
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOpto-digital molecular analytics
Chelsea Violita
Stanley
 ab,
Yi
Xiao
ab,
Tong
Ling
a,
Dong-Sheng
Li
c and
Peng
Chen
*abd
ab,
Yi
Xiao
ab,
Tong
Ling
a,
Dong-Sheng
Li
c and
Peng
Chen
*abd
aSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 637457, Singapore. E-mail: chenpeng@ntu.edu.sg
bLee Kong Chian School of Medicine, Institute for Digital Molecular Analytics and Science, Nanyang Technological University, 636921, Singapore
cCollege of Materials and Chemical Engineering, Key Laboratory of Inorganic Nonmetallic Crystalline and Energy Conversion Materials, China Three Gorges University, Yichang, 443002, P. R. China
dSkin Research Institute of Singapore, 308232, Singapore
First published on 4th March 2025
Abstract
In contrast to conventional ensemble-average-based methods, opto-digital molecular analytic approaches digitize detection by physically partitioning individual detection events into discrete compartments or directly locating and analyzing the signals from single molecules. The sensitivity can be enhanced by signal amplification reactions, signal enhancement interactions, labelling by strong signal emitters, advanced optics, image processing, and machine learning, while specificity can be improved by designing target-selective probes and profiling molecular dynamics. With the capabilities to attain a limit of detection several orders lower than the conventional methods, reveal intrinsic molecular information, and achieve multiplexed analysis using a small-volume sample, the emerging opto-digital molecular analytics may be revolutionarily instrumental to clinical diagnosis, molecular chemistry and science, drug discovery, and environment monitoring. In this article, we provide a comprehensive review of the recent advances, offer insights into the underlying mechanisms, give comparative discussions on different strategies, and discuss the current challenges and future possibilities.
 Chelsea Violita Stanley | Chelsea Violita Stanley received her bachelor's degree in chemistry and biological chemistry from Nanyang Technological University. She is currently a PhD candidate at the School of Chemistry, Chemical Engineering and Biotechnology and Institute for Digital Molecular Analytics and Science of Nanyang Technological University. Her research primarily focuses on developing enzyme-free digital biosensors for ultrasensitive detection of biological analytes. |
 Peng Chen | Peng Chen is a Professor of bioengineering at the School of Chemistry, Chemical Engineering and Biotechnology, Institute for Digital Molecular Analytics and Science, and Lee Kong Chian School of Medicine at Nanyang Technological University. He also serves as the chief engineer at the Skin Research Institute of Singapore. Working at the interface between biomedicine, chemistry, materials, engineering and nanotechnology, his research team endeavors to develop interdisciplinary solutions for biosensing and medical treatment, resulting in about 300 publications in reputable journals. He is a fellow of the Royal Society of Chemistry and a global highly cited researcher (Clavirate). |
1. Introduction
Ultrasensitive molecular detection and analysis techniques are instrumental to biology and medicine,1,2 molecular chemistry and science,3 environment monitoring4 and clinical diagnosis.5,6 Conventional methods are based on ensemble-averaged measurements; hence, the signal-to-noise ratio (SNR) drops as the target concentration decreases, resulting in poor limit of detection (LOD). In addition, conventional detection also suffers from low spatiotemporal resolution, unsatisfactory specificity, and a loss of information about sample heterogeneity. Many disease biomarkers and pathogens are present in trace concentrations (aM to pM) that are often several orders of magnitude lower than the LOD of conventional ensemble assays.7–11 Moreover, a large sample volume is typically necessitated for ensemble assays, making it difficult to analyze biofluids with limited volume (e.g., skin interstitial fluid, cerebrospinal fluid, ocular fluid, and blood from small animal models). Increasing the volume by dilution makes the target concentration even lower. The emerging opto-digital molecular analytics addresses the abovementioned limitations of conventional detection by optically enabling a binary signal read-out (1 or 0) at each discrete reaction site or probing single molecules with a high spatiotemporal resolution, thereby improving the LOD by several orders and obtaining more molecular information with high specificity from only a small-volume sample. In opto-digital analytics, the signal generated by individual molecules is concentrated within a tiny detection volume with a low background, giving a high SNR. And both signal and background noise (hence SNR) are independent of the target concentration; counting the number of signals reveals the target concentration. Based on two different signal read-out modalities, we classify the opto-digital molecular analytics into (i) photon emission methods which require emissive labeling and (ii) photon scattering methods which enable direct interrogation in a label-free manner (Fig. 1). | ||
| Fig. 1 Opto-digital molecular analytic approaches based on photon emission and scattering. | ||
Although ultrasensitive molecular detection relying on optical and non-optical read-outs has been discussed in several excellent review articles,12–14 an update in this rapidly evolving field is necessary. Furthermore, the concept of opto-digital molecular analytics is proposed here for the first time, with the emphasis on digitizing the detection based on optical signals. In this article, we introduce the basic concepts of opto-digital molecular analytics, provide comprehensive coverage of the latest innovations, and offer insights into the underlying mechanisms. Digitization strategies are described, including physically or chemically compartmentalizing the individual molecular binding events, or directly locating and analyzing the emission or scattering signals from single molecules. We emphasize how signals from specific binding events can be confidently distinguished from background signals (sensitivity) and non-specific binding events (specificity). Specifically, sensitivity can be enhanced by signal amplification reactions, signal enhancement interactions (e.g., plasmonic effect, photothermal effect), labelling by strong signal emitters, advanced optics, image processing, and machine learning (Fig. 1); while high specificity is conferred by target-selective probes, extraction of more molecular information (e.g., molecular weight, charge, binding affinity), and novel data analysis methods. One advantage of opto-digital molecular analytics is the amenability to leverage the powerful image-based (static) or video-based (dynamic) analyses. For example, the latter allows profiling the molecular dynamics (binding–unbinding kinetics, motion patterns) to distinguish specific binding from non-specific interaction. Towards real-world applications, various innovations in assay automation, device miniaturization, and machine learning-powered analysis are also highlighted. Different opto-digital analytical methods are compared in terms of sensitivity, specificity, multiplexing capability, deployability in point-of-care (POC) settings (e.g., through miniaturization), assay simplicity, and equipment complexity. We also provide succinct comparisons with electronic digital molecular analytics. Lastly, the current challenges and future possibilities for opto-digital molecular analytics are discussed. With this, we hope to inspire a diverse scientific community and stimulate further collaborative development of this exciting emerging field.
2. Photon emission-based detection
Optical detection often relies on emissive dye molecules which are excited into a higher energy state by light (fluorescence) or a chemical reaction (chemiluminescence) and then emit photons during the radiative relaxation process. However, the signal from a single dye molecule is too weak to be detected from background noises with conventional microscopes. Therefore, amplification reactions can be designed to recruit a large number of dye molecules to report the binding of a single target molecule. Alternatively, recent advances in microscopy may enable signal read-out from individual dye molecules. Although these methods are capable of observing single molecular binding, their LODs may not be a single molecule because of background interference and binding dissociation.2.1. Amplification-based detection
Signal amplification often involves target-binding-triggered enzymatic reactions to enable the emission of many dye molecules (signal reporter amplification) or replication of the target molecules to allow the binding of more dye molecules (target amplification). To digitize the detection of individual molecular binding events, it is necessary to confine the freely diffusing molecules generated from the amplification reaction within individual compartments. This can be realized by using microwells and microfluidic droplet-generating devices (physical compartmentalization), or the chemical clustering of the dye molecules via intermolecular bonds (chemical compartmentalization) (Fig. 2). Both compartmentalization strategies greatly reduce the detection volume, thereby enhancing SNR. And, a physically compartmentalized tiny reaction volume ensures accelerated reaction kinetics due to the high surface-to-volume ratio. | ||
| Fig. 2 Compartmentalization strategies for emission-based digital molecular analytics. | ||
To further reduce the reaction volume for SNR enhancement and enable massively parallel reactions, dPCR was performed in a microwell array chip comprising thousands of nL to pL-sized wells.18–20 In addition, microfluidic devices have been employed to better control solution loading into the tiny wells.16,17,21,22 Considering Poisson statistics, single molecule occupancy within each compartment only applies at low target concentration. When the target concentration goes beyond a certain point, some compartments will be occupied by more than one molecule.16,23 In this case, the average number of target nucleic acids in each compartment (λ) can be estimated with eqn (1), where p is the fraction of compartments with positive signal. The target concentration can be calculated with eqn (2). However, when p > 0.7, eqn (1) can no longer give accurate estimations,24 defining the upper detection limit of dPCR.
| λ = −ln[1 − p] | (1) |
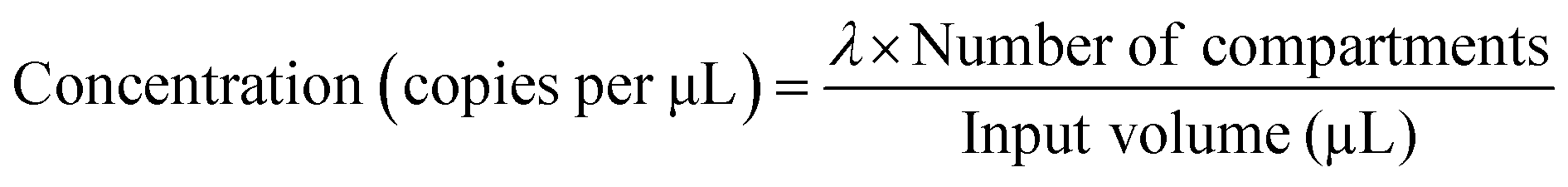 | (2) |
The conventional nucleic acid detection based on ensemble-averaged real-time PCR requires external calibrators for quantification and suffers from limited accuracy due to the variability in amplification efficiency of the polymerase. In contrast, for dPCR, the binary end-point read-out is much less affected by such variability, and the absolute number of target nucleic acids can be counted without the need for calibration.25 The ultimate sensitivity, i.e., detection of single nucleic acid molecules, can even be achieved by dPCR.26,27
Digital assays for nucleic acids via direct target amplification have also been devised using other nucleic acid replication strategies such as recombinase polymerase amplification (RPA) and loop-mediated isothermal amplification (LAMP).28–32 Furthermore, both the target and signal reporter can be amplified by combining nucleic acid replication and CRISPR-Cas systems. Specifically, binding of the Cas-crRNA complexes to the abundantly replicated nucleic acids triggers the trans-cleavage of nearby fluorophore-quencher-labeled single-stranded DNA or RNA reporters, thus freeing the fluorophores to enable emission. The CRISPR-Cas system has been used in digital LAMP (dLAMP),33–35 and digital RPA (dRPA)-based36–39 detection of DNAs and micro RNAs (miRNAs). Using a Cas-crRNA complex with ultra-high turnover rate such as Cas13a and Cas12a, sufficient fluorescent signal can be yielded from a single nucleic acid molecule without replication (signal reporter amplification only) (Fig. 3(A)).40–43
 | ||
| Fig. 3 Signal amplification and physical compartmentalization enabled digitization. (A) CRISPR for digital RNA detection in pL-sized droplets. Adapted with permission from ref. 40. Copyright 2021 American Chemical Society. (B) Single molecule array (Simoa) system with antibody-immobilized magnetic beads being compartmentalized in fL-sized wells after target binding. Signal amplification occurs inside microwells, enabling binary read-out. Adapted with permission from Macmillan Publishers Ltd, Nature Biotechnology, ref. 11, copyright 2010. (C) Multiplexed dLAMP using encoded primers, followed by machine learning-assisted decoding and analysis. Reprinted with permission from ref. 44. Copyright 2023 American Chemical Society. | ||
Rissin et al. translated the idea of digitization for protein detection through digital enzyme-linked immunosorbent assay (dELISA), which they termed as the single molecule array (Simoa) system (Fig. 3(B)).11 Unlike dPCR, dLAMP or dRPA which rely on specific target amplification reactions, the specificity of Simoa is conferred by a capture and detection antibody pair, while a signal reporter amplification reaction is catalyzed by β-galactosidase (β-gal) conjugated on the detection antibody to convert non-fluorescent substrates (e.g., resorufin-β-D-galactopyranoside-RGP) into fluorescent products (e.g., resorufin). Simoa used antibody-coated magnetic beads to capture the target analytes, which were subsequently bound with β-gal-conjugated detection antibodies to form sandwich immunocomplexes. The capture beads were in large excess with respect to the target molecules, thus the association–dissociation equilibrium of the binding event was driven kinetically towards the target-bound state to maximize capture. Each bead captured either one or no target because the binding followed Poisson distribution and capture beads were in large excess. The sandwich immunocomplexes were then magnetically separated from the free β-gal-conjugated detection antibodies, followed by introduction into an array of 50-fL microwells. Each microwell acted as a micro-reaction-chamber where β-gal catalyzed the fluorescence-generating reaction. The microwells were then imaged and the fraction of beads with positive signals (fON) was quantified to give a calibration curve. Unlike polymerase-amplification-based digital detection of nucleic acids, calibration is necessary for protein quantification because complete capture of all target proteins is not possible due to the presence of association–dissociation equilibrium between the affinity-based probe and the target protein. Simoa conferred an improvement in immunoassay LOD from pM down to the aM range. Simoa was also compatible with DNA detection, offering a LOD of 150 aM.
The dynamic range of detection for Simoa is narrow, however, because each bead can catch multiple target molecules when the target concentration is high. To extend the detection range, analog analysis was applied for high concentrations.24 Specifically, the average number of targets per bead or enzyme per bead (AEB) was used to plot the calibration curve. Each bead carrying at most one target was valid only when the fraction of beads giving a positive signal was <10% (fON < 0.1). Therefore, by counting fON digitally and converting it into AEB based on Poisson statistics, the actual target concentration was more accurately reflected. The AEB was calculated differently in the digital (fON < 0.7, eqn (3)) and analog regimes (fON > 0.7, eqn (4)). When fON > 0.7, statistically every bead was bound to multiple target molecules. As such, AEB was derived from the average signal intensity of all active beads (Ībead) and the average signal from the bead bound with only one target (Īsingle) (eqn (4)). Īsingle was determined separately from a sample giving fON < 0.1. Through this combined approach, the linear dynamic range of Simoa was extended to more than 4 logs of target concentration for the detection of prostate-specific antigen (PSA) in serum (∼250 aM–3 pM).
| AEBdigital = −ln[1 − fON] | (3) |
 | (4) |
To obtain fON, bright-field and fluorescence imaging are required to quantify the total beads and signal-yielding beads respectively.11 If fluorescent beads are used, only fluorescence imaging is necessary.45 Automated image analysis to obtain the AEB can be done using a proprietary algorithm from Quanterix.24 Alternatively, Yang et al. proposed the use of mean fluorescence intensity increase (MFII) instead of AEB to derive the calibration curve.45 By analyzing the change in fluorescence intensity of each microwell before and after the enzymatic amplification reaction, MFII can be calculated through dividing the total brightness increase from all microwells by the total number of beads. A Python-based algorithm was developed to automate the image analysis process.
Based on kinetic modelling and the assumption that background AEB is independent of the bead number, Simoa sensitivity can be further enhanced by using fewer capture beads to raise the AEB value.46,47 However, given that the bead loading efficiency into microwells is low (∼5%), using fewer beads means an even smaller number of beads are available for analysis. This compromises the sensitivity and precision in the presence of background noise. Higher bead loading efficiency (∼50%) was realized with the magnetic-meniscus sweeping (MMS) technique which used magnetic force to recruit magnetic beads into microwells and hold them in place.46 Combining the use of fewer beads (<1% of beads in original Simoa), MMS technique and an extended target incubation time (24 h), >400-fold improvement in sensitivity has been achieved with a LOD in the sub-aM range. Digital microfluidics- and vacuum-assisted loading have also been used for active loading, and hydrophilic-in-hydrophobic microwells have been devised to drive bead-loading via liquid-surface adhesive and repelling forces.48–50
Other strategies for opto-digital analysis of proteins have also been devised. Kim et al. developed a digital homogeneous entropy-driven biomolecular assay (dHEBA) to detect proteinaceous targets.51 dHEBA is an enzyme-free one-pot assay with a fast single recognition and signal amplification step (10 min). Specifically, dHEBA used two antibody-conjugated single-stranded DNA oligonucleotides (ssDNAs) as the recognition element and double-stranded DNA oligonucleotides (dsDNAs) conjugated with a fluorophore and quencher on each strand as the signal reporter. The binding of the target to two recognition ssDNA sequences brought them together and triggered a series of ssDNA displacement reactions to separate the dsDNA, leading to the release of the fluorophore-linked ssDNA from its complementary quencher-linked ssDNA. Single-molecule compartmentalization in dHEBA relies on random settlement following Poisson distribution. Each microwell in an array of 3364 statistically contains either 0 or 1 target molecule when its concentration is below 1 fM. Beyond this concentration, direct digital counting is no longer reliable.
Using relatively large microwells, digital detection of E. coli bacteria at a single-cell level has been demonstrated.52 6-Chloro-4-methylumbelliferyl-β-D-glucuronide (6c-MUG), the substrate of β-D-glucuronidase enzyme found in E. coli, was used as the probe. The hydrolysis of 6c-MUG into fluorescein in the presence of E. coli resulted in a positive read-out. Digital detection of mammalian cells (hybridoma cells) was similarly realized using an antigen–DNA probe pair to specifically recognize the target antibody secreted by the cells.53 Upon target binding, rolling circle amplification (RCA) was triggered by the DNA probe and many pyrophosphate anions were generated as the by-products. A chemiluminescence-based detection was then realized through signal reporter amplification. In particular, copper(II)-inhibited horseradish peroxidase (HRP) was activated in the presence of pyrophosphate anion, which subsequently catalyzed the oxidation of luminol in the presence of H2O2 and led to the generation of a chemiluminescent signal confined within the individual wells. These approaches allow high-throughput analysis of individual cells within a heterogeneous population of cells.
Microfluidic devices have often been employed to automate sample loading into the microwells. Due to the demand for portable and easy-to-use diagnostic devices, power-free poly(dimethylsiloxane) (PDMS) microfluidic chips have been devised to omit the need for an external power source, pump and control electronics.54–56 During degassing in a vacuum, the dissolved air was evacuated from the PDMS substrate and redissolved back through the microchannel walls when returned to atmospheric pressure. This process provided kinetic energy for fluid movement through the microchannels. Using this system, Zhu et al. introduced the self-priming compartmentalization (SPC) microfluidic device, which is a power-free self-digitization chip for dLAMP and dPCR.31,57 The SPC device has undergone improvements over the years, such as reduced operational complexity and time,58–61 reduced thickness to improve heat transfer efficiency,62 and integration with smartphones.63 For dRPA, an integrated and power-free microfluidic device enabling one-step nucleic acid analysis from unprocessed blood has been developed by Yeh et al., which relied on a unique design to inhibit the entry of blood cells into the microwells.64 The device used a vacuum battery system, which comprised on-chip compartments with pre-created vacuum, to provide a more robust and controlled flow of blood plasma and reagents into the microfluidics and compartmentalize them into the microwell array. Self-digitization devices without relying on vacuum systems have also been designed. Named the SlipChip, the device for dPCR, dRPA and dLAMP allows the generation of a large number of microdroplets on-chip using simple manual operation.34,65–67 The SlipChip is composed of two microwell array-containing chips, one placed atop another, with the top side facing each other. The microwell array is designed in a way that the fluidic paths between the microwells can connect or disconnect by a simple slipping motion between the two chips. The connection and disconnection of fluidic paths enable fluid manipulation for reagent loading into the microwells.
Alternatively, microdroplets have been utilized to digitize detection (Fig. 2, top right). Microdroplets rely on an oil–water interface to establish a physical barrier. Each droplet acts as a tiny reactor for the signal amplification reaction and a confiner to the fluorescent product. One way to generate a microdroplet array involves passing liquid over a hydrophobic surface with an array of hydrophilic spots where the aqueous microdroplets form on top.68 Microfluidic devices have also been widely utilized to generate monodisperse droplets (fL to pL), by dispensing water-in-oil emulsion droplets,27,33,69–71 or direct droplet printing on-surface.72–74 Inkjet printing has also been used to make an array of monodisperse microdroplets on a surface75,76 or within a capillary.77 Generation of larger microdroplets (diameter ∼40 μm) allowed imaging with a mobile phone.78 To better cater to point-of-care (POC) needs, microdroplets can be generated without microfluidic or inkjet devices by simple shaking the water/oil mixture, albeit with some degree of polydispersity in size.79 More recently, monodisperse pL-sized microdroplet generation was realized by simply centrifuging aqueous solution in the presence of emulsifying reagents through an ultra-partitioning droplet generator column.80 The microdroplets were collected into a microcentrifuge tube. As more than 30 million droplets can be generated from a 50 μL sample, by Poisson statistics, single-molecule occupancy within each droplet can be ensured even at high target concentrations. In addition, the optically clear and stable droplets can be imaged three-dimensionally in a microcentrifuge tube using a light sheet microscope.
Typical image analysis of droplet-based assays differs slightly from the microwell counterpart. An image analysis algorithm has been developed to identify the empty droplets from the bright-field image, and differentiate bead-containing signal-negative droplets and bead-containing signal-yielding droplets from the fluorescence image.71 An edge detection algorithm was applied to locate the droplets, while a watershed algorithm was applied to separate the individual droplets. Empty droplets were determined by overlapping the bright-field and fluorescent images, and their median fluorescence intensity was used for background correction.
Multiplex detection systems have been developed to enable simultaneous analysis of multiple targets.21,81–86 However, it is typically challenging to increase the target number beyond six because of the overlapping emission bandwidth of the fluorophores. To address this, color encoding strategies, together with machine-learning powered color analysis, have been developed. Combining two to three fluorophores, up to 49 unique color codes can be generated (Fig. 3(C)).44,45,87 However, the multiplexing capability may also be limited by the number of microwells or microdroplets available. A trade-off must be made between the number of targets and the sample size of each target allocated from the total compartment number. Furthermore, a high degree of multiplexing comes with an increased probability of cross-reaction between the different capture and detection antibodies, which increases non-specific signals and therefore reduces the assay sensitivity and accuracy.
For protein detection, supramolecular constructs have been designed to enable hierarchical labeling of the target and give a strong signal.90 Specifically, the detection antibody was conjugated onto a large scaffold molecule made of multi-biotinylated protein and biopolymer (e.g., dextran), which was capable of recruiting a large number of multi-fluorophore-labeled streptavidin. A bead-based droplet-free digital assay for proteinaceous targets has also been designed using tyramide signal amplification (TSA) in a method termed CARD-dELISA. It involves HRP-catalyzed binding of ample fluorophore-conjugated tyramide molecules with tyrosine and tryptophan residues of the immunocomplex via radical-mediated reaction.88,91 Without a physical partition, the diffusion and deposition of a fluorescent product from one bead to another can be minimized at a sufficiently low bead concentration, because the inter-bead distance is large enough such that the generated short-lived tyramide radical cannot reach another bead. With bound signal reporter molecules, beads can be directly drop-cast as a monolayer film for imaging.91,92 TSA has also been used in digital electrochemiluminescence (ECL) droplet-free assays.93 With ECL probe [Ru(bpy)3]2+ being conjugated onto tyramide, poly-HRP on the immunocomplex catalyzed the deposition of [Ru(bpy)3]2+ onto the beads. An ECL signal was subsequently generated from the charge transfer process between [Ru(bpy)3]2+ luminophore and tri-n-propylamine co-reactant on an indium tin oxide electrode.
For nucleic acid detection, the hybridization chain reaction (HCR) has been leveraged as a chemical compartmentalization strategy. The HCR is an enzyme-free signal amplification strategy which utilizes two DNA hairpin molecules to generate a long chain of DNA concatemer from an initiator single-stranded nucleic acid molecule (Fig. 4(A)).94,95 The target nucleic acid can either be used as the initiator directly or be recognized by a complementary initiator ssDNA. Two fluorescently labeled DNA hairpins (H1 and H2) act as the signal reporter. The presence of an initiator triggers the opening of H1. One end of H1 hybridizes with the target molecule, while the other end hybridizes with H2. H2 then opens another H1. This cascade of hybridization reaction cycle between the metastable DNA hairpins proceeds to generate a long fluorescent DNA concatemer. When multiple initiator molecules are used, a larger branched fluorescent DNA reporter can be produced.96 A split-initiator pair has been designed to reduce the background signal arising from non-specific bindings.97 It is made of two strands of ssDNA complementary to the adjacent sites within the target nucleic acid. Only when both initiator ssDNAs bind adjacently, will HCR be triggered, thereby ensuring high detection specificity. HCR can also be used as a signal amplification mechanism for protein detection by using an initiator-labeled antibody.94 In principle, this strategy can be further developed into digital analytics.
 | ||
| Fig. 4 Signal amplification and chemical compartmentalization enabled digitization. (A) Mechanism of hybridization chain reaction (HCR) to generate a long DNA polymer from an RNA target or HCR initiator-labeled antibody. Adapted from ref. 94 according to Creative Commons Attribution 4.0 International License (CC BY 4.0). (B) Digital RCA (dRCA) to generate a large nucleic acid molecule (1 μm in hydrodynamic diameter) for droplet-free digital nucleic acid detection. Reprinted with permission from Macmillan Publishers Ltd. Nature Methods, ref. 98, copyright 2006. (C) On-bead dRCA followed by digital counting using flow cytometry. Reprinted with permission from ref. 99. Copyright 2022 American Chemical Society. | ||
Chemical compartmentalization can also be realized by RCA reaction. For nucleic acid detection, DNA polymerase elongates the pre-circularized target nucleic acid into a micron-sized concatemer, which can be fluorescently labeled to yield a single-molecule read-out. It has been implemented in solution (Fig. 4(B)),98,100 on-bead (Fig. 4(C)),99 and on-surface.101 Several improvements to digital RCA (dRCA) have been made to increase SNR, for instance, by using compaction nucleotides or polyethylene glycol (PEG) to cause compaction of DNA concatemer and hence increase of fluorescence intensity.101,102 Branched DNA can also be obtained from two consecutive RCA reactions, allowing each target nucleic acid to grow into tens of GigaDalton in size within a relatively short time.100 The large RCA product can be stained and counted easily with flow cytometry. For protein detection, a detection antibody with circular RCA template binds with surface- or bead-captured target protein.89,92,99 DNA polymerase then catalyzes the elongation of the target-bound circular RCA template into a long concatemer. After fluorescence labeling, the surface-captured target can be visualized under the microscope, while the bead-captured target can be counted directly using a flow cytometer.
For dRCA, multiplex detection can be realized by labeling different target-grown concatemers using distinct fluorophores.103,104 To allow multiplex detection without signal cross-talks between the different fluorophores, a strand displacement strategy has been developed to allow sequential staining and destaining with different fluorophore-labeled detection oligonucleotides.101 As discussed in Section 2.1.1, the multiplexing capability of compartment-based assays is limited by the number of microwells or microdroplets available, because reduction of the sample size compromises the reliability of the detection. Flow cytometry analysis of bead-surface-confined detection can overcome this issue as there is essentially no limitation in the number of beads that can be analyzed. The Molecular On-bead Signal Amplification for Individual Counting (MOSAIC) technique has shown an 8-plex detection down to low-fM sensitivity using an on-bead signal amplification via dRCA and flow cytometry analysis while maintaining low cross-reactivity.99 Specifically, capture antibody-conjugated beads encoded with different fluorophore combinations were used to catch different targets. The bead-captured target was subsequently recognized by RCA template-conjugated detection antibody. The subsequent elongation of the RCA template on-bead and fluorescent-labeling of the concatemer gave amplified signals. Recent improvement in MOSAIC technology enabled a further reduction of LOD to low-aM levels.105 Instead of using differently colored beads but the same fluorescent probe for differentiating different targets, the improved design relied on distinct bead–probe pairs. Specifically, each detection antibody was conjugated to a unique RCA template, which was paired with a specific, colored bead. The RCA concatemer resulting from a unique RCA template permits specific labelling by different fluorophores. Simultaneous analysis of both color-encoded RCA concatemers and capture beads reduced non-specific signals, thereby enhancing the LOD in a multiplexed system.
In addition to proteins and nucleic acids, the detection of single extracellular vesicles (EVs) can also be instrumental to diagnostics. Due to their small size (∼100 nm) and low abundance during early disease development, ensemble-averaged detection of EVs using ELISA or Western blotting is often ineffective. Digitized assays offer enhanced sensitivity and allow identification of unique subpopulations of EVs which are characterized by distinct compositions of proteins and nucleic acids. Colocalized detection of both protein and nucleic acid (e.g., miRNA) markers on a single EV for early cancer detection has been enabled by RCA.106 Individual EVs were captured by a magnetic nanorod conjugated with capture antibodies, followed by recognition of the EV surface markers using nucleic acid probes and signal amplification via RCA. Magnetic agitation of the nanorods accelerated EV capture and biomarker recognition. In addition, as each nanorod can only accommodate one EV, aggregation of EVs was prevented, which could otherwise lead to miscounting. Digital counting of EVs carrying both protein and miRNA markers provided enhanced detection specificity. The HCR has also been used to amplify the signals from single EVs, enabling convenient counting of various EV sub-populations using flow cytometry.107
2.2. Amplification-free detection
While signal amplification-based detection has shown great success, it often requires non-trivial compartmentalization procedures and time-consuming reactions. Hence, direct and amplification-free techniques for digital detection have been a topic of interest. Single molecular binding events can be directly observed by advanced confocal laser scanning microscopy (CLSM) and total internal reflection fluorescence microscopy (TIRFM) which can largely avoid the interference from background noise by optical mechanism and image processing algorithm. Alternatively, nanoparticles or nanostructured surfaces can be used to greatly enhance the fluorescence signal, thereby enabling single-molecule detection even with a standard microscope.For a sufficiently diluted sample, a transient burst of photons will be detected when a single fluorophore transits through the detection volume. Recently, the use of a photon burst search algorithm for the in-solution digital detection of protein biomarkers by CLSM was reported (Fig. 5(A)).108 To differentiate analyte-bound and free fluorescence probes, an electrophoretic-based separation mechanism built on a microfluidic chip was employed based on their difference in electrophoretic mobility (charge/volume ratio). In addition, based on the inter-photon interval, random fluctuation of photons (shot noise) can be discerned from the fluorophore signal. Alternatively, a commercialized single-molecule immunoassay system by Singulex (Erenna) analyzes the number of photons passing through the confocal volume within 1 ms time frames.109 To avoid shot noise interference, detection is counted only if the photon count is >5× the background fluctuation. Analog analysis has been employed to extend the detection range for higher protein concentrations by summing the total number of photons from all detection time frames.
 | ||
| Fig. 5 Direct digital detection without signal amplification or enhancement. (A) Digital immunosensor assay (DigitISA) based on electrophoretic separation of target-bound probes from free probes followed by a photon burst search algorithm for the identification of individual detection events from background fluctuations. Adapted from ref. 108 according to Creative Commons Attribution 4.0 International License (CC BY 4.0). (B) Single-molecule colocalization assay (SiMCA) based on two-color coincidence detection (TCCD). Adapted from ref. 110 according to Creative Commons Attribution 4.0 International License (CC BY 4.0). (C) Single-molecule recognition through equilibrium Poisson sampling (SiMREPS) for specific digital detection of single miRNA molecules based on kinetic fingerprinting. Adapted with permission from Macmillan Publishers Ltd. Nature Biotechnology, ref. 111, copyright 2015. | ||
Although the photon-burst-counting method is sensitive, it is prone to false positive detection due to non-specific bindings and fluorescent impurities. A dual-color fluorescence colocalization method, also known as two-color coincidence detection (TCCD), has been designed to improve detection specificity.112,113 In this method, the target was recruited by two fluorescently labeled probes of differing colors. Specific binding events were determined as temporal coincidence of photon bursts at both color channels. Apart from detection in solution, TCCD is also compatible with the detection of surface-captured targets based on spatial colocalization of the capture and detection probes (Fig. 5(B)).110 In these cases, photons are continuously emitted from the fluorescently labeled targets rather than appearing as transient bursts. Image analysis generally involves locating the center position (centroid) of individual probes through Gaussian fitting, and subsequent colocalization analysis by calculating the distance between the centroids of the two probes. TCCD has been used for the detection of small molecules,114 nucleic acids115 and proteins.110,116
Specific detection on-surface has also been enabled by video-based binding kinetic analysis. In a method named SiMREPS (single-molecule recognition through equilibrium Poisson sampling), a low-affinity protein probe or short oligonucleotide probe was used to allow repetitive binding to the target (Fig. 5(C)).111,117 Specific binding events yielded a distinguishable kinetic fingerprint from non-specific binding events, enabling direct specific detection without washing. SiMREPS is capable of detecting one nucleic acid in a background containing one million other molecules,118 detecting proteins at aM to fM levels,117 and discriminating single base mutation in miRNA at the single-copy-level.111
As well as providing the target analyte concentration, video-based analysis can also reveal additional molecular information such as hydrodynamic radius and binding affinity with other molecules.119 Protein hydrodynamic radii can be derived from its diffusion pattern across the confocal detection volume by fitting it to an advection–diffusion model. Binding affinity can be obtained by monitoring binding–unbinding events based on changes in diffusion pattern. Considering rapid Brownian motion limits the observation time of a molecule to 1 ms, an anti-Brownian electrokinetic (ABEL) trap has been developed to enable extended observation time (a few seconds) for analysis of single molecules.120 The motion pattern of the trapped molecules can be used to infer their diffusion coefficient and hydrodynamic size.121 In addition, the extended observation time permits spectroscopic characterization of fluorescently-labeled single molecules. Squires and co-workers leveraged this possibility to achieve multiplexed detection of nucleic acids and proteins (up to 27-plex).121 In particular, a series of DNA-based probes were designed, which comprised a Cy3 fluorescence resonance energy transfer (FRET) donor on one end and a Cy5 FRET acceptor on the other end, separated by DNA of varying lengths. Therefore, many spectroscopically distinct FRET-based probes were generated.
Aside from organic fluorescent dyes, inorganic quantum dots (QDs) can also be used as a label. Given its larger size (several nm) and stronger emission, direct detection of QD-labeled molecules can be realized using simpler optics.122 Furthermore, QDs generally show higher photostability than fluorescent dyes and thus can be advantageous for video-based detection which requires longer observation times. Typical assays rely on detection probe-conjugated QDs, which form sandwich structures with the target and capture probe-modified beads,123 surfaces,122 nanoparticles,124 or QDs.125 Compared to capturing on the surface, capturing by well-dispersed beads is more efficient. However, the Brownian motion of the beads imposes challenges in imaging. Therefore, particle sedimentation was used to bring the beads closer to the surface and thus restrict their movement.123 The high efficiency (>90%) and uniformity of particle sedimentation on the surface allowed more particles to be detected accurately, giving a LOD as low as 3.9 aM. In another study, the sandwich complexes were homogeneously dispersed in solution, and colocalization of the scattering signal from gold nanoparticles on the capture probe and fluorescence signals from QDs on the detection probe enabled wash-free homogeneous detection with high specificity.124 Homogeneous digital assays have also been developed based on aggregation-dependent blue-shift of QDs.124–126 Specifically, when the sandwich immunocomplex forms between a QD-conjugated capture and detection antibody pair bridged by a target molecule, the resulting QD aggregate leads to a split in its first-order diffraction streak, which can be observed under a transmission grating-based spectral microscope and used to differentiate the complex from free QD-conjugated antibodies.
Plasmon-enhanced digital detection has been demonstrated by a plasmonic fluor-linked immunosorbent assay (p-FLISA) (Fig. 6(A)).128 The target molecule initially bound to a capture antibody functionalized on a glass surface. Subsequently, a detection antibody conjugated with a fluorophore-coated plasmonic nanoparticle (plasmonic-fluor) bound to the target molecule, enabling fast read-out without the need for amplification reactions. Plasmonic-fluors offer signal enhancement by several thousand fold as compared to common fluorophores, thereby enabling single molecule detection with just 20× magnification and a LOD at a sub-fM level. Further signal enhancement has been enabled by capturing the target on a photonic crystal surface instead of a glass substrate, attributed to photonic–plasmonic hybrid coupling.129 Plasmon-enhanced emission has also been realized by capturing the target molecule on the surface of a plasmonic nanostructure. For instance, a gold nanoparticle array assembled into a 2D honeycomb lattice has been fabricated for antibody immobilization.130 The capture of the target molecule followed by binding of a fluorescently labelled probe enabled single molecule detection at 10× magnification. A multilayered plasmonic surface made of a silver sheet, dielectric sheet and silver nanoparticles has also been designed to enable single molecule detection with a low excitation power.131
 | ||
| Fig. 6 Fluorescence signal enhancement through plasmonic coupling or waveguiding for digital detection. (A) Plasmonic-fluor labeling enables direct single molecule detection using epifluorescence microscopy. Adapted with permission from ref. 128. Copyright 2024 American Chemical Society. (B) Single molecule fluorescence enhancement with a light-guiding nanowire. Adapted with permission from ref. 132. Copyright 2019 American Chemical Society. (C) Combination of plasmonic coupling and waveguiding in zero-mode waveguide enables label-free single protein fluorescence detection. Adapted with permission from ref. 133. Copyright 2019 American Chemical Society. | ||
Aside from plasmonic coupling, fluorescence signals can be enhanced by a waveguiding micro- or nanostructure which guides the emitted photon to the detector with minimal loss in propagation and minimal background noise. A microfluidic device possessing a waveguiding effect, named anti-resonant reflecting optical waveguide (ARROW), has been utilized for digital detection.134 The low propagation loss was attributed to the use of a highly reflective material. The magnetic-bead-captured target was isolated and stained fluorescently on-chip. The fluidic channel carrying the target intersected the optical channel that guided the excitation light, and a small detection volume (∼10 fL) was formed at the intersection to enable single molecule detection with a high SNR. A combination of digital and analog analyses enabled a detection range of thirteen orders of magnitude. Vertical semiconductor GaP nanowires with high refractive index (>3.1) have been fabricated to function as waveguides which collect emission from surface-bound fluorophores and re-emit directionally at the tip, enabling single-molecule detection (Fig. 6(B)).132
Fluorescence enhancement can also be realized through a combination of plasmonic and waveguiding effects. Zero-mode waveguide (ZMW) comprises sub-wavelength nanowells within a plasmonic metal film coated on a glass cover slip (Fig. 6(C)).133,135 As light enters the nanowells from below, a highly localized evanescent field (20 to 30 nm thick) is generated. The resulting small detection volume (aL to zL) in combination with the plasmonic coupling effect yields a high SNR for the observation of single molecules. In addition, the nanowells serve the purpose of compartmentalization. Typically, occupancy of target molecules within ZMW is Poissonian distributed, hence single molecule occupancy and digitized read-out are limited to a low concentration range when the total occupancy is low (<37%). To make full use of the nanowell array, non-Poissonian distribution of molecules can be attained by making nanowells sufficiently small and shallow such that only one molecule can occupy it at a time even at high total occupancy (∼90%).136 A careful design of ZMW is important because the detection SNR is highly dependent on its dimensions.137 So far, the quantitation of membrane-bound proteins on cells,138 label-free detection of single proteins,133 and analysis of target binding kinetics have been demonstrated using ZMW.139
3. Photon scattering-based detection
Photon emission-based detection can be plagued by photobleaching, photoblinking, or the short lifetime of emissive dyes. Therefore, detecting photon scattering directly from the target analyte or from a bound nanoparticle can sometimes be advantageous. Photon scattering can be elastic (Rayleigh scattering) or inelastic (Raman scattering). The latter is much weaker, but it provides chemical information about the molecule.3.1. Rayleigh scattering
In Rayleigh scattering, the scattering cross-section is proportional to the sixth power of the diameter of a non-plasmonic target. Hence, Rayleigh scattering-based detection can sensitively differentiate molecules of different sizes. On the other hand, it also means that detecting small molecules is challenging. Hence, plasmonic nanoparticles, which scatter light more strongly, are often used to label the target molecules. In addition, the signal contrast can be improved by reducing the background or enhancing the scattering signal from the target. Although all Rayleigh scattering-based methods probe single molecules, their LODs may not be single molecule because of the uncertainty caused by background interference.3.1.1.1. Dark-field imaging. DFM achieves background suppression using a unique ring-shaped light for illuminating the sample (Fig. 7(B)). This enables only the elastically scattered light from the sample to enter the objective while rejecting the illumination light, resulting in a high-SNR image. As an opto-digital detection tool, DFM is capable of capturing the actual color of the particle. Hence, a color change induced by target binding can be sensed. For instance, a pair of probes (e.g., antibodies, aptamers) labeled with plasmonic nanoparticles of differing colors have been used to recruit the target analyte (Fig. 8(A)).140–142 The formation of a sandwich complex brought the two plasmonic nanoparticles close for plasmon coupling to occur, resulting in enhanced scattering and spectral shifts of the scattered light from both nanoparticles. Alternatively, the difference in colors between uncoated and coated plasmonic nanoparticles can be leveraged to signal target binding. For instance, gold nanoparticles that were coated with manganese oxide (MnO2) have been used as a probe to detect alkaline phosphatase (ALP) enzyme143 or its inhibitor.144 Both assays exploited the enzymatic activity of ALP which generated a reducing agent to decompose the MnO2 shell and consequently shifted the color of the scattered light.
 | ||
| Fig. 7 Scattering based detection. (A) Bright-field microscopy. (B) Dark-field microscopy. (C) Interferometry imaging. (D) Photothermal-enhanced scattering imaging. | ||
 | ||
| Fig. 8 Dark field microscopy (DFM) for scattering-based digital detection. (A) Plasmon-enhanced scattering for colorimetric detection of extracellular vesicles on the surface. Adapted with permission from Macmillan Publishers Ltd. Nature Biomedical Engineering, ref. 141, copyright 2017. (B) Dynamic detection based on single particle motion analysis within discrete microwells. Adapted with permission from ref. 145. Copyright 2019 American Chemical Society. | ||
Non-colorimetric detection using DFM has been demonstrated through the analysis of the particle's Brownian motion (Fig. 8(B)).145 Detection-antibody-coated magnetic nanoparticles recruited the target molecules and were subsequently pulled magnetically into individual microwells. The capture antibodies were conjugated at the microwell bottom through long flexible linkers. Upon binding, the target-bound nanoparticle underwent a Brownian motion that was distinct from both freely diffusing and non-specifically bound nanoparticles. Detection based on Brownian motion analysis has also been realized using a different strategy without microwells.146 Specifically, detection-antibody-coated microbeads were tethered to a capture-antibody-coated glass surface via a long flexible linker. The binding of the target analyte created a sandwich complex (detection antibody–target–capture antibody), thereby largely restricting the microbead movement.
A total internal reflection system can be incorporated into dark-field microscopy (TIRDFM) to reduce out-of-plane scattering and enhance image contrast. Using TIRDFM, the label-free detection of a single influenza virus has been demonstrated.147 Additional modifications to a TIRDFM set-up have also been employed to further suppress residual stray light. For instance, the placing of a circular stop after an objective has been utilized to further exclude illumination light to achieve an extreme background rejection ratio of >107 and successful imaging of single gold nanoparticles down to 10 nm in size.148 In another work, two micromirrors were used to couple the illumination light into the objective and redirect the total-internally reflected illumination light away from the detection optics to suppress the background signal.149 The system has demonstrated ultrafast tracking (temporal resolution ∼10 μs) of gold nanoparticles down to 20 nm in size. Although these set-ups have only been applied in particle imaging, they show promising capability for digital molecular analysis. Furthermore, even though the detection of nanoparticle-labeled targets is challenging at high concentrations due to the light diffraction limit, the use of machine learning has shown promising performance in resolving individual nanoparticles from DFM images.150
Typically, the detection of unlabeled biomolecules in solution is challenging due to their small scattering cross-section and rapid Brownian motion which prevent sufficient signal accumulation for detection. More recently, label-free detection of single biomolecule (>66 kDa) using DFM has been conceived by the incorporation of a nanofluidic device.151 Named nanofluidic scattering microscopy (NSM), this device works by allowing the molecules to diffuse within a nanochannel to restrict their motion within the microscope's focal plane. To reveal the weakly scattering molecule, the image of an empty nanochannel was subtracted from the images taken in the presence of the molecule. In addition, the extraction of additional molecular information such as the analyte's molecular weight based on scattering contrast, as well as hydrodynamic radius and molecular conformation based on diffusivity, have been demonstrated.
3.1.1.2. Interferometry imaging. Interferometry-based microscopes such as interferometric scattering microscopy (iSCAT), interferometric plasmonic microscopy (iPM) and evanescent scattering systems (ESMs) construct an image from the interference between a reference light and the analyte scattering (Fig. 7(C)).152–155 For iSCAT, part of the illumination light reflected/transmitted at the glass/water interface is used as the reference. In iPM, plasmonic material is coated on the glass surface for signal enhancement, and surface plasmonic waves are generated when illumination comes at a particular angle, which acts as an evanescent illumination (thickness 100–200 nm) to minimize background scattering. An image is constructed from the interference between the reflected plasmonic wave and the scattered light from the target. As the plasmonic field generates heat at a high power of illumination, the iPM is not suitable for imaging temperature-sensitive analytes. To tackle this issue, ESM was developed. It relies on the interference between the evanescent scattering by the surface roughness of the cover glass and the target. High-intensity illumination can be used to enhance the signal without causing significant heating.
To reduce the influence of background scattering, image processing can be applied to subtract the image of the background, which is separately taken in the absence of the target. Alternatively, if a moving particle is analyzed, the background can be reduced by subtracting the previous frame. In interferometry imaging, the scattering signal has a linear dependence on the particle's volume, hence, molecular weight;156–158 therefore molecular binding events can be detected by analyzing the enhancement in signal contrast. Typically, when the target analytes are captured by a probe-functionalized surface, high-contrast spots appear (Fig. 9(A)). This has enabled specific and label-free detection of single proteins,154,159,160 exosomes,153 and virus particles161 in complex samples. Young and Hundt et al. leveraged the mass dependency of scattering contrast to gauge the molecular weight of single proteins between 50 and 800 kDa with high precision (∼2%);158 hence, single-molecule identification can be achieved probe-free. For small molecules (<40 kDa), detection is challenging due to their weak scattering signal. However, the highly precise and sensitive mass determination enabled the detection of small molecules (>1 kDa) when they bind with a large probe.158 Alternatively, machine learning can be employed to extract target molecules in low-SNR images, enabling the accurate detection of proteins down to 9 kDa.162 Given their strong scattering, plasmonic nanoparticles have also been used to label small molecules. For instance, one design relied on a gold nanoparticle-conjugated DNA probe that was functionalized on-surface for miRNA detection, based on the change of its motion pattern upon miRNA binding (Fig. 9(B)).154
 | ||
| Fig. 9 Interferometric digital detection. (A) Specific detection of IgA binding events on an anti-IgA-functionalized surface. Adapted with permission from Macmillan Publishers Ltd. Nature Methods, ref. 160, copyright 2020. (B) miRNA detection by motion analysis of a gold nanoparticle label. Adapted from ref. 154 according to Creative Commons Attribution 4.0 International License (CC BY 4.0). | ||
The high spatiotemporal resolution of the interferometric system has also enabled video-based detection by analyzing the lifetime of molecular binding events.163 Specific binding events which have longer lifetime than non-specific binding events can be accurately discerned, giving a LOD of sub-fM. To ensure sufficient binding events occur at low target concentrations by improving mass transport, Yu and co-workers leveraged electrical and magnetic manipulation techniques to accelerate the movement of detection probe-conjugated gold nanoparticles and detection probe-conjugated magnetic nanoparticles, respectively. Video-based analysis also permits binding affinity determination by counting the binding and unbinding events, as well as single virus tracking at <3 nm spatial resolution and μs temporal resolution.154,161 As the binding of a virus to its detection probe reduces its diffusion mobility, motion analysis has been applied to unambiguously identify the specific binding events.164 Ma et al. demonstrated that, in the presence of an alternating electric field, monitoring the change of the induced oscillation of surface-tethered antibody gave information about the binding, size, and charge of the target protein.165 Specifically, the antibody was tethered to the surface via a flexible polymer linker, and a fast Fourier Transform (FFT) was applied on each pixel of the interferometric image to derive the oscillation amplitude of the antibody–target complex. The hydrodynamic diameter and charge of the protein can be determined based on the relationship between oscillation amplitude and electric field intensity. Similarly, the binding/unbinding of ions (e.g., Ca2+) with a surface-tethered protein was resolved based on monitoring the charge alteration.
While the application of PTM as an opto-digital analysis tool has not yet been explored much, it can be advantageous to enhance detection specificity. Different molecules have characteristic infrared absorption wavelengths due to the presence of characteristic chemical groups. Scattering can be selectively enhanced by matching the heating light with the absorption peak of the target molecule. Using this strategy, the differentiation of drug molecules, lipids and proteins has been demonstrated.166 Additionally, by probing the photothermally enhanced scattering signal across a range of wavelengths, signal fingerprints unique to different types of viruses have been obtained, enabling their specific detection in a probe-free manner.169
3.2. Raman scattering
Given the low probability (∼1 in 108 photons) of Raman scattering events, a highly sensitive detector, long integration time and a high-power excitation source are typically required to detect the weak Raman signals. To improve the detection sensitivity, Raman signals can be amplified by several orders of magnitude using metallic nanostructures via a surface-enhanced Raman scattering (SERS) effect. Specifically, the oscillation of surface electrons excited by incident photons creates a strong local electromagnetic field, which in turn enhances Raman scattering of nearby molecules.171 The electromagnetic enhancement effect by plasmonic nanostructures is anisotropic, meaning certain regions (known as hot spots, such as sharp edges or nanogaps) can generate a tremendously high field strength which even enables signal detection at a single molecule level.Conventional SERS detection is based on target concentration-dependent ensemble-averaged measurement. It typically suffers from poor reproducibility due to the signal fluctuations arising from non-uniform hot spot distribution and differences in the position or orientation of the target molecules. Digitization addresses these limitations by counting the signals above the threshold. Digital detection can be achieved by mapping the Raman spectrum across a surface to identify surface-captured target molecules (static detection), typically using a confocal Raman microscope. At low concentrations, each pixel (<1 μm) is occupied by none or one target molecule. A binary read-out is obtained at each pixel by applying an intensity threshold for a characteristic Raman peak (wavenumber) originated from the target or the reporter molecule. Freely diffusing target molecules can be detected digitally as they pass through the confocal detection volume (dynamic detection).
Due to their small Raman scattering cross-sections and complex chemical structures, direct analysis of proteins and nucleic acids is challenging. Hence, detection is typically achieved using an easily identifiable molecule with strong Raman scattering as a reporter, such as Nile Blue, Rhodamine B, Rhodamine 6G, etc. Plasmonic nanoparticles decorated with a Raman reporter and detection probes have been used as nanotags to generate strong and uniform hot spots around the bound target protein.172–175 Capture probes are typically immobilized on a plasmonic metal-coated surface for further signal enhancement. Capture probes can also be immobilized on nanopillar arrays, which confine each target molecule within a discrete pixel (Fig. 10(A)).172,174,176 This kind of digital SERS assay design has enabled the detection of cytokines and viral proteins down to the sub-fM level,172,173,175 small EVs down to 12 particles μL−1,174 as well as dopamine down to the pM level.176 By conjugating unique Raman reporters with specific detection antibodies on the nanoparticles, multiplexed detection of proteins has been demonstrated (Fig. 10(A)).172,174 To enhance the detection specificity, a dual reporter system has been devised.177 Specifically, the capture aptamer-functionalized plasmonic surface and detection aptamer-conjugated plasmonic nanoparticle were functionalized with different Raman reporters. Only the colocalized signals from both reporters were counted as positive events.
 | ||
| Fig. 10 Digital SERS detection. (A) Static digital detection of cytokines captured on nanopillars, with multiplexed detection enabled by SERS nanotags made of different Raman reporter molecules. Adapted from ref. 172 according to Creative Commons Attribution 4.0 International License (CC BY 4.0). (B) Dynamic digital SERS within a capillary using metallic nanocolloids. Adapted with permission from Macmillan Publishers Ltd. Nature, ref. 178, copyright 2024. | ||
Label-free protein detection has been achieved using nanocavities coated with metal–insulator–metal film, which generated hot spots within the nanocavity in both longitudinal and transverse directions, resulting in more significant field enhancement.179 In addition, the nanocavities can compartmentalize individual proteins. Label-free digital SERS detection has also been reported for larger targets such as exosomes,180,181 yeasts,182 and bacteria.183,184 An array of nanopyramids were fabricated to generate uniform hot spots.180,181 Microcavities with an inverted pyramidal shape have also been etched on plasmonic surfaces to isolate single particles, thereby avoiding miscounting due to aggregation or variation in particle size.182,184 Biomolecules exhibit complex and overlapping Raman signals. Machine learning has been employed to decipher the complex Raman fingerprints from the targets.181,185
Dynamic digital SERS detection recently emerged. Specifically, a flow cell has been designed to allow the target to flow on the surface of a plasmonic nanostructured substrate.186,187 The Raman signatures of the target were detected as it passed by the detection volume. To generate a strong hot spot precisely at the detection volume, a manipulatable nanogap has been constructed in solution using optical tweezers.188 In particular, two microbeads coated with silver nanoparticles were positioned at the detection volume, separated by a nanogap through which the target molecule could pass through. A nanocavity has also been constructed from three silver nanoparticles using optical tweezers for further electromagnetic field enhancement.189 Nanocolloids have also been used as hot spot generators (Fig. 10(B)).178 Specifically, the target molecule together with silver nanocolloids were flowed through a capillary. Detection was done at multiple voxels along the capillary, and the characteristic signal above the threshold was counted as a positive event.
4. Electronic digital molecular analytics
Although this article focuses on optical methods, digital detection can also be electronically realized. For example, nanopore technology enables digital electrical read-out of single molecules because the transient or permanent presence of a molecule inside the nanopore causes a measurable blockage of ionic current.190–192 As a nanopore can only be occupied by one molecule at a time, the concentration of the target molecule can be determined by simply counting from the blockage events from an array of nanopores. A nanopore even allows digital read-out of the molecular sequence of a nucleic acid or protein while they are electrically driven through the nanopore because their different monomers (nucleotides or amino acids) generate characteristic current blockage signals.193–195 The analysis from an array of nanopores increases the throughput and accuracy. Considering the challenges involving unfolding the proteins for sequencing and their non-uniform charge distribution, a DNA barcoded probe has been used to capture the target protein and act as a specific label whose sequence can be read by the nanopore.196 Nanopore sensors can also reveal additional molecular information such as size, charge, binding affinity and conformation.197 Aside from nanopores, electrical detection of single nucleic acids, proteins and small molecules has also been realized based on the conductance change of a molecular junction (a single-molecule wide gap between two electrodes).198,199 Specifically, the capture probe is used to bridge the two electrodes, permitting a current flow under a voltage bias. The binding of the target molecule to the probe results in a detectable current change. Alternatively, the capture probes are functionalized on both electrodes, and a current arises upon target binding. It has also been demonstrated that electrolyte-gated organic-field-effect-transistors (EGOFET) can attain the ultimate LOD (single molecule).200,201 In principle, digital analysis can be realized using an array of EGOFET.As compared to optical methods, electronic detection does not require bulky equipment and the detection signal is more stable. But electronic detection is often only applicable to charged molecules, and the number of sensing units in the array is limited because of the intimidating complexity for electronics when the number of the multiplexing channels goes beyond several hundred. This greatly limits the detection range and the information on molecular heterogeneity. In contrast, an enormous number of pixels can be analyzed by opto-digital methods.
5. Discussion and outlook
Significant research efforts have been devoted to breaking the limits of conventional molecular analytic methods, driven by the persistent demand in healthcare to detect, quantify, and analyze the biomarkers and pathogens existing in trace concentrations. Opto-digital analytics offers superior performance in terms of LOD, spatiotemporal resolution and specificity compared with the conventional ensemble averaging-based methods. Opto-digital biosensors also enable the interrogation of sample heterogeneity, high throughput analyses with small sample volume and ease of multiplex detection, and the possibility to provide additional molecular information such as molecular weight, conformation, binding affinity with its probe, and charge.Opto-digital molecular analytics are based on either photon emission or scattering. Compared to scattering-based methods, emission-based techniques equipped with advanced optics (high-power laser source and sensitive detector) generally provide higher SNR because of the lower background interference. Benchtop fluorescence microscopes may be sufficient for emission-based digital detection when signal amplification or enhancement strategies are rationally devised, albeit at the price of a more tedious workflow and non-trivial fabrication of micro- or nano-devices. Additionally, emission-based detection is more amenable to multiplexed detection due to the wide variety of fluorescent probes available. It is also more amenable to miniaturization due to the smaller background interference, hence less demanding requirement for complex optics. On the other hand, with advanced image processing strategies, scattering-based techniques permit label-free probing of the presence of single molecules and their inherent properties without or with minimal sample preparation. Moreover, long-term observation without photobleaching is enabled. In addition to emission and scattering-based read-outs, other optical signals could be used for digital detection. Very recently, label-free digital detection based on transmission and reflection was reported.202 Single proteins down to 1.2 kDa were detected within a high-finesse fibre-based Fabry–Pérot microcavity, which can confine light waves to enhance light–molecule interactions. The presence of proteins within the optical cavity induces on-resonance temperature changes; and thermo-optic resonance shifts are amplified by Pound–Drever–Hall (PDH) frequency locking to give high SNR.
The development of opto-digital analytics must address two key considerations, namely: (i) digitizing the detection, and (ii) reliable distinction of individual binding events from the background (sensitivity) and non-specific binding (specificity). Digitization can be realized either by physically partitioning the binding and amplification reactions into discrete compartments (e.g., microwells, microdroplets) or by using a microscope with a sufficiently high spatiotemporal resolution to resolve single molecules directly. For the former, the development of one-pot reaction systems and automated microfluidic devices can greatly simplify the workflow; while for the latter, machine learning has been increasingly relied on for more robust image acquisition, single-molecule localization and analysis.203,204 Specifically, machine learning can automate image acquisition by finding the correct focal plane from a stack of images with different focus depths (autofocus) and determining the suitable frame of view. And single molecules can be localized from background interference and photon fluctuations based on signal-time trajectory. A machine learning algorithm can also be devised for automated post-acquisition image processing, such as contrast enhancement, and image analysis to obtain molecular information.
High sensitivity can be achieved by signal enhancement/amplification, background suppression/subtraction, or their combination. The ultimate sensitivity is limited by the shot noise due to random fluctuations of photons collected by the detector. Research has shown the possibility of obtaining sub-shot noise resolution using quantum imaging and machine learning for effective denoising.205–207 Specificity is ensured by target-selective probes (e.g., antibodies, aptamers). It can be improved by using multiple recognition probes, analyzing binding kinetics and motion dynamics, and extracting specific molecular information. Continuous effort should be made to design novel probes with enhanced binding affinity, better stability, and even the ability to catalyze signal amplification.
Some challenges in the development of ultrasensitive opto-digital diagnostic tools should be highlighted. Firstly, due to the light diffraction limit, optical microscopes can spatially resolve individual molecules only when they are separated >200 nm apart in a diluted sample. This limits the dynamic range of detection. The issue can be partially circumvented by combining digital detection for the low-concentration range with ensemble-averaged measurement for the high-concentration range. A more complex data processing and analysis algorithm is however required. Super-resolution fluorescence microscopy powered by special optical designs and computational algorithms has emerged to break the diffraction limit, but its practicality for molecular digital analysis is hampered by the extravagant cost. In contrast to ensemble-averaged measurement, ideally opto-digital detection methods should be able to report the exact number of the target through direct counting without the need for a calibration curve. For most assays, however, only a portion of the target analytes are detected due to the association–dissociation equilibrium between the target and the probe, hence necessitating a prior-established calibration curve based on the standard samples. In principle, direct counting could be realized through a probe-free detection strategy, for instance, by directly probing the intrinsic properties of the analyte such as size, molecular weight, charge, mobility and characteristic chemical groups. Ensemble-averaged detection methods without relying on recognition probes have been demonstrated based on target-triggered chemical reactions to transduce a readable signal.208,209 Conceivably, similar strategies could be designed for opto-digital analytics.
In addition, there are some practical limitations of opto-digital analytics. At low target concentrations, locating the scarce target molecules within the limited field-of-view becomes challenging. For image-based analysis, hundreds of frames may need to be taken to locate sufficient numbers of molecules and achieve an acceptable level of precision. This also applies to video-based analysis, which necessitates a long observation time to detect a sufficient number of molecules passing through the small detection volume, or sufficient number of binding events on the surface. Some ways to overcome the diffusion-limited detection include pre-concentrating the sample,109 and using molecule manipulation techniques to actively bring the target molecules to the sensing interface.123,163 Another consideration is the sample volume. At 1 aM, only 0.6 molecules are present per μL, thus several hundred μLs may be required to avoid false negative readings. While this does not pose an issue to assays that can handle large sample volumes (e.g., bead-based assays with flow cytometry detection, on-surface wide-field detection), microchip-based methods, which typically can only handle a few μLs, demand repetitive assays or sample pre-concentration before assay on-chip. Alternatively, with continuous perfusion of the sample solution through the microfluidic channel, the target molecules can be enriched in the detection chamber by surface-immobilized capture antibodies prior to imaging.
Several opto-digital sensors have been successfully commercialized and demonstrated excellent utility for research and healthcare,11,109,210 while some proof-of-concept developments show promising potential for practical use. Looking forward, there are several considerations for future advances. Firstly, the balance consideration between sensitivity, time-to-result and cost is required, as often there exists a trade-off between these attributes. Secondly, the ease of deployability in common healthcare and even POC settings can be achieved by devising lab-on-chip devices,48,65,211 smartphone-based analysis,63,212 and miniaturization of optical systems.117,213 Furthermore, it is desirable to develop versatile platforms that can detect a wide range of analytes (proteins, nucleic acids, small biomolecules, extracellular vesicles, pathogens, etc.) in complex biofluids. In view of all the challenges, it is apparent that collaboration between multiple disciplines is indispensable to bring exciting breakthroughs to this exciting emerging field which will bring enormous benefits to medical diagnostics, precision medicine, drug discovery, environmental monitoring, and molecular science.
Author contributions
CVS, YX and PC conceived this review article and planned its contents. The manuscript was mainly written by CVS and PC with valuable inputs from TL, YX and DL.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by IDMxS (Institute for Digital Molecular Analytics and Science) funded by Singapore Ministry of Education (MOE) under the Research Centres of Excellence scheme, an AcRF Tier 2 grant (MOE-T2EP30124-0010) from Singapore MOE, and the 111 Project (D20015) from Hubei Province (China).References
- Y. Tao, L. Chen, M. Pan, F. Zhu and D. Zhu, ACS Sens., 2021, 6, 3146–3162 Search PubMed.
- L. Wu, Y. Wang, X. Xu, Y. Liu, B. Lin, M. Zhang, J. Zhang, S. Wan, C. Yang and W. Tan, Chem. Rev., 2021, 121, 12035–12105 CrossRef CAS PubMed.
- M. R. Wasielewski, M. D. E. Forbes, N. L. Frank, K. Kowalski, G. D. Scholes, J. Yuen-Zhou, M. A. Baldo, D. E. Freedman, R. H. Goldsmith, T. Goodson, M. L. Kirk, J. K. McCusker, J. P. Ogilvie, D. A. Shultz, S. Stoll and K. B. Whaley, Nat. Rev. Chem., 2020, 4, 490–504 CrossRef CAS PubMed.
- Y. Zhang, Y. Zhu, Z. Zeng, G. Zeng, R. Xiao, Y. Wang, Y. Hu, L. Tang and C. Feng, Coord. Chem. Rev., 2021, 431, 213681 CrossRef CAS.
- C. D. Flynn, D. Chang, A. Mahmud, H. Yousefi, J. Das, K. T. Riordan, E. H. Sargent and S. O. Kelley, Nat. Rev. Bioeng., 2023, 1, 560–575 Search PubMed.
- G. A. Kwong, S. Ghosh, L. Gamboa, C. Patriotis, S. Srivastava and S. N. Bhatia, Nat. Rev. Cancer, 2021, 21, 655–668 CrossRef CAS PubMed.
- J. M. Barletta, D. C. Edelman and N. T. Constantine, Am. J. Clin. Pathol., 2004, 122, 20–27 Search PubMed.
- D. A. Giljohann and C. A. Mirkin, Nature, 2009, 462, 461–464 CrossRef CAS PubMed.
- K. Blennow and H. Zetterberg, Nat. Med., 2015, 21, 217–219 CrossRef CAS PubMed.
- V. Riitho, R. Connon, A. Gwela, J. Namusanje, R. Nhema, A. Siika, M. Bwakura-Dangarembizi, V. Musiime, J. A. Berkley, A. J. Szubert, D. M. Gibb, A. S. Walker, N. Klein and A. J. Prendergast, Nat. Commun., 2024, 15, 5492 CrossRef CAS PubMed.
- D. M. Rissin, C. W. Kan, T. G. Campbell, S. C. Howes, D. R. Fournier, L. Song, T. Piech, P. P. Patel, L. Chang, A. J. Rivnak, E. P. Ferrell, J. D. Randall, G. K. Provuncher, D. R. Walt and D. C. Duffy, Nat. Biotechnol., 2010, 28, 595–599 CrossRef CAS PubMed.
- Y. Wu, R. D. Tilley and J. J. Gooding, J. Am. Chem. Soc., 2019, 141, 1162–1170 Search PubMed.
- J. J. Gooding and K. Gaus, Angew. Chem., Int. Ed., 2016, 55, 11354–11366 CrossRef CAS PubMed.
- L. Priest, J. S. Peters and P. Kukura, Chem. Rev., 2021, 121, 11937–11970 CrossRef CAS PubMed.
- B. Vogelstein and K. W. Kinzler, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9236–9241 CrossRef CAS PubMed.
- L. Warren, D. Bryder, I. L. Weissman and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17807–17812 CrossRef CAS PubMed.
- H. C. Fan and S. R. Quake, Anal. Chem., 2007, 79, 7576–7579 CrossRef CAS PubMed.
- H. Nagai, Y. Murakami, Y. Morita, K. Yokoyama and E. Tamiya, Anal. Chem., 2001, 73, 1043–1047 CrossRef CAS PubMed.
- H. Nagai, Y. Murakami, K. Yokoyama and E. Tamiya, Biosens. Bioelectron., 2001, 16, 1015–1019 CrossRef CAS PubMed.
- T. Morrison, J. Hurley, J. Garcia, K. Yoder, A. Katz, D. Roberts, J. Cho, T. Kanigan, S. E. Ilyin, D. Horowitz, J. M. Dixon and C. J. H. Brenan, Nucleic Acids Res., 2006, 34, e123 CrossRef PubMed.
- E. A. Ottesen, J. W. Hong, S. R. Quake and J. R. Leadbetter, Science, 2006, 314, 1464–1467 CrossRef CAS PubMed.
- K. A. Heyries, C. Tropini, M. VanInsberghe, C. Doolin, O. I. Petriv, A. Singhal, K. Leung, C. B. Hughesman and C. L. Hansen, Nat. Methods, 2011, 8, 649–651 CrossRef CAS PubMed.
- E. T. Lagally, I. Medintz and R. A. Mathies, Anal. Chem., 2001, 73, 565–570 CrossRef CAS PubMed.
- D. M. Rissin, D. R. Fournier, T. Piech, C. W. Kan, T. G. Campbell, L. Song, L. Chang, A. J. Rivnak, P. P. Patel, G. K. Provuncher, E. P. Ferrell, S. C. Howes, B. A. Pink, K. A. Minnehan, D. H. Wilson and D. C. Duffy, Anal. Chem., 2011, 83, 2279–2285 CrossRef CAS PubMed.
- R. A. White, P. C. Blainey, H. C. Fan and S. R. Quake, BMC Genomics, 2009, 10, 116 CrossRef PubMed.
- N. R. Beer, B. J. Hindson, E. K. Wheeler, S. B. Hall, K. A. Rose, I. M. Kennedy and B. W. Colston, Anal. Chem., 2007, 79, 8471–8475 Search PubMed.
- B. J. Hindson, K. D. Ness, D. A. Masquelier, P. Belgrader, N. J. Heredia, A. J. Makarewicz, I. J. Bright, M. Y. Lucero, A. L. Hiddessen, T. C. Legler, T. K. Kitano, M. R. Hodel, J. F. Petersen, P. W. Wyatt, E. R. Steenblock, P. H. Shah, L. J. Bousse, C. B. Troup, J. C. Mellen, D. K. Wittmann, N. G. Erndt, T. H. Cauley, R. T. Koehler, A. P. So, S. Dube, K. A. Rose, L. Montesclaros, S. Wang, D. P. Stumbo, S. P. Hodges, S. Romine, F. P. Milanovich, H. E. White, J. F. Regan, G. A. Karlin-Neumann, C. M. Hindson, S. Saxonov and B. W. Colston, Anal. Chem., 2011, 83, 8604–8610 CrossRef CAS PubMed.
- Z. Li, Y. Liu, Q. Wei, Y. Liu, W. Liu, X. Zhang and Y. Yu, PLoS One, 2016, 11, e0153359 CrossRef PubMed.
- S. Santiago-Felipe, L. A. Tortajada-Genaro, R. Puchades and Á. Maquieira, Microchim. Acta, 2016, 183, 1195–1202 CrossRef CAS.
- A. Gansen, A. M. Herrick, I. K. Dimov, L. P. Lee and D. T. Chiu, Lab Chip, 2012, 12, 2247–2254 RSC.
- Q. Zhu, Y. Gao, B. Yu, H. Ren, L. Qiu, S. Han, W. Jin, Q. Jin and Y. Mu, Lab Chip, 2012, 12, 4755–4763 RSC.
- L. Cao, X. Guo, P. Mao, Y. Ren, Z. Li, M. You, J. Hu, M. Tian, C. Yao, F. Li and F. Xu, ACS Sens., 2021, 6, 3564–3574 CrossRef CAS PubMed.
- H. Wu, X. Cao, Y. Meng, D. Richards, J. Wu, Z. Ye and A. J. deMello, Biosens. Bioelectron., 2022, 211, 114377 CrossRef CAS PubMed.
- Z. Yu, L. Xu, W. Lyu and F. Shen, Lab Chip, 2022, 22, 2954–2961 RSC.
- X. Wu, C. Chan, S. L. Springs, Y. H. Lee, T. K. Lu and H. Yu, Anal. Chim. Acta, 2022, 1196, 339494 CrossRef CAS PubMed.
- X. Wu, J. K. Tay, C. K. Goh, C. Chan, Y. H. Lee, S. L. Springs, D. Y. Wang, K. S. Loh, T. K. Lu and H. Yu, Biomaterials, 2021, 274, 120876 CrossRef CAS PubMed.
- J. S. Park, K. Hsieh, L. Chen, A. Kaushik, A. Y. Trick and T.-H. Wang, Adv. Sci., 2021, 8, 2003564 CrossRef CAS PubMed.
- J. Q. Cui, F. X. Liu, H. Park, K. W. Chan, T. Leung, B. Z. Tang and S. Yao, Biosens. Bioelectron., 2022, 202, 114019 CrossRef CAS PubMed.
- F. X. Liu, J. Q. Cui, H. Park, K. W. Chan, T. Leung, B. Z. Tang and S. Yao, Anal. Chem., 2022, 94, 5883–5892 CrossRef CAS PubMed.
- T. Tian, B. Shu, Y. Jiang, M. Ye, L. Liu, Z. Guo, Z. Han, Z. Wang and X. Zhou, ACS Nano, 2021, 15, 1167–1178 CrossRef CAS PubMed.
- H. Shinoda, Y. Taguchi, R. Nakagawa, A. Makino, S. Okazaki, M. Nakano, Y. Muramoto, C. Takahashi, I. Takahashi, J. Ando, T. Noda, O. Nureki, H. Nishimasu and R. Watanabe, Commun. Biol., 2021, 4, 1–7 CrossRef PubMed.
- H. Yue, B. Shu, T. Tian, E. Xiong, M. Huang, D. Zhu, J. Sun, Q. Liu, S. Wang, Y. Li and X. Zhou, Nano Lett., 2021, 21, 4643–4653 CrossRef CAS PubMed.
- D. Wang, X. Wang, F. Ye, J. Zou, J. Qu and X. Jiang, ACS Nano, 2023, 17, 7250–7256 CrossRef CAS PubMed.
- K. Wu, Q. Fang, Z. Zhao and Z. Li, Anal. Chem., 2023, 95, 5069–5078 CrossRef CAS PubMed.
- S.-M. Yang, Q. Bi, W. J. Zhang, X. Cui, Y. Zhou, C. Yuan and Y. Cui, Lab Chip, 2022, 22, 3015–3024 RSC.
- C. W. Kan, C. I. Tobos, D. M. Rissin, A. D. Wiener, R. E. Meyer, D. M. Svancara, A. Comperchio, C. Warwick, R. Millington, N. Collier and D. C. Duffy, Lab Chip, 2020, 20, 2122–2135 RSC.
- L. Chang, D. M. Rissin, D. R. Fournier, T. Piech, P. P. Patel, D. H. Wilson and D. C. Duffy, J. Immunol. Methods, 2012, 378, 102–115 CrossRef CAS PubMed.
- D. Witters, K. Knez, F. Ceyssens, R. Puers and J. Lammertyn, Lab Chip, 2013, 13, 2047–2054 RSC.
- S. H. Kim, S. Iwai, S. Araki, S. Sakakihara, R. Iino and H. Noji, Lab Chip, 2012, 12, 4986–4991 RSC.
- D. Decrop, G. Pardon, L. Brancato, D. Kil, R. Zandi Shafagh, T. Kokalj, T. Haraldsson, R. Puers, W. van der Wijngaart and J. Lammertyn, ACS Appl. Mater. Interfaces, 2017, 9, 10418–10426 CrossRef CAS PubMed.
- D. Kim, O. B. Garner, A. Ozcan and D. Di Carlo, ACS Nano, 2016, 10, 7467–7475 CrossRef CAS PubMed.
- W. Wu, B. T. T. Nguyen, P. Y. Liu, G. Cai, S. Feng, Y. Shi, B. Zhang, Y. Hong, R. Yu, X. Zhou, A. Q. Liu, Y. Zhang, E. P. H. Yap and L. K. Chin, Biosens. Bioelectron., 2022, 215, 114594 CrossRef CAS PubMed.
- H. Ao, W. Chen, J. Wu, W. Xiao and H. Ju, Commun. Biol., 2022, 5, 1–9 CrossRef PubMed.
- K. Hosokawa, K. Sato, N. Ichikawa and M. Maeda, Lab Chip, 2004, 4, 181–185 RSC.
- K. Hosokawa, M. Omata, K. Sato and M. Maeda, Lab Chip, 2006, 6, 236–241 RSC.
- I. K. Dimov, L. Basabe-Desmonts, J. L. Garcia-Cordero, B. M. Ross, A. J. Ricco and L. P. Lee, Lab Chip, 2011, 11, 845–850 RSC.
- Q. Zhu, L. Qiu, B. Yu, Y. Xu, Y. Gao, T. Pan, Q. Tian, Q. Song, W. Jin, Q. Jin and Y. Mu, Lab Chip, 2014, 14, 1176–1185 RSC.
- Q. Song, Y. Gao, Q. Zhu, Q. Tian, B. Yu, B. Song, Y. Xu, M. Yuan, C. Ma, W. Jin, T. Zhang, Y. Mu and Q. Jin, Biomed. Microdevices, 2015, 17, 64 CrossRef PubMed.
- X. Cui, L. Wu, Y. Wu, J. Zhang, Q. Zhao, F. Jing, L. Yi and G. Li, Anal. Chim. Acta, 2020, 1107, 127–134 CrossRef CAS PubMed.
- X. Cui, T. Hu, Q. Chen, Q. Zhao, Y. Wu, T. Xie, P. Liu, X. Su and G. Li, Talanta, 2021, 233, 122589 CrossRef CAS PubMed.
- G. Xu, H. Si, F. Jing, P. Sun and D. Wu, Biosensors, 2021, 11, 158 CrossRef CAS PubMed.
- C.-J. Lee and Y.-H. Hsu, Microfluid. Nanofluid., 2022, 26, 1–13 CrossRef.
- T. Gou, J. Hu, W. Wu, X. Ding, S. Zhou, W. Fang and Y. Mu, Biosens. Bioelectron., 2018, 120, 144–152 CrossRef CAS PubMed.
- E.-C. Yeh, C.-C. Fu, L. Hu, R. Thakur, J. Feng and L. P. Lee, Sci. Adv., 2017, 3, e1501645 CrossRef PubMed.
- F. Shen, W. Du, J. E. Kreutz, A. Fok and R. F. Ismagilov, Lab Chip, 2010, 10, 2666–2672 RSC.
- L. Xu, H. Qu, D. G. Alonso, Z. Yu, Y. Yu, Y. Shi, C. Hu, T. Zhu, N. Wu and F. Shen, Biosens. Bioelectron., 2021, 175, 112908 CrossRef CAS PubMed.
- Z. Yu, W. Lyu, M. Yu, Q. Wang, H. Qu, R. F. Ismagilov, X. Han, D. Lai and F. Shen, Biosens. Bioelectron., 2020, 155, 112107 CrossRef CAS PubMed.
- A. H. Kunding, L. L. Busk, H. Webb, H. W. Klafki, M. Otto, J. P. Kutter and M. Dufva, Lab Chip, 2018, 18, 2797–2805 RSC.
- Z. Guan, Y. Zou, M. Zhang, J. Lv, H. Shen, P. Yang, H. Zhang, Z. Zhu and C. James Yang, Biomicrofluidics, 2014, 8, 014110 CrossRef PubMed.
- F. Schuler, M. Trotter, M. Geltman, F. Schwemmer, S. Wadle, E. Domínguez-Garrido, M. López, C. Cervera-Acedo, P. Santibáñez, F. von Stetten, R. Zengerle and N. Paust, Lab Chip, 2015, 16, 208–216 RSC.
- L. Cohen, N. Cui, Y. Cai, P. M. Garden, X. Li, D. A. Weitz and D. R. Walt, ACS Nano, 2020, 14, 9491–9501 CrossRef CAS PubMed.
- Y. Zhu, Y.-X. Zhang, W.-W. Liu, Y. Ma, Q. Fang and B. Yao, Sci. Rep., 2015, 5, 9551 CrossRef PubMed.
- Y. Pan, T. Ma, Q. Meng, Y. Mao, K. Chu, Y. Men, T. Pan, B. Li and J. Chu, Talanta, 2020, 211, 120680 CrossRef CAS PubMed.
- W. Bu, W. Li, J. Li, T. Ao, Z. Li, B. Wu, S. Wu, W. Kong, T. Pan, Y. Ding, W. Tan, B. Li, Y. Chen and Y. Men, Sens. Actuators, B, 2021, 348, 130678 CrossRef CAS.
- Y. Sun, X. Zhou and Y. Yu, Lab Chip, 2014, 14, 3603–3610 RSC.
- Z. Fan, Y. Sun and J.-M. Lin, Chemosensors, 2022, 10, 247 CrossRef CAS.
- W. Zhang, N. Li, D. Koga, Y. Zhang, H. Zeng, H. Nakajima, J.-M. Lin and K. Uchiyama, Anal. Chem., 2018, 90, 5329–5334 CrossRef CAS PubMed.
- V. Yelleswarapu, J. R. Buser, M. Haber, J. Baron, E. Inapuri and D. Issadore, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4489–4495 CrossRef CAS PubMed.
- S. A. Byrnes, T. Huynh, T. C. Chang, C. E. Anderson, J. J. McDermott, C. I. Oncina, B. H. Weigl and K. P. Nichols, Anal. Chem., 2020, 92, 3535–3543 CrossRef CAS PubMed.
- E. Y. Shum, J. H. Lai, S. Li, H. G. Lee, J. Soliman, V. K. Raol, C. K. Lee, S. P. A. Fodor and H. C. Fan, Anal. Chem., 2022, 94, 17868–17876 CrossRef CAS PubMed.
- A. J. Rivnak, D. M. Rissin, C. W. Kan, L. Song, M. W. Fishburn, T. Piech, T. G. Campbell, D. R. DuPont, M. Gardel, S. Sullivan, B. A. Pink, C. G. Cabrera, D. R. Fournier and D. C. Duffy, J. Immunol. Methods, 2015, 424, 20–27 CrossRef CAS PubMed.
- D. H. Wilson, D. M. Rissin, C. W. Kan, D. R. Fournier, T. Piech, T. G. Campbell, R. E. Meyer, M. W. Fishburn, C. Cabrera, P. P. Patel, E. Frew, Y. Chen, L. Chang, E. P. Ferrell, V. von Einem, W. McGuigan, M. Reinhardt, H. Sayer, C. Vielsack and D. C. Duffy, SLAS Technol., 2016, 21, 533–547 CrossRef CAS PubMed.
- Y. Bai, Y. Qu, Z. Wu, Y. Ren, Z. Cheng, Y. Lu, J. Hu, J. Lou, J. Zhao, C. Chen and H. Mao, Biosens. Bioelectron., 2019, 142, 111523 CrossRef CAS PubMed.
- H. Yin, Z. Wu, N. Shi, Y. Qi, X. Jian, L. Zhou, Y. Tong, Z. Cheng, J. Zhao and H. Mao, Biosens. Bioelectron., 2021, 188, 113282 CrossRef CAS PubMed.
- Y.-L. Tan, A.-Q. Huang, L.-J. Tang and J.-H. Jiang, Chem. Sci., 2021, 12, 8445–8451 RSC.
- J. W. Choi, W. H. Seo, T. Kang, T. Kang and B. G. Chung, Lab Chip, 2023, 23, 2389–2398 RSC.
- J. Yi, Z. Gao, Q. Guo, Y. Wu, T. Sun, Y. Wang, H. Zhou, H. Gu, J. Zhao and H. Xu, Sens. Actuators, B, 2022, 369, 132214 CrossRef CAS.
- K. Akama, K. Shirai and S. Suzuki, Anal. Chem., 2016, 88, 7123–7129 CrossRef CAS PubMed.
- J. Park, M. Park, J. Kim, Y. Heo, B. H. Han, N. Choi, C. Park, R. Lee, D.-G. Lee, S. Chung and J. Y. Kang, Biosens. Bioelectron., 2023, 232, 115316 CrossRef CAS PubMed.
- Q. Ruan, P. J. Macdonald, K. M. Swift and S. Y. Tetin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2025033118 CrossRef CAS PubMed.
- A. M. Maley, P. M. Garden and D. R. Walt, ACS Sens., 2020, 5, 3037–3042 CrossRef CAS PubMed.
- C. Wu, P. M. Garden and D. R. Walt, J. Am. Chem. Soc., 2020, 142, 12314–12323 CrossRef CAS PubMed.
- K. Ito, K. Y. Inoue, K. Ino and H. Shiku, Biosens. Bioelectron.: X, 2023, 13, 100312 CAS.
- M. Schwarzkopf, M. C. Liu, S. J. Schulte, R. Ives, N. Husain, H. M. T. Choi and N. A. Pierce, Dev. Camb. Engl., 2021, 148, dev199847 CAS.
- J. Wu, J. Lv, X. Zheng and Z.-S. Wu, Talanta, 2021, 234, 122637 CrossRef CAS PubMed.
- S. Shah, E. Lubeck, M. Schwarzkopf, T.-F. He, A. Greenbaum, C. H. Sohn, A. Lignell, H. M. T. Choi, V. Gradinaru, N. A. Pierce and L. Cai, Development, 2016, 143, 2862–2867 CrossRef CAS PubMed.
- H. M. T. Choi, M. Schwarzkopf, M. E. Fornace, A. Acharya, G. Artavanis, J. Stegmaier, A. Cunha and N. A. Pierce, Development, 2018, 145, dev165753 CrossRef PubMed.
- J. Jarvius, J. Melin, J. Göransson, J. Stenberg, S. Fredriksson, C. Gonzalez-Rey, S. Bertilsson and M. Nilsson, Nat. Methods, 2006, 3, 725–727 CrossRef CAS PubMed.
- C. Wu, T. J. Dougan and D. R. Walt, ACS Nano, 2022, 16, 1025–1035 CrossRef CAS PubMed.
- L. Chen, A. Eriksson, S. Weström, T. Pandzic, S. Lehmann, L. Cavelier and U. Landegren, Nat. Commun., 2022, 13, 4033 CrossRef CAS PubMed.
- J. Björkesten, S. Patil, C. Fredolini, P. Lönn and U. Landegren, Nucleic Acids Res., 2020, 48, e73 Search PubMed.
- C.-M. Clausson, L. Arngården, O. Ishaq, A. Klaesson, M. Kühnemund, K. Grannas, B. Koos, X. Qian, P. Ranefall, T. Krzywkowski, H. Brismar, M. Nilsson, C. Wählby and O. Söderberg, Sci. Rep., 2015, 5, 12317 CrossRef PubMed.
- M. Kühnemund, I. Hernández-Neuta, M. I. Sharif, M. Cornaglia, M. A. M. Gijs and M. Nilsson, Nucleic Acids Res., 2017, 45, e59 Search PubMed.
- S. Ciftci, F. Neumann, S. Abdurahman, K. S. Appelberg, A. Mirazimi, M. Nilsson and N. Madaboosi, J. Mol. Diagn., 2020, 22, 272–283 CrossRef CAS PubMed.
- S. J. Zhang, C. Wu and D. R. Walt, ACS Nano, 2024, 18, 29891–29901 CrossRef CAS PubMed.
- Z. Li, K. Guo, Z. Gao, J. Chen, Z. Ye, M. Cao, S. E. Wang, Y. Yin and W. Zhong, Sci. Adv., 2024, 10, eadh8689 CrossRef CAS PubMed.
- W. Shen, K. Guo, G. B. Adkins, Q. Jiang, Y. Liu, S. Sedano, Y. Duan, W. Yan, S. E. Wang, K. Bergersen, D. Worth, E. H. Wilson and W. Zhong, Angew. Chem., Int. Ed., 2018, 57, 15675–15680 CrossRef CAS PubMed.
- G. Krainer, K. L. Saar, W. E. Arter, T. J. Welsh, M. A. Czekalska, R. P. B. Jacquat, Q. Peter, W. C. Traberg, A. Pujari, A. K. Jayaram, P. Challa, C. G. Taylor, L.-M. van der Linden, T. Franzmann, R. M. Owens, S. Alberti, D. Klenerman and T. P. J. Knowles, Nat. Commun., 2023, 14, 653 CrossRef CAS PubMed.
- J. Todd, B. Freese, A. Lu, D. Held, J. Morey, R. Livingston and P. Goix, Clin. Chem., 2007, 53, 1990–1995 CrossRef CAS PubMed.
- A. A. Hariri, S. S. Newman, S. Tan, D. Mamerow, A. M. Adams, N. Maganzini, B. L. Zhong, M. Eisenstein, A. R. Dunn and H. T. Soh, Nat. Commun., 2022, 13, 5359 CrossRef CAS PubMed.
- A. Johnson-Buck, X. Su, M. D. Giraldez, M. Zhao, M. Tewari and N. G. Walter, Nat. Biotechnol., 2015, 33, 730–732 CrossRef CAS PubMed.
- H. Li, L. Ying, J. J. Green, S. Balasubramanian and D. Klenerman, Anal. Chem., 2003, 75, 1664–1670 CrossRef CAS PubMed.
- A. Agrawal, C. Zhang, T. Byassee, R. A. Tripp and S. Nie, Anal. Chem., 2006, 78, 1061–1070 CrossRef CAS PubMed.
- H. Zhang, Y. Liu, K. Zhang, J. Ji, J. Liu and B. Liu, Anal. Chem., 2018, 90, 9315–9321 CrossRef CAS PubMed.
- A. Agrawal, R. Deo, G. D. Wang, M. D. Wang and S. Nie, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3298–3303 CrossRef CAS PubMed.
- J. Liu, X. Yang, K. Wang, Q. Wang, W. Liu and D. Wang, Nanoscale, 2013, 5, 11257–11264 RSC.
- T. Chatterjee, A. Knappik, E. Sandford, M. Tewari, S. W. Choi, W. B. Strong, E. P. Thrush, K. J. Oh, N. Liu, N. G. Walter and A. Johnson-Buck, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 22815–22822 CrossRef CAS PubMed.
- S. L. Hayward, P. E. Lund, Q. Kang, A. Johnson-Buck, M. Tewari and N. G. Walter, J. Am. Chem. Soc., 2018, 140, 11755–11762 CrossRef CAS PubMed.
- G. Krainer, R. P. B. Jacquat, M. M. Schneider, T. J. Welsh, J. Fan, Q. A. E. Peter, E. A. Andrzejewska, G. Šneiderienė, M. A. Czekalska, H. Ausserwoeger, L. Chai, W. E. Arter, K. L. Saar, T. W. Herling, T. M. Franzmann, V. Kosmoliaptsis, S. Alberti, F. U. Hartl, S. F. Lee and T. P. J. Knowles, Nat. Commun., 2024, 15, 7740 CrossRef CAS PubMed.
- Q. Wang, R. H. Goldsmith, Y. Jiang, S. D. Bockenhauer and W. E. Moerner, Acc. Chem. Res., 2012, 45, 1955–1964 CrossRef CAS PubMed.
- J. Chu, A. Ejaz, K. M. Lin, M. R. Joseph, A. E. Coraor, D. A. Drummond and A. H. Squires, Nat. Nanotechnol., 2024, 19, 1150–1157 CrossRef CAS PubMed.
- Q. Zhang, J. Li, X. Pan, X. Liu and H. Gai, Anal. Chem., 2021, 93, 12848–12853 CrossRef CAS PubMed.
- Q. Zhang, W. Chai, X. Pan and H. Gai, Anal. Chem., 2024, 96, 3054–3061 CAS.
- X. Liu, X. Lin, X. Pan and H. Gai, Anal. Chem., 2022, 94, 5830–5837 CrossRef CAS PubMed.
- X. Liu, C. Huang, X. Dong, A. Liang, Y. Zhang, Q. Zhang, Q. Wang and H. Gai, Chem. Commun., 2018, 54, 13103–13106 RSC.
- X. Shi, S. Dong, M. Li, X. Liu, Q. Zhang, W. Zhao, C. Zong, Y. Zhang and H. Gai, Chem. Commun., 2015, 51, 2353–2356 RSC.
- J. B. Khurgin and G. Sun, J. Opt. Soc. Am. B, 2009, 26, B83–B95 CrossRef CAS.
- A. Seth, Y. Liu, R. Gupta, Z. Wang, E. Mittal, S. Kolla, P. Rathi, P. Gupta, B. A. Parikh, G. M. Genin, S. Gandra, G. A. Storch, J. A. Philips, I. A. George and S. Singamaneni, Nano Lett., 2024, 24, 229–237 CrossRef CAS PubMed.
- P. Barya, Y. Xiong, S. Shepherd, R. Gupta, L. D. Akin, J. Tibbs, H. Lee, S. Singamaneni and B. T. Cunningham, Small, 2023, 19, 2207239 CrossRef CAS PubMed.
- A. Minopoli, B. Della Ventura, B. Lenyk, F. Gentile, J. A. Tanner, A. Offenhäusser, D. Mayer and R. Velotta, Nat. Commun., 2020, 11, 6134 CrossRef CAS PubMed.
- K. Ray, R. Badugu, H. Szmacinski and J. R. Lakowicz, Chem. Commun., 2015, 51, 15023–15026 RSC.
- D. Verardo, B. Agnarsson, V. P. Zhdanov, F. Höök and H. Linke, Nano Lett., 2019, 19, 6182–6191 CrossRef CAS PubMed.
- A. Barulin, J.-B. Claude, S. Patra, N. Bonod and J. Wenger, Nano Lett., 2019, 19, 7434–7442 CrossRef CAS PubMed.
- H. Cai, J. W. Parks, T. A. Wall, M. A. Stott, A. Stambaugh, K. Alfson, A. Griffiths, R. A. Mathies, R. Carrion, J. L. Patterson, A. R. Hawkins and H. Schmidt, Sci. Rep., 2015, 5, 14494 CrossRef CAS PubMed.
- P. Zhu and H. G. Craighead, Annu. Rev. Biophys., 2012, 41, 269–293 CrossRef CAS PubMed.
- T. Plénat, S. Yoshizawa and D. Fourmy, ACS Appl. Mater. Interfaces, 2017, 9, 30561–30566 CrossRef PubMed.
- T. Tanii, R. Akahori, S. Higano, K. Okubo, H. Yamamoto, T. Ueno and T. Funatsu, Phys. Rev. E, 2013, 88, 012727 CrossRef PubMed.
- S. Yang, N. Klughammer, A. Barth, M. E. Tanenbaum and C. Dekker, ACS Nano, 2023, 17, 20179–20193 CrossRef CAS PubMed.
- M. P. Goldschen-Ohm, D. S. White, V. A. Klenchin, B. Chanda and R. H. Goldsmith, Angew. Chem., Int. Ed., 2017, 56, 2399–2402 CrossRef CAS PubMed.
- C.-Y. Poon, L. Wei, Y. Xu, B. Chen, L. Xiao and H.-W. Li, Anal. Chem., 2016, 88, 8849–8856 CrossRef CAS PubMed.
- K. Liang, F. Liu, J. Fan, D. Sun, C. Liu, C. J. Lyon, D. W. Bernard, Y. Li, K. Yokoi, M. H. Katz, E. J. Koay, Z. Zhao and Y. Hu, Nat. Biomed. Eng., 2017, 1, 1–11 CrossRef.
- J. Li, Y. Jiao, Q. Liu and Z. Chen, Anal. Chim. Acta, 2018, 1028, 66–76 CrossRef CAS PubMed.
- F. Wang, Y. Li, Y. Han, Z. Ye, L. Wei, H.-B. Luo and L. Xiao, Anal. Chem., 2019, 91, 6329–6339 CrossRef CAS PubMed.
- M. Huang, Y. Fan, X. Yuan and L. Wei, Sens. Actuators, B, 2022, 353, 131135 CrossRef CAS.
- K. Akama, N. Iwanaga, K. Yamawaki, M. Okuda, K. Jain, H. Ueno, N. Soga, Y. Minagawa and H. Noji, ACS Nano, 2019, 13, 13116–13126 CrossRef CAS PubMed.
- E. W. A. Visser, J. Yan, L. J. van IJzendoorn and M. W. J. Prins, Nat. Commun., 2018, 9, 2541 CrossRef PubMed.
- S. Enoki, R. Iino, N. Morone, K. Kaihatsu, S. Sakakihara, N. Kato and H. Noji, PLoS One, 2012, 7, e49208 CrossRef CAS PubMed.
- A. Weigel, A. Sebesta and P. Kukura, ACS Photonics, 2014, 1, 848–856 CrossRef CAS.
- X. Meng, A. Sonn-Segev, A. Schumacher, D. Cole, G. Young, S. Thorpe, R. W. Style, E. R. Dufresne and P. Kukura, ACS Photonics, 2021, 8, 3111–3118 CrossRef CAS PubMed.
- M. K. Song, Y. P. Ma, H. Liu, P. P. Hu, C. Z. Huang and J. Zhou, Anal. Chem., 2022, 94, 4610–4616 CrossRef CAS PubMed.
- B. Špačková, H. Klein Moberg, J. Fritzsche, J. Tenghamn, G. Sjösten, H. Šípová-Jungová, D. Albinsson, Q. Lubart, D. van Leeuwen, F. Westerlund, D. Midtvedt, E. K. Esbjörner, M. Käll, G. Volpe and C. Langhammer, Nat. Methods, 2022, 19, 751–758 CrossRef PubMed.
- R. W. Taylor and V. Sandoghdar, Nano Lett., 2019, 19, 4827–4835 CrossRef CAS PubMed.
- Y. Yang, G. Shen, H. Wang, H. Li, T. Zhang, N. Tao, X. Ding and H. Yu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10275–10280 CrossRef CAS PubMed.
- P. Zhang, L. Zhou, R. Wang, X. Zhou, J. Jiang, Z. Wan and S. Wang, Nat. Commun., 2022, 13, 2298 CrossRef CAS PubMed.
- X. Zhou, A. Chieng and S. Wang, ACS Sens., 2024, 9, 543–554 CrossRef CAS PubMed.
- J. Ortega-Arroyo and P. Kukura, Phys. Chem. Chem. Phys., 2012, 14, 15625–15636 RSC.
- M. Piliarik and V. Sandoghdar, Nat. Commun., 2014, 5, 4495 CrossRef CAS PubMed.
- G. Young, N. Hundt, D. Cole, A. Fineberg, J. Andrecka, A. Tyler, A. Olerinyova, A. Ansari, E. G. Marklund, M. P. Collier, S. A. Chandler, O. Tkachenko, J. Allen, M. Crispin, N. Billington, Y. Takagi, J. R. Sellers, C. Eichmann, P. Selenko, L. Frey, R. Riek, M. R. Galpin, W. B. Struwe, J. L. P. Benesch and P. Kukura, Science, 2018, 360, 423–427 CrossRef CAS PubMed.
- D. Cole, G. Young, A. Weigel, A. Sebesta and P. Kukura, ACS Photonics, 2017, 4, 211–216 CrossRef CAS PubMed.
- P. Zhang, G. Ma, W. Dong, Z. Wan, S. Wang and N. Tao, Nat. Methods, 2020, 17, 1010–1017 CrossRef CAS PubMed.
- Y.-F. Huang, G.-Y. Zhuo, C.-Y. Chou, C.-H. Lin, W. Chang and C.-L. Hsieh, ACS Nano, 2017, 11, 2575–2585 CrossRef CAS PubMed.
- M. Dahmardeh, H. Mirzaalian Dastjerdi, H. Mazal, H. Köstler and V. Sandoghdar, Nat. Methods, 2023, 20, 442–447 CrossRef CAS PubMed.
- Q. Zeng, X. Zhou, Y. Yang, Y. Sun, J. Wang, C. Zhai, J. Li and H. Yu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2120379119 CrossRef CAS PubMed.
- S. Alhaddad, H. Bey, O. Thouvenin, P. Boulanger, C. Boccara, M. Boccara and I. Izeddin, Biophys. Rep., 2023, 3, 100119 CAS.
- G. Ma, Z. Wan, Y. Yang, P. Zhang, S. Wang and N. Tao, Nat. Commun., 2020, 11, 4768 CrossRef CAS PubMed.
- D. Zhang, C. Li, C. Zhang, M. N. Slipchenko, G. Eakins and J.-X. Cheng, Sci. Adv., 2016, 2, e1600521 CrossRef PubMed.
- Q. Li, Z. Shi, L. Wu and H. Wei, Nanoscale, 2020, 12, 8397–8403 RSC.
- S. Berciaud, L. Cognet, G. A. Blab and B. Lounis, Phys. Rev. Lett., 2004, 93, 257402 CrossRef PubMed.
- Y. Zhang, C. Yurdakul, A. J. Devaux, L. Wang, X. G. Xu, J. H. Connor, M. S. Ünlü and J.-X. Cheng, Anal. Chem., 2021, 93, 4100–4107 CrossRef CAS PubMed.
- P. Zijlstra, P. M. R. Paulo and M. Orrit, Nat. Nanotechnol., 2012, 7, 379–382 CrossRef CAS PubMed.
- L. A. Lane, X. Qian and S. Nie, Chem. Rev., 2015, 115, 10489–10529 CrossRef CAS PubMed.
- J. Li, A. Wuethrich, A. A. I. Sina, H.-H. Cheng, Y. Wang, A. Behren, P. N. Mainwaring and M. Trau, Nat. Commun., 2021, 12, 1087 CrossRef CAS PubMed.
- J.-E. Shim, Y. J. Kim, J.-H. Choe, T. G. Lee and E.-A. You, ACS Appl. Mater. Interfaces, 2022, 14, 38459–38470 CrossRef CAS PubMed.
- J. Li, A. A. I. Sina, F. Antaw, D. Fielding, A. Möller, R. Lobb, A. Wuethrich and M. Trau, Adv. Sci., 2023, 10, 2204207 CrossRef CAS PubMed.
- A. Li, X. Mo, Y. Lu, G. Zhu, C. Liu, X. Yang, Y. Huang, J. Sheng, H. Zhang, D. Meng and X. Zhao, Biosens. Bioelectron., 2025, 270, 116973 CrossRef CAS PubMed.
- W. Nam, W. Kim, W. Zhou and E.-A. You, Nanoscale, 2021, 13, 17340–17349 RSC.
- T. T. Chuong, A. Pallaoro, C. A. Chaves, Z. Li, J. Lee, M. Eisenstein, G. D. Stucky, M. Moskovits and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9056–9061 CrossRef CAS PubMed.
- X. Bi, D. M. Czajkowsky, Z. Shao and J. Ye, Nature, 2024, 628, 771–775 CrossRef CAS PubMed.
- D. Li, W. Yue, Q. He, P. Gao, T. Gong, Y. Luo, C. Wang and X. Luo, Talanta, 2024, 278, 126494 CrossRef CAS PubMed.
- Z. Yan, S. Dutta, Z. Liu, X. Yu, N. Mesgarzadeh, F. Ji, G. Bitan and Y.-H. Xie, ACS Sens., 2019, 4, 488–497 CrossRef CAS PubMed.
- Z. Liu, M. Ng, S. Srivastava, T. Li, J. Liu, T. A. Phu, B. Mateescu, Y.-T. Wang, C.-F. Tsai, T. Liu, R. L. Raffai and Y.-H. Xie, PLoS One, 2024, 19, e0305418 CrossRef CAS PubMed.
- P. Wen, F. Yang, H. Zhao, Y. Xu, S. Li and L. Chen, Anal. Chem., 2024, 96, 1454–1461 CrossRef CAS PubMed.
- D. Yang, H. Zhou, C. Haisch, R. Niessner and Y. Ying, Talanta, 2016, 146, 457–463 CrossRef CAS PubMed.
- P. Wen, F. Yang, H. Zhao, S. Li, Y. Xu and L. Chen, ACS Sens., 2024, 9, 6167–6173 CrossRef CAS PubMed.
- H. Shin, B. H. Choi, O. Shim, J. Kim, Y. Park, S. K. Cho, H. K. Kim and Y. Choi, Nat. Commun., 2023, 14, 1644 CrossRef CAS PubMed.
- L. Xiao, C. Wang, C. Dai, L. E. Littlepage, J. Li and Z. D. Schultz, Angew. Chem., Int. Ed., 2020, 59, 3439–3443 CrossRef CAS PubMed.
- H. C. Schorr and Z. D. Schultz, Analyst, 2024, 149, 3711–3715 RSC.
- X. Dai, W. Fu, H. Chi, V. S. D. Mesias, H. Zhu, C. W. Leung, W. Liu and J. Huang, Nat. Commun., 2021, 12, 1292 CrossRef CAS PubMed.
- W. Fu, H. Chi, X. Dai, H. Zhu, V. S. D. Mesias, W. Liu and J. Huang, Nat. Commun., 2023, 14, 6996 CrossRef CAS PubMed.
- L. Xue, H. Yamazaki, R. Ren, M. Wanunu, A. P. Ivanov and J. B. Edel, Nat. Rev. Mater., 2020, 5, 931–951 CrossRef CAS.
- K. Chuah, Y. Wu, S. R. C. Vivekchand, K. Gaus, P. J. Reece, A. P. Micolich and J. J. Gooding, Nat. Commun., 2019, 10, 2109 CrossRef PubMed.
- Y. Wu, Y. Yao, S. Cheong, R. D. Tilley and J. J. Gooding, Chem. Sci., 2020, 11, 12570–12579 RSC.
- A. Sauciuc, B. Morozzo della Rocca, M. J. Tadema, M. Chinappi and G. Maglia, Nat. Biotechnol., 2023, 1–7 Search PubMed.
- H. Brinkerhoff, A. S. W. Kang, J. Liu, A. Aksimentiev and C. Dekker, Science, 2021, 374, 1509–1513 CrossRef CAS PubMed.
- F. L. R. Lucas, R. C. A. Versloot, L. Yakovlieva, M. T. C. Walvoort and G. Maglia, Nat. Commun., 2021, 12, 5795 CrossRef CAS PubMed.
- C. Koch, B. Reilly-O’Donnell, R. Gutierrez, C. Lucarelli, F. S. Ng, J. Gorelik, A. P. Ivanov and J. B. Edel, Nat. Nanotechnol., 2023, 18, 1483–1491 CrossRef CAS PubMed.
- N. Varongchayakul, J. Song, A. Meller and M. W. Grinstaff, Chem. Soc. Rev., 2018, 47, 8512–8524 RSC.
- E. M. Dief, P. J. Low, I. Díez-Pérez and N. Darwish, Nat. Chem., 2023, 15, 600–614 CrossRef CAS PubMed.
- Y. Li, J. M. Artés, B. Demir, S. Gokce, H. M. Mohammad, M. Alangari, M. P. Anantram, E. E. Oren and J. Hihath, Nat. Nanotechnol., 2018, 13, 1167–1173 CrossRef CAS PubMed.
- E. Macchia, K. Manoli, B. Holzer, C. Di Franco, M. Ghittorelli, F. Torricelli, D. Alberga, G. F. Mangiatordi, G. Palazzo, G. Scamarcio and L. Torsi, Nat. Commun., 2018, 9, 3223 CrossRef PubMed.
- E. Macchia, L. Sarcina, C. Driescher, Z. Gounani, A. Tewari, R. Osterbacka, G. Palazzo, A. Tricase, Z. M. Kovacs Vajna, F. Viola, F. Modena, M. Caironi, F. Torricelli, I. Esposito and L. Torsi, Adv. Electron. Mater., 2021, 7, 2100304 CrossRef CAS.
- L.-M. Needham, C. Saavedra, J. K. Rasch, D. Sole-Barber, B. S. Schweitzer, A. J. Fairhall, C. H. Vollbrecht, S. Wan, Y. Podorova, A. J. Bergsten, B. Mehlenbacher, Z. Zhang, L. Tenbrake, J. Saimi, L. C. Kneely, J. S. Kirkwood, H. Pfeifer, E. R. Chapman and R. H. Goldsmith, Nature, 2024, 629, 1062–1068 CrossRef CAS PubMed.
- X. Liu, Y. Jiang, Y. Cui, J. Yuan and X. Fang, Chem. Sci., 2022, 13, 11964–11980 RSC.
- X. Feng, C. Zhai, J. Xu, Y. Yang and H. Yu, View, 2023, 4, 20230004 CrossRef.
- G. Brida, M. Genovese and I. Ruo Berchera, Nat. Photonics, 2010, 4, 227–230 CrossRef CAS.
- E. Knyazev, F. Y. Khalili and M. V. Chekhova, Opt. Express, 2019, 27, 7868–7885 CrossRef CAS PubMed.
- X. Li, Y. Li, Y. Zhou, J. Wu, Z. Zhao, J. Fan, F. Deng, Z. Wu, G. Xiao, J. He, Y. Zhang, G. Zhang, X. Hu, X. Chen, Y. Zhang, H. Qiao, H. Xie, Y. Li, H. Wang, L. Fang and Q. Dai, Nat. Biotechnol., 2023, 41, 282–292 CrossRef CAS PubMed.
- Y. Xiong, X. Zeng, L. Yan, Y. Wang, Y. Lin, K. Ao, P. Feng, Y. Xie and P. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 27687–27695 CrossRef CAS PubMed.
- O. Rifaie-Graham, J. Pollard, S. Raccio, S. Balog, S. Rusch, M. A. Hernández-Castañeda, P.-Y. Mantel, H.-P. Beck and N. Bruns, Nat. Commun., 2019, 10, 1369 CrossRef PubMed.
- C. I. Tobos, S. Kim, D. M. Rissin, J. M. Johnson, S. Douglas, S. Yan, S. Nie, B. Rice, K.-J. Sung, H. D. Sikes and D. C. Duffy, J. Immunol. Methods, 2019, 474, 112643 CrossRef CAS PubMed.
- J. Shim, R. T. Ranasinghe, C. A. Smith, S. M. Ibrahim, F. Hollfelder, W. T. S. Huck, D. Klenerman and C. Abell, ACS Nano, 2013, 7, 5955–5964 CrossRef CAS PubMed.
- P. N. Hedde, ACS Sens., 2021, 6, 2654–2663 CrossRef CAS PubMed.
- L. Skolrood, Y. Wang, S. Zhang and Q. Wei, Sens. Actuators Rep., 2022, 4, 100063 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |