
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in thermocatalytic acetylene selective hydrogenation
Xiaocheng
Lan
a,
Jingguang G.
Chen
 *b and
Tiefeng
Wang
*a
*b and
Tiefeng
Wang
*a
aDepartment of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: wangtf@tsinghua.edu.cn
bDepartment of Chemical Engineering, Columbia University, New York, NY 10027, USA. E-mail: jgchen@columbia.edu
First published on 15th July 2025
Abstract
Selective acetylene hydrogenation is a crucial reaction for purifying ethylene in the petroleum industry and presents a promising non-oil route for producing ethylene by integrating acetylene production from natural gas and coal. Despite significant advancements in catalyst development, achieving both high catalytic activity and ethylene selectivity remains challenging due to competing side reactions, including over-hydrogenation to ethane, C–C coupling leading to oligomers, and C–C bond cleavage resulting in coke formation. This review provides a comprehensive overview of recent progress in the development of catalysts and understanding of reaction mechanism for acetylene hydrogenation to ethylene. Firstly, benchmarks for conversion and selectivity calculation are critically discussed. Then, research on active site design is categorized into monometallic sites, disordered alloy sites, intermetallic compound (IMC) sites, and single-atom (SA) sites, with a distinction between Pd-based and non-Pd-based catalysts. This categorization highlights the active site design strategies and summarizes state-of-the-art performance metrics. Emphasis is placed on the structure–performance relationships and the role of different active metals in enhancing ethylene selectivity and catalytic activity. In addition, the roles of catalyst support and modifiers are reviewed. Finally, we discuss challenges and future research directions in mechanistic understanding and catalyst design, aiming to guide further innovations in this important field.
 Xiaocheng Lan | Dr Xiaocheng Lan is an Assistant Researcher in the Department of Chemical Engineering at Tsinghua University. He received his bachelor's and doctoral degrees in Chemical Engineering from Tsinghua University in 2014 and 2019, respectively. He worked as a postdoctoral researcher at Tsinghua University from 2019 to 2021. His research focuses on heterogeneous catalysis and slurry bed reactors for fine chemistry and renewable energy. |
 Jingguang G. Chen | Dr Jingguang G. Chen is the Thayer Lindsley Professor of Chemical Engineering at Columbia University, with a joint appointment at Brookhaven National Laboratory. His research interests include fundamental understanding of carbides, nitrides and bimetallic catalysts for applications in thermocatalysis and electrocatalysis. His research group utilizes a combination of experimental studies, in situ characterization and density functional theory calculations. |
 Tiefeng Wang | Dr Tiefeng Wang is a Professor and former Chair of the Department of Chemical Engineering at Tsinghua University. He received his PhD from Tsinghua University in 2004 and was a visiting professor at the University of Delaware from 2010 to 2011. His research focuses on heterogeneous catalysis, multiphase flow reactors, and clean energy chemical engineering. He has published over 280 papers and holds more than 40 patents. He has developed several industrialized technologies, including a partial oxidation reactor for natural gas and a slurry reactor for methyl methacrylate (MMA) synthesis, which have been successfully implemented in industry. |
1. Introduction
The selective hydrogenation of acetylene to ethylene is a catalytic reaction with both significant industrial value and academic interest. In industrial applications, this reaction serves as a crucial ethylene purification technology in the petrochemical industry, selectively removing trace amounts of acetylene impurities from ethylene feedstock to ensure the stability of downstream polymerization processes.1–3 Moreover, its integration with acetylene production from natural gas or coal-based processes offers an alternative route for ethylene production beyond petroleum dependence.4–6 From an academic perspective, this reaction is widely regarded as a model system for selective hydrogenation, providing valuable insights into the structure–activity relationship between active site configurations, surface adsorption behaviors, and reaction selectivity.7–9The primary target product of acetylene hydrogenation is ethylene, while undesired side reactions mainly include the over-hydrogenation to ethane, C–C coupling reactions forming oligomers, and C–C bond cleavage leading to coke formation. Suppressing these side reactions to enhance ethylene selectivity remains a key challenge in catalyst design. Palladium has long been recognized as an outstanding active metal due to its superior activity and ethylene selectivity. In 2006 and 2008, Borodziński and Bond reviewed Pd catalysts for acetylene hydrogenation in ethylene-rich streams, addressing critical aspects such as Pd phase evolution (PdCx and PdHx formation), hydrogenation mechanisms over Pd sites, reaction kinetics, and the effects of Pd particle size, carbon monoxide, and promoters.1,2 However, monometallic Pd catalysts alone fail to fully meet the stringent industrial requirements for acetylene selective hydrogenation, necessitating continuous efforts to develop more advanced catalysts. The early introduction of the “site isolation” concept, which minimizes neighboring Pd active sites by incorporating a secondary metal, significantly improved the catalytic performance. The most commonly studied bimetallic catalyst, PdAg alloy, remains widely used in modern petrochemical industries for removing trace acetylene from ethylene-rich streams.2 To further enhance catalytic performance, researchers have explored various active site configurations, including disordered alloy sites, ordered intermetallic compound (IMC) phases, and single-atom (SA) sites. Additionally, to reduce the catalyst costs, the development of non-Pd-based catalysts incorporating more abundant and cost-effective elements has become an important research direction. Furthermore, the influence of supports and modifiers on the overall catalytic performance has also been extensively investigated.
Since 2008, significant efforts have been devoted to developing highly efficient catalysts for acetylene hydrogenation. Previous reviews have summarized progress by categorizing catalysts into Pd-based, Ni-based, IB metal-based, bimetallic/trimetallic, and other types, with some focusing exclusively on non-noble metal-based catalysts.3,10 However, a review based on active site types—monometallic sites, disordered alloy sites, IMC phases, and SA sites—would provide more depth in mechanistic understanding and guide further catalyst design. In addition, the roles of supports and modifiers are also crucial for achieving superior catalytic performance, yet a systematic review of these aspects is still lacking. Furthermore, the use of inconsistent calculation methods to report selectivity in the literature renders direct comparisons of catalytic performance across studies difficult. Therefore, it is timely to establish a comprehensive framework that not only systematically addresses recent advancements in acetylene selective hydrogenation but also defines standardized benchmarks for performance evaluation.
This review classifies various active sites and elucidates their roles in acetylene hydrogenation. Additionally, the effects of catalyst support and modifiers are thoroughly examined to guide rational catalyst design (Fig. 1). Section 2 provides an overview of industrial processes and reaction pathways, with a particular focus on defining benchmark for selectivity calculations to establish a standardized basis for comparing catalytic performance reported in the literature. In Section 3, the active sites are structurally categorized into monometallic sites, disordered alloy sites, IMC phases, and SA sites, further distinguishing between Pd-based and non-Pd-based catalysts to analyze the influence of different active metals on acetylene hydrogenation performance. This section presents the overall development trends and the state-of-the-art in active site design, emphasizing the structure–performance relationship and key research directions for enhancing ethylene selectivity and activity. Density functional theory (DFT) calculations on acetylene hydrogenation over different active sites are discussed in Section 4 to provide mechanistic insights and guide the rational design of active sites. Sections 5 and 6 summarize the role of catalyst supports and modifiers in improving catalytic performance, with an emphasis on their effects on selectivity and activity. Section 7 discusses the varying catalyst design requirements across different industrial processes for acetylene hydrogenation, highlighting the challenges and future perspectives in mechanistic understanding and catalyst development to anticipate future trends in this field. In addition, this section discusses the extension of catalytic strategies to the selective hydrogenation of other alkynes and dienes, as well as the emerging alternatives to thermocatalysis, including electrocatalytic and photocatalytic acetylene hydrogenation, highlighting their opportunities and limitations.
 | ||
| Fig. 1 Overview of the scope of the review. | ||
2. Overview of acetylene hydrogenation reaction
2.1 Industrial processes for acetylene hydrogenation
The primary application of acetylene selective hydrogenation is in the modern petrochemical industry, where trace amounts of acetylene need to be effectively removed from ethylene-rich streams. To meet the requirement of acetylene content (<10 ppm or even lower) in polymer-grade ethylene, an extremely high conversion of acetylene (>99.5% based on 0.2% acetylene feed concentration) must be achieved while limiting ethylene loss to no more than 1%. There are two main processes for acetylene hydrogenation in ethylene-rich streams, namely the tail-end and the front-end processes. The characteristics, advantages, and disadvantages of these two processes have been well summarized by Borodziński and Bond.1 The primary difference between these processes lies in the feed composition (Table 1). The tail-end process typically operates with a H2/C2H2 ratio of 1.2, while the front-end process exhibits a H2/C2H2 ratio of 110.Another potential industrial application of acetylene hydrogenation is the increased utilization of non-oil alternative resources to produce ethylene, such as coal and natural gas. This potential route for producing ethylene involves first producing acetylene, followed by its hydrogenation to ethylene. Several commercial processes have been developed to produce acetylene from these resources, including partial oxidation of natural gas (POX), thermal plasma pyrolysis of coal (PPY), and the calcium carbide process. The typical product concentrations for these processes are summarized in Table 1. For the gas products of POX and PPY, the concentrations of both acetylene and CO are several times higher than those from naphtha cracking. Similar to conventional acetylene hydrogenation in the petrochemical route, the tail-end process can be used by initially separating the large amounts of CO and H2. However, this process incurs higher costs due to the separation and operational complexities, as the conventional solvent extraction process for separating C2H2 from CO is both costly and energy-intensive. On the other hand, direct hydrogenation of acetylene under high concentrations of CO and H2, similar to the front-end process, simplifies the overall process. This approach is particularly beneficial when the downstream process involves CO, such as in hydroformylation of ethylene, as it eliminates the need to reintroduce CO. Nevertheless, designing highly efficient catalysts for acetylene hydrogenation under these harsh feed conditions remains a challenge. The large amounts of CO present can poison the metal active sites and accelerate the formation of oligomers, complicating the catalytic process.
2.2 Reaction pathway of acetylene hydrogenation
The reaction pathways for acetylene hydrogenation are illustrated in Fig. 2. Acetylene and atomic hydrogen adsorb onto the catalyst surface, where subsequent reactions at the active sites lead to the formation of ethylene, representing the primary reaction pathway. However, three main side reactions may occur during the hydrogenation process. The first one involves over-hydrogenation, where adsorbed ethylene or other C2 unsaturated intermediates further react with active hydrogen to form ethane. The second side reaction consists of coupling, where adsorbed acetylene and ethylene species undergo C–C coupling, leading to chain growth and the generation of C4+ products such as 1,3-butadiene and butene. These coupling by-products typically exhibit strong polymerization tendencies on the catalyst surface, often resulting in the formation of polymeric deposits (referred to as green oil, GO), which can block the active sites and lead to catalyst deactivation. The third side reaction involves C–C bond cleavage, leading to coke deposition that typically modifies the active phase, including the formation of PdCx on Pd-based catalysts under reaction conditions.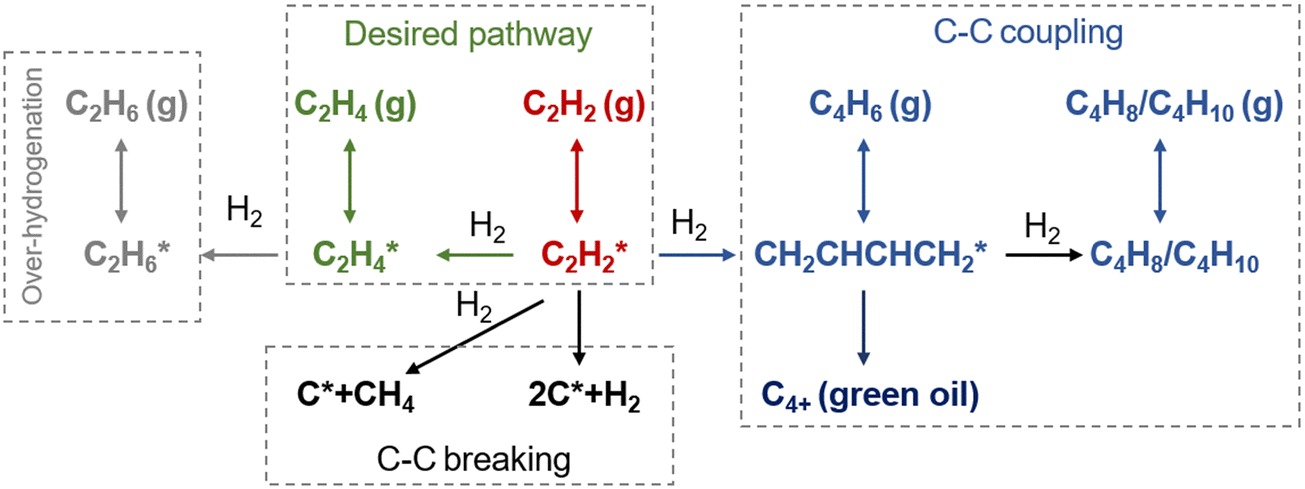 | ||
| Fig. 2 Reaction pathways for acetylene hydrogenation. | ||
The hydrogenation of unsaturated carbon–carbon bonds is an exothermic reaction. The standard molar enthalpy changes (ΔH) of the main and side reactions in acetylene selective hydrogenation are shown in eqn (1)–(5):
| C2H2 + H2 → C2H4 ΔHθ298 = −172 kJ mol−1 | (1) |
| C2H4 + H2 → C2H6 ΔHθ298 = −136 kJ mol−1 | (2) |
| 2C2H2 + H2 → C4H6 ΔHθ298 = −343 J mol−1 | (3) |
| C2H2 → 2C2 + H2 ΔHθ298 = −433 kJ mol−1 | (4) |
| C2H2 → C + CH4 ΔHθ298 = −125 kJ mol−1 | (5) |
2.3 Benchmark for calculating acetylene conversion and ethylene selectivity
Research on acetylene hydrogenation is generally carried out in a fixed-bed or stirred reactor under continuous flow conditions. The inlet and outlet gases are typically analysed by gas chromatography (GC) equipped with a gas sample loop. The GC provides the peak areas of each product in the gas sample, which can be used to further calculate molar concentrations. However, it should be noted that the peak area ratio from chromatogram should not be directly used as the molar concentration. The accurate formulas for calculating the conversion (X) and selectivity (S) are as follows: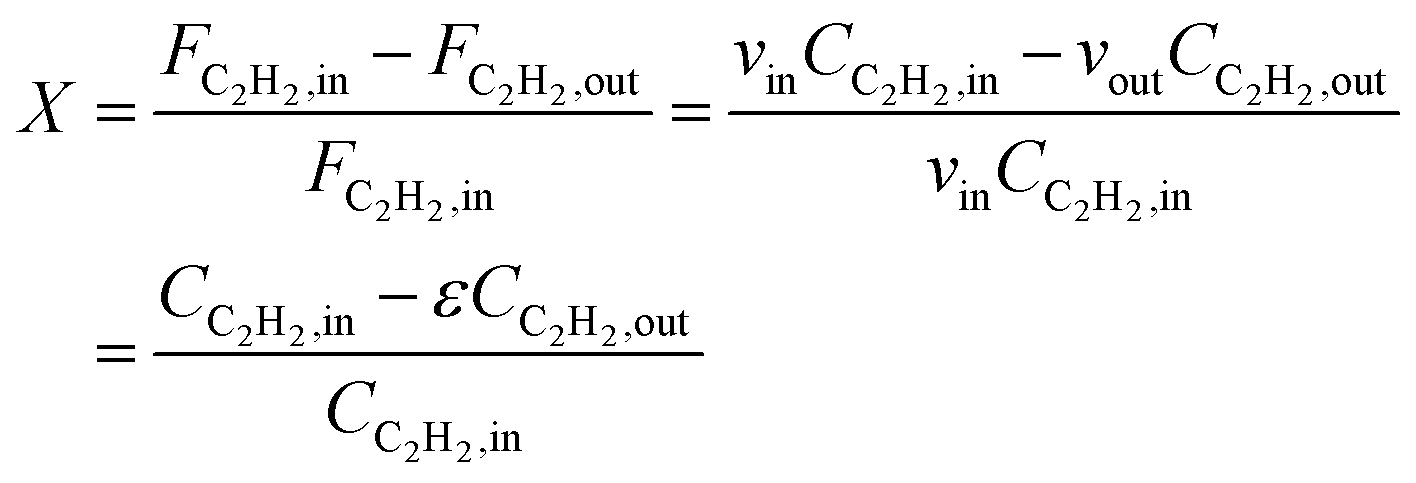 | (6) |
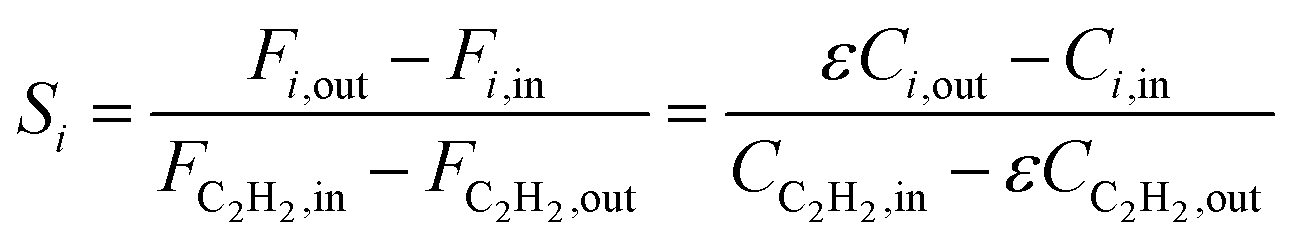 | (7) |
Ethylene selectivity is one of the most important indicators for evaluating catalyst performance. In the feed conditions of the petrochemical industry, the concentration of acetylene is usually below 0.5%, therefore the volume change after the reaction can be neglected (ε ≈ 1) when excess ethylene in the feed does not undergo over-hydrogenation. Therefore, ethylene selectivity can be expressed as:
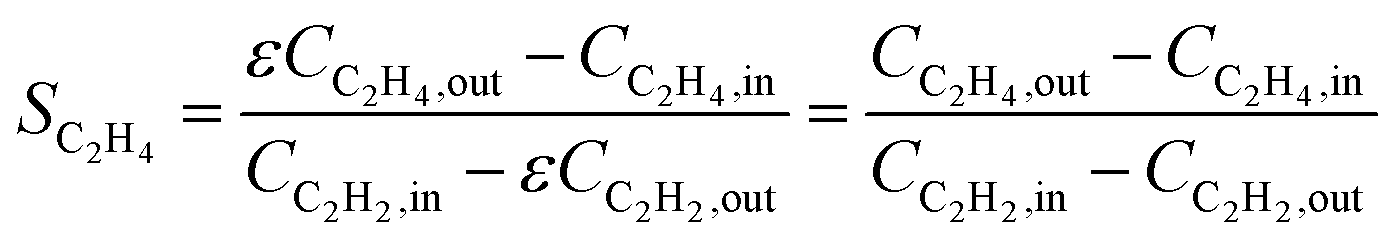 | (8) |
Over the past decade, several alternative methods have been proposed for the indirect calculation of ethylene selectivity under feed conditions involving ethylene-rich streams, as summarized in Table 2. Methods A, B, C, and D estimate ethylene selectivity indirectly by measuring ethane and C4 components, since these species can be accurately quantified due to their absence in the feed. However, acetylene hydrogenation is often accompanied by oligomerization, leading to the formation of GO, which consists of deposited long-chain hydrocarbons. GC used for quantitative analysis primarily detects light hydrocarbons such as acetylene, ethylene, ethane, and C4, while GO components remain undetected. The omission of GO in Methods A, B, C, and D may result in an overestimation of the intrinsic selectivity toward ethylene. Notably, Methods D consider only ethylene and ethane as products, disregarding C4 by-products and GO formation, which can further overestimate the intrinsic ethylene selectivity. Another strategy to estimate the ethylene selectivity in ethylene-rich stream is to carried out isotope labeling experiments, where Method E was used to determine SC2H4 by using two different isotopes, 13C and 12C, for ethylene and acetylene, respectively.13
| Method | Formula of SC2H4 | Advantages or limitations |
|---|---|---|
| Indirect method (in ethylene-rich feed stream) | ||
| A |

|
• Selectivity definition differs from the conventional approach |
| • Overestimate the selectivity due to the neglect of C4 and GO | ||
| B |
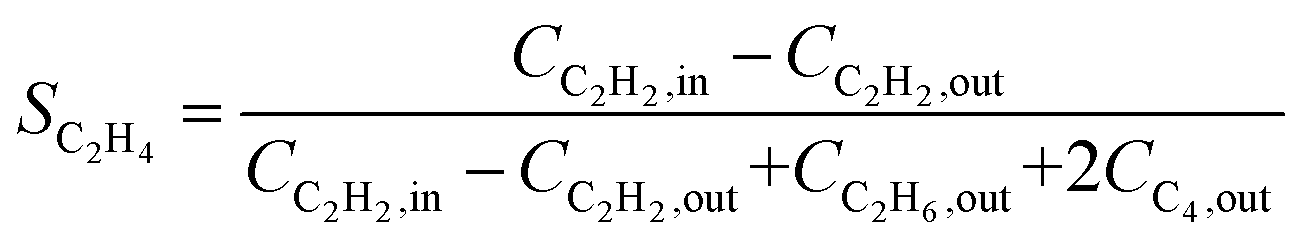
|
• Selectivity definition differs from the conventional approach |
| • Overestimate the selectivity due to the neglect of GO | ||
| C |
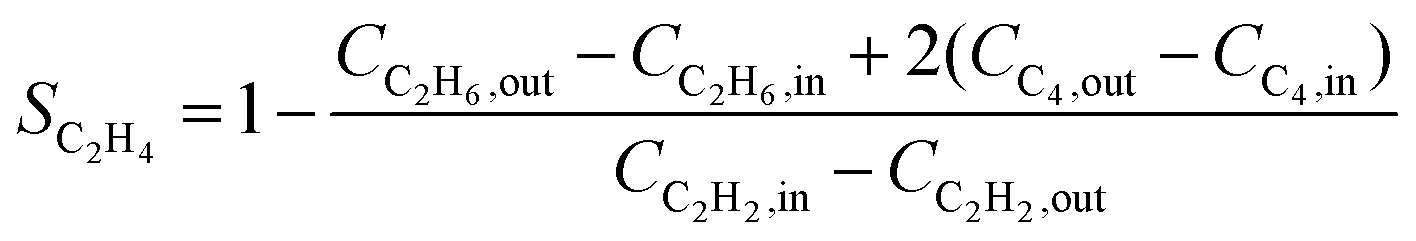
|
• Overestimate the selectivity due to the neglect of GO |
| D |
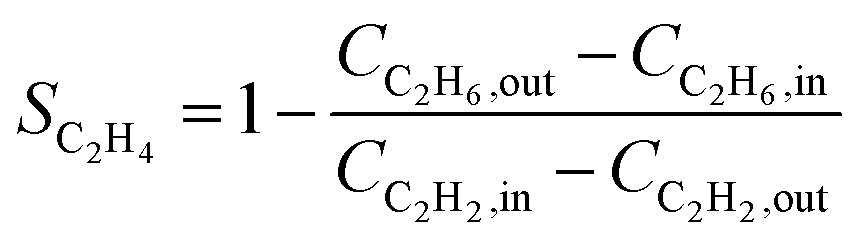
|
• Overestimate the selectivity due to the neglect of C4 and GO |
| • Direct method (in ethylene-free feed stream) | ||
| E |

|
• Not suitable for ethylene-rich feed streams |
| • Accurately defines selectivity for ethylene-free feed streams | ||
| F |

|
• Overestimate the selectivity due to the neglect of GO |
| G |
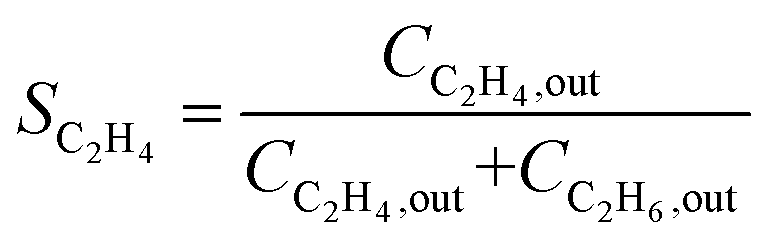
|
• Overestimate the selectivity due to the neglect of C4 and GO |
For acetylene hydrogenation in an ethylene-free feed stream, the change in ethylene concentration can be accurately measured, enabling direct calculation of SC2H4 using Method E. Additionally, Methods F and G have been proposed as simplified approaches for calculating ethylene selectivity. However, Method F neglects the formation of GO, while Method G excludes both C4 and GO by-products, potentially leading to an overestimation of ethylene selectivity.
Based on the above discussion, the following recommendations are proposed for calculating ethylene selectivity:
(i) When comparing state-of-the-art catalyst performance, a standardized ethylene selectivity calculation method should be adopted.
(ii) For testing new catalysts for industrial processes involving ethylene-rich feed streams, the primary objective is to reduce trace acetylene to the ppm level while minimizing ethylene loss. Therefore, the residual acetylene concentration and the absolute ethylene concentration in the reaction effluent are key performance indicators. To evaluate both catalytic activity and selectivity, it is recommended to follow the strategy proposed by Borodziński and Bond,2 as illustrated in Fig. 3.
 | ||
| Fig. 3 Acetylene hydrogenation test for catalysts in ethylene-rich streams. Reprinted with permission from ref. 2. Copyright 2008 Taylor & Francis. The difference between T1 and T2 is called the operating window (OW). Catalyst with a large OW is more thermally stable at the temperature necessary to remove acetylene from the feed. | ||
(iii) To investigate the acetylene hydrogenation mechanism over specific active sites, using an ethylene-free feed stream can provide a more accurate determination of the intrinsic selectivity of the products.
(iv) For testing new catalysts for the production of ethylene from coal and natural gas, the volume change after the reaction cannot be neglected due to the high acetylene concentration. Therefore, ε must be taken into account when calculating the conversion and selectivity.
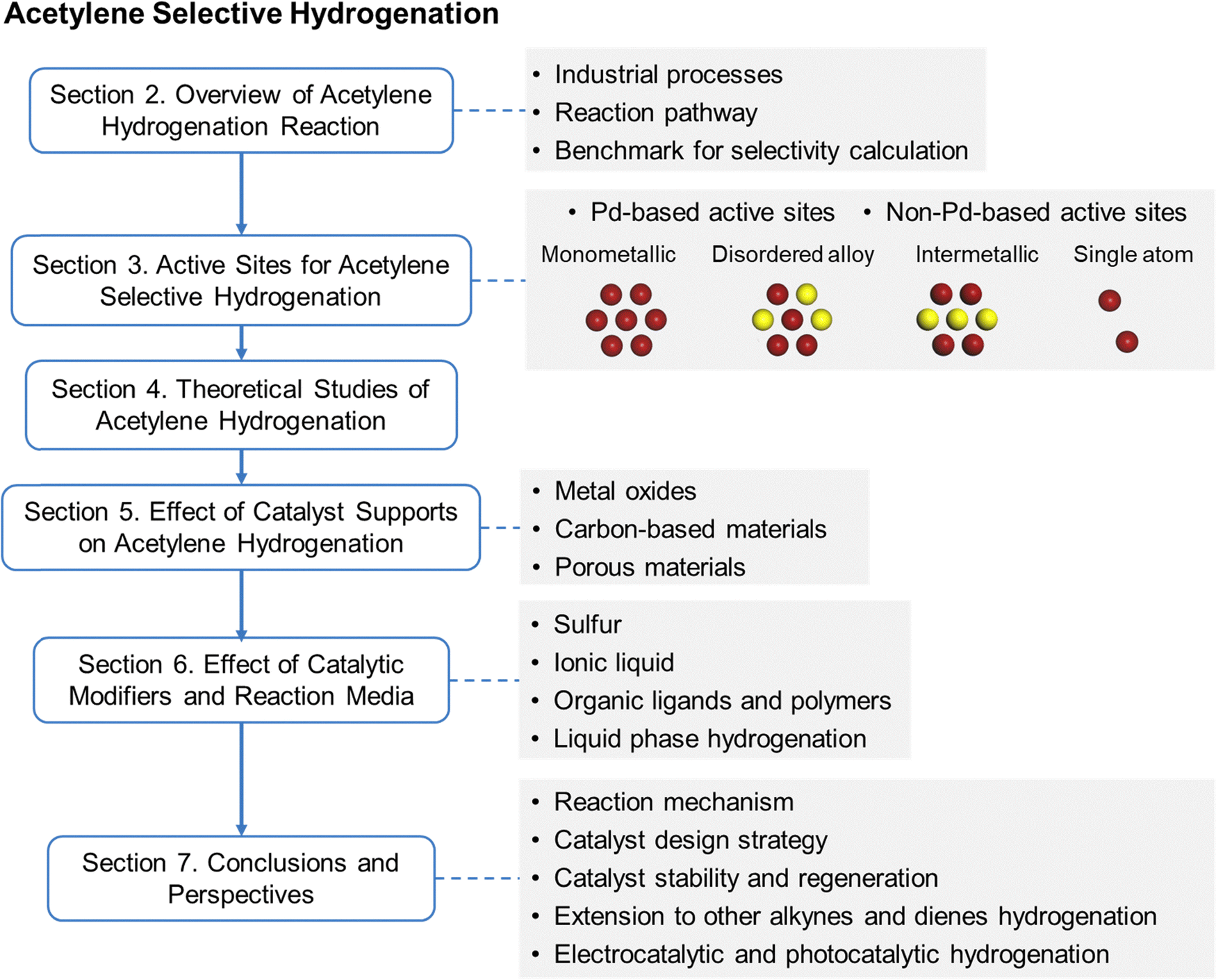
3. Active sites for acetylene selective hydrogenation
In acetylene selective hydrogenation, the active metal sites are typically hydrogenation-active transition metals, such as Pd, Au, Ni, and Cu. Based on their structural characteristics, active sites for acetylene hydrogenation can be classified into monometallic, disordered alloy, IMC, and SA sites. This section provides a comprehensive summary and discussion of research progress based on active site structures. Section 3.1 presents the development trends and the current advances in active site design. Given that Pd is the most extensively studied active metal for acetylene hydrogenation, Sections 3.2 and 3.3 further distinguish between Pd-based and non-Pd-based active sites, respectively.3.1 Overview and state-of-the-art of active site design
Acetylene selective hydrogenation requires the conversion of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds to C
C bonds to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C while simultaneously suppressing both over-hydrogenation to ethane and C–C coupling reactions that lead to oligomerization. The key challenge in this reaction lies in the design of active sites with high performance. It has been reported that active sites composed of neighbouring active metal ensembles preferentially stabilize di-σ-bonded acetylene and adsorbed ethylene, thereby promoting both ethylene over-hydrogenation and C–C coupling reactions.2,14 Despite its relatively high ethylene selectivity among monometallic catalysts, Pd nanoparticles (NPs) inherently feature adjacent active sites that facilitate side reactions and thus compromise ethylene selectivity.
C while simultaneously suppressing both over-hydrogenation to ethane and C–C coupling reactions that lead to oligomerization. The key challenge in this reaction lies in the design of active sites with high performance. It has been reported that active sites composed of neighbouring active metal ensembles preferentially stabilize di-σ-bonded acetylene and adsorbed ethylene, thereby promoting both ethylene over-hydrogenation and C–C coupling reactions.2,14 Despite its relatively high ethylene selectivity among monometallic catalysts, Pd nanoparticles (NPs) inherently feature adjacent active sites that facilitate side reactions and thus compromise ethylene selectivity.
An effective strategy to enhance ethylene selectivity is the incorporation of a secondary metal to construct selective hydrogenation active sites (Fig. 4a). The electronic interaction between the secondary metal and the active metal modulates the adsorption strength of CC and CC bonds, thereby enhancing ethylene selectivity. In addition, site isolation by the secondary metal or a single-atom structure spatially separates the active metal sites, preventing C–C coupling reactions and ethylene over-hydrogenation. Guided by these active site design principles, research over the past decade has focused on enhancing catalyst performance. Beyond monometallic sites, the active sites in acetylene hydrogenation catalysts can be classified into three distinct types (Fig. 4b):
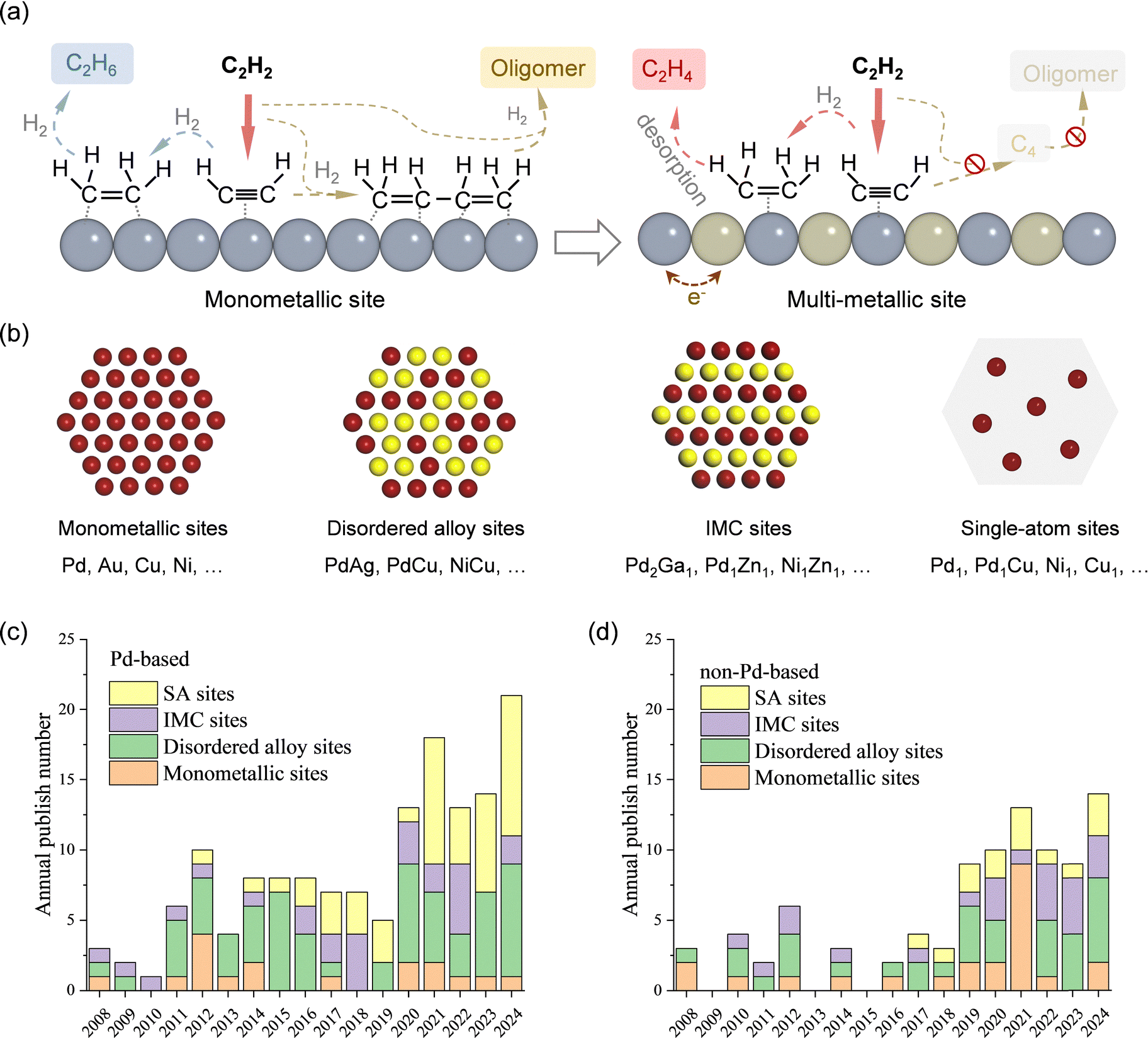 | ||
| Fig. 4 (a) Schematic illustration of the active sites design strategy. (b) Four distinct types of active sites for acetylene hydrogenation. Annual published papers for the corresponding active sties of (c) Pd-based catalyst and (d) non-Pd-based catalysts. | ||
(1) Multi-metallic sites with disordered alloy structures. A representative class of multi-metallic catalysts consists of disordered alloy phases. In such disordered alloys, the surface atomic arrangement is unpredictable, leading to the presence of adjacent active metal sites.
(2) IMC sites with well-defined structures. IMCs possess precisely defined atomic compositions and ordered structures, in which metal atoms are arranged in a highly specific manner.
(3) SA sites. By maximizing the spatial separation of metal centers, SA structures completely eliminate neighboring active metal sites.
Fig. 4c and d illustrate the number of annual publications (since 2008) related to these active site categories for both Pd-based and non-Pd-based catalysts, respectively. For Pd-based catalysts, secondary metal modification has been widely explored to enhance selectivity and stability. Additionally, since the emergence of the SA concept, there has been a growing interest in developing high-performance Pd SA catalysts, particularly in the past five years. Researchers have also increasingly focused on replacing Pd with alternative metal sites, particularly non-precious metals. Over the past five years, efforts have primarily centered on the development of multi-metallic alloy and IMC active sites.
Tables 3–10 summarize the state-of-the-art catalytic performance for acetylene selective hydrogenation, categorized according to the active site classifications discussed above. In Section 2.3, we highlighted the importance of using benchmark methods for calculating ethylene selectivity. Correspondingly, we also include the specific calculation methods used in each study in Tables 3–10. To ensure a fair comparison, the performance of different catalysts is analyzed based on results obtained using the same calculation method.
| Catalyst | Description | T, °C | Feeda, % | X, % | S, % | Activityb | Methodc | Ref |
|---|---|---|---|---|---|---|---|---|
| a The feed composition listed in the table corresponds to C2H2/H2/C2H4. b Activity is calculated as the number of moles of acetylene converted per hour per total molar Pd active metal (molC2H2 molPd−1 h−1). c Method refers to the calculation method specified in Table 2. E* indicates that Method E was applied under ethylene-rich feed conditions. | ||||||||
| Pd/CNF | 8 nm Pd | 150 | 2/4/0 | 36 | 1 | 4434.2 | E | 15 |
| Pd/CNF | 11 nm Pd | 150 | 2/4/0 | 47 | 59 | 5789.1 | E | |
| Pd/CNF | 13 nm Pd | 150 | 2/4/0 | 43 | 62 | 5296.4 | E | |
| Pd/TiO2 | 31 | 1.1/3.3/95.6 | 43 | 50 | 204.1 | D | 16 | |
| Pd/TiO2 | H RF plasma treat | 31 | 1.1/3.3/95.6 | 37 | 74 | 175.6 | D | |
| Pd/TiO2 | O RF plasma treat | 31 | 1.1/3.3/95.6 | 30 | 90 | 142.4 | D | |
| Pd/TiO2 | Ar RF plasma treat | 31 | 1.1/3.3/95.6 | 27 | 94 | 128.1 | D | |
| Pd/LDH | Spherical Pd | 50 | 0.3/0.6/32.9 | 20 | 85 | 53.1 | E* | 17 |
| Pd/MgAl-LDH | Tetra Pd | 50 | 0.3/0.6/32.9 | 48 | 97 | 127.4 | E* | |
| Pd/MgAl-LDH | Octa Pd | 50 | 0.3/0.6/32.9 | 47 | 89 | 129.5 | E* | |
| Pd/MgAl-LDH | L-Wires Pd | 40 | 0.3/0.6/32.9 | 50 | 82 | 76.2 | E* | 18 |
| Pd/MgAl-LDH | S-Wires Pd | 40 | 0.3/0.6/32.9 | 45 | 84 | 68.3 | E* | |
| Pd/MgAl-LDH | Cuboctahedral Pd | 40 | 0.3/0.6/32.9 | 45 | 84 | 68.0 | E* | |
| Pd/CNF | Cubic Pd | 120 | 1.5/20/0 | 13 | 62 | — | E | 19 |
| Pd/CNF | Cuboctahedral Pd | 120 | 1.5/20/0 | 13 | 64 | — | E | |
| Pd/CNF | Octahedral Pd | 120 | 1.5/20/0 | 13 | 65 | — | E | |
| Pd/Al2O3 | Cubic Pd | 60 | 0.6/1.2/49.3 | 75 | 32 | 133.1 | E* | 20 |
| Pd/Al2O3 | Spherical Pd | 60 | 0.6/1.2/49.3 | 75 | −5 | 51.2 | E* | |
| Pd/ZnO | Octahedra Pd | 100 | 2/4/0 | 25 | 90 | 6681.6 | — | 21 |
| Pd/ZnO | Cubes Pd | 100 | 2/4/0 | 30 | 80 | 6472.8 | — | |
| Pd/SiO2 | Octahedra Pd | 100 | 2/4/0 | 25 | 90 | 4554.0 | — | |
| Pd/SiO2 | Cubes Pd | 100 | 2/4/0 | 20 | 80 | 2070.0 | — | |
| Pd/Al2O3 | 140 | 2/20/0 | 60 | 62 | 3420.6 | E | 22 | |
| Pd/C | 75 | 1/1/0 | 92 | 50 | — | — | 23 | |
| Pd–B/C | Subsurface B atom | 75 | 1/1/0 | 81 | 78 | — | — | |
| Pd–Li/C | Subsurface Li atom | 75 | 1/1/0 | 83 | 79 | — | — | |
| Pd/msAC | 2 nm Pd | 260 | — | 100 | ∼90 | — | — | 24 |
| Pd/CNTs | PdCx | 200 | 0.9/8.8/88.2 | 82 | 40 | — | B | 25 |
| Pd-ac/Al2O3 | PdCx | 70 | 0.3/0.7/33 | 95 | 60 | — | E* | 26 |
| PdAl-120 | PdCx | 80 | 2/2.2/80 | 90 | 93 | — | D | 27 |
| Catalyst | Description | T, °C | Feeda, % | X, % | S, % | Activityb | Methodc | Ref |
|---|---|---|---|---|---|---|---|---|
| a The feed composition listed in the table corresponds to C2H2/H2/C2H4. b Activity is calculated as the number of moles of acetylene converted per hour per total molar Pd active metal (molC2H2 molPd−1 h−1). c Method refers to the calculation method specified in Table 2. E* indicates that Method E was applied under ethylene-rich feed conditions. | ||||||||
| PdAg | ||||||||
| PdAg/Al2O3 | 35 | 1/1.4/30 | 70 | ∼67 | C | 28 | ||
| PdAg/Al2O3 | 60 | 0.9/1.8/97.2 | 90 | 20 | 316.1 | — | 29 | |
| NiPdAg/Al2O3 | 60 | 1.1/2.2/96.8 | 87 | 25 | 355.9 | — | 30 | |
| Pd@Ag/TiO2 | 60 | 0.73/1.46/52.6 | 82 | 79 | 215.5 | C | 31 | |
| PdAg/SiO2 | 65 | 1/5/20 | 18 | 79 | 116.6 | B | 32 | |
| PdAg/Al2O3 | Egg-shell | 30 | 1/1/30 | 44 | 75 | 325.7 | — | 33 |
| PdAg/MgAl-LDH | Mesocrystal | 77 | 0.35/0.6/32.8 | 90 | 82 | 1.8 | E* | 34 |
| Ag@PdAg/LDHs | Core–shell | 100 | 0.31/0.6/32 | 99.5 | 97 | 7.6 | E* | 35 |
| PdAg/carbon material | 45 | 4/excess/0 | 35 | 76 | 15963.0 | E | 36 | |
| 100Pd0Ag/Al2O3 | 50 | 1.47/6.95/29.5 | ∼70 | 50 | 1400.0 | B | 37 | |
| 90Pd10Ag/Al2O3 | 50 | 1.47/6.95/29.5 | ∼70 | 60 | B | |||
| 75Pd25Ag/Al2O3 | 50 | 1.47/6.95/29.5 | ∼70 | 70 | B | |||
| 50Pd50Ag/Al2O3 | 50 | 1.47/6.95/29.5 | ∼70 | 75 | B | |||
| PdAg/Al2O3 | 150 | 14.3/71.4/14.3 | 95 | 62 | 5416.0 | E* | 38 | |
| Pd/K+-b-zeolite | 30 | 2.5/10/2.5 | −5 | E | 39 | |||
| PdAg/K+-b-zeolite | 75 | 2.5/10/2.5 | 70 | E | ||||
| PdCu | ||||||||
| PdCu/Al2O3 | 60 | 0.9/1.8/97.2 | 100 | 5 | 351.2 | — | 40 | |
| PdCu/Al2O3 | 90 | 0.6/1.8/0 | 70 | 62 | E | 41 | ||
| PdCu/Al2O3 | 80 | 0.59/1.76/0 | 100 | 72 | E | 42 | ||
| CuPd/Al2O3 | Copper rich | 50 | 0.59/1.76/0 | 35.3 | 63.4 | E | 43 | |
| PdCu/MgAl-cHT | 100 | 0.3/0.6/32.9 | 100 | 84 | 367.3 | E* | 44 | |
| AgPd0.64/TiO2 | 40 | 1/25/25 | <3 | 99 | 54.3 | D | 45 | |
| CuPd0.08/TiO2 | 40 | 1/25/25 | <3 | 100 | 92.6 | D | ||
| CuPd0.02/TiO2 | 40 | 1/25/25 | <3 | 98.5 | 108.5 | D | ||
| Pd/TiO2 | 40 | 1/25/25 | <3 | 95 | 47.9 | D | ||
| Pd/oCNTs | 200 | 0.5/5/20.1 | 97.1 | 39 | 237.5 | C | 46 | |
| Pd3Cu/oCNT | 200 | 0.5/5/20.1 | 98.5 | 55 | 228.0 | C | ||
| Pd2Cu/oCNT | 200 | 0.5/5/20.1 | 99.2 | 68 | 205.6 | C | ||
| PdCu/oCNT | 200 | 0.5/5/20.1 | 98.8 | 72 | 152.0 | C | ||
| PdCu3/oCNT | 200 | 0.5/5/20.1 | 98.5 | 71 | 106.0 | C | ||
| Pd/BF-500/250 | Pd–Cu–Zn | 80 | 0.625/6.25/31.25 | 25 | 85 | C | 47 | |
| PdCu/C | FCC phase | 100 | 0.33/0.66/33 | 100 | 61 | 502.4 | E* | 48 |
| PdCu/C | BCC phase | 100 | 0.33/0.66/33 | 100 | 86 | 460.9 | E* | |
| Pd1Cu2/C | Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu = 48:51 :Cu = 48:51 |
96 | 0.33/0.66/33 | 88 | 82 | 163.4 | E* | 49 |
| Pd1Cu3/C | Pd:Cu = 38:62 |
116 | 0.33/0.66/33 | 85 | 87 | 180.4 | E* | |
| Pd1Cu4/C | Pd:Cu = 34:66 |
135 | 0.33/0.66/33 | 80 | 98 | 145.7 | E* | |
| PdCu/siloxene | 200 | 1/10/0 | 91 | 93 | 4323.3 | E | 50 | |
| Pd40Cu60 | Disorder phase | 200 | 0.5/5/50 | 90 | 74 | B | 51 | |
| Pd40Cu60 | Order phase | 200 | 0.5/5/50 | 90 | 92 | B | ||
| PdCu/SiO2 | B2 phase | 25 | 1/2/1 | 43 | 78 | — | 52 | |
| PdCu/SiO2 | FCC phase | 25 | 1/2/1 | 13 | 71 | — | ||
| CuPd-2/Al2O3 | B2 phase | 90 | 0.5/3/0 | 100 | 95 | E | 53 | |
| Pd/SiO2(NS) | 70 | 12.5/37.5/0 | 55 | 47 | E | 54 | ||
| Pd1Cu1/SiO2(NS) | 70 | 12.5/37.5/0 | 40 | 55 | E | |||
| Pd1Cu3/SiO2 | 70 | 12.5/37.5/0 | 42 | 47 | E | |||
| Au@PdCu/C | 122 | 0.33/0.66/32.98 | 100 | 92 | 210.0 | E* | 55 | |
| PdAu | ||||||||
| PdAu/MgAlOx | Nanoflowers | 100 | 0.3/0.3/31.7 | 94 | 53 | 376.0 | E* | 56 |
| AuPd/TiO2 | 250 | 0.97/2.91/96.1 | 50 | 93 | 3632.4 | D | 57 | |
| Au@Pd/TiO2 | Core–shell | 90 | 1.2/2.2/0 | 100 | 53 | E | 58 | |
| m-PdAuAg2/HT | 140 | 0.33/0.6/32.8 | 99.5 | 76.2 | 391.9 | E* | 59 | |
| TiOx/Au8Pd@SBA | 0.5/5/10 | 92 | 75 | C | 60 | |||
| PdZn | ||||||||
| Pd/γ-Al2O3 | 60 | 1.2/2.5/96.3 | 47.2 | 31.3 | 118.8 | A | 61 | |
| Pd–Zn/γ-Al2O3 | 60 | 1.2/2.5/96.3 | 56.1 | 74.4 | 140.4 | A | ||
| Pd/α-Al2O3 | 60 | 1.2/2.5/96.3 | 64.9 | 21.2 | 21.2 | A | ||
| PdZn/α-Al2O3 | 60 | 1.2/2.5/96.3 | 67.1 | 88.6 | 23.0 | A | ||
| PdZn/carbon material | 45 | 4/excess/0 | 40 | 73 | 18243.4 | E | 62 | |
| Pd/ZnO/SMFs | PdZn | 140 | 1.5/20/0 | 15 | 82 | E | 63 | |
| Pd/CNFs/SMFs | 140 | 1.5/20/0 | 15 | 70 | E | |||
| PdCo | ||||||||
| CoPd/MgO | 110 | 0.5/1/70 | 78 | 59 | 334.9 | C | 64 | |
| Pd/MgO | 110 | 0.5/1/70 | 80 | 5 | 316.7 | C | ||
| PdCo/Al2O3 | 85 | 0.33/0.66/33 | 99.5 | 88 | 522.6 | E* | 65 | |
| Pd/C | 45 | 4/96/0 | 72 | 60 | 10946.1 | E | 66 | |
| PdCo(1:0.5)/C |
45 | 4/96/0 | 65 | 63 | 9881.9 | E | ||
| PdCo(1:1)/C |
45 | 4/96/0 | 50 | 66 | 7601.4 | E | ||
| PdCo(1:2)/C |
45 | 4/96/0 | 30 | 67 | 4560.9 | E | ||
| PdCo(1:4)/C |
45 | 4/96/0 | 7 | 67 | 1064.2 | E | ||
| PdX | ||||||||
| SnPd/MWNTs | 160 | 10/20/0 | 100 | 93 | E | 67 | ||
| Pd/MWNTs | 160 | 10/20/0 | 100 | 87 | E | |||
| PdTi/SiO2-T | 110 | 1/4/0 | 100 | 89 | E | 68 | ||
| Pd/SiO2 | 110 | 1/4/0 | 100 | 64 | E | |||
| PdGa/MgO–Al2O3 | 45 | 0.3/0.6/32.9 | 85.9 | 87.2 | 518.4 | E* | 69 | |
| Pd/MgO–Al2O3 | 45 | 0.3/0.6/32.9 | 89.5 | 54.7 | E* | |||
| PdNi/MgAlO | 70 | 0.3/0.6/32.9 | 92 | 70 | E* | 70 | ||
| PdIn/Al2O3 | 60 | 0.6/1.8/0 | 95 | 70 | 779.9 | C | 71 | |
| PdMn-2-300/Al2O3 | 40 | 1.05/1.94/5.01 | 87 | 81 | 449.1 | E* | 72 | |
| Pd NPs@Bi0.5/Al2O3 | 120 | 1/20/20 | 70 | 99 | 1995.4 | D | 73 | |
| Pd0.25La0.25/N-pgBC | 70 | 1/2/0 | 94 | 53 | E | 74 | ||
| Catalyst | Description | T, °C | Feeda, % | X, % | S, % | Activityb | Methodc | Ref. |
|---|---|---|---|---|---|---|---|---|
| a The feed composition listed in the table corresponds to C2H2/H2/C2H4. b Activity is calculated as the number of moles of acetylene converted per hour per total molar Pd active metal (molC2H2 molPd−1 h−1). c Method refers to the calculation method specified in Table 2. E* indicates that Method E was applied under ethylene-rich feed conditions. | ||||||||
| PdGa | ||||||||
| PdGa etched | Bulk | 200 | 0.5/5/50 | 91 | 56 | — | B | 75 |
| Pd3Ga7 etched | Bulk | 200 | 0.5/5/50 | 65 | 48 | — | B | |
| Pd/Al2O3 | Pd | 200 | 0.5/5/50 | 43 | 17 | — | B | |
| Pd2Ga/Al2O3 | Pd2Ga | 200 | 0.5/5/50 | 88 | 67 | 6615.1 | B | 76 |
| Pd/Al2O3 | Pd | 200 | 0.5/5/50 | 42 | 49 | 2451.9 | B | |
| Pd2Ga/MgGa2O4 | Pd2Ga | 200 | 0.5/5/50 | 98 | 70 | 28600.4 | B | 77 |
| GaPd/Al2O3 | Nano-PdGa | 200 | 0.5/5/50 | 83.9 | 82 | 4140.0 | B | 78 |
| GaPd2/Al2O3 | Nano-Pd2Ga | 200 | 0.5/5/50 | 87.6 | 66 | 7120.0 | B | |
| Pd/Al2O3 | Pd | 200 | 0.5/5/50 | 44.9 | 20 | 2450.0 | B | |
| GaPd2/a-Al2O3 | Pd2Ga | 200 | 0.5/5/50 | 92 | 76 | 480.0 | B | 79 |
| Sputtered GaPd2 | Pd2Ga | 200 | 0.5/5/50 | 41.1 | 72 | 3930.0 | B | 80 |
| Pd–Ga2O3 | Pd2Ga–Pd1 | 200 | 0.5/10/20 | 88 | 80 | — | C | 81 |
| Pd–Ga2O3 | Pd2Ga–Pd3 | 140 | 0.5/10/20 | 97 | 60 | — | C | |
| PdIn | ||||||||
| InPd2 powder | InPd2 | 200 | 0.5/5/50 | 90 | 80 | — | B | 82 |
| PdIn/MgAl2O4 | PdIn | 90 | 0.5/5/50 | 95 | 90 | 325.0 | B | 83 |
| Pd3In/MgAl2O4 | Pd3In | 90 | 0.5/5/50 | 100 | 20 | 342.1 | ||
| PdIn/CNT | Pd3In | 50 | 0.5/2.5/30 | 90 | 10 | 256.5 | C | 84 |
| PdIn/CNT | PdIn | 50 | 0.5/2.5/30 | 50 | 90 | 142.5 | C | |
| PdIn/CNT | Pd2In3 | 50 | 0.5/2.5/30 | 18 | 80 | 51.3 | C | |
| Pd/In2O3/SiO2 | PdIn | 100 | 1/10/0 | 80 | 68 | — | E | 85 |
| PdZn | ||||||||
| Pd/ZnO | Pd1Zn1 | 60 | 2/20/0 | 98 | 70 | 1676.1 | E | 86 |
| Pd/ZnO | Pd1Zn1 | 80 | 2/20/40 | 92 | 89 | — | D | |
| Pd8Zn44 | Pd8Zn44 | 100 | 1/31/18 | 100 | 82 | — | E | 13 |
| Pd9Zn43 | Pd9Zn43 | 100 | 1/31/18 | 100 | 70 | — | E | |
| Pd8AuZn43 | Pd8AuZn43 | 100 | 1/31/18 | 100 | 63 | — | E | |
| Pd3.9Zn6.1/CNS | Pd3.9Zn6.1 | 60 | 1/20/19 | 40 | 90 | — | D | 87 |
| PdZn | PdZn | 60 | 1/20/19 | 15 | 45 | — | D | |
| Pd3ZnCx | Pd3ZnCx | 120 | 2/2.2/80 | 89 | 92 | 528.5 | D | 88 |
| Others | ||||||||
| Bulk-Pd11Bi2Se2 | 200 | 0.5/5/50 | 75 | 93 | — | B | 89 | |
| PdBi/Calcite | Pd1Bi1 | 120 | 1/20/20 | 60 | >99 | 380.1 | D | 90 |
| PdBi/SiO2 | Pd3Bi | 40 | 1/4/0 | 78 | 80 | 277.9 | G | 91 |
| Sn–Pd/MWNTs | Pd2Sn | 160 | 10/20/0 | 100 | 93 | — | E | 67 |
| Pd2Sn/C | Pd2Sn | 160 | 0.33/0.66/32.8 | 100 | 92 | 17.3 | E* | 92 |
| Pd3P/TiO2 | Pd3P | 225 | 1.2/2.4/0 | 100 | 78 | — | E | 93 |
| PdP2/TiO2 | PdP2 | 225 | 1.2/2.4/0 | 100 | 84 | — | E | |
| Ga1−xSnxPd2 | Bulk-Ga1Pd2 | 200 | 0.5/5/50 | 91 | 83 | 5.5 | B | 94 |
| Ga1−xSnxPd2 | Bulk-Ga0.7Sn0.3Pd2 | 200 | 0.5/5/50 | 91 | 83 | 9.5 | B | |
| CaPdH2 | 100 | 1/10/0 | 85 | 81 | — | E | 95 | |
| Catalyst | T, °C | Feeda, % | X, % | S, % | Activityb | Methodc | Ref. |
|---|---|---|---|---|---|---|---|
| a The feed composition listed in the table corresponds to C2H2/H2/C2H4. b Activity is calculated as the number of moles of acetylene converted per hour per total molar Pd active metal (molC2H2 molPd−1 h−1). c Method refers to the calculation method specified in Table 2. E* indicates that Method E was applied under ethylene-rich feed conditions. | |||||||
| Carbon material supported | |||||||
| Pd1/C3N4 | 100 | 0.5/1/25 | 40 | 95 | D | 96 | |
| Pd1/ND@G | 120 | 1/10/20 | 15 | 95 | 388.7 | D | 97 |
| Pd1/Graphene | 50 | 1/2/0 | 64 | 78 | 364.9 | E | 98 |
| Pd1/C3N4 | 50 | 1/2/0 | 30 | 96 | 171.0 | E | |
| Pd-SAs-900 | 100 | 0.5/5/50 | 75 | 95 | 13.4 | B | 99 |
| Pd1/MPNC | 110 | 0.5/5/50 | 84 | 82 | 483.9 | B | 100 |
| Pd1–N8/CNT | 40 | 2/4/50 | 62.6 | 96.4 | 2163.0 | E* | 101 |
| Pd1Cu1/ND@G | 90 | 1/10/20 | 67 | 93 | 2122.1 | C | 102 |
| Pd1–N1C3 | 100 | 0.65/5/50 | 92 | 80 | 968.5 | — | 103 |
| Pd/GDY | 160 | 0.33/0.66/32.8 | 75 | 100 | E* | 104 | |
| Pd1/N-graphene | 125 | 1/10/20 | 99 | 93.5 | D | 105 | |
| Pd1/N,S-C | 150 | 0.5/5/50 | 100 | 80 | 2024.5 | D | 106 |
| Pd1-Ndicy/hNCNC | 160 | 1/215 | 90 | 87 | 3644.2 | D | 107 |
| Metal oxide supported | |||||||
| Pd1/ZnO | 80 | 2/20/40 | 99 | 84 | 33864.4 | D | 108 |
| Pd1/MgO | 150 | 1/10/0 | 25 | 74 | 49884.4 | E | 109 |
| Pd1/MgO | 120 | 1/10/20 | 30 | 94 | D | 110 | |
| Pd1–Fe1/Fe2O3(012) | 40 | 1/10/20 | 85 | 97 | 227.2 | G | 111 |
| Pd1/MgO | 120 | 1/2/0 | 39 | 42 | 1208.4 | E | 112 |
| Pd1/CeO2 | 140 | 2/20/40 | 35 | 90 | 299.3 | D | 113 |
| Pd1/α-Fe2O3 | 120 | 2/20/40 | 50 | 85 | 427.6 | D | |
| Single atom alloy | |||||||
| AuPd0.01/SiO2 | 120 | 1/20/20 | 50 | 80 | 6334.5 | D | 114 |
| AgPd0.01/SiO2 | 160 | 1/20/20 | 60 | 80 | 3694.0 | D | 115 |
| CuPd0.006/SiO2 | 160 | 1/20/20 | 100 | 85 | 5770.3 | D | 116 |
| Pd1Ni/SiO2 | 80 | 1/10/20 | 90 | 88 | 10126.9 | D | 117 |
| PdCu-SAA | 160 | 0.45/0.9/45 | 60 | 96 | 529.1 | E* | 118 |
| Pd0.1Ni2/SiO2-GR-500R | 130 | 1/10/20 | 60 | 90 | 2565.5 | D | 119 |
| PdxAg/Al2O3 | 60 | 1/10/20 | 96 | 94 | 1368.3 | — | 120 |
| Pd30 ppm/Fe5C2 | 80 | 1/3/0 | 38 | 82 | E | 121 | |
| Pd1-NiGa | 110 | 0.5/2.5/40 | 100 | 96.8 | 593.9 | C | 122 |
| Pd1Ag10/Al2O3 | 75 | 0.5/2.65/45.1 | 65 | 97 | 395.3 | D | 123 |
| Pd1Cun/Al2O3 | 120 | 0.33/0.66/33 | 100 | 82 | D | 124 | |
| Others | |||||||
| Pd1@Cu-SiW | 80 | 0.5/5/50 | 12 | 92 | 2.8 | B | 125 |
| Pd1@NENU-1 | 80 | 0.5/5/50 | 75 | 88 | 0.2 | B | |
| Pd1@Y | 80 | 0.5/5/50 | 35 | 60 | 7.6 | B | |
| T-Pd1/UiO-66-NH2 | 150 | 0.33/0.66/34.5 | 45 | 93 | 582.0 | E* | 126 |
| BmimBF4-0.021Pd/HAP | 100 | 2/20/40 | 95 | 88 | D | 127 | |
| Pd/Ni(OH)2 | 105 | 0.65/5/50 | 80 | 75 | 14822.8 | B | 128 |
| 0.8 Pd/WS2 | 25 | 1/20/0 | 65 | 65 | C | 129 | |
| Catalyst | Description | T, °C | Feeda, % | X, % | S, % | Activityb | Methodc | Ref. |
|---|---|---|---|---|---|---|---|---|
| a The feed composition listed in the table corresponds to C2H2/H2/C2H4. b Activity is calculated as the number of moles of acetylene converted per hour per total molar active metal (molC2H2 molmetal−1 h−1). c Method refers to the calculation method specified in Table 2. E* indicates that Method E was applied under ethylene-rich feed conditions. | ||||||||
| Au | ||||||||
| Au/CeO2 | Au-2 nm | 270 | 0.1/0.7/0 | 75 | 100 | 7.4 | G | 130 |
| Au/Fe2O3 | Au-3.5 | 169 | 0.11/15.4/0 | 50 | 95.9 | 3.0 | E | 131 |
| Au/SiO2-R250 | O2 plasma | 150 | 0.8/16/83.2 | 80 | 76 | D | 132 | |
| Au/C-TiO2-R250 | 150 | 1.0/10.2/88.7 | 62 | 43 | 334.8 | D | 133 | |
| Au/C-TiO2-R250 | 150 | 1.1/5.4/93.5 | 40 | 79 | 227.6 | D | ||
| Au/ZnO–F–O | 200 | 1/20/0 | 66 | 80 | 63.3 | E | 134 | |
| Au/CNA | 250 | 0.6/3/0 | 14 | 98 | 81.0 | E | 135 | |
| Au/Ce0.9Zr0.1O2 | 250 | 0.6/3/0 | 9.4 | E | 136 | |||
| Au/C-TiO2-am | 185 | 1.1/2.2/96.7 | 98 | 78 | D | 137 | ||
| Ni | ||||||||
| Ni/MCM-41 | 250 | 1:3:0 |
100 | 40 | 524.1 | E | 138 | |
| Ni/AC-N-0.5 | 200 | 1:3:0 |
96 | 46 | E | 139 | ||
| Ni/MCM-41 | 200 | 1:2:0 |
82 | 85 | E | 140 | ||
| Cu | ||||||||
| Cu(OH)2-derived | CuC | 110 | 0.45/10/88.8 | 98 | 82 | 0.1 | D | 141 |
| Cu-derived | CuxC–Cu | 110 | 0.47/7/91.8 | 100 | 80 | 0.1 | D | 142 |
| Cu2O derived | CuxC | 110 | 0.45/10/0 | 100 | 52.4 | E | 143 | |
| Catalyst | T, °C | Feeda, % | X, % | S, % | Activityb | Methodc | Ref. |
|---|---|---|---|---|---|---|---|
| a The feed composition listed in the table corresponds to C2H2/H2/C2H4. b Activity is calculated as the number of moles of acetylene converted per hour per total molar active metal (molC2H2 molmetal−1 h−1). c Method refers to the calculation method specified in Table 2. E* indicates that Method E was applied under ethylene-rich feed conditions. | |||||||
| NiX alloy | |||||||
| NiZn2/MgAl2O4 | 100 | 2/20/0 | 75 | 60 | 84.2 | E | 144 |
| Ni/MgAl2O4 | 100 | 2/20/0 | 75 | 47 | 29.5 | E | |
| Ni6In/SiO2 | 180 | 1/10/0 | 100 | 60 | 11.8 | E | 145 |
| Ni10In/SiO2 | 180 | 1/10/0 | 100 | 60 | 11.8 | E | |
| AgNi0.25/SiO2 | 120 | 1/20/20 | 50 | 50 | 97.1 | D | 146 |
| NiGa/α-Al2O3 | 160 | 15/58.2/0.8 | 37 | 72 | 46.5 | E | 147 |
| NiZn/TiO2 | 150 | 2/20/0 | 67 | 81 | 842.8 | — | 148 |
| Ni/Al2O3 | 200 | 0.5/10/20 | 100 | −60 | 12.6 | D | 149 |
| NiCo/Al2O3 | 200 | 0.5/10/20 | 55 | 87 | 6.9 | D | |
| NiFe/Al2O3 | 200 | 0.5/10/20 | 99 | 79 | 12.5 | D | |
| NiMn/Al2O3 | 200 | 0.5/10/20 | 98 | 65 | 12.3 | D | |
| NiMo/Al2O3 | 200 | 0.5/10/20 | 64 | 95 | 8.1 | D | |
| Ni5Ga/SiO2 | 180 | 1/5/0 | 100 | 78 | 11.8 | E | 150 |
| Ni7Sn/ZSM-12 | 250 | 1/2/0 | 100 | 84 | E | 151 | |
| Ni/ZSM-12 | 250 | 1/2/0 | 100 | 70 | E | ||
| Ni/SiO2 | 250 | 1/2/0 | 97 | 55 | E | 152 | |
| Ni7Mg/SiO2 | 250 | 1/2/0 | 100 | 31 | E | ||
| Ni7Sn/SiO2 | 250 | 1/2/0 | 100 | 65 | E | ||
| Ni/8%Sn–CeO2 | 250 | 1/2/0 | 100 | 83 | 153 | ||
| NiCu alloy | |||||||
| NiCu/Al2O3 | 115 | 0.31/3.1/30.4 | 80 | 86 | 4.5 | E | 154 |
| pre-NiCu/MMO | 160 | 0.33/0.66/34.5 | 100 | 70 | 9.1 | — | 155 |
| Ni&Cu-NP/Al2O3 | 130 | 1/2/0 | 100 | 71 | E | 156 | |
| CuNi0.125/SiO2 | 160 | 1/20/0 | 95 | 62 | 364.3 | E | 157 |
| NiCu0.125/MCM-41 | 250 | 1/3/0 | 100 | 62 | E | 158 | |
| CuNi7/ZSM-12 | 250 | 33.3/66.7/0 | 100 | 55 | E | 159 | |
| NiCu/ZrO2 | 220 | 1/10/20 | 100 | 93 | 1.1 | D | 160 |
| Ni2.5Co2.5Cu5@C/Al2O3 | 120 | 0.5/10/0 | 98 | 80 | E | 161 | |
| CuNiCs/TiO2 | 180 | 0.33/6.6/33 | 98 | 92 | E* | 162 | |
| Au–Ni alloy | |||||||
| AuNi/γ-Al2O3 | 58 | 4.3/8.7/87.0 | 64 | >99.99 | 17.6 | — | 163 |
| AuNi0.5/SiO2 | 100 | 1/20/0 | 48 | 39 | 1.7 | E | 164 |
| Others | |||||||
| PtCoOx/Z | 140 | 1/20/20 | 85 | 88 | 185.0 | C | 165 |
| PtCuOx/Z | 140 | 1/20/20 | 85 | 95 | 185.0 | C | |
| PtCu/Al2O3 | 150 | 1.1/11.1/22.2 | 65 | 85 | 17.8 | C | 166 |
| Cu0.8Zn0.2(OH) | 90 | 0.48/4/94.75 | 80 | 96 | D | 167 | |
| CuZnCo/HEOs | — | 0.71/2.86/70.72 | 100 | 90 | — | 168 | |
| AuAg/SiO2 | 250 | 0.8/16/83.2 | 82 | 55 | D | 169 | |
| Catalyst | Description | T, °C | Feeda, % | X, % | S, % | Activityb | Methodc | Ref |
|---|---|---|---|---|---|---|---|---|
| a The feed composition listed in the table corresponds to C2H2/H2/C2H4. b Activity is calculated as the number of moles of acetylene converted per hour per total molar active metal site (molC2H2 molmetal−1 h−1). c Method refers to the calculation method specified in Table 2. E* indicates that Method E was applied under ethylene-rich feed conditions. | ||||||||
| NiX IMC | ||||||||
| Ni3Ga-MIHMs | Ni3Ga | 120 | 0.65/5/50 | 70 | 80 | 0.6 | B | 170 |
| Ni/MHMs | Ni | 120 | 0.65/5/50 | 23 | 70 | 0.2 | B | |
| NiGa/MgAlO | Ni5Ga3 | 190 | 1/10/20 | 80 | 75 | 30.2 | C | 171 |
| NiGa/MgAlO | Ni5Ga3 | 90 | 1/10/0 | 75 | 78 | 28.3 | C | |
| NiGa/MgAlO | NiGa | 190 | 1/10/20 | 70 | 10 | 26.4 | C | |
| NiGa/MgAlO | NiGa | 90 | 1/10/0 | 25 | 82 | 9.4 | C | |
| Ni | Ni | 90 | 1/10/0 | 65 | 78 | 24.5 | C | |
| Ni3ZnC0.7/oCNT | Ni3ZnC0.7 | 200 | 0.5/4.5/20 | 98 | 95 | 61.6 | D | 172 |
| Ni/oCNT | Ni | 200 | 0.5/4.5/20 | 70 | 88 | 29.4 | D | |
| Ni3ZnC0.7/C | 160 | 1/15/0 | 95 | 85 | E | 173 | ||
| NiFeCuGaGe/SiO2 | CsCl0-type | 150 | 1.7/16.7/16.7 | 39 | 85 | 61.3 | C | 174 |
| NiGa/SiO2 | Ni1Ga1 | 150 | 1.7/16.7/16.7 | 54 | 90 | 14.2 | C | |
| Crystalline CeNi2 | CeNi2 | 100 | 2/10/0 | 3 | 85 | G | 175 | |
| Amorphous CeNi2Hx | CeNi2Hx | 100 | 2/10/0 | 90 | 26 | G | ||
| NiGa/MgAlO | Ni3Ga | 110 | 0.5/2.5/10 | 100 | 65.1 | C | 176 | |
| Ni3GaC0.5 | 110 | 0.5/2.5/10 | 100 | 89.1 | C | |||
| Ni | 110 | 0.5/2.5/10 | 100 | 46.7 | C | |||
| NiSb/MgAlO | P63/mmc NiSb | 180 | 0.5/2.5/10 | 50 | 93 | 0.4 | C | 177 |
| Ni3Zn/ZnO | Ni3Zn | 100 | 1/20/0 | 98 | 80 | 9.1 | E | 178 |
| Ni3Zn/ZnO | Ni3Zn | 125 | 1/20/20 | 20 | 70 | 1.8 | D | |
| NiZn/ZnO | NiZn | 100 | 1/20/0 | 16 | 12 | 1.5 | E | |
| NiZn/ZnO | NiZn | 125 | 1/20/20 | 54 | 75 | 5.0 | D | |
| Ni3Ga/TiO2 | NI3Ga | 150 | 1.8/18.2/9.1 | 30 | 90 | 59.0 | C | 179 |
| (Ni0.8Cu0.2)3Ga/TiO2 | 130 | 1.8/18.2/9.1 | 40 | 95 | 78.6 | C | ||
| (Ni0.67Cu0.33)3Ga/TiO2 | 110 | 1.8/18.2/9.1 | 60 | 92 | 117.9 | C | ||
| NiMn0.88Fe0.12Ge | 210 | 1/20/20 | 30 | 95 | 0.0 | C | 180 | |
| Ni3GaC0.5/oCNT | Ni3GaC0.5 | 160 | 0.5/5/20 | 89 | 75 | C | 181 | |
| PtX IMC | ||||||||
| PtGe/MCM-41 | Pt1Ge1 | 200 | 14/29/0 | 72 | 64 | — | E | 182 |
| PtSn1.0@MSW | Pt1Sn1 | 200 | 0.5/5.0/49.8 | 20 | 83 | 58.5 | B | 183 |
| Pt3Fe/SiO2 | L1-Pt3Fe | 180 | 1/2/0 | 99 | 83 | 27.4 | E | 184 |
| Pt3Fe/SiO2 | A1-Pt3Fe-alloy | 180 | 1/2/0 | 50 | 77 | 13.8 | E | |
| Others | ||||||||
| BiRh | Bulk-Rh1Bi1 | 200 | 0.5/5/50 | 95 | 88 | 0.6 | — | 185 |
| RhZn/SiO2 | Rh1Zn1 | 150 | 1/15/0 | 50 | 92 | 275.9 | E | 186 |
| RhZn/SiO2 | Rh1Zn1 | 217 | 1/15/0 | 100 | 91 | E | ||
| Al13Fe4 | 200 | 0.5/5/50 | 78 | 82 | B | 187 | ||
| Al13Fe4 | 200 | 2/10/88 | 16.5 | 75 | — | 188 | ||
| CoSb | 240 | 0.5/2.5/40 | 92 | 92 | C | 189 | ||
| Catalyst | Description | T, °C | Feeda, % | X, % | S, % | Activityb | Methodc | Ref. |
|---|---|---|---|---|---|---|---|---|
| a The feed composition listed in the table corresponds to C2H2/H2/C2H4. b Activity is calculated as the number of moles of acetylene converted per hour per total molar active metal (molC2H2 molmetal−1 h−1). c Method refers to the calculation method specified in Table 2. E* indicates that Method E was applied under ethylene-rich feed conditions. | ||||||||
| Ni SA | ||||||||
| Ni1/N–C | 150 | 0.5/10/50 | 70 | 95 | — | 190 | ||
| cal-Ni1@CHA | 100 | 1/16/0 | 60 | 95 | 5.4 | G | 191 | |
| Ni1MoS/Al2O3 | 100 | 0.15/3.03/15 | 50 | ∼100 | 1.1 | E* | 192 | |
| 120 | 0.15/3.03/15 | 100 | 90 | 2.2 | E* | |||
| Ni1Cu2/g-C3N4 | Ni1–Cu2 | 160 | 0.5/5/25 | 80 | 90 | 4.1 | C | 193 |
| Ni1/g-C3N4 | 160 | 0.5/5/25 | 13 | 85 | 0.7 | C | ||
| Nin/ND@G | Fully exposed | 190 | 1/10/20 | 100 | 85 | 91.0 | C | 194 |
| Cu SA | ||||||||
| Cu1/ND@G | 180 | 1/10/20 | 40 | 98 | 13.6 | C | 195 | |
| Cu1/ND@G | 200 | 1/10/20 | 90 | 98 | 30.6 | C | ||
| Cun/ND@G | Cu cluster | 200 | 1/10/20 | 20 | 93 | 6.8 | C | |
| 0.5Cu/Al2O3 | Single atom | 178 | 1/10/20 | 82 | 93 | 18.6 | C | 196 |
| 0.8Cu/Al2O3 | 3.4 nm Cu NPs | 172 | 1/10/20 | 60 | 92 | 13.6 | C | |
| Cu1/ND@G | 200 | 1/10/20 | 100 | 92 | 38.7 | C | 197 | |
| CuZn/NC2 | Cu1–Zn1 | 160 | 0.33/0.66/33 | 73 | 98 | E* | 198 | |
| Pt SA | ||||||||
| Pt1–In2O3 | 135 | 0.5/2.5/5 | 95 | 90 | C | 199 | ||
Fig. 5a and d illustrate the ethylene selectivity of Pd-based and non-Pd-based catalysts under ethylene-free feed conditions, with SC2H4 calculated using Method E. A monometallic Pd active site typically achieves ∼60% ethylene selectivity (E) due to ethane formation and C4+ byproducts. Incorporating a secondary metal, forming either a disordered alloy or an IMC phase, typically enhances ethylene selectivity to 70–80% (E). For Ni-based catalysts, monometallic Ni generally exhibits poor ethylene selectivity due to the formation of C4 species and GO. Ni alloys and Ni-based IMC structures can improve ethylene selectivity to some extent.
 | ||
| Fig. 5 Ethylene selectivity versus acetylene conversion over (a)–(c) Pd-based and (d)–(f) non-Pd-based catalysts. Methods C, D, and E in the figures correspond to the ethylene calculation methods specified in Table 2. The data are summarized from Tables 3–10. | ||
Under ethylene-rich feed conditions, ethylene selectivity is predominantly calculated using Methods C and D (Fig. 5b, c, e and f). Method D provides insights into the ability to suppress the ethylene over-hydrogenation pathway. Notably, Pd SA sites exhibit exceptional resistance to ethane formation even at nearly 100% acetylene conversion (Fig. 5c). Similarly, Cu single-atom catalysts effectively suppress both ethane and C4 formation, achieving SC2H4 above 90% (C) (Fig. 5e).
The catalytic activity is defined as the moles of acetylene converted per hour per active metal site. It should be noted that the activity is strongly influenced by reaction temperature and acetylene concentration. These factors must be taken into account when comparing catalytic performance from different studies. Fig. 6 presents a comparative analysis of the hydrogenation activity of different types of active sites under an acetylene feed concentration of ∼1%. Pd-based active sites generally demonstrate significantly higher activity compared to non-Pd-based active sites. For instance, Pd SA sites exhibit an activity that is more than two orders of magnitude higher than that of Cu SA sites.
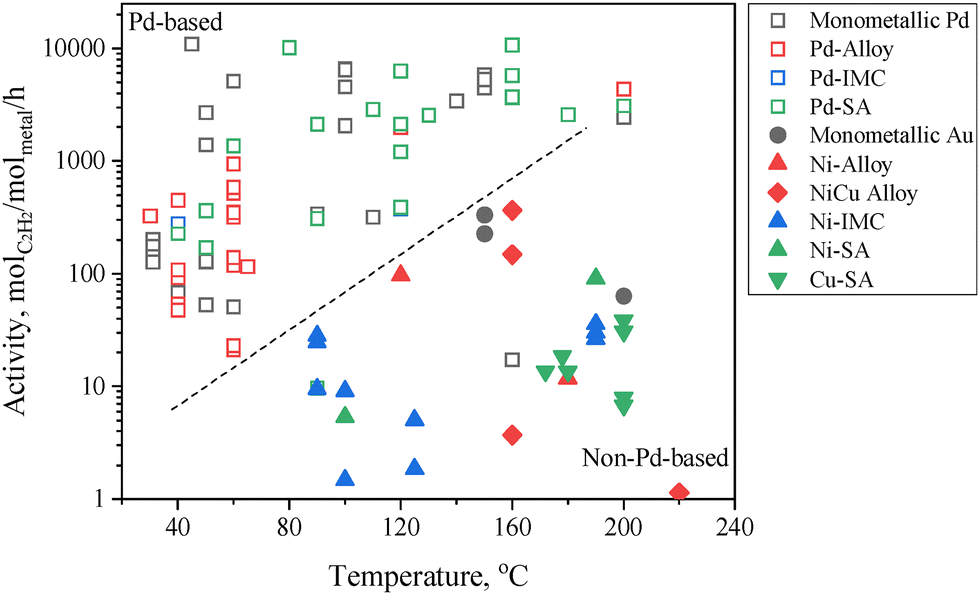 | ||
| Fig. 6 Acetylene hydrogenation activity over Pd-based and non-Pd-based active sites. Data are summarized from Tables 3–10. | ||
3.2 Pd-based active sites
Pd-based active sites are the most extensively studied catalysts for acetylene selective hydrogenation and are the primary catalysts currently used in industrial acetylene hydrogenation processes. Compared to non-Pd-based catalysts, Pd-based catalysts exhibit a significant advantage in catalytic activity. The evolution of Pd-based active sites has progressed from monometallic Pd to Pd-based disordered alloys, followed by Pd IMC sites and Pd SA sites. In addition, Pd-based active sites coated with carbon or metal oxide layer are reported as an efficient strategy to enhance the catalytic performance. In this section, we summarize the catalytic performance and research progress of Pd-based active sites across the different structural categories.The formation of β-PdHx and PdCx phases has also been reviewed.1,2 The formation of both β-PdHx and PdCx contributes to enhanced activity in acetylene hydrogenation, while the β-PdHx phase reduces ethylene selectivity. Since 2008, researchers have continued to investigate the effect of PdCx. It has been reported that the PdCx phase promotes higher ethylene selectivity. Compared to Pd/α-Al2O3 catalysts, Pd/α-Al2O3-AAS catalysts containing the PdCx phase showed improved ethylene selectivity, increasing from approximately 70% to 85% (calculated using Method E).200 Furthermore, researchers have identified that metal precursors, additives, and carbonization conditions significantly influence the formation of a stable PdCx phase. The introduction of silane coupling agents and the use of Pd(acac)2 as the metal precursor have been identified as effective strategies for promoting the formation of the PdCx phase.26,201
Advancements in nanocrystal synthesis have led researchers to investigate the influence of the Pd crystal structure on catalytic performance. By synthesizing Pd NPs in various shapes, including cubic (exposing only (100) faces), octahedral (exposing only (111) faces), cuboctahedral (exposing both (100) and (111) faces), and spherical Pd (primarily exposing (111) faces), researchers have investigated how the exposed Pd crystal facets affect acetylene hydrogenation activity and selectivity. For instance, Kim et al.20 synthesized cubic and spherical Pd NPs supported on alumina. Their results demonstrated that Pd sites on the (100) face of cubic NPs exhibited a TOF more than twice that of Pd sites on the (111) face of spherical NPs. Additionally, cubic Pd NPs displayed higher ethylene selectivity. However, other studies have drawn contrasting conclusions. It is commonly believed that the Pd(111) facet exhibits higher hydrogenation activity and selectivity.17,21 Yarulin et al.19 demonstrated that atoms on the (111) face are approximately 1.5 times more active than those on the (100) face. Recently, Xing et al.202 combined experimental results with computational methods, including the atomic pair distribution function and reverse Monte Carlo method. Their work revealed the presence of surface Pd sites with compressive strain in spherical Pd NPs (Fig. 7). This strain downshifts the local d-band and weakens the strength of ethylene adsorption, leading to an enhanced ethylene selectivity.
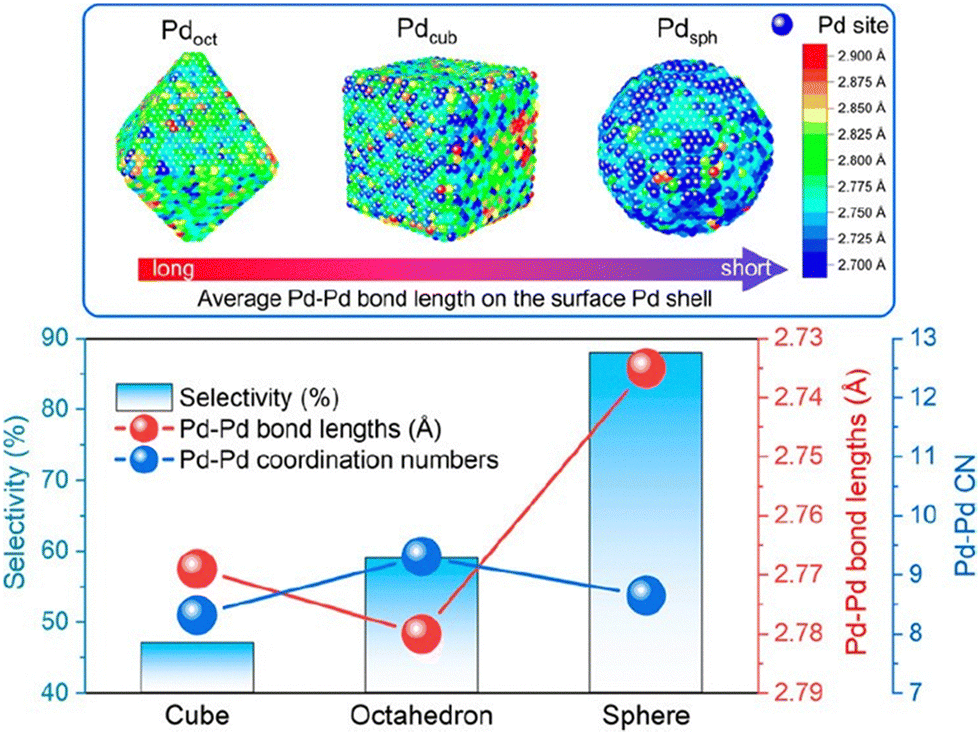 | ||
| Fig. 7 Comparison of partial Pd–Pd interatomic distances and Pd–Pd coordination numbers at the surface of Pd NPs with their selectivity for acetylene hydrogenation. Reprinted with permission from ref. 202. Copyright 2023 American Chemical Society. | ||
3.2.2.1 PdAg alloy. The PdAg alloy is the most commonly studied bimetallic catalyst for acetylene hydrogenation and remains widely used in the modern petrochemical industry. The incorporation of Ag enhances the ethylene selectivity of Pd-based catalysts through both electronic and geometric effects. Electronically, the interaction between Ag and Pd weakens ethylene adsorption on the surface, thereby suppressing its further hydrogenation to ethane.203 Geometrically, the presence of Ag reduces the number of neighboring Pd surface atoms, which in turn lowers the formation of oligomers.
An optimal Ag/Pd ratio was observed in the catalytic performance of PdAg catalysts. An excessive amount of Ag can significantly decrease hydrogenation activity, due to the poor H2 dissociation ability of metallic Ag. Delgado et al.37 reported that PdAg catalysts prepared by one-pot synthesis exhibited the best catalytic performance in terms of ethylene selectivity and stability at a Pd/Ag atomic ratio of 1.0. However, other studies have reported that Pd/Ag atomic ratios either below or above 1.0 also achieve optimal performance.28,38 This discrepancy can be attributed to differences in catalyst preparation methods, which significantly affect the microscopic mixing and structural configuration of PdAg NPs. PdAg bimetallic alloys exist as solid solutions, where the Pd and Ag atom randomly distributed within the NPs. Identical overall composition may produce a mixture of monometallic and bimetallic NPs with ill-defined compositions. Surface segregation of Pd during reactions further complicates the structure–property relationships. Williams et al.32 reported an enhanced ethylene selectivity of ∼80% (E) by synthesizing PdAg/SiO2 catalysts via a galvanic displacement method, which achieved near-atomic dispersion of Pd on Ag surfaces. Thermal treatment under H2 flow at 400–500 °C enabled Ag intercalation into Pd lattices, generating a Pd0.6Ag0.4 solid solution with ∼76% (E) ethylene selectivity.36
The surface atomic composition and arrangement of NPs play a crucial role in catalytic performance. The incorporation of Ag effectively isolates neighboring Pd active sites. To investigate the geometric effect of Ag, researchers have directly deposited Ag atoms onto Pd NP surfaces using electroless deposition (ED) or surface redox (SR) methods.29,204,205 Their results showed that both TOF and ethylene selectivity increased with Ag coverage. Higher Ag coverage leads to the formation of smaller Pd ensembles, which causes acetylene to adsorb as a π-bonded species, favoring selective hydrogenation to ethylene. Additionally, Pd@Ag core–shell catalysts have been proposed to enhance catalytic performance.31,33,35 Compared with PdAg/TiO2 catalysts prepared by co-impregnation, Pd@Ag catalysts synthesized via sequential photodeposition increased ethylene selectivity from ∼20% to ∼80% (C).31
3.2.2.2 PdCu alloy. Different from Ag, which primarily acts as a promoter, metallic Cu exhibits intrinsic activity for acetylene hydrogenation. However, its hydrogen dissociation capability is relatively weak compared to the Pd active sites. The activation energy for H2 dissociation on a Cu(111) surface has been calculated to be 0.46 eV, significantly higher than that on Pd(111) (0.20 eV).206
To enhance the catalytic activity, Pd has been incorporated into Cu-based catalysts at Cu/Pd atomic ratios ranging from 10 to 50.41–45,50 The PdCu catalyst with a Cu/Pd ratio of 43.6 exhibited a TOF that was an order of magnitude higher than that of monometallic Cu.44 At high Cu/Pd ratios, Pd atoms can be isolated within Cu particles, where hydrogen dissociation occurs on Pd sites, followed by hydrogen spillover to adjacent Cu sites, thereby enhancing the overall hydrogenation activity.41 The Cu/Pd ratio also plays a crucial role in determining ethylene selectivity. A PdCu catalyst with a Cu/Pd ratio of 50 exhibited an ethylene selectivity of 70–80% (E), while further decreasing the Cu/Pd ratio to 10 led to excessive hydrogenation of ethylene to ethane.41,42
PdCu catalysts with lower Cu/Pd atomic ratios (∼1.0) have also been investigated for acetylene hydrogenation.40,46,49,54 In these cases, Pd and Cu form a bimetallic alloy phase with a solid-solution structure, where Pd and Cu atoms are randomly distributed. Similar to PdAg catalysts, an optimal balance between catalytic activity and selectivity is closely related to the PdCu ratio. At low Cu/Pd ratios (e.g., Pd3Cu), the catalyst exhibited large Pd ensembles with minimal Pd isolation, which promoted the over-hydrogenation of ethylene to ethane. Conversely, at high Cu/Pd ratios (e.g., PdCu3), the catalysts were limited by H2 activation and thus showed higher selectivity to polymerization byproducts.46 The crystal structure of PdCu significantly influences acetylene hydrogenation performance.48,52,53,207 It has been reported that modifying the Cu60Pd40 structure from a disordered phase to a partially ordered CsCl-type structure increased ethylene selectivity from 74% to 92% (B).51 Moreover, PdCu alloys with a ordered body-centered cubic (B2) structure have shown superior ethylene selectivity compared to those with a face-centered cubic (FCC) structure.48,52,53
3.2.2.3 PdX alloy. In addition to widely investigated PdAg and PdCu, other bimetallic PdX catalysts have been developed for acetylene selective hydrogenation. The secondary metal (X) includes Au, Zn, Co, Sn, Ga, Ti, In, La, and Mn.57,64,208,209 Due to variations in reaction evaluation conditions and selectivity benchmarks across studies, direct comparisons of catalytic performance among these bimetallic catalysts remain difficult. To assess the effectiveness of bimetallic catalysts, researchers typically provide performance data for the corresponding monometallic Pd catalysts as references. Fig. 8 compares the catalytic performance of monometallic and bimetallic catalysts under identical reaction conditions, using a similar Pd metal loading within the same study. In these investigations, the catalysts were primarily synthesized via the impregnation or colloidal solution methods, resulting in bimetallic NPs that generally form an alloy phase without a well-defined structure. Introducing a secondary metal (X) consistently improves ethylene selectivity, though its effect on catalytic activity depends on the specific element. Notably, metals like Zn, Sn, In, La, and Mn enhance both selectivity and activity, outperforming monometallic Pd catalysts.
 | ||
| Fig. 8 Catalytic performance of monometallic and bimetallic Pd-based alloy catalysts for acetylene hydrogenation. Letters in parentheses indicate the selectivity calculation method as specified in Table 2. E* indicates that Method E was applied under ethylene-rich feed conditions. | ||
The structure of PdX bimetallic NPs significantly influences their catalytic performance in acetylene hydrogenation. Core–shell bimetallic catalysts, such as Au@Pd,210 Pd@Pt,211 and Ru@Pd,212 have been extensively developed for acetylene hydrogenation. Core–shell architectures offer a distinct approach to modulating the electronic and geometric properties of catalysts due to their unique strain effects and coordination environments, while maintaining a continuous and abundant exposure of shell–metal ensembles. The shell thickness plays a critical role in determining catalytic performance. A monolayer of the shell metal exhibits the strongest electronic interaction with the core metal, maximizing its electronic effects. Studies have shown that a thinner shell generally provides higher hydrogenation activity than a thicker one. For example, a Ru(5.1 nm)@0.7ML-Pd catalyst with a 0.7 monolayer Pd shell exhibited more than twice the hydrogenation activity compared to Ru(5.1 nm)@2.3ML-Pd with a 2.3 monolayer Pd shell, while ethylene selectivity increased from 51% to 71% (D).212
Trimetallic catalysts have also been investigated to enhance the performance of acetylene hydrogenation, with examples including Ni–Pd–Ag, Pd–Au–Ag, Al–Pd–Ru, and Pd–Cu–Zn.30,47,59,213 Compared to the corresponding bimetallic systems, trimetallic catalysts often exhibit better performance. For instance, under conditions ensuring 100% acetylene conversion, the PdAuAg2 catalyst achieved an ethylene selectivity of over 75% (E*), whereas monometallic Pd and bimetallic PdAu and PdAg catalysts all exhibited selectivity below 65% (E*).59 However, the introduction of a third metal increases the structural complexity of the catalysts, making it more difficult to identify active sites and hindering mechanistic understanding. Unraveling the structure–performance relationships in trimetallic systems remains a challenge.
 | ||
| Fig. 9 Catalytic performance of Pd-based IMC catalysts for acetylene hydrogenation. Letters in parentheses indicate the selectivity calculation method as specified in Table 2. | ||
3.2.3.1 PdGa IMC. PdGa IMCs are among the most extensively studied IMC catalysts for acetylene hydrogenation.79,80,214 Schlögl et al. first proposed that the well-defined crystal structure of PdGa IMCs could effectively isolate Pd atoms, thereby modifying their catalytic properties.14,76,77 They synthesized bulk PdGa and Pd3Ga7via the metal melting method and evaluated their performance for acetylene hydrogenation. These bulk samples exhibited excellent long-term stability and significantly higher ethylene selectivity (70–75%, B) compared to the Pd/Al2O3 reference (17%, B).75 However, the catalytic activity was relatively low due to the presence of surface oxide species, which formed during the grinding or milling process. Surface etching, which removed these oxide species, enhanced the surface activity (g mcat−2 h−1) to a level comparable to that of Pd/Al2O3. However, the ethylene selectivity decreased to ∼50% (B), likely due to partial decomposition of the IMC phase, leading to Pd-enriched surfaces.76
To improve the dispersion of PdGa IMCs and enhance their catalytic activity, researchers have focused on synthesizing well-defined PdGa IMC NPs. Two-step synthesis and precursor-based methods have proven effective in producing GaPd and GaPd2 NPs with particle sizes below 7 nm.78,215 Investigations of nanoscale PdGa IMC catalysts have demonstrated their intrinsically higher activity compared to monometallic Pd in acetylene hydrogenation. For instance, nano-GaPd2@Al2O3 and nano-GaPd@Al2O3 exhibited hydrogenation activities of 7120 and 4140 molC2H4 molPd−1 h−1, which were 2.9 and 1.7 times higher than that of Pd/Al2O3, respectively.78 Moreover, the ethylene selectivity over nano-GaPd@Al2O3 increased to 82% (B). The significantly enhanced catalytic performance of PdGa IMCs was attributed to the presence of isolated Pd atoms within their intermetallic structure. This conclusion was further validated through precise atomic-level engineering of the catalyst surface.81 The exposed surface structures of Pd2Ga IMC were tailored via reactive metal–support interactions, where increasing the reduction temperature induced a transition from the energetically favorable (013)/(020) facets to (011)/(002). This structural evolution resulted in two distinct configurations (Fig. 10): Pd2Ga(Pd1), characterized by isolated Pd atoms (Pd1), and Pd2Ga(Pd3), characterized by consecutive Pd trimers (Pd3). Notably, Pd2Ga(Pd1) exhibited a higher ethylene selectivity (80%, C) compared to Pd2Ga(Pd3) (60%, C).
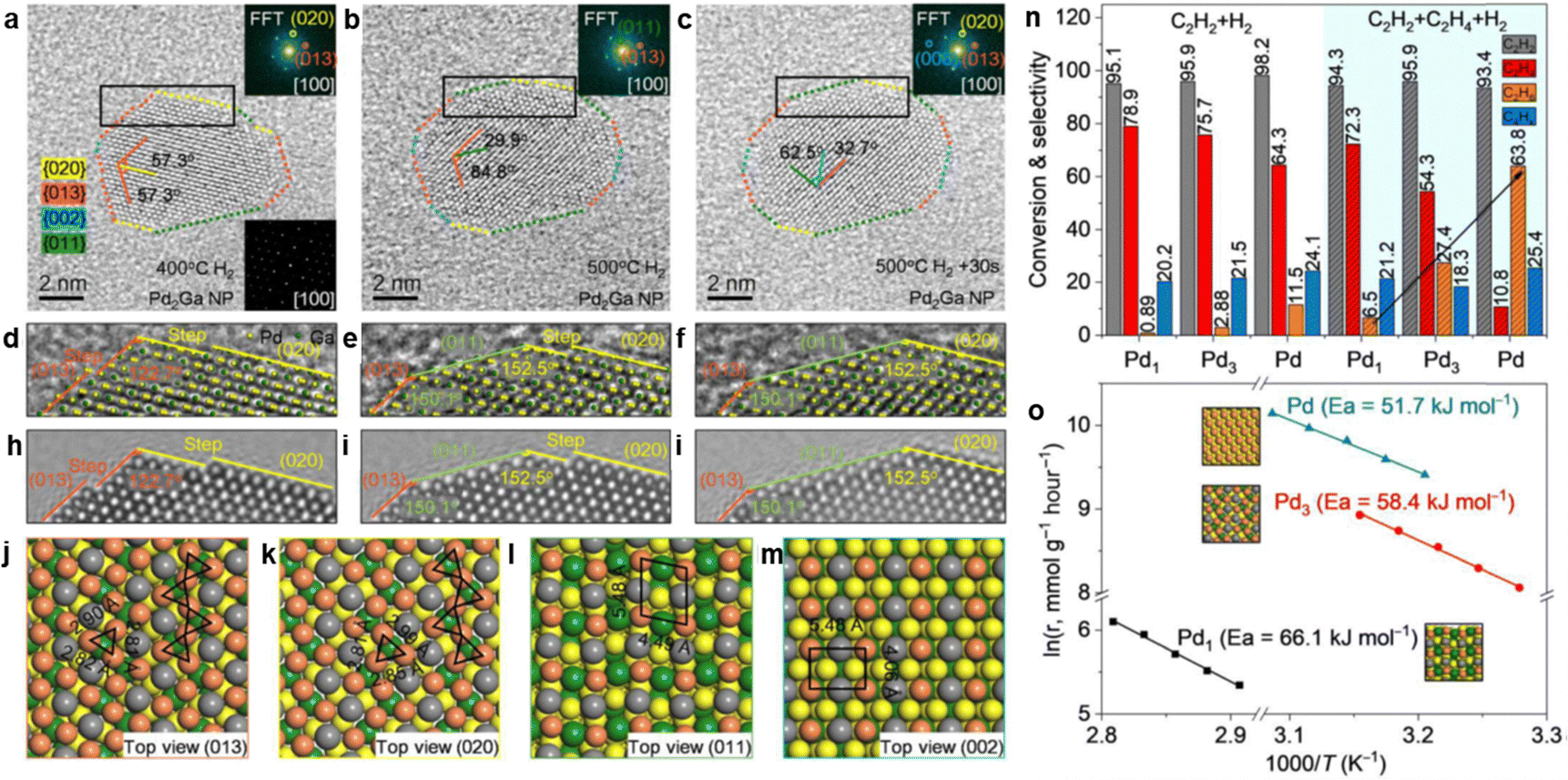 | ||
| Fig. 10 (a)–(m) Surface atom arrangement and variation during Pd2Ga refacetting, and (n) and (o) Hydrogenation properties of different surface Pd geometries. Reprinted with permission from ref. 81 Copyright 2022 The American Association for the Advancement of Science. | ||
3.2.3.2 PdZn IMC. PdZn bimetallic catalysts exhibit both ill-defined alloy phases and well-defined IMC phases. The formation of a well-defined structure is highly dependent on the catalyst preparation method. Various PdZn IMC catalyst, including Pd1Zn1, Pd3.9Zn6.1, and the (Pd,Zn) γ-brass phase, have been successfully synthesized for acetylene hydrogenation.13,86,87,216 Pd/ZnO, which predominantly contained the Pd1Zn1 phase (Fig. 11a), achieved a high ethylene selectivity of 89% (D) under ethylene-rich feed conditions.86 The high selectivity suggests that the Pd1Zn1 phase effectively suppresses ethylene over-hydrogenation to ethane. DFT calculations revealed that the Pd–Zn–Pd ensembles in the Pd1Zn1 phase exhibited a weak π-bonding interaction with ethylene (Fig. 11b), consistent with the experimental findings. However, it is noteworthy that the Pd/ZnO catalyst does not effectively suppress the formation of oligomers. Under ethylene-free feed conditions, Pd/ZnO showed an ethylene selectivity of ∼70% (E), while the selectivity toward oligomer (C4+) products exceeded 20% (E).86
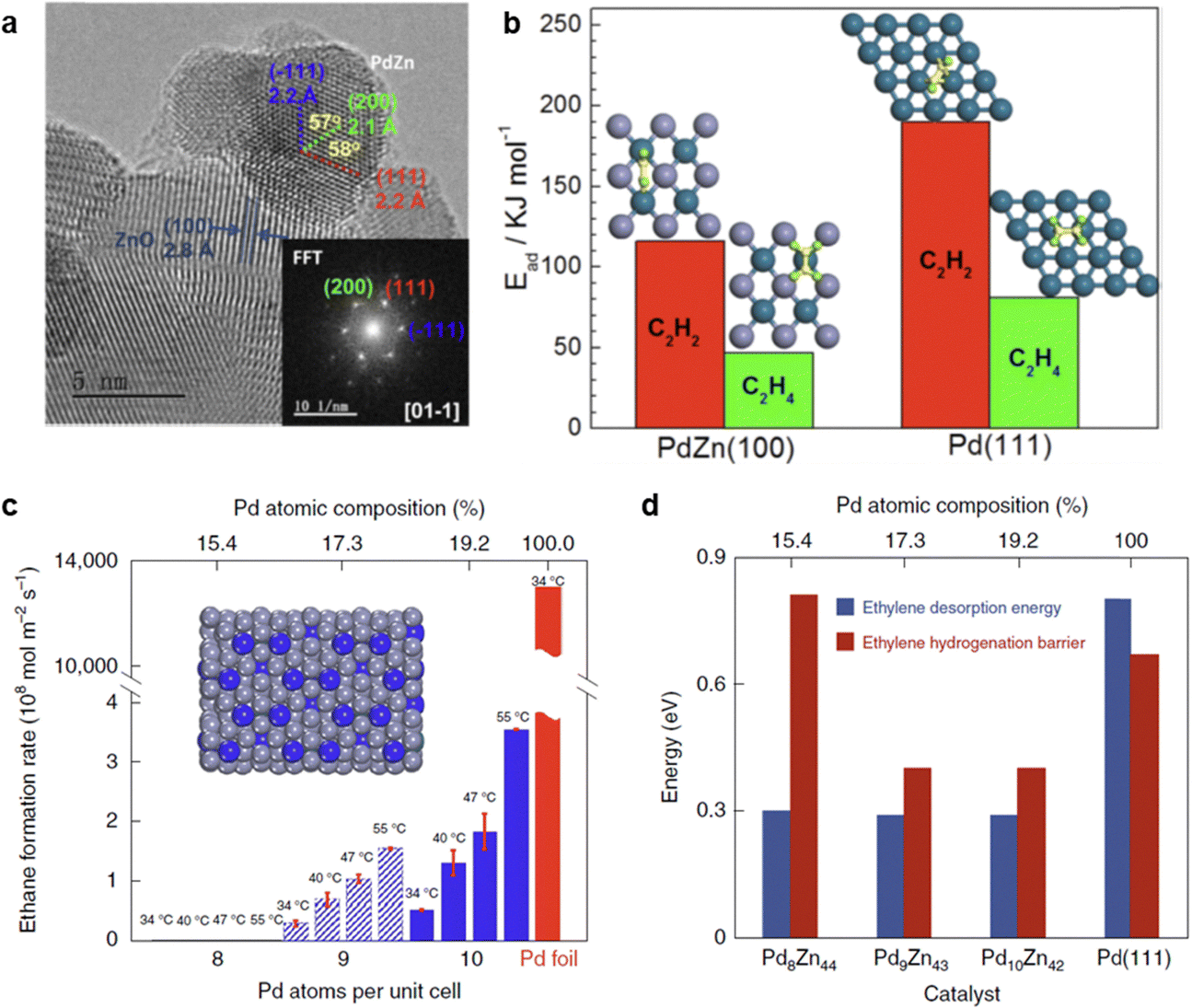 | ||
| Fig. 11 (a) HRTEM image of the Pd/ZnO catalyst with the formation of Pd1Zn1 phase; (b) DFT calculations of acetylene and ethylene adsorption on PdZn(100) or Pd(111) surfaces. Reprinted with permission from ref. 86. Copyright 2016 American Chemical Society. (c) Ethylene hydrogenation activity at different temperatures over Pd8Zn44, Pd9Zn43, Pd10Zn42 and Pd foil; (d) DFT-calculated ethylene hydrogenation barrier and desorption energy. Reprinted with permission from ref. 13. Copyright 2022 Springer Nature. | ||
The site-isolation effect in acetylene hydrogenation was further examined using the intermetallic (Pd, Zn) γ-brass phase. Pd8Zn44, which featured completely isolated Pd sites, exhibited an extremely low formation rate of ethane (Fig. 11c and d). Isotope labeling experiments enabled a direct calculation of the intrinsic ethylene selectivity (Method E) under ethylene-rich feed conditions, revealing an ethylene selectivity of ∼80%. However, it should be noted that the acetylene hydrogenation rate over Pd8Zn44 was significantly lower than that of Pd9Zn43, which contained Pd3 trimer sites on the surface. This difference is attributed to the limited ability of Pd8Zn44 to activate H2 dissociation, highlighting a trade-off between site isolation and H2 activation in PdZn IMCs.
3.2.3.3 PdX IMC. Given its structural similarity to Pd2Ga, Pd2In (Pnma) has been proposed as a potential IMC catalyst for acetylene hydrogenation. DFT calculations revealed that Pd2In and Pd2Ga exhibited only minor differences in their electronic structures, as reflected in their density of states (DOS).82 Catalytic performance evaluations revealed that Pd2In demonstrated good stability and a slightly higher ethylene selectivity of 80% (B) compared to GaPd2 (75%, B).82 To further explore PdIn IMCs for acetylene hydrogenation, compositions with varying In/Pd ratios, such as Pd3In, PdIn, and Pd2In3, have been developed. Generally, an increase in the In/Pd ratio enhanced ethylene selectivity but reduced acetylene hydrogenation activity. For example, as the In/Pd ratio increased from 1/3 to 1/1, the selectivity to ethylene significantly improved from 10% to 90% (C), whereas acetylene conversion decreased from 90% to 50%.82
Pd1Bi1 has been reported to exhibit highly dispersed Pd sites, which result in an extremely weak adsorption of ethylene.90 This unique electronic structure effectively suppressed the formation of β-PdHx and inhibited over-hydrogenation of ethylene to ethane. Consequently, the PdBi/Calcite catalyst (Pd1Bi1 phase) achieved an exceptionally high ethylene selectivity (>99%, D) under ethylene-rich feed conditions. Pd3Bi1 was employed for acetylene hydrogenation under ethylene-free feed conditions, where it exhibited an ethylene selectivity of ∼80% (E) at full acetylene conversion.91
Several other IMCs, including Pd2Sn, Pd3P, PdP2, and CaPdH2, have been investigated for acetylene hydrogenation.92,93,217 These IMC phases effectively modify the Pd ensemble structure and tune its electronic properties, leading to enhanced catalytic performance. In addition, ternary IMC catalysts have been developed to further improve the selective hydrogenation of acetylene to ethylene.89,94,218 Notable examples include Pd2Ga1−xSnx (0 ≤ x ≤ 1) and Pd11Bi2Se2. The introduction of Sn into Pd2Ga was found to enhance catalytic activity. Specifically, bulk-Ga0.7Sn0.3Pd2 exhibited an acetylene hydrogenation rate of 9.5 molC2H2 molPd−1 h−1, nearly doubling that of bulk-Ga1Pd2 (5.5 molC2H2 molPd−1 h−1), while maintaining the same ethylene selectivity.94
3.2.4.1 Pd SA sites supported on carbon materials. Carbon-based materials are among the most widely used supports for anchoring SA sites (Fig. 12). Through defect sites or nitrogen doping, atomically dispersed Pd species can be stabilized via Pd–N or Pd–C bonds.96,100,103,105 The unique structure of single-atom Pd sites effectively inhibits the formation of non-selective subsurface hydrogen species while facilitating the desorption of surface C2H4 species, thereby preventing the over-hydrogenation of ethylene to ethane. Under ethylene-rich feed conditions, Pd1/ND@G achieved a high ethylene selectivity of 90% (D) at full acetylene conversion.97 Li et al.99 developed a Pd SA catalyst via the atomization of Pd NPs, achieving an ethylene selectivity of 95% (B) at ∼75% acetylene conversion.
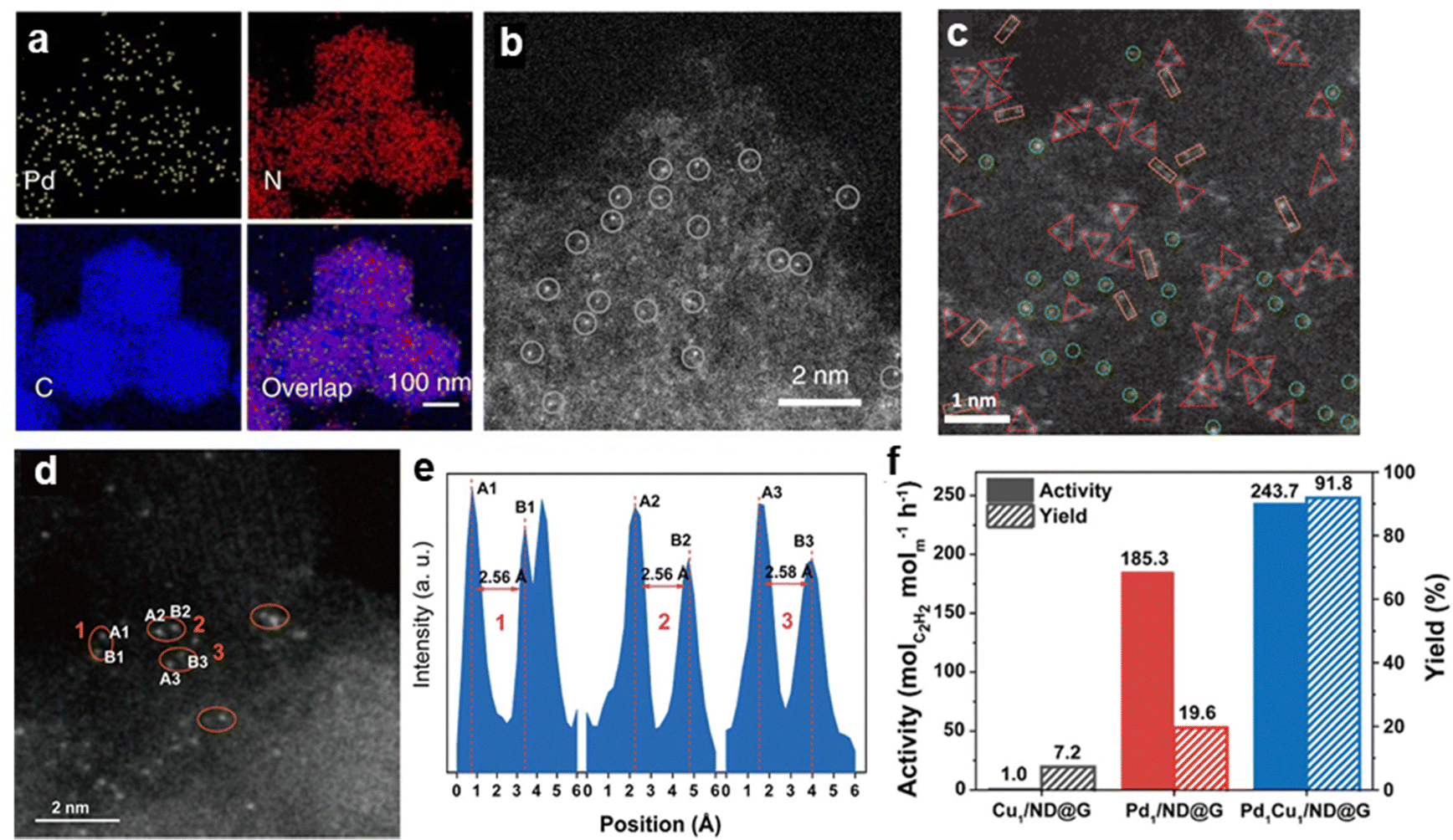 | ||
| Fig. 12 (a) EDS mapping and (b) HAADF-STEM image of Pd1/CN-900. Reprinted with permission from ref. 99. Copyright 2018 Springer Nature. (c) HAADF-STEM image of Pd3 trimer active site. Reprinted with permission from ref. 104. Copyright 2022 Springer Nature. (d) HAADF-STEM image and (e) intensity profile of Pd1Cu1/ND@G; (f) activity and C2H4 yields of the different catalysts. Reprinted with permission from ref. 102. Copyright 2022 American Chemical Society. | ||
The formation of Pd–N or Pd–C bonds weakened ethylene adsorption and reduced H2 activation, which leaded to sluggish H2 dissociation kinetics and alters the reaction pathway due to the absence of metal ensembles in Pd SA.102 In the acetylene hydrogenation over Pd SA sites, the support atoms forming bonds with Pd were considered to participate in H2 activation.98,99,101,219 However, the inherently low H2 dissociation ability of isolated Pd atoms limits hydrogenation activity, necessitating significantly higher reaction temperatures to achieve nearly 100% conversion compared to Pd NP catalysts (180 °C vs. 20 °C).97 To overcome this limitation, dual-atom102,220 and triatomic104 catalysts have been proposed to introduce cooperative sites that facilitate H2 dissociation and acetylene hydrogenation. Ma et al.102 designed a Pd1–Cu1 dual-atom catalyst (Pd1Cu1/ND@G), which exhibited a fivefold increase in catalytic activity compared to Pd1/ND@G (Fig. 12d–f). The proposed mechanism suggests that Cu sites serve as active centers for H2 activation, while Pd sites catalyze acetylene hydrogenation, thereby significantly improving catalytic performance.
3.2.4.2 Pd SA sites supported on metal oxide and other materials. Metal oxides, such as ZnO, MgO, Fe2O3, CeO2, and Al2O3, have been utilized as supports for anchoring Pd single atoms in acetylene hydrogenation.108,110,111,113,221 These catalysts have demonstrated good performance in suppressing the over-hydrogenation of ethylene to ethane, achieving ethylene selectivity exceeding 80% (D) under ethylene-rich feed conditions (Table 6). Similar to carbon-supported Pd SA, Pd SA sites on metal oxides exhibit limited efficiency in cleaving the H–H bond, thereby restricting hydrogenation activity. The reduced hydrogenation activity may accelerate the polymerization pathways. A 20% (E) selectivity to C4 products was observed over the Pd1/MgO catalyst. Atomic Pd anchored on α-Fe2O3(012) exhibited a unique Pd–Fe pair structure, which enhanced d-electron dominance near the Fermi level, thereby accelerating H2 dissociation.111
Beyond metal oxides, Pd single atoms have also been stabilized on other supports, including MOFs, hydroxyapatite, and Ni(OH)2.125,126,128 These catalysts typically enhance the ethylene selectivity to some extent and leverage the intrinsic properties of the supports to further enhance acetylene hydrogenation. For instance, the abundant hydroxyl groups in Ni(OH)2 promoted acetylene hydrogenation activity.128
3.2.4.3 Pd single atom alloy. Pd1Cu single-atom alloy (SAA) is among the earliest reported SAA catalysts for acetylene hydrogenation. While monometallic Cu catalysts exhibit intrinsic activity for acetylene hydrogenation, their weak H2 dissociation ability limits overall hydrogenation efficiency. To overcome this limitation, researchers introduced isolated Pd single atoms onto Cu surfaces as active sites for efficient hydrogen dissociation. Through a combination of DFT calculations and ultrahigh vacuum (UHV) surface experiments, Sykes et al.222 demonstrated that Pd1/Cu(111) exhibits efficient H2 dissociation and highly selective hydrogenation of acetylene (Fig. 13a). Zheng et al.223 further revealed that Pd single atoms anchored on Cu(100) displayed a higher hydrogen spillover rate and enhanced hydrogenation activity compared to those on Cu(111). Supported Pd1Cu catalysts were synthesized with an ultralow Pd content, achieving a Cu/Pd atomic ratio of up to 160.116,224 The isolation of Pd by Cu weakened C2H4 adsorption (Fig. 13b), leading to a high ethylene selectivity of 85% (D) at nearly 100% acetylene conversion. Beyond Pd1Cu, analogous Pd SAA incorporating Au,114 Ag,115,120,225,226 Ni,117,119 and Fe5C2121 have also been evaluated for acetylene hydrogenation. Compared to Pd1Au (<70%, D), Pd1Ag exhibited higher ethylene selectivity (∼80%, D), particularly at acetylene conversion levels exceeding 70%.115
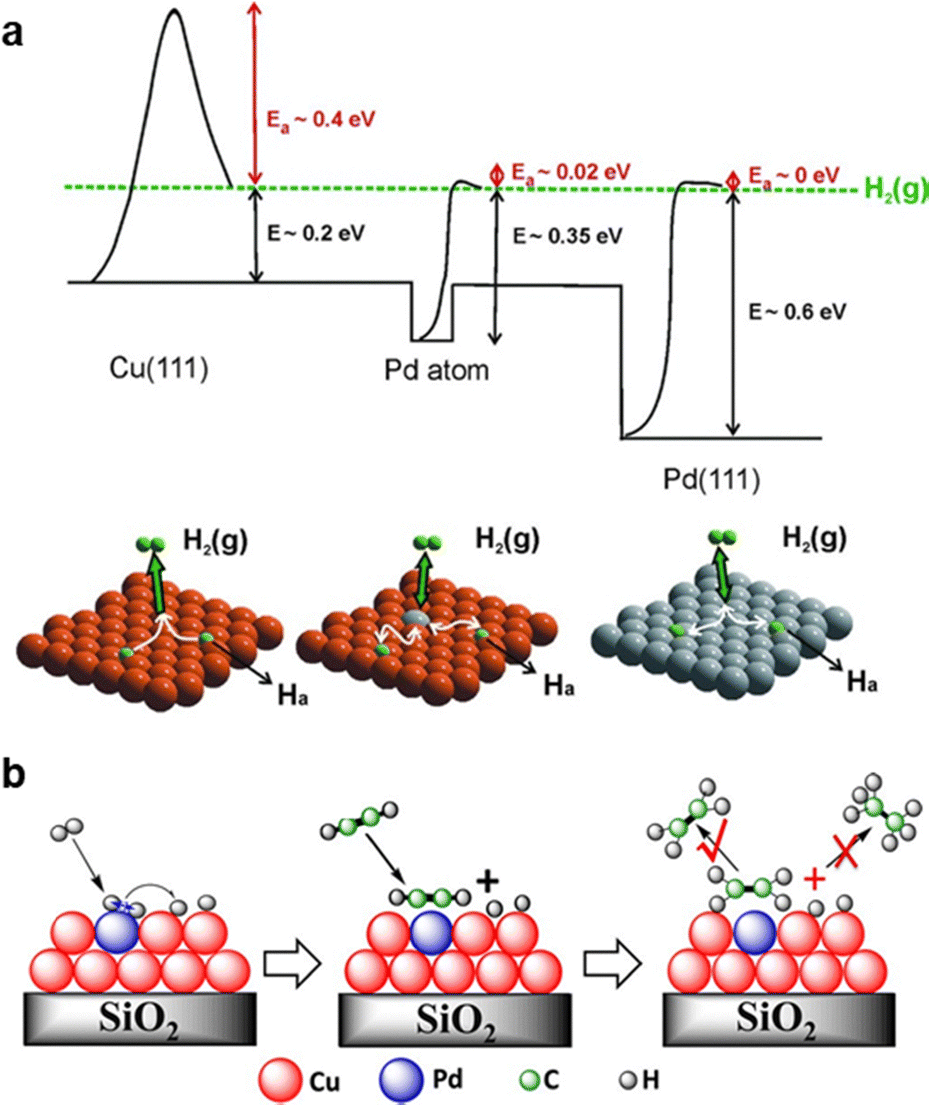 | ||
| Fig. 13 (a) Potential energy diagram of H2 dissociation over the Pd1/Cu(111) surface, where the orange and gray atoms are Cu and Pd, respectively. Reprinted with permission from ref. 222. Copyright 2012 The American Association for the Advancement of Science. (b) Proposed reaction pathway for semihydrogenation of acetylene over silica-supported Cu-alloyed Pd SA. Reprinted with permission from ref. 116. Copyright 2017 American Chemical Society. | ||
Metal oxides such as Bi2O3 and Ga2O3 have been deposited onto metal cores to enhance acetylene hydrogenation performance.73,233 These metal oxides can selectively decorate specific regions of the metal core and improve both selectivity and catalytic activity. Through precise structural design, researchers have successfully enhanced catalytic performance. For instance, atomic layer deposition (ALD) was employed to deposit controlled amounts of Ga2O3 adlayers on Pd NPs.234 This selective modification preferentially covered the edges and open facets of Pd, effectively suppressing oligomer formation (Fig. 14a). Lu et al.233 developed a Ga2O3 coating of Ag@Pd core–shell NP catalysts, which significantly enhanced the selectivity and activity (Fig. 14b). Additionally, Pd/Bi2O3 catalysts featuring nanometer-scale metal/oxide interfaces were designed to facilitate intra-cluster electron transfer, weakening ethylene adsorption and improving selectivity (Fig. 14c).
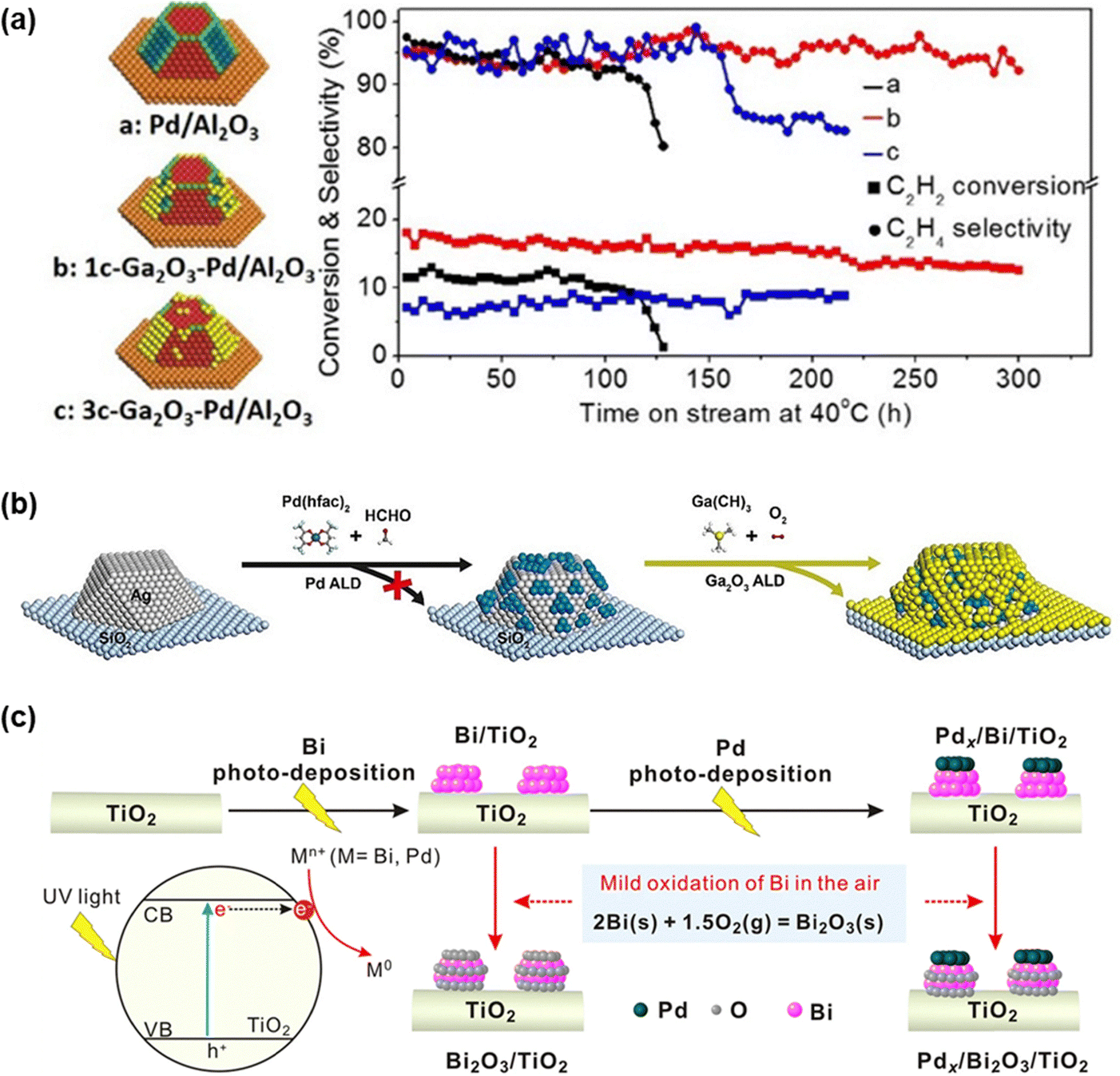 | ||
| Fig. 14 (a) Acetylene hydrogenation over Ga2O3-coated Pd particles. Reprinted with permission from ref. 234. Copyright 2016 American Chemical Society. (b) Ga2O3-coated Pd@Ag/SiO2 catalysts prepared by the ALD method. Reprinted with permission from ref. 233. Copyright 2021 Wiley-VCH GmbH. (c) Grafting nanometer metal/oxide interface of Pdx/Bi2O3/TiO2 catalyst. Reprinted with permission from ref. 235. Copyright 2021 Springer Nature. | ||
3.3 Non-Pd-based active sites
Non-Pd-based metals, such as Au, Ni, and Cu, had already been reported to exhibit selective hydrogenation activity for acetylene. Although their hydrogenation activity is significantly lower than that of Pd-based catalysts, ongoing research continues to develop catalysts based on less expensive and more abundant metals. The development of non-Pd-based catalysts follows a similar trajectory to Pd-based catalysts, where the design of active sites evolve from monometallic sites to disordered alloy structures, and further to well-defined IMC structures and SA sites. This section discusses the research progress of non-Pd-based active sites across these categories. Additionally, a non-metallic type of active site, known as frustrated Lewis pairs (FLPs), has also been applied in acetylene selective hydrogenation and is further discussed in Section 3.3.5.Both Pd and Au are precious metals, and from the perspective of reducing catalyst costs, non-precious metals such as Ni138,238,239 and Cu143,240 have been explored for acetylene hydrogenation. Among them, Ni is considered one of the most promising active metals due to its high efficiency in H2 dissociation. However, acetylene can easily decompose on the sites of three-nickel-atom arrangements to form ethylidine species.241 A major challenge associated with monometallic Ni catalysts is the formation of substantial amounts of C4 and GO during hydrogenation.138–140 The selectivity toward oligomers exceeded 40%, leading to rapid catalyst deactivation, with acetylene conversion dropping from 100% to below 50% within a 5-hour test.138 In comparison, monometallic Cu catalysts suffer from intrinsically low activity due to their poor ability to dissociate H2. Compared to Au, the activity of Cu can be lower by up to two orders of magnitude.141,143 Additionally, other transition metals such as Co242 and Fe243 have also been investigated for acetylene hydrogenation, but these catalysts typically show low activity and selectivity for acetylene selective hydrogenation.
3.3.2.1 NiCu alloy. Monometallic Ni and Cu each exhibit distinct advantages in terms of catalytic activity and selectivity, respectively. To take advantage of both metals, researchers have proposed Ni–Cu alloy catalysts as a strategy to enhance acetylene hydrogenation performance.154,155,160 Compared to monometallic Ni, NiCu alloy catalysts showed improved ethylene selectivity. Long-term stability tests revealed that pre-NiCu/MMO (derived from NiCuMgAl layered double hydroxide) maintained an ethylene selectivity above 50%, whereas imp-Ni/MMO (prepared by impregnation method) experienced a decline in selectivity to ∼10%.155 To further optimize catalytic performance, the Cu/Ni atomic ratio was varied156–159 and the optimal Cu/Ni ratio was identified to be in the range of 0.05–1. For instance, while the ethylene selectivity over NiCu1 and NiCu0.05 was ∼55% (E), it increased to 62% (E) over NiCu0.125.158 However, suppressing C–C coupling reactions on NiCu catalysts remains a significant challenge.
3.3.2.2 NiX alloy. In addition to NiCu alloys, other Ni-based bimetallic catalysts have been studied for acetylene hydrogenation. Using DFT calculations, Nørskov et al.244 predicted that Ni–Zn alloys should exhibit selectivity comparable to Pd–Ag systems. Experimental evaluations confirmed that a 25%Ni–75%Zn catalyst displayed remarkably low selectivity to ethane, even lower than that of a 25%Pd–75%Ag catalyst, under ethylene-rich feed conditions. Subsequent research efforts have focused on developing highly selective Ni–Zn alloy catalysts and evaluating their intrinsic ethylene selectivity under ethylene-free feed conditions.144,148 Compared to the ethylene selectivity of 47% (E) over Ni/MgAl2O4, the selectivity was enhanced to 60% (E) over NiZn2/MgAl2O4.144 Further increasing the Zn/Ni atomic ratio increased ethylene selectivity, reaching 80% (E) over NiZn5.148 However, rapid deactivation was observed during long-term stability tests. Beyond Ni–Zn, several other secondary metals have been explored in Ni-based bimetallic catalysts for acetylene hydrogenation, including In, Ag, Co, Fe, Mo, Mn, Ga, and Sn.145–147,150–152,245 Notably, NiGa catalysts exhibited enhanced ethylene selectivity of 70–80% (E) at nearly 100% acetylene conversion.147,150 Among the Ni-based bimetallic catalysts with Fe, Co, Mn, and Mo as the secondary metals, NiMo demonstrated the most effective suppression of ethylene over-hydrogenation to ethane, achieving an ethylene selectivity of 95% (D).149
3.3.2.3 Others multi-metallic alloy. Pt-based bimetallic catalysts have been explored for acetylene hydrogenation (Table 9). Chen et al.246 investigated acetylene hydrogenation over a Ni/Pt(111) model surface in a batch reactor, demonstrating that PtNi bimetallic catalysts exhibited both higher hydrogenation activity and improved selectivity than monometallic Ni and Pt catalysts. Further research has been conducted on supported Pt-based bimetallic catalysts, including PtAg, PtNi, PtCo, PtFe, and PtCu, for acetylene hydrogenation.165,166,247–249 Notably, PtCo and PtCu catalysts achieved ethylene selectivity >85% (C).165,166 In contrast, Au-based bimetallic catalysts, such as AuAg and AuNi, showed limited improvement in ethylene selectivity.163,164,169 Under ethylene-free feed conditions, the ethylene selectivity over AuNi remained below 80% (E).164 Other metal alloy catalysts, including CuZn167 and ternary NiCoCu161 alloys, demonstrated the ability to suppress ethylene over-hydrogenation to ethane. However, they still face challenges in effectively suppressing the oligomerization reaction.
3.3.3.1 NiX IMC. Ni-based IMC catalysts are the most extensively studied non-Pd-based IMC catalysts for acetylene hydrogenation. Fig. 15 summarizes the catalytic performance in terms of activity and selectivity compared with benchmark catalysts.
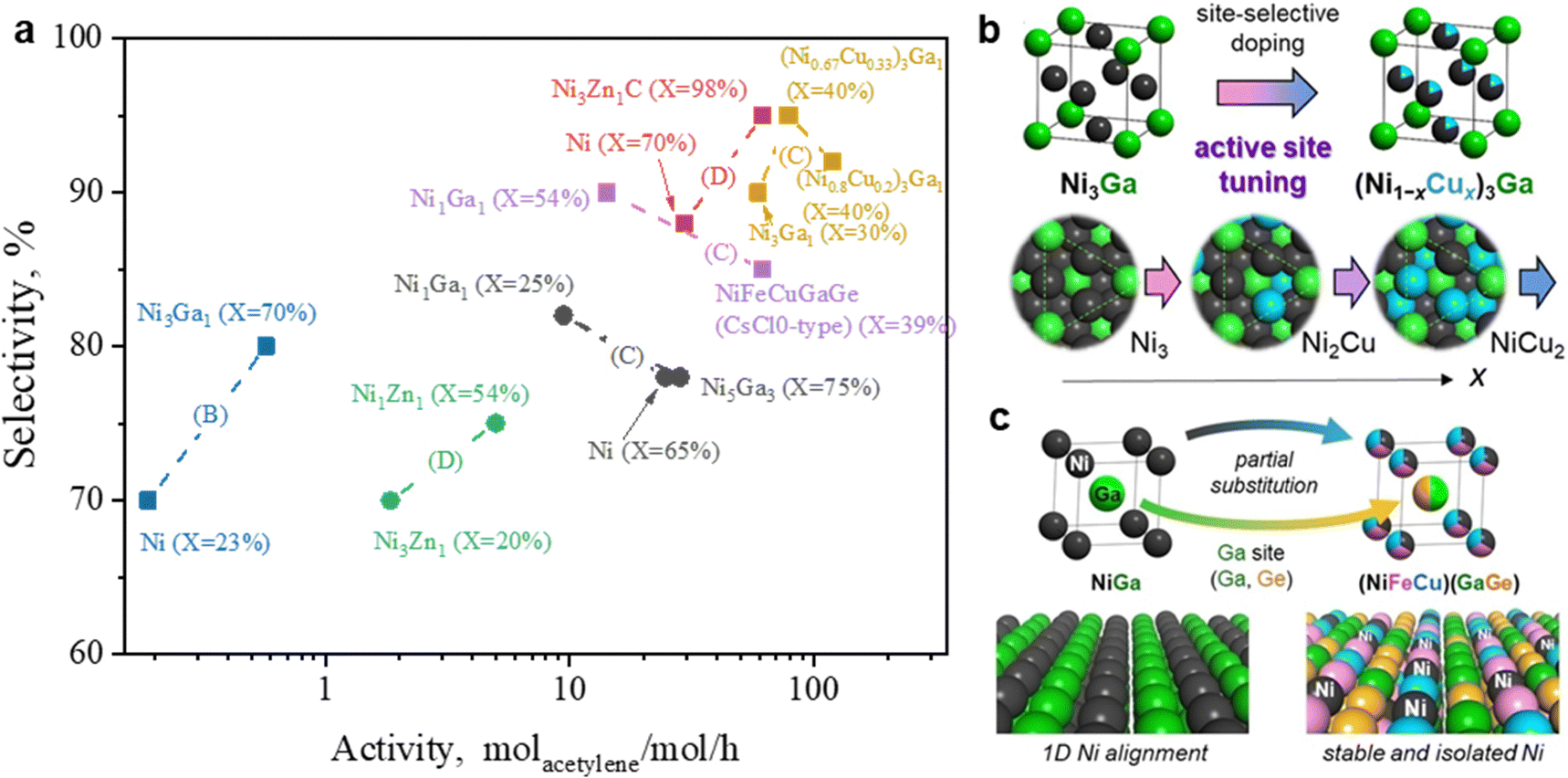 | ||
| Fig. 15 (a) Catalytic performance of Ni based IMC catalysts for acetylene hydrogenation. (b) Site-selective doping of Cu to the Ni sites of Ni3Ga IMC. Reprinted with permission from ref. 179. Copyright 2024 Royal Society of Chemistry. (c) Multi-metallization of intermetallic NiGa to (NiFeCu)(GaGe) HEI. Reprinted with permission from ref. 174. Copyright 2022 Wiley-VCH GmbH. Letters in parentheses indicate the selectivity calculation method as specified in Table 2. | ||
Initially, intermetallic NiZn was identified as a cost-effective alternative to PdAg catalysts through DFT descriptor-based analysis across late 3d transition metal alloys.244 However, early studies primarily synthesized NiZn as a disordered alloy phase rather than a well-defined IMC phase, limiting the ability to establish clear structure–performance relationship. Rioux et al.250 successfully synthesized a series of bulk Ni–Zn IMC phases, including Ni4Zn, Ni1Zn1, and Ni5Zn21, and evaluated their catalytic performance using isotopic labeling in a batch reactor. As the Zn/Ni ratio increased, the intrinsic ethylene selectivity (E) increased from <10% (Ni) to ∼20% (Ni4Zn & Ni1Zn1) and further to ∼50% (Ni5Zn21). Despite these advancements, a major challenge remains in effectively suppressing the formation of oligomer products.178
NiGa IMCs have also been investigated for acetylene hydrogenation, exhibiting a similar trend between the Ga/Ni ratio and ethylene selectivity as observed in the NiZn IMC system.170,171 As the Ga/Ni ratio increased from 0 to 1, ethylene selectivity increased from 78% to 82% (C).171 Atomically isolating Ni sites has been proposed as a strategy to mitigate oligomer formation. Furthermore, coupling this approach with the electronic modification effect of a secondary metal can further enhance catalytic performance. For instance, Sb, which has a higher electronegativity than Ni, was incorporated into the Ni lattice, forming Ni1Sb1. This IMC catalyst exhibited high ethylene selectivity (93%, C) at nearly 100% acetylene conversion.177
Incorporating interstitial carbon atoms into the lattice of NiX IMCs has been demonstrated as an effective strategy to simultaneously enhance both catalytic activity and selectivity. Representative intermetallic carbides include Ni3ZnC0.7 and Ni3GaC0.5.172,173,176 Compared with Ni/CNT, the Ni3ZnC0.7/CNT catalyst exhibited a twofold increase in activity, while the ethylene selectivity improved from 88% to 95% (D).172 Under ethylene-free feed conditions, the ethylene selectivity was enhanced from ∼70% (E) over Ni3Zn/CNT to 85% (E) over Ni3ZnC0.7/C.173
Recently, multi-metallic IMC and high-entropy intermetallic (HEI) catalysts have been proposed for acetylene hydrogenation (Fig. 15b and c). Multi-metallic IMCs offer additional opportunities for tuning the electronic properties of the Ni active sites while preserving the geometric structure essential for reactant adsorption and activation. For instance, partial substitution of Ni in the Ni3Ga IMC phase with Cu ((Ni0.67Cu0.33)3Ga/TiO2) nearly doubled the hydrogenation activity.179 Moreover, the HEI NiFeCuGaGe/SiO2 catalyst exhibited more than a fourfold increase in activity compared to Ni1Ga1/SiO2.174
3.3.3.2 Other IMCs. PtX IMC catalysts, including Pt1Ge1, Pt1Sn1, and Pt3Fe (L12), have been evaluated for acetylene hydrogenation.182–184 The transformation of disordered Pt3Fe (A1) particles into an ordered Pt3Fe (L12) phase resulted in the formation of isolated three-fold hollow Pt sites, with electronic properties finely tuned by neighboring Fe single atoms (Fig. 16). This structural modification led to a twofold increase in acetylene hydrogenation activity, while ethylene selectivity increased from 77% to 83% (E).184
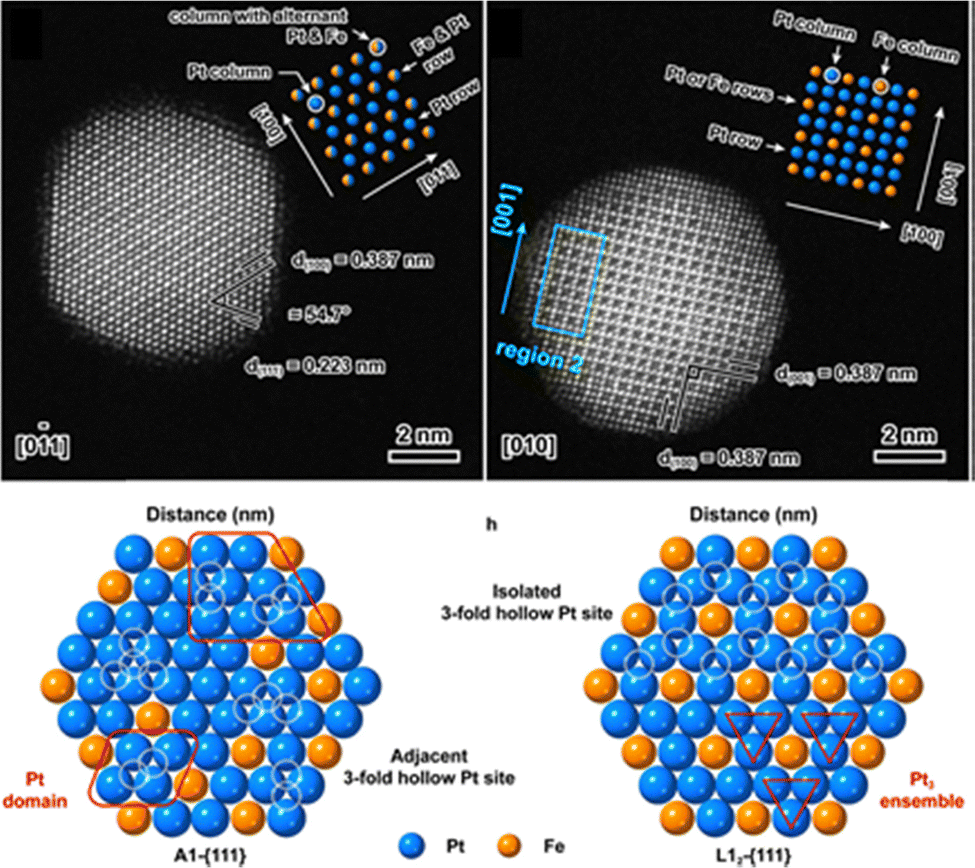 | ||
| Fig. 16 Geometric arrangements of Pt and Fe atoms in the Pt3Fe A1 and L12 NPs. Reprinted with permission from ref. 184. Copyright 2023 Elsevier. | ||
Rh-based IMCs, such as Rh1Bi1 and Rh1Zn1, have demonstrated enhanced performance for acetylene hydrogenation as well.185,186 Lan et al.186 proposed a Zn atom emitting–trapping–ordering (Zn-ETO) strategy, which enabled the synthesis of highly pure-phase IMC catalysts. Compared with monometallic Rh/SiO2, the Rh1Zn1/SiO2 catalyst synthesized via the Zn-ETO method significantly enhanced ethylene selectivity from 50% to 92% (E).
Another approach in IMC catalyst development involves replacing noble metals with cost-effective alternatives. MoP and Al13Fe4 have been explored for acetylene hydrogenation, exhibiting moderate hydrogenation activity and selectivity.187,188,251 Specifically, Al13Fe4 achieved an ethylene selectivity of 82% (B) at a conversion of 78%.187 However, challenges remain in terms of limited activity and rapid deactivation.
Karim et al.255 investigated the electronic and catalytic properties of TiO2 supported Pt SA and subnanometer clusters (Fig. 17a). Their results demonstrated that as the size of Pt species decreased from 2.1 nm to single atoms, the TOF significantly dropped from 2.5 × 10−2 s−1 to 6.0 × 10−5 s−1 (at 70 °C), while ethylene selectivity within the C2 products increased from 50% to 100% (G). A comparable trend was observed for Cu-based SA catalyst (Fig. 17b). As the size of Cu in Cu/Al2O3 decreased from 9.3 nm to single atoms, the TOF dropped from 147 h−1 to 4 h−1, while ethylene selectivity improved from 84% to 91% (C).196 In addition to enhancing selectivity, SACs also demonstrated a certain degree of suppression of undesired coupling side reactions, leading to significantly improved catalyst stability.194,195,197 Compared with Cu NP catalysts, Cu SACs exhibited a much lower coking rate after ∼5 h on stream, achieving a steady state following an initial deactivation.196
 | ||
| Fig. 17 Size effect of (a) Pt active site and (b) Cu active site on the activity and selectivity for acetylene hydrogenation. Reprinted with permission from refs. 255 and 196. Copyright 2019 American Chemical Society and 2020 American Chemical Society. (c) and (d) Structural characterization of Ni1Cu2/g-C3N4 catalyst. Reprinted with permission from ref. 193. Copyright 2021 Springer Nature. | ||
To further enhance catalytic performance in acetylene hydrogenation, multi-heteronuclear SA catalysts have been proposed, such as Ni1Cu2/g-C3N4193 and dual-atom CuZn/NC2.198 Compared with Ni1/g-C3N4, Ni1Cu2/g-C3N4 exhibited higher catalytic activity, ethylene selectivity, and catalyst stability. Characterization and computational studies revealed that the Ni active site, confined within two stable hydroxylated Cu grippers (Fig. 17c and d), underwent dynamic structural rearrangements. These rearrangements involved the breaking of interfacial Ni-support bonds upon reactant adsorption and their reformation upon product desorption, thereby optimizing the overall catalytic efficiency.193
| Catalyst | T, °C | Feeda, % | X, % | S, % | Activityb | Methodc | Ref. |
|---|---|---|---|---|---|---|---|
| a The feed composition shown corresponds to the ratio of C2H2/H2/C2H4. b Activity is expressed as the amount of acetylene converted per hour per gram of catalyst mass (mmolC2H2 gcat−1 h−1); values marked with an asterisk (*) are calculated per unit volume of catalyst (mmolC2H2 mLcat−1 h−1). c Method refers to the selectivity calculation method defined in Table 2. E* denotes Method E applied under ethylene-rich feed conditions. | |||||||
| CeO2 | 250 | 1/30/0 | 80 | 85 | 11.86* | E | 257 |
| Graphenes | 110 | 1.05/3.2/95.7 | 81 | — | 56.63 | 258 | |
| In3O5 | 300 | 1/30/0 | 70 | 84 | 3.63* | E | 259 |
| Ni–CeO2 | 75 | 0.5/2.0/0 | 46 | 63 | 2.05 | D | 260 |
| CrOx/γ-Al2O3 | 120 | 0.48/36.5/9.3 | 55 | — | 2.53 | 261 | |
| r-CeO2-500 | 200 | 2.5/75/0 | 13 | 74 | 2.09 | E | 262 |
| h-Al2O3/FeO1.25 | 130 | 0.7/13.5/52.7 | 83 | 85 | — | E* | 263 |
| Cerial-VO | 200 | 2.5/75/0 | 3 | 12 | 0.12 | E | 264 |
| Cerial-H | 200 | 2.5/75/0 | 2 | 20 | 0.08 | E | |
| Cerial-O | 200 | 2.5/75/0 | 7 | 75 | 0.28 | E | |
FLP catalysts for acetylene hydrogenation include both metal oxide and carbon materials. Carbon materials such as graphene, graphene oxides, and multi-walled carbon nanotubes generally exhibited higher hydrogenation activity, with rates exceeding 1.0 gC2H2 gcat−1 h−1.258,265–267 However, these materials typically showed low selectivity for ethylene, due to over-hydrogenation of ethylene to ethane and the formation of a significant amount of oligomers.
Metal oxides, including CeO2,257,260,262,268 In3O5,259 CrOx,261 and FeOx,263,269 have also been investigated for acetylene hydrogenation, where the intrinsic selectivity to ethylene can be enhanced to >70% (E). It has been demonstrated that ceria catalysts with stoichiometric surface OH groups (Ceria-O) were crucial for enhancing ethylene selectivity. Compared to ceria with surface oxygen vacancies (Ceria-VO) or hydride species (Ceria-H), the Ceria-O catalyst increased ethylene selectivity from <20% to 75%.264 However, it should be noted that metal oxides face significant challenges in terms of hydrogenation activity. Even at high reaction temperatures of 200 °C, the catalytic activity was lower than 0.1 gC2H2 gcat−1 h−1.
4. Theoretical studies of acetylene hydrogenation
Experimental studies have demonstrated that the composition and structure of active sites significantly influence the performance of acetylene hydrogenation. In earlier studies, researchers could only infer possible reaction mechanisms based on the adsorption behavior and kinetic fitting models from experimental results. With the rapid advancement of computational quantum chemistry, researchers now have a powerful toolkit to investigate the structure–performance relationships of active sites at the atomic level. In the past two decades over 100 studies have focused on the reaction mechanisms of acetylene selective hydrogenation from a theoretical perspective. These computational studies have not only rationalized experimental observations but also offered theoretical insights to guide the design of improved active sites.To summarize these theoretical investigations, we categorize these studies according to the structural characteristics of the studied active sites. The computational models are classified into Pd-based and non-Pd-based categories and further subdivided into monometallic, disordered alloy, IMC, and SA site types, following the framework established in Sections 3.2 and 3.3 for experimental studies. The key research themes and major findings are summarized in Tables 12 and 13. DFT studies on the reaction mechanism of acetylene selective hydrogenation typically focus on three key aspects:
| Active site | Investigation theme | Key observations | Ref. |
|---|---|---|---|
| Monometallic Pd | Pdn (n = 2–12) cluster size effect | • Pd2 cluster showed more selectivity while Pd12 showed higher activity. | 270 and 271 |
| Exposed Pd crystal surface | • Clean Pd(111) surface exhibits higher selectivity and activity compared to Pd(100). | 272–274 | |
| • Pd(100) is more prone to forming PdCx phase, and in its carbide form, Pd(100) demonstrates higher activity than Pd(111). | |||
| Subsurface or interstitial atoms of C, B, H, et al. | • Subsurface H increase the overall activity while decrease in the ethylene selectivity. | 275–280 | |
| • Subsurface C increase the ethylene selectivity. | |||
| • B-modified Pd surfaces show similar properties with C-modified Pd. | |||
| Molecule coverage effect | • Coverage-dependent microkinetic model gives a much more reasonable TOF (1.41 s−1). | 281 and 282 | |
| PdX (X = coinage metals of Ag, Cu, Au) | Promotion effect of Ag | • Ag promotes ethylene and hydrogen desorption, reducing their further reaction. | 283–285 |
| • Pd–Ag(100) hinders carbon deposit migration, leading to low selectivity due to hydrogenation of carbonaceous species. | |||
| Ratio of Ag to Pd and PdxAgy cluster structure | • Pd1Ag3 is proposed as a potential phase with optimized catalytic performance | 286–289 | |
| Surface restructure | • Surface Pd contents in PdAg and PdAu tend to increase under acetylene hydrogenation conditons | 290 and 291 | |
| Core–shell structure | • Cu@Pd core–shell structure showed better activity and selectivity than Au@Pd and Ag@Pd | 292 and 293 | |
| Screen of PdX alloy | • Coinage metals in Pd promote the ethylene selectivity and hinder the formation 1,3-butadiene. | 294–301 | |
| • Pd1Cu3, Pd1Ag1 and Pd1Au1 catalysts show better activity and ethylene selectivity and inhibit the formation of oligomer. | |||
| Pd-based IMC | PdGa IMC | • PdGa IMC surface terminations significantly impact catalytic pathways, activity and selectivity. | 302–307 |
| • Ga-enriched GaPd2(010) surfaces showed enhanced ethylene selectivity. | |||
| Pd2Sn, and AlPd IMC | • The B20-type AlPd compound exhibits potential as an active and selective hydrogenation catalyst. | 308,309 | |
| • High-coverage model calculations show comparable hydrogenation barriers on Pd(111) and Pd3Sn(111). | |||
| Pd SA | Pd1/gC3N4, Pd1/TiO2, Pd1/Fe3O4 | • H diffusion is significantly modified by the type of single metal atoms | 310–312 |
| Pd SAA | Screen the Pd-based SAA | • Pd1M(100) (M = Cu, Ag, Au) with surface Pd coordination number of 8 exhibits the best catalytic performance | 313 and 314 |
| Pd1Ag | • H2 and C2H2 can simultaneously bind with a single Pd doping atom in Ag clusters | 225, 315 and 316 | |
| Pd1Cu | • High selectivity on Pd1Cu(111) arises from facile ethylene desorption and moderate H2 dissociation activity. | 317–320 | |
| Active site | Investigation theme | Key observations | Ref. |
|---|---|---|---|
| Monometallic | Cu | • B- or N-doped Cu(111) has excellent C2H4 activity and selectivity and greatly inhibits green oil. | 321–324 |
| • CuGCN4.8 with defect site is screened out to inhibit green oil and presents excellent C2H4 activity and selectivity | |||
| Ni | • Ni(111) surface with defect sites showed better catalytic activity and selectivity of ethylene | 325 and 326 | |
| Pt | • Pt/α-WC(0001)–W enables selective C2H2 hydrogenation to C2H4 (−0.44 eV) while preventing over-hydrogenation and catalyst deactivation by suppressing CCH3 formation. | 327,328 | |
| Others (Au, transition metal carbide, Rh) | • α-MoC(101) exhibits Pt-like electronic properties with enhanced C2H4 activity, selectivity, and stability against green oil formation. | 329–333 | |
| Multi-metallic | Cu-based | • PdCu(211) and AgCu(211) surfaces favor C2H4 desorption, indicating Pd and Ag enhance selectivity for C2H4 formation. | 334 and 335 |
| Ni-based | • Au, Ag, and Cu doping on Ni modulates acetylene adsorption, enhancing catalytic activity. | 149 and 336 | |
| Pt-based | • PtCu3 IMC is screened out as the optimal catalyst among Pt–M (M = Au, Ag, Cu) for acetylene hydrogenation | 337 | |
| IMC | NiX IMC (X = Ga, In, Sn, Ti) | • A targeted subspace of IMC enables access to new Brønsted–Evans–Polanyi relationships. | 338–341 |
| • NiIn and Ni2In3 suppressed ethane formation but promoted oligomerization. | |||
| Al13Co4 and Al5Co2 | • Active sites are pentagonal CoAl5 clusters but not isolated Co atoms. | 342–345 | |
| • The rate-limiting activation energies on CoAl5 are comparable to those of conventional Pd or PdAg. | |||
| Metal phosphide | • FeP(101)–I exhibits Pd-like performance with enhanced performance compared to Pd and other MxPγ catalysts (M = Mo, W, Fe, Co, Ni, and Pd). | 346 | |
| SA | Supported Ni single atom | • Single-Ni-doped catalysts with four-N-modified C34 universally exhibit high ethylene selectivity | 347–350 |
| • Ni1(OH)2/TiO2(101) favors acetylene adsorption and promotes the heterolytic dissociation of H2 | |||
| Pd1/TiO2 and Pt1/Cu | • Pd1/TiO2 shows the highest ethylene selectivity but lowest activity among Ptn/TiO2, | 351–353 | |
| • Pt1–Cu(111) exhibits the highest selectivity and stability among Pt1–X(111) (X = Cu, Ag, Au), | |||
| FLPs | CeO2 and TiO2 | • Ce/O FLPs facilitate heterolytic H2 dissociation and the resulting O–H and Ce–H species drive acetylene hydrogenation. | 354–359 |
| • Doped metals such as Ga, Cu, Ni, Pd, Pt, or Co promote surface oxygen vacancy formation and generate diverse FLP types with surface O atoms. | |||
| Perovskite oxyhydride (POHs) and carbon nitride | • POHs are capable of selective hydrogenation due to their rich hydrogen surface-redox chemistry | 360 and 361 | |
(1) Adsorption energetics and geometries. This is the common type of DFT calculation, where researchers model the adsorption behavior of reactants on different active sites, analyzing their binding energy and geometric configurations.
(2) Energy profiles of reaction pathways. These studies go beyond adsorption and map out the transition state pathways for elementary reaction steps, providing a deeper understanding of reaction barriers and intermediates.
(3) Activity and selectivity analysis based on DFT-calculated energy profiles. Several models have been proposed to evaluate catalytic activity and selectivity, helping to rationalize experimental observations and guide the design of more efficient catalysts.
This section will discuss the progress of DFT studies on acetylene selective hydrogenation from the above three perspectives.
4.1 Adsorption energetics and catalyst structural evolution
DFT calculations are widely used to model adsorption energetics and geometries of reactants and intermediates. Surface science experiments have postulated possible adsorbed species and adsorption geometries in acetylene hydrogenation (Fig. 18a), including π-adsorbed acetylene, di-σ-adsorbed acetylene, vinyl species, ethylidyne, ethylidene, top-adsorbed acetylene, and vinylidene.362 DFT calculations can determine the adsorption energies (Ead) of these surface intermediates on different model surfaces. An effective catalyst should exhibit relatively strong acetylene adsorption (Ead-C2H2 as negative as possible) to enhance acetylene hydrogenation and weak ethylene adsorption (Ead-C2H4 as positive as possible) to improve selectivity. Nørskov et al.244 proposed scaling relations between adsorption energies of reactants and intermediates and identifying a descriptor for catalytic activity and selectivity, which should enable efficient screening of high-performance acetylene hydrogenation catalysts (Fig. 18b and c). Charge analysis and density of states (DOS) calculations further quantify charge transfer and electronic properties of adsorption models, providing additional insights into the electronic interactions between active sites and adsorbed species.363,364 | ||
| Fig. 18 (a) Proposed surface intermediates in hydrogenation of acetylene. Reprinted with permission from ref. 362. Copyright 2001 Elsevier. (b) Heats of adsorption for C2H2 and C2H4 and (c) Price (in 2006) of 70 binary IMC plotted against the heat of adsorption for methyl. Reprinted with permission from ref. 244. Copyright 2008 The American Association for the Advancement of Science. (d) Computed two-dimensional Gibbs free energy maps for the accumulation and dissolution of C atoms on the two outermost Pd(111) interlayers. Reprinted with permission from ref. 276. Copyright 2013 American Chemical Society. | ||
DFT calculations are also used to analyze catalyst structure evolution during acetylene hydrogenation. Experiments have reported dynamic changes in the structure and composition of active sites, such as PdCx phase formation and surface-segregated Pd in PdAg catalysts, which have been investigated and confirmed by DFT calculations.273,290 Sautet et al.276 combined DFT calculations with atomistic ab initio thermodynamics to study the influence of reaction conditions on PdCx phase formation (Fig. 18d). They introduced the Gibbs free energy of formation(Δγf) as a criterion to evaluate the likelihood and stability of carbide phase formation.
4.2 Energy profile of reaction pathways
Acetylene hydrogenation generally proceeds via the Horiuti-Polanyi mechanism,320,365 with the reaction network (Fig. 19) classified into three main pathways: C2 hydrogenation, C–C bond breaking, and C–C coupling. In the C2 hydrogenation pathway, acetylene undergoes sequential hydrogenation to ethylene, which can be further hydrogenated to ethane under excess hydrogen conditions. In the C–C breaking pathway, intermediates such as CH, CCH, CCH2, and CCH3 are formed through C–C and/or C–H bond cleavage, which can lead to PdCx formation and coke deposition.283,317,339 In the C–C coupling pathway, reactive species like CCH, CCH2, CHCH, and CHCH2 can couple to form 1,3-butadiene, a known precursor to green oil.297,299 When calculating the energy profiles of each pathway, the transition state (TS) of each step is identified, and the activation energy (ΔEa) is determined as the difference between the transition state energy (ETS) and the corresponding initial state energy (EIS).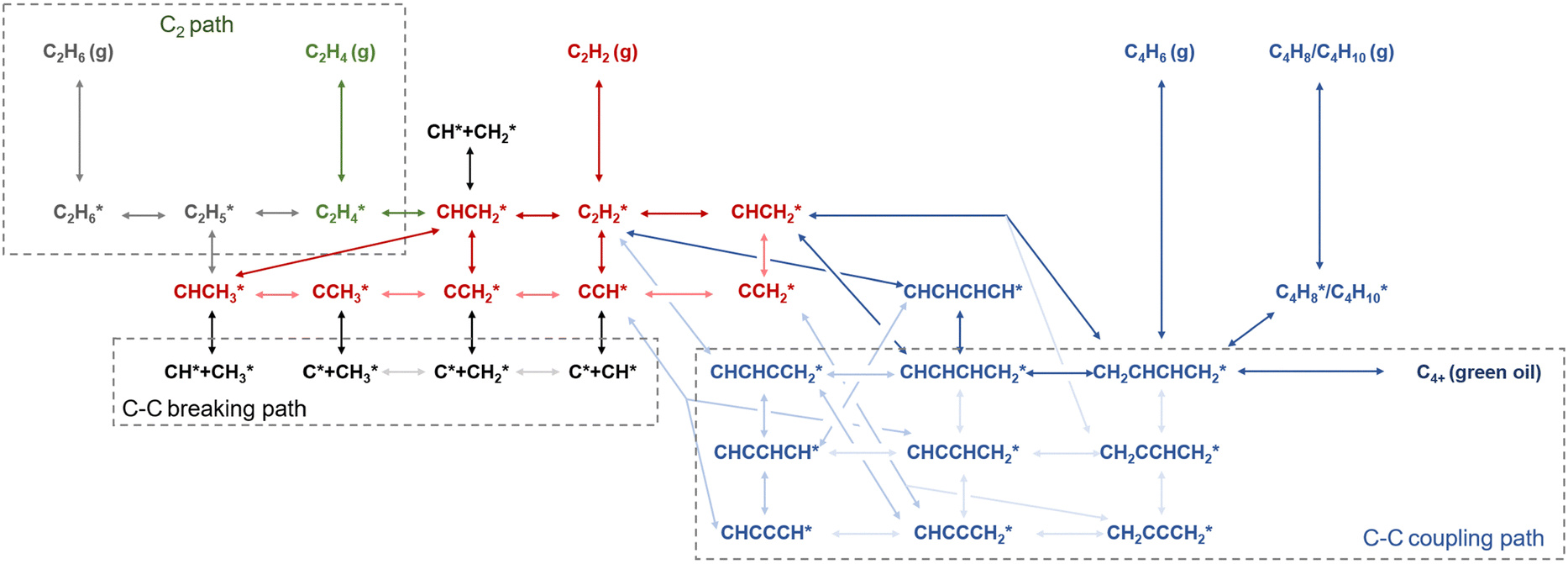 | ||
| Fig. 19 Reaction network of acetylene hydrogenation for DFT calculation. | ||
Many studies evaluate catalytic performance based solely on the C2 hydrogenation pathway, where catalytic efficiency is determined by the energy barrier of the rate-determining step. For ethylene selectivity, ethylene from the gas phase can re-adsorb onto the surface and undergo further hydrogenation to ethane. A selective active site should have a higher activation barrier for ethylene hydrogenation (ΔEa-C2H4) than for ethylene desorption (ΔEdes-C2H4). On Pd(111), ΔEa-C2H4 and ΔEdes-C2H4 were comparable, leading to ethylene over-hydrogenation to ethane. In contrast, PdAg catalysts exhibited a much higher activation barrier for ethylene hydrogenation, thereby enhancing ethylene selectivity (Fig. 20a). Similarly, NiZn has been predicted to suppress ethane formation due to a much higher ΔEa-C2H4 than ΔEdes-C2H4 (Fig. 20b).
 | ||
| Fig. 20 (a) and (b) Energy diagrams of C2 path for acetylene hydrogenation on Pd(111), PdAg(111) and NiZn(110). Reprinted with permission from ref. 244. Copyright 2008 The American Association for the Advancement of Science. (c) Energy diagram for the oligomerization of acetylene on Ni(111) and NiZn(101). Reprinted with permission from ref. 250. Copyright 2014 Elsevier. (d) Energy diagram for the possible oligomerization paths over Cu(111). Reprinted with permission from ref. 323. Copyright 2022 American Chemical Society. | ||
DFT calculations limited to the C2 hydrogenation pathway reveal the ability of a catalyst to inhibit ethylene over-hydrogenation, but they do not address the competing C–C coupling pathway. Experimental studies have shown that some catalysts produce significant amounts of oligomers, sometimes even exceeding ethane formation. Thus, incorporating C–C coupling pathway analysis is essential for a comprehensive evaluation of catalytic performance. 1,3-Butadiene formation is often used as a representative descriptor of the C–C coupling pathway in DFT calculations. Previous studies suggest that oligomerization on Ni surfaces occurs via C–C bond formation between adsorbed acetylene and vinyl, with the reaction pathway beginning with partial acetylene hydrogenation to vinyl, followed by C4H5 formation and subsequent hydrogenation to butadiene (Fig. 20c). Zhang et al.297,323,324,366 proposed three primary C–C coupling routes, including CHCH–CHCH coupling, CHCH–CHCH2 coupling, and CHCH2–CHCH2 coupling (Fig. 20d). DFT results further reveal that Group 1B metals in Pd-based catalysts, such as Cu3Pd1, can effectively suppress the C–C coupling pathway and consequently green oil formation.
Experimental studies revealed that the PdCx phase forms during acetylene hydrogenation, with carbon species derived from acetylene decomposition. DFT calculations have been employed to investigate the C–C breaking pathway, showing that reducing the size of Pd ensembles can inhibit C–C bond cleavage. Specifically, carbon deposition on the 0.01 ML Pd/Cu(111) surface exhibited a significantly higher activation energy (2.00 eV) compared to the 1.00 ML Pd/Cu(111) surface (1.30 eV), suggesting that smaller Pd ensembles suppress carbon accumulation by making C–C cleavage less favorable.317
4.3 Activity and selectivity analysis from DFT calculations
The energy profile results provide activation energies for each step, which can be used to calculate elementary reaction rates based on transition state theory (TST). Several models have been proposed to further evaluate the overall activity of reaction pathways, including the energetic span model, the two-step model, and the microkinetic model.The energetic span model, originally introduced by Amatore and Jutand367 and further developed by Kozuch and Shaik,368 quantifies the efficiency of catalytic cycles through TOF, which is closely related to the highest transition state and the most stable intermediate in the energy profile. By applying this model, researchers can analyze acetylene hydrogenation activity over different catalyst structures.270,286,336,340 For instance, Pd2Ag2C exhibited the highest TOF among Pd4, Pd3Ag1, Pd1Ag3, Pd2Ag2, and Pd2Ag2C catalysts,286 while Au/Ni(111) demonstrated higher activity than Ni(111), Cu/Ni(111), Ag/Ni(111), and Au(111).336
The two-step model, proposed by Hu et al.,254,272,289 treats acetylene hydrogenation as a kinetic process consisting of two steps: adsorption and desorption. The adsorption rate (rad) and desorption rate (rde) are determined by the effective energy barriers for reactant adsorption and product desorption, respectively. Under steady-state conditions, the overall reaction rate (r) is obtained from r = rad = rde.272 Using this model, reaction rates can be predicted, as demonstrated by the case of Pd6Ag7, which exhibited the highest reaction rate among Pd13−mAgm clusters.289
The microkinetic model for acetylene selective hydrogenation was developed based on the Langmuir–Hinshelwood (L–H) mechanism, where the adsorption and desorption of C2H2 and H2 were assumed to be in equilibrium, and TST was applied to determine rate constants for each elementary step based on the energy profile.281,288,299,351 Additionally, the model assumes that the sum of the total coverage of all intermediates and free adsorption sites equals to unity. The steady-state approximation is employed by setting the time derivatives of all intermediate coverages to zero. The microkinetic model provides insights into each step of various reaction pathways as a function of reaction temperature and reactant ratio. For instance, studies based on the microkinetic model have identified CHCH and CHCH2 as the main fragments responsible for C–C coupling on PdM(111) (M = Cu, Ag, Au), and revealed that high temperatures and high H2-to-C2H2 ratios inhibited of green oil formation.299 Furthermore, coverage-dependent microkinetic models have been proposed to incorporate the effects of surface coverage on Ead for all intermediates. For example, a model was used to quantify the coverage (θ) effect on Ead, demonstrating that the coverage-dependent microkinetic model obtained a significantly more reasonable TOF (1.41 s−1 at 300 K) compared to the coverage-independent model (3.16 × 10−24 s−1).281
Ethylene selectivity was also analyzed based on the energy profiles.7,250,335 A simplified metric for ethylene selectivity was defined as the difference between the activation energy for C2H4 hydrogenation and the desorption energy of C2H4:
| ΔEselect = ΔEa-C2H4 − ΔEdes-C2H4 | (9) |
5. Effect of catalyst supports on acetylene hydrogenation
Acetylene hydrogenation catalysts are typically metal catalysts supported on solid materials, making the selection of support material crucial to overall catalytic performance. On one hand, the inherent properties of the support can directly affect acetylene hydrogenation. For instance, the acid–base characteristics of the support can influence C–C coupling reaction pathways. On the other hand, the support can modulate the properties of metal active sites, further influencing the overall catalytic performance. For example, high-surface-area supports enhance metal dispersion, while reducible supports (e.g., TiOx) exhibit the strong metal–support interactions (SMSI) effect. The most commonly used supports for acetylene hydrogenation catalysts are metal oxides and carbon-based materials. In addition, porous materials, such as zeolites and metal–organic frameworks (MOFs), have also been explored. The performance of active sites on different supports for acetylene hydrogenation is summarized in Tables 14 and 15. This section summarizes the effect of supports on acetylene hydrogenation.| Catalyst | Description | T, °C | Feeda, % | X, % | S, % | Activityb | Methodc | Ref. |
|---|---|---|---|---|---|---|---|---|
| a The feed composition listed in the table corresponds to C2H2/H2/C2H4. b Activity is calculated as the number of moles of acetylene converted per hour per total molar active metal (molC2H2 molmetal−1 h−1). c Method refers to the calculation method specified in Table 2. E* indicates that Method E was applied under ethylene-rich feed conditions. | ||||||||
| Al2O3-based oxides | ||||||||
| Pd/γ-Al2O3 | 40 | 1.5/1.7/96.8 | 52 | 78 | — | 369 | ||
| Pd/α-Al2O3 | 40 | 1.5/1.7/96.8 | 70 | 82 | — | |||
| Pd/α&γ-Al2O3 | 65% α-Al2O3 | 40 | 1.5/1.7/96.8 | 80 | 88 | — | ||
| Pd/γ,θ,δ-Al2O3 | 60 | 1/1.5/60 | 16 | 18 | C | 370 | ||
| Pd/θ,δ-Al2O3 | 60 | 1/1.5/60 | 78 | 65 | C | |||
| Pd/α-Al2O3 | 55 | 0.45/0.71/92.4 | 99 | 36 | E | 371 | ||
| Pd/α-Al2O3@SiC | 65 | 0.45/0.71/92.4 | 95 | 48 | E | |||
| Pd–Ag/α-Al2O3 | 55 | 0.45/0.71/92.4 | 100 | 41 | E | |||
| Pd–Ag/α-Al2O3@SiC | 65 | 0.45/0.71/92.4 | 99 | 57 | E | |||
| 0.05Pd-Cat-600/400 | 70 | 0.33/0.66/33.3 | 55 | 62 | 180.5 | C | 372 | |
| 0.05Pd-Cat-100/400 | 70 | 0.33/0.66/33.3 | 100 | 36 | 313.6 | C | ||
| Pd/meso-Al2O3 | 130 | 0.33/0.66/33.3 | 100 | 85 | D | 373 | ||
| Pd/com-Al2O3 | 130 | 0.33/0.66/33.3 | 98 | 25 | 4008.1 | D | ||
| Pd/α-Al2O3-C | 50 | 0.3/1.5/6 | 45 | 61 | 188.5 | C | 374 | |
| Pd/Al2O3-1400 | 50 | 0.3/1.5/6 | 64 | 63 | 320.4 | C | ||
| Pd/α-Al2O3 | 1.5/1.7/96.8 | 82 | 65 | E | 375 | |||
| Pd/Fe-modified α-Al2O3 | 1.5/1.7/96.8 | 84 | 72 | E | ||||
| Au/Al2O3 | 250 | 0.6/3/0 | 4 | 87 | 17.5 | E | 376 | |
| Au/0.3%CeO2–Al2O3 | 250 | 0.6/3/0 | 7 | 100 | 30.7 | E | ||
| Pd/Zn/0.1Ce/Al2O3 | 100 | 2/2.2/80(1MPa) | 84 | 93 | 19969.7 | D | 377 | |
| Pd/Al2O3 | 40 | 0.3/0.6/33.1 | 12 | 88 | 44.6 | D | 234 | |
| Pd/Al2O3 | 100 | 0.3/0.6/33.1 | 90 | <0 | 334.6 | D | ||
| 1c-Ga2O3–Pd/Al2O3 | Ga2O3-ALD | 40 | 0.3/0.6/33.1 | 15 | 95 | 55.8 | D | |
| 1c-Ga2O3–Pd/Al2O3 | Ga2O3-ALD | 100 | 0.3/0.6/33.1 | 100 | 0 | 371.8 | D | |
| Pd/Al2O3 | 80 | 0.91/1.82/97.3 | 68 | 83 | E* | 378 | ||
| Pd/MgO–Al2O3 | 80 | 0.91/1.82/97.3 | 95 | 95 | E* | |||
| Pd/HT | 55 | 0.35/0.6/0 | 97 | 75 | 195.3 | E | 379 | |
| Pd/MgO | 80 | 0.35/0.6/0 | 96 | 48 | 198.1 | E | ||
| Pd/Al2O3 | 100 | 0.35/0.6/0 | 85 | 38 | 177.6 | E | ||
| Au/Al2O3 | 250 | 0.6/3/0 | 5 | 92 | 7.3 | E | 380 | |
| Au/3%MgOx–Al2O3 | Reduced at 550 | 250 | 0.6/3/0 | 12 | 92 | 20.9 | E | |
| Pd/A-Pal | Air roasting | 70 | 1/2/0 | 98 | 52 | 3491.9 | E | 381 |
| Pd/N-Pal | NH3-roasting | 70 | 1/2/0 | 50 | 58 | 890.8 | E | |
| 3D-Al-Pd/MMO | 55 | 0.33/0.66/34.5 | 95 | 88 | 126.5 | F | 382 | |
| Pd/Al2O3 | 55 | 0.33/0.66/34.5 | 30 | 90 | 40.2 | F | ||
| Pd/Al2O3 | 100 | 0.33/0.66/34.5 | 98 | 10 | F | |||
| Pd/MgAl2O4-400 | 80 | 1/5/20 | 48 | 92 | 5473.0 | D | 383 | |
| Pd/MgAl2O4-600 | 80 | 1/5/20 | 17 | 98 | 1938.4 | D | ||
| Cu/MgAlOx | 225 | 0.33/1.02/32.86 | 64 | 95 | E* | 384 | ||
| Cu/Fe0.12MgOx | 215 | 0.33/1.02/32.86 | 79 | 98 | E* | |||
| Pd/MgO | 50 | 4/96/0 | 79 | 49 | 18015.4 | — | 385 | |
| Ti-containing oxides | ||||||||
| Pd–SiO2/300 | 60 | 1/10/0 | 87 | 5 | — | 386 | ||
| Pd–1La–0.2Ti/300 | 60 | 1/10/0 | 87 | 40 | — | |||
| Pd–0.2Ti/300 | 60 | 1/10/0 | 87 | 5 | — | |||
| Pd–1Nb–0.2Ti/300 | 60 | 1/10/0 | 94 | 48 | — | |||
| Pd–Ti/SiO2 | 60 | 0.98/1.96/97.05 | 95 | 34 | — | 387 | ||
| Pd/SiO2 | 60 | 0.98/1.96/97.05 | 93 | 15 | — | |||
| PdAg/TiO2 | 85 | 0.32/0.6/32.9 | 90 | 75 | 238.7 | E* | 388 | |
| PdAg/NiTi-LDH | 70 | 0.32/0.6/32.9 | 90 | 82 | 242.1 | E* | ||
| PdAg/TiO2 | 65 | 0.33/0.66/32.8 | 50 | 68 | 76.4 | E* | 389 | |
| PdAg/Mg0.3Ti0.7Oy | 65 | 0.33/0.66/32.8 | 67 | 71 | 102.4 | E* | ||
| PdAg/Mg0.5Ti0.5Oy | 65 | 0.33/0.66/32.8 | 92 | 86 | 140.6 | E* | ||
| PdAg/Mg0.7Ti0.3Oy | 65 | 0.33/0.66/32.8 | 81 | 79 | 123.8 | E* | ||
| PdAg/MgO | 65 | 0.33/0.66/32.8 | 64 | 62 | 97.8 | E* | ||
| Pd/1c-TiO2/MCM-41 | Reduced at 500 °C | 40 | 0.1/0.2/2.5 | 50 | 75 | E* | 390 | |
| Si containing oxides | ||||||||
| I-Pd/M–SiO2 | 50 | 1/7/0 | 87 | 38 | E | 391 | ||
| Pd/C–SiO2 | 80 | 1/7/0 | 95 | 66 | E | |||
| PdAg/Na + -β-zeolite | 50 | 2.5/10/2.5 | 35 | 52 | 807.0 | E* | 392 | |
| PdAg/K + -β-zeolite | 50 | 2.5/10/2.5 | 18 | 68 | 415.0 | E* | ||
| PdAg/Rb + -β-zeolite | 50 | 2.5/10/2.5 | 47 | 50 | 1083.6 | E* | ||
| Pd(V)/FG | Fiberglass | 85 | 0.49/0.74/59.4 | 99 | 60 | 356.8 | B | 393 |
| Pd-HNT-az-Ac | Halloysite nanotube | 30 | 1.05/1.94/5.01 | 27 | 76 | E* | 394 | |
| Others | ||||||||
| Au/CeO2(R)–air | 250 | 0.6/3/0 | 17 | 100 | 18.2 | E | 395 | |
| Au/CeO2(R)–H2 | 250 | 0.6/3/0 | 6 | 93 | 6.4 | E | ||
| Ni–Cu/CeO2-NP | 90–180 | 0.6/2.4/5.4 | 85 | 84 | E | 396 | ||
| Ni–Cu/CeO2-NC | 90–180 | 0.6/2.4/5.4 | 85 | 50 | E | |||
| Pd1.0/Bi2O3/TiO2 | 40 | 1/20/20 | 61 | 93 | 151.2 | D | 235 | |
| Pd–SrTiO3 | 100 | 1/10/20 | 96 | 90 | 1094.6 | C | 397 | |
| Catalyst | T, °C | Feeda, % | X, % | S, % | Activityb | Methodc | Ref. |
|---|---|---|---|---|---|---|---|
| a The feed composition listed in the table corresponds to C2H2/H2/C2H4. b Activity is calculated as the number of moles of acetylene converted per hour per total molar active metal (molC2H2 molmetal−1 h−1). c M refers to the calculation method specified in Table 2. E* indicates that Method E was applied under ethylene-rich feed conditions. | |||||||
| Carbon material as support | |||||||
| Ni10Co90@C | 0.5/10/20 | 95 | 81 | D | 398 | ||
| Pd/C | 85 | 0.49/20.77/34.27 | 80 | 30 | 376.3 | E* | 399 |
| Pd/CNTs-out | 80 | 0.87/0.87/42.26 | 97.7 | 83.1 | 30.3 | E* | 400 |
| Pd/CNTs-in | 80 | 0.87/0.87/42.26 | 95.4 | 73.3 | 29.6 | E* | |
| Pd/CNT | 80 | 1/3/20 | 80 | 15 | 684.1 | C | 401 |
| Pd/N2.1-CNT | 80 | 1/3/20 | 100 | 40 | 855.2 | C | |
| Pd@NMC-850 | 100 | 0.35/0.70/30.5 | 65 | 83 | C | 402 | |
| Pd/Al2O3 | 60 | 0.35/0.70/30.5 | 90 | 30 | C | ||
| chrGO/Pd-NP | 90 | 1/2/80 | 89.68 | 41.72 | 12.2 | E | 403 |
| Pd/CMK-3 | 180 | 11.25/45/0 | 95 | 84 | E | 404 | |
| Porous material | |||||||
| PdZn-1.2@ZIF-8C | 120 | 0.65/5/50 | 88 | 79 | 186.3 | B | 405 |
| PdZn-2.7/ZIF-8C | 60 | 0.65/5/50 | 88 | 84 | 26.1 | B | |
| Pd@SOD | 80 | 0.6/6/0 | 20 | 95 | E | 406 | |
| Pd/SOD | 80 | 0.6/6/0 | 60 | 23 | E | ||
| Other supports | |||||||
| Pd/TiO2 | 80 | 1/2/0 | 98 | 65 | 172.4 | E | 407 |
| Pd/SiC | 80 | 1/2/0 | 90 | 85 | 160.3 | E | |
| 1.06-Pd/Si3N4 | 160 | 0.66/6.6/32.9 | 100 | 95 | 91.3 | E* | 408 |
| Pd/C | 160 | 0.66/6.6/32.9 | 100 | 77 | 89.6 | E* | |
| Pd/β-Mo2C-600 | 120 | 1/10/0 | 70 | 80 | E | 409 | |
5.1 Metal oxides
Al2O3 exists in several crystalline phases, including β, γ, η, χ, κ, ρ, δ, θ, and α, each of which exhibits distinct structural, physical, and chemical properties.369,370,410–413 The choice of phase has a significant impact on catalytic performance for acetylene hydrogenation. Using Pd/Al2O3 as an example, the α-Al2O3 phase had a low surface area (0.1–2 m2 g−1), which limited Pd dispersion and consequently reduced catalytic activity. In contrast, the γ-Al2O3 phase typically exhibited a much higher surface area, promoting metal dispersion, however, it also introduced a greater number of acidic sites, which accelerated the formation of oligomer products.369 It is reported that oligomer formation was about four to five times faster on Pd/γ-Al2O3 compared with the α-Al2O3-supported catalyst.414 The phase transformation of Al2O3 is highly dependent on the calcination temperature. As the heating temperature increased from 900 °C to 1100 °C, the phase transition proceeded as follows: γ → (γ+ δ + θ) at 900 °C → (δ + θ) at 1000–1050 °C → α at 1100 °C. During this transformation, the surface area of Al2O3 decreased significantly from 244 m2 g−1 to 16.4 m2 g−1.370 Catalytic evaluations have shown that Pd/Al2O3 calcined at 1050 °C (containing δ and θ phases) exhibited the highest ethylene selectivity. However, some studies have reported that α-Al2O3 generally resulted in the lowest hydrocarbon deposition and the highest ethylene selectivity, while at the cost of catalytic activity.410 Although some variations exist in reported results, a common conclusion is that reducing the acidity of the Al2O3 support can effectively suppress the formation of oligomer products, thereby improving ethylene selectivity.
Basic oxides, such as MgO, have been introduced to reduce the acidity of Al2O3 while preserving its high surface area, thereby balancing both acetylene hydrogenation activity and ethylene selectivity. MgO can be directly deposited onto the Al2O3 surface via impregnation with a magnesium precursor.380,415 Alternatively, it can be incorporated into the support during synthesis to form Mg–Al composite oxides, such as Mg–Al-layered double hydroxide (MgAl-LDH),378,382,416 hydrotalcite (HT),379 palygorskite,381 and spinel.383 The incorporation of MgO significantly improves ethylene selectivity. For instance, Pd supported on HT (Pd/HT) exhibited an ethylene selectivity of 75% (E), considerably higher than that of Pd/MgO (48%, E) and Pd/Al2O3 (38%, E).379 Additionally, MgO-Al2O3 composite oxides derived from MgAl-LDH exhibited a larger surface area and lower surface acidity, which not only enhanced ethylene selectivity but also improved catalytic activity compared with Pd/Al2O3.378,416
The properties of Al2O3 can also be tuned by incorporating second metal species such as Zn, Fe, and Ce.375–377,417–419 Studies have shown that doping α-Al2O3 with Zn and Fe via the sol–gel method effectively reduced the acidity of the support, which mitigated deactivation caused by oligomer formation and enhanced ethylene selectivity.375,417 Similarly, impregnating Al2O3 with a Ce precursor decreased its acidity, but an optimal Ce loading was necessary to achieve the best catalytic performance.376 In addition to acidity regulation, electronic interactions between CeOx and the active metal were observed, which also contributed to the catalytic activity enhancement.376,377
Al2O3-based oxide supports with unique structures have been designed to improve catalytic performance, including α-Al2O3@SiC core–shell structures, microfibrous AlOOH/Al-fiber, microrod Al2O3, and array-modified molded alumina.371–373,420,421 These tailored structures exhibited optimized acidic properties, enhanced the interaction between the support and active metal to improve metal dispersion, and modified the electronic properties of the active metal via electronic effects. These synergistic effects contribute to enhanced catalytic activity and selectivity.
To further optimize catalytic performance, secondary metal oxides such as La, Ni, and Mg have been incorporated into Ti-containing oxides.386,388,389 These additional oxides further modified the geometric and electronic properties of the active metal while also tuned the surface acid–base characteristics, contributing to improved catalytic activity and selectivity.
Beyond SiO2, other Si-containing materials have also been explored for the design of highly efficient acetylene hydrogenation catalysts, including zeolites, fiberglass, halloysite and porous hollow silica nanoparticles.392–394,429–431 These materials offered unique properties that promoted small particle sizes and uniform dispersion of Pd NPs.430,431 Additionally, these supports have also been reported to regulate the adsorption of reaction intermediates to improve the catalytic activity and selectivity.393,430
5.2 Carbon-based materials
Carbonaceous materials represent another widely used class of supports for heterogeneous catalysis. Owing to their varied sp2 carbon atom arrangements and structures, these materials exhibit unique electronic properties that can effectively modify active metal sites. Studies have shown that Pd supported on carbonaceous materials demonstrated higher ethylene selectivity and reduced oligomer formation compared to Pd/Al2O3 catalysts.399,433 Various carbonaceous materials have been explored for acetylene hydrogenation, including carbon nanotubes (CNTs), carbon nanofibers (CNFs), and graphene oxide.400,403,434,435 Notably, compared with CNTs, CNFs with a stacked structure provided a more finely dispersed Pd phase, leading to enhanced hydrogenation activity.435Nitrogen doping in carbonaceous materials has been shown to further improve catalytic performance.401,402,436–438 Specifically, graphitic nitrogen modulates the electronic properties of active sites, facilitating the desorption of ethylene and thereby increasing ethylene selectivity. For example, Pd supported on nitrogen-doped CNTs (Pd/N1.4–CNT) exhibited 1.5 times higher hydrogenation activity compared to Pd/CNTs, with ethylene selectivity improving from 40% to 55% (C) at 90% conversion.401
5.3 Porous materials
In acetylene hydrogenation reactions, porous materials such as zeolites,406,439 MOFs,405,440–443 and covalent triazine frameworks444 have also been explored as catalyst supports. The confinement effect of these materials facilitates the formation of smaller active metal NPs, while the electronic interactions and spatial constraints between the porous support and the active metal further enhance catalytic performance. Additionally, novel reaction mechanisms have been proposed. For instance, encapsulating Pd within sodalite (SOD) zeolite prevented the direct interaction of acetylene and ethylene with Pd active sites, while H2 could diffuse into the SOD pores and be activated by the encapsulated Pd nanoclusters. A hydrogen spillover process subsequently generated OH species on the SOD surface, which then reacted with acetylene, leading to selective hydrogenation. As a result, Pd@SOD exhibited significantly higher ethylene selectivity compared to Pd/SOD.4066. Effect of catalytic modifiers and reaction media on acetylene hydrogenation
In addition to catalytic active sites and catalyst supports, modifiers also play a crucial role in influencing reaction performance. Among them, CO has been employed as a modifier to suppress the over-hydrogenation of ethylene to ethane. In industrial ethylene purification, small amounts of CO are intentionally introduced into the feed gas to enhance hydrogenation selectivity and mitigate the risk of temperature runaway. Alkali metals are effective catalyst modifiers that enhance catalytic performance of acetylene hydrogenation. The mechanism and effects of CO and alkali modification have been reviewed in previous work.1,2 DFT calculations revealed that CO exhibited a comparable adsorption strength to acetylene but bound more strongly than ethylene on the Pd(111) surface,445,446 reducing the number of available active sites for hydrogenation. Moreover, the remaining free sites on the CO-covered Pd(111) surface led to significantly weaker ethylene adsorption, further suppressing ethylene over-hydrogenation.445 However, it should be noted that CO also accelerated green oil formation on Pd catalysts, particularly in tail-end process. A similar phenomenon has been observed on Ni-based catalysts.144,447,448 For instance, it was reported that the addition of 550 ppm CO reduced ethane selectivity from 20% to 10%, while increasing oligomer selectivity from 35% to 51%.448Beyond CO and alkali, other modifiers, such as sulfur, organic ligands, and ionic liquids, have been employed as catalyst additives, which will be further discussed in Section 6.1. Additionally, the liquid phase has been reported as a process modifier, as it influences the reactant concentration around active sites due to the solubility differences of reactant gases in the liquid solvent. This aspect will be further discussed in Section 6.2.
6.1 Catalytic modifiers
Beyond the solubility effects, ILs also modify the electronic properties of active metals. It has been reported that certain ILs formed unique carbene structures around Pd active sites, facilitating the desorption of ethylene.463 This electronic effect enhances ethylene selectivity. However, catalyst deactivation due to oligomer formation remains a challenge for IL-coated catalysts.462,464 The use of Pd@S-1 core–shell materials coated with ILs has been reported, showing improved stability for acetylene hydrogenation.461
 | ||
| Fig. 21 (a) Schematic illustration of the semihydrogenation of alkyne on Cu2O–C16. Reprinted with permission from ref. 473. Copyright 2024 American Chemical Society. (b) Reversibility of Pd–PPS interaction and proposed acetylene hydrogenation scheme over Pd/PPS. Reprinted with permission from ref. 474. Copyright 2020 The American Association for the Advancement of Science. | ||
The concept of dynamic metal–polymer interaction was proposed by Choi et al for acetylene hydrogenation.474–478 In their study, polyphenylene sulfide (PPS) chains were utilized to encapsulate Pd particles through strong metal–polymer interactions, effectively suppressing the over-hydrogenation of ethylene to ethane. The proposed mechanism (Fig. 21b) suggested that the Pd–PPS interface selectively facilitated H2 activation only in the presence of acetylene, which strongly bound to Pd and disrupted the Pd–PPS interaction. Once acetylene underwent hydrogenation to form ethylene, which exhibited weaker binding on Pd, the PPS chains rapidly re-adsorbed onto the Pd surface. This dynamic re-adsorption repelled ethylene, preventing its further hydrogenation to ethane and thereby enhancing ethylene selectivity.474,479
6.2 Acetylene hydrogenation in liquid phase
The introduction of a liquid phase medium in acetylene hydrogenation was first reported by Edvinsson et al. in 1995.480 In their study, heptane was used to form a liquid film on the catalyst layer within a monolith reactor, which improved catalyst stability by efficiently removing green oil deposits. Later, Shitova et al.481 employed N-methylpyrrolidone (NMP) as a liquid solvent in a shaker-type flow reactor, achieving high ethylene selectivity. Acetylene exhibits high solubility in polar solvents such as acetone, NMP, dimethylformamide (DMF), and certain ionic liquids, whereas ethylene has much lower solubility in these solvents.482–484 For instance, the selective solubility factor (α) of acetylene to ethylene in NMP exceeded 11 at temperatures below 100 °C.482 As a result, the actual reactant concentration in the vicinity of the active sites is significantly modified by the liquid solvent, effectively suppressing the over-hydrogenation of ethylene to ethane (Fig. 22a). Compared to the gas-phase process over Pd/SiO2 catalysts, the introduction of the liquid phase media increased ethylene selectivity from <60% to >80% (E).482 | ||
| Fig. 22 (a) Schematic illustration of the liquid phase hydrogenation. (b) Reactor models for gas phase (PBR) and liquid phase (CSTR) hydrogenation. Reprinted with permission from ref. 485. Copyright 2024 Elsevier. | ||
The liquid phase hydrogenation is typically conducted in a stirred-tank reactor (CSTR) (Fig. 22b), where the dissolved composition in the solvent remains nearly constant and closely matches the outlet composition once a steady state is reached. At high acetylene conversions, acetylene rapidly and uniformly dissolves within the liquid phase upon entering the reactor, maintaining a relatively low acetylene concentration. This characteristic reduces the formation of oligomerization products. A theoretical model was developed for the liquid phase hydrogenation process, integrating reaction kinetics, mass transfer, and reactor modeling under different reaction conditions.485 The simulation results suggested that the liquid phase process operated at a lower acetylene concentration and a higher H2/C2H2 ratio, particularly at high acetylene conversions, effectively inhibiting the formation of oligomer byproducts.
The choice of the liquid solvent has a significant impact on the performance in liquid phase hydrogenation. A solvent with a higher α value is expected to enhance ethylene selectivity. A typical comparison was made between NMP and decane. Decane exhibited a lower α value of 0.8, meaning that acetylene and ethylene had comparable solubilities in this solvent. As a result, when decane was used as the liquid phase over Pd/SiO2, the ethylene selectivity remained below 70%, lower than that observed with NMP (>80%, E).482 ILs exhibited extremely high α values, making them promising candidates for acetylene selective hydrogenation. For instance, 1,3-dimethylimidazolium methylphosphite ([DMIM][MeHPO3]) had an α value of 45, suggesting its potential as an effective liquid solvent. However, [DMIM][MeHPO3] also possessed a high viscosity (363 mPa·s), which was much larger than that of NMP (1.65 mPa s). The high viscosity leaded to severe mass transfer resistance, significantly reducing the overall reaction activity when ILs are directly used as the liquid phase.462
Building on the liquid phase hydrogenation process, researchers have further investigated the catalytic performance of different bimetallic catalysts, including PdGa,486–488 PdFe,489 PdZn,490,491 and PdAg.485,492 It has been reported that the intrinsic selectivity of the bimetallic active sites can be partially combined with the benefits of liquid phase hydrogenation. For example, PdAg/C catalysts exhibited higher ethylene selectivity than PdZn/C and PdGa/C in the liquid phase process.486,493 However, optimizing the combination of specific catalysts with liquid phase hydrogenation processes requires further study.
7. Conclusions and perspectives
Acetylene selective hydrogenation has undergone extensive development over the past two decades. This review attempts to establish a standardized framework for evaluating catalytic performance and summarizes trends and advances in active site design. Multiple classes of active sites, including monometallic, disordered alloy, IMC, and SA sites, have been strategically engineered to achieve high activity and selectivity. In addition, catalyst supports and modifiers have emerged as effective strategies for enhancing catalytic performance. Despite these advancements, further research is needed to improve both the intrinsic activity and ethylene selectivity of catalysts. Key challenges and future opportunities are discussed below to guide the continued development of efficient catalytic systems for acetylene hydrogenation.7.1 Reaction mechanism and structure–performance relationship
Understanding the reaction mechanism and structure–performance relationship is crucial for designing high-performance catalysts. However, this remains challenging for acetylene hydrogenation due to difficulty in identifying the true active sites under reaction conditions and the structural evolution of these sites during the reaction. For instance, monometallic Pd active sites may form PdCx and PdHx phases under reaction conditions, while certain Pd-based active site structures, such as Pd1Ga1 IMC and Pd SA, have been reported to suppress the formation of PdCx and PdHx. The Pd species in PdAg catalysts tend to segregate to the surface under reaction conditions, while some IMCs, such as Ni3Zn, gradually transform into a Ni3ZnC phase. These structural evolutions can have varying effects on catalytic performance. For example, the formation of the Ni3ZnC phase has been shown to enhance ethylene selectivity, whereas PdAg segregation may promote side reactions, leading to catalyst deactivation. Therefore, unraveling the real-time structural evolution of active sites under reaction conditions and its correlation with catalytic performance is essential for the rational design of improved catalysts. Furthermore, acetylene hydrogenation involves multiple reaction pathways, each consisting of various elementary steps and intermediate species (Fig. 19). The specific elementary reactions at different active sites require further investigation. A deeper understanding of these aspects will facilitate theoretical modeling and computational studies to elucidate the reaction mechanism, ultimately providing valuable guidance for the design and optimization of catalytic active sites.In situ characterization technologies play a crucial role in gaining the above-mentioned insights. These techniques have been widely employed to determine the structure of active sites under reaction conditions.494–496 For instance, in situ XANES and EXAFS have been used to investigate carbide formation and decomposition in Pd NPs under different gas atmospheres. It was observed that a mixture of acetylene and hydrogen induced the formation of a hydride phase, whereas exposure to pure acetylene leaded to the decomposition of the hydride phase and rapid formation of carbide species.497,498In situ TEM revealed that under reaction conditions, Pd(100) facets transformed into stepped facets, resulted in a decline in ethylene selectivity.499 Surface-sensitive techniques such as in situ XPS and in situ prompt gamma activation analysis (PGAA) have provided valuable insights into the surface chemical properties of active sites under reaction conditions. These studies demonstrated that PdGa exhibited high stability, effectively inhibiting both alloy segregation and subsurface chemical interactions involving C and/or H atoms, which were phenomena commonly observed in monometallic Pd.500 Additionally, various in situ spectroscopic techniques, including polarization-dependent reflection absorption infrared spectroscopy (RAIRS),495 diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS),501 inelastic neutron scattering spectroscopy (INS),502 and Raman spectroscopy,503 have been employed to identify reaction intermediates and probe their adsorption geometries on active sites.
DFT calculations serve as a powerful tool for investigating the reaction mechanism of acetylene selective hydrogenation and have been widely applied to elucidate the structure–activity relationships of different active sites (Section 4). However, DFT calculations are inherently limited by their static nature, as they require assumptions about surface structures and reaction mechanisms. In this regard, in situ characterization techniques provide crucial information about active site structures and reaction intermediates under realistic conditions, which greatly benefits for constructing more accurate DFT models and reaction pathways. Moreover, DFT calculations should comprehensively consider all possible reaction pathways, especially the C–C coupling reaction pathway. Many DFT studies have primarily focused on the C2 hydrogenation pathway, which have contributed to understanding the mechanism of ethylene over-hydrogenation. However, in many cases, the formation of C–C coupling products is non-negligible and can even constitute the major side reactions. Understanding the oligomerization mechanism is therefore crucial for designing catalysts with high selectivity and stability. It is important to note that the elementary reaction network of C–C coupling is significantly more complex than that of the C2 hydrogenation pathway. Establishing a simplified yet accurate elementary reaction scheme remains a key challenge in DFT calculations of acetylene hydrogenation. In addition, combining DFT calculations with microkinetic modeling provides a more effective and accurate approach (Section 4.3). By incorporating coverage effects and real reaction conditions, microkinetic models enable the evaluation of intrinsic catalytic performance for different active sites under practical conditions, thereby enhancing the predictive power of DFT calculations.
7.2 Catalyst design for acetylene selective hydrogenation
The ideal catalyst for acetylene selective hydrogenation should efficiently adsorb and activate H2 and C2H2, facilitate the conversion of acetylene to ethylene, and exhibit weak adsorption of ethylene to prevent its over-hydrogenation to ethane.244 To achieve this, the site isolation concept has been proposed and widely applied in active site design. In this review, the active sites are categorized into monometallic, disordered alloy, IMC, and SA sites (Section 3). The introduction of a secondary metal has proven to be an effective strategy for developing active sites with enhanced ethylene selectivity. Additionally, engineering bimetallic active sites into ordered phases has been demonstrated to modulate the adsorption strength of key intermediates, thereby improving ethylene selectivity. Another critical approach is to maximize the spatial separation of metal centers, forming SA sites, which effectively minimize the side reactions.A trade-off between activity and selectivity exists due to the linear free energy relationships and BEP scaling on metal surfaces. Achieving higher ethylene selectivity often comes at the cost of reduced hydrogenation activity, as weakening ethylene adsorption also decreases the ability to activate H2. To overcome the trade-off between activity and selectivity, further efforts are required to design novel active sites. Dual-atom sites and fully exposed atomic sites have been proposed as strategies to retain the site isolation concept while simultaneously providing additional sites for H2 activation and intermediate stabilization. Additionally, multi-metallic active sites, such as trimetallic IMCs, offer an alternative approach to balancing activity and selectivity. Recently, machine learning (ML) has emerged as a promising tool for catalyst-design and for uncovering the atomic-scale reaction mechanisms of acetylene hydrogenation.504 The integration of DFT calculations and ML has provided deeper insights into the PdAg catalytic system, guiding the identification of the optimal Pd1Ag3 alloy crystal active sites.287,505 Furthermore, ML has been employed to screen and predict potential single-atom alloys,506 metal oxides,507 and Ni-based IMCs508 for acetylene hydrogenation. ML holds great promise for accelerating the development of highly efficient acetylene hydrogenation catalysts. Further research is necessary to elucidate the reaction mechanisms and establish key catalytic descriptors, particularly for the complex reaction pathways involved in acetylene hydrogenation.
For catalyst active site design, the choice between Pd-based and non-precious metals is also an important consideration. In Section 3, we have summarized the performance of Pd-based and non-Pd-based active sites for acetylene hydrogenation. Non-Pd-based active sites are primarily based on non-precious metals such as Ni and Cu. Pd-based catalysts generally exhibit activities that are more than two orders of magnitude higher than those of non-precious metal catalysts. Additionally, Pd-based catalysts typically offer superior ethylene selectivity and stability compared to Ni- and Cu-based catalysts. However, from the perspective of cost and sustainability, non-precious metal catalysts have significant advantages over Pd-based systems. Although Pd-based catalysts have been widely applied in industrial acetylene hydrogenation, developing advanced non-precious metal catalysts remains a key research direction. For Pd-based active sites, enhancing intrinsic activity and reducing Pd loading, such as through SA or cluster sites, are important strategies, while catalyst stability and regenerability should be considered. For non-precious metal systems, active site design regarding metal composition and atomic arrangement is essential to improve activity and selectivity, especially by suppressing C–C coupling reactions to achieve long-term stability.
Catalyst performance is also closely related to the support and modifiers (Sections 5 and 6). Supports play a crucial role in stabilizing active sites and providing synergistic effects, while modifiers can enhance catalyst selectivity and stability by tuning the electronic structure of active sites and regulating the local reaction environment. Beyond designing high-performance active sites, optimizing suitable supports and modifiers is essential for further improving catalytic performance and advancing industrial catalyst development. In addition, the development of high-performance catalysts also relies on effective synthesis strategies. For example, traditional impregnation methods often lead to heterogeneous metal distributions in the synthesis of targeted active sites, resulting in a mixture of monometallic species, disordered alloys, and IMC at the nanoscale. Such structural complexity hinders the establishment of a clear structure–activity relationship. Therefore, developing strategies for precise catalyst synthesis to achieve desired active sites with high-phase purity and scalability is crucial for both fundamental mechanistic understanding and catalyst design for industrial applications.
7.3 Effect of feeding composition in acetylene selective hydrogenation
The selective hydrogenation of acetylene is primarily applied in two industrial processes: (1) the removal of trace acetylene impurities in ethylene production via naphtha cracking and (2) the hydrogenation of acetylene derived from non-oil alternative resources such as natural gas and coal. It is important to note that the composition of feed gases in these two processes differs significantly (as detailed in Section 2.1 and Table 1). The feed composition has a profound impact on the catalytic performance. For instance, a high concentration of acetylene tends to promote C4 hydrocarbon formation, while an increased hydrogen concentration can suppress oligomerization to some extent but potentially promotes the side reaction of ethylene over-hydrogenation. Therefore, when developing industrial catalysts, it is crucial to distinguish between the targeted processes and select representative feed compositions accordingly.In the petrochemical industry, acetylene hydrogenation requires the removal of trace acetylene to a residual concentration below 10 ppm, or even lower, while minimizing ethylene loss. Therefore, an ideal catalyst must achieve an exceptionally high conversion (>99.5%) while preventing ethylene loss as much as possible. When evaluating catalysts under ethylene-rich feed conditions, particular attention should be paid to the benchmarks for selectivity and conversion calculations (Section 2.3). Direct measurement of ethylene concentration changes to calculate ethylene selectivity is not recommended unless isotope labeling experiments are conducted. Given the stringent requirement for acetylene removal, researchers are more concerned with catalyst performance at high acetylene conversions. Due to the challenges in direct calculation of selectivity, it is suggested to evaluate the individual concentration changes of acetylene and ethylene instead (Section 2.3 and Fig. 3). Another critical parameter for catalyst evaluation is the operating window (OW), as a wider OW ensures better catalyst stability at the required temperature for acetylene removal. Currently, industrial catalysts for this process are Pd-based catalysts, with lifespans exceeding one year. To reduce catalyst costs, the development of non-precious metal catalysts or more efficient Pd-based catalysts is of great significance. When developing alternative catalysts, researchers should consider catalyst stability to ensure long-term industrial viability.
For the potential application of acetylene hydrogenation in ethylene production from natural gas or coal, the feed gas is ethylene-free, and the acetylene concentration is significantly higher (Table 1). Most reported catalysts are designed for feeds with acetylene concentration of ≤2%, whereas higher acetylene concentrations impose more stringent requirements on catalyst performance. Under such conditions, C–C coupling reactions become more pronounced, with selectivity to coupling products exceeding 10%.38,54,147,159,182 Notably, over monometallic Pd and bimetallic PdCu catalysts, the selectivity to GO can even surpass 40%.54 Therefore, suppressing C–C coupling reactions remains a major challenge in catalyst development for high-acetylene-concentration hydrogenation. An alternative strategy involves reactor and process design to regulate the actual acetylene concentration adjacent to the catalytic active sites. For example, replacing the PBR with the CSTR can help mitigate high local acetylene concentrations (Fig. 22b). Acetylene hydrogenation in liquid phase reactors has been reported to effectively suppress oligomer formation. Additionally, acetylene hydrogenation is a highly exothermic reaction, and higher acetylene concentrations further intensify heat transfer challenges. Efficient heat removal is essential to maintain catalyst stability and prevent runaway reactions. In this regard, liquid phase hydrogenation offers distinct advantages due to its superior heat transfer properties. Therefore, the development of catalysts tailored for high-acetylene-concentration liquid phase hydrogenation represents a promising research direction.
The gas products derived from partial oxidation (POX) and pyrolysis (PPY) processes contain large amounts of CO and H2. Direct acetylene hydrogenation under such CO- and H2-rich conditions can simplify the overall process. CO can effectively inhibit the over-hydrogenation of ethylene to ethane, however, excessive CO concentrations significantly reduce catalyst activity and promote C–C coupling reactions. Additionally, gas products from POX and PPY often contain sulfur species, which are highly toxic to catalysts. Although sulfur species can be removed via adsorption or other desulfurization pretreatments, their complete removal significantly increases operational costs. In addition, CO is also often present in the feed that can lead to the poison of metal active sites from competitive adsorption. Therefore, developing high-performance catalysts with tolerance to sulfur and CO is essential for such systems. One effective approach is tuning the intrinsic electronic properties of active sites to enhance sulfur tolerance. For instance, trace sulfur can actually improve the selectivity of Pd catalysts (as discussed in Section 6.1.1), but maintaining active sites in a sulfur-lean phase, such as Pd4S, is critical, as excess sulfur can severely poison catalytic activity. Other Pd-based active sites, including Pd5P2 and Pd2Si, have also demonstrated good sulfur tolerance in hydrogenation reactions.509,510 Furthermore, confinement strategies, such as encapsulating active sites within N-doped graphene shells or zeolites, can effectively prevent sulfur poisoning.511,512 Future catalyst development for these systems must carefully balance sulfur tolerance with high hydrogenation activity and selectivity. To improve CO tolerance, tailoring the electronic structure of active sites to weaken CO adsorption is critical. Studies have shown that SA sites and tightly coupled metallic Ru–Ni clusters exhibit weakened CO binding, thereby enhancing CO tolerance in hydrogenation.513,514 From the perspective of balancing the adsorption energies of CO and reactants, combining DFT calculations with ML to screen active sites offers a promising approach.
7.4 Catalyst stability and regeneration for acetylene selective hydrogenation
Catalyst stability plays a crucial role in industrial applications. It is widely accepted that catalyst deactivation during acetylene hydrogenation mainly arises from the formation of GO and coke. A deactivation process has been proposed for monometallic Pd catalysts.515,516 In the initial stage, light green oil accumulates on the Pd surface, resulting in a slight decline in catalytic activity. As deactivation progresses, 1,3-butadiene undergoes polymerization, forming heavy green oil that blocks catalyst pores and restricts hydrogen diffusion, leading to a substantial loss in activity.The formation of coke and GO primarily arises from side reactions, including C–C bond cleavage and C–C coupling. Section 3 summarizes how active site design strategies can mitigate these pathways. Since C–C coupling generally requires adjacent active metal sites, one effective approach is to dilute the active metal using secondary metals to adjust active site geometry, thereby suppressing oligomer formation. In multi-metallic systems, two typical geometric structures exist: disordered alloys and IMC. Disordered alloy structures tend to feature adjacent active metal sites and, under reaction conditions, active metal segregation can occur at the nanoparticle surface (e.g., in PdAg alloys), limiting their ability to inhibit C–C coupling. In comparison, the ordered arrangement of secondary metals in IMC structures spatially isolates adjacent active sites, providing a unique geometry that effectively suppresses C–C coupling. Another approach is the use of SA structures, which completely eliminate neighbouring active sites. Although both IMC and SA structures geometrically favour the suppression of C–C coupling, active site design must also account for H2 activation. It is important to recognize that the C2 path and C–C coupling path are competitive parallel reactions. Insufficient H2 activation can lead to increased selectivity toward C–C coupling products, as activated C2H2 species tend to couple in the absence of adequate hydrogenation sites. Although site isolation is designed to eliminate neighbouring active metal sites and suppress C–C coupling reactions, experimental results showed that a certain extent of oligomerization can still occur, even on IMC or SA sites. The exact reaction mechanism for C–C coupling pathways remains unclear, and interactions between active metal sites and support materials, such as acidic-basic properties, may also contribute to oligomer formation.
Beyond the geometry of active sites, catalyst supports and reaction conditions also play critical roles in determining catalyst stability. Metal oxides are the most widely used supports for acetylene hydrogenation, with Al2O3 being commonly employed in industrial PdAg catalysts. It is well established that support acidity promotes C–C coupling reactions; thus, reducing support acidity can enhance catalyst stability. However, this must be balanced against other important support properties, such as surface area, which is crucial for effective metal dispersion. Regarding reaction conditions, the ratio of H2/C2H2 significantly affects catalyst stability. In industrial practice, acetylene hydrogenation is typically performed via two main processes: the tail-end and the front-end processes. The front-end process offers better catalyst stability due to a much higher H2/C2H2 ratio compared to the tail-end process.1,2 For industrial acetylene hydrogenation, these typical reaction compositions are generally fixed by upstream processes and are difficult to adjust. However, for potential applications of acetylene hydrogenation in ethylene production from natural gas or coal, optimizing reaction conditions remains essential for achieving high ethylene selectivity and catalyst stability. While a high H2/C2H2 ratio improves catalyst stability, it also promotes over-hydrogenation of ethylene to ethane. Striking a balance between these factors is a key challenge in future process design. Additionally, the introduction of solvents can help modulate the local concentrations of reactive species near the catalyst surface.
Regeneration is another crucial aspect for the industrial application of catalysts. Under practical reaction conditions, it is nearly impossible to completely avoid the formation of GO and coke. As these deposits accumulate, catalytic activity gradually declines. A common approach to compensate for this decline is to incrementally increase the reaction temperature until catalyst activity drops below acceptable production levels. At that point, catalytic activity is typically restored through high-temperature calcination (up to 350 °C) in an oxygen-rich atmosphere. However, this regeneration process can induce metal sintering, causing irreversible loss of catalytic activity.517–520 Since sintering originates from the high-temperature hydrothermal environment of regeneration, optimizing the regeneration process and developing milder regeneration conditions remain challenging yet essential for extending catalyst lifetime. Some studies have addressed this by adjusting parameters such as regeneration temperature and O2/steam feed rates to reduce sintering.521,522 Others have explored alternative regeneration strategies, such as using supercritical carbon dioxide instead of conventional oxidative treatments.523,524 It is worth noting that most current regeneration studies focus on the industrially established PdAg/Al2O3 catalysts. Although the active site design strategies discussed in Section 3 can partially mitigate the formation of GO and coke, the regeneration ability of these advanced catalysts remains relatively underexplored. Further efforts are needed to evaluate and improve catalyst regeneration performance for broader industrial applications.
7.5 Extension to selective hydrogenation of other alkynes and dienes
Selective hydrogenation is a critical process for removing trace alkynes and dienes from alkene streams to meet polymer-grade requirements (<10 ppm).525–527 In addition to acetylene hydrogenation for polymer-grade ethylene production, selective hydrogenation is also applied to propyne (for polymer-grade propylene), 1,3-butadiene and butynes (for polymer-grade C4 olefins), and phenylacetylene (for polymer-grade styrene). Similar to acetylene, these alkynes undergo hydrogenation pathways including semi-hydrogenation to target alkenes, over-hydrogenation to alkanes, and side reactions such as C–C coupling to form oligomers.526,527For terminal alkynes such as propyne, 1-butyne, 1-pentyne, and phenylacetylene, the adsorption configurations on active sites are in general analogous to those of acetylene, including end-on (vinyl or vinylidene) and flat-lying (π- or di-σ-bonded) configurations.1,525 Therefore, the design principles of active sites for acetylene selective hydrogenation should also be applicable to these systems. Catalyst development for terminal alkynes has followed a similar trend, where active sites evolve from monometallic sites to disordered alloy structures, and further to well-defined IMC structures and SA sites. Pd-based systems (e.g., PdAg, PdCu, PdZn) are the most widely investigated,528–531 among which PdAg catalysts are already industrially employed for propyne removal from propylene-rich streams. IMC catalysts such as Pd2Ga, which enable electronic and geometric tuning of CC and CC groups adsorption, have shown promising performance in phenylacetylene hydrogenation.532 To replace precious metal Pd, non-precious metal IMCs such as Ni1Sb1 and Ni3Sn2 have been developed for propyne hydrogenation, achieving improved propylene selectivity.533,534 Additionally, the site isolation strategy for constructing SA sites have been effective in suppressing over-hydrogenation and C–C coupling pathways for alkynes hydrogenation.535–539 While terminal alkynes have mechanistic similarities with acetylene, the effect of substituent groups on the CC bond introduces additional complexity. For instance, phenylacetylene, with its bulky phenyl group, exhibits different adsorption behavior from acetylene. DFT calculations revealled that phenylacetylene preferentially adsorbed on top of isolated Pd atoms, whereas acetylene favored bridge sites at the Pd–Ag interface on Pd1/Ag(111) surfaces.539
For 1,3-butadiene selective hydrogenation, the reaction pathway is inherently linked to that of acetylene hydrogenation (Fig. 19), as 1,3-butadiene serves as a key intermediate in the C–C coupling of acetylene, which contributes to the formation of GO. Several catalysts that demonstrated promising performance in acetylene hydrogenation have also been investigated for 1,3-butadiene selective hydrogenation. These include disordered alloy (e.g., PdAg and PdCu)540–542 and IMC (e.g., Ni3ZnC and Ni3In)543,544 catalysts, which exhibited enhanced 1-butene selectivity. More recently, SA sites engineering has emerged as an effective strategy to further improve 1-butene selectivity.545–548 The mechanism by which SA sites enhance 1-butene selectivity is largely attributed to the distinct adsorption configurations of 1,3-butadiene. On conventional Pd(111) surfaces, the most stable adsorption configuration for 1,3-butadiene was the 1,2,3,4-tetra-σ mode, in which the molecule interacts with four adjacent Pd atoms.541 When Pd was maximally isolated into SA sites, the adsorption mode shifted toward mono-π interaction,546,547 which favored the formation of 1-butene while significantly suppressing its further hydrogenation to butane due to steric hindrance, ultimately leading to improved 1-butene selectivity.546
7.6 Emerging alternatives to thermocatalytic acetylene selective hydrogenation
Electrocatalytic and photocatalytic acetylene hydrogenation (EAH and PAH) have recently emerged as promising alternatives to traditional thermocatalytic process. EAH can be conducted in aqueous media using water as the proton source and powered by renewable electricity under ambient conditions.549,550 PAH utilizes visible light as the energy input and typically employs water or other non-H2 proton sources.551–553 PAH has demonstrated exceptionally high ethylene selectivity (≥99%), but still faces significant challenges in terms of overall process efficiency and the production of polymer-grade ethylene.551–553 Compared with PAH, EAH has been more extensively studied and holds greater potential for replacing traditional thermocatalytic routes in industrial applications.In addition to conventional performance indicators such as acetylene conversion, ethylene selectivity, and catalyst stability, EAH introduces several electrochemical metrics, including overpotential, current density, and faradaic efficiency (FE), which are critical for evaluating electrocatalytic performance.550 Besides the typical side reactions of over-hydrogenation and C–C coupling observed in thermocatalysis, EAH faces a unique challenge from the competitive hydrogen evolution reaction (HER), which significantly diminishes the FE toward ethylene (FEC2H4). For practical and industrial relevance, achieving high FEC2H4 at elevated current densities is essential. Zhang et al. have shown that ethylene production via EAH became economically viable when FEC2H4 exceeded 85% at current densities ≥200mA cm−2.554
EAH was first reported in 1964 using a palladized cathode in an alkaline aqueous solution, demonstrating the feasibility of this approach.555 However, the inherently low solubility of acetylene in aqueous media limited the performance of early EAH systems operated in batch cell reactors, resulting in low FEC2H4 (<70%) and restricting operation to low current densities (<3.5 mA cm−2), even when using precious metal-based electrocatalysts such as Pd, Pt, and Ag.549,555,556 The development of flow cells and membrane electrode assemblies has successfully overcome mass transport limitations caused by poor acetylene solubility, significantly enhancing the feasibility of EAH using water as the proton source. Over the past decade, substantial efforts have been devoted to designing advanced electrocatalysts for EAH in flow-cell configurations.5,557 Among them, Cu-based catalysts have received the most attention due to their earth abundance and theoretically predicted suitability for EAH catalysis.558 Various Cu-based architectures, including Cu dendrites,559 microparticles,5 single atoms,560,561 and nanodots,562 have been developed to improve EAH performance. For instance, Cu dendrites supported on gas diffusion electrodes achieved a high FEC2H4 of 97% at −0.7 V vs. RHE and an ethylene partial current density of ∼130 mA cm−2 under a pure C2H2 feed.559 Cu nanodots have also exhibited excellent performance, delivering a FEC2H4 of ∼95.9% at −0.15 V vs. RHE and current density exceeding 450 mA cm−2.562 Bimetallic Cu catalysts, such as Cu NP/Fe-NC,563 Cu–Cd,564 Cu–Pd,565 and Cu–Zn,566 have been explored to tailor adsorption properties. Notably, Zn doping enhanced acetylene adsorption while increasing the *H–*H binding energy barrier, effectively lowering the overpotential and suppressing both hydrogen evolution and over-hydrogenation side reactions.566 Beyond Cu-based systems, Ni-, Co-, and Ag-based electrocatalysts have also been developed.567–570 For example, Ag nanocubes exposing (100) facets achieved a high partial current density of 337 mA cm−2 at −1.5 V vs. RHE, significantly outperforming (111)-facet analogues.567 Recently, 2-thiolimidazole, a metal-free catalyst, was reported to achieve ethylene partial current density and FEC2H4 values comparable to those of metal-based systems such as Cu and Pd.571
Although significant progress has been made in the development of advanced electrocatalysts for EAH, challenges remain before this technology can be translated into industrial-scale applications. Current research in EAH mainly focuses on two feed conditions: ethylene-rich feeds (aimed at removing trace acetylene impurities) and ethylene-free feeds (targeting ethylene production from acetylene). For ethylene-rich feed conditions, several catalysts have demonstrated the ability to reduce residual acetylene concentrations to below 5 ppm while achieving high FEC2H4.559,572 In particular, a membrane flow cell equipped with Cu dendrite catalysts achieved <5 ppm residual acetylene with a specific ethylene selectivity of 97% at a high space velocity of 9.6 × 104 mL gcat−1 h−1, comparable to those used in thermocatalysis.559 However, the current density was relatively low (1.6 mA cm−2), far from the level required for industrial-scale operation. In contrast, industrial current densities have been used under ethylene-free feed conditions.572,573 For example, a fluorine-modified Cu catalyst achieved a partial current density of up to 760 mA cm−2 with an FEC2H4 exceeding 90% using a 70% C2H2/Ar feed.572 Nevertheless, the acetylene conversion under these conditions remained low (∼32.6%), highlighting the inherent difficulty in simultaneously achieving high ethylene current density and high acetylene conversion. This trade-off is further reflected by the inverse correlation between acetylene concentration and both FEC2H4 and ethylene partial current density. As the acetylene feeding concentration decreases from 50% to 1%, FEC2H4 declines from 90% to 20%, while the current density drops from ∼300 to ∼100 mA cm−2.572 Therefore, significant efforts are still required to develop EAH processes that can simultaneously deliver high activity, ethylene selectivity, and durability under industrially relevant current densities and acetylene conversion. Moreover, the scalable synthesis of high-performance electrocatalysts and the scale-up of electrochemical reactors are critical steps toward the commercial implementation of the EAH technology.550
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
X. C. L. and T. F. W. acknowledge the support from the National Natural Science Foundation of China (no. 22378224, 22108148, and 22178195). J. G. C. acknowledges support from the United States Department of Energy, Division of Chemical Sciences, Geosciences, & Biosciences, Office of Basic Energy Sciences (contract number DE-SC0012704).References
- A. Borodziński and G. C. Bond, Catal. Rev., 2006, 48, 91–144 CrossRef.
- A. Borodziński and G. C. Bond, Catal. Rev., 2008, 50, 379–469 CrossRef.
- J. Guo, Y. Lei, H. Liu, Y. Li, D. Li and D. He, Catal. Sci. Technol., 2023, 13, 4045–4063 RSC.
- Y. Gao, L. Neal, D. Ding, W. Wu, C. Baroi, A. M. Gaffney and F. Li, ACS Catal., 2019, 9, 8592–8621 CrossRef CAS.
- S. H. Wang, K. Uwakwe, L. Yu, J. Y. Ye, Y. Z. Zhu, J. T. Hu, R. X. Chen, Z. Zhang, Z. Y. Zhou, J. F. Li, Z. X. Xie and D. H. Deng, Nat. Commun., 2021, 12, 7072 CrossRef CAS PubMed.
- E. Morais, F. Cameli, G. D. Stefanidis and A. Bogaerts, EES Catal., 2025, 3, 475–487 RSC.
- M. Krajčí and J. Hafner, ChemCatChem, 2016, 8, 34–48 CrossRef.
- X. Deng, J. Wang, N. Guan and L. Li, Cell Rep. Phys. Sci., 2022, 3, 101017 CrossRef CAS.
- G. Tiwari, S. Sarkar and B. R. Jagirdar, ChemCatChem, 2024, 16, e202400586 CrossRef CAS.
- L. Zhang, J. Lin, Z. Liu and J. Zhang, Sci. China: Chem., 2023, 66, 1963–1974 CrossRef CAS.
- Y. Liu, T. Wang, Q. Li and D. Wang, Chin. J. Chem. Eng., 2011, 19, 424–433 CrossRef CAS.
- B. Yan, Y. Cheng, Y. Jin and C. Y. Guo, Fuel Process. Technol., 2012, 100, 1–10 CrossRef CAS.
- A. Dasgupta, H. R. He, R. S. Gong, S. L. Shang, E. K. Zimmerer, R. J. Meyer, Z. K. Liu, M. J. Janik and R. M. Rioux, Nat. Chem., 2022, 14, 523 CrossRef CAS PubMed.
- J. Osswald, R. Giedigkeit, R. E. Jentoft, M. Armbrüster, F. Girgsdies, K. Kovnir, T. Ressler, Y. Grin and R. Schlögl, J. Catal., 2008, 258, 210–218 CrossRef CAS.
- M. Ruta, N. Semagina and L. Kiwi-Minsker, J. Phys. Chem. C, 2008, 112, 13635–13641 CrossRef CAS.
- Y. N. Li and B. W. L. Jang, Appl. Catal., A, 2011, 392, 173–179 CrossRef CAS.
- J. T. Feng, X. Y. Ma, Y. F. He, D. G. Evans and D. Q. Li, Appl. Catal., A, 2012, 413, 10–20 CrossRef.
- Y. F. He, J. T. Feng, Y. Y. Du and D. Q. Li, ACS Catal., 2012, 2, 1703–1710 CrossRef CAS.
- A. E. Yarulin, R. M. Crespo-Quesada, E. V. Egorova and L. L. K. Minsker, Kinet. Catal., 2012, 53, 253–261 CrossRef CAS.
- S. K. Kim, C. Kim, J. H. Lee, J. Kim, H. Lee and S. H. Moon, J. Catal., 2013, 306, 146–154 CrossRef CAS.
- M. Crespo-Quesada, S. Yoon, M. S. Jin, Y. N. Xia, A. Weidenkaff and L. Kiwi-Minsker, ChemCatChem, 2014, 6, 767–771 CrossRef CAS.
- L. P. L. Gonçalves, J. G. Wang, S. Vinati, E. Barborini, X. K. Wei, M. Heggen, M. Franco, J. P. S. Sousa, D. Y. Petrovykh, O. Soares, K. Kovnir, J. Akola and Y. V. Kolen'ko, Int. J. Hydrogen Energy, 2020, 45, 1283–1296 CrossRef.
- I. T. Ellis, E. H. Wolf, G. Jones, B. Lo, M. M. J. Li, A. P. E. York and S. C. E. Tsang, Chem. Commun., 2017, 53, 601–604 RSC.
- Q. Q. Guan, J. Zhang, L. He, R. R. Miao, Y. Z. Shi and P. Ning, ACS Sustainable Chem. Eng., 2020, 8, 11638–11648 CrossRef CAS.
- L. D. Shao, B. S. Zhang, W. Zhang, D. Teschner, F. Girgsdies, R. Schlögl and D. S. Su, Chem. – Eur. J., 2012, 18, 14962–14966 CrossRef CAS PubMed.
- T. X. Yang, M. Zhao, X. Wang, R. Ma, Y. A. Liu, Y. F. He and D. Q. Li, Catal. Lett., 2022, 152, 227–238 CrossRef CAS.
- H. Chen, Z. N. Yu, B. Yang, Y. F. Zhang, C. X. Che, X. Y. Liu, F. Zhang, W. Han, H. Wen, A. Q. Wang and T. Zhang, Chin. J. Catal., 2024, 60, 190–200 CrossRef CAS.
- A. Pachulski, R. Schödel and P. Claus, Appl. Catal., A, 2011, 400, 14–24 CrossRef CAS.
- J. H. Lee, S. K. Kim, I. Y. Ahn, W. J. Kim and S. H. Moon, Catal. Commun., 2011, 12, 1251–1254 CrossRef CAS.
- J. H. Lee, S. K. Kim, I. Y. Ahn, W. J. Kim and S. H. Moon, Korean J. Chem. Eng., 2012, 29, 169–172 CrossRef CAS.
- Y. X. Han, D. Peng, Z. Y. Xu, H. Q. Wan, S. R. Zheng and D. Q. Zhu, Chem. Commun., 2013, 49, 8350–8352 RSC.
- Y. Y. Zhang, W. J. Diao, J. R. Monnier and C. T. Williams, Catal. Sci. Technol., 2015, 5, 4123–4132 RSC.
- M. Kuhn, M. Lucas and P. Claus, Ind. Eng. Chem. Res., 2015, 54, 6683–6691 CrossRef CAS.
- Y. F. He, Y. N. Liu, P. F. Yang, Y. Y. Du, J. T. Peng, X. Z. Cao, J. Yang and D. Q. Li, J. Catal., 2015, 330, 61–70 CrossRef CAS.
- R. Ma, Y. F. He, J. T. Feng, Z. Y. Hu, G. Van Tendeloo and D. Q. Li, J. Catal., 2019, 369, 440–449 CrossRef CAS.
- D. V. Glyzdova, T. N. Afonasenko, E. V. Khramov, N. N. Leont'eva, M. V. Trenikhin, A. M. Kremneva and D. A. Shlyapin, Mol. Catal., 2021, 511, 111717 CAS.
- J. A. Delgado, O. Benkirane, S. Lachaux, C. Claver, J. Ferré, D. Curulla-Ferré and C. Godard, ChemNanoMat, 2022, 8, e202200058 CrossRef CAS.
- K. S. Kley, J. De Bellis and F. Schueth, Catal. Sci. Technol., 2023, 13, 119–131 RSC.
- W. Huang, W. Pyrz, R. F. Lobo and J. G. G. Chen, Appl. Catal., A, 2007, 333, 254–263 CrossRef CAS.
- S. K. Kim, J. H. Lee, I. Y. Ahn, W. J. Kim and S. H. Moon, Appl. Catal., A, 2011, 401, 12–19 CrossRef CAS.
- A. J. McCue, C. J. McRitchie, A. M. Shepherd and J. A. Anderson, J. Catal., 2014, 319, 127–135 CrossRef CAS.
- A. J. McCue, A. M. Shepherd and J. A. Anderson, Catal. Sci. Technol., 2015, 5, 2880–2890 RSC.
- A. J. McCue and J. A. Anderson, J. Catal., 2015, 329, 538–546 CrossRef CAS.
- Y. N. Liu, Y. F. He, D. R. Zhou, J. T. Feng and D. Q. Li, Catal. Sci. Technol., 2016, 6, 3027–3037 RSC.
- M. R. Ball, K. R. Rivera-Dones, E. B. Gilcher, S. F. Ausman, C. W. Hullfish, E. A. Lebrón and J. A. Dumesic, ACS Catal., 2020, 10, 8567–8581 CrossRef CAS.
- S. Y. Liu, Y. M. Niu, Y. Z. Wang, J. N. Chen, X. P. Quan, X. Zhang and B. S. Zhang, Chem. Commun., 2020, 56, 6372–6375 RSC.
- S. Wang, J. Zhu, J. Q. Si, G. F. Zhao, Y. Liu and Y. Lu, J. Catal., 2020, 382, 295–304 CrossRef CAS.
- T. X. Yang, Y. L. Feng, R. Ma, Q. Li, H. Yan, Y. N. Liu, Y. F. He, J. T. Miller and D. Q. Li, ACS Appl. Mater. Interfaces, 2021, 13, 706–716 CrossRef CAS PubMed.
- F. Xue, Q. Li, M. X. Lv, S. X. Weng, T. Y. Li, Y. Ren, Y. N. Liu, D. Q. Li, Y. F. He, Q. H. Li, X. Chen, Q. H. Zhang, L. Gu, J. X. Deng, J. Chen, L. H. He, X. J. Kuang, J. Miao, Y. L. Cao, K. Lin and X. R. Xing, Nano Lett., 2024, 24, 6269–6277 CrossRef CAS PubMed.
- X. Pei, D. Zhang, R. Tang, S. Wang, C. Zhang, W. Yuan and W. Sun, Nanoscale, 2024, 16, 12411–12419 RSC.
- M. Friedrich, S. A. Villaseca, L. Szentmiklósi, D. Teschner and M. Armbrüster, Materials, 2013, 6, 2958–2977 CrossRef PubMed.
- S. Liu, Y. Li, X. J. Yu, S. B. Han, Y. Zhou, Y. Q. Yang, H. Zhang, Z. Jiang, C. W. Zhu, W. X. Li, C. Wöll, Y. M. Wang and W. J. Shen, Nat. Commun., 2022, 13, 4559 CrossRef CAS PubMed.
- Q. Gao, Z. H. Yan, W. J. Zhang, H. S. Pillai, B. Q. Yao, W. J. Zang, Y. Q. Liu, X. Han, B. Min, H. Zhou, L. Ma, B. Anaclet, S. Zhang, H. L. Xin, Q. He and H. Y. Zhu, J. Am. Chem. Soc., 2023, 145, 19961–19968 CrossRef CAS PubMed.
- J. Pei, Y. Xu, Q. Yang and R. Hou, Catal. Sci. Technol., 2024, 14, 3638–3651 RSC.
- F. Xue, Q. Li, W. H. Ji, M. X. Lv, H. K. Xu, J. R. Zeng, T. Y. Li, Y. Ren, L. H. Zhou, X. Chen, J. X. Deng, K. Lin and X. R. Xing, Chem. Sci., 2024, 15, 11837–11846 RSC.
- C. Ma, Y. Y. Du, J. T. Feng, X. Z. Cao, J. Yang and D. Q. Li, J. Catal., 2014, 317, 263–271 CrossRef CAS.
- S. Zhang, C. Y. Chen, B. W. L. Jang and A. M. Zhu, Catal. Today, 2015, 256, 161–169 CrossRef CAS.
- A. J. McCue, R. T. Baker and J. A. Anderson, Faraday Discuss., 2016, 188, 499–523 RSC.
- J. T. Feng, Y. N. Liu, M. Yin, Y. F. He, J. Y. Zhao, J. H. Sun and D. Q. Li, J. Catal., 2016, 344, 854–864 CrossRef CAS.
- T. Sun, B. Ge, S. Huang, X. Wang, Y. Tian, X. Cai, W. Ding and Y. Zhu, Angew. Chem., Int. Ed., 2025, 64, e202420274 CrossRef CAS PubMed.
- I. S. Mashkovskii, O. P. Tkachenko, G. N. Baeva and A. Y. Stakheev, Kinet. Catal., 2009, 50, 768–774 CrossRef CAS.
- D. V. Glyzdova, T. N. Afonasenko, E. V. Khramov, N. N. Leont'eva, M. V. Trenikhin, I. P. Prosvirin, A. V. Bukhtiyarov and D. A. Shlyapin, Top. Catal., 2020, 63, 139–151 CrossRef CAS.
- E. Esmaeili, A. M. Rashidi, Y. Mortazavi, A. A. Khodadadi and M. Rashidzadeh, J. Energy Chem., 2013, 22, 717–725 CrossRef CAS.
- W. G. Menezes, L. Altmann, V. Zielasek, K. Thiel and M. Bäumer, J. Catal., 2013, 300, 125–135 CrossRef CAS.
- R. Ma, T. X. Yang, J. H. Sun, Y. F. He, J. T. Feng, J. T. Miller and D. Q. Li, Chem. Eng. Sci., 2019, 210, 115216 CrossRef.
- D. V. Yurpalova, T. N. Afonasenko, I. P. Prosvirin, A. V. Bukhtiyarov, M. A. Panafidin, Z. S. Vinokurov, M. V. Trenikhin, E. Y. Gerasimov, T. I. Gulyaeva, L. M. Kovtunova and D. A. Shlyapin, Catalysts, 2023, 13, 735 CrossRef.
- E. Esmaeili, Y. Mortazavi, A. A. Khodadadi, A. M. Rashidi and M. Rashidzadeh, Appl. Surf. Sci., 2012, 263, 513–522 CrossRef CAS.
- L. Y. Zhao, Z. Wei, M. Y. Zhu and B. Dai, J. Ind. Eng. Chem., 2012, 18, 45–48 CrossRef CAS.
- Y. F. He, L. L. Liang, Y. N. Liu, J. T. Feng, C. Ma and D. Q. Li, J. Catal., 2014, 309, 166–173 CrossRef CAS.
- Q. Jin, Y. F. He, M. Y. Miao, C. Y. Guan, Y. Y. Du, J. T. Feng and D. Q. Li, Appl. Catal., A, 2015, 500, 3–11 CrossRef CAS.
- Y. Q. Cao, Z. J. Sui, Y. Zhu, X. G. Zhou and D. Chen, ACS Catal., 2017, 7, 7835–7846 CrossRef CAS.
- D. Melnikov, V. Stytsenko, E. Saveleva, M. Kotelev, V. Lyubimenko, E. Ivanov, A. Glotov and V. Vinokurov, Catalysts, 2020, 10, 624 CrossRef CAS.
- H. Q. Kang, J. Z. Wu, B. H. Lou, Y. Wang, Y. L. Zhao, J. J. Liu, S. H. Zou and J. Fan, Molecules, 2023, 28, 2335 CrossRef CAS PubMed.
- Y. Chen, P. Ning, R. R. Miao, Y. Z. Shi, L. He and Q. Q. Guan, New J. Chem., 2020, 44, 20812–20822 RSC.
- J. Osswald, K. Kovnir, M. Armbrüster, R. Giedigleit, R. E. Jentoft, U. Wild, Y. Grin and R. Schlögl, J. Catal., 2008, 258, 219–227 CrossRef CAS.
- M. Armbrüster, K. Kovnir, M. Behrens, D. Teschner, Y. Grin and R. Schlögl, J. Am. Chem. Soc., 2010, 132, 14745–14747 CrossRef PubMed.
- A. Ota, M. Armbrüster, M. Behrens, D. Rosenthal, M. Friedrich, I. Kasatkin, F. Girgsdies, W. Zhang, R. Wagner and R. Schlögl, J. Phys. Chem. C, 2011, 115, 1368–1374 CrossRef CAS.
- M. Armbrüster, G. Wowsnick, M. Friedrich, M. Heggen and R. Cardoso-Gil, J. Am. Chem. Soc., 2011, 133, 9112–9118 CrossRef PubMed.
- M. Siebert, R. R. Zimmermann, M. Armbrüster and R. Dittmeyer, ChemCatChem, 2017, 9, 3733–3742 CrossRef CAS.
- R. R. Zimmermann, M. Siebert, S. Ibrahimkutty, R. Dittmeyer and M. Armbrüster, Z. Anorg. Allg. Chem., 2020, 646, 1218–1226 CrossRef CAS.
- Y. M. Niu, Y. Z. Wang, J. N. Chen, S. Y. Li, X. Huang, M. G. Willinger, W. Zhang, Y. F. Liu and B. S. Zhang, Sci. Adv., 2022, 8, eabq5751 CrossRef CAS PubMed.
- Y. Luo, S. A. Villaseca, M. Friedrich, D. Teschner, A. Knop-Gericke and M. Armbrüster, J. Catal., 2016, 338, 265–272 CrossRef CAS.
- Q. C. Feng, S. Zhao, Y. Wang, J. C. Dong, W. X. Chen, D. S. He, D. S. Wang, J. Yang, Y. M. Zhu, H. L. Zhu, L. Gu, Z. Li, Y. X. Liu, R. Yu, J. Li and Y. D. Li, J. Am. Chem. Soc., 2017, 139, 7294–7301 CrossRef CAS PubMed.
- X. H. Ge, Y. Q. Cao, K. L. Yan, Y. R. Li, L. H. Zhou, S. Dai, J. Zhang, X. Q. Gong, G. Qian, X. G. Zhou, W. K. Yuan and X. Z. Duan, Angew. Chem., Int. Ed., 2022, 61, e202215225 CrossRef CAS PubMed.
- Q. L. Wu, C. Y. Shen, K. H. Sun, M. H. Liu and C. J. Liu, Chem. Eng. J., 2024, 486, 150333 CrossRef CAS.
- H. R. Zhou, X. F. Yang, L. Li, X. Y. Liu, Y. Q. Huang, X. L. Pan, A. Q. Wang, J. Li and T. Zhang, ACS Catal., 2016, 6, 1054–1061 CrossRef CAS.
- X. X. Cao, R. J. Tong, S. Y. Tang, B. L. Jang, A. Mirjalili, J. Y. Li, X. N. Guo, J. Y. Zhang, J. X. Hu and X. Meng, Molecules, 2022, 27, 5736 CrossRef CAS PubMed.
- H. Chen, L. L. Li, Z. J. Zhao, B. Yang, Y. F. Zhang, X. Y. Liu, Q. Q. Gu, Z. N. Yu, X. F. Yang, J. L. Gong, A. Q. Wang and T. Zhang, Nat. Commun., 2024, 15, 9850 CrossRef CAS PubMed.
- I. G. Aviziotis, A. Götze, F. Göhler, H. Kohlmann and M. Armbrüster, Z. Anorg. Allg. Chem., 2018, 644, 1777–1781 CrossRef CAS.
- B. H. Lou, H. Q. Kang, W. T. Yuan, L. Ma, W. X. Huang, Y. Wang, Z. Jiang, Y. H. Du, S. H. Zou and J. Fan, ACS Catal., 2021, 11, 6073–6080 CrossRef CAS.
- W. Q. Zhang, X. B. Zhang, J. Y. Wang, A. Ghosh, J. Zhu, N. J. LiBretto, G. H. Zhang, A. K. Datye, W. Liu and J. T. Miller, ACS Catal., 2022, 12, 10531–10545 CrossRef CAS.
- R. Li, Y. Yue, Z. Chen, X. Chen, S. Wang, Z. Jiang, B. Wang, Q. Xu, D. Han and J. Zhao, Appl. Catal., B, 2020, 279, 119348 CrossRef CAS.
- Y. N. Liu, A. J. McCue, C. L. Miao, J. T. Feng, D. Q. Li and J. A. Anderson, J. Catal., 2018, 364, 406–414 CrossRef CAS.
- O. Matselko, R. R. Zimmermann, A. Ormeci, U. Burkhardt, R. Gladyshevskii, Y. Grin and M. Armbrüster, J. Phys. Chem. C, 2018, 122, 21891–21896 CrossRef CAS.
- Q. Guo, R. T. Chen, J. P. Guo, C. Qin, Z. T. Xiong, H. X. Yan, W. B. Gao, Q. J. Pei, A. A. Wu and P. Chen, J. Am. Chem. Soc., 2021, 143, 20891–20897 CrossRef CAS PubMed.
- X. H. Huang, Y. J. Xia, Y. J. Cao, X. S. Zheng, H. B. Pan, J. F. Zhu, C. Ma, H. W. Wang, J. J. Li, R. You, S. Q. Wei, W. X. Huang and J. L. Lu, Nano Res., 2017, 10, 1302–1312 CrossRef CAS.
- F. Huang, Y. C. Deng, Y. L. Chen, X. B. Cai, M. Peng, Z. M. Jia, P. J. Ren, D. Q. Xiao, X. D. Wen, N. Wang, H. Y. Liu and D. Ma, J. Am. Chem. Soc., 2018, 140, 13142–13146 CrossRef CAS PubMed.
- X. H. Huang, H. Yan, L. Huang, X. H. Zhang, Y. Lin, J. J. Li, Y. J. Xia, Y. F. Ma, Z. H. Sun, S. G. Wei and J. L. Lu, J. Phys. Chem. C, 2019, 123, 7922–7930 CrossRef CAS.
- S. J. Wei, A. Li, J. C. Liu, Z. Li, W. X. Chen, Y. Gong, Q. H. Zhang, W. C. Cheong, Y. Wang, L. R. Zheng, H. Xiao, C. Chen, D. S. Wang, Q. Peng, L. Gu, X. D. Han, J. Li and Y. D. Li, Nat. Nanotechnol., 2018, 13, 856 CrossRef CAS PubMed.
- Q. C. Feng, S. Zhao, Q. Xu, W. X. Chen, S. B. Tian, Y. Wang, W. S. Yan, J. Luo, D. S. Wang and Y. D. Li, Adv. Mater., 2019, 31, 1901024 CrossRef PubMed.
- M. C. Hu, Z. Y. Wu, Z. H. Yao, J. S. Young, L. L. Luo, Y. G. Du, C. M. Wang, Z. Iqbal and X. Q. Wang, J. Catal., 2021, 395, 46–53 CrossRef CAS.
- F. Huang, M. Peng, Y. L. Chen, X. B. Cai, X. T. Qin, N. Wang, D. Q. Xiao, L. Jin, G. Q. Wang, X. D. Wen, H. Y. Liu and D. Ma, J. Am. Chem. Soc., 2022, 144, 18485–18493 CrossRef CAS PubMed.
- S. J. Wei, X. W. Liu, C. Wang, X. C. Liu, Q. H. Zhang and Z. Li, ACS Nano, 2023, 17, 14831–14839 CrossRef CAS PubMed.
- R. R. Li, Y. X. Yue, X. L. Chen, R. Q. Chang, J. X. Zhang, B. Zhao, J. Y. Zhang, D. Cai, Y. H. Zhu, D. M. Han, J. Zhao and X. N. A. Li, Nano Res., 2023, 16, 6167–6177 CrossRef CAS.
- S. Q. Zhou, L. Shang, Y. X. Zhao, R. Shi, G. I. N. Waterhouse, Y. C. Huang, L. R. Zheng and T. R. Zhang, Adv. Mater., 2019, 31, 1900509 CrossRef PubMed.
- Q. H. Li, S. J. Liu, J. C. Liu, Z. Li and Y. D. Li, J. Am. Chem. Soc., 2024, 147, 5615–5623 CrossRef PubMed.
- Y. Zeng, M. Q. Xia, F. J. Gao, C. K. Zhou, X. Y. Cheng, L. W. Liu, L. Jiao, Q. Wu, X. Z. Wang, L. J. Yang, Y. N. Fan and Z. Hu, Nano Res., 2024, 17, 8243–8249 CrossRef CAS.
- H. R. Zhou, X. F. Yang, A. Q. Wang, S. Miao, X. Y. Liu, X. L. Pan, Y. Su, L. Li, Y. Tan and T. Zhang, Chin. J. Catal., 2016, 37, 692–699 CrossRef CAS.
- C. Qin, Q. Guo, J. P. Guo and P. Chen, Chem. – Asian J., 2021, 16, 1225–1228 CrossRef CAS PubMed.
- Y. L. Guo, H. F. Qi, Y. Su, Q. K. Jiang, Y. T. Cui, L. Li and B. T. Qiao, ChemNanoMat, 2021, 7, 526–529 CrossRef CAS.
- R. J. Gao, J. S. Xu, J. Wang, J. Lim, C. Peng, L. Pan, X. W. Zhang, H. M. Yang and J. J. Zou, J. Am. Chem. Soc., 2022, 144, 573–581 CrossRef CAS PubMed.
- X. Tao, B. Nan, Y. A. Li, M. Du, L. L. Guo, C. Tian, L. Z. Jiang, L. Shen, N. N. Sun and L. N. Li, ACS Appl. Energy Mater., 2022, 5, 10385–10390 CrossRef CAS.
- Y. L. Guo, Y. Y. Li, X. R. Du, L. Li, Q. K. Jiang and B. T. Qiao, Nano Res., 2022, 15, 10037–10043 CrossRef CAS.
- G. X. Pei, X. Y. Liu, A. Q. Wang, L. Li, Y. Q. Huang, T. Zhang, J. W. Lee, B. W. L. Jang and C. Y. Mou, New J. Chem., 2014, 38, 2043–2051 RSC.
- G. X. Pei, X. Y. Liu, A. Q. Wang, A. F. Lee, M. A. Isaacs, L. Li, X. L. Pan, X. F. Yang, X. D. Wang, Z. J. Tai, K. Wilson and T. Zhang, ACS Catal., 2015, 5, 3717–3725 CrossRef CAS.
- G. X. Pei, X. Y. Liu, X. F. Yang, L. L. Zhang, A. Q. Wang, L. Li, H. Wang, X. D. Wang and T. Zhang, ACS Catal., 2017, 7, 1491–1500 CrossRef CAS.
- M. Q. Zhong, J. F. Zhao, Y. X. Fang, D. F. Wu, L. N. Zhang, C. C. Du, S. X. Liu, S. F. Yang, S. L. Wan, Y. B. Jiang, J. Y. Huang and H. F. Xiong, Appl. Catal., A, 2023, 662, 119288 CrossRef CAS.
- X. J. Xu, Q. Wang, L. L. Xie, Y. N. Liu, D. Q. Li, J. T. Feng and X. Duan, AIChE J., 2023, 69, e18042 CrossRef CAS.
- S. X. Liu, D. F. Wu, F. Yang, K. Chen, Z. B. Luo, J. W. Li, Z. Zhang, J. F. Zhao, L. N. Zhang, Y. C. Zhang, H. L. Zhang, S. L. Wan, Y. K. Peng, K. H. L. Zhang and H. F. Xiong, Chem. Eng. J., 2024, 481, 113043 Search PubMed.
- S. S. Chai, D. L. Gao, J. Xia, Y. P. Yang and X. Wang, ChemCatChem, 2023, 15, e202300217 CrossRef CAS.
- M. S. Sun, F. M. Wang, G. J. Lv and X. B. Zhang, Ind. Eng. Chem. Res., 2022, 61, 13341–13353 CrossRef CAS.
- X. H. Ge, Y. D. Jing, N. N. Fei, K. L. Yan, Y. J. Liang, Y. Q. Cao, J. Zhang, G. Qian, L. N. Li, H. Jiang, X. G. Zhou, W. K. Yuan and X. Z. Duan, Angew. Chem., Int. Ed., 2024, 63, e202410979 CAS.
- I. S. Mashkovsky, A. V. Bukhtiyarov, P. V. Markov, G. O. Bragina, G. N. Baeva, N. S. Smirnova, M. A. Panafidin, I. A. Chetyrin, E. Y. Gerasimov, Y. V. Zubavichus and A. Y. Stakheev, Appl. Surf. Sci., 2025, 681, 161516 CrossRef CAS.
- L. Y. Xu, Y. X. Qin, Q. J. Zhang, J. Zhou, J. Zhao, F. Feng, T. L. Sun, X. L. Xu, Y. H. Zhu, C. S. Lu, Q. F. Zhang, Q. T. Wang and X. N. Li, Chem. Eng. J., 2024, 495, 150582 Search PubMed.
- Y. W. Liu, B. X. Wang, Q. Fu, W. Liu, Y. Wang, L. Gu, D. S. Wang and Y. D. Li, Angew. Chem., Int. Ed., 2021, 60, 22522–22528 CrossRef CAS PubMed.
- W. Ru, Y. N. Liu, B. Fu, F. Z. Fu, J. T. Feng and D. Q. Li, Small, 2022, 18, 2103852 CrossRef CAS PubMed.
- S. P. Ding, Y. L. Guo, M. J. Hülsey, B. Zhang, H. Asakura, L. M. Liu, Y. Han, M. Gao, J. Y. Hasegawa, B. T. Qiao, T. Zhang and N. Yan, Chem, 2019, 5, 3207–3219 CAS.
- M. Z. Hu, J. Zhang, W. Zhu, Z. Chen, X. Gao, X. J. Du, J. W. Wan, K. B. Zhou, C. Chen and Y. D. Li, Nano Res., 2018, 11, 905–912 CrossRef CAS.
- W. W. Zhang, K. Uwakwe, J. T. Hu, Y. Wei, J. T. Zhu, W. Zhou, C. Ma, L. Yu, R. Huang and D. H. Deng, Nat. Commun., 2024, 15, 9457 CrossRef CAS PubMed.
- Y. Azizi, C. Petit and V. Pitchon, J. Catal., 2008, 256, 338–344 CrossRef CAS.
- A. Sárkány, Z. Schay, K. Frey, É. Széles and I. Sajó, Appl. Catal., A, 2010, 380, 133–141 CrossRef.
- X. Y. Liu, C. Y. Mou, S. Lee, Y. N. Li, J. Secrest and B. W. L. Jang, J. Catal., 2012, 285, 152–159 CrossRef CAS.
- X. L. Yan, J. Wheeler, B. Jang, W. Y. Lin and B. R. Zhao, Appl. Catal., A, 2014, 487, 36–44 CrossRef CAS.
- H. R. Zhou, B. X. Li, Y. X. Zhang, X. Y. Yan, W. X. Lv, X. B. Wang, B. B. Yuan, Y. Liu, Z. X. Yang and X. D. Lou, ACS Appl. Mater. Interfaces, 2021, 13, 40429–40440 CrossRef CAS PubMed.
- Y. Zhang, X. Sun, Y. L. Zhao, H. J. Su, T. Murayama and C. X. Qi, Top. Catal., 2021, 64, 197–205 CrossRef CAS.
- X. Y. Hua, Y. H. Zheng, Z. X. Yang, L. B. Sun, H. J. Su, T. Murayama and C. X. Qi, Top. Catal., 2021, 64, 206–214 CrossRef CAS.
- X. L. Yan, J. H. Bao, C. Yuan, J. Wheeler, W. Y. Lin, R. F. Li and B. W. L. Jang, J. Catal., 2016, 344, 194–201 CrossRef CAS.
- S. Z. Zhou, L. H. Kang, Z. Xu and M. Y. Zhu, RSC Adv., 2020, 10, 1937–1945 RSC.
- Z. Xu, S. Z. Zhou and M. Y. Zhu, Catal. Commun., 2021, 149, 106241 CrossRef CAS.
- J. Y. Zhao, L. He, J. D. Yu, Y. Z. Shi, R. R. Miao, Q. Q. Guan and P. Ning, New J. Chem., 2021, 45, 1054–1062 RSC.
- C. Y. Lu, A. N. Zeng, Y. Wang and A. J. Wang, ACS Omega, 2021, 6, 3363–3371 CrossRef CAS PubMed.
- C. Y. Lu, A. N. Zeng, Y. Wang and A. J. Wang, Eur. J. Inorg. Chem., 2021, 997–1004 CrossRef CAS.
- C. Y. Lu, Y. Wang, R. G. Zhang, B. J. Wang and A. J. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 46027–46036 CrossRef CAS PubMed.
- D. L. Trimm, N. W. Cant and I. O. Y. Liu, Catal. Today, 2011, 178, 181–186 CrossRef CAS.
- Y. J. Chen and J. X. Chen, Appl. Surf. Sci., 2016, 387, 16–27 CrossRef CAS.
- G. X. Pei, X. Y. Liu, A. Wang, Y. Su, L. Li and T. Zhang, Appl. Catal., A, 2017, 545, 90–96 CrossRef CAS.
- Q. Y. Li, Y. X. Wang, G. Skoptsov and J. L. Hu, Ind. Eng. Chem. Res., 2019, 58, 20620–20629 CrossRef CAS.
- Z. Wang, G. Wang, C. Louis and L. Delannoy, Res. Chem. Intermed., 2021, 47, 91–116 CrossRef CAS.
- H. D. Yao, L. Y. Pan, L. W. Cui, J. F. Wang, X. Y. Wu, H. Y. Wang, Z. C. Na, D. Li and W. H. Li, Appl. Surf. Sci., 2024, 648, 159079 CrossRef CAS.
- L. Wang, F. X. Li, Y. J. Chen and J. X. Chen, J. Energy Chem., 2019, 29, 40–49 CrossRef.
- D. Wang, R. Ye, C. Zhang, C. Jin, Z.-H. Lu, M. Shakouri, B. Han, T. Wang, Y. Zhang, R. Zhang, Y. Hu, J. Zhou and G. Feng, Energy Fuels, 2023, 37, 13305–13318 CrossRef CAS.
- C. Zhang, L. X. Wu, R. P. Ye, G. Feng and R. B. Zhang, Catal. Lett., 2024, 154, 3619–3627 CrossRef CAS.
- X. M. Sun, R. D. Wu, M. A. Nawaz, S. Meng, T. Guan, C. Zhang, C. Y. Sun, Z. H. Lu, R. B. Zhang, G. Feng and R. P. Ye, Inorg. Chem., 2024, 63, 24313–24330 CrossRef CAS PubMed.
- Y. F. Song, S. X. Weng, F. Xue, A. J. McCue, L. R. Zheng, Y. F. He, J. T. Feng, Y. A. Liu and D. Q. Li, ACS Catal., 2023, 13, 1952–1963 CrossRef CAS.
- Y. N. Liu, J. Y. Zhao, J. T. Feng, Y. F. He, Y. Y. Du and D. Q. Li, J. Catal., 2018, 359, 251–260 CrossRef CAS.
- N. M. Hu, C. H. Yang, L. He, Q. Q. Guan and R. R. Miao, New J. Chem., 2019, 43, 18120–18125 RSC.
- H. Liu, M. Q. Chai, G. X. Pei, X. Y. Liu, L. Li, L. L. Kang, A. Q. Wang and T. Zhang, Chin. J. Catal., 2020, 41, 1099–1108 CrossRef CAS.
- S. Z. Zhou, L. H. Kang, X. N. Zhou, Z. Xu and M. Y. Zhu, Nanomaterials, 2020, 10, 509 CrossRef CAS PubMed.
- S. Hu, C. Zhang, M. Y. Wu, R. P. Ye, D. P. Shi, M. J. Li, P. Zhao, R. B. Zhang and G. Feng, Catalysts, 2022, 12, 1072 CrossRef CAS.
- Z. W. Li, J. P. Zhang, J. M. Tian, K. Feng, Z. Jiang and B. H. Yan, Chem. Eng. J., 2022, 450, 138244 CrossRef CAS.
- S. H. Zhou, C. Y. Lu, Y. Bi, C. L. Zhou, Q. Li, L. X. Tan and L. C. Dong, Chem. Eng. J., 2023, 476, 146594 CrossRef CAS.
- C. X. Jin, P. Gao, S. W. Zhou, Y. X. Yue, S. S. Wang, R. Q. Chang, C. Y. Li, T. L. Sun, S. P. Zhang, Y. H. Cai, Y. H. Zhu, J. Zhao and X. N. Li, Chem. Eng. J., 2025, 507, 160617 CrossRef CAS.
- S. A. Nikolaev, V. V. Smirnov, A. Y. Vasil'kov and V. L. Podshibikhin, Kinet. Catal., 2010, 51, 375–379 CrossRef CAS.
- M. Q. Chai, X. Y. Liu, L. Li, G. X. Pei, Y. J. Ren, Y. Su, H. K. Cheng, A. Q. Wang and T. Zhang, Chin. J. Catal., 2017, 38, 1338–1346 CrossRef CAS.
- O. B. Ayodele, T. D. Shittu, O. S. Togunwa, D. Yu and Z. Y. Tian, Chem. Eng. J., 2024, 479, 147496 CrossRef CAS.
- O. B. Ayodele, R. S. Cai, J. G. Wang, Y. Ziouani, Z. F. Liang, M. C. Spadaro, K. Kovnir, J. Arbiol, J. Akola, R. E. Palmer and Y. V. Kolen'ko, ACS Catal., 2020, 10, 451–457 CrossRef CAS.
- C. Y. Lu, A. N. Zeng, Y. Wang and A. J. Wang, Ind. Eng. Chem. Res., 2022, 61, 18696–18702 CrossRef CAS.
- F. Y. Zhang, Y. J. Zhang, J. Y. Wang, Q. Wang, H. X. Xu, D. Q. Li, J. T. Feng and X. Duan, Angew. Chem., Int. Ed., 2024, 63, e202412637 CrossRef CAS PubMed.
- X. Y. Liu, Y. N. Li, J. W. Lee, C. Y. Hong, C. Y. Mou and B. W. L. Jang, Appl. Catal., A, 2012, 439, 8–14 CrossRef.
- M. Z. Hu, W. J. Yang, S. J. Liu, W. Zhu, Y. Li, B. T. Hu, Z. Chen, R. A. Shen, W. C. Cheong, Y. Wang, K. B. Zhou, Q. Peng, C. Chen and Y. D. Li, Chem. Sci., 2019, 10, 614–619 RSC.
- Y. Q. Cao, H. Zhang, S. F. Ji, Z. J. Sui, Z. Jiang, D. S. Wang, F. Zaera, X. G. Zhou, X. H. Duan and Y. D. Li, Angew. Chem., Int. Ed., 2020, 59, 11647–11652 CrossRef CAS PubMed.
- Y. M. Niu, X. Huang, Y. Z. Wang, M. Xu, J. A. Chen, S. L. Xu, M. G. Willinger, W. Zhang, M. Wei and B. S. Zhang, Nat. Commun., 2020, 11, 3324 CrossRef CAS PubMed.
- Y. Wang, B. Y. Liu, X. C. Lan and T. F. Wang, ACS Catal., 2021, 11, 10257–10266 CrossRef CAS.
- J. M. Ma, F. L. Xing, Y. Nakaya, K. I. Shimizu and S. Furukawa, Angew. Chem., Int. Ed., 2022, 61, e202200889 CrossRef CAS PubMed.
- R. Tsukuda, S. Ohhashi, Y. Xu, C. Nishimura and S. Kameoka, Mater. Trans., 2022, 63, 343–350 CrossRef CAS.
- X. H. Ge, Z. H. Ren, Y. Q. Cao, X. Liu, J. Zhang, G. Qian, X. Q. Gong, L. W. Chen, X. G. Zhou, W. K. Yuan and X. Z. Duan, J. Mater. Chem. A, 2022, 10, 19722–19731 RSC.
- X. H. Ge, M. Y. Dou, Y. Q. Cao, L. Xi, Y. W. Qiang, Z. Jing, Q. Gang, X. Q. Gong, X. G. Zhou, L. W. Chen, W. K. Yuan and X. Z. Duan, Nat. Commun., 2022, 13, 5534 CrossRef CAS PubMed.
- H. R. Zhou, H. G. Fu, B. X. Li, X. Y. Yan, Y. Su, X. L. Pan, Q. H. Tang, X. B. Wang, X. H. Zhao, Y. Liu, Z. X. Yang, Z. S. Lu, X. D. Lou and L. Li, ACS Sustainable Chem. Eng., 2023, 11, 11052–11066 CrossRef CAS.
- J. M. Ma, F. L. Xing, K. Shimizu and S. Furukawa, Chem. Sci., 2024, 15, 4086–4094 RSC.
- Y. L. Liu, C. J. Xu, B. Yang, X. Y. Meng, G. W. Qin and S. Li, Catal. Sci. Technol., 2023, 13, 5345–5350 RSC.
- S. M. Dong, Y. M. Niu, Y. H. Pu, Y. Z. Wang and B. S. Zhang, Chin. Chem. Lett., 2024, 35, 109525 CrossRef CAS.
- T. Komatsu, K. Sou and K. Ozawa, J. Mol. Catal. A: Chem., 2010, 319, 71–77 CrossRef CAS.
- R. V. Maligal-Ganesh, Y. C. Pei, C. X. Xiao, M. D. Chen, T. W. Goh, W. J. Sun, J. S. Wu and W. Y. Huang, ChemCatChem, 2020, 12, 3022–3029 CrossRef CAS.
- D. Zhou, G. H. Zhang, Y. Li, S. Liu, S. B. Han, Y. Zhou and W. J. Shen, Chem. Eng. J., 2023, 472, 144875 CrossRef CAS.
- D. Köhler, M. Heise, A. I. Baranov, Y. Luo, D. Geiger, M. Ruck and M. Armbrüster, Chem. Mater., 2012, 24, 1639–1644 CrossRef.
- X. C. Lan, Y. Wang, B. Y. Liu, Z. Y. Kang and T. F. Wang, Chem. Sci., 2024, 15, 1758–1768 RSC.
- M. Armbrüster, K. Kovnir, M. Friedrich, D. Teschner, G. Wowsnick, M. Hahne, P. Gille, L. Szentmiklósi, M. Feuerbacher, M. Heggen, F. Girgsdies, D. Rosenthal, R. Schlögl and Y. Grin, Nat. Mater., 2012, 11, 690–693 CrossRef PubMed.
- I. G. Aviziotis, T. Duguet, K. Soussi, M. Heggen, M. C. Lafont, F. Morfin, S. Mishra, S. Daniele, A. G. Boudouvis and C. Vahlas, Phys. Status Solidi A, 2018, 215, 1700692 CrossRef.
- X. H. Ge, J. Q. Ming, Y. D. Jing, N. N. Fei, L. L. Guo, Y. Q. Cao, H. Jiang, J. Zhang, G. Qian, X. G. Zhou and X. Z. Duan, ACS Catal., 2025, 15, 2282–2291 CrossRef CAS.
- X. Y. Dai, Z. Chen, T. Yao, L. R. Zheng, Y. Lin, W. Liu, H. X. Ju, J. F. Zhu, X. Hong, S. Q. Wei, Y. E. Wu and Y. D. Li, Chem. Commun., 2017, 53, 11568–11571 RSC.
- Y. C. Chai, G. J. Wu, X. Y. Liu, Y. J. Ren, W. L. Dai, C. M. Wang, Z. K. Xie, N. J. Guan and L. D. Li, J. Am. Chem. Soc., 2019, 141, 9920–9927 CrossRef CAS PubMed.
- B. A. Fu, A. J. McCue, Y. A. Liu, S. X. Weng, Y. F. Song, Y. F. He, J. T. Feng and D. Q. Li, ACS Catal., 2022, 12, 607–615 CrossRef CAS.
- J. Gu, M. Z. Jian, L. Huang, Z. H. Sun, A. W. Li, Y. Pan, J. Z. Yang, W. Wen, W. Zhou, Y. Lin, H. J. Wang, X. Y. Liu, L. L. Wang, X. X. Shi, X. H. Huang, L. N. Cao, S. Chen, X. S. Zheng, H. B. Pan, J. F. Zhu, S. Q. Wei, W. X. Li and J. L. Lu, Nat. Nanotechnol., 2021, 16, 1141 CrossRef CAS PubMed.
- C. Q. Sui, H. Ma, F. Huang, M. L. Wang, X. B. Cai, J. Y. Diao, P. J. Ren, X. D. Wen, L. Jin, G. Q. Wang, D. Ma and H. Y. Liu, ACS Catal., 2024, 14, 14689–14695 CrossRef CAS.
- F. Huang, Y. C. Deng, Y. L. Chen, X. B. Cai, M. Peng, Z. M. Jia, J. L. Xie, D. Q. Xiao, X. D. Wen, N. Wang, Z. Jiang, H. Y. Liu and D. Ma, Nat. Commun., 2019, 10, 4431 CrossRef PubMed.
- X. X. Shi, Y. Lin, L. Huang, Z. H. Sun, Y. Yang, X. H. Zhou, E. Vovk, X. Y. Liu, X. H. Huang, M. Sun, S. Q. Wei and J. L. Lu, ACS Catal., 2020, 10, 3495–3504 CrossRef CAS.
- F. Huang, M. Peng, Y. L. Chen, Z. R. Gao, X. B. Cai, J. L. Xie, D. Q. Xiao, L. Jin, G. Q. Wang, X. D. Wen, N. Wang, W. Zhou, H. Y. Liu and D. Ma, ACS Catal., 2022, 12, 48–57 CrossRef CAS.
- Y. X. Yue, B. L. Wang, C. X. Jin, K. X. Huang, Q. Zhou, R. Q. Chang, S. S. Wang, Z. Y. Pan, J. Zhao and X. N. Li, ACS Catal., 2024, 14, 3900–3911 CrossRef CAS.
- Y. R. Li, Y. Q. Cao, X. H. Ge, H. Zhang, K. L. Yan, J. Zhang, G. Qian, Z. Jiang, X. Q. Gong, A. M. Li, X. G. Zhou, W. K. Yuan and X. Z. Duan, J. Catal., 2022, 407, 290–299 CrossRef CAS.
- M. S. Sun, F. M. Wang, J. Q. Hu, G. J. Lv and X. B. Zhang, Chem. Eng. Sci., 2022, 247, 116939 CrossRef CAS.
- M. S. Sun, F. M. Wang, G. J. Lv and X. B. Zhang, Appl. Surf. Sci., 2022, 589, 153021 CrossRef CAS.
- F. Xue, Q. Li, M. X. Lv, Y. F. Song, T. X. Yang, X. G. Wang, T. Y. Li, Y. Ren, K. Ohara, Y. F. He, D. Q. Li, Q. H. Li, X. Chen, K. Lin and X. R. Xing, J. Am. Chem. Soc., 2023, 145, 26728–26735 CrossRef CAS PubMed.
- C. X. Che, G. L. Gou, H. Wen, Y. L. Liang, W. Han, F. Zhang and X. X. Cai, Inorg. Nano-Met. Chem., 2021, 51, 70–77 CrossRef CAS.
- Y. Y. Zhang, W. J. Diao, C. T. Williams and J. R. Monnier, Appl. Catal., A, 2014, 469, 419–426 CrossRef CAS.
- S. Riyapan, Y. Y. Zhang, A. Wongkaew, B. Pongthawornsakun, J. R. Monnier and J. Panpranot, Catal. Sci. Technol., 2016, 6, 5608–5617 RSC.
- Q. Fu and Y. Luo, ACS Catal., 2013, 3, 1245–1252 CrossRef CAS.
- P. Zhai, D. A. Cullen and K. L. Ding, Chem. Eng. J., 2024, 480, 145028 CrossRef.
- J. G. Ma, C. Y. Yang, X. Ye, X. L. Pan, S. Y. Nie, X. Cao, H. A. Li, H. Matsumoto, L. Wu and C. Chen, Chem. Sci., 2024, 15, 8363–8371 RSC.
- V. D. Stytsenko, D. P. Melnikov, A. P. Glotov and V. A. Vinokurov, Mol. Catal., 2022, 533, 112750 CAS.
- A. Sárkány, O. Geszti and G. Sáfrán, Appl. Catal., A, 2008, 350, 157–163 CrossRef.
- P. Zhai, Y. F. Shi, Q. X. Wang, Y. N. Xia and K. L. Ding, Nanoscale, 2021, 13, 18498–18506 RSC.
- C. W. Zhu, W. L. Xu, F. Liu, J. Luo, J. L. Lu and W. X. Li, Angew. Chem., Int. Ed., 2023, 62, e202300110 CrossRef CAS PubMed.
- K. Abe, R. Tsukuda, N. Fujita and S. Kameoka, RSC Adv., 2021, 11, 15296–15300 RSC.
- K. Kovnir, J. Osswald, M. Armbrüster, D. Teschner, G. Weinberg, U. Wild, A. Knop-Gericke, T. Ressler, Y. Grin and R. Schlögl, J. Catal., 2009, 264, 93–103 CrossRef CAS.
- A. Ota, J. Kröhnert, G. Weinberg, I. Kasatkin, E. L. Kunkes, D. Ferri, F. Girgsdies, N. Hamilton, M. Armbrüster, R. Schlögl and M. Behrens, ACS Catal., 2014, 4, 2048–2059 CrossRef CAS.
- F. Meunier, M. Maffre, Y. Schuurman, S. Colussi and A. Trovarelli, Catal. Commun., 2018, 105, 52–55 CrossRef CAS.
- Q. Guo, C. Qin, J. P. Guo and P. Chen, Chem. Commun., 2023, 59, 2259–2262 RSC.
- K. Bader, A. Dorner and P. Gille, J. Cryst. Growth, 2020, 532, 125401 CrossRef CAS.
- M. C. Hu and X. Q. Wang, Catal. Today, 2016, 263, 98–104 CrossRef CAS.
- S. M. Hosseini, M. Ghiaci and H. Farrokhpour, Struct. Chem., 2021, 32, 2087–2097 CrossRef CAS.
- S. Hock, M. Lucas, E. Kolle-Görgen, M. Mellin, J. P. Hofmann and M. Rose, ChemCatChem, 2023, 15, e202201479 CrossRef CAS.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- L. Z. Jiang, K. L. Liu, S. F. Hung, L. Y. Zhou, R. X. Qin, Q. H. Zhang, P. X. Liu, L. Gu, H. M. Chen, G. Fu and N. F. Zheng, Nat. Nanotechnol., 2020, 15, 848 CrossRef CAS PubMed.
- G. X. Pei, X. Y. Liu, M. Q. Chai, A. Q. Wang and T. Zhang, Chin. J. Catal., 2017, 38, 1540–1548 CrossRef CAS.
- C. Hartwig, K. Schweinar, T. E. Jones, S. Beeg, F. P. Schmidt, R. Schlögl and M. Greiner, J. Chem. Phys., 2021, 154, 184703 CrossRef CAS PubMed.
- D. L. Molina, M. Muir, M. K. Abdel-Rahman and M. Trenary, J. Chem. Phys., 2021, 154, 184701 CrossRef CAS PubMed.
- L. Y. Zhang, Y. X. Ding, K. H. Wu, Y. M. Niu, J. J. Luo, X. K. Yang, B. S. Zhang and D. S. Su, Nanoscale, 2017, 9, 14317–14321 RSC.
- Y. N. Liu, F. Z. Fu, A. McCue, W. Jones, D. M. Rao, J. T. Feng, Y. F. He and D. Q. Li, ACS Catal., 2020, 10, 15048–15059 CrossRef CAS.
- S. H. Zhou, C. Y. Lu, W. Y. Zhou, Y. Bi, C. L. Zhou, A. N. Zeng, A. J. Wang, L. X. Tan and L. C. Dong, Chem. Commun., 2022, 58, 11398–11401 RSC.
- C. Y. Lu, S. H. Zhou, W. Y. Zhou, C. L. Zhou, Q. Li, A. A. Zeng, A. J. Wang, L. X. Tan and L. C. Dong, Chem. Eng. J., 2023, 464, 142609 CrossRef CAS.
- L. N. Yang, Y. S. Guo, J. Long, L. X. Xia, D. Li, J. P. Xiao and H. Y. Liu, Chem. Commun., 2019, 55, 14693–14696 RSC.
- Q. Fu, F. Wu, B. X. Wang, Y. X. Bu and C. Draxl, ACS Appl. Mater. Interfaces, 2020, 12, 39352–39361 CrossRef CAS PubMed.
- F. Liu, Y. J. Xia, W. L. Xu, L. N. Cao, Q. Q. Guan, Q. Q. Gu, B. Yang and J. L. Lu, Angew. Chem., Int. Ed., 2021, 60, 19324–19330 CrossRef CAS PubMed.
- L. B. Ding, H. Yi, W. H. Zhang, R. You, T. Cao, J. L. Yang, J. L. Lu and W. X. Huang, ACS Catal., 2016, 6, 3700–3707 CrossRef CAS.
- S. H. Zou, B. H. Lou, K. R. Yang, W. T. Yuan, C. Z. Zhu, Y. H. Zhu, Y. H. Du, L. F. Lu, J. J. Liu, W. X. Huang, B. Yang, Z. M. Gong, Y. Cui, Y. Wang, L. Ma, J. Y. Ma, Z. Jiang, L. P. Xiao and J. Fan, Nat. Commun., 2021, 12, 5770 CrossRef CAS PubMed.
- A. Sarkany, React. Kinet. Catal. Lett., 2009, 96, 43–54 CrossRef CAS.
- H. J. Chen, Z. M. Li, Z. X. Qin, H. J. Kim, H. Abroshan and G. Li, ACS Appl. Nano Mater., 2019, 2, 2999–3006 CrossRef CAS.
- T. Fu, T. Wang, H. F. Sun, Y. D. Xu, Z. Dong, X. K. Guo, L. M. Peng, Y. Zhu, Z. X. Chen and W. P. Ding, Sci. China: Chem., 2018, 61, 1014–1019 CrossRef CAS.
- Z. J. Yuan, A. Kumar, D. J. Zhou, J. T. Feng, B. Liu and X. M. Sun, J. Catal., 2022, 414, 374–384 CrossRef CAS.
- J. Liu, A. Zeng, B. Xu, Y. Wang, Z. Sun, Y. Liu, W. Wang and A. Wang, Catal. Lett., 2024, 154, 5171–5183 CrossRef CAS.
- J. C. Rodríguez, A. J. Marchi, A. Borgna and A. Monzón, J. Catal., 1997, 171, 268–278 CrossRef.
- A. Aitugan, S. Tanirbergenova, Y. Tileuberdi, O. Yucel, D. Tugelbayeva, Z. Mansurov and Y. Ongarbayev, React. Kinet., Mech. Catal., 2021, 133, 277–292 CrossRef CAS.
- S. Hock, C. V. Reichel, A. M. Zieschang, B. Albert and M. Rose, ACS Sustainable Chem. Eng., 2021, 9, 16570–16576 CrossRef CAS.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sorensen, C. H. Christensen and J. K. Norskov, Science, 2008, 320, 1320–1322 CrossRef CAS PubMed.
- Y. A. Liu, B. A. Fu, F. Z. Fu, Y. F. He and D. Q. Li, Chem. Eng. Sci., 2022, 260, 117852 CrossRef CAS.
- Y. Y. Shu, L. E. Murillo, J. P. Bosco, W. Huang, A. I. Frenkel and J. G. Chen, Appl. Catal., A, 2008, 339, 169–179 CrossRef CAS.
- E. Kockrick, F. Schmidt, K. Gedrich, M. Rose, T. A. George, T. Freudenberg, R. Kraehnert, R. Skomski, D. J. Sellmyer and S. Kaskel, Chem. Mater., 2010, 22, 1624–1632 CrossRef CAS.
- L. C. Jones, Z. Buras and M. J. Gordon, J. Phys. Chem. C, 2012, 116, 12982–12988 CrossRef CAS.
- L. C. Jones and M. J. Gordon, J. Phys. Chem. C, 2012, 116, 23472–23476 CrossRef CAS.
- C. S. Spanjers, J. T. Held, M. J. Jones, D. D. Stanley, R. S. Sim, M. J. Janik and R. M. Rioux, J. Catal., 2014, 316, 164–173 CrossRef CAS.
- G. L. Zhou, P. G. Wang, Z. X. Jiang, P. L. Ying and C. Li, Chin. J. Catal., 2011, 32, 27–30 CrossRef CAS.
- Y. X. Zhao, Ö. Bozkurt, S. F. Kurtoglu-Öztulum, M. S. Yordanli, A. S. Hoffman, J. Y. Hong, J. E. Perez-Aguilar, A. Saltuk, D. Akgül, O. Demircan, T. A. Atesin, V. Aviyente, B. C. Gates, S. R. Bare and A. Uzun, J. Catal., 2024, 429, 115196 CrossRef CAS.
- O. B. Ayodele, S. Vinati, E. Barborini, L. Boddapati, K. El Hajraoui, J. Kröhnert, F. L. Deepak, A. Trunschke and Y. V. Kolen'ko, Catal. Sci. Technol., 2020, 10, 7471–7475 RSC.
- J. Cheng, P. Hu, P. Ellis, S. French, G. Kelly and C. M. Lok, J. Phys. Chem. C, 2008, 112, 1308–1311 CrossRef CAS.
- C. T. Kuo, Y. B. Lu, L. Kovarik, M. Engelhard and A. M. Karim, ACS Catal., 2019, 9, 11030–11041 CrossRef CAS.
- F. Zhang, Q. J. Zhang, Y. C. Chen, L. Y. Xu, Z. L. Li, Q. T. Wang, Q. F. Zhang and X. N. Li, Appl. Catal., A, 2023, 667, 119447 CrossRef CAS.
- J. Carrasco, G. Vilé, D. Fernández-Torre, R. Pérez, J. Pérez-Ramírez and M. V. Ganduglia-Pirovano, J. Phys. Chem. C, 2014, 118, 5352–5360 CrossRef CAS.
- A. Primo, F. Neatu, M. Florea, V. Parvulescu and H. Garcia, Nat. Commun., 2014, 5, 5291 CrossRef CAS PubMed.
- D. Albani, M. Capdevila-Cortada, G. Vilé, S. Mitchell, O. Martin, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2017, 56, 10755–10760 CrossRef CAS PubMed.
- C. Riley, A. De La Riva, S. L. Zhou, Q. Wan, E. Peterson, K. Artyushkova, M. D. Farahani, H. B. Friedrich, L. Burkemper, N. V. Atudorei, S. Lin, H. Guo and A. Datye, ChemCatChem, 2019, 11, 1526–1533 CrossRef CAS.
- Y. B. Wang, J. Yang, R. T. Gu, L. M. Peng, X. F. Guo, N. H. Xue, Y. Zhu and W. P. Ding, ACS Catal., 2018, 8, 6419–6425 CrossRef CAS.
- T. Cao, R. You, Z. R. Li, X. Y. Zhang, D. Li, S. L. Chen, Z. H. Zhang and W. X. Huang, Appl. Surf. Sci., 2020, 501, 144120 CrossRef CAS.
- G. L. Gou, C. X. Che, H. Wen, J. H. Qin, X. Cao, W. Han, F. Zhang, Y. Long and J. T. Ma, J. Catal., 2022, 408, 128–132 CrossRef CAS.
- Z. R. Li, L. Chen, Z. F. Wu, A. P. Jia, S. C. Shi, H. Zhang, J. Wang, Z. Liu, W. P. Shao, F. Yang, X. P. Wu, X. Q. Gong and W. X. Huang, ACS Catal., 2023, 13, 5213–5224 CrossRef CAS.
- I. B. Bychko, A. A. Abakumov, N. V. Lemesh and P. E. Strizhak, ChemCatChem, 2017, 9, 4470–4474 CrossRef CAS.
- A. A. Abakumov, I. B. Bychko, O. V. Selyshchev, D. R. T. Zahn, X. H. Qi, J. G. Tang and P. E. Strizhak, Carbon, 2020, 157, 277–285 CrossRef CAS.
- A. A. Abakumov, I. B. Bychko, O. V. Selyshchev, D. R. T. Zahn, M. Chen, J. Tang and P. E. Strizhak, Materialia, 2021, 18, 277–285 CrossRef.
- C. Riley, S. L. Zhou, D. Kunwar, A. De La Riva, E. Peterson, R. Payne, L. Y. Gao, S. Lin, H. Guo and A. Datye, J. Am. Chem. Soc., 2018, 140, 12964–12973 CrossRef CAS PubMed.
- M. Tejeda-Serrano, M. Mon, B. Ross, F. Gonell, J. Ferrando-Soria, A. Corma, A. Leyva-Pérez, D. Armentano and E. Pardo, J. Am. Chem. Soc., 2018, 140, 8827–8832 CrossRef CAS PubMed.
- J. N. Li, M. Pu, C. C. Ma, Y. Tian, J. He and D. G. Evans, J. Mol. Catal. A: Chem., 2012, 359, 14–20 CrossRef CAS.
- T. Abdollahi and D. Farmanzadeh, Appl. Surf. Sci., 2018, 433, 513–529 CrossRef CAS.
- B. Yang, R. Burch, C. Hardacre, G. Headdock and P. Hu, J. Catal., 2013, 305, 264–276 CrossRef CAS.
- M. Crespo-Quesada, S. Yoon, M. S. Jin, A. Prestianni, R. Cortese, F. Cárdenas-Lizana, D. Duca, A. Weidenkaff and L. Kiwi-Minsker, J. Phys. Chem. C, 2015, 119, 1101–1107 CrossRef CAS.
- B. Yang, R. Burch, C. Hardacre, P. Hu and P. Hughes, Surf. Sci., 2016, 646, 45–49 CrossRef CAS.
- P. Tiruppathi, J. J. Low, A. S. Y. Chan, S. R. Bare and R. J. Meyer, Catal. Today, 2011, 165, 106–111 CrossRef CAS.
- D. Torres, F. Cinquini and P. Sautet, J. Phys. Chem. C, 2013, 117, 11059–11065 CrossRef CAS.
- B. Yang, R. Burch, C. Hardacre, P. Hu and P. Hughes, J. Phys. Chem. C, 2014, 118, 3664–3671 CrossRef CAS.
- P. Wang and B. Yang, Chin. J. Catal., 2018, 39, 1493–1499 CrossRef CAS.
- J. F. Wang, W. S. Hao, L. J. Ma, J. F. Jia and H. S. Wu, Comput. Theor. Chem., 2019, 1170, 112636 CrossRef CAS.
- Y. Y. Wu, W. T. Zhao, Y. Wang, B. J. Wang, M. H. Fan and R. G. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 56743–56757 CrossRef CAS PubMed.
- W. B. Xie, J. Y. Xu, Y. X. Ding and P. Hu, ACS Catal., 2021, 11, 4094–4106 CrossRef CAS.
- W. B. Xie and P. Hu, Catal. Sci. Technol., 2021, 11, 5212–5222 RSC.
- D. H. Mei, M. Neurock and C. M. Smith, J. Catal., 2009, 268, 181–195 CrossRef CAS.
- J. H. Liu, L. D. Meng, C. Q. Lv and G. C. Wang, RSC Adv., 2016, 6, 14593–14601 RSC.
- Q. Li, Y. C. Qin, D. P. Tan, Y. Xie, M. L. Lv and L. J. Song, New J. Chem., 2018, 42, 19827–19836 RSC.
- L. D. Meng and G. C. Wang, Phys. Chem. Chem. Phys., 2014, 16, 17541–17550 RSC.
- X. T. Li, L. Chen, C. Shang and Z. P. Liu, J. Am. Chem. Soc., 2021, 143, 6281–6292 CrossRef CAS PubMed.
- J. Y. Wang, H. X. Xu, C. X. Che, J. Q. Zhu and D. J. Cheng, ACS Catal., 2023, 13, 433–444 CrossRef CAS.
- P. P. Wei, J. Zheng, Q. Li, Y. C. Qin, H. M. Guan, D. P. Tan and L. J. Song, Inorg. Chem. Front., 2022, 9, 5169–5180 RSC.
- E. Vignola, S. N. Steinmann, B. D. Vandegehuchte, D. Curulla and P. Sautet, J. Phys. Chem. C, 2016, 120, 26320–26327 CrossRef CAS.
- Z. J. Wang, J. Y. Chen, Y. G. Huang and Z. X. Chen, Mater. Today Commun., 2021, 28, 102475 CrossRef CAS.
- R. G. Zhang, M. F. Xue, B. J. Wang, L. X. Ling and M. H. Fan, J. Phys. Chem. C, 2019, 123, 16107–16117 CrossRef CAS.
- Y. M. Qi, X. X. Shao, B. J. Wang, D. B. Li, L. X. Ling and R. G. Zhang, Chem. Eng. Sci., 2021, 245, 116786 CrossRef.
- R. G. Zhang, B. Zhao, L. X. Ling, A. J. Wang, C. K. Russell, B. J. Wang and M. H. Fan, ChemCatChem, 2018, 10, 2424–2432 CrossRef CAS.
- C. X. Yang, G. Q. Wang, A. M. Liang, Y. Yue, H. Peng and D. J. Cheng, Catal. Commun., 2019, 124, 41–45 CrossRef CAS.
- Y. Wang, W. J. Zheng, B. J. Wang, L. X. Ling and R. G. Zhang, Chem. Eng. Sci., 2021, 229, 116131 CrossRef CAS.
- W. J. Zheng, L. X. Ma, B. J. Wang, J. G. Wang and R. G. Zhang, Mol. Catal., 2021, 510, 111660 CAS.
- W. J. Zheng, Y. Wang, B. J. Wang, M. H. Fan, L. X. Ling and R. G. Zhang, J. Phys. Chem. C, 2021, 125, 15251–15261 CrossRef CAS.
- Y. G. Huang and Z. X. Chen, Appl. Surf. Sci., 2022, 575, 151513 CrossRef CAS.
- Y. G. Huang, H. L. Lu and Z. X. Chen, Phys. Chem. Chem. Phys., 2022, 24, 3182–3190 RSC.
- A. C. Foucher, H. T. Ngan, T. Shirman, A. Filie, K. Duanmu, M. Aizenberg, R. J. Madix, C. M. Friend, J. Aizenberg, P. Sautet and E. A. Stach, ACS Appl. Nano Mater., 2023, 6, 22927–22938 CrossRef CAS.
- M. Krajcí and J. Hafner, J. Catal., 2012, 295, 70–80 CrossRef.
- J. Prinz, C. A. Pignedoli, Q. S. Stöckl, M. Armbrüster, H. Brune, O. Gröning, R. Widmer and D. Passerone, J. Am. Chem. Soc., 2014, 136, 11792–11798 CrossRef CAS PubMed.
- M. Krajci and J. Hafner, J. Catal., 2014, 312, 232–248 CrossRef CAS.
- M. Krajcí and J. Hafner, J. Phys. Chem. C, 2014, 118, 12285–12301 CrossRef.
- N. Kumar and P. Ghosh, J. Phys. Chem. C, 2016, 120, 28654–28663 CrossRef CAS.
- M. Sandoval, P. Bechthold, V. Orazi, E. A. González, A. Juan and P. V. Jasen, Appl. Surf. Sci., 2018, 435, 568–573 CrossRef CAS.
- M. Krajcí and J. Hafner, J. Phys. Chem. C, 2012, 116, 6307–6319 CrossRef.
- J. B. Zhao, S. J. Zha, R. T. Mu, Z. J. Zhao and J. L. Gong, J. Phys. Chem. C, 2018, 122, 6005–6013 CrossRef CAS.
- Y. Zhao, M. Y. Zhu and L. H. Kang, Catal. Lett., 2018, 148, 2992–3002 CrossRef CAS.
- S. F. Yuk, G. Collinge, M. T. Nguyen, M. S. Lee, V. A. Glezakou and R. Rousseau, J. Chem. Phys., 2020, 152, 154703 CrossRef CAS PubMed.
- S. Zhao, Y. Tang, X. H. Yu and J. Li, Sci. China: Mater., 2023, 66, 3912–3921 CAS.
- H. Thirumalai and J. R. Kitchin, Top. Catal., 2018, 61, 462–474 CrossRef CAS.
- Y. M. Qi, B. J. Wang, M. H. Fan, D. B. Li and R. G. Zhang, Chem. Eng. Sci., 2021, 243, 116786 CrossRef CAS.
- D. Liu, Appl. Surf. Sci., 2016, 386, 125–137 CrossRef CAS.
- D. L. Molina, M. Muir, M. K. Abdel-Rahman and M. Trenary, J. Chem. Phys., 2021, 154, 184701 CrossRef CAS PubMed.
- L. L. Ma, C. Q. Lv and G. C. Wang, Appl. Surf. Sci., 2017, 410, 154–165 CrossRef CAS.
- R. G. Zhang, J. Zhang, B. Zhao, L. L. He, A. J. Wang and B. J. Wang, J. Phys. Chem. C, 2017, 121, 27936–27949 CrossRef CAS.
- R. G. Zhang, M. F. Xue, B. J. Wang and L. X. Ling, Appl. Surf. Sci., 2019, 481, 421–432 CrossRef CAS.
- M. Jorgensen and H. Grönbeck, J. Am. Chem. Soc., 2019, 141, 8541–8549 CrossRef PubMed.
- B. Zhao, R. G. Zhang, Z. X. Huang and B. J. Wang, Appl. Catal., A, 2017, 546, 111–121 CrossRef CAS.
- T. Abdollahi and D. Farmanzadeh, J. Alloys Compd., 2018, 735, 117–130 CrossRef CAS.
- Y. Wang, Y. Wu, X. Guo, B. Wang, M. Fan and R. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 41896–41911 CrossRef CAS PubMed.
- Y. Wang, B. J. Wang, M. H. Fan, L. X. Ling and R. G. Zhang, Chem. Eng. Sci., 2022, 251, 117494 CrossRef CAS.
- T. Abdollahi and D. Farmanzadeh, C. R. Chim, 2018, 21, 484–493 CrossRef CAS.
- P. Yin, Y. Jie, X. J. Zhao, Y. L. Feng, T. Sun, D. M. Rao, M. Pu and H. Yan, Phys. Chem. Chem. Phys., 2021, 23, 27340–27347 RSC.
- Z. S. Wang, A. Garg, L. X. Wang, H. R. He, A. Dasgupta, D. Zanchet, M. J. Janik, R. M. Rioux and Y. Román-Leshkov, ACS Catal., 2020, 10, 6763–6770 CrossRef CAS.
- C. Jimenez-Orozco, A. A. Koverga, E. Florez and J. A. Rodriguez, Surf. Sci., 2023, 728, 122197 CrossRef CAS.
- K. R. Yang and B. Yang, Faraday Discuss., 2021, 229, 50–61 RSC.
- Z. F. Wu, F. Wang, G. H. Sun, F. Xiong, B. T. Teng and W. X. Huang, J. Phys. Chem. Lett., 2022, 13, 7667–7672 CrossRef CAS PubMed.
- L. Wang, B. J. Wang, M. H. Fan, L. X. Ling and R. G. Zhang, Fuel, 2023, 336, 127131 CrossRef CAS.
- J. Y. Li, Z. H. Yao, J. Y. Zhao, S. W. Deng, S. B. Wang and J. G. Wang, Mol. Catal., 2023, 535, 112845 CAS.
- K. H. Sun, R. Zou, C. Y. Shen and C. J. Liu, Catal. Sci. Technol., 2024, 14, 3041–3049 RSC.
- R. G. Zhang, J. Zhang, Z. Jiang, B. J. Wang and M. H. Fan, Chem. Eng. J., 2018, 351, 732–746 CrossRef CAS.
- R. G. Zhang, B. Zhao, L. L. He, A. J. Wang and B. J. Wang, Phys. Chem. Chem. Phys., 2018, 20, 17487–17496 RSC.
- B. Yang, R. Burch, C. Hardacre, G. Headdock and P. Hu, ACS Catal., 2012, 2, 1027–1032 CrossRef CAS.
- L. Wang, B. J. Wang, M. H. Fan, L. X. Ling and R. G. Zhang, Fuel, 2022, 321, 124118 CrossRef CAS.
- D. M. Rao, S. T. Zhang, C. M. Li, Y. D. Chen, M. Pu, H. Yan and M. Wei, Dalton Trans., 2018, 47, 4198–4208 RSC.
- Y. J. Song and S. Laursen, J. Catal., 2019, 372, 151–162 CrossRef CAS.
- D. M. Rao, T. Sun, Y. S. Yang, P. Yin, M. Pu, H. Yan and M. Wei, Phys. Chem. Chem. Phys., 2019, 21, 1384–1392 RSC.
- Z. Almisbaa, H. A. Aljama, K. Almajnouni, L. Cavallo and P. Sautet, ACS Catal., 2023, 13, 7358–7370 CrossRef CAS.
- M. Krajcí and J. Hafner, Philos. Mag., 2011, 91, 2904–2912 CrossRef.
- M. Krajcí and J. Hafner, J. Catal., 2011, 278, 200–207 CrossRef.
- D. Kandaskalov, V. Fournée, J. Ledieu and E. Gaudry, J. Phys. Chem. C, 2017, 121, 18738–18745 CrossRef CAS.
- M. Meier, J. Ledieu, V. Fournée and É. Gaudry, J. Phys. Chem. C, 2017, 121, 4958–4969 CrossRef CAS.
- L. Wang, B. J. Wang, M. H. Fan, L. X. Ling and R. G. Zhang, J. Catal., 2022, 416, 112–128 CrossRef CAS.
- Y. Wang and L. H. Kang, Catalysts, 2020, 10, 115 CrossRef.
- Y. Wang and L. H. Kang, Chem. Phys. Lett., 2020, 757, 137871 CrossRef CAS.
- H. Y. Zhuo, X. H. Yu, Q. Yu, H. Xiao, X. Zhang and J. Li, Sci. China: Mater., 2020, 63, 1741–1749 CAS.
- M. Z. Jian, J. X. Liu and W. X. Li, Chem. Sci., 2021, 12, 10290–10298 RSC.
- H. Y. Ma and G. C. Wang, ACS Catal., 2020, 10, 4922–4928 CrossRef CAS.
- D. L. Gao, D. Yi, F. Lu, S. Li, L. Pan, Y. Xu and X. Wang, Chem. Eng. Sci., 2021, 240, 116664 CrossRef CAS.
- X. X. Shao, B. J. Wang, M. H. Fan, L. X. Ling and R. G. Zhang, Appl. Surf. Sci., 2023, 611, 155720 CrossRef CAS.
- Z. Q. Huang, L. P. Liu, S. T. Qi, S. Zhang, Y. Q. Qu and C. R. Chang, ACS Catal., 2018, 8, 546–554 CrossRef CAS.
- M. C. Padole, B. P. Gangwar, A. Pandey, A. Singhal, S. Sharma and P. A. Deshpande, Phys. Chem. Chem. Phys., 2017, 19, 14148–14159 RSC.
- S. L. Zhou, L. Y. Gao, F. F. Wei, S. Lin and H. Guo, J. Catal., 2019, 375, 410–418 CrossRef CAS.
- S. L. Zhou, Q. Wan, S. Lin and H. Guo, Phys. Chem. Chem. Phys., 2022, 24, 11295–11304 RSC.
- X. Y. Guo, H. J. Han, B. J. Wang, L. X. Ling, M. H. Fan and R. G. Zhang, Chem. Eng. Sci., 2024, 296, 120263 CrossRef CAS.
- Q. Wan, Y. Chen, S. L. Zhou, J. Lin and S. Lin, J. Mater. Chem. A, 2021, 9, 14064–14073 RSC.
- K. W. Romero, F. Polo-Garzon, Z. L. Wu, A. Savara and D. E. Jiang, ACS Catal., 2023, 13, 9213–9221 CrossRef.
- Q. Wan, J. Li, R. Jiang and S. Lin, Phys. Chem. Chem. Phys., 2021, 23, 24349–24356 RSC.
- Á. Molnár, A. Sárkány and M. Varga, J. Mol. Catal. A: Chem., 2001, 173, 185–221 CrossRef.
- Q. Li, L. J. Song, L. H. Pan, X. L. Zhuang, M. L. Ling and L. H. Duan, Phys. Chem. Chem. Phys., 2013, 15, 20345–20353 RSC.
- X. J. Xie, X. L. Song, W. Y. Dong, Z. H. Liang, C. M. Fan and P. D. Han, Chin. J. Chem., 2014, 32, 631–636 CrossRef CAS.
- I. Horiuti and M. Polanyi, Trans. Faraday Soc., 1934, 30, 1164–1172 RSC.
- Y. Y. Wu, X. Y. Guo, X. F. Shi, B. J. Wang, M. H. Fan and R. G. Zhang, Appl. Surf. Sci., 2023, 637, 157906 CrossRef CAS.
- C. Amatore and A. Jutand, J. Organomet. Chem., 1999, 576, 254–278 CrossRef CAS.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- S. Komhom, O. Mekasuwandumrong, P. Praserthdam and J. Panpranot, Catal. Commun., 2008, 10, 86–91 CrossRef CAS.
- S. Komeili, M. T. Ravanchi and A. Taeb, Appl. Catal., A, 2015, 502, 287–296 CrossRef CAS.
- H. L. Zhang, J. L. Cao, B. J. Wu, W. Dai, Z. H. Chen and M. J. Ma, RSC Adv., 2016, 6, S7174–S7182 Search PubMed.
- S. Wang, G. F. Zhao, Y. Liu and Y. Lu, Ind. Eng. Chem. Res., 2019, 58, 16431–16441 CrossRef CAS.
- L. Y. Xu, S. Y. Hua, J. Zhou, Y. Q. Xu, C. S. Lu, F. Feng, J. Zhao, X. L. Xu, Q. T. Wang, Q. F. Zhang and X. N. Li, Appl. Catal., A, 2022, 642, 118690 CrossRef CAS.
- Z. R. Yang, Y. Li, Y. Q. Cao, X. Q. Zhao, W. Y. Chen, J. Zhang, G. Qian, C. Peng, X. Q. Gong and X. Z. Duan, Chem. Eng. J., 2022, 445, 136681 CrossRef CAS.
- T. Sangkhum, O. Mekasuwandumrong, P. Praserthdam and J. Panpranot, React. Kinet. Catal. Lett., 2009, 97, 115–123 CrossRef CAS.
- L. B. Sun, L. Jiang, X. Y. Hua, Y. H. Zheng, X. Sun, M. Zhang, H. J. Su and C. X. Qi, J. Alloys Compd., 2019, 811, 152052 CrossRef CAS.
- H. Chen, B. Yang, Y. F. Zhang, C. X. Che, F. Zhang, W. Han, H. Wen, A. Q. Wang and T. Zhang, ChemCatChem, 2024, 16, e202400566 CrossRef CAS.
- J. T. Feng, X. Y. Ma, D. G. Evans and D. Q. Li, Ind. Eng. Chem. Res., 2011, 50, 1947–1954 CrossRef CAS.
- Y. F. He, J. X. Fan, J. T. Feng, C. Y. Luo, P. F. Yang and D. Q. Li, J. Catal., 2015, 331, 118–127 CrossRef CAS.
- X. Sun, F. F. Li, J. J. Shi, Y. H. Zheng, H. J. Su, L. B. Sun, S. Peng and C. X. Qi, Appl. Surf. Sci., 2019, 487, 625–633 CrossRef CAS.
- H. Dai, X. Xiao, L. H. Huang, C. J. Zhou and J. Deng, Appl. Clay Sci., 2021, 211, 106173 CrossRef CAS.
- Z. J. Yuan, L. Liu, W. Ru, D. J. Zhou, Y. Kuang, J. T. Feng, B. Liu and X. M. Sun, Nano Res., 2022, 15, 6010–6018 CrossRef CAS.
- Z. W. Li, G. Lin, Y. X. Chen, Q. Q. Xue, K. Feng and B. H. Yan, Catal. Today, 2023, 423, 114253 CrossRef CAS.
- F. Z. Fu, Y. A. Liu, Y. W. Li, B. A. Fu, L. R. Zheng, J. T. Feng and D. Q. Li, ACS Catal., 2021, 11, 11117–11128 CrossRef CAS.
- E. V. Ilyina, D. V. Yurpalova, D. A. Shlyapin, G. B. Veselov, D. M. Shivtsov, V. O. Stoyanovskii, A. V. Bukhtiyarov and A. A. Vedyagin, Mol. Catal., 2024, 560, 114151 CAS.
- E. Kim, E. W. Shin, C. W. Bark, I. Chang, W. J. Yoon and W. J. Kim, Appl. Catal., A, 2014, 471, 80–83 CrossRef CAS.
- J. E. Um, W. J. Yoon, H. W. Choi and W. J. Kim, J. Ind. Eng. Chem., 2014, 20, 4183–4187 CrossRef CAS.
- Y. N. Liu, J. T. Feng, Y. F. He, J. H. Sun and D. Q. Li, Catal. Sci. Technol., 2015, 5, 1231–1240 RSC.
- Y. N. Liu, J. Y. Zhao, Y. F. He, J. T. Feng, T. Wu and D. Q. Li, J. Catal., 2017, 348, 135–145 CrossRef CAS.
- T. Gong, Y. Huang, L. J. Qin, W. L. Zhang, J. G. Li, L. F. Hui and H. Feng, Appl. Surf. Sci., 2019, 495, 143495 CrossRef CAS.
- K. J. Wang, Y. Y. Chen, X. S. Li and H. X. Ding, Catal. Lett., 2009, 127, 392–399 CrossRef CAS.
- W. Huang, A. Li, R. F. Lobo and J. G. Chen, Catal. Lett., 2009, 130, 380–385 CrossRef CAS.
- Y. K. Gulyaeva, V. V. Kaichev, V. I. Zaikovskii, A. P. Suknev and B. S. Bal'zhinimaev, Appl. Catal., A, 2015, 506, 197–205 CrossRef CAS.
- D. Melnikov, M. Reshetina, A. Novikov, K. Cherednichenko, A. Stavitskaya, V. Stytsenko, V. Vinokurov, W. Huang and A. Glotov, Appl. Clay Sci., 2023, 232, 106763 CrossRef CAS.
- S. Peng, X. Sun, L. B. Sun, M. Zhang, Y. H. Zheng, H. J. Su and C. X. Qi, Catal. Lett., 2019, 149, 465–472 CrossRef CAS.
- Y. N. Liu, A. J. McCue, P. F. Yang, Y. F. He, L. R. Zheng, X. Z. Cao, Y. Man, J. T. Feng, J. A. Anderson and D. Q. Li, Chem. Sci., 2019, 10, 3556–3566 RSC.
- Z. W. Li, J. J. Zhang, J. M. Tian, K. Feng, Y. X. Chen, X. Li, Z. H. Zhang, S. R. Qian, B. Yang, D. Su, K. H. Luo and B. H. Yan, ACS Catal., 2024, 14, 1514–1524 CrossRef CAS.
- Z. H. Tao, L. Y. Pan, H. D. Yao, L. Chao, L. W. Cui, C. P. Du, Y. X. Wang, Y. S. Xi, D. Li and W. H. Li, Surf. Interfaces, 2024, 52, 104923 CrossRef CAS.
- A. D. Benavidez, P. D. Burton, J. L. Nogales, A. R. Jenkins, S. A. Ivanov, J. T. Miller, A. M. Karim and A. K. Datye, Appl. Catal., A, 2014, 482, 108–115 CrossRef CAS.
- H. M. Lu, B. L. Xu, X. Z. Wang, Z. Hu and Y. N. Fan, Catal. Lett., 2014, 144, 2198–2203 CrossRef CAS.
- Y. Q. Cao, W. Z. Fu, Z. H. Ren, Z. J. Sui, J. H. Zhou, J. Luo, X. Z. Duan and X. G. Zhou, AIChE J., 2020, 66, e16857 CrossRef CAS.
- Q. T. Wang, J. Y. Zhao, L. Y. Xu, L. Yu, Z. H. Yao, G. J. Lan, L. L. Guo, J. Zhao, C. S. Lu, Z. Y. Pan, J. G. Wang, Q. F. Zhang and X. N. A. Li, Appl. Surf. Sci., 2021, 562, 150141 CrossRef CAS.
- A. Khannanov, I. Il'yasov, A. Kiiamov, I. Vakhitov, A. Kirgizov, A. Lamberov and A. M. Dimiev, New J. Chem., 2019, 43, 19035–19043 RSC.
- S. Hwang, Y. B. Hwang, H. W. Hwang, D. Kim, J. Y. Lee, J. Hwang, E. D. Park and S. K. Kim, Adv. Energy Sustainability Res., 2025 DOI:10.1002/aesr.202400341.
- M. Z. Hu, S. Zhao, S. J. Liu, C. Chen, W. X. Chen, W. Zhu, C. Liang, W. C. Cheong, Y. Wang, Y. Yu, Q. Peng, K. B. Zhou, J. Li and Y. D. Li, Adv. Mater., 2018, 30, e201801878 Search PubMed.
- S. Wang, Z. J. Zhao, X. Chang, J. B. Zhao, H. Tian, C. S. Yang, M. R. Li, Q. Fu, R. T. Mu and J. L. Gong, Angew. Chem., Int. Ed., 2019, 58, 7668–7672 CrossRef CAS PubMed.
- Z. L. Guo, Y. F. Liu, Y. Liu and W. Chu, Appl. Surf. Sci., 2018, 442, 736–741 CrossRef CAS.
- R. R. Li, Y. X. Yue, Y. K. Li, X. L. Chen, R. Q. Chang, J. X. Zhang, B. Zhao, X. Ying, Z. J. Wang, J. Zhao and X. N. Li, Inorg. Chem. Front., 2022, 9, 5969–5981 RSC.
- Q. L. Wu, C. Y. Shen and C. J. Liu, Mol. Catal., 2024, 569, 114529 CAS.
- M. T. Ravanchi, S. Fadaeerayeni and M. R. Fard, Res. Chem. Intermed., 2016, 42, 4797–4811 CrossRef.
- M. T. Ravanchi, S. Sahebdelfar and M. R. Fard, Int. J. Chem. React. Eng., 2016, 14, 1035–1046 CrossRef.
- K. Ravindran, G. Madhu, V. R. Renjith and S. Rugmini, React. Kinet., Mech. Catal., 2021, 134, 867–882 CrossRef CAS.
- K. Ravindran, G. Madhu, V. R. Renjith and S. Rugmini, J. Indian Chem. Soc., 2023, 100, 867–882 CrossRef.
- S. Asplund, J. Catal., 1996, 158, 267–278 CrossRef CAS.
- C. X. Che, B. K. Wang, C. F. Shan, H. Chen, W. S. Liu and Y. Tang, Catal. Lett., 2017, 147, 483–490 CrossRef CAS.
- X. Y. Ma, Y. Y. Chai, D. G. Evans, D. Q. Li and J. T. Feng, J. Phys. Chem. C, 2011, 115, 8693–8701 CrossRef CAS.
- S. Chinayon, O. Mekasuwandumrong, P. Praserthdam and J. Panpranot, Catal. Commun., 2008, 9, 2297–2302 CrossRef CAS.
- X. P. Gao, Y. A. Zhou, F. L. Jing, J. J. Luo, Q. S. Huang and W. Chu, Chin. J. Chem., 2017, 35, 1009–1015 CrossRef CAS.
- S. Komeili, M. T. Ravanchi and A. Taeb, Res. Chem. Intermed., 2018, 44, 1335–1349 CrossRef CAS.
- C. L. Miao, L. Y. Cai, Y. F. Wang, X. J. Xu, J. R. Yang, Y. F. He, D. Q. Li and J. T. Feng, Ind. Eng. Chem. Res., 2021, 60, 8362–8374 CrossRef CAS.
- D. R. Aireddy, H. R. Yu, D. A. Cullen and K. L. Ding, ACS Appl. Mater. Interfaces, 2022, 14, 24290–24298 CrossRef CAS PubMed.
- S. Riyapan, Y. Boonyongmaneerat, O. Mekasuwandumrong, P. Praserthdam and J. Panpranot, Catal. Today, 2015, 245, 134–138 CrossRef CAS.
- X. P. Gao, Z. L. Guo, Y. N. Zhou, F. L. Jing and W. Chu, Acta Phys.-Chim. Sin., 2017, 33, 602 CAS.
- Y. P. Yang, J. R. Yang, S. C. Shi, Y. H. Luo, X. J. Xu, Y. A. Liu, D. Q. Li and J. T. Feng, AIChE J., 2023, 69, e18078 CrossRef CAS.
- Z. L. Guo, Q. S. Huang, S. Z. Luo and W. Chu, Top. Catal., 2017, 60, 1009–1015 CrossRef CAS.
- W. J. Kim, I. Y. Ahn, J. H. Lee and S. H. Moon, Catal. Commun., 2012, 24, 52–55 CrossRef CAS.
- W. J. Kim and S. H. Moon, Catal. Today, 2012, 185, 2–16 CrossRef CAS.
- W. F. Simanullang, J. M. Ma, K. Shimizu and S. Furukawa, Catal. Sci. Technol., 2021, 11, 4016–4020 RSC.
- Z. W. Liu, Q. F. Zhu, N. Hou, Y. Fu, L. X. Wen and J. F. Chen, Catal. Today, 2013, 216, 205–210 CrossRef CAS.
- Y. K. Gulyaeva, V. V. Kaichev, V. I. Zaikovskii, E. V. Kovalyov, A. P. Suknev and B. S. Bal'zhinimaev, Catal. Today, 2015, 245, 139–146 CrossRef CAS.
- L. H. Kang, B. Z. Cheng and M. Y. Zhu, R. Soc. Open Sci., 2019, 6, 191155 CrossRef PubMed.
- M. Q. Chai, Y. Tan, G. X. Pei, L. Li, L. L. Zhang, X. Y. Liu, A. Q. Wang and T. Zhang, J. Phys. Chem. C, 2017, 121, 19727–19734 CrossRef CAS.
- W. G. Augustyn, R. I. McCrindle and N. J. Coville, Appl. Catal., A, 2010, 388, 1–6 CrossRef CAS.
- H. Bazzazzadegan, M. Kazemeini and A. M. Rashidi, Appl. Catal., A, 2011, 399, 184–190 CrossRef CAS.
- V. V. Chesnokov, O. Y. Podyacheva and R. M. Richards, Mater. Res. Bull., 2017, 88, 78–84 CrossRef CAS.
- X. L. Chen, Q. Q. Xu, B. Zhao, S. B. Ren, Z. H. Wu, J. P. Wu, Y. X. Yue, D. M. Han and R. R. Li, Catal. Lett., 2021, 151, 3372–3380 CrossRef CAS.
- F. Dodangeh, A. Rashidi, H. Aghaie and K. Zare, J. Phys. Chem. Solids, 2021, 158, 110219 CrossRef CAS.
- H. R. Zhou, B. X. Li, H. G. Fu, X. H. Zhao, M. M. Zhang, X. B. Wang, Y. Liu, Z. X. Yang and X. D. Lou, ACS Sustainable Chem. Eng., 2022, 10, 4849–4861 CrossRef CAS.
- Q. S. Luo, H. Wang, L. Wang and F. S. Xiao, ACS Mater. Au, 2022, 2, 313–320 CrossRef CAS PubMed.
- J. Yang, F. J. Zhang, H. Y. Lu, X. Hong, H. L. Jiang, Y. Wu and Y. D. Li, Angew. Chem., Int. Ed., 2015, 54, 10889–10893 CrossRef CAS PubMed.
- L. R. Redfern, Z. Y. Li, X. Zhang and O. K. Farha, ACS Appl. Nano Mater., 2018, 1, 4413–4417 CrossRef CAS.
- M. R. Mian, L. R. Redfern, S. M. Pratik, D. Ray, J. Liu, K. B. Idrees, T. Islamoglu, L. Gagliardi and O. K. Farha, Chem. Mater., 2020, 32, 3078–3086 CrossRef CAS.
- J. J. Wang, H. T. Xu, C. C. Ao, X. B. Pan, X. K. Luo, S. J. Wei, Z. Li, L. D. Zhang, Z. L. Xu and Y. D. Li, iScience, 2020, 23, 101233 CrossRef CAS PubMed.
- J. Q. Zhang, G. Y. Zhang, L. He, Y. Z. Shi, R. R. Miao, Y. Z. Zhu and Q. Q. Guan, Appl. Surf. Sci., 2021, 570, 150881 CrossRef CAS.
- N. López, B. Bridier and J. Pérez-Ramírez, J. Phys. Chem. C, 2008, 112, 9346–9350 CrossRef.
- X. X. Shao, X. Y. Guo, X. F. Shi, B. J. Wang, M. H. Fan and R. G. Zhang, Fuel, 2024, 358, 130053 CrossRef CAS.
- D. L. Trimm, I. O. Y. Liu and N. W. Cant, J. Mol. Catal. A: Chem., 2009, 307, 13–20 CrossRef CAS.
- D. L. Trimm, I. O. Y. Liu and N. W. Cant, Appl. Catal., A, 2010, 374, 58–64 CrossRef CAS.
- A. J. McCue, A. Guerrero-Ruiz, I. Rodríguez-Ramos and J. A. Anderson, J. Catal., 2016, 340, 10–16 CrossRef CAS.
- D. Albani, M. Shahrokhi, Z. P. Chen, S. Mitchell, R. Hauert, N. López and J. Pérez-Ramírez, Nat. Commun., 2018, 9, 2634 CrossRef PubMed.
- Y. N. Liu, A. J. McCue, J. T. Feng, S. L. Guan, D. Q. Li and J. A. Anderson, J. Catal., 2018, 364, 204–215 CrossRef CAS.
- L. Wang, B. Zhao, C. K. Russell, M. H. Fan, B. J. Wang, L. X. Ling and R. G. Zhang, Chem. Eng. Sci., 2021, 246, 116984 CrossRef CAS.
- Y. Wang, Y. Zhang, B. J. Wang, M. H. Fan, L. X. Ling and R. G. Zhang, Fuel, 2022, 315, 123180 CrossRef CAS.
- X. X. Shao, B. J. Wang, M. H. Fan, L. X. Ling and R. G. Zhang, Chem. Eng. Sci., 2023, 270, 118494 CrossRef CAS.
- Y. Y. Wu, B. J. Wang, M. H. Fan, L. X. Ling and R. G. Zhang, Fuel, 2023, 331, 125661 CrossRef CAS.
- M. Ruta, G. Laurenczy, P. J. Dyson and L. Kiwi-Minsker, J. Phys. Chem. C, 2008, 112, 17814–17819 CrossRef CAS.
- T. Herrmann, L. Rössmann, M. Lucas and P. Claus, Chem. Commun., 2011, 47, 12310–12312 RSC.
- T. Zhou, K. Jang and B. W. L. Jang, Catal. Today, 2013, 211, 147–155 CrossRef CAS.
- Y. C. Chen, Q. J. Zhang, F. Zhang, Z. L. Li, Y. K. Zhou, Y. X. Qin, L. Y. Xu, F. Feng, Q. T. Wang, Q. F. Zhang and X. N. Li, Appl. Catal., A, 2024, 681, 11977 CrossRef.
- Q. F. Zhang, Y. Q. Xu, Q. T. Wang, W. M. Huang, J. Zhou, Y. S. Jiang, H. Xu, L. L. Guo, P. Z. Zhang, J. Zhao, F. Feng and X. N. Li, Chem. Commun., 2019, 55, 14910–14913 RSC.
- Q. T. Wang, Y. Q. Xu, J. Zhou, L. Y. Xu, L. Yu, D. H. Jiang, C. S. Lu, Z. Y. Pan, Q. F. Zhang and X. N. Li, J. Ind. Eng. Chem., 2021, 93, 448–460 CrossRef CAS.
- R. J. Hou, X. C. Lan and T. F. Wang, Catal. Today, 2015, 251, 47–52 CrossRef CAS.
- Y. Q. Xu, Y. S. Jiang, H. Xu, Q. T. Wang, W. M. Huang, H. H. He, Y. Y. Zhai, S. X. Di, L. L. Guo, X. L. Xu, J. Zhao, F. Feng, Q. F. Zhang and X. N. Li, Appl. Catal., A, 2018, 567, 12–19 CrossRef CAS.
- Y. Kim, T. Kim, K. H. Kang and I. Ro, ChemCatChem, 2023, 15, e202201428 CrossRef CAS.
- F. M. McKenna and J. A. Anderson, J. Catal., 2011, 281, 231–240 CrossRef CAS.
- F. M. McKenna, R. P. K. Wells and J. A. Anderson, Chem. Commun., 2011, 47, 2351–2353 RSC.
- F. M. McKenna, L. Mantarosie, R. P. K. Wells, C. Hardacre and J. A. Anderson, Catal. Sci. Technol., 2012, 2, 632–638 RSC.
- A. J. McCue, F. M. McKenna and J. A. Anderson, Catal. Sci. Technol., 2015, 5, 2449–2459 RSC.
- L. Altmann, X. D. Wang, J. Stöver, M. Klink, V. Zielasek, K. Thiel, J. Kolny-Olesiak, K. Al-Shamery, H. Borchert, J. Parisi and M. Bäumer, ChemCatChem, 2013, 5, 1803–1810 CrossRef CAS.
- G. Tiwari, G. Sharma, R. Verma, P. Gakhad, A. K. Singh, V. Polshettiwar and B. R. Jagirdar, Chem. – Eur. J., 2023, 29, e202301932 CrossRef CAS PubMed.
- Q. L. Wu, C. Y. Shen and C. J. Liu, Appl. Surf. Sci., 2023, 607, 154976 CrossRef CAS.
- E. Wasim, N. U. Din, D. Le, X. M. Zhou, G. E. Sterbinsky, M. S. Pape, T. S. Rahman and S. L. Tait, J. Catal., 2022, 413, 81–92 CrossRef CAS.
- T. Liu, J. Q. Xiong, Q. Luo, S. J. Mao and Y. Wang, ACS Catal., 2024, 14, 5838–5846 CrossRef CAS.
- S. Lee, S. J. Shin, H. Baek, Y. Choi, K. Hyun, M. Seo, K. Kim, D. Y. Koh, H. Kim and M. Choi, Sci. Adv., 2020, 6, eabb7369 CrossRef CAS PubMed.
- K. Hyun, Y. Park, S. Lee, J. Lee, Y. Choi, S. J. Shin, H. Kim and M. Choi, Angew. Chem., Int. Ed., 2021, 60, 12482–12489 CrossRef CAS PubMed.
- Y. Park, S. Lee, K. Hyun, J. Lee, J. Y. Park, R. Ryoo and M. Choi, J. Catal., 2021, 404, 716–725 CrossRef CAS.
- K. Hyun, Y. Park and M. Choi, J. Catal., 2022, 416, 267–276 CrossRef CAS.
- K. Hyun, S. H. Yun and M. Choi, ACS Catal., 2024, 14, 2938–2948 CrossRef CAS.
- Y. Park, K. Hyun, S. Yun and M. Choi, ChemCatChem, 2024, 16, e202301378 CrossRef CAS.
- R. K. Edvinsson, A. M. Holmgren and S. Irandoust, Ind. Eng. Chem. Res., 1995, 34, 94–100 CrossRef CAS.
- N. B. Shitova, D. A. Shlyapin, T. N. Afonasenko, E. N. Kudrya, P. G. Tsyrul'nikov and V. A. Likholobov, Kinet. Catal., 2011, 52, 251–257 CrossRef CAS.
- R. J. Hou, T. F. Wang and X. C. Lan, Ind. Eng. Chem. Res., 2013, 52, 13305–13312 CrossRef CAS.
- B. B. Huang, T. Wang, C. Lei, W. Q. Chen, G. M. Zeng and F. Maran, J. Catal., 2016, 339, 14–20 CrossRef CAS.
- H. Shariff and M. H. Al-Dahhan, Ind. Eng. Chem. Res., 2024, 63, 8899–8905 CrossRef CAS.
- Z. Kang, Y. Wang, B. Liu, Z. Huang, X. Lan and T. Wang, Chem. Eng. J., 2024, 491, 151755 CrossRef CAS.
- D. V. Glyzdova, N. S. Smirnova, N. N. Leont'eva, E. Y. Gerasimov, I. P. Prosvirin, V. I. Vershinin, D. A. Shlyapin and P. G. Tsyrul'nikov, Kinet. Catal., 2017, 58, 140–146 CrossRef CAS.
- T. N. Afonasenko, V. L. Temerev, D. A. Shlyapin and P. G. Tsyrul'nikov, Russ. J. Appl. Chem., 2019, 92, 128–134 CrossRef CAS.
- T. N. Afonasenko, V. L. Temerev, D. V. Glyzdova, N. N. Leont'eva, M. V. Trenikhin, I. P. Prosvirin and D. A. Shlyapin, Mater. Lett., 2021, 305, 130843 CrossRef CAS.
- B. B. Huang, T. Wang, Z. Yang, W. T. Qian, J. M. Long, G. M. Zeng and C. Lei, ACS Sustainable Chem. Eng., 2017, 5, 1668–1674 CrossRef CAS.
- D. V. Glyzdova, N. S. Smirnova, V. L. Temerev, E. V. Khramov, T. I. Gulyaeva, M. V. Trenikhin, D. A. Shlyapin and P. G. Tsyrul'nikov, Russ. J. Appl. Chem., 2017, 90, 1908–1917 CrossRef CAS.
- D. V. Glyzdova, T. N. Afonasenko, E. Khramov, M. Trenikhin, I. P. Prosvirin and D. A. Shlyapin, ChemCatChem, 2022, 14, e202200893 CrossRef CAS.
- D. V. Glyzdova, T. N. Afonasenko, E. V. Khramov, N. N. Leont'eva, I. P. Prosvirin, A. V. Bukhtiyarov and D. A. Shlyapin, Appl. Catal., A, 2020, 600, 117627 CrossRef CAS.
- D. V. Glyzdova, A. A. Vedyagin, A. M. Tsapina, V. V. Kaichev, A. L. Trigub, M. V. Trenikhin, D. A. Shlyapin, P. G. Tsyrulnikov and A. V. Lavrenov, Appl. Catal., A, 2018, 563, 18–27 CrossRef CAS.
- J. D. Krooswyk, I. Waluyo and M. Trenary, ACS Catal., 2015, 5, 4725–4733 CrossRef CAS.
- C. S. Spanjers, R. S. Sim, N. P. Sturgis, B. Kabius and R. M. Rioux, ACS Catal., 2015, 5, 3304–3315 CrossRef CAS.
- O. B. Ayodele and G. Jacobs, Chem. Eng. J., 2024, 498, 155168 CrossRef CAS.
- A. L. Bugaev, A. A. Guda, A. Lazzarini, K. A. Lomachenko, E. Groppo, R. Pellegrini, A. Piovano, H. Emerich, A. V. Soldatov, L. A. Bugaev, V. P. Dmitriev, J. A. van Bokhoven and C. Lamberti, Catal. Today, 2017, 283, 119–126 CrossRef CAS.
- A. L. Bugaev, O. A. Usoltsev, A. A. Guda, K. A. Lomachenko, I. A. Pankin, Y. V. Rusalev, H. Emerich, E. Groppo, R. Pellegrini, A. V. Soldatov, J. A. van Bokhoven and C. Lamberti, J. Phys. Chem. C, 2018, 122, 12029–12037 CrossRef CAS.
- Y. M. Niu, B. S. Zhang, J. J. Luo, L. Y. Zhang, C. M. Chen and D. S. Su, ChemCatChem, 2017, 9, 3435–3439 CrossRef CAS.
- K. Kovnir, M. Armbrüster, D. Teschner, T. V. Venkov, L. Szentmiklósi, F. C. Jentoft, A. Knop-Gericke, Y. Grin and R. Schlögl, Surf. Sci., 2009, 603, 1784–1792 CrossRef CAS.
- T. Cao, R. You, X. Y. Zhang, S. L. Chen, D. Li, Z. H. Zhang and W. X. Huang, Phys. Chem. Chem. Phys., 2018, 20, 9659–9670 RSC.
- J. Moon, Y. Q. Cheng, L. L. Daemen, M. J. Li, F. Polo-Garzon, A. J. Ramirez-Cuesta and Z. L. Wu, ACS Catal., 2020, 10, 5278–5287 CrossRef CAS.
- C. S. Wondergem, J. J. G. Kromwijk, M. Slagter, W. L. Vrijburg, E. J. M. Hensen, M. Monai, C. Vogt and B. M. Weckhuysen, ChemPhysChem, 2020, 21, 625–632 CrossRef CAS PubMed.
- X.-T. Li, L. Chen, G.-F. Wei, C. Shang and Z.-P. Liu, ACS Catal., 2020, 10, 9694–9705 CrossRef CAS.
- L. Chen, X. T. Li, S. C. Ma, Y. F. Hu, C. Shang and Z. P. Liu, ACS Catal., 2022, 12, 14872–14881 CrossRef CAS.
- H. S. Feng, H. Ding, S. Wang, Y. J. Liang, Y. Deng, Y. S. Yang, M. Wei and X. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 25288–25296 CrossRef CAS PubMed.
- R. B. Fang, Q. J. Zhang, C. Yao, H. J. Wu, S. K. Xie, X. H. Zhang, Q. T. Wang, J. Lyu, F. Feng, C. S. Lu, Q. F. Zhang and X. N. Li, Appl. Catal., A, 2024, 687, 119969 CrossRef CAS.
- X. H. Ge, J. Yin, Z. H. Ren, K. L. Yan, Y. D. Jing, Y. Q. Cao, N. N. Fei, X. Liu, X. N. Wang, X. G. Zhou, L. W. Chen, W. K. Yuan and X. Z. Duan, J. Am. Chem. Soc., 2024, 146, 4993–5004 CrossRef CAS PubMed.
- K. Yang, X. Chen, J. Qi, Z. Bai, L. Zhang and C. Liang, J. Catal., 2019, 369, 363–371 CrossRef CAS.
- Z. Guo, R. Wang, Y. Guo, J. Jiang, Z. Wang, W. Li and M. Zhang, ACS Catal., 2022, 12, 15193–15206 CrossRef CAS.
- H. Yang, H. Chen, J. Chen, O. Omotoso and Z. Ring, J. Catal., 2006, 243, 36–42 CrossRef CAS.
- X. Zhang, Y. Zhou, G. Li, L. Zhang, C. Yin, Y. Yang, H. Wang, F. Feng, L. Wei, Q. Zhang, F. Yang, L. Lin, C. Lu and X. Li, Appl. Catal., B, 2022, 315, 121566 CrossRef CAS.
- L. Lin, S. Yao, R. Gao, X. Liang, Q. Yu, Y. Deng, J. Liu, M. Peng, Z. Jiang, S. Li, Y.-W. Li, X.-D. Wen, W. Zhou and D. Ma, Nat. Nanotechnol., 2019, 14, 354–361 CrossRef CAS PubMed.
- Z. Wang, C. Dong, X. Tang, X. Qin, X. Liu, M. Peng, Y. Xu, C. Song, J. Zhang, X. Liang, S. Dai and D. Ma, Nat. Commun., 2022, 13, 4404 CrossRef CAS PubMed.
- I. Y. Ahn, J. H. Lee, S. K. Kim and S. H. Moon, Appl. Catal., A, 2009, 360, 38–42 CrossRef CAS.
- M. T. Ravanchi and S. Sahebdelfar, Appl. Catal., A, 2016, 525, 197–203 CrossRef.
- R. J. Liu, P. A. Crozier, C. M. Smith, D. A. Hucul, J. Blackson and G. Salaita, Appl. Catal., A, 2005, 282, 111–121 CrossRef CAS.
- A. A. Lamberov, S. R. Egorova, I. R. Il’yasov, K. K. Gil’manov, S. V. Trifonov, V. M. Shatilov and A. S. Ziyatdinov, Kinet. Catal., 2007, 48, 136–142 CrossRef CAS.
- A. Pachulski, R. Schödel and P. Claus, Appl. Catal., A, 2011, 400, 14–24 CrossRef CAS.
- H.-X. Su, Y. Jiao, J.-G. Shi, Z.-W. Yuan, D. Zhang, X.-P. Wang, J. Ren, D. Liu, J.-Z. Gui, H.-Y. Gao and X.-L. Xu, Pet. Sci., 2024, 21, 1405–1414 CrossRef CAS.
- M. R. Rahimpour, O. Dehghani, M. R. Gholipour, M. S. Shokrollahi Yancheshmeh, S. Seifzadeh Haghighi and A. Shariati, Chem. Eng. J., 2012, 198–199, 491–502 CrossRef CAS.
- M. Mosafer, A. Hafizi, M. R. Rahimpour and A. Bolhasani, J. Nat. Gas Sci. Eng., 2016, 34, 1382–1391 CrossRef CAS.
- T. R. Bilalov, F. M. Gumerov, F. R. Gabitov, K. E. Kharlampidi, G. I. Fedorov, A. A. Sagdeev, R. S. Yarullin and I. A. Yakushev, Russ. J. Phys. Chem. B, 2009, 3, 1093–1105 CrossRef.
- B. T. Burganov, K. E. Kharlampidi, F. M. Gumerov and V. F. Khairutdinov, Russ. J. Phys. Chem. B, 2016, 10, 1147–1152 CrossRef CAS.
- Y. Liu, Y. Li, J. A. Anderson, J. Feng, A. Guerrero-Ruiz, I. Rodríguez-Ramos, A. J. McCue and D. Li, J. Catal., 2020, 383, 51–59 CrossRef CAS.
- S. A. Nikolaev, N. Z. Leonid, V. V. Smirnov, A. A. Vyacheslav and K. L. Zanaveskin, Russ. Chem. Rev., 2009, 78, 231 CrossRef CAS.
- M. R. Wang, Y. Wang, X. L. Mou, R. H. Lin and Y. J. Ding, Chin. J. Catal., 2022, 43, 1017–1041 CrossRef CAS.
- A. J. McCue, A. Gibson and J. A. Anderson, Chem. Eng. J., 2016, 285, 384–391 CrossRef CAS.
- Z. Q. Wang, L. Yang, R. Zhang, L. Li, Z. M. Cheng and Z. M. Zhou, Catal. Today, 2016, 264, 37–43 CrossRef CAS.
- Z. S. Wang, C. L. Yang, S. L. Xu, H. Nan, S. C. Shen and H. W. Liang, Inorg. Chem., 2020, 59, 5694–5701 CrossRef CAS PubMed.
- Z. X. Li, M. L. Hu, B. W. Liu, J. H. Liu, P. Wang, J. S. Yao, X. Zhang, M. He and W. Y. Song, ChemCatChem, 2021, 13, 868–873 CrossRef CAS.
- G. Wowsnick, D. Teschner, M. Armbrüster, I. Kasatkin, F. Girgsdies, Y. Grin, R. Schlögl and M. Behrens, J. Catal., 2014, 309, 221–230 CrossRef CAS.
- K. L. Yan, X. H. Ge, W. H. Li, Y. J. Liang, W. J. Xiong, J. Zhang, G. Qian, D. Chen, Y. Q. Cao, X. G. Zhou and X. Z. Duan, J. Mater. Chem. A, 2024, 12, 16482–16490 RSC.
- J. H. Xi, K. L. Yan, N. C. Zhu, X. H. Ge, Y. W. Qiang, M. M. Chen, H. Jiang, Y. Q. Cao, X. G. Zhou and X. Z. Duan, AIChE J., 2024, 70, e18416 CrossRef CAS.
- M. K. Abdel-Rahman and M. Trenary, ACS Catal., 2020, 10, 9716–9724 CrossRef CAS.
- Q. Yuwen, K. L. Yan, X. H. Ge, G. Qian, J. Zhang, Y. Q. Cao, X. G. Zhou and X. Z. Duan, Ind. Eng. Chem. Res., 2023, 62, 16280–16290 CrossRef.
- L. M. Zhao, X. T. Qin, X. R. Zhang, X. B. Cai, F. Huang, Z. M. Jia, J. Y. Diao, D. Q. Xiao, Z. Jiang, R. F. Lu, N. Wang, H. Y. Liu and D. Ma, Adv. Mater., 2022, 34, e202110455 Search PubMed.
- S. C. Wang, T. Liu, Y. A. Zhu, X. K. Liu, Q. Q. Luo, M. Z. Zhu, T. Ding and T. Yao, J. Phys. Chem. C, 2023, 127, 5911–5919 CrossRef CAS.
- H. H. Ibrahim, T. Weckman, D. Y. Murzin and K. Honkala, J. Catal., 2024, 434, 115523 CrossRef CAS.
- Q. C. Yang, R. J. Hou and K. N. Sun, J. Catal., 2019, 374, 12–23 CrossRef CAS.
- H. W. Ma, X. P. Xu, H. X. Xu, H. X. Feng, Y. Xie and D. J. Cheng, Catal. Commun., 2021, 149, 106255 CrossRef CAS.
- O. E. B. Corstius, H. L. Nolten, G. F. Tierney, Z. Xu, E. J. Doskocil, J. E. S. van der Hoeven and P. E. de Jongh, Catal. Today, 2024, 441, 114877 CrossRef.
- Z. B. Chen, X. T. Chen, Y. L. Lv, X. L. Mou, J. H. Fan, J. W. Li, L. Yan, R. H. Lin and Y. J. Ding, Chin. J. Catal., 2024, 60, 304–315 CrossRef CAS.
- Z. B. Chen, Y. L. Lv, X. T. Chen, X. L. Mou, J. W. Li, L. Yan, R. H. Lin and Y. J. Ding, Catal. Sci. Technol., 2024, 14, 137–144 RSC.
- Y. X. Feng, L. S. Zhou, Q. Wan, S. Lin and H. Guo, Chem. Sci., 2018, 9, 5890–5896 RSC.
- H. Yan, H. F. Lv, H. Yi, W. Liu, Y. J. Xia, X. H. Huang, W. X. Huang, S. Q. Wei, X. J. Wu and J. L. Lu, J. Catal., 2018, 366, 70–79 CrossRef CAS.
- Z. Yan, L. X. Cui, Z. Pang, K. Shi, M. Y. Zhang, J. W. Guo, R. Gao and H. G. Hao, Appl. Surf. Sci., 2023, 617, 156585 CrossRef CAS.
- M. R. Hossain and M. Trenary, J. Phys. Chem. C, 2024, 128, 19204–19209 CrossRef CAS.
- B. B. Huang, C. Durante, A. A. Isse and A. Gennaro, Electrochem. Commun., 2013, 34, 90–93 CrossRef CAS.
- Z. Liu, L. Zhang, Z. Ren and J. Zhang, Chem. – Eur. J., 2023, 29, e202202979 CrossRef CAS PubMed.
- F. Arcudi, L. Dordevic, N. Schweitzer, S. I. Stupp and E. A. Weiss, Nat. Chem., 2022, 14, 1007 CrossRef CAS PubMed.
- A. Stone, L. Dordevic, S. I. Stupp, E. A. Weiss, F. Arcudi and J. T. Hupp, ACS Energy Lett., 2023, 8, 4684–4693 CrossRef CAS.
- A. Stone, A. Fortunato, X. J. Wang, E. Saggioro, R. Q. Snurr, J. T. Hupp, F. Arcudi and L. Dordevic, Adv. Mater., 2025, 37, e202408658 CrossRef PubMed.
- B.-H. Zhao, F. Chen, M. Wang, C. Cheng, Y. Wu, C. Liu, Y. Yu and B. Zhang, Nat. Sustainability, 2023, 6, 827–837 CrossRef.
- L. D. Burke, F. A. Lewis and C. Kemball, Trans. Faraday Soc., 1964, 60, 919–929 RSC.
- M. Bełtowska-Brzezinska, T. Łuczak, M. Mączka, H. Baltruschat and U. Müller, J. Electroanal. Chem., 2002, 519, 101–110 CrossRef.
- L. Zhang, Z. Chen, Z. P. Liu, J. Bu, W. X. Ma, C. Yan, R. Bai, J. Lin, Q. Y. Zhang, J. Z. Liu, T. Wang and J. Zhang, Nat. Commun., 2021, 12, 6574 CrossRef CAS PubMed.
- Z. Chen, C. Cai and T. Wang, J. Phys. Chem. C, 2022, 126, 3037–3042 CrossRef CAS.
- J. Bu, Z. P. Liu, W. X. Ma, L. Zhang, T. Wang, H. P. Zhang, Q. Y. Zhang, X. L. Feng and J. Zhang, Nat. Catal., 2021, 4, 557–564 CrossRef CAS.
- Z. P. Wang, L. Shang, H. Z. Yang, Y. X. Zhao, G. I. N. Waterhouse, D. Li, R. Shi and T. R. Zhang, Adv. Mater., 2023, 35, e202400122 Search PubMed.
- X. L. Jiang, L. Tang, L. Dong, X. D. Sheng, W. F. Zhang, Z. Liu, J. H. Shen, H. L. Jiang and C. Z. Li, Angew. Chem., Int. Ed., 2023, 62, e202307848 CrossRef CAS PubMed.
- W. Q. Xue, X. Y. Liu, C. X. Liu, X. Y. Zhang, J. W. Li, Z. W. Yang, P. X. Cui, H. J. Peng, Q. Jiang, H. L. Li, P. P. Xu, T. T. Zheng, C. Xia and J. Zeng, Nat. Commun., 2023, 14, 2137 CrossRef CAS PubMed.
- X. Gao, D. S. Wang, R. Bai, D. Lin, J. Zhang, Z. J. Li, Y. Hou, L. C. Lei and B. Yang, Adv. Funct. Mater., 2025, 35, e202415384 Search PubMed.
- Z. P. Wang, C. Y. Li, G. Peng, R. Shi, L. Shang and T. R. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202400122 CrossRef CAS PubMed.
- X. H. Lv, H. Huang, L. T. Cui, Z. Y. Zhou, W. K. Wu, Y. C. Wang and S. G. Sun, ACS Appl. Mater. Interfaces, 2024, 16, 8668–8678 CrossRef CAS PubMed.
- C. Dou, Y. M. Huang, B. H. Zhao, W. W. Lei, B. Zhang and Y. F. Yu, Angew. Chem., Int. Ed., 2025, 64, e202423381 CrossRef CAS PubMed.
- B. Miao, F. Chen, C. Cheng, M. Tao, B.-H. Zhao and B. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202502757 CrossRef CAS PubMed.
- Z. P. Liu, Z. Chen, J. Bu, W. X. Ma, L. Zhang, H. Zhong, L. Cheng, S. M. Li, T. Wang and J. Zhang, Chem. Eng. J., 2022, 431, 134129 CrossRef CAS.
- W. X. Ma, Z. Chen, J. Bu, Z. P. Liu, J. J. Li, C. Yan, L. Cheng, L. Zhang, H. P. Zhang, J. C. Zhang, T. Wang and J. Zhang, J. Mater. Chem. A, 2022, 10, 6122–6128 RSC.
- M. Yuan, H. Jiang, R. Jiang, Z. Wang, Z.-H. Zhao, B.-L. Su and J. Zhang, ACS Catal., 2024, 14, 18385–18396 CrossRef CAS.
- L. Zhang, R. Bai, J. Lin, J. Bu, Z. P. Liu, S. Y. An, Z. H. Wei and J. Zhang, Nat. Chem., 2024, 16, 893–900 CrossRef CAS PubMed.
- L. Bai, Y. Wang, Z. Han, J. B. Bai, K. Y. Leng, L. R. Zheng, Y. T. Qu and Y. Wu, Nat. Commun., 2023, 14, 8384 CrossRef CAS PubMed.
- L. S. Huang, D. Y. Bao, Y. L. Jiang, Y. Zheng and S. Z. Qiao, Angew. Chem., Int. Ed., 2024, 63, e202405943 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |