
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLanthanide(III)-binding peptides and proteins: coordination properties and applications
Enrico
Falcone†
 *,
Emilie
Mathieu†
* and
Christelle
Hureau
*,
Emilie
Mathieu†
* and
Christelle
Hureau
LCC-CNRS, Université de Toulouse, CNRS, Toulouse, France. E-mail: enrico.falcone@lcc-toulouse.fr; emilie.mathieu@lcc-toulouse.fr
First published on 22nd September 2025
Abstract
Lanthanides play a crucial role in modern medicine and technology as well as in the metabolism of methylotrophic bacteria. In this context, the research on lanthanide-binding peptides and proteins is an active and rapidly developing field. This comprehensive and critical review focuses on the structural, thermodynamic (affinity and selectivity) and kinetic parameters governing the interaction of Ln3+ ions with different peptides and proteins, including both naturally occurring and de novo-designed scaffolds. It thus provides guidelines and future directions for the rational design of Ln-binding peptides and proteins with suitable features for the main applications explored to date, including luminescent sensing, magnetic resonance imaging, Ln separation and recovery and Ln-based (photo)-catalysis.
1. Introduction
1.1 Context and scope
Lanthanides (Ln) are f-block elements known for their unique magnetism and luminescence properties. They are used in a wide variety of fields: as contrast agents in medicine, in laser technology, in superconductors and in permanent magnets.1 Rare earth elements (REE), which include Ln, lanthanum (La), yttrium (Y) and scandium (Sc), have been identified as strategic resources by the European Union (EU).2 Indeed, many advanced technologies depend on them, and China remains the main exporter of these resources, supplying over 98% of REE importation to the EU. Of note, the mining and extraction of REE have a deleterious impact on the environment.3Even before the emergence of interest in Ln biochemistry due to the discovery of the first Ln-utilizing bacteria in the 2010s,4–7 the design of Ln3+-binding peptides was an intensive research field aimed at exploiting the unique physical properties of Ln3+ ions for applications in sensing or imaging.8
The discovery of Ln-utilizing bacteria has been accompanied by the identification of natural Ln3+-binding proteins and Ln3+–enzymes, paving the way for a better description of the lanthanome, which corresponds to the proteins involved in Ln3+-uptake, trafficking and utilisation.9–12 These findings have inspired a large community of biologists and chemists, resulting in a proliferation of studies on the rational design of Ln-binding peptides and proteins, and their applications in biomedicine,13 biohydrometallurgy,14,15 and catalysis.16–21
Previous reviews in the field focused on the rational design of lanthanide-binding peptides,8 and a general presentation of the coordination of f-block elements to bio-relevant ligands, including peptides and proteins.22 Here, we first discuss some critical parameters of Ln3+ aqueous chemistry and the design of peptides and proteins with a high affinity for Ln3+ ions. Then, we provide guidelines on how selectivity against metal ions (Ca2+, actinides (An3+), d-block metal ions) or among Ln3+ can be achieved, and on how to include kinetic considerations in the description of these systems. Last, we highlight recent applications for sensing, MRI, Ln separation and catalysis. In this review, we focus on Ln3+-binding peptides and proteins using natural and/or unnatural amino acids, excluding peptides and proteins functionalized with macrocycles such as DOTA, some of which were reviewed in ref. 23. This choice was driven by the focus on the interplay between the structure and the coordination properties (affinity, selectivity and kinetics) of Ln3+-binding peptides and proteins scaffold, which do not apply to macrocycles. Nevertheless, small chelators will be occasionally discussed to introduce key features and concepts (e.g. selectivity) useful for Ln-binding peptides and proteins, and peptides with appended macrocycles will be briefly mentioned among the applications, as they represent useful models for the design of luminescent and MRI probes.
1.2 Chemistry of Ln ions in aqueous solution
The most stable oxidation state for Ln in aqueous solution is +3 (electronic configuration [Xe]4fn, with n = 0–14 from La3+ to Lu3+). Other oxidation states are also accessible for some Ln ions in aqueous solution, such as +2 for Eu and +4 for Ce, due to the enhanced stability associated with a half-filled 4f sub-shell ([Xe]4f7 for Eu2+) and with closed shell ([Xe]4f0 for Ce4+) configuration (Fig. 1A). Of note, in organic solvents, the entire series of Ln (except for the radioactive Pm) could be stabilised in the +2 oxidation state, while for the +4 oxidation state only Ce4+, Tb4+ and Pr4+ complexes were isolated.24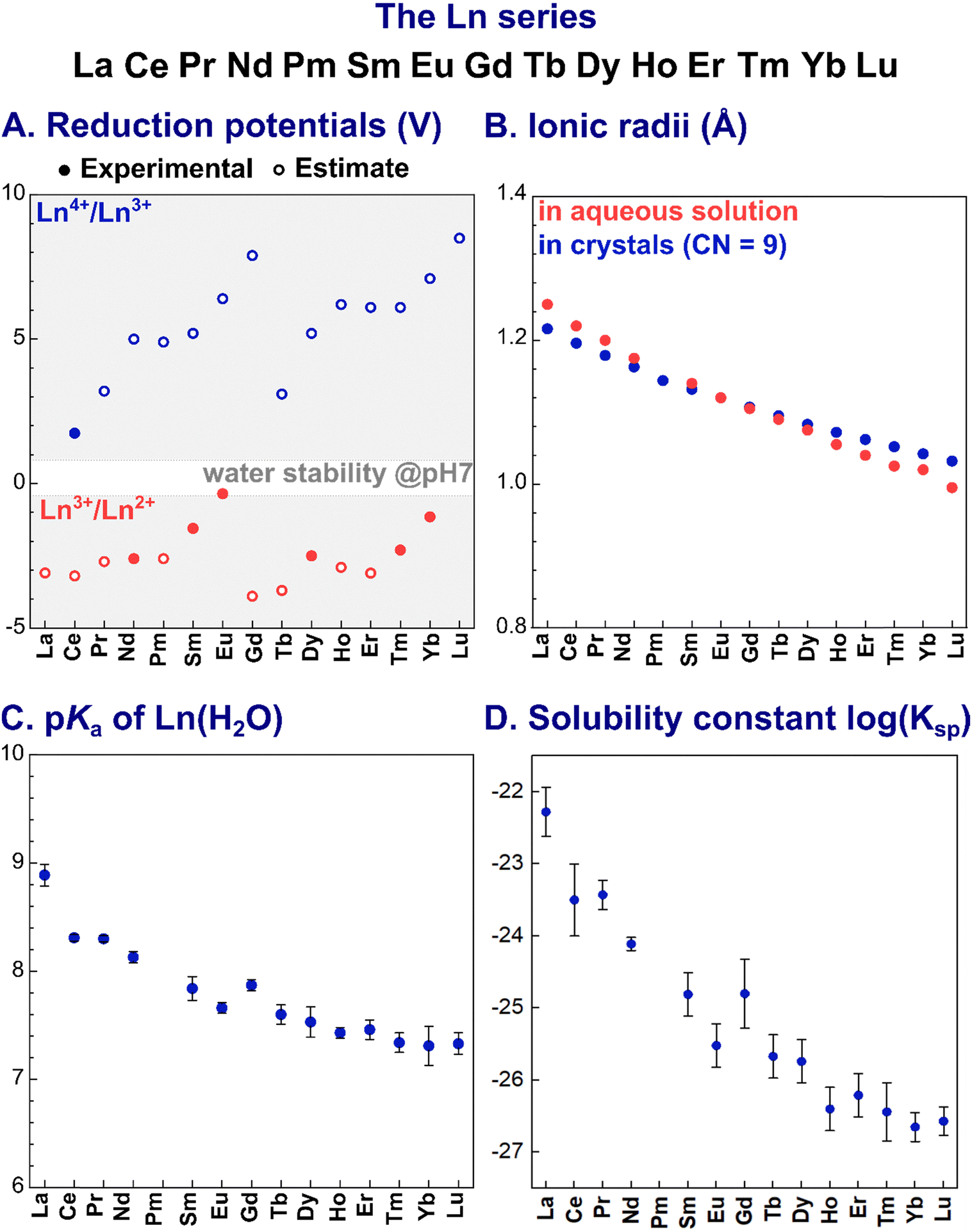 | ||
Fig. 1 Trends of key parameters along the Ln series: (A) reduction potentials;26 (B) ionic radii in aqueous solution27 and in crystals (CN = 9);28 (C) acidity constants (pKa) for Ln3+-bound water molecule; and (D) solubility constants (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ksp).29 The values plotted are reported in Table 14. Ksp).29 The values plotted are reported in Table 14. | ||
Ln3+ ions are hard Lewis acids. Their Lewis acidity slightly increases along the series as a consequence of the decrease of the ionic radius, known as lanthanide contraction (Fig. 1B). Noteworthy, Ln3+ ions have a similar size to Ca2+ (∼1.0 Å), but are larger than d-block metal ions. As a consequence, Ln3+ have higher coordination numbers (CN) than d-block elements.
Due to their hard character, according to Pearson's HSAB principle,25 Ln3+ ions prefer hard negatively charged ligands (e.g. carboxylate and phosphate). As a result of the core nature of the f orbitals, the nature of the Ln3+–ligand bond is mainly ionic, and coordination geometries are hence mostly dictated by electrostatic and steric repulsions between the ligands.
In water, the larger Ln3+ ions (La3+–Nd3+) are coordinated by 9 water molecules and adopt a tri-capped trigonal prismatic coordination geometry, whereas smaller Ln3+ (Gd3+–Lu3+) are bound to 8 water molecules in a square-antiprismatic arrangement. For Pm3+ to Eu3+, [Ln(H2O)8]3+ and [Ln(H2O)9]3+ are in equilibrium.30
Importantly, as Ln3+ aqua ions display Brønsted acidity in aqueous solution, Ln3+ speciation depends on the pH, with the acidity constant pKa (eqn (1)) decreasing along the series (Fig. 1C):
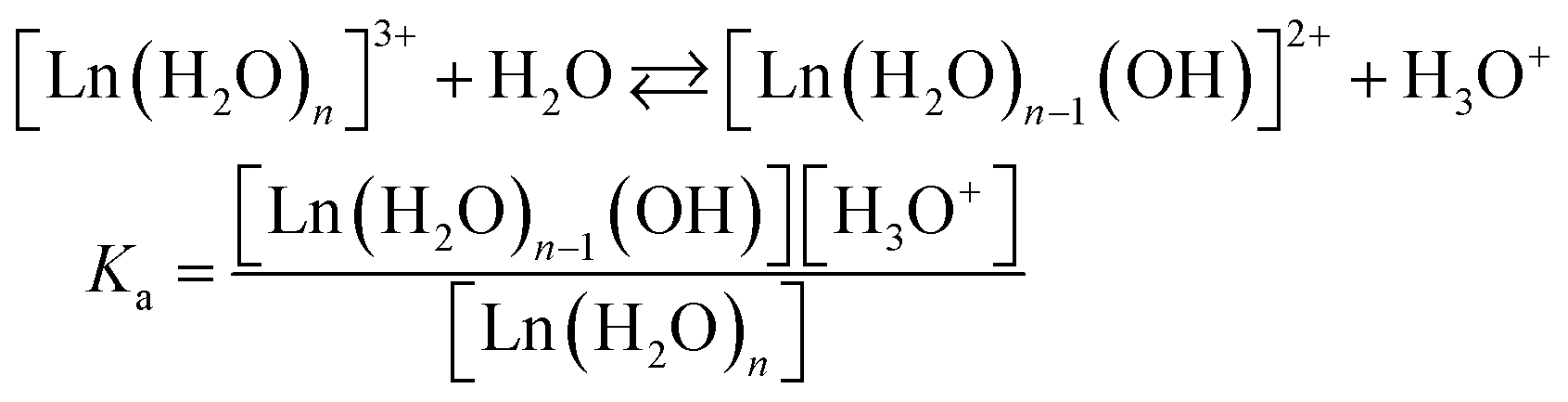 | (1) |
| Ln(OH)3 (s) ⇄ Ln3+(aq) + 3HO− |
| Ksp = [Ln3+][HO−]3 | (2) |
2. Scaffolds structure & Ln3+ coordination
2.1 Generalities
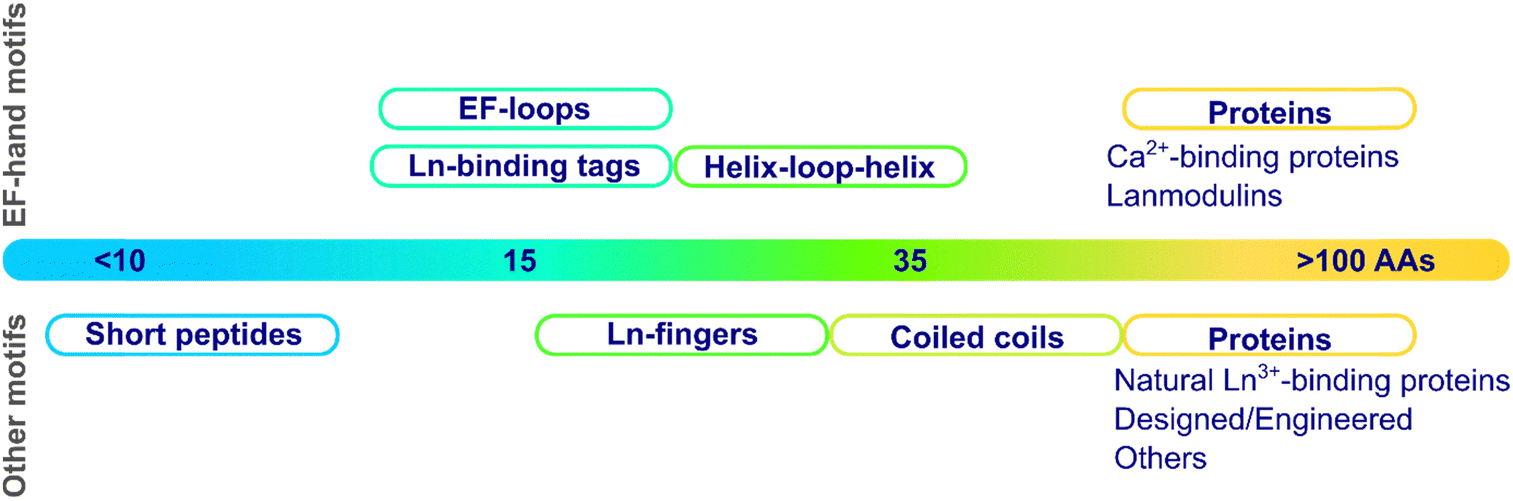 | ||
| Fig. 2 Categories of scaffolds by size (number of amino acids). | ||
These scaffolds can be also differentiated based on their structure in the apo-form, i.e. in the absence of metal ions (Fig. 3). Whereas some are disordered in the apo-form and fold upon metal binding, others are pre-folded even in the absence of metal ions. In this latter case, the binding site can be (i) pre-disposed, i.e. the coordinating residues are placed at appropriate positions for metal-binding, or (ii) pre-organised, i.e. the coordinating residues are not only at an appropriate position but also with a good orientation that requires minimal reorganisation upon metal-binding.31 To our knowledge, there is no example of a pre-organised metal binding site in Ln3+-binding peptides and proteins, and hence, in the following, we will only describe unfolded scaffolds, or folded ones with a predisposed binding site. In order to discuss the scaffold structure and the Ln3+-coordination, we grouped the scaffolds into four sub-categories: (i) scaffolds relying on the well-described EF-hand motifs, which are found in short peptides such as lanthanide-binding tags (LBT), in helix-loop-helix (HLH) peptides, and naturally-occurring proteins such as calmodulin and lanmodulin; (ii) short sequences of ∼10 amino acids; (iii) peptides of intermediate size, spanning from Ln fingers (LF) to coiled coils (CC); (iv) Ln3+-binding proteins either naturally occurring or engineered, in which the Ln3+ ion is not coordinated in an EF-hand motif.
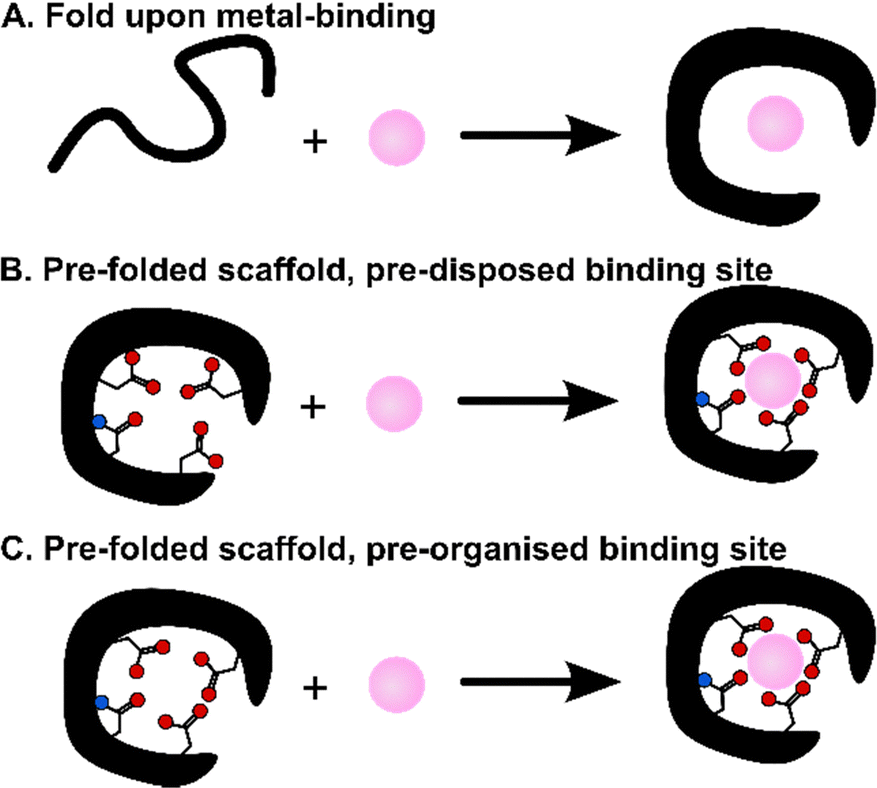 | ||
| Fig. 3 Scaffolds that fold upon metal-binding (A), or are folded in the apo-form with a pre-disposed (B) or a pre-organized (C) binding site. | ||
Ln3+ coordination sphere. X-ray crystallography is the most accurate way to determine the Ln3+ coordination sphere, especially the CN and the mode of binding of certain amino acids (e.g. mono- or bi-dentate for carboxylates in Asp and Glu residues). However, it requires the formation of crystals, which are often difficult to obtain, in particular when working with peptides.
Nevertheless, information on the mono- or bi-dentate binding mode of carboxylates can also be obtained in solution through FT-IR measurements.32,33 Moreover, the CN may be also investigated in solution through X-ray absorption spectroscopy, specifically EXAFS (extended X-ray absorption fine structure),34–37 which has not been applied to Ln3+-binding peptides and proteins so far.
In addition, insights into the Ln3+ coordination sphere can be gained by luminescence measurements. Eu3+-hypersensitive transitions can report on the geometry of the coordination environment.38 The hydration number q, which corresponds to the number of Ln3+-bound water molecules, is also easily determined by measuring Ln3+ luminescence lifetimes (Ln = Eu3+, Tb3+, Yb3+) for a given complex in H2O and D2O and using empirical relationships,39,40 which have been established based on a library of complexes with q ranging from 0 to 6. This is the most solid method and should be favoured compared to other empirical equations that rely only on lifetimes measured in water. Importantly, when working with peptides and proteins it may be difficult to work in H2O-free conditions. Instead, measurements can be performed for different H2O:D2O ratios in order to extrapolate the value in 100% D2O.41
Information beyond the number of Ln3+-bound water molecules can be gained from NMR measurements, by taking advantage of the magnetic properties of lanthanides.42–46 Comparison of spectra obtained with diamagnetic Ln3+ (La, Lu) and paramagnetic ones (all the other) can be used to identify residues in the first coordination sphere or further away from the Ln3+-binding site. For well-folded scaffolds, paramagnetic Ln can even help to refine the protein solution structure. This has been accomplished either by incorporating Ln3+ in an intrinsic metal-binding site, such as in Ca2+-binding proteins,47,48 or by introducing Ln-binding tags (LBT, see Section 2.2.2) in the sequence of the targeted protein.46
Scaffolds stability. Information on scaffold stability can be gained from denaturation experiments, either using a temperature ramp or chemical denaturants, followed by circular dichroism. By comparison to a “native” scaffold, these measurements can inform on the impact of engineering a Ln3+-binding site into the scaffold and the range of temperatures for which the peptide/protein is folded. It is also helpful to quantitatively assess the stabilisation induced by rational optimisations of the scaffold and by Ln3+ binding.
2.2 EF-hand motifs
EF-hand motifs represent the largest and best-known family of Ln3+-binding sites in peptides/proteins. They are found in most Ca2+-binding proteins, including calmodulin (CaM), parvalbumin and troponin C (TnC), and in a recently discovered selective Ln-binding protein, lanmodulin. The EF-hand consists of two α-helices (almost perpendicular to each other, a topology that reminds the spread thumb and forefinger of human hands) linked by a 12-residues loop, which is responsible for Ca2+ and Ln3+ binding through the amino acids in positions 1, 3, 5, 7, 9 and 12 (Fig. 4).49–53 In the following paragraphs, EF-hand proteins and peptides are described starting from long-known Ca2+-binding proteins (Section 2.2.1), which have inspired the design of optimized Ln3+-binding peptides (see 2.2.2 LBTs and 2.2.3 HLHs), and finishing by a recently discovered family of Ln3+-selective proteins (2.2.4 Lanmodulins).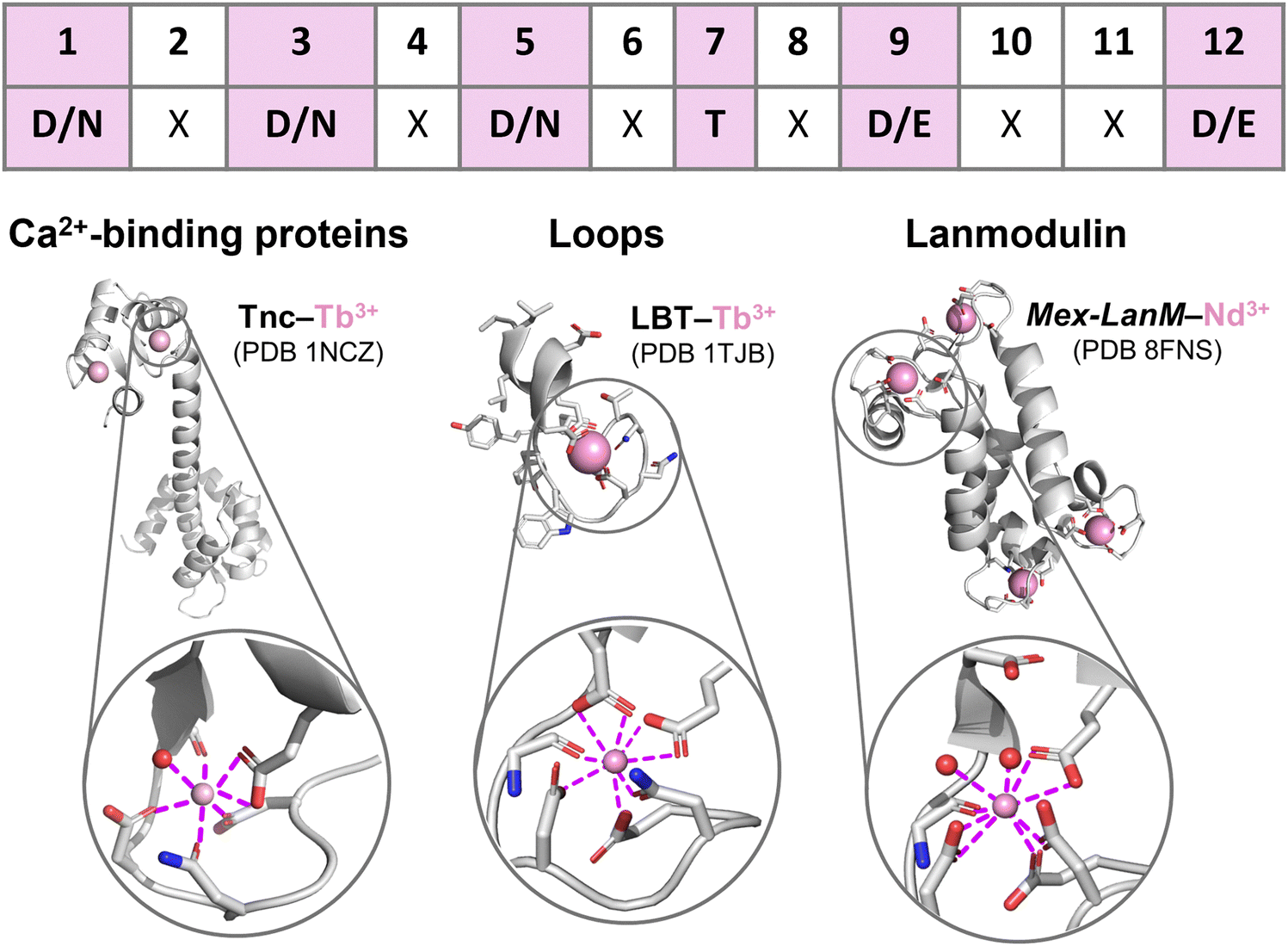 | ||
| Fig. 4 EF-hand peptides and proteins. Top: EF-loop sequence showing the most common Ln3+-binding amino acids (in bold and highlighted in pink) in each position. Bottom: Protein structures and Ln3+ coordination spheres in: left, troponin C (TnC) bound to two Tb3+ ions (PDB 1NCZ); middle, a lanthanide-binding tag (LBT) bound to Tb3+ (PDB 1TJB); right, Mex-LanM bound to four Nd3+ ions (PDB 8FNS). Figures were generated using Pymol. | ||
|
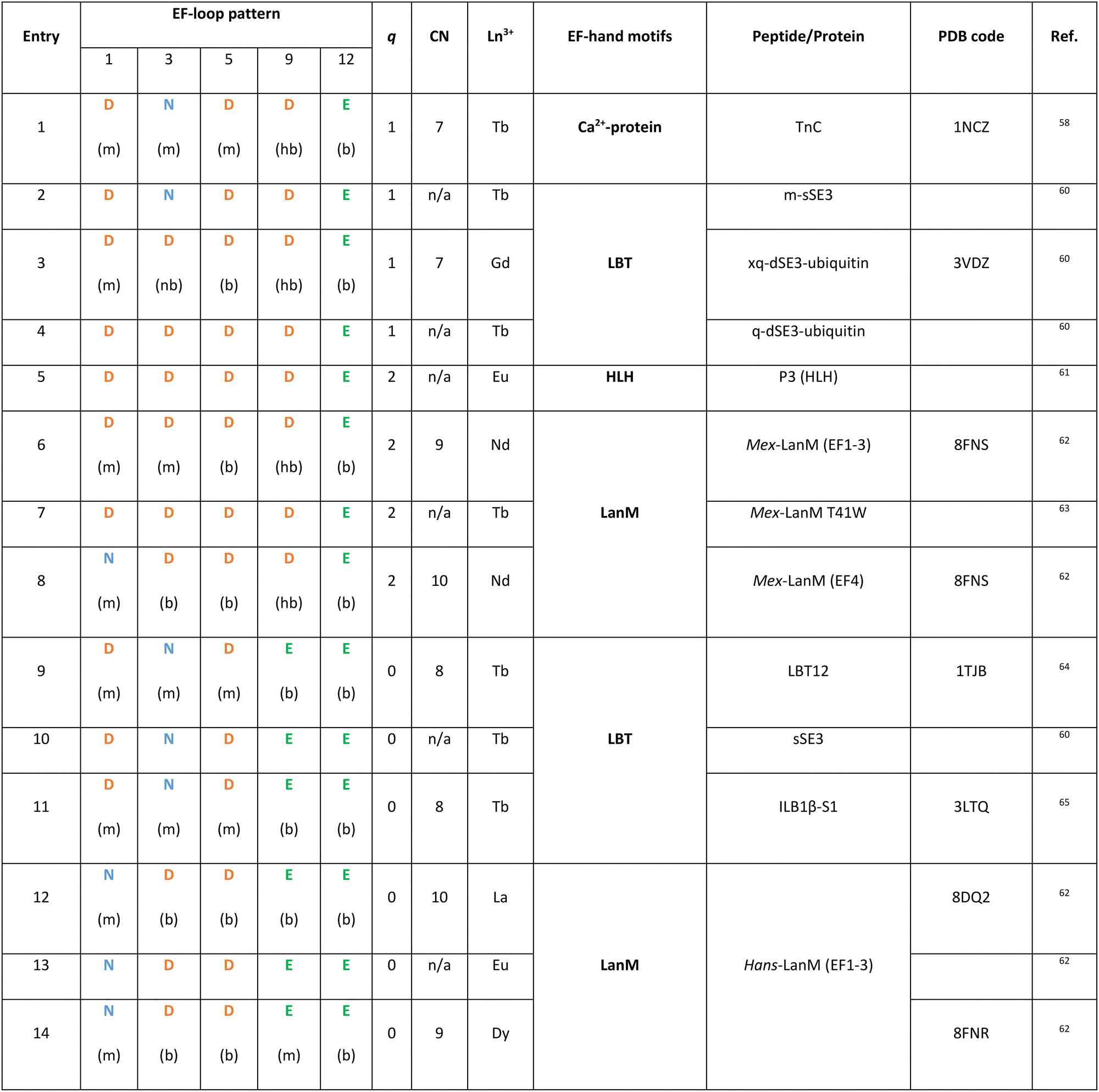
Since Ln3+ ions have similar ionic radius to Ca2+, the luminescent properties of some Ln3+ ions, mostly Tb3+ and Eu3+, have been exploited to probe the metal binding sites in Ca2+-binding proteins,54–57 showing that the same coordination sphere with CN = 7 and q = 1 is observed for Ln3+ ions bound to Ca2+–proteins such as TnC (Fig. 4 and entry 1 in Table 1).52,58,59
LBTs are 15- to 20-residue-long peptides that were designed by elongating the core 12-residue EF-loop at both termini with apolar amino acids that stabilize the Ln3+-bound conformation via hydrophobic interactions. The primary sequence of LBTs was optimized to feature high affinity for Tb3+ (see 3.2 EF-hand motifs),72,73 and to control the hydration number for specific applications. For instance, LBT featuring a Ln3+-bound water molecule (entries 2–4, in Table 1) were explored for applications as MRI contrast agents (see 6.2 MRI). The crystal structure of Gd3+ bound to a double LBT-ubiquitin fusion construct showed a CN = 7, which is as low as that observed in Ca2+-binding proteins.60
As Ln3+-bound water molecules quench its luminescence emission (see 6.1 Luminescent tags and probes), the coordination sphere was further optimized to afford efficient luminescent LBT by replacing the hydrogen-bonded Asp9 with bidentate Glu9. Thus, Tb3+–LBT complexes with q = 0 and CN = 8 were obtained (Fig. 4 and entries 9–11 in Table 1).64 Moreover, in order to sensitize Tb3+ emission a Trp residue serving as the antenna was introduced in the 7th position of the EF-hand motif (see 6.1 Luminescent tags and probes).66,74
Compared to Mex-LanM, no Ln3+-bound water molecules are found in Hans-LanM binding sites due to replacement of Asp9 with Glu9 (entries 6–8 vs. 12–14 in Table 1), as already observed for LBT (entries 9 vs. 2 in Table 1). In Hans-LanM, the increased CN relative to Ca2+-binding proteins results from the presence of a bidentate Asp3 (entries 12 and 14) rather than monodentate Asn3 (entries 1, 9–11) and a monodentate-to-bidentate shift of Asp5 (entries 12 and 14 vs. 9–11 in Table 1). Furthermore, a decrease in the CN is observed when La3+ is replaced by the smaller Dy3+ stemming from a bidentate-to-monodentate switch of Glu9 (entries 12 vs. 14 in Table 1).62
2.3 Short peptides
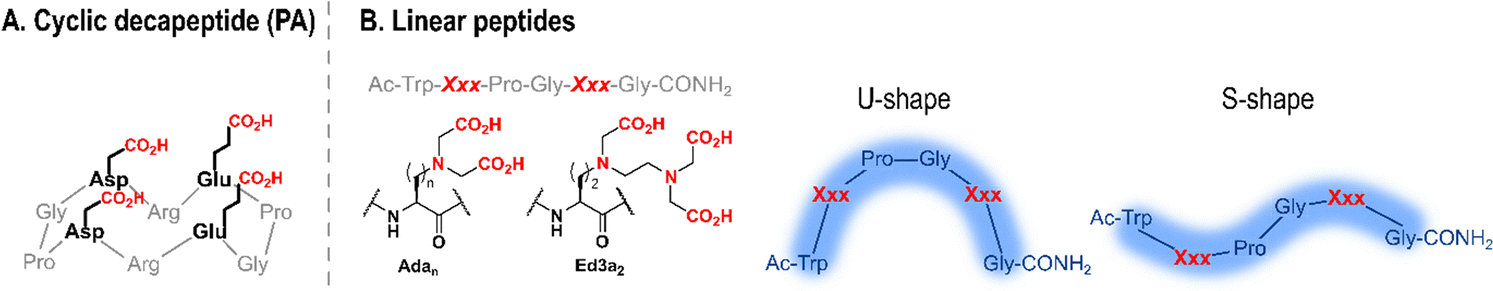 | ||
| Fig. 5 Schematic structures of short cyclic (A) and linear (B) Ln-binding peptides. In (B) the structures of unnatural amino acids Xxx = Adan and Ed3a2, and shapes adopted by linear peptides are shown. Adapted from ref. 8. | ||
| Peptide | Speciation | Ln3+-binding AAs | Coordination mode | Structure | q | Ref. |
|---|---|---|---|---|---|---|
| P11 | 1:1 and multimetallic species |
Ada1 | Tri- and tetra-dentate | S-shape | 1 | 84 |
| P22 | 1:1 |
Ada2 | Tridentate | U-shape | 3 | 85 |
| P33 | 1:1 and multimetallic species |
Ada3 | Tridentate | n.d. | 3 | 85 |
| P12 | 1:1 |
Ada1 and Ada2 | Ada1: tetradentate | S-shape | 0 | 86 |
| P21 | 1:1 and multimetallic species |
Ada2 and Ada1 | Ada1: tridentate | U-shape | 3 | 86 |
| PHD2 | 1:1 |
Ada2 and Ed3a2 | Ed3a2: pentadentate | n.d. | 0 | 87 |
| PHD5 | 1:1 and multimetallic species |
Ed3a2 and Ada2 | Ed3a2: pentadentate | n.d. | 0 | 87 |
All the peptides formed 1:1 ligand:metal complexes below 1 equivalent of Ln3+ relative to the peptide, however, in the presence of more equivalents of Ln3+ the formation of multimetallic species was also observed (Table 2).
Changes in Adan side chain length impacted its coordination mode to Ln3+ (Table 2). Ada2 and Ada3 bind Ln3+ in a tridentate manner,85 whereas Ada1 is tri- or tetra-dentate depending on its position in the sequence (fifth or second, respectively).84 The additional chelating group was proposed to be the backbone carbonyl of the Ada1 main chain, which can form an additional chelate ring. This change of coordination mode impacted the number of water molecules in the first coordination sphere of Tb3+, which dropped from 3 (P22, P21 and P33) to 1 and 0 in peptides where Ada1 is tetradentate (P11 and P12, respectively). It also impacted the structure of Ln3+–peptide complexes in solution: in the presence of Ln3+, P22 and P21 adopted a U-shape conformation, likely further stabilised by the formation of a type-II β-turn,85 while coordination of Ada1 backbone carbonyl to Ln3+ did not allow for the formation of a β-turn and resulted instead in an S-shape conformation of P11 and P12 (Fig. 5).84
Building upon this knowledge, Delangle and co-workers combined the pentadentate Ed3a2 unnatural amino acid with Ada2 either by placing Ed3a2 in the second (PHD2) or fifth (PHD5) position in the sequence (Table 2).87 Again, the speciation of the complexes depended on the position of the Ln3+-binding amino acids in the sequence. For both peptides, the pentadentate Ed3a2 excluded water molecules from the Tb3+ coordination sphere (q = 0).
More recently, short Ln3+-binding peptides rich in Asp or Glu residues were reported. Veliscek-Carolan and co-workers studied peptides made of two to four Glu residues, either with an L- or D-stereochemistry, and coupled to a naphthalene antenna.88 Simoni and co-workers studied the structure–affinity relationships of three Asp-rich pentapeptides (ADPDA, DPDPD, DGDGD) with actinide ions and Eu3+ and they predicted that the Eu3+ coordination sphere contained two carboxylates, one backbone carbonyl, and 4–5 water molecules.89
2.4 Peptide scaffolds of intermediate size
In the late 1980s, chemists began to dissect the complexity of proteins' fold and function, with the ultimate goal of being able to predict the primary sequence–structure–activity relationship.90 This led to the development of de novo proteins, which refers to proteins designed from first principles. The design of α-helices was soon mastered, and the study of α-helical coiled coil made of the assembly of several α-helices has helped unravel design principles to obtain well-folded coiled coil assemblies. Since then, de novo proteins have been implemented with metal-binding site(s) and functions such as catalytic properties or electron transfer capacities.91 Regarding Ln-binding peptides, a large amount of work has been dedicated to the design of Ln-binding coiled coils (2.4.1 Coiled coils),92–102 and a seminal work described the design of Ln-fingers103 (2.4.2 Ln fingers).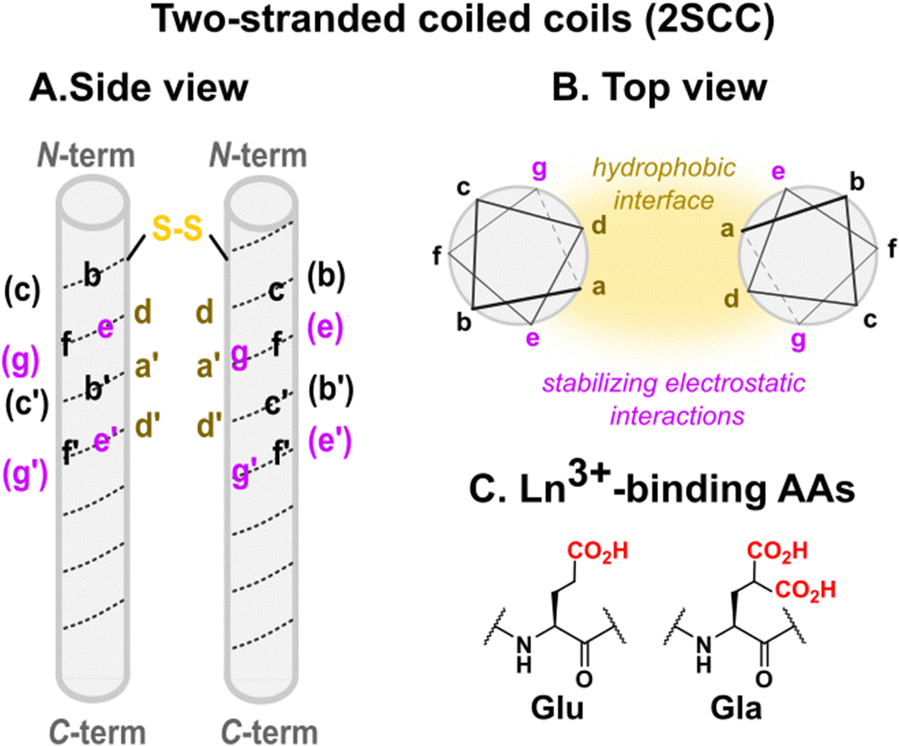 | ||
| Fig. 6 Two-stranded coiled coils scaffolds designed by Hodges and coworkers: (A) side view, (B) top view, (C) Ln-binding sites are placed at the helical interface using Glu or Gla AAs in e and g positions. In (B) stabilising non-covalent interactions (hydrophobic, electrostatic) responsible for the folding and assembly of coiled coils are highlighted. | ||
Three strategies were employed to insert an Ln3+-binding site in such scaffolds: (i) in two-stranded coiled coils (2SCC, Fig. 6 and Table 3), positions e and g were used to introduce chelating moieties and place the Ln3+-binding site at the helical interface; in three-stranded coiled coils (3SCC, Fig. 8), the binding site was (ii) either placed at the N-terminal (Table 4) or (iii) buried in the hydrophobic core by replacing core amino acids in position a and d by Ln3+-binding residues (Table 5).

|

|

|
Two-stranded coiled coils (2SCC). Pioneering works on Ln3+-binding 2SCCs were done by Hodges and coworkers.94,95 They used as a starting point a de novo α-helical peptide made of the repetition of 5 heptads (35 amino acids, Table 3). The formation of a disulphide bridge ensured the obtention of a parallel 2SCC (Fig. 6). The native sequence was modified to include two (E2) or three (E3) Glu residues in e or g positions to form two Ln3+-binding sites per coiled coil.94 The stability of the apo-peptides in their oxidized form was quantitatively investigated by urea denaturation followed by circular dichroism (Fig. 7). The destabilisation induced by the mutation of Gln to Glu was the greatest for the E2(15,20)ox construct, in which Glu in positions 15 and 20 generates the highest interchain repulsion. The introduction of a third Glu residue in the sequence (E3(13,15,20) and E3(15,20,22)) further destabilised the coiled coil.
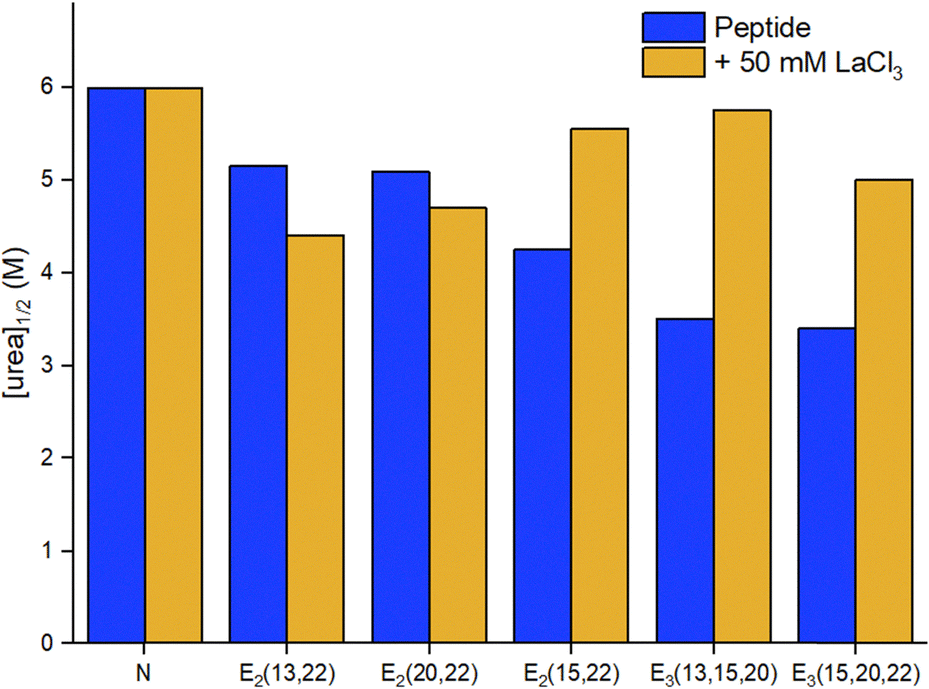 | ||
| Fig. 7 Urea denaturation of 2SCC at pH 7 followed by circular dichroism.94 Peptide sequences are given in Table 3. Experiments performed at 20 °C in 50 mM TRIS, 100 mM KCl, [pep] = 70–140 μM, without or with 50 mM LaCl3. [urea]1/2 corresponds to the concentration at which 50% of the peptide is unfolded. | ||
A stabilising effect of La3+ on peptide unfolding was noted for E2(15,20) and E3 constructs, in which the Glu residues are well positioned to bind La3+ at the interhelical interface, the resulting interhelical bridges being responsible for the stability enhancement. The higher stabilisation measured for the E3 sites may also indicate that the third Glu could participate in binding.
In addition, the authors evaluated the stability of the reduced peptides. These were less stable than their oxidised counterparts, highlighting the importance of the disulphide bridge for the stability of the scaffold. The addition of LaCl3 resulted in an important stabilisation, which was attributed not only to La3+-binding but also to a La3+-driven dimerization. Hence, the most stable Ln3+-binding constructs were achieved thanks to the presence of a disulphide bridge to form exclusively a 2SCC, and to Ln3+-binding residues at the helical interface.
In a following work, Hodges and co-workers turned to γ-carboxyglutamic acid (Gla, Fig. 6), with two carboxylate functions, in order to (i) enhance the Ln3+-affinity of the coiled coil thanks to the higher denticity of Gla vs. Glu, and (ii) design a system that folds upon metal-binding as the higher electrostatic repulsion induced by Gla was expected to lead an unfolded apo-peptide.95 In order to further destabilise the apo-peptides, they mutated a Val residue (V) in position 23 by an Asn (N) (Table 3). The resulting peptide in its oxidised form, Gla2(15,20)N, was fully unfolded at 20 °C pH 7. The peptide folded upon the addition of 0.5 mM LaCl3, the amount of coiled coil formation increasing from ∼3 to 100% to match the one of the native Asn.
Three-stranded coiled coils (3SCC). A few years later, Kashiwada and coworkers used the same Gla residue for Ln3+-binding, but inserted it in a three-stranded coiled coil (3SCC) scaffold.96 In their design, the Gla residue was placed in an a layer of Pep1 (Table 4) so that the Ln3+-binding site would be buried in the hydrophobic core rather than placed at the helical interface. Due to the insertion of the Gla residue in the sequences, the resulting peptide Pep3 was unfolded at 20 °C, pH 7. Circular dichroism experiments showed that the addition of 1 equivalent of Ln3+ (Ce3+, Eu3+, Tb3+) per trimer drove the assembly and folding of the coiled coil.
Ashkenazy and coworkers investigated an alternative strategy, by placing the Ln3+-binding site at the N-terminal of the 3SCC (Fig. 8 and Table 4), which has the advantage of not perturbing the folding and assembly of 3SCC.97 They coupled a hydroxyphenol oxazoline moiety (HPO) to the peptide through a triazole linker. HPO acted both as a chelating moiety and as a sensitizing antenna.
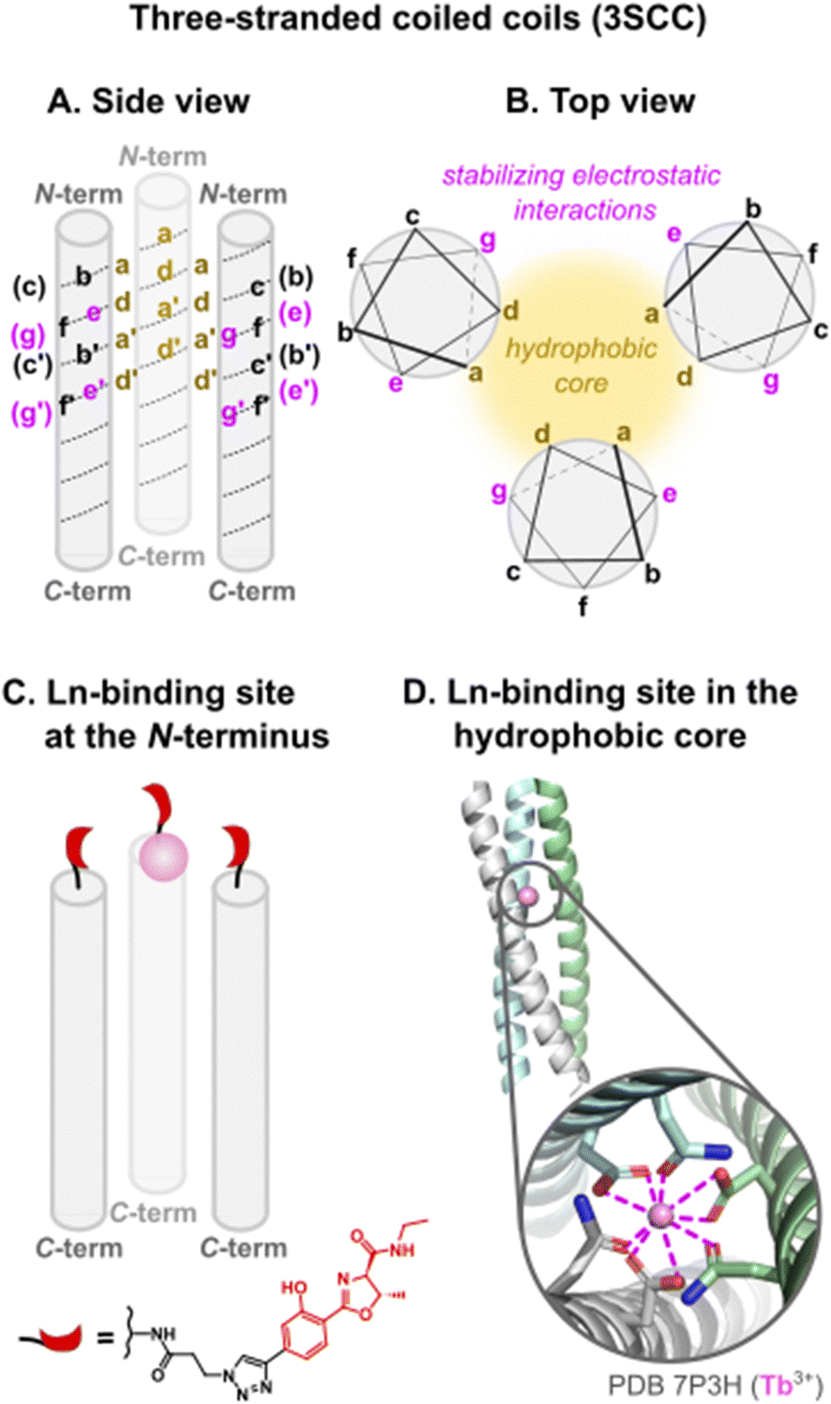 | ||
| Fig. 8 Three-stranded coiled coils scaffolds: (A) side view, (B) top view, (C) Ln-binding site placed at the N-terminal with a hydroxyphenol oxazoline (HPO) ligand, (D) Ln-binding site in the hydrophobic core (a and d positions). In (B) stabilising non-covalent interactions responsible for the folding and assembly of coiled coils are highlighted. | ||
The group of Peacock contributed to the design of Ln3+-binding 3SCCs and provided a detailed understanding of the influence of Ln3+-binding site position, Ln3+-binding residues, second sphere effects, and scaffold size on Ln3+-affinity, selectivity, and hydration number (Table 5).98–102,104 They also reported the design of a heterobimetallic 3SCC bearing an Ln3+-binding site and a Hg-binding site.92
The first peptide designed by the Peacock group, MB1-2, was a parallel 3SCC made of the repetition of 5 heptads and with an (Asn)3(Asp)3 metal binding site with Asn residues in a d layer, and Asp residues in the following a layer (Table 5).98 The peptide also contains Trp residues to sensitize Tb3+ emission (see 6.1 Luminescent tags and probes).
The positioning of the Ln3+-binding site within the hydrophobic core of the 3SCC destabilised the peptide, which was unfolded in the apo-state but assembled and folded upon Ln3+-binding (Fig. 9). The hydrophobic environment and the adequate coordination sphere ((Asn)3(Asp)3) provided a suitable CN to the Ln3+ ion, preventing water from binding directly to the metal ion (q = 0).
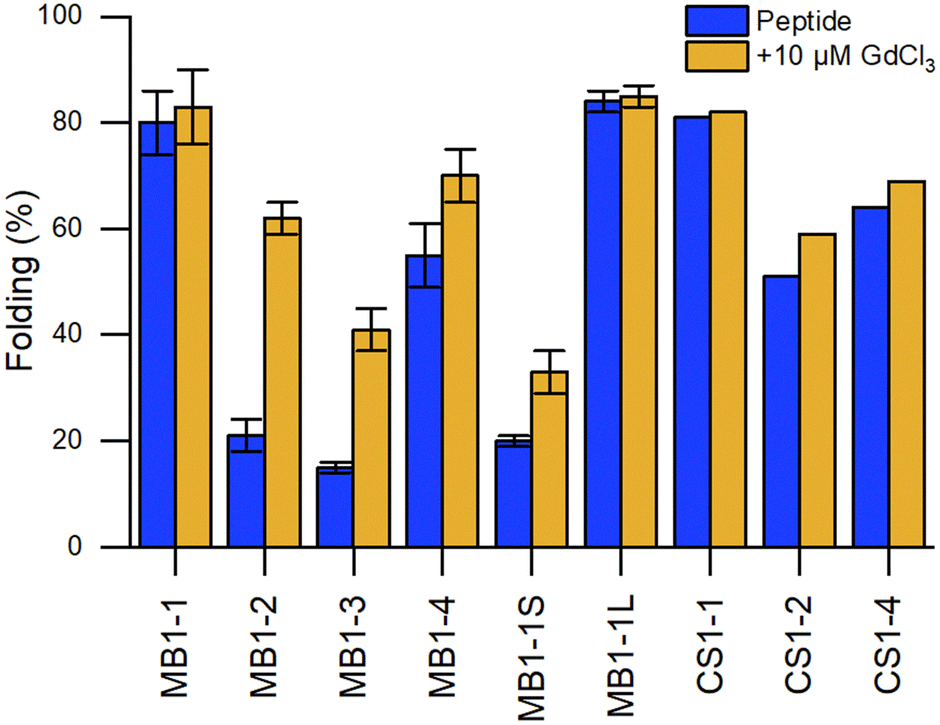 | ||
Fig. 9 Comparison of the percentage of folding of 3SCC.99,102 Peptide sequences are given in Table 5. Experiments were performed in 5 mM HEPES, pH 7, 293 K, [pep] = 30 μM, without or with 10 μM GdCl3. The percentage folding was calculated based on: 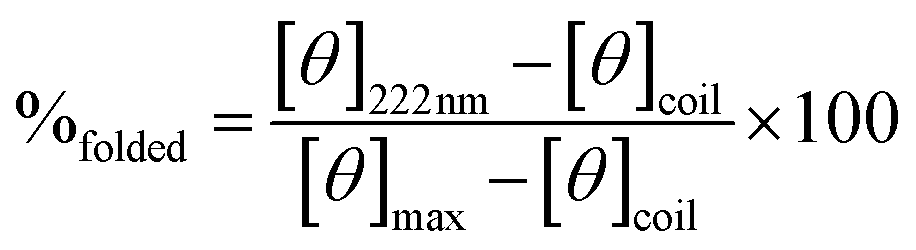 , with [θ]222nm the molar ellipticity at 222 nm, [θ]coil the molar ellipticity for a random coil, and , with [θ]222nm the molar ellipticity at 222 nm, [θ]coil the molar ellipticity for a random coil, and 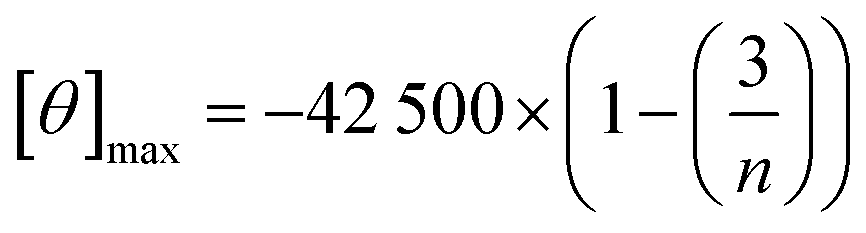 the theoretical maximum molar ellipticity, where n is the number of residues in the sequence.105 the theoretical maximum molar ellipticity, where n is the number of residues in the sequence.105 | ||
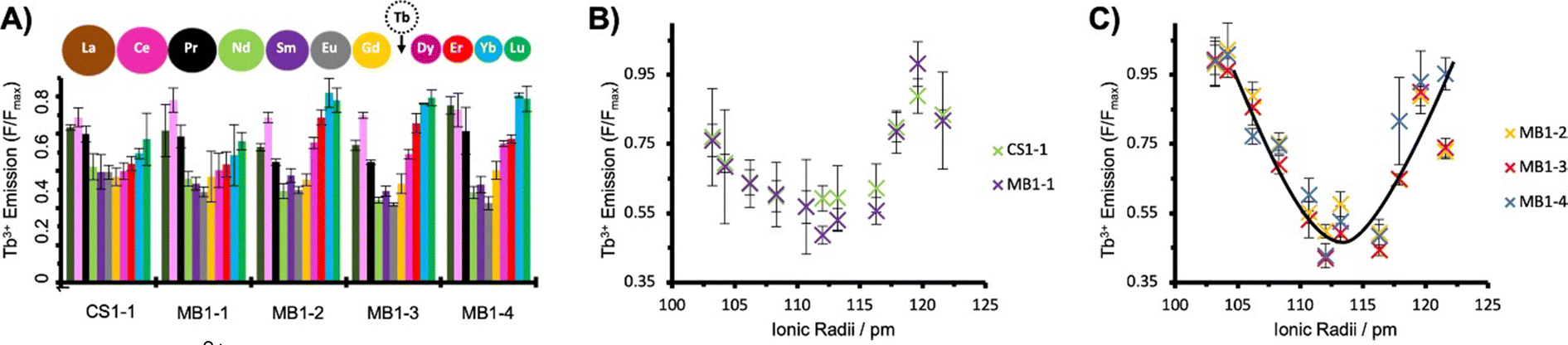
Building on this work, the group of Peacock investigated how the position of the binding site in the 3SCC scaffold and the type of binding site ((Asn)3(Asp)3vs. (Asp)3) would impact Ln3+ coordination and hydration number.99 With this aim, they extended the MB1 series containing an (Asn)3(Asp)3 binding site with peptides MB1-1, MB1-3 and MB1-4, and designed the CS1 series containing an (Asp)3 binding site with peptides CS1-1, CS1-2 and CS1-4 (Table 5). In the MB1 series, the position of the metal-binding site impacted differently the stability of the apo-peptides (Fig. 9). Whereas positioning the metal-binding site in the middle of the scaffold destabilized the apo-peptides (21% and 15% folded for MB1-2 and MB1-3, respectively), positioning at the C-terminal was better tolerated (55% folded), and when placed close to the N-terminal the apo-peptide seemed almost unaffected (80% folded). For the CS1 series, in which there is no top (Asn)3 layer, the apo-peptides were always more stable than their MB1 analogues (Fig. 9). As was previously observed for MB1-2,98 Gd3+ stabilised the 3SCC assembly and led to the formation of 1:3 Ln3+:peptide complex. However, the authors noted that for CS1-2 and CS1-4, there was only a small improvement of folding upon Tb3+-binding compared to their MB analogues (Fig. 9), and only a small increase in Tb3+-emission which suggested that these peptides may bind Tb3+ through non-specific interactions.
Depending on the position of the binding site, the number of Tb3+-bound water molecules varied from 4 (CS1-1) and 3 (MB1-1) for the more solvent-exposed sites at the N-terminal, to 2 (MB1-4) for the site at the C-terminal, to 0 (MB1-2 and MB1-3) for sites more buried into the scaffold (Table 5). These results demonstrated that it is possible to control the number of Ln3+-bound water molecules by controlling the position of the metal binding site in the 3SCC. The more solvent-exposed sites at the N- and C-termini had the higher hydration number, which suggested a change in Asp coordination mode (monodentate vs. bidentate) or that not all Asp and Asn were involved in Ln3+-coordination. On the other hand, the sites that were more buried within the hydrophobic core of the 3SCC were shielded from the solvent. This is consistent with the (Asn)3(Asp)3 site fulfilling the preference of Ln3+ for a high coordination number. The recent publication of the crystal structure of HC02 (PDB 7P3H), a 3SCC similar to MB1-2, confirmed that Tb3+ is nona-coordinated by three Asn (monodentate) and three Asp (bidentate) as shown in Fig. 8.101
In later works, Peacock and coworkers investigated both the effect of the peptide length on the overall stability of the assembly102 and of second-sphere effects on the hydration number (Table 5).100 Whereas the shorter MB1-1S was too destabilised to bind GdCl3, there was no marked difference in peptide folding or hydration number between MB1-1 and the longer MB1-1L (Fig. 9), and only a qualitative enhancement of stability for MB1-1L compared to MB1-1. One explanation proposed was that the binding site is too far (4–5 nm) from the sixth heptad to detect any positive effect on folding, Ln3+ affinity and hydration number.
In order to investigate 2nd sphere effects on the hydration number, core Ile (I) residues placed in the a layer above the metal-binding site of MB1-1 were mutated to Ala (A), Phe (F), Tyr (Y) or Trp (W) (MB1-1(2X) series, Table 5). Tuning of the steric hindrance provided control of the hydration number of Tb3+ that varied from q = 3 (MB1-1, MB1-1(2A)), to 2 (MB1-1(2F)), to 1 (MB1-1(2Y)), to 0 (MB1-1(2W)). This strategy was also successfully applied at the C-terminal for MB1-4 site, which was mutated to MB1-4(37W) in order to shield Tb3+ from the solvent, which decreased the hydration number from 2 to 0.
The results obtained by Peacock and coworkers highlight that with the same set of coordinating AAs, the number of Ln3+-bound water molecules can be controlled either by controlling the position of the Ln3+-binding site within the scaffold or by tuning second sphere residues thanks to steric effects. The replacement of core amino acids by Ln3+-binding residues resulted in the destabilisation of peptide folding and assembly that depended on the position of the site within the scaffold and could be in part compensated for by Ln3+-binding.
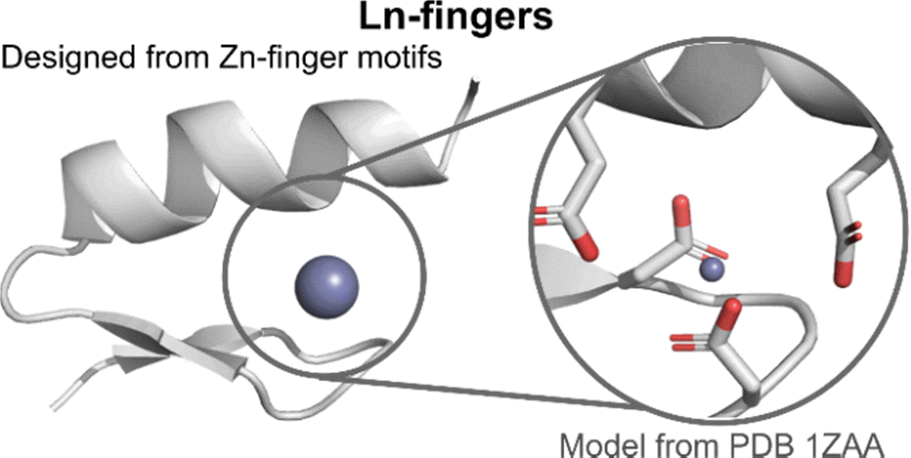 | ||
| Fig. 10 Lanthanide fingers. A model of the hypothetic structure and binding site of Ln-fingers was generated from a closely related zinc finger structure (PDB 1ZAA) using Pymol. Zn is represented as a grey sphere. | ||

|
The (His)2(Cys)2 Zn2+-binding site of Zn-fingers was replaced by an (Asp)2(Glu)2 binding site, more suited for Ln3+-binding. The LF scaffold was redesigned to favour the formation of secondary structures, leading to LF1. Modifications included notably the use of α-helix or β-sheet inducer amino acids and of a type II′ β-turn (DPro-Ser) to favour the formation of the β-hairpin. The addition of an N-cap (LF2) and a C-cap (LF3), which are motifs favouring the formation of α-helices,106–108 improved the folding. The extent of stabilisation of peptides fold following these modifications was qualitatively assessed by comparing CD spectra obtained for apo-peptides. The influence of the size of the binding site on Ln3+-binding was investigated by opening up the site thanks to the deletion of two AAs in the sequence (LF4), as well as the type of Ln3+-coordinating residues (LF7). Genetically encodable LFs were designed by replacing the type II′ β-turn (DPro-Ser) with Thr-Ile (LF5 and LF6).
A detailed NMR analysis of LF4 without and with Lu3+ showed rearrangements throughout the peptide upon Lu3+-binding, which suggested that, as for Zn-fingers, Ln-fingers fold upon metal binding. For all the sequences, Ln3+ binding improved the folding of the peptides and the data obtained were consistent with a 1:1 peptide:Ln3+ ratio. Hydration numbers were not determined, but H2O molecules are likely to participate in Ln3+ coordination sphere in addition to the Ln3+-binding AAs.
2.5 Other Ln-binding protein scaffolds
Ln-ADH. The discovery of Ln-utilizing bacteria has sparked a great interest in the identification of the lanthanome.9–12 The first Ln-binding protein discovered in the 2010s was an enzyme: a pyrroloquinoline quinone (PQQ)-dependent methanol dehydrogenases (MDH) encoded by the gene xoxF, whose active site also contains an Ln3+ taking part in the catalysis.4–7 A characteristic of this enzyme is that it is directly involved in the metabolism of the organisms that produce it, mostly methylotrophic bacteria, which are hence the sole organisms known so far for which Ln3+ can be essential elements.9 Following this discovery, several Ln-MDH were isolated and their crystal structures were reported with either La3+,109,110 Ce3+,7 Nd3+,111 or Eu3+
112 (ref. 112) in the active site (Fig. 11). In addition, the family of Ln-dependent enzymes was extended to ethanol dehydrogenases (EDH) following the identification of ExaF113 and PedH114 in methylotrophs and non-methylotrophs.115 The crystal structure obtained for a Pr3+-EDH showed that the active site of this enzyme shared a lot of similarities with the one of Ln-MDH, including conserved residues involved in Ln3+- and PQQ-binding.116 Due to these similarities, Ln-MDH and Ln-EDH are often grouped under the term Ln-alcohol dehydrogenases (Ln-ADH).
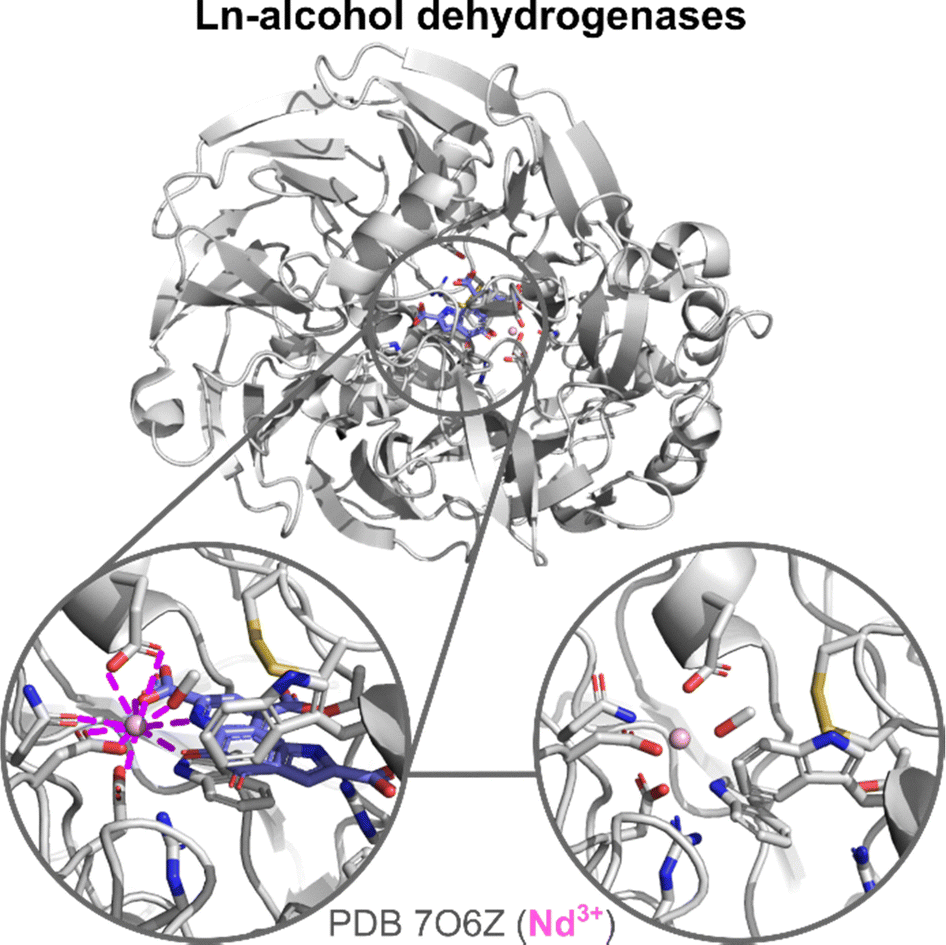 | ||
| Fig. 11 Ln-ADH structure and active site. Zoom on the active site and Ln-coordination sphere of crystals obtained with the PQQ cofactor (blue) in the active site (left) and without it (right). In addition to Ln-binding AAs and PQQ, a methanol molecule is also bound to Nd3+. | ||
The active site of Ln-ADH is closely related to other PQQ-dependent alcohol dehydrogenases, whose active site contains a Ca2+ ion, but which are encoded by different genes (mxaF for Ca-MDH, exaA and pedE for Ca-EDH).9 The main difference between the active sites of Ca- and Ln-ADH is the presence of an additional Asp residue in the Ln3+ coordination sphere. Crystal structures showed that in Ln-ADH, the Ln3+ ion is nona-coordinated by two Asp (one monodentate, one bidentate), a Glu (bidentate), an Asn (monodentate), and the PQQ cofactor (tridentate), as shown in Fig. 11. For some structures, an additional ligand (PEG,7 MeOH111) is also found coordinated to the Ln3+, which suggests that there is a free site for substrate coordination to the metal ion, giving a total coordination number of 10.
It is not clear how the mature Ln-ADHs are formed, especially how they acquire their two cofactors, PQQ and Ln3+, and how these impact the stability of the scaffold. Based on the obtention of crystal structures with only Ln3+ (and not PQQ) in the active site,110,111 it has been proposed that Ln3+ could be loaded independently of PQQ. However, it is also possible that PQQ leaks out of the enzyme pocket during the crystallization steps. The comparison of the holo-MDH (with Ln and PQQ) and Ln-only-MDH (no PQQ) shows nearly identical structures (Fig. 11).110,111 Ln is coordinated by the same residues in both cases. Residues in interaction with PQQ show minimal rearrangement in the holo-MDH relative to the Ln-only MDH, which suggests that the MDH active site may be predisposed for PQQ binding.
On the other hand, ADH enzymes can also be obtained with PQQ-only in their active site. Indeed, several papers report on the purification of Ln-ADH with 0.4 to 1 equivalent of Ln per protein.6,7,109–113,117,118 Moreover, Daumann and coworkers demonstrated that upon incubation of partially metallated MDH with Ln3+ the activity of the enzyme could be improved, which suggests the in situ reconstitution of a functional enzyme in which PQQ was already present.112,119 Martinez-Gomez and coworkers also observed the partial metallation of a Nd-MDH, without change in the PQQ content of the enzyme.117 However, with their conditions, the reconstitution of a holo-enzyme by incubation with Nd3+ was unsuccessful.
Working on a parent enzyme, the PQQ- and Ca2+-dependent soluble glucose dehydrogenase (sGDH), Stines-Chaumeil, Limoges and coworkers demonstrated that reconstitution of the holo-enzyme could follow either a path where PQQ is added first to the active site or one where Ca2+ is bound first.120 Their results evidenced that the kinetic of reconstitution depended on the order of addition of the two cofactors. The reconstitution was fast when the protein was metallated with Ca2+ before the binding of PQQ, and slow if PQQ binding happened first. Although little is known about the mechanism and kinetics of Ln-ADH reconstitution, the two reconstitution paths proposed for sGDH could also be considered for Ln-ADH.
The data on Ln-ADH stability are scarce. Some data show that Ln-MDHs are sensitive to temperature and denature at temperatures higher than 50 °C.111,112 However, the exact value at which denaturation occurs may be influenced, among other factors, by the organism Ln-MDH is purified from, as the enzyme obtained from the extremophile M. fumariolicum SolV has an optimum temperature for catalytic activity at 60 °C.7 In addition, the thermal stability of Ln-MDH could be Ln-sensitive, as suggested by Nakagawa and coworkers who compared the thermal stability of a La- and a Nd-MDH and found that the latter denatured at lower temperatures although it is not clear what could cause such a difference.118
Lanpepsy (LanP). Following the discovery of ADH, other Ln3+-binding proteins were identified: lanmodulins (LanM, see above), lanpepsy (LanP), and landiscernin (LanD also referred to as LutD, see below).121 LanP is composed of two PepSY domain.122 Studies on deletion strains suggested that LanP function is not directly linked to Ln3+ sensing or uptake. The 3D structure predicted by AlphaFold suggested that the folded protein forms a cavity with several negatively charged residues. Competitive titration experiments determined that LanP could bind up to 3–4 Ln3+ ions, which was confirmed by ICP-MS, although a higher number of sites was determined by ITC titration (∼6). The exact binding sites and coordination environments of Ln3+ in LanP still need to be uncovered.
Landiscernin (LanD). A role for LanD in the acquisition of Ln3+ ions was first suggested in 2019.123,124 Following these preliminary works, the protein was isolated and crystallized by Cotruvo and coworkers.125 It forms a three-helix bundle stabilised by a disulphide bridge and hydrophobic interactions at its core. Several negatively charged residues are found at its surface, close to the N-terminal. Crystal structures obtained with 0.5 equivalents of Ln3+ (La, Ce, Eu, and Ho) per protein showed the formation of a Ln3+-binding site at the interface between two proteins made of three Glu residues from each protein (Glu70, Glu73, Glu75), which except for this coordination site do not interact with each other (Fig. 12). Interestingly, as the Ln3+ ionic radius decreases, the coordination number decreases from 9 (La3+, Ce3+) to 8 (Eu3+, Ho3+), due to the loss of a Ln3+-bound water molecule for the latter (Fig. 12).
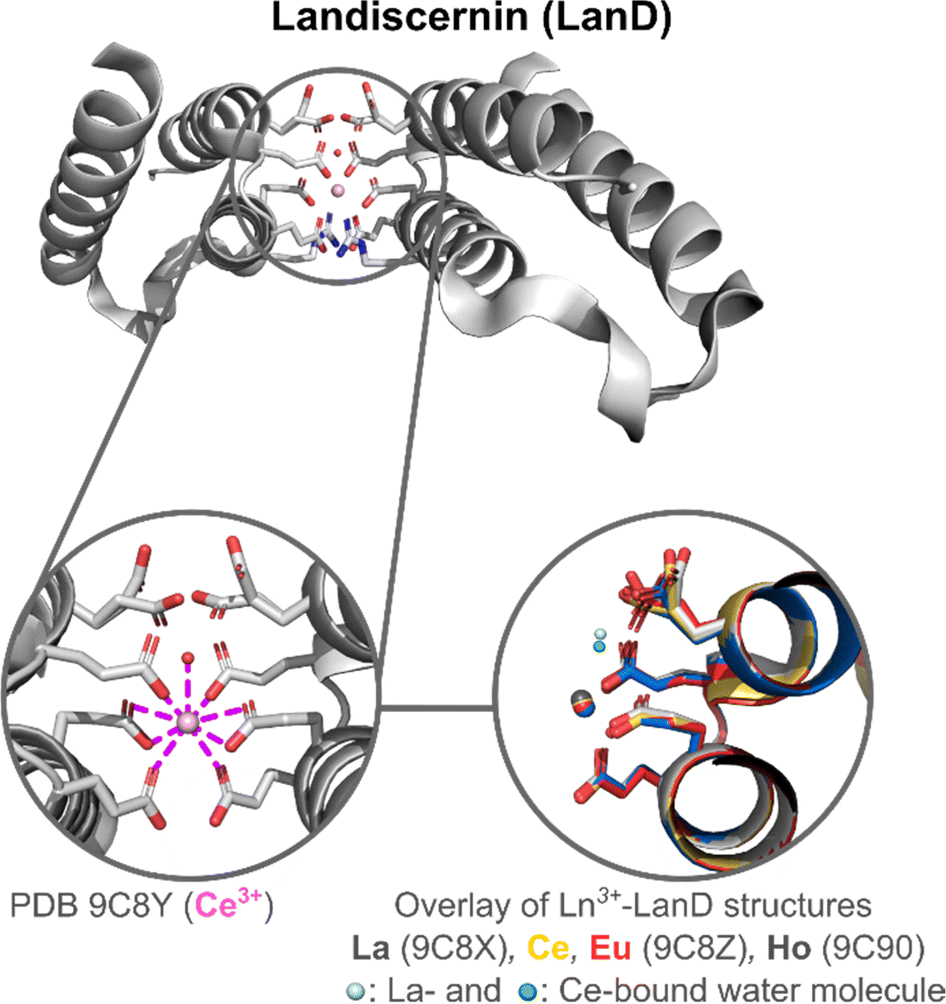 | ||
| Fig. 12 Landiscernin (LanD) structure and active site. Zoom on the Ln3+ coordination sphere (left). Overlay of Ln3+–LanD structures (Ln3+ = La, Ce, Eu, Ho) was generated with Pymol (right). A Ln3+-bound water molecule is only present in the structure of La3+–LanD and Ce3+–LanD. Two glutamate residues adopt different orientations in the crystal structures and their rotamers are represented. They do not participate in Ln3+-binding but are important for dimer formation. | ||
In solution, the protein was shown to be monomeric in the apo-state with a dissociation constant for the dimer of Kdimer = 610 μM but was differently influenced by metal ions. Whereas La3+, Ce3+ and Nd3+ seemed to favour the dimerization of the protein (Kdimer = 117, 200 and 253 μM, respectively), this was not the case with Eu3+ and Ho3+ (Kdimer = 1400 and 700 μM, respectively). Based on the crystal structures obtained, the authors proposed that as the ionic radius of the metal ion decreases, electrostatic repulsions due to the presence of negative charges on the surface of the proteins increase and disfavour the dimerization.
PqqT. The discovery of Ln-utilising bacteria has also inspired the field of artificial enzyme design, with two lead examples recently published. Olshansky and coworkers aimed at designing a Ln-ADH mimic, which could serve as a model to study the mechanism of the enzymes.20 Analysis of the X-ray structure of PqqT, a PQQ-binding protein, showed that several residues near PQQ may be predisposed for Ln-binding, including Glu111, Asp141 and Asn143 (Fig. 13). In addition, a lysine (Lys142) interacts through electrostatic interactions with PQQ with its side chain positioned in the PQQ pocket where Ln3+ binds. Mutation of Lys142 (K142) to an Asp afforded the mutant PqqT–K142D that binds 1 PQQ (model structure in Fig. 13) and 1 Ln3+ ion. In the engineered active site, PQQ was required for the binding of La3+ and hence participated in its coordination sphere. In addition to Asp142, other residues (Glu111, Asp141, Asn143) may also participate in Ln-binding, which still needs to be confirmed. The mutant containing La3+ and PQQ, noted La3+–PQQ⊂PqqT–K142D, was able to oxidize benzyl alcohol under aqueous conditions (see 6.4 Catalysis).
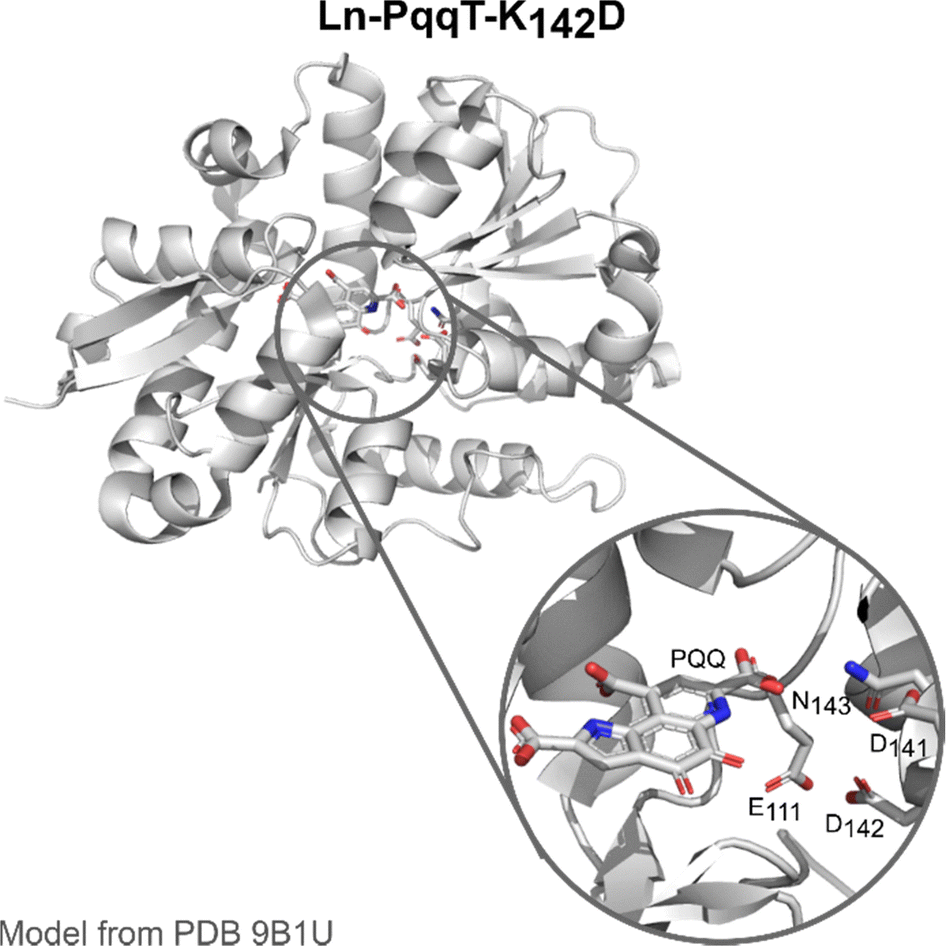 | ||
| Fig. 13 Ln–PqqT–K142D mutant bound to PQQ and its possible Ln3+-binding site (zoom). Model structure was generated from the crystallographic structure of PQQ-bound PqqT (PDB 9B1U) using the mutagenesis tool in PyMOL. | ||
TIM barrel. The second example of an Ln artificial enzyme came from Zeymer and coworkers. Starting from a computationally designed de novo TIM barrel scaffold,126 they aimed to design an enzyme for Ce4+ photoredox catalysis (see 6.4 Catalysis).19
The initial scaffold was designed by combining a TIM domain made of eight parallel β-strands surrounded by eight α-helices, and a ferredoxin (FD) insert domain, either with a (His)2(Glu)2 or with a (Glu)4 binding site.126 The stability of the scaffolds with these two binding sites was assessed by thermal and chemical denaturation. The two scaffolds showed a high stability with temperature (Tm > 95 °C), and unfolded in two steps with increasing concentration of guanidinium chloride, each event corresponding to the unfolding of one domain (TIM and FD). X-ray structure of the apo-protein scaffold showed a pre-disposed (Glu)4 binding site. The hydration number of Tb3+ was determined using a third mutant with a Trp for Tb3+ sensitisation. In this mutant, Tb3+ was bound by one water molecule. X-ray structure showed a CN = 9 for Tb3+, with the four Glu acting as bidentate ligands (Fig. 14).
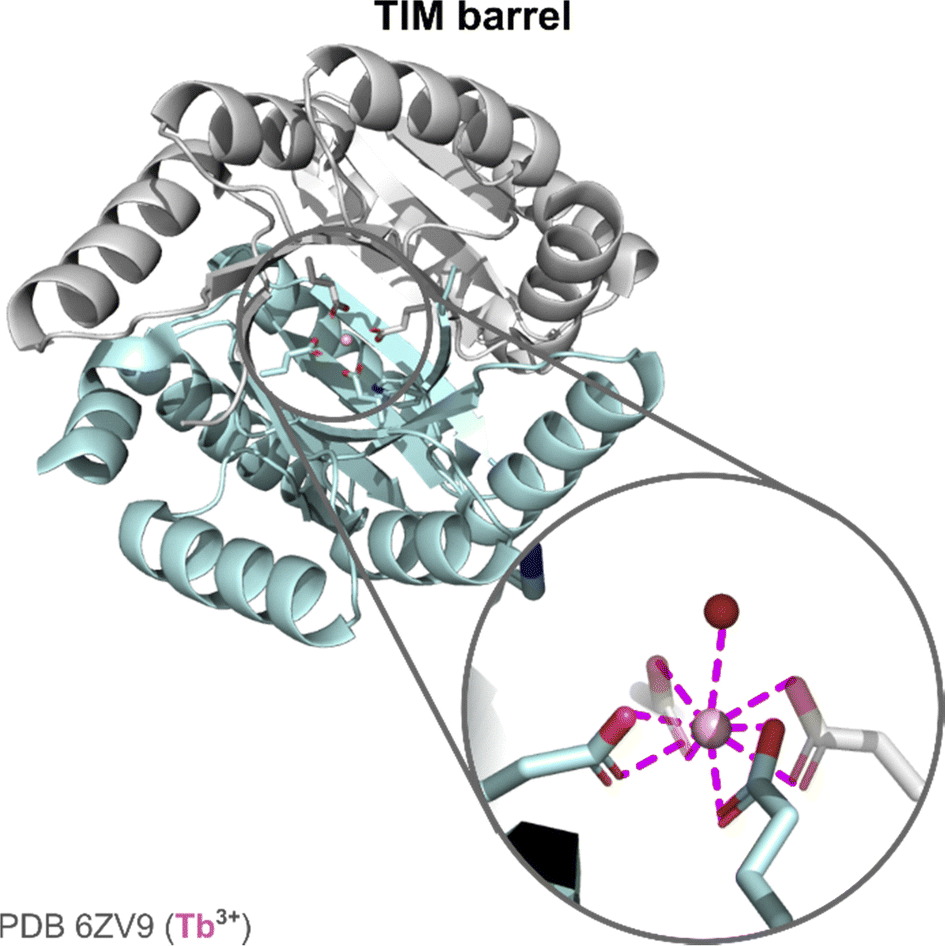 | ||
| Fig. 14 TIM barrel structure and active site. Zoom on Ln-coordination sphere showing the presence of a Ln3+-bound water molecule. | ||
RTX. RTX corresponds to the block V of the repeats-in-toxins (RTX) domain of adenylate cyclase.127–129 It is a Ca2+-binding protein that can bind up to eight Ca2+ ions. In the apo-state, the protein is intrinsically disordered and folds upon metal binding. The folded protein adopts a parallel β-helix structure. Ca2+-binding sites are located between turns of successive layers and are composed of one Asp residue and other donor atoms from the main chain to provide CN = 7. Recently, the capacity of RTX to bind Ln3+ was investigated more thoroughly for potential applications in Ln3+ recycling (see 6.3 Ln recovery and separation).129 Circular dichroism experiments demonstrated that Ln3+ induced the folding of the protein. However, it also suggested that the protein might adopt a different structure relative to the Ca2+-induced folding, and the exact Ln-coordination sphere in this protein is still unknown.
PTE-R18. PTE-R18 is a mutant from a Zn2+-dependent phosphotriesterase enzyme (PTE) designed for the hydrolysis of 2-naphthyl hexanoate. Jackson and coworkers investigated the use of this scaffold for Ln3+-binding to design an Ln–enzyme and exploit the Lewis acid properties of Ln3+ (see Catalysis).21 Crystal structures obtained by co-incubation of the apo-protein and Ln3+ (Eu, Gd) showed the positioning of the Ln3+ ion in the Zn2+-binding site coordinated by two His, one Asp (bidentate), a water molecule and a chloride ion (CN = 6). This low CN is surprising given the preference of Ln3+ for a higher coordination number, and could explain the low occupancy (∼20%) of the metal-site by Ln3+.
2.6 Discussion and guidelines
A large variety of scaffolds were reported for Ln3+-binding, which range from peptide sequences of a few AAs to proteins, that either fold upon metal-binding or that are pre-folded. For unfolded scaffolds or pre-folded scaffolds of intermediate size, additional stabilising interactions beyond the binding site are required to obtain a stable assembly, which in turn may improve Ln affinity (see Thermodynamic stability). In particular, the design of pre-folded scaffolds with a predisposed Ln3+-binding site using short or intermediate-size peptides requires finding a balance between the stability of the structure, which relies mostly on hydrophobic interactions, and the introduction of metal-binding residues that can destabilise the scaffold. With this aim, two strategies were employed, sometimes in combination: (i) to put the metal binding site at a position that ensures a minimal perturbation of the scaffold (e.g. on the surface vs. inside the hydrophobic core of coiled coils, or at different positions within the hydrophobic core of coiled coils); (ii) to select amino acids known for their propensity to favour the formation of specific secondary structures. Overall, only a few studies investigated in detail the stability of their constructs and the impact of Ln3+-binding site incorporation and Ln3+ binding. These data could be helpful to guide the optimisation of scaffolds and validate the strategies followed.All Ln3+ coordination sites reported meet Ln3+ preference for a high coordination number (CN 8–10) and hard Lewis bases. This includes either natural (Asp, Glu, Gla) or unnatural (Adan, Ed3a2) negatively charged AAs and other AAs with O-donor atoms either in their side chains (Asn, Gln) or main chain (carbonyl of peptide bond). When such high CN cannot be achieved solely by the peptide or protein residues, water molecules are found coordinated to Ln3+.
Control over the hydration number is important for applications in imaging (see 6. Applications). A few general principles can be drawn upon the analysis of the CN and q of EF-hand motifs with distinct amino acid patterns (identity and position of residues 1, 3, 5, 9 and 12), which constitute the larger database on this topic. First, in EF-hand the presence of Glu9 (rather than Asp9) warrants a coordination sphere devoid of water molecules (q = 0), which, as mentioned above, can be sought to optimize luminescent emission (see 6.1 Luminescent tags and probes). Instead, Asp9 is never directly involved in Ln3+-binding, but instead hydrogen-bonded to an Ln3+-bound water molecule. Furthermore, higher CN (9–10) are observed in LanM, which are proteins evolved by Nature to bind Ln3+ in physiological conditions, relative to Ca2+-binding proteins or engineered LBTs (CN = 7–8). This does not stem merely from a change in the pattern of coordinating amino acids, but rather from second-sphere factors including a fine-tuning of backbone conformation and orientation of amino acid side chains. From the analysis of the data obtained with non-EF-hand scaffolds, two additional general principles can be underlined. First, the denticity of Ln-binding residues impacts q, as is seen with short peptides (e.g. Adanvs. Ed3a2) and coiled coils (e.g. Glu vs. Gla). Second, the environment in which the Ln-binding site is placed participates in the tuning of q, as evidenced in 3SCC, for which Ln3+-hydration state depends on the position of Ln3+-binding site in different regions (e.g. the more solvent-exposed termini vs. the buried hydrophobic core of the coiled coils), and on second-sphere effects (e.g. tuning of steric hindrance of second sphere residues). Thus, to summarize, control over the hydration number can be achieved by tuning three key parameters: (i) the denticity of Ln-binding residues and the length of their side chain; (ii) the position of the binding site in the scaffold; and (iii) the steric hindrance of second sphere residues. In addition, works on EF-hand motifs underline the influence of a fourth key parameter, more difficult to control, which is the spatial positioning of AAs side chains that allows AAs to bind in a mono- or bi-dentate fashion, and so participate in the fine-tuning of Ln3+ hydration number.
3. Thermodynamic stability
3.1 Generalities
 | (3) |
Nevertheless, conditional stability constants, cK, at a given pH can be derived from the stability constant K taking into account the protonation constants (Ka,n) of the ligand (eqn (4)).
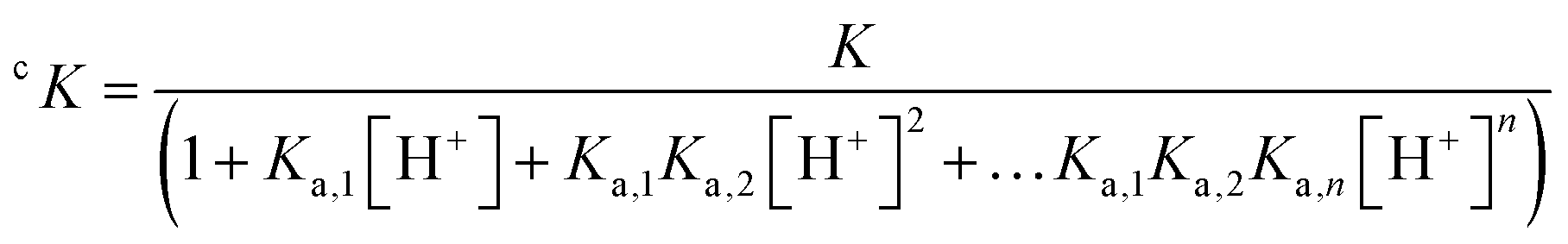 | (4) |
| cK = Kapp(1 + cKcompCcomp + cKbufferCbuffer) | (5) |
Another important point to keep in mind is that when working with peptides and proteins, the techniques used to determine the apparent stability constant can monitor the metal binding and/or the induced folding of the scaffold. For instance, CD (circular dichroism) can only inform indirectly on metal binding, since this technique measures metal-induced changes in the scaffold folding. Variations of chemical shifts in NMR spectroscopy are due to both metal-binding and subsequent conformational changes; similarly, ITC (isothermal titration calorimetry) measures the heat exchanged for all events happening during the titration, including conformational changes and metal-binding. To some extent, luminescence titration is also dependent on folding and metal binding. Ln3+–luminescence increases upon Ln3+-binding due to the substitution of water molecules. Thus, in general, Ln3+–luminescence is also sensitive to induced local folding events (i.e. the formation of the binding site), but not to conformational changes that are more distant.
It is important to underscore that Kapp values should be compared only if they were determined in the same conditions (metal and ligand concentration, buffer, pH, etc.) and through the same technique, and refers to complexes with the same metal–ligand stoichiometry. For a more accurate comparison of complexes with different metal–ligand stoichiometry or different metal/ligand concentrations, the use of pM (eqn (6)) would be recommended.132,133
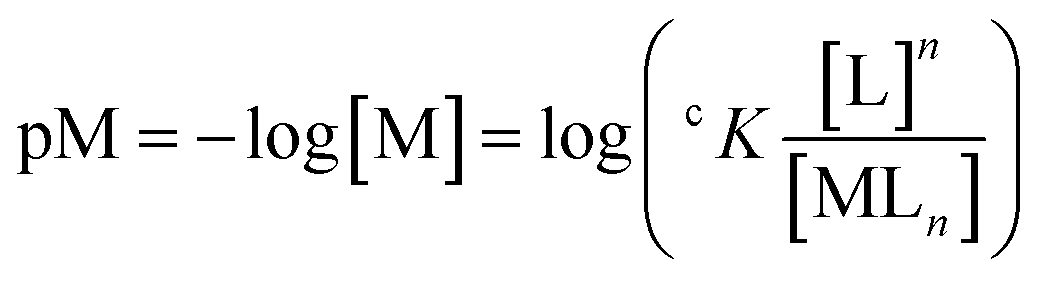 | (6) |
:1, 1:2 Ln3+–buffer complexes as well as [Ln(buffer)(OH)n] species. The stability constants (logβ) reported for 1:1 Ln3+–buffer complexes range between 3 and 4, but contrasting and hence no reliable values have been reported for the same Ln3+–buffer system.
β) of 1:1 Ln3+–buffers systems
| Ln3+ | Buffer | |||||
|---|---|---|---|---|---|---|
| MOPSO | MES | PIPES | HEPES | POPSO | MOPS | |
|
a Ref. 134.
b Ref. 135.
c Ref. 136.
d Values have been calculated by correcting the logβ reported in ref. 137 for the buffer pKas (logβ + pKa): MES, 6.27; PIPES, 7.14; HEPES, 7.56; MOPS, 7.18.138
|
||||||
| La3+ | 3.34a | 3.40a | n.d. | 3.33a | n.d. | n.d. |
| Ce3+ | 3.31a | 3.36a | n.d. | 3.40a | n.d. | n.d. |
| Pr3+ | 3.36a | 3.39a | 4.11b | 3.44a | n.d. | n.d. |
| 4.18b | 4.26b | |||||
| Eu3+ | 3.39a | 3.38a | 4.22b | 3.43a | 3.28c | 3.68d |
| 4.24b | 4.27b | 2.51c | 3.04c | 3.68d | ||
| 2.57d | 3.04d | 3.4d | ||||
| Gd3+ | 3.27b | 3.8b | 3.57b | n.d. | n.d. | n.d. |
| Dy3+ | 4.09b | 4.19b | 4.03b | n.d. | n.d. | n.d. |
More recently, the interaction of some buffers with Eu3+ was investigated employing NMR and time-resolved laser-induced fluorescence spectroscopy (TRLFS).137 However, it is not clear how the values found in this study were determined. Overall, there is little agreement on the stability constants of Ln3+–buffers systems, for which reliable values need to be determined. Notwithstanding, given the range of their stability constants, buffers could compete with peptides and proteins with low affinity for Ln3+ ions. Therefore, the choice of the pH buffer and its concentration should be considered carefully.
Furthermore, buffer molecules could replace loosely bound water in the coordination sphere of Ln3+ complexes, leading to the formation of ternary species, which has been investigated only in a few cases. For instance, Delangle and coworkers compared the number of Ln3+-bound water molecules (hydration number, q) of a Tb3+-cyclic decapeptide complex in HEPES buffer (10 mM, pH 7) and in the absence of buffer at pH 6.5.41 In both cases, they obtained similar Tb3+ luminescence lifetimes and hydration numbers (τH2O = 1.65 ms, τD2O = 6.99 ms, and q = 2.0 in HEPES buffer; τH2O = 1.68 ms, τD2O = 6.28 ms and q = 1.9 in absence of buffer), suggesting the absence of buffer molecules in the first coordination sphere of Tb3+.
Kapp ∼ 2–13; Fig. 15). Table 8 gives some examples of logKapp for Tb3+ around neutral pH. Most of the scaffolds, regardless of their origin (natural or artificial) and size, have an affinity in the micro- to nanomolar range (logKapp = 5–9). The highest affinity is exhibited by large scaffolds, such as the natural LanM81 and the de novo designed TIM barrel protein (TFD-EE N6W),126 and by short peptides (6 amino acids) featuring multidentate unnatural amino acids.87 In the following paragraphs, several factors influencing the affinity of each scaffold will be described and discussed.
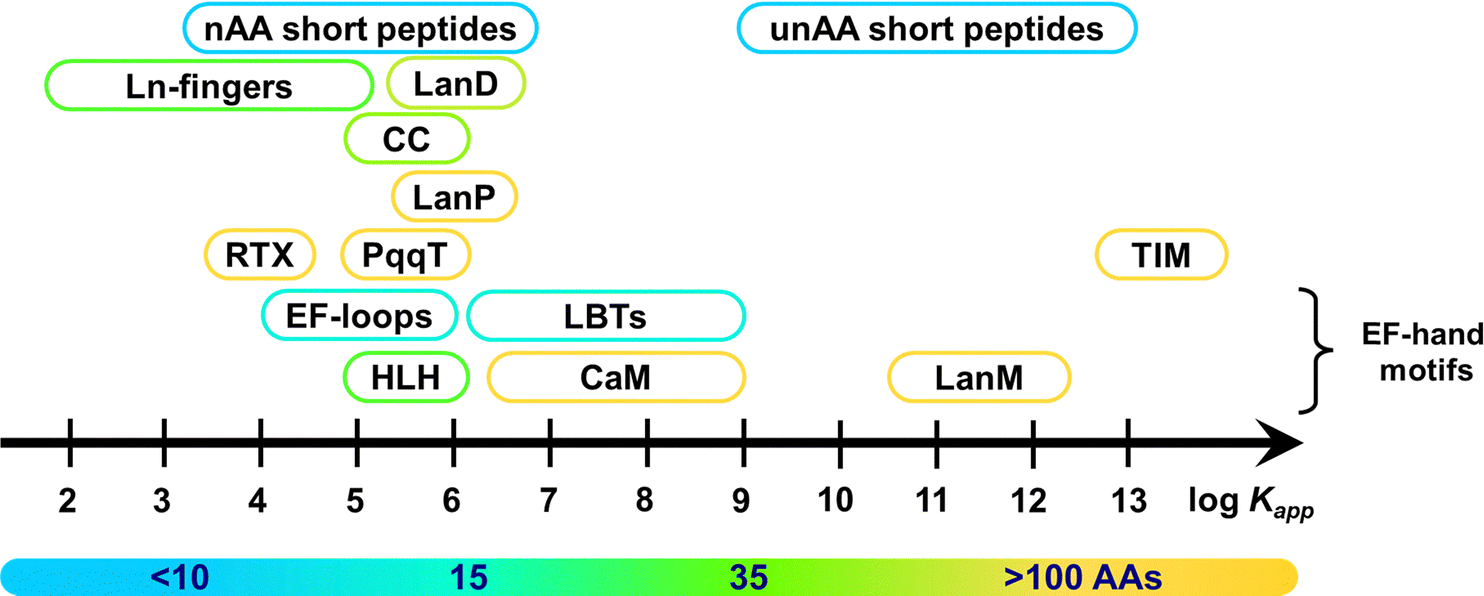 | ||
| Fig. 15 Affinity range of different Ln3+-binding peptide and protein scaffolds around neutral pH (6–7.5). Colour code is based on the scaffold size (as in Fig. 2). Abbreviations: nAA, natural amino acids; unAA, unnatural amino acids; CC, coiled coils; LBT: Ln-binding tag; HLH, helix-loop-helix. | ||
| Scaffold (peptide/protein) | logKapp (Tb3+) |
Ref. |
|---|---|---|
| a Bis-Tris 20 mM pH 6, NaCl 25 mM. b HEPES 10 mM pH 7.5, NaCl 10 mM. c HEPES 10 mM pH 7, NaCl 100 mM. d HEPES 10 mM pH 7. e HEPES 10 mM pH 6.9. f HEPES 10 mM pH 7. g PIPES 25 mM pH 6.8, KCI 100 mM. h HEPES 10 mM pH 7, NaCl 100 mM. i MOPS 30 mM pH 7.2, KCl 100 mM. j HEPES 10 mM pH 7, NaCl 100 mM. k HEPES 25 mM pH 7.5, NaCl 100 mM. | ||
| RTX | 4.6a | 129 |
| Ln-fingers (LF4) | 4.9b | 103 |
| EF-hand (LBT1) | 5.1c | 139 |
| 3SCC (MB1-1) | 5.3d | 99 |
| HLH (P3W) | 5.4e | 80 |
| Cyclic decapeptide (PA) | 6.3f | 41 |
| CaM (bovine, site I) | 8.8g | 82 |
| LBT–protein conjugate (ILB1β-L3) | 8.9h | 65 |
| LanM (Mex-LanM) | 11.1i | 63 |
| unAA short peptides (PHD2) | 12.7j | 87 |
| TIM barrel (TFD-EE N6W) | 13.1k | 126 |
3.2 EF-hand motifs
Thanks to the large variety of natural and engineered EF-hand motifs, it is possible to evaluate the effect of several structural factors influencing their affinity for Ln3+ ions.Kapp ≈ 5). Natural Ca2+-binding proteins and LBT–protein conjugates bind Ln3+ ions with at best logKapp ≈ 8–9 at pH ∼ 7. LanMs display the highest affinity (logKapp ≈ 10–12 at pH ∼ 7) among naturally-occurring Ln3+-binding scaffolds. The structural determinants of such enhanced affinity are not clear. Based on the current literature,62,81,83 it seems that it results from second-sphere effects, including different loop backbone conformation and hydrogen bonding networks that ensure an optimal orientation of coordinating amino acids within the loop.
The loss of 5 orders of magnitude in Tb3+ affinity when EF1-3 loops are isolated from their native Mex-LanM scaffold (ΔlogKapp ≈ 5, see entries 1 and 2, Table 9) highlights the momentous impact of a suitably folded, but not necessarily pre-disposed (note the LanM is disordered in the apo-state) scaffold on the affinity.
The important role of the core helical bundle in LanM has been further underscored by the recent study of a Mex-LanM fragment encompassing the highest-affinity and cooperative EF2-3 domains devoid of flanking helices, which showed much weaker affinity (logKapp < 5) than the whole protein.140 Besides, another recent study has explored the impact of replacing EF-loops in CaM with those found in Mex-LanM.141 Interestingly, this chimaera (LanM-GCaMP) showed a weak conformational response to Ln3+ (from the micro- to the milli-molar range, depending on the Ln3+). Nevertheless, the authors were able to obtain a modified CaM scaffold (LanTERN) with a conformational transition similar to LanM (logKapp = 10.3 for La3+) by introducing key mutations in CaM loops that are considered responsible for enhanced Ln3+ selectivity in LanM (i.e. Pro2 and Asp9, see Section 3.2). Altogether, this evidence suggests that to further boost the Ln3+-binding affinity of EF-hand proteins, the loop sequence, including non-coordinating amino acids, and the protein folding must be optimised simultaneously.

|
Furthermore, as already mentioned for natural proteins, the insertion of LBT within a protein scaffold increases the affinity up to logKapp ≈ 8–9 (entries 8–9 in Table 9). Of note, the rigidity of the regions flanking the loops is also crucial to achieving such an enhancement of the affinity, which is not accomplished, for instance, in the chimeric helix-loop-helix domains (logKapp ≈ 5–6) likely due to the high flexibility of the flanking helices (entry 11 in Table 9).80
The pattern of coordinating amino acids in the EF loop also influences the affinity. Notably, the affinity increases of more than 1 order of magnitude (entries 5–6 vs. 4 in Table 9) when (i) Asn3 is mutated to Asp (entries 5 vs. 4 in Table 9), likely due to the more negative charge of Asp relative to Asn, and (ii) Asp9 is replaced by Glu (entries 6 vs. 4 in Table 9), which serves as bidentate ligands displacing the Ln3+-bound water hydrogen-bonded to Asp9.
In order to develop kinase/phosphatase- and nitration-responsive probes (see 6.1.5 Probe design and selected applications), the group of Zondlo has also widely explored EF-hand motifs featuring phosphorylated and nitrated amino acids, including phosphotyrosine (pTyr), phosphoserine (pSer), phosphothreonine (pThr) and 3-nitrotyrosine (nTyr). These studies have shown that Glu9 can be replaced by pSer, pTyr and nTyr with minor impact (ΔlogKapp < ±1) of the EF-loop affinity (entries 1–5 in Table 10).144–146 A similar effect was also observed when Asp residues in positions 1, 3 or 5 were replaced by Cys sulfinic acid (entries 7–9 in Table 10).147 Remarkably, an affinity gain of about 1 order of magnitude was reported when Asp9 was replaced by pThr (entry 6 in Table 10).148 Interestingly, a similar affinity gain was also observed for C-terminally truncated 9-residues loops bearing pThr9 or pSer9 (entries 10–12 in Table 10).148 Unfortunately, the lack of structural studies prevents a detailed description of the Ln3+ coordination sphere in these modified EF-hand motifs. Noteworthy, the impact of phosphorylated or nitrated amino acids in natural EF-hand proteins has not been investigated yet.

|
With respect to Ln3+ binding to phosphorylated peptides, it is also worth pointing out that a micromolar affinity for Tb3+ (logKapp = 6.5 at pH 7) has been reported for a fragment of the protein α-synuclein (119–132) with an EF-hand-like amino acid pattern and bearing a pTyr residue (Asp1, Asp3, Glu5, pTyr7, Glu12).149
3.3 Other scaffolds
Among the short peptides reported in the literature (cyclic or linear), the one with the best affinity was obtained when using unnatural amino acids for Ln3+-binding (PHD2 Ada2 and Ed3a2, logKapp = 12.7, Fig. 5).87 Compared to Glu and Asp, these unnatural amino acids have a higher denticity, which is more favourable for Ln3+ binding. Furthermore, the peptide backbone designed to adopt a β-turn-fold upon metal binding provides additional stabilising interactions that make the peptide a better ligand than aminodiacetate groups separated by long alkyl chains (n > 2), but not as good as ligands such as EDTA for which the formation of 5- and 6-membered chelate rings result in a large stabilising chelate effect.85
The influence of the coordination sphere on the apparent affinity constants is evidenced for LF scaffolds, in which modification of the coordination sphere from (Asp)2(Glu)2 (LF4) to AspAsn(Glu)2 (LF7) decreased the apparent affinity constant for Eu3+ from logKapp = 4.6 to logKapp = 4.0. It is also apparent in the 3SCC designed by Peacock and coworkers when comparing the affinity of MB1-1 and CS1-1 for Tb3+ (logKapp = 5.3 vs. 4.6, respectively).99 The additional (Asn)3 layer in MB1-1 contributes to the slightly higher affinity observed relative to CS1-1. However, a change of hydration number for a given coordination sphere does not impact the apparent affinity constant, as can be seen in the 3SCC MB1-n series (n = 1–4), which have comparable affinities for Tb3+ (logKapp = 5.2–5.5) and hydration number ranging from q = 0 (MB1-2 and MB1-3) to q = 3 (MB1-1).99
The influence of denticity on Ln-affinity is well illustrated by the 2SCC described by Hodges when using Gla instead of Glu.95 The affinity of E2(15,20)ox has a logKapp ≈ 2 for La3+, whereas the one of Gla2(15,20)Nox has a logKapp = 6.2 for La3+, which can be linked to the higher denticity of the latter.
Finally, the link between scaffold stability and Ln-affinity was investigated in the design of the Ln-finger series. Changes in the sequence to improve the formation of secondary structures resulted in improved apparent affinity constants from logKapp < 2 for Eu3+ of LF1, to logKapp = 3.7 of LF3.103 Increasing the size of the binding site by deletion of two AAs (LF4) gave the best apparent affinity constant for Eu3+ with logKapp = 4.6. In the MB1 series shifting the position of the binding site within the 3SCC scaffold impacted the stability of the assembly (see 2.4.1 Coiled coils).99 However, this did not significantly impact the apparent affinity constants for Tb3+, which were all in the range logKapp = 5.2–5.5. The addition of an additional heptad in MB1-1L also did not impact the apparent affinity constant for Tb3+ (logKapp = 5.0).102
Kapp ranging from 4 to 13. This large range of affinities can be explained by Ln3+-binding sites that are either not fully optimised for Ln3+-binding (e.g. in Ca2+–proteins), or found in natural proteins whose biological function may require affinity in an intermediate range (e.g. LanP, LanD).
The Ca2+-binding protein RTX has an apparent affinity constant for Ln3+ in the range logKapp = 4.1–4.6 at pH 6, and may bind 4 to 7 Ln3+ per protein.129 Note that these values were obtained for a fusion protein, in which RTX is fused with two fluorescent proteins for detection of folding upon metal binding by FRET. The affinity of LanP for Ln3+ is logKapp = 6.0 and this protein binds 3 to 4 Ln3+.122 It was identified in Ln-utilizing organisms and due to its Ln3+-binding ability, it was hypothesized that LanP could be of importance in Ln-dependent methylotrophy. Similarly, the apparent affinity constant of the monomeric protein LanD for Ln3+ ranges from logKapp(La3+) = 5.7 to logKapp(Nd3+) = 6.6.125 Because LanD and LanM are part of the same gene cluster involved in Ln3+ transport and utilisation, it was proposed that LanD could be responsible for transferring Ln3+ to LanM. This hypothesis was supported by the metalation of the modified fluorescent LaMP1123 in the presence of La3+–LanD, and by ITC titration experiments that showed a specific 1:1 interaction between apo-Mex-LanD and apo-Mex-LanM with a dissociation constant of 4 μM of physiological relevance. As of now, little is known about the coordination environment of Ln3+ in these proteins, except for LanD, and the stability of these scaffolds. This does not allow the identification of specific features that could explain the affinity range observed.
When the protein scaffold and Ln3+-binding site are optimised, high affinity can be obtained by design, as is the case for the computationally designed TIM barrel that displayed the highest affinity for Ln3+.126,150 The affinity of the mutant with a (Glu)4 binding site and Trp for Tb3+-sensitisation (TFD-EE N6W) was first suggested to be logKapp ≈ 15 by competitive titration with EGTA.126 However, when the same experiments were reproduced with a higher concentration of Tb3+–EGTA relative to TFD-EE N6W and longer equilibration times, the apparent affinity constant for Tb3+ was rather found to be logKapp = 13.1.150 Of note, this stressed the importance of taking into account kinetic considerations (see 5. Kinetic stability), since when protein metalation, or metal ion exchange with a given competitor, is slow, reaching the thermodynamic equilibrium may take days as it is the case for this example. Nevertheless, it also shows that in a rigid scaffold (Tm > 95 °C) with a predisposed binding site suitable for Ln3+-binding, high affinity can be achieved by rational design.
Ln-ADH and its mimics stand apart since Ln-binding depends not only on the protein but also on the presence of the cofactor PQQ. Early work on Ln-ADH extrapolated apparent affinity constant based on activity assays and predicted an affinity in the low μM range for Ln3+ in the presence of PQQ (logKapp ∼ 6).112,119 More recently, Zeymer and coworkers studied the affinity of a PedH mutant for Ln3+ in the absence of PQQ in the active site.19 They determined an apparent affinity constant logKapp = 7.0 for Tb3+ by luminescence titration, and logKapp = 6.2 for Ce3+ based on competition with Tb3+. Similarly, for the engineered PqqT scaffold, a logKapp = 6.2 was obtained for the mutant PqqT–K142D containing PQQ (noted PQQ⊂PqqT–K142D).20 This represents a 10-fold improvement compared to the wild-type scaffold (PQQ⊂PqqT) and to the PQQ⊂PqqT–K142A mutant. The presence of PQQ inside the protein was required for Ln3+-binding, as demonstrated by controlled experiments performed with the apo-scaffolds (PqqT, PqqT–K142A, and PqqT–K142D).
3.4 Effect of the pH
Ln recovery requires ligands with high affinity and selectivity for Ln3+ ions at pH ranging from pH < 1 to pH = 5–6 depending on the source (see 6. Applications).151,152 The capacity to bind Ln3+ ions in acidic conditions is an overlooked feature of peptides and proteins, mainly because (i) protein folding is generally sensitive to acidic pH and (ii) typical Ln3+-binding residues (Asp, Glu) become protonated below pH 3–4, weakening Ln3+ binding.Nonetheless, some works investigated the pH sensitivity of their scaffolds. 3SCC loose their Ln3+-binding ability below pH ∼ 4–5.92 CaM and LBTs start releasing Ln3+ below pH 6 and show no binding below pH 4.153,154 Similarly to small chelators such as EDTA and DTPA, LanMs are able to bind Ln3+ ions down to pH ∼ 2.5, retaining approximately nanomolar affinity at pH 4 (logKapp ≈ 8–9) and 5 (logKapp ≈ 9–10).155 The reason behind the higher pH stability of LanM relative to CaM and LBTs is not understood. Interestingly, the RTX scaffold has been recently reported to retain partial Ln3+-binding below pH 2. The protein could recover up to 20% of Nd3+ from a synthetic NdFeB magnet solution containing Nd, Dy, Fe and Co at pH < 1.129
On the other end of the pH scale, studies on 3SCC scaffolds also showed that such moderate-affinity ligands undergo partial de-complexation of Ln3+ at basic pH (> ∼8.5), likely due to competing formation of lanthanide hydroxides.92
More systematic studies will be necessary to understand the different pH-dependence of Ln3+ binding to different peptide and protein scaffolds.
3.5 Discussion and guidelines
Among natural peptide and protein scaffolds, LanM stands out for its logKapp ≈ 10–12. Although the structural factors for this remarkably high affinity have not been fully elucidated yet, the constraint imposed by the global protein scaffold, even if not pre-folded, is paramount. Indeed, isolated EF-loops with the same potential coordinating residues are much weaker ligands.
All rationally designed scaffolds with natural amino acids (i.e. short peptides, LFs and CCs), except for the TIM barrel (logKapp(Tb3+) = 13.1), have lower or at best comparable, affinity than natural scaffolds. As was noted earlier, these small to intermediate scaffolds are sensitive to the insertion of a metal-binding site, which destabilises the structure and in turn, may decrease Ln3+-affinity. Peptides tend to be flexible and explore a large conformational space. Ln3+-binding may improve the folding of the peptide; however, it is generally not sufficient by itself to compensate for the entropic cost of restricting the peptide conformational space. Optimisations of the peptide sequence to improve the formation of secondary structures can help fold the peptide, and hence, increase the affinity for Ln3+. This is observed qualitatively by the successive optimisations performed on Ln-fingers. It is also the case for the de novo designed TIM barrel, whose packing was optimised to rigidify the structure, and which displays a high stability (Tm > 95 °C) and affinity for Ln3+. Similarly, even though LanMs are not pre-folded, their affinity for Ln3+ is coupled to the formation of a thermally stable (Tm > 95 °C) folded protein. Nonetheless, there seem to be limitations to this, as increasing the size and stability of 3SCC (MB1-1 vs. MB1-1L) did not result in an impact on Ln3+-affinity.102 This could be due to the position of the binding site at the N-terminal, whereas the elongation was at the C-terminal. Interestingly, the correlation between the stability of the scaffold and metal-binding affinity has been observed for other metal-binding peptides. Studies performed on β-sheet WW-domain-like metal binding mini-proteins showed an improved affinity for Zn2+, Cu2+ and Ni2+ by improving the stability of the scaffold.156 Moreover, a study on Hg- and Cd-binding coiled coils demonstrated a direct correlation between the stability of the coiled coil scaffold and metal ion affinity.157 Altogether, these data validate the strategy of optimising the scaffold structure in order to improve Ln3+-affinity.
An alternative strategy is to use unnatural amino acids. Indeed, affinities comparable to that of LanM or TIM barrel were achieved in very short peptides (6 AAs) by combining Adan and Ed3a2 (PHD2, logKapp(Tb3+) = 12.7). Even in this case, the primary sequence played an important part since residues that favour the formation of a β-turn upon metal-binding were chosen to bring additional stabilising interactions.
4. Selectivity
The selectivity of peptide and protein scaffolds for Ln3+vs. other metal ions is a key aspect for their applications in both environmental and biological samples, as well as for understanding Ln biochemistry. In particular, the selectivity among Ln3+ and against actinides (An3+) is of interest for Ln3+ mining and separation, while the selectivity for Ln3+ over Ca2+ and other physiological cations (e.g. Zn2+ and Cu2+) is particularly relevant in the biological context.The selectivity between two different metal ions can be quantified by calculating the selectivity factor, ΔlogK (or Δlogβ), which corresponds to the difference between the affinities of a certain ligand for distinct metal ions. In the following, we will express the Ln3+-selectivity, ΔlogKLn (or ΔlogβLn) as the difference between the lowest and the highest logKLn along the series (eqn (7)).
| ΔlogKLn = logKLnmax − logKLnmin | (7) |
KCa (ΔlogβCa), is expressed as the difference between the logKLn for a certain Ln3+ and logKCa (eqn (8)).| ΔlogKCa = logKLn − logKCa | (8) |
4.1 Selectivity among Ln3+
Prior to peptides and proteins, it is useful to discuss the selectivity of small chelators (Fig. 16 and 17A), which can be classified into four types.158–161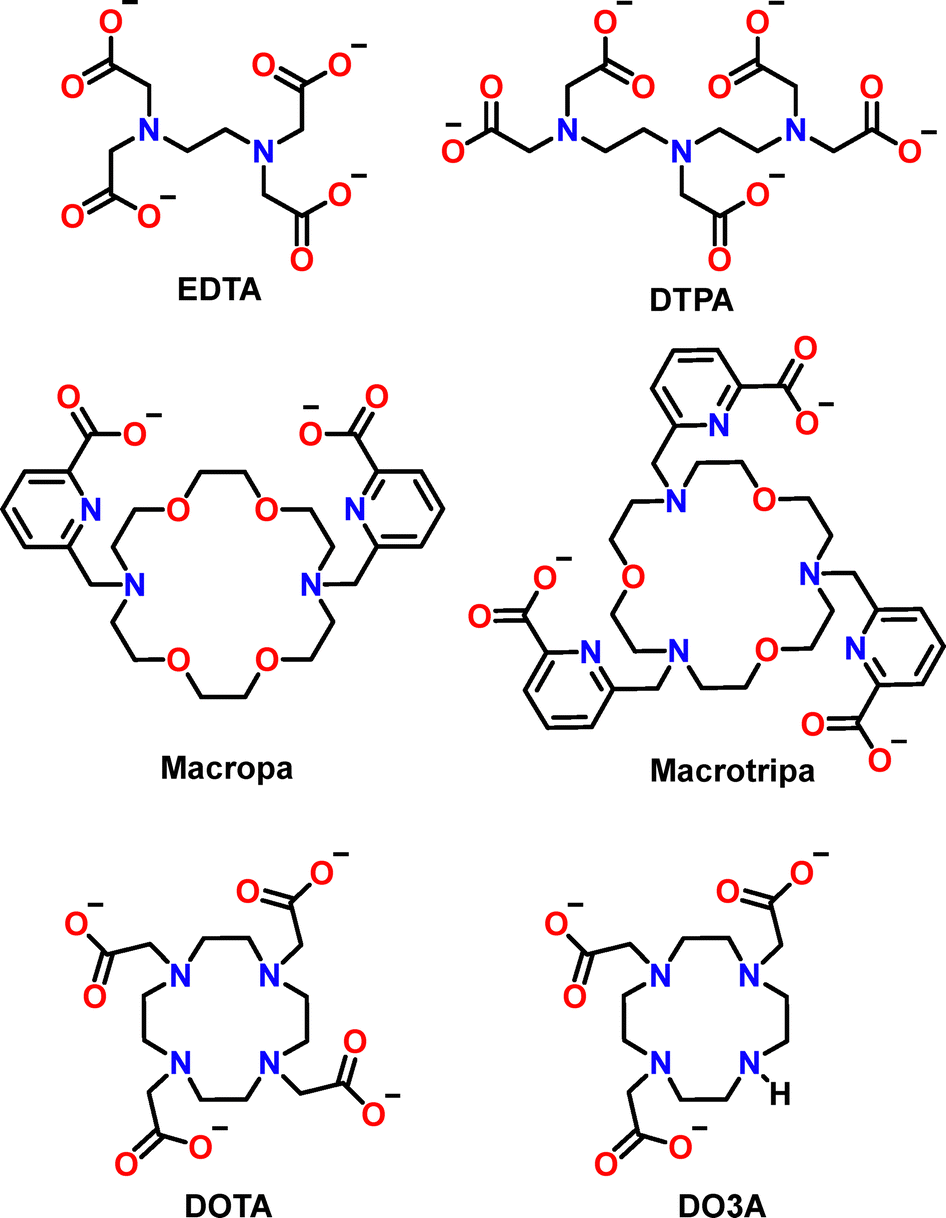 | ||
| Fig. 16 Chemical structure of the Ln3+ chelators mentioned in the text. | ||
 | ||
| Fig. 17 Selectivity of different ligands across the Ln3+ series. (A) Small chelators and (B) peptides and protein scaffolds. Note that logβ and logKapp are reported in (A) and (B), respectively. Values for LanM are referred to (i) Mex-LanM at pH 7.2 measured by CD spectroscopy; (ii) Mex-LanM at pH 5 measured by CD spectroscopy; (iii) Hans-LanM at pH 5 measured by CD spectroscopy; (iv) Mex-LanM at pH 5 measured by UV-vis spectroscopy. The values plotted are reported in Table 15. | ||
Most ligands, including EDTA (Fig. 16), show increasing stability constants across the Ln series (type I, black dots, Fig. 17A), commensurate with the increase of Ln3+ Lewis acidity. For other ligands, such as DTPA and DOTA (Fig. 16), the affinity increases along the first part of the series, reaches a maximum and then remains constant or even decreases for late Ln3+ ions (type II, red dots, Fig. 17A). This is the result of the ligand steric hindrance, which impairs to accommodate the smaller Ln3+ ions.158,159,162
An uncommon behaviour is shown by a few ligands, including Macropa (Fig. 16), for which a reverse-size selectivity is observed due to a fairly rigid scaffold better suited for larger than smaller Ln3+ ions (type III, blue dots, Fig. 17A).160 Finally, a biphasic selectivity trend (type IV, green dots, Fig. 17A) has been recently reported with ligands such as Macrotripa (Fig. 16) that are better suited for both early and late Ln3+ due to a switch between a 10-coordinated conformation accommodating larger Ln3+ ions and an 8-coordinated conformation stabilizing smaller Ln3+ ions.161,163
Overall, the selectivity trend among Ln3+ ions results from a compromise between electronic and steric effects, whose balance varies depending on the properties of the ligand, such as its denticity and rigidity. Moreover, the behaviour of these small chelators points out a common misconception: Ln3+ are not all the same, especially when bound to a chelator. Indeed, a remarkable difference can be observed in the stability constants of certain ligands for distinct Ln3+ ions: for instance, EDTA and Macropa show selectivity for Lu3+vs. La3+ (ΔlogβLn) of ∼5 and ∼7 orders of magnitude, respectively.
The Ln3+-selectivity of only a few peptides and protein scaffolds has been reported (Fig. 17B). LBTs, Ln-fingers, RTX and LanD display a type II selectivity (Fig. 17B). The affinity of LBTs increases by almost 2 orders of magnitude from La3+ up to Ln3+ in the middle of the series (Eu3+–Tb3+) and then slightly decreases towards the end of the series.64 Since LBTs were derived from Ca2+-binding loops, their preference for middle-sized Ln3+ ions (1.04–1.07 Å, CN = 8 in LBTs) could be explained by the closer similarity to Ca2+ ionic radius (1.06 Å, CN = 7 in EF loops).
Ishida and coworkers employed molecular dynamics simulations, as well as ITC and NMR measurements, in order to elucidate the selectivity trend of LBTs.164 The authors found that one or two water molecules can be accommodated in the coordination sphere of large Ln3+ ions (La3+–Nd3+); this weakens the binding between the Ln3+ and Asn5 and enhances the flexibility of the complex, resulting in reduced affinity. For Ln3+ from Sm3+ to Lu3+, water binding is rarely observed, correlating with the higher affinity for these smaller ions.
For LF4, the affinity increases by nearly 1 order of magnitude from La3+ to Er3+, then decreases back towards Lu3+, which was found to have the same affinity as La3+.103 A similar trend is shown by RTX and LanD proteins, but in this case the ΔlogKLn measured is much lower (<1).125,129
The relative selectivity of 3SCC among Ln3+ has been also reported based on luminescence measurements of Tb3+ displacement by other Ln3+ ions.101 A bell-shaped selectivity trend was found for several 3SCC scaffolds that differ for the location of the Ln3+-binding site along the helices (MB1 series, Fig. 18 and Table 5). In particular, no significant discrimination was observed among medium-sized Ln3+ ions (Nd3+–Tb3+) for all scaffolds. For competing ions smaller than Tb3+, size-dependent discrimination was observed with scaffolds where the binding site is located around the centre or the C-terminal of the coiled coil. The higher promiscuity of the N-terminal binding site was attributed by the authors to its greater flexibility, which hence allows it to better accommodate also smaller Ln3+ ions.101
 | ||
| Fig. 18 Tb3+–displacement experiments followed by luminescence in three-stranded coiled coils showing a bell-shape selectivity within the Ln3+ series (MB1 series, Table 5). (A) Luminescence displacement, (B) Comparison of CS1-1 and MB1-1, (C) Comparison of MB1-2, MB1-3, and MB1-4. Reproduced from ref. 101. | ||
LanP122 and PqqT20 do not display significant selectivity among early Ln3+ ions (La3+–Gd3+; late Ln3+ were not studied). The TIM barrel TDF-EE N6W binds Eu3+, Gd3+, and Tb3+ with comparable affinity, while ∼10-fold weaker binding was observed with Ce3+.126
Among Ln3+-binding peptides and proteins, LanM represents a peculiar and controversial case. In most reports by the groups of Cotruvo and Daumann, an unusual preference of LanM for larger (La3+–Eu3+) over smaller Ln3+ ions has been underscored based on CD and luminescence measurements,81,123,165 which are responsive to both Ln3+ binding and Ln3+-induced conformational change (Fig. 17B, LanM(i) and LanM(ii)). It must be noted again, that similarly to RTX and 3SCC, Mex-LanM shows a very modest Ln3+-discrimination (ΔlogKLn ∼ 0.5) relative to small chelators and LBTs (Fig. 17B). An enhanced selectivity (ΔlogKLn ≈ 1.6) for early (La3+, Nd3+) vs. late Ln3+ (Dy3+) was found for Hans-LanM (Fig. 17B, LanM(iii)), thanks to a Ln3+ size-dependent dimerization.62
Curiously, the opposite trend, i.e. an affinity increase across the end of the Ln3+ series, was observed when the intrinsic Ln3+-affinity of Mex-LanM was determined via UV-vis-NIR spectrophotometric competition experiments (Fig. 17B, LanM(iv)).155 As suggested by the authors,166 this could highlight a decoupling between Ln3+-binding and conformational change in LanM, which warrants further investigations, and underscores the importance of the method and the conditions chosen to determine and compare affinity values. Nonetheless, higher retention of early vs. late Ln3+ ions was observed for immobilized LanM upon pH-induced desorption.62,167,168 Such (at least apparent) inconsistency between the affinity values measured via different techniques and in different conditions is worth further systematic investigations.
Finally, it is also worth mentioning that LanM showed higher affinity for the rare-earth element Sc3+ relative to Ln3+ ions (3-fold higher relative to Nd3+),167 commensurate with its smaller ionic radius and higher Lewis acidity.
4.2 Selectivity over Ca2+
Peptide and protein scaffolds generally show selectivity for Ln3+ relative to Ca2+ (and Mg2+) which is classically attributed to their higher charge and coordination numbers (Fig. 19). In addition, as Ln3+ have higher CN than Ca2+ in water, Ln3+-binding releases a higher number of water molecules, resulting in a more favourable entropic change.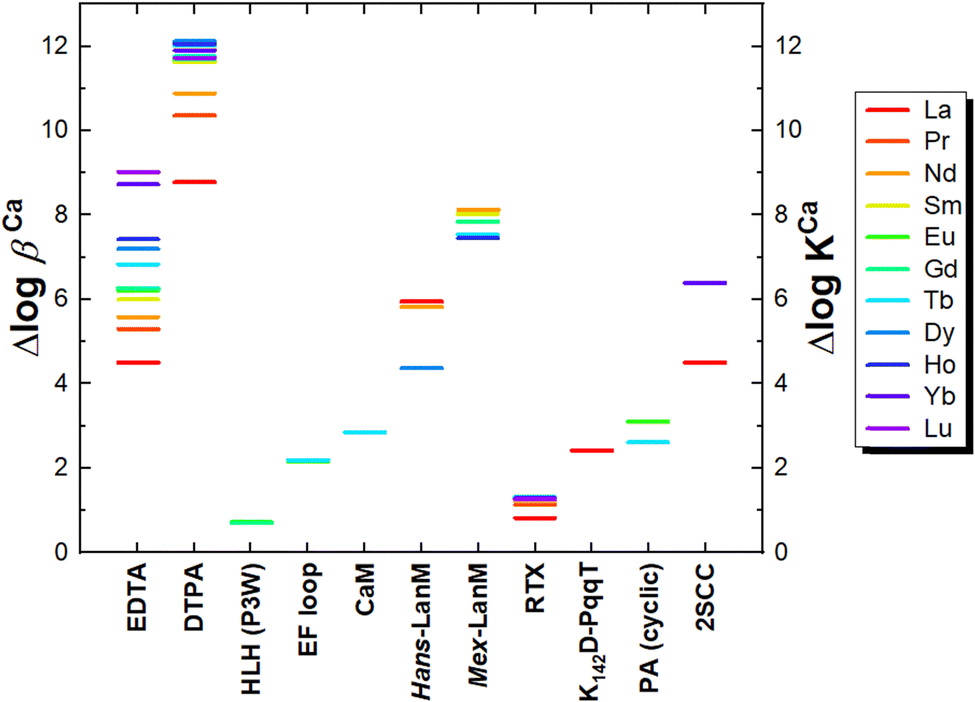 | ||
| Fig. 19 Selectivity of small chelators and some peptide/protein scaffolds for Ln3+ over Ca2+. Δlogβ and ΔlogKapp are reported for small chelators and peptide/protein scaffolds, respectively. Values plotted were calculated from values reported in Table 16. | ||
Among the EF-hand motifs, LanMs show a remarkably higher conformational selectivity against Ca2+ (ΔlogKCa ≈ 8 for Mex-LanM)81,155 relative to both natural and engineered EF loops (ΔlogKCa ≈1–3).59,82,169 In this respect, it is worth noting that Ca2+ seems to bind to LanM with logKCa = 5.5 without inducing a conformational change, which occurs upon further Ca2+ binding events with logKCa ≈ 3.81
The Pro2 residue found in all EF loops of Mex-LanM is crucial for the conformational selectivity of LanM for Ln3+ over Ca2+. Notably, mutation of all four Pro2 residues to Ala reduces the selectivity of Mex-LanM for Ln3+ against Ca2+ by strengthening the conformational response of Ca2+-binding to LanM of more than 2 order of magnitudes.81 Moreover, Pro2 also had a key role in the Ln3+-specific conformational response of LanTERN chimera already discussed above (see 3.2.1 Influence of the protein scaffold).141 However, it is noteworthy that high selectivity over Ca2+ (ΔlogKCaa ≈ 4–6) is also observed in Hans-LanM, in which a Pro2 residue is found only in one of the four EF-hand loops. Hence, it is likely that the unique selectivity of LanM for Ln3+vs. Ca2+ stems from minor differences in the second coordination sphere and backbone conformation.62
The main difference between Ca-dependent and Ln-dependent alcohol dehydrogenases is the presence of an additional Asp in the coordination sphere of Ln3+. This residue appears essential for Ln3+-coordination and selectivity relative to Ca2+. Indeed, mutation of this AA to Ala in MDH and EDH enzymes led to the production of enzymes metallated only with Ca2+.110 However, the mutation did not lead to catalytically competent Ca-enzymes, which underscores that differences between Ca-dependent and Ln-dependent ADH beyond the metal coordination sphere are important for catalysis.
Among the de novo designed Ln3+-binding proteins, 2SCC scaffolds bearing non-canonical multidentate carboxyglutamic (Gla, Fig. 6) residues show a selectivity over Ca2+ comparable to that of small chelators and LanMs.95
4.3 Selectivity over An3+
The groups of Deblonde and Daumann have explored the capacity of Mex-LanM to bind An3+ ions.165,170–172 LanM exhibits a higher affinity for An3+ over Ln3+ ions and Y3+. In particular, a ∼5-fold stronger affinity was found for Am3+ and Cm3+ relative to Ln3+ of similar size (Pr3+, Nd3+, Sm3+),170 and for Ac3+ over Y3+.171 This trend is similar to that of small chelators such as EDTA and DTPA, which show slightly higher selectivity for Am3+ over Nd3+ (Δlogβ ∼ 1.5).173
For small complexes, the non-negligible covalency of An3+–ligand bonds has been generally considered accountable for the higher stability of An3+-complexes over their Ln3+-analogues, but this remains to be ascertained for An3+–protein complexes.170
An improved selectivity for An3+ over Ln3+ ions at low pH (∼3) was obtained by Deblonde and coworkers via Mex-LanM engineering with softer Asn ligands in place of native Asp9 in each loop (LanM 3D9N).174
A slightly higher affinity for Cm3+ over Eu3+ has also been reported for the EF-hand loops isolated from Mex-LanM.143 Am3+ binding to an LBT was shown to be ∼6-fold stronger than Nd3+ and comparable to smaller Ln3+ (Tb3+, Eu3+). A higher (∼10-fold) selectivity for Am3+ over Nd3+ could be obtained by a modified LBT bearing a Cys5 residue functionalized with a soft 2-methylene-pyridine group in place of Asp5.175
Daumann and coworkers also demonstrated that An3+ ions such as Am3+ and Cm3+ can replace early Ln3+ ions in the catalytic activity of the Ln3+-dependent MDH enzyme and ensure the growth of Ln3+-dependent bacteria in the absence of Ln3+ ions.176
4.4 Selectivity over d-block metal ions
Despite its relevance for biomedical and bio-metallurgic applications, the selectivity of Ln3+-binding peptides and proteins over d-block metal ions is a largely neglected aspect. Generally, a higher selectivity for Ln3+ over d-block metal ions is obtained by leveraging their preference towards O- (carboxylates, carbonyls) over N-donors (e.g. histidines), and higher coordination numbers (8–10 vs. 4–6). For instance, LanP displayed no binding to Fe2+, Fe3+, or Zn2+.122 A 3SCC scaffold (MB1-2) engineered with Asn and Asp residues for Ln3+ binding, shows ∼6-fold higher affinity for Tb3+ over Cu2+.98,177 A remarkably high selectivity for Nd3+ ions over Cu2+ (ΔlogK ∼ 6) and Zn2+ (ΔlogK ∼ 8) has been reported for Mex-LanM at pH 5.155 On the contrary, EDTA shows higher selectivity for Cu2+ over Nd3+ (i.e. 100-fold higher affinity for Cu2+ over Nd2+) and no selectivity between Nd3+ and Zn2+, while DTPA has no selectivity between Nd3+ and Cu2+, but a ∼1000-fold higher affinity for Nd3+ relative to Zn2+.155,173 It is worth noting that EDTA has a higher affinity for Cu2+ (logβCu = 18.8) than most Ln3+ ions (La3+–Ho3+) and DTPA has a higher affinity for Cu2+ (logβCu = 21.4) than the early Ln3+ (La3+–Pr3+). Altogether, these data point out that peptides and proteins benefit from a higher selectivity for the whole Ln3+ series against d-block metal ions relative to small chelators.
5. Kinetic stability
Beyond thermodynamic parameters, kinetic aspects are also important for a complete characterization of Ln3+-binding peptides and proteins, especially for further applications, such as MRI or Ln recovery (see 6. Applications).5.1 Lability and inertness: exchange reaction
Due to the shielding by 5s and 5p orbitals, 4f orbitals do not undergo strong ligand field stabilisation, making Ln3+ ions kinetically labile.The lability/inertness is witnessed by the rate of the self-exchange reaction. Such a reaction can proceed according to two extreme pathways: associative or dissociative, corresponding to an increase or a decrease in the coordination number (CN) of the intermediate species, respectively. In turn, this depends on the CN of the starting complexes: for instance, Ln3+ complexes with low CN (CN ≤ 8, Gd3+–Lu3+) are expected to proceed through an associative pathway with a nine-coordinate intermediate, while those with high CN (CN ≥ 9, La3+–Eu3+) by a dissociative pathway with an eight-coordinated intermediate (Fig. 20A).30 The rates of self-exchange for a certain Ln3+ ion depend on the relative stabilities of the eight- and nine-coordinated species for that Ln3+.
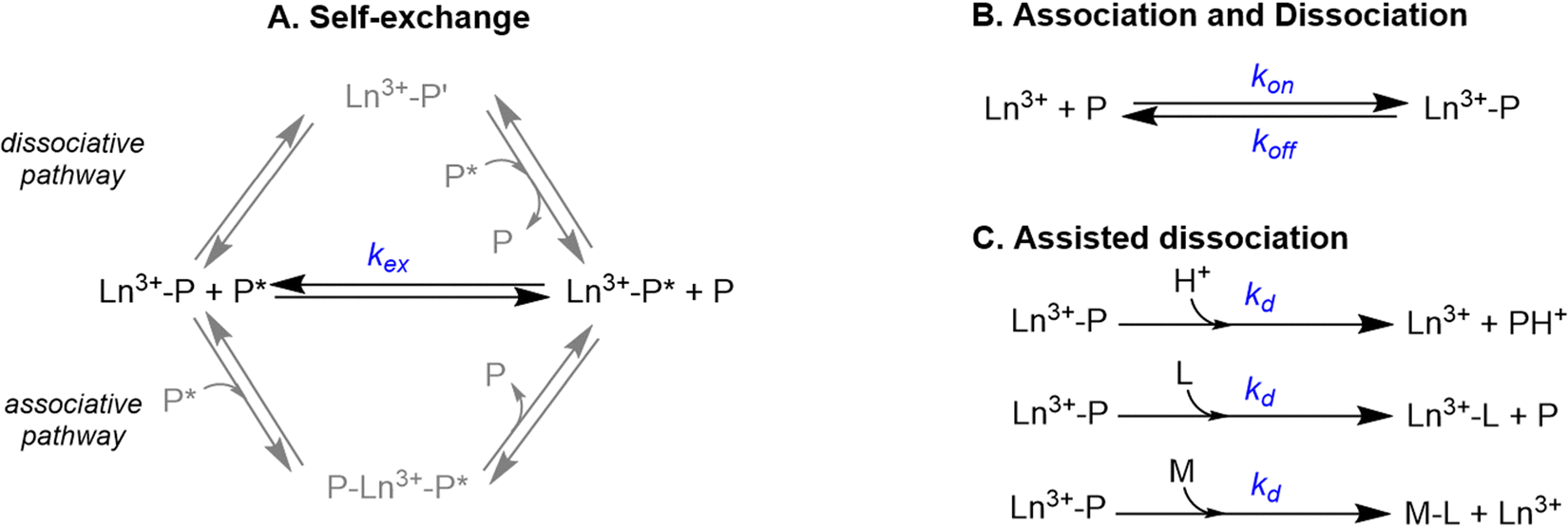 | ||
| Fig. 20 (A) Self-exchange reaction characterized by the rate constant kex and related pathways. P and P* are two molecules of the same peptide. In the case of the dissociative pathway, P′ corresponds to peptide P which has some of its binding residues de-coordinated from the Ln. Hence, Ln3+ has a lower CN when bound to peptide P′ than P. (B) Association and dissociation reactions with their respective rate constants kon and koff. (C) Simplified scheme of proton-, ligand- and metal-assisted dissociation pathways (rate constants kd). | ||
Recently, Peacock and coworkers measured the activation parameters for water exchange in the Gd3+ complex with a 3SCC scaffold (MB1-1, q = 3) and proposed an associative mechanism.102 Furthermore, key insight can be deduced from the comparative NMR study of La3+vs. Lu3+ binding to the LBT3 peptide.164 In particular, Ln3+ binding to LBT3 showed a fast and slow exchange regime for La3+ and Lu3+, respectively,164 which may be explained through the different structural features of the complexes. Indeed, the La3+–LBT3 complex has CN = 9, with q = 1 and is structurally flexible, hence the de-coordination of the weakly-bound (relative to the peptide ligands) H2O molecule can promote the self-exchange reaction through a dissociative pathway. Instead, the Lu3+–LBT3 complex has CN = 8, with q = 0, and poor solvent accessibility, hindering the possibility of undergoing an associative mechanism.
As a small number of data is available, it is difficult to generalize whether the exchange reaction proceeds via a dissociative or an associative mechanism in other Ln3+-binding peptides and proteins, and which parameters would influence the preference between one or the other pathways.
Furthermore, the self-exchange rate kex can be easily inferred by NMR experiments. For most unfolded or poorly folded peptides, a fast exchange regime characterized by a broadening and shift of the signals was reported at sub-stoichiometric Ln3+:P ratio76,164,169,178 except for a 2SCC scaffold95 and an HLH scaffold18 for which a slow exchange regime was observed. This is in line with the intrinsic lability of Ln3+ ions and the absence of constraints induced by the peptide scaffolds.
5.2 Association and dissociation rates
Beyond the exchange rate kex, key parameters to be defined are the association and dissociation rates, noted kon and koff, respectively. These two parameters are related to the thermodynamic stability constants K according to eqn (9):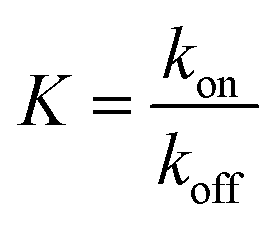 | (9) |
The koff can be then calculated from the kon and stability constant K values, using eqn (9). Indeed, the koff cannot be directly determined experimentally since the release of the Ln3+ ion from a polydentate ligand, such as peptides and proteins, to form the fully solvated species is unlikely. Nevertheless, the complete dissociation of a Ln3+ from a polydentate ligand can be assisted by (i) protonation of the ligands (proton-assisted dissociation), (ii) capture by another ligand(s) (ligand-assisted dissociation), which can also be the buffer, (iii) trans-metalation by another metal ion (metal-assisted dissociation), or any combination of these three processes. It is worth noting that in these processes, the otherwise disfavoured Ln3+ release occurs through the alteration of the system thermodynamics, e.g. by weakening the ligand affinity (acid-assisted dissociation) or by inducing metal-transfer/exchange reaction with competing ligand or cations. Thus, although the concept of complex inertness is often equated with that of “assisted dissociation” by language abuse, they refer to intrinsically distinct processes.
Ligand-assisted dissociation rate constants (kd), which differs from koff, were reported by Falke and coworkers for the complex of Tb3+ with an EF-hand protein (galactose-binding protein, GBP) using excess EDTA as the competing ligand.179 Interestingly, by measuring the dissociation rates of several mutants, these authors showed that the 9th residue of the EF-hand motif acts as a gateway that tunes the kinetics of Ln3+ dissociation. Notably, the Glu9 mutant displayed the slowest dissociation rate (∼0.006 s−1) compared to Asp9 (∼0.002 s−1) and Asn9 (∼1 s−1).
The chelator-free koff of Tb3+-bound to Mex-LanM (∼0.02–0.05 s−1) was estimated by extrapolation from kobs values obtained by stopped-flow spectrofluorimetry in the presence of different concentrations of EGTA.63 However, this approach is questionable, since the koff value relative to a dissociative mechanism is inferred from ligand-assisted dissociation rates, for which a different, namely associative, mechanism is at play. Moreover, it is worth noting that, as mentioned for kon values, dissociation rates may also take into account the contribution of protein unfolding induced by Ln3+ release.
Daumann and coworkers monitored over time the metal exchange reactions between a pre-formed Eu3+–LanM complex and the other Ln3+ ions. Interestingly, equilibration times varied from minutes for the late Ln3+ to hours for the early Ln3+, correlating with the size and Lewis acidity of the competing ions.165
5.3 Discussion and guidelines
The kinetic aspects of Ln3+ coordination by peptides and proteins have been largely neglected and overlooked even though they are important in applications for imaging and Ln recovery. NMR is the method of choice to get a better picture of the dynamics of the system. A simple 1H NMR titration experiment can help gain qualitative insights into the exchange rates using the diamagnetic La3+ and Lu3+, and thus determine the exchange regime on the NMR timescale. Comparison with paramagnetic Ln3+ can bring additional information on Ln3+ coordination site, and even peptides/protein solution structure.Although it is difficult to predict the kinetic behaviour, based on the results and concepts described above, it is anticipated that the spontaneous dissociation of Ln3+ ions from polydentate peptides and proteins is negligible, especially in the case of buried sites within well-folded scaffolds. Notwithstanding, Ln3+ dissociation can be triggered upon acidification or the addition of competing ligands or cations. In the case of ligand-assisted dissociation, the presence of weakly bound water molecules in the Ln3+ coordination sphere is expected to promote dissociation since its replacement facilitates the formation of ternary intermediate complexes with the competing ligand.
6. Applications
Ln3+-binding peptides and proteins have been employed for a number of applications spanning from luminescence sensing and magnetic resonance imaging to metal ion separation and catalysis (Table 11). In the following paragraphs, applications in these four major fields will be discussed with respect to the peculiar properties of each type of Ln3+–peptide and protein scaffold.| Scaffold | Luminescence imaging/sensing | MRI | Metal ion separation | Catalysis (reaction) | |
|---|---|---|---|---|---|
| EF-hand motifs | LBT | X | X | X | |
| HLH | X | X (hydrolysis) | |||
| LanM | X | X | X | ||
| Other Ln-binding sites | Cyclic decapeptide | X | |||
| 3SCC | X | ||||
| LanD | X | ||||
| RTX | X | ||||
| TIM barrel | X (alcohols photooxidation) | ||||
| ADH or ADH mimics | X (alcohols oxidation) |
6.1 Luminescent tags and probes
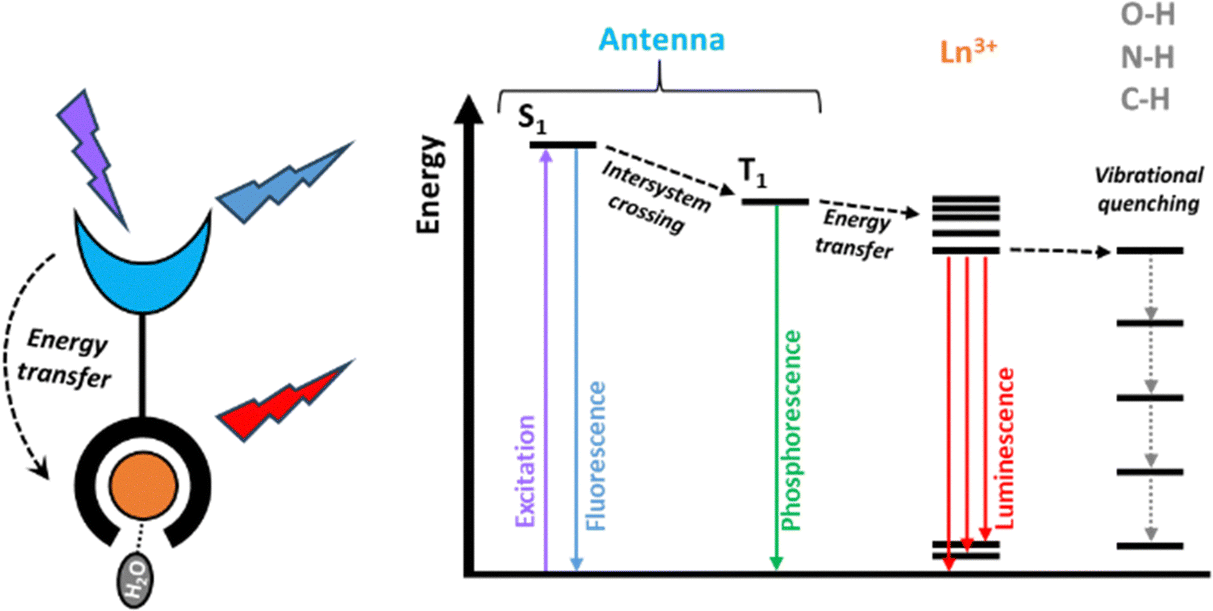 | ||
| Fig. 21 Scheme of the “antenna” effect: upon irradiation, a chromophore called “antenna” (blue, which can be part of or conjugated to a Ln-chelator, black) is excited to a singlet state (S1); then, the energy is transferred to an excited triplet state (T1) via intersystem crossing and finally transferred to an excited level of the Ln3+ (orange) from which emission takes place. Vibrational de-excitation by high-energy oscillators such as O–H groups is also depicted. | ||
The efficiency of the energy transfer from the antenna to the Ln3+ depends on (i) the energy gap between an antenna triplet state and the Ln3+ excited acceptor state, which depends on the antenna–Ln3+ couple, and (ii) the antenna–Ln3+ distance. Several non-radiative pathways compete with Ln3+ luminescence:189 (i) the antenna fluorescence and phosphorescence emissions, (ii) quenching of the antenna T1 state by 3O2, (iii) back energy transfer from Ln3+ excited state to the antenna triplet state when the energy levels are close, (iv) photo-induced electron transfer (PeT) from the excited antenna to the Ln3+, to which the most reducible Ln3+ (Eu3+, Yb3+, Sm3+) are particularly sensitive,190 (v) non-radiative vibrational de-excitation of Ln3+ excited state by high-energy oscillators such as O–H, N–H and C–H groups.39,40 As a result, Ln3+-bound H2O molecules quench Ln3+ emission. Thus, the type and position of the antenna as well as the hydration number are key parameters influencing the luminescence of Ln3+ complexes, including those with peptide and protein ligands.
Despite its straightforward incorporation in peptide and protein sequences, Trp is not an ideal antenna for applications in bioimaging, as its excitation wavelength in the UV spectral region challenges applications in samples with high absorption and fluorescence background, such as biological media.194
To overcome such limitations, different unnatural antenna amino acids have been introduced. Gunnlaugsson and coworkers incorporated at the N-terminal of an EF-loop isolated from parvalbumin a 1,8-naphthalimide chromophore (λex = 345 nm) able to sensitise Tb3+ and Eu3+ emission (Fig. 22).169,178 The HPO ligand introduced into 3SCCs scaffolds (see 2.4.1 Coiled coils) was able to sensitize Tb3+ and Eu3+ upon excitation at 380 nm. Imperiali and co-workers have developed LBTs with unnatural amino acids featuring a carbostyryl-124 (Cs124) or an acridone (Acd) antenna able to sensitize Tb3+ (Cs124) and Eu3+ (Cs124 and Acd) at longer wavelengths, namely ∼340 and 390 nm, respectively (Fig. 22).195 In particular, the Cs124 antenna was able to sensitise Tb3+ better than Trp, while Acd sensitised Eu3+ better than Cs124 (Fig. 22).
 | ||
| Fig. 22 Antenna amino acids found in Ln3+-binding peptide scaffolds discussed in this review (top) and in peptides with appended polydentate Ln3+-chelators (bottom). The excitation wavelength and the Ln3+ ions that each antenna is able to sensitize are indicated. The antenna moieties are highlighted in blue. Abbreviations: Trp, tryptophan; Glu, glutamate; Cs124, carbostyryl-124; Naph, 1,8-naphthalimide; HPO, hydroxyphenol oxazoline; Acd, acridone; Nal, naphthyl-alanine; Phen, phenanthroline; Anthra, anthryl-alanine; NBD, nitrobenzodiazole. | ||
Lately, Acd and its sulfur-substituted SAcd (Fig. 22) were incorporated into Mex-LanM via genetic code expansion, achieving long-lived sensitized Eu3+ emission upon excitation at 390 nm.196 Interestingly, SAcd showed to be a more efficient antenna than Acd due to a prolonged triplet state lifetime.
These unnatural antenna amino acids were not able to sensitise the emission from other Ln3+ ions including Nd3+, Er3+, Tm3+ and Yb3+, which are NIR emitters. Actually, no Ln3+-binding peptide or protein scaffolds have been reported bearing suitable antenna amino acids for NIR-emitting Ln3+ ions, which are particularly sensitive to quenching by O–H, N–H and even C–H oscillators. Thus, in addition to a suitable antenna, the design of Ln–peptides and Ln–proteins as NIR emitters will also require avoiding O–H oscillators in their first coordination sphere, and likely also N–H oscillators. To the best of our knowledge, these antenna amino acids are the sole used in Ln3+-binding peptide and protein scaffolds.8 Other antennae were described in peptide and proteins functionalised with polydentate Ln3+-chelators such as EDTA and DOTA as reviewed in part in ref. 23. In these cases, the antenna has been introduced through either (i) ligand derivatization, often with conjugated pyridine or picolinate groups,197–199 or (ii) unnaturall amino acids (Fig. 22), including naphthalene (for Eu3+, λex = 280 nm),200 NBD (for Nd3+, λex = 480 nm),201,202 anthracene (for Nd3+ and Yb3+, λex = 330–400 nm)203 and phenanthroline (for Tb3+ and Eu3+, λex = 300 nm).204 Similarly to HPO, phenanthroline served as both Ln3+ ligand and antenna. Such an Ln3+-coordinating antenna is generally preferable because its proximity to the ion enhances the energy transfer efficiency.
Szabo and coworkers have thoroughly investigated how the position of aromatic amino acids (Trp, Phe or Tyr) within the EF-hand motif sequence influences Tb3+ emission.66 In particular, they compared the excitation and emission spectra of Tb3+ bound to EF-hand peptides containing aromatic amino acids in positions 2, 4, 7 or 10. Very interestingly, they found that, regardless of which amino acid occupied positions 2, 4 and 10, the amino acid in the 7th position is responsible for Tb3+ sensitization due to its closer distance to the ion (∼5 Å). For instance, despite Phe and Tyr having a lower extinction coefficient than Trp, Tb3+ was mainly sensitized by either Phe or Tyr in the 7th position even when Trp was simultaneously present in the 2nd position. Moreover, the authors reported a favourable impact of Tyr in the 2nd and 4th, but not 10th, position on the Tb3+ emission in peptides having Trp in the 7th position. In particular, the results suggested that this occurs thanks to an energy transfer between excited Tyr and Trp, which in turn sensitises Tb3+ emission. Based on these studies, Trp7 has been incorporated as the Tb3+ sensitizer in many EF-hand motif scaffolds, including LBTs72,73 and Mex-LanM mutants.63 The 7th loop position has also been used to incorporate unnatural Cs124 and Acd antenna amino acids in LBTs195 and Mex-LanM (in the latter case after screening of 42 incorporation sites).196 Zondlo and coworkers also showed that Tb3+ emission can be sensitized using Trp at the 8th position of EF-loop-like peptides.148
Besides, peptide and protein scaffolds with suitable antennae may serve as luminescent probes for Ln3+ ions. For instance, a Mex-LanM Trp mutant (T90W) has been successfully employed to measure Tb3+ in acid mine drainage at very low pH (∼3),63 while LanM-Acd mutants were applied for Eu3+ detection in cell culture.196
Alternatively, Walker and coworkers reported on the design of fluorescent proteins with a high concentration of negative charges at their surface for the detection of Ln3+ (Ln = Sm, Eu, Tb, Dy, Tm, Yb) by LRET in the micro- to millimolar concentration range.206 Instead of exciting an antenna, it is the Ln3+ that is excited and transfers its energy to the fluorescent proteins, which resulted in a twofold higher fluorescence intensity and longer emission lifetimes.
As reviewed in ref. 23 and 180, Ln3+-based responsive probes, whose luminescent signal changes upon the interaction with a given target, can be conceived based on the above-mentioned factors influencing Ln3+ luminescence, such as (i) chemical modifications of the antenna, (ii) variation of the Ln3+–antenna distance, for instance through conformational changes induced upon target-binding or via proteolytic cleavage, and (iii) modification of the Ln3+ coordination sphere and hydration number.
These principles have been used to develop several responsive probes based on peptides with appended Ln3+–chelator complexes.23 For instance, Vazquez and coworkers designed a peptide probe that folds upon RNA binding, inducing the coordination of a phenanthroline antenna to an appended Tb3+–EDTA complex.204 Similarly, Sénèque and coworkers developed Zn2+-201 and RNA-responsive207 peptide probes based on conformational changes that decrease the distance between the antenna and a Ln3+–DOTA complex attached to the peptide. Exploiting the distinct emission bands of Ln3+ couples such as Tb3+–Eu3+ in the visible and Nd3+–Yb3+ in the NIR, the group of Sénèque also developed ratiometric probes (i.e. based on the ratio between two signals) introducing two suitably positioned Ln3+–chelator complexes.200,203 In order to synthesize such regio-selectively hetero-metallated complexes, they used native chemical ligation to assemble two peptides functionalized with different Ln3+ complexes. Of note, this strategy relies on the kinetic inertness of the Ln3+-complexes grafted on the peptides and has never been applied so far on peptide scaffolds with “intrinsic” Ln3+–binding sites.
Lately, LanM-Acd mutants have been developed as protease sensors based on the impairment of the antenna effect upon protein cleavage. To this end, protease recognitions sequences were incorporated in suited position with the LanM scaffold.196
Pazos and coworkers developed peptides with appended Tb3+–DO3A complex (Fig. 15) responsive to (i) Ser phosphorylation, based on a decrease of the hydration number,208 and (ii) to Tyr nitration, relying on the quenching of Tb3+ emission by 3-nitro-Tyr.209 Quenching of Tb3+ emission was also exploited in the design of Cu2+-selective peptide probes.194,210
Zondlo and coworkers also developed Tb3+ complexes with EF-hand-like peptides (Table 10) responsive to (de)phosphorylation of Tyr, Ser and Thr,144,145,148 Tyr nitration146 and Cys oxidation.147,211 In particular, Cys, Tyr, Ser or Thr residues were introduced in place of Asp or Glu residues within EF-loops (Table 10), drastically reducing Tb3+-binding (logKapp < 4), while their derivatization with phosphate or sulphinyl groups restored a logKapp ≈ 4–5. Hence, in this case, the response was mainly due to a change in the metallopeptide affinity upon amino acid modification.
6.2 Magnetic resonance imaging
Clinical contrast agents (CAs) for magnetic resonance imaging (MRI), reviewed by Caravan and coworkers in ref. 212, are mostly Gd3+-complexes with polyaminocarboxylate chelators, such as DOTA and DTPA (Fig. 16). Gd3+-based CAs (GBCAs) enhance image contrast by shortening the longitudinal (T1) and transverse (T2) relaxation time of proximal water protons, to an extent that depends on their longitudinal (r1) and transverse (r2) relaxivity, as defined by eqn (10): | (10) |
| Scaffold | Peptide/protein | MW (kDa) | q | τ R (ns) | τ m (ns) | r 1 (mM−1 s−1) | r 2 (mM−1 s−1) | Magnetic field (T) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Values obtained by the measurement of Tb3+ or Eu3+ luminescence lifetimes. b Values measured at 298 K. | |||||||||
| DOTA | n/a | ∼0.5 | 1 | ∼0.08 | 244 | 4.3 | 5.7 | 7 | 215 |
| n/a | n/a | 4.7 | n/a | 0.47 | 216 | ||||
| DTPA | n/a | ∼0.5 | 1 | ∼0.06 | 300 | n/a | n/a | n/a | 215 |
| n/a | n/a | 4.7 | n/a | 0.47 | 216 | ||||
| n/a | n/a | 3.5 | n/a | 1.4 | 77 | ||||
| n/a | n/a | 5.4 | 8 | 1.5 | 217 | ||||
| n/a | n/a | 4.2 | 6.8 | 3 | 217 | ||||
| n/a | n/a | 5.1 | 9.4 | 7 | 218 | ||||
| HLH | P3W | ∼4 | 2 | n/a | n/a | 16.2 | n/a | 0.47 | 77 |
| 21.2 | n/a | 1.4 | |||||||
| LBT | m-sSE3 | ∼2 | 1 | n/a | n/a | 5.5 | n/a | 11.75 | 60 |
| sSE3 | 0 | n/a | n/a | 1.2 | n/a | 11.75 | 60 | ||
| dLBT | dSE3 | ∼4 | 0.08 | n/a | n/a | 5.9 | n/a | 11.75 | 60 |
| dLBT–protein | dSE3-Ubiquitin | ∼12 | 0.08 | n/a | n/a | 2.3 | n/a | 11.75 | 60 |
| q-dSE3-Ubiquitin | 1 | n/a | n/a | 4.2 | n/a | 11.75 | 60 | ||
| xq-dSE3-Ubiquitin | 0; 1 | n/a | n/a | 5.0 | n/a | 11.75 | 60 | ||
| Cyclic decapeptide | PA | ∼1.3 | 2 | 0.4 | n/a | ∼30 | ∼40 | 4.7 | 41 and 219 |
| ∼20 | ∼35 | 9.4 | |||||||
| ∼20 | ∼35 | 11.75 | |||||||
| 3SCC | MB1-1 | ∼12 | 3.1 | 7 | 1.56 | 10 | 89.3 | 7 | 102 |
| 64.3 | 87.9 | 1 | |||||||
| MB1-1 (2W) | 0 | n/a | n/a | 3.9 | 24.2 | 7 | 100 | ||
| MB1-2 | 0 | n/a | n/a | 4.2 | 21.3 | 7 | 99 and 100 | ||
| MB1-3 | 0 | n/a | n/a | 4 | 20.9 | 7 | 99 | ||
| MB1-4 | 2 | n/a | n/a | 7.5 | 37.9 | 7 | 99 and 100 | ||
| MB1-1L | ∼15 | 3.7 | 10 | n/a | 10.9 | 81.8 | 7 | 102 | |
| 67.4 | 96.5 | 1 | |||||||
| Proteins | CA1.CA2 | ∼12 | 2 | 9.1 | n/a | 117 | 129 | 1.5 | 217 |
| 48 | 88 | 3 | |||||||
| 6 | 50 | 9.4 | |||||||
| ProCA32 | ∼12 | 0.5 | n/a | n/a | 33.4 | 44.6 | 1.4 | 218 | |
| 21.9 | 56.9 | 4.7 | |||||||
| 18.9 | 48.6 | 7 | |||||||
| LanM | ∼12 | 2 | n/a | n/a | 12.1 | 17 | 3 | 220 | |
| LanND | ∼12 | 2 | n/a | n/a | 13.2 | 25.1 | 3 | ||
| 6.9 | 31.2 | 7 | |||||||
In order to increase MRI sensitivity, reduce the injected GBCA dose and hence limit toxic side-effects, GBCAs with higher relaxivity are currently sought.212 The approaches developed to achieve this goal have been reviewed in ref. 214. The main molecular factors affecting the inner-sphere relaxivity of GBCAs are the hydration number q, the mean residency time τm of Gd-bound water and the rotational correlation time τR (Fig. 23).212 Hereafter, these factors are discussed with respect to the Gd3+ complexes with peptides and proteins that have been investigated as GBCAs (Table 12).
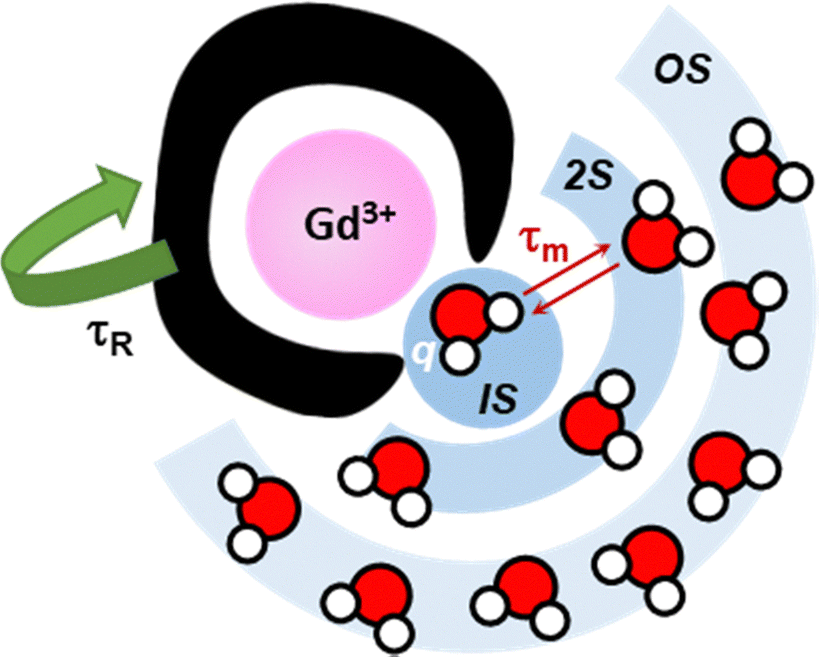 | ||
| Fig. 23 Scheme of a GBCA and graphical description of the parameters that influence its relaxivity, including inner-sphere (IS) contributions (hydration number, q; water residency time, τm; rotational correlation time, τR) as well as second (2S) and outer (OS) sphere effects. | ||
Peacock and co-workers have investigated the relaxivity of Gd3+–3SCC scaffolds, showing a significant dependence on the location of the Ln3+-binding site along the coiled coil and the second sphere layer, which both affect the hydration number (see 2.4.1 Coiled coils).98–100,102,104 Remarkably, scaffolds with high hydration numbers, such as MB1-1 (q = 3), MB1-4 and the recently reported LanM derivative LanND (q = 2), display significantly higher relaxivity compared to clinical GBCAs, especially at low clinical field (1 T, Table 12).
 | ||
| Fig. 24 Experimental 1H NMRD profiles showing the dependence of the relaxivity (per Gd3+ ion) as a function of the magnetic field strength for Gd-DTPA derivatives with short (A, ∼0.1 ns), intermediate (B), and long (C, ∼10 ns) rotational correlation times. Reprinted with permission from ref. 212. Copyright 2019 American Chemical Society. | ||
To improve the relaxivity, a τR increase can obtained by increasing the molecular weight and the rigidity of GBCAs.214
For instance, the Gd3+ complex with the rigid cyclodecapeptide scaffold PA (see 2.3.1 Cyclic decapeptide) showed remarkably high relaxivity (20–40 mM−1 s−1, Table 12) at high magnetic fields (4.7–11.75 T) thanks to a medium-range molecular weight (∼1.3 kDa) implying an optimal τR ≈ 0.4 ns.41
Besides, Yang and co-workers have developed protein-based CAs by engineering protein scaffolds with Gd3+-binding sites, achieving much higher relaxivity relative to small GBCAs at both low and high fields (Table 12).217,218 In CA1.CD2 (∼12 kDa), where a de novo designed rigid Gd3+-binding site was engineered into a compact CD2 (a cell adhesion protein) domain, the enhanced relaxivity relative to small GBCAs is mainly the result of a higher hydration number (q = 2) and a remarkably longer τR (∼9 ns) relative to commercial GBCAs.217 It is worth noting that the local rigidity of the Gd3+ complex, and not only the molecular weight, is crucial to achieving high relaxivity. As an example, Yang and coworkers also showed that Gd3+ bound to a CD2 domain engineered with a flexibly conjugated EF-loop from CaM showed a relaxivity comparable to that of small commercial GBCAs (∼3.4 mM−1 s−1 at 3 T).217 Analogously, the mere conjugation of double LBT (dLBT) on a protein scaffold (ubiquitin) did not improve their relaxivity (Table 12).60
A long τR (7–10 ns) was also reported for Gd3+–3SCC complexes;102 however, it is noteworthy that lengthening the coiled coil from five (MB1-1, ∼12 kDa) to six heptads (MB1-1L, ∼15 kDa) and the consequent τR increase had negligible impact on the relaxivity (Table 12), suggesting that the tumbling rate is not a limiting factor for such scaffolds.
This is the case, for instance, of some Gd3+–3SCC scaffolds (e.g. MB1-2 and MB1-3) that showed a relaxivity comparable to that of commercial GBCAs at high field (7 T), despite having q = 0 (Table 11). Indeed, the authors hypothesized that this could be mainly related to an OS mechanism involving hydrogen bonding and proton exchange networks between the peptide surface and bulk water.99
Delangle and coworkers measured the IS and OS relaxivities of the Gd3+–PA complex and indirectly determined the 2S contribution, which accounts for about 25% of the total relaxivity.41,219 By means of MD simulations and the fit of the experimental data with appropriate models, the authors provided a molecular interpretation of the 2S contribution, identifying three 2S water molecules with a high residence time (≈1 ns).
Important 2S and OS contributions were also evidenced for the protein-based GBCAs reported by Yang and coworkers.218
However, most peptide and protein scaffolds display a lower affinity for Gd3+ (logKapp ≤ ∼13, see 3. Thermodynamic stability) than currently used GBCAs (logcK ≈ 15–19 at pH 7.4212) and their kinetic inertness has been poorly explored, challenging their in vivo applications. Nevertheless, some protein-based GBCAs, including ProCA32 and LanND-Gd benefit from higher selectivity against physiological metal ions (e.g. Ca2+, Cu2+, Zn2+) than small chelators (see 4.4 Selectivity over d-block metal ions), and showed good biocompatibility, resulting as a promising alternative as next-generation GBCAs. Indeed, ProCA32 showed a half-life of ∼4 hours in the blood plasma of mice, which increased to ∼10 hours upon PEGylation. At 14 days postinjection of ProCA32, a 3- to 10-fold lower amount of Gd3+ was detected in different organs compared to the administration of Gd bound to a DTPA derivative.221 Hence, ProCA32 has been conjugated with different targeting moieties, including PSMA (prostate-specific membrane-associated antigen), CXCR4 (chemokine receptor 4) and a collagen-targeting peptide, to selectively target prostate cancer, liver metastasis or lung fibrosis.222–226 Similarly to clinical GBCAs, LanND concentration in mice blood dropped below detection level within 1 hour, and renal clearance was observed within 3 hours after LanND injection. Moreover, LanND did not show cellular and neural toxicity and did not elicit an evident immune response. Based on these favourable properties, LanND allowed the high-resolution visualization of brain vessels and the monitoring of kidney dysfunction in mice.220
Beyond the research of safer and more sensitive GBCAs, there is a growing interest in the development of responsive GBCAs, which have been reviewed in ref. 227–231. For instance, GBCAs responsive to the pH, molecular targets (cations, anions, metabolites, nucleic acids), biomarkers, or enzyme activity can be designed based on a change of the relaxivity, that is on the variation of hydration number (q-based) or the tumbling rate (τR-based), or both.
In the early 2000s, the HLH scaffold P3W (see 2.2.3 Chimeric helix-loop-helix motifs) was the first, and currently unique, Ln3+-binding peptide investigated as a responsive GBCA. Gd3+–P3W showed a 4- to 6-fold higher relaxivity than Gd3+–DTPA at low clinical magnetic fields (Table 11), as a result of higher hydration number (q = 2) and slower tumbling rate, whose further decrease upon DNA binding yielded a ∼100% relaxivity increase.77 Lately, Sénèque and coworkers have also developed Zn2+-responsive peptides with appended Gd3+–DOTA/DO3A derivatives that show a Zn2+-dependent relaxivity increase based on a change of the system rigidity and/or hydration number upon Zn2+ binding.232,233 These examples illustrate how responsive peptide/protein-based GBCAs could be designed.
6.3 Ln recovery and separation using Ln-binding peptides and proteins
 | ||
| Fig. 25 Overview of the most developed Industrial uses of Ln: rechargeable batteries, permanent magnets, lightings, catalysis and refinery, and medicine. Pm is a radioactive element and does not occur in Nature. | ||
The REE market is dominated by China, which supplies 98% of REE used in the EU.2 Due to the expected increasing demand for REE in the coming years and to the lack of high concentrated REE mining sites in Europe, REEs have been identified as critical elements by the EU.2,235 Thus, there is a strong interest in developing REE recovery technologies from secondary sources, such as the enormous amounts of waste from electrical and electronic equipment generated each year (WEEE 1–30% wt REE).234 Additionally, sources with low concentrations of REE such as phosphogypsum (0.01–2% wt REE), red mud (500–2500 ppm REE), mine tailings (3–5% wt REE), coal ashes (0.1–1% wt REE), or acid mine drainages (200–900 μg L−1 REE) are available in such amounts that the total content of REE they contained could be worth exploiting. The industrial and economic feasibility of the valorisation of such wastes was discussed in ref. 236.
Beyond economic and geopolitical aspects, it is urgent to develop new and more sustainable REE recovery technologies to limit damages caused by mining and reduce the greenhouse gas emissions and the detrimental impact on the environment of actual processes that rely heavily on pyrometallurgy (high energy consumption, release of toxic gases, volatile metals and dust) and hydrometallurgy (use of strong acids such as HCl, HNO3, or H2SO4; solvents such as kerosene or hexane; large amount of liquid wastes and sludges; risk of heavy metal pollution, including by radionuclides).3 The separation of REE is an additional challenge, which is performed by multiple separation cycles involving liquid/liquid extraction, or precipitation, and relying on the use of organic ligands.14,237
In this context, biometallurgy is emerging as a sustainable alternative that could reduce the energy consumption, waste production, and volumes of strong acids and solvents used, even though it has not yet been translated at the industrial scale.14,238–243 This approach relies on the use of micro-organisms or biomaterials for the (selective) recovery of critical metal ions and is particularly suited for low-grade REE feedstocks with low REE concentrations.
Several bio-inspired strategies have been investigated for the recovery of REE from secondary sources which were discussed in recent reviews: (i) non-modified microorganisms: microorganisms were used either for biosorption of REE on their surface, or for their (active) bioaccumulation as reviewed in ref. 239–243. Notably, the Ln-dependent methylotrophic strain M. fumariolicum SolV was recently described for its ability to recover REE from various low-grade sources;244(ii) display of REE-binding motifs: micro-organisms were also modified to display Ln3+-binding peptides on their surface in order to improve REE biosorption, which was recently reviewed.15,242 Examples include LBT displayed on phages,245,246 or E. coli,154,247–251 and LanM displayed on Y. lipolytica and E. coli;252,253(iii) Ln-binding peptides and proteins: Ln3+-binding peptides and proteins were also used directly for recycling (Table 13) as reviewed in ref. 15, 241, 242 and 254 and discussed in more detail below.
| Scaffold | pH range | REE3+ | Sorption (S) | Intra-REE selectivity | Ref. |
|---|---|---|---|---|---|
| Competing metal ions | Desorption (D) | ||||
| (source, pH) | |||||
| LBT | >5 | Ln | S: filtration on functionalised fibres | Yes | 255 |
| Al, Ca, Cu, Fe, Ni | D: HNO3, pH 2 | ||||
| (mine tailings, pH 7) | |||||
| ∼4 | Tb | S: filtration on functionalised fibres | n.d. | 256 | |
| Ca or Cu or Fe or Zn | D: HCl, pH 1.7 | ||||
| — | |||||
| RTX | >0.5 | Nd, Dy | S: filtration on 3 kDa cutoff ultrafilter | n.d. | 129 |
| Fe, Co | D: n/a | ||||
| (NdFeB magnet solution, pH 6) | |||||
| LanM | >2.5 | Sc, Y, Ln | S: filtration on 3 kDa cutoff ultrafilter (Mex-LanM) | no | 155 |
| Li, Be, Na, Mg, Ca, Sr, Al, Si, Mn, Fe, Co, Ni, Cu, Zn, U | D: n/a | ||||
| (lignite leachate, pH 3.7) | |||||
| >2.4 | Sc, Y, Ln | S: filtration on functionalized agarose beads (Mex-LanM) | Yes | 257 | |
| Li, Na, K, Rb, Mg, Ca, Sr, Ba, Al, Si, Pb, V, Mn, Fe, Co, Ni, Cu, Zn, Se, Th, U | D: pH steps (HCl, pH 2.3, then pH 1.5) | ||||
| (coal fly ash leachate, pH 5) | |||||
| — | Sc, Y, Ln | S: filtration on functionalized agarose beads (Mex-LanM) | Yes | 167 | |
| — | D: two-cycle process: 1. Malonate, then pH ramp (2.3-2.1-1.9), 2. Citrate, then pH 1.5 | ||||
| (coal combustion, mine tailings, pH 3) | |||||
| >2 | Y, Ln | S: liquid–liquid phase separation (Mex-LanM) | Yes | 258 and 259 | |
| Mg, Na, Ca | D: phosphate-citrate buffer, pH 2.2 | ||||
| (coal fly ash leachate, pH 4.5) | |||||
| >3 | Sc, Y, Ln | S: functionalised magnetic nanoparticles (Mex-LanM) | n.d. | 260 | |
| Li, Na, K, Rb, Mg, Ca, Ba, Al, Si, Pb, V, Mn, Fe, Co, Ni, Cu, Zn, As, Se, Cs | D: HCl, pH 1.7 | ||||
| (coal fly ash leachate, pH 5) | |||||
| — | Dy, Nd | S: filtration on functionalized agarose beads (Hans-LanM, Hans-LanM-R100K) | Yes | 62 | |
| — | D: 1. Malonate, 2. HCl, pH 1.5 | ||||
| (pH 5) | |||||
| LanD | n.d. | La, Ce, Pr, Nd | S: filtration on 10 kDa cutoff ultrafilter (LanD-E75Q/E78A) | Yes | 125 |
| — | D: n/a | ||||
| (pH 6) | |||||
| Ln3+ | E3+/2+ (V)26 | E4+/3+ (V)26 | r (Å)27 | pKa28 | logKsp29 |
|---|---|---|---|---|---|
| La3+ | −3.1 | 1.216 | 8.89 | −22.3 | |
| Ce3+ | −3.2 | 1.7 | 1.196 | 8.31 | −23.5 |
| Pr3+ | −2.7 | 3.4 | 1.179 | 8.3 | −23.4 |
| Nd3+ | −2.6 | 4.6 | 1.163 | 8.13 | −24.1 |
| Pm3+ | −2.6 | 4.9 | 1.144 | n/a | n/a |
| Sm3+ | −1.55 | 5.2 | 1.132 | 7.84 | −24.8 |
| Eu3+ | −0.34 | 6.4 | 1.12 | 7.66 | −25.5 |
| Gd3+ | −3.9 | 7.9 | 1.107 | 7.87 | −24.8 |
| Tb3+ | −3.7 | 3.3 | 1.095 | 7.6 | −25.7 |
| Dy3+ | −2.5 | 5 | 1.083 | 7.53 | −25.7 |
| Ho3+ | −2.9 | 6.2 | 1.072 | 7.43 | −26.4 |
| Er3+ | −3.1 | 6.1 | 1.062 | 7.46 | −26.2 |
| Tm3+ | −2.3 | 6.1 | 1.052 | 7.34 | −26.4 |
| Yb3+ | −1.05 | 7.1 | 1.042 | 7.31 | −26.7 |
| Lu3+ | 8.5 | 1.032 | 7.33 | −26.6 |
| Ln3+ | EDTAa | DTPAa | Macropab | Macrotripac | LBT12d | LF4e | RTXf | LanDg | LanMh | LanMi | LanMj | LanMk |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a logβML from ref. 173.
b logβML from ref. 160.
c logβML from ref. 161.
d logKapp in HEPES 10 mM pH 7, NaCl 100 mM, ref. 64.
e logKapp in HEPES 10 mM pH 7.5, NaCl 10 mM, ref. 103.
f logKapp in bis-Tris 20 mM pH 6, NaCl 25 mM, ref. 129.
g logKapp in MOPS 30 mM pH 7, KCl 100 mM, ref. —.
h logKapp for Mex-LanM in MOPS 30 mM pH 7.2, KCl 100 mM, measured by CD spectroscopy, ref. 81.
i logKapp for Mex-LanM in acetate buffer 20 mM pH 5, KCl 100 mM, measured by CD spectroscopy, ref. 155.
j logKapp for Hans-LanM acetate buffer 20 mM pH 5, KCl 100 mM, measured by CD spectroscopy, ref. 280.
k logKapp for Mex-LanM KCl/KCH3COO− buffer 100 mM pH 5.0, measured by UV-vis spectroscopy, ref. 155.
|
||||||||||||
| La3+ | 15.5 | 19.5 | 15.0 | 12.6 | 5.5 | 4.2 | 4.1 | 5.7 | 11.3 | 10.2 | ||
| Ce3+ | 16.1 | 20.5 | 15.1 | 12.8 | 6.0 | 6.0 | ||||||
| Pr3+ | 16.3 | 21.1 | 14.7 | 12.7 | 4.5 | 6.3 | 10.2 | 11.0 | ||||
| Nd3+ | 16.6 | 21.6 | 14.4 | 12.3 | 6.6 | 4.5 | 6.6 | 11.3 | 10.0 | 10.7 | ||
| Sm3+ | 17.0 | 22.3 | 13.8 | 11.4 | 6.5 | 11.2 | 10.6 | |||||
| Eu3+ | 17.2 | 22.4 | 13.0 | 10.9 | 7.2 | 4.6 | 4.6 | 6.5 | ||||
| Gd3+ | 17.3 | 22.5 | 13.0 | 10.2 | 7.1 | 4.6 | 6.2 | 11.0 | 10.0 | |||
| Tb3+ | 17.8 | 22.7 | 11.8 | 10.2 | 7.2 | 4.9 | 4.6 | 10.7 | ||||
| Dy3+ | 18.2 | 22.8 | 11.7 | 10.5 | 7.2 | 4.6 | 9.7 | 8.6 | 10.8 | |||
| Ho3+ | 18.4 | 22.8 | 10.6 | 10.7 | 4.9 | 10.6 | 9.6 | 11.1 | ||||
| Er3+ | 18.9 | 22.7 | 10.1 | 11.1 | 7.1 | 5.0 | 11.9 | |||||
| Tm3+ | 19.6 | 22.7 | 9.6 | 11.4 | 12.4 | |||||||
| Yb3+ | 19.7 | 22.6 | 8.9 | 11.8 | 7.0 | 4.4 | 4.6 | |||||
| Lu3+ | 20.0 | 22.4 | 8.3 | 11.9 | 6.9 | 4.2 | 4.6 | |||||
| Ln3+ | EDTAa | DTPAa | P3Wb | EF loopc | CaMd | Hans-LanMe | Mex-LanMf | RTXg | PqqTh | PAi | 2SCCj |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
a logβML from ref. 173.
b logKapp in TRIS 5 mM pH 7.8, from ref. 76.
c logKapp in HEPES 10 mM pH 7, NaCl 100 mM, from ref. 169.
d logKapp in PIPES 25 mM pH 6.8, KCI 100 mM, from ref. 82.
e logKapp for Hans-LanM acetate buffer 20 mM pH 5, KCl 100 mM, measured by CD spectroscopy, ref. 280.
f logKapp for Mex-LanM in MOPS 30 mM pH 7.2, KCl 100 mM, measured by CD spectroscopy, ref. 81.
g logKapp in bis-Tris 20 mM pH 6, NaCl 25 mM, ref. 129.
h logKapp in HEPES 30 mM pH 7, NaCl 100 mM, from ref. 20.
i logKapp in HEPES 10 mM pH 7, from ref. 41.
j logKapp in Imidazole 30 mM pH 6.9, KCl 50 mM, from ref. 95.
|
|||||||||||
| La3+ | 15.5 | 19.5 | 5.20 | 10.2 | 11.3 | 4.1 | 6.2 | 6.2 | |||
| Ce3+ | 16.1 | 20.5 | |||||||||
| Pr3+ | 16.3 | 21.1 | 4.5 | ||||||||
| Nd3+ | 16.6 | 21.6 | 10.0 | 11.3 | 4.5 | ||||||
| Sm3+ | 17.0 | 22.3 | 11.2 | ||||||||
| Eu3+ | 17.2 | 22.4 | 5.2 | 6.8 | 8.00 | 4.6 | 6.8 | ||||
| Gd3+ | 17.3 | 22.5 | 5.2 | 11.0 | |||||||
| Tb3+ | 17.8 | 22.7 | 6.8 | 8.8 | 10.7 | 4.6 | 6.3 | ||||
| Dy3+ | 18.2 | 22.8 | 8.6 | 4.6 | |||||||
| Ho3+ | 18.4 | 22.8 | 10.6 | ||||||||
| Er3+ | 18.9 | 22.7 | |||||||||
| Tm3+ | 19.6 | 22.7 | |||||||||
| Yb3+ | 19.7 | 22.6 | 4.6 | 6.4 | |||||||
| Lu3+ | 20.0 | 22.4 | 4.6 | ||||||||
| Ca2+ | 11.0 | 10.7 | 4.5 | 4.6 | 6 | 4.2 | 3.2 | 3.3 | 3.8 | 3.7 | 1.7 |
Joshi and coworkers used LBT to develop filters for REE recovery from dilute acid wash.255 They genetically engineered a Curli fibre produced in E. coli biofilm and known to form amyloid fibres extracellularly, to fuse it with two or four repeats of an LBT sequence. Following a similar strategy, Kaplan and co-workers developed filters based on silk nanofibrils (SNF) functionalized with LBT peptides (Fig. 26), either by chemical coupling or by genetically fusing LBT to a silk-elastin-like protein (SELPS).256
 | ||
| Fig. 26 Overview of the two approaches used to fabricate biobased filter membranes. Reprinted with permission from ref. 256. Copyright 2024 American Chemical Society. | ||
Following the discovery of LanM, their potential for the recovery of REE (including Y3+ and Sc3+) was investigated mostly with Mex-LanM, and more recently with Hans-LanM (Table 13). The strategies employed relied on size-exclusion filtration, on LanM-functionalised agarose beads,257 on functionalized magnetic nanoparticles (MNP-LanM, Fig. 27),260 on polymeric membranes functionalized with LanM-derived peptides,261 and on liquid–liquid phase separation made by fusing LanM to a thermo-responsive genetically encoded polypeptide RELP.258
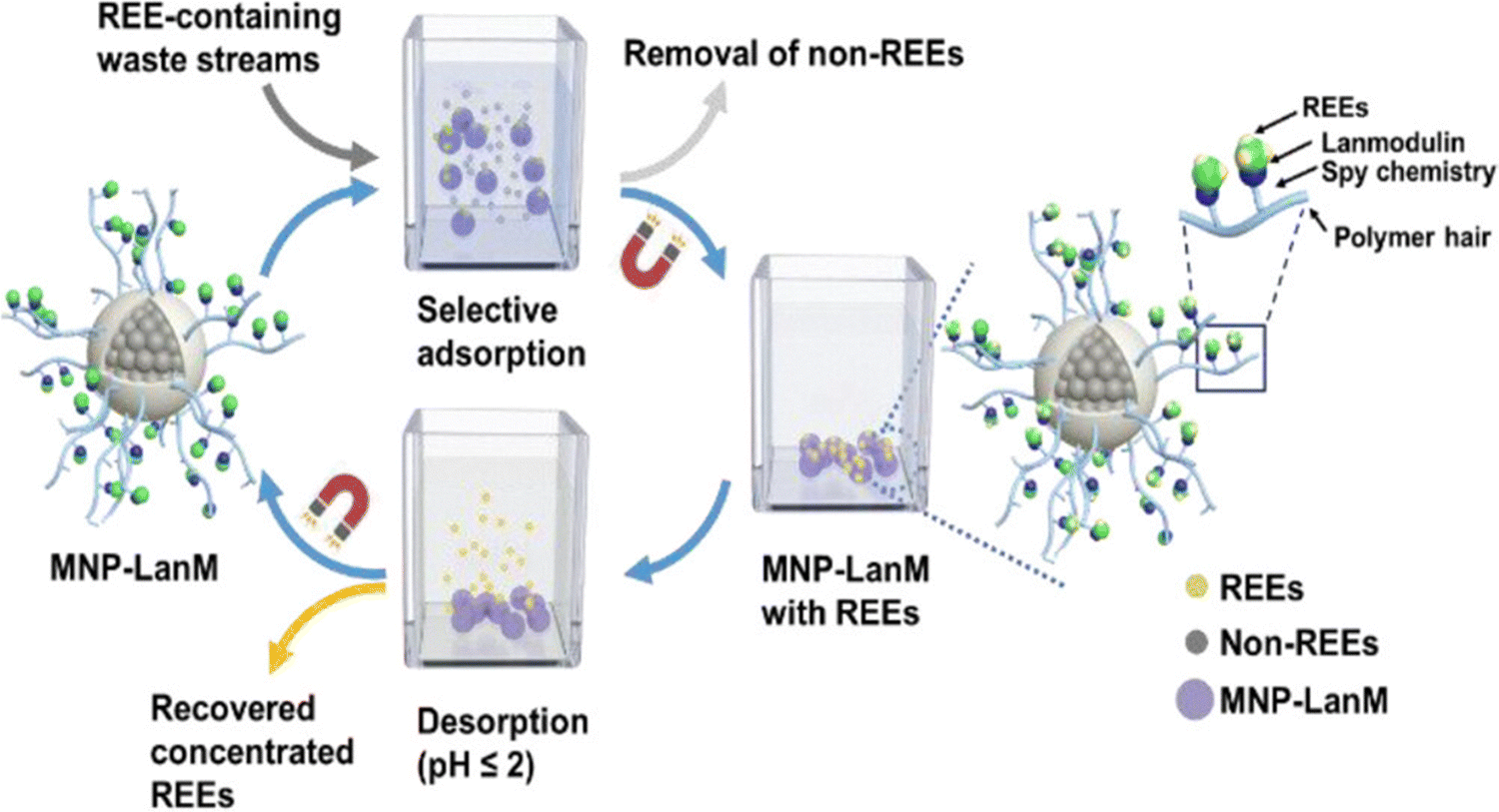 | ||
| Fig. 27 Use of LanM-functionalized magnetic nanoparticles for REE recovery.258,259 Reprinted with permission from ref. 260. Copyright 2023 American Chemical Society. | ||
Cotruvo and coworkers investigated the use of LanD for large Ln-separation.125 They hypothesized that by combining mutations of LanD to lower the affinity of its monomer for Ln3+ (E75Q) and favour the dimer formation by removing repulsive electrostatic interactions (E78A), they could obtain a dimer that preferentially binds Pr3+ and Nd3+ over La3+ and Ce3+.
Still with the aim of developing a method for REE recovery, Banta and coworkers proposed to use the RTX protein, which can bind up to 8 lanthanides at pH 5.5.129
LBT-based fibres displayed a similar pH-sensitivity than observed in solution.255 The fibres could bind Tb3+ down to pH 5, and their Tb3+-sorption capacity was decreased to the level of non-functionalized fibres at pH 3. Park and Cotruvo showed that Mex-LanM functionalised agarose beads retained their ability to bind REE at pH 3,257 which was consistent with Mex-LanM capacity to bind Ln3+ down to pH 2.5 in solution.155 RTX is the protein that seems to be the more tolerant to pH as it retains some Ln3+-binding capacity at pH < 1.5. However, the proteins precipitate at low pH, which complicated the analysis.
Although the different scaffolds described to date present various pH-sensitivity, most of them were assayed for their ability to bind Ln on slightly acidic solutions (pH 4–7, Table 12) corresponding to REE sources obtained from mine tailings, or leachates. Desorption of REE was only investigated for LBT- and LanM-based materials and could be achieved with acidic solutions (HCl, HNO3) at pH < 2. This is a consequence of the protonation of Ln3+-binding amino acids (e.g. Asp, Glu), which results in a lower affinity for Ln3+.
REE vs. non-REE. Working with low-grade, secondary sources of REE that have a low content in REE (WEEE, phosphogypsum, bauxite, mine tailings, coal ashes, mine drainages) requires a good selectivity for REE vs. non-REE in order to minimize the purification steps. Usual competing metal ions consist notably of alkaline-earth (Ca, Mg), and d-block (Mn, Fe, Co, Ni, Cu, Zn) elements (Table 12). The ability of the different scaffolds or derived materials to retain selectively REE vs. non-REE has been assessed in most studies.
For the LBT-based fibres described by Joshi and coworkers,255 REE sorption fell in the presence of 10 to 100 times more concentrated non-REE metal ions (Al, Cu, Fe, Ni), whereas Ca only had a small impact. Similarly, Kaplan and coworkers reported that the Tb3+-binding capacity of silk nanofibrils functionalised with LBT was decreased in the presence of 10 to 100 times more concentrated Cu or Fe, but not by Ca and Zn.256 LBT has a selectivity against Ca2+ of ΔlogKCa ≈ 2–3 (see 4. Selectivity), which appears sufficient to compensate for the difference in concentrations. However, from the observations made in these two studies, it is anticipated that LBT selectivity against d-block elements is quite low.
LanMs display a very high selectivity against Ca (ΔlogKCa ≈ 8 for Mex-LanM,81,155 ΔlogKCaa ≈ 4–6 for Hans-LanM62) and d-block elements (ΔlogKCu ≈ 6 and ΔlogKZn ≈ 8 for Mex-LanM155) (see 4. Selectivity). This can explain the impressive ability of LanM to selectively recover REE among other metal ions in complex solutions (Table 12) with high recovery yields and purity when using size-exclusion separation,155 functionalised agarose beads,257 or liquid–liquid phase separation.258,259 Moreover, Gao, Wei and coworkers demonstrated that even with concentrations of non-REE 10 to 1000 times higher than of REE, LanM-functionalized magnetic nanoparticles could selectively bind REE over competing metal ions and achieve a REE purity of 31% wt (compared to the 0.03% wt of the initial solution) corresponding to a 967-fold improvement.260
RTX is interesting due to its ability to bind several Ln per protein over a broad pH range.129 However, one limitation to its use is the precipitation of the protein at low pH. To overcome precipitation issues, Banta and coworkers coupled the RTX protein to a maltose-binding protein (MBP). Working at pH 6, they showed that RTX-MBP could selectively recover up to ∼85% of Nd3+ and Dy3+ in a solution containing also Fe2+ and Co2+, which suggests a good selectivity of this modified protein for Ln over d-block metal ions.
Intra-REE. The design of Ln-binding peptides and proteins with good intra-REE selectivity would be highly appealing with the aim to reduce the number of steps required to obtain pure and isolated REE, but it is an outstanding challenge given the similarities of REE (see 1. Introduction) and the moderate intra-REE selectivity displayed so far by Ln-binding peptides and proteins (see 4. Selectivity). The LBT-fibers reported by Joshi and coworkers showed a moderate preference for the binding of Eu3+, Tb3+, and Dy3+ relative to other Ln3+.255 Cotruvo and coworkers noted an intra-REE selectivity between heavy (HREE: Tb3+–Lu3+, Y3+) and light (LREE: La3+–Gd3+) when using LanM-functionalised on agarose beads similar to the one observed in solution.257
To overcome the moderate intra-REE selectivity of Ln-binding peptides and proteins, some studies have proposed innovative strategies. The separation process based on LanM-functionalised on agarose beads was optimised by using mild competitors for Ln desorption (malonate, citrate).167 A two-cycle process was proposed in order to recover five distinct fractions: Sc3+, Y3+, HREE (Gd3+–Lu3+), middle REE (MREE: Pr3+–Eu3+), and LREE (La3+, Ce3+). A solution reproducing metal ions concentrations found in coal combustion products or mine tailings was adsorbed onto the column. The first desorption cycle used malonate in order to desorb up to 99% of Sc with >99% purity. Next, pH desorption was performed by decreasing the pH successively to 2.3, 2.1 and 1.9, which enabled a first separation of REE into HREE and Y3+, LREE, and MREE. These fractions were further purified using citrate desorption, which led to the recovery of HREE with 88–100% purity, >95% recovery of Y3+ with 92% purity, recovery of MREE with ∼80% purity, and recovery of LREE with 98% purity.
As an alternative strategy, Cotruvo and coworkers investigated how intra-REE selectivity could be improved by controlling the dimerization state of Hans-LanM to separate Dy3+ from Nd3+ working with a synthetic leachate solution of NdFeB magnets.62 With Hans-LanM functionalised agarose beads, Dy3+ could be desorbed from the column with malonate and obtained with 83% purity without significant desorption of Nd3+, which was then desorbed at pH 1.5 and obtained at 99.8% purity. In a similar approach, the LanD-E75Q/E78A mutant optimised to form dimer with medium REE, was able to retain Pr3+ and Nd3+ better than the larger Ce3+ and La3+.
REE recovery using Ln-binding peptides and proteins requires to meet several criteria. The first is the selectivity of the peptides and proteins for REE vs. non-REE, and ideally also for intra-REE separation. The discovery of LanM, which has a high selectivity vs. non-REE has represented a breakthrough compared to LBT-based strategies that are more sensitive to high concentrations of d-block metal ions. Nonetheless, Ln-binding peptides and proteins still present a moderate intra-REE selectivity (see 4. Selectivity) that will need to be improved. The second criterion is to improve the pH range in which the scaffold is stable and retains Ln-binding capacity. Last, different strategies were proposed for the sorption and recovery of REE. Scaling up to meet industrial needs will require a facile and low-cost production of the scaffold and its derived material. The strategy employed for functionalizing magnetic nanoparticles with LanM using Spy chemistry is interesting for this purpose, as it enabled the selective coupling of the protein directly from cell lysates, thus avoiding protein purification steps.262
Importantly, despite the considerable efforts made to develop innovative, more sustainable technologies to exploit secondary REE sources, the main challenge to their industrial implementation remains their economic viability. The economic feasibility of the biosorption process for the recovery of REE from different low-grade sources was evaluated and strongly depends on REE market prices. Only an increase of REE prices back to their level of 2011 would make actual biosorption processes profitable.263 The fluctuating demands in REE and its impact on the development at the industrial scale of more environmentally friendly technologies have been discussed in ref. 235.
6.4 Catalysis
The field of Ln–proteins and Ln–peptides catalysis is emerging, and a few examples have been published. Ln3+ Lewis acidity has been exploited for hydrolysis reactions and in alcohol dehydrogenase (ADH)-like reactions (Section 6.2.1). Ln3+ photoredox properties have been exploited for the homolytic cleavage and oxidation of diols (Section 6.2.2).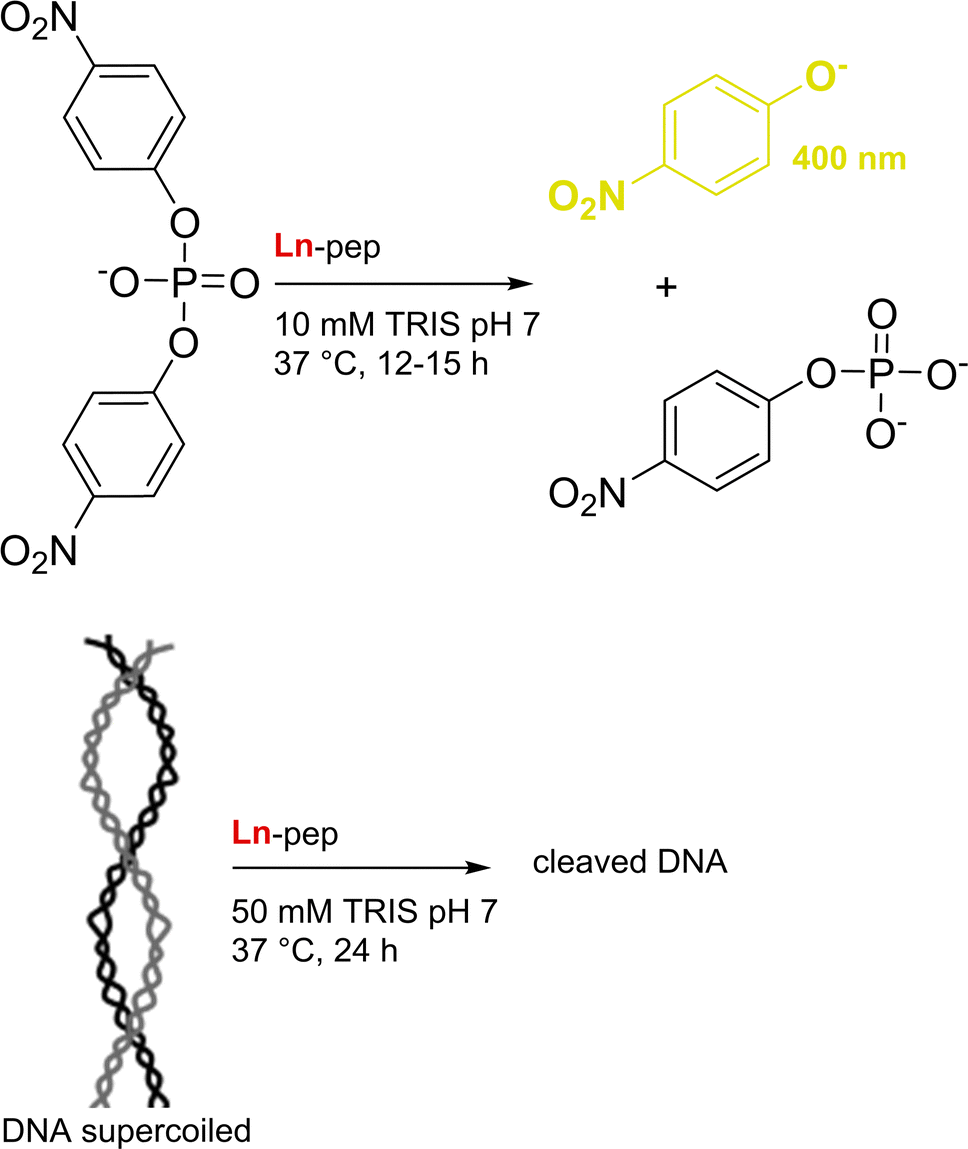 | ||
| Fig. 28 Phosphate ester and DNA cleavage activities of Ln-pep (Ln = Eu3+, Ce4+). | ||
The Ce4+–peptides were more active than their Eu3+ counterparts, consistent with the increased Lewis acidity and nucleophilicity of the Ln3+-bound water molecules.16 The effect of the pH on Ln–peptides reactivity was evaluated in the range 6.9–9.0.266 The rate increased at pH 8.2, which is close to the pKa of Ln3+-bound water molecule (Fig. 1), indicating that it is involved in DNA hydrolytic cleavage. In addition, the cleavage catalysed by Ln–peptides was site-selective, suggesting that DNA sequences could be recognized and cleaved preferentially.16
Ln3+ Lewis acidic properties were also used for the catalysis of the hydrolysis of 2-naphthyl hexanoate (Fig. 29). Jackson and coworkers used the PTE-R18 protein, which has a Zn2+-binding site, and showed that the demetallated protein could bind Ln3+.21 The activity of the resulting Ln–protein was pH-dependent, which was linked to the deprotonation of a Ln-bound water molecule.
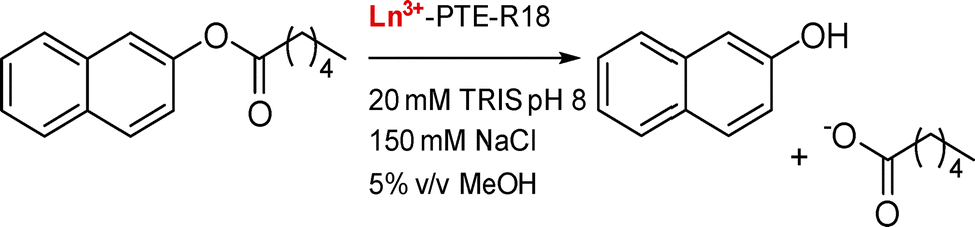 | ||
| Fig. 29 Hydrolysis of 2-naphthyl hexanoate by Ln-PTE-R18. | ||
The first and sole known Ln–enzymes, Ln-alcohol dehydrogenases (Ln-ADH), are an example of how the fine-tuning of Ln3+ Lewis acid properties along the series may impact reactivity. In the enzyme, Ln3+ is essential for catalysis because it tunes the redox potential of the redox cofactor, the pyrroloquinoline quinone (PQQ). Interestingly, the activity of the XoxF-methanol dehydrogenase (MDH) is strongly dependent on the Ln3+ in the active site, the lighter and less Lewis acidic Ln3+ giving the higher activity while no activity is detected for Ln3+ heavier than Er3+ (Fig. 30).176 A similar trend has been also recently observed for an Ln-ADH from Pseudomonas putida KT2440 (PedH).267 Several hypotheses have been proposed and are still under investigation to explain this dramatic impact on Ln-ADH reactivity. They consider not only PQQ redox properties, but also changes in the coordination number, or exchange rate constants of Ln3+-bound substrate/product.
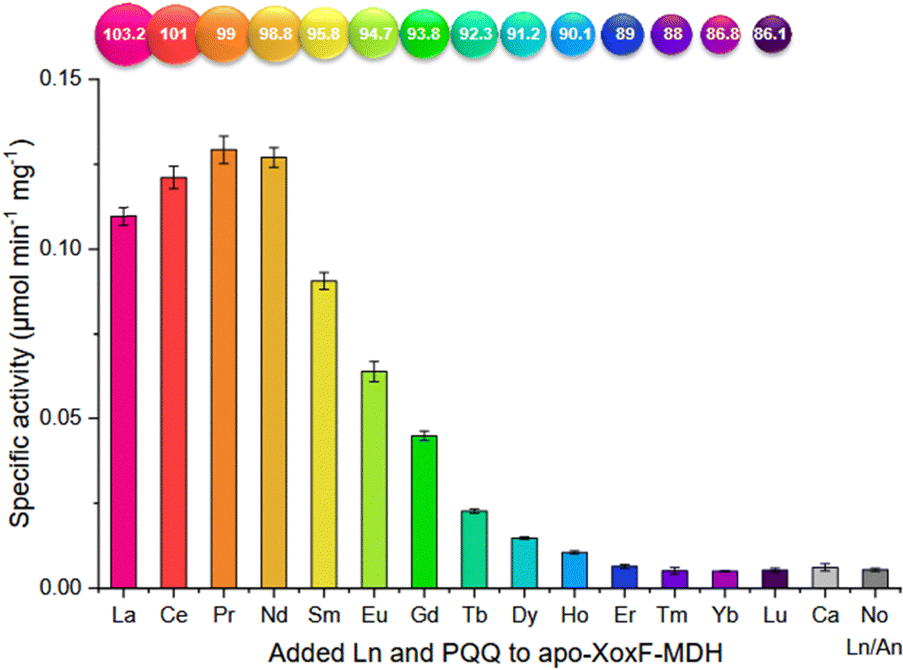 | ||
| Fig. 30 Ln-dependent activity of XoxF-MDH. The enzyme was reconstituted from the apo-enzyme by incubation with PQQ and metal ion for 72 h at 4 °C. Reproduced from ref. 176 (Creative Commons Attribution Non-Commercial License 2023). | ||
Working towards bringing new insights into Ln-MDH mechanism, Olshansky and coworkers built a structural mimic of the enzyme.20 Starting from the PQQ-binding protein PqqT, they engineered the binding site with a single mutation (K142D) in order to introduce a Ln3+-binding site. They tested the ability of this structural mimic of Ln-MDH to oxidize benzyl alcohol, using a chemical trap (O-((perfluorophenyl)methyl)hydroxylamine (PFBHA)) to capture the benzaldehyde formed and identify it by LC-MS (Fig. 31). Only the La3+–PQQ⊂K142D–PqqT mutant was able to perform this reaction, and not the WT protein or K142A mutant both lacking an Ln3+-binding site, nor the mutants lacking La3+, or PQQ.
 | ||
| Fig. 31 Benzyl alcohol oxidation by La3+-bound PQQ⊂K142D–PqqT. | ||
The ability of ADH to oxidize alcohols has been exploited further by Bange, Klebensberger and coworkers for foreseen application in bio-based plastic production.116 Starting from the Pr-PedH from P. putida KT2440, they constructed libraries of mutants of the enzyme expressed in E. coli and screened each colony for in vivo enzymatic activity towards the oxidation of HMFA (Fig. 32). This compound is an important intermediate for the obtention of furan-2,5-dicarboxylic acid (FDCA), a synthon that could serve as a plant-based alternative in the production of polymers. Out of 30000 clones, 3 mutants were identified and their activity was confirmed by testing the purified enzymes. They shared mutations of the same residues, F412 and W561, positioned in the substrate channel and on a lid loop, respectively. These mutations enable bulkier substrates, such as HMFA, to access the active site. The mutant with the best catalytic activity for HMFA oxidation, PedHF412V/W561A, could also successfully oxidize ethanol, HMF and FFF (Fig. 32), which opens up the route to the whole-cell biocatalytic production of FDCA.
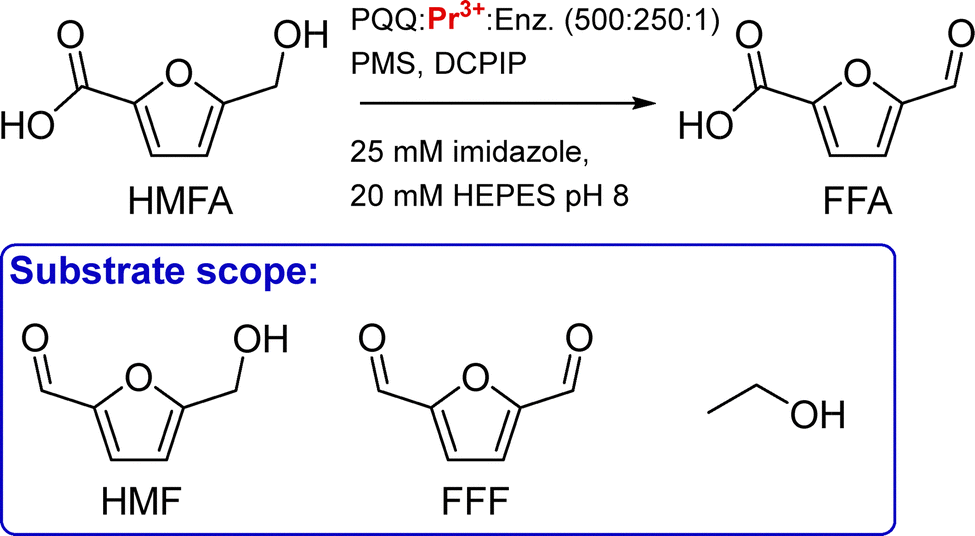 | ||
| Fig. 32 Oxidation of 5-(hydroxymethyl)furoic acid (HMFA) and derivatives by mutants of PedH. FFA: 5-formylfuroic acid; HMF: 5-(hydroxymethyl)furfural; FFF: 5-formylfurfural; PMS: phenazine methosulfate; DCPIP: 2,6-dichlorophenol indophenol. | ||
The scaffold TFD-EE N6W was the starting point for the following catalysis study (Fig. 33).19 Changes were made to the scaffold in order to introduce asymmetry in the binding site (single chain variant), to limit photodamage (removal of Trp residues), and to prevent non-specific metal binding to the protein surface (removal of clusters of negatively charged residues by mutating Asp to Asn and Glu to Gln), leading to the optimised scaffold PLZ1.4.
 | ||
| Fig. 33 Photoredox catalysis of aromatic diols homolytic cleavage by Ce-artificial enzymes. | ||
Upon irradiation, the artificial enzyme Ce-PLZ1.4 was able to catalyse the homolytic cleavage of homobenzoin into benzaldehyde (Fig. 33). Compared to molecular catalysts, the interest of the protein scaffold is to be able to control the stereoselectivity of the reaction. The authors showed that Ce-PLZ1.4 catalysed better the cleavage of (R,R)- and (S,S)-hydrobenzoin compared to the meso-isomer, and so displayed some diastereoselectivity. Investigation of the substrate scope of the enzyme also evidenced chemoselectivity, with aromatic diols being better cleaved than aliphatic ones. Last, the PLZ1.1 protein was expressed at the surface of E. coli cells, which were subsequently treated with CeCl3 and light and were able to cleave a lignin surrogate (2-phenoxy-1-phenylpropane-1,3-diol) opening the way to photocontrolled whole-cell catalysis.
To date, the scope of Ln–peptides and Ln–proteins catalysts has been limited mainly to reactions exploiting Ln3+ Lewis acidity properties, with the exception of an original work demonstrating that photoredox catalysis is also achievable.19 In addition, the peptide scaffold of the catalysts has been used to select the substrate, whether by using DNA recognition sequence, or by engineering the catalytic pocket,116 or by favouring one stereoisomer over others.19
In the medium term, recent advances obtained with molecular catalysts should be transposable to Ln artificial enzymes. Ln-complexes have been used as Lewis acids catalysts,268,269 in particular for C–C bond formation in aqueous media.268 Ln-complexes are also powerful photocatalysts, that can be used for photo-reduction or photo-oxidation.270–274 Notably, the reduction of CO2, N2 and NOx was achieved in mild aqueous conditions.273,274 Similar reactions could be implemented in artificial Ln–enzymes, which operate in aqueous media and whose peptide scaffold allows modulation of the hydrophilicity of the active site. Combining the chemical properties of Ln with the modularity of the peptide scaffold will help control the reactivity of the metal centre, the selectivity of the reaction and fine-tuning of second sphere interactions, and is expected to broaden the reactivities accessible with artificial enzyme.275
7. Conclusions & perspectives
In the last decade, significant progress has been made in the study of natural Ln-dependent proteins as well as in the design and engineering of Ln-binding peptides and proteins for a variety of relevant applications. Notwithstanding, numerous challenges remain to be addressed for the advancement of this flourishing field, starting with a sound and quantitative understanding of Ln coordination by pH buffers to refine the accuracy of available data and allow reliable comparisons.The current limitations in designing Ln-binding systems with high affinity, selectivity, and inertness underscore the complexity of controlling key factors such as the coordination sphere (i.e. coordination and hydration number), scaffold stability, and the influence of long-range effects.
The control over the hydration number q is well understood and achievable for EF-hand motifs, and it can be tuned for other scaffolds by adjusting the denticity and positioning of the binding site within a hydrophobic environment. However, certain long-range outer-sphere factors, such as the local conformation of the main chain and the orientation of amino acids can influence q in ways that are not yet fully controllable.
The majority of peptide and protein scaffolds exhibit an Ln-binding affinity in the micro- to nanomolar range (logKapp ∼ 6–9), with only a limited number extending to the picomolar range (logKapp ∼ 12), which is significantly lower than the affinity achievable with small chelators. Similarly to the tuning of the hydration number q, long-range outer-sphere factors, which are not yet understood, strongly influence the Ln affinity, as in the case of LanM compared to its isolated EF-hand motifs. It is worth noting that this gap between the Ln-affinity of peptide/proteins and small organic chelators is not observed for d-block metal ions, for which proteins and chelators of similar affinity, up to logKapp ∼ 21, are known. The underlying reason for this remains to be elucidated. One possible explanation is that, considering that we are at the dawn of lanthanome research, the tightest Ln-binding proteins have yet to be discovered. Besides, due to the ionic nature of Ln bonding and their preference for higher CN than d-block metal ions, peptides and proteins might need more demanding constraints to bind Ln3+ as efficiently as d-block metal ions.
Achieving higher affinities requires careful optimisation of both the Ln-binding site and scaffold stability by fine-tuning key parameters such as the type and denticity of the coordinating amino acids, as well as local and long-range structural constraints in the scaffold. To this end, computational design appears as a powerful tool to obtain a well-structured and stable protein scaffold, as is the case for the Ln-binding TIM barrel. In addition, the pH-dependency of Ln-binding by peptides and proteins has only been seldom looked at but is an important parameter for applications in REE recovery.
The selectivity relative to Ca2+ is generally favourable due to the higher coordination number and higher charge of Ln3+. Nevertheless, the remarkable selectivity exhibited by LanM remains to be fully elucidated. Again, this is likely attributable to the influence of outer sphere effects.
With respect to d-block elements, the selectivity of peptides and proteins for Ln3+ often surpasses that of small chelators, a phenomenon attributable to distinct preferences for ligand types, coordination number and geometry.
In comparison to Ln3+, peptide and protein scaffolds demonstrate a slightly higher affinity for An3+, presumably due to enhanced covalency. However, further investigation is warranted to fully elucidate the underlying mechanisms and to boost the selectivity between Ln3+ and An3+. Since An3+ can be (radioactive) contaminants of common Ln3+ sources, this will be crucial in the context of Ln recovery and radioactive decontamination.
The intra-Ln selectivity remains an outstanding challenge due to their similar ionic radii and Lewis acidity. The intra-Ln selectivity exhibited by peptide and protein scaffolds is not yet comparable to that observed for small chelators. This discrepancy may be attributed to the greater flexibility of peptides, which consequently enables a higher degree of adaptability to Ln3+ ions differences in ionic radius and coordination number. Further studies are required to ascertain whether size discrimination can be enhanced based on both electronic and steric considerations, for instance by modulating the rigidity of the coordination site and its position (buried or surface-exposed) within the scaffold.
The kinetic inertness of Ln coordinated to peptides and proteins is a largely neglected aspect that merits greater attention and research focus in the upcoming years, especially with respect to the development of efficient imaging agents and tools for Ln separation. In order to understand how to control the kinetic inertness of Ln–peptide and protein complexes, it is also essential to accumulate a substantial body of experimental data, which is currently lacking.
Overall, it appears that the elusive contributions beyond the first coordination sphere are of paramount importance in tuning properties like affinity, selectivity, and inertness, and must be studied further to achieve optimized designs. It is expected that emerging technologies based on machine learning and artificial intelligence will soon be beneficial in unravelling the influence of such long-range interactions, provided that solid experimental knowledge is established.276–278 In this context, significant contributions are also expected from approaches beyond rational design, such as directed evolution.279
By deepening our understanding of these long-range effects and their impact on the coordination environment, we will be able to address the key challenges in the field: (i) developing high-affinity, intra-Ln selective peptides and proteins for enhanced Ln-recovery; (ii) achieving kinetically inert Ln-binding scaffolds for medical imaging and therapy; and (iii) optimizing the design of Ln-based (photo)catalysts. These advancements hold the potential to revolutionize a wide range of applications, from environmental sustainability to cutting-edge medical technologies.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Appendices
Acknowledgements
The authors acknowledge financial support from the French National Research Agency (ANR) through the ANR-23-CE07-0002 (LnZYME) and ANR-22-CE29-0026 (DREAMY) projects.References
- S. V. Eliseeva and J.-C. G. Bünzli, New J. Chem., 2011, 35, 1165–1176 RSC.
- Directorate-General for Internal Market, Industry, Entrepreneurship and SMEs (European Commission), M. Grohol and C. Veeh, Study on the critical raw materials for the EU 2023: final report, Publications Office of the European Union, 2023.
- V. Balaram, Geosci. Front., 2019, 10, 1285–1303 CrossRef CAS.
- N. A. Fitriyanto, M. Fushimi, M. Matsunaga, A. Pertiwiningrum, T. Iwama and K. Kawai, J. Biosci. Bioeng., 2011, 111, 613–617 CrossRef CAS PubMed.
- Y. Hibi, K. Asai, H. Arafuka, M. Hamajima, T. Iwama and K. Kawai, J. Biosci. Bioeng., 2011, 111, 547–549 CrossRef CAS.
- T. Nakagawa, R. Mitsui, A. Tani, K. Sasa, S. Tashiro, T. Iwama, T. Hayakawa and K. Kawai, PLoS One, 2012, 7, e50480 CrossRef CAS.
- A. Pol, T. R. M. Barends, A. Dietl, A. F. Khadem, J. Eygensteyn, M. S. M. Jetten and H. J. M. O. den Camp, Environ. Microbiol., 2014, 16, 255–264 CrossRef CAS.
- L. Ancel, A. Niedźwiecka, C. Lebrun, C. Gateau and P. Delangle, C. R. Chim., 2013, 16, 515–523 CrossRef CAS.
- E. Skovran, C. Raghuraman and N. C. Martinez-Gomez, Curr. Issues Mol. Biol., 2019, 101–116 CrossRef CAS.
- L. J. Daumann, Angew. Chem., Int. Ed., 2019, 58, 12795–12802 CrossRef CAS.
- E. R. Featherston and J. A. Cotruvo, Biochim. Biophys. Acta, Mol. Cell Res., 2021, 1868, 118864 CrossRef CAS PubMed.
- L. J. Daumann, A. Pol, H. J. M. Op Den Camp and N. C. Martinez-Gomez, Advances in Microbial Physiology, Elsevier, 2022, vol. 81, pp. 1–24 Search PubMed.
- A. M. Webster and A. F. A. Peacock, Chem. Commun., 2021, 57, 6851–6862 RSC.
- J. A. Mattocks and J. A. Cotruvo, Chem. Soc. Rev., 2020, 49, 8315–8334 RSC.
- Q. Ye, D. Wang and N. Wei, Trends Biotechnol., 2024, 42, 575–590 CrossRef CAS PubMed.
- R. T. Kovacic, J. T. Welch and S. J. Franklin, J. Am. Chem. Soc., 2003, 125, 6656–6662 CrossRef CAS.
- S. Lim and S. J. Franklin, Cell. Mol. Life Sci., 2004, 61, 2184–2188 CrossRef CAS.
- S. J. Franklin and J. T. Welch, Comments Inorg. Chem., 2005, 26, 127–164 CrossRef CAS.
- A. S. Klein, F. Leiss-Maier, R. Mühlhofer, B. Boesen, G. Mustafa, H. Kugler and C. Zeymer, J. Am. Chem. Soc., 2024, 146(38), 25976–25985 CrossRef CAS.
- P. J. Thompson, D. G. Boggs, C. A. Wilson, A. T. Bruchs, U. Velidandla, J. Bridwell-Rabb and L. Olshansky, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2405836121 CrossRef CAS.
- C. W. Breeze, Y. Nakano, E. C. Campbell, R. L. Frkic, D. W. Lupton and C. J. Jackson, Acta Crystallogr., Sect. D:Struct. Biol., 2024, 80, 289–298 CrossRef CAS.
- L. Götzke, G. Schaper, J. März, P. Kaden, N. Huittinen, T. Stumpf, K. K. K. Kammerlander, E. Brunner, P. Hahn, A. Mehnert, B. Kersting, T. Henle, L. F. Lindoy, G. Zanoni and J. J. Weigand, Coord. Chem. Rev., 2019, 386, 267–309 CrossRef.
- R. Sánchez-Fernández, I. Obregon-Gomez, A. Sarmiento, M. E. Vázquez and E. Pazos, Chem. Commun., 2024, 60, 12650–12661 RSC.
- M. A. Hay and C. Boskovic, Chem. – Eur. J., 2021, 27, 3608–3637 CrossRef CAS PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- L. R. Morss, Chem. Rev., 1976, 76, 827–841 CrossRef CAS.
- P. D’Angelo, A. Zitolo, V. Migliorati, G. Chillemi, M. Duvail, P. Vitorge, S. Abadie and R. Spezia, Inorg. Chem., 2011, 50, 4572–4579 CrossRef PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- C. Ekberg and P. L. Brown, Hydrolysis of Metal Ions, John Wiley & Sons, Ltd, 2016 Search PubMed.
- H. C. Aspinall, f-Block Chemistry, Oxford University Press, 2020 Search PubMed.
- L. Ruckthong, M. L. Zastrow, J. A. Stuckey and V. L. Pecoraro, J. Am. Chem. Soc., 2016, 138, 11979–11988 CrossRef CAS.
- S. C. Edington, S. Liu and C. R. Baiz, in Methods in Enzymology, ed. J. A. Cotruvo, Academic Press, 2021, vol. 651, pp. 157–191 Search PubMed.
- S. Liu, E. R. Featherston, J. A. Cotruvo and C. R. Baiz, Phys. Chem. Chem. Phys., 2021, 23, 21690–21700 RSC.
- S. A. Cotton, C. R. Chim., 2005, 8, 129–145 CrossRef CAS.
- A. Smerigan, S. Biswas, F. D. Vila, J. Hong, J. Perez-Aguilar, A. S. Hoffman, L. Greenlee, R. B. Getman and S. R. Bare, Inorg. Chem., 2023, 62, 14523–14532 CrossRef CAS PubMed.
- T. Summers, D. Zhang, J. Sobrinho, A. de Bettencourt-Dias, R. Rousseau, V.-A. Glezakou and D. Cantu, ChemRxiv, 2024, preprint DOI:10.26434/chemrxiv-2024-lgkjh.
- H. Lumpe, A. Menke, C. Haisch, P. Mayer, A. Kabelitz, K. V. Yusenko, A. Guilherme Buzanich, T. Block, R. Pöttgen, F. Emmerling and L. J. Daumann, Chem. – Eur. J., 2020, 26, 10133–10139 CrossRef CAS.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504 RSC.
- R. M. Supkowski and W. D. W. Horrocks, Inorg. Chim. Acta, 2002, 340, 44–48 CrossRef CAS.
- C. S. Bonnet, P. H. Fries, S. Crouzy, O. Sénèque, F. Cisnetti, D. Boturyn, P. Dumy and P. Delangle, Chem. – Eur. J., 2009, 15, 7083–7093 CrossRef CAS PubMed.
- G. Pintacuda, M. John, X.-C. Su and G. Otting, Acc. Chem. Res., 2007, 40, 206–212 CrossRef CAS PubMed.
- K. N. Allen and B. Imperiali, Curr. Opin. Chem. Biol., 2010, 14, 247–254 CrossRef CAS PubMed.
- W.-M. Liu, M. Overhand and M. Ubbink, Coord. Chem. Rev., 2014, 273–274, 2–12 CrossRef CAS.
- C. Nitsche and G. Otting, Prog. Nucl. Magn. Reson. Spectrosc., 2017, 98–99, 20–49 CrossRef PubMed.
- D. Joss and D. Häussinger, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 114–115, 284–312 CrossRef.
- I. Baig, I. Bertini, C. Del Bianco, Y. K. Gupta, Y.-M. Lee, C. Luchinat and A. Quattrone, Biochemistry, 2004, 43, 5562–5573 CrossRef PubMed.
- I. Bertini, C. Del Bianco, I. Gelis, N. Katsaros, C. Luchinat, G. Parigi, M. Peana, A. Provenzani and M. A. Zoroddu, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6841–6846 CrossRef PubMed.
- N. C. J. Strynadka and M. N. G. James, Annu. Rev. Biochem., 1989, 58, 951–999 CrossRef.
- A. Lewit-Bentley and S. Réty, Curr. Opin. Struct. Biol., 2000, 10, 637–643 CrossRef PubMed.
- Z. Grabarek, J. Mol. Biol., 2006, 359, 509–525 CrossRef PubMed.
- J. L. Gifford, M. P. Walsh and H. J. Vogel, Biochem. J., 2007, 405, 199–221 CrossRef.
- D. B. Halling, B. J. Liebeskind, A. W. Hall and R. W. Aldrich, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(9), 1216–1225 CrossRef PubMed.
- H. G. Brittain, F. S. Richardson and R. B. Martin, J. Am. Chem. Soc., 1976, 98, 8255–8260 CrossRef PubMed.
- B. Martin and F. S. Richardson, Q. Rev. Biophys., 1979, 12, 181–209 CrossRef PubMed.
- W. D. Horrocks and D. R. Sudnick, Acc. Chem. Res., 1981, 14, 384–392 CrossRef.
- M. A. Valentini and J. C. Wright, Anal. Biochem., 1985, 150, 47–57 CrossRef PubMed.
- C.-L. A. Wang, P. C. Leavis, W. D. Horrocks and J. Gergely, Biochemistry, 1981, 20, 2439–2444 CrossRef PubMed.
- J. Gariepy, B. D. Sykes and R. S. Hodges, Biochemistry, 1983, 22, 1765–1772 CrossRef PubMed.
- K. D. Daughtry, L. J. Martin, A. Sarraju, B. Imperiali and K. N. Allen, ChemBioChem, 2012, 13, 2567–2574 CrossRef PubMed.
- S. Jain, J. T. Welch, W. D. Horrocks and S. J. Franklin, Inorg. Chem., 2003, 42, 8098–8104 CrossRef PubMed.
- J. A. Mattocks, J. J. Jung, C.-Y. Lin, Z. Dong, N. H. Yennawar, E. R. Featherston, C. S. Kang-Yun, T. A. Hamilton, D. M. Park, A. K. Boal and J. A. Cotruvo, Nature, 2023, 618, 87–93 CrossRef PubMed.
- E. R. Featherston, E. J. Issertell and J. A. Cotruvo, J. Am. Chem. Soc., 2021, 143, 14287–14299 CrossRef PubMed.
- M. Nitz, M. Sherawat, K. J. Franz, E. Peisach, K. N. Allen and B. Imperiali, Angew. Chem., Int. Ed., 2004, 43, 3682–3685 CrossRef.
- K. Barthelmes, A. M. Reynolds, E. Peisach, H. R. A. Jonker, N. J. DeNunzio, K. N. Allen, B. Imperiali and H. Schwalbe, J. Am. Chem. Soc., 2011, 133, 808–819 CrossRef PubMed.
- J. P. MacManus, C. W. Hogue, B. J. Marsden, M. Sikorska and A. G. Szabo, J. Biol. Chem., 1990, 265, 10358–10366 CrossRef.
- J. Wöhnert, K. J. Franz, M. Nitz, B. Imperiali and H. Schwalbe, J. Am. Chem. Soc., 2003, 125, 13338–13339 CrossRef PubMed.
- L. J. Martin, M. J. Hähnke, M. Nitz, J. Wöhnert, N. R. Silvaggi, K. N. Allen, H. Schwalbe and B. Imperiali, J. Am. Chem. Soc., 2007, 129, 7106–7113 CrossRef PubMed.
- N. R. Silvaggi, L. J. Martin, H. Schwalbe, B. Imperiali and K. N. Allen, J. Am. Chem. Soc., 2007, 129, 7114–7120 CrossRef.
- D. Barthelmes, M. Gränz, K. Barthelmes, K. N. Allen, B. Imperiali, T. Prisner and H. Schwalbe, J. Biomol. NMR, 2015, 63, 275–282 CrossRef PubMed.
- T. W. Victor, K. H. O’Toole, L. M. Easthon, M. Ge, R. J. Smith, X. Huang, H. Yan, Y. S. Chu, S. Chen, D. Gursoy, M. Ralle, B. Imperiali, K. N. Allen and L. M. Miller, J. Am. Chem. Soc., 2020, 142, 2145–2149 CrossRef.
- M. Nitz, K. J. Franz, R. L. Maglathlin and B. Imperiali, ChemBioChem, 2003, 4, 272–276 CrossRef.
- L. J. Martin, B. R. Sculimbrene, M. Nitz and B. Imperiali, QSAR Comb. Sci., 2005, 24, 1149–1157 CrossRef.
- L. J. Martin and B. Imperiali, in Peptide Libraries: Methods and Protocols, ed. R. Derda, Springer, New York, NY, 2015, pp. 201–220 Search PubMed.
- Y. Kim, J. T. Welch, K. M. Lindstrom and S. J. Franklin, JBIC, J. Biol. Inorg. Chem., 2001, 6, 173–181 CrossRef PubMed.
- J. T. Welch, W. R. Kearney and S. J. Franklin, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3725–3730 CrossRef CAS.
- P. Caravan, J. M. Greenwood, J. T. Welch and S. J. Franklin, Chem. Commun., 2003, 2574–2575 RSC.
- S. Lim and S. J. Franklin, Protein Sci., 2006, 15, 2159–2165 CrossRef PubMed.
- S. W. Wong-Deyrup, Y. Kim and S. J. Franklin, JBIC, J. Biol. Inorg. Chem., 2006, 11, 17–25 CrossRef PubMed.
- K. L. Harris, S. Lim and S. J. Franklin, Inorg. Chem., 2006, 45, 10002–10012 CrossRef PubMed.
- J. A. Cotruvo, E. R. Featherston, J. A. Mattocks, J. V. Ho and T. N. Laremore, J. Am. Chem. Soc., 2018, 140, 15056–15061 CrossRef PubMed.
- C.-L. A. Wang, R. R. Aquaron, P. C. Leavis and J. Gergely, Eur. J. Biochem., 1982, 124, 7–12 CrossRef.
- E. C. Cook, E. R. Featherston, S. A. Showalter and J. A. Cotruvo, Biochemistry, 2019, 58, 120–125 CrossRef PubMed.
- F. Cisnetti, C. Lebrun and P. Delangle, Dalton Trans., 2010, 39, 3560–3562 RSC.
- F. Cisnetti, C. Gateau, C. Lebrun and P. Delangle, Chem. – Eur. J., 2009, 15, 7456–7469 CrossRef PubMed.
- A. Niedźwiecka, F. Cisnetti, C. Lebrun, C. Gateau and P. Delangle, Dalton Trans., 2012, 41, 3239–3247 RSC.
- A. Niedźwiecka, F. Cisnetti, C. Lebrun and P. Delangle, Inorg. Chem., 2012, 51, 5458–5464 CrossRef PubMed.
- J. Veliscek-Carolan, T. L. Hanley and K. A. Jolliffe, RSC Adv., 2016, 6, 75336–75346 RSC.
- S. Safi, A. Jeanson, J. Roques, P. L. Solari, F. Charnay-Pouget, C. Den Auwer, G. Creff, D. J. Aitken and E. Simoni, Inorg. Chem., 2016, 55, 877–886 CrossRef CAS PubMed.
- I. V. Korendovych and W. F. DeGrado, Q. Rev. Biophys., 2020, 53, e3 CrossRef CAS PubMed.
- K. J. Koebke, T. B. J. Pinter, W. C. Pitts and V. L. Pecoraro, Chem. Rev., 2022, 122, 12046–12109 CrossRef CAS.
- P. Teare, C. F. Smith, S. J. Adams, S. Anbu, B. Ciani, L. J. C. Jeuken and A. F. A. Peacock, Dalton Trans., 2018, 47, 10784–10790 RSC.
- D. N. Woolfson, Advances in Protein Chemistry, Academic Press, 2005, vol. 70, pp. 79–112 Search PubMed.
- W. D. Kohn, C. M. Kay and R. S. Hodges, J. Pept. Res., 1998, 51, 9–18 CrossRef CAS PubMed.
- W. D. Kohn, C. M. Kay, B. D. Sykes and R. S. Hodges, J. Am. Chem. Soc., 1998, 120, 1124–1132 CrossRef CAS.
- A. Kashiwada, K. Ishida and K. Matsuda, Bull. Chem. Soc. Jpn., 2007, 80, 2203–2207 CrossRef CAS.
- M. Samiappan, S. Alasibi, R. Cohen-Luria, A. Shanzer and G. Ashkenasy, Chem. Commun., 2012, 48, 9577–9579 RSC.
- M. R. Berwick, D. J. Lewis, A. W. Jones, R. A. Parslow, T. R. Dafforn, H. J. Cooper, J. Wilkie, Z. Pikramenou, M. M. Britton and A. F. A. Peacock, J. Am. Chem. Soc., 2014, 136, 1166–1169 CrossRef CAS.
- M. R. Berwick, L. N. Slope, C. F. Smith, S. M. King, S. L. Newton, R. B. Gillis, G. G. Adams, A. J. Rowe, S. E. Harding, M. M. Britton and A. F. A. Peacock, Chem. Sci., 2016, 7, 2207–2216 RSC.
- L. N. Slope, M. G. Hill, C. F. Smith, P. Teare, F. J. de Cogan, M. M. Britton and A. F. A. Peacock, Chem. Commun., 2020, 56, 3729–3732 RSC.
- L. N. Slope, O. J. Daubney, H. Campbell, S. A. White and A. F. A. Peacock, Angew. Chem., Int. Ed., 2021, 60, 24473–24477 CrossRef CAS PubMed.
- S. L. Newton, A. Franke, A. Zahl, G. Molinaro, A. Kenwright, D. J. Smith, I. Ivanovic-Burmazovic, M. M. Britton and A. F. A. Peacock, Dalton Trans., 2023, 52, 15665–15668 RSC.
- C. W. A. Ende, H. Y. Meng, M. Ye, A. K. Pandey and N. J. Zondlo, ChemBioChem, 2010, 11, 1738–1747 CrossRef.
- A. F. A. Peacock, J. Inorg. Biochem., 2025, 268, 112903 CrossRef CAS PubMed.
- J. K. Myers, C. N. Pace and J. M. Scholtz, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2833–2837 CrossRef CAS PubMed.
- J. S. Richardson and D. C. Richardson, Science, 1988, 240, 1648–1652 CrossRef CAS PubMed.
- L. G. Presta and G. D. Rose, Science, 1988, 240, 1632–1641 CrossRef CAS.
- L. Serrano and A. R. Fersht, Nature, 1989, 342, 296–299 CrossRef CAS.
- Y. W. Deng, S. Y. Ro and A. C. Rosenzweig, JBIC, J. Biol. Inorg. Chem., 2018, 23, 1037–1047 CrossRef CAS PubMed.
- N. M. Good, M. Fellner, K. Demirer, J. Hu, R. P. Hausinger and N. C. Martinez-Gomez, J. Biol. Chem., 2020, 295, 8272–8284 CrossRef CAS.
- R. A. Schmitz, N. Picone, H. Singer, A. Dietl, K.-A. Seifert, A. Pol, M. S. M. Jetten, T. R. M. Barends, L. J. Daumann and H. J. M. Op den Camp, mBio, 2021, 12, e01708–21 CrossRef PubMed.
- B. Jahn, A. Pol, H. Lumpe, T. R. M. Barends, A. Dietl, C. Hogendoorn, H. J. M. Op den Camp and L. J. Daumann, ChemBioChem, 2018, 19, 1147–1153 CrossRef PubMed.
- N. M. Good, H. N. Vu, C. J. Suriano, G. A. Subuyuj, E. Skovran and N. C. Martinez-Gomez, J. Bacteriol., 2016, 198, 3109–3118 CrossRef.
- M. Wehrmann, P. Billard, A. Martin-Meriadec, A. Zegeye and J. Klebensberger, mBio, 2017, 8, e00570–17 CrossRef.
- J. Huang, Z. Yu, J. Groom, J.-F. Cheng, A. Tarver, Y. Yoshikuni and L. Chistoserdova, ISME J., 2019, 13, 2005–2017 CrossRef PubMed.
- M. Wehrmann, E. M. Elsayed, S. Köbbing, L. Bendz, A. Lepak, J. Schwabe, N. Wierckx, G. Bange and J. Klebensberger, ACS Catal., 2020, 10, 7836–7842 CrossRef.
- N. M. Good, R. S. Moore, C. J. Suriano and N. C. Martinez-Gomez, Sci. Rep., 2019, 9, 1–12 CrossRef PubMed.
- L. Wang, A. Hibino, S. Suganuma, A. Ebihara, S. Iwamoto, R. Mitsui, A. Tani, M. Shimada, T. Hayakawa and T. Nakagawa, Enzyme Microb. Technol., 2020, 136, 109518 CrossRef.
- H. Lumpe, A. Pol, H. J. M. Op den Camp and L. J. Daumann, Dalton Trans., 2018, 47, 10463–10472 RSC.
- C. Stines-Chaumeil, F. Mavré, B. Kauffmann, N. Mano and B. Limoges, ACS Omega, 2020, 5, 2015–2026 CrossRef PubMed.
- P. Roszczenko-Jasińska, H. N. Vu, G. A. Subuyuj, R. V. Crisostomo, J. Cai, N. F. Lien, E. J. Clippard, E. M. Ayala, R. T. Ngo, F. Yarza, J. P. Wingett, C. Raghuraman, C. A. Hoeber, N. C. Martinez-Gomez and E. Skovran, Sci. Rep., 2020, 10, 12663 CrossRef.
- J. L. Hemmann, P. Keller, L. Hemmerle, T. Vonderach, A. M. Ochsner, M. Bortfeld-Miller, D. Günther and J. A. Vorholt, J. Biol. Chem., 2023, 299, 102940 CrossRef.
- J. A. Mattocks, J. V. Ho and J. A. Cotruvo, J. Am. Chem. Soc., 2019, 141, 2857–2861 CrossRef PubMed.
- A. M. Ochsner, L. Hemmerle, T. Vonderach, R. Nüssli, M. Bortfeld-Miller, B. Hattendorf and J. A. Vorholt, Mol. Microbiol., 2019, 111, 1152–1166 CrossRef PubMed.
- W. B. Larrinaga, J. J. Jung, C.-Y. Lin, A. K. Boal and J. A. Cotruvo, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2410926121 CrossRef PubMed.
- S. J. Caldwell, I. C. Haydon, N. Piperidou, P.-S. Huang, M. J. Bick, H. S. Sjöström, D. Hilvert, D. Baker and C. Zeymer, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 30362–30369 CrossRef.
- M. A. Blenner, O. Shur, G. R. Szilvay, D. M. Cropek and S. Banta, J. Mol. Biol., 2010, 400, 244–256 CrossRef PubMed.
- W. Abdallah, K. Solanki and S. Banta, ACS Catal., 2018, 8, 1602–1613 CrossRef.
- F. Khoury, Z. Su and S. Banta, Inorg. Chem., 2024, 63(29), 13223–13230 CrossRef CAS PubMed.
- J. A. Mattocks, J. L. Tirsch and J. A. Cotruvo, in Methods in Enzymology, ed. J. A. Cotruvo, Academic Press, 2021, vol. 651, pp. 23–61 Search PubMed.
- G. Deblonde, Methods in Enzymology, Academic Press, 2021, vol. 651, pp. 1–22 Search PubMed.
- W. R. Harris, C. J. Carrano and K. N. Raymond, J. Am. Chem. Soc., 1979, 101, 2722–2727 CrossRef CAS.
- E. Falcone and P. Faller, Dalton Trans., 2023, 52, 2197–2208 RSC.
- Z. M. Anwar and H. A. Azab, J. Chem. Eng. Data, 2001, 46, 613–618 CrossRef CAS.
- H. A. Azab, S. S. Al-Deyab, Z. M. Anwar, I. I. Abd El-Gawad and R. M. Kamel, J. Chem. Eng. Data, 2011, 56, 2613–2625 CrossRef CAS.
- H. A. Azab, Z. M. Anwar and R. M. Kamel, J. Solution Chem., 2016, 45, 1516–1528 CrossRef CAS.
- P. Mandal, J. Kretzschmar and B. Drobot, JBIC, J. Biol. Inorg. Chem., 2022, 27, 249–260 CrossRef CAS PubMed.
- R. N. Goldberg, N. Kishore and R. M. Lennen, J. Phys. Chem. Ref. Data, 2002, 31, 231–370 CrossRef CAS.
- K. J. Franz, M. Nitz and B. Imperiali, ChemBioChem, 2003, 4, 265–271 CrossRef CAS.
- E. A. Alasadi, W. Choi, X. Chen, J. A. J. Cotruvo and C. R. Baiz, ACS Chem. Biol., 2024, 19, 1056–1065 CrossRef CAS PubMed.
- E. M. Jones, Y. Su, C. Sander, Q. A. Justman, M. Springer and P. A. Silver, ACS Synth. Biol., 2024, 13, 958–962 CrossRef CAS.
- L. L. Clainche, M. Figuet, V. Montjardet-Bas, S. Blanchard and C. Vita, Biotechnol. Bioeng., 2006, 95, 29–36 CrossRef.
- S. M. Gutenthaler, S. Tsushima, R. Steudtner, M. Gailer, A. Hoffmann-Röder, B. Drobot and L. J. Daumann, Inorg. Chem. Front., 2022, 9, 4009–4021 RSC.
- S. C. Zondlo, F. Gao and N. J. Zondlo, J. Am. Chem. Soc., 2010, 132, 5619–5621 CrossRef CAS PubMed.
- S. Balakrishnan and N. J. Zondlo, J. Am. Chem. Soc., 2006, 128, 5590–5591 CrossRef CAS.
- A. R. Urmey and N. J. Zondlo, Biochemistry, 2019, 58, 2822–2833 CrossRef CAS.
- A. R. Urmey and N. J. Zondlo, Free Radical Biol. Med., 2020, 152, 166–174 CrossRef CAS.
- F. Gao, B. S. Thornley, C. M. Tressler, D. Naduthambi and N. J. Zondlo, Org. Biomol. Chem., 2019, 17, 3984–3995 RSC.
- L. L. Liu and K. J. Franz, J. Am. Chem. Soc., 2005, 127, 9662–9663 CrossRef CAS PubMed.
- J. A. Mattocks, J. L. Tirsch and J. A. Cotruvo, Methods in Enzymology, Elsevier, 2021, vol. 651, pp. 23–61 Search PubMed.
- B. Mwewa, M. Tadie, S. Ndlovu, G. S. Simate and E. Matinde, J. Environ. Chem. Eng., 2022, 10, 107704 CrossRef CAS.
- S. Pramanik, S. Kaur, I. Popovs, A. S. Ivanov and S. Jansone-Popova, Eur. J. Inorg. Chem., 2024, e202400064 CrossRef CAS.
- J. Haiech, C. B. Klee, J. G. Demaille and J. Haiech, Biochemistry, 1981, 20, 3890–3897 CrossRef CAS.
- D. M. Park, D. W. Reed, M. C. Yung, A. Eslamimanesh, M. M. Lencka, A. Anderko, Y. Fujita, R. E. Riman, A. Navrotsky and Y. Jiao, Environ. Sci. Technol., 2016, 50, 2735–2742 CrossRef CAS.
- G. J.-P. Deblonde, J. A. Mattocks, D. M. Park, D. W. Reed, J. A. Cotruvo and Y. Jiao, Inorg. Chem., 2020, 59, 11855–11867 CrossRef CAS PubMed.
- T. L. Pham, M. R. Conde González, S. Fazliev, A. Kishore, P. Comba and F. Thomas, ChemBioChem, 2024, 25, e202300715 CrossRef CAS PubMed.
- D. Ghosh, K.-H. Lee, B. Demeler and V. L. Pecoraro, Biochemistry, 2005, 44, 10732–10740 CrossRef CAS.
- J. A. Peters, K. Djanashvili, C. F. G. C. Geraldes and C. Platas-Iglesias, Coord. Chem. Rev., 2020, 406, 213146 CrossRef CAS.
- R. K. Wilharm, S.-Y. Huang, I. J. Gugger and V. C. Pierre, Inorg. Chem., 2021, 60, 15808–15817 CrossRef CAS.
- A. Roca-Sabio, M. Mato-Iglesias, D. Esteban-Gómez, É. Tóth, A. de Blas, C. Platas-Iglesias and T. Rodríguez-Blas, J. Am. Chem. Soc., 2009, 131, 3331–3341 CrossRef CAS.
- A. Hu, S. N. MacMillan and J. J. Wilson, J. Am. Chem. Soc., 2020, 142, 13500–13506 CrossRef CAS.
- S. L. Wu and W. D. W. Horrocks, J. Chem. Soc., Dalton Trans., 1997, 1497–1502 RSC.
- A. Hu, E. Aluicio-Sarduy, V. Brown, S. N. MacMillan, K. V. Becker, T. E. Barnhart, V. Radchenko, C. F. Ramogida, J. W. Engle and J. J. Wilson, J. Am. Chem. Soc., 2021, 143, 10429–10440 CrossRef CAS PubMed.
- T. Hatanaka, N. Kikkawa, A. Matsugami, Y. Hosokawa, F. Hayashi and N. Ishida, Sci. Rep., 2020, 10, 19468 CrossRef CAS.
- H. Singer, B. Drobot, C. Zeymer, R. Steudtner and L. J. Daumann, Chem. Sci., 2021, 12, 15581–15587 RSC.
- J. A. Mattocks, J. L. Tirsch and J. A. Cotruvo, Methods in Enzymology, Elsevier, 2021, vol. 651, pp. 23–61 Search PubMed.
- Z. Dong, J. A. Mattocks, J. A. Seidel, J. A. Cotruvo and D. M. Park, Sep. Purif. Technol., 2024, 333, 125919 CrossRef CAS.
- J. Seidel, P. Diep, Z. Dong, J. A. Cotruvo and D. M. Park, JACS Au, 2024, 4(11), 4273–4284 CrossRef CAS PubMed.
- C. S. Bonnet, M. Devocelle and T. Gunnlaugsson, Org. Biomol. Chem., 2012, 10, 126–133 RSC.
- G. J.-P. Deblonde, J. A. Mattocks, H. Wang, E. M. Gale, A. B. Kersting, M. Zavarin and J. A. Cotruvo, J. Am. Chem. Soc., 2021, 143, 15769–15783 CrossRef CAS.
- G. J.-P. Deblonde, J. A. Mattocks, Z. Dong, P. T. Wooddy, J. A. Cotruvo and M. Zavarin, Sci. Adv., 2021, 7, eabk0273 CrossRef CAS PubMed.
- G. J.-P. Deblonde, K. Morrison, J. A. Mattocks, J. A. Cotruvo, M. Zavarin and A. B. Kersting, Environ. Sci. Technol., 2023, 57, 20830–20843 CrossRef CAS.
- R. M. Smith and A. E. Martell, Sci. Total Environ, 1987, 64, 125–147 CrossRef CAS.
- J. A. Mattocks, J. A. Cotruvo and G. J.-P. Deblonde, Chem. Sci., 2022, 13, 6054–6066 RSC.
- S. Özçubukçu, K. Mandal, S. Wegner, M. P. Jensen and C. He, Inorg. Chem., 2011, 50, 7937–7939 CrossRef.
- H. Singer, R. Steudtner, A. S. Klein, C. Rulofs, C. Zeymer, B. Drobot, A. Pol, N. Cecilia Martinez-Gomez, H. J. M. Op den Camp and L. J. Daumann, Angew. Chem., Int. Ed., 2023, 62, e202303669 CrossRef PubMed.
- A. Shah, M. J. Taylor, G. Molinaro, S. Anbu, M. Verdu, L. Jennings, I. Mikulska, S. Diaz-Moreno, H. E. L. Mkami, G. M. Smith, M. M. Britton, J. E. Lovett and A. F. A. Peacock, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2219036120 CrossRef CAS.
- C. S. Bonnet, M. Devocelle and T. Gunnlaugsson, Chem. Commun., 2008, 4552–4554 RSC.
- S. K. Drake and J. J. Falke, Biochemistry, 1996, 35, 1753–1760 CrossRef CAS.
- M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496–4539 CrossRef CAS.
- A. J. Amoroso and S. J. A. Pope, Chem. Soc. Rev., 2015, 44, 4723–4742 RSC.
- E. Mathieu, A. Sipos, E. Demeyere, D. Phipps, D. Sakaveli and K. E. Borbas, Chem. Commun., 2018, 54, 10021–10035 RSC.
- U. Cho and J. K. Chen, Cell Chem. Biol., 2020, 27, 921–936 CrossRef CAS PubMed.
- J. H. S. K. Monteiro, Molecules, 2020, 25, 2089 CrossRef CAS PubMed.
- R. Lengacher, K. E. Martin, D. Śmiłowicz, H. Esseln, P. Lotlikar, A. Grichine, O. Maury and E. Boros, J. Am. Chem. Soc., 2023, 145, 24358–24366 CrossRef CAS PubMed.
- J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- S. V. Eliseeva, Angew. Chem., Int. Ed., 2015, 54, 8598 CrossRef CAS.
- A. D’Aléo, F. Pointillart, L. Ouahab, C. Andraud and O. Maury, Coord. Chem. Rev., 2012, 256, 1604–1620 CrossRef.
- J.-C. G. Bünzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef.
- D. Kovacs and K. E. Borbas, Coord. Chem. Rev., 2018, 364, 1–9 CrossRef CAS.
- J. De Jersey, P. J. Morley and R. B. Martin, Biophys. Chem., 1981, 13, 233–243 CrossRef CAS.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodríguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- W. D. Horrocks, J. P. Bolender, W. D. Smith and R. M. Supkowski, J. Am. Chem. Soc., 1997, 119, 5972–5973 CrossRef CAS.
- E. Falcone, P. Gonzalez, L. Lorusso, O. Sénèque, P. Faller and L. Raibaut, Chem. Commun., 2020, 56, 4797–4800 RSC.
- A. M. Reynolds, B. R. Sculimbrene and B. Imperiali, Bioconjugate Chem., 2008, 19, 588–591 CrossRef CAS.
- J. Wang, X. Liu, K. Li, T. Shi, Q. Xu, T. Peng, Q. Huang, Z. Gao, H. Zhou, W. Lu and J. Wang, J. Am. Chem. Soc., 2025, 147, 15205–15215 CrossRef CAS PubMed.
- M. Starck, J. D. Fradgley, S. Di Vita, J. A. Mosely, R. Pal and D. Parker, Bioconjugate Chem., 2020, 31, 229–240 CrossRef CAS PubMed.
- H.-F. Chau, Y. Wu, W.-Y. Fok, W. Thor, W. C.-S. Cho, P. Ma, J. Lin, N.-K. Mak, J.-C. G. Bünzli, L. Jiang, N. J. Long, H. L. Lung and K.-L. Wong, JACS Au, 2021, 1, 1034–1043 CrossRef CAS PubMed.
- J.-H. Choi, G. Fremy, T. Charnay, N. Fayad, J. Pécaut, S. Erbek, N. Hildebrandt, V. Martel-Frachet, A. Grichine and O. Sénèque, Inorg. Chem., 2022, 61, 20674–20689 CrossRef CAS PubMed.
- C. Cepeda, S. A. Denisov, D. Boturyn, N. D. McClenaghan and O. Sénèque, Inorg. Chem., 2021, 60, 17426–17434 CrossRef CAS.
- M. Isaac, L. Raibaut, C. Cepeda, A. Roux, D. Boturyn, S. V. Eliseeva, S. Petoud and O. Sénèque, Chem. – Eur. J., 2017, 23, 10992–10996 CrossRef CAS.
- Y.-H. Yeung, H.-F. Chau, H.-Y. Kai, W. Zhou, K. H.-Y. Chan, W. Thor, L. J. Charbonnière, F. Zhang, Y. Fan, Y. Wu and K.-L. Wong, Adv. Opt. Mater., 2024, 12, 2302070 CrossRef CAS.
- C. Cepeda, L. Raibaut, G. Fremy, S. V. Eliseeva, A. Romieu, J. Pécaut, D. Boturyn, S. Petoud and O. Sénèque, Chem. – Eur. J., 2020, 26, 13476–13483 CrossRef CAS.
- C. Penas, J. L. Mascareñas and M. E. Vázquez, Chem. Sci., 2016, 7, 2674–2678 RSC.
- B. R. Sculimbrene and B. Imperiali, J. Am. Chem. Soc., 2006, 128, 7346–7352 CrossRef CAS PubMed.
- K. Y. Huang, L. Cardenas, A. D. Ellington and D. J. F. Walker, Nat. Commun., 2024, 15, 9200 CrossRef CAS PubMed.
- L. Raibaut, W. Vasseur, G. D. Shimberg, C. Saint-Pierre, J.-L. Ravanat, S. L. J. Michel and O. Sénèque, Chem. Sci., 2017, 8, 1658–1664 RSC.
- E. Pazos, M. Goličnik, J. L. Mascareñas and M. E. Vázquez, Chem. Commun., 2012, 48, 9534–9536 RSC.
- R. Sánchez-Fernández, A. Sánchez-Temprano, D. Esteban-Gómez and E. Pazos, ChemBioChem, 2023, 24, e202300072 CrossRef.
- E. Falcone, B. Vileno, M. Hoang, L. Raibaut and P. Faller, J. Inorg. Biochem., 2021, 221, 111478 CrossRef CAS PubMed.
- M. J. Scheuermann, C. R. Forbes and N. J. Zondlo, Biochemistry, 2018, 57, 6956–6963 CrossRef CAS PubMed.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- M. E. Ladd, P. Bachert, M. Meyerspeer, E. Moser, A. M. Nagel, D. G. Norris, S. Schmitter, O. Speck, S. Straub and M. Zaiss, Prog. Nucl. Magn. Reson. Spectrosc., 2018, 109, 1–50 CrossRef CAS PubMed.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512–523 RSC.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- D. H. Powell, O. M. N. Dhubhghaill, D. Pubanz, L. Helm, Y. S. Lebedev, W. Schlaepfer and A. E. Merbach, J. Am. Chem. Soc., 1996, 118, 9333–9346 CrossRef CAS.
- J. J. Yang, J. Yang, L. Wei, O. Zurkiya, W. Yang, S. Li, J. Zou, Y. Zhou, A. L. W. Maniccia, H. Mao, F. Zhao, R. Malchow, S. Zhao, J. Johnson, X. Hu, E. Krogstad and Z.-R. Liu, J. Am. Chem. Soc., 2008, 130, 9260–9267 CrossRef CAS PubMed.
- S. Xue, H. Yang, J. Qiao, F. Pu, J. Jiang, K. Hubbard, K. Hekmatyar, J. Langley, M. Salarian, R. C. Long, R. G. Bryant, X. P. Hu, H. E. Grossniklaus, Z.-R. Liu and J. J. Yang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6607–6612 CrossRef CAS.
- C. S. Bonnet, P. H. Fries, S. Crouzy and P. Delangle, J. Phys. Chem. B, 2010, 114, 8770–8781 CrossRef CAS PubMed.
- Y. Liu, D. Gao, Y. He, J. Ma, S. Y. Chong, X. Qi, H. J. Ting, Z. Luo, Z. Yi, J. Tang, C. Chang, J. Wang, Z. Sheng, H. Zheng and X. Liu, Nat. Commun., 2024, 15, 9834 CrossRef CAS PubMed.
- D. Li, M. Kirberger, J. Qiao, Z. Gui, S. Xue, F. Pu, J. Jiang, Y. Xu, S. Tan, M. Salarian, O. Ibhagui, K. Hekmatyar and J. J. Yang, Invest. Radiol., 2024, 59, 170–186 CrossRef CAS PubMed.
- F. Pu, M. Salarian, S. Xue, J. Qiao, J. Feng, S. Tan, A. Patel, X. Li, K. Mamouni, K. Hekmatyar, J. Zou, D. Wu and J. J. Yang, Nanoscale, 2016, 8, 12668–12682 RSC.
- M. Salarian, H. Yang, R. C. Turaga, S. Tan, J. Qiao, S. Xue, Z. Gui, G. Peng, H. Han, P. Mittal, H. E. Grossniklaus and J. J. Yang, Biomaterials, 2019, 224, 119478 CrossRef CAS PubMed.
- S. Tan, H. Yang, S. Xue, J. Qiao, M. Salarian, K. Hekmatyar, Y. Meng, R. Mukkavilli, F. Pu, O. Y. Odubade, W. Harris, Y. Hai, M. L. Yushak, V. M. Morales-Tirado, P. Mittal, P. Z. Sun, D. Lawson, H. E. Grossniklaus and J. J. Yang, Sci. Adv., 2020, 6, eaav7504 CrossRef CAS PubMed.
- H. Yang, S. Tan, J. Qiao, Y. Xu, Z. Gui, Y. Meng, B. Dong, G. Peng, O. Y. Ibhagui, W. Qian, J. Lu, Z. Li, G. Wang, J. Lai, L. Yang, H. E. Grossniklaus and J. J. Yang, Cancer Gene Ther., 2022, 29, 1827–1839 CrossRef CAS PubMed.
- O. Y. Ibhagui, D. Li, H. Han, G. Peng, M. L. Meister, Z. Gui, J. Qiao, M. Salarian, B. Dong, Y. Yuan, Y. Xu, H. Yang, S. Tan, G. Satyanarayana, S. Xue, R. C. Turaga, M. Sharma, Y. Hai, Y. Meng, K. Hekmatyar, P. Sun, G. Sica, X. Ji, Z.-R. Liu and J. J. Yang, Chem. Biomed. Imaging, 2023, 1, 268–285 CrossRef CAS.
- Q. N. Do, J. S. Ratnakar, Z. Kovács and A. D. Sherry, ChemMedChem, 2014, 9, 1116–1129 CrossRef CAS PubMed.
- G. Angelovski and É. Tóth, Chem. Soc. Rev., 2017, 46, 324–336 RSC.
- J. Lux and A. D. Sherry, Curr. Opin. Chem. Biol., 2018, 45, 121–130 CrossRef CAS.
- C. S. Bonnet and É. Tóth, Curr. Opin. Chem. Biol., 2021, 61, 154–169 CrossRef CAS PubMed.
- H. Lu, A. Chen, X. Zhang, Z. Wei, R. Cao, Y. Zhu, J. Lu, Z. Wang and L. Tian, Nat. Commun., 2022, 13, 7948 CrossRef CAS PubMed.
- M. Isaac, A. Pallier, F. Szeremeta, P.-A. Bayle, L. Barantin, C. S. Bonnet and O. Sénèque, Chem. Commun., 2018, 54, 7350–7353 RSC.
- K. P. Malikidogo, A. Pallier, F. Szeremeta, C. S. Bonnet and O. Sénèque, Dalton Trans., 2023, 52, 6260–6266 RSC.
- M. Sethurajan, E. D. van Hullebusch, D. Fontana, A. Akcil, H. Deveci, B. Batinic, J. P. Leal, T. A. Gasche, M. Ali Kucuker, K. Kuchta, I. F. F. Neto, H. M. V. M. Soares and A. Chmielarz, Crit. Rev. Environ. Sci. Technol., 2019, 49, 212–275 CrossRef CAS.
- K. Binnemans, P. T. Jones, T. Müller and L. Yurramendi, J. Sustainable Metall., 2018, 4, 126–146 CrossRef.
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven and Y. Pontikes, J. Cleaner Prod., 2015, 99, 17–38 CrossRef CAS.
- S. Pramanik, S. Kaur, I. Popovs, A. S. Ivanov and S. Jansone-Popova, Eur. J. Inorg. Chem., 2024, e202400064 CrossRef CAS.
- N. C. Martinez-Gomez, H. N. Vu and E. Skovran, Inorg. Chem., 2016, 55, 10083–10089 CrossRef CAS PubMed.
- S. Dev, A. Sachan, F. Dehghani, T. Ghosh, B. R. Briggs and S. Aggarwal, Chem. Eng. J., 2020, 397, 124596 CrossRef CAS.
- R. M. Brown, A. Mirkouei, D. Reed and V. Thompson, Renewable Sustainable Energy Rev., 2023, 173, 113099 CrossRef CAS.
- M. Chakankar, F. Lederer, R. Jain, S. Matys, S. Kutschke and K. Pollmann, Management of Electronic Waste, John Wiley & Sons, Ltd, 2024, pp. 375–405 Search PubMed.
- X. Qian, C. Ma, H. Zhang and K. Liu, Bioorg. Chem., 2024, 143, 107040 CrossRef CAS PubMed.
- M. Vítová and D. Mezricky, World J. Microbiol. Biotechnol., 2024, 40, 189 CrossRef.
- H. Singer, R. Steudtner, I. Sottorff, B. Drobot, A. Pol, H. J. M. O. den Camp and L. J. Daumann, Chem. Commun., 2023, 59, 9066–9069 RSC.
- I. Chae, A. Shivkumar, F. M. Doyle and S.-W. Lee, Nano Lett., 2024, 24, 9946–9952 CrossRef CAS PubMed.
- I. Chae, F. M. Doyle and S.-W. Lee, TMS 2025 154th Annual Meeting & Exhibition Supplemental Proceedings, Springer, Cham, 2025, pp. 1139–1148 Search PubMed.
- D. M. Park, A. Brewer, D. W. Reed, L. N. Lammers and Y. Jiao, Environ. Sci. Technol., 2017, 51, 13471–13480 CrossRef CAS PubMed.
- A. Brewer, E. Chang, D. M. Park, T. Kou, Y. Li, L. N. Lammers and Y. Jiao, Environ. Sci. Technol., 2019, 53, 7714–7723 CrossRef CAS PubMed.
- A. Brewer, A. Dohnalkova, V. Shutthanandan, L. Kovarik, E. Chang, A. M. Sawvel, H. E. Mason, D. Reed, C. Ye, W. F. Hynes, L. N. Lammers, D. M. Park and Y. Jiao, Environ. Sci. Technol., 2019, 53, 13888–13897 CrossRef CAS PubMed.
- D. Park, A. Middleton, R. Smith, G. Deblonde, D. Laudal, N. Theaker, H. Hsu-Kim and Y. Jiao, Sep. Purif. Technol., 2020, 241, 116726 CrossRef CAS.
- E. Chang, A. W. Brewer, D. M. Park, Y. Jiao and L. N. Lammers, Appl. Geochem., 2020, 112, 104478 CrossRef CAS.
- X. Xie, K. Yang, Y. Lu, Y. Li, J. Yan, J. Huang, L. Xu, M. Yang and Y. Yan, J. Hazard. Mater., 2022, 438, 129561 CrossRef CAS PubMed.
- M. Gut, T. Wilhelm, O. Beniston, S. Ogundipe, C.-C. Kuo, K. Nguyen and A. Furst, Adv. Mater., 2025, 37, 2412607 CrossRef CAS PubMed.
- F. F. Khoury, B. S. Heater, D. R. Marzolf, S. S. Abeyrathna, J. W. Picking, P. Kumar, S. A. Higgins, R. Jones, A. T. Lewis, K. H. Kucharzyk and S. Banta, Chem. Sci., 2025, 16, 15333–15346 RSC.
- P. K. R. Tay, A. Manjula-Basavanna and N. S. Joshi, Green Chem., 2018, 20, 3512–3520 RSC.
- R. A. Scheel, J. K. Sahoo, L. D. Morton, Z. Xia, J. R. Blum, Z. Martin-Moldes and D. L. Kaplan, ACS ES&T Eng., 2024, 4(9), 2135–2144 Search PubMed.
- Z. Dong, J. A. Mattocks, G. J.-P. Deblonde, D. Hu, Y. Jiao, J. A. Cotruvo and D. M. Park, ACS Cent. Sci., 2021, 7, 1798–1808 CrossRef CAS PubMed.
- Z. Hussain, S. Kim, J. Cho, G. Sim, Y. Park and I. Kwon, Adv. Funct. Mater., 2022, 32, 2109158 CrossRef CAS.
- Z. Hussain, D. Dwivedi and I. Kwon, Front. Bioeng. Biotechnol., 2024, 12 DOI:10.3389/fbioe.2024.1385845.
- Q. Ye, X. Jin, B. Zhu, H. Gao and N. Wei, Environ. Sci. Technol., 2023, 57, 4276–4285 CrossRef CAS PubMed.
- L. Johnson and C. E. Duval, Langmuir, 2025, 41, 9581–9589 CrossRef CAS PubMed.
- Q. Ye, X. Jin, B. Zhu, H. Gao and N. Wei, Environ. Sci. Technol., 2023, 57, 4276–4285 CrossRef CAS PubMed.
- H. Jin, D. M. Park, M. Gupta, A. W. Brewer, L. Ho, S. L. Singer, W. L. Bourcier, S. Woods, D. W. Reed, L. N. Lammers, J. W. Sutherland and Y. Jiao, ACS Sustainable Chem. Eng., 2017, 5, 10148–10155 CrossRef CAS.
- J. T. Welch, M. Sirish, K. M. Lindstrom and S. J. Franklin, Inorg. Chem., 2001, 40, 1982–1984 CrossRef CAS PubMed.
- M. Sirish and S. J. Franklin, J. Inorg. Biochem., 2002, 91, 253–258 CrossRef CAS PubMed.
- Y. Kim and S. J. Franklin, Inorg. Chim. Acta, 2002, 341, 107–112 CrossRef CAS.
- L. Wang, K. Liu, Z. Song, H. Do, L. Yang, J. Wu, L. Jiang and H. Yu, Colloids Surf., B, 2025, 251, 114596 CrossRef CAS PubMed.
- M. J. Allen, Synlett, 2016, 1310–1317 CrossRef CAS PubMed.
- H. Pellissier, Coord. Chem. Rev., 2017, 336, 96–151 CrossRef CAS.
- Y. Qiao and E. J. Schelter, Acc. Chem. Res., 2018, 51, 2926–2936 CrossRef CAS PubMed.
- A. Prieto and F. Jaroschik, Curr. Org. Chem., 2022, 26, 6–41 CrossRef CAS.
- N. Mahieu, J. Piątkowski, T. Simler and G. Nocton, Chem. Sci., 2023, 14, 443–457 RSC.
- R. Bhimpuria, R. Charaf, K. Ye, A. Thapper, H. Sathyan, M. Ahlquist, L. Hammarström and K. E. Borbas, Chem, 2025, 102547 CAS.
- R. Bhimpuria, M. Tomar, A. Thapper, M. Ahlquist and K. E. Borbas, Chem, 2025, 102450 CAS.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed.
- P. Kouba, P. Kohout, F. Haddadi, A. Bushuiev, R. Samusevich, J. Sedlar, J. Damborsky, T. Pluskal, J. Sivic and S. Mazurenko, ACS Catal., 2023, 13, 13863–13895 CrossRef CAS PubMed.
- P. Notin, N. Rollins, Y. Gal, C. Sander and D. Marks, Nat. Biotechnol., 2024, 42, 216–228 CrossRef CAS PubMed.
- A. Winnifrith, C. Outeiral and B. L. Hie, Curr. Opin. Struct. Biol., 2024, 86, 102794 CrossRef CAS PubMed.
- K. K. Yang, Z. Wu and F. H. Arnold, Nat. Methods, 2019, 16, 687–694 CrossRef CAS PubMed.
- J. A. Mattocks, J. J. Jung, C.-Y. Lin, Z. Dong, N. H. Yennawar, E. R. Featherston, C. S. Kang-Yun, T. A. Hamilton, D. M. Park, A. K. Boal and J. A. Cotruvo, Nature, 2023, 618, 87–93 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |