+(CuICuII2) is greatly responsible for the improved catalytic performance, charge separation, and improved performance of Ni sites.
To further explore the role of linkage chemistry, a fully conjugated multivariate –CC– linked donor-π–acceptor MCOF, namely UJN-1, based on Cu cyclic trimer (Cu-CTU) sites was fabricated. UJN-1 was synthesized by the integration of 1,4-phenyldiacetonitrile (PDAN) with two different aldehyde precursors, electron-rich tris(4-formylphenyl)amine (TFPA) and an electron-deficient Cu3L3 cluster [1H-pyrazole-4-carbaldehyde (HL)]. This resulted in a robust structure with strong intramolecular charge separation due to the donor–acceptor moieties, extended conjugation, and abundant active sites. The meticulous design results in a high photocatalytic CO production rate of 114.8 μmol g−1 in 4 h without additional co-catalysts or sacrificial reagents.101 The mechanistic investigation revealed that the high performance is due to incorporating electron-deficient Cu-CTU that facilitates charge separation, accelerating photogenerated charge transfer from the electron-rich triphenylamine to Cu-CTU. A similar MCOF, UJN-2, with imine-linkage, and vinylene-linked COF UJN-3, lacking Cu-CTU moieties, show much lesser CO production of 28.9 μmol g−1 and 50.0 μmol g−1 underscoring the importance of charge separation driving force and the linkage in guiding the photocatalytic performance.
Furthermore, as discussed previously, a dual metalation strategy can be used to improve the catalytic performance of the COFs. A series of crystalline benzothiazole COFs (Co, Ni COF-n, n = 0,1,2,3, denoting the number of carbonyls in the as-synthesized COFs) were obtained by the reaction with benzene-1,3,5-tricarbaldehyde derivatives bearing various carbonyl groups to tune the photophysical properties.103 DFT calculations revealed that the metal ions were coordinated in the interlayer spacing with metal-nitrogen interaction, and a spacing of 3.5 Å was the optimized spacing. The photocatalytic measurements revealed that CoNi COF-3 had the highest CO production rate of 2567 μmol g−1 h−1 with ∼92% selectivity. The best performance is owing to the synergistic effect of the dual metallic sites and the formation of the β-ketoenamine structure in the COF that facilitates CO2 adsorption, stabilizes the CO2 reduction intermediate, and promotes charge separation.
In other materials, dual metalation can be done on the COF backbone and the interlayer spacing. The Co/Cu3-TPA-COF was engineered by incorporating trinuclear Cu clusters in the COF structure linked by covalent bonds, and the single atomic Co sites are intercalated between the COF layers by anchoring on the imine-N moieties.104 The Co centres are not only well dispersed and exposed within the COF channels but also promote oriented charge transfer from dual electron donors to Co sites, facilitating CO2 reduction performance. The electron-withdrawing CuI/CuII centres in the Cu cluster and electron-rich amine site promote charge separation upon visible light illumination. This leads to effective induction of oriented electron transfer from the COF backbone to the intercalated Co-imine N sites, resulting in a high CO evolution rate of 25247.7 μmol g−1 h−1 with ∼80% selectivity.
Metal ions can interact synergistically with the COF structure, leading to new reaction pathways, enhanced stability, and increased durability under reaction conditions. However, like other metal-based COF photocatalysts, they are also prone to leaching of metal ions, degrading the performance. Also, achieving the structure necessitates complex design to promote intercalation between layers.
6.2. Encapsulating metallic catalysts in the COF pores
The high porosity, well-defined pore channels, and tunable microenvironments of COFs make them ideal scaffolds for confining various systems, including nanoparticles, polyoxometalates, and other homogeneous catalysts. Such confinement can significantly influence the chemical and physical properties of these systems through the synergistic effects between the confined catalyst and the porous frameworks.105 The close spatial proximity between substrates and catalysts within COF pores facilitates increased reaction rates, improves the stability of homogeneous catalysts, and supports long term recyclability under the reaction conditions.106 The catalysts can be confined in COF cavities through various interactions, including electrostatic, van der Waals, hydrogen, coordination, and covalent bonding.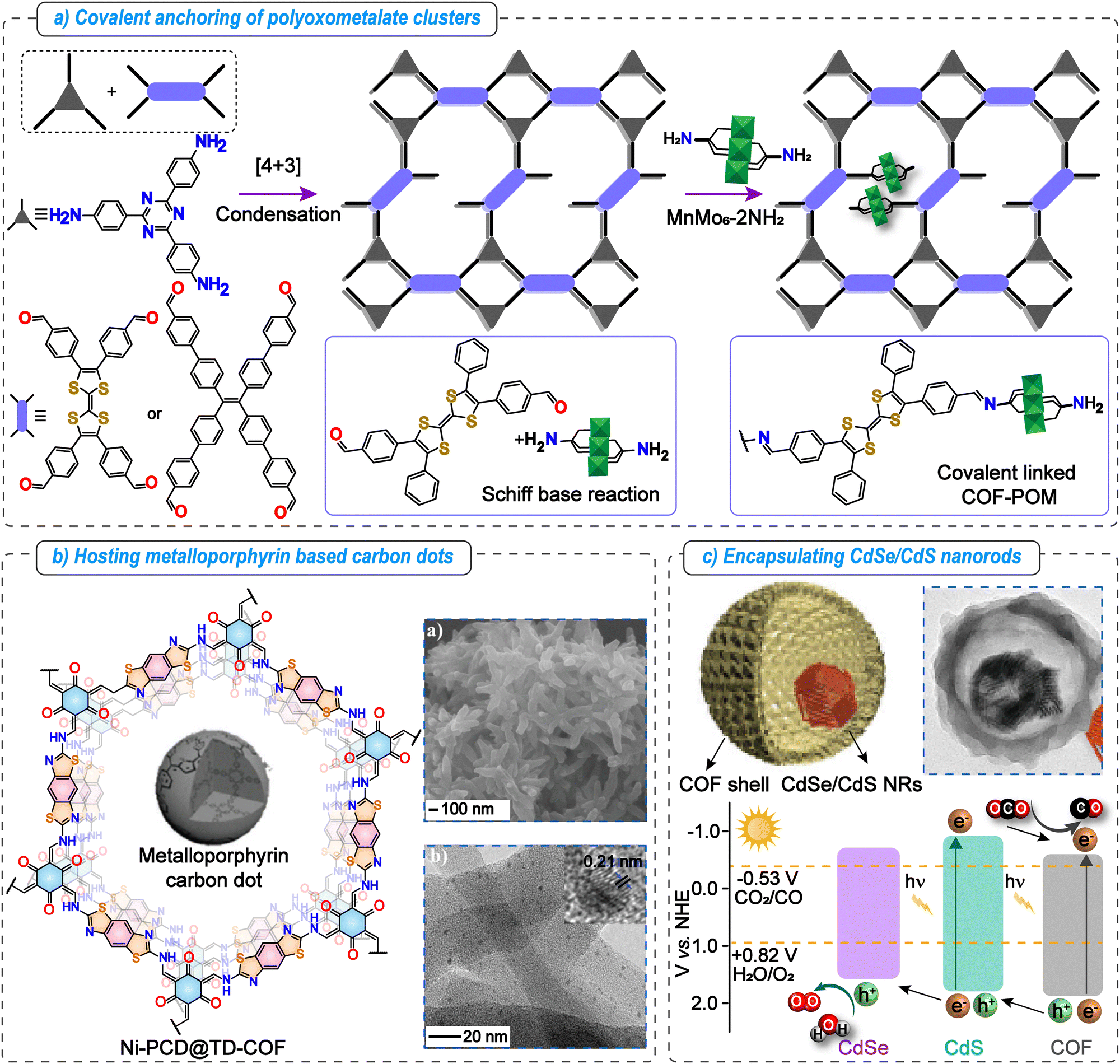 | ||
| Fig. 7 Metalated COFs beyond single atom catalysts for photocatalytic CO2 reduction. Key strategies to use the nanoconfinement effect of COFs. (a) Schematic illustration of covalent anchoring of polyoxometalate clusters in COF pores. (b) Hosting of metalloporphyrin-based carbon dots in a β-ketoenamine linked TD-COF. The SEM image indicates COF morphology and the TEM image indicates the uniformly dispersed carbon dots in the COF matrix. Adapted with permission from ref. 109, © 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (c) Schematic illustration and TEM image of the double-shelled structure formed by encapsulating CdSe/CdS nanorods in the COF. Band-structure diagram of TAPT-DMTA, CdS, and CdSe illustrating the plausible electron transfer pathway. Adapted with permission from ref. 110, Copyright © 2022, Hui Li et al. | ||
Another example includes poly aryl ether (PAE) linked COF-316 and COF-318 bearing EWGs susceptible to nucleophilic attack by –OH groups. Utilizing this, they are covalently anchored with oxide semiconductors (TiO2, Bi2WO6, and α-Fe2O3) having surfaces functionalized with hydroxy functionalities.111 These inorganic catalysts are capable of water oxidation, while COF-316 and COF-318 can perform CO2 photoreduction. Among all the synthesized catalysts, the COF-318-TiO2 heterojunction catalyst showed the highest CO production rate of 69.67 μmol g−1 h−1 under gas–solid conditions without any external photosensitizer or SED, much superior to the pristine COF or TiO2 catalysts or their physical mixture. Mechanistic investigations using DFT revealed that photogenerated charges are transferred from the semiconductor to the COF via covalent linkages and accumulate on the –CN/pyridine active sites of the COF backbone, which facilitates CO2 reduction. Meanwhile, the holes remaining on the semiconductor drive water oxidation.
Leveraging the photocatalytic properties of both MOFs and COFs, a covalently tethered MOF–COF hetero-nanosheet (NZZ/TTCOF) was synthesized via a seed-mediated strategy.112 The TTCOF was formed through an imine condensation reaction between TP and TAPT precursors, while the NZZ MOF nanosheets were prepared by coordinating 2-methylimidazole and 5-amino-1H-tetrazole with Zn nodes. The resulting heterostructure was achieved by covalently anchoring the COF onto the MOF nanosheets through C–N bonds within the keto-enamine framework of the TTCOF. NZZ exhibits UV responsiveness, while the TTCOF is responsive to IR radiation, enabling the heterostructure to achieve broad-spectrum light absorption capabilities. As expected, the NZZ/TTCOF hetero-nanosheet exhibits superior photocatalytic CO2 reduction ability (RCO = 7.01 μmol g−1 h−1 and RCH4 = 2.50 μmol g−1 h−1) compared to both the pristine TTCOF and NZZ MOF, highlighting the role of forming heterostructures in improving the catalytic performance. Notably, this was achieved under gas–solid conditions without any external photosensitizer or co-catalyst.
Compared to the metallic active sites anchored through non-covalent linkages, the catalysts presented in the reports discussed above possess significant advantages. The metal atoms tend to leach over several cycles and are also known to coagulate into larger particles degrading the catalytic activity over time. In contrast, covalent anchoring of the semiconductor is an important development in the direction of overcoming this limitation. However, this requires sophisticated designs which can be sometimes complex to synthesize.
The selective photocatalytic reduction of CO2 to HCOOH was obtained by immobilizing Ru nanoparticles (∼8 nm) in a β-ketoenamine-linked Tp-Pa1 COF.115 Like the previous cases, forming an organic–inorganic composite structure facilitated broader light absorption and electron transfer through the Ru NPs. 3.0 wt% Ru/TpPa-1 demonstrated the highest HCOOH yield of 108.8 μmol g−1 h−1, superior to Tp-Pa1 (32.4 μmol g−1 h−1). Likewise, the role of the semiconductor facet in determining the photocatalytic efficiency was highlighted by synthesizing 101-TiO2/COF and 001-TiO2/COF heterostructures.116 It was found that 101-TiO2/COF, having an electron-rich nature and a relatively lower conduction band than 001-TiO2/COF, showed a higher CO2 reduction efficiency with a CO production rate of 11.6 μmol h−1. In contrast, the CO production rates of 101-TiO2 and 001-TiO2/COF were 0.8 and 2.5 μmol h−1, respectively.
The immobilization of inorganic nanoparticles with organic COF supports presents an interesting platform for enhancing CO2 photoreduction efficiency. However, with long-term use, the crystallinity of the COFs tends to degrade, leading to potential metal leaching. Additionally, the nanoparticles are susceptible to coagulation over multiple cycles, which can undermine the overall performance. To address this critical challenge, further research is required to improve the design.
Thus, metalated COF-based catalysts for photocatalytic CO2 reduction offer several advantages, making them highly effective in converting CO2 into value-added products. The well-defined pore structures and tunable linkers, linkages, and morphologies of COFs allow precise incorporation of metal active sites, enhancing catalytic efficiency and providing greater synthetic flexibility. This structural control not only improves metal accessibility, reducing the overall metal requirement, but also facilitates catalyst recyclability. When incorporated into porous materials like COFs, these metal sites become more accessible, reducing the overall metal requirement and enhancing recyclability. Additionally, the protective environment of COFs stabilizes the metal centres, prolonging the catalyst lifetime and maintaining activity over multiple cycles. However, metal-based catalysts also have notable limitations. One major drawback is metal leaching, which leads to a decline in catalytic performance over time. Furthermore, the reliance on noble metals, such as Pt or Ru, increases costs and reduces the sustainability of large-scale applications.
6.3. Metal-free COFs as heterogeneous catalysts for photocatalytic CO2 reduction
COFs constructed from π-conjugated building blocks offer an excellent platform for developing metal-free photocatalysts for CO2 reduction reactions (CO2RR). These frameworks can be enriched with heteroatoms, such as nitrogen (N), sulfur (S), and phosphorus (P), which serve as active sites for CO2 adsorption and subsequent conversion. The heteroatoms facilitate CO2 activation by inducing polarization through dipole–quadrupole interactions.Furthermore, the extended π-conjugation in these materials enhances visible-light absorption, while their donor–acceptor architecture promotes efficient charge separation and prolongs the lifetime of photoexcited electrons. Various photoactive moieties, such as eosin Y, pyrene, and perylene, have been incorporated into the COF or porous organic polymer (POP) backbones to enhance visible-light responsiveness.117,118 Additionally, triazine- based linkers, which feature abundant nitrogen sites, have shown significant potential for CO2 activation. This section highlights key design strategies to develop metal-free COF-based systems for photocatalytic CO2 reduction.
 | ||
| Fig. 8 Key strategies to develop metal-free COFs for photocatalytic CO2 reduction. (a) Formation of a donor–acceptor structure, (b) design of 3D structure, adapted with permission from ref. 121, Copyright © 2022, American Chemical Society. (c) Fabricating COF membranes with the triazine–imine–triazine linkage, (d) incorporation of heteroatoms, and (e) triazole-based system. | ||
Another study demonstrated the effect of bromine moiety incorporation on modulating the bandgap for improving photocatalytic performance. COF-366, designed from the condensation of 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (TAPP) and terephthalaldehyde, doesn’t have an appropriate band position for the water oxidation reaction. To overcome this limitation, bromine was incorporated into the building unit to design the TAPBB-COF by condensing TAPP with 2,5-dibromo-terephthalaldehyde (BDB).124 The TAPBB-COF demonstrated a CO generation rate of 24.6 μmol g−1 h−1 with greater than 95% selectivity, about 3 times higher than COF-366 and 1.5 times over g-C3N4. Introducing Br sites shifts the VB to a more positive position, promoting the water oxidation reaction. DFT calculations support the role of the N atoms of the porphyrin ring and imine group and the Br atoms in enhancing CO2 activation and conversion with water in the photocatalytic reaction.
:1 MeCN:H2O solvent. Even in direct sunlight, an excellent CH4 production rate of 61.62 μmol g−1 h−1 was achieved. DFT calculations showed that the presence of 1,2,4-triazole sites in the COF backbone stabilizes the *CO intermediate through electron donation, thereby assisting the multielectron reduction of CO2. The triazole species exists in an equilibrium between tautomeric forms 1H-triazole and 4H-triazole that can be interconverted via weak bases, like, in this case, TEA. This is also supported experimentally as the absence of TEA and using only BNAH as a sacrificial agent lead to poor catalytic performance. 1H-Triazole plays an active role in stabilizing the *CO intermediate.Building on the previous discussion, COFs offer exceptional structural tunability, which enables the design and optimization of systems for photocatalytic CO2 reduction. Their environmental friendliness and sustainability further enhance their potential for future applications. Recent efforts have focused on performing these reactions in an aqueous medium, which significantly increases their practical appeal. The structural tunability of COFs allows for the design of materials with high selectivity, minimizing the competing hydrogen evolution reaction. Reticular chemistry is employed to adjust the band gap and facilitate charge transfer, addressing the challenge of water oxidation. Additionally, the reduced reliance on sacrificial agents helps prevent the overestimation of CO2 reduction products. Many studies have incorporated isotopic labeling with 13CO2 to confirm the source of reduction products and eliminate the possibility of products stemming from secondary electron donors (SEDs). In situ techniques like DRIFTS provide valuable insights into the active intermediates formed during the reduction process. As previously mentioned, the presence of extended conjugation in COFs positions them as promising candidates for overcoming the reliance on metals. However, the field is still in its early stages, and the efficiency of metal-free catalysts must be further improved to meet industrial standards.
7. Structure–property relationship in reticular materials for modulating photocatalytic CO2 reduction
The photocatalytic CO2 reduction performance of a material, characterized by its efficiency and selectivity, is governed by a complex interplay of various material properties, including CO2 uptake capacity, intermediate stabilization, light-harvesting ability, band alignments, structural stability, and accessibility to active sites. Understanding the structure–property relationship is crucial for optimizing photocatalytic activity. For instance, the accessibility of active sites significantly influences catalytic efficiency; however, despite high porosity, strong interlayer stacking in certain COFs may hinder access to deeply buried active sites.127 Developing strategies to enhance active site accessibility remains a critical area of research.Covalent organic frameworks (COFs) offer exceptional structural tunability, where minor structural modifications can drastically impact reaction rates, product selectivity, and catalytic efficiency. Researchers can optimize critical parameters such as adsorption, activation, charge transport, and product specificity by systematically leveraging this tunability. Although understanding the structure–property relationship in COFs is still in their nascency, significant efforts have been made to identify the parameters affecting photocatalytic performance. This section provides an overview of how subtle structural modifications in reticular materials guide their photocatalytic performance, offering insights into the design and development of advanced catalysts for CO2 reduction.
7.1. Spin-state manipulation of the metal active sites
The N4 binding pockets in the porphyrin are apt for binding various metal ions and are capable of modulating the catalytic performance. Jiang and co-workers have shown that for the same metal ion, manipulation of the oxidation state, and hence the spin state, can profoundly impact selectivity and efficiency. To demonstrate this, COF-367-bearing cobalt-porphyrin groups with CoII and CoIII having a spin state of S = 1/2 and 0 spin ground states, respectively, were synthesized (Fig. 9a).128 The evaluation of photocatalytic CO2 reduction depicts almost similar production rates (in μmol g−1 h−1) of HCOOH (48.6 ± 9.2), CO (16.5 ± 1.9), and CH4 (12.8 ± 1.9) in the material bearing CoII (COF-367-CoII). In contrast, COF-367-CoIII demonstrates a highly selective HCOOH production rate of 93.0 ± 4.63 μmol g−1 h−1. At the same time, a negligible amount of CO and CH4 is produced, indicating the role of spin state modulation in guiding the photocatalytic activity and selectivity. COF-367 doesn’t show any photocatalytic CO2 reduction, highlighting the role of Co active sites. Notably, the precursors CoII-TAP and CoIII-TAP show much lower activity towards HCOOH production, suggesting activity enhancement during the periodic assembly of the multiple Co-TAP units in the COF. The photoelectrochemical measurement shows that the spin state is essential in determining charge separation efficiency, which follows the order of COF-367-CoIII > COF-367-CoII > COF-367. Theoretical insights using DFT suggest that strong binding of CO2 and HCOOH in COF-367-CoIII over COF-367-CoII ultimately translates into high selectivity and activity. The first step involves CO2 binding through the formation of a Co–O bond, and the calculations show higher binding energy and a smaller bond length in COF-367-CoIII over COF-367-CoII, suggesting stronger binding with COF-367-CoIII. The more favorable binding energy results from the spin state of the central cobalt ion, wherein in CoII with S = 1/2, Co-3dxz or Co-3dyz couples with O-2p. Meanwhile, for COF-367-CoIII, Co-3dz2 overlaps with O-2p, resulting in a much stronger orbital interaction. The further conversion of adsorbed CO2 to HCOOH proceeds through two hydrogenation steps, which are more favorable for COF-367-CoIII over COF-367-CoII, supporting its higher activity towards HCOOH production. The subsequent conversion of HCOOH to CO and CH4 is energetically more favorable in COF-367-CoII, accounting for the poor selectivity. The work highlights the role of reticular chemistry in promoting structural changes that can reciprocate towards guiding the product selectivity and catalytic activity. | ||
| Fig. 9 Structure–property relationship in reticular materials for modulating photocatalytic CO2 reduction. (a) Schematic illustration to show the effect of spin state modulation of the active metal centre to tune the photocatalytic efficiency and selectivity. (b) Delamination of bulk COFs into ultrathin nanosheets to modulate photocatalytic efficiency and selectivity. (c) Postsynthetic annulation to modulate the photophysical properties to improve photocatalytic performance. Adapted with permission from ref. 121, Copyright © 2023, American Chemical Society. | ||
7.2. Dimensionality modulation
The rigid and planar building blocks of 2D COFs lead to strong interlayer stacking, limiting active site accessibility. Likewise, most 3D COFs are based on tetrahedral building blocks restricting structural diversity and functional regulation. Besides, interpenetration in these materials leads to decreased porosity and smaller pore sizes, which are detrimental to catalysis. In this context, Fang et al. synthesized an stp-topologized 3D COF JUC-640-M (M = Co, Ni, or H) based on D3h triptycene and metalloporphyrin building blocks.129 The large surface areas and interconnected pore channels in this 3D COF make the active porphyrin sites more accessible and exposed. Inspired by this, the photocatalytic CO2 reduction studies were performed in an MeCN:H2O = 2:3 mixture utilizing BIH as the SED and Ru(bpy)3Cl2·6H2O as the photosensitizer. This results in high photocatalytic CO2 conversion to CO 15.1 mmol g−1 h−1 with more than 94% selectivity in JUC-640-Co.
The 2D imine-linked COFs suffer from poor charge transfer efficiency due to the polarization of the imine linkage, which increases the energy barrier for electron delocalization and leads to low efficiency without external photosensitizers. To overcome this limitation, Cao and co-workers developed 1D pyrene-based COFs bearing dual-chain-like edge architectures. The 1D PyTTA-COF-Co demonstrated a CO evolution of 1003 μmol g−1 in 8 h, over 59 times higher than the 2D congener, highlighting the role of dimensionality in determining the catalytic efficiency.130 Insights from the experimental observations and theoretical calculations show that 1D structures reduce exciton dissociation and thermal relaxation of photoelectrons, thereby improving photocatalytic efficiency.
However, research on 3D and 1D COFs for photocatalytic CO2 reduction is still in its nascency and more research is required to unlock their full potential.
7.3. Improving the active site accessibility
As discussed earlier, 2D COFs suffer from mass transfer limitations due to their strong interlayer stacking. A way to overcome this limitation is to increase the interlayer stacking distance. A series of 2D porphyrin-decorated COFs were synthesized by the reaction of porphyrin aldehyde MPor-CHO (M = H2, Co, and Ni) with 3,8-diamino-6-phenylphenanthridine (DPP).131 The presence of a bulky phenyl linker in the DPP amine leads to a high interlayer distance of 6 Å in the MPor-DPP-COFs. Consequently, the CoPor-DPP-COF showed a high Co generation rate of 10200 μmol g−1 h−1 with 82% selectivity. In contrast, COF-367-Co, having a similar Co active site but smaller interlayer spacing, showed a much inferior CO production rate of 5560 μmol g−1 h−1 with reduced selectivity of 78%, highlighting the importance of interlayer spacing in guiding the performance.
Another vital strategy in this context is the delamination of bulk COFs to produce ultrathin covalent organic nanosheets (CONs). Compared to the stacked 2D counterparts, CONs with minuscule thickness boast higher accessible sites, leading to shorter diffusion pathways. Besides, they are more processable than bulk COFs and can be dispersed well in several organic solvents. Taking advantage of these features in CONs, Jiang et al. developed a series of CONs with thickness <2.1 nm through a bottom-up imine exchange strategy. To achieve this, an excess amount of bulky 2,4,6-trimethylbenzaldehyde was added to the reaction mixture that reacted along the edges during COF crystallization, hindering the π–π stacking and growth along the z direction.132 Notably, COF-367-Co NSs demonstrate a high CO production rate of 10162 μmol g−1 h−1 with 78% selectivity, exceeding the bulk counterpart that showed a far inferior CO production rate of 124 μmol g−1 h−1 with a selectivity of only 13% under similar conditions, highlighting the importance of NSs in improvising the performance (Fig. 9b). Likewise, Cooper and co-workers designed a series of CONs with Co active sites coordinated through the iminopyridine functionality for photocatalytic CO2 reduction. The Co-FPy-CONs with fluorine to enhance the CO2 uptake showed CO production of 10.1 μmol with 76% selectivity in 6 h, 4.3 times higher than the bulk Co-FPy-COF, which might be partially due to lower Co loading.133 The Co-Py-CON produced a less CO of 7.4 μmol in 6 h but was again higher than the bulk material.
7.4. Post-synthetic annulation
The prerequisite of photocatalytic activity is efficient light harnessing. Introducing highly conjugated building units into the COF backbone is instrumental in light absorption, but it is limited by the poor solubility of the precursors hindering COF crystallization. Post-synthetic annulation is an intriguing approach to enhance the light harnessing and stability, thereby enhancing the photocatalytic activity of the resulting material. Compared to 2D COFs, 3D COFs serve as suitable platforms for post-synthetic functionalization owing to their more exposed sites and interconnected pore channels. A 3D COF, NJU-319Fe, was synthesized by connecting hexaphenyl-triphenylene and Fe-porphyrin-based building units.134 Interestingly, hexaphenyl-triphenylene was post-synthetically cyclized into higher π-conjugated hexabenzo-trinaphthylene, generating the pNJU-319Fe COF, using intramolecular oxidative cyclodehydrogenation, commonly known as the Scholl reaction under mild conditions (Fig. 9c). As expected, the PSM enhanced visible light absorption reflected in the photocatalytic CO2 reduction performance. The pNJU-319Fe COF showed a CO evolution of 688 μmol g−1 in 10 h, 2.5-fold higher than the pristine NJU-319Fe (268 μmol g−1) under similar conditions. The study highlights the role of small structural tunability that can profoundly impact the overall material performance.7.5. Electronic property modulation
Imine chemistry has been widely explored for developing COF photocatalysts for the CO2RR. Interestingly, the linkage direction, i.e., whether the imine linkage is forward (f) or reversed (r) in isomeric COF structures, can have a profound impact on determining the catalytic performance. Four imine-linked COFs incorporating bipyridine and triazine groups were designed, both in their metal-free and Re-metalated forms. These COFs featured distinct linkage orientations designated as the f-COF, Re-f-COF, r-COF, and Re-r-COF.135 The isomeric Re-r-COF (7.4% Re) and Re-f-COF (6.2% Re) have distinct photocatalytic performance. The Re-f-COF shows a CO generation rate of 6.3 ± 1.2 mmol g−1 in 8 h, while the Re-r-COF gives only 0.30 ± 0.05 mmol g−1 in the same duration. The drastic change in the behavior is attributed to the change in the photophysical properties of the structure. 2,2′-Bipyridine behaves as an electron-donating group in the r-imine, while it switches to an electron acceptor in the f-imine. This, in turn, reverses the direction of intramolecular charge transfer (ICT). The imine reversal is further manifested in the orbital overlap of the Re and bpy in the metalated COFs, significantly affecting the excited state and charge separation pathway. The excited state was more prolonged in the Re-f-COF than in the Re-r-COF. These differences brought about by slight structural changes had a huge impact on the overall catalytic performance, making the isomeric Re-f-COF a better photocatalyst over the Re-r-COF.The choice of building units can influence the local electron density and charge separation, determining the catalytic efficiency. High local electron density can be obtained using building units with electron-rich and electron-deficient properties (ERP and EDP) linked through an efficient charge transfer channel. Keeping this in perspective, VL-MCOF-1 was designed by Knoevenagel condensation of the electron-rich metal cluster Cu3(PyCA)3 ED 3,5-dicyano-2,4,6-trimethylpyridine (DCTMP) linked via robust –CC– linkage.136 As expected, VL-MCOF-1 with a well-defined molecular junction showed a high CO2 to HCOOH reduction efficiency of 283.41 μmol g−1 h−1 with 97.1% selectivity. However, replacing any one of the strong ED or ER with weaker moieties led to much inferior photocatalytic performance. For instance, VL-MCOF-2 bearing the same metal cluster and linkage but a weaker ED, 2,4,6-trimethyl-1,3,5-triazine, gave a HCOOH generation rate of 195.57 μmol g−1 h−1 with 96.34% selectivity, and the g-C34N6-COF, formed with a weak ER and a strong ED DCTMP, gave a much lower HCOOH production of 31.53 μmol g−1 h−1 with 82.09% selectivity. Light utilization was significantly enhanced in VL-MCOF-1 due to the synergistic effect of ER and ED groups and the appropriate CTC, highlighting the importance of modulating local electron density in the overall catalytic performance.
8. Conclusions and future outlook
The molecular engineering of COFs with structural and functional modulation to boost the photocatalytic CO2 reduction efficiency is currently emerging as a promising avenue. In this review, we have summarized the key developments in this field and tried to evaluate the key parameters instrumental for improving the catalytic properties. We have classified COF-based photocatalysts into metalated and metal-free categories and provided an in-depth analysis of the various synthetic protocols developed in these areas. Our discussion encompasses COFs with intrinsic single-atom metallic active sites as well as those functionalized through post-synthetic modifications. Additionally, we have highlighted significant systems designed to leverage the nanoconfinement effects of COF pores. Key design strategies for developing metal-free photocatalysts have also been thoroughly examined. Throughout the discussion, we have emphasized the strategies researchers have employed to address the challenges associated with photocatalytic CO2 reduction using covalent organic framework photocatalysts.The development of futuristic catalytic systems requires taking the pros and cons of the process into account simultaneously. While high efficiency and selectivity have been obtained with COFs, the rampant use of sacrificial agents like TEOA and organic solvents is a cause of concern. On the one hand, incorporating these additives renders the process economically unsustainable; more critically, their degradation under reaction conditions could lead to overestimating the product, hindering future advancements. Efforts need to be made to increase the sustainability of the process by developing efficient catalysts that can perform in the water medium. Design strategies must be developed to increase the local concentration of CO2, promote the challenging 4e− water oxidation process, and minimize the competitive hydrogen evolution reaction in an aqueous medium.
Another organic photocatalyst, such as graphitic carbon nitride (g-C3N4), can undergo rapid self-decomposition under reaction conditions, contributing to gaseous byproducts that might be misinterpreted as CO2 reduction products. Therefore, comprehensive isotopic studies are crucial to eliminate such ambiguities. Furthermore, understanding the catalytic mechanism in COF-based catalysts remains challenging due to their heterogeneous nature. However, with advancements in the field, detailed in situ mechanistic investigations are essential to elucidate the reaction pathways and identify the intermediates involved. Although COFs are stable for several cycles, consistent efforts are required to improve their photostability under harsh conditions.
COFs are predominantly synthesized as insoluble powdered materials, subsequently dispersed and utilized for catalysis. However, for practical applications, it is essential to control the macroscopic architecture of COFs, where alternative forms such as films, membranes, and granules could offer greater viability. While the chemical tunability of COFs has been extensively explored, studies on their nanomorphologies and their influence on bulk-scale properties remain limited. Moreover, nanomorphologies significantly affect catalytic performance; for example, COF nanosheets, with their higher density of exposed active sites compared to bulk COFs, facilitate faster diffusion kinetics and enhanced catalytic efficiency. Despite notable progress in improving COF processability, further efforts are needed to advance this area. A comprehensive understanding of the morphology–structure–function interplay is crucial for optimizing their practical applications. The large-scale synthesis of COFs and their cost also need to be considered.
The ability to fine-tune the molecular structure underscores the potential of reticular chemistry to play a pivotal role in designing next-generation CO2 reduction systems. However, the field is still in its infancy, and unlocking its full potential necessitates significant effort to open new avenues toward practical solutions for photocatalytic CO2 reduction.
Author contributions
Shibani Mohata: conceptualization, writing – original draft, writing – reviewing and editing; Poulami Majumder: global checking; and Rahul Banerjee: conceptualization, writing – reviewing and editing.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. M. and P. M. acknowledge the IISER-Kolkata IPhD program for fellowship support. R. B. acknowledges the funding from the DST Project; DST/C3E/MI2.0/CCUS/2K23/CALL/2023/153, submitted against FOA R&D in the area of CCUS with Mission Innovation and ANRF SUPRA [SPR/2021/000020] for funding.References
- W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. He and S.-E. Park, Chem. Soc. Rev., 2020, 49, 8584–8686 RSC.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS.
- S. Navarro-Jaén, M. Virginie, J. Bonin, M. Robert, R. Wojcieszak and A. Y. Khodakov, Nat. Rev. Chem., 2021, 5, 564–579 CrossRef.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS.
- Q.-J. Wu, J. Liang, Y.-B. Huang and R. Cao, Acc. Chem. Res., 2022, 55, 2978–2997 CrossRef CAS.
- S. Bierbaumer, M. Nattermann, L. Schulz, R. Zschoche, T. J. Erb, C. K. Winkler, M. Tinzl and S. M. Glueck, Chem. Rev., 2023, 123, 5702–5754 CrossRef CAS PubMed.
- S. Roy, A. Cherevotan and S. C. Peter, ACS Energy Lett., 2018, 3, 1938–1966 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- B. Chang, H. Pang, F. Raziq, S. Wang, K.-W. Huang, J. Ye and H. Zhang, Energy Environ. Sci., 2023, 16, 4714–4758 RSC.
- S. Fang, M. Rahaman, J. Bharti, E. Reisner, M. Robert, G. A. Ozin and Y. H. Hu, Nat. Rev. Methods Primers, 2023, 3, 61 CrossRef CAS.
- W. Gao, H. Li, J. Hu, Y. Yang, Y. Xiong, J. Ye, Z. Zou and Y. Zhou, Chem. Sci., 2024, 15, 14081–14103 RSC.
- L. Yang, Z. Chen, Q. Cao, H. Liao, J. Gao, L. Zhang, W. Wei, H. Li and J. Lu, Adv. Mater., 2024, 36, 2306758 CrossRef CAS PubMed.
- P. Zhou, M. Luo and S. Guo, Nat. Rev. Chem., 2022, 6, 823–838 CrossRef CAS.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- B. Mishra, A. Alam, A. Chakraborty, B. Kumbhakar, S. Ghosh, P. Pachfule and A. Thomas, Adv. Mater., 2024, 2413118 CrossRef.
- A. Basak, S. Karak and R. Banerjee, J. Am. Chem. Soc., 2023, 145, 7592–7599 CrossRef CAS.
- A. Rodríguez-Camargo, K. Endo and B. V. Lotsch, Angew. Chem., Int. Ed., 2024, 63, e202413096 CrossRef.
- Q. Lin, Y. Yusran, J. Xing, Y. Li, J. Zhang, T. Su, L. Yang, J. Suo, L. Zhang, Q. Li, H. Wang, Q. Fang, Z.-T. Li and D.-W. Zhang, ACS Appl. Mater. Interfaces, 2024, 16, 5869–5880 CrossRef CAS.
- G.-F. Liu, Z.-W. Li, Z.-J. Huang, Z. Zhou, Y.-X. Li, A. Huang, Z. Cai, G. Ouyang, B.-H. Ye and Y.-B. Zhang, J. Am. Chem. Soc., 2025, 147, 1840–1850 CrossRef CAS.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS.
- M. Kim, J. Yi, S.-H. Park and S. S. Park, Adv. Mater., 2023, 35, 2203791 CrossRef CAS.
- N. Keller and T. Bein, Chem. Soc. Rev., 2021, 50, 1813–1845 RSC.
- Z. Chen, J. Wang, M. Hao, Y. Xie, X. Liu, H. Yang, G. I. N. Waterhouse, X. Wang and S. Ma, Nat. Commun., 2023, 14, 1106 CrossRef CAS PubMed.
- S. Bag, H. S. Sasmal, S. P. Chaudhary, K. Dey, D. Blätte, R. Guntermann, Y. Zhang, M. Položij, A. Kuc, A. Shelke, R. K. Vijayaraghavan, T. G. Ajithkumar, S. Bhattacharyya, T. Heine, T. Bein and R. Banerjee, J. Am. Chem. Soc., 2023, 145, 1649–1659 CrossRef CAS.
- A. Dhakshinamoorthy, S. Navalon, A. Corma and H. Garcia, Energy Environ. Sci., 2012, 5, 9217–9233 RSC.
- J. Di, C. Chen, S.-Z. Yang, S. Chen, M. Duan, J. Xiong, C. Zhu, R. Long, W. Hao, Z. Chi, H. Chen, Y.-X. Weng, J. Xia, L. Song, S. Li, H. Li and Z. Liu, Nat. Commun., 2019, 10, 2840 CrossRef PubMed.
- J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar, D. Ma and J. Tang, Nat. Catal., 2018, 1, 889–896 CrossRef CAS.
- O. Stroyuk, A. Raevskaya and N. Gaponik, Chem. Soc. Rev., 2018, 47, 5354–5422 RSC.
- H. Mai, D. Chen, Y. Tachibana, H. Suzuki, R. Abe and R. A. Caruso, Chem. Soc. Rev., 2021, 50, 13692–13729 RSC.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- C. D. Windle and R. N. Perutz, Coord. Chem. Rev., 2012, 256, 2562–2570 CrossRef CAS.
- C. Costentin and J.-M. Savéant, Nat. Rev. Chem., 2017, 1, 0087 CrossRef CAS.
- H. Kumagai, Y. Tamaki and O. Ishitani, Acc. Chem. Res., 2022, 55, 978–990 CrossRef CAS PubMed.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- H. Xiong, Y. Dong, D. Liu, R. Long, T. Kong and Y. Xiong, J. Phys. Chem. Lett., 2022, 13, 1272–1282 CrossRef CAS PubMed.
- D. Li, M. Kassymova, X. Cai, S.-Q. Zang and H.-L. Jiang, Coord. Chem. Rev., 2020, 412, 213262 CrossRef CAS.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- T. D. Bennett, F.-X. Coudert, S. L. James and A. I. Cooper, Nat. Mater., 2021, 20, 1179–1187 CrossRef CAS PubMed.
- J. D. Wuest, Nat. Commun., 2020, 11, 4652 CrossRef CAS PubMed.
- A. Giri, Y. Khakre, G. Shreeraj, T. K. Dutta, S. Kundu and A. Patra, J. Mater. Chem. A, 2022, 10, 17077–17121 RSC.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. García, Chem. Rev., 2023, 123, 445–490 CrossRef.
- J.-D. Xiao and H.-L. Jiang, Acc. Chem. Res., 2019, 52, 356–366 CrossRef CAS PubMed.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168–190 CrossRef CAS.
- T.-X. Wang, H.-P. Liang, D. A. Anito, X. Ding and B.-H. Han, J. Mater. Chem. A, 2020, 8, 7003–7034 RSC.
- J. Y. Choi, B. Check, X. Fang, S. Blum, H. T. B. Pham, K. Tayman and J. Park, J. Am. Chem. Soc., 2024, 146, 11319–11327 CAS.
- Z. Meng and K. A. Mirica, Chem. Soc. Rev., 2021, 50, 13498–13558 RSC.
- N.-N. Vu, S. Kaliaguine and T.-O. Do, Adv. Funct. Mater., 2019, 29, 1901825 CrossRef.
- Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- J. Yang, Z. Chen, L. Zhang and Q. Zhang, ACS Nano, 2024, 18, 21804–21835 CrossRef CAS PubMed.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- G. M. Tomboc, S. Choi, T. Kwon, Y. J. Hwang and K. Lee, Adv. Mater., 2020, 32, 1908398 CrossRef CAS.
- Ž. Kovačič, B. Likozar and M. Huš, ACS Catal., 2020, 10, 14984–15007 CrossRef.
- N.-Y. Huang, J.-Q. Shen, X.-W. Zhang, P.-Q. Liao, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2022, 144, 8676–8682 CrossRef CAS PubMed.
- J. Li, G. Chen, Y. Zhu, Z. Liang, A. Pei, C.-L. Wu, H. Wang, H. R. Lee, K. Liu, S. Chu and Y. Cui, Nat. Catal., 2018, 1, 592–600 CrossRef CAS.
- Y. Tamaki and O. Ishitani, ACS Catal., 2017, 7, 3394–3409 CrossRef CAS.
- H. Shen, T. Peppel, J. Strunk and Z. Sun, Sol. RRL, 2020, 4, 1900546 CrossRef CAS.
- W. Xie, J. Xu, U. Md Idros, J. Katsuhira, M. Fuki, M. Hayashi, M. Yamanaka, Y. Kobori and R. Matsubara, Nat. Chem., 2023, 15, 794–802 CrossRef CAS.
- Q. Guo, F. Liang, X.-B. Li, Y.-J. Gao, M.-Y. Huang, Y. Wang, S.-G. Xia, X.-Y. Gao, Q.-C. Gan, Z.-S. Lin, C.-H. Tung and L.-Z. Wu, Chem, 2019, 5, 2605–2616 CAS.
- R. Das, S. Chakraborty and S. C. Peter, ACS Energy Lett., 2021, 6, 3270–3274 CrossRef CAS.
- R. N. Sampaio, D. C. Grills, D. E. Polyansky, D. J. Szalda and E. Fujita, J. Am. Chem. Soc., 2020, 142, 2413–2428 CrossRef CAS PubMed.
- Z. Liu, J. Li, Z. Chen, M. Li, L. Wang, S. Wu and J. Zhang, Appl. Catal., B, 2023, 326, 122338 CrossRef CAS.
- P. Chen, X. a Dong, M. Huang, K. Li, L. Xiao, J. Sheng, S. Chen, Y. Zhou and F. Dong, ACS Catal., 2022, 12, 4560–4570 CrossRef CAS.
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, Introduction to Reticular Chemistry, 2019, pp. 267–283 DOI:10.1002/9783527821099.ch11.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS.
- B. B. Rath, S. Krause and B. V. Lotsch, Adv. Funct. Mater., 2024, 34, 2309060 CrossRef CAS.
- J. Guo and D. Jiang, ACS Cent. Sci., 2020, 6, 869–879 CrossRef CAS.
- H. S. Sasmal, A. Kumar Mahato, P. Majumder and R. Banerjee, J. Am. Chem. Soc., 2022, 144, 11482–11498 CrossRef CAS PubMed.
- K. Dey, S. Mohata and R. Banerjee, ACS Nano, 2021, 15, 12723–12740 CrossRef CAS.
- J. Yang, X. Zhang, W. Si, Y. Cao, J. Qian, Y. Li, B. Li and W. Qin, ACS Catal., 2024, 14, 2022–2030 CrossRef CAS.
- S. Mohata, K. Dey, S. Bhunia, N. Thomas, E. B. Gowd, T. G. Ajithkumar, C. M. Reddy and R. Banerjee, J. Am. Chem. Soc., 2022, 144, 400–409 CrossRef CAS.
- D. Rodríguez-San-Miguel, C. Montoro and F. Zamora, Chem. Soc. Rev., 2020, 49, 2291–2302 RSC.
- C.-H. Hsueh, C. He, J. Zhang, X. Tan, H. Zhu, W.-C. M. Cheong, A.-Z. Li, X. Chen, H. Duan, Y. Zhao and C. Chen, J. Am. Chem. Soc., 2024, 146, 33857–33864 CrossRef CAS.
- X. Kang, X. Wu, X. Han, C. Yuan, Y. Liu and Y. Cui, Chem. Sci., 2020, 11, 1494–1502 RSC.
- F. Haase and B. V. Lotsch, Chem. Soc. Rev., 2020, 49, 8469–8500 RSC.
- K. Koner, S. Mohata, Y. Ogaeri, Y. Nishiyama, M. A. Addicoat and R. Banerjee, Angew. Chem., Int. Ed., 2024, 63, e202316873 CrossRef CAS.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS.
- S. Xu, M. Richter and X. Feng, Acc. Mater. Res., 2021, 2, 252–265 CrossRef CAS.
- Y.-N. Gong, X. Guan and H.-L. Jiang, Coord. Chem. Rev., 2023, 475, 214889 CrossRef CAS.
- R. Wang, Y. Yuan, K.-T. Bang and Y. Kim, ACS Mater. Au, 2023, 3, 28–36 CrossRef CAS.
- L. Yao, A. M. Pütz, H. Vignolo-González and B. V. Lotsch, J. Am. Chem. Soc., 2024, 146, 9479–9492 CrossRef CAS.
- P. M. Stanley, J. Haimerl, N. B. Shustova, R. A. Fischer and J. Warnan, Nat. Chem., 2022, 14, 1342–1356 CrossRef CAS PubMed.
- S. Mohata, R. Das, K. Koner, M. Riyaz, K. Das, S. Chakraborty, Y. Ogaeri, Y. Nishiyama, S. C. Peter and R. Banerjee, J. Am. Chem. Soc., 2023, 145, 23802–23813 CrossRef CAS PubMed.
- D. A. Cagan, D. Bím, N. P. Kazmierczak and R. G. Hadt, ACS Catal., 2024, 14, 9055–9076 CrossRef CAS PubMed.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS.
- Z. Fu, X. Wang, A. M. Gardner, X. Wang, S. Y. Chong, G. Neri, A. J. Cowan, L. Liu, X. Li, A. Vogel, R. Clowes, M. Bilton, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Sci., 2020, 11, 543–550 RSC.
- Y.-Z. Cheng, W. Ji, P.-Y. Hao, X.-H. Qi, X. Wu, X.-M. Dou, X.-Y. Bian, D. Jiang, F.-T. Li, X.-F. Liu, D.-H. Yang, X. Ding and B.-H. Han, Angew. Chem., Int. Ed., 2023, 62, e202308523 CrossRef CAS PubMed.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS.
- Q. Zhang, S. Gao, Y. Guo, H. Wang, J. Wei, X. Su, H. Zhang, Z. Liu and J. Wang, Nat. Commun., 2023, 14, 1147 CrossRef CAS PubMed.
- E. Nikoloudakis, I. López-Duarte, G. Charalambidis, K. Ladomenou, M. Ince and A. G. Coutsolelos, Chem. Soc. Rev., 2022, 51, 6965–7045 RSC.
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J.-L. Wang, D.-Q. Yuan and Y.-Q. Lan, Angew. Chem., Int. Ed., 2019, 58, 12392–12397 CrossRef CAS PubMed.
- Y. H. Kim, J.-P. Jeon, Y. Kim, H.-J. Noh, J.-M. Seo, J. Kim, G. Lee and J.-B. Baek, Angew. Chem., Int. Ed., 2023, 62, e202307991 CrossRef CAS.
- T.-X. Luan, J.-R. Wang, K. Li, H. Li, F. Nan, W. W. Yu and P.-Z. Li, Small, 2023, 19, 2303324 CrossRef CAS.
- L. Ran, Z. Li, B. Ran, J. Cao, Y. Zhao, T. Shao, Y. Song, M. K. H. Leung, L. Sun and J. Hou, J. Am. Chem. Soc., 2022, 144, 17097–17109 CrossRef CAS PubMed.
- Y. Yang, H.-Y. Zhang, Y. Wang, L.-H. Shao, L. Fang, H. Dong, M. Lu, L.-Z. Dong, Y.-Q. Lan and F.-M. Zhang, Adv. Mater., 2023, 35, 2304170 CrossRef CAS PubMed.
- W. Zhou, X. Wang, W. Zhao, N. Lu, D. Cong, Z. Li, P. Han, G. Ren, L. Sun, C. Liu and W.-Q. Deng, Nat. Commun., 2023, 14, 6971 CrossRef CAS PubMed.
- Y. Yang, Y. Lu, H.-Y. Zhang, Y. Wang, H.-L. Tang, X.-J. Sun, G. Zhang and F.-M. Zhang, ACS Sustainable Chem. Eng., 2021, 9, 13376–13384 CrossRef CAS.
- J. Zhou, J. Li, L. Kan, L. Zhang, Q. Huang, Y. Yan, Y. Chen, J. Liu, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2022, 13, 4681 CrossRef CAS PubMed.
- X. Wang, X. Ding, Y. Jin, D. Qi, H. Wang, Y. Han, T. Wang and J. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202302808 CrossRef CAS PubMed.
- S. Li, C. Gao, H. Yu, Y. Wang, S. Wang, W. Ding, L. Zhang and J. Yu, Angew. Chem., Int. Ed., 2024, 63, e202409925 CrossRef CAS PubMed.
- M. Lu, Q. Li, J. Liu, F.-M. Zhang, L. Zhang, J.-L. Wang, Z.-H. Kang and Y.-Q. Lan, Appl. Catal., B, 2019, 254, 624–633 CrossRef.
- J. Wang, W. Zhu, F. Meng, G. Bai, Q. Zhang and X. Lan, ACS Catal., 2023, 13, 4316–4329 CrossRef CAS.
- X. Lan, H. Li, Y. Liu, Y. Zhang, T. Zhang and Y. Chen, Angew. Chem., Int. Ed., 2024, 63, e202407092 CrossRef CAS.
- L. Liu and A. Corma, Nat. Rev. Mater., 2021, 6, 244–263 CrossRef CAS.
- P. Majumder, S. Mohata, H. S. Sasmal, B. Chandra, H. Kuiry and R. Banerjee, Angew. Chem., Int. Ed., 2024, 63, e202412122 CrossRef CAS PubMed.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- M. Lu, M. Zhang, J. Liu, T. Y. Yu, J. N. Chang, L. J. Shang, S. L. Li and Y. Q. Lan, J. Am. Chem. Soc., 2022, 144, 1861–1871 CrossRef CAS.
- H. Zhong, R. Sa, H. Lv, S. Yang, D. Yuan, X. Wang and R. Wang, Adv. Funct. Mater., 2020, 30, 2002654 CrossRef CAS.
- H. Li, C. Cheng, Z. Yang and J. Wei, Nat. Commun., 2022, 13, 6466 CrossRef CAS.
- M. Zhang, M. Lu, Z.-L. Lang, J. Liu, M. Liu, J.-N. Chang, L.-Y. Li, L.-J. Shang, M. Wang, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 6500–6506 CrossRef CAS.
- Y. Qiu, C. Jiang, X. Xin, Y.-Y. Li, H. Wang, J. Xu, H. Lin, L. Wang and V. Turkevich, ACS Appl. Nano Mater., 2024, 7, 11234–11247 CrossRef CAS.
- M. Xu, C. Lai, X. Liu, B. Li, M. Zhang, F. Xu, S. Liu, L. Li, L. Qin, H. Yi and Y. Fu, J. Mater. Chem. A, 2021, 9, 24148–24174 RSC.
- L. Zou, R. Sa, H. Zhong, H. Lv, X. Wang and R. Wang, ACS Catal., 2022, 12, 3550–3557 CrossRef CAS.
- K. Guo, X. Zhu, L. Peng, Y. Fu, R. Ma, X. Lu, F. Zhang, W. Zhu and M. Fan, Chem. Eng. J., 2021, 405, 127011 CrossRef CAS.
- X. An, J. Bian, K. Zhu, R. Liu, H. Liu and J. Qu, Chem. Eng. J., 2022, 442, 135279 CrossRef CAS.
- X. Yu, Z. Yang, B. Qiu, S. Guo, P. Yang, B. Yu, H. Zhang, Y. Zhao, X. Yang, B. Han and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 632–636 CrossRef CAS.
- S. Wang, X. Hai, X. Ding, S. Jin, Y. Xiang, P. Wang, B. Jiang, F. Ichihara, M. Oshikiri, X. Meng, Y. Li, W. Matsuda, J. Ma, S. Seki, X. Wang, H. Huang, Y. Wada, H. Chen and J. Ye, Nat. Commun., 2020, 11, 1149 CrossRef PubMed.
- O. P. Dimitriev, Chem. Rev., 2022, 122, 8487–8593 CrossRef CAS PubMed.
- K. Lei, D. Wang, L. Ye, M. Kou, Y. Deng, Z. Ma, L. Wang and Y. Kong, ChemSusChem, 2020, 13, 1725–1729 CrossRef CAS.
- Z. Shan, M. Wu, D. Zhu, X. Wu, K. Zhang, R. Verduzco and G. Zhang, J. Am. Chem. Soc., 2022, 144, 5728–5733 CrossRef CAS.
- S. Gao, Q. Zhang, X. Su, X. Wu, X.-G. Zhang, Y. Guo, Z. Li, J. Wei, H. Wang, S. Zhang and J. Wang, J. Am. Chem. Soc., 2023, 145, 9520–9529 CrossRef CAS.
- J.-X. Cui, L.-J. Wang, L. Feng, B. Meng, Z.-Y. Zhou, Z.-M. Su, K. Wang and S. Liu, J. Mater. Chem. A, 2021, 9, 24895–24902 RSC.
- L.-j Wang, R.-l Wang, X. Zhang, J.-l Mu, Z.-y Zhou and Z.-m Su, ChemSusChem, 2020, 13, 2973–2980 CrossRef CAS.
- S. Biswas, F. A. Rahimi, R. K. Saravanan, A. Dey, J. Chauhan, D. Surendran, S. Nath and T. K. Maji, Chem. Sci., 2024, 15, 16259–16270 RSC.
- X. Yu, K. Gong, S. Tian, G. Gao, J. Xie and X.-H. Jin, J. Mater. Chem. A, 2023, 11, 5627–5635 RSC.
- J. Ren, C. Ji, B. Du, Q. Liu, K. Yu, D. Ahn, Z. Zhang, Y. Ye, C. R. Göb and D. Zhao, J. Am. Chem. Soc., 2024, 146, 30784–30789 CrossRef CAS PubMed.
- K. Sun, Y. Huang, Q. Wang, W. Zhao, X. Zheng, J. Jiang and H.-L. Jiang, J. Am. Chem. Soc., 2024, 146, 3241–3249 CrossRef CAS.
- J. Ding, X. Guan, J. Lv, X. Chen, Y. Zhang, H. Li, D. Zhang, S. Qiu, H.-L. Jiang and Q. Fang, J. Am. Chem. Soc., 2023, 145, 3248–3254 CrossRef CAS PubMed.
- L. Zou, Z.-A. Chen, D.-H. Si, S.-L. Yang, W.-Q. Gao, K. Wang, Y.-B. Huang and R. Cao, Angew. Chem., Int. Ed., 2023, 62, e202309820 CrossRef CAS.
- X. Wang, X. Ding, T. Wang, K. Wang, Y. Jin, Y. Han, P. Zhang, N. Li, H. Wang and J. Jiang, ACS Appl. Mater. Interfaces, 2022, 14, 41122–41130 CrossRef CAS PubMed.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS.
- X. Wang, Z. Fu, L. Zheng, C. Zhao, X. Wang, S. Y. Chong, F. McBride, R. Raval, M. Bilton, L. Liu, X. Wu, L. Chen, R. S. Sprick and A. I. Cooper, Chem. Mater., 2020, 32, 9107–9114 CrossRef CAS.
- P. Dong, X. Xu, R. Luo, S. Yuan, J. Zhou and J. Lei, J. Am. Chem. Soc., 2023, 145, 15473–15481 CrossRef CAS.
- D. H. Streater, E. R. Kennehan, D. Wang, C. Fiankor, L. Chen, C. Yang, B. Li, D. Liu, F. Ibrahim, I. Hermans, K. L. Kohlstedt, L. Luo, J. Zhang and J. Huang, J. Am. Chem. Soc., 2024, 146, 4489–4499 CrossRef CAS PubMed.
- M. Zhang, P. Huang, J.-P. Liao, M.-Y. Yang, S.-B. Zhang, Y.-F. Liu, M. Lu, S.-L. Li, Y.-P. Cai and Y.-Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202311999 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |