
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reactor operating parameters and their effects on the local reaction environment of CO(2) electroreduction
Xiao Kun
Lu
 and
Linsey C.
Seitz
*
and
Linsey C.
Seitz
*
Department of Chemical and Biological Engineering, Northwestern University, Evanston, IL, USA. E-mail: linsey.seitz@northwestern.edu
First published on 22nd May 2025
Abstract
Low temperature aqueous electrochemical CO(2) reduction (ECR) emerged as a pathway to close the carbon cycle with the integration of renewable energy. However, activity, selectivity, and stability barriers prevent ECR from entering industrial scale operation. While catalyst design has made meaningful progress towards selective and active production of many products including CO, formate, and ethylene, operating conditions during catalyst testing have not been standardized. Operational parameters drastically impact the local reaction environment of the ECR and thus the performance of ECR. Herein, we summarize the prevailing operational variability of ECR and their interconnectedness. We first analyze reactant availability via tuning of cell geometry and CO(2) pressures. Then, optimization towards electrolyzer components including electrolyte, electrodes, and bipolar plates is discussed. We further assess the electrochemical protocols to enhance the performance or accelerate the degradation of ECR and the considerations required to scale up ECR to pilot scale. Finally, we provide perspectives on the current challenges of ECR and their promising solutions.
 Xiao Kun Lu | Xiao Kun Lu received his BS degree in Chemical and Biomolecular Engineering from the University of California, Berkeley in 2019. He then obtained his PhD in Chemical and Biological Engineering from Northwestern University under guidance of Prof. Linsey C. Seitz in 2025. His research interests include electrocatalytic systems and CO2 removal technologies. |
 Linsey C. Seitz | Linsey Seitz joined the faculty of the Chemical and Biological Engineering Department at Northwestern University in 2018. She earned her BS (2010) in Chemical Engineering from Michigan State University, followed by MS (2013) and PhD (2015) in Chemical Engineering from Stanford University. Linsey was also previously a Helmholtz Postdoctoral Fellow at the Karlsruhe Institute for Technology with the Institute of Photon Science and Synchrotron Radiation. Her research uses tools at the interface of electrocatalysis and spectroscopy to investigate dynamic catalyst materials and reaction environments towards the sustainable production of fuels and chemicals, as well as upconversion of waste streams. |
1. Introduction
Anthropogenic CO2 emissions due to burning of fossil fuels post industrial revolution have disturbed the Earth's natural carbon cycles, causing atmospheric CO2 to reach >420 ppm.1,2 CO2 is a known cause to ocean acidification and the greenhouse effect, which leads to potentially irreversible changes in sea levels, global temperature, and ecosystems.3 As the energy and materials demands continue to increase, CO2 emissions are projected to reach >1100 ppm by 2100 in the absence of interference or protective measures.4–6 Hence, there is an urgent need to develop technologies to enable a transition to renewable energy and cleaner fuels and chemicals.Low temperature electrochemical CO(2) reduction (ECR) has emerged as a promising pathway to enable a storage mechanism for intermittent renewable energy (e.g., wind, hydro, and solar) while producing carbon-based fuels and chemicals that are easily integrable into contemporary infrastructures. However, the performance of ECR in terms of activity, selectivity, and stability has hindered its commercialization. To be economically viable, CO2 electrolyzers are expected to meet current density requirements of >200 mA cm−2 at <3 Vcell while achieving >80% faradaic efficiency (FE) towards a desired product with stability up to 3000–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h.7,8 A lower full cell potential decreases the operating cost, while being able to operate at high current densities for longer durations decreases the capital cost.
000 h.7,8 A lower full cell potential decreases the operating cost, while being able to operate at high current densities for longer durations decreases the capital cost.
Over the past decade, extensive research has been focused on developing more active and selective catalysts. Methods such as size and shape control,9,10 porosity control,11,12 faceting,13,14 creating defects,15,16in situ restructuring,17,18 alloying,19,20 and utilizing ligand effects21 have been adopted to enhance the intrinsic and extrinsic activity of catalysts. However, the performance of catalysts in the literature is evaluated in drastically different systems ranging from H-type cells22 to membrane electrode assemblies,23 with the electrolyte bulk pH ranging from acidic24 to alkaline,25 and catalyst supports ranging from metal foils26 to polytetrafluoroethylene (PTFE) membranes.27 While this wide range of testing parameters has been beneficial in probing the flexibility and performance of these systems, it has also made it difficult to assess the overall progress in the field, in particular because ECR is extremely sensitive to local reaction environments, which are widely perturbed in these systems and not directly probed in most studies. For example, employing identical catalyst modifications or other enhancement approaches such as adoption of polymer additives often exhibits different electrochemical performances when evaluated in different reactor geometries.28,29 Therefore, to collectively advance the field of ECR towards commercialization, effects of bulk reactor operating conditions on local reaction environments must be elucidated to standardize testing protocols and enable proper comparison between catalyst materials such that we can collectively achieve enhanced ECR performance by engineering these operation control variables.
This review provides a systematic overview of various operation and local reaction environment effects including cell geometry, CO(2) concentration, local pH, hydrophobicity, conductivity of support, cation and anion effects, salt precipitation, temperature, and pulsing/accelerated stress test protocols (Fig. 1). Due to convolutions and intricacies between reaction environment effects, a simple survey of literature does not sufficiently bridge together this complex network. This review aims to offer insightful analyses of essential tuning knobs for ECR through exhaustive investigation of recent literature to draw useful connections among diverse factors. Furthermore, current challenges regarding each enhancement effect are addressed to encourage further exploration.
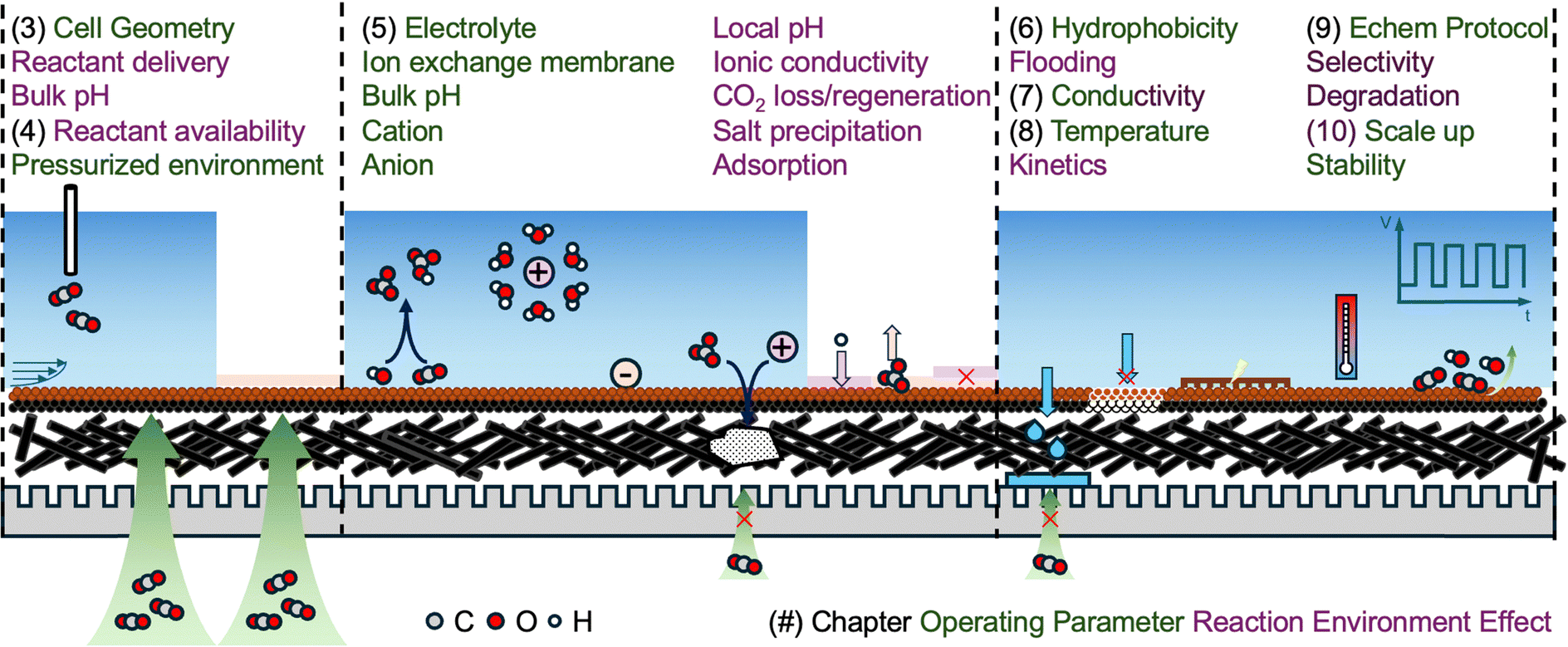 | ||
| Fig. 1 Illustrations of important operating parameters and their effects on local reaction environments. | ||
2. Background
2.1. Thermodynamics and kinetics
Aqueous ECR is a cathodic reaction where protons from water are used to reduce CO(2).| xCO2 + nH+ + ne− → product + yH2O | (1) |
The commonly reported products and their thermodynamic redox potentials are listed in Table 1. The counter electrode oxidation reaction is typically the oxidation of abundant water present in the system, namely the oxygen evolution reaction (OER).
| 2H2O → O2 + 4H+ + 4e− | (2) |
| Product | Reaction | E° (vs. RHE) |
|---|---|---|
| Carbon Monoxide | CO2 + H2O + 2e− → CO + 2OH− | −0.10 |
| Formate | CO2 + H2O + 2e− → HCOO− + OH− | −0.25 |
| Methanol | CO2 + 5H2O + 6e− → CH3OH + 6OH− | 0.03 |
| Methane | CO2 + 6H2O + 8e− → CH4 + 8OH− | 0.17 |
| Acetate | 2CO2 + 5H2O + 8e− → CH3COO− + 7OH− | 0.11 |
| Ethylene | 2CO2 + 8H2O + 12e− → C2H4 + 12OH− | 0.08 |
| Ethanol | 2CO2 + 9H2O + 12e− → C2H5OH + 12OH− | 0.09 |
| Propylene | 3CO2 + 12H2O + 18e− → C3H6 + 18OH− | 0.13 |
| n-Propanol | 3CO2 + 18H2O + 18e− → C3H7OH + 18OH− | 0.10 |
| Acetate | 2CO + 3H2O + 4e− → CH3COO− + 3OH− | 0.45 |
| Methane | CO + 5H2O + 6e− → CH4 + 6OH− | 0.26 |
| Ethylene | 2CO + 6H2O + 8e− → C2H4 + 8OH− | 0.17 |
| Ethanol | 2CO + 7H2O + 8e− → C2H5OH + 8OH− | 0.18 |
| n-Propanol | 3CO + 10H2O + 12e− → C3H7OH + 12OH− | 0.20 |
| Hydrogen | 2H+ + 2e− → H2 | 0 |
| Oxygen | 2H2O → O2 + 4H+ + 4e− | 1.23 |
Since water is both the source of protons and electrons, this process does not require reductants of high global warming potential (e.g., H2). However, proton reduction, or the hydrogen evolution reaction (HER), is a naturally occurring competing cathodic reaction in aqueous media. The HER is heavily dependent on local pH since protons are the only reactant; therefore, a way of suppressing the HER is to increase the local pH.30
As shown in Table 1, although it appears that most ECR products are thermodynamically favorable compared to those of HER, due to the complex kinetics of ECR, more energy beyond the equilibrium potential is required to drive ECR compared to the HER. Therefore, ECR catalysts are utilized to adjust for favorable binding energy of *H and *CO intermediates to minimize the HER (Fig. 2).31 For noble metals such as Pt and Pd that bind to both *H and *CO too strongly, the HER will be the predominant cathodic reaction. For metals that bind to both *H and *CO too weakly, CO2 is only reduced by 2e− and subsequently desorbs before further conversion can take place, forming CO (Au, Ag, and Zn) or formate (Pb, In, and Bi) as the primary products. In the unique case of Cu, which has an intermediary binding energy for *H and *CO, it is possible to form further reduced products that require transfer of more than 2e−. Since both CO2 and CO reduction primarily proceed through a *CO intermediate, enhancement effects regarding *CO are often viewed as transferrable between the two reactions.32 While thermodynamic descriptors such as *H and *CO binding energies serve well as a general guideline for product classification, it is insufficient to capture the trends in product formation for multi-carbon products. In a practical experiment for Cu-catalyzed CO2 reduction (CO2R), certain Cu sites are responsible for the reduction of CO2 to *CO, while others are responsible for subsequent reduction to multi-carbon products.33 The mechanisms towards a specific multi-carbon product depend on subsequent reaction steps such as C–O bond cleavage that bifurcates into ethylene or ethanol, which require detailed theoretical studies and catalyst functionalization to steer.34,35 Kinetic barriers and the presence of water molecules further complicate the theoretical simulations of such mechanisms. For example, mechanisms proposed by Nie et al. and Xiang et al. eleven years apart, with and without considering the effect of water, show CO2 reduction to methane proceeding through different intermediates over Cu(111).36,37 Overall, the wide spectrum of ECR products gives rise to broad selectivity challenges that must be addressed.
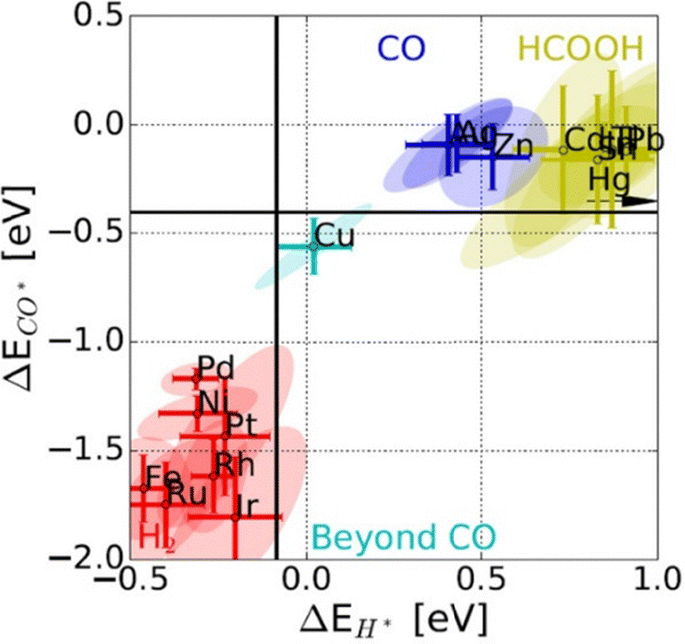 | ||
| Fig. 2 Binding energies of *CO and *H adsorbed intermediates as descriptors for trends in primary product formation over various ECR catalysts.31 Reprinted (adapted) with permission from the Wiley materials. | ||
2.2. Figures of merit
Similar to conventional catalytic reactions, electrocatalytic reactions are evaluated based on the activity, selectivity, and stability. The activity, or formation rate of a product, is directly proportional to the current, I, or a normalized current density, j. | (3) |
There are a wide variety of normalization approaches that can be adopted for current density. The most common approach is to calculate the geometric current density that is suitable for scale up as it would indicate the size of the bipolar plates; however, it does not reflect the number of active sites. A mass normalized current density can indicate the amount of catalyst material required, which is especially important for precious metal catalysts such as Ir-based anodes. Active site specific normalization can include electrochemically active surface area (ECSA) normalization via capacitance measurements or deposition methods such as CO stripping or hydrogen and mercury underpotential deposition.38–40 With knowledge of active site density, Γ, the turnover frequency (TOF) as an intrinsic activity measure can be calculated from site-normalized current density jsite, Avogadro number NA, number of electrons, n, Faraday's constant, F, and Γ.
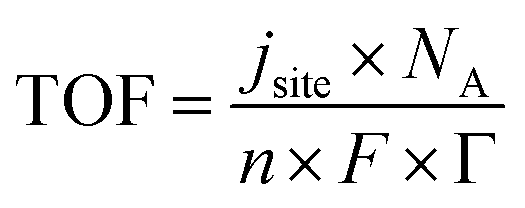 | (4) |
The selectivity is represented by faradaic efficiency (FE) or current efficiency, which is the ratio of the amount of charge passed towards the desired product, Qi, and the total charge passed, Qtotal. FE can also be simplified to the ratio of partial current towards a product, Ii, and the total current, Itotal.
 | (5) |
For gaseous products quantified by gas chromatography, the FE is calculated with the injection result in ppm, x, the flow rate of electrolyzer effluent, ambient temperature and pressure, T and P, ideal gas constant, R, number of electrons, n, Faraday's constant, F, and the total current, Itotal.
 | (6) |
It is important to note that even though the inlet gas flow rate is almost always controlled by a flow meter, electrochemical reactions and CO2 capture by an electrolyte would consume and/or generate gas, leading to a difference between the electrolyzer inlet and outlet flow rates. The flow rate used for FE calculation must be the measured outlet flow rate to ensure accurate carbon balance.41 The liquid products can be quantified by nuclear magnetic resonance spectroscopy or high performance liquid chromatography. The liquid product FE is computed with concentration, C, total volume of electrolyte, V, n, F, and Qtotal.
 | (7) |
Since the driving force for the reaction is electrical potential, it is important to measure the applied potential, Eapplied, to calculate an overpotential, η, beyond the thermodynamic redox potential, Etheoretical for the various products.
| η = |Eapplied − Etheoretical| | (8) |
The applied potential may be measured in a two-electrode setup, or a three-electrode setup with a reference electrode to report a half-cell potential. Half-cell potential is useful to isolate ECR from the OER and allows for better control of the applied potential, while the full cell potential indicates the total energy requirement of the system. Reference electrode potentials are often converted to the reversible hydrogen electrode (RHE) potential via calibration to hydrogen evolution and oxidation reactions to exclude pH effects.
| ERHE = Emeasured + Erefcalibration + 0.0591 × pH | (9) |
Taking into account both activity and selectivity, the energy efficiency (EE) describes the ratio between the actual electrical energy input and the minimum thermodynamic requirement.
 | (10) |
Conversion in ECR is typically calculated based on single pass carbon efficiency (SPCE). In contrast to many other examples of heterogeneous catalysis, the SPCE of CO2R is rather low (<50%) in most systems. This is due to CO2 loss to the alkaline electrolyte as (bi)carbonate.42 Strategies for mitigating CO2 loss will be outlined in Section 5.
The stability of the system is defined as the amount of time under continuous electrolysis. While other fields of electrocatalysis such as the OER have adopted additional specific metrics (e.g., S-number43,44 and decay rate in potential required (V/time)45), there is no general consensus on a stability metric for ECR. The instability of ECR systems arises from a variety of sources, including but not limited to multiple materials components of the electrodes, chemicals used for the electrolyte, mechanical parts, and operational procedures. Stability is a critical metric to consider after meeting activity and selectivity requirements, which have not yet been broadly met for ECR towards multi-carbon products.
3. Reactor geometry
Electrochemical reactors, or electrolyzers, are typically specialized to facilitate the electrochemical reactions taking place. However, fundamental components present in almost all reactors include electrodes, electrolytes, separators, and reactants. The electrolyzer configuration then dictates how the CO(2) reactant is fed, what interfaces with the catalyst, and how products are collected. The four most commonly used configurations are H-type cells, rotating disk electrode (RDE) cells, gas diffusion electrode (GDE) flow cells, and membrane electrode assemblies (MEAs) (Fig. 3). The main difference between liquid immersed electrode geometries (H-type and RDE) and gas diffusion electrode geometries (GDE flow cell and MEA) is that the gaseous reactant is delivered in the aqueous phase versus gas phase. Dissolving the CO2 into an aqueous phase impacts the availability of reactants at the catalyst surface such that the current density is limited. The dissolved CO2 buffering equilibrium also renders the electrolyte neutral, limiting the tunability of local pH. | ||
| Fig. 3 Schematics of (A) an H-type cell, (B) an RDE, (C) a GDE flow cell, and (D) an MEA. | ||
3.1. Liquid immersed cathodes: H-type cells and rotating (ring) disk electrode cells
H-type cells are excellent for fundamental studies due to their simple operation and control over configuration. Fig. 3A illustrates a typical H-type cell. In an H-type cell, the cathode is fully immersed in the catholyte where CO(2) gas is dissolved via a dispersion tube or bubbler. A reference electrode is typically placed in the cathodic compartment to identify the operating half-cell potential. The cathodic compartment is separated from the anodic compartment by an ion exchange membrane (IEM) to prevent re-oxidation of ECR products and plating of ions leached out from the anode. Despite the presence of IEM separators, organic and ionic species may still cross over the IEM due to swelling and electrostatic drag. To accurately calculate FEs, liquid products in both catholyte and anolyte need to be quantified.46 However, there would be still products lost to re-oxidation in the anolyte, of which the amount will depend on the choice of the anode catalyst.47The solubility of CO2 and CO is low in water, at just 34 mM and 1 mM, respectively. Through modeling, Burdyny et al. concluded that the amount of dissolved CO2 is only sufficient for current densities up to 25 mA cm−2, which would require high capital expenditure (CapEx) to reach the same desired production rate compared to GDE-based geometries (Fig. 4A).48 The transport model in this work also revealed that a more concentrated electrolyte also hinders the transport of CO2 to the catalyst surface. Therefore, there is a conflict between increasing the ionic conductivity of the electrolyte to reduce the ohmic drop of the system vs. allowing for better CO2(aq) diffusion.
 | ||
| Fig. 4 (A and B) CO2 concentration and local pH of the electrolyte at the electrode surface as a function of current density in an H-type cell.48 (C) CO2 concentration of the electrolyte as a function of current density using a GDL.48 (D) Illustration of TPB in GDL-based cells.49 Reprinted (adapted) with permission. Copyright 2022 Springer Nature. (E) Local pH of the electrolyte at the electrode surface as a function of current density with a GDL.48 Reproduced with permission from the Royal Society of Chemistry. | ||
In addition, there exists a buffering equilibrium between dissolved CO2 and water.
| CO2(g) + 2OH−(aq) ⇌ HCO3−(aq) + OH−(aq) + ⇌ 2CO32−(aq) + H2O | (11) |
The pKa values for the above (bi)carbonate equilibrium are 6.4 and 10.3, respectively. The pH of H-type cells is naturally regulated by this set of buffering reactions to be near-neutral regardless of starting pH (Fig. 4B).48 Hence, bicarbonate electrolytes became the popular electrolytes of choice for systems with dissolved CO2. The inability to use alkaline electrolytes leads to one fewer tuning parameter to suppress the HER via decreasing the concentration of H+. On the other hand, the pH of systems driving CO reduction (COR) in H-type cells can be adjusted, where alkaline media are a popular choice for suppressing the HER.50,51
Although the current density limit prevents H-type cells from being scaled up for ECR applications, they are suitable for fundamental studies. They are also critical for in situ characterization techniques that require the backside of the catalyst support to interface with the instrument. For instance, differential electrochemical mass spectroscopy constantly pulls a vacuum on a catalyst-deposited pervaporation membrane that only allows CO2 to be delivered via the electrolyte.52 H-type cells can also be pressurized since they will not suffer from liquid-gas pressure imbalances. H-type pressurized spectro-electrochemical cells can be used for in situ attenuated total reflection infrared spectroscopy that requires catalyst deposited on crystals to reveal important reaction orders with respect to surface adsorbates.53,54
Another cell geometry that requires immersion of the working electrode is the rotating (ring) disk electrode (R(R)DE) setup. This classic setup has well-controlled mass transport due to the convection from rotation.55,56 Since the geometry and system parameters are defined, R(R)DE systems enable the use of dimensionless numbers such as the Sherwood and Damköhler numbers for analysis of CO2 transport and consumption to decouple the complexity of mass transport effects and reaction kinetics.57 As a result of these well-defined mass transport conditions, R(R)DE can be used to isolate the effects of mass transport from the reaction kinetics.58 It was also noted that R(R)DE supports sufficient time resolution to mitigate the effects of homogeneous liquid-phase reactions for product detection methods such as gas chromatography.59 Although the small electrode area prevents R(R)DE systems from achieving high currents, this platform is useful to study catalyst materials that undergo unique structural changes under low applied potentials and current density.55,56,60
3.2. GDE flow cells
To overcome the solubility limit of CO(2) in aqueous environments, a gas diffusion layer (GDL) is often used as a porous catalyst support to allow CO(2) to be directly supplied in the gas phase, drastically decreasing the CO(2) diffusion distance by three orders of magnitude and allowing for operation at much higher current densities (Fig. 4C).48 The cathodic and anodic compartments are again separated by ion exchange membranes; no GDL is required for the anodic OER since the reaction occurs in a liquid phase (Fig. 3C). A large fraction of research on ECR transitioned from H-type cells to GDE-based cells post 2015 to evaluate catalyst performance at closer to industrially-relevant current densities. To efficiently utilize gas phase CO(2), GDE flow cells require maintenance of a delicate triple-phase boundary (TPB) for the gas-fed CO(2) – solid catalyst, gaseous reactant, and liquid electrolyte (Fig. 4D).49 If the TPB is disrupted, CO(2) would have to dissolve into the aqueous catholyte before reaching the catalyst surface, yielding a similar solubility limit to liquid immersed cathode geometries.An imbalance of pressure from either the gas or the liquid phase leads to species crossover, disrupting the TPB.61 In general, the CO(2) crossover at the GDE is acceptable, as GDE flow cells may be operated in a flow-through configuration.62 Indeed, Duarte et al. demonstrated that for CO2R to CO on Ag nanoparticles, a CO2 flow-through configuration reached higher current density while maintaining the same FE and potential compared to the CO2 flow-by configuration. However, this was at the cost of higher ohmic drop and worse stability due to bubbles and salt precipitation in the GDL.
Liquid crossover across the GDL into the gaseous compartment, also known as flooding, is a much more common failure mechanism of GDE flow cells. Liquid intrusion in the GDL decreases the efficiency of gas transport due to longer diffusion path lengths through aqueous electrolyte, as well as introduction of ions such as K+ and OH− to form carbonate salts with CO2 that obstruct the porous channels of the GDL.63 Flooding arises from several physical phenomena.64 Capillary action due to the porous GDL together with porous salt formation due to electrolyte salts naturally draws liquid into the GDL. When operating under a negative applied potential that is far from the potential of zero charge (PZC), the electrode surface also experiences electrowetting, which is the process of excessive negative charge on the electrode surface drawing cations and liquid to the surface to cause wetting. The various effects of the electrolyte on the GDE PZC and methods to mitigate flooding will be discussed in the coming sections.
Since much of the CO2 supplied to the reaction is in the gas phase, the bulk electrolyte pH is less affected by the (bi)carbonate equilibrium and CO(2) transport is no longer hindered by the concentrated electrolyte in GDEs as compared to CO(2)-purged systems. Therefore, GDE flow cell research is typically conducted in KOH ranging from 1–10 M.14,27 The local pH in a GDE flow cell is predicted to remain above 12 due to the OH− being produced as a byproduct during ECR and alkaline HER (Fig. 4E).14,27,48
The GDE flow cell is also excellent for collecting gas phase products from the tail gas of either the gas or liquid cathodic compartments, depending on flow geometry. Conversely, liquid products are mixed with a salty catholyte that imposes extra operation expenditure for downstream separations.65 The added catholyte layer also contributes to higher ohmic drop in the cell compared to a zero-gap geometry, lowering the EE of the overall process.
3.3. MEA
MEA is a zero-gap configuration where the two electrodes are compressed against an IEM by the flow fields (Fig. 3D). This cell design also employs porous transport layers such as the GDE for improved mass transport of reactants and products. The ohmic resistance is minimized to merely originate from electrodes and the membrane, due to the elimination of a catholyte electrolyte layer between the electrodes. The liquid products are instead collected via cold traps at the gas effluent and in the anolyte.66 Therefore, it is the ideal cell design to achieve high current densities while minimizing the cell potential. Additionally, the scalability of MEAs into electrolyzer stacks makes them most suitable for commercialization.MEAs are also a robust platform for evaluating catalyst performance. Stability tests have reached >1000 h in MEA-type electrolyzers in single- and multi- stack configurations.67–69 Commercialized electrolyzers are available for purchase from vendors globally to reduce the variability in cell geometry, as ECR performance tests using other platforms are often conducted in custom manufactured cells. Knowledge gained from mature electrocatalytic processes such as polymer exchange membrane fuel cells and water electrolyzers (PEMFCs and PEMWEs) can be implemented for ECR. Although cell-to-cell variability is decreased, operating conditions can still noticeably impact the performance; hence, explicating and untangling effects of operating conditions on local reaction environments are key to advancing the field.
3.4. Flow field
Flow fields are required for GDEs in scaled up devices to facilitate the transport of reactants and mechanically support the porous transport layers. There are four types of commonly used flow patterns: (1) pin-type, (2) parallel, (3) serpentine, and (4) integrated (Fig. 5). The pin-type and parallel patterns are suitable for high viscosity fluids such as water for the anodic OER due to their low pressure drop; therefore, they cannot be used to deliver gas-phase reactants in ECR. The fluid is allowed to travel through different parallel paths and would naturally distribute itself to the path of least resistance, resulting in lower pressure drop compared to serpentine design with a single path.70 The integrated pattern has blockages between the inlet and outlet, which convectively forces the reactant to pass through the porous transport layer. This pattern also causes high pressure drop due to fluids traveling through the GDL and would force liquid out at the GDE–liquid or GDE–membrane interface when gases exit the GDL. This may be paradoxical as flushing liquid through the GDL helps with removal of precipitated electrolyte salts to prevent blockage but would also disrupt the TPB and cause instability.71,72 | ||
| Fig. 5 Illustrations of flow field patterns: (A) pin-type, (B) parallel, (C) serpentine, and (D) integrated. | ||
The most adopted flow pattern for ECR in the literature is serpentine. Subramanian et al. tested failure mechanisms in CO2 electrolyzers including loss of CO2 access, flooding, and salt precipitation and concluded that the serpentine pattern is more robust than parallel and integrated patterns.73 The serpentine pattern allows for more complete coverage of the GDE area, but induces higher pressure drop along the path of the channel. In devices with geometric area >10 cm2, the multi-serpentine patterns where multiple serpentine channels run in parallel are used to alleviate excessive pressure drop. Large pressure drop down the serpentine channel causes flooding of the GDE and it is typical to maintain a certain positive Δp between the gas and liquid phase through back pressure regulation (Fig. 6A).74–76 Yuan et al. showed that a multi-serpentine pattern outperforms both regular and dense serpentine patterns at 500 mA cm−2 in terms of full cell potential and CO2-to-CO FE (Fig. 6B and C).77 The observed reactant concentration decreases along the serpentine channel as it is being consumed while the product distribution shifts to less reduced products (CH4 and H2) in a 25 cm2 COR electrolyzer (Fig. 6D).76 Exploiting this phenomenon, tandem reactions can be designed to occur within the serpentine flow channel by calculating the distance required to fully convert an intermediate product. Zhang et al. layered a segment of CO-producing catalyst on top of a Cu catalyst to perform CO2R to CO and utilized the in situ produced CO to undergo subsequent reduction along the channel on the Cu catalyst which allowed for 90% multi-carbon product FE at 1 A cm−2 (Fig. 6E and F).78 However, the consumption of the reactant along a plug flow type path generally causes a conversion–selectivity trade-off where lower concentration of a reactant leads to promotion of the side reaction, HER, that in turn decreases the economic viability.79
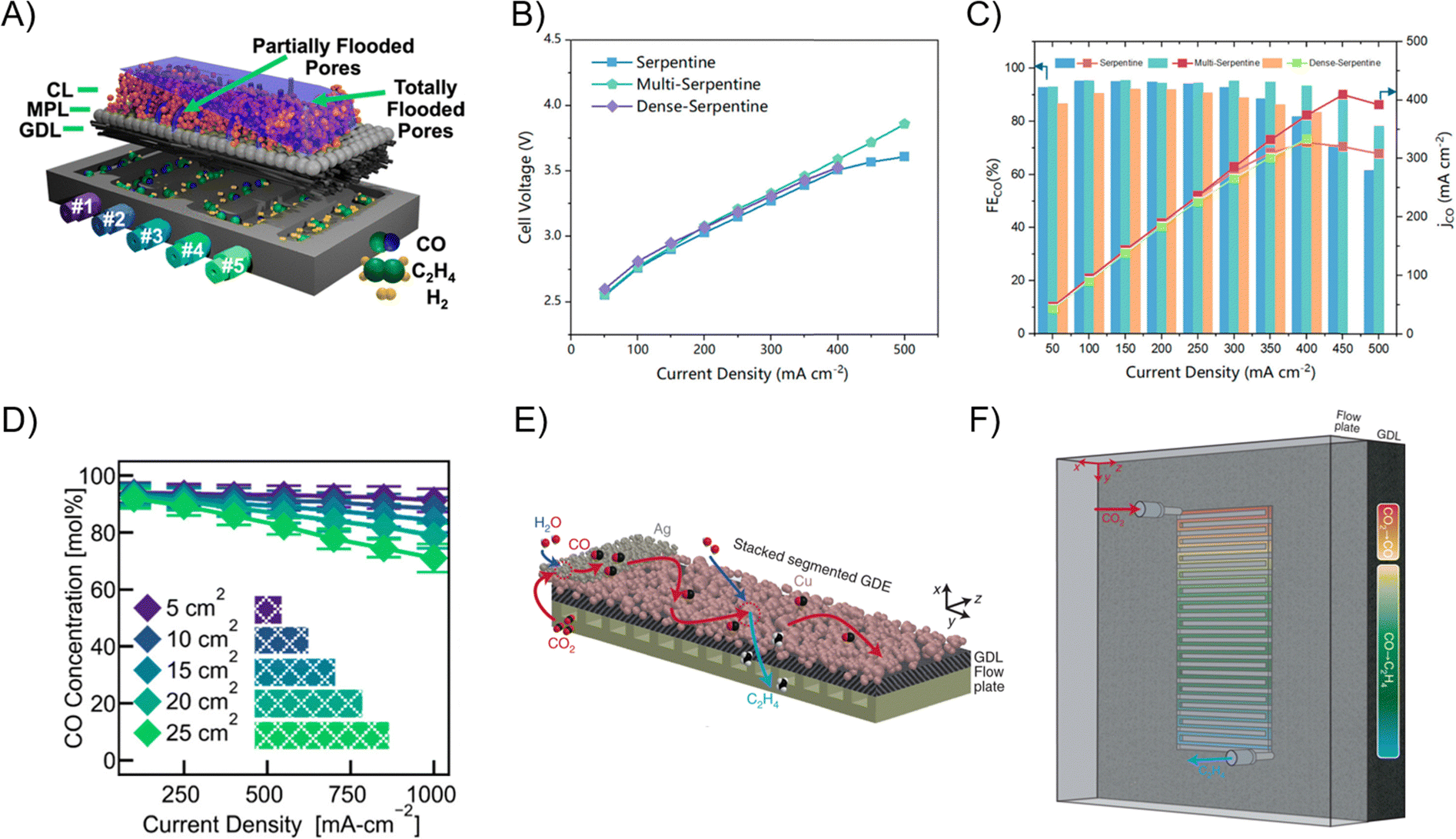 | ||
| Fig. 6 (A) Schematic of the COR on Cu catalyst along the serpentine channel. CO partial pressure decreases along the channel, leading to higher H2 production and transport resistance which causes flooding down the channel.76 (B and C) Full cell potential and CO FE of CO2R over Ag catalyst in a MEA device with serpentine, multi-serpentine, and dense-serpentine flow patterns.77 Reprinted (adapted) with permission. Copyright 2024 American Chemical Society. (D) Relative CO concentration during the COR over a Cu catalyst in a 25 cm2 MEA measured at points #1–5 on Fig. 5A.76 Reprinted (adapted) with permission. Copyright 2023 American Chemical Society. (E and F) Schematic of tandem CO2-to-CO-to-multi-carbon products reactions using Ag segment layered on Cu GDE in a flow cell with serpentine flow channel.78 Reprinted (adapted) with permission. Copyright 2022 Springer Nature. | ||
4. CO(2) availability
4.1. CO(2) flow
While the literature summarized in Section 3 demonstrated superior CO(2) transport for GDEs over H-type cells, H-type cells nonetheless have important design parameters. H-type cells are also configurable to flow cells. Billy and Co studied the effects of gas and liquid flow rates in an H-type flow cell and showed that, at low gas/solution flow ratios, the C2H4 FE declines due to insufficient transport of CO2, causing the ECR product profile to shift to CH4 (Fig. 7A).80 In additional to faster gas flow rate to improve CO2 transport, Lobaccaro et al. integrated a glass frit to reduce the bubble size of CO2(g) streams in the H-type cell.81 A noticeable increase in FE directed towards ECR versus HER is achieved when the CO2 bubble size is decreased (Fig. 7B). The other extreme of using elongated bubbles and an ultra-thin (1 μm) layer of electrolyte to form Taylor-flow has been proposed to improve CO2 availability.82 Bagemihl et al. employed a model to demonstrate that, under Taylor-flow, 1 bar of CO2 is capable of sustaining ECR above 200 mA cm−2 (Fig. 7C).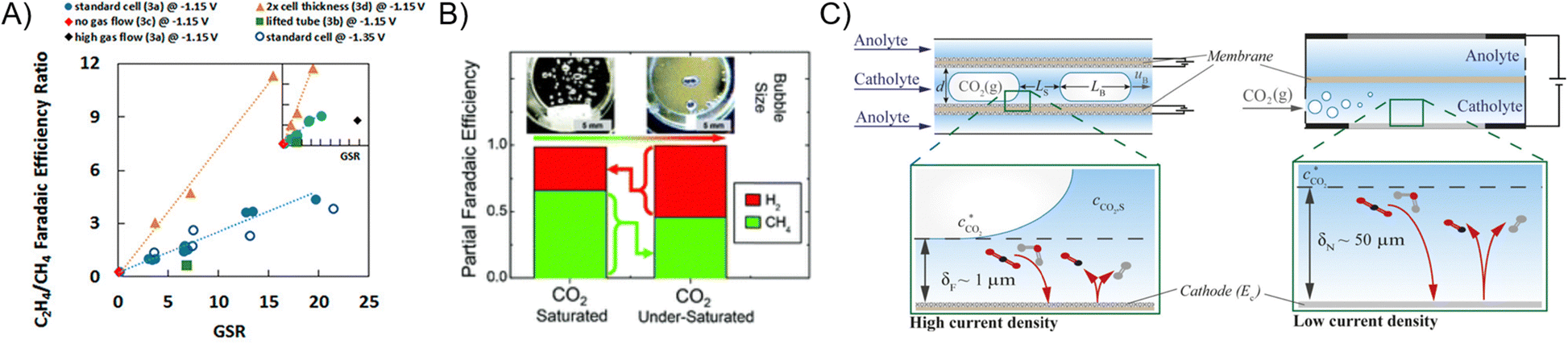 | ||
| Fig. 7 (A) C2H4-to-CH4 FE ratio of CO2 reduction at various gas to liquid flow rate ratios in an H-type flow cell.80 Reprinted (adapted) with permission. Copyright 2018 American Chemical Society. (B) ECR FE with small (left) and large (right) bubbles of CO2.83 Reproduced with permission from the Royal Society of Chemistry. (C) Illustration of Taylor flow tubular cell.82 Reprinted (adapted) with permission. Copyright 2022 American Chemical Society. | ||
The CO(2) flow rate also plays a pivotal role in GDE systems. Gabardo et al. tested CO2R flow rates across two orders of magnitude, ranging from 2 to 100 s.c.c.m (Fig. 8A).66 The general trend observed is that as the CO2 flow rate increases, the CO2R FE increased while the CO2 conversion decreased. In their 5 cm2 electrolyzer, C2H4 FE plateaus at flow rates >10 s.c.c.m. As shown in Fig. 8B, a high CO2 flow rate leads to low CO2 conversion and CO2R product concentration in the exit stream, which incurs additional downstream separation cost. Somewhat in contrast, studies aiming to improve the SPCE tend to decrease the reactant flow rate. Da Cunha et al. have analyzed the recent studies that aimed at either optimizing for FE or optimizing for SPCE.84 While product concentration in the outlet stream and SPCE can be improved via decreasing the CO2 inlet flow rate (Fig. 8C), the FE towards products, especially multi-carbon products, is lower compared to operation that is aimed at maximizing product selectivity (Fig. 8D). For instance, Pan et al. reached ∼90% SPCE in a CO2-to-CO MEA at 1 s.c.c.m. CO2 flow rate, but at the expense of sacrificing CO FE to ∼40%.85 Therefore, it is suggested that ECR research should not focus on maximizing SPCE via feeding stoichiometrically insufficient CO2 to compromise the selectivity.
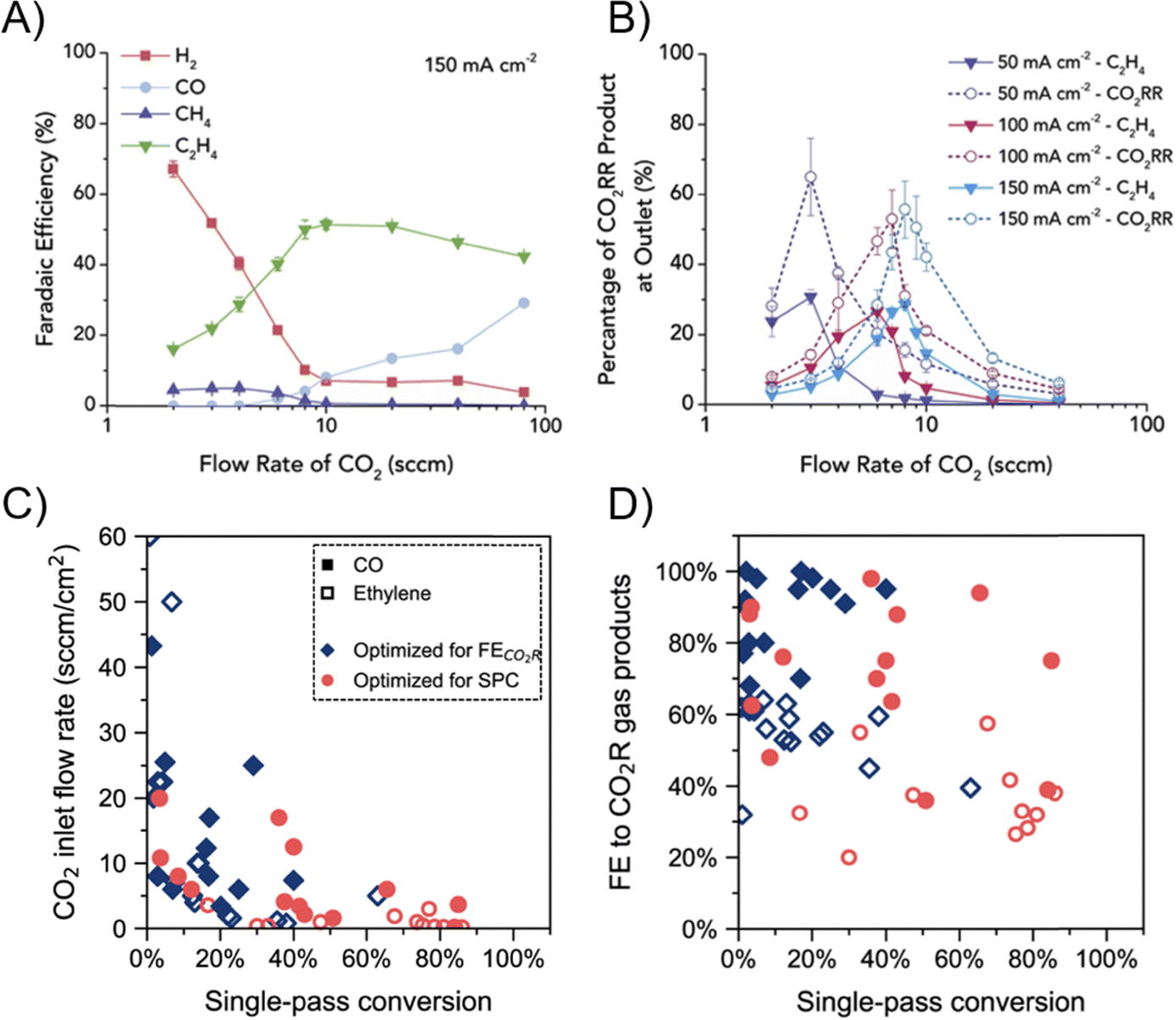 | ||
| Fig. 8 (A) FE as a function of the CO2 inlet flow rate over the Cu catalyst in an MEA at 150 mA cm−2.66 (B) Product stream concentration as a function of CO2 inlet flow rate over the Cu catalyst at 50 and 100 mA cm−2.66 Reprinted (adapted) with permission. Copyright 2019 Elsevier. (C) Literature CO2 SPCE at various CO2 inlet flow rates.84 (D) Literature ECR cathodic tail gas composition at various CO2 SPCE.84 Reprinted (adapted) with permission under Creative Commons License. | ||
4.2. CO(2) partial pressure and contaminants
The inlet CO(2) partial pressure (i.e. utilizing <1 bar CO(2) with N2 as the balance gas) is often studied to obtain kinetic insights or optimize selectivity for ECR. Chang et al. varied the pCO between 0.1 and 1 atm and used in situ surface-enhanced infrared spectroscopy to identify that the rate determining step of COR is likely the hydrogenation of CO rather than the C–C coupling step.86 In an H-type cell, a moderate pCO2of 0.4–0.6 atm reduces the buffer capacity of (bi)carbonate and increases the pH of the bulk electrolyte, therefore suppressing the HER (Fig. 9A).87 For CO2-to-CO conversion in GDE flow cells, pCO2 of 0.45 is found to still maintain high FECO (>80%) while achieving improved SPCE compared to a pure CO2 inlet stream, but at the expense of lower current density (Fig. 9B and C).88 As expected, for CO2 conversion to hydrocarbons in GDE flow cells on Cu-based catalysts, a decrease in pCO2 leads to lower production of multi-carbon products.89 The main ECR product becomes CH4 and an optimized pCO2 of 0.84 atm achieves a FECH4 of 56% (Fig. 9D).90 | ||
| Fig. 9 (A) C2H4 FE for CO2R as a function of pCO2 over a Cu catalyst in an H-type cell.87 Reprinted (adapted) with permission. Copyright 2019 Elsevier. (B and C) CO FE and product stream CO:CO2 ratio for CO2R over a Ag catalyst in a GDE flow cell.88 Reprinted (adapted) with permission. Copyright 2019 Elsevier. (D) CH4 FE for CO2R over a dilute Au/Cu catalyst in a GDE flow cell.90 Reprinted (adapted) with permission under Creative Commons License. | ||
Studies discussed thus far have employed an inert balance gas. In practical upstream feeds such as flue gas, a variety of contaminants including O2, NOx, SOx, and H2S may introduce side reactions or modify the catalyst structures. The concentration of O2 can reach up to 15 v/v% in flue gas; with a reduction redox potential of 1.23 V vs. RHE (∼1 V more favorable than ECR), the O2 reduction reaction (ORR) can easily displace ECR and occupy up to 99% of the FE at concentrations as low as 4 v/v% O2 (Fig. 10A).91 Nevertheless, it was shown by He et al. that despite having lower ECR FE, the partial current density of multi-carbon ECR products (e.g., C2H4) increased by up to 216-fold at −0.8 V vs. RHE when CO2 is co-reduced with 20 v/v% O2 due to the presence of surface-oxidized Cu species (Fig. 10B).92 Both SOx and NOx react with water to form acids that could lower the electrolyte pH and lead to higher HER activity.93 Although typical SOx concentration in flue gas is only on the order of hundreds of ppm,94 SOx reacts with electrocatalysts and induces S-doping that irreversibly shifts the product distribution toward formate/formic acid (Fig. 10C).95,96 ECR selectivity also decreases in the presence of NOx due to NOx reduction, but in contrast to SOx contaminants, this change is reversible once the NOx contaminant source is removed (Fig. 10D).97
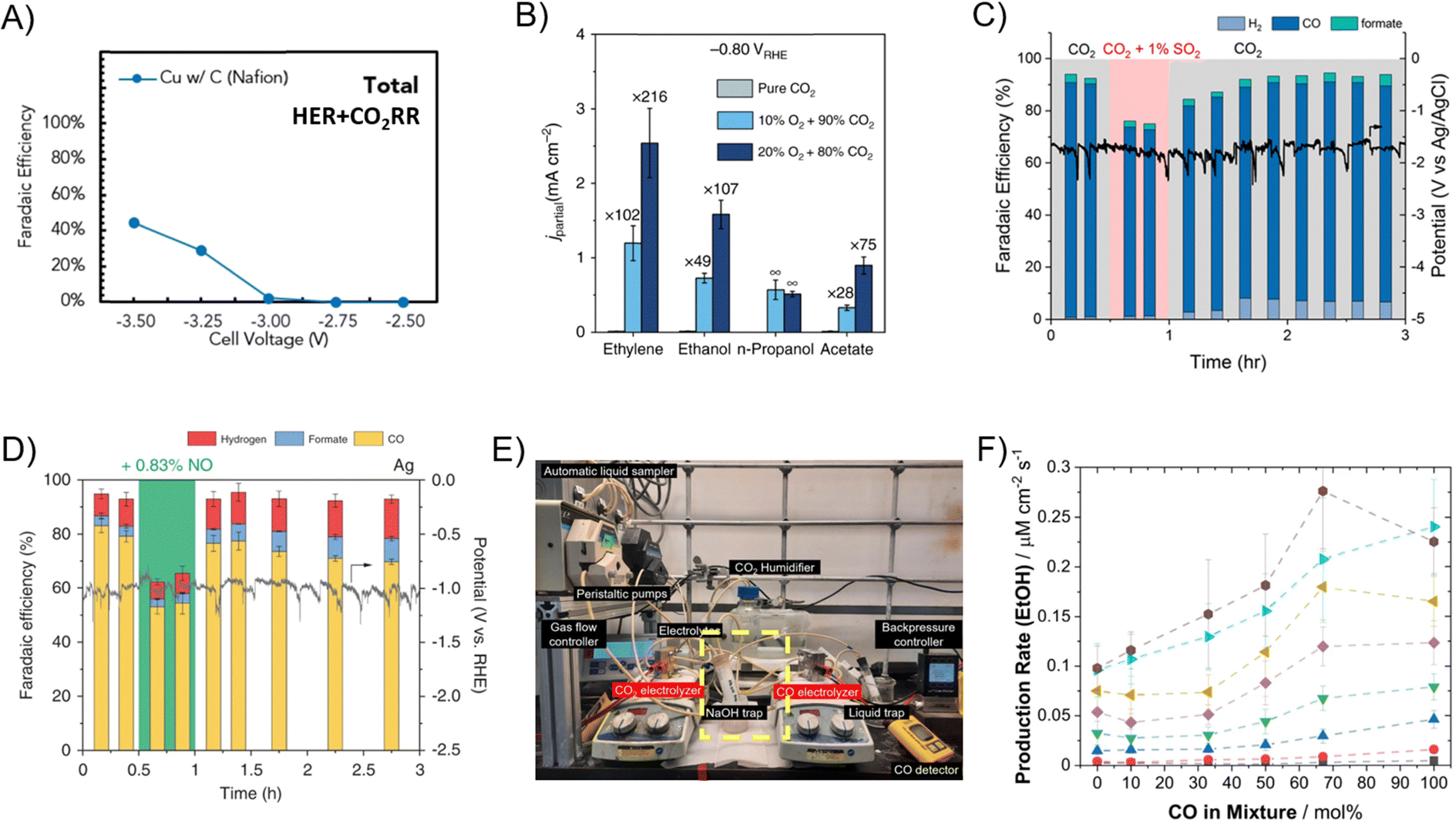 | ||
| Fig. 10 (A) Sum of HER and CO2R FE over a Cu catalyst with simulated flue gas containing 4 v/v% O2 and 15 v/v% CO2 in a GDE flow cell.91 Reproduced with permission from the Royal Society of Chemistry. (B) Multi-carbon product FEs for CO2R at 0, 10, and 20% O2 with CO2 balance over a Cu catalyst in an H-type cell.92 Reprinted (adapted) with permission under Creative Commons License. (C) Various product FEs and applied potential during CO2 + 1% SO2 electrolysis at 100 mA cm−2 over a Ag catalyst in a GDE flow cell.95 Reprinted (adapted) with permission. Copyright 2019 American Chemical Society. (D) Various product FEs and applied potential during CO2 + 0.83% NO electrolysis at 100 mA cm−2 over a Ag catalyst in a GDE flow cell.97 (E) Photo of tandem CO2 and CO reduction setup for acetate and C2H4 production. A NaOH trap is setup to remove unconverted CO2 from the first electrolyzer.98 Reprinted (adapted) with permission. Copyright 2024 Springer Nature. (F) Ethanol production rate as a function of CO concentration in CO/CO2 co-feed electrolysis over a Cu catalyst in a GDE flow cell.74 Reprinted (adapted) with permission under Creative Commons License. | ||
A mixture of CO2 and CO creates a peculiar case when investigating the impacts of lower reactant partial pressures. For applications such as acetate/acetic acid production, a pure CO inlet is desired. Crandall et al. performed tandem CO2-to-CO-to-acetic acid production starting with a pure CO2 stream and noted that a NaOH trap is required to remove all CO2 from the effluent of the first electrolyzer to maintain high acetic acid selectivity from COR (Fig. 10E).98 For production of other multi-carbon products, a co-feed of CO2 and CO increases the yield of C2H4 by ∼50% and alcohols by up to 100%, benefiting from the lower energy barrier of CO2–CO cross-coupling (Fig. 10F).74,99
4.3. Pressurized CO(2)
The inlet CO(2) can also be pressurized to alter the product profile due to increased CO(2) solubility in aqueous systems. The literature has presented mixed results regarding pressurized CO2 reactions. On a metallic Cu foil catalyst, an increase in pressure up to 100 atm of CO2 shifts the dominant ECR product to CH4 in an unstirred reactor, or to formate when the reactor is stirred.100 A balance of CO2 supply to the electrode surface and electron supply is required to improve hydrocarbon selectivity, as high CO2 mass transport (high stir rate and pressure) leads to increased 2e− ECR products, and high electron supply rate (high current density) leads to an increased HER (Fig. 11A). This is also analogous to ambient temperature systems where CO or formate is the main product at low applied potential due to insufficient supply of electrons, and CH4 is the main product at high applied potential due to low CO2 concentration to perform C–C coupling.101 Similar results have been observed by Huang et al., where they utilized a polymer-functionalized Cu catalyst to achieve stable and efficient formate production.102 This effect is also noticeable on other metallic catalysts, regardless of whether they are suitable for ECR under ambient conditions. Group 8–10 metals including Fe, Co, Rh, Ni, Pd, and Pt, which typically catalyze the HER under atmospheric CO2 pressure, shift to producing CO and/or formate under pressurized CO2.103 Mildly pressurizing CO2 to just 2 bar also increases the local CO2 concentration, making CO2-to-CO conversion more efficient (Fig. 11B).104 | ||
| Fig. 11 (A) Dependence of product distribution on electron supply and CO2 supply.100 Reprinted (adapted) with permission. Copyright 1994 IOP Publishing. (B) CO FE for CO2R at 1 and 2 bar CO2 pressure over a Ag catalyst in a GDE flow cell.104 Reprinted (adapted) with permission under Creative Commons License. (C) Acetate FE for COR at −0.57 VRHE over a Cu/Ag catalyst in a pressurized GDE flow cell.105 Reprinted (adapted) with permission. Copyright 2023 Springer Nature. (D) Chronopotentiometry of pressure swings during alkaline water electrolysis at 55 mA cm−2 (4 bar/1 bar) for bubble removal.106 Reprinted (adapted) with permission. Copyright 2019 Elsevier. | ||
Pressurization of CO(2) is also known to increase the coverage of *CO, an important intermediate to achieve C–C coupling.54 Qiu et al. utilized this *CO coverage effect to steer the primary liquid CO2R product to ethanol on Cu2O@Cu hollow spheres when operating with 30 atm CO2.107 For COR to acetic acid, Jin et al. through computational modeling concluded that a high *CO coverage (4/9 monolayer) has lower energy barrier to divert the ECR pathway to acetate production compared to lower coverages.105 Building off this conclusion, a record high acetate FE of 91% is achieved at 10 atm CO (Fig. 11C). High CO pressures and *CO coverage also enable catalysts that otherwise are poor at performing COR. For example, Raaijman et al. showed that COR on Ag at 60 bar CO produces a variety of alcohols and oxygenates including ethanol, acetic acid, and ethylene glycol, whereas Ag typically produces only CO under atmospheric pressure of CO2.108
Increased operating pressure and controlled pressure swings are also associated with improved bubble removal from electrode/transport layers and as a result, a decrease in kinetic and mass transport resistance (Fig. 11D).106,109,110 However, it is important to note that elevated O2 permeation across the IEM during pressurized operation could lead to a decrease in ECR performance at elevated pressures, despite the fact that this aspect is not commonly considered in the ECR literature.111
5. Electrolyte effects
5.1. Polymer electrolyte membranes and ionomers
Polymer electrolyte membranes or IEMs are employed as separators in H-type and GDE flow cells, as well as the sole ion conductor in the zero-gap configuration MEA. There are three types of membranes, which are classified by the charged species passed: the cation exchange membrane (CEM), anion exchange membrane (AEM), and bipolar membrane (BPM) (Fig. 12A–D). | ||
| Fig. 12 Schematic illustrations of (A) a CEM, (B) an AEM, (C) a forward-BPM, and (D) a reverse-BPM. | ||
CEMs, for instance Nafion, have negatively charged functional groups, such as sulfonates (SO3−); they offer an acidic electrode environment for proton exchange and benefit from the uniquely high ionic conductivity of protons. In contrast, AEMs have positively charged functional groups, such as imidazolium and phosphonium, to allow for hydroxide exchange. Since (bi)carbonate is often present in the system, it also serves as an anion conductor. AEMs are widely used for ECR to support an alkaline reaction environment that suppresses HER, although at the cost of higher ohmic drop across the membrane.
BPMs are made from laminating CEMs and AEMs to prevent ionic species from crossing over the membrane. BPMs can be employed in forward or reverse configuration, which results in the formation or dissociation of water at the CEM/AEM interface, respectively. Since a BPM is practically two membranes, the additional ohmic drop across it is also much higher than a CEM. Furthermore, in the reverse bias mode, an applied potential is required to drive water dissociation at the CEM/AEM interface. Incorporation of a water dissociation catalyst between the CEM and AEM has resulted in lower water dissociation overpotential in BPM systems.112–114 In the forward bias mode, formation of water at the CEM/AEM interface often leads to delamination of BPM and stability challenges. For best membrane stability, the CEM and AEM should not be intercalated as water formation at the CEM/AEM interface would cause delamination, and additional punctures on the CEM may be needed to allow for water drainage.67,115,116
The selective permeability of IEMs limits the types of electrolytes used. CEMs are inherently acidic and AEMs are alkaline. Using them as the ionic conductor in MEAs dictates the local pH at the electrodes. Altering the pH of IEMs with liquid electrolyte is unfavorable; for example, using an alkaline solution (e.g., KOH) in cation exchange membrane system negates the benefit of high proton ionic conductivity due to low proton concentration.
Although not a popular choice, porous separators adopted from mature alkaline water electrolysis technology serve as an alternative to IEMs for separating cathodic and anodic compartments in liquid electrolyte systems.117,118 Perazio et al. compared a PEM and a Zirfon diaphragm (polysulfone matrix and zirconium oxide filler) in a GDE flow cell with acidic electrolyte to conclude that the Zirfon diaphragm resulted in a lower cell potential due to lower ohmic resistance (Fig. 13).117 Diaphragms are not ion conductive alone, their conductivities are determined by the type of electrolyte used that pass through their pores.119 A major disadvantage of this property is that diaphragms are not selective for species crossover, so liquid ECR products may also cross to the anodic compartment and be lost via reoxidation.117,118
 | ||
| Fig. 13 Comparison of the voltage drop between two identical CO2R setups featuring either a Nafion membrane or a porous Zirfon separator in 3 M KCl/0.05 M H2SO4 electrolyte.117 Reprinted (adapted) with permission under Creative Commons License. | ||
5.2. Bulk and local pH
Liquid electrolytes are used in all ECR experiments. Liquid electrolytes present opportunities for various factors to affect ECR performance, including but not limited to pH, cation, anion, and hydrophobicity effects, which will be discussed in depth in this and the following sections. An alkaline reaction environment can be used as a tool to suppress the HER by reducing the proton concentration.30 The local pH at the catalyst surface is modulated by a complex interplay of proton consumption, (bi)carbonate buffering equilibrium, and cation hydration. Quantifying the local pH of the electrode surface has been extensively studied using methods such as pH-sensing RRDE,120–122in situ Raman spectroscopy,123–126 and confocal microscopy.123–125,127Both the ECR and the competing HER consume protons for reduction, and since water is the reductant in the system, water dissociation generates OH− as electrolysis proceeds. Therefore, the electrolyte at the catalyst surface has a higher local pH (∼8) than the bulk 0.1 M KHCO3 electrolyte (∼6.8, CO2 saturated) in a liquid immersed cathode system (Fig. 14A).122,128 In a GDE flow cell where high current density operation is possible, the rate of OH− production is higher, leading to a local pH difference of >3 (i.e., pH >10) in the 0.1–1 M KHCO3 catholyte.123,127 In contrast, when an alkaline starting electrolyte such as 1 M KOH (pH 14) is used in GDE flow cells, the local pH (7–9) is lower than the bulk pH since OH− produced by electrochemical reactions and from the electrolyte reaction with CO2 to form (bi)carbonates at the TPB.123,125
 | ||
| Fig. 14 (A) Computationally modelled and experimentally measured surface pH in an RRDE setup during CO2R over a Au catalyst in 0.5 M KHCO3 electrolyte.122 Reprinted (adapted) with permission. Copyright 2022 American Chemical Society. (B) Anode gas composition analysis in an AEM MEA.129 (C) Schematic of CO2 recovery with protons in solid electrolyte layer in double MEA.129 Reprinted (adapted) with permission. Copyright 2022 Springer Nature. (D) FE and current density of CO2R over a Cu catalyst in 0.05M H2SO4 electrolyte in a GDE flow cell.130 Reprinted (adapted) with permission under Creative Commons License. (E) FE of COR over the Cu/Ag catalyst in a pressurized flow cell with varying concentrations of KOH electrolyte.105 Reprinted (adapted) with permission. Copyright 2023 Springer Nature. (F) Partial current density of multi-carbon products from COR over a Cu catalyst at −1.5 VSHEversus OH− concentration in an H-type cell with a fixed 1.0 M cation concentration.131 (G) Cell voltage versus local pH indicating of the pH gradient and the benefits of alkaline conditions.132 Reprinted (adapted) with permission. Copyright 2024 American Chemical Society. | ||
In fact, an alkaline reaction environment (alkaline liquid electrolyte or AEM) is difficult to maintain because of the CO2 loss to the electrolyte at pH > 10. In ECR, for every two electron transferred, two OH− will be formed to generate one CO32−, with the exception of anionic products which form less CO32− (formate and acetate).129 Such CO2 loss decreases the pH of the electrolyte, leading to higher H2 FE and 3-d transition metal-based anode dissolution. Chen et al. electrodeposited a film of polyamine to shift the buffering reaction to protonation of amine functional groups which has a higher pKa than the (bi)carbonate equilibrium to increase the local pH.124 Lost CO2 in the form of (bi)carbonate will be oxidized at the anode to form CO2, and it is economically taxing to regenerate the CO2 from anode tail gas (Fig. 14B). To mitigate CO2 loss in alkaline systems, Kim et al. used an acidic solid electrolyte layer in a dual MEA to regenerate 90% of the lost CO2 from (bi)carbonate without altering the local reaction environment of the catalyst to maintain 90 + % FE towards CO at 200 mA cm−2 (Fig. 14C).129
Alternatively, an acidic reaction environment (acidic liquid electrolyte, CEM, or BPM) can be used to suppress (bi)carbonate formation. However, the most crucial challenge in acidic ECR is that HER becomes the sole cathodic reaction in mineral acids, such as H2SO4, due to the absence of cations that stabilize the *CO2− intermediate (Fig. 14D).24,130,133
Several studies utilized high KOH concentrations (>2 M) for COR to reach remarkable FEs of >80% to acetate or multi-carbon products in general (Fig. 14E).134,135 Nevertheless, since both K+ and OH− concentrations were increased, the enhancement effect due to electrolyte concentration cannot be attributed to OH− alone. It was proven by Li et al. that if the cation concentration remained constant, higher concentrations of OH− do not promote C–C coupling, but still increase the ECR FE due to HER suppression (Fig. 14F).131
Briefly considering the pH effects at the anode, the OER is also sensitive to pH due to proton-coupled electron transfer steps. Under acidic conditions, H2O molecules are oxidized whereas under alkaline conditions, OH− ions are oxidized (eqn (12) and (13)).
| 2H2O → O2 + 4H+ + 4e− (acidic OER 1.23 vs. SHE) | (12) |
| 4OH− → O2 + 2H2O + 4e− (alkaline OER 0.40 vs. SHE) | (13) |
The efficiency of the OER is observed to increase under alkaline conditions due to the *OH intermediate requiring no water dissociation.136 Non-precious metal catalysts are also enabled under alkaline conditions, decreasing the capital expenditure for electrocatalysts.137,138
In both acidic and alkaline environments, the OER will decrease the local pH via either production of H+, or consumption of OH−. Under concentrated alkaline anolyte (e.g., 1 M KOH or high pH), the OH− consumption does not meaningfully impact the local pH at the anode (<1). In the case of neutral anolyte, the local pH gradient could be up to 3–4 pH units, with an absolute pH value of 3–4 (Fig. 14G).139 These pH gradients translate to a Nernstian potential penalty of <0.059 V and ∼0.5 V.132,140 Therefore, operating with a neutral pH electrolyte results in a significantly higher minimum thermodynamic potential requirement.
5.3. Cations
Cations have a crucial influence on ECR reaction environments. Due to electric potential gradients, cations in the system will be drawn to the electrochemical double layer of the cathode. A commonly observed and modeled trend of cation enhancement effects for ECR is observed as Cs+ > K+ > Na+ > Li+ (Fig. 15A), with electrolytes utilizing Cs+ resulting in the best ECR performance.141,142 There are various computational explanations for this enhancement effect, including but not limited to (i) cations stabilize ECR intermediates such as *CO2, *CO, *COOH, and *OHCCHO through electrostatic interactions in the outer Helmholtz plane,141,143 (ii) cations provide higher surface charge to modify the interfacial electric field,144 and (iii) cations hydrolyze near the electrode surface to buffer the local pH.141,143,145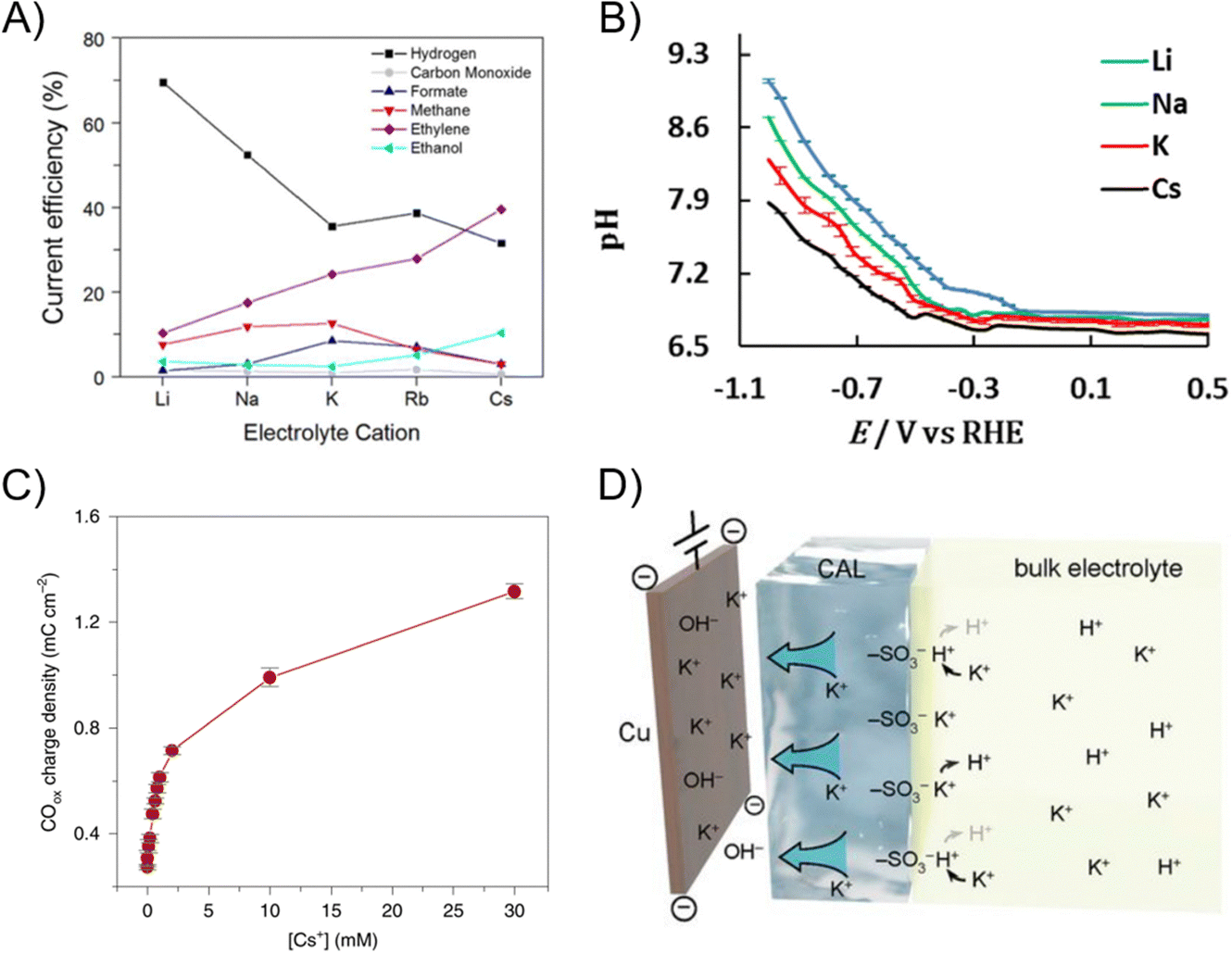 | ||
| Fig. 15 (A) FE of CO2 reduction over a Cu catalyst at −1.0 VRHE as a function of cation size in an H-type cell.141 Reprinted (adapted) with permission. Copyright 2017 American Chemical Society. (B) pH of the electrode surface measured by in situ infrared spectroscopy during CO2R on a Au catalyst.146 Reprinted (adapted) with permission. Copyright 2017 American Chemical Society. (C) CO produced (measured by oxidation at the RRDE ring) after polarization to –1V versus RHE in the presence of different concentrations of Cs+ in a Li2SO4 background electrolyte. The total cation concentration in solution was kept at 0.1M and the pH = 3.133 Reprinted (adapted) with permission. Copyright 2021 Springer Nature. (D) Schematic illustration of the ionic environment and transport near the catalyst surface functionalized by the ionomer.24 Reprinted (adapted) with permission. Copyright 2021 American Association for the Advancement of Science. | ||
Metal cations have been shown to be solvated, that is they are surrounded by the solvent molecules.147 Among classes or groups of cations, those with a smaller ionic radius have larger charge density, resulting in a larger hydration shell or hydration radius due to having multiple layers of solvated water molecules.148 Larger hydration shells mean that the cation is less favorable to be in the Helmholtz plane and stabilize ECR; consequently, the Cs+ to Li+ ranking relationship also reflects the energy to place a hydrated cation in the vicinity of the electrode surface.141 Organic cations such as alkylammonium and 1-ethyl-3-methylimidazolium also stabilize the intermediates through coordination effects.149–152 The size of organic cations play an important role, as Li et al. have shown that alkylammonium with short side chains (tetramethyl and tetraethyl ammonium) facilitates hydrogen binding to stabilize *CO, while longer side chains (tetrapropyl and tetrabutyl ammonium) are too bulky and block the *CO from accessing interfacial water.
Experimental evidence has provided evidence for pH modulation via cation effects in various cell geometries. Zhang et al. and Ayemoba et al. verified a local pH increase ranking in Li+ > Na+ > K+ > Cs+ using neutral MHCO3 electrolytes with a pH-sensitive ring on an RRDE and in situ infrared spectroscopy (Fig. 15B).128,146,153 The trend in pH is explained by the pKa of hydrolysis for solvated cations, which decreases as the cation size increases.145 As the local pH increases in neutral systems, local CO2 is lost as (bi)carbonates, leading to lower ECR activity, such that electrolytes using Cs+ result in the best ECR performance.
Under acidic conditions, ECR commences when the local concentration of cations is sufficient. Liu et al. noted that proton reduction is dominant at pH 3 and low operating potential, but as the applied overpotential increases, H+ depletes and the local pH is less acidic which enables ECR to occur.154 Similarly, as the bulk cation concentration in acidic electrolyte increases, ECR onset will begin (Fig. 15C).155,156 The local cation concentration needs to be higher than the local H+ concentration to significantly impede the mass transport of H+ and suppress proton reduction. Therefore, ECR performed in acidic environments often requires a high dose of K+ (e.g., 0.5 M K2SO4 with ≤0.1 M H2SO4).157 In MEAs, a common strategy is to coat various positively charged groups such as ionomers and solid electrolyte resins to the electrode surface to reduce the local H+ (Fig. 15D).154,158–160
In AEM or reverse-BPM MEAs, the presence of concentrated cations improves the selectivity of ECR. Due to imperfect ion selectivity in AEMs, researchers have noticed unintended cation crossover through AEMs due to electroosmotic drag.161,162 While unintended crossover is troublesome for product collection, cation crossover allows for cation effects to enhance ECR performance, even when no catholyte is used. El-Nagar et al. observed a higher degree of K+ crossover when 1 M KOH was used as the catholyte, but also higher multi-carbon FE on Cu-based catalysts compared to 0.05 M KOH (Fig. 16A).163 It was also noted that a more concentrated KOH anolyte (1 M) caused an oxide-derived Cu catalyst to be fully reduced to the metallic state, whereas the surface of oxide-derived Cu catalyst operating under dilute (<0.005 M) anolyte maintained 80 + % Cu1+ species. Likewise, in a reverse-BPM system where the acidic CEM hinders ECR, Yang et al. reported that a higher concentration of K+ (3 M) in the anolyte improved the CO2-to-CO selectivity by 3-fold due to K+ crossover in BPM (Fig. 16B).164
 | ||
| Fig. 16 (A) FE of CO2R and amount of K+ extracted from the cathode flow field as a function of anolyte concentration over a Cu catalyst at a full cell potential of 3.2V in an AEM MEA.165 Reprinted (adapted) with permission. Copyright 2021 American Chemical Society. (B) FE of CO2R on a Cu catalyst as a function of KOH anolyte concentration at 200 mA cm−2 in a BPM MEA.164 Reprinted (adapted) with permission. Copyright 2021 American Chemical Society. (C) CO2/O2 ratio in the anodic tail gas and photographs of cathode flow fields taken after 1 h CO2R at 200 mA cm−2 in an AEM MEA.166 Reproduced with permission from the Royal Society of Chemistry. (D) Proposed interfacial structure near the electrode surface during direct reduction of amine-captured CO2 without and with K+.167 Reprinted (adapted) with permission. Copyright 2021 Springer Nature. (E) CO partial current density from CO2R on a Ni-N-C catalyst in 1–5 M ethanolamine in H-type cell.168 Reproduced with permission from the Royal Society of Chemistry. (F) Potential cycling recorded for CO2R on a Au catalyst in CO2-saturated, unpurified 0.1 M NaHCO3 electrolyte solution in an H-type cell.169 Reprinted (adapted) with permission. Copyright 2023 American Chemical Society. | ||
While cations enhance catalytic performance, higher concentrations of cations also lead to faster solid carbonate salt precipitation in the pores of GDEs, which hinders CO(2) transport across the GDE. The rate at which M2CO3 carbonate salt precipitates form is related to their solubility in water, which trends as Li+ < Na+ < K+ < Cs+ (Table 2).170 A general measure of the degree of salt precipitation is then the CO2/O2 ratio of the anode tail gas where a high CO2/O2 ratio indicates little loss as carbonate salt, but more loss as bicarbonate ions (Fig. 16C).166 There is no visual identification of CsCO3 precipitate on the flow field when Cs+ is the cation; on the other hand, in the case of Li+, Li2CO3 precipitate forms rapidly at the electrode surface, before penetrating the GDE and reaching the flow field. Several operational strategies such as actively flushing the GDL with solvent, or pulsed electrolysis to periodically decrease the cation concentration at the GDE have been implemented to mitigate salt precipitation.72,171
| Salt | Solubility at 20 °C (M) |
|---|---|
| NaHCO3 | 1.14 |
| KHCO3 | 2.24 |
| CsHCO3 | 3.94 |
| Na2CO3 | 2.06 |
| K2CO3 | 7.93 |
| Cs2CO3 | 8.01 |
Another emerging branch of ECR is the direct reduction of reactive captured CO2 to avoid regeneration of CO2via heat cycling.172 Ethanolamine (EA) is a popular capture agent that has been made into an electrolyte for electroreduction of captured CO2. Nevertheless, EAH+ is a bulky cation in the electrolyte and congregates at the cathode surface. EA-captured CO2 (EACOO−) is too far away from the cathode surface and the primary reduction product is H2 on Ag catalysts (Fig. 16D).167 With the addition of alkali metal cations such as K+, CO2R is enabled, as K+ ions are appropriately sized for EACOO− to be reduced. Another approach to overcome EA cation effects is to adjust the potential of zero charge (PZC) of the electrode. Cation effects are present when the applied potential deviates away from the PZC of the electrode. By using a Ni single atom catalyst that has more positive PZC than Ag, Kim et al. were able to mitigate cation effects even at high EA concentrations and obtained high CO2-to-CO FE with reactive captured CO2 solutions (Fig. 16E).168
Finally, it is important to highlight that liquid electrolytes contain impurities such as trace metal cations that could disable ECR catalysts (Fig. 16F).169,173 To combat this effect, metal ion complexation or chelation has been proposed to prevent metal deposition on ECR cathodes and has shown promise for prolonged catalyst stability. Another source of metal ions comes from dissolution of anode catalysts. In acidic PEM systems, Ir is commonly employed as the anode catalyst; Ir-deposition on the cathode catalyst has been reported after performing acidic OER which shifts the cathode product selectivity towards H2.174 In alkaline AEM systems with KOH anolyte, 3d-transition metals are variably employed as the anode catalyst, which can undergo more significant leaching during operation due to the decrease in alkaline anolyte pH from (bi)carbonate equilibrium upon CO2 loss.175 Xu et al. noticed a smaller increase in the H2 FE under ECR operation with a Ni-based anode compared to Ir-based anodes in 0.1 M KOH. The cathode was also free of deposited Ni after 3 h of operation.176 However, under rare instances, contaminants can be beneficial. For example, the anodic OER reaction under alkaline conditions over 3d-metal transition catalysts such as Ni and Co can be enhanced by trace Fe contaminants that are common in KOH, which is the reason plain Ni foam is adopted as an ECR anode.27,98,177,178
5.4. Anions
It is critical to note first that the ubiquitous anions in neutral or alkaline, aqueous CO2R, CO32− and HCO3− are not inert. On Cu surfaces, *CO32− also adsorbs competitively with *CO, giving rise to an overpotential to exchange *CO32− with *CO.179 Similarly, HCO3− has been identified to poison Co-phthalocyanine catalysts at less reductive potentials.180 The HCO3− in the electrolyte is a CO2 source that can be reduced through exchange with CO2(aq).181,182 There are two ways through which HCO3− reduction can proceed. The first is through direct reduction, which has been demonstrated in a pressurized stagnant H-cell with a maximum formate partial current density of 3.2 mA cm−2 in 2.8 M KHCO3 solution (Fig. 17A).183 The other pathway makes use of the acidity from the CEM component of a BPM to regenerate CO2 from HCO3− within the electrolyzer, which is then reduced; this route has been shown to reach current densities >100 mA cm−2 (Fig. 17B).184–186 | ||
| Fig. 17 (A) Illustration of direct HCO3− reduction via HCO3−/CO2 equilibrium.183 Reprinted (adapted) with permission. Copyright 2015 American Chemical Society. (B) Illustration of in situ release of CO2 from HCO3−via H+ produced with reverse-BPM.187 Reprinted (adapted) with permission. Copyright 2019 Elsevier. (C) Total CO2R FE on a Au catalyst with various halide anions in an H-type cell.188 (D) Total CO2R FE on a Au catalyst with various carboxylate anions in an H-type cell.188 Reprinted (adapted) with permission. Copyright 2024 American Chemical Society. (E) Formate FE over Bi oxyhalide catalysts in a GDE flow cell.189 Reprinted (adapted) with permission under Creative Commons License. | ||
Anions, such as HCO3− and H2PO4−, are also proton donors. Based on pKa values, HCO3− (10.3) is a weaker proton donor than H2PO4−.181 When H2PO4− is present in ECR systems, it would transiently increase the FE of H2 and CH4 due to its proton donating effect until local H2PO4− has been depleted.190 Yoo et al. showed that adsorbed carboxylate anions were identified to enhance ECR, and adsorbed propionate yielded a peak CO2-to-CO FE of 99% at ∼3 mA cm−2 by hindering the transport of H+/OH− to negatively impact HER kinetics (Fig. 17C and D).188 Halide adsorption also induces morphological changes to the catalysts that expose ECR-favoring facets to enhance the performance (Fig. 17E).189,191,192 The degree to which halides enhance ECR is in the following order: I− > Br− > Cl−, which is ranked by increasing adsorption strength on the cathode surface. More broadly, in contrast to cations which are drawn to the cathode surface, anions migrate towards the anode in AEM liquid electrolyte systems, leaving the catholyte with as little as <10% of its original anion concentration, as shown by Zheng et al. in a GDE flow cell.193
Overall, there are a multitude of changes that complicate the electrolyte effects as electrolysis proceeds, namely pH changes, (bi)carbonate formation, and species migration that could lead to changes in activity, selectivity, ohmic drop, and stability.
6. Hydrophobicity effects
Since high current density operation in aqueous ECR depends heavily on delivery of CO(2) in the gas phase, the TPB must be maintained with suitable wetting conditions. This is exceptionally difficult when a liquid catholyte is present due to gas–liquid pressure gradient and electrowetting that causes GDE flooding.64 This section summarizes the methods to mitigate flooding via tuning of the GDL, catalyst layer (CL), and electrolyte composition.6.1. GDL modifications
Carbon-based GDLs are the most prevalent choice for cathodes in ECR. As shown in Fig. 18A, they are made of a macroporous carbon fiber backbone (CFB) and an optional microporous layer (MPL).194 The carbon fiber backbone holds the bulk structure and provides conductivity. It governs the mechanical properties (e.g., strength, response to compression) of GDL. The MPL, typically made of conductive carbon particles and PTFE, ensures smooth contact with the CL, and provides water management to avoid flooding of the GDL. Although PTFE incorporation in MPL creates a highly hydrophobic environment (typical water contact angles >150°), after CL deposition and under ECR conditions, flooding still occurs within ∼3 h of electrolysis in GDE flow cells (Fig. 18B).27,71,124,195 A main cause of GDL flooding is that commercial GDLs are most appropriate for fuel cell and water electrolyzer applications where the GDL does not interface with the liquid electrolyte. Hence, GDLs are generally optimized for gas transport with microcracks in the MPL. For example, in Sigracet's line of GDL models, the BB models that were developed as successors to BC models contain much higher microcrack density compared to the older BC models (Fig. 18C).196 | ||
| Fig. 18 (A) Illustration of a GDE containing the carbon fiber substrate (macroporous layer), MPL, and CL. Figure adapted from ref. 197. Reprinted (adapted) with permission. Copyright 2020 American Chemical Society. (B) Current density and FE comparison of the CO2R stability of Bi/C GDEs with and without PTFE modification in a flow cell.195 Reprinted (adapted) with permission under Creative Commons License. (C) SEM images of MPL of (top) Sigracet 22BB and (bottom) Sigracet 39BB carbon-based GDLs.196 (D) Illustration of flooding mechanism in a GDL with bimodal pore size distribution. Large pores are filled with electrolyte while smaller pores allow for gas transport.196 Reprinted (adapted) with permission. Copyright 2022 American Chemical Society. (E) Illustration of a Cu/C-coated PTFE membrane as a GDE.27 Reprinted (adapted) with permission. Copyright 2018 American Association for the Advancement of Science. (F) Partial current density of products on Cu/C-coated PTFE as a function of C layer thickness in a GDE flow cell.198 Reprinted (adapted) with permission. Copyright 2022 American Chemical Society. | ||
The failure mechanism of GDL flooding is that the pores of the CFB are filled with water, thereby hindering delivery of gas phase CO(2) and forming carbonate salts. Baumgartner et al. tested carbon-based GDLs with various thickness and pore structure and concluded that having bimodal pore size distribution allows electrolyte to fill up the larger pores while leaving the smaller pores available for gas transport (Fig. 18D).196 To prevent liquid from entering the CFB, PTFE can be infiltrated through the CFB to embed an auxiliary hydrophobic layer between the CFB and MPL that could mitigate 90% of electrolyte seepage into the CFB.199 Other researchers have implemented additional hydrophobic layers between MPL and CL to avoid electrolyte penetration and salt precipitation.200
Hydrophobic membranes have also been used to replace carbon-based GDLs for their exceptional resistance towards flooding. Numerous polymeric materials including nanoporous polyethylene membranes,201 PTFE membranes,25,27 and even 3D printed perfluoropolyether202 have shown prolonged stability for high current density operations (Fig. 18E). Nonetheless, too much hydrophobicity, as Kong et al. showed, causes reductant water diffusion limitations.198 In this study, a 6.6 μm layer of carbon black deposited on a Cu-based GDE yielded the highest multi-carbon FE via modulation of local water availability (Fig. 18F). Furthermore, since most polymeric membranes are electrically insulating, supplementary current collectors need to be incorporated, and they are discussed in detail in Section 7.
6.2. CL modifications
Even though hydrophobic GDLs prevent flooding of GDL pores, catalyst particles under ECR conditions are also fully wetted.195,197 This again results in lower local [CO(2)] and longer diffusion path lengths for CO(2) molecules. Catalyst structure engineering has been employed to mimic nature's superhydrophobic surfaces such as lotus leaves203 and aquatic arachnids204 to resist CL flooding; these three-dimensional catalyst structures have been proven to surpass their planar counterparts in terms of hydrophobicity.195,197,205,206Others have also integrated fluoropolymer treatment into the CL (Fig. 18B).71,195,207 Industry standard ionomer Nafion is a perfluorosulfonic acid that also contains a fluorinated backbone. As the US Environmental Protection Agency and European Commission roll out restrictions to prevent polyfluoroalkyl substances contamination, other hydrophobic substances such as lipid ligands and N-heterocyclic carbenes polymers have also been employed.208–211 When hydrophobicity modifiers are added to the system, their stability must also be considered to warrant actual improvement of ECR stability.
6.3. Electrolyte modifications
Electrolyte ions exhibit the aforementioned electric field effects depending on the hydration of the cation. Weakly hydrated cations such as K+ and Cs+ are more concentrated near the electrode surface and lower the liquid tension to cause flooding.64 ECR liquid products also have lower surface tension compared to water and are likely to wet the surface.212 In summary, redesign of the GDL and catalyst structure help prevent GDE flooding and prolong operation stability. Future studies could focus on developing oleophobic GDEs for concentrated liquid product collection, as well as optimization of flow-through cell geometry to address the fundamental underlying cause of flooding.7. Conductivity effects
Establishing high conductivity throughout the electrolyzer decreases the operating voltage and energy consumption of ECR. As ECR moves towards commercialization, savings in electrical energy consumption is critical, not only at the single cell level, but multiplied by the stack number. Ohmic drop originates from all components of the electrolyzer, including the electrolyte, catalyst, substrate, flow field, and current collector.7.1. Electrolyte conductivity
Ion conductivity in liquid electrolyte systems dictates the solution series resistance. The molar ion conductivity values of common ions present in ECR systems are listed in Table 3. The ideal ion conductor is H+ due to its high molar conductivity, but high H+ concentration shifts the product selectivity to H2; thus, the best ECR ion conductor in most applications is OH−. (By)products from ECR such as (bi)carbonate and oxygenates, which can also become ion conductors. They increase the liquid conductivity, but may hinder the transport of more conductive ions, such as OH−, through AEMs. In the same way, when cations such as K+ are used in liquid electrolyte systems, they also become ion conductors and increase the ohmic drop across CEMs due to competitive transport with H+. The concentration of electrolyte solutions also affects the conductivity. Typical electrolyte conductivity increases as the concentration increases until the solution becomes too viscous. The peak conductivity for KOH for low temperature ECR is at 6–8 M for low temperature ECR (Fig. 19A).213 Nevertheless, high concentration of ions causes electrowetting and salt precipitation, as noted previously; electrolyte concentrations above 1 M are not regularly used in the ECR literature.| Ion | Molar ionic conductivity (S cm−2 mol−1) |
|---|---|
| H+ | 350.1 |
| Na+ | 50.1 |
| K+ | 73.5 |
| Cs+ | 77.3 |
| OH− | 198 |
| HCO3− | 44.5 |
| CO32− | 138.6 |
| COOH− | 54.2 |
| CH3COO− | 40.9 |
 | ||
| Fig. 19 (A) KOH and its specific conductivity as a function of molarity at 30, 60, and 100 °C.213 Reprinted (adapted) with permission. Copyright 2007 Elsevier. (B) WU and IEC of Nafion N115 membrane as received, wet, boiled in 3 wt% H2O2 and 1 M H2SO4, and dried after treatment.216 Reprinted (adapted) with permission. Copyright 2016 American Chemical Society. (C) Polarization curves of PEMWE showing performance with Nafion NR212, N115, and N117 at 80 °C.217 (D) Polarization curves of PEMWE with Nafion NR212, N115, and N117 after 0, 72 h, and 114 h of constant operation at 3.0 A cm−2 and 80 °C.217 Reprinted (adapted) with permission. Copyright 2024 Elsevier. | ||
IEM conductivity is measured in terms of ion exchange capacity (IEC, concentration of charged functional groups within the membrane), or equivalent weight (EW, weight of membrane per mole of ion exchange sites). Water uptake (WU) also affects the ion conductivity, as ion transport is facilitated through the membrane. The ionic conductivity of the Nafion 117 membrane is 0.13 Ω−1 cm−1 at 100% relative humidity (RH) and 75 °C, but at 80% RH and the same temperature, the conductivity is halved.218 The Nafion membrane WU improves from H2SO4 and H2O2 treatment, but if it dries out before use, the WU and IEC reverts to the untreated state (Fig. 19B).216 IEMs are generally sold based on their functional groups, thickness, EW, and polymer reinforcement.
While sulfonic acid is the standard functional group for CEMs, AEM brands such as Sustainion and Piperion utilize different functional groups to either stabilize CO2− intermediates or allow for high carbonate conductance.219,220 The thickness of IEMs trends inversely with through-plane resistance. For instance, Nafion series NR212, N115, and N117 have thicknesses of 50.8, 127, and 177.8 μm, respectively; the high frequency resistance (HFR), which mainly represents the ionic resistance of the IEM, of this Nafion series is as follows: NR212 < N115 < N117 (Fig. 19C).217,221 However, as the membranes become thinner, they are more prone to H2/O2 gas crossover, and their HFR increases more drastically over time (Fig. 19D). Unintuitively, a thinner AEM decreases the CO2 loss by facilitating the OH− transport from the cathode surface to the anode to prevent OH− reaction with CO2.222 However, this comes at the cost of mechanical stability as thinner membranes tear more easily. Polymer impregnation with expanded PTFE (ePTFE) is often done to reinforce the mechanical stability of IEMs.223–225 At the same thickness, ePTFE-reinforced composite IEMs typically have lower water uptake and higher resistance than regular IEMs, but due to enhanced mechanical stability, composite IEMs can be made thinner to compensate for the increased resistance.
7.2. Electrode conductivity
The addition of PTFE particles or using PTFE membranes as a substrate both give rise to electrode conductivity challenges. This issue will be exacerbated by larger electrode areas. As the PTFE content of MPL increases from 10 to 50 wt%, the charge transfer resistance of the GDE also increases from ∼5 Ω to ∼8 Ω (Fig. 20A).226 A tradeoff between hydrophobicity and ohmic loss is present; it is important to obtain the most flooding resistance from the same amount of PTFE loading by decreasing the PTFE particle size which could stand higher breakthrough pressure indicated by the Young-Laplace equation.195 Likewise, the PTFE membrane is largely inert, so either a thick layer of catalyst (>1 mg cm−2 of particles or >200 nm of sputtered materials) needs to be deposited,227,228 or a conductive overcoat such as graphite and/or an additional catalyst-ionomer mixture needs to be applied.27,69,229 In worse scenarios, a lack of conductivity causes ECR to only take place near the current collector, inducing nonuniform local heating of the GDE.230 A non-invasive approach to improve PTFE-based GDE conductivity is to use grids of metal current collector, either embedded in or on top of the catalyst layer (Fig. 20B and C).27,69,229–232 | ||
| Fig. 20 (A) Impedance spectra of Ag-GDEs with varying PTFE content in the MPLs at a full cell potential of −2.0 V.226 Reprinted (adapted) with permission. Copyright 2016 Elsevier. (B) Schematic and photograph of the non-invasive current collector (NICC) deposited on a Cu-PTFE GDE.230 (C) C2H4 FE comparison between 50 nm Cu, 500 nm Cu, and 50 nm Cu with NICC on a PTFE GDE in a flow cell.230 Reprinted (adapted) with permission under Creative Commons License. (D) Current density of a PEMWE at a full cell voltage of 1.9 V over time using platinized (dark gray) and non-platinized (light gray) Ti PTLs.233 Reprinted (adapted) with permission under Creative Commons License. (E) Polarization curves of IrOx anodes at various loadings with or without PtOx conductive additive in PEMWEs.234 Reprinted (adapted) with permission. Copyright 2024 American Chemical Society. (F) Polarization curves measured during alkaline water electrolysis using flow fields that are made of 3D-printed polylactic acid (PLA), Ni, and Ni plated 3D-printed PLA.235 Reproduced with permission from the Royal Society of Chemistry. | ||
The most popular PTL used for anodes in neutral bicarbonate anolytes is Ti-based.236 Since Ti oxidizes to insulating TiO2, a protective noble metal layer with Pt is required to protect Ti PTL against oxidation (Fig. 20D).233 Conductive additives are also applicable to the anode OER catalysts. Ferner et al. demonstrated that with 1.21 mg cm−2 of conductive PtOx support, 0.2 mg cm−2 IrOx achieves OER performance comparable to that of 1.88 mg cm−2 of IrOx at ∼ 2 A cm−2 (Fig. 20E).234
7.3. Device conductivity
Bipolar plates comprise up to 80% of the weight in electrolyzer stacks, and contribute to 53% of electrolyzer cost, even in PEMWE systems which contain expensive noble metal catalysts.237 It is crucial to maximize the mass transport and current conduction of bipolar plates. 3D printing allows for rapid prototyping of flow fields to screen through the most efficient flow pattern; nonetheless, 3D-printed flow fields are not conductive and require metal coating for current collection (Fig. 20F).235,238 Graphite is the standard material for PEMWE and PEMFC applications when water is the only liquid present in the system for their high electrical conductivity and thermal stability.239 Ti flow fields are selective for conductivity and corrosion against acid and base; Ni flow fields could only be used on the anode with alkaline anolyte due to reaction with CO.239–242 Gold coating can be applied to further decrease the contact resistance of bipolar plates.109,2438. Temperature effects
Although “low temperature” ECR only operates between 20 and 80 °C due to IEM thermal stability,244,245 temperature can significantly impact the ECR performance by changing the reactant solubility, reactant and product diffusion, reaction thermodynamics and kinetics, and electrical conductivity of the system (Fig. 21A). Henry's law states that the solubility of gas in the liquid decreases as the temperature increases.81,246 In liquid immersed cathode systems, where the reaction rate heavily depends on dissolved reactant concentration, temperature control to maintain cell temperature from ohmic heating needs to be implemented. | ||
| Fig. 21 (A) Variation of different ECR system parameters with temperature as normalized to 20 °C.132 Reprinted (adapted) with permission. Copyright 2024 American Chemical Society. (B) C2H4 FE from CO2R on a Cu catalyst as a function of temperature in a heated H-type cell.247 Reprinted (adapted) with permission. Copyright 2023 American Chemical Society. (C) Proton conductivity of the Nafion N115 membrane at various temperatures during heat cycles.248 Reprinted (adapted) with permission under Creative Commons License. | ||
Gas-fed GDE systems generally benefit from higher operating temperatures, though it is difficult to decouple diffusion and reaction effects. Both kinetic and diffusion coefficients follow exponential growth with temperature, thereby decreasing the kinetic and diffusion overpotentials.249 Vos et al. showed experimentally that an intermediate operating temperature of 40–50 °C yields the highest multi-carbon product FE (Fig. 21B).66,247,250 Enhancement of C–C coupling arises from increased *CO coverage and local pH.
Operation at >90 °C accelerates degradation of IEMs. Water management, or drying, is the most significant contributor to membrane degradation.251 Specific functional groups also experience chemical degradation.252 For example, sulphonic acid on Nafion with H3O+ will degrade to form ˙OH radicals that attack the fluorinated backbone to release CF2 molecules.253–255 At 100%RH, the peak Nafion membrane conductivity is between 70 and 90 °C (Fig. 21C);248 to balance thermodynamics of electrochemical reactions and membrane degradation, 80 °C is often chosen as the maximum operating temperature of Nafion membranes where the degradation rate is only 3 μV h−1.256 Below 80 °C, water mobility increases with increasing temperature, causing a higher WU which generally benefits ionic conductivity.257,258
9. Electrochemical techniques
Standard ECR operation includes electrochemical impedance spectroscopy (EIS), conditioning, and chronoamperometry (CA, constant potential hold) or chronopotentiometry (CP, constant current hold) for resistance quantification and product analysis.259 Conditioning steps prior to product distribution tests can include cyclic voltammetry (CV) sweeps, or CA/CP holds at low operating potential/current to activate the system.260,261 Although the goal for industrial operation is to maintain constant current, it is important to evaluate the performance across different catalyst loading levels using CAs and accounting for difference in ECSA to yield a fair comparison.262 Catalyst dissolution and redeposition may occur during conditioning, although not resulting in significant changes to the ECSA.263 Cu-based catalysts may undergo immediate dissolution upon contact with the electrolyte at open circuit potential that leads to irreversible loss in ECR selectivity. When catalyst particles are small (∼5 nm), a slightly reductive potential is required to stabilize the catalyst particles.264 For product distribution testing, CA or CP holds are performed with gradually increasing intensity to step towards the highest operational condition that system stability permits.62,265 During longer term operation, the electrolyte needs to be periodically replaced, for the reasons that were mentioned previously that contribute to system instability.9.1. Pulsed electrolysis
Pulsed electrolysis is an electrochemical technique where the applied potential of the system is cycled between two values Ea and Ec at time intervals ta and tc. Pulsed electrolysis has been employed to enhance ECR performance in both liquid-immersed cathode systems and gas-fed systems.266 There are two types of pulsed electrolysis studied in ECR, one with Ea set to a mildly cathodic potential with no noticeable amount of reaction, and the other with Ea that intentionally induces electrochemical oxidation of the catalyst.Cycling between “on” and “off” states during ECR has a few major benefits. Kim et al. performed pulsed electrolysis in a differential electrochemical mass spectrometry cell for real time detection of reactant and product concentration. It was reported that pulsing between Ea = −0.8 VRHE and Ec = −1.15 VRHE could enrich the local CO concentration during Ea to enhance multi-carbon FE during Ec (Fig. 22A and B).267 On further utilizing the CO2 enrichment effect of Sustainion ionomers and OH− trapping of Nafion ionomers, a multi-carbon FE of 90% was achieved under pulsed electrolysis.268 Cation effects are also amplified with pulsed electrolysis. While Li+, Na+, K+, and Cs+ cations showed increasing ratios of multi-carbon product to CH4 FEs spanning from 0.7 to 1.9, the same series of cations under pulsed electrolysis had FE ratios ranging between 0.1 and 8.4 due to improved *CO coverage as a result of cation enrichment.269 Although a high local concentration of cations is reported during Ec, time spent at Ea avoids prolonged saturation with cations and prevents salt precipitation.171 Lastly, pulsed electrolysis can also allow for Cu-based catalyst regeneration by re-oxidation (Fig. 22C and D).270,271
 | ||
| Fig. 22 (A) CO and H2 mass spectrometry signal intensity and (B) FE for CO2R products from pulsed electrolysis on a Cu catalyst operated at different tc at Ec = −1.15 VRHE in H-type cell. ta was 10 s and Ea = −0.8 VRHE for all cases of pulsed electrolysis in this work.267 Reprinted (adapted) with permission. Copyright 2020 American Chemical Society. (C) Schematic illustrations involving the catalyst during galvanostatic and pulsed electrolysis.270 (D) C2H4 FE of pulsed electrolysis over a Cu catalyst as a function of time operated at jc = −100 mA cm−2, Ea = open circuit potential, tc = 15 min, and ta = 5 or 15 min in a GDE flow cell.270 Reprinted (adapted) with permission. Copyright 2020 American Chemical Society. (E) Schematic depiction of Cu2O catalyst structures and compositions during pulsed electrolysis.272 Reprinted (adapted) with permission under Creative Commons License. (F) Current density and FE for CO2R products from pulsed electrolysis on Cu2O catalysts operated at Ea = 0.9 and 1.2 VRHE and Ec = −0.7 VRHE in a GDE flow cell.273 Reprinted (adapted) with permission. Copyright 2021 American Chemical Society. | ||
Going further past the oxidation redox potential in the positive regime (0.6 VRHE) during Ea oxidizes Cu to Cu2O and CuO. Short pulses (<2 s) between oxidized states and the metallic state creates and replenishes multivalent Cu sites that boost ethanol formation, whereas longer pulses and more anodic Ea (1.2 V) formed Cu moieties selective towards CH4 (Fig. 22E and F).272,274–276 The key intermediate binding energy that is altered in this operation mode is *OH where ethanol formation is due to increased *OH coverage compared to galvanostatic operation, yet at high *OH coverage, the catalyst is poisoned hindering C–C coupling.
9.2. Accelerated stress tests
Accelerated stress tests (ASTs) are electrochemical protocols developed to assess the durability of an electrolyzer in a shorter amount of time since performing galvanostatic holds to reach the actual stability requirements, e.g., 3000 h or 4.1 months, is impractical when screening through new control variables. ASTs apply harsh conditions to electrolzyer components and thereby expediate degradation processes; for electrochemical processes, rapid potential and/or current cycling accelerates degradation of catalysts, ionomer binders, and conductive carbon support (Fig. 23A–D).277 Cofell et al. showed that Ag-based catalysts undergo agglomeration and carbonate deposition during AST that caused a decrease in ECR FE (Fig. 24A–D).278 Ionomers such as Nafion that contain CFx groups are prone to oxidation to C–O or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups.279 Conductive carbon, if present in ECR systems, will also surface oxidize to insulating C–O/CO groups or evolve CO2 at half-cell potential >1.8 VRHE.280 Note that pulsed electrolysis is essentially cycling through potentials or currents, which resembles ASTs;281,282 the long-term stability effects (>1000 h) of pulsed electrolysis has yet to been assessed.
O groups.279 Conductive carbon, if present in ECR systems, will also surface oxidize to insulating C–O/CO groups or evolve CO2 at half-cell potential >1.8 VRHE.280 Note that pulsed electrolysis is essentially cycling through potentials or currents, which resembles ASTs;281,282 the long-term stability effects (>1000 h) of pulsed electrolysis has yet to been assessed.
 | ||
| Fig. 23 (A) Cell potential during an AST in an AEM MEA with the 0.1 M CsHCO3 electrolyte. (B) Cs signal intensity (could be used towards quantification) measured by in situ x-ray fluorescence. (C) Water signal intensity measured by wide angle x-ray scattering. (D) CO2R FE for CO and H2 during the AST.277 Reprinted (adapted) with permission under Creative Commons License. | ||
 | ||
| Fig. 24 (A) SEM image of an as-prepared Ag-GDE. (B) Current and cell potential of an “AST 1” protocol in a GDE flow cell. (C) SEM of GDE after the “AST 1” protocol. (D) CO FE and current as a function of time during the “AST 1” protocol.278 Reprinted (adapted) with permission. Copyright 2022 American Chemical Society. | ||
10. Scale up
Scaling up of ECR not only means operating at higher current density, but also at larger capacity and with more diversified products. Currently, literature reports include several examples of >100 cm2 operation,67,98,283–288 and start-up companies have disclosed 2500 cm2 operation.289Fig. 25 shows the various target molecules that companies aim to produce from scaled up ECR process.290–292 Tandem thermal reactions of the electrolyzer gaseous effluent containing a mixture of single- and multi-carbon products are also a promising pathway to broaden the product profile.293–295 | ||
| Fig. 25 Summary of pathways and target products of companies based on ECR technologies.290 Reprinted (adapted) with permission. Copyright 2024 American Chemical Society. | ||
Scale up of ECR requires altering the preparation of several main components including catalyst layers, transport layers, IEM, and bipolar plates. Starting with the catalyst layer, it must be uniform to avoid local hot spots that could melt the IEMs due to resistive heating. Catalyst inks are usually sonicated for homogenization, and a larger volume of inks will require longer sonication time that could lead to heating of up to 50 °C in an hour.296 A combination of ultrasonic bath and probe sonication has been shown to prevent agglomerates and achieve temperature control to prevent catalyst ink overheating.297 Catalyst can either be deposited on the membrane (catalyst coated membrane, CCM), or on the GDL (catalyst coated substrate, CCS) (Fig. 26A and B).298 CCS is typically fabricated by ultrasonic spray coating the GDL with catalyst ink, and CCM can be done by either direct deposition onto the IEM or decal transfer with a smooth substrate via spray coating or film casting. In a PEMWE device, CCM has been shown to outperform CCS or partial CCM (CCM anode and CCS cathode) due to larger interfacial contact area between the GDL and IEM (Fig. 26C).299 Due to the mechanical stability and swelling of AEMs, scaled up AEM-based CO(2) electrolyzers in the literature have exclusively employed CCS while PEM-based CO(2) electrolyzers such as the Opus system by Twelve have opted for CCM.300 A common high throughput method to scale up the coating process is to use roll-to-roll continuous fabrication.301,302 The CCM deposition method must leave the IEM deformation free to ensure normal operation.
 | ||
| Fig. 26 Schematic illustrations of (A) CCS and (B) CCM for MEAs.303 Reprinted (adapted) with permission under Creative Commons License. (C) Comparison of CCM and half CCM/half CCS water electrolysis with the same catalysts in a PEMWE device under the same operating conditions.299 Reprinted (adapted) with permission under Creative Commons License. | ||
The transport layers for CO(2) electrolyzers take a larger role in mass transport when the device is scaled up. As previously discussed, the TPB needs to be stable for gaseous reactants to benefit from being delivered in the gas phase. In a lab scale 5 cm2 device, a 10–20 mbar of Δpliquid–gas is suitable to prevent gas/liquid crossover in a GDE flow cell (GDL type unspecified).75 When scaled up to 1526 cm2 GDE flow cell, the Δpgas–liquid now needs to be >120 mbar to prevent flooding using a carbon cloth with MPL (Fig. 27A).284 Therefore, carbon-based GDL needs hydrophobic treatment that may include coating of a hydrophobic polymer and calcination to remove the surfactants that are in the polymer solution. Both Jiao and Sinton groups have demonstrated operation with these fluoropolymer-modified carbonaceous GDLs at 300 A and 800 A, respectively, at >200 h.98,200 Anode PTLs also benefit from a MPL made from sintered titanium particles for decreased cell resistance.304,305
 | ||
| Fig. 27 (A) CO2-to-formate FE over the Sn catalyst in a 3052 cm2 GDE flow cell at various gas and liquid operating back pressures.284 Reprinted (adapted) with permission under Creative Commons License. (B) Anolyte pH, anolyte conductivity, and full cell voltage of CO2-to-CO conversion over the Ag catalyst in a MEA as a function of electrolysis time.306 Reprinted (adapted) with permission. Copyright American Chemical Society. Full cell voltage and CO FE of CO2-to-CO conversion over Ag catalyst with (C) 0.1 M and (D) 0.01 M KHCO3 anolyte during stability tests.307 Reprinted (adapted) with permission. Copyright 2025 Springer Nature. | ||
The bipolar plates for scale up operation need to reconsider flow patterns. For an 800 cm2 cell, Nelson et al. scaled up the width of multi-serpentine flow channels to 5 mm compared to the 0.8 mm single-serpentine channels for 5 cm2 research-grade cells to maintain a 3.5% channel width to side length ratio.308 The depth of multi-serpentine channels was optimized to an average of 0.68 mm to ensure no stagnation zones and a higher mass transport coefficient.
At the stack scale, any source of voltage increase is multiplied by the stack number, so it is crucial to minimize all sources of overpotential. Edwards et al. reported a series resistance of 9.23 Ω cm2 at 25 mA cm−2 in their 800 cm2 electrolyzer with 0.1 M KHCO3, which is decreased to 7.41 Ω cm2 at 75 mA cm−2 due to the production of OH− with higher ionic conductivity.309 Although OH− has higher ionic conductivity, it is impractical to run large scale reactors with KOH due to the formation of (bi)carbonate from carbon loss. In a 5 cm2 electrolyzer operating at 150 mA cm−2, 50 mL of 1 M KOH would be converted to KHCO3 within 2 h, leading to a conductivity decay from 220 to 70 S cm−1 (Fig. 27B).306 Increasing the concentration of the electrolyte can also reduce the cell potential, but higher concentration of K+ results in faster formation of salt precipitates (Fig. 27C and D) to tolerate higher anolyte K+ concentration, Hao et al. coated the flow field with a hydrophobic polymer to facilitate removal of penetrated electrolyte droplets.307 As discussed in Section 5.2, the inherent nature of CO(2)R and OER increases and decreases the local pH, respectively. Nernstian overpotential due to pH gradient within the electrolyzer amounts to 0.51 V.309
In short, scaling up ECR calls for modifications that maximize performance and stability. However, there exist various trade-offs when choosing operating parameters. High resistive overpotential can be reduced via a concentrated electrolyte that leads to faster salt precipitation. Carbon loss can be mitigated via the use of an acidic electrolyte that promotes side HER. Careful lifecycle assessment and technoeconomic analyses are necessary to optimize and balance around these trade-offs. According to Da Cunha et al., the electrolyzer CapEx decreases when the production rate is increased and is calculated to be outweighed by separations CapEx at ∼500 mA cm−2 for CO production, which is tied to selectivity.79 The largest contributor to operating expenditure (OpEx) is the electricity cost; operating the electrolyzer makes up 78% of the energy consumption with anode tail gas downstream separation due to CO2 crossover only contributing to a small fraction.
11. Conclusion and outlook
This review described the effect of operational parameters including reactant availability, electrolyte choice, hydrophobicity treatment, conductivity modification, operating temperature, and electrochemical protocols on ECR performance in H-type cells and GDE-based flow cells or MEAs. Key takeaways from Sections 3–9 are summarized as follows:(1) Liquid-immersed cathode geometries (H-type cell and RRDE) are useful to conduct fundamental studies, but the solubility limit of reactants hinder operation to <30 mA cm−2. GDEs allow gas phase delivery of reactants and based on the need for electrolyte tunability, both the flow cell and MEA geometries offer operating current densities >200 mA cm−2.
(2) CO(2) partial pressures <1 atm can be used to perform kinetic studies. A decrease in multi-carbon FE is often observed when pCO(2)< 1 atm. Impurities in the reactant stream may cause irreversible changes to catalysts or may become the primary species to be reduced. CO(2) pressures >1 atm facilitate C–C coupling by increasing the *CO coverage and can render transition metal catalysts active for ECR, even if they are otherwise only suitable for the HER.
(3) Electrolyte bulk pH and buffering capacity affect the HER FE of the system. Alkaline electrolytes cause CO2 loss in the form of (bi)carbonate. Acidic electrolytes can be used to improve SPCE but need cations to stabilize ECR intermediates. Cation hydration changes the local pH to more alkaline in the order of Li+ > Na+ > K+ > Cs+. Bicarbonate is in equilibrium with CO2 and therefore can be used as a carbon source.
(4) Maintaining the TPB of a GDE is critical for gas fed reactants. Hydrophobicity is important to prevent flooding of GDEs and can be achieved via catalyst structure tuning, additive engineering, and using polymeric membranes as GDLs.
(5) Improved electrical conductivity decreases energy lost due to ohmic resistance. Adjusting the electrolyte concentration and maintaining WU of the IEM improve electrolyte ion conductivity. Deposition of conductive metal grids on GDEs enables more uniform ECR activity on larger scale GDEs. Metal coatings on bipolar plates also lower the contact resistance of the system.
(6) Increased temperature causes lower reactant solubility in the aqueous phase but enhances diffusion and kinetics of ECR. In gas-fed systems, moderately elevated temperature (up to 80 °C) enhances performance without sacrificing stability.
(7) Pulsed electrolysis improves performance and stability via catalyst regeneration and saturation of local reactants and cations. Universal AST protocols have not been developed for ECR, but potential/current cycling accelerates degradation of catalysts, ionomer binders, and conductive carbon.
(8) Scale up of ECR devices requires redesign to ensure uniformity. Any source of overpotential will also be exacerbated by the stack number and therefore needs to be minimized. Nevertheless, methods to improve performance usually come at the expense of decreased durability. Technoeconomic analyses are necessary to identify the best operating parameters for scale up.
Optimizing ECR towards commercialization requires high activity, selectivity, carbon efficiency, and durability. To collectively progress the ECR field, the following challenges and suggestions may be worth considering:
11.1. Activity
Since breaking CO bonds in CO(2) is so energetically unfavorable, the EE of low temperature electrolysis must be high to compete with high temperature electrolysis or thermal chemical routes.310–312 The majority of ECR studies done in GDE flow cells and MEAs use the CCS method to deposit the anode catalysts. CCM systems have been proven in PEMWE, PEMFC, and ECR MEA to reduce the contact resistance.298,313 Depending on the anolyte used (e.g., KHCO3), ECR may require OER catalysts that outperform Ir under mildly acidic conditions.314 The anodic reaction could also be replaced with organic oxidation reactions that have lower oxidative redox potential, and could bring additional economic value-add to the system.298,313,315,316 However, the stability of the system may be jeopardized with organic solvents, and the current density is often limited in aqueous media.
11.2. Selectivity
Both C1 products, CO and formate, have been shown to reach >95% FE through catalyst design. Multi-carbon product FEs have reached a sticking point where the record for producing a single multi-carbon product (in this case C2H4) was 75% FE in 2020 over the Cu–Al alloyed catalyst.19 While this was a promising breakthrough, testing of this catalyst was in a GDE flow cell on a PTFE membrane with concentrated KOH and KI, which is atypical for ECR product distribution testing. The field must develop standardized cell geometry and operating conditions to yield fair comparison among catalysts. Product collection and FE analyses also require rigorous assessment, as the tail gas flow rate changes during the reaction and product crossover or evaporation may occur.46,317 FEs are also measured based on electrons consumed towards a specific product whereas the actual product concentration in the product stream is far lower than 50% unless SPCE was optimized, requiring high downstream separation cost. Tandem catalysis could be used also to improve multi-carbon FE since COR yields C2H4 and acetate as the sole ECR gas or liquid product under the correct conditions.98,31811.3. Carbon efficiency
Although SPCE of alkaline ECR systems remains low, optimizing SPCE may not be the most cost efficient due to lower EE in alternative systems.84 Technoeconomic analysis should be performed to assess the optimization of SPCE versus spending additional energy regenerating CO2. Direct reduction of CO2 in the captured state, as (bi)carbonate or carbamate, should also be considered as it interfaces with the carbon capture sector, as well as prevents carbon loss to the electrolyte.18411.4. Stability
Thus far only ECR performed in PEM MEAs have been tested beyond 1000 h.69 AEMs, which are generally developed for AEMWE and AEMFC, have not been optimized for durable operation under ECR conditions (high alkalinity and organic molecules). This calls for membrane development tailored towards ECR. The electrolyte requires periodic replacement, or potentially purification to remove contaminants. Although GDE flooding is largely mitigated in MEAs, hydrophobic modifications would further improve the stability.98,319 The field also needs to investigate universal AST protocols not limited to electrochemical methods, but also humidity and thermal cycling for stability testing as ECR moves towards higher technology readiness levels.320,32111.5. Device
The field has largely shifted its focus from catalyst design to reaction environment and system levels analysis over the past decade.322 This is indeed a good indication that ECR technologies have moved from initial materials discovery and design to optimization for scaling up. The incorporation of GDL in ECR devices enabled industrially relevant current density operation but also created a stability concern at the TPB. Designing GDLs that are resistant to flooding and salt precipitation, thin AEMs that remain mechanically stable and conductive for ECR devices, and anodes that are suitable for mildly acidic conditions are critical next steps to increase performance and durability for pilot scale operation.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the NSF Engineering Research Center (ERC), Center for Innovative and Strategic Transformation of Light Alkane Resources (CISTAR; NSF award number EEC-1647722).References
- NOAA, Trends in Atmospheric Carbon Dioxide (CO2), https://gml.noaa.gov/ccgg/trends/, (accessed 10/07/2024).
- NETL, Carbon Dioxide 101, https://netl.doe.gov/carbon-management/carbon-storage/faqs/carbon-dioxide-101, (accessed 10/18/2024).
- S. Solomon, G.-K. Plattner, R. Knutti and P. Friedlingstein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef CAS PubMed.
- NOAA, Climate Change: Atmospheric Carbon Dioxide, https://www.climate.gov/news-features/understanding-climate/climate-change-atmospheric-carbon-dioxide, (accessed 10/07/2024).
- S.-K. Kim, J. Shin, S.-I. An, H.-J. Kim, N. Im, S.-P. Xie, J.-S. Kug and S.-W. Yeh, Nat. Clim. Change, 2022, 12, 834–840 CrossRef CAS.
- J. Pastor and W. M. Post, Nature, 1988, 334, 55–58 CrossRef.
- B. Seger, M. Robert and F. Jiao, Nat. Sustainability, 2023, 6, 236–238 CrossRef.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Nat. Rev. Mater., 2021, 7, 55–64 CrossRef.
- X. Deng, D. Alfonso, T.-D. Nguyen-Phan and D. R. Kauffman, ACS Catal., 2022, 12, 5921–5929 CrossRef CAS.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef PubMed.
- W. Luo, J. Zhang, M. Li and A. Züttel, ACS Catal., 2019, 9, 3783–3791 CrossRef CAS.
- C. Hahn, T. Hatsukade, Y. G. Kim, A. Vailionis, J. H. Baricuatro, D. C. Higgins, S. A. Nitopi, M. P. Soriaga and T. F. Jaramillo, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5918–5923 CrossRef CAS PubMed.
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang, C. M. Gabardo, C.-T. Dinh, T.-T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
- Q. Wu, C. Liu, X. Su, Q. Yang, X. Wu, H. Zou, B. Long, X. Fan, Y. Liao, L. Duan, Z. Quan and S. Luo, ACS Nano, 2023, 17, 402–410 CrossRef CAS PubMed.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4606–4609 CrossRef CAS PubMed.
- X. Wu, Y. Guo, Z. Sun, F. Xie, D. Guan, J. Dai, F. Yu, Z. Hu, Y.-C. Huang, C.-W. Pao, J.-L. Chen, W. Zhou and Z. Shao, Nat. Commun., 2021, 12, 660 CrossRef CAS PubMed.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman, J. Feijóo, P.-C. Chen, H. Wang, C. J. Pollock, X. Huang, Y.-T. Shao, C. Wang, D. A. Muller, H. D. Abruña and P. Yang, Nature, 2023, 614, 262–269 CrossRef CAS PubMed.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C. T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C. S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S. C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- H. Shang, S. K. Wallentine, D. M. Hofmann, Q. Zhu, C. J. Murphy and L. R. Baker, Chem. Sci., 2020, 11, 12298–12306 RSC.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- E. Huang Jianan, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, K. Miao Rui, C.-T. Dinh, D. Sinton and H. Sargent Edward, Science, 2021, 372, 1074–1078 CrossRef PubMed.
- C.-T. Dinh, F. P. García de Arquer, D. Sinton and E. H. Sargent, ACS Energy Lett., 2018, 3, 2835–2840 CrossRef CAS.
- S. Y. Lee, H. Jung, N. K. Kim, H. S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2018, 140, 8681–8689 CrossRef CAS PubMed.
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 781 CrossRef PubMed.
- Y. Liu and C. C. L. McCrory, Nat. Commun., 2019, 10, 1683 CrossRef PubMed.
- L. Yao, C. Yin, K. E. Rivera-Cruz, C. C. L. McCrory and N. Singh, ACS Appl. Mater. Interfaces, 2023, 15, 31438–31448 CrossRef CAS PubMed.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed.
- H. Peng, M. T. Tang, X. Liu, P. Schlexer Lamoureux, M. Bajdich and F. Abild-Pedersen, Energy Environ. Sci., 2021, 14, 473–482 RSC.
- W. Gao, Y. Xu, H. Xiong, X. Chang, Q. Lu and B. Xu, Angew. Chem., Int. Ed., 2023, 62, e202313798 CrossRef CAS PubMed.
- H. Zhang, X. Wang, Y. Sun, X. Wang, Z. Tang, S. Li, X. Gao, J. Wang, Z. Hou, K. Nie, J. Xie, Z. Yang and Y.-M. Yan, Appl. Catal., B, 2024, 351, 123992 CrossRef CAS.
- Z. Liu, L. Song, X. Lv, M. Liu, Q. Wen, L. Qian, H. Wang, M. Wang, Q. Han and G. Zheng, J. Am. Chem. Soc., 2024, 146, 14260–14266 CrossRef CAS PubMed.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS PubMed.
- S.-Q. Xiang, J.-L. Shi, S.-T. Gao, W. Zhang and L.-B. Zhao, ACS Catal., 2021, 11, 2422–2434 CrossRef CAS.
- S. Jung, C. C. L. McCrory, I. M. Ferrer, J. C. Peters and T. F. Jaramillo, J. Mater. Chem. A, 2016, 4, 3068–3076 RSC.
- T. Binninger, E. Fabbri, R. Kötz and T. J. Schmidt, J. Electrochem. Soc., 2014, 161, H121 CrossRef CAS.
- J. Edgington, S. Vispute, R. Li, A. Deberghes and L. C. Seitz, Sci. Adv., 2024, 10, eadp8911 CrossRef CAS PubMed.
- G. O. Larrazábal, M. Ma and B. Seger, Acc. Mater. Res., 2021, 2, 220–229 CrossRef.
- G. O. Larrazábal, P. Strøm-Hansen, J. P. Heli, K. Zeiter, K. T. Therkildsen, I. Chorkendorff and B. Seger, ACS Appl. Mater. Interfaces, 2019, 11, 41281–41288 CrossRef PubMed.
- S. Geiger, O. Kasian, M. Ledendecker, E. Pizzutilo, A. M. Mingers, W. T. Fu, O. Diaz-Morales, Z. Li, T. Oellers, L. Fruchter, A. Ludwig, K. J. J. Mayrhofer, M. T. M. Koper and S. Cherevko, Nat. Catal., 2018, 1, 508–515 CrossRef CAS.
- J. Edgington and L. C. Seitz, ACS Catal., 2023, 13, 3379–3394 CrossRef CAS.
- H. Liu, Z. Zhang, J. Fang, M. Li, M. G. Sendeku, X. Wang, H. Wu, Y. Li, J. Ge, Z. Zhuang, D. Zhou, Y. Kuang and X. Sun, Joule, 2023, 7, 558–573 CrossRef CAS.
- Z.-Z. Niu, L.-P. Chi, R. Liu, Z. Chen and M.-R. Gao, Energy Environ. Sci., 2021, 14, 4169–4176 RSC.
- S. Overa, B. S. Crandall, B. Shrimant, D. Tian, B. H. Ko, H. Shin, C. Bae and F. Jiao, Nat. Catal., 2022, 5, 738–745 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Nat. Rev. Mater., 2022, 7, 55–64 CrossRef CAS.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. L. Stephens, M. W. Kanan and I. Chorkendorff, JACS Au, 2015, 137, 9808–9811 CrossRef CAS PubMed.
- L. Wang, S. A. Nitopi, E. Bertheussen, M. Orazov, C. G. Morales-Guio, X. Liu, D. C. Higgins, K. Chan, J. K. Nørskov, C. Hahn and T. F. Jaramillo, ACS Catal., 2018, 8, 7445–7454 CrossRef CAS.
- E. L. Clark, M. R. Singh, Y. Kwon and A. T. Bell, Anal. Chem., 2015, 87, 8013–8020 CrossRef CAS PubMed.
- X. Chang, H. Xiong, Q. Lu and B. Xu, JACS Au, 2023, 3, 2948–2963 CrossRef CAS PubMed.
- J. Hou, X. Chang, J. Li, B. Xu and Q. Lu, J. Am. Chem. Soc., 2022, 144, 22202–22211 CrossRef CAS PubMed.
- A. Giaccherini, M. Al Khatib, S. Cinotti, E. Piciollo, E. Berretti, P. Giusti, M. Innocenti, G. Montegrossi and A. Lavacchi, Sci. Rep., 2020, 10, 13433 CrossRef CAS PubMed.
- H. Chen, E. Kätelhön and R. G. Compton, Anal. Chem., 2023, 95, 12826–12834 CrossRef CAS PubMed.
- J. Jang, M. Rüscher, M. Winzely and C. G. Morales-Guio, AIChE J., 2022, 68, e17605 CrossRef CAS.
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. M. Koper, J. Am. Chem. Soc., 2020, 142, 4154–4161 CrossRef CAS PubMed.
- B. A. Zhang, T. Ozel, J. S. Elias, C. Costentin and D. G. Nocera, ACS Cent. Sci., 2019, 5, 1097–1105 CrossRef CAS PubMed.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- L. M. Baumgartner, C. I. Koopman, A. Forner-Cuenca and D. A. Vermaas, ACS Sustainable Chem. Eng., 2022, 10, 4683–4693 CrossRef CAS PubMed.
- X. Wang, K. Klingan, M. Klingenhof, T. Moller, J. Ferreira de Araujo, I. Martens, A. Bagger, S. Jiang, J. Rossmeisl, H. Dau and P. Strasser, Nat. Commun., 2021, 12, 794 CrossRef CAS PubMed.
- Q. Bai, L. Xiong, Y. Zhang, M. Ma, Z. Jiao, F. Lyu, Z. Deng and Y. Peng, EES Catal., 2024, 2, 1228–1237 RSC.
- M. Li, M. N. Idros, Y. Wu, T. Burdyny, S. Garg, X. S. Zhao, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2021, 9, 19369–19409 RSC.
- C. Xia, P. Zhu, Q. Jiang, Y. Pan, W. Liang, E. Stavitski, H. N. Alshareef and H. Wang, Nat. Energy, 2019, 4, 776–785 CrossRef CAS.
- C. M. Gabardo, C. P. O’Brien, J. P. Edwards, C. McCallum, Y. Xu, C.-T. Dinh, J. Li, E. H. Sargent and D. Sinton, Joule, 2019, 3, 2777–2791 CrossRef CAS.
- X. She, L. Zhai, Y. Wang, P. Xiong, M. M.-J. Li, T.-S. Wu, M. C. Wong, X. Guo, Z. Xu, H. Li, H. Xu, Y. Zhu, S. C. E. Tsang and S. P. Lau, Nat. Energy, 2024, 9, 81–91 CrossRef CAS.
- H. Yang, J. J. Kaczur, S. D. Sajjad and R. I. Masel, J. CO2 Util., 2020, 42, 101349 CrossRef CAS.
- W. Fang, W. Guo, R. Lu, Y. Yan, X. Liu, D. Wu, F. M. Li, Y. Zhou, C. He, C. Xia, H. Niu, S. Wang, Y. Liu, Y. Mao, C. Zhang, B. You, Y. Pang, L. Duan, X. Yang, F. Song, T. Zhai, G. Wang, X. Guo, B. Tan, T. Yao, Z. Wang and B. Y. Xia, Nature, 2024, 626, 86–91 CrossRef CAS PubMed.
- A. Zaffora, O. Barbera, E. Gallo, M. Santamaria and G. Giacoppo, J. Power Sources, 2024, 616, 235129 CrossRef CAS.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed.
- B. De Mot, M. Ramdin, J. Hereijgers, T. J. H. Vlugt and T. Breugelmans, ChemElectroChem, 2020, 7, 3839–3843 CrossRef CAS.
- S. Subramanian, K. Yang, M. Li, M. Sassenburg, M. Abdinejad, E. Irtem, J. Middelkoop and T. Burdyny, ACS Energy Lett., 2023, 8, 222–229 CrossRef CAS PubMed.
- T. Möller, M. Filippi, S. Brückner, W. Ju and P. Strasser, Nat. Commun., 2023, 14, 5680 CrossRef PubMed.
- H.-P. Iglesias van Montfort, S. Subramanian, E. Irtem, M. Sassenburg, M. Li, J. Kok, J. Middelkoop and T. Burdyny, ACS Energy Lett., 2023, 4156–4161, DOI:10.1021/acsenergylett.3c01561.
- H. Simonson, W. E. Klein, D. Henckel, S. Verma, K. C. Neyerlin and W. A. Smith, ACS Energy Lett., 2023, 8, 3811–3819 CrossRef CAS.
- S. Yuan, R. Wang, R. Xue, L. Wu, G. Zhang, H. Li, Q. Wang, J. Yin, L. Luo, S. Shen, L. An, X. Yan and J. Zhang, ACS Energy Lett., 2024, 5945–5954, DOI:10.1021/acsenergylett.4c02534.
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS.
- S. C. da Cunha and J. Resasco, ACS Energy Lett., 2024, 9, 5550–5561 CrossRef CAS.
- J. T. Billy and A. C. Co, ACS Catal., 2017, 7, 8467–8479 CrossRef CAS.
- P. Lobaccaro, M. R. Singh, E. L. Clark, Y. Kwon, A. T. Bell and J. W. Ager, Phys. Chem. Chem. Phys., 2016, 18, 26777–26785 RSC.
- I. Bagemihl, C. Bhatraju, J. R. van Ommen and V. van Steijn, ACS Sustainable Chem. Eng., 2022, 10, 12580–12587 CrossRef CAS PubMed.
- P. Lobaccaro, M. R. Singh, E. L. Clark, Y. Kwon, A. T. Bell and J. W. Ager, Phys. Chem. Chem. Phys., 2016, 18, 26777–26785 RSC.
- S. C. da Cunha and J. Resasco, Nat. Commun., 2023, 14, 5513 CrossRef CAS PubMed.
- B. Pan, J. Fan, J. Zhang, Y. Luo, C. Shen, C. Wang, Y. Wang and Y. Li, ACS Energy Lett., 2022, 7, 4224–4231 CrossRef CAS.
- X. Chang, J. Li, H. Xiong, H. Zhang, Y. Xu, H. Xiao, Q. Lu and B. Xu, Angew. Chem., Int. Ed., 2022, 61, e202111167 CrossRef CAS PubMed.
- M. Moradzaman, C. S. Martínez and G. Mul, Sustainable Energy Fuels, 2020, 4, 5195–5202 RSC.
- B. Kim, S. Ma, H.-R. Molly Jhong and P. J. A. Kenis, Electrochim. Acta, 2015, 166, 271–276 CrossRef CAS.
- X. Wang, A. Xu, F. Li, S. F. Hung, D. H. Nam, C. M. Gabardo, Z. Wang, Y. Xu, A. Ozden, A. S. Rasouli, A. H. Ip, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 3525–3531 CrossRef CAS PubMed.
- X. Wang, P. Ou, J. Wicks, Y. Xie, Y. Wang, J. Li, J. Tam, D. Ren, J. Y. Howe, Z. Wang, A. Ozden, Y. Z. Finfrock, Y. Xu, Y. Li, A. S. Rasouli, K. Bertens, A. H. Ip, M. Graetzel, D. Sinton and E. H. Sargent, Nat. Commun., 2021, 12, 3387 CrossRef CAS PubMed.
- Y. Xu, J. P. Edwards, J. Zhong, C. P. O’Brien, C. M. Gabardo, C. McCallum, J. Li, C.-T. Dinh, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2020, 13, 554–561 RSC.
- M. He, C. Li, H. Zhang, X. Chang, J. G. Chen, W. A. Goddard, 3rd, M. J. Cheng, B. Xu and Q. Lu, Nat. Commun., 2020, 11, 3844 CrossRef CAS PubMed.
- U. Legrand, U. P. Apfel, D. C. Boffito and J. R. Tavares, J. CO2 Util., 2020, 42, 101315 CrossRef CAS.
- R. J. Barla, S. Raghuvanshi and S. Gupta, in Waste and Biodiesel, ed. B. Singh and A. Guldhe, Elsevier, 2022, pp. 191–215 DOI:10.1016/B978-0-12-823958-2.00007-0.
- W. Luc, B. H. Ko, S. Kattel, S. Li, D. Su, J. G. Chen and F. Jiao, J. Am. Chem. Soc., 2019, 141, 9902–9909 CrossRef CAS PubMed.
- W. Ma, S. Xie, X.-G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D.-Y. Wu and Y. Wang, Nat. Commun., 2019, 10, 892 CrossRef PubMed.
- B. H. Ko, B. Hasa, H. Shin, E. Jeng, S. Overa, W. Chen and F. Jiao, Nat. Commun., 2020, 11, 5856 CrossRef CAS PubMed.
- B. S. Crandall, B. H. Ko, S. Overa, L. Cherniack, A. Lee, I. Minnie and F. Jiao, Nat. Chem. Eng., 2024, 1, 421–429 CrossRef.
- X. Wang, J. F. de Araujo, W. Ju, A. Bagger, H. Schmies, S. Kuhl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 Search PubMed.
- K. Hara, A. Tsuneto, A. Kudo and T. Sakata, J. Electrochem. Soc., 1994, 141, 2097 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050 RSC.
- L. Huang, G. Gao, C. Yang, X.-Y. Li, R. K. Miao, Y. Xue, K. Xie, P. Ou, C. T. Yavuz, Y. Han, G. Magnotti, D. Sinton, E. H. Sargent and X. Lu, Nat. Commun., 2023, 14, 2958 CrossRef CAS PubMed.
- K. Hara, A. Kudo and T. Sakata, J. Electroanal. Chem., 1995, 391, 141–147 CrossRef.
- Q. Xu, A. Xu, S. Garg, A. B. Moss, I. Chorkendorff, T. Bligaard and B. Seger, Angew. Chem., Int. Ed., 2023, 62, e202214383 CrossRef CAS PubMed.
- J. Jin, J. Wicks, Q. Min, J. Li, Y. Hu, J. Ma, Y. Wang, Z. Jiang, Y. Xu, R. Lu, G. Si, P. Papangelakis, M. Shakouri, Q. Xiao, P. Ou, X. Wang, Z. Chen, W. Zhang, K. Yu, J. Song, X. Jiang, P. Qiu, Y. Lou, D. Wu, Y. Mao, A. Ozden, C. Wang, B. Y. Xia, X. Hu, V. P. Dravid, Y.-M. Yiu, T.-K. Sham, Z. Wang, D. Sinton, L. Mai, E. H. Sargent and Y. Pang, Nature, 2023, 617, 724–729 CrossRef CAS PubMed.
- M. M. Bakker and D. A. Vermaas, Electrochim. Acta, 2019, 319, 148–157 CrossRef CAS.
- R. Qiu, J. Jia, L. Peng, R. Li, S. Yan, J. Li, J. Zhang, D. T. Sun, Z. Lan, T. Xue, G. Xu, L. Cui, Z. Lv, C. Li, Y. Hong, Y. Guo, B. Ren, S. Yang, J. Li and B. Han, Green Chem., 2023, 25, 684–691 RSC.
- S. J. Raaijman, M. P. Schellekens, P. J. Corbett and M. T. M. Koper, Angew. Chem., Int. Ed., 2021, 60, 21732–21736 CrossRef CAS PubMed.
- M. Bernt and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, F3179 CrossRef CAS.
- M. Suermann, T. J. Schmidt and F. N. Büchi, ECS Trans., 2015, 69, 1141 CrossRef CAS.
- M. Schalenbach, M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 14921–14933 CrossRef CAS.
- S. Z. Oener, M. J. Foster and S. W. Boettcher, Science, 2020, 369, 1099–1103 CrossRef CAS PubMed.
- L. Chen, Q. Xu, S. Z. Oener, K. Fabrizio and S. W. Boettcher, Nat. Commun., 2022, 13, 3846 CrossRef CAS PubMed.
- F. Meng, J. Qin, Q. Wu, H. Dai, P. Zhu, T. Tang, L. Zhang, Z. B. Zhang and K. Zuo, ACS Energy Lett., 2024, 5444–5451, DOI:10.1021/acsenergylett.4c02677.
- J. Disch, S. Ingenhoven and S. Vierrath, Adv. Energy Mater., 2023, 13, 2301614 CrossRef CAS.
- L. Hu, J. A. Wrubel, C. M. Baez-Cotto, F. Intia, J. H. Park, A. J. Kropf, N. Kariuki, Z. Huang, A. Farghaly, L. Amichi, P. Saha, L. Tao, D. A. Cullen, D. J. Myers, M. S. Ferrandon and K. C. Neyerlin, Nat. Commun., 2023, 14, 7605 CrossRef CAS PubMed.
- A. Perazio, M. W. Schreiber, C. E. Creissen and M. Fontecave, ChemElectroChem, 2024, 11, e202400045 CrossRef CAS.
- K.-J. Yim, D.-K. Song, C.-S. Kim, N.-G. Kim, T. Iwaki, T. Ogi, K. Okuyama, S.-E. Lee and T.-O. Kim, RSC Adv., 2015, 5, 9278–9282 RSC.
- M. T. de Groot and A. W. Vreman, Electrochim. Acta, 2021, 369, 137684 CrossRef CAS.
- M. C. O. Monteiro, X. Liu, B. J. L. Hagedoorn, D. D. Snabilié and M. T. M. Koper, ChemElectroChem, 2022, 9, e202101223 CrossRef CAS.
- B. N. Ruggiero, A. R. Weidner, J. M. Notestein and L. C. Seitz, J. Phys. Chem. C, 2023, 127, 20640–20651 CrossRef CAS.
- B. M. Tackett, D. Raciti, N. W. Brady, N. L. Ritzert and T. P. Moffat, J. Phys. Chem. C, 2022, 126, 7456–7467 CrossRef CAS.
- D. A. Henckel, M. J. Counihan, H. E. Holmes, X. Chen, U. O. Nwabara, S. Verma, J. Rodríguez-López, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2021, 11, 255–263 CrossRef CAS.
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2020, 4, 20–27 CrossRef.
- X. Lu, C. Zhu, Z. Wu, J. Xuan, J. S. Francisco and H. Wang, J. Am. Chem. Soc., 2020, 142, 15438–15444 CrossRef CAS PubMed.
- Z. Zhang, L. Melo, R. P. Jansonius, F. Habibzadeh, E. R. Grant and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 3101–3107 CrossRef CAS.
- A. J. Welch, A. Q. Fenwick, A. Böhme, H.-Y. Chen, I. Sullivan, X. Li, J. S. DuChene, C. Xiang and H. A. Atwater, J. Phys. Chem. C, 2021, 125, 20896–20904 CrossRef CAS.
- F. Zhang and A. C. Co, Angew. Chem., Int. Ed., 2020, 59, 1674–1681 CrossRef CAS PubMed.
- J. Y. T. Kim, P. Zhu, F.-Y. Chen, Z.-Y. Wu, D. A. Cullen and H. Wang, Nat. Catal., 2022, 5, 288–299 CrossRef CAS.
- Z. Ma, Z. Yang, W. Lai, Q. Wang, Y. Qiao, H. Tao, C. Lian, M. Liu, C. Ma, A. Pan and H. Huang, Nat. Commun., 2022, 13, 7596 CrossRef CAS PubMed.
- J. Li, D. Wu, A. S. Malkani, X. Chang, M.-J. Cheng, B. Xu and Q. Lu, Angew. Chem., Int. Ed., 2020, 59, 4464–4469 CrossRef CAS PubMed.
- C. P. O’Brien, R. K. Miao, A. Shayesteh Zeraati, G. Lee, E. H. Sargent and D. Sinton, Chem. Rev., 2024, 124(7), 3648–3693 CrossRef PubMed.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS.
- S. Guo, Y. Liu, Y. Huang, H. Wang, E. Murphy, L. Delafontaine, J. L. Chen, I. V. Zenyuk and P. Atanassov, ACS Energy Lett., 2023, 8, 935–942 CrossRef CAS.
- M. Jouny, G. S. Hutchings and F. Jiao, Nat. Catal., 2019, 2, 1062–1070 CrossRef CAS.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- J. Wang, Y. Gao, H. Kong, J. Kim, S. Choi, F. Ciucci, Y. Hao, S. Yang, Z. Shao and J. Lim, Chem. Soc. Rev., 2020, 49, 9154–9196 RSC.
- M. Plevová, J. Hnát and K. Bouzek, J. Power Sources, 2021, 507, 230072 CrossRef.
- T. Nishimoto, T. Shinagawa, T. Naito and K. Takanabe, J. Catal., 2020, 391, 435–445 CrossRef CAS.
- H. Zeng, J. Chen, C. Wang, J. Qi, Z. Liu, M. Li, L. Gu, J. Wang, E. Hong, Y. Zhang, J. Xu and C. Yang, ACS Mater. Lett., 2024, 6, 2295–2303 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed.
- A. Murata and Y. Hori, Bull. Chem. Soc. Jpn., 1991, 64, 123–127 CrossRef CAS.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager, III and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS PubMed.
- O. Ayemoba and A. Cuesta, ACS Appl. Mater. Interfaces, 2017, 9, 27377–27382 CrossRef CAS PubMed.
- D. Strmcnik, D. F. van der Vliet, K. C. Chang, V. Komanicky, K. Kodama, H. You, V. R. Stamenkovic and N. M. Marković, J. Phys. Chem. Lett., 2011, 2, 2733–2736 CrossRef CAS.
- A. S. Malkani, J. Li, N. J. Oliveira, M. He, X. Chang, B. Xu and Q. Lu, Sci. Adv., 2020, 6, eabd2569 CrossRef CAS PubMed.
- M. Urushihara, K. Chan, C. Shi and J. K. Nørskov, J. Phys. Chem. C, 2015, 119, 20023–20029 CrossRef CAS.
- J. Li, X. Li, C. M. Gunathunge and M. M. Waegele, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9220–9229 CrossRef CAS PubMed.
- S. Weng, W. L. Toh and Y. Surendranath, J. Am. Chem. Soc., 2023, 145, 16787–16795 CrossRef CAS PubMed.
- L. Xue, Z. Gao, T. Ning, W. Li, J. Li, J. Yin, L. Xiao, G. Wang and L. Zhuang, Angew. Chem., Int. Ed., 2023, 62, e202309519 CrossRef CAS PubMed.
- M. H. Hicks, W. Nie, A. E. Boehme, H. A. Atwater, T. Agapie and J. C. Peters, J. Am. Chem. Soc., 2024, 146, 25282–25289 CrossRef CAS PubMed.
- X. Liu and M. T. M. Koper, J. Am. Chem. Soc., 2024, 146, 5242–5251 CrossRef CAS PubMed.
- Y. Yao, E. P. Delmo and M. Shao, Angew. Chem., Int. Ed., 2024, e202415894 Search PubMed.
- H.-G. Qin, F.-Z. Li, Y.-F. Du, L.-F. Yang, H. Wang, Y.-Y. Bai, M. Lin and J. Gu, ACS Catal., 2023, 13, 916–926 CrossRef CAS.
- L.-P. Chi, Z.-Z. Niu, Y.-C. Zhang, X.-L. Zhang, J. Liao, Z.-Z. Wu, P.-C. Yu, M.-H. Fan, K.-B. Tang and M.-R. Gao, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2312876120 CrossRef CAS PubMed.
- M. Fan, J. E. Huang, R. K. Miao, Y. Mao, P. Ou, F. Li, X.-Y. Li, Y. Cao, Z. Zhang, J. Zhang, Y. Yan, A. Ozden, W. Ni, Y. Wang, Y. Zhao, Z. Chen, B. Khatir, C. P. O’Brien, Y. Xu, Y. C. Xiao, G. I. N. Waterhouse, K. Golovin, Z. Wang, E. H. Sargent and D. Sinton, Nat. Catal., 2023, 6, 763–772 CrossRef CAS.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- B. Wu, B. Wang, B. Cai, C. Wu, W. W. Tjiu, M. Zhang, Z. Aabdin, S. Xi and Y. Lum, J. Am. Chem. Soc., 2024, 146(43), 29801–29809 CrossRef CAS PubMed.
- S. Garg, C. A. Giron Rodriguez, T. E. Rufford, J. R. Varcoe and B. Seger, Energy Environ. Sci., 2022, 15, 4440–4469 RSC.
- F. Habibzadeh, P. Mardle, N. Zhao, H. D. Riley, D. A. Salvatore, C. P. Berlinguette, S. Holdcroft and Z. Shi, Electrochem. Energy Rev., 2023, 6, 26 CrossRef CAS.
- G. A. El-Nagar, F. Haun, S. Gupta, S. Stojkovikj and M. T. Mayer, Nat. Commun., 2023, 14, 2062 CrossRef CAS PubMed.
- K. Yang, M. Li, S. Subramanian, M. A. Blommaert, W. A. Smith and T. Burdyny, ACS Energy Lett., 2021, 6, 4291–4298 CrossRef CAS PubMed.
- G. A. El-Nagar, F. Haun, S. Gupta, S. Stojkovikj and M. T. Mayer, Nat. Commun., 2023, 14, 2062 CrossRef CAS PubMed.
- S. Garg, Q. Xu, A. B. Moss, M. Mirolo, W. Deng, I. Chorkendorff, J. Drnec and B. Seger, Energy Environ. Sci., 2023, 16, 1631–1643 RSC.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. Sedighian Rasouli, F. Li, M. Luo, A. H. Ip, Y.-C. Joo and E. H. Sargent, Nat. Energy, 2021, 6, 46–53 CrossRef CAS.
- J. H. Kim, H. Jang, G. Bak, W. Choi, H. Yun, E. Lee, D. Kim, J. Kim, S. Y. Lee and Y. J. Hwang, Energy Environ. Sci., 2022, 15, 4301–4312 RSC.
- Z. Cui, M. A. Marx, M. N. Tegomoh and A. C. Co, ACS Energy Lett., 2023, 8, 5201–5205 CrossRef CAS.
- M. Sassenburg, M. Kelly, S. Subramanian, W. A. Smith and T. Burdyny, ACS Energy Lett., 2023, 8, 321–331 CrossRef CAS PubMed.
- Y. Xu, J. P. Edwards, S. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O’Brien, J. Li, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 809–815 CrossRef CAS.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 RSC.
- A. Wuttig and Y. Surendranath, ACS Catal., 2015, 5, 4479–4484 CrossRef CAS.
- J.-P. B. Haraldsted, Z. Révay, R. Frydendal, A. Verdaguer-Casadevall, J. Rossmeisl, J. Kibsgaard and I. Chorkendorff, Mater. Today Energy, 2019, 14, 100352 CrossRef.
- Á. Vass, B. Endrődi, G. F. Samu, Á. Balog, A. Kormányos, S. Cherevko and C. Janáky, ACS Energy Lett., 2021, 6, 3801–3808 CrossRef PubMed.
- Q. Xu, S. Garg, A. B. Moss, M. Mirolo, I. Chorkendorff, J. Drnec and B. Seger, Nat. Catal., 2023, 6, 1042–1051 CrossRef CAS.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- A. Wuttig, J. Ryu and Y. Surendranath, J. Phys. Chem. C, 2021, 125, 17042–17050 CrossRef CAS.
- J. S. Zeng, N. Corbin, K. Williams and K. Manthiram, ACS Catal., 2020, 10, 4326–4336 CrossRef CAS.
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef CAS PubMed.
- M. Dunwell, Q. Lu, J. M. Heyes, J. Rosen, J. G. Chen, Y. Yan, F. Jiao and B. Xu, J. Am. Chem. Soc., 2017, 139, 3774–3783 CrossRef CAS PubMed.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS PubMed.
- G. Lee, A. S. Rasouli, B.-H. Lee, J. Zhang, D. H. Won, Y. C. Xiao, J. P. Edwards, M. G. Lee, E. D. Jung, F. Arabyarmohammadi, H. Liu, I. Grigioni, J. Abed, T. Alkayyali, S. Liu, K. Xie, R. K. Miao, S. Park, R. Dorakhan, Y. Zhao, C. P. O’Brien, Z. Chen, D. Sinton and E. Sargent, Joule, 2023, 7, 1277–1288 CrossRef CAS.
- T. Li, E. W. Lees, Z. Zhang and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2624–2630 Search PubMed.
- E. W. Lees, M. Goldman, A. G. Fink, D. J. Dvorak, D. A. Salvatore, Z. Zhang, N. W. X. Loo and C. P. Berlinguette, ACS Energy Lett., 2020, 5, 2165–2173 Search PubMed.
- T. Li, E. W. Lees, M. Goldman, D. A. Salvatore, D. M. Weekes and C. P. Berlinguette, Joule, 2019, 3, 1487–1497 Search PubMed.
- J. M. Yoo, J. Ingenmey, M. Salanne and M. R. Lukatskaya, J. Am. Chem. Soc., 2024, 146(46), 31768–31777 CrossRef CAS PubMed.
- S. Yang, H. An, S. Arnouts, H. Wang, X. Yu, J. de Ruiter, S. Bals, T. Altantzis, B. M. Weckhuysen and W. van der Stam, Nat. Catal., 2023, 6, 796–806 CrossRef CAS.
- D. Hochfilzer, A. Xu, J. E. Sørensen, J. L. Needham, K. Krempl, K. K. Toudahl, G. Kastlunger, I. Chorkendorff, K. Chan and J. Kibsgaard, ACS Catal., 2022, 12, 5155–5161 CrossRef CAS.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, ACS Catal., 2016, 6, 2136–2144 Search PubMed.
- T. Yuan, T. Wang, G. Zhang, W. Deng, D. Cheng, H. Gao, J. Zhao, J. Yu, P. Zhang and J. Gong, Chem. Sci., 2022, 13, 8117–8123 Search PubMed.
- Z. Zheng, Y. Yao, W. Yan, H. Bu, J. Huang and M. Ma, ACS Catal., 2024, 14, 6328–6338 Search PubMed.
- F. Lapicque, M. Belhadj, C. Bonnet, J. Pauchet and Y. Thomas, J. Power Sources, 2016, 336, 40–53 CrossRef CAS.
- Z. Xing, X. Hu and X. Feng, ACS Energy Lett., 2021, 6, 1694–1702 Search PubMed.
- L. M. Baumgartner, C. I. Koopman, A. Forner-Cuenca and D. A. Vermaas, ACS Appl. Energy Mater., 2022, 5, 15125–15135 Search PubMed.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2020, 6, 33–40 CrossRef.
- X. Kong, C. Wang, Z. Xu, Y. Zhong, Y. Liu, L. Qin, J. Zeng and Z. Geng, Nano Lett., 2022, 22, 8000–8007 CrossRef CAS PubMed.
- Y. Wu, L. Charlesworth, I. Maglaya, M. N. Idros, M. Li, T. Burdyny, G. Wang and T. E. Rufford, ACS Energy Lett., 2022, 7, 2884–2892 Search PubMed.
- C. P. O’Brien, D. McLaughlin, T. Böhm, Y. C. Xiao, J. P. Edwards, C. M. Gabardo, M. Bierling, J. Wicks, A. Sedighian Rasouli, J. Abed, D. Young, C.-T. Dinh, E. H. Sargent, S. Thiele and D. Sinton, Joule, 2024, 8, 2903–2919 Search PubMed.
- J. Li, G. Chen, Y. Zhu, Z. Liang, A. Pei, C.-L. Wu, H. Wang, H. R. Lee, K. Liu, S. Chu and Y. Cui, Nat. Catal., 2018, 1, 592–600 CrossRef CAS.
- J. Wicks, M. L. Jue, V. A. Beck, J. S. Oakdale, N. A. Dudukovic, A. L. Clemens, S. Liang, M. E. Ellis, G. Lee, S. E. Baker, E. B. Duoss and E. H. Sargent, Adv. Mater., 2021, 33, 2003855 CrossRef CAS PubMed.
- W. Zhang, A. Yu, H. Mao, G. Feng, C. Li, G. Wang, J. Chang, D. Halat, Z. Li, W. Yu, Y. Shi, S. Liu, D. W. Fox, H. Zhuang, A. Cai, B. Wu, F. Joshua, J. R. Martinez, L. Zhai, M. D. Gu, X. Shan, J. A. Reimer, Y. Cui and Y. Yang, J. Am. Chem. Soc., 2024, 146, 21335–21347 CrossRef PubMed.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed.
- M. Li, M. N. Idros, Y. Wu, S. Garg, S. Gao, R. Lin, H. Rabiee, Z. Li, L. Ge, T. E. Rufford, Z. Zhu, L. Li and G. Wang, React. Chem. Eng., 2021, 6, 345–352 RSC.
- L. Zhao, Z. Yuan, B. Ma, X. Ding, Y. Tian and X. Yang, Appl. Surf. Sci., 2023, 628, 157369 CrossRef CAS.
- T. H. M. Pham, J. Zhang, M. Li, T. H. Shen, Y. Ko, V. Tileli, W. Luo and A. Züttel, Adv. Energy Mater., 2022, 12, 2103663 CrossRef CAS.
- Y.-J. Ko, C. Lim, J. Jin, M. G. Kim, J. Y. Lee, T.-Y. Seong, K.-Y. Lee, B. K. Min, J.-Y. Choi, T. Noh, G. W. Hwang, W. H. Lee and H.-S. Oh, Nat. Commun., 2024, 15, 3356 CrossRef CAS PubMed.
- Q. Luo, H. Duan, M. C. McLaughlin, K. Wei, J. Tapia, J. A. Adewuyi, S. Shuster, M. Liaqat, S. L. Suib, G. Ung, P. Bai, S. Sun and J. He, Chem. Sci., 2023, 14, 9664–9677 RSC.
- U. EPA, Key EPA Actions to Address PFAS, https://www.epa.gov/pfas/key-epa-actions-address-pfas, (accessed 10/18/2024).
- European Commission, Commission restricts use of a sub-group of PFAS chemicals to protect human health and the environment, https://ec.europa.eu/commission/presscorner/detail/en/ip_24_4763, (accessed 10/18/2025).
- M. E. Leonard, M. J. Orella, N. Aiello, Y. Román-Leshkov, A. Forner-Cuenca and F. R. Brushett, J. Electrochem. Soc., 2020, 167, 124521 CrossRef CAS.
- R. J. Gilliam, J. W. Graydon, D. W. Kirk and S. J. Thorpe, Int. J. Hydrogen Energy, 2007, 32, 359–364 CrossRef CAS.
- M. Bončina, A. Apelblat and M. Bešter-Rogač, J. Chem. Eng. Data, 2010, 55, 1951–1957 Search PubMed.
- M. J. Kadhim and M. I. Gamaj, J. Chem. Rev., 2020, 2, 182–188 Search PubMed.
- B. Jiang, L. Yu, L. Wu, D. Mu, L. Liu, J. Xi and X. Qiu, ACS Appl. Mater. Interfaces, 2016, 8, 12228–12238 Search PubMed.
- T. T. Phan, S.-K. Kim, J. Islam, M.-J. Kim and J.-H. Lee, Int. J. Hydrogen Energy, 2024, 49, 875–885 Search PubMed.
- F. Bauer, S. Denneler and M. Willert-Porada, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 786–795 CrossRef CAS.
- B. Endrődi, E. Kecsenovity, A. Samu, T. Halmágyi, S. Rojas-Carbonell, L. Wang, Y. Yan and C. Janáky, Energy Environ. Sci., 2020, 13, 4098–4105 RSC.
- S. D. Sajjad, Y. Gao, Z. Liu, H. Yang and R. Masel, ECS Trans., 2017, 77, 1653 CrossRef CAS.
- T. Lin, L. Hu, W. Wisely, X. Gu, J. Cai, S. Litster and L. B. Kara, Energy AI, 2021, 3, 100045 CrossRef.
- C. McCallum, C. M. Gabardo, C. P. O’Brien, J. P. Edwards, J. Wicks, Y. Xu, E. H. Sargent and D. Sinton, Cell Rep. Phys. Sci., 2021, 2, 100522 CrossRef CAS.
- X. Luo, D. I. Kushner and A. Kusoglu, J. Membr. Sci., 2023, 685, 121945 CrossRef CAS.
- X. Zhu, H. Zhang, Y. Liang, Y. Zhang, Q. Luo, C. Bi and B. Yi, J. Mater. Chem., 2007, 17, 386–397 RSC.
- F. Liu, B. Yi, D. Xing, J. Yu and H. Zhang, J. Membr. Sci., 2003, 212, 213–223 CrossRef CAS.
- B. Kim, F. Hillman, M. Ariyoshi, S. Fujikawa and P. J. A. Kenis, J. Power Sources, 2016, 312, 192–198 CrossRef CAS.
- M. Filippi, T. Möller, L. Liang and P. Strasser, Energy Environ. Sci., 2023, 16, 5265–5273 RSC.
- F. Huq, I. Sanjuán, S. Baha, M. Braun, A. Kostka, V. Chanda, J. R. C. Junqueira, N. Sikdar, A. Ludwig and C. Andronescu, ChemElectroChem, 2022, 9, e202101279 CrossRef CAS.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- H.-P. Iglesias van Montfort, M. Li, E. Irtem, M. Abdinejad, Y. Wu, S. K. Pal, M. Sassenburg, D. Ripepi, S. Subramanian, J. Biemolt, T. E. Rufford and T. Burdyny, Nat. Commun., 2023, 14, 6579 CrossRef CAS PubMed.
- M. Filippi, T. Möller, R. Pastusiak, E. Magori, B. Paul and P. Strasser, ACS Energy Lett., 2024, 9, 1361–1368 CrossRef CAS PubMed.
- S. Yamaguchi, H. Ebe, T. Minegishi and M. Sugiyama, ACS Appl. Mater. Interfaces, 2024, 16, 17371–17376 CrossRef CAS PubMed.
- S. Alkhaldi, M. Aziz, A. Amrite and A. K. Prasad, J. Appl. Electrochem., 2024, 55, 327–343 CrossRef.
- K. J. Ferner and S. Litster, ACS Appl. Energy Mater., 2024, 7, 8124–8135 CrossRef CAS PubMed.
- J. R. Hudkins, D. G. Wheeler, B. Peña and C. P. Berlinguette, Energy Environ. Sci., 2016, 9, 3417–3423 RSC.
- T. L. Doan, H. E. Lee, S. S. H. Shah, M. Kim, C.-H. Kim, H.-S. Cho and T. Kim, Int. J. Energy Res., 2021, 45, 14207–14220 CrossRef CAS.
- S. K. Kamarudin, W. R. W. Daud, A. Md.Som, M. S. Takriff and A. W. Mohammad, J. Power Sources, 2006, 157, 641–649 CrossRef CAS.
- G. Chisholm, P. J. Kitson, N. D. Kirkaldy, L. G. Bloor and L. Cronin, Energy Environ. Sci., 2014, 7, 3026–3032 RSC.
- P. Liu and S. Xu, Int. J. Hydrogen Energy, 2023, 48, 8216–8246 CrossRef CAS.
- R. Wlodarczyk, D. Zasada, S. Morel and A. Kacprzak, Int. J. Hydrogen Energy, 2016, 41, 17644–17651 Search PubMed.
- S. Lædre, O. E. Kongstein, A. Oedegaard, H. Karoliussen and F. Seland, Int. J. Hydrogen Energy, 2017, 42, 2713–2723 CrossRef.
- J. Engbæk, O. Lytken, J. H. Nielsen and I. Chorkendorff, Surf. Sci., 2008, 602, 733–743 CrossRef.
- H.-Y. Jung, S.-Y. Huang, P. Ganesan and B. N. Popov, J. Power Sources, 2009, 194, 972–975 CrossRef CAS.
- S. R. Samms, S. Wasmus and R. F. Savinell, J. Electrochem. Soc., 1996, 143, 1498 CrossRef CAS.
- R. B. Kutz, Q. Chen, H. Yang, S. D. Sajjad, Z. Liu and I. R. Masel, Energy Technol., 2017, 5, 929–936 CrossRef CAS.
- T. Hatsukade, PhD, Stanford University, 2016.
- R. E. Vos, K. E. Kolmeijer, T. S. Jacobs, W. van der Stam, B. M. Weckhuysen and M. T. M. Koper, ACS Catal., 2023, 13, 8080–8091 CrossRef CAS PubMed.
- N. A. Nazir, T. Kyu, A. M. Reinsel, M. Espe, M. Nosaka, H. Kudo and T. Nishikubo, Polymer, 2011, 3, 2018–2038 CAS.
- A. Löwe, C. Rieg, T. Hierlemann, N. Salas, D. Kopljar, N. Wagner and E. Klemm, ChemElectroChem, 2019, 6, 4497–4506 Search PubMed.
- R. E. Vos, J. P. Smaak and M. T. M. Koper, J. Catal., 2024, 436, 115613 CrossRef CAS.
- A. Zhegur, N. Gjineci, S. Willdorf-Cohen, A. N. Mondal, C. E. Diesendruck, N. Gavish and D. R. Dekel, ACS Appl. Polym. Mater., 2020, 2, 360–367 CrossRef CAS.
- A. Amel, S. B. Smedley, D. R. Dekel, M. A. Hickner and Y. Ein-Eli, J. Electrochem. Soc., 2015, 162, F1047 Search PubMed.
- Q. Feng, X. Z. Yuan, G. Liu, B. Wei, Z. Zhang, H. Li and H. Wang, J. Power Sources, 2017, 366, 33–55 CrossRef CAS.
- E. Kuhnert, V. Hacker and M. Bodner, Int. J. Energy Res., 2023, 2023, 3183108 Search PubMed.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- S. H. Frensch, F. Fouda-Onana, G. Serre, D. Thoby, S. S. Araya and S. K. Kær, Int. J. Hydrogen Energy, 2019, 44, 29889–29898 CrossRef CAS.
- M. A. Barique, E. Tsuchida, A. Ohira and K. Tashiro, ACS Omega, 2018, 3, 349–360 Search PubMed.
- Q. Duan, S. Ge and C.-Y. Wang, J. Power Sources, 2013, 243, 773–778 CrossRef CAS.
- A. C. Lazanas and M. I. Prodromidis, ACS Meas. Sci. Au, 2023, 3, 162–193 CrossRef CAS PubMed.
- B. Pribyl-Kranewitter, A. Beard, T. Schuler, N. Diklić and T. J. Schmidt, J. Electrochem. Soc., 2021, 168, 043506 CrossRef CAS.
- C. Azenha, C. Mateos-Pedrero, T. Lagarteira and A. M. Mendes, J. CO2 Util., 2023, 68, 102368 CrossRef CAS.
- H. Xiong, J. Li, D. Wu, B. Xu and Q. Lu, Chem. Commun., 2023, 59, 5615–5618 RSC.
- P. Chauhan, K. Hiekel, J. S. Diercks, J. Herranz, V. A. Saveleva, P. Khavlyuk, A. Eychmüller and T. J. Schmidt, ACS Mater. Au, 2022, 2, 278–292 CrossRef CAS PubMed.
- D. Hochfilzer, J. E. Sørensen, E. L. Clark, S. B. Scott, I. Chorkendorff and J. Kibsgaard, ACS Energy Lett., 2021, 6, 1879–1885 CrossRef CAS.
- M. Abdinejad, S. Subramanian, M. K. Motlagh, M. Noroozifar, S. Duangdangchote, I. Neporozhnii, D. Ripepi, D. Pinto, M. Li, K. Tang, J. Middelkoop, A. Urakawa, O. Voznyy, H.-B. Kraatz and T. Burdyny, Adv. Energy Mater., 2023, 13, 2300402 CrossRef CAS.
- R. Casebolt, K. Levine, J. Suntivich and T. Hanrath, Joule, 2021, 5, 1987–2026 CrossRef CAS.
- C. Kim, L.-C. Weng and A. T. Bell, ACS Catal., 2020, 10, 12403–12413 CrossRef CAS.
- C. Kim, J. C. Bui, X. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS.
- Z. Li, L. Wang, L. Sun and W. Yang, J. Am. Chem. Soc., 2024, 146, 23901–23908 CrossRef CAS PubMed.
- J. Kok, J. de Ruiter, W. van der Stam and T. Burdyny, J. Am. Chem. Soc., 2024, 146, 19509–19520 CrossRef CAS PubMed.
- T. N. Nguyen, Z. Chen, A. S. Zeraati, H. S. Shiran, S. M. Sadaf, M. G. Kibria, E. H. Sargent and C. T. Dinh, J. Am. Chem. Soc., 2022, 144, 13254–13265 CrossRef CAS PubMed.
- J. Timoshenko, A. Bergmann, C. Rettenmaier, A. Herzog, R. M. Arán-Ais, H. S. Jeon, F. T. Haase, U. Hejral, P. Grosse, S. Kühl, E. M. Davis, J. Tian, O. Magnussen and B. Roldan Cuenya, Nat. Catal., 2022, 5, 259–267 CrossRef CAS.
- H. S. Jeon, J. Timoshenko, C. Rettenmaier, A. Herzog, A. Yoon, S. W. Chee, S. Oener, U. Hejral, F. T. Haase and B. Roldan Cuenya, J. Am. Chem. Soc., 2021, 143, 7578–7587 CrossRef CAS PubMed.
- K. W. Kimura, K. E. Fritz, J. Kim, J. Suntivich, H. D. Abruña and T. Hanrath, ChemSusChem, 2018, 11, 1781–1786 Search PubMed.
- A. Herzog, M. Lopez Luna, H. S. Jeon, C. Rettenmaier, P. Grosse, A. Bergmann and B. Roldan Cuenya, Nat. Commun., 2024, 15, 3986 CrossRef CAS PubMed.
- H. S. Jeon, J. Timoshenko, C. Rettenmaier, A. Herzog, A. Yoon, S. W. Chee, S. Oener, U. Hejral, F. T. Haase and B. Roldan Cuenya, J. Am. Chem. Soc., 2021, 143, 7578–7587 CrossRef CAS PubMed.
- J. Drnec, Q. Xu, J. Z. Zeledón, B. Ó. Joensen, L. Trotochaud, A. Sartori, L. Kaas, A. Moss, M. Mirolo and L. Mairena, 2024.
- E. R. Cofell, Z. Park, U. O. Nwabara, L. C. Harris, S. S. Bhargava, A. A. Gewirth and P. J. A. Kenis, ACS Appl. Energy Mater., 2022, 5(10), 12013–12021 CrossRef CAS.
- U. O. Nwabara, M. P. de Heer, E. R. Cofell, S. Verma, E. Negro and P. J. A. Kenis, J. Mater. Chem. A, 2020, 8, 22557–22571 Search PubMed.
- J. Edgington, A. Deberghes and L. C. Seitz, ACS Appl. Energy Mater., 2022, 5, 12206–12218 CrossRef CAS.
- C. Spöri, C. Brand, M. Kroschel and P. Strasser, J. Electrochem. Soc., 2021, 168, 034508 CrossRef.
- M. Zlatar, D. Escalera-López, M. G. Rodríguez, T. Hrbek, C. Götz, R. Mary Joy, A. Savan, H. P. Tran, H. N. Nong, P. Pobedinskas, V. Briega-Martos, A. Hutzler, T. Böhm, K. Haenen, A. Ludwig, I. Khalakhan, P. Strasser and S. Cherevko, ACS Catal., 2023, 13, 15375–15392 CrossRef CAS.
- M. Ramdin, A. R. T. Morrison, M. de Groen, R. van Haperen, R. de Kler, E. Irtem, A. T. Laitinen, L. J. P. van den Broeke, T. Breugelmans, J. P. M. Trusler, W. D. Jong and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2019, 58, 22718–22740 CrossRef CAS.
- A. G. Fink, F. Navarro-Pardo, J. R. Tavares and U. Legrand, ChemCatChem, 2024, 16, e202300977 Search PubMed.
- J. J. Kaczur, H. Yang, Z. Liu, S. D. Sajjad and R. I. Masel, C, 2020, 6, 33 CAS.
- R. Krause, D. Reinisch, C. Reller, H. Eckert, D. Hartmann, D. Taroata, K. Wiesner-Fleischer, A. Bulan, A. Lueken and G. Schmid, Chem. Ing. Tech., 2020, 92, 53–61 Search PubMed.
- L. Li, A. Ozden, S. Guo, F. P. García de Arquer, C. Wang, M. Zhang, J. Zhang, H. Jiang, W. Wang, H. Dong, D. Sinton, E. H. Sargent and M. Zhong, Nat. Commun., 2021, 12, 5223 CrossRef CAS PubMed.
- A. Kaur, B. Kim, R. Dinsdale, A. Guwy, E. Yu and G. Premier, J. Chem. Technol. Biotechnol., 2021, 96, 2461–2471 CrossRef CAS.
- Y. Cheng, P. Hou, X. Wang and P. Kang, Acc. Chem. Res., 2022, 55, 231–240 Search PubMed.
- B. Belsa, L. Xia and F. P. García de Arquer, ACS Energy Lett., 2024, 9, 4293–4305 CrossRef CAS PubMed.
- Lydian, Lydian Technology, https://www.lydianlabs.com/technology, (accessed 02/22, 2025).
- Twelve, E-Jet SAF, https://www.twelve.co/saf, (accessed 2/19, 2025).
- A. N. Biswas, L. R. Winter, Z. Xie and J. G. Chen, JACS Au, 2023, 3, 293–305 CrossRef CAS PubMed.
- S. Garg, Z. Xie and J. G. Chen, Nat. Chem. Eng., 2024, 1, 139–148 CrossRef.
- M. G. Lee, X.-Y. Li, A. Ozden, J. Wicks, P. Ou, Y. Li, R. Dorakhan, J. Lee, H. K. Park, J. W. Yang, B. Chen, J. Abed, R. dos Reis, G. Lee, J. E. Huang, T. Peng, Y.-H. Chin, D. Sinton and E. H. Sargent, Nat. Catal., 2023, 6, 310–318 CrossRef CAS.
- B. G. Pollet and J. T. E. Goh, Electrochim. Acta, 2014, 128, 292–303 CrossRef CAS.
- M. Wang, J. H. Park, S. Kabir, K. C. Neyerlin, N. N. Kariuki, H. Lv, V. R. Stamenkovic, D. J. Myers, M. Ulsh and S. A. Mauger, ACS Appl. Energy Mater., 2019, 2, 6417–6427 CrossRef.
- B. H. Lim, E. H. Majlan, A. Tajuddin, T. Husaini, W. R. Wan Daud, N. A. Mohd Radzuan and M. A. Haque, Chin. J. Chem. Eng., 2021, 33, 1–16 CrossRef CAS.
- M. Bühler, P. Holzapfel, D. McLaughlin and S. Thiele, J. Electrochem. Soc., 2019, 166, F1070 CrossRef.
- K. Kuhl, E. Cave, N. Flanders and L. Wang, Electrochemical conversion of coal-derived CO2 into fuels and chemicals using a modified PEM electrolyzer, Final report, United States, 2021 Search PubMed.
- Y. Devi, P.-J. Huang, W.-T. Chen, R.-H. Jhang and C.-H. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 9231–9239 CrossRef CAS PubMed.
- J. Park, Z. Kang, G. Bender, M. Ulsh and S. A. Mauger, J. Power Sources, 2020, 479, 228819 Search PubMed.
- S. A. Lee, S. E. Jun, S. H. Park, K. C. Kwon, J. H. Kang, M. S. Kwon and H. W. Jang, EES Catal., 2024, 2, 49–70 Search PubMed.
- J. K. Lee, G. Y. Lau, F. Shen, A. Bergeson-Keller, X. Peng and M. C. Tucker, J. Electrochem. Soc., 2024, 171, 064505 Search PubMed.
- B. Hasa, U. R. Aryal, S. Higashi, N. E. Tolouei, J. T. Lang, B. Erb, A. Smeltz, I. V. Zenyuk and G. Zhu, Appl. Catal., B, 2025, 361, 124616 Search PubMed.
- H. Haspel and J. Gascon, ACS Appl. Energy Mater., 2021, 4, 8506–8516 Search PubMed.
- S. Hao, A. Elgazzar, N. Ravi, T.-U. Wi, P. Zhu, Y. Feng, Y. Xia, F.-Y. Chen, X. Shan and H. Wang, Nat. Energy, 2025, 10, 266–277 Search PubMed.
- V. E. Nelson, C. P. O’Brien, J. P. Edwards, S. Liu, C. M. Gabardo, E. H. Sargent and D. Sinton, ACS Appl. Mater. Interfaces, 2024, 16, 50818–50825 Search PubMed.
- J. P. Edwards, T. Alerte, C. P. O’Brien, C. M. Gabardo, S. Liu, J. Wicks, A. Gaona, J. Abed, Y. C. Xiao, D. Young, A. Sedighian Rasouli, A. Sarkar, S. A. Jaffer, H. L. MacLean, E. H. Sargent and D. Sinton, ACS Energy Lett., 2023, 8, 2576–2584 Search PubMed.
- R. Küngas, J. Electrochem. Soc., 2020, 167, 044508 Search PubMed.
- D. Xu, I. Sullivan, C. Xiang and M. Lin, ACS Sustainable Chem. Eng., 2022, 10, 13945–13954 Search PubMed.
- J. Gao, S. Choo Sze Shiong and Y. Liu, Chem. Eng. J., 2023, 472, 145033 CrossRef CAS.
- W. Luc, J. Rosen and F. Jiao, Catal. Today, 2017, 288, 79–84 CrossRef CAS.
- N. Jiang, Z. Zhu, W. Xue, B. Y. Xia and B. You, Adv. Mater., 2022, 34, e2105852 CrossRef PubMed.
- Á. Vass, A. Kormányos, Z. Kószó, B. Endrődi and C. Janáky, ACS Catal., 2022, 12, 1037–1051 CrossRef PubMed.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 Search PubMed.
- M. Ma, E. L. Clark, K. T. Therkildsen, S. Dalsgaard, I. Chorkendorff and B. Seger, Energy Environ. Sci., 2020, 13, 977–985 Search PubMed.
- A. Ozden, Y. Wang, F. Li, M. Luo, J. Sisler, A. Thevenon, A. Rosas-Hernández, T. Burdyny, Y. Lum, H. Yadegari, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, Joule, 2021, 5, 706–719 Search PubMed.
- B. N. Ruggiero, X. K. Lu, K. Adonteng, J. Dong, J. M. Notestein and L. C. Seitz, Chem. Eng. J., 2024, 486, 150246 Search PubMed.
- K. Panha, M. Fowler, X.-Z. Yuan and H. Wang, Appl. Energy, 2012, 93, 90–97 Search PubMed.
- M. Casciola, A. Donnadio and P. Sassi, J. Power Sources, 2013, 235, 129–134 Search PubMed.
- P. Christopher, ACS Energy Lett., 2024, 9, 5539–5540 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |