
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA surface chemistry perspective on SERS: revisiting the basics to push the field forward
Chiara
Deriu
 * and
Laura
Fabris
* and
Laura
Fabris
Department of Applied Science and Technology, Politecnico di Torino, 10129 Turin, Italy. E-mail: chiara.deriu@polito.it
First published on 26th March 2025
Abstract
Surfaces are well known to be complex entities that are extremely difficult to study, and any phenomenon that is related to them is consequently challenging to approach. Moving from the bulk to the nanoscale adds a further layer of complexity to the problem. Because SERS relies on surfaces at the nanoscale, a rigorous understanding of the chemical phenomena that concur in the observation of the SERS signal is still limited or disorganized at best. Specifically, the lack of understanding of the chemical properties of nanoparticle surfaces has direct consequences on the development of SERS-based devices, causing a widespread belief that SERS is an inherently unreliable and fundamentally irreproducible analytical technique. Herein, we discuss old and new literature from SERS and related fields to accompany the reader through a journey that explores the chemical nature and architecture of colloidal plasmonic nanoparticles as the most popular SERS-active surfaces. By examining the chemistry of the surface landscape of the most common SERS colloids and the thermodynamic equilibria that characterize it, we aim to paint a chemically realistic picture of what a SERS analyst deals with on a daily basis. Thus, our goal for this review is to provide a centralized compilation of key, state-of-the-art surface chemistry information that can guide the rational development of analytical protocols and contribute an additional path through which our community can continue to advance SERS as a reliable and robust analytical tool.
 Chiara Deriu | Chiara Deriu obtained her PhD in Chemistry from Florida International University (FIU), Miami, USA, in 2020, where she worked on tailoring the stabilization of nanomaterials for the detection of drugs by SERS. Following, she received a postdoctoral appointment at FIU to work on the computational modeling of adsorbates on bimetallic clusters. She then joined Politecnico di Torino (Italy) in September 2021, where she is a postdoctoral researcher. Her scientific interests lie at the intersection of physical chemistry and the analytical sciences, with particular attention to nanoscale surface chemistry and its effects on both nanofabrication and SERS analytical performance. |
 Laura Fabris | Laura Fabris received her BS/MS degree in Chemistry in 2001 from the University of Padova, where she was awarded a PhD in 2006. She was then a postdoc at the University of California Santa Barbara. In 2009 she moved to Rutgers University in the Department of Materials Science and Engineering as an Assistant and then Associate Professor. Upon receiving an ERC Consolidator Grant, she moved to Politecnico di Torino (Italy) in the Department of Applied Science and Technology where she is a Full Professor. Her research combines fundamental notions of nanomaterials chemistry and spectroscopy for the development of new technologies addressing important challenges in medicine, biology, and energy. |
1. Introduction
1.1 The origin of a bad reputation
Since its first observation in 19741 and explanation in 1977,2 surface enhanced Raman scattering (SERS) has dramatically evolved from being primarily studied on roughened metallic electrodes to becoming a phenomenon for which a plethora of enhancing nanostructured substrates is designed and developed. Within this timeframe, the mechanistic description of SERS has focused primarily on understanding signal enhancement, reaching a consensus on the coexistence of an electromagnetic mechanism (EM), based primarily on plasmon-induced generation of enhanced scattered electric fields at the metal–dielectric interface, and of a chemical mechanism (CM), in which charge transfer states, created upon energy level hybridization between the nanoparticle and the chemisorbed analyte, can be occupied and contribute to signal enhancement. For many years, the former of the two mechanisms has attracted the interest of the majority of SERS and plasmonics scientists—an interest perhaps fueled by the signal enhancement values obtainable with it, vastly surpassing those achievable through the chemical counterpart. In both cases, the events occurring at the nanoparticle surface are key to explaining the SERS effect, albeit for different reasons. In the EM, it is the interplay between the metal core and the dielectric outer environment to modulate the SERS signal intensity.3 On the other hand, in the simplest instance of the CM, it is the interaction between the Fermi level of the metal nanostructure and the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO, respectively) of the analyte that causes the signal enhancement.4 Still, both types of event are surface-confined and surface-modulated.After the initial years since its discovery, during which SERS has remained relegated to the physicist and physical-chemist optical table, at the onset of the new millennium SERS regained momentum as a promising, easy to implement analytical technique, capable of detecting in principle any analyte by simply mixing it with a nanoparticle suspension and an aggregating agent, such as an inorganic salt. This popularity upsurge and connected experimental approach, while producing readily achievable results and great enthusiasm, have also produced an unwanted effect. Because of the lack of control in nanoparticle aggregation that was achievable in those years, it was not possible to obtain reproducible results among different experiments, leading SERS to obtain a bad reputation for being an extremely unreliable and irreproducible technique.
This attribute could have been somehow warranted in those days; back then, the lack of control over the aggregation process was not the only challenge that SERS scientists had to face, as the fine control over colloidal synthesis as we know it today was still far from being achieved. At that time, our inability to reproducibly synthesize suitably homogeneous silver nanoparticles was such that, to still exploit the superior SERS enhancement properties of silver, a process commonly referred to as “silver growth” was in use. More controllable gold nanoparticles were synthesized first and utilized to mix the analyte in, after which a solution of AgNO3—the so-called silver growth—was reduced in situ to create a thick, uneven layer of metallic silver. While the silver coating did contribute to increasing the enhancement, the lack of control over the analyte entrapment during the silver growth contributed to a very high variability of the resulting SERS output.
Since those days, the field has seen impressive progress in many aspects, including unfathomable advances in the world of controlled substrate fabrication. Yet, SERS is still thought of as an analytical technique that is extremely unreliable and fundamentally irreproducible. Concomitantly, each of us can also quickly assess that within the scientific community or in industry, a very enthusiastic set of beliefs around SERS coexists with the previously mentioned pessimistic view: SERS is often also presented as an “academic sensation”, with alluringly low limits of detection that are used as the primary focus of many application papers, and claims that trace and ultratrace detection can be achieved by simply mixing analytes with any nanoparticle.
This duality is rather puzzling and invites us to dig deeper on what is causing such a different range of experiences and “attitudes” towards SERS. In our opinion, the common denominator of this wide range of experiences is something intrinsic to SERS, that is, its surface spectroscopic nature and its dependence on the adsorption or close proximity of analytes to plasmonic surfaces. The active involvement of surfaces and interfaces in SERS implies that the thermodynamics of the system will control whether an analyte will approach the surface and benefit from the electromagnetic field that enables signal enhancement, ultimately leading to its observation at trace concentrations. Therefore, SERS detection methods that blindly mix components (i.e., nanoparticles and analytes) together are bound to produce intermittently working analytical protocols—adsorption and chemistry at large are based on affinity, and affinity is not universal. The very same enthusiastic attitude towards SERS method development, or more frankly, a lack thereof, together with a progressive detachment from the surface-dependent nature of SERS itself, still fuels negative attitudes towards the technique, contributing to slowing down its progression as an established analytical tool.
1.2 The future: a new necessary interpretation and revival
Most publications on SERS focus on the exceptionally low limits of detection of a given target analyte or class of molecules, with little to no explication of which experimental conditions make those outstanding enhancements possible and why. So far in 2024, around 58% of the total number of publications on SERS have sensitivity as the main subject, while only 2.3% focus on the fundamental aspects of surface chemistry and analyte-metal adsorption (Fig. 1a). Despite the surging interest towards surfaces and their role in SERS, in some cases pushed by emerging hot topics in ultrafast spectroscopy that have exposed how the metal surface is never static but gets rearranged upon interaction with adsorbed molecules and light,5,6 or even without,7 in the last ten years there has been globally little change in the overall SERS literature trends (Fig. 1b). So, can a focus on surface chemistry and a back-to-basics approach to the fundamentals of signal production be the key to releasing SERS—especially direct SERS—from the “potential well” in which it is currently trapped? Our view is that this is indeed one of the key approaches that will promote the translation of SERS from academic laboratories to routine applications. | ||
| Fig. 1 Keyword frequencies (web of science core collection) within SERS publications (a) for the year 2024 (January–November) and (b) in the time window from 1990 to 2024. The inset shows a 174% increase of SERS publications having a focus on surface phenomena over the last decade. This increase, although significant, is minimal compared to the global SERS literature production. | ||
If we analyze direct SERS in the more general context of instrumental analysis, and we compare it to what is now considered the gold standard in analytical practice – i.e., mass spectrometry – some interesting considerations can be made. If we look at the history of mass spectrometry, we easily notice that what made the fortune of these instruments was their effective coupling with upstream separation techniques. In particular, the invention of electrospray ionization (ESI) in the 1980s8 enabled its efficient coupling with liquid chromatography, thus unleashing its full potential for targeted and untargeted derivatization-free analysis of a vast array of samples, including complex mixtures and biological specimens.9 An originally bulk, difficult to interpret technique became the reliable technique for routine analysis of complex samples in many application fields, from clinical diagnostics to therapeutic drug monitoring, from environmental analysis to forensics. While the parallel with mass spectrometry does not suggest that the same development solutions are valid for all analytical platforms, direct SERS, like mass spectrometry, is too a bulk analytical technique. Net of differences in SERS cross sections, anything that is adsorbed on the surface of the plasmonic substrate will produce an enhanced Raman signal, conflating in a single, often hard to interpret SERS spectrum. Conceptualizing (direct) SERS as a bulk technique suggests that not only it would greatly benefit from coupling with separation techniques, as already implemented in a number of applications,10–16 albeit not ubiquitously, but it also further suggests that insisting on the surface to selectively observe what we are interested in, and augmenting in this way the interpretability and the physical meaning of the SERS spectrum, can be an advantageous developmental solution towards its routinization and accessibility.
The necessity of a non-ambiguous interpretation of the spectral response is particularly important in all those applications that result in potentially life-altering decisions for individuals, such as clinical diagnostics and forensics, where SERS has often been projected to become a leading technology for routine analysis. In these cases, the likelihood of adoption of a new method over others that are already established also passes from the trust that the method itself can offer in terms of clarity of results interpretation and, of course, reproducibility. We believe that the latter is also strongly dependent on surface chemistry considerations, in that they control the analyte–substrate interaction. By intimately understanding the analyte-substrate interaction, knowledge on how to control it, and thus reliably reproduce it, can be achieved. Once more, it is evident that surface chemistry considerations and the thermodynamics of the SERS sample components are central to the implementation of a rational and robust SERS measurement protocol.
In this review, we would like to bring to the reader's attention old and new SERS literature through a surface chemistry lens. By revisiting established concepts and connecting them to current hot topics, we intend to contribute an additional path through which our community can continue to advance SERS as a reliable and robust analytical tool. Our review will accomplish this goal by guiding the reader through a journey into a colloidal SERS sample, exploring each component of its architecture: The plasmonic metal nanoparticle, the rich variety of possible adsorbed species that can populate its native, as-synthesized surface, the thermodynamic hierarchy of these species, and the changes that can occur at both the metal core and the adsorbed crown due to the nanofabrication process itself or the aging of the system. These elements will provide the essential colors that are needed to paint a realistic picture of the surfaces that we utilize in our analytical SERS applications, and together with them, a set of foundational methodological cornerstones with which to systematize and rationalize our SERS analytical protocol development. Because the field of SERS substrates is expanding to incorporate materials beyond the traditional plasmonic metals gold and silver,17,18 some brief considerations on the surface chemistry of inorganic semiconductors is also discussed at the end.
2. The anatomy of a colloidal plasmonic nanoparticle
SERS substrates are varied, and several extensive reviews have been written on the subject.19–26 Herein, we will limit our treatment of enhancing substrates to colloidal nanoparticles. Besides their popularity, the main reason for this focus is that the influence of surface phenomena is most prominent and relevant to the outcome of a SERS measurement when colloidal nanoparticles are used and measurements in solution are performed. As an example, colloidal stability issues often preclude surface property modifications such as ligand exchange, which might be indispensable to a given detection need, evidencing that it is first and foremost in colloidal systems that the surface plays a key role in our ability to carry out SERS analyses.By definition, colloidal nanoparticles are solid nanoscale particles that are dispersed in a solution medium thanks to the chemical features of their surface. A surface charge that is either developed in solution or acquired via the adsorption of charged species (vide infra), as well as the possible presence of adsorbed bulky species, provide electrostatic repulsions and/or steric hindrance, respectively, thus ensuring nanoparticle dispersion and stabilization. However, for the sake of an easy schematization, colloidal nanoparticles are often depicted as solid entities of a given shape, and ligands are seldom represented, except in those cases in which conjugation chemistry of functionalized nanoparticles must be explicitly shown (Fig. 2a and b, respectively). Another common representation that lays in between the two extremes involves adding a surface charge to the solid body entity (Fig. 2c). Regardless of the cartoon depiction chosen, these representations are far away from describing in detail the chemical species present and the events occurring on the surface.
 | ||
| Fig. 2 Commonly encountered depictions of metallic (M) colloidal nanoparticles. (a) Representation of colloidal nanoparticles as solid bodies of a given shape, e.g., a sphere. (b) Complex representation of a SERS nanotag (i.e., functionalized nanoparticle for indirect SERS) in which the conjugation chemistry is highlighted. (c) Representation of colloidal nanoparticles as solid bodies bearing a surface charge. | ||
The development of a surface charge at the solid/liquid interface may be caused by a variety of phenomena, and strongly depends on the nature of the surface itself, as represented in Fig. 3. For noble metal nanoparticles, depictions showing two distinguishable domains within the nanostructure—an inner region in which the metal is in its bulk, zero-valent state, and an outer, superficial layer in which the metal is positively charged (bottom drawing in Fig. 2c)—are encountered in the literature,27,28 suggesting that the charging mechanism in solution is nothing other than the intrinsic presence of a net charge given by the material itself, which attracts opposite charged species via electrostatic attraction. Such two-domain depictions are typically explained to result from an incomplete reduction of the metal precursor during synthesis. However, unless specific Au–Au(I) clusters are synthesized,29 one must recall that reduction in conditions of (large) excess of reducing agent are frequently adopted in plasmonic nanoparticle synthesis. Even when milder reducing agents (i.e., L-ascorbic acid) are utilized and incomplete reduction is experimentally ascertained, such as in the case of the early fabrication of gold nanorods,30 excess metal precursor is not found to constitute the outermost layer of the otherwise metallic nanoparticle.31,32 Rather, unreacted precursors have been reported to take part to nanoparticle growth via self-catalyzed and disproportionation reactions in various fabrication protocols,33,34 or react with other species in solution (e.g., AgCl35,36), rendering the presence of a stable, positively charged Au+ or Ag+ layer on the surface of the nanoparticle something unrealistic. Studies on the oxidation state of colloidal noble metal nanoparticles are sparse, but they all seem to agree that the surface of the metals is in their zero-valent states,31,32 and that chemisorption of charged species is the mechanism of surface charge formation in plasmonic colloidal sols (Fig. 3f).31,32 With the term chemisorption we here indicate species-specific adsorption in which the formation of the final surface complex (i.e., metal + capping ligand) is non-electrostatically driven.
 | ||
| Fig. 3 Schematic representation of possible solid-solution interface phenomena at the basis of the development of surface charge and onset of Coulombic repulsions: (a) ionization of acid groups constituting the surface; (b) ionization of basic group constituting the surface; (c) and (d) differential dissolution of sparingly soluble compounds; (e) isomorphous substitution in a surface; (f) specific adsorption of an ionic compound. Figure inspired by D.H. Everett.37 | ||
Plasmonic colloidal nanoparticles can thus remain suspended in a solution medium thanks to the species that populate their surfaces, providing electrostatic repulsions and/or steric hindrance for their dispersion and stabilization. Albeit certainly complicated to schematize in a figure, a more accurate representation should therefore include not only the metallic core of the nanoparticle ensemble, but also its immediate chemical environment and the main composition of the suspension medium. This entity, as a whole, is what interacts with our target analyte when we prepare our samples for SERS measurement. In the perspective of rationally designing colloidal substrates for SERS, it is therefore useful to think about these three components and how they interact with each other.
2.1 From bulk solution to adsorbed species at the solid/liquid interface: what is left after reaction completion?
The most common approach to the fabrication of colloidal plasmonic nanoparticles for SERS is bottom-up, wet chemical synthesis. In its simplest declination, this process typically involves the reduction of a precursor salt of one or more plasmonic metals (e.g., tetrachloroauric acid for gold and silver nitrate for silver) by a reducing agent of varying strength (e.g., sodium borohydride, sodium citrate, L-ascorbic acid, hydroxylamine, etc.). As with any redox reaction, the reduction of the precursor salt to metallic nanoparticles is accompanied by the oxidation of the reducing agent; however, this step is often neglected when accounting for the population of adsorbed and non-adsorbed species in colloidal sols, and the reducing agent is often assumed to cap the final nanoparticles—unchanged.An example of this assumption is given by the term “borohydride-capped” nanoparticles that is sometimes utilized to refer to sodium borohydride-reduced silver or gold nanoparticles;38–40 borohydride ions are oxidized during the reaction as they transfer electrons to the metal precursor species, reducing it. Moreover, borohydride ions are known to spontaneously decompose in water to produce several species of boron hydroxides [BHx(OH)4−x]−, while evolving molecular hydrogen.41–44 While such a decomposition process is self-limited due to the rate limiting effects provided by the alkaline environment produced by the spontaneously generated boron hydroxides,42,45,46 it is unlikely that it will cause the final plasmonic nanoparticle sol to be capped by unchanged borohydride ions. The concentrations at which sodium borohydride is utilized in plasmonic nanoparticle syntheses are quite low, in the order of millimoles to sub-millimoles per liter.36,47,48 Thus, the typical pH of borohydride-reduced colloidal nanoparticles is around 6 to 7, whereas the previously mentioned decomposition rate limiting alkaline conditions are generally reached at very basic pH (i.e., pH > 12–14), such as those reached in the presence of 8 M NaOH.46 In fact, at pH 7.0, the hydrogen evolution reaction has been reported to be 90% complete after only 5 minutes.49 Moreover, the presence of acids (e.g., gold precursor tetrachloroauric acid) or noble metal nanoparticles themselves are also known to be a factor accelerating the decomposition kinetics of the borohydride ions.44,49 Interestingly, recent density functional theory (DFT) studies with complementing experimental measurements43 have shown that also hydrated silver ions catalyze the decomposition of borohydride anions in water, and that the reduction of silver cations to silver nanoparticles actually occurs as a side-reaction to the decomposition process of sodium borohydride.
In defense of the belief that reducing agents cap the produced nanoparticles as unchanged species, not much attention has been invested by the SERS community into elucidating the oxidation products of wet chemical nanofabrication processes, either because they are traditionally deemed unimportant and information must be sought from other scientific areas (e.g., energy storage literature for sodium borohydride oxidation), or due to a lack of adequately sensitive surface characterization techniques. Keeping our discussion circumscribed to borohydride-reduced nanoparticles, the identification and measurement of boron capping species is a challenging task, due to the combined effects of their short-lived nature and their trace concentration. For this reason, among the boron hydroxides generated by the borohydride decomposition to hydrogen gas, only [B(OH)4]− and trace [BH(OH)3]− have been reported experimentally, although their formation passes through the short-lived intermediates [BH3(OH)]− and [BH2(OH)2]−.42,43,50 This difficulty may be further exacerbated by the characterization technique of choice, which could suffer from intrinsic limitations due to the small cross-section of the boron element, such as in the case of X-ray photoelectron spectroscopy (XPS).51 For example, other boron oxide compounds have been proposed to populate the surface of sodium borohydride-reduced gold nanoparticles, such as hydrated (Na+)BO2−, but their unambiguous confirmation by XPS is challenging.51,52 Lastly, a popular reference in the plasmonic nanoparticle literature reports the formation of diborane,53 but experimental evidence for its presence in aqueous solution upon silver nitrate reduction still needs to find a consensus.
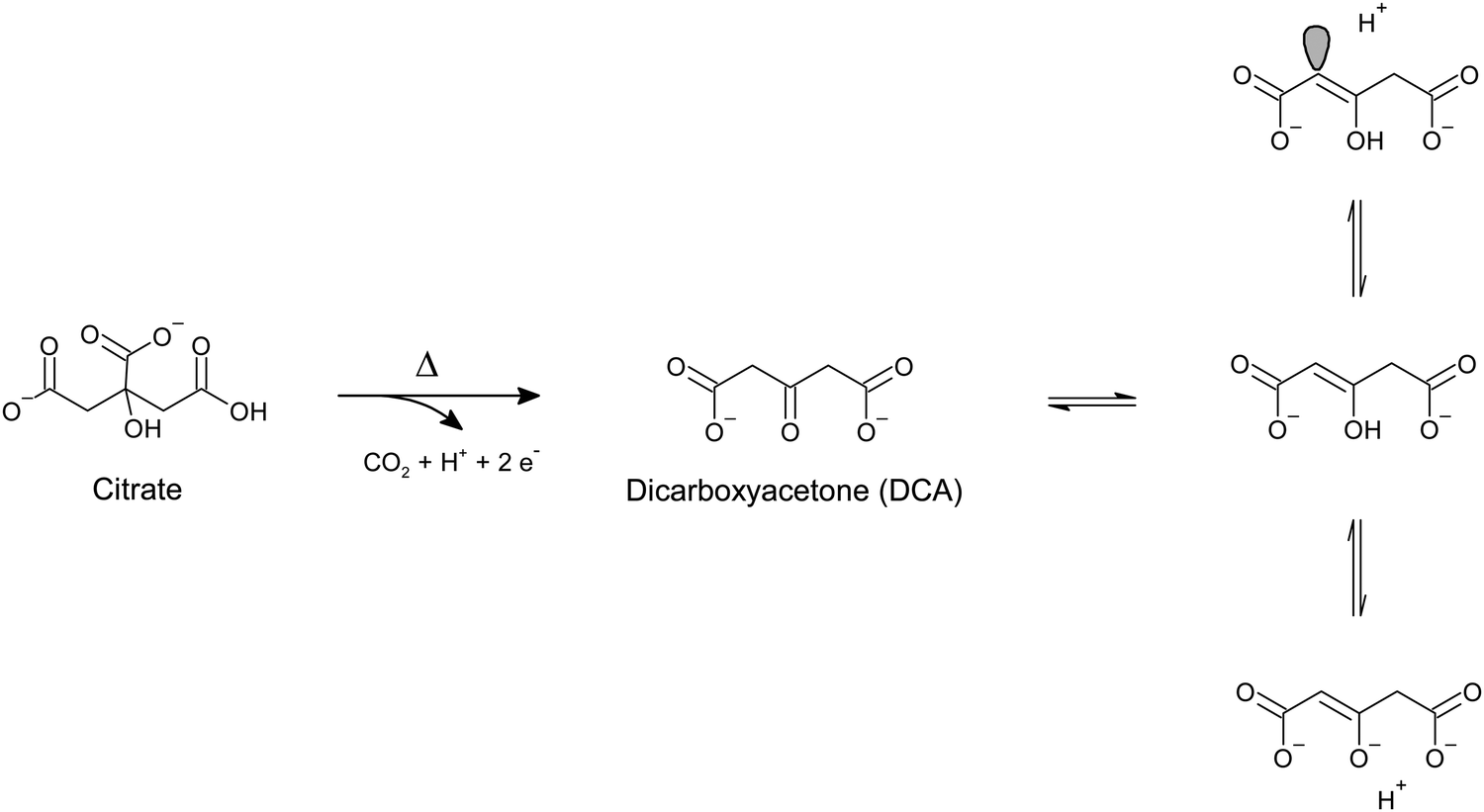
To avoid falling for the simplistic depiction of the unchanged reducing agent as the capping species surrounding our colloidal nanoparticles, it is important to always keep in mind what is going on during our wet chemistry synthesis, how is the redox reaction proceeding, and what are the chemically plausible end-products of it. Besides sodium borohydride (i.e., the Creighton method),47 another common reducing agent for the synthesis of colloidal plasmonic nanoparticles is sodium citrate. This fabrication approach is also known as the Turkevich-Frens method for gold nanoparticles,54,55 or the Lee-Meisel method for silver nanoparticles,56 and because of its popularity, it has been extensively investigated by the SERS community. Net of the debate on the mechanistic details of the redox reaction and the fine control of the morphological and dimensional uniformity of the final nanoparticles, a consensus has been reached on the major by-products of the reaction. Because this method and its variations always involve heating the reaction solution to boil,54–59 citrate thermally decomposes by successive decarboxylation steps into dicarboxyacetone, acetoacetate, and smaller molecules such as glyoxylate, acetate, formate, acetone, and of course, CO2.54,60–64 This notwithstanding, HPLC studies57 have shown that only about 7% of citrate is thermally decomposed, and the final colloid still has a large (∼93%57) excess of unreacted citrate. Opposite to what was discussed for sodium borohydride, this abundance of stable, unreacted citrate would justify the widespread habit to call “citrate-capped” the resulting gold or silver nanoparticles; citrate is quantitatively and stably present at the end of the reaction and it has been shown to populate the surface of the resulting nanoparticles,60,65,66 to the point that it is also frequently utilized as a stabilizer of nanoparticles obtained via other fabrication routes (e.g., sodium borohydride reduction,31 or L-ascorbic acid-reduced seedless, surfactant-free gold nanostars32).
Recent studies by Oliveira de Souza et al.66 highlighted how the surface of gold nanospheres obtained with the Turkevich method is also populated by trace dicarboxyacetone (DCA). DCA has traditionally be considered not present on the surface of citrate-reduced gold or silver nanoparticles due to its labile nature and consequent quick decomposition to acetoacetate. Early studies on citrate-reduced silver nanoparticles by Munro et al.60 reported that citrate oxidation species are not detectable by SERS upon aggregation of the as-synthesized colloid and correlated this finding to the absence of citrate oxidation products on the nanoparticle surface. By analogy, citrate-reduced gold nanoparticles are also traditionally considered to display the same behavior. Through more modern lenses, it can be pointed out that this conclusion excludes the possibility that aggregation alters the adsorption equilibria that exist in the colloid's native state; upon aggregation, citrate oxidative decomposition products might be displaced from the nanoparticle surface, or minimized in such a way that their concentration falls below the limit of detection (LOD) of the utilized analytical method. In the experiments reported by Oliveira de Souza et al.,66 drop-cast citrate-reduced gold nanoparticles were illuminated using very low laser power (i.e., 0.04 mW, λexc 633 nm) and the variation of their SERS signal was monitored over time. While the low laser power was not such to cause photo-induced chemical reactions at the surface of the drop-cast colloid, it was in fact enough to lead to thermally-induced reconfigurations of the hotspots location and arrangement (e.g., modification of interparticle distance due to water evaporation) that enabled the detection of previously elusive DCA. The inquisitive reader might object that the analysis of dried residues of non-purified, drop-cast colloids is not representative of the nanoparticle surface in the colloid's native state, as the drying process might force the adsorption of species that are otherwise present as free molecules in the solution bulk. However, the presence of DCA was also interestingly observed by electrospray ionization mass spectrometry (ESI-MS) of the same citrate-reduced nanoparticles in their native state (i.e., without prior laser illumination and drop-casting), confirming not only the non-photoinduced nature of this species, but also its likely existence on the native nanoparticle surface.
An earlier work by Grasseschi et al.67 also reports the SERS and mass spectrometric detection of DCA in citrate-reduced gold nanoparticles. The authors attributed the presence of DCA to a species-stabilizing effect caused by the synthetic conditions in which the citrate-reduced gold nanoparticles were obtained, that is, using near stoichiometric citrate-to-gold ratios (i.e., 1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and fast reactant addition kinetics. The latter was hypothesized to cause a rapid nucleation of nanoparticles, on which the enolate tautomer of DCA (Scheme 1) immediately adsorbs by formation of a charge transfer complex with gold.67 The resulting enolate-gold surface complex is in resonance at 633 nm, and thus, it can be observed by SERS measurements at or close to that excitation wavelength, without any prior aggregation (Fig. 4a). On the other hand, control experiments in which the citrate-to-gold ratio was higher (i.e., 2.5:1), and thus closer to the traditional Turkevich54 protocol, and in which the kinetics of reagents addition was slower (i.e., drop by drop), show no presence of m/z peaks ascribable to DCA. This absence was explained in terms of a slower nucleation kinetics and a DCA decomposition rate that was faster than the time scale at which DCA adsorbs onto gold.67 SERS measurements of the resulting colloid show the typical, mostly flat spectrum profile when analyzed as such, while they exhibit citrate bands after prior colloid aggregation (Fig. 4b), consistent with the seminal data on citrate-reduced silver nanoparticles published by Munro et al.60
:1) and fast reactant addition kinetics. The latter was hypothesized to cause a rapid nucleation of nanoparticles, on which the enolate tautomer of DCA (Scheme 1) immediately adsorbs by formation of a charge transfer complex with gold.67 The resulting enolate-gold surface complex is in resonance at 633 nm, and thus, it can be observed by SERS measurements at or close to that excitation wavelength, without any prior aggregation (Fig. 4a). On the other hand, control experiments in which the citrate-to-gold ratio was higher (i.e., 2.5:1), and thus closer to the traditional Turkevich54 protocol, and in which the kinetics of reagents addition was slower (i.e., drop by drop), show no presence of m/z peaks ascribable to DCA. This absence was explained in terms of a slower nucleation kinetics and a DCA decomposition rate that was faster than the time scale at which DCA adsorbs onto gold.67 SERS measurements of the resulting colloid show the typical, mostly flat spectrum profile when analyzed as such, while they exhibit citrate bands after prior colloid aggregation (Fig. 4b), consistent with the seminal data on citrate-reduced silver nanoparticles published by Munro et al.60
 | ||
| Fig. 4 SERS spectra of gold nanoparticles (AuNPs) capped by citrate and dicarboxyacetone (DCA). (a) SERS spectra of AuNPs capped by DCA (Ket-AuNPs), obtained using an integration time of 120 s, laser power of 50 mW, and different excitation sources: 785 nm (top spectrum), 633 nm (middle spectrum), 532 nm (bottom spectrum). (b) Comparison between the SERS spectrum of aggregated citrate-capped gold nanoparticles (Cit-AuNPs, top spectrum) and that of Ket-AuNP (bottom spectrum). Colloid aggregation was achieved by adding 100 μL of 1 M NaCl to the sol and the utilized excitation source was 785 nm. Modified with permission of the Royal Society of Chemistry, from Grasseschi et al., RSC Adv., 2015, 5, 5716–5724;67 permission conveyed through Copyright Clearance Center, Inc. | ||
A SERS spectrum with a mostly flat background in both unaggregated and aggregated conditions is typical of hydroxylamine-reduced silver colloids. In this method, also known as Leopold and Lendl's,68 an alkalinized hydroxylamine hydrochloride solution is utilized as the reducing environment for silver ions, generating nitrogen gas and water as the hydroxylamine oxidation products.68–70 The SERS background signal of this colloid is indeed characterized by the sole presence of an intense band at ∼243–247 cm−1, which is assigned to ν(Ag–Cl) from adsorbed chloride.71–73 It must be recalled that this band is also present in virtually all bottom-up fabricated gold nanoparticles as a result of the ubiquitous use of tetrachloroauric acid as the gold precursor salt, and in other preparations that involve the use of chloride-containing species, such as borohydride-reduced silver nanoparticles obtained in the presence of spermine tetrahydrochloride.48
Bottom-up fabrication protocols for the synthesis of colloidal plasmonic nanoparticles are nowadays innumerable, and an extensive treatment of each and every redox reaction would be impossible in the space of a section in a review paper. Not only hydroquinone or hydrogen peroxide,74–78 but also diverse extracts from plants or other organisms have been used to obtain plasmonic colloidal nanoparticles.79–83 For the sake of brevity, we have chosen to treat only the most commonly used reducing agents for the fabrication of nanospheres, that is, sodium borohydride, citrate, and hydroxylamine, and hope to have equipped the reader with a methodological approach that they will be able to translate to other systems. One last mention that we think should be given, and that is outside the listed popular reducing agent triad, is L-ascorbic acid.
Also known as vitamin C, L-ascorbic acid is a mild reducing agent that is widely employed in the growth of anisotropic colloidal nanoparticles, such as nanorods, nanostars, and nanotriangles.36,84–88 The redox behavior of this molecule has long been studied because it is an enzyme cofactor in both animals and plants, as well as an important nutrient and antioxidant additive in the food industry.89–91 The redox behavior of L-ascorbic acid entails the transfer of two electrons per molecule, which can be transferred one at a time, generating a free radical intermediate, semidehydro-L-ascorbate, followed by the fully oxidized species, dehydro-L-ascorbic acid (DHA, Scheme 2).89,92–94 At reaction completion, L-ascorbic acid, or more likely, its anionic form L-ascorbate (pKa1 3.69–4.4595), and DHA will both be present in solution, with ratios that depend on the stoichiometry of the nanofabrication process. While L-ascorbate likely adsorbs on the surface of gold and silver, albeit weakly,96,97 no dedicated study has been found on the adsorption properties of DHA on plasmonic metals. Infrared characterization of ascorbate-reduced nanospheres of a less common plasmonic metal, copper, has evidenced the presence of polyhydroxyl species on the surface, which have been hypothesized by the authors to be a possible hydrolysis product of DHA.98 This species is indeed reported to undergo hydrolysis in aqueous environment (Scheme 2), with a half-life that is a function of pH and solution composition.94,99–101 For example, the half-life of DHA in aqueous solution is around 30 hours at pH 5 and shortens to 30 minute when increasing the pH to 7.4;99 at the same pH but in phosphate buffered saline, the half-life increases to 90 minutes,101 while in presence of carbonate ions it has been reported to decrease below 2 minutes.100 It is therefore apparent that more in-depth studies on both end-of-reaction solution species and surface adsorbed species of ascorbate-reduced nanoparticles should be performed, to elucidate their actual capping population and the role of the solution environment and the nanoparticle themselves in determining it.
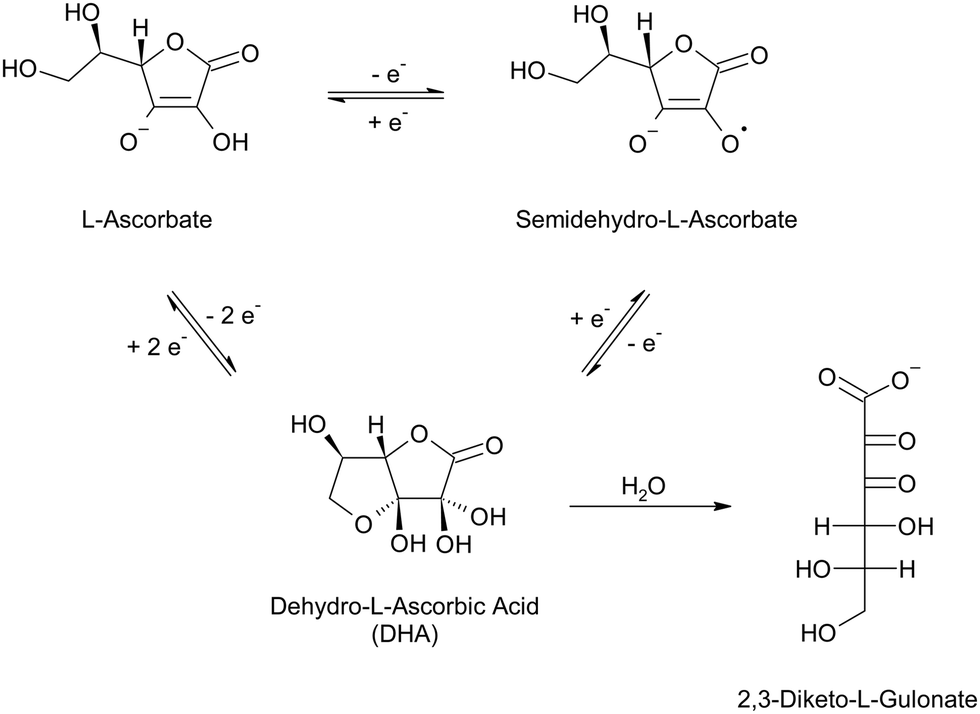 | ||
| Scheme 2 One-electron and two-electron oxidation–reduction of L-ascorbate in aqueous solution, and hydrolysis of dehydro-L-ascorbic acid (DHA) into 2,3-diketo-L-gulonate. The hydrolytic product 2,3-diketo-L-gulonate can further degrade to multiple 5-carbon and 4-carbon species.94 The reaction diagram reports the best accepted structures of the species.92–94,102–105 | ||
2.2 The bulk of the solution and the unchanged surface population: surfactant and polymeric shape directors
In contrast with the chemically changing nature that we have discussed for the reducing agents, are species that take active part in the synthesis mechanism, but do not necessarily chemically change during it. These species are mainly ascribable to surfactants and polymers that are utilized as shape directors in anisotropic nanoparticle synthesis; by adsorbing with different affinity and coverage on different crystal facets, surfactants and polymers can direct the growth along one or more preferential directions, leading to the fabrication of nanorods, nanostars, nanoplates, etc. Because of the role of these molecules as shape directors, they do interact with the surface, and their presence, adsorption energy, and coverage must be considered when representing a nanoparticle architecture and planning its use as a SERS substrate.While it is beyond the scope of this review to discuss the interactions of surfactants during synthesis (the reader is redirected to other reviews106), it is important to understand how they might be interacting with the surface at reaction completion and understand that, while they might not be typically involved in the redox process that leads to the nucleation and growth of plasmonic nanoparticles (i.e., they are not subjected to a chemical transformation during nanofabrication), they might in fact undergo supramolecular rearrangements that might be crucial to subsequent applications in SERS. Different supramolecular arrangements might indeed imply different surface–ligand energetics and resulting coverage.
For example, molecular dynamics simulations by Meena and Sulpizi107 and Da Silva and Meneghetti108 showed that, towards reaction completion, cetyltrimethylammonium bromide (CTAB) in gold nanorods passes from a micellar arrangement to a bilayer arrangement, influenced by interface phenomena governed by co-adsorbed species, such as bromide. The surfactant's counterion has indeed the ability to complex gold;109 in molecular dynamics simulations, gold-bromide complexes travel along the intermicellar channels formed by the cetyltrimethylammonium cation (CTA+), leading not only to a faster deposition of metallic gold along the surfaces that are curved and less densely packed by the surfactant,108,110 but also to an increase in the concentration of bromide at the metal–surfactant interface.107,108 These processes ultimately cause the micelles to collapse into the densely packed CTAB bilayer that is experimentally observed in mature nanorods.108,111,112
As expected for halides with weak ion-solvent interactions (in the order, I− < Br− < Cl−),113 bromide strongly adsorbs on gold, forming a chemical bond that has a more covalent than ionic character,114 and whose stretching mode is observable in SERS spectra at ∼176 cm−1.115 The energy of this interaction, together with the strong electrostatic attraction of bromide with CTA+, are such that they alter the native surfactant conformation in solution, stabilizing a densely packed bilayer on the gold nanorod. As evidenced by recent small-angle X-ray and neutron scattering (SAXS and SANS) measurements,116 the strong electrostatic interaction between bromide and CTA+ enhances the screening of the cationic head groups, causing an increase in the packing density of the surfactant at the gold surface. Patchier, micellar aggregates of CTA+ are instead reported for nanorods obtained using chloride as a counterion,116 consistent with the previously mentioned halide–solvent interaction trends, and thus, metal–halide bond energy and coverage trends.113
When talking about ions and, more in general, species in solution, it is important to stress that their presence does not necessarily imply that they adsorb on the nanoparticle surface, and if they do, they might not do so strongly enough to be relevant under a SERS perspective. It must also be recalled that adsorption ultimately depends on the electronic structure of the surface,117 and while gold and silver are often comparable, they do have different electronic structures due to which they can exhibit a different behavior towards adsorbing molecules.118–121 The complexity and interdependence of such statements can be illustrated using the polyatomic anion nitrate as an example. Nitrate is known to be a generally weakly-bound species,117 to the point that it can be approximated to a non-adsorbing species on gold surfaces.117,122 Fundamental studies for catalytic purposes show how the adsorption strength of nitrate is still very low on pure silver, albeit higher than gold; however, nitrate's affinity for the surface can be modulated via surface silver alloying of gold, which exhibits maximized adsorption strength on geometries that are rich of defects and have sub-monolayer silver alloying on the outermost layer of a gold surface.117 Therefore, in nitrate-containing colloids, such as most silver nanospheres as well as gold nanorods84,85 and some gold nanostars,36,86,87,123 the majority of this ion is generally expected to populate the solution bulk,32 rather than the surface, and the surface coverage of the anion is expected to be best in gold–silver alloyed systems that are predominantly gold and bear a sub-monolayer of alloyed silver on the surface.117
Following on this topic, the coverage with which adsorbates cap nanoparticles is certainly a function of their binding affinity towards the metal, with densely packed crystal facets typically being associated with higher adsorbate binding energy113 and lower steric effects.124 Because steric hindrance in adsorbate–surface interactions can be seen as a form of nonbonding interaction similar to the one existing among neighboring ligands complexed on the same metal center in traditional organometallics, it can be stated that the local surface geometry, and in particular, its curvature, directly contributes to the steric effects that result in a given adsorbate surface configuration.124 The effects of surface curvature on the topographical adsorbate distribution across a nanoparticle have been indirectly observed for decades,125–127 starting with early reports on the anisotropic reactivity of CTAB-capped gold nanorods,128,129 which were indeed hypothesized to be caused by a lower capping density at the curved tips. Pioneering studies involving the application of aberration-corrected scanning transmission electron microscopy coupled with electron energy loss spectroscopy (STEM-EELS) allowed for the direct visualization and quantitation of the organic adsorbate distribution across both gold nanorod surfaces deposited on graphene substrates.130 The distribution of the CTAB bilayer across as-synthesized gold nanorods was reported to be 40% less dense at the tips compared to flatter regions along the longitudinal axis of the nanoparticle (Fig. 5),130 and successive independent molecular dynamics simulations110 showed curvature-specific packing of CTAB also in its micellar conformation during synthesis.
 | ||
| Fig. 5 STEM/EELS analysis of the organic shell of gold nanorods deposited on graphene substrates. (a) EELS carbon compositional map of an isolated CTAB-capped gold nanorod, after graphene background subtraction. Low heat map values are observed for the carbon signal at the tips of the nanorod, indicating lower packing density of CTAB at those locations. The scale bar is 10 nm. (b) Relative binding density of CTAB as a function of position along the boundary of multiple gold nanorods. The solid line represents the Savitsky−Golay filtered median of the individual datapoints in the scatter plot (positive x-axis side). The scatter plot and median are mirrored across x = 0 for ease of visualization. The capping density of CTAB decreases at the tips of the nanorods. Figures adapted from Janicek B. E. et al., Nano Lett. 2019, 19 (9), 6308–6314; figures licensed under ACS Editor's Choice via CC BY 4.0 International Public License, https://pubs.acs.org/page/policy/authorchoice_ccby_termsofuse.html. | ||
These results might seem in contradiction with the common knowledge that nanocrystals display higher binding reactivity at their step edges (i.e., high energy surface sites), which are typically identified as the tips in anisotropic nanostructures. For example, analogous measurements performed on gold nanorods natively capped by (16-mercaptohexadecyl)trimethylammonium bromide (MTAB), show a ∼10% increase of ligand density at the tips.130 STEM-EELS studies performed on gold nanostars also show higher capping density at the tips; while no information on the as-synthesized system was provided (i.e., identity of the native surface capping), gold nanostars that were post-synthetically functionalized with unmethylated CpG DNA strands display a higher capping density in correspondence of higher convex curvature when compared to the coverage on flat nanoparticle domains.131 Jiang et al.132 reconcile the two opposite curvature-capping scenarios on the basis of capping agent concentration, and thus, coverage. At low coverage, preferential adsorption occurs at high-curvature sites (i.e., tips), while at high coverage, that is, in the presence of excess adsorbing species, sparser packing at the high-curvature domains occurs.132 In other words,124 under a high coverage regime, nonbonding interactions at high-curvature sites prevail. These considerations are particularly important when utilizing surfactant-capped nanoparticles for SERS applications. The washing procedures to which these colloids are typically subjected133 decrease the concentration of the surfactant, thus altering the adsorbate/free molecule equilibrium at the surface. Therefore, when working under these conditions, we must be cognizant of the possible regioselective surface packing rearrangements that might occur as a consequence of the diminished global coverage of the nanoparticles.
As discussed, surfactants aggregate into supramolecular structures whose architecture, size, and packing density are not only a function of the chemical identity of the constituting individual molecules, but are also a function of pH, temperature, ionic strength, and composition of the solvent system in which they exist,134 which includes the nanoparticle itself, with its possible co-adsorbed species. Like surfactants, polymers are sensitive to solvent parameters, as reported for example by Qin's group135 for polyvinylpyrrolidone (PVP) on silver nanocubes. While the interaction of PVP with the metal is not modified upon changing the nanoparticle dispersion solvent and adsorption occurs via the π-donor pyrrolidone's carbonyl in all investigated instances,135–137 changes in the dispersion solvent cause reversible, SERS-observable conformational changes due to the different degree of hydrogen bonding between the polymer and the solvent.135 Consequently, the packing density of the polymer on the surface is altered.135 Being cognizant of these types of conformational rearrangements at the surface is extremely important to tailor the surface accessibility for ligand exchange reactions and SERS tag fabrication, as well as for direct SERS measurements.
While for predominantly historical reasons the bulk of the scientific literature on surfactant interactions on plasmonic nanoparticles entails CTAB and its supramolecular arrangement on gold nanorods during and after synthesis, the range of surfactants and polymers that a SERS user can encounter in everyday practice is the most disparate,73,138–141 and CTAB represents only a minimal, albeit notable, portion of this very complex world. Unfortunately, owing to the intrinsic complexity of modeling large supramolecular systems at plasmonic surfaces and the general difficulties associated with the implementation of adequate solution-phase characterization techniques, knowledge of the conformation of other surfactants or polymers on plasmonic nanoparticles, including facet- and curvature-directed patchiness, is sparse at best, when not completely lacking. For example, Triton X-100 is utilized for the synthesis of high-yield six-branch nanostars,36,139 and it is known from early SERS literature142 to interact with silver via its polyethylene oxide tail, when utilized above its critical micellar concentration (CMC). However, the higher-level architecture of the micelles on specific crystal facets or possible supramolecular rearrangements at reaction completion are at present unknown, and a similar scarcity of knowledge can be observed for several other bulky shape directors. However, in the same way in which we have tried to equip the reader with a methodological approach on how to think about the complexities related to the reducing agents, we hope to invite considerations also about the complexities related to the architecture of the so-called “organic shell” of nanoparticles at reaction completion and in the successive steps of their use as enhancing substrates for SERS, as these details are determining towards a full understanding of the surface for a true rational design of SERS-based experiments.
It must be pointed out that, while it is true that shape-directing surfactants and polymers can be generally regarded as chemically unchanged at reaction completion, there might be instances in which these species do undergo chemical changes. For example, some polymers and surfactants have mild reducing properties that are sometimes exploited to achieve specific morphological control during synthesis, such as in the case of polyols like polyethylene glycol (PEG), ethylene glycol, and glycerol.143 Other examples are PVP144 or the poloxamers commercially known as the Pluronic series,145,146 which are hypothesized to exert their mild reducing action via their terminal hydroxyl groups,144 or their degradation into reactive oxygen species,147 respectively. These chemical transformations might introduce surface-interacting moieties in the system such as carbonyl functional groups, which contribute to diversifying the surface adsorbate population. While it is beyond the scope of this review to detail these types of changes, the reader should be warned that, albeit not predominant in common SERS-relevant nanosystems, these mechanisms could also take place, and thus, an aprioristic exclusion of their existence is not advisable.
2.3 The dynamic nature of a plasmonic surface: reconstructions and rearrangements at the nanoscale
Except for the considerations made on surfactants and polymers rearrangements on the surface, we might have so far painted a rather static picture of a plasmonic nanoparticle surface. Besides the obvious reminders that anything at a solid/solution interface exists in a dynamic equilibrium, we must additionally stress that the metallic domain of that interface is also highly mobile and subjected to changes over different time scales. Gold surfaces are prime examples of highly mobile systems, and this is particularly evident when we observe the strong restructuring that highly anisotropic gold nanoparticles undergo over time, producing duller features and ultimately more thermodynamically stable morphologies with aging.87,148 Mono- or multilayer shells of other metals or the presence of chemisorbed inorganic and organic species can modulate this property by either enhancing or limiting it. For example, silver has lower mobility compared to gold, and increasing content of atomic silver on the surface of six-branch gold nanostars has been reported to halt their surface reconstruction and preserve the morphology during aging;36 on the contrary, the partial π character of the dative charge transfer of halides and thiols to gold surfaces weakens the bond energy among surface atoms, thus increasing their diffusion coefficient and creating local adatom features on the surface.119,124 Interaction with light such as during a SERS measurement can exacerbate this adatom feature by further pulling these weakened atoms, generating transient single-atom defects (i.e., picocavities).149,150 On a less localized nanoscale, chemisorbed species can be seen as agents that actually stabilize the overall morphology of a nanoparticle. This process is observed in highly anisotropic plasmonic nanoparticles that retain their shape over time when they are capped by chemisorbing surfactants or small molecules.32,87Changes in the surface due to adatom reconstructions might also trigger different binding motifs for the adsorbed species, and thus, their susceptibility to displacement by another capping ligand or a SERS analyte. Grys et al.5 showed that gold nanospheres obtained with the Turkevich-Frens method undergo surface restructuring via thermally activated adatom formation during aging. Gold sols were stored at different temperature conditions (T = 5, 18, and 28 °C) for 35 days, and investigated by SERS at different time points, after prior aggregation with molecular spacer cucurbit[5]uril (CB[5]). During aging, the capping citrate molecules were observed to transition from a bidentate coordination via the central carboxylate (ν(CC) at 1020 cm−1) to a double bidentate coordination involving both central and terminal carboxylate moieties (ν(CC) at 1080 cm−1), indicating the formation of adatoms on the surface, as supported by DFT calculations (Fig. 6a).5 This change in the surface configuration of adsorbing citrates was more evident the higher the temperature of storage of the gold colloid. The concomitant observation of an intensity decrease of the CB[5] ring breathing mode at 830 cm−1 also suggested a variation of the binding energy with time, in line with the expected strength of a single bidentate coordination complex vs. a double bidentate complex (Fig. 6b).5 This binding energy modulation is particularly important as it signals that colloids prepared in the same exact way but differently stored and differently aged can show different surface affinities for a given identical species (CB[5], in the case presented by Grys et al.5), which is extremely important when evaluating the reproducibility of SERS results across different laboratories, or even by the same group but on different days. Grys et al.5 also demonstrated that etching and subsequent boiling of the aged gold sols lead to a complete refreshing of the surfaces, thus improving their reliable application in sensing.5 We must however point out that such processes might not be valid for anisotropic shapes, as the overall morphological, and thus optical, properties might not be retained post-treatment, as a consequence of the high adatom mobility reported above.
 | ||
| Fig. 6 Adatom-induced changes during aging of citrate-reduced gold nanoparticles. (a) Proposed citrate coordination on fresh citrate-reduced gold nanoparticles (left) and adatom-induced citrate reconfiguration upon aging (right), as hypothesized from SERS spectra obtained after prior aggregation with CB[5]. (b) SERS intensity changes over time of the marker bands of CB[5] at 830 cm−1 (left) and bridging coordination of citrate at 1080 cm−1 (right), at three different temperatures. Changes are more prominent at higher storage temperatures, indicating a role of thermally activated adatoms in the aging process that causes the citrate coordination mode to rearrange and CB[5] to experience an affinity decrease for the surface. Figures adapted from Grys D. et al., ACS Nano 2020, 14 (7), 8689–8696; figures licensed under ACS AuthorChoice via CC BY 4.0 International Public License, https://pubs.acs.org/page/policy/authorchoice_ccby_termsofuse.html. | ||
Time-dependent changes due to the aging of the adsorbate population must also be considered. As briefly mentioned in Section 2.1 when discussing L-ascorbate-reduced surfaces,94,99–101 molecules that are adsorbed or in solution might degrade over time, by action of oxidative, photo-initiated, or hydrolytic processes, just to name a few. Consequently, the identity of the adsorbate(s) might change over time, leading to the formation of degradation products on the surface that have different (i.e., stronger or weaker) interactions with the plasmonic metal, compared to those established by the original, unaltered adsorbates. In the same way, species that were initially nonadsorbing can potentially degrade into species with an increased affinity for the metal and populate the nanoparticle surface over time, changing its chemical reactivity. Because literature on this specific topic is lacking, a data-driven weighing of adsorbate degradation and aging in the overall pool of factors that determine SERS reproducibility cannot be made. However, because of this very lack of data, efforts must be made not to lose sight of the dynamic nature of SERS-active surfaces and what it implies on different timescales.
Expanding on the subject of timescales, it must be recalled that surface rearrangements do not exclusively pertain to the timescale of what is commonly referred to as the aging processes (i.e., days, months), but they also occur transiently, at the ultra-short timescale. As mentioned at the beginning of this section, random, light-, and/or thermally induced surface reconstruction events can also happen transiently, and their interplay is associated to the typical fluctuations that are observed in single molecule and high-speed acquisition experiments.151,152 A multitude of timescales should be therefore considered to gather a full picture of the extent and type of surface reconstructions that characterize plasmonic metal nanoparticles and that ultimately have an effect on the observed SERS response. Additionally, rearrangements could also involve the electronic structure of the surface; these changes can be induced by plasmon-mediated electron transfer events that lead to the formation of charged radical species within the adsorbate population.153,154 Finally, local heating during illumination might not only lead to a deformation and rearrangement of the original nanostructure surface, but it could also induce temperature-dependent enrichment-depletion gradients at the adsorbate level,155 thus contributing to the complexity of the generated illumination- and time-dependent SERS spectral profiles.
3. Utilizing surface chemistry and thermodynamics to understand SERS-relevant adsorption hierarchies
Now that the main elements that go into assembling a realistic depiction of a colloidal plasmonic surface have been discussed, a question must be asked: Is this surface conducive to our analytical objectives? It might happen that the answer is no, and suitable ligand exchange reactions must be performed. In certain lucky cases, where the nucleation and growth processes are unaffected or negligibly perturbed by the presence of our added surface-modifier, proper ligand exchange reactions can be avoided and the right capping species for our applications can be added during synthesis, such as in the incorporation of spermine for the fabrication of cationic silver nanospheres.48 In most other cases, however, ligand exchange cannot be avoided. The process of tuning the surface chemistry for a given analytical application is not trivial, and it requires extensive work that should be considered an essential and non-negotiable part of SERS method development. Even though a trial-and-error approach will eventually produce the desired practical result, it is evident that knowledge of the surface chemistry and thermodynamics of the system to be modified could greatly simplify the task. Additionally, important information on the dynamic equilibria established during SERS sample preparation can be more clearly derived: Which adsorption hierarchies are established upon analyte addition to the colloidal sol? Is the analyte displacing the capping species on the surface? Is the capping species complexing the analyte? Are other species in solution modulating the adsorption equilibria? Ultimately, why does the generated spectrum profile look the way it does?In this section we will present some new techniques to quantitatively determine the energetics involved in the adsorption of common plasmonic nanoparticle capping agents, as well as computational studies that elucidate the thermodynamic parameters among common capping agents and plasmonic surfaces and how they can be utilized to the advantage of a SERS protocol development pipeline, involving either ligand exchange reactions or direct SERS measurements. Finally, we will discuss emerging N-heterocyclic carbenes (NHCs) and their role in incrementing the variety of persistent functionalization species that can be utilized in applications that require low susceptibility to adsorbate displacement.
3.1 Quantifying affinities to elucidate and tailor analyte adsorption: displacement vs. multilayer adsorption vs. co-adsorption
The lack of widely available and established in situ characterization techniques for the quantification of the binding strength of adsorbate systems on colloidal nanoparticles in their native state makes this task particularly challenging. As we have outlined in Section 2, colloidal nanoparticles are complex, heterogeneous systems that dynamically change in response to external stimuli, inevitably making ex situ characterizations an approximation of their properties in their native state. Over the years, first-principles computational studies such as DFT calculations have come to the nanoscientist's aid and advanced the understanding of nanoscale phenomena that were (or are still) not experimentally accessible, providing insight into the thermochemistry of the nanoscale adsorption of molecules on specific metal surfaces or clusters.31,120,156–158 It must be recalled that, in their simplest implementations, DFT calculations describe isolated systems in the gas phase;159 while this approach is certainly valid for certain applications, such as the calculation of Raman spectra of analytes in the solid phase,160 condensed phases such as colloidal nanoparticles are more accurately described by the introduction of explicit, implicit, or hybrid explicit–implicit solvents that account for their native conditions and properties. The latter approach, which combines the modeling of an explicit first solvation shell and the application of a dielectric continuum,161 allowed to gain insight into the participation of water molecules to the surface coordination of doubly deprotonated citrate, on both monometallic Au20 and bimetallic Au19Ag (111) clusters representing the alloyed surfaces of 18:1 AuAg nanostars.120 The theoretical adsorption free energies in solution of both directly bound and water-mediated citrate/metal systems were in line with experimental values (vide infra), and ranged from −7.7 to −14.3 kcal mol−1, indicating weak chemisorptive interactions that are likely established via lone-pair-driven charge transfer complex formation.120
As previously addressed, being cognizant of the type and magnitude of the binding interactions between a stabilizer and the metal surface on which it is chemisorbed is of utmost importance when developing a ligand exchange or a SERS protocol, as they will determine the final configuration of the species on the plasmonic surface. For example, it was recently shown that amines interact with gold surfaces via their lone pair, with energies that are positively correlated to their basicity, net of corrections for steric hindrance.162 The most basic primary amine that was investigated, ethylamine, was reported to adsorb on Au(111) with an energy of −0.673 eV, which corresponds to −15.5 kcal mol−1.162 Because the basicity of amines increases with alkyl substitution, and because the theoretical free energies of adsorption discussed for citrate120 and ethylamine162 can be in principle utilized as benchmarking points, we could reasonably expect that a tertiary alkylamine, either utilized as a ligand in an exchange reaction or a target analyte in a SERS sample, would displace citrate on gold surfaces, winning the competition for adsorption sites. Such type of quantitative, benchmarking information is crucial to understand whether a SERS sample system is a monolayer of our analyte, a mixed monolayer of our analyte and one or more additional species, or a multilayer system in which our analyte is not the species that is closest to the metal surface. Because SERS is a near field tensorial effect, these adsorption hierarchies are key to understanding and interpreting SERS spectra (Fig. 7).
 | ||
| Fig. 7 Schematic representation of some of the possible adsorbate architectures that can generate at the surface of a colloidal nanoparticle upon analyte addition during SERS sample preparation, provided that for all instances Kad[analyte/metal] > Kad[analyte/analyte]. If Kad[analyte/metal] > Kad[capping agent/metal], the analyte can displace the capping agent and (a) form a chemical bond with the substrate or (b) physically interact with it. (c) Analyte addition to a substrate with pre-adsorbed species having Kad[capping agent/metal] ≈ Kad[analyte/metal] might result in co-adsorption with no displacement. If Kad[analyte/metal] < Kad[capping agent/metal], and there is no ensuing repulsive interaction between the capping environment and the analyte, a layered surface architecture can form instead. (d) Depending on the physicochemical properties of the capping agent, the analyte can (d) form an ionic pair, (e) form a chemical bond, or interact via dispersion forces, and reside at a distance that is proportional to the steric bulk of the capping agent itself. | ||
As the computational cost of DFT calculations scales with the number of electrons composing the system,159 a detailed modeling of complex surfaces with large, adsorbed molecules may not be feasible on a reasonable timescale. While the use of pseudopotentials renders the treatment of metals such as gold and silver computationally affordable,159 large assemblies of organic molecules or individual macromolecules, such as surfactant micelles or polymers, also heavily contribute to both computational cost and degrees of freedom of the system, greatly complicating its DFT modeling. For these reasons, such large organic systems are often simplified to their individual constituents, segmented into their relevant functional units, or reduced to oligomers, as exemplified by a recent work on divinylpyrrolidone (DVP), which was utilized as a model compound to study the adsorption behavior of PVP on silver.163 The DFT-calculated binding energy of the dimer was 1.13 eV on Ag(100) and 1.06 eV on Ag(111), corresponding to about −26.1 and −24.4 kcal mol−1, respectively.163 Interestingly, the authors report that the presence of co-adsorbed chloride on the surface was positively correlated with the binding strength of DVP, and that chloride preferentially binds to Ag(100) facets, thus determining the facet-selectivity of the dimer, and as an extension, of the polymer, too. While these studies were originally conducted to examine the role of trace chloride in reagent grade PVP, SERS users know that chloride is a common anion that can be introduced when utilizing inorganic aggregation salts (NaCl, KCl, LiCl, MgCl2, …) or performing the so-called “chloride passivation” (or “activation”). Knowledge of the described interplay of chloride and DVP adsorption energies could therefore be utilized to elucidate possible stabilization of PVP on silver nanoparticles in the presence of such salts, as it has been shown to happen for CTA+ and Br− (Section 2.2).
Experimentally, if the adsorbate to be studied is charged, and its adsorption behavior can be modeled by a thermodynamically derived isotherm function, electrophoretic light scattering (ELS) titrations can be utilized to derive ζ potential values at different adsorbate concentrations, which can in turn be leveraged to calculate the associated adsorption constant and free energy, directly on the nanoparticles, in their native colloidal state.32,164 This approach was demonstrated for the characterization of citrate's thermodynamic quantities and cooperativity when adsorbed on sparingly-capped, L-ascorbate-reduced gold–silver (18:1) nanostars, concluding that it weakly chemisorbs in a positively cooperative way (nHill ≫ 1), with an affinity constant in the order of 103 M−1 and energies of −4.36 ± 0.08 and −4.58 ± 0.05 kJ mol−1 at 10 and 20 °C, respectively.32 These results are in agreement with the previously cited DFT-simulated data of hybrid implicitly–explicitly solvated citrate on both pure gold and Au19Ag clusters.120 Elucidating adsorption cooperativity is important not only to obtain a clearer depiction of how the adsorbate layer organizes itself (e.g., number of molecules constituting the adsorbate layer, preference for the solvent rather than the metal, etc.), but also because it provides a way to corroborate free energy adsorption trends, as cooperativity tends to weaken and approach 1 (i.e., absence of cooperativity, pure Langmuir adsorption) the stronger the adsorbate/surface interactions are.97
Although ELS is affordable and easily implementable, it only provides an average picture of the assembly that is subjected to the applied voltage. In other words, it is a bulk measurement. We have seen, however, that adsorption might differ from nanoparticle domain to nanoparticle domain, as a function of crystallography and surface curvature (Section 2.2), and in certain cases those differences might need to be assessed. To directly measure such differences, new in situ characterization techniques with sub-particle resolution are being implemented. For example, energetics and cooperativity information at the sub-nanoparticle level can be derived via a competition-enabled imaging technique with super-resolution—COMPEITS.165 COMPEITS makes use of a solid particle-catalyzed fluorogenic reaction to derive affinity constants and cooperativity information relative to a ligand of interest, which competes for adsorption sites with the auxiliary fluorogenic reaction, thus modulating its fluorogenic kinetics. Because COMPEITS is based on fluorescence, the product of the auxiliary reaction, and the competition-induced fluorescence suppression thereof, can be imaged with nanometer resolution by single-molecule fluorescence localization microscopy, achieving concomitant nanometer-scale spatial information on the nonfluorescent adsorption process of interest.165 This technique has been utilized to characterize in situ the energy involved in the interaction of common noble metal nanoparticle adsorbates (i.e., CTAB, PVP, halides, a small thiol) on different crystallographic domains of large (micrometers wide, ∼14 nm thick) gold nanoplates, coated with a mesoporous silica shell for stability, as well as on small (Ø = 5 nm) gold nanospheres sparingly capped with sub-μM citrate.97Fig. 8 shows the COMPEITS image relative to the CTAB studies, which indicate adsorption differences across the different crystallographic domains of the nanoparticle—Au(111) at flat regions and Au(110) at side facets—with abundance increasing from the edges to the corners.97 In particular, it was possible to measure that the affinity constants are in the order of high to mid 105 M−1 and trended as corner (110) > edge (110) > flat-facets (111), with cooperativities varying from (1.7 ± 0.2) and (1.8 ± 0.2) on (110) facets to (2.2 ± 0.2) on (111) facets.97 These results are in accordance with previously discussed gold nanorod EELS mapping results130 showing that CTAB more densely packs on the sides of nanorods than on the tips, which are characterized by Au(111) surfaces. It must be specified that these studies were conducted below the CMC of the surfactant, to avoid complications due to the presence of micelles.
 | ||
| Fig. 8 Super resolution images of COMPEITS fluorogenic auxiliary reaction products on an individual gold nanoplate coated with a mesoporous silica shell. (a) SEM image of an individual gold nanoplate coated with a mesoporous silica shell. The bright spots are small mesoporous silica particles adsorbed onto the shell; the authors report no observable interfering effects on the COMPEITS experiments. (b) Super resolution image of the COMPEITS fluorogenic auxiliary reaction products on an individual gold nanoplate coated with a mesoporous silica shell. (c) Super resolution image of the COMPEITS fluorogenic auxiliary reaction products on an individual gold nanoplate coated with a mesoporous silica shell and [CTAB] = 0.50 μM. The pixel size of the super resolution images is 40 × 40 nm2, while n is the number of COMPEITS fluorogenic auxiliary reaction products detected over 45minutes. All scale bars correspond to 500 nm. Figures adapted from Ye, R. et al. Nat. Commun. 2019, 12, 4287; figures licensed under CC BY 4.0 International Public License, https://creativecommons.org/licenses/by/4.0/. | ||
On 5-nm gold nanospheres, where crystallographic-specific information could not be resolved (COMPEITS resolution is around 20 to 40 nm),97 the COMPEITS-determined adsorption constant for CTAB was of the same order of magnitude, 105 M−1, with positive cooperativity of about 2, consistent with the known formation of an interfacial bilayer. Interestingly, bromide affinity measurements made with KBr as the anion source showed that the adsorption constant of this halide is in the order of the low 103 M−1 range, which would comparatively imply that the contribution of bromide to the overall CTAB adsorption constant is only 0.2%, thus making its role apparently negligible.97 However, comparison with COMPEITS-derived adsorption constant of CTAB analogs CTAC and CTAOH indicate that synergistic effects due to the nature of the surfactant counterion are in fact present,97 in line with a wide variety of literature on the modulating effects of CTA+ adsorption on gold by different counterions, such as chloride or iodide.
Another interesting finding that emerges from COMPEITS reports is that PVP with standard molecular weight for nanofabrication (i.e., 55 k) showed an average adsorption constant to 5-nm gold nanospheres in the order of 109 M−1, a value that is two orders of magnitude higher than that measured for β-mercaptoethanol on the same system (Kad(βME) = 2.6 ± 0.1 × 107 M−1).97 However, this adsorption constant decreased with decreasing polymer molecular weight, reaching (2.9 ± 0.3) × 108 M−1 for 10 k PVP and showing no significant adsorption at the investigated concentrations when monomer vinylpyrrolidone was probed, indicating chain length-dependent multivalency effects.97 In addition, negative cooperativity was observed for all investigated molecular weights of the polymer, suggesting the existence of either repulsive electrostatic interactions between charged PVP chains (pKb of N-alkylpyrrolidone is around 3.5, while the COMPEITS titrations were performed at pH of about 7.4) or strong solvation effects that might cause PVP to prefer interactions with water rather than with itself.97
It is important to reiterate that quantitative, in situ information on the adsorption of stabilizers on noble metal nanoparticles is unfortunately generally lacking—the notable quantities that we were able to retrieve from the literature are relatively few and are reported in Table 1. Knowledge of affinities and changes in free energy of adsorption is important because it enables a rationalization of the “interaction architecture” that is established on the nanoparticle surface when a SERS sample is prepared, and with it, an increased understanding of the physical meaning of the SERS spectrum itself.166 It is also worthy to stress that, in the absence of a thermodynamically-guided design of a surface–analyte pair, it is statistically wise and realistic to believe that our SERS sample will likely contain non-negligible co-adsorbates. Recently, Zhang et al.166 highlighted how common halide “passivating” (or “activating”) techniques that are traditionally achieved by addition of halide salts and deemed to form full anion monolayers and displace citrate in its entirety, do not actually form complete monolayers on gold. Rather, they alter the adsorption equilibria of the species in solution, generating halide-specific adsorbate landscapes. In fact, patchy adsorbate layers are formed, with spacings that are hypothesized to be mainly dictated by the steric bulk of the anion itself. As a result, a well-packed, near full monolayer is formed by chloride, while bromide and iodide organize on the gold surface as increasingly incomplete monolayers, regardless of the incubation time. Albeit in a non-quantitative way, the authors spectroscopically demonstrate that these differences determine varying changes in the chemistry of the resulting nanoparticle surface, and thus, in the solid/solution equilibria; for example, a selective desorption of citrate in favor of the adsorption of its thermal decomposition byproduct acetoacetate (Section 2.1) was observed for “chloride-activated” citrate-reduced gold nanoparticles, generating a mixed ligand surface of chloride and acetoacetate on gold that is observable by SERS itself, after colloid aggregation with MgSO4.166
| Adsorbate | Nanoparticle system | Measuring technique | K ad (M−1) | Cooperativity | ΔGad (kcal mol−1) | Bibliographic details |
|---|---|---|---|---|---|---|
| a K 1/2 is the apparent adsorption constant derived from fitting the ascorbate/Au nanoparticle titration data with an empirical kinetic saturation equation of the form v0 = (aK1/2[Ligand])/(1+ K1/2[Ligand]) + b,168 where a and b are empirical constants and the reaction rate constant is 50% of the saturation rate at [Ligand]=1/K1/2.97 | ||||||
| 3-Mercaptopropionic acid | Commercial, 5 nm citrate-capped Au nanospheres | Isothermal titration calorimetry, 25 C° | (3.3 ± 0.5) × 106 | 0.37 ± 0.01 | –8.9 ± 0.4 | Ref. 167 |
| 6-Mercaptohexanoic acid | Commercial, 5 nm citrate-capped Au nanospheres | Isothermal titration calorimetry, 25 C° | (2.7 ± 0.4) × 106 | 0.20 ± 0.01 | –8.8 ± 0.2 | Ref. 167 |
| Ascorbate | 5 nm Au nanospheres | Bulk fluorimetry | K 1/2 ∼ 6.4 × 102.a | N/A | N/A | Ref. 97 |
| Bromide | 5 nm Au nanospheres | Bulk fluorimetry | (1.5 ± 0.3) × 103 | 1 | N/A | Ref. 97 |
| 5 nm Au nanospheres | COMPEITS, particle averaged | (1.2 ± 0.3) × 103 | 1 | N/A | ||
| Au nanoplates, corners | COMPEITS, particle averaged | (1.3 ± 0.1) × 103 | 1 | N/A | ||
| Au nanoplates, edges | COMPEITS, particle averaged | (1.6 ± 0.1) × 103 | 1 | N/A | ||
| Au nanoplates, flat facets | COMPEITS, particle averaged | (2.8 ± 0.2) × 103 | 1 | N/A | ||
| Citrate | AuAg (18:1) surfactant-free nanostars |
ELS titrations, 10 °C | (2.0 ± 0.3) × 103 | 2.8 ± 0.3 | –4.36 ± 0.08 | Ref. 32 |
| AuAg (18:1) surfactant-free nanostars |
ELS titrations, 20 °C | (2.3 ± 0.2) × 103 | 3.4 ± 0.4 | –4.58 ± 0.05 | ||
| CTAB | 5 nm Au nanospheres | Bulk fluorimetry | (6.0 ± 1.2) × 105 | 1.8 ± 0.4 | N/A | Ref. 97 |
| 5 nm Au nanospheres | COMPEITS, particle averaged | (6.6 ± 0.2) × 105 | 2.0 ± 0.1 | N/A | ||
| Au nanoplates, corners | COMPEITS, particle averaged | (9.3 ± 0.8) × 105 | 1.7 ± 0.2 | N/A | ||
| Au nanoplates, edges | COMPEITS, particle averaged | (7.6 ± 0.6) × 105 | 1.8 ± 0.2 | N/A | ||
| Au nanoplates, flat facets | COMPEITS, particle averaged | (5.8 ± 0.3) × 105 | 2.2 ± 0.2 | N/A | ||
| CTAC | 5 nm Au nanospheres | Bulk fluorimetry | (3.5 ± 1.1) × 106 | 1.2 ± 0.1 | N/A | Ref. 97 |
| 5 nm Au nanospheres | COMPEITS, particle averaged | (4.7 ± 0.1) × 106 | 1.1 ± 0.1 | N/A | ||
| Iodide | 5 nm Au nanospheres | Bulk fluorimetry | (1.8 ± 0.5) × 107 | 1 | N/A | Ref. 97 |
| 5 nm Au nanospheres | COMPEITS, particle averaged | (1.8 ± 0.5) × 107 | 1 | N/A | ||
| Au nanoplates, corners | COMPEITS, particle averaged | (5.2 ± 0.3) × 106 | 1 | N/A | ||
| Au nanoplates, edges | COMPEITS, particle averaged | (5.8 ± 0.5) × 106 | 1 | N/A | ||
| Au nanoplates, flat facets | COMPEITS, particle averaged | (6.4 ± 0.5) × 106 | 1 | N/A | ||
| PVP (MWave ∼ 55 k) | 5 nm Au nanospheres | Bulk fluorimetry | (2.5 ± 0.8) × 109 | 0.81 ± 0.11 | N/A | Ref. 97 |
| 5 nm Au nanospheres | COMPEITS, particle averaged | (2.0 ± 0.2) × 109 | 0.75 ± 0.03 | N/A | ||
| Au nanoplates, corners | COMPEITS, particle averaged | (1.4 ± 0.1) × 109 | 0.83 ± 0.02 | N/A | ||
| Au nanoplates, edges | COMPEITS, particle averaged | (6.4 ± 0.2) × 108 | 0.75 ± 0.01 | N/A | ||
| Au nanoplates, flat facets | COMPEITS, particle averaged | (4.0 ± 0.4) × 108 | 0.68 ± 0.05 | N/A | ||
| PVP (MWave ∼ 40 k) | 5 nm Au nanospheres | Bulk fluorimetry | (1.6 ± 0.8) × 109 | 0.71 ± 0.09 | N/A | Ref. 97 |
| 5 nm Au nanospheres | COMPEITS, particle averaged | (1.4 ± 0.1) × 109 | 0.68 ± 0.02 | N/A | ||
| PVP (MWave ∼ 10 k) | 5 nm Au nanospheres | Bulk fluorimetry | (4.2 ± 1.4) × 108 | 0.63 ± 0.07 | N/A | Ref. 97 |
| 5 nm Au nanospheres | COMPEITS, particle averaged | (2.9 ± 0.3) × 108 | 0.64 ± 0.02 | N/A | ||
| Au nanoplates, corners | COMPEITS, particle averaged | (5.1 ± 0.3) × 108 | 0.89 ± 0.02 | N/A | ||
| Au nanoplates, edges | COMPEITS, particle averaged | (4.6 ± 0.4) × 108 | 0.84 ± 0.03 | N/A | ||
| Au nanoplates, flat facets | COMPEITS, particle averaged | (3.1 ± 0.6) × 108 | 0.76 ± 0.05 | N/A | ||
| β-Mercaptoethanol | 5 nm Au nanospheres | Bulk fluorimetry | (3.5 ± 0.5) × 107 | 1 | N/A | Ref. 97 |
| 5 nm Au nanospheres | COMPEITS, particle averaged | (2.6 ± 0.1) × 107 | 1 | N/A | ||
| Au nanoplates, corners | COMPEITS, particle averaged | (5.1 ± 0.4) × 107 | 1 | N/A | ||
| Au nanoplates, edges | COMPEITS, particle averaged | (4.8 ± 0.3) × 107 | 1 | N/A | ||
| Au nanoplates, flat facets | COMPEITS, particle averaged | (4.8 ± 0.2) × 107 | 1 | N/A | ||
Incomplete halide monolayers on halide-aggregated citrate-reduced gold and silver nanoparticles and modulation of analyte (co-)adsorption were also reported by Xie et al.,169 who investigated the synergistic effects that take place between analyte, halide aggregating agent, and gold or silver surfaces during the preparation of a SERS sample. Everyday SERS practice shows that the order of addition of such components can have a dramatic influence on the intensity of the resulting SERS spectrum, often producing differences also in terms of band position and band presence or absence (Fig. 9); such behavior was explained by the authors as a result of competitive adsorption and synergistic effects between analytes and halides and the establishment of mixing conditions that favor direct analyte adsorption over ion pairing (i.e., multilayer adsorption). Again, while no quantitative measure of the binding affinities of the species at play were provided, it is interesting to highlight that despite the small size of the capping halides and the consequent residence of the analytes within 1 nm from the surface in either case, differences between direct adsorption and co-adsorption by ion pairing were evident in terms of intensity.169
 | ||
| Fig. 9 Effect of sample components addition on the SERS spectrum profile of indole alkaloid koumine (1 mg L−1), obtained utilizing citrate-reduced gold nanoparticles and either NaBr or NaI as the aggregating agent. The effect of the order of addition of the three components (i.e., analyte, nanoparticle, aggregating agent) and the resulting chemical environment is evident. Adapted with permission from Xie, L. et al. J. Phys. Chem. Lett. 2020, 11 (3), 1022–1029. Copyright 2020 American Chemical Society. | ||
3.2 When displacement is unwanted: N-heterocyclic carbene chemistry for persistent adsorbates
As we have discussed so far, (purposely) populating the surface of a plasmonic nanoparticle with displaceable ligands is a sought-after characteristic when implementing direct SERS applications. On the contrary, the fabrication of nanotags148,170 for indirect SERS applications or nanoparticle oligomers171,172 for engineered hotspot formation might instead require the use of adsorbates that strongly bind to the metallic surface and exhibit a high desorption barrier. Typically, organosulfur ligands like thiols and α–ω dithiols are utilized to this scope, due to their known high affinity for gold and silver and their ability to self-assemble as densely packed monolayers. While thiol-gold and thiol-silver bonds are strong (e.g., bond enthalpies in the order of ∼125 kJ mol−1 for alkenethiol–gold systems173), they are not inert, and might instead be labile and lead to ligand desorption when submitted to relatively harsh conditions, such as biologically relevant oxidizing environments and high temperatures,174,175 or resonant illumination.176Ligands based on N-heterocyclic carbene (NHC) chemistry have emerged as powerful thiol replacements as they form stronger, less labile bonds with plasmonic metals.177,178 NHCs are stable heterocyclic compounds characterized by the presence of a divalent carbon with a six-electron valence shell and at least one adjacent nitrogen within the ring (Fig. 10a).179,180 The presence of the nitrogen(s) adjacent to the carbene carbon provide electronic stability to the ligand by both lowering the energy of the carbene's occupied σ orbital (stabilization by inductive effect) and simultaneously behaving as π electron-donors, providing added electron density to the carbene's p orbital (stabilization by mesomeric effect).180 Additionally, the N-substituents (“wingtip groups”) lie on the same plane as the binding site (i.e., the carbene carbon), thus directly determining its steric environment and contributing to the kinetic stabilization of the whole NHC by sterically disfavoring the typical dimerization equilibrium of stable carbenes.179–182 All of these stability features are retained by the complexes of NHC ligands with their substrates, including gold, and ensure higher stability performances of the resulting adsorbate systems compared to common self-assembled monolayers based on thiol chemistry.177,180,183
 | ||
| Fig. 10 Structures of common N-heterocyclic carbenes (NHC) and their orientation on gold surfaces according to experimental and theoretical studies. (a) Ground state electronic structure of a minimalistic NHC and most common backbones and N-substituents. Figure inspired by Hopkinson, M. et al.180 (b) STM images of the surface patterning produced by differently N-substituted 1,3-dialkylbenzimidazol-2-ylidenes NHCs adsorbed on Au (111) at saturation coverage, and their respective DFT-simulated surface configurations: (I–III) the symmetric tert-butyl-N-substituted species self-assembles into short closely packed lines and is calculated to lie perpendicularly to the gold surface, (IV–VI) while the less bulky, asymmetric ethyl- and isopropyl-N-substituted species forms flat-lying (NHC)2Au complexes. Figure adapted from Inayeh, A. et al. Nat. Commun. 2021, 12, 4034; figure licensed under CC BY 4.0 International Public License, https://creativecommons.org/licenses/by/4.0/. (c) DFT-simulated (top, blue and red profiles) and experimental SERS spectra (bottom, black profiles) of two differently N-substituted benzymidazolylidenes. Despite the DFT-calculated energies for the vertically aligned and the flat-lying orientations indicate a more favorable formation of the latter, both orientations are experimentally observed. The bands that are uniquely associated with the flat-lying orientation are marked with an asterisk. Adapted with permission from Thimes, R.L. et al. J. Phys. Chem. Lett. 2023, 14 (18), 4219–4224. Copyright 2023 American Chemical Society. | ||
In addition to an improved stability of the functionalized surface towards pH, thermal, and chemical oxidation treatments,183 NHC chemistry can also offer a platform to explore regioselective adsorption. As seen in Section 2.2, the steric environment at an adsorbate binding site generates nonbonding interactions between the adsorbate and the surface, which result in curvature-specific regioselectivity.124 Consequently, because the steric environment of NHC ligands is controlled via the identity of the N-substituents,180 regioselective binding can be modulated by carefully choosing their steric hindrance,124 whereby bulky groups achieve preferential binding at adatom and high curvature sites, such as step edges and tips.124,184,185 In Section 2.2, we have seen that some level of regioselectivity and anisotropic adsorbate packing is observed across a multitude of adsorbates, due to the interplay between surface curvature, different crystallographic-dependent affinities and packing, and overall surface coverage.124–127,130–132 However, a finer control and modulation of regioselectivity via the identity of the substituents flanking the ligand binding site as enabled by NCH chemistry is not achievable with common α–ω adsorbates, such as the typical alkylthiols utilized in SERS-relevant nanofabrication (e.g., α–ω mercaptoamines or mercaptocarboxylates).
In common α–ω thiol ligands, the position of substituents is opposite to the ligand binding direction, and prevents the establishment of the nonbonding interactions between the surface and the binding site, which indeed appear to be the crucial contributors to NHC regioselectivity control.124 On this subject, it must be recalled that the same nonbonding contributions to surface interaction generate a trade-off between regioselectivity and bond strength, because the NHC–metal bond strength decreases with increasing steric bulk of the so-called wingtip groups.124,185 Consequently, the orientation of the NHC ligand to the surface, as well as its mobility and propensity to deform the outermost atomic layer of the metal surface, can also be controlled via a careful choice of the identity of the N-substituents.185,186 Recent scanning tunnelling microscopy (STM) observations and DFT simulations on differently substituted 1,3-dialkylbenzimidazol-2-ylidenes NHCs (Fig. 10b) have highlighted this trend, where the presence of non-bulky wingtip groups such as asymmetric isopropyl and ethyl N-substituents result in a higher mobility of the ligand compared to minimally bulkier tert-butyl substituents.185 Moreover, they have thermodynamically preferred flat conformations at the surface, and tend to cause adatom abstraction and complexation as desorbed free molecules when they interact with Au(111) surfaces.185
The orientation on the surface of colloidal gold nanoparticles is however more complex than what has been elucidated for non-colloidal gold surfaces, and recent studies based on SERS and accompanying DFT calculations demonstrated that both flat and upright conformations are present on spherical nanogold across different benzimidazolium NHCs of varying steric bulk of the wingtip group (i.e., methyl, ethyl, hexyl, and tert-butyl).187 DFT calculations of putative upright- and flat-oriented complexes on Au58 clusters show that flat configurations at the surface are clearly preferred for all structures, with adsorption energies that are 10 to 30 kcal mol−1 more negative compared to those calculated for upright model complexes (Fig. 10c, top); however, the SERS spectra show the coexistence of the two orientations (Fig. 10c, bottom), probably resulting from the stabilization of a small population of the less thermodynamically favored upright adsorbates via kinetic trapping or solvent effects.187 The comparison among the experimental SERS spectrum and the DFT-simulated spectra obtained by averaging the contributions of upright- and flat-oriented populations with varying relative abundancies showed that the N-methyl NHC/Au nanoparticle system is consistent with a 1:25 upright:flat ratio,187 in line with previously discussed results on Au(111) surfaces and NHCs bearing N-substituents with low steric hindrance.185 On the contrary, the N-tert-butyl NHC/Au nanoparticle system exhibited opposite behavior to what has been reported for Au(111) surfaces,185 with a best match of the experimental SERS spectrum of the functionalized nanoparticle system with the DFT-simulated one that was obtained by modeling a 1:10 upright:flat orientation ratio, and presence of experimental SERS bands that are unambiguously related to the flat NHC configuration, such as those centered at 929 and 1448 cm−1 (Fig. 10c).187 This behavior could be explained by a possible adatom effect, which could accommodate bulky substituents that would otherwise be forced in an upright position on a perfectly flat surface.187 The general importance of elucidating adsorption configuration trends in addition to overall regioselective trends stems from the fact that SERS is intrinsically less sensitive to modes that lie parallel to the plasmon-sustaining surface, thus making the surface orientation preferences of a given ligand non-decouplable from sensing performance when a functionalized system is designed for a defined, analytical scope.
The way NHC ligands can be adsorbed onto colloidal gold nanoparticles is not trivial. As demonstrated by Dominique et al.,188 the known transmetalation reactions commonly utilized in organometallic chemistry to transfer an NHC ligand from a starting metal center (e.g., silver or copper) to gold have more complex dynamics when applied to colloidal gold nanoparticles than in simple organometallic complexes. The computationally-supported multianalytical (XPS, SERS, laser desorption MS) approach utilized by the authors highlighted that proper NHC transmetalation on citrate-capped gold nanoparticles occurs as a complex function of the NHC scaffold (benzimidazole vs. imidazole), the steric bulk of the N-substituents, and the metal center of the starting NHC complex.188 For example, copper complexes of N-diisopropylphenyl-substituted imidazole-NHC are conducive to transmetalation, with transfer of the NHC ligand to a nanoparticle gold adatom, while silver complexes of the same NHC ligand do not undergo transmetalation, but form adducts via the incorporation of the silver center onto the surface of the gold nanoparticle as an alloyed adatom. Alloying of silver into the gold nanoparticle was also observed with silver complexes of N-isopropyl-substituted imidazole-NHC, but in this case proper transfer of the NHC to gold and formation of an NHC–Au bond occurred.188 Readers interested in NHC-functionalization of SERS-active colloidal nanoparticles should therefore be aware that this chemistry might not only alter the original adsorbate landscape at the nanoparticle surface, but it can also change the nature, and thus, the electronic structure of the metallic surface domain.
It is probably evident at this point that the fine mechanistic details of NHC chemistry for plasmonic nanoparticle engineering offers tremendous room for exploration, and research must be pushed such that we can paint a more rounded and realistic picture of the strengths and limitations of this class of noble metal modifiers. For example, the groups of Camden, Jenkins, and - Arroyo-Currás189,190 have recently highlighted that the various domains in which NHC adsorbates outperform thiol should not include electrochemical stability. NHC monolayers were in fact shown to exhibit an electrochemical behavior that is similar to thiols’ in the −1.0 to 0.5 V vs. Ag|AgCl voltage range,190 while outside it, NHC molecules cathodically desorb at lower bias than thiols, and anodically desorb at analogous bias.189 Moreover, susceptibility to degradation via continuous cycling and consequent operational lifetime were found to be a function of the NHC backbone.190 These findings evidence limitations that must be taken into account for a robust implementation of these surface architectures in applications such as electrochemical SERS.
4. Alternative surface chemistries: Semiconductors
In bioanalytical SERS, it is not infrequent to find gold nanoparticles or SERS nanotags that are encapsulated by a silica shell, with the scope of enhancing the stability and biocompatibility of the system itself in biological media.148 Obviously, the presence of a SiO2 shell makes it such that the chemical environment at the solid/solution interface is different from that of the native, plasmonic-only nanoparticle—and the differences are substantial. The first that comes to mind is the way metal oxides develop a charge in solution. As opposed to what has been discussed for noble metal nanoparticles, which are zero-valent and develop a charge in solution as a consequence of the charged adsorbed species on their surface, metal oxide surfaces can be seen as a network of undercoordinated metal cations and oxygen anions; as a result, most metal oxides are amphoteric in solution, and their charge can be switched upon pH modulation.37 Certainly, in a colloidal environment, adsorbed species will populate the surface of metal oxides, and they will modulate the overall surface charge of the nanomaterial depending on their identity. However, it is important to remember that the thermodynamic trends that are observed for plasmonic nanomaterials, and thus, the types and strengths of the interactions between the surface and the adsorbates that are commonly found in SERS applications, might not translate identically on the two materials. For example, the electronegativity of the anionic component of the metal oxide lattice (i.e., oxygen) is such that, among halides, only fluoride is able to form strong bonds with the surface metal atoms.113 This feature is distinctive of metal oxides and is sensibly different from the halide binding trends reported for traditional plasmonic nanomaterials like gold and silver (see Section 3.1).When considering emerging semiconductor materials for SERS substrates, for example metal chalcogenides like MoS2191 and CuTe,192 differences in the electronegativity of the anionic component of their lattices compared to oxygen in metal oxides makes it such that these surfaces do form strong bonds with all halides via their cationic surface lattice component.113 While it is beyond the scope of this review to discuss the details of the surface chemistries of all of the many semiconductor nanomaterials that are nowadays starting to find application in SERS,148 this section wants to reiterate that, the control and exploitation to full potential of any surface-specific phenomenon must pass by the understanding of the surface chemistry that enables it, and SERS is not an exception to this rule of thumb. When semiconductors are exploited as SERS substrates, either as a shell or decoration in a composite material or as the sole constituent, this necessity becomes even more stringent, because the chemical mechanism will be the preponderant phenomenon in enabling the signal enhancement of the analyte. Whenever an interposed semiconductor shell or decoration is present on top of a plasmonic core, and even more so, when a semiconductor is utilized as a SERS substrate in the absence of a plasmonic constituent, the chemical enhancement mechanism relies on the interaction between the conduction band of the semiconductor and the HOMO and LUMO of the analyte, dictating the necessity for analyte-semiconductor chemisorption. Consequently, the exploitation of semiconductor and composite semiconductor-plasmonic materials as SERS substrates and the rational development of analytical methods based on them cannot disregard the study of their surface chemistry. On the contrary, the understanding of the surface interactions that occur between the nanoparticle and the molecular analyte is essential and must come first and foremost, and a careful, ad hoc surface-analyte match is essential.
5. Conclusions and perspectives
Surfaces are well known to be complex entities that are extremely difficult to study, and consequently, any phenomenon that is somewhat related to them inherits similar challenges in fully accessing and understanding it. When we transpose this concept to SERS, we must also deal with the challenges imposed by the nanoscale dimension, which are not only difficult to envision, but often also extremely complex to study. However, we often find ourselves stuck by constraints that are dictated by an unchallenging attitude towards the status quo, rather than an actual lack of experimental or theoretical means to access new or more extensive information on nanoscale surfaces. For example, the dynamics with which a capping agent interacts with the substrate in the presence of an analyte is a very complex matter, and typically, over the course of the years, contrasting or contradicting hypotheses have been formulated, especially when experimental techniques to directly measure the phenomena have remained unavailable or unestablished. Nevertheless, despite the actual and substantial improvement of our knowledge in the field, some of these hypotheses seem to have passed on from scientist to scientist in the form of a ground truth, contributing to further contradictions along the line. This “old wives’ tale” type of approach has in turn been fueling the reputation of unreliable technique that SERS seems to still have among scientists, oddly enough even within the SERS scientific community. In this review, we aimed at revitalizing old SERS literature in the light of our current understanding of the nanoscale, and connecting it to literature collected from neighboring fields, centralizing the aspects of surface chemistry to create a methodological paradigm that can be utilized to rationally approach method development and quantitatively dispel the myth. In our view, a centralization of surface chemistry considerations can tangibly contribute to robust, reproducible, and physically meaningful analytical SERS results, thus contrasting the backlash that this technique has accumulated over the years.Because SERS is a highly multidisciplinary technique, it is important to keep in mind that sister sciences, such as energy and catalysis, or even apparently unrelated ones, such as food chemistry, can sometimes be explored to obtain crucial pieces of information, as demonstrated, for instance, for sodium borohydride and L-ascorbic acid degradation, respectively. For example, catalysis literature should be seen as an important source from which to gather specific information on the surface chemistry of metal oxides and semiconductors in general, as surface structures, defects, and adsorption of reactants on these substrates are core topics in the field. This information can be then suitably translated to a SERS context and wisely leveraged to extrapolate fundamental key points in the study of surface–molecule interactions among an analyte and a semiconductor-based SERS substrate. Similarly, keeping on with the comparison among different scientific fields, the SERS community should look in detail at the history of the growth of mass spectrometry as the reliable analytical technique that it is today. If one looks closely, in fact, differently generated SERS spectra, obtained from different interactions of the analyte under study with the SERS substrate, are different in the same way that differently generated fragmentation patterns in mass spectrometry are different. We have seen that this has been a non-problem in mass spectrometry: so, why should it be a problem in SERS?
Reciprocally, we could leverage our knowledge in SERS to benefit sister sciences and processes, such as nanomaterials chemistry and the rational bottom-up synthesis of new SERS colloidal nanoparticles. Knowledge of interaction modes and energetics, and their role in defining the relative velocity of deposition and diffusion of as-reduced metallic atoms during the growth of a plasmonic nanoparticle would allow us to tailor the synthesis process from the start, instead of performing ligand exchange, hoping for it to be successful without incurring in destabilization and precipitation of the nanoparticle during the process. The approach of learning from and informing other fields should be purposely leveraged to generate a feedback loop enhancement of knowledge in the materials chemistry and analytical chemistry sides of SERS research, thus allowing for a more rapid solution of long-standing issues, as well as an effective, productive translation of SERS in the industrial and medical fields.
Finally, while looking sideways in neighboring fields, we should also look backwards into them, exploring the rich knowledge collected by our colleagues in the past. One of such instances is the field of self-assembled monolayers (SAM), an area of science that has been extremely productive in the 1990s.193 SAMs are by nature ubiquitous in nanoparticles, even though nowadays we tend not to call them as such anymore. Rapidly assembling from molecules in solution on a metallic surface, they rearrange over time to reach their thermodynamically stable state, if allowed, or local potential minima in constrained situations. Their stability, or lack thereof, on the surface of metal nanoparticles determines successful colloidal suspension, functionalization, ligand exchange, and all the other phenomena that we have described throughout this review. Over the years, mixed SAMs have attracted much attention and have been studied in depth because of their technological potential.194–196 However, in SERS, SAMs, and most importantly mixed SAMs, while actively engineered when fabricating tags,197 are only seldom taken into consideration when interpreting direct SERS results or when hypothesizing a plausible adsorbate population in direct SERS samples. This approach appears almost as if it were dictated by a fear of acknowledging the surface complexity of SERS samples, as if its existence could detract from the validity of the measurement and the technique as a whole, and as if the need of addressing it could “scare away” possible investors interested in bringing the technology to the market. In our opinion, this vision is underselling the SERS community and its expertise. In fact, while it is true that several issues need to be addressed to effectively bring SERS to the market, most of which related to our understanding of the surface, as we have seen, it is also true that we, as a community, have the means and knowledge to collectively address them. All we need to do is to define these issues, understand them, and overcome them.
We believe that now is the moment to go back to the fundamentals, revisit SERS with a new lens, and make it more robust and reliable, ultimately enabling its true translatability to routine industrial and clinical applications. With this review, we thus ultimately hope to promote the incorporation of surface chemistry considerations into SERS method development and its use as a tool to explore future directions of SERS that are progressively less and less based on trial and error.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no 865819). CD and LF acknowledge photographers Cristián Garnier and Paolo Properzi, respectively, for the headshots in the biography section.References
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem. Interfacial Electrochem., 1977, 84, 1–20 CAS.
- S. Atta, T. V. Tsoulos and L. Fabris, J. Phys. Chem. C, 2016, 120, 20749–20758 CAS.
- J. R. Lombardi, R. L. Birke, T. Lu and J. Xu, J. Chem. Phys., 1986, 84, 4174–4180 CAS.
- D.-B. Grys, B. de Nijs, A. R. Salmon, J. Huang, W. Wang, W.-H. Chen, O. A. Scherman and J. J. Baumberg, ACS Nano, 2020, 14, 8689–8696 CrossRef CAS PubMed.
- N. C. Lindquist and A. G. Brolo, J. Phys. Chem. C, 2021, 125, 7523–7532 CrossRef CAS.
- M. Cioni, D. Polino, D. Rapetti, L. Pesce, M. Delle Piane and G. M. Pavan, J. Chem. Phys., 2023, 158, 124701 CrossRef CAS PubMed.
- M. Yamashita and J. B. Fenn, J. Phys. Chem., 1984, 88, 4451–4459 CrossRef CAS.
- J. Griffiths, Anal. Chem., 2008, 80, 5678–5683 CrossRef CAS PubMed.
- K. T. Carron and B. J. Kennedy, Anal. Chem., 1995, 67, 3353–3356 CrossRef CAS.
- B. Zachhuber, C. Carrillo-Carrión, B. M. Simonet Suau and B. Lendl, J. Raman Spectrosc., 2012, 43, 998–1002 CrossRef CAS.
- W. W. Yu and I. M. White, Analyst, 2013, 138, 3679 RSC.
- M. V. Cañamares, D. A. Reagan, J. R. Lombardi and M. Leona, J. Raman Spectrosc., 2014, 45, 1147–1152 CrossRef.
- A. Subaihi, D. K. Trivedi, K. A. Hollywood, J. Bluett, Y. Xu, H. Muhamadali, D. I. Ellis and R. Goodacre, Anal. Chem., 2017, 89, 6702–6709 CrossRef CAS PubMed.
- Y.-H. Lo and H. Hiramatsu, Anal. Chem., 2020, 92, 14601–14607 CAS.
- L. Xiao, C. Wang, C. Dai, L. E. Littlepage, J. Li and Z. D. Schultz, Angew. Chem., Int. Ed., 2020, 132, 3467–3471 CrossRef.
- S. Jin, D. Zhang, B. Yang, S. Guo, L. Chen and Y. M. Jung, Analyst, 2024, 149, 11–28 RSC.
- A. Ten, V. Lomonosov, C. Boukouvala and E. Ringe, ACS Nano, 2024, 18, 18785–18799 CrossRef CAS PubMed.
- Z. Li, X. Huang and G. Lu, J. Mater. Chem. C, 2020, 8, 3956–3969 RSC.
- C. Li, Y. Huang, X. Li, Y. Zhang, Q. Chen, Z. Ye, Z. Alqarni, S. E. J. Bell and Y. Xu, J. Mater. Chem. C, 2021, 9, 11517–11552 RSC.
- Á. I. López-Lorente, Anal. Chim. Acta, 2021, 1168, 338474 CrossRef PubMed.
- S. Yadav and J. Satija, J. Mater. Chem. B, 2021, 9, 267–282 RSC.
- X. Wang, E. Zhang, H. Shi, Y. Tao and X. Ren, Analyst, 2022, 147, 1257–1272 CAS.
- O. Guselnikova, H. Lim, H. Kim, S. H. Kim, A. Gorbunova, M. Eguchi, P. Postnikov, T. Nakanishi, T. Asahi, J. Na and Y. Yamauchi, Small, 2022, 18(25), 2107182 CAS.
- G. A. Vinnacombe-Willson, Y. Conti, A. Stefancu, P. S. Weiss, E. Cortés and L. Scarabelli, Chem. Rev., 2023, 123, 8488–8529 CAS.
- Y. Ying, Z. Tang and Y. Liu, Nanoscale, 2023, 15, 10860–10881 CAS.
- P. Mulvaney, Metal Nanoparticles: Double Layers, Optical Properties, and Electrochemistry, in Nanoscale Materials in Chemistry, ed. K. J. Klabunde, John Wiley & Sons, Inc., New York, USA, 2001, pp. 121–167 Search PubMed.
- R. Aroca, Surface-Enhanced Vibrational Spectroscopy, John Wiley & Sons, Ltd, Chichester, UK, 2006 Search PubMed.
- G. Corthey, L. J. Giovanetti, J. M. Ramallo-López, E. Zelaya, A. A. Rubert, G. A. Benitez, F. G. Requejo, M. H. Fonticelli and R. C. Salvarezza, ACS Nano, 2010, 4, 3413–3421 CAS.
- C. J. Orendorff and C. J. Murphy, J. Phys. Chem. B, 2006, 110, 3990–3994 CAS.
- H. Al-Johani, E. Abou-Hamad, A. Jedidi, C. M. Widdifield, J. Viger-Gravel, S. S. Sangaru, D. Gajan, D. H. Anjum, S. Ould-Chikh, M. N. Hedhili, A. Gurinov, M. J. Kelly, M. El Eter, L. Cavallo, L. Emsley and J. M. Basset, Nat. Chem., 2017, 9, 890–895 Search PubMed.
- C. Deriu, A. Bracho and B. McCord, J. Phys. Chem. C, 2022, 126, 2023–2040 Search PubMed.
- H. Xia, Y. Xiahou, P. Zhang, W. Ding and D. Wang, Langmuir, 2016, 32, 5870–5880 CAS.
- L. Scarabelli, A. Sánchez-Iglesias, J. Pérez-Juste and L. M. Liz-Marzán, J. Phys. Chem. Lett., 2015, 6, 4270–4279 CAS.
- M. R. Langille, M. L. Personick, J. Zhang and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 14542–14554 CrossRef CAS PubMed.
- S. Atta, M. Beetz and L. Fabris, Nanoscale, 2019, 11, 2946–2958 CAS.
- D. H. Everett, Basic Principles of Colloid Science, RSC Paperbacks, Royal Society of Chemistry, Cambridge, 1st edn, 1988 Search PubMed.
- N. E. Larm, J. A. Thon, Y. Vazmitsel, J. L. Atwood and G. A. Baker, Nanoscale Adv., 2019, 1, 4665–4668 CAS.
- M. R. Mirghafouri, S. Abbasi-Moayed, F. Ghasemi and M. R. Hormozi-Nezhad, Anal. Methods, 2020, 12, 5877–5884 CAS.
- A. Sharma, S. Sarmah, A. S. Roy and K. S. Ghosh, Luminescence, 2022, 37, 1200–1207 CrossRef CAS PubMed.
- A. D’Ulivo, Spectrochim. Acta, Part B, 2010, 65, 360–375 CrossRef.
- J. Andrieux, U. B. Demirci, J. Hannauer, C. Gervais, C. Goutaudier and P. Miele, Int. J. Hydrogen Energy, 2011, 36, 224–233 CrossRef CAS.
- T. Mondal, A. Sermiagin, D. Meyerstein, T. Zidki and H. Kornweitz, Nanoscale, 2020, 12, 1657–1672 RSC.
- H. N. Abdelhamid, Int. J. Hydrogen Energy, 2021, 46, 726–765 CrossRef CAS.
- R. Retnamma, A. Q. Novais and C. M. Rangel, Int. J. Hydrogen Energy, 2011, 36, 9772–9790 CrossRef CAS.
- D. Alligier, E. Petit and U. B. Demirci, Int. J. Hydrogen Energy, 2022, 47, 23310–23315 CrossRef CAS.
- J. A. Creighton, C. G. Blatchford and M. G. Albrecht, J. Chem. Soc., Faraday Trans. 2, 1979, 75, 790–798 RSC.
- D. Van Lierop, Ž. Krpetić, L. Guerrini, I. A. Larmour, J. A. Dougan, K. Faulds and D. Graham, Chem. Commun., 2012, 48, 8192–8194 Search PubMed.
- H. I. Schlesinger, H. C. Brown, A. E. Finholt, J. R. Gilbreath, H. R. Hoekstra and E. K. Hyde, J. Am. Chem. Soc., 1953, 75, 215–219 CrossRef CAS.
- G. Guella, B. Patton and A. Miotello, J. Phys. Chem. C, 2007, 111, 18744–18750 CrossRef CAS.
- S. Atta, S. Rangan and L. Fabris, ChemNanoMat, 2020, 6, 53–57 CrossRef CAS.
- A. Sermiagin, D. Meyerstein, G. S. Rolly, T. Mondal, H. Kornweitz and T. Zidki, Int. J. Hydrogen Energy, 2022, 47, 3972–3979 CrossRef CAS.
- L. Mulfinger, S. D. Solomon, M. Bahadory, A. V. Jeyarajasingam, S. A. Rutkowsky and C. Boritz, J. Chem. Educ., 2007, 84, 322 CrossRef.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- G. Frens, Nature (London), Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- I. Ojea-Jiménez, N. G. Bastús and V. Puntes, J. Phys. Chem. C, 2011, 115, 15752–15757 CrossRef.
- N. G. Bastus, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098–11105 CrossRef CAS PubMed.
- F. Schulz, T. Homolka, N. G. Bastús, V. Puntes, H. Weller and T. Vossmeyer, Langmuir, 2014, 30, 10779–10784 CrossRef CAS PubMed.
- C. H. Munro, W. E. Smith, M. Garner, J. Clarkson and P. C. White, Langmuir, 1995, 11, 3712–3720 Search PubMed.
- S. Kumar, K. S. Gandhi and R. Kumar, Ind. Eng. Chem. Res., 2007, 46, 3128–3136 CrossRef CAS.
- C. Xue, G. S. Métraux, J. E. Millstone and C. A. Mirkin, J. Am. Chem. Soc., 2008, 130, 8337–8344 Search PubMed.
- I. Ojea-Jiménez, F. M. Romero, N. G. Bastús and V. Puntes, J. Phys. Chem. C, 2010, 114, 1800–1804 CrossRef.
- M. Doyen, K. Bartik and G. Bruylants, J. Colloid Interface Sci., 2013, 399, 1–5 CrossRef CAS PubMed.
- J.-W. Park and J. S. Shumaker-Parry, J. Am. Chem. Soc., 2014, 136, 1907–1921 CrossRef CAS PubMed.
- D. Oliveira de Souza, J. Girardon, D. J. Hoffmann and E. Berrier, Chem. Phys. Chem., 2023, 24, e202200744 CrossRef CAS PubMed.
- D. Grasseschi, R. A. Ando, H. E. Toma and V. M. Zamarion, RSC Adv., 2015, 5, 5716–5724 RSC.
- N. Leopold and B. Lendl, J. Phys. Chem. B, 2003, 107, 5723–5727 CrossRef CAS.
- T. H. James, J. Am. Chem. Soc., 1939, 61, 2379–2383 CrossRef CAS.
- C. P. Lloyd and W. F. Pickering, J. Inorg. Nucl. Chem., 1967, 29, 1907–1914 CAS.
- A. Torres-Nuñez, K. Faulds, D. Graham, R. A. Alvarez-Puebla and L. Guerrini, Analyst, 2016, 141, 5170–5180 Search PubMed.
- S. Sánchez-Cortés and J. V. García-Ramos, J. Raman Spectrosc., 1998, 29, 365–371 CrossRef.
- Q. Huang, D. Qin and Y. Xia, Chem. Sci., 2024, 15, 6321–6330 RSC.
- S. D. Perrault and W. C. W. Chan, J. Am. Chem. Soc., 2009, 131, 17042–17043 CrossRef CAS PubMed.
- J. Li, J. Wu, X. Zhang, Y. Liu, D. Zhou, H. Sun, H. Zhang and B. Yang, J. Phys. Chem. C, 2011, 115, 3630–3637 CrossRef CAS.
- D. Kumar, I. Mutreja and P. Sykes, Nanotechnology, 2016, 27, 355601 CrossRef PubMed.
- T. K. Sarma and A. Chattopadhyay, Langmuir, 2004, 20, 3520–3524 CrossRef CAS PubMed.
- K. Pacławski and K. Fitzner, Metall. Mater. Trans. B, 2006, 37, 703–714 Search PubMed.
- S. A. Akintelu, S. C. Olugbeko and A. S. Folorunso, Int. Nano Lett., 2020, 10, 237–248 CAS.
- F. Sahin, N. Celik, A. Camdal, M. Sakir, A. Ceylan, M. Ruzi and M. S. Onses, ACS Appl. Nano Mater., 2022, 5, 13112–13122 CAS.
- R. D. Ávila-Avilés, M. A. Camacho-López, I. G. Becerril-Juárez, E. Castro-Longoria and A. R. Vilchis-Nestor, MRS Adv., 2022, 7, 12–17 Search PubMed.
- V. Dzhagan, N. Mazur, O. Smirnov, O. Yeshchenko, O. Isaieva, M. Kovalenko, M. Vuichyk, M. Skoryk, Y. Pirko, A. Yemets, V. Yukhymchuk and M. Valakh, J. Nanopart. Res., 2023, 25, 37 CAS.
- X. Li, X. Mao, W. Xie, B. Liu and F. Chen, ACS Sustainable Chem. Eng., 2022, 10, 4872–4880 CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, J. Phys. Chem. B, 2001, 105, 4065–4067 CAS.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CAS.
- H. Yuan, C. G. Khoury, H. Hwang, C. M. Wilson, G. A. Grant and T. Vo-Dinh, Nanotechnology, 2012, 23, 075102 Search PubMed.
- S. He, M. W. C. Kang, F. J. Khan, E. K. M. Tan, M. A. Reyes and J. C. Y. Kah, J. Opt., 2015, 17, 114013–114026 Search PubMed.
- C. Wu, X. Zhou and J. Wei, Nanoscale Res. Lett., 2015, 10, 354 Search PubMed.
- C. S. Johnston, F. M. Steinberg and R. B. Rucker, in Handbook of Vitamins, ed. R. B. Rucker, J. W. Suttie, D. B. McCormick and L. J. Machlin, Marcel Dekker, Inc., Basel, 3rd edn, 2001, pp. 529–554 Search PubMed.
- J. Mandl, A. Szarka and G. Bánhegyi, Br. J. Pharmacol., 2009, 157, 1097–1110 CAS.
- A. C. Caritá, B. Fonseca-Santos, J. D. Shultz, B. Michniak-Kohn, M. Chorilli and G. R. Leonardi, Nanomedicine, 2020, 24, 102117 CrossRef PubMed.
- R. C. Kerber, J. Chem. Educ., 2008, 85, 1237 CrossRef CAS.
- D. Njus, P. M. Kelley, Y.-J. Tu and H. B. Schlegel, Free Radical Biol. Med., 2020, 159, 37–43 CrossRef CAS PubMed.
- B. A. Wagner and G. R. Buettner, Adv. Redox Res., 2023, 9, 100077 CrossRef CAS PubMed.
- L. Stojanov and V. Mirčeski, J. Electrochem. Soc., 2023, 170, 065504 CrossRef CAS.
- L. Malassis, R. Dreyfus, R. J. Murphy, L. A. Hough, B. Donnio and C. B. Murray, RSC Adv., 2016, 6, 33092–33100 RSC.
- R. Ye, M. Zhao, X. Mao, Z. Wang, D. A. Garzón, H. Pu, Z. Zhao and P. Chen, Nat. Commun., 2021, 12, 4287 CAS.
- J. Xiong, Y. Wang, Q. Xue and X. Wu, Green Chem., 2011, 13, 900 CAS.
- J. Velíšek, J. Davídek and G. Janíček, Collect. Czech. Chem. Commun., 1972, 37, 1465–1470 Search PubMed.
- I. Koshiishi, Y. Mamura and T. Imanari, Biochim. Biophys. Acta, Gen. Subj., 1998, 1379, 257–263 CAS.
- J. Pullar, S. Bayer and A. Carr, Antioxidants, 2018, 7, 29 Search PubMed.
- K. Pfeilsticker, F. Marx and M. Bockisch, Carbohydr. Res., 1975, 45, 269–274 CAS.
- B. M. Tolbert and J. B. Ward, Dehydroascorbic Acid, ed. P. A. Seib and B. M. Tolbert, American Chemical Society, 1982, pp. 101–123 Search PubMed.
- T. Kurata and Y. Nishikawa, Biosci., Biotechnol., Biochem., 2000, 64, 1651–1655 CAS.
- Y.-J. Tu, D. Njus and H. B. Schlegel, Org. Biomol. Chem., 2017, 15, 4417–4431 CAS.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS PubMed.
- S. K. Meena and M. Sulpizi, Angew. Chem., Int. Ed., 2016, 55, 11960–11964 CrossRef CAS PubMed.
- J. A. Da Silva and M. R. Meneghetti, Langmuir, 2018, 34, 366–375 CrossRef CAS PubMed.
- J. A. Edgar, A. M. McDonagh and M. B. Cortie, ACS Nano, 2012, 6, 1116–1125 CrossRef CAS PubMed.
- J. A. da Silva, P. A. Netz and M. R. Meneghetti, Langmuir, 2020, 36, 257–263 CrossRef CAS PubMed.
- B. Nikoobakht and M. A. El-Sayed, Langmuir, 2001, 17, 6368–6374 CrossRef CAS.
- S. Gómez-Graña, F. Hubert, F. Testard, A. Guerrero-Martínez, I. Grillo, L. M. Liz-Marzán and O. Spalla, Langmuir, 2012, 28, 1453–1459 CrossRef PubMed.
- S. Ghosh and L. Manna, Chem. Rev., 2018, 118, 7804–7864 CrossRef CAS PubMed.
- A. Migani and F. Illas, J. Phys. Chem. B, 2006, 110, 11894 CAS.
- P. Gao and M. J. Weaver, J. Phys. Chem., 1986, 90, 4057–4063 CAS.
- T. Zech, T. Schmutzler, D. M. Noll, M. S. Appavou and T. Unruh, Langmuir, 2022, 38, 2227–2237 CAS.
- F. Calle-Vallejo, M. Huang, J. B. Henry, M. T. M. Koper and A. S. Bandarenka, Phys. Chem. Chem. Phys., 2013, 15, 3196 CAS.
- J. P. Palafox-Hernandez, Z. Tang, Z. E. Hughes, Y. Li, M. T. Swihart, P. N. Prasad, T. R. Walsh and M. R. Knecht, Chem. Mater., 2014, 26, 4960–4969 CAS.
- S. Haseena, R. M. Kumar, V. Rajapandian and V. Subramanian, Appl. Surf. Sci., 2019, 487, 1409–1419 CAS.
- C. Deriu, A. N. Morozov and A. M. Mebel, J. Phys. Chem. A, 2022, 126, 5236–5251 CAS.
- K. M. R. Scher, Z. Wang, A. Nair, Y. Wu, M. Bartoli, M. Rovere, A. Tagliaferro, S. Rangan, L. Wang and L. Fabris, J. Phys. Chem. C, 2022, 126, 16499–16513 CAS.
- F. Silva and A. Martins, J. Electroanal. Chem., 1999, 467, 335–341 CAS.
- D. W. Mulder, M. M. Phiri, A. Jordaan and B. C. Vorster, R. Soc. Open Sci., 2019, 6, 190160 CAS.
- Y. Kim, S. Ji and J.-M. Nam, Acc. Chem. Res., 2023, 56, 2139–2150 CrossRef CAS PubMed.
- Z. Nie, D. Fava, M. Rubinstein and E. Kumacheva, J. Am. Chem. Soc., 2008, 130, 3683–3689 CrossRef CAS PubMed.
- X. Kou, Z. Sun, Z. Yang, H. Chen and J. Wang, Langmuir, 2009, 25, 1692–1698 CrossRef CAS PubMed.
- J. G. Hinman, J. R. Eller, W. Lin, J. Li, J. Li and C. J. Murphy, J. Am. Chem. Soc., 2017, 139, 9851–9854 CrossRef CAS PubMed.
- N. R. Jana, L. Gearheart, S. O. Obare and C. J. Murphy, Langmuir, 2002, 18, 922–927 CrossRef CAS.
- K. K. Caswell, J. N. Wilson, U. H. F. Bunz and C. J. Murphy, J. Am. Chem. Soc., 2003, 125, 13914–13915 CAS.
- B. E. Janicek, J. G. Hinman, J. J. Hinman, S. Hyun Bae, M. Wu, J. Turner, H.-H. Chang, E. Park, R. Lawless, K. S. Suslick, C. J. Murphy and P. Y. Huang, Nano Lett., 2019, 19, 6308–6314 CAS.
- S. Hyun Bae, P. Huang, K. Lee and T. Odom, Microsc. Microanal., 2021, 27, 3320–3322 Search PubMed.
- T. Jiang, J. Zong, Y. Feng and H. Chen, Precis. Chem., 2023, 1, 94–99 CAS.
- R. A. Alvarez-Puebla and L. M. Liz-Marzán, Chem. Soc. Rev., 2012, 41, 43–51 CAS.
- P. A. Hassan and S. L. Gawali, Langmuir, 2019, 35, 9635–9646 CAS.
- T.-H. Yang, J. Ahn, S. Shi and D. Qin, ACS Nano, 2021, 15, 14242–14252 CAS.
- Y. Gao, P. Jiang, D. F. Liu, H. J. Yuan, X. Q. Yan, Z. P. Zhou, J. X. Wang, L. Song, L. F. Liu, W. Y. Zhou, G. Wang, C. Y. Wang, S. S. Xie, J. M. Zhang and D. Y. Shen, J. Phys. Chem. B, 2004, 108, 12877–12881 CAS.
- W. A. Saidi, H. Feng and K. A. Fichthorn, J. Phys. Chem. C, 2013, 117, 1163–1171 CAS.
- C.-H. Kuo, T.-F. Chiang, L.-J. Chen and M. H. Huang, Langmuir, 2004, 20, 7820–7824 CAS.
- P. Pallavicini, A. Donà, A. Casu, G. Chirico, M. Collini, G. Dacarro, A. Falqui, C. Milanese, L. Sironi and A. Taglietti, Chem. Commun., 2013, 49, 6265–6267 CAS.
- P. Vijayaraghavan, C.-H. Liu and K. C. Hwang, ACS Appl. Mater. Interfaces, 2016, 8, 23909–23919 CAS.
- J. J. Velázquez-Salazar, L. Bazán-Díaz, Q. Zhang, R. Mendoza-Cruz, L. Montaño-Priede, G. Guisbiers, N. Large, S. Link and M. José-Yacamán, ACS Nano, 2019, 13, 10113–10128 Search PubMed.
- P. Matejka, B. Vlckova, J. Vohlidal, P. Pancoska and V. Baumruk, J. Phys. Chem., 1992, 96, 1361–1366 CrossRef CAS.
- F. Fiévet, S. Ammar-Merah, R. Brayner, F. Chau, M. Giraud, F. Mammeri, J. Peron, J.-Y. Piquemal, L. Sicard and G. Viau, Chem. Soc. Rev., 2018, 47, 5187–5233 RSC.
- I. Washio, Y. Xiong, Y. Yin and Y. Xia, Adv. Mater., 2006, 18, 1745–1749 CrossRef CAS.
- D. S. B. Gomes, L. G. Paterno, A. B. S. Santos, A. V. Garay, D. Mertz, S. M. Freitas and M. A. G. Soler, J. Mol. Liq., 2018, 268, 181–189 CrossRef CAS.
- T. L. Braga, P. C. V. Conrado, L. G. Z. Silva, E. V. Bergmann, A. C. P. da Silva, P. de Souza Bonfim de Mendonça, J. M. G. Mikcha, L. S. Herculano, L. C. Malacarne, E. C. I. Muniz, C. F. de Freitas and W. Caetano, J. Mol. Liq., 2023, 383, 122111 CrossRef CAS.
- M. Sokolsky-Papkov and A. Kabanov, Polymers, 2019, 11, 1553 CrossRef CAS PubMed.
- C. Deriu, S. Thakur, O. Tammaro and L. Fabris, Nanoscale Adv., 2023, 5, 2132–2166 CAS.
- F. Benz, M. K. Schmidt, A. Dreismann, R. Chikkaraddy, Y. Zhang, A. Demetriadou, C. Carnegie, H. Ohadi, B. de Nijs, R. Esteban, J. Aizpurua and J. J. Baumberg, Science, 1979, 2016(354), 726–729 Search PubMed.
- J. J. Baumberg, Nano Lett., 2022, 22, 5859–5865 CAS.
- N. C. Lindquist, C. D. L. de Albuquerque, R. G. Sobral-Filho, I. Paci and A. G. Brolo, Nat. Nanotechnol., 2019, 14, 981–987 CAS.
- N. C. Lindquist, A. T. Bido and A. G. Brolo, J. Phys. Chem. C, 2022, 126, 7117–7126 CAS.
- S. Sloan-Dennison, C. M. Zoltowski, P. Z. El-Khoury and Z. D. Schultz, J. Phys. Chem. C, 2020, 124, 9548–9558 CAS.
- C. D. L. de Albuquerque, C. M. Zoltowski, B. T. Scarpitti, D. N. Shoup and Z. D. Schultz, ACS Nanosci. Au, 2021, 1, 38–46 CAS.
- T. Kang, S. Hong, Y. Choi and L. P. Lee, Small, 2010, 6, 2649–2652 CAS.
- J. M. Gisbert-González, W. Cheuquepán, A. Ferre-Vilaplana, E. Herrero and J. M. Feliu, J. Electroanal. Chem., 2020, 875, 114015 Search PubMed.
- V. Liamtsau, G. Liu, A. N. Morozov, A. M. Mebel and Y. Cai, Talanta, 2022, 250, 123688 CAS.
- S. Kumaran, V. Vetrivelan, S. Muthu and A. A. Al-Saadi, Heliyon, 2024, 10, e24475 CAS.
- C. J. Cramer, Essentials of computational chemistry: Theory and Models, John Wiley & Sons Ltd, Chichester, 2002 Search PubMed.
- C. Deriu, I. Conticello, A. M. Mebel and B. McCord, Anal. Chem., 2019, 91, 4780–4789 CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CAS.
- Y. Lyu, L. M. Becerril, M. Vanzan, S. Corni, M. Cattelan, G. Granozzi, M. Frasconi, P. Rajak, P. Banerjee, R. Ciancio, F. Mancin and P. Scrimin, Adv. Mater., 2024, 36, 2211624 CAS.
- H. Xu, Z. Chen, S. Hao, K. A. Fichthorn and B. J. Wiley, Nanoscale, 2023, 15, 5219–5229 CAS.
- Y. Liu, J. Chem. Eng. Data, 2009, 54, 1981–1985 CAS.
- X. Mao, C. Liu, M. Hesari, N. Zou and P. Chen, Nat. Chem., 2019, 11, 687–694 CAS.
- Y. Zhang, S. Prabakar and E. C. Le Ru, J. Phys. Chem. C, 2022, 126, 8692–8702 CAS.
- V. Ravi, J. M. Binz and R. M. Rioux, Nano Lett., 2013, 13, 4442–4448 CAS.
- N. Zou, X. Zhou, G. Chen, N. M. Andoy, W. Jung, G. Liu and P. Chen, Nat. Chem., 2018, 10, 607–614 CAS.
- L. Xie, J. Lu, T. Liu, G. Chen, G. Liu, B. Ren and Z. Tian, J. Phys. Chem. Lett., 2020, 11, 1022–1029 CAS.
- L. Fabris, J. Opt., 2015, 17, 114002 CrossRef.
- I. Izquierdo-Lorenzo, J. Kubackova, D. Manchon, A. Mosset, E. Cottancin and S. Sanchez-Cortes, J. Phys. Chem. C, 2013, 117, 16203–16212 CrossRef CAS.
- K. Sokolowska, E. Hulkko, L. Lehtovaara and T. Lahtinen, J. Phys. Chem. C, 2018, 122, 12524–12533 CAS.
- D. J. Lavrich, S. M. Wetterer, S. L. Bernasek and G. Scoles, J. Phys. Chem. B, 1998, 102, 3456–3465 CAS.
- M. Borzenkov, G. Chirico, L. D’Alfonso, L. Sironi, M. Collini, E. Cabrini, G. Dacarro, C. Milanese, P. Pallavicini, A. Taglietti, C. Bernhard and F. Denat, Langmuir, 2015, 31, 8081–8091 CAS.
- C. M. Crudden, J. H. Horton, M. R. Narouz, Z. Li, C. A. Smith, K. Munro, C. J. Baddeley, C. R. Larrea, B. Drevniok, B. Thanabalasingam, A. B. McLean, O. V. Zenkina, I. I. Ebralidze, Z. She, H.-B. Kraatz, N. J. Mosey, L. N. Saunders and A. Yagi, Nat. Commun., 2016, 7, 12654 CAS.
- E. E. Coughlin, J. Hu, A. Lee and T. W. Odom, J. Am. Chem. Soc., 2021, 143, 3671–3676 CAS.
- C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 Search PubMed.
- S. Engel, E.-C. Fritz and B. J. Ravoo, Chem. Soc. Rev., 2017, 46, 2057–2075 CAS.
- W. Kirmse, Angew. Chem., Int. Ed., 2010, 49, 8798–8801 CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CAS.
- H.-W. Wanzlick and E. Schikora, Angew. Chem., 1960, 72, 494 CAS.
- V. P. W. Böhm and W. A. Herrmann, Angew. Chem., 2000, 39, 4036–4038 Search PubMed.
- M. J. MacLeod and J. A. Johnson, J. Am. Chem. Soc., 2015, 137, 7974–7977 CAS.
- M. J. Trujillo, S. L. Strausser, J. C. Becca, J. F. DeJesus, L. Jensen, D. M. Jenkins and J. P. Camden, J. Phys. Chem. Lett., 2018, 9, 6779–6785 CAS.
- A. Inayeh, R. R. K. Groome, I. Singh, A. J. Veinot, F. C. de Lima, R. H. Miwa, C. M. Crudden and A. B. McLean, Nat. Commun., 2021, 12, 4034 CAS.
- A. Bakker, A. Timmer, E. Kolodzeiski, M. Freitag, H. Y. Gao, H. Mönig, S. Amirjalayer, F. Glorius and H. Fuchs, J. Am. Chem. Soc., 2018, 140, 11889–11892 CAS.
- R. L. Thimes, A. V. B. Santos, R. Chen, G. Kaur, L. Jensen, D. M. Jenkins and J. P. Camden, J. Phys. Chem. Lett., 2023, 14, 4219–4224 CrossRef CAS PubMed.
- N. L. Dominique, R. Chen, A. V. B. Santos, S. L. Strausser, T. Rauch, C. Q. Kotseos, W. C. Boggess, L. Jensen, D. M. Jenkins and J. P. Camden, Inorg. Chem. Front., 2022, 9, 6279–6287 RSC.
- M. A. Pellitero, I. M. Jensen, N. L. Dominique, L. C. Ekowo, J. P. Camden, D. M. Jenkins and N. Arroyo-Currás, ACS Appl. Mater. Interfaces, 2023, 15, 35701–35709 CrossRef CAS PubMed.
- N. L. Dominique, A. Chandran, I. M. Jensen, D. M. Jenkins and J. P. Camden, Chem. – Eur. J., 2024, 30, e202303681 CrossRef CAS PubMed.
- J. E. Sanchez, S. A. Jaramillo, E. Settles, J. J. Velazquez Salazar, A. Lehr, J. Gonzalez, C. Rodríguez Aranda, H. R. Navarro-Contreras, M. O. Raniere, M. Harvey, D. M. Wagner, A. Koppisch, R. Kellar, P. Keim and M. Jose Yacaman, RSC Adv., 2021, 11, 25788–25794 RSC.
- W. Li, R. Zamani, P. Rivera Gil, B. Pelaz, M. Ibáñez, D. Cadavid, A. Shavel, R. A. Alvarez-Puebla, W. J. Parak, J. Arbiol and A. Cabot, J. Am. Chem. Soc., 2013, 135, 7098–7101 CrossRef CAS PubMed.
- A. R. Bishop and R. G. Nuzzo, Curr. Opin. Colloid Interface Sci., 1996, 1, 127–136 CrossRef CAS.
- M. J. Wirth, R. W. P. Fairbank and H. O. Fatunmbi, Science, 1979, 1997(275), 44–47 Search PubMed.
- R. K. Smith, P. A. Lewis and P. S. Weiss, Prog. Surf. Sci., 2004, 75, 1–68 CrossRef CAS.
- G. D. Kong, S. E. Byeon, S. Park, H. Song, S. Kim and H. J. Yoon, Adv. Electron. Mater., 2020, 6, 1901157 CrossRef CAS.
- S. Sloan-Dennison, M. R. Bevins, B. T. Scarpitti, V. K. Sauvé and Z. D. Schultz, Analyst, 2019, 144, 5538–5546 RSC.
| This journal is © The Royal Society of Chemistry 2025 |